

Abstract
In recent years, human serum albumin (HSA) has been characterized as an ideal drug carrier in the cancer arena. Caveolin-1 (Cav-1) has been established as the principal structural protein of caveolae and, thus, critical for caveolae-mediated endocytosis. Cav-1 has been shown to be overexpressed in cancers of the lung and pancreas, among others. We found that Cav-1 expression plays a critical role in both HSA uptake and response to albumin-based chemotherapies. As such, developing a novel albumin-based chemotherapy that is more selective for tumors with high Cav-1 expression or high levels of caveolar-endocytosis could have significant implications in biomarker-directed therapy. Herein, we present the development of a novel and effective HSA-SN-38 conjugate (SSH20). We find that SSH20 uptake decreases significantly by immunofluorescence assays and western blotting after silencing of Cav-1 expression through RNA interference. Decreased drug sensitivity occurs in Cav-1-depleted cells using cytotoxicity assays. Importantly, we find significantly reduced sensitivity to SSH20 in Cav-1-silenced tumors compared to Cav-1-expressing tumors in vivo. Notably, we show that SSH20 is significantly more potent than irinotecan in vitro and in vivo. Together, we have developed a novel HSA-conjugated chemotherapy that is potent, effective, safe, and demonstrates improved efficacy in high Cav-1-expressing tumors.
Keywords: SSH20, lung cancer, pancreatic cancer, caveolin-1, albumin, SN-38
Graphical abstract
In this study, the authors have developed and established a novel albumin-SN-38 conjugate (SSH20) and tested it in pre-clinical models of pancreatic and lung cancer. The advantageous properties inherent to SN-38 conjugation to albumin and targeted delivery make SSH20 an attractive candidate for further preclinical and potential clinical study.
Introduction
Human serum albumin (HSA) is the most abundant protein found in human blood. Several properties of HSA make this protein an attractive candidate as a drug carrier in developing novel chemotherapeutics.1,2 Owing to its strong hydrophilic nature, chemical conjugation to this protein allows otherwise-insoluble hydrophobic compounds to be dissolved in clinically compatible solvents.3,4 As a robustly stable protein with an unusually long half-life of 19 days, HSA has been reported to extend the pharmacokinetic timeline of compounds bound to it.5,6 Furthermore, it has been well documented that HSA naturally targets tumors, accumulating in particular abundance at highly proliferative sites within tumors due to the enhanced permeability and retention effect (EPR), in part related to impaired tumor angiogenesis and leaky vascular fenestrae.7, 8, 9 In addition to this extracellular tumor localization, HSA is also readily internalized by tumor cells via caveolae-mediated endocytosis.6,10,11
Caveolae are flask-shaped invaginations in the plasma membrane. Previous publications, including work from our group, have established caveolin-1 (Cav-1) as the principal structural protein of caveolae and, thus, necessary for caveolae-mediated endocytosis.12 Cav-1 is upregulated in many cancer types, including pancreatic and non-small cell lung cancers.13, 14, 15, 16 Although tumor-type context dependency has been found, Cav-1 upregulation has been largely reported to be associated with cancer progression.14,17 We have also shown previously that Cav-1 expression plays a critical role in mediating albumin uptake as well as response to albumin-based chemotherapies.11
Irinotecan, a cytotoxic anticancer agent, is widely used for numerous patients worldwide, including those with non-small cell lung cancer and pancreatic, advanced gastric, and cervical cancer.18 SN-38 (7-ethyl-10-hydroxy-camptothecin), the active metabolite of irinotecan, inhibits DNA topoisomerase I (Top1), disrupting DNA replication and transcription, which results in cell death.19, 20, 21 SN-38 is 100- to 1,000-fold more potent than irinotecan itself but is virtually insoluble in any pharmaceutical solvent, limiting its clinical application.
Here, after testing over 30 different formulations of HSA to cytotoxic chemotherapies, we have identified a compound with high potency. This novel compound, in which we covalently conjugated HSA to SN-38, is designated SSH20. We show that SSH20 is robustly stable and soluble in aqueous solvents, while maintaining high potency relative to irinotecan. SSH20 demonstrates significant efficacy in both in vitro and in vivo pancreatic and lung cancer models. Furthermore, our results reveal that SSH20 targets cancer cells that express high levels of Cav-1, potentially providing a predictive biomarker to determine which patients would benefit most from SSH20.
Results
Characterization of SSH20
The reaction scheme for the synthesis of SSH20 is shown in Figure 1A. The SN-38 and HSA concentrations were determined by UV absorption at 360 nm and bicinchoninic acid (BCA) protein assay (Thermo Scientific, Waltham, MA, USA), respectively. The results showed that the SSH20 conjugate contained an average of 6.9 SN-38s per HSA molecule. Similar results were also obtained by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Figure 1B), which indicated an average of 7.75 SN-38s per HSA molecule. To examine the linkage between SN-38 and HSA, SDS-PAGE (Figure 1C) and organic solvent extraction (Figure 1D) were performed to analyze HSA protein and SN-38, respectively. As shown in Figure 1C, the major band of SSH20 shifted upward compared to that of HSA, suggesting that SN-38 molecules were covalently bound to HSA, resulting in a higher molecular weight. Similar results were obtained by organic solvent extraction. The extraction percentage of SN-38 from SSH20 was <1%, normalized to an equal concentration of standard (Figure 1D), suggesting that SN-38 molecules were bound to HSA through covalent ester bonds and could not be extracted out into an organic solvent. To determine the drug-releasing effect of plasma esterases on SSH20, SSH20 serum stability was analyzed. The results indicated the release of SN-38 from SSH20 occurred in a time-dependent manner with a half-life of ∼64.2 h (Figure S1).
Figure 1.

Characterization of SSH20
(A) SSH20 synthesis scheme. The process consists of SN-38 (compound 1), SN-38-suc (compound 2), SN-38-suc-NHS (compound 3), and SSH20 (compound 4). (B) MALDI-TOF mass spectrometry demonstrated an average of 7.75 SN38 molecules per HSA. The linkage between SN-38 and HSA was tested by SDS-PAGE/Coomassie stain (C) and organic solvent extraction (D), respectively, showing slower migration of SSH20 compared to free HSA and resistance of SSH20 to organic solvent extraction.
Cav-1 expression mediates uptake of HSA in vitro
In order to first verify the role of Cav-1 expression on albumin uptake into cells, we compared uptake between our stable shCtrl and shCav-1 in MIA-PaCa-2 (MP2) and H23 cell lines (previously generated11). We first confirmed that Cav-1 expression was depleted in our shCav-1 cells relative to shCtrl cells via immunoblotting (Figure 2A). Then, we pulsed cells with fluorescein isothiocyanate (FITC)-HSA (0.01%) for 1 h in regular growth medium to compare uptake of albumin in shCav-1 versus shCtrl cells by direct immunofluorescence (Figure 2B). We then measured the FITC channel fluorescence intensity of individual cells and observed significant reduction of albumin uptake in shCav-1 cells in both MP2 and H23 groups (Figure 2C). As an initial test of whether this pattern of HSA uptake was maintained after covalent conjugation to SN-38, we performed immunoblotting for human-specific albumin after cells were pulsed for 1 h with increasing doses of SSH20. Albumin levels were found to be higher in shCtrl than shCav-1 cells (Figure 2D). These results support our previous finding that cancer cells with higher expression of Cav-1 are able to internalize albumin more abundantly than low Cav-1-expressing cancer cells. Furthermore, the properties of HSA resulting in uptake through caveolae-mediated endocytosis are maintained in our SN-38 conjugate, SSH20.
Figure 2.

Cav-1 expression mediates uptake of HSA in vitro
(A) Cav-1 expression in stable MP2-shCtrl/shCav-1 and H23-shCtrl/shCav-1 cells by immunoblotting. (B) Direct fluorescence images of internalized FITC-labeled HSA demonstrate reduced HSA uptake in stable Cav-1 knockdown MP2/H23 cells. (C) Quantification of internalized FITC-labeled HSA in stable MP2 and H23 cells, reported as the mean of average intensity per cell (with SEM), measured in no less than 50 individual cells for each column. ∗∗∗∗p < 0.0001. (D) Uptake and expression of HSA detected by immunoblotting in shCtrl and shCav-1 cells treated with increasing doses of SSH20 for 1 h. GAPDH shown as equal loading control for (A) and (D). Scale bar 20 microns.
SSH20 targets cancer cells with high Cav-1 expression in vitro
Next, we tested the ability of SSH20 to target cells with high (shCtrl) versus low Cav-1 (shCav-1) expression in our isogenic cell lines. Taking advantage of the autofluorescent property of SN-38, we pulsed cells with SN-38 or SSH20 for 2 h and determined the amount of drug that was internalized by direct fluorescence microscopy. Upon imaging, we observed a marked difference in SN-38 autofluorescent signal between shCtrl and shCav-1 SSH20-treated cells, but no discernable difference in SN-38-treated cells (Figures 3A and 3B). This was confirmed quantitatively by comparative analysis of the fluorescence intensity. The intensity measured in the shCav-1 cells was significantly less than that in the shCtrl cells, in both MP2 and H23 isogenic cell lines (Figures 3C and 3D). Furthermore, we co-cultured shCtrl and shCav-1 cells at a 1:1 ratio and, as before, pulsed the cells with SN-38 or SSH20 for 2 h. We performed direct fluorescence for SN-38 and indirect immunofluorescence for Cav-1 protein with a secondary antibody conjugated with AlexaFluor-568. After acquiring images (Figures 3E and 3F), we measured the unicellular fluorescent intensity of both the SN-38 autofluorescent signal (green) and Cav-1-AF568 (red) channels and generated a scatterplot using these values (Figures 3G and 3H). In both MIA-PaCa-2 (Figures 3E and 3G) and H23 (Figures 3F and 3H) co-culture experiments, Pearson correlation coefficient analysis revealed a highly significant correlation between Cav-1 expression and SSH20, but not SN-38. Taken together, these data strongly suggest that cancer cells with higher Cav-1 expression uptake SSH20 more readily and further support the hypothesis that SSH20 targets cancer cells with increased levels of caveolae-mediated endocytosis.
Figure 3.

SSH20 targets cancer cells with high Cav-1 expression in vitro
(A and B) Direct fluorescence images of internalized SN-38 in shCtrl and shCav-1 cells pulsed with 250 nM of either SN-38/SSH20 for 2 h prior to cell fixation. (C and D) Quantitation of internalized SN-38 in shCtrl and shCav-1 cells. Quantitation is reported as the mean average intensity per cell (with SEM), measured in no less than 50 individual cells for each column. ∗∗∗∗p < 0.0001; n.s., p > 0.05. (E and F) Representative fluorescence images of Cav-1 expression (red) and internalized SN-38 (green) in shCtrl and shCav-1 cells, co-cultured at a 1:1 ratio and treated with 250 nM SSH20 for 2 h prior to fixation. (G and H) Correlation between Cav-1 intensity and SN-38 intensity in co-cultured shCtrl and shCav-1 cells. Each data point represents an individual cell. No less than 50 cells were measured to generate Pearson correlation plots. Line of best fit is indicated by the red line with 95% confidence bands (dashed lines). p and R2 values reported. Scale bar 20 microns.
Knocking down Cav-1 expression reduces sensitivity to SSH20 in vitro
As differences in Cav-1 expression resulted in significantly different SSH20 uptake, we tested whether this would directly translate to a similar trend in sensitivity to SSH20. Using cytotoxicity assays, we found that Cav-1 depletion resulted in decreased sensitivity of the cells to SSH20, but not to SN-38, after 72 h treatment (Figures 4A and 4B). In order to evaluate whether the observed difference in sensitivity was, at least in part, a result of the rate of drug internalization, we pulse-treated the cells for shorter time intervals of 6 and 12 h. An increased differential of SSH20 sensitivity between shCav-1 and shCtrl cells was observed as the treatment time decreased, as noted by separation of the curves (Figures 4A–4C). In addition, Cav-1 depletion markedly reduced SSH20-induced apoptosis in MP2 cells (Figure 4D). Taken together, these results indicate that reduction of Cav-1 expression decreases sensitivity of cells to SSH20, at least in part through reduced induction of apoptotic cell death, and support that caveolae-mediated endocytosis is a critical factor of SSH20 sensitivity.
Figure 4.

Cav-1 depletion reduces sensitivity to SSH20 in vitro
(A and B) Cell cytotoxicity was determined in MP2-shCtrl/shCav-1 and H23-shCtrl/shCav-1 stable cells with different doses of SN-38 and SSH20 for different periods of drug exposure (6, 12, 72 h), using alamarBlue assay. ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05. (C) Half-maximal inhibitory concentration (IC50) of each treatment group was calculated and compared in MP2-shCtrl/shCav-1 and H23-shCtrl/shCav-1 cells. Fold change (IC50 shCav-1/IC50 shCtrl) shows increasing fold-change difference with shorter time periods of exposure. (D) SSH20-induced apoptosis was confirmed by immunoblotting for cleaved PARP (cPARP) and cleaved caspase 3 (cCas3). GAPDH shown as equal loading control.
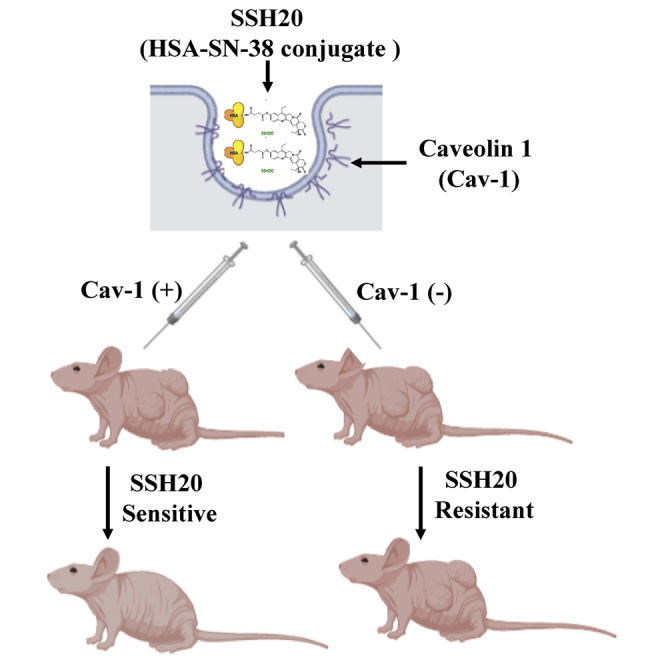
SSH20 demonstrates higher potency than irinotecan and efficacy in Cav-1-expressing tumor models in vivo
To explore the impact of Cav-1 expression on SSH20 sensitivity in vivo, we injected MP2-shCtrl/shCav-1 and H23-shCtrl/shCav-1 cells into the flanks of athymic nude mice, then treated the mice with or without SSH20 as indicated in Figure 5A. SSH20 treatment significantly reduced tumor growth rate in shCtrl (Cav-1 proficient) groups (Figures 5B and 5D, left panels). However, no significant difference of tumor growth rate was found in shCav-1 xenografts with or without SSH20 treatment (Figures 5C and 5E, left panels). In addition, tumor growth rate was not reduced by equivalent doses of irinotecan treatment in shCtrl-tumor-bearing mice as expected, given the lower potency of irinotecan at equivalent doses (Figures 5B and 5D, left panels). Kaplan-Meier survival curves based on percent of mice free from tumor doubling showed that SSH20 treatment markedly extended tumor doubling time in shCtrl groups, while no difference was detected in shCav-1 groups (Figures 5B–5E, right panels). Notably, mice tolerated treatment with SSH20 well, with no observable clinical signs of toxicity and minimal weight changes detected with doses of 5 or 10 mg/kg of SSH20 (Figure S2).
Figure 5.

High sensitivity and potency of SSH20 in tumors with high Cav-1 expression in vivo
(A) Experimental treatment schema. MP2-shCtrl/shCav-1 and H23-shCtrl/shCav-1 cells were injected into the flanks of mice. Once tumors reached approximately 100–200 mm3 in size, 0.9% saline (vehicle), SSH20 (10 mg/kg), or the molar equivalent of irinotecan was administered intravenously via retro-orbital injection. Tumor growth rate was significantly reduced in the SSH20-treated groups compared to the control and irinotecan groups in MP2-shCtrl and H23-shCtrl groups (B and D), but not in MP2-shCav-1 and H23-shCav-1 groups (C and E). Similarly, tumor doubling time was significantly prolonged in SSH20-treated groups compared to the control and irinotecan groups in MP2-shCtrl and H23-shCtrl groups (B and D), but not in MP2-shCav-1 and H23-shCav-1 groups (C and E). n = 10/group, ∗p < 0.01.
Discussion
Major issues related to success of novel therapeutics include improving drug delivery/solubility and predicting which tumors will respond best to the therapy. Irinotecan has long been used as a cancer therapeutic, and irinotecan is metabolized to the much more potent metabolite SN-38, which interacts with the nuclear enzyme topoisomerase I, resulting in irreversible double-strand breaks and cell death.22,23 However, SN-38 cannot be solubilized into a form that is deliverable to humans previously. Here, we describe the development of a novel and stable HSA-conjugated SN-38 protein-drug nanoparticle conjugate termed SSH20 that demonstrates significant in vitro and in vivo efficacy in pancreatic and lung cancer models. Mice tolerate SSH20 treatment well, with minimal signs of toxicity. Furthermore, SSH20 treatment demonstrates improved efficacy over irinotecan and is most effective in tumors with high Cav-1 expression.
As reported by previous studies, strategic conjugation with hydrophilic groups can solubilize SN-38.24,25 Due to the covalent conjugation of SN-38 to HSA, SSH20 remains stable and soluble in water or saline solution. This allows for intravenous administration, providing a method of drug delivery not viable with free SN-38. SN-38 could be released from albumin conjugate to exhibit its activity through esterases, which are generally overexpressed by tumor cells, and our data indicated that SN-38 was slowly released during the 120-h incubation (Figure S1). Previous publications have indicated the advantage of HSA conjugation: biocompatible breakdown, increased pharmacokinetic half-life, and preferential tumor localization.5,26,27 Based on these, development of HSA drug conjugates has been considered an attractive approach to increase drug targeting. The success of nab-paclitaxel (Abraxane) in the clinic is a prime example of a US Food and Drug Administration (FDA)-approved albumin-chemotherapeutic for the treatment of cancer.28,29 However, nab-paclitaxel is loosely bound to albumin by virtue of not being directly chemically conjugated, likely resulting in increased dissociation of paclitaxel from albumin before the drug reaches its intended tumor cell targets. Consequences of this are that more free paclitaxel would lead to increased normal tissue toxicity and less specificity for Cav-1-expressing tumor cells.
Numerous research groups have attempted to synthesize albumin-conjugated compounds.30, 31, 32 Interestingly, Sepehri et al.32 also developed an SN-38-albumin conjugate and reported the in vitro cytotoxicity and in vivo biodistribution and blood cytotoxicity in colon cancer. Our conjugate is distinct, as we conjugated the albumin to the phenol –OH of the SN-38 compound, while Sepehri et al. conjugated it to the aliphatic –OH. The difference in –OH attachment between SSH20 and the conjugate from Sepehri et al. is enough to establish that SSH20 is a novel compound. In addition, SSH20 has a clear advantage, as the conjugation synthesis is simpler without sacrificing purity. Furthermore, more molecules of SN-38 are conjugated to HSA in SSH20 compared to the Sepehri et al. strategy (∼7–8 versus ∼2–4). Finally, it achieves at least 99.99% purity, which is significantly higher than the conjugate from Sepehri et al. Protein-drug conjugates must strike a balance between esterase stability and drug releasability. Our studies support that esterases may function to degrade SSH20 and account for reduced efficacy in our models. Interestingly, mice have several-fold higher plasma esterase activity than humans. Despite this, however, we still observed a dramatic effect in vivo, and we speculate that the therapeutic index could be widened in an esterase-depleted environment. In fact, this is one of the key issues in the design and development of antibody-drug conjugates, which typically have stayed away from highly stable linkers. The optimization of linker chemistry and characterization of ester linkage for SSH20 will be further established in our future studies.
It has been difficult to identify predictive biomarkers of irinotecan treatment effectiveness. In agreement with our previous work, we found that a further property attributed to using HSA as drug carrier for SN-38 is potential Cav-1-mediated specificity. We have shown that both free HSA and SSH20 are internalized to a greater degree in cancer cells relative to the level of Cav-1 protein. Our results also indicate that the increased uptake is accompanied by an increased SSH20 sensitivity in lung and pancreatic cancer cells expressing high levels of Cav-1. Taken together, these results suggest that SSH20 likely has improved efficacy in high Cav-1-expressing tumors. Our study provides preliminary support for the potential of SSH20 as a novel treatment approach against lung and pancreatic cancers with high Cav-1 expression. As Cav-1 is commonly overexpressed among these cancer types, using Cav-1 as a predictive biomarker would allow for improved likelihood of success in clinical trials and facilitate implementation of a more personalized approach for SSH20 treatment.
As we know, albumin is a nutrient source for tumor cells and is especially accumulated in the tumor microenvironment (TME) due to the EPR effect, which is likely an advantage of this novel SSH20 as well. It is also important to note that we found SSH20 was significantly more potent than irinotecan in vivo (in Cav-1-expressing tumors). Importantly, there was no apparent difference in toxicity between them in vivo. These findings, in addition to the tumor-targeting properties resulting from the HSA group, provide evidence for SSH20 being a more effective alternative strategy for patients who would receive irinotecan according to the current standard of care.
Certainly, it is possible that tumor cells have additional uptake mechanisms of HSA-conjugated drugs aside from caveolae-mediated endocytosis. Indeed, in our in vitro studies, we found that the highest degree of separation between Cav-1-proficient (Ctrl) and Cav-1-deficient (shCav-1) cells in our IC50 experiments occurred with shorter periods of time of exposure of cells to SSH20 (i.e., 6 or 12 h versus 72 h). This suggests that SSH20 is preferentially taken up first by caveolae-mediated endocytosis but that other unidentified mechanisms of uptake may predominate at later time points. Nevertheless, the marked separation in efficacy in our in vivo experiments between Cav-1-proficient and Cav-1-deficient tumors clearly demonstrates that caveolae-mediated endocytosis is a profound mechanism of uptake for SSH20, and that Cav-1 expression is likely to be a very useful predictive biomarker. This degree of separation was not observed with nab-paclitaxel in our previous publication,11 which may be related to the instability and frequent dissociation of paclitaxel from albumin and subsequent free diffusion of paclitaxel through the cell membrane (independent of Cav-1/caveolae-mediated endocytosis).
It is important to highlight the limitations of our study. One of the advantages of albumin-bound drugs is based on the EPR effect, which we did not directly test in this study. Additionally, in our in vivo models, we have used subcutaneous flank mouse models for pancreatic cancer (and lung cancer), which do not recapitulate the stroma-rich and native TME of pancreatic cancer. To better reflect that intrinsic TME, orthotopic implantation of human patient-derived pancreatic cancer or autochthonous genetically engineered mouse models can serve as better models for more clinically relevant testing. Similar studies can be done for lung cancer.
In conclusion, we have developed and established the initial safety and efficacy of a novel albumin-SN-38 conjugate (SSH20) in pre-clinical models of pancreatic and lung cancer. Our results confirm that (1) SSH20 is effective in lung and pancreatic cancer in vitro and in vivo with high Cav-1 expression, and (2) SSH20 is more potent than irinotecan with no apparent difference in overall toxicity in mouse models. Taken together, the advantageous properties inherent to SN-38 conjugation to albumin and targeted delivery make SSH20 an attractive candidate for further preclinical and potentially clinical study.
Materials and methods
Treatments, reagents, antibodies
HSA solution (25%) was purchased from Octapharma (Paramus, NJ, USA). SN-38 was purchased from Ark Pharm, (Arlington Heights, IL, USA). Succinic anhydride, N, N′-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), and 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) were purchased from Sigma-Aldrich Chemical (St. Louis, MO, USA). Solvents, buffers, and salts, including dimethylformamide (DMF), acetone, chloroform, sodium carbonate, HEPES, and phosphate-buffered saline (PBS), were purchased from Fisher Scientific (Hampton, NH, USA). Irinotecan (West-Ward Pharmaceuticals; Eatontown, NJ, USA) and SSH20 were dissolved in 0.9% saline. SN-38 was dissolved in DMSO. RPMI 1640 media, minimum essential media (MEM), penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.25% w/v trypsin/1 mM EDTA were purchased from Gibco Life Technologies (Grand Island, NY, USA). Dulbecco’s modified Eagle’s medium (DMEM) and PBS were purchased from GE Healthcare Bio-Sciences (Pittsburg, PA, USA). Fetal bovine serum (FBS) and lyophilized powder HSA was purchased from Millipore-Sigma (St. Louis, MO, USA). Cleaved caspase 3, cleaved PARP, human albumin, and GAPDH primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Caveolin-1 primary antibody (N-20) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-rabbit immunofluorescent secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE, USA). Alexa Fluor secondary antibodies were purchased from Invitrogen (Waltham, MA, USA).
SSH20 synthesis and purification
SN-38 and succinic anhydride were dissolved in 6 mL DMF at a molar ratio of 1:1.1. Equimolar ratio of DBU to SN-38 was then added to the solution. The mixture was covered by aluminum foil and stirred overnight at room temperature to obtain SN-38-suc. Next, a molar ratio of 1:1.1:1.1 of SN-38 to DCC and NHS were added to the same mixture and stirred at room temperature for 6 h to obtain SN-38-suc-NHS. For HSA conjugation, a 25% HSA solution was diluted in 50 mM HEPES buffer to 10%, and pH was titrated to pH 8.5 using 1 M sodium carbonate. Then, a 20-fold equivalent of the above-synthesized SN-38-suc-NHS was added to the HSA/HEPES solution dropwise, and the mixture was stirred at room temperature for 4 h to obtain SSH20. The solution pH was monitored and maintained at ∼pH 8.5 using 1 M sodium carbonate. After HSA conjugation, the isourea (DCU) reaction byproduct was removed by centrifugation at 10,000 × g for 15 min. The supernatant was collected in polypropylene conical tubes with 10 mL per tube. 40 mL of pre-chilled acetone (at −20°C) was added to each tube to form SSH20 precipitate. The protein/acetone mixture was incubated at −80°C for 1 h, and SSH20 precipitate was spun down at 4,000 × g for 20 min at 4°C. The supernatant was removed, and the protein pellet was dried in a desiccator under vacuum for 10 min to remove residual acetone. The pellet was resuspended in PBS to obtain a crude SSH20 solution. The crude SSH20 was dialyzed against PBS overnight using 10 kDa dialysis cassettes to remove residual acetone, SN-38, and byproducts. The purified SSH20 was sterile-filtered through 0.45 μm polyethersulfone membrane to remove invisible aggregates. The final product was stored at −20°C.
SSH20 characterization
For MALDI-TOF mass spectrometry, SSH20 and HSA were desalted using a PD-10 column (GE Healthcare Life Science) and buffer-exchanged to deionized water. The protein concentrations were measured by BCA assay from Thermo Scientific (Waltham, MA, USA) and diluted to 1 mg/mL using deionized water. The protein samples were then analyzed on a Bruker ultrafleXtreme MALDI-TOF-TOF MS in the Campus Chemical Instrument Center (CCIC) Mass Spectrometry and Proteomics Facility at The Ohio State University. For SDS-PAGE characterization, SSH20 was diluted to 2 mg/mL using PBS. HSA standard was prepared in PBS at 2 mg/mL (calibrated by NanoDrop OD280 using its extinction coefficient of 35,700 M−1cm−1). Sample buffer (BioRad, Hercules, CA, USA) with 5% β-mercaptoethanol (β-ME) was added to the protein samples, and the mixtures were incubated at 100°C for 5 min. The denatured protein samples were then run on a 4%–15% TGX precast protein gel (BioRad) along with Precision Plus Protein dual-color standard (BioRad) at a constant voltage of 100 V for 75 min. The gel was fixed with 40% ethanol/10% acetic acid for 15 min, rinsed with deionized water, and stained with QC Colloidal Coomassie Stain (BioRad) overnight. The stained gel was destained with deionized water and imaged on a ProteinSimple Fluorochem M Imager. For organic solvent extraction, 100 μL chloroform was added to an equal volume of SSH20 and mixed on a high-speed vortex for 1 min. The chloroform layer was separated by centrifugation and analyzed by high-performance liquid chromatography (HPLC). The analysis was run with an isocratic 40% acetonitrile/60% water mobile phase and on a C18 column (Kromasil 100-5-C18, 4.6 × 150 mm). The photodiode array detector was set at 365 nm for detection.
Cell culture
MIA-PaCa-2 and H23 cells were obtained from ATCC (Manassas, VA, USA) and authenticated (via short tandem repeat profiling). Stable shRNA Cav-1 knockdown MIA-PaCa-2 and H23 cells were generated as described previously.11 Cells were maintained at 37°C in 5% CO2 in DMEM (MIA-PaCa-2) or RPMI 1640 (H23) media supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were cultured for no more than 3 months continuously before re-thawing cells from an early passage. Cells were routinely tested for Mycoplasma. SN-38, SSH20, irinotecan, and methyl-beta-cyclodextrin were added to media with a final vehicle concentration of no more than 0.1%.
Cellular proliferation assay
alamarBlue proliferation assay was performed according to the manufacturer’s instructions (BioRad Antibodies, Oxford, UK). Briefly, cells were seeded in 96-well plates in 4 replicates at a density of 1,000–3,000 cells per well in 100 μL medium and treated as described. Seventy-two hours after plating, alamarBlue reagent was added and incubated at 37°C for 4–8 h, and absorbance was measured at 570 and 600 nm.
Immunofluorescence
Cells were plated on coverslips and treated with or without FITC-HSA, SN-38, or SSH20. Cells were pulsed either 1 h with 0.1% FITC-HSA or 2 h with 250 nM SN-38/SSH20 prior to fixation. When applicable, in order to remove membrane-bound albumin, two acid/salt washes were performed with 0.1 M glycine and 0.1 M NaCl (pH 3.02) on ice for 2 min each, followed by two washes with PBS. Cells were then fixed with 2% paraformaldehyde for 15 min at room temperature and washed with 1 × PBS 2 times for 5 min each. Cells were incubated in 0.1% Triton X-100 for 10 min on ice to permeabilize, then washed twice with 1 × PBS prior to blocking with 3% bovine serum albumin in 1× PBS overnight at 4°C. In a humidified chamber, primary antibodies (1:50) in blocking buffer were added and incubated for 1 h at 4°C, followed by three 10-min rinses with blocking buffer. Secondary antibody (conjugated to Alexa Fluor 568, 1:1,000) was added along with DAPI for 1 h at room temperature. Cells were then rinsed, and coverslips were mounted onto slides and then sealed. Cells were then imaged with a confocal microscope with locked settings across the control and respective experimental conditions. For auto-fluorescence experiments with SN-38 or SSH20, images were taken at an emission wavelength of 543 nm. ImageJ was used to measure the fluorescence intensity of individual cells.
Immunoblotting
For assessment of Cav-1 expression, octyl-b-D-glucopyranoside (Millipore-Sigma; St. Louis, MO, USA) was added at 60 mM final concentration to RIPA buffer (Thermo Fisher Scientific; Waltham, MA, USA) containing protease and phosphatase inhibitor cocktails (Roche; Basel, Switzerland). Protein concentration was determined with a Dc Protein Assay Kit (BioRad; Hercules, CA, USA). For albumin immunoblots, 2 acid/salt washes with 0.1 M glycine and 0.1 M NaCl (pH 3.02) were performed on ice for 2 min each, followed by a PBS wash prior to cell lysis. Proteins were resolved by SDS/PAGE and transferred to nitrocellulose membranes. Membranes were incubated in 5% BSA in TBS-Tween blocking buffer for 1 h at room temperature. Primary antibodies were allowed to bind overnight at 4°C and used at a dilution of 1:1,000. After washing in TBS-Tween three times for 10 min each, the membranes were incubated with immunofluorescent secondary antibodies at a 1:5,000 dilution for 1 h at room temperature. Membranes were washed with TBS-Tween prior to imaging via LI-COR Odyssey CLx Imaging System (Lincoln, NE, USA).
In vivo studies
Animal studies were conducted in accordance with an approved protocol adhering to the Institutional Animal Care and Use Committee policies and procedures at The Ohio State University (Columbus, OH, USA). Eight- to ten-week-old male athymic nude mice (Taconic Farms) were caged in groups of five or less and fed a diet of animal chow and water ad libitum. MIA-PaCa-2 or H23 cells (2 × 106) with stable control shRNA (shCtrl) or shCav-1 were injected subcutaneously into the flanks of athymic nude mice. Treatment regimens were started once tumors reached approximately 100–200 mm3 in size (typically 1–3 weeks post-injection). SSH20 (10 mg/kg) and the molar equivalent of irinotecan in 0.9% saline was administered intravenously via retro-orbital injection accordingly. To obtain a tumor growth curve, perpendicular diameter measurements of each tumor were measured every 1–5 days from the first day of injection with digital calipers, and volumes were calculated using the formula (L × W × W)/2. Weight and clinical signs of toxicity were monitored several times per week.
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM) for proliferation assays, immunofluorescence intensity, and tumor growth experiments. The group comparisons of the percent change in tumor volume were performed at individual time points. Statistical comparisons were made between the control and experimental conditions using the unpaired two-tailed Student t test with significance assessed at p < 0.05. Log-rank (Mantel-Cox) test was performed for comparison of survival (Kaplan-Meier) curves between control and SSH20 treatment groups with significance assessed at p < 0.05. GraphPad Prism (GraphPad Software) was used to perform the statistical analyses.
Acknowledgments
This work was supported by grant National Institutes of Health (NIH) R01 CA198128-01 (to T.M. Williams). Research reported in this article was also supported by The Ohio State University Comprehensive Cancer Center (OSU-CCC) and the NIH (P30 CA016058).
Author contributions
R.R.: Conceptualization, methodology, investigation, validation, formal analysis, writing – original draft, writing – review and editing. J.C.K.: Methodology, investigation, formal analysis, validation, writing – original draft, writing – review and editing. Y.L.: Validation, methodology, writing – review. S.C.-G.: Validation, methodology, writing – original draft, writing – review and editing. T.C.: Formal analysis, writing – review and editing. A.H.: Methodology, validation, formal analysis, review and editing. G.N.: Methodology, validation, formal analysis, review and editing. R.L.: Resources, supervision, writing – review and editing. T.M.W.: Conceptualization, resources, formal analysis, supervision, funding acquisition, validation, investigation, methodology, writing – original draft, writing – review and editing.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.07.013.
Contributor Information
Robert J. Lee, Email: lee.1339@osu.edu.
Terence M. Williams, Email: terwilliams@coh.org.
Supplemental information
References
- 1.Garmann D., Warnecke A., Kalayda G.V., Kratz F., Jaehde U. Cellular accumulation and cytotoxicity of macromolecular platinum complexes in cisplatin-resistant tumor cells. J. Control. Release. 2008;131:100–106. doi: 10.1016/j.jconrel.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Yang F., Liang H. Editorial: HSA-based drug development and drug delivery systems. Curr. Pharm. Des. 2015;21:1784. doi: 10.2174/138161282114150326101436. [DOI] [PubMed] [Google Scholar]
- 3.Kratz F. A clinical update of using albumin as a drug vehicle - a commentary. J. Control. Release. 2014;190:331–336. doi: 10.1016/j.jconrel.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Kratz F., Elsadek B. Clinical impact of serum proteins on drug delivery. J. Control. Release. 2012;161:429–445. doi: 10.1016/j.jconrel.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 5.Sleep D., Cameron J., Evans L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta. 2013;1830:5526–5534. doi: 10.1016/j.bbagen.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 6.Hoogenboezem E.N., Duvall C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018;130:73–89. doi: 10.1016/j.addr.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi H., Watanabe R., Choyke P.L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81–89. doi: 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ojha T., Pathak V., Shi Y., Hennink W.E., Moonen C.T.W., Storm G., Kiessling F., Lammers T. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv. Drug Deliv. Rev. 2017;119:44–60. doi: 10.1016/j.addr.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golombek S.K., May J.N., Theek B., Appold L., Drude N., Kiessling F., Lammers T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018;130:17–38. doi: 10.1016/j.addr.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mo Y., Barnett M.E., Takemoto D., Davidson H., Kompella U.B. Human serum albumin nanoparticles for efficient delivery of Cu, Zn superoxide dismutase gene. Mol. Vis. 2007;13:746–757. [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee M., Ben-Josef E., Robb R., Vedaie M., Seum S., Thirumoorthy K., Palanichamy K., Harbrecht M., Chakravarti A., Williams T.M. Caveolae-Mediated Endocytosis Is Critical for Albumin Cellular Uptake and Response to Albumin-Bound Chemotherapy. Cancer Res. 2017;77:5925–5937. doi: 10.1158/0008-5472.CAN-17-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams T.M., Lisanti M.P. The Caveolin genes: from cell biology to medicine. Ann. Med. 2004;36:584–595. doi: 10.1080/07853890410018899. [DOI] [PubMed] [Google Scholar]
- 13.Sunaga N., Miyajima K., Suzuki M., Sato M., White M.A., Ramirez R.D., Shay J.W., Gazdar A.F., Minna J.D. Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer. Cancer Res. 2004;64:4277–4285. doi: 10.1158/0008-5472.CAN-03-3941. [DOI] [PubMed] [Google Scholar]
- 14.Chatterjee M., Ben-Josef E., Thomas D.G., Morgan M.A., Zalupski M.M., Khan G., Robinson C.A., Griffith K.A., Chen C.S., Ludwig T. Caveolin-1 is Associated with Tumor Progression and Confers a Multi-Modality Resistance Phenotype in Pancreatic Cancer. Sci. Rep. 2015;5:10867. doi: 10.1038/srep10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H.X., Shen H.L., Zhang Y., Zhong F., Liu Y.K., Qin L.X., Yang P.Y. CAV1 Promotes HCC Cell Progression and Metastasis through Wnt/beta-Catenin Pathway. PloS One. 2014;9:e106451. doi: 10.1371/journal.pone.0106451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato K., Hida Y., Miyamoto M., Hashida H., Shinohara T., Itoh T., Okushiba S., Kondo S., Katoh H. Overexpression of caveolin-1 in esophageal squamous cell carcinoma correlates with lymph node metastasis and pathologic stage. Cancer. 2002;94:929–933. [PubMed] [Google Scholar]
- 17.Kim Y.J., Kim J.H., Kim O., Ahn E.J., Oh S.J., Akanda M.R., Oh I.J., Jung S., Kim K.K., Lee J.H. Caveolin-1 enhances brain metastasis of non-small cell lung cancer, potentially in association with the epithelial-mesenchymal transition marker SNAIL. Cancer Cell Int. 2019;19:171. doi: 10.1186/s12935-019-0892-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailly C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019;148:104398. doi: 10.1016/j.phrs.2019.104398. [DOI] [PubMed] [Google Scholar]
- 19.Ueno M., Nonaka S., Yamazaki R., Deguchi N., Murai M. SN-38 induces cell cycle arrest and apoptosis in human testicular cancer. Eur. Urol. 2002;42:390–397. doi: 10.1016/s0302-2838(02)00321-4. [DOI] [PubMed] [Google Scholar]
- 20.Maurya D.K., Ayuzawa R., Doi C., Troyer D., Tamura M. Topoisomerase I inhibitor SN-38 effectively attenuates growth of human non-small cell lung cancer cell lines in vitro and in vivo. J. Environ. Pathol. Toxicol. Oncol. 2011;30:1–10. doi: 10.1615/jenvironpatholtoxicoloncol.v30.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura N., Hirano K., Kishino K., Hashimoto K., Amano O., Shimada J., Sakagami H. Analysis of type of cell death induced by topoisomerase inhibitor SN-38 in human oral squamous cell carcinoma cell lines. Anticancer Res. 2012;32:4823–4832. [PubMed] [Google Scholar]
- 22.Rothenberg M.L. Topoisomerase I inhibitors: review and update. Ann. Oncol. 1997;8:837–855. doi: 10.1023/a:1008270717294. [DOI] [PubMed] [Google Scholar]
- 23.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 24.Salmanpour M., Yousefi G., Samani S.M., Mohammadi S., Anbardar M.H., Tamaddon A. Nanoparticulate delivery of irinotecan active metabolite (SN38) in murine colorectal carcinoma through conjugation to poly (2-ethyl 2-oxazoline)-b-poly (L-glutamic acid) double hydrophilic copolymer. Eur. J. Pharm. Sci. 2019;136:104941. doi: 10.1016/j.ejps.2019.05.019. [DOI] [PubMed] [Google Scholar]
- 25.Tsuchihashi Y., Abe S., Miyamoto L., Tsunematsu H., Izumi T., Hatano A., Okuno H., Yamane M., Yasuoka T., Ikeda Y., Tsuchiya K. Novel Hydrophilic Camptothecin Derivatives Conjugated to Branched Glycerol Trimer Suppress Tumor Growth without Causing Diarrhea in Murine Xenograft Models of Human Lung Cancer. Mol. Pharm. 2020;17:1049–1058. doi: 10.1021/acs.molpharmaceut.9b00249. [DOI] [PubMed] [Google Scholar]
- 26.Carter D.C., Ho J.X. Structure of serum albumin. Adv. Protein Chem. 1994;45:153–203. doi: 10.1016/s0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- 27.Larsen M.T., Kuhlmann M., Hvam M.L., Howard K.A. Albumin-based drug delivery: harnessing nature to cure disease. Mol. Cell. Ther. 2016;4:3. doi: 10.1186/s40591-016-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Socinski M.A., Bondarenko I., Karaseva N.A., Makhson A.M., Vynnychenko I., Okamoto I., Hon J.K., Hirsh V., Bhar P., Zhang H. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial. J. Clin. Oncol. 2012;30:2055–2062. doi: 10.1200/JCO.2011.39.5848. [DOI] [PubMed] [Google Scholar]
- 30.Shen Z., Wei W., Zhao Y., Ma G., Dobashi T., Maki Y., Su Z., Wan J. Thermosensitive polymer-conjugated albumin nanospheres as thermal targeting anti-cancer drug carrier. Eur. J. Pharm. Sci. 2008;35:271–282. doi: 10.1016/j.ejps.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Choi S.H., Byeon H.J., Choi J.S., Thao L., Kim I., Lee E.S., Shin B.S., Lee K.C., Youn Y.S. Inhalable self-assembled albumin nanoparticles for treating drug-resistant lung cancer. J. Control. Release. 2015;197:199–207. doi: 10.1016/j.jconrel.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Sepehri N., Rouhani H., Ghanbarpour A.R., Gharghabi M., Tavassolian F., Amini M., Ostad S.N., Ghahremani M.H., Dinarvand R. Human serum albumin conjugates of 7-ethyl-10-hydroxycamptothecin (SN38) for cancer treatment. BioMed Res. Int. 2014;2014:963507. doi: 10.1155/2014/963507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.