

Abstract
Recently, multitargeted drugs are considered a potential approach in treating cancer. In this study, twelve in-house indole-based derivatives were preliminary evaluated for their inhibitory activities over VEGFR-2, CDK-1/cyclin B and HER-2. Compound 15l showed the most inhibitory activities among the tested derivatives over CDK-1/cyclin B and HER-2. Compound 15l was tested for its selectivity in a small kinase panel. It showed dual selectivity for CDK-1/cyclin B and HER-2. Moreover, in vitro cytotoxicity assay was assessed for the selected series against nine NCI cell lines. Compound 15l showed the most potent inhibitory activities among the tested compounds. A deep in silico molecular docking study was conducted for compound 15l to identify the possible binding modes into CDK-1/cyclin B and HER-2. The docking results revealed that compound 15l displayed interesting binding modes with the key amino acids in the binding sites of both kinases. In vitro and in silico studies demonstrate the indole-based derivative 15l as a selective dual CDK-1 and HER-2 inhibitor. This emphasizes a new challenge in drug development strategies and signals a significant milestone for further structural and molecular optimization of these indole-based derivatives in order to achieve a drug-like property.
Keywords: CDK-1/cyclin B, HER-2, anti-proliferative, anti-cancer, molecular docking, drug repurposing
1. Introduction
Cancer is a multifactorial disease defined by a number of uncontrolled cellular growth and signaling pathways. CDK-1 (cyclin-dependent kinase type 1) and HER-2 (epidermal growth factor receptor type 2) are considered important key elements in human cells since they play fundamental roles in signal transduction and cellular growth [1,2].
CDKs are among the protein kinase family which have a prominent and significant impact in cell cycle regulation [3]. They are serine–threonine kinases, which phosphorylate their substrate on serine and threonine amino acid residues [4]. CDK-1 (one member of the CDK family) builds an active form with cyclin B (CDK-1/cyclin B), which in turn lead to cell cycle progression at M phase [5,6]. Mutation and overexpression of this active complex (CDK-1/cyclin B) can lead to abnormal cell growth and proliferation [7,8]. In recent years, kinase inhibition has become a major area for therapeutic intervention to treat abnormal cell proliferation [9]. A number of CDK inhibitors (CDIs) have been developed (Figure 1). Among them, flavopiridol (1, AlvocidibTM, Sanofi) [10], a broad spectrum CDK inhibitor with activity against CDK-1, CDK-2 and CDK-4, is currently in clinical trial phase 2 as an anti-proliferative agent [11]. It was reported that CDIs compete with ATP (adenosine triphosphate) at its binding site in the CDK-1/cyclin B complex causing inhibition of phosphorylation and further suppression of other signaling components, leading finally to blocking of the other important transcriptional activators for M phase [12].
Figure 1.

CDK-1/cyclin B inhibitors.
HER-2 (ErbB-2, epidermal growth factor receptor-2) is one member of the ErbB receptor family and has an essential role in cell proliferation, survival, migration, adhesion and differentiation [13,14]. Overexpression of HER-2 leads to uncontrolled activation of the receptor and hence to cell growth in tumors [15,16]. Therefore, targeting human epidermal growth factor-2 (HER-2) is a common approach for cancer therapy [17,18]. Tyrosine kinase domain (TK) of HER-2 has been identified as an efficient target in inhibiting and suppressing overexpressed HER-2 cancer cells [19,20]. A number of inhibitors have been reported and approved by the FDA (Food and Drug Administration) to inhibit the TK domain of HER-2 through binding to the adenosine triphosphate (ATP) binding site and blocking the key triggering phosphorylation step in the cell growth signaling cascade [21] (Figure 2). Gefitinib (7, IressaTM, AstraZeneca) [22], erlotinib (8, TarcevaTM, Genentech) [23] and lapatinib (9, TykerbTM, GlaxoSmithKline) [24,25] are examples of reported HER-2 TK inhibitors. However, acquired resistance has been developed for these drugs after long term treatment [24,26]. Therefore, multitarget anticancer therapies have emerged as a promising treatment strategy through simultaneous modulation of a network of tumor-related targets, to overcome both acquired drug resistance and drug toxicity [27,28].
Figure 2.

HER-2 TK inhibitors.
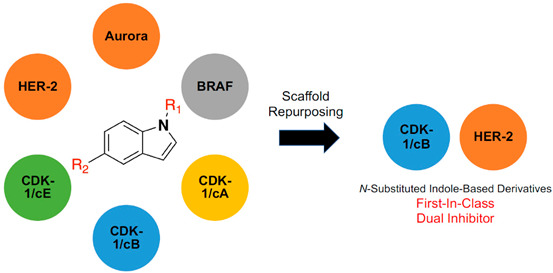
Lately, drug repurposing has been gaining significance in addressing novel clinical uses of existing therapeutic agents [29,30,31]. In the present study, our research group applied the concept of drug repurposing to twelve in-house N-substituted indole-based analogues (reported as potent inhibitors of MAO-B enzyme; monoamine oxidase type B) [32]. A literature search revealed that indole-based derivatives displayed broad kinase inhibitory activities [33,34]. In vitro enzyme assays and cytotoxicity evaluation were carried out for selected candidates to investigate their kinase inhibitory activities. In addition, a deep in silico molecular docking was conducted to investigate the binding modes into the binding site of the selected proteins.
2. Results and Discussion
2.1. Chemistry
The scale-up synthesis of the tested compounds was designed and carried out as depicted in Scheme 1. 5-nitroindole (12) was treated with dimethyl carbonate (methyl donor) in the presence of potassium carbonate as basic catalyst to afford the N-methyl derivative (13a) [35], while compounds 13b–d were synthesized by treating 5-nitroindole with appropriate aryl halide in presence of sodium hydride as a base [36,37]. Reduction of compounds 13a–d was achieved by iron in acidic medium to afford amino-based derivatives (14a–d) [38]. Final target compounds (15a–l) (Table 1 and Supplementary Table S1) were synthesized by treating amino-based derivatives (14a–d) with appropriate heteroaryl carboxylic acid using HATU as coupling agent in presence of Hunig’s base (DIPEA).
Scheme 1.

Reagents and conditions. a, dimethyl carbonate, K2CO3, DMF, reflux, 3 h (13a); b, appropriate aryl halide, NaH, DMF, 0–100 °C, 24 h (13b–d); c, Fe, NH4Cl, EtOH/H2O, reflux, 1 h; d, appropriate aryl carboxylic acid, HATU, DIPEA, DMF, mw, 116 °C, 45 min.
Table 1.
Key structure of compounds 15a–l.
| Compound | X | R | Ar |
|---|---|---|---|
| 15a | CH2 | H |
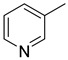
|
| 15b | CH2 | H |
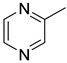
|
| 15c | CH2 | H |
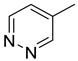
|
| 15d | CH2 |
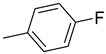
|
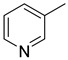
|
| 15e | CH2 |
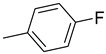
|
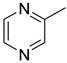
|
| 15f | CH2 |
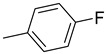
|
|
| 15g | CH2 |
|
|
| 15h | CH2 |
|
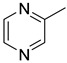
|
| 15i | CH2 |
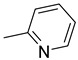
|
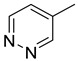
|
| 15j | C=O |
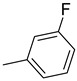
|
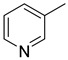
|
| 15k | C=O |
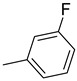
|
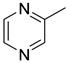
|
| 15l | C=O |
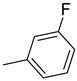
|
|
2.2. Biological Evaluation
2.2.1. In Vitro Kinase Inhibitory Assay
Preliminary biological screening was designed to test the kinase inhibitory activity of the selected library (15a–l) over three kinases (VEGFR-2; vascular endothelial growth factor receptor type 2, CDK-1/cyclin B and HER-2). The selected derivatives were tested in a single dose duplicate mode at a concentration of 10 μM. Control compound, staurosporine, was tested in 10-dose IC50 (half maximal inhibitory concentration) mode with 4-fold serial dilution starting at 20 μM. Reactions were carried out at 10 μM ATP.
The results show that the selected indole-based derivatives (15a–k) did not display any inhibitory activities over 3 kinases. However, Compound 15l (pyridazine-bearing derivative with carbonyl linker and terminal F-substituted phenyl ring) exhibited potent inhibitory activities over both CDK-1/cyclin B and HER-2 (Act% = 51% and 52%, respectively) compared to that of the tested compounds 15a–k (Act% > 83%) (Table 2).
Table 2.
Kinase activity (%) of the tested compounds (15a–l) over VEGFR-2, CDK-1/cyclin B and HER-2.
| Compound | % Activity | ||
|---|---|---|---|
| VEGFR-2 | CDK-1/cyclin B | HER-2 | |
| 15a | 96 | 91 | 95 |
| 15b | 100 | 100 | 93 |
| 15c | 100 | 91 | 99 |
| 15d | 91 | 85 | 90 |
| 15e | 98 | 88 | 86 |
| 15f | 96 | 91 | 100 |
| 15g | 100 | 92 | 83 |
| 15h | 100 | 95 | 97 |
| 15i | 85 | 89 | 87 |
| 15j | 90 | 91 | 93 |
| 15k | 90 | 92 | 92 |
| 15l | 92 | 51 | 52 |
The results suggest that the carbonyl-bearing linker along with the terminal substituted phenyl ring with F group may have a prominent role in binding into the active site of the kinase domain of CDK-1/cyclin B, as well as HER-2. This suggestion can be explained by the degree of similarity in the chemical group distribution around the main scaffold (Figure 3); HBD (hydrogen bond donor)/HBA (hydrogen bond acceptor)-bearing spacer in CDK-1/cyclin B inhibitors (2, roscovitine and 3, NU-6102) and the HBD/HBA-bearing spacer along with terminal phenyl ring with halogen substitution in HER-2 inhibitors (7, gefitinib). These two factors (HBD/HBA-bearing spacer and terminal phenyl ring with halogen substitution) can participate in the dual inhibition of both CDK-1/cyclin B and HER-2 kinase domain.
Figure 3.

Structural correlation between the most active compound (15l) and CDK-1/cyclin B inhibitors (2, roscovitine and 3, NU-6102) and HER-2 inhibitor (7, gefitinib).
To investigate the selectivity profile of the most active compound (15l), we treated this compound as in the previous protocol with a small kinase panel (aurora, BRAF V600A; V600A mutated rapidly accelerated fibrosarcoma kinase type B, HER-2, CDK-1/cyclin A, CDK-1/cyclin B and CDK-1/cyclin E) (Table 3). The results revealed that compound 15l reserved its dual and selective inhibitory activities over CDK-1/cyclin B and HER-2. It did not show any inhibitory activities over the rest of the kinase panel.
Table 3.
Kinase activity (%) of 6 kinase panel with compound 15l.
| Kinase | % Activity |
|---|---|
| Aurora | 86 |
| BRAF V600A | 100 |
| HER-2 | 52 |
| CDK-1/cyclin A | 100 |
| CDK-1/cyclin B | 51 |
| CDK-1/cyclin E | 100 |
| |
2.2.2. In Vitro Cytotoxicity Assay
All compounds under study (15a–l) were submitted to the National Cancer Institute (NCI) and selected for one-dose-testing over NCI human cancer cell lines (leukemia, non-small cell lung cancer, colon cancer, CNS (central nervous system) cancer, melanoma, ovarian cancer, renal cancer, prostate cancer and breast cancer). A few tested derivatives exhibited moderate activities over human renal cancer cell lines (Table 4 and Figure S1). Most of the tested compounds showed poor cytotoxic activities over NCI human cancer cell lines (Table S2 and Figures S2–S13). The results revealed that the A498 sub-panel renal cancer cell line was the most sensitive (G%A498 = 23%, 15d; 39%, 15f and 41%, 15h).
Table 4.
Growth % of NCI renal cancer cell lines treated with tested compounds (15a–l).
| Compound | Growth% | ||||
|---|---|---|---|---|---|
| Renal Cancer Cell Line | |||||
| A498 | CAKI-1 | RXF 393 | TK-10 | UO-31 | |
| 15a | 76 | 96 | 100 | 98 | 97 |
| 15b | 76 | 93 | 100 | 84 | 95 |
| 15c | 68 | 69 | 65 | 73 | 65 |
| 15d | 23 | 56 | 37 | 62 | 53 |
| 15e | 53 | 48 | 57 | 53 | 54 |
| 15f | 39 | 45 | 63 | 59 | 42 |
| 15g | 55 | 70 | 53 | 72 | 61 |
| 15h | 41 | 66 | 48 | 62 | 57 |
| 15i | 57 | 66 | 59 | 60 | 59 |
| 15j | 59 | 41 | 70 | 56 | 58 |
| 15k | 87 | 71 | 68 | 65 | 72 |
| 15l | 56 | 63 | 82 | 60 | 75 |
Compound 15l (the most active compound in the in vitro kinase inhibitory assay) showed moderate cytotoxic activity over renal cancer cell lines (G%A498 = 56%). The deprived cytotoxic activity of tested compounds can be referred to the poor cell permeability or drug efflux mechanism, washing the compound out rapidly. The relative sensitivity of the human renal cancer cell line can suggest a new correlation between the dual inhibition of CDK-1/cyclin B and HER-2 and their role in treating renal cancer.
2.3. In Silico Molecular Docking
A molecular docking experiment was conducted to predict the binding modes for the most active compound (15l) into the active site of the kinase domain of both CDK-1/cyclin B and HER-2 (PDB ID: 5LQF [39] and 3RCD [40], respectively). The docking experiment was carried out for the reference ligands of both kinase domains (NU-6102 (3) for CDK-1/cyclin B and TAK-285 (11) for HER-2) to validate the docking protocol. The validation step revealed that both reference ligands (3 and 11) exhibited identical binding modes, as well as binding forces with the key amino acid residues inside the binding site, for both CDK-1/cyclin B and HER-2, respectively, as reported in the protein data bank [39,40]. A virtual docking screening was conducted for compound 15l in both kinase domains.
The results revealed that compound 15l showed hypothetical binding modes with the key binding areas inside the active site (Figure 4 and Figure 5). In addition, it was seen that the main scaffold (indole-based scaffold) occupied the hinge region of both active sites with hydrophobic arene and H-bonding interactions. The right-handed aryl halide (3-fluorophenyl) moiety was directed into the back room of both kinase, while the left-handed heteroaryl ring (pyridazine ring) was revealed to the solvent-exposed surface.
Figure 4.

Binding mode of compound 15l into CDK-1/cyclin B active site (PDB ID: 5LQF); (a): 3D visualization shows the orientation of compound 15l binding motifs in the main regions of the active site; (b): graphical illustration of orientation of compound 15l binding motifs in the CDK-1/cyclin B active site. Indole-based central motif occupied hinge binding region (green color), 3-fluorophenyl ring aligned into the back groove (red color) and terminal pyridazine ring travelled out towards solvent-exposed surface (blue color).
Figure 5.

Binding mode of compound 15l into HER-2 active site (PDB ID: 3RCD); (a): 3D visualization showed the orientation of compound 15l binding motifs in the main regions of the active site; (b): graphical illustration of orientation of compound 15l binding motifs in the HER-2 active site. Indole-based central motif occupied hinge binding region (green color), 3-fluorophenyl ring aligned into the back groove (red color) and terminal pyridazine ring travelled out towards solvent-exposed surface (blue color).
It was suggested that the two linkers in compound 15l (carbonyl and amide spacers that connect the central indole motif, the terminal aryl halide ring and terminal pyridazine ring, respectively) have a significant role in modulating the position of these pharmacophoric motifs in their corresponding grooves. For example, the carbonyl spacer will force the 3-fluorophenyl ring to be directed to the back pocket. In addition, it exhibited an additional hydrogen bond interaction with Asp 86 (aspartic acid) amino acid residue in CDK-1/cyclin B active site with a distance of 2.41 Å. Furthermore, the amide spacer (2-atoms-linker) gives a suitable distance between pyridazine and indole motif to stabilize the central scaffold in the hinge region, keeping the terminal pyridazine waving out to the solvent exposed area (Table 5).
Table 5.
Virtual binding interactions of compound 15l into CDK-1/cyclin B and HER-2 kinase domain binding sites.
| Kinase Domain | Compound 15l Binding Motifs | ||
|---|---|---|---|
| Pyridazine | Indole | 3-Fluorophenyl | |
| CDK-1/cyclin B | Val 18 Lys 33 |
Ile 10 Phe 83 Leu 135 |
Met 85 Lys 89 |
| HER-2 | Gly 804 Cyc 805 Leu 852 Phe 1004 |
Val 734 Ala 751 Lys 753 Leu 796 |
Ser 783 Leu 785 Asp 863 |
Val, valine; Lys, lysine; Ile, isoleucine; Phe, phenylalanine; Leu, leucine; Met, methionine; Gly, glycine; Cyc, cysteine; Ala, alanine; Ser, serine; Asp, Aspartic acid.
3. Materials and Methods
3.1. Chemistry
The tested compounds were synthesized as reported in our previous publication [32]. The experimental method, along with spectral analysis for both key intermediates (13a–d and 14a–d) and final compounds (15a–l), are cited in the Supplementary Materials. General methods for the chemical experiments were carried out as previously reported [41,42].
3.2. Biological Evaluation
3.2.1. In Vitro Kinase Inhibitory Assay
The in vitro kinase inhibitory assay was conducted at Reaction Biology Corp. Kinase HotSpot service was used for screening the tested derivatives. The experiment protocol is reported in Reaction Biology Corp. website (www.reactionbiology.com, accessed on 10 March 2021) and described in detail in our previous reports [43,44].
3.2.2. In Vitro Cytotoxicity Assay
The in vitro cytotoxic screening over nine NCI human cancer cell lines was conducted at the National Cancer Institute (NCI), Bethesda, MD, USA (dtp.cancer.gov), applying their standard protocol (dtp.cancer.gov/discovery_development/nci-60/methodology.htm, accessed on 5 April 2021), which was followed in earlier reports [45,46].
3.3. In Silico Molecular Docking
Molecular docking study was conducted using Discovery Studio Client 21.1.0.20298 (Biovia Corp) (www.3ds.com, accessed on 7 April 2021). The X-ray structure of kinase domain of both CDK-1/cyclin B and HER-2 was downloaded from protein data bank (www.rcsb.org, assessed on 7 April 2021) (PDB ID: 5LQF [39] and 3RCD [40], respectively). Non-essential chains as well as water molecules were removed keeping only one kinase domain with its corresponding reference ligand. The bonds and bond orders of the amino acid chain were checked and corrected. The terminal residues were checked and adjusted.
CHARMm (chemistry at Harvard macromolecular mechanics) forcefield was applied to the protein domain. Momany-Rone forcefield was selected for the partial charge. The binding site was defined using the coordinates of the reference ligand. CDOCKER algorithm (CHARMm-based molecular dynamic scheme) [47] was used in this study to generate the most stable conformers with the binding sites. Random ligand conformations were generated (10 conformers) from the initial ligand structure through dynamic target temperature (100 K) followed by random rotations. The generated conformers were refined by grid-based (GRID-1) simulated annealing.
The docking protocol was validated by running an initial docking experiment (pre-docking) for the reference ligands (3, NU-6102 for PDB ID: 5lQF and 11, TAK-285 for PDB ID: 3RCD). The docking experiment of compound 15l was conducted to generate the 10 most possible conformers with the binding site. The generated conformers were visualized to investigate the binding interaction between the tested compound (15l) and key amino acid residues in the active site of both kinase domains (CDK-1/cyclin B and HER-2).
4. Conclusions
A group of indole-based in-house derivatives (15a–l) was screened biologically over a small kinase panel. Compound 15l (with terminal 3-fluorophenyl and pyridazine rings along with carbonyl and amide connected linkers to indole moiety, respectively) showed the most potent dual inhibitory activities among the tested compounds over CDK-1/cyclin B and HER-2. In vitro cytotoxic activity was evaluated to the selected compounds over NCI human cancer cell lines. Limited lipophilicity of the tested compounds may account for the poor cell induction, and hence leads to the low cytotoxic activities. However, the tested series showed moderate cytotoxic activities over human renal cancer cell lines (A498). An in silico molecular docking study was carried out to investigate the possible hypothetical binding modes of compound 15l (the most active compound among the tested series) into both CDK-1/cyclin B and HER-2. Compound 15l exhibited interesting binding mode in the kinase binding sites. The indole central motif occupied the hinge regions of both kinases. Terminal 3-fluorophenyl ring was directed back to the hydrophobic pocket. Pyridazine ring was pointed out to the solvent exposure area. In vitro and in silico studies afforded the indole-based derivatives as a selective dual CDK-1 and HER-2 inhibitor. This can be considered as a potential scaffold for further drug development strategies including both structural and molecular optimization.
Acknowledgments
Ahmed Elkamhawy extends his appreciation to Korea Institute of Science and Technology (KIST) for supporting this work through “2021 KIST School partnership project” and in the accomplishment of this project, he would like to thank the Technology Innovation Commercial Office (TICO) at Mansoura University for their highly effective contribution. Magda H. Abdellattif thanks Taif University Researchers Project number (TURSP-2020/91), Taif University, Taif, Saudi Arabia.
Supplementary Materials
1HNMR, 13CNMR, purity and HRMS data of the compounds tested in this study, in addition to the detailed in vitro cytotoxicity data and NCI reports. Table S1: SMILES codes for the final target compounds 15a–l, Table S2: Mean growth % of tested compounds 15a–l over NCI human cancer cell lines, Figure S1: Graphical representation of in vitro cytotoxic evaluation of compounds 15a–l over NCI human cancer cell lines. Y-axis, mean % inhibition; X-axis, target compounds. The graph shows that the renal cancer cell lines (deep blue) is more sensitive to the tested compounds compared to other NCI cancer cell lines, Figure S2: One dose mean growth % of compound 15a over NCI human cancer cell lines, Figure S3: One dose mean growth % of compound 15b over NCI human cancer cell lines, Figure S4: One dose mean growth % of compound 15c over NCI human cancer cell lines, Figure S5: One dose mean growth % of compound 15d over NCI human cancer cell lines, Figure S6: One dose mean growth % of compound 15e over NCI human cancer cell lines, Figure S7: One dose mean growth % of compound 15f over NCI human cancer cell lines, Figure S8: One dose mean growth % of compound 15g over NCI human cancer cell lines, Figure S9: One dose mean growth % of compound 15h over NCI human cancer cell lines, Figure S10: One dose mean growth % of compound 15i over NCI human cancer cell lines, Figure S11: One dose mean growth % of compound 15j over NCI human cancer cell lines, Figure S12: One dose mean growth % of compound 15k over NCI human cancer cell lines, Figure S13: One dose mean growth % of compound 15l over NCI human cancer cell lines.
Author Contributions
Conceptualization, A.E. and U.M.A.; methodology, A.E., U.M.A., S.P. and M.H.E.; validation, M.H.A.; K.L. and E.J.R.; formal analysis, A.E., U.M.A., S.P. and M.H.E.; investigation, M.H.A.; K.L. and E.J.R.; resources, K.L. and E.J.R.; data curation, A.E. and U.M.A.; writing—original draft preparation, U.M.A.; writing—review and editing, A.E. and U.M.A.; visualization, U.M.A.; supervision, E.J.R.; project administration, A.E.; funding acquisition, K.L. and E.J.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the KIST Institutional programs (Grant No. 2E31140) from Korea Institute of Science and Technology, the Creative Fusion Research Program through the Creative Allied Project funded by the National Research Council of Science & Technology (CAP-12-1-KIST), and the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2018R1A5A2023127).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 15a–l are available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Reddy V.G., Reddy T.S., Nayak V.L., Prasad B., Reddy A.P., Ravikumar A., Taj S., Kamal A. Design, synthesis and biological evaluation of N-((1-benzyl-1 H-1,2,3-triazol-4-yl)methyl)-1,3-diphenyl-1 H -pyrazole-4-carboxamides as CDK1/Cdc2 inhibitors. Eur. J. Med. Chem. 2016;122:164–177. doi: 10.1016/j.ejmech.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 2.Sun M., Jia J., Sun H., Wang F. Design and synthesis of a novel class EGFR/HER2 dual inhibitors containing tricyclic oxazine fused quinazolines scaffold. Bioorg. Med. Chem. Lett. 2020;30:127045. doi: 10.1016/j.bmcl.2020.127045. [DOI] [PubMed] [Google Scholar]
- 3.Harper J., Adams P. Cyclin-dependent kinases. Chem. Rev. 2001;101:2511–2526. doi: 10.1021/cr0001030. [DOI] [PubMed] [Google Scholar]
- 4.Hall M., Peters G. Genetic Alterations of Cyclins, Cyclin-Dependent Kinases, and Cdk Inhibitors in Human Cancer. Adv. Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 5.Fischer P.M., Gianella-Borradori A. Recent progress in the discovery and development of cyclin-dependent kinase inhibitors. Expert Opin. Investig. Drugs. 2005;14:457–477. doi: 10.1517/13543784.14.4.457. [DOI] [PubMed] [Google Scholar]
- 6.Benson C., Kaye S., Workman P., Garrett M., Walton M., De Bono J. Clinical anticancer drug development: Targeting the cyclin-dependent kinases. Br. J. Cancer. 2005;92:7–12. doi: 10.1038/sj.bjc.6602229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cicenas J., Valius M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011;137:1409–1418. doi: 10.1007/s00432-011-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esposito L., Indovina P., Magnotti F., Conti D., Giordano A. Anticancer Therapeutic Strategies Based on CDK Inhibitors. Curr. Pharm. Des. 2013;19:5327–5332. doi: 10.2174/13816128113199990377. [DOI] [PubMed] [Google Scholar]
- 9.Spanò V., Pennati M., Parrino B., Carbone A., Montalbano A., Cilibrasi V., Zuco V., Lopergolo A., Cominetti D., Diana P., et al. Preclinical Activity of New [1,2]Oxazolo[5,4-e]isoindole Derivatives in Diffuse Malignant Peritoneal Mesothelioma. J. Med. Chem. 2016;59:7223–7238. doi: 10.1021/acs.jmedchem.6b00777. [DOI] [PubMed] [Google Scholar]
- 10.Carlson B.A., Dubay M.M., Sausville E.A., Brizuela L., Worland P.J. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56:2973–2978. [PubMed] [Google Scholar]
- 11.Zhai S., Senderowicz A.M., Sausville E.A., Figg W.D. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in clinical development. Ann. Pharmacother. 2002;36:905–911. doi: 10.1345/aph.1A162. [DOI] [PubMed] [Google Scholar]
- 12.Sedlacek H. Mechanisms of action of flavopiridol. Crit. Rev. Oncol. 2001;38:139–170. doi: 10.1016/S1040-8428(00)00124-4. [DOI] [PubMed] [Google Scholar]
- 13.Ciardiello F., Tortora G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 14.Citri A., Yarden Y. EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 15.Zou M., Li J., Jin B., Wang M., Chen H., Zhang Z., Zhang C., Zhao Z., Zheng L. Design, synthesis and anticancer evaluation of new 4-anilinoquinoline-3-carbonitrile derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg. Chem. 2021;114:105200. doi: 10.1016/j.bioorg.2021.105200. [DOI] [PubMed] [Google Scholar]
- 16.Yin S., Tang C., Wang B., Zhang Y., Zhou L., Xue L., Zhang C. Design, synthesis and biological evaluation of novel EGFR/HER2 dual inhibitors bearing a oxazolo[4,5-g]quinazolin-2(1H)-one scaffold. Eur. J. Med. Chem. 2016;120:26–36. doi: 10.1016/j.ejmech.2016.04.072. [DOI] [PubMed] [Google Scholar]
- 17.Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 18.Traxler P. Tyrosine kinases as targets in cancer therapy–successes and failures. Expert Opin. Ther. Targets. 2003;7:215–234. doi: 10.1517/14728222.7.2.215. [DOI] [PubMed] [Google Scholar]
- 19.Gschwind A., Fischer O.M., Ullrich A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 20.Reid A., Vidal L., Shaw H., de Bono J. Dual inhibition of ErbB1 (EGFR/HER1) and ErbB2 (HER2/neu) Eur. J. Cancer. 2007;43:481–489. doi: 10.1016/j.ejca.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Camidge D.R., Pao W., Sequist L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014;11:473–481. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 22.Barker A.J., Gibson K.H., Grundy W., Godfrey A.A., Barlow J.J., Healy M.P., Woodburn J.R., Ashton S.E., Curry B.J., Scarlett L., et al. Studies leading to the identification of ZD1839 (iressa™): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001;11:1911–1914. doi: 10.1016/S0960-894X(01)00344-4. [DOI] [PubMed] [Google Scholar]
- 23.Pao W., Miller V.A., Politi K.A., Riely G.J., Somwar R., Zakowski M.F., Kris M.G., Varmus H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valabrega G., Montemurro F., Aglietta M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007;18:977–984. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 25.Rusnak D.W., Lackey K., Affleck K., Wood E.R., Alligood K.J., Rhodes N., Keith B.R., Murray D.M., Knight W.B., Mullin R.J., et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 26.Kang S.H., Kang K.W., Kim K.-H., Kwon B., Kim S.-K., Lee H.-Y., Kong S.-Y., Lee E.S., Jang S.-G., Yoo B.C. Upregulated HSP27 in human breast cancer cells reduces Herceptin susceptibility by increasing Her2 protein stability. BMC Cancer. 2008;8:286. doi: 10.1186/1471-2407-8-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma H., Huang B., Zhang Y. Recent advances in multitarget-directed ligands targeting G-protein-coupled receptors. Drug Discov. Today. 2020;25 doi: 10.1016/j.drudis.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Sanea M.M., Elkamhawy A., Paik S., Lee K., El Kerdawy A.M., Abbas B.S.N., Roh E.J., Eldehna W.M., Elshemy H.A., Bakr R.B., et al. Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorg. Med. Chem. 2020;28:115525. doi: 10.1016/j.bmc.2020.115525. [DOI] [PubMed] [Google Scholar]
- 29.Aubé J. Drug Repurposing and the Medicinal Chemist. ACS Med. Chem. Lett. 2012;3:442–444. doi: 10.1021/ml300114c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oprea T.I., Bauman J.E., Bologa C.G., Buranda T., Chigaev A., Edwards B.S., Jarvik J.W., Gresham H.D., Haynes M.K., Hjelle B., et al. Drug repurposing from an academic perspective. Drug Discov. Today Ther. Strat. 2011;8:61–69. doi: 10.1016/j.ddstr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang S., Lee J.M., Jeon B., Elkamhawy A., Paik S., Hong J., Oh S.-J., Paek S.H., Lee C.J., Hassan A.H., et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018;151:186–198. doi: 10.1016/j.ejmech.2018.03.055. [DOI] [PubMed] [Google Scholar]
- 32.Elkamhawy A., Paik S., Kim H.J., Park J.-H., Londhe A.M., Lee K., Pae A.N., Park K.D., Roh E.J. Discovery of N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide: A novel, selective, and competitive indole-based lead inhibitor for human monoamine oxidase B. J. Enzym. Inhib. Med. Chem. 2020;35:1568–1580. doi: 10.1080/14756366.2020.1800666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaware N., Kisliuk R., Bastian A., Ihnat M.A., Gangjee A. Synthesis and evaluation of 5-(arylthio)-9 H -pyrimido[4,5- b ]indole-2,4-diamines as receptor tyrosine kinase and thymidylate synthase inhibitors and as antitumor agents. Bioorg. Med. Chem. Lett. 2017;27:1602–1607. doi: 10.1016/j.bmcl.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pecnard S., Hamze A., Pozzo J.-L., Alami M., Provot O. Synthesis of Oxazino[4,3-a]indoles and biological applications. Eur. J. Med. Chem. 2021;224:113728. doi: 10.1016/j.ejmech.2021.113728. [DOI] [PubMed] [Google Scholar]
- 35.Jiang X., Tiwari A., Thompson M., Chen Z., Cleary T.P., Lee T.B.K. A Practical Method forN-Methylation of Indoles Using Dimethyl Carbonate. Org. Process. Res. Dev. 2001;5:604–608. doi: 10.1021/op0102215. [DOI] [Google Scholar]
- 36.Smaill J.B., Gonzales A.J., Spicer J.A., Lee H., Reed J.E., Sexton K., Althaus I.W., Zhu T., Black S.L., Blaser A., et al. Tyrosine Kinase Inhibitors. 20. Optimization of Substituted Quinazoline and Pyrido[3,4-d]pyrimidine Derivatives as Orally Active, Irreversible Inhibitors of the Epidermal Growth Factor Receptor Family. J. Med. Chem. 2016;59:8103–8124. doi: 10.1021/acs.jmedchem.6b00883. [DOI] [PubMed] [Google Scholar]
- 37.Li B., Ma J., Xie W., Song H., Xu S., Wang B. Regioselective C2 Oxidative Olefination of Indoles and Pyrroles through Cationic Rhodium(III)-Catalyzed C-H Bond Activation. Chem. A Eur. J. 2013;19:11863–11868. doi: 10.1002/chem.201301987. [DOI] [PubMed] [Google Scholar]
- 38.Ramadas K., Srinivasan N. Iron-Ammonium Chloride—A Convenient and Inexpensive Reductant. Synth. Commun. 1992;22:3189–3195. doi: 10.1080/00397919208021132. [DOI] [Google Scholar]
- 39.Coxon C.R., Anscombe E., Harnor S.J., Martin M.P., Carbain B., Golding B.T., Hardcastle I.R., Harlow L.K., Korolchuk S., Matheson C.J., et al. Cyclin-Dependent Kinase (CDK) Inhibitors: Structure–Activity Relationships and Insights into the CDK-2 Selectivity of 6-Substituted 2-Arylaminopurines. J. Med. Chem. 2017;60:1746–1767. doi: 10.1021/acs.jmedchem.6b01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishikawa T., Seto M., Banno H., Kawakita Y., Oorui M., Taniguchi T., Ohta Y., Tamura T., Nakayama A., Miki H., et al. Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo[3,2-d]pyrimidine Scaffold. J. Med. Chem. 2011;54:8030–8050. doi: 10.1021/jm2008634. [DOI] [PubMed] [Google Scholar]
- 41.Farag A.K., Elkamhawy A., Londhe A.M., Lee K.-T., Pae A.N., Roh E.J. Novel LCK/FMS inhibitors based on phenoxypyrimidine scaffold as potential treatment for inflammatory disorders. Eur. J. Med. Chem. 2017;141:657–675. doi: 10.1016/j.ejmech.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Elkamhawy A., Farag A.K., Viswanath A.N.I., Bedair T., Leem D.G., Lee K.-T., Pae A.N., Roh E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg. Med. Chem. Lett. 2015;25:5147–5154. doi: 10.1016/j.bmcl.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 43.Elkamhawy A., Park J.-E., Cho N.-C., Sim T., Pae A.N., Roh E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzym. Inhib. Med. Chem. 2015;31:158–166. doi: 10.3109/14756366.2015.1004057. [DOI] [PubMed] [Google Scholar]
- 44.Elkamhawy A., Paik S., Hassan A.H., Lee Y.S., Roh E.J. Hit discovery of 4-amino- N -(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: A novel EGFR inhibitor from a designed small library. Bioorg. Chem. 2017;75:393–405. doi: 10.1016/j.bioorg.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Al-Sanea M.M., Elkamhawy A., Zakaria A., Park B.S., Kwon Y., Lee S.H., Lee S.W., Kim I.T. Synthesis and in Vitro Screening of Phenylbipyridinylpyrazole Derivatives as Potential Antiproliferative Agents. Molecules. 2015;20:1031–1045. doi: 10.3390/molecules20011031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elkamhawy A., Al-Sanea M.M., Song C., Sim T., Roh E.J. Design and Synthesis of New [1,2,3]Triazolo[4,5-d]pyrimidine Derivatives as Potential Antiproliferative Agents. Bull. Korean Chem. Soc. 2015;36:1863–1873. doi: 10.1002/bkcs.10363. [DOI] [Google Scholar]
- 47.Wu G., Robertson D.H., Brooks C.L., III, Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.