

Abstract
Activated protein C (APC) is an anticoagulant plasma serine protease which exhibits potent cytoprotective and antiinflammatory activities. Here, we studied protective effects of APC on the proinflammatory function of polyinosinic-polycytidylic acid [poly(I:C)], a synthetic analog of viral double-stranded RNA, in cellular and animal models. Poly(I:C) induced histone H3 extranuclear translocation via interaction with Toll-like receptor 3 in two established endothelial cell lines. Furthermore, poly(I:C) induced histone H3 extranuclear translocation in J774A.1 macrophages and human neutrophils and formation of macrophage (M) and neutrophil (N) extracellular traps (ET). Mechanistically, poly(I:C) was found to upregulate expression of peptidylarginine deiminase 4 and enhance its interaction with histone H3, thereby leading to increased histone citrullination and NET formation. Poly(I:C) elicited proinflammatory signaling responses by inducing NF-κB activation and disrupting endothelial cell permeability. In vivo, poly(I:C) enhanced cell surface expression of Mac-1 on neutrophils in mice and facilitated their infiltration to lung tissues. Poly(I:C) also downregulated thrombomodulin expression in mouse tissues and reduced its circulating soluble level in plasma. We demonstrate in this study that APC and a signaling-selective mutant of APC effectively inhibits proinflammatory signaling effects of poly(I:C) in both cellular and animal models. We further demonstrate that unlike the requirement for EPCR on endothelial cells, the integrin Mac-1 is involved in the PAR1-dependent APC inhibition of MET formation in J774A.1 cells. Taken together, these results support a key role for APC signaling in inhibiting the viral mimetic-induced proinflammatory signaling responses and histone translocation-associated formation of ETs by innate immune cells.
Keywords: activated protein C, viral mimetic, poly(I:C), inflammation, extracellular traps
Introduction
Activated protein C (APC) exerts its cytoprotective and antiinflammatory signaling function by endothelial protein C receptor (EPCR)-dependent activation of protease-activated receptor 1 (PAR1) on endothelial and most other cell types1–4. We recently demonstrated that APC inhibits bacterial endotoxin lipopolysaccharide (LPS)-induced cytoplasmic translocation and extracellular release of high mobility group box 1 (HMGB1) by modulating the acetylation status of the nuclear protein5. APC also inhibits extracellular HMGB1-mediated proinflammatory signaling6. Like HMGB1, histones function as damage-associated molecular patterns (DAMPs) when released from the nucleus to extracellular spaces by both damaged and activated cells (e.g., neutrophils and macrophages), exhibiting significant cytotoxic and proinflammatory activities as demonstrated by in vitro and in vivo studies7, 8. Extracellular histones bind to several cell surface receptors specific for DAMPs including Toll-like receptors (TLRs) and the receptor for advanced glycation end-products (RAGE), thereby triggering activation of multiple signaling pathways in a coordinated manner9, 10. HMGB1 and histones levels are elevated in animals and in patients with infection, inflammation and cancer, suggesting extracellular roles for these nuclear proteins in human diseases11. Circulating levels of HMGB1, histones and in particular the citrullinated form of histone H3 (H3Cit) have become reliable blood biomarkers for diagnosis and prognosis of a variety of inflammatory diseases12, 13. We and others have demonstrated a key cytoprotective role for APC in inhibiting the expression and/or inflammatory signaling function of nuclear proteins in both cellular and animal models5–8, 14, 15.
Activated neutrophils can release their nuclear content during infection by a process termed NETosis under which decondensed chromatin fibers consisting of DNA, histones, and granular content are used as extracellular traps (ETs) to kill invading microorganisms16, 17. Although the process was first discovered for neutrophils, later studies indicated that ETs are also released from activated monocytes and macrophages17–19. It has been demonstrated that citrullination of histones is catalyzed by the nuclear peptidylarginine deiminase 4 (PAD4), which results in chromatin decondensation and formation of NETs20–23. The expression of macrophage antigen-1 (Mac-1, CD11b/CD18), a β2 integrin expressed on neutrophils and macrophages, contributes to the formation of both macrophage ETs (METs) and neutrophil ETs (NETs) during infection24, 25. NETs are also generated during viral infection26–29 and may be associated with the disease pathology in coronavirus 2019 (COVID-19) patients30, 31. APC is known to inhibit NETosis mediated by activated platelets and phorbol 12-myristate 13-acetate32.
RNA viruses infect cells primarily through interaction with TLR3, TLR7 and TLR8 transmembrane receptors, with TLR3 specifically recognizing double-stranded RNA (dsRNA) viruses33. Synthetic polyinosinic-polycytidylic acid [poly(I:C)], an analog of viral dsRNA, has been widely utilized as a mimetic to simulate viral infection and to understand the mechanism of TLR3-dependent infection by these viruses under different conditions34, 35. In addition to its TLR3-dependent potent inflammatory signaling function through activation of the transcription factors NF-κB and AP-1, and activation of mitogen activated protein kinases33–36, poly(I:C) also induces TLR3-dependent NETs formation and acute lung injury in vivo37. In our present study, we investigated the proinflammatory signaling mechanism of poly(I:C) and the protective antiinflammatory effect of APC and its signaling-selective variant against the viral mimetic in both cellular and animal models. We demonstrate that APC exerts a PAR1-dependent antiinflammatory effect against poly(I:C) and PAD4-dependent citrullination and translocation of histone H3, thereby inhibiting inflammation and formation of ETs by innate immune cells.
Methods
Reagents and antibodies
Recombinant proteins including activated protein C (APC), the signaling-selective mutant of APC (APC-2Cys), were prepared as described38. Anti-histone H3 was a kind gift from Dr. Charles T. Esmon (Oklahoma Medical Research Foundation, OK). Poly(I:C) was from InvivoGen (San Diego, CA, USA). SYTOX® Green Nucleic Acid Stain was from Invitrogen (Carlsbad, CA, USA). Cell Death Detection ELISA kit (#11544675001) was from Roche (Indianapolis-Marion County, Indiana, USA). Anti-EPCR (#MABS1271, clone JRK1535) and anti-PAR1 (#MABF244, clone ATAP2) function-blocking antibodies were from Millipore (Burlington, MA, USA). Anti-Mac-1 (#550282, clone M1/70) function-blocking antibody was from BD Biosciences (San Jose, CA, USA). PE-conjugated Mac-1 (#101208, clone M1/70) and FITC-conjugated Ly-6G (#127606, clone 1A8) antibodies were from BioLegend (San Diego, CA, USA). Anti-myeloperoxidase (#ab208670), anti-PAD4 (#ab50247), anti-histone H3 (citrulline R2 + R8 + R17) (#ab5103) and anti-TLR3 [TLR3.7] (#ab12085) antibodies were from Abcam (Boston, MA, USA). Anti-NF-κB p65 (#8242), anti-Phospho-NF-κB p65 (Ser536) (#3033) and anti-β-actin (#4967) antibodies were from Cell Signaling Technology (Beverly, MA, USA). Anti-fibrin(ogen) antibody was from DAKO (Santa Clara, CA, USA). Anti-ICAM-1 (#107), anti-neutrophil elastase (#55548), anti-PAD4 (#166645) and anti-GAPDH (glyceraldehyde-3-phoshate dehydrogenase, #47724) were from Santa Cruz Biotechnologies Inc. (Santa Cruz, CA, USA). Anti-thrombomodulin (#MAB3894) was from R&D Systems (Minneapolis, MN, USA). FITC-conjugated ICAM-1 monoclonal antibody (MEM-111) (#MHCD5401), PE-conjugated VCAM-1 (#12–1069-42) and anti-VCAM-1 (#MA5–11447) antibodies were from Thermo Fisher Scientific (Waltham, MA, USA). Cy3-conjugated goat anti-rabbit IgG (#111–166-144) was from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Alexa Fluor 488-conjugated goat anti-mouse IgG (#A11029) and Alexa Fluor 488-conjugated goat anti-rabbit IgG (#A11008) were from Invitrogen Corporation (Carlsbad, CA, USA).
Cell lines and culture
Transformed HUVECs (EA.hy926 cells), human dermal microvascular endothelial cells immortalized with hTERT (immortalized HDMECs), HUVECs immortalized with hTERT (immortalized HUVEC) and J774A.1 cells were obtained from ATCC (Manassas, VA, USA). EA.hy926 cells were maintained in complete Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS, 100 μg/mL penicillin, 100 μg/mL streptomycin, 1X HAT supplement (#25–046 from Mediatech Inc. (Manassas, VA, USA) and 2 mM L-glutamine. Immortalized HDMECs and HUVECs were maintained in complete growth medium (#PCS-100–300) supplemented with endothelial cell growth components (#PCS-100–040), 100 μg/mL penicillin and 100 μg/mL streptomycin. The murine macrophage cell line J774A.1 were maintained in complete DMEM containing 10% FBS, 100 μg/mL penicillin, 100 μg/mL streptomycin and 2 mM L-glutamine. All cells were cultured at 37°C in a humidified incubator with 5% CO2.
Confocal imaging
Following treatments, all cell lines were fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100/PBS, followed by blocking for 1h with normal goat serum. For analysis of histone H3 translocation, cells were incubated with mouse or rabbit anti-histone H3 overnight at 4°C followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse or anti-rabbit secondary antibody for 1h at room temperature in the dark. For co-localization of histone H3 with PAD4, cells were incubated with mouse anti-histone H3 and rabbit anti-PAD4 overnight at 4°C followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse and fluorescent Cyanine 3 (Cy3, red)-conjugated goat anti-rabbit secondary antibodies for 1h at room temperature in the dark. Cells were then washed and stained with DAPI or Hoechst (1 μg/mL), and mounted with anti-fade mounting media (DAKO products, Santa Clara, CA, USA). For analysis of extracellular trap formation in neutrophils, cells were incubated with rabbit anti-Myeloperoxidase (MPO) and then incubated with Cy3-conjugated goat anti-rabbit secondary antibody, and stained with SYTOX® Green Nucleic Acid Stain, Photomicrographs were obtained using a Nikon C2 Confocal Microscope. For quantitative analysis of histone H3 translocation, cells with diffused histone H3 in the extranuclear space were defined as translocation-positive, and were counted in at least 10 randomly selected fields from three independent samples. The translocation efficiency was calculated on the basis of the ratio of translocation-positive cells to total cells in these fields.
Extracellular traps analysis in neutrophils and macrophages
Blood was collected from healthy adult volunteers (following an approved institutional review board protocol) in EDTA buffer. Blood neutrophils were isolated according to the Polymorphprep™ (Alere Technologies AS, Oslo, Norway) protocol. Briefly, whole undiluted blood was carefully layered over an equal volume of Polymorphprep™ and centrifuged at 500g for 30 min at room temperature. The plasma and mononuclear cells (upper band of cells) were removed, and the lower band of PMNs containing neutrophils were harvested and washed with HBSS solution. Red blood cells were removed using a lysis buffer (Invitrogen; Carlsbad, CA, USA). Isolated neutrophils were suspended in RPMI1640 containing 0.5% BSA and plated on FBS-coated coverslips for 20 min at 37° C. Neutrophils were incubated along with poly(I:C) in either the presence or absence of APC or the signaling-selective mutant of APC (APC-2Cys). After 4 h of incubation, cells were fixed with 4% paraformaldehyde and processed for staining. For ETs analysis, neutrophils or macrophages were incubated with rabbit anti-Myeloperoxidase (MPO) or citrullinated histone H3 (H3Cit) or histone H3 and then incubated with Cy3-conjugated goat anti-rabbit secondary antibody, and stained with SYTOX® Green Nucleic Acid Stain, Photomicrographs were obtained using a Nikon C2 Confocal Microscope. ET-forming (elongated or extruded chromatin fiber) cells in neutrophils or macrophages were counted and quantified from 10 randomly selected fields from three independent samples.
Histological examination and immunofluorescence analysis of mouse tissue
All animal studies were performed in compliance with institutional guidelines, and were approved by the Institutional Animal Care and Use Committee of Oklahoma Medical Research Foundation. Male C57BL/6J mice, aged 8–12 weeks were injected i.p. with either saline or poly(I:C) (110μg/mouse). In some experiments, mice were first injected i.p. with APC or APC-2Cys (1μg/g body weight) followed by i.p. injection of poly(I:C) after 1h. At indicated time points mice were sacrificed and blood was collected via heart puncture and whole body perfused with 30 mL of PBS-EDTA before collecting of the organs.
For histological analysis, mouse tissues were freshly harvested, fixed with 10% neutral buffered formalin (ThermoFisher Scientific), embedded in paraffin and sectioned. All 5 μm sections were stained with hematoxylin and eosin (H&E). Pictures were obtained with an Eclipse E1000 microscope (Nikon). Histologic severity of inflammation was assessed in a blinded fashion. For immunofluorescence, tissues were fixed with 4% paraformaldehyde overnight at 4°C. Tissue samples were washed in PBS, cryoprotected in 20% sucrose in PBS at 4°C overnight, embedded in 50% tissue freezing medium/50% OCT compound, and cryosectioned (5–10 μm sections).
Flow cytometry analysis
HDMECs and HUVECs were serum deprived when 90% confluent, and were then detached using HBSS containing 10 mM EDTA, washed and resuspended in HBSS containing 2 mM EDTA and 0.1% human serum albumin (HSA). Cells were stained using FITC-conjugated anti-ICAM-1 and PE-conjugated anti-VCAM-1 antibodies. Cell surface levels of ICAM-1 and VCAM-1 were detected using a FACS Celesta flow cytometer and data analyzed using FlowJo software (BD Life Sciences). For detecting changes in Mac1 surface expression on blood neutrophils in mice, whole blood was incubated with FITC-conjugated Ly6G and PE-conjugated Mac-1 antibodies for 45 min on ice. Red blood cells were then lysed with 150 mM NH4Cl, 10 mM NaHCO3, and 1 mM EDTA, and cells were washed and resuspended in HBSS with EDTA and HSA and analyzed by flow cytometry.
Western blotting
Treated cells were lysed with RIPA buffer (1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholic acid, 5 mM tetrasodium pyrophosphate, 50 mM sodium fluoride, 5 mM EDTA, 150 mM NaCl, 25 mM Tris-HCl (pH 7.5), 5 mM Na3VO4 and protease inhibitor cocktail). All lysate samples were boiled in loading buffer with 5% β-mercaptoethanol and resolved on 10–15% SDS-polyacrylamide gels. The resolved proteins were then electro-blotted onto a polyvinylidene difluoride (PVDF) membrane, blocked with 5% skim milk (wt/vol) and incubated with primary antibodies. The immunoblotted PVDF membranes were then incubated with the respective horseradish peroxidase-conjugated secondary antibody for 1h and bands were detected using enhanced chemiluminescence reagent. Protein bands were quantified using NIH Image J software.
Cell permeability assay
Cell permeability in response to poly(I:C) was monitored by spectrophotometric analysis of the leakage of Evans blue-bound albumin across endothelial cell monolayer in a modified two-compartment chamber model. HDMECs and HUVECs (2 × 105 cells/well) were seeded on trans-well inserts (3.0 μm pore size) in complete growth medium and allowed to become confluent. Thereafter, cells were treated with APC (20 nM) in serum free DMEM media containing 0.5% BSA for 3h followed by stimulation of cells with poly(I:C) (10 μg/mL). After 4h, Evans blue dye (0.67 mg/mL) and BSA (4%) were added to the upper chamber. Permeability was measured by collecting media from the lower chamber and measuring the absorbance of the leaked Evans blue dye at 650 nm. Final values were plotted as fold change to untreated controls.
Statistical analysis
All data are presented as mean ± SE from ≥3 independent experiments. Data were analyzed by the Student t-test, and group data were analyzed using ANOVA followed by Bonferroni post hoc test using Graph Pad Prism7 (Graph Pad Prism, CA). A p value of <0.05 was considered statistically significant.
Results
Poly(I:C) induces histone extranuclear translocation via TLR3
To investigate whether viral infection can modulate subcellular localization/translocation of histones, we chose poly(I:C) as a synthetic analog of viral double-stranded RNA (dsRNA) and investigated its signaling effect on two endothelial cell lines: immortalized human dermal microvascular endothelial cells (HDMECs) and human umbilical vein endothelial cells (HUVECs). Poly(I:C) has been widely used to simulate dsRNA viral infection in cellular and animal models33–35. Since poly(I:C) enters cells primarily via interaction with TLR3, we first examined the basal cell surface expression level of this receptor. Flow cytometry analysis revealed both cell lines express TLR3 at considerable levels (Fig. 1A,B). Next, we monitored poly(I:C)-mediated subcellular localization/translocation of histone H3 in both HDMECs and HUVECs. Confocal immunofluorescence (IF) analysis indicated that poly(I:C) induces translocation of histone H3 from the nucleus to the extranuclear space in HDMECs in a time-dependent manner and that a significant fraction of histone H3 translocates to the extranuclear space after 1h (Suppl. Fig. S1). We used this treatment conditions and showed that the anti-TLR3 antibody effectively inhibits poly(I:C)-mediated translocation of histone H3 from the nucleus to the extranuclear space in both cell lines (Fig. 1C,D). For quantitative analysis, the translocation of histone H3 in poly(I:C)-treated cells was analyzed in randomly selected areas of the coverslips harboring 50 cells. Histone H3 translocation from the nucleus to the extranuclear space was detected in approximately 60% of poly(I:C)-treated cells, which were effectively inhibited by the TLR3 neutralizing antibody in both HDMEC and HUVEC cell lines (Fig. 1E,F), suggesting that TLR3 is essential for poly(I:C)-induced histone H3 extranuclear translocation in endothelial cells.
Figure 1. Poly(I:C) induces histone extranuclear translocation via TLR3 in endothelial cells.
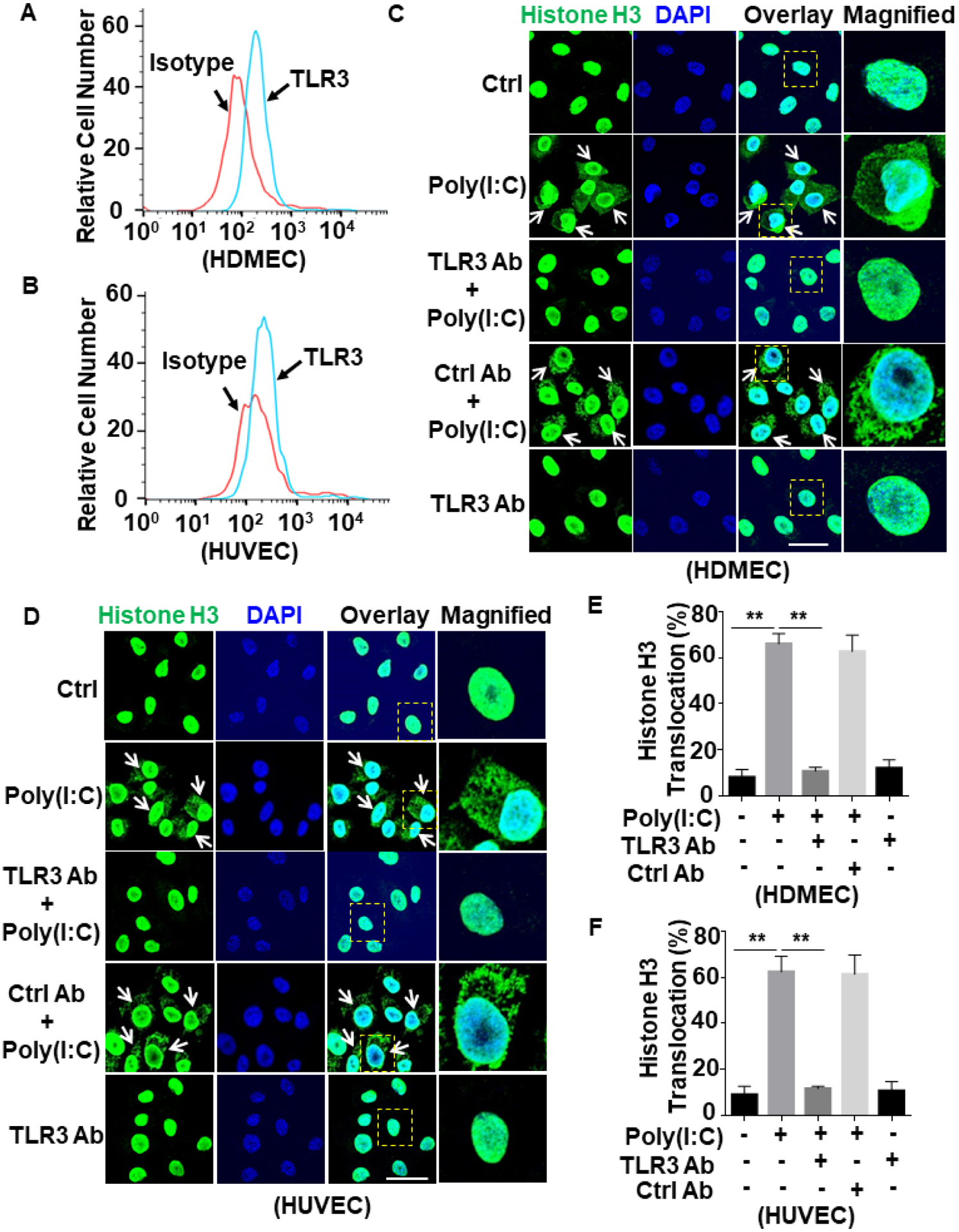
(A and B) Confluent HDMECs (A) or HUVECs (B) were stained with mouse anti-TLR3 antibody and FITC-conjugated goat anti-mouse IgG. The cell surface level of TLR3 was measured by flow cytometry. (C and D) HDMECs (C) or HUVECs (D) were pretreated with anti-TLR3 antibody or a control antibody (5 μg/mL for 1h) followed by stimulation with poly(I:C) (10 μg/mL for 1h). Cells were fixed and permeabilized followed by staining for histone H3 with mouse anti-histone H3 antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG. The nucleus was stained with DAPI. Immunofluorescence images were taken by confocal microscopy. Arrows indicate extranuclear translocation of histone H3. The magnified insets correspond to cells marked with yellow dashed boxes. (E and F) The quantitation of poly(I:C)-mediated translocated cells from the nucleus to the extranuclear space for HDMECs (C) and HUVECs (D). Scale bar: 20 μm. Results are shown as means ± standard error from at least 3 independent experiments. **p < 0.01. Ctrl, control.
APC inhibits poly(I:C)-induced histone H3 translocation and citrullination
Confocal IF analysis of poly(I:C)-stimulated HDMECs and HUVECs in the absence and presence of APC showed that APC significantly inhibits poly(I:C)-induced histone H3 extranuclear translocation in both cell lines (Fig. 2A–D). Because of its elevated plasma levels, citrullinated histone H3 (H3Cit) has been proposed as a possible blood biomarker in a number of inflammatory diseases including sepsis and cancer12, 13. Thus, we decided to determine whether histone H3 citrullination level is altered by the viral mimetic. By using an antibody specific to H3Cit in the Western blots, we discovered that poly(I:C) induces histone H3 citrullination in both HDMECs and HUVECs, which was effectively inhibited by APC (Fig. 2E,F). These results support a novel protective and antiinflammatory role for APC against viral mimetic-mediated proinflammatory effect through inhibition of extranuclear translocation and posttranslational modification of histone H3 in endothelial cells. As expected, the inhibitory effect of APC on histone H3 extranuclear translocation by poly(I:C) was mediated through EPCR-dependent activation of PAR1 since function-blocking antibodies to both receptors inhibited this protective signaling function of APC in endothelial cells (Suppl. Fig. S2).
Figure 2. APC inhibits poly(I:C)-induced citrullination and extranuclear translocation of histone H3 in endothelial cells.
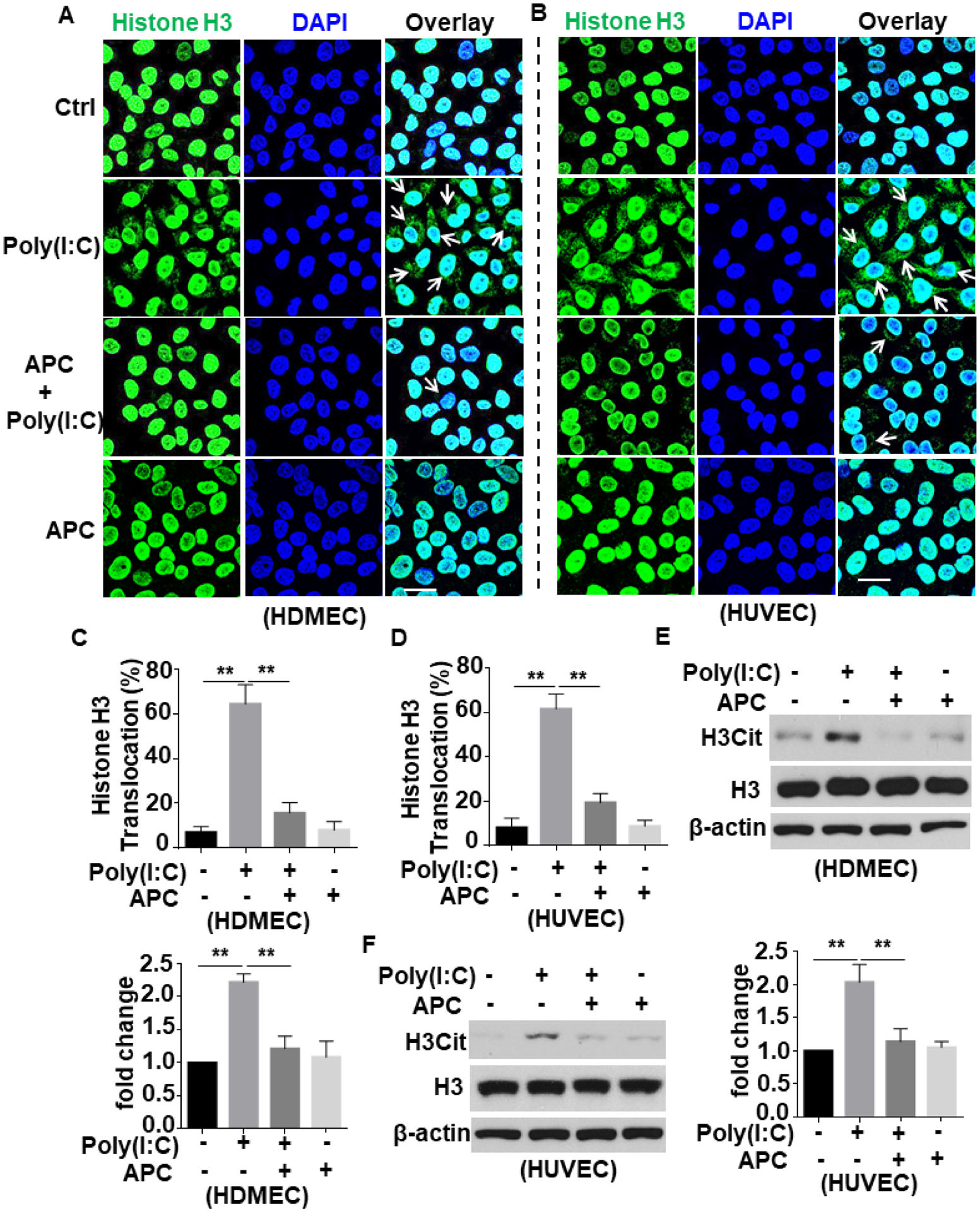
(A and B) HDMECs (A) or HUVECs (B) were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 1h) (APC remained in the media after addition of poly(I:C)). Cells were fixed and permeabilized followed by staining for histone H3 with mouse anti-histone H3 antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG. The nucleus was stained with DAPI. Immunofluorescence images were taken by confocal microscopy. Arrows indicate extranuclear translocation of histone H3. (C and D) The quantification of poly(I:C)-mediated translocated cells from the nucleus to the extranuclear space for HDMECs (A) and HUVECs (B). (E and F) HDMECs (C) or HUVECs (D) were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 1h). Cell lysates were immunoblotted and probed with an antibody specific for unmodified or citrullinated histone H3 (H3Cit). β-actin was used as a loading control. Scale bar: 20 μm. Results are shown as means ± standard error from at least 3 independent experiments. **p < 0.01. Ctrl, control.
APC inhibits poly(I:C)-induced histone translocation and extracellular trap formation in macrophages
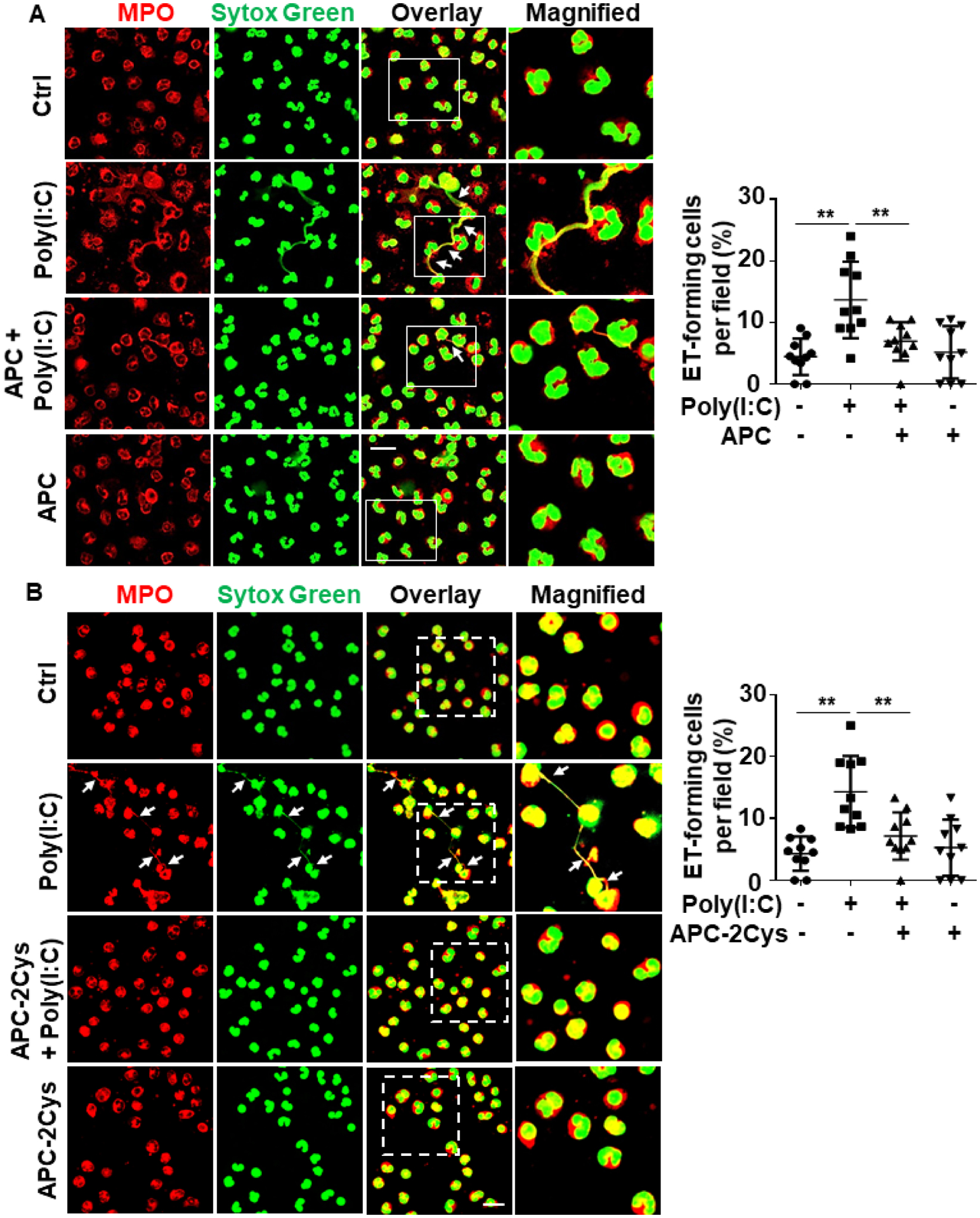
Nuclear proteins may be actively released from activated innate immune cells (e.g., macrophages and neutrophils) as extracellular traps (ETs)16, 20. To determine whether poly(I:C) can induce histone extranuclear translocation in macrophagesm, we stimulated J774A.1 murine macrophages with poly(I:C) for 1h and then monitored histone H3 translocation by confocal IF. Results showed that poly(I:C) induces histone H3 extranuclear translocation and APC significantly inhibits this response (Fig. 3A,B). Next, we extended the poly(I:C) treatment time for up to 4h and performed co-staining of histone H3 with Sytox Green nucleus DNA staining dye. Confocal IF analysis revealed that poly(I:C) triggers formation of ETs, displaying elongated chromatin fibers composed of DNA and histones and APC inhibited this process as well (Fig. 3C,D). APC exerts its cytoprotective and antiinflammatory effects through EPCR-dependent activation of PAR1 in endothelial cells1–4. Interestingly, analysis of ET formation in J774A.1 macrophages by the use of anti-PAR1, anti-Mac-1 or anti-EPCR function-blocking antibodies revealed that PAR1 and Mac-1, but not EPCR, are involved in the APC’s inhibitory effect against poly(I:C)-mediated ET formation in macrophages (Fig. 4). This is consistent with previous results showing that APC exerts its protective effect through PAR1 and Mac-1 in human macrophages39.
Figure 3. APC inhibits poly(I:C)-induced histone extranuclear translocation and extracellular trap formation in macrophages.
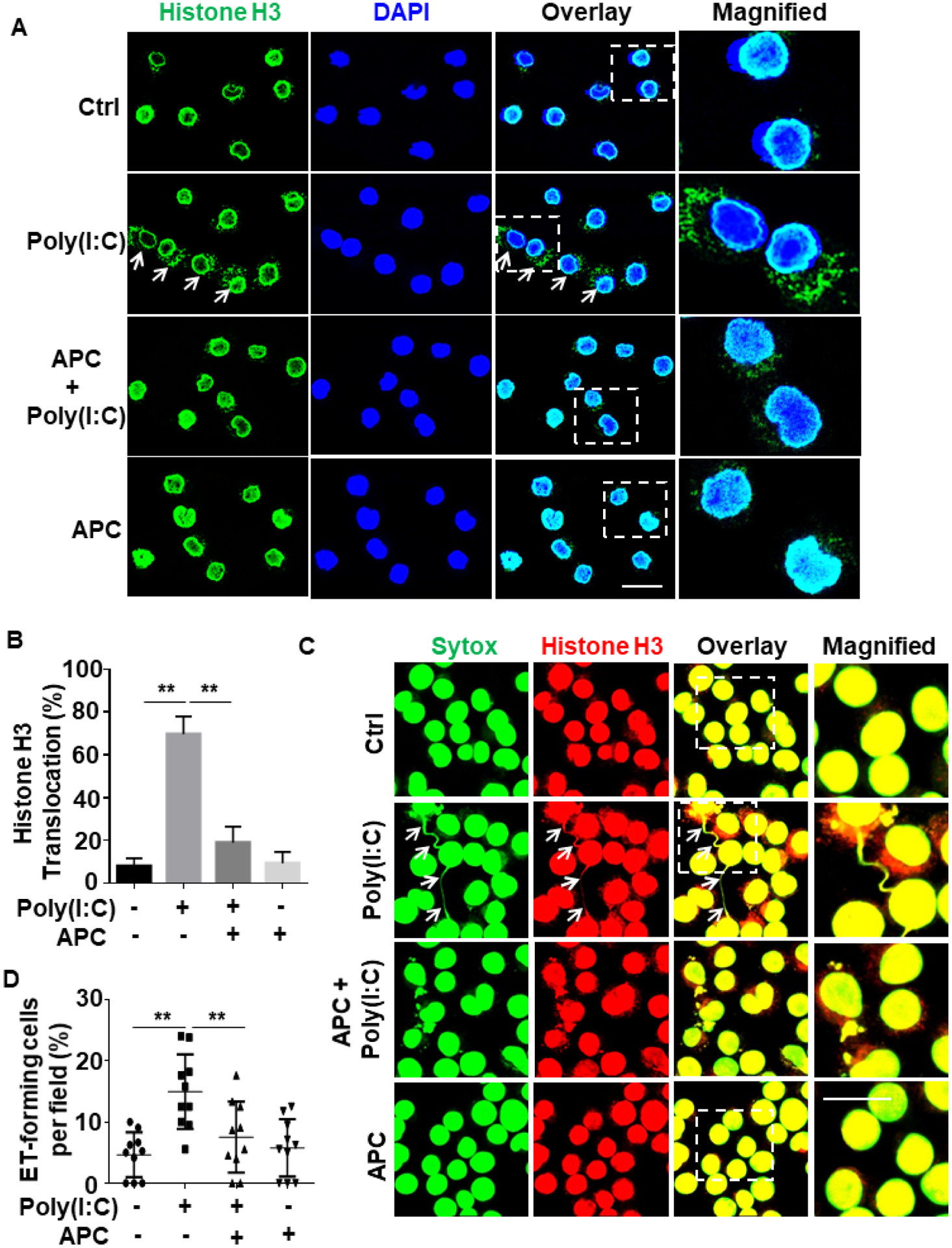
(A) J774A.1 macrophages were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 1h) (APC remained in the media after addition of poly(I:C)). Cells were fixed and permeabilized, followed by staining for histone H3 with mouse anti-histone H3 antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG. The nucleus was stained with DAPI. Immunofluorescence images were taken by confocal microscopy. Arrows indicate extranuclear translocation of histone H3. (B) The quantification of poly(I:C)-mediated translocated cells from the nucleus to the extranuclear space for J774A.1 macrophages. (C) J774A.1 macrophages were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 4h). Cells were fixed and permeabilized, followed by staining for histone H3 with mouse anti-histone H3 antibody and Alexa Fluor 555-conjugated goat anti-mouse IgG. DNA was stained with Sytox Green. Immunofluorescence images were taken by confocal microscopy. Arrows indicate cells with extracellular trap formation. The inset boxes from each group are magnified. (D) The quantitation of ET-forming cells in (C). All experiments were repeated at least three times. **p < 0.01. Ctrl, control. Scale bar: 20 μm.
Figure 4. Mac-1 and PAR1, but not EPCR, are required for APC inhibition of extracellular traps in macrophages.
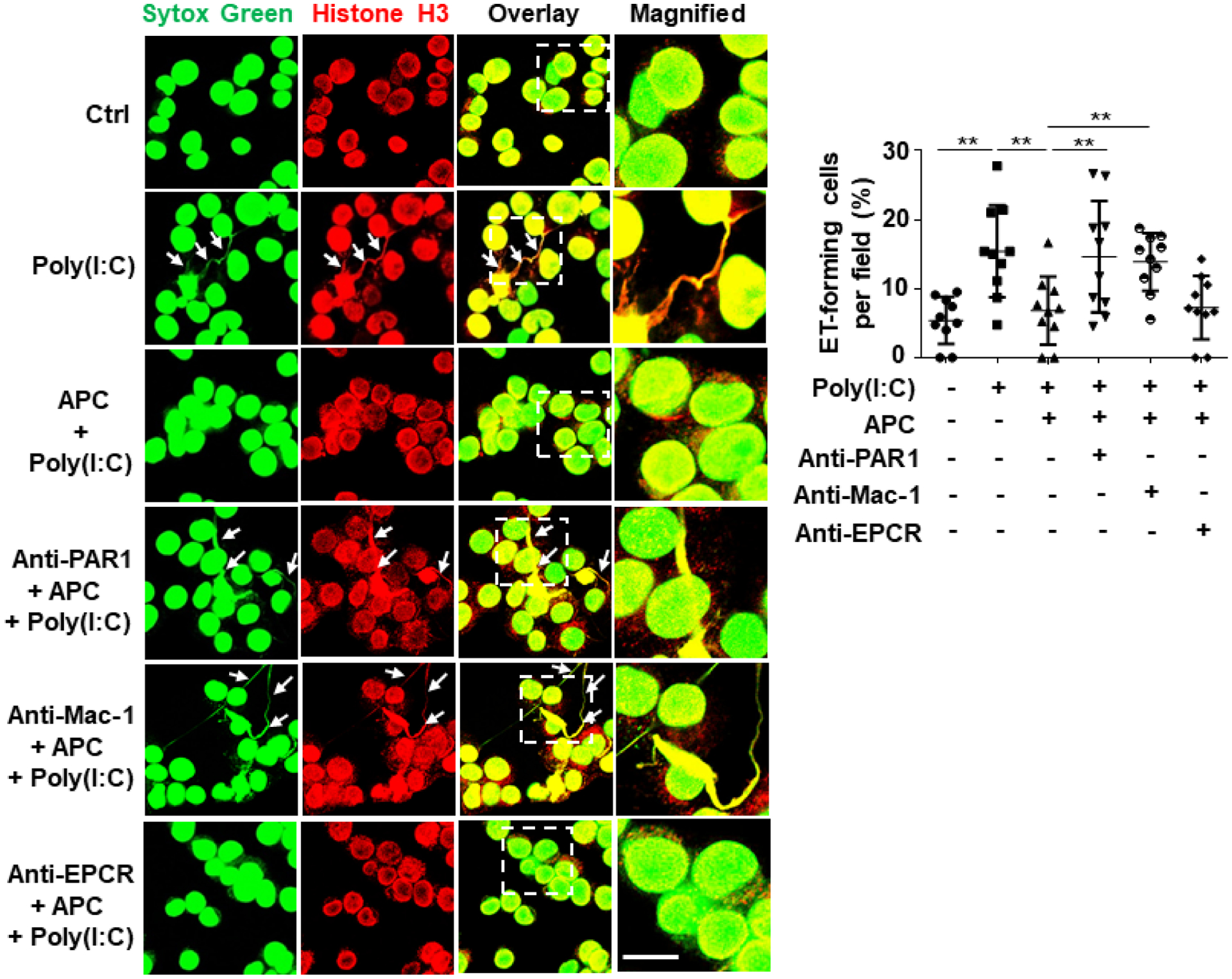
J774A.1 macrophages were pretreated with PAR1, Mac-1 or EPCR function-blocking antibodies (15–20 μg/mL for 1h) followed by treatment with APC (20 nM for 3h) before stimulation with poly(I:C) (10 μg/mL for 4h) (APC remained in the media after addition of poly(I:C)). Cells were fixed and permeabilized, followed by staining for histone H3 with mouse anti-histone H3 antibody and Alexa Fluor 555-conjugated goat anti-mouse IgG. DNA was stained with Sytox Green. Immunofluorescence images were taken by confocal microscopy. Arrows indicate cells with extracellular trap formation. The inset boxes from each group are magnified. Dot plot shows quantification of ET-forming cells. Experiments were repeated at least three times. **p < 0.01. Ctrl, control. Scale bar: 10 μm.
Poly(I:C) induces extracellular trap formation in human neutrophils
Recent results have indicated that, similar to bacterial infection, viral infection can also induce NET formation26–29. To investigate the effect of poly(I:C) on human neutrophils, we isolated fresh human peripheral blood neutrophils and treated them with poly(I:C) at different concentrations (10, 25, 100 μg/mL) for 4h. Confocal IF analysis of co-staining for MPO, a neutrophil marker, and DNA revealed that poly(I:C) effectively induces NET formation in neutrophils (Fig. 5A).
Figure 5. Poly(I:C) induces NETs probably by increasing PAD4 expression and its interaction with histones.
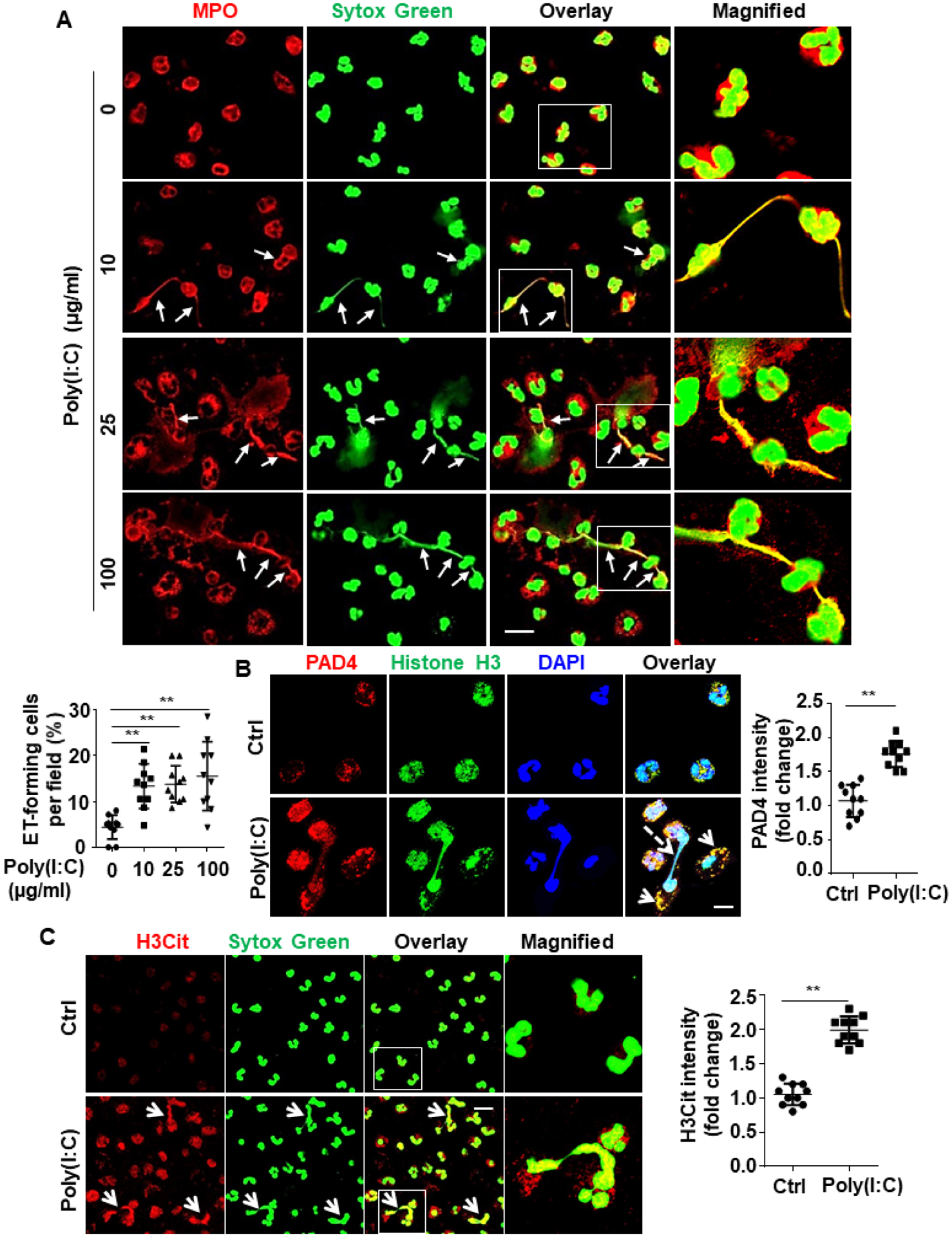
(A) Human blood neutrophils were isolated, cultured and stimulated with poly(I:C) for 4h at designated concentrations (10, 25, 100 μg/mL). Cells were then fixed, permeabilized and Myeloperoxidase (MPO), a neutrophil marker, was stained with rabbit anti-MPO antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG. DNA was stained with Sytox Green. Immunofluorescence images were taken by confocal microscopy. Arrows indicate cells with extracellular trap formation. The inset boxes from each group are magnified. Dot plot shows quantification of NET-forming cells. (B) Human blood neutrophils were isolated, cultured and stimulated with poly(I:C) (10 μg/mL for 4h). Cells were then fixed, permeabilized and histone H3 was stained with mouse anti-histone H3 antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG. Peptidylarginine deiminase 4 (PAD4) was stained with rabbit anti-PAD4 antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG. Immunofluorescence images were taken by confocal microscopy. Arrows indicate the colocalization of PAD4 with histone H3. Dashed arrows indicate cells with NET formation. Dot plot shows quantitation of the relative expression levels of PAD4. (C) Human blood neutrophils were isolated, cultured and stimulated with poly(I:C) (10 μg/mL for 4h). Cells were then fixed, permeabilized and citrullinated histone H3 (H3Cit) was stained with rabbit anti-H3Cit antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG. DNA was stained with Sytox Green. Immunofluorescence images were taken by confocal microscopy. Arrows indicate cells with NET formation. The magnified insets correspond to the cells marked with white boxes. Dot plot shows quantitation of the relative expression levels of H3Cit. All experiments were repeated at least three times. **p < 0.01. Ctrl, control. Scale bar: 10 μm (B), 20 μm (A and C).
Poly(I:C) upregulates PAD4 and enhances its interaction with histone H3
Peptidylarginine deiminase 4 (PAD4) is known to citrullinate histone H3 and mediate NET formation21, 23. We discovered that poly(I:C) upregulates PAD4 expression and enhances its interaction with histone H3 during NET formation (Fig. 5B). Confocal IF analysis after co-staining of H3Cit with DNA revealed that poly(I:C) increased histone H3 citrullination during NET formation by human neutrophils (Fig. 5C). These data suggest that poly(I:C) induces NET formation most likely by upregulating PAD4 expression and promoting PAD4-mediated histone H3 citrullination. Confocal IF analysis showed that APC effectively inhibits poly(I:C)-induced NET formation by human neutrophils (Fig. 6A). The signaling-selective mutant of APC (APC-2Cys), which exhibits PAR1-dependent signaling activity but lacks an anticoagulant activity38, also inhibited NET formation by neutrophils (Fig. 6B).
Figure 6. APC inhibits poly(I:C)-induced extracellular traps in human neutrophils.
Human blood neutrophils were pretreated with APC (A) or a signaling-selective mutant of APC (APC-2Cys) (20 nM for 3h) (B), followed by stimulation with poly(I:C) (10 μg/mL for 4h) (APC remained in the media after addition of poly(I:C)). Cells were then fixed, permeabilized and Myeloperoxidase (MPO) was stained with rabbit anti-MPO antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG. DNA was stained with Sytox Green. Immunofluorescence images were taken by confocal microscopy. Arrows indicate cells with extracellular trap formation. The inset boxes from each group are magnified. Dot plot shows quantitation of NET-forming cells in (A) or (B). All experiments were repeated at least three times. **p < 0.01. Ctrl, control. Scale bar: 20 μm.
Poly(I:C) activates NF-κB signaling to promote inflammation
Confocal IF and Western blot analyses revealed that poly(I:C) activates NF-κB signaling by inducing NF-κB p65 translocation to the nucleus in HDMECs (Fig. 7A). Flow cytometry assay further showed increased cell surface expression of adhesion molecules (ICAM-1 and VCAM-1) in poly(I:C)-treated endothelial cells (Fig. 7B–E). In addition to enhanced expression of proinflammatory signaling molecules, a functional permeability assay revealed that poly(I:C) dose-dependently enhances cellular permeability in both endothelial cell lines (Fig. 7F).
Figure 7. Poly(I:C) activates NF-κB signaling to promote inflammation.
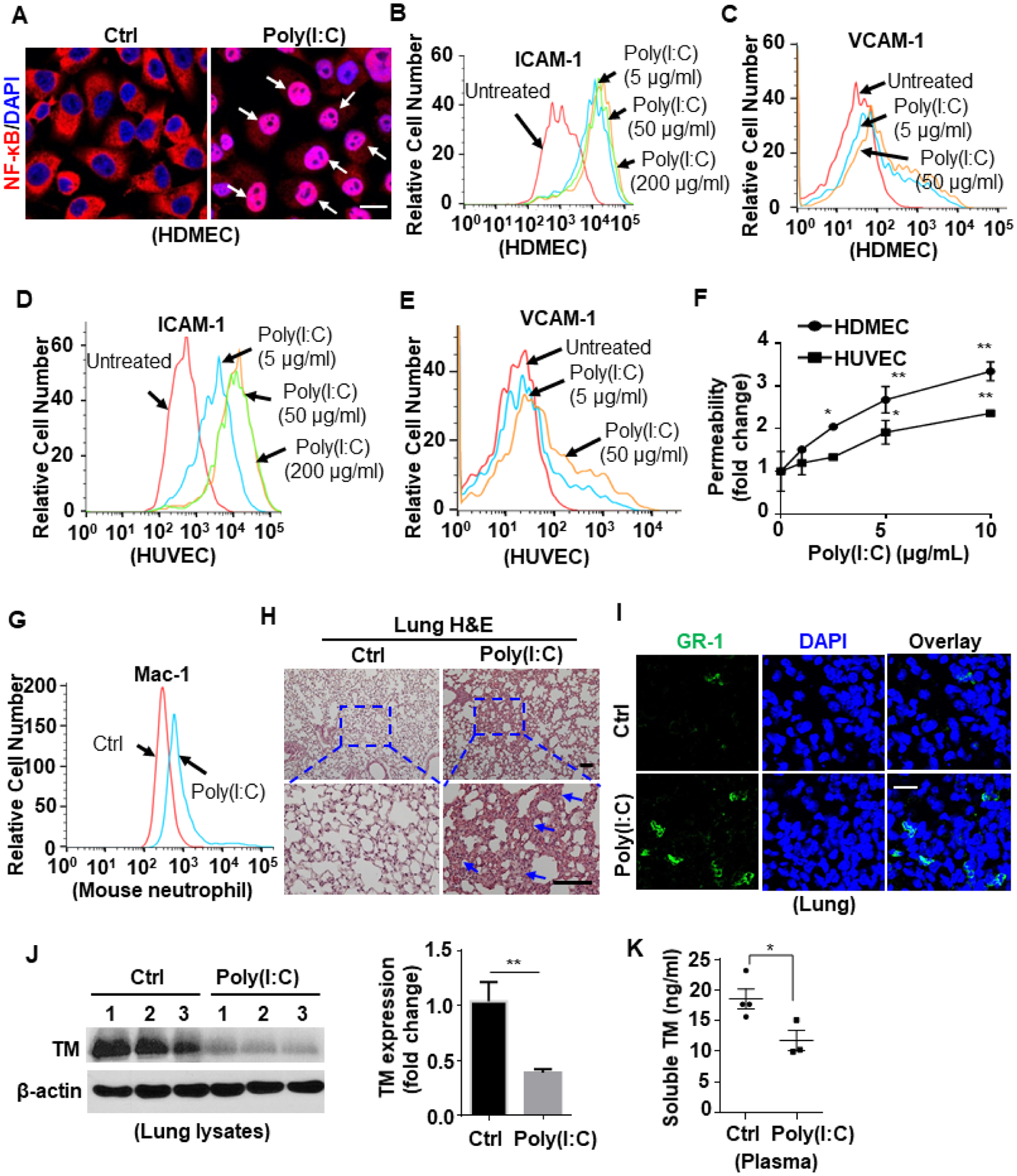
(A) HDMECs were stimulated with poly(I:C) (10 μg/mL for 1h). Cells were then fixed, permeabilized and NF-κB p65 was stained with rabbit anti-NF-κB antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG. The nucleus was stained with DAPI. Immunofluorescence images were taken by confocal microscopy. Arrows indicate nuclear translocation of NF-κB. (B-E) Confluent HDMECs (B and C) or HUVECs (D and E) were stimulated with poly(I:C) at 5, 50 or 200 μg/mL for 6h. The cell surface levels of ICAM-1 (B and D) or VCAM-1 (C and E) were measured by flow cytometry. (F) HDMECs or HUVECs were stimulated with poly(I:C) at designated concentrations (1, 2.5, 5, 10 μg/mL) for 4h. The amount of Evans blue dye that leaked into the lower chamber in the Trans-well assay plates was measured. (G) Mice were injected i.p. with poly(I:C) for 3h and blood samples were collected and stained for Ly6G-FITC & Mac-1-PE. The cell surface expression of Mac-1 in Ly6G-FITC-positive neutrophil population was measured by flow cytometry. Representative data were obtained from 3–5 mice per group (n = 3–5). (H) Mice were injected i.p. with poly(I:C) for 3h, and lung tissue was collected and processed for histological analyses. Paraffin-embedded sections of lung tissue were stained with H&E. Representative images were obtained from 5 mice per group (n = 5). The inset boxes from each group are magnified. The arrows indicate inflammatory foci. (I) Mice were injected i.p. with poly(I:C) for 3h, and lung tissue was collected and processed. Cryosections of lung tissue were fixed and permeabilized. GR1 was stained with rat anti-GR-1 antibody and Alexa Fluor 488-conjugated goat anti-rat IgG. The nucleus was stained with DAPI. Immunofluorescence images were taken by confocal microscopy. Representative images were obtained from 3–5 mice per group (n = 3–5). (J) Mice were injected i.p. with poly(I:C) for 24h, and lung tissue was harvested for lysis. Tissue lysates were immunoblotted for thrombomodulin (TM) and β-actin. The relative expression levels of TM were quantified. (K) Soluble TM level in the plasma of saline control or poly(I:C)-injected mice was measured by ELISA according to the manufacturer’s instructions. Dot plot shows quantification of plasma TM levels from Ctrl mice (n = 4) and poly(I:C)-treated mice (n = 3). Scale bar: 10 μm (A), 200 μm (H) and 40 μm (I). All experiments were repeated at least three times. Results are shown as mean ± standard error. *p < 0.05, **p < 0.01.
Poly(I:C) promotes inflammation in vivo
Next, we examined if poly(I:C) induces proinflammatory signaling responses in vivo. Thus, mice were injected with saline or poly(I:C) intraperitoneally (i.p.). Analysis of blood samples, collected at different time points (3, 6, and 24h) and co-stained for Ly6G-FITC & Mac-1-PE by flow cytometry revealed that poly(I:C) upregulates cell surface expression of Mac-1 on mouse neutrophils (Fig. 7G; Suppl. Fig. S3), suggesting that neutrophils are activated by poly(I:C) for subsequent adhesion to the vascular endothelium. After 3h, lung, liver and kidney tissues were collected and processed for histological analyses. Paraffin-embedded sections of lung tissue were stained with H&E. Data showed that poly(I:C) increased inflammatory foci in lungs (Fig. 7H) and confocal IF further revealed that poly(I:C) enhances leukocyte infiltration to the lung tissue (Fig. 7I).
Thrombomodulin (TM) exhibits a range of physiologically important antiinflammatory and anticoagulant properties40–42 and poly(I:C) is known to modulate TM expression43. We examined TM expression in the harvested tissues and found that poly(I:C) downregulates TM expression in liver, kidney and lung tissues (Suppl. Fig. S4). Western blot analysis of lung tissue lysates revealed a decreased TM expression level in poly(I:C)-treated mice (Fig. 7J). ELISA of the mouse plasma samples after 24h revealed that poly(I:C) reduces the circulating level of the soluble TM in the mouse plasma (Fig. 7K), which is likely due to a decreased level of cellular TM expression. We also examined EPCR expression in lung tissue lysates and did not observe significant changes after poly(I:C) treatment (Suppl. Fig. S5).
APC inhibits poly(I:C)-induced proinflammatory signaling both in vitro and in vivo
Both confocal IF and Western blot analyses showed that APC significantly inhibits poly(I:C)-mediated NF-κB activation (Fig. 8A–D) and barrier-disruptive hyper-permeability responses in both endothelial cell lines (Fig. 8E,F). As expected this function of APC was mediated through EPCR-dependent activation of PAR1 since function-blocking antibodies inhibited the barrier-protective effect of APC in endothelial cells (Suppl. Fig. S6). Flow cytometry analysis further revealed that APC and its signaling-selective APC-2Cys mutant both attenuate poly(I:C)-mediated increase in cell surface expression of Mac-1 on neutrophils in mice (Fig. 8G). H&E staining of the lung tissue demonstrated that APC inhibits poly(I:C)-induced inflammatory cell infiltration (Fig. 8H). Western blot analysis of the lung tissue lysates showed that poly(I:C) increases the expression of the neutrophil markers, including MPO and neutrophil elastase (NE) (Fig. 8I). The expression of the endothelial cell activation marker, VCAM-1, and the NETosis marker H3Cit were also significantly increased in the lung tissue lysates (Fig. 8I) and APC significantly inhibited VCAM-1 upregulation and histone H3 citrullination (Fig. 8I). These results suggest that the protective signaling function of APC protects against poly(I:C)-mediated neutrophil infiltration, endothelial cell activation and ET formation in vivo.
Figure 8. APC inhibits poly(I:C)-induced proinflammatory signaling.
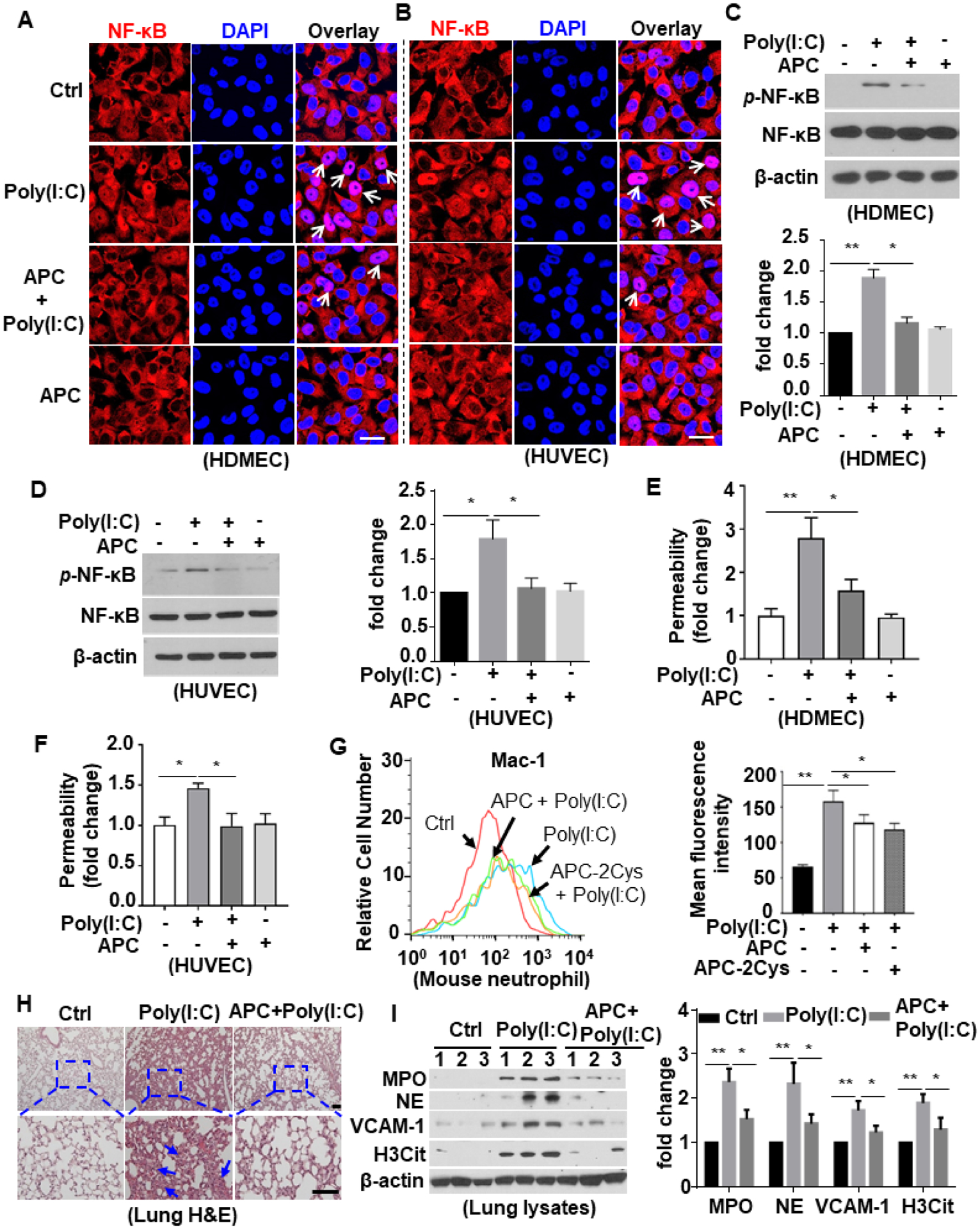
(A and B) HDMECs (A) or HUVECs (B) were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 1h) (APC remained in the media after addition of poly(I:C)). Cells were then fixed, permeabilized and NF-κB p65 was stained with rabbit anti-NF-κB antibody and Alexa Fluor 555-conjugated goat anti-rabbit IgG. The nucleus was stained with DAPI. Immunofluorescence images were taken by confocal microscopy. Arrows indicate nuclear translocation of NF-κB. (C and D) HDMECs (C) or HUVECs (D) were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 1h). Cell lysates were immunoblotted and probed with an antibody specific for phosphorylated NF-κB (p-NF-κB). β-actin was used as a loading control. (E and F) HDMECs (E) or HUVECs (F) were pretreated with APC (20 nM for 3h) followed by stimulation with poly(I:C) (10 μg/mL for 4h). The amount of Evans blue dye that leaked into the lower chamber in the Trans-well assay plates was measured. (G) Mice were injected i.p. with APC or the signaling-selective mutant of APC (APC-2Cys) 1h prior to poly(I:C) i.p. injection for 3h and blood was collected and stained for Ly6G-FITC & Mac-1-PE. The cell surface expression of Mac-1 in Ly6G-FITC-positive neutrophil population was measured by flow cytometry. Representative data were obtained from 3–5 mice per group (n = 3–5). The mean fluorescence intensity was quantified. (H and I) Mice were pretreated with APC for 1h followed by i.p. injection with poly(I:C) for 3h, and lung tissue was collected and processed for histological analyses. Paraffin-embedded sections of lung tissue were stained with H&E (H). Representative images were obtained from 5 mice per group (n = 5). The inset boxes from each group are magnified. The arrows indicate inflammatory foci. The lung tissue was harvested for lysis. Tissue lysates were immunoblotted for myeloperoxidase (MPO), neutrophil elastase (NE), VCAM-1, citrullinated H3 (H3Cit) and β-actin. The relative expression levels of these proteins were quantified (I). Scale bar: 20 μm (A and B), 200 μm (H). All experiments were repeated at least three times. Results are shown as mean ± standard error. *p < 0.05, **p < 0.01.
Discussion
In this study, we used poly (I:C) as a surrogate for dsRNA viral infection and investigated the mechanism by which viral infection may induce inflammatory responses in cellular and animal models. Moreover, since there is an unmet need for new therapeutics against viral infection, we also evaluated the beneficial effect of the antiinflammatory serine protease APC in downregulating the pathogenic effects of the viral dsRNA mimetic poly(I:C). Our results support previous findings that poly(I:C) promotes both NETs formation and inflammation in vivo. Furthermore, our results for the first time demonstrate that poly(I:C) induces histone H3 citrullination and its extranuclear translocation in vascular endothelial cells and promotes formation of extracellular traps by both macrophages and neutrophils. Our results further demonstrate that APC effectively inhibits poly(I:C)-mediated histone translocation, ET formation and inflammation in both in vitro and in vivo systems.
Toll-like receptors (TLRs) are closely related members of the pattern recognition receptor (PRR) family, which are known to be widely expressed in endothelial cells, macrophages, neutrophils and other cell types9–11. Viral dsRNA and its mimetic poly(I:C) are primarily recognized by TLR3, however, they are also ligands for RIG-I-like receptors (RLRs) including melanoma differentiation-associated gene 5 (MDA5) and retinoic acid-inducible gene 1 (RIG-1)33. Through interaction with its receptors, poly(I:C) can activate transcriptional factors NF-κB and interferon regulatory factor 3 (IRF3) signaling pathways to induce expression of inflammatory cytokines33–35. Our results demonstrated that poly(I:C) through interaction with TLR3 induces NF-κB activation, enhances expression of cell adhesion molecules and disrupts barrier-permeability function of endothelial cells and APC effectively inhibits these inflammatory responses. More importantly, our results for the first time demonstrated that poly(I:C), through interaction with TLR3, induces extranuclear translocation of histone H3 in endothelial cells. Western blot analysis of poly (I:C)-treated endothelial cell lysates detected higher levels of citrullinated histone H3 in both cell lines, suggesting that this mechanism of posttranslational modification of histones, which is known to be associated with chromatin decondensation20, 21, is responsible for the extranuclear histone translocation in these cells. APC effectively inhibited both processes of histone H3 citrullination and its extranuclear translocation in endothelial cells. The protective inhibitory function of APC was mediated through EPCR-dependent activation of PAR1. A crosstalk between PAR1 and TLR3 signaling has been reported during viral interaction33. Further studies will be required to determine whether APC, in addition to its inhibition of TLR3-dependent histone citrullination and translocation, can also downregulate expression of TLR3 in endothelial cells.
Another interesting observation of this study is the finding that poly(I:C) induces ET formation in both macrophages and neutrophils. The formation of ETs by innate immune cells is a natural defense mechanism in response to infection by pathogenic microorganisms16, 17. The process is initiated when inflammatory signaling through PRRs induces decondensation of the chromatin structure and release of the nuclear content as ETs into the extracellular spaces by activated innate immune cells, recruited to the site of infection7, 21, 44, 45. In addition to decondensed chromatin fibers, ETs are also rich in histones and proteases, like MPO and elastase which exhibit potent anti-bacterial and anti-viral activity20, thereby immobilizing and killing invading microorganisms. However, in addition to their essential role in fighting infection, excessive ET formation can also damage host tissue and are implicated in many human diseases if not regulated by physiological mechanisms18, 46–48. The observation that APC inhibited ET formation by innate immunes cells suggests that the APC pathway is possibly one such natural mechanism which is involved in regulating this process. Results of several studies have indicated that citrullination of histones regulates chromatin decondensation, thereby leading to ET formation21, 44. Our results suggest that poly(I:C) induces ET formation by citrullination of histones as evidenced by significantly elevated levels of citrullinated histone H3 both in freshly isolated human peripheral blood neutrophils. Results further suggest that APC effectively inhibits poly(I:C)-mediated extranuclear translocation of histones and MET formation in murine macrophages. Interestingly, unlike endothelial cells in which both EPCR and PAR1 receptors were required for the inhibition of histone H3 citrullination, results with function-blocking antibodies revealed that APC inhibits poly(I:C)-mediated histone H3 citrullination and subsequent macrophage ET formation through Mac-1 (CD11b/CD18)-dependent but not EPCR-dependent activation of PAR1. This result is consistent with a previous report demonstrating that Mac-1, but not EPCR is the required receptor for the PAR1-dependent protective effect of APC on human macrophages39. The nuclear enzyme PAD4 is known to be responsible for catalyzing histone citrullination that leads to unfolding of the chromatin structure and formation of NETs by neutrophils during infection21, 44. Poly(I:C) markedly upregulated expression of PAD4 in human neutrophils and induced its co-localization with histone H3. We hypothesize that the PAR1-dependent signaling function of APC inhibits histone citrullination and its subsequent NET formation by downregulating nuclear PAD4 expression in neutrophils. Nevertheless, further studies will be required to validate this hypothesis. These findings are in line with other reports demonstrating a key role for APC in downregulating inflammatory responses by epigenetic regulation of nuclear molecules5, 49.
Finally, the in vivo relevance of poly(I:C)-mediated inflammatory responses was investigated by the i.p. injection of the viral mimetic to mice with or without pre-injection with APC. Results suggest that poly(I:C) increases cell surface expression of Mac-1 on mouse neutrophils and their infiltration into the lung tissue. Interestingly, Western blot analysis of the lung tissue lysates revealed that poly(I:C) may induce NET formation in vivo as evidenced by markedly elevated levels of MPO, neutrophil elastase, VCAM1, citrullinated histone H3 and elevated expression of Mac-1 on neutrophils. In support of this hypothesis, it has been demonstrated in vivo that Mac-1 expression on neutrophils induces NET formation during hantavirus-induced inflammation50. APC effectively inhibited all of these inflammatory responses. The signaling function of APC was responsible for inhibiting NET formation since the signaling-selective APC-2Cys, similar to APC-WT, inhibited expression of Mac-1 on mice neutrophils. Taken together, these results suggest that the viral dsRNA mimetic promotes both inflammation in cellular and animal models and APC has a potent protective effect in both systems. Thus, the therapeutic potential of APC in protecting against different types of viral infection may warrant further investigation.
Supplementary Material
What is known about this topic?
APC exerts its cytoprotective signaling function by EPCR-dependent activation of PAR1 on endothelial cells and most other cell types.
APC inhibits LPS-mediated cytoplasmic translocation and extracellular release of HMGB1 from the nucleus.
Poly(I:C) is a synthetic analog of viral dsRNA that induces characteristic inflammatory responses associated with the viral infection.
What does this paper add?
Poly(I:C) induces histone H3 extranuclear translocation via interaction with TLR3 in endothelial cells and promotes extracellular trap formation in macrophages and neutrophils.
APC inhibits poly(I:C)-mediated histone H3 translocation in a EPCR- and PAR1-dependent manner in endothelial cells.
APC inhibits poly(I:C)-induced extracellular trap formation in macrophages and neutrophils.
APC inhibits poly(I:C)-mediated inflammation in mice.
Acknowledgments
We would like to thank Cindy Carter for technical assistance and Audrey Rezaie for editorial work on the manuscript.
Funding
This work was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institute of Health HL 101917 and HL 62565 to ARR.
Footnotes
Conflict of Interest
None declared.
References
- 1.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 2002;296(5574):1880–1882 [DOI] [PubMed] [Google Scholar]
- 2.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood 2007;109(8):3161–3172 [DOI] [PubMed] [Google Scholar]
- 3.Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood 2014;124(10):1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rezaie AR. Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost 2014;112(5):876–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai X, Biswas I, Panicker SR, Giri H, Rezaie AR. Activated protein C inhibits lipopolysaccharide-mediated acetylation and secretion of high-mobility group box 1 in endothelial cells. J Thromb Haemost 2019;17(5):803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bae JS, Rezaie AR. Activated protein C inhibits high mobility group box 1 signaling in endothelial cells. Blood 2011;118(14):3952–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost 2015;13 Suppl 1:S82–91 [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med 2009;15(11):1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fritz G RAGE: a single receptor fits multiple ligands. Trends Biochem Sci 2011;36(12):625–632 [DOI] [PubMed] [Google Scholar]
- 10.Ibrahim ZA, Armour CL, Phipps S, Sukkar MB. RAGE and TLRs: relatives, friends or neighbours? Mol Immunol 2013;56(4):739–744 [DOI] [PubMed] [Google Scholar]
- 11.Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) 2014;92(5):465–472 [DOI] [PubMed] [Google Scholar]
- 12.Paues Goranson S, Thalin C, Lundstrom A, et al. Circulating H3Cit is elevated in a human model of endotoxemia and can be detected bound to microvesicles. Sci Rep 2018;8(1):12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grilz E, Mauracher LM, Posch F, et al. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br J Haematol 2019;186(2):311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinarvand P, Hassanian SM, Qureshi SH, et al. Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood 2014;123(6):935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nazir S, Gadi I, Al-Dabet MM, et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 2017;130(24):2664–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303(5663):1532–1535 [DOI] [PubMed] [Google Scholar]
- 17.Chow OA, von Kockritz-Blickwede M, Bright AT, et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010;8(5):445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okubo K, Kurosawa M, Kamiya M, et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med 2018;24(2):232–238 [DOI] [PubMed] [Google Scholar]
- 19.Liu P, Wu X, Liao C, et al. Escherichia coli and Candida albicans induced macrophage extracellular trap-like structures with limited microbicidal activity. PLoS One 2014;9(2):e90042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009;184(2):205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leshner M, Wang S, Lewis C, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 2012;3:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 2013;110(21):8674–8679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 2010;207(9):1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behnen M, Leschczyk C, Moller S, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac-1. J Immunol 2014;193(4):1954–1965 [DOI] [PubMed] [Google Scholar]
- 25.Rossaint J, Herter JM, Van Aken H, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014;123(16):2573–2584 [DOI] [PubMed] [Google Scholar]
- 26.Wardini AB, Guimaraes-Costa AB, Nascimento MT, et al. Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol 2010;91(Pt 1):259–264 [DOI] [PubMed] [Google Scholar]
- 27.Narasaraju T, Yang E, Samy RP, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 2011;179(1):199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saitoh T, Komano J, Saitoh Y, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012;12(1):109–116 [DOI] [PubMed] [Google Scholar]
- 29.Schonrich G, Raftery MJ. Neutrophil Extracellular Traps Go Viral. Front Immunol 2016;7:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020;5(11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med 2020;217(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Healy LD, Puy C, Fernandez JA, et al. Activated protein C inhibits neutrophil extracellular trap formation in vitro and activation in vivo. J Biol Chem 2017;292(21):8616–8629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antoniak S, Mackman N. Multiple roles of the coagulation protease cascade during virus infection. Blood 2014;123(17):2605–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001;413(6857):732–738 [DOI] [PubMed] [Google Scholar]
- 35.Zimmer S, Steinmetz M, Asdonk T, et al. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ Res 2011;108(11):1358–1366 [DOI] [PubMed] [Google Scholar]
- 36.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci 2008;1143:1–20 [DOI] [PubMed] [Google Scholar]
- 37.Gan T, Yang Y, Hu F, et al. TLR3 Regulated Poly I:C-Induced Neutrophil Extracellular Traps and Acute Lung Injury Partly Through p38 MAP Kinase. Front Microbiol 2018;9:3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bae JS, Yang L, Manithody C, Rezaie AR. Engineering a disulfide bond to stabilize the calcium-binding loop of activated protein C eliminates its anticoagulant but not its protective signaling properties. J Biol Chem 2007;282(12):9251–9259 [DOI] [PubMed] [Google Scholar]
- 39.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest 2010;120(6):1971–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esmon CT. Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J 1995;9(10):946–955 [DOI] [PubMed] [Google Scholar]
- 41.Conway EM. Thrombomodulin and its role in inflammation. Semin Immunopathol 2012;34(1):107–125 [DOI] [PubMed] [Google Scholar]
- 42.Giri H, Cai X, Panicker SR, Biswas I, Rezaie AR. Thrombomodulin Regulation of Mitogen-Activated Protein Kinases. Int J Mol Sci 2019;20(8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shibamiya A, Hersemeyer K, Schmidt Woll T, et al. A key role for Toll-like receptor-3 in disrupting the hemostasis balance on endothelial cells. Blood 2009;113(3):714–722 [DOI] [PubMed] [Google Scholar]
- 44.Anzilotti C, Pratesi F, Tommasi C, Migliorini P. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev 2010;9(3):158–160 [DOI] [PubMed] [Google Scholar]
- 45.Funchal GA, Jaeger N, Czepielewski RS, et al. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS One 2015;10(4):e0124082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Shikh MEM, El Sayed R, Nerviani A, et al. Extracellular traps and PAD4 released by macrophages induce citrullination and auto-antibody production in autoimmune arthritis. J Autoimmun 2019;105:102297. [DOI] [PubMed] [Google Scholar]
- 47.Radermecker C, Sabatel C, Vanwinge C, et al. Locally instructed CXCR4(hi) neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat Immunol 2019;20(11):1444–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129(10):1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bock F, Shahzad K, Wang H, et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A 2013;110(2):648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raftery MJ, Lalwani P, Krautkrmer E, et al. beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J Exp Med 2014;211(7):1485–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.