

Abstract
Background
Many studies have recently been conducted to assess the antidepressant efficacy of glutamate modification in mood disorders. This is an update of a review first published in 2015 focusing on the use of glutamate receptor modulators in unipolar depression.
Objectives
To assess the effects ‐ and review the acceptability and tolerability ‐ of ketamine and other glutamate receptor modulators in alleviating the acute symptoms of depression in people with unipolar major depressive disorder.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), Ovid MEDLINE, Embase and PsycINFO all years to July 2020. We did not apply any restrictions to date, language or publication status.
Selection criteria
Double‐ or single‐blinded randomised controlled trials (RCTs) comparing ketamine, memantine, esketamine or other glutamate receptor modulators with placebo (pill or saline infusion), other active psychotropic drugs, or electroconvulsive therapy (ECT) in adults with unipolar major depression.
Data collection and analysis
Three review authors independently identified studies, assessed trial quality and extracted data. The primary outcomes were response rate (50% reduction on a standardised rating scale) and adverse events. We decided a priori to measure the efficacy outcomes at different time points and run sensitivity/subgroup analyses. Risk of bias was assessed using the Cochrane tool, and certainty of the evidence was assessed using GRADE.
Main results
Thirty‐one new studies were identified for inclusion in this updated review. Overall, we included 64 studies (5299 participants) on ketamine (31 trials), esketamine (9), memantine (5), lanicemine (4), D‐cycloserine (2), Org26576 (2), riluzole (2), atomoxetine (1), basimglurant (1), citicoline (1), CP‐101,606 (1), decoglurant (1), MK‐0657 (1), N‐acetylcysteine (1), rapastinel (1), and sarcosine (1).
Forty‐eight studies were placebo‐controlled, and 48 were two‐arm studies. The majority of trials defined an inclusion criterion for the severity of depressive symptoms at baseline: 29 at least moderate depression; 17 severe depression; and five mild‐to‐moderate depression. Nineteen studies recruited only patients with treatment‐resistant depression, defined as inadequate response to at least two antidepressants.
The majority of studies investigating ketamine administered as a single dose, whilst all of the included esketamine studies used a multiple dose regimen (most frequently twice a week for four weeks). Most studies looking at ketamine used intravenous administration, whilst the majority of esketamine trials used intranasal routes.
The evidence suggests that ketamine may result in an increase in response and remission compared with placebo at 24 hours odds ratio (OR) 3.94, 95% confidence interval (CI) 1.54 to 10.10; n = 185, studies = 7, very low‐certainty evidence). Ketamine may reduce depression rating scale scores over placebo at 24 hours, but the evidence is very uncertain (standardised mean difference (SMD) ‐0.87, 95% CI ‐1.26 to ‐0.48; n = 231, studies = 8, very low‐certainty evidence). There was no difference in the number of participants assigned to ketamine or placebo who dropped out for any reason (OR 1.25, 95% CI 0.19 to 8.28; n = 201, studies = 6, very low‐certainty evidence).
When compared with midazolam, the evidence showed that ketamine increases remission rates at 24 hours (OR 2.21, 95% CI 0.67 to 7.32; n = 122,studies = 2, low‐certainty evidence). The evidence is very uncertain about the response efficacy of ketamine at 24 hours in comparison with midazolam, and its ability to reduce depression rating scale scores at the same time point (OR 2.48, 95% CI 1.00 to 6.18; n = 296, studies = 4,very low‐certainty evidence). There was no difference in the number of participants who dropped out of studies for any reason between ketamine and midazolam (OR 0.33, 95% CI 0.05 to 2.09; n = 72, studies = 1, low‐certainty evidence).
Esketamine treatment likely results in a large increase in participants achieving remission at 24 hours compared with placebo (OR 2.74, 95% CI 1.71 to 4.40; n = 894, studies = 5, moderate‐certainty evidence). Esketamine probably results in decreases in depression rating scale scores at 24 hours compared with placebo (SMD ‐0.31, 95% CI ‐0.45 to ‐0.17; n = 824, studies = 4, moderate‐certainty evidence). Our findings show that esketamine increased response rates, although this evidence is uncertain (OR 2.11, 95% CI 1.20 to 3.68; n = 1071, studies = 5, low‐certainty evidence). There was no evidence that participants assigned to esketamine treatment dropped out of trials more frequently than those assigned to placebo for any reason (OR 1.58, 95% CI 0.92 to 2.73; n = 773, studies = 4,moderate‐certainty evidence).
We found very little evidence for the remaining glutamate receptor modulators.
We rated the risk of bias as low or unclear for most domains, though lack of detail regarding masking of treatment in the studies reduced our certainty in the effect for all outcomes.
Authors' conclusions
Our findings show that ketamine and esketamine may be more efficacious than placebo at 24 hours. How these findings translate into clinical practice, however, is not entirely clear. The evidence for use of the remaining glutamate receptor modulators is limited as very few trials were included in the meta‐analyses for each comparison and the majority of comparisons included only one study.
Long term non‐inferiority RCTs comparing repeated ketamine and esketamine, and rigorous real‐world monitoring are needed to establish comprehensive data on safety and efficacy.
Plain language summary
Ketamine and other glutamate receptor modulators for depression in adults
Why is this review important?
Depression is one of the most common mental disorders, estimated to affect 350 million people worldwide. Antidepressant medication tends to be given as a first treatment for people with major depression. These drugs are however only effective in about one in four people at one year. Effective alternative medications to treat depression are needed, especially for rapid treatment. A new group of medications is called ‘glutamate receptor modulators’, which act on the glutamergic system. This group includes the medicine ketamine. In this review we examined the evidence for glutamate receptor modulators, including ketamine, as a treatment for depression.
Who will be interested in this review?
‐ People with depression, their friends and families.
‐ General practitioners, psychiatrists, psychologists and pharmacists.
‐ Professionals working in adult mental health services.
What questions does this review aim to answer?
1. Is treatment with ketamine and other glutamate receptor modulators more effective than treatment with placebo (dummy pill) or other drugs?
2. Is treatment with ketamine and other glutamate receptor modulators more acceptable than placebo or other drugs?
Which studies were included in the review?
We searched medical databases to find all relevant studies (specifically randomised controlled trials) completed up to 30 July 2020. To be included in the review, studies had to compare ketamine or other glutamate receptor modulators with placebo, other medicines or electroconvulsive therapy (ECT) for depression in adults (aged 18 and over). The studies also had to be single‐blind (the participant does not know which treatment they are receiving) or double‐blind (neither the participant or researcher know which treatment the participant is receiving), to attempt to reduce bias. We included 64 studies in the review, involving a total of 5299 people. The studies investigated16 different glutamate receptor modulator medications. The majority of participants had treatment‐resistant depression (depression which had not responded to two or more medications) at the start of the studies. Most studies were two‐armed, where the glutamate receptor modulator was compared with one other intervention.
What does the evidence from the review tell us?
Among the 16 drugs included in this review, only ketamine and esketamine were more effective than placebo at reducing symptoms of depression. The effects of ketamine lasted no more than one week after treatment and clearly disappeared after two weeks. Ketamine did, however, cause more side effects than placebo. The effects of esketamine were seen at 24 hours and could last up to four weeks with repeated doses. Esketamine caused a lot more side effects than placebo. The certainty of evidence varied considerably.
There was no evidence of a difference between the other glutamate receptor modulators included in this review and placebo or other medications.
What should happen next?
Ketamine and esketamine appear to reduce the symptoms of depression. However, the trials were all short term so we do not know about the long‐term effects. It is important to note that in some trials attempts to prevent participants and investigators from knowing what medicine was being given were not successful and this may have inflated the positive effects of the active drugs.
Future studies should examine what happens when people are repeatedly given the drug, with the aim to mimic the real‐world practice and assess longer‐term effects. More non‐inferiority trials should be conducted, where glutamate receptor modulators are compared with other active medications rather than placebo to find out whether they are better than alternative treatments.
In most of the ketamine trials in this review, participants were given the drug by injection into a vein. This restricts the wide‐scale application of ketamine in clinical settings, so trials of ketamine by other routes are needed. Esketamine trials usually used nasal sprays, which are easier to use and could potentially be taken at home if further monitoring and trials found that it was safe to do so. Further studies assessing administration are needed in order to draw more reliable and firm conclusions.
Summary of findings
Background
Description of the condition
Major depressive disorder is among the most commonly encountered psychiatric disorder, with reported lifetime and one‐year prevalence rates of 10.8% and 7.2%, respectively (Lim 2018) . Although an episode of depression may happen only once over a person's span, more commonly it is a recurrent condition. During an episode, symptoms are present most of the day, nearly every day, and may include feelings of sadness, emptiness, or unhappiness; loss of interest and pleasure in normal activities; sleep disturbances; tiredness and lack of energy; changes in appetite; frequent thoughts of death; suicidal thoughts; cognitive impairment; and unexplained physical problems. Major depressive disorder is diagnosed clinically by the presence of one or more major depressive episodes, in the absence of manic or hypomanic symptoms, and is also referred to as ’unipolar depression’ (APA 2013; WHO 2008a). Currently estimated to affect 350 million people worldwide, the disorder has been increasingly recognised as a major global health concern (De Leo 2014; WHO 2012), leading to substantial disability (WHO 2008b), impaired quality of life (Rapaport 2005), and considerable economic burden (Donohue 2007). Moreover, depressive illness is associated with an increased risk of suicide (Hawton 2009). Despite the clinical importance of depression, its underlying pathophysiology is still incompletely understood, with various factors suggested to be involved, as well as to serve as potential targets for treatment (Hasler 2010a). One of the most well‐researched theories of previous decades has been the monoamine hypothesis of depression, implying a dysregulation of the 5‐hydroxytryptamine (5‐HT, serotonin), noradrenaline, and dopamine systems (Coppen 1967; Hirschfeld 2000). However, even though robust evidence supports the idea that monoamine neurotransmitters, and serotonin in particular, have a role in the pathophysiology of depression, it appears that simple monoamine depletion is insufficient to account for the development of the disorder (Ruhe 2007).
Description of the intervention
The mainstay of the pharmacological treatment of depression for the last 40 or more years has been monoamine potentiating antidepressants. Tricyclic antidepressants (TCAs) were introduced in the 1950s, the first being imipramine (NICE 2009). The mode of action thought to be responsible for the mood‐elevating properties of this class of drugs is their ability to block the synaptic reuptake of noradrenaline and 5‐HT, exerting re‐uptake of these neurotransmitters at different levels. Although the introduction of the TCAs was welcome, their ability to blockade cholinergic, histaminergic, and other receptor systems resulted in side effects that reduced the acceptability of the drugs. Most TCAs were also potentially lethal in overdose. In response to this, new classes of antidepressants have been developed, including the selective serotonin reuptake inhibitors and related drugs; and also a range of other pharmacologically unrelated antidepressants, like mirtazapine or trazodone. The side effect profile of these agents varies considerably, although their mood‐elevating effects are again thought to be mediated through increasing intrasynaptic levels of monoamines, some primarily affecting noradrenaline, some 5‐HT, and others affecting both noradrenaline and 5‐HT to varying degrees and in different ways (NICE 2009). Generally, they have an improved safety profile relative to TCAs. In addition to monoamines, various other neurotransmitters have been implicated in the pathogenesis of depression, including the amino acid neurotransmitters, ƴ‐aminobutyric acid (GABA) and glutamate. While decreased plasma levels of GABA have been demonstrated in depressed patients (Petty 1984), results from studies using magnetic resonance spectroscopy (MRS) to measure GABA levels in the brain have been less consistent. Overall, however, it seems likely that brain GABA levels in depression measured by MRS are decreased in depressed patients (Godfrey 2018). Generally, drugs that increase GABA activity, for example, benzodiazepines (Birkenhager 1995) are not thought to be effective antidepressants. Nevertheless, there is recent interest in the rapid antidepressant effect of a GABA‐modulating neurosteroid, brexanolone, which has been licensed for the treatment of post‐partum depression (Zheng 2019). The discovery of the rapid antidepressant effects of the N‐methyl‐D‐aspartate (NMDA) receptor antagonist, ketamine, has been a great stimulus for investigations into the role of glutamatergic mechanisms in the pathophysiology of depression and its treatment. Magnetic resonance spectroscopy studies of glutamate again are somewhat inconsistent. Overall there may be a decrease in glutamate levels in frontal brain regions in depressed patients (Moriguchi 2019). However, some patient groups appear to have elevated glutamate metabolism in subcortical regions (Godlewska 2018). Post‐mortem findings of glutamate levels in people dying with depression are also inconsistent (Moriguchi 2019). Post‐mortem and in vivo positron emission tomography (PET) imaging evidence more reliably indicate a reduction in cortical and subcortical binding of the mGluR5 receptor, a metabotropic glutamate receptor (Moriguchi 2019). In addition, post‐mortem studies also reveal loss of glial cells in the medial frontal cortex of patients with depression. Glial cells play a key role in the metabolism and synthesis of neuronal glutamate and their loss would have a significant impact on glutamate cycling (Cotter 2001). The first randomised cross‐over trial demonstrating antidepressant efficacy of a sub‐anaesthetic dose of ketamine (0.5mg/kg) took place in seven depressed patients with evidence of a fast (onset within 24 hours) antidepressant effect (Berman 2000). Since then, researchers have attempted to supplement these findings, mainly by increasing the size of the study population, as well as studying longer‐term effects like durability of benefit following repeated infusions (Diamond 2014; Murrough 2013; Valentine 2011; Zarate 2006a). Esketamine, the s‐enantiomer of ketamine, has recently been licensed for the treatment of resistant depression, following the completion of both acute and maintenance treatment trials. In these studies, nasal esketamine was usually administered once or twice weekly. Similar to intravenous ketamine, nasal esketamine requires administration in a supervised clinical setting.
How the intervention might work
The main pharmacological mechanism of action of ketamine is non‐competitive blockade of the ion channel associated with NMDA receptor complex. However, other drugs with an apparently similar pharmacological profile, for example, memantine, are not apparently effective antidepressants (Zarate 2006b). Therefore, other factors must be involved in ketamine’s antidepressant effect. The currently favoured hypothesis is that blockade of NMDA receptors on inhibitory GABA neurones leads to a glutamate ‘surge’ which then activates 2‐amino‐3‐ (5‐methyl‐3‐oxo‐1,2‐oxazol‐4‐yl)propanoic acid (AMPA) receptors. Simulation of AMPA receptors leads to increased neuroplasticity, with elevated levels of brain‐derived neurotrophic factor (BDNF) and phosphorylation of tropomyosin receptor kinase B (TrkB) (Wilkinson 2019). Another suggested downstream effector of ketamine is the mammalian target of rapamycin (mTOR) pathway (Li 2010). Activation of the mTOR pathway by ketamine in a rat model has resulted in both an antidepressant effect and formation of spine synapses in the prefrontal cortex, whereas blockade of this pathway abolished this response (Li 2010). An unexplained, contradictory finding which has not yet been replicated is that in depressed patients, blockade of mTOR with rapamycin actually enhanced the antidepressant response to ketamine (Abdallah 2018). Ketamine also has some effects on opiate receptors and one study has shown that pre‐treatment with the opiate receptor blocker, naltrexone, prevented the antidepressant effect of ketamine, suggesting a possible role for opiate mechanisms in its antidepressant action (Williams 2018), although contradictory evidence has also been found in a pilot study (Yoon 2019). Thus, the precise way in which ketamine relieves depressive symptoms is not clear. Ketamine also has several disadvantages in its clinical use as an antidepressant, such as the risk of transient dissociative states following acute administration. There are also concerns about potential adverse effects during longer‐term maintenance treatment; for example, tolerance, dependence, adverse cognitive effects and bladder toxicity. The surprising antidepressant efficacy of ketamine together with its disadvantages had led to the search for other glutamate modifying drugs as antidepressants. This includes well‐known compounds such as riluzole and d‐cycloserine, as well as agents newly discovered by Industry such as rapastinel and lanicemine. In this respect it is worth noting that the NMDA receptor has several binding sites that can be targeted pharmacologically. In addition, drugs working at the AMPA receptor or metabotropic glutamate autoreceptors may have clinical utility in depression (Wilkinson 2019).
Why it is important to do this review
This review is an update of the previous Cochrane Review (Caddy 2015) and is one of a pair; the other Ketamine and other glutamate receptor modulators for depression in bipolar disorder in adults is currently being updated (Dean 2021; McCloud 2015). Reliable information about ketamine and other glutamate receptor modulators in unipolar depression (including modes of administration, comparative efficacy, duration of effect, and safety) is not only clinically useful but also urgently needed because such evidence can improve patients’ outcomes in the treatment of depression and provide a basis for future clinical research and treatment guidelines.
Objectives
To assess the effects of ketamine and other glutamate receptor modulators in comparison to placebo (pill or saline infusion), other pharmacologically active agents, or electroconvulsive therapy (ECT) in alleviating the acute symptoms of depression in people with unipolar major depressive disorder.
To review the acceptability of ketamine and other glutamate receptor modulators in comparison to placebo (pill or saline infusion), other pharmacologically active agents, or ECT in people with unipolar major depressive disorder.
Methods
Criteria for considering studies for this review
Types of studies
We included only double‐blind or single‐blind randomised controlled trials (RCTs) (both published and unpublished) comparing ketamine, memantine, or other glutamate receptor modulators with other active psychotropic drugs or placebo (pill or saline infusion) in people with unipolar major depression.
For trials with a cross‐over design, we considered only results from the first period prior to cross‐over (see Unit of analysis issues for further details).
We planned to include cluster randomised trials (CRTs) where the effect of clustering was or could be accounted for in the statistical analysis (see Unit of analysis issues). However, no CRTs were identified.
We excluded quasi‐randomised trials, such as those allocating by using alternate days of the week, as well as trials that did not explicitly describe the method of allocation as randomised.
Types of participants
Participant characteristics and diagnosis
We considered for inclusion people of both sexes aged 18 years or older with a primary diagnosis of unipolar major depressive disorder according to any of the following standard operational criteria: Feighner criteria (Feighner 1972), Research Diagnostic Criteria (Spitzer 1978), DSM‐III (APA 1980), DSM‐III‐R (APA 1987), DSM‐IV (APA 1994), DSM‐IV‐TR (APA 2000), DSM‐5 (APA 2013), or ICD‐10 (WHO 1992). We included studies using operational diagnostic criteria essentially similar to the above.
We excluded studies using ICD‐9 ((International Classification of Diseases, 9th erevision), as it has only disease names and no diagnostic criteria. We also excluded studies that define depression as scoring above a certain cut‐off on a screening questionnaire.
We included studies recruiting participants with treatment‐resistant depression, and examined this in a sensitivity analysis.
Comorbidities
We included studies in which less than 20% of participants were diagnosed with bipolar depression, and thus at least 80% of participants had unipolar depression, and examined the validity of this decision in a sensitivity analysis. We did not consider concurrent secondary diagnosis of another psychiatric disorder an exclusion criterion. However, we excluded studies in which all participants had concurrent primary diagnosis of another Axis I or II disorder. We also excluded participants with a serious concomitant medical illness or with postpartum depression.
Setting
We applied no restriction on setting.
Subset data
We also included in the analysis studies with a subset of participants that met the review inclusion criteria, provided that we could extract data for this subset from the study report.
Types of interventions
Experimental interventions
Ketamine: any dose and pattern of administration
Riluzole: any dose and pattern of administration
Amantadine: any dose and pattern of administration
Dextromethorphan (alone or in combination with quinidine)
Quinolinic acid: any dose and pattern of administration
Memantine: any dose and pattern of administration
Atomoxetine: any dose and pattern of administration
Tramadol: any dose and pattern of administration
Lanicemine: any dose and pattern of administration
MK‐0657: any dose and pattern of administration
Any other glutamate receptor modulators (for example, D‐cycloserine, GLYX‐13)
Comparator interventions
Placebo (pill or saline infusion)
Any pharmacologically active agent (either conventional, like midazolam, or non‐conventional, like scopolamine or Hypericum) or agent included to mimic the psychotropic side effects of the glutamate agent
Electroconvulsive therapy (ECT)
All interventions could be either as monotherapy or combined with other treatments. We applied no restrictions on dose, frequency, intensity, route, and duration. We included trials that allowed rescue medications (as required, short‐term, infrequent use of medications aimed at emergent symptom relief only, for example short‐term use of hypnotics) as long as these medications were equally distributed among the randomised arms.
We did not include lamotrigine among the list of comparisons because the randomised evidence about this drug has been synthesised elsewhere (Goh 2019; Solmi 2016).
Types of outcome measures
We included studies that met the above inclusion criteria regardless of whether they reported on the following outcomes.
Primary outcomes
Efficacy outcome (dichotomous): number of participants who respond to treatment, where treatment response is defined as (1) a reduction of at least 50% compared to baseline on the Hamilton Rating Scale for Depression (HRSD) (Hamilton 1960), Montgomery‐Åsberg Depression Rating Scale (MADRS) (Montgomery 1979), or any other depression scale, depending on the study authors' definition, or (2) 'much or very much improved' (score 1 or 2) on the Clinical Global Impression‐Improvement scale (CGI‐S) (Guy 1976). Where both scales were provided we preferred the former criteria for judging response. We used the response rate instead of a continuous symptom score for the primary efficacy analysis in order to make the interpretation of results easier for clinicians (Guyatt 1998). To avoid possible outcome reporting bias, we did not use the original authors' definitions of response or remission, if different from above (Furukawa 2007a).
-
Adverse events outcome (dichotomous): We evaluated adverse events using the following outcome measures.
Total number of participants experiencing at least one side effect.
-
Total number of participants experiencing the following specific side effects:
agitation/anxiety;
constipation;
delusions;
diarrhoea;
dissociative symptoms;
dizziness;
dry mouth;
hallucinations;
headache;
hypo/hypertension;
insomnia;
mania/hypomania;
nausea;
seizure;
sleepiness/drowsiness;
urination problems;
vomiting;
tremor.
In order to avoid missing any relatively rare or unexpected, yet important side effects (for instance, sexual side effects), in the data extraction phase we collected information on all side effects data reported in the studies and discussed ways to summarise them post hoc. We extracted descriptive data regarding adverse‐effect profiles from all available studies. Due to a lack of consistent reporting of adverse effects (which came primarily from the study authors' descriptions), we combined terms describing similar side effects. For example, we combined 'dry mouth', 'reduced salivation', and 'thirst' into 'dry mouth'. We then grouped all adverse effect categories by organ system, such as neuropsychiatric, gastrointestinal, respiratory, sensory, genitourinary, dermatological, and cardiovascular.
Secondary outcomes
Efficacy outcome (dichotomous): number of participants who achieve remission. Remission is defined as (1) a score of less than 7 on the HRSD‐17 (Furukawa 2007b), or less than 8 for all the other longer versions of the HRSD, or less than 11 on the MADRS (Bandelow 2006), or less than 6 on the Quick Inventory of Depressive Symptomatology (16‐Item) (Self Report) (http://www.ids-qids.org/); or (2) participants who were 'not ill or borderline mentally ill' (score 1 or 2) on the Clinical Global Impression‐Severity scale out of the total number of randomised participants. Where both are provided, we used the former criterion for judging remission.
Efficacy outcome (continuous): mean endpoint scores or mean change scores in depression severity from baseline to the time point in question. We allowed a looser form of intention‐to‐treat (ITT) analysis, whereby all the participants with at least one post‐baseline measurement were represented by their last observations carried forward (LOCF), but in any pooled analysis we planned to examine the impact of the LOCF in a sensitivity analysis).
Suicidality, including suicidal ideation, suicide attempts (nonfatal self‐harm), and deaths by suicide. We examined suicidality and suicide ideation according to the outcome measures reported in the original studies (either as spontaneously reported or as a score on a standardised rating scale).
Cognition. We examined this according to the outcome measures reported in the original studies.
Loss of hope and other health‐related quality of life measures. We included data on any validated quality of life instruments.
Costs to healthcare services. We collected data according to what was reported in the original studies.
-
Acceptability (dichotomous), evaluated using the following outcome measures:
overall number of participants who dropped out during the trial as a proportion of the total number of randomised participants;
number of participants who dropped out due to lack of efficacy during the trial as a proportion of the total number of randomised participants
number of participants who dropped out due to side effects during the trial as a proportion of the total number of randomised participants.
Timing of outcome assessment
As study authors report response rates at various time points of trials, we decided a priori to subdivide the treatment indices as follows.
Ultra‐rapid response: at 24 hours, ranging between 12 and 36 hours (primary efficacy outcome).
Rapid response: at 72 hours, ranging between 37 and less than 96 hours.
Early response: at one week, ranging between four and 10 days.
Acute response: at two weeks, ranging between 11 days and less than three weeks.
Medium response: at four weeks, ranging between three and six weeks.
Long‐term response: at three months, ranging between seven weeks and six months.
Hierarchy of outcome measures
When several possible outcome measures are reported for the same outcome, we used the primary outcome according to the original study.
Search methods for identification of studies
Electronic searches
1. Bibliographic databases
For the second version of this review (first published in September 2015 (Caddy 2015)), the Information Specialist with the Cochrane Common Mental Disorders Group (CCMD) conducted update searches (30 July 2020) directly on the core bibliographic databases, from 2015 onwards (Appendix 1):
Cochrane Central Register of Controlled Trials (CENTRAL; 2020, Issue 7) in the Cochrane Library (searched 30 July 2020);
MEDLINE Ovid (2015 to July 28 2020);
Embase Ovid (2015 to 2020 Week 30);
PsycINFO Ovid (2015 to July Week 3).
Earlier searches of these databases was conducted via the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (all years to 9 January 2015) (Appendix 2).
2. International trial registries
International trial registries were searched via CENTRAL on the Cochrane Library and directly via the World Health Organization's trials portal (ICTRP) and ClinicalTrials.gov to identify unpublished or ongoing studies (30 July 2020).
3. Adverse events search
The information Specialist with CCMD also conducted a companion search for adverse events data (30 July 2020) on Ovid MEDLINE, Embase and PsycINFO (Appendix 3), although we have not incorporated these data into this version of the review.
We applied no restrictions on language or publication status to the searches.
Searching other resources
Grey literature
We conducted complementary searches on the websites of the following drug regulatory authorities for additional unpublished data: the US Food and Drug Administration (FDA), the Medicines and Healthcare products Regulatory Agency in the UK, the European Medicines Agency in the EU, the Pharmaceuticals and Medical Devices Agency in Japan, and the Therapeutic Goods Administration in Australia.
Reference lists
We checked the reference lists of all included studies and relevant systematic reviews and major textbooks of affective disorder written in English to identify additional studies missed from the original electronic searches (for example, unpublished or in‐press citations).
Correspondence
We contacted trialists and subject experts for information on unpublished or ongoing studies, or to request additional trial data.
Data collection and analysis
Selection of studies
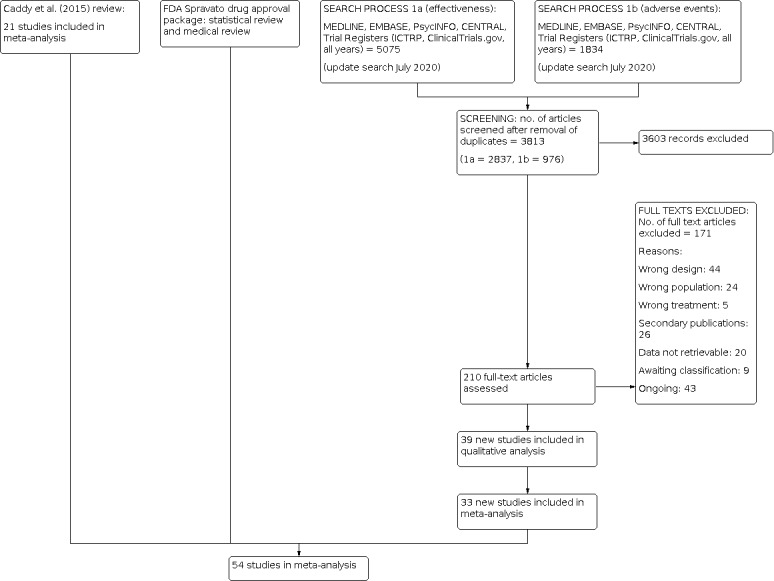
Two review authors (RD, SH, SS, RS, AB) independently screened titles and abstracts for inclusion of all the potential studies we identified as a result of the search and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the full‐text study reports/publication, and two review authors (RD, SH, SS, RS, AB) independently screened the full text and identified studies for inclusion, and identified and recorded reasons for exclusion of the ineligible studies. Any disagreements were resolved through discussion or, if required, by consulting a third person (CH, AC). We identified and removed duplicate records and collated multiple reports that related to the same study so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA (Moher 2009) flow diagram (Figure 1) and Characteristics of excluded studies tables.
1.
Study flow diagram.
Data extraction and management
We used a data collection form to extract study characteristics and outcome data that had been piloted on at least one study in the review. Two review authors (RD, SH, SS, RS, AB) extracted study characteristics and outcome data from included studies. We extracted the following study characteristics.
Participant characteristics (age, sex, depression diagnosis, comorbidity, depression severity, antidepressant treatment history for the index episode, study setting).
Intervention details (intended dosage range, mean daily dosage actually prescribed, cointervention if any, ketamine as investigational drug or as comparator drug, sponsorship).
Outcome measures of interest from the included studies.
Depression severity was defined using the same criteria set out by Cipriani 2012, with severe depression defined by a baseline score of 25 or more on the HRSD and 31 or more on the MADRS (Dozois 2004; Muller 2003).
We noted in the Characteristics of included studies tables if outcome data were not reported in a usable way. We resolved disagreements by consensus or by involving a third person (AC, CH). Two review authors (RD, SH, SS, RS, AB) transferred data into the Review Manager 5 (Revman 2020) file. We double‐checked that data were entered correctly by comparing the data presented in the systematic review with the study reports. A third review author (RD) checked study characteristics for accuracy against the trial report. The comparisons were done by individual drug (see Types of interventions).
Main comparisons
Ketamine versus placebo
Ketamine versus other glutamate receptor modulators
Ketamine versus other pharmacologically active agents (either conventional, like midazolam, or nonconventional, like scopolamine or Hypericum)
Other glutamate receptor modulators versus placebo
Other glutamate receptor modulators versus other pharmacologically active agents (either conventional, like midazolam, or nonconventional, like scopolamine or Hypericum)
Ketamine versus ECT
Other glutamate receptor modulators versus ECT
Other glutamate receptor modulators will be considered individually as separate comparisons. All interventions could be either as monotherapy or combined with other treatments. We applied no restrictions on dose, frequency, intensity, route, and duration.
Assessment of risk of bias in included studies
Five review authors (RD, SH, SS, RS, AB) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020). Any disagreements were resolved by discussion or by involving another review author (AC, CH). We assessed the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
We judged each potential source of bias as high, low, or unclear and provide a supporting quotation from the study report together with a justification for our judgement in the risk of bias tables. We summarised the risk of bias judgements across different studies for each of the domains listed. We considered blinding separately for different key outcomes where necessary (for example, for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different than for a participant‐reported mood scale). Where information on risk of bias relates to unpublished data or correspondence with a trialist, we noted this in the risk of bias table.
When considering treatment effects, we took into account the risk of bias for the studies that contribute to that outcome.
Measures of treatment effect
Dichotomous data
We calculated the odds ratio (OR) with corresponding 95% confidence interval (95% CI) for dichotomous or event‐like outcomes. We calculated response rates out of the total number of randomised participants. We applied ITT analysis whereby all dropouts not included in the analysis were considered as nonresponders. For statistically significant results, we calculated the number needed to treat to benefit (NNTB) and the number needed to treat to harm (NNTH).
Continuous data
We calculated the mean difference (MD) or standardised mean difference (SMD) along with corresponding 95% CI for continuous outcomes. We used the MD where the same scale was used to measure an outcome. We employed the SMD where different scales were used to measure the same underlying construct.
For both continuous and dichotomous data, we undertook meta‐analyses only where this was meaningful, that is if the treatments, participants, and the underlying clinical question were similar enough for pooling to make sense. We described narratively skewed data reported as medians and interquartile ranges.
Where multiple trial arms were reported in a single trial, we included only the relevant arms.
Unit of analysis issues
Cluster‐randomised trials
No cluster‐randomised trials were found in the search, however we would have included cluster‐randomised trials if either of the two methods below were possible.
\if the cluster‐randomised trial was correctly analysed in the original report, we would have entered the effect estimate and standard error using the generic inverse variance method in RevMan 5 (Revman 2020).
-
If the original report failed to adjust for cluster effects, we planned to include such a trial in the meta‐analysis if we were able to extract the following information:
number of clusters randomised to each intervention or the average size of each cluster;
outcome data ignoring the cluster design for the total number of participants;
estimate of the intracluster correlation coefficient (ICC).
The ICC could be borrowed from similarly‐designed studies when such were available. We then conducted the approximately correct analysis following the procedures described in section 16.3.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020).
Cross‐over trials
A major concern of cross‐over trials is the potential of carry‐over effects, which occur when an effect (for example, pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase, the participants can differ systematically from their initial state, despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in major depression, we only used data from the first phase of cross‐over studies. However, we are aware that cross‐over trials for which only first period data are available should be considered to be at risk of bias (Higgins 2020).
Studies with multiple treatment groups
Where a study involved more than two treatment arms, we included all relevant treatment arms in comparisons. If data were binary, we simply added and combined them into one group or divided the comparison arm into two (or more) as appropriate. If data were continuous, we combined data following the formula in section 6.5.2.10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020).
Dealing with missing data
Dichotomous data
We calculated treatment responders and treatment remitters on a strict ITT basis; we included dropouts in the analysis. Where participants were excluded from the trial before the endpoint, we assumed that they experienced a negative outcome (for example, failure to respond to treatment). We examined the validity of this decision in sensitivity analyses by applying worst‐ and best‐case scenarios (that is we assumed missing data to be responders or nonresponders in the corresponding sensitivity analyses). When dichotomous outcomes were not reported but baseline mean, endpoint mean, and corresponding standard deviations (SDs) of the HRSD (or other depression scale) were reported, we converted continuous outcome data expressed as mean and SD into the number of responding and remitted participants, based on a validated imputation method (Furukawa 2005). If a more sophisticated and arguably more valid imputation method was reported in the original study (for example mixed‐effects model), we used these numbers to impute the number of responders. We examined the validity of this imputation in sensitivity analyses.
Continuous data
When there were missing continuous data and the method of LOCF was used to perform an ITT analysis, we used the LOCF data.
Missing data
We contacted the original study authors for missing data.
Missing statistics
When only the standard error or t‐test or P values were reported, we calculated SDs as suggested by Altman 1996. Where SDs were not reported, we contacted trial authors and asked them to supply the data. In the absence of a response from the trial authors, we borrowed SDs from other studies in the review, if possible, or calculated the SDs according to a validated imputation method (Furukawa 2006). We examined the validity of this imputation in sensitivity analyses.
Assessment of heterogeneity
We first investigated heterogeneity between studies by visual inspection of the forest plots. If the 95% confidence intervals (CIs) of the ORs for each study in the pooled analysis did not include means of other studies, we investigated potential sources of heterogeneity. We also calculated the I2 statistic (Higgins 2020). We used the Cochrane Handbook for Systematic Reviews of Interventions' rough guide to its interpretation as follows: 0% to 40% might not be important; 30% to 60% may represent moderate heterogeneity; 50% to 90% may represent substantial heterogeneity; and 75% to 100%, considerable heterogeneity. We also kept in mind that the importance of the observed value of I2 depends on (i) the magnitude and direction of effects and (ii) the strength of evidence for heterogeneity (for example P value from the Chi2 test, or a CI for I2). If the I2 value was below 50%, but the direction and magnitude of treatment effects were suggestive of important heterogeneity, we investigated the potential sources of heterogeneity. Finally, we planned to perform subgroup analyses to investigate heterogeneity. We reported I2 values in all analyses including two or more studies.
Assessment of reporting biases
We planned to enter data from included studies into a funnel plot (trial effect against trial variance) to investigate small‐study effects (Sterne 2000), but none of our analyses contained sufficient studies to allow this. In future updates of this review, we plan to use the test for funnel plot asymmetry only when at least 10 studies are included in the meta‐analysis, as per protocol. In the event of using a funnel plot, we will interpret results cautiously, with visual inspection of the funnel plots (Higgins 2011a). If we identify evidence of small‐study effects, we will investigate possible reasons for funnel plot asymmetry, including publication bias (Egger 1997).
Data synthesis
For the primary analysis, we calculated the pooled OR with corresponding 95% CI for dichotomous outcomes. We calculated the pooled MD or SMD as appropriate with corresponding 95% CIs for continuous outcomes. We presented any skewed data and non quantitative data descriptively. An outcome that has a minimum score of zero could be considered skewed when the mean is smaller than twice the SD. However, the skewness of change scores is difficult to depict as the possibility of negative values exists. We therefore used change scores for meta‐analysis of mean difference MDs. We considered a P value of less than 0.05 and a 95% CI that does not cross the line of no effect statistically significant. In forest plots with two or more studies we used a random‐effects model for both dichotomous and continuous variables. We adopted the random‐effects model under these circumstances because it has the highest generalisability for empirical examination of summary effect measures in meta‐analyses (Furukawa 2002). However, as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (10.4.4.1), to assess the influence of small‐study effects on the results of a meta‐analysis with between‐study heterogeneity, we routinely examined the robustness by comparing the fixed‐effect model and the random‐effects model. We reported any material differences between the models.
Subgroup analysis and investigation of heterogeneity
As multiple analyses will lead to false‐positive and false‐negative conclusions, subgroup analyses should be performed and interpreted with caution (Brookes 2001; Brookes 2004). We planned the following subgroup analyses, where possible, for the following variables.
Depression severity (severe major depression, moderate or mild major depression): 'Severe major depression' was defined by a threshold baseline severity score for entry of 25 or more for the 17‐item HRSD (Dozois 2004), and 31 or more for MADRS (Muller 2003).
Treatment settings (psychiatric inpatients, psychiatric outpatients, primary care): As depressive disorder in primary care has a different profile than that of psychiatric inpatients or outpatients (Suh 1997), it is possible that results obtained from either of these settings may not be applicable to the other settings (Arroll 2009).
Older people (greater than 65 years of age), separately from other adult participants: Older people may be more vulnerable to adverse effects associated with antidepressants, and a decreased dosage is often recommended. We planned to pool groups whose mean age was more than 65 years.
Sensitivity analysis
We originally planned the following sensitivity analyses for primary outcomes.
Excluding trials with unclear allocation concealment or unclear double blinding.
Excluding studies that included participants with bipolar depression or psychotic features.
Excluding studies that recruited participants with treatment‐resistant depression (defined as inadequate response to at least two antidepressants).
Excluding studies with unfair dose comparisons (Cipriani 2009).
Excluding trials with a dropout rate greater than 20%.
Excluding trials for which the response rates had to be calculated based on an imputation method (Furukawa 2005), and for which the SD had to be borrowed from other trials (Furukawa 2006).
We decided post‐hoc to conduct the following additional sensitivity analyses for primary outcomes.
Excluding trials with add‐on ECT
Excluding multiple doses
Our routine comparisons of random‐effects and fixed‐effect models, as well as our secondary outcomes of remission rates and continuous severity measures, may be considered additional forms of sensitivity analyses.
Summary of findings and assessment of the certainty of the evidence
We constructed a summary of findings table for the main comparisons (ketamine versus placebo, ketamine versus midazolam, and esketamine versus placebo), with regard to the following five outcomes. Where possible, we presented data at all four prespecified time points for the primary outcomes. For secondary outcomes, we selected a primary time point of 24 hours as this was considered the most clinically relevant, and presented the data closest to this time point only.
Efficacy: number of participants who respond to treatment.
Acceptability: total dropouts.
Efficacy: number of participants who achieve remission.
Severity of depression at end of trial.
Acceptability: dropouts due to adverse effects.
In the summary of findings tables, we used GRADEproGDT software (GradePro GDT 2020) and the principles of the GRADE (Schünemann 2013) approach, which assesses the quality of a body of evidence based on the extent to which there can be confidence that the obtained effect estimate reflects the true underlying effect. The quality of a body of evidence is judged on the basis of the included studies’ risks of bias, the directness of the evidence, unexplained heterogeneity, imprecision, and the risk of publication bias. We used the average rate in all the arms of the included trials as the 'assumed risk' for each outcome because we did not expect salient differences in such risks among different agents. We therefore did not target any particularly high‐ or low‐risk populations; all the tables are for medium‐risk populations.
Results
Description of studies
Results of the search
CCMD’s Information Specialist ran update searches using two separate strategies, one for effectiveness (CENTRAL, Ovid MEDLINE, Embase, PsycINFO, Trial Registers 2015 to 30 July 2020) (n = 5075 refs), and one for adverse effects data (Ovid MEDLINE, Embase, PsycINFO, Trial Registers, 2015 to 30 July 2020) (n = 1834). This has been reported in the PRISMA (Moher 2009) diagram (Figure 1).
From a total of 6909 records retrieved from the searches, we removed 3096 duplicate records and excluded a further 3603 on the basis of the title and abstract. We retrieved the full‐text articles for 210 records, yielding 39 new studies.
Included studies
See: Characteristics of included studies; Figure 1.
The initial version of this Cochrane Review (Caddy 2015) identified 25 studies (corresponding to 23 primary references and 61 references overall; 1242 participants) which met the inclusion criteria for this review (Berk 2014; Berman 2000; Ghasemi 2013; Heresco‐Levy 2006; Heresco‐Levy 2013; Huang 2013; Ibrahim 2012a; Ibrahim 2012b; Jarventausta 2013; Lapidus 2014; Loo 2012; Michelson 2007; Murrough 2013; Nations 2012 (part I); Nations 2012 (part II); Omranifard 2014; Preskorn 2008; Sanacora 2014 (a); Sanacora 2014 (b); Smith 2013; Sos 2013; Yoosefi 2014; Zarate 2006a; Zarate 2006b; Zarate 2013). Of these 25 studies, eight RCTs assessed the efficacy of ketamine (Berman 2000; Ghasemi 2013; Lapidus 2014; Loo 2012; Murrough 2013; Sos 2013; Yoosefi 2014; Zarate 2006a); three assessed memantine (Omranifard 2014; Smith 2013; Zarate 2006b); three assessed AZD6765 (Sanacora 2014 (a); Sanacora 2014 (b); Zarate 2013); two assessed D‐cycloserine (Heresco‐Levy 2006; Heresco‐Levy 2013); two assessed Org26576 (Nations 2012 (part I); Nations 2012 (part II)); and one each assessed atomoxetine (Michelson 2007), CP‐101,606 (Preskorn 2008), MK‐0657 (Ibrahim 2012b), N‐acetylcysteine (Berk 2014), riluzole (Ibrahim 2012a), and sarcosine (Huang 2013). One study which was previously included in the ketamine comparison, was re‐evaluated as assessing esketamine (Jarventausta 2013).
Thirty‐nine new studies met the inclusion criteria for this updated review (Abbasinazari 2015; Amidfar 2016; Anderson 2017; Arabzadeh 2018; Canuso 2018; Carspecken 2018; Chen 2017; Chen 2018; Correia‐Melo 2020; Daly 2018; Downey 2016; Fava 2018; Fedgchin 2019; Fernie 2017; Fu 2020; Gálvez 2018; Grunebaum 2018; Hu 2016; Ionescu 2018; Ionescu 2020; Jagtiani 2014; Kuşçu 2015; Li 2016; Ochs‐Ross 2020; Preskorn 2015; Popova 2019; Quiroz 2016; Roohi‐Azizi 2017; Salardini 2016; Salehi 2015; Sanacora 2017; Shams Alizadeh 2015; Shiroma 2020; Singh 2016 a; Singh 2016 b; Su 2017; Sumner 2020; Tiger 2020; Umbricht 2020).
The new search identified an additional 22 RCTs for inclusion assessing the efficacy of ketamine (Anderson 2017; Arabzadeh 2018; Carspecken 2018; Chen 2017; Chen 2018; Correia‐Melo 2020; Downey 2016; Fava 2018; Fernie 2017; Gálvez 2018; Grunebaum 2018; Hu 2016; Ionescu 2018; Jagtiani 2014; Kuşçu 2015; Li 2016; Salehi 2015; Shams Alizadeh 2015; Shiroma 2020; Singh 2016 a; Su 2017; Sumner 2020; Tiger 2020); eight assessing esketamine (Canuso 2018; Correia‐Melo 2020; Daly 2018; Fedgchin 2019; Fu 2020; Ionescu 2020; Ochs‐Ross 2020; Popova 2019; Singh 2016 b); two assessing memantine (Abbasinazari 2015; Amidfar 2016); two assessing lanicemine (Downey 2016; Sanacora 2017), one assessing basimglurant (Quiroz 2016), one assessing citicoline (Roohi‐Azizi 2017); one assessing decoglurant (Umbricht 2020); one assessing rapastinel (Preskorn 2015); one assessing riluzole (Salardini 2016).
The majority of included studies were placebo‐controlled trials (48 out of 64, 75%), with the remaining 16 studies directly comparing a glutamate receptor modulator with an active comparison (citalopram, electroconvulsive therapy (ECT), esketamine, midazolam, methohexital, remifentanil hydrochloride, thiopental). The majority were two‐arm studies (48 out of 64, 75%), whilst nine of the remaining studies (Chen 2018; Fedgchin 2019; Li 2016; Nations 2012 (part II); Quiroz 2016; Sanacora 2014 (b); Sanacora 2017; Singh 2016 b; Su 2017) employed a three‐arm methodology, comparing differing doses of an active drug to placebo. Two studies utilised three‐arm methodologies to compare ketamine with both an active comparator and placebo (Downey 2016; Kuşçu 2015). Four used four‐ and five‐arm methodologies to test differing doses of ketamine versus placebo, respectively (Daly 2018; Fava 2018; Preskorn 2015; Umbricht 2020). Another used a four‐arm methodology to test differing treatment regimens (either two or three times weekly) for ketamine against placebo (Singh 2016 a).
Design
All of the studies were double‐blind randomised controlled trials (RCTs), with the exception of Ghasemi 2013, which was single‐blind study, and Kuşçu 2015 in which blinding was at least single‐blind but unclear on double‐blinding (for full details about study blinding, please refer to Characteristics of included studies). Eight of the 64 studies had a cross‐over design (Berman 2000; Heresco‐Levy 2006; Ibrahim 2012b; Lapidus 2014; Sos 2013; Sumner 2020; Zarate 2006a; Zarate 2013). The treatment period ranged from one single administration to 12 weeks.
Sample sizes
The total number of participants from the 64 studies was 5299, with a minimum sample size of five (Ibrahim 2012b; Gálvez 2018) and a maximum of 357 (Umbricht 2020).
Setting
The majority of trials treated patients on an outpatient basis (24 studies), inpatient basis (20 studies), or both (five studies), whilst in the remaining trials the setting was unclear (15 studies). Twenty‐three out of the 64 trials took place in the USA, 16 in Asia, eight in Europe, three in Australia, one in New Zealand one in South America, nine cross‐continental, and in three the study was unclear. Thirty‐one out of the 64 trials were single‐centre studies; 23 were multi‐centre and in the remaining 10 trials it was unclear whether the studies were single‐centred or multi‐centred.
Participants
All studies reported the demographic and/or clinical characteristics of patients, with the exception of Ibrahim 2012b, where no details were reported. The proportion of women ranged from 0% (Gálvez 2018) to 87.5% (Su 2017). Two studies (Omranifard 2014; Ochs‐Ross 2020) recruited older adults above age 60, whilst in the remaining studies mean age ranged from 25.7 to 58.7 years.
The majority of studies defined an inclusion criterion specifying the severity of depression: 29 of these studies specified at least moderate depression; 17 of these studies specified severe depression and five specified mild‐moderate depression. Nineteen studies (Carspecken 2018; Daly 2018; Fava 2018; Fedgchin 2019; Heresco‐Levy 2013; Ibrahim 2012a; Ibrahim 2012b; Jarventausta 2013; Kuşçu 2015; Murrough 2013; Popova 2019; Preskorn 2008; Salehi 2015; Sanacora 2014 (a); Sanacora 2014 (b); Shiroma 2020; Singh 2016 a; Singh 2016 b; Zarate 2006a) recruited only treatment‐resistant patients, which we defined as inadequate response to at least two antidepressants.
In 59 of the 64 studies patients had a diagnosis of unipolar major depression based on the DSM‐IV, DSM‐IV‐TR, or DSM‐V criteria. The remaining five studies (Berman 2000; Ghasemi 2013; Loo 2012; Anderson 2017; Gálvez 2018), recruited mixed samples of major depressive disorder and bipolar depression, with 11.11%, 5.56%, 19.57%, 15.7%, and 25% of the sample diagnosed with bipolar disorder, respectively. One study (Jarventausta 2013) recruited patients with recurrent severe or psychotic major depressive disorder, with 10 out of the 32 participants suffering from psychotic major depressive disorder.
Interventions
A total of 31 studies included ketamine as the experimental intervention; 16 compared ketamine with placebo (Anderson 2017; Arabzadeh 2018; Berman 2000; Chen 2017; Chen 2018; Hu 2016; Ionescu 2018; Lapidus 2014; Li 2016; Loo 2012; Shams Alizadeh 2015; Singh 2016 a; Sos 2013; Su 2017; Tiger 2020; Zarate 2006a); five compared ketamine with midazolam (Fava 2018; Gálvez 2018; Grunebaum 2018; Murrough 2013; Shiroma 2020); four compared ketamine with thiopental (Jagtiani 2014; Kuşçu 2015; Salehi 2015; Yoosefi 2014); one compared ketamine with esketamine (Correia‐Melo 2020); one compared ketamine with lanicemine (Downey 2016); one compared ketamine with methohexital (Carspecken 2018); one compared ketamine with propofol (Fernie 2017); one compared ketamine with remifentanil hydrochloride (Sumner 2020); and one compared ketamine with ECT (Ghasemi 2013).
Fifteen different glutamate receptor modulators were compared with placebo in 32 studies: esketamine (Canuso 2018; Daly 2018; Fedgchin 2019; Fu 2020; Ionescu 2020; Jarventausta 2013; Ochs‐Ross 2020; Popova 2019; Singh 2016 b); memantine (Abbasinazari 2015; Amidfar 2016; Omranifard 2014; Smith 2013; Zarate 2006b); lanicemine (Sanacora 2014 (a); Sanacora 2014 (b); Sanacora 2017; Zarate 2013); D‐cycloserine (Heresco‐Levy 2006; Heresco‐Levy 2013); Org 26576 (Nations 2012 (part I); Nations 2012 (part II)); riluzole (Ibrahim 2012a; Salardini 2016); atomoxetine (Michelson 2007); basimglurant (Quiroz 2016); citicoline (Roohi‐Azizi 2017); CP‐101,606 (Preskorn 2008); decoglurant (Umbricht 2020); MK‐0657 (Ibrahim 2012b); N‐acetylcysteine (Berk 2014); rapastinel (Preskorn 2015). Sarcosine was compared with an active comparator, citalopram, in one study (Huang 2013).
Ketamine was administered intravenously in all studies except three, of which two were administered intranasally (Gálvez 2018; Lapidus 2014), and one was administered orally (Arabzadeh 2018). Esketamine was administered intranasally in all studies except for three in which the drug was administered intravenously (Correia‐Melo 2020; Jarventausta 2013; Singh 2016 b). The majority of the remaining glutamate receptor modulators were administered orally, with the exception of CP‐101,606 (Preskorn 2008) and AZD6765 (Sanacora 2014 (a); Sanacora 2014 (b); Sanacora 2017; Zarate 2013), which were administered intravenously. All comparator interventions matched the administration method of the glutamate receptor modulator, with the exception of ECT versus ketamine (Ghasemi 2013).
In the majority of studies, patients received concomitant medication for their depression alongside the experimental intervention. However, in five studies this information was unclear (Abbasinazari 2015; Chen 2017; Michelson 2007; Sanacora 2014 (a); Yoosefi 2014).
Outcomes
Most studies reported on at least one dichotomous efficacy outcome of response and remission rate. There were eight exceptions (Carspecken 2018; Downey 2016; Fernie 2017; Jagtiani 2014; Salehi 2015; Shams Alizadeh 2015; Singh 2016 b; Sumner 2020).
The continuous efficacy outcome in all included studies was measured on MADRS or HRSD. We imputed missing response and remission rates for 10 studies (Berman 2000; Ghasemi 2013; Loo 2012; Michelson 2007; Murrough 2013; Nations 2012 (part I); Nations 2012 (part II); Sos 2013; Yoosefi 2014; Sanacora 2017) using a validated method reported by Furukawa 2005. We imputed the combined group depression rating scale scores for groups using the same glutamate receptor modulator at different doses (Chen 2018; Fedgchin 2019; Li 2016; Quiroz 2016; Sanacora 2017) using the validated method of Higgins 2011d. We imputed missing SDs for one study (Yoosefi 2014) using P values and a method validated by Altman 1996.
Five comparisons did not include any data about adverse events (namely ketamine versus esketamine, ketamine versus methohexital, ketamine versus propofol, ketamine versus remifentanil hydrochloride, and MK‐0657 versus placebo), while increase in systolic blood pressure and heart rate was the only adverse event with usable information in the comparison ketamine versus ECT. For acceptability outcomes, 23 studies reported data on total dropout rates, six on dropouts due to adverse events, and one on dropouts due to lack of efficacy.
Excluded studies
(See: Characteristics of excluded studies and Figure 1)
We excluded 174 studies. The main reasons for exclusions were study design (44), secondary publications (26), or wrong population (24).
Ongoing studies
(See Characteristics of ongoing studies and Figure 1)
After screening retrieved records and checking full‐texts, we identified 43 ongoing studies.
Studies awaiting classification
(See Characteristics of studies awaiting classification and Figure 1)
We identified 9 studies awaiting classification.
Risk of bias in included studies
See Characteristics of included studies for the risk of bias judgement for each study. A summary of the overall risk of bias is presented in Figure 2 and Figure 3.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
We cannot rule out the potential bias introduced by inadequate blinding procedures. For instance, saline infusion does not necessarily provide adequate blinding for ketamine, as both patients and personnel can probably guess which treatment a patient has received based on differences during the infusion; for example, psychotomimetic side effects. The assessment of bias reported below is based on the adequacy of blinding attempts as described in each papers’ methods, not on the actual degree of blinding achieved. We rated studies as 'low risk' when all measures used to blind study participants and personnel from knowledge of which intervention a participant received was described. We rated studies as 'unclear risk' when there was a lack of information on blinding procedures. Of the 31 included studies assessing the efficacy of ketamine, five tested the blinding and provided information relating to whether the intended blinding was effective (Anderson 2017; Fava 2018; Shiroma 2020; Sumner 2020; Tiger 2020). Blinding was found to be ineffective in all of these studies, with the exception of one study in which participants received concomitant ECT (Anderson 2017).
Allocation
Random sequence generation
The majority of included studies (Abbasinazari 2015; Amidfar 2016; Anderson 2017; Arabzadeh 2018; Canuso 2018; Carspecken 2018; Chen 2017; Chen 2018; Correia‐Melo 2020; Daly 2018; Fava 2018; Fedgchin 2019; Fernie 2017; Fu 2020; Gálvez 2018; Grunebaum 2018; Heresco‐Levy 2013; Huang 2013; Ionescu 2018; Ionescu 2020; Jagtiani 2014; Lapidus 2014; Li 2016; Loo 2012; Michelson 2007; Murrough 2013; Nations 2012 (part I); Ochs‐Ross 2020; Omranifard 2014; Popova 2019; Preskorn 2015; Quiroz 2016; Roohi‐Azizi 2017; Salardini 2016; Salehi 2015; Sanacora 2017; Shams Alizadeh 2015; Shiroma 2020; Singh 2016 a; Singh 2016 b; Smith 2013; Sos 2013; Sumner 2020; Tiger 2020; Umbricht 2020; Yoosefi 2014; Zarate 2006a; Zarate 2013) reported detail on the method of random sequence generation and we classified them as 'low risk'. The remaining 16 studies (Berk 2014; Berman 2000; Downey 2016; Ghasemi 2013; Heresco‐Levy 2006; Hu 2016; Ibrahim 2012a; Ibrahim 2012b; Jarventausta 2013; Kuşçu 2015; Nations 2012 (part II); Preskorn 2008; Sanacora 2014 (a); Sanacora 2014 (b); Su 2017; Zarate 2006b) described the trials as randomised, but gave no details of the methods used to achieve random allocation, so we classified them as 'unclear risk'.
Allocation concealment
Thirty‐eight of the studies (Abbasinazari 2015; Amidfar 2016; Anderson 2017; Arabzadeh 2018; Canuso 2018; Carspecken 2018; Chen 2017; Chen 2018; Correia‐Melo 2020; Downey 2016; Fava 2018; Fedgchin 2019; Fernie 2017; Fu 2020; Huang 2013; Ibrahim 2012a; Ionescu 2018; Ionescu 2020; Jagtiani 2014; Lapidus 2014; Li 2016; Loo 2012; Murrough 2013; Nations 2012 (part I); Ochs‐Ross 2020; Omranifard 2014; Popova 2019; Preskorn 2008; Preskorn 2015; Quiroz 2016; Roohi‐Azizi 2017; Salardini 2016; Sanacora 2017; Shiroma 2020; Singh 2016 a; Smith 2013; Tiger 2020; Umbricht 2020) reported details on allocation concealment and we classified them as 'low risk'. We classified one study (Yoosefi 2014) as 'high risk' due to randomisation being conducted by one of the trial investigators. We classified the remaining 25 studies (Berk 2014; Berman 2000; Daly 2018; Gálvez 2018; Ghasemi 2013; Grunebaum 2018; Heresco‐Levy 2006; Heresco‐Levy 2013; Hu 2016; Ibrahim 2012b; Jarventausta 2013; Kuşçu 2015; Michelson 2007; Nations 2012 (part II); Salehi 2015; Sanacora 2014 (a); Sanacora 2014 (b); Shams Alizadeh 2015; Singh 2016 b; Sos 2013; Su 2017; Sumner 2020; Zarate 2006a; Zarate 2006b; Zarate 2013) as 'unclear risk' as they did not provide details of the methods used to achieve allocation concealment.
Blinding
Blinding of participants and personnel
Twenty‐two out of 64 studies reported detail on the blinding of participants and personnel, and we classified them as 'low risk' (Anderson 2017; Berk 2014; Fernie 2017; Ghasemi 2013; Heresco‐Levy 2006; Heresco‐Levy 2013; Huang 2013; Ibrahim 2012a; Ibrahim 2012b; Jagtiani 2014; Lapidus 2014; Loo 2012; Michelson 2007; Omranifard 2014; Preskorn 2008; Salardini 2016; Sanacora 2017; Smith 2013; Umbricht 2020; Yoosefi 2014; Zarate 2006a; Zarate 2013). Four studies were classified as high risk due to high numbers of participants guessing their treatment allocation, suggesting that blinding was not effective (Fava 2018; Shiroma 2020; Sumner 2020; Tiger 2020). We classified the remaining 38 studies as 'unclear risk' as they did not provide full details of the methods used to blind participants and personnel (Abbasinazari 2015; Amidfar 2016; Arabzadeh 2018; Berman 2000; Canuso 2018; Carspecken 2018; Chen 2017; Chen 2018; Correia‐Melo 2020; Daly 2018; Downey 2016; Fedgchin 2019; Fu 2020; Gálvez 2018; Grunebaum 2018; Hu 2016; Ionescu 2018; Ionescu 2020; Jarventausta 2013; Kuşçu 2015; Li 2016; Murrough 2013; Nations 2012 (part I); Nations 2012 (part II); Ochs‐Ross 2020; Popova 2019; Preskorn 2015; Quiroz 2016; Roohi‐Azizi 2017; Salehi 2015; Sanacora 2014 (a); Sanacora 2014 (b); Shams Alizadeh 2015; Singh 2016 a; Singh 2016 b; Sos 2013; Su 2017; Zarate 2006b).
Blinding of outcome assessment
Twenty‐one studies reported details on the methods used in the blinding of outcome assessment and we classified them as 'low risk' (Anderson 2017; Correia‐Melo 2020; Fernie 2017; Ghasemi 2013; Hu 2016; Huang 2013; Ionescu 2018; Jagtiani 2014; Jarventausta 2013; Lapidus 2014; Murrough 2013; Omranifard 2014; Preskorn 2008; Salardini 2016; Sanacora 2017; Shams Alizadeh 2015; Singh 2016 b; Smith 2013; Sumner 2020; Yoosefi 2014; Zarate 2013). Two studies were classified as high risk; one due to the high numbers of correct guesses of treatment assignment, suggesting that blinding of the outcome assessment was not effective (Fava 2018; Tiger 2020), and another due to conflicting information concerning blinding (Salehi 2015). We classified 40 studies as 'unclear risk' as they did not provide full details of the methods used in the blinding of outcome assessment (Abbasinazari 2015; Amidfar 2016; Arabzadeh 2018; Berk 2014; Berman 2000; Canuso 2018; Carspecken 2018; Chen 2017; Chen 2018; Daly 2018; Downey 2016; Fedgchin 2019; Fu 2020; Gálvez 2018; Grunebaum 2018; Heresco‐Levy 2006; Heresco‐Levy 2013; Ibrahim 2012a; Ibrahim 2012b; Ionescu 2020; Kuşçu 2015; Li 2016; Loo 2012; Michelson 2007; Nations 2012 (part I); Nations 2012 (part II); Ochs‐Ross 2020; Popova 2019; Preskorn 2015; Quiroz 2016; Roohi‐Azizi 2017; Sanacora 2014 (a); Sanacora 2014 (b); Shiroma 2020; Singh 2016 a; Sos 2013; Su 2017; Umbricht 2020; Zarate 2006a; Zarate 2006b).
Incomplete outcome data
We rated five studies as 'high risk' in terms of attrition bias (Berman 2000; Fedgchin 2019; Fernie 2017; Hu 2016; Loo 2012), and 11 as 'unclear' (Downey 2016; Jarventausta 2013; Michelson 2007; Nations 2012 (part I); Nations 2012 (part II); Salehi 2015; Sanacora 2014 (a); Shiroma 2020; Smith 2013; Yoosefi 2014; Zarate 2006a).
Selective reporting
As no protocol was available for studies or authors could not provide us with supplementary information, we judged 36 trials to have 'unclear' risk of bias (Abbasinazari 2015; Amidfar 2016; Arabzadeh 2018; Berk 2014; Chen 2017; Chen 2018; Downey 2016; Gálvez 2018; Grunebaum 2018; Heresco‐Levy 2006; Heresco‐Levy 2013; Hu 2016; Huang 2013; Ibrahim 2012b; Ionescu 2018; Jagtiani 2014; Jarventausta 2013; Lapidus 2014; Loo 2012; Michelson 2007; Murrough 2013; Nations 2012 (part I); Nations 2012 (part II); Preskorn 2008; Sanacora 2014 (a); Sanacora 2014 (b); Shams Alizadeh 2015; Shiroma 2020; Smith 2013; Sos 2013; Sumner 2020; Tiger 2020; Yoosefi 2014; Zarate 2006a; Zarate 2006b; Zarate 2013). We considered eight trials as having 'high risk' of reporting bias (Berman 2000; Daly 2018; Fernie 2017; Ibrahim 2012a; Kuşçu 2015; Li 2016; Omranifard 2014; Sanacora 2017) because the protocol was unavailable and some outcome measures or time points were not reported.
Other potential sources of bias
We rated 14 studies as high risk due to being funded by pharmaceutical companies and authors having the potential to financially benefit from positive findings (Canuso 2018; Daly 2018; Downey 2016; Fava 2018; Fedgchin 2019; Fu 2020; Ionescu 2018; Ionescu 2020; Ochs‐Ross 2020; Popova 2019; Preskorn 2015; Quiroz 2016; Sanacora 2017; Umbricht 2020).
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings 1. Ketamine compared to placebo for adults with unipolar major depressive disorder.
| Ketamine compared to Placebo for adults with unipolar major depressive disorder | ||||||
| Patient or population: adults (aged 18 years+) with unipolar major depressive disorder Setting: any setting (outpatient, inpatient, or both) Intervention: ketamine Comparison: placebo | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of the evidence (GRADE) | What happens | ||
| Without ketamine | With ketamine | Difference | ||||
| Efficacy: number of participants who respond to treatment ‐ at 24 hours (Response) assessed with: HDRS, HDRS‐17, MADRS № of participants: 185 (7 RCTs) | OR 3.94 (1.54 to 10.10) | Study population | ⊕⊝⊝⊝ VERY LOW 1 2 | |||
| 8.8% | 27.4% (12.9 to 49.2) | 18.7% more (4.1 more to 40.4 more) | ||||
| Efficacy: number of participants who achieve remission ‐ at 24 hours (Remission) assessed with: MADRS, HDRS № of participants: 75 (3 RCTs) | OR 5.60 (1.07 to 29.46) | Study population | ⊕⊝⊝⊝ VERY LOW 3 4 | |||
| 2.4% | 12.0% (2.5 to 41.8) | 9.6% more (0.2 more to 39.4 more) | ||||
| Depression rating scale score ‐ at 24 hours assessed with: HDRS, HDRS‐17, MADRS № of participants: 231 (8 RCTs) | ‐ | ‐ | ‐ | SMD 0.87 lower (1.26 lower to 0.48 lower) | ⊕⊝⊝⊝ VERY LOW 1 2 5 | |
| Acceptability: total dropouts № of participants: 201 (6 RCTs) | OR 1.25 (0.19 to 8.28) | Study population | ⊕⊝⊝⊝ VERY LOW 1 2 6 | |||
| 34.0% | 39.1% (8.9 to 81) | 5.2% more (25.1 fewer to 47 more) | ||||
| Acceptability: dropouts due to adverse effects ‐ not reported | ‐ | ‐ | ‐ | ‐ | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HDRS: Hamilton Depression Rating Scale; MADRS: Montgomery‐Asberg Depression Rating Scale; OR: Odds ratio;RCT: randomised controlled trial;SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by one point due to the low number of participants available for this outcome and the associated width of the confidence intervals.
2 Downgraded by two points due to the majority of trials being unclear or high risk regarding the blinding of outcome assessments.
3 Downgraded by two points due to the very low number of participants available for this outcome and the associated width of the confidence intervals.
4 Downgraded by one point due to the majority of trials being unclear regarding blinding of outcome assessments.
5 Downgraded by one point due to moderately large heterogeneity (I2 value = 30% to 60%).
6 Downgraded by two points due to substantially large heterogeneity (I2 value = 50% to 90%).
Summary of findings 2. Ketamine compared to midazolam for adults with unipolar major depressive disorder.
| Ketamine compared to Midazolam for adults with unipolar major depressive disorder | ||||||
| Patient or population: adults (aged 18 years+) with unipolar major depressive disorder Setting: any setting (outpatient, inpatient, or both) Intervention: ketamine Comparison: midazolam | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of the evidence (GRADE) | What happens | ||
| Without ketamine | With ketamine | Difference | ||||
| Efficacy: number of participants who respond to treatment ‐ at 24 hours assessed with: HAM‐D‐6, HAM‐D‐17, MADRS № of participants: 296 (4 RCTs) | OR 2.48 (1.00 to 6.18) | Study population | ⊕⊝⊝⊝ VERY LOW 1 2 3 | |||
| 25.9% | 46.5% (25.9 to 68.4) | 20.5% more (0 fewer to 42.5 more) | ||||
| Efficacy: number of participants who achieve remission ‐ at 24 hours assessed with: MADRS № of participants: 122 (2 RCTs) | OR 2.21 (0.67 to 7.32) | Study population | ⊕⊕⊝⊝ LOW 2 3 | |||
| 18.0% | 32.7% (12.8 to 61.6) | 14.7% more (5.2 fewer to 43.6 more) | ||||
| Depression rating scale score ‐ at 24 hours assessed with: MADRS № of participants: 297 (4 RCTs) | ‐ | ‐ | ‐ | SMD 0.49 lower (0.87 lower to 0.1 lower) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | |
| Acceptability: total dropouts № of participants: 72 (1 RCT) | OR 0.33 (0.05 to 2.09) | Study population | ⊕⊕⊝⊝ LOW 4 | |||
| 12.0% | 4.3% (0.7 to 22.2) | 7.7% fewer (11.3 fewer to 10.2 more) | ||||
| Acceptability: dropouts due to adverse effects ‐ not reported | ‐ | ‐ | ‐ | ‐ | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval;HAM‐D: Hamilton Depression Rating Scale; MADRS: Montgomery‐Asberg Depression Rating Scale;OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by two points due to the majority of trials being unclear or high risk regarding the blinding of outcome assessments.
2 Downgraded by one point due to moderately large heterogeneity (I2 value = 30% to 60%).
3 Downgraded by one point due to the low number of participants available for this outcome and the associated width of the confidence intervals.
4 Downgraded by two points due to the very low number of participants available for this outcome and the associated width of the confidence intervals.
Summary of findings 3. Esketamine compared to placebo for adults with unipolar major depressive disorder.
| Esketamine compared to Placebo for adults with unipolar major depressive disorder | ||||||
| Patient or population: adults (aged 18 years+) with unipolar major depressive disorder Setting: any setting (outpatient, inpatient, or both) Intervention: esketamine Comparison: placebo | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of the evidence (GRADE) | What happens | ||
| Without esketamine | With esketamine | Difference | ||||
| Efficacy: number of participants who respond to treatment ‐ at 24 hours (Response) assessed with: MADRS № of participants: 1071 (5 RCTs) | OR 2.11 (1.20 to 3.68) | Study population | ⊕⊕⊝⊝ LOW 1 2 | |||
| 15.0% | 27.1% (17.5 to 39.4) | 12.1% more (2.5 more to 24.4 more) | ||||
| Efficacy: number of participants who achieve remission ‐ at 24 hours (Remission) assessed with: MADRS № of participants: 894 (5 RCTs) | OR 2.74 (1.71 to 4.40) | Study population | ⊕⊕⊕⊝ MODERATE 1 | |||
| 7.2% | 17.5% (11.7 to 25.4) | 10.3% more (4.5 more to 18.2 more) | ||||
| Depression rating scale score ‐ at 24 hours assessed with: MADRS № of participants: 824 (4 RCTs) | ‐ | ‐ | ‐ | SMD 0.31 lower (0.45 lower to 0.17 lower) | ⊕⊕⊕⊝ MODERATE 1 | |
| Acceptability: total dropouts № of participants: 773 (5 RCTs) | OR 1.58 (0.92 to 2.73) | Study population | ⊕⊕⊕⊝ MODERATE 1 | |||
| 8.5% | 12.9% (7.9 to 20.3) | 4.3% more (0.6 fewer to 11.8 more) | ||||
| Acceptability: dropouts due to adverse effects ‐ not reported | ‐ | ‐ | ‐ | ‐ | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; MADRS: Montgomery‐Asberg Depression Rating Scale; OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by one point due to the majority of trials being unclear regarding blinding of outcome assessments.
2 Downgraded by one point due to moderately large heterogeneity (I2 value = 30% to 60%).
We found data for ketamine versus placebo, ketamine versus pharmacologically active agents, ketamine versus placebo, glutamate receptor modulators versus placebo, and one glutamate receptor modulator (sarcosine) versus a pharmacologically active agent (citalopram). We also found data for ketamine versus another glutamate receptor modulator (esketamine). We did not find any data for glutamate receptor modulators (other than ketamine) versus electroconvulsive therapy (ECT).
From our prespecified time points outlined in the methodology, the majority of comparisons had data provided for up to four weeks, with few studies measuring outcomes at three months. For ketamine versus methohexital data were only provided for 72 hours, and the ketamine versus esketamine and basimglurant versus placebo comparisons only had data up to one week. The ketamine versus ECT and ketamine versus remifentanil hydrochloride comparisons had data up to two weeks. Both CP‐101,606 and MK‐0657 comparisons against placebo had data for two weeks, and citicoline and decoglurant comparisons only had data for four weeks. For the atomoxetine comparison we found data only at the three‐month time point.
For adverse events, we reported all findings in the forest plots, but below we only report findings that were statistically significant. Unless otherwise specified, we report data here below using a fixed‐effect model due to the majority of forest plots including only one study.
A. Ketamine versus placebo
1. Ketamine versus placebo
Ten new studies contributed to this comparison (Anderson 2017; Arabzadeh 2018; Chen 2017; Chen 2018; Hu 2016; Ionescu 2018; Li 2016; Singh 2016 a; Su 2017; Tiger 2020) making a total of 14 including four from the previous review (Berman 2000; Loo 2012; Sos 2013; Zarate 2006a). Outcome data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months. See also Table 1.
Thirteen of these studies provided ketamine intravenously, and one administered ketamine orally (Arabzadeh 2018). Nine studies infused a dose of 0.5mg/kg ketamine (Anderson 2017; Berman 2000; Hu 2016; Ionescu 2018; Loo 2012; Singh 2016 a; Sos 2013; Tiger 2020; Zarate 2006a), three studies allocated either 0.2mg/kg or 0.5mg/kg (Chen 2018; Li 2016; Su 2017), and one infused 0.3mg/kg (Chen 2017). The one study that used oral administration routes prescribed doses of 50 mg (Arabzadeh 2018).
In eight studies ketamine was administered intravenously as a single infusion (Berman 2000; Chen 2018; Hu 2016; Li 2016; Sos 2013; Su 2017; Tiger 2020; Zarate 2006a). Two studies infused participants twice weekly (Anderson 2017; Ionescu 2018), two studies infused three times per week (Chen 2017; Loo 2012), and one randomised participants to either two or three infusions weekly (Singh 2016 a). One study using oral administration dosed once daily for six weeks (Arabzadeh 2018).
The majority of studies allowed concomitant medications (Anderson 2017; Chen 2017; Chen 2018; Hu 2016; Ionescu 2018; Li 2016; Loo 2012; Singh 2016 a; Sos 2013; Su 2017). Four studies did not allow concomitant medications (Arabzadeh 2018; Berman 2000; Tiger 2020; Zarate 2006a). In three studies, participants received ECT treatments alongside ketamine (Anderson 2017; Chen 2017; Loo 2012).
1.1 Efficacy: number of participants who respond to treatment
Ketamine was more efficacious than placebo in terms of the number of participants who responded to treatment at 24 hours (random‐effects odds ratio (OR) 3.94, 95% confidence interval (CI )1.54 to 10.10; P = 0.004; participants = 185; studies = 7; I2 = 14%), very low‐certainty evidence,, at 72 hours (random‐effects OR 15.84, 95% CI 3.68 to 68.12; P = 0.0002; participants = 83; studies = 4; I2 = 0%), and at one week (random‐effects OR 3.76, 95% CI 0.98 to 14.42; P = 0.05; participants = 196; studies = 5; I2 = 43%) (Analysis 1.1). Results at two weeks, four weeks, and three months had high levels of heterogeneity, so no conclusions could be made about the response efficacy of ketamine versus placebo at these time points: two weeks (random‐effects OR 2.92, 95% CI 0.48 to 17.78; P = 0.24; participants = 206; studies = 4; I2 = 83%); four weeks (random‐effects OR 1.37, 95% CI 0.50 to 3.77; P = 0.54; participants = 202; studies = 4; I2 = 59%); three months (random‐effects OR 1.95, 95% CI 0.24 to 15.69; P = 0.53; participants = 117; studies = 3; I2 = 80%). Figure 4.
1.1. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 1: Response rate
4.

Forest plot of comparison: 1 Ketamine versus Placebo, outcome: 1.1 Response rate.
1.2 Adverse events
Participants assigned to treatment with ketamine were more likely to report the following adverse events than those who received placebo: agitation/anxiety (random‐effects OR 3.44, 95% CI 1.07 to 11.04; P = 0.04; participants = 143; studies = 3; I2 = 9%; Analysis 1.3), confusion (random‐effects OR 3.76, 95% CI 1.13 to 12.47; P = 0.03; participants = 76; studies = 2; I2 = 4%; Analysis 1.6), dissociative symptoms (random‐effects OR 7.72, 95% CI 1.31 to 45.51; P = 0.02; participants = 145; studies = 3; I2 = 0%; Analysis 1.7).
1.3. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 3: AE Agitation/anxiety
1.6. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 6: AE Confusion
1.7. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 7: AE Dissociative symptoms
We found no difference in terms of other adverse events (Analysis 1.2; Analysis 1.4; Analysis 1.5; Analysis 1.8; Analysis 1.9; Analysis 1.10; Analysis 1.11; Analysis 1.12; Analysis 1.13; Analysis 1.14; Analysis 1.15; Analysis 1.16; Analysis 1.17; Analysis 1.18; Analysis 1.19; Analysis 1.20; Analysis 1.21; Analysis 1.22; Analysis 1.23; Analysis 1.24; Analysis 1.25).
1.2. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 2: AE Abdominal Pain
1.4. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 4: AE Blurred vision
1.5. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 5: AE Change in blood pressure
1.8. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 8: AE Dizziness
1.9. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 9: AE Emotional blunting
1.10. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 10: AE Euphoria
1.11. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 11: AE Hallucinations
1.12. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 12: AE Headache
1.13. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 13: AE Infections and Infestations
1.14. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 14: AE Loss of Appetite
1.15. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 15: AE Mania/hypomania
1.16. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 16: AE Musculoskeletal and connective tissue disorders
1.17. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 17: AE Nausea/vomiting
1.18. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 18: AE Nervousness
1.19. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 19: AE Nervous system disorders
1.20. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 20: AE Palpitations
1.21. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 21: AE Psychiatric disorders
1.22. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 22: AE Restlessness
1.23. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 23: AE Skin and subcutaneous tissue disorders
1.24. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 24: AE Suicidal Ideas
1.25. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 25: AE Tremor
1.3 Efficacy: number of participants who achieve remission
Ketamine resulted in increased remission rates over placebo at 24 hours (random‐effects OR 5.60, 95% CI 1.07 to 29.46; P = 0.04; participants = 75; studies = 3; I2 = 0%), very low‐certainty evidence, at 72 hours (random‐effects OR 6.60, 95% CI 1.51 to 28.92; P = 0.01 participants = 83; studies = 4; I2 = 0%), and at one week (random‐effects OR 4.64, 95% CI 1.37 to 15.68; P = 0.01; participants = 196; studies = 5; I2 = 0%) (Analysis 1.26). We found no difference in remission between ketamine and placebo at two weeks (random‐effects OR 1.67, 95% CI 0.38 to 7.27; P = 0.50; participants = 206; studies = 4; I2 = 63%), at four weeks (random‐effects OR 1.46, 95% CI 0.54 to 3.95; P = 0.46; participants = 202; studies = 4; I2 = 35%), or at three months (random‐effects OR 1.09, 95% CI 0.45 to 2.67; participants = 90; studies = 2; I2 = 0%).
1.26. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 26: Remission rate
1.4 Change scores on depression scale from baseline
We found evidence that ketamine was more effective than placebo at reducing depression rating scale scores from baseline at 24 hours (random‐effects standardised mean difference (SMD) ‐0.87, 95% CI ‐1.26 to ‐0.48; P < 0.0001; participants = 231; studies = 8; I2 = 41%), very low‐certainty evidence, at 72 hours (random‐effects SMD ‐0.68, 95% CI ‐1.28 to ‐0.07; P = 0.03; participants = 148; studies = 6; I2 = 62%), at one week (random‐effects SMD ‐0.72, 95% CI ‐1.10 to ‐0.33; P = 0.0003; participants = 143; studies = 6; I2 = 13%), and at four weeks (random‐effects SMD ‐0.68, 95% CI ‐1.07 to ‐0.29; P = 0.0006; participants = 107; studies = 2; I2 = 0%) (Analysis 1.27). We found no conclusive evidence of reduction in depression rating scale scores between ketamine and placebo at two weeks due to substantial heterogeneity (random‐effects SMD ‐0.43, 95% CI ‐0.90 to 0.04; P = 0.07; participants = 236; studies = 5; I2 = 65%).
1.27. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 27: Depression rating scale score
1.5 Suicidality
We found no difference in suicidal ideation scores between ketamine and placebo at any time point: at 24 hours (random‐effects mean difference (MD) 0.02, 95% CI ‐0.78 to 0.82; P = 0.96; participants = 48; studies = 1; I2 = 0%), at 72 hours (random‐effects MD 0.34, 95% CI ‐0.25 to 0.93; P = 0.26; participants = 68; studies = 2; I2 = 10%), at one week (random‐effects MD ‐0.30, 95% CI ‐1.56 to 0.96; P = 0.64; participants = 19; studies = 1; I2 = 0%), and at two weeks (random‐effects MD ‐0.20, 95% CI ‐1.46 to 1.06; P = 0.76; participants = 19; studies = 1; I2 = 0%) (Analysis 1.28).
1.28. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 28: Suicidal ideation composite
1.6 Cognition
Ketamine was associated with high cognition scores over placebo in immediate‐term memory (random‐effects MD 0.80, 95% CI 0.12 to 1.48; P = 0.02; participants = 127; studies = 1; I2 = 0%), short‐term memory (random‐effects MD 6.90, 95% CI 5.01 to 8.79; P < 0.00001; participants = 127; studies = 1; I2 = 0%), and long‐term memory (random‐effects MD 4.50, 95% CI 2.79 to 6.21; P < 0.00001; participants = 127; studies = 1; I2 = 0%) (Analysis 1.29).
1.29. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 29: Cognition scores
1.7 Quality of life
There were no differences in quality of life between ketamine and placebo (random‐effects MD 0.11, 95% CI ‐0.05 to 0.27; P = 0.19; participants = 64; studies = 1; I2 = 0%) (Analysis 1.30).
1.30. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 30: Quality of Life
1.8 Cost to healthcare services
No data were available for this outcome.
1.9 Acceptability: total dropouts
We found no difference between ketamine and placebo in terms of participants who dropped out due to any cause (random‐effects OR 1.25, 95% CI 0.19 to 8.28; P = 0.81; participants = 201; studies = 6; I2 = 75%), very low‐certainty evidence (Analysis 1.31).
1.31. Analysis.

Comparison 1: Ketamine versus Placebo, Outcome 31: Acceptability
B. Ketamine versus other pharmacologically active agents
2. Ketamine versus midazolam
Five studies contributed data to this comparison, including four newly identified ones (Fava 2018; Gálvez 2018; Grunebaum 2018; Shiroma 2020) and one from the previous review (Murrough 2013). Data were available for 24 hours, 72 hours, one week, two weeks, four weeks, and three months. Table 2
2.1 Efficacy: number of participants who respond to treatment
Ketamine had higher response efficacy over midazolam at 24 hours (random‐effects OR 2.48, 95% CI 1.00 to 6.18; P = 0.05; participants = 296; studies = 4; I2 = 58%) very low‐certainty evidence, at one week (random‐effects OR 3.11, 95% CI 1.38 to 7.04; P = 0.006; participants = 126; studies = 2; I2 = 0%), and at two weeks (random‐effects OR 4.89, 95% CI 1.49 to 16.10; P = 0.009; participants = 53; studies = 1; I2 = 0%) (Analysis 2.1)At 72 hours the OR favoured ketamine over midazolam, but wide confidence intervals and heterogeneity created uncertainty about this effect (random‐effects OR 2.20, 95% CI 0.92 to 5.28; P = 0.08; participants = 218; studies = 3; I2 = 46%), No difference was found for response at four weeks (random‐effects OR 0.50, 95% CI 0.01 to 19.56; P = 0.71; participants = 5; studies = 1; I2 = 0%), or three months (random‐effects OR 3.00, 95% CI 0.08 to 115.34; P = 0.56; participants = 5; studies = 1; I2 = 0%). Figure 5.
2.1. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 1: Response rate
5.

Forest plot of comparison: 2 Ketamine versus Midazolam, outcome: 2.1 Response rate.
2.2 Adverse events
Patients receiving midazolam were less likely to report blurred vision over ketamine 24 hours post‐infusion (random‐effects OR 8.52, 95% CI 1.80 to 40.39; P = 0.007; participants = 72; studies = 1; I2 = 0%), but not at one week (random‐effects OR 1.03, 95% CI 0.23 to 4.70; P = 0.97; participants = 126; studies = 2; I2 = 0%) (Analysis 2.5). Ketamine increased the incidence of dizziness at 24 hours post‐infusion over midazolam (random‐effects OR 3.42, 95% CI 1.44 to 8.14; P = 0.005; participants = 224; studies = 2; I2 = 0%), but not at one week (random‐effects OR 1.05, 95% CI 0.43 to 2.56; P = 0.91; participants = 283; studies = 4; I2 = 0%) (Analysis 2.14). Midazolam was more likely than ketamine to cause general malaise at 24 hours post‐infusion (random‐effects OR 0.18, 95% CI 0.04 to 0.75; P = 0.02; participants = 72; studies = 1; I2 = 0%), however this difference disappeared at one week (random‐effects OR 1.90, 95% CI 0.32 to 11.44; P = 0.48; participants = 131; studies = 3; I2 = 53%) (Analysis 2.18). Ketamine was associated with increased blood pressure or heart rate over midazolam at one week (random‐effects OR 9.37, 95% CI 2.49 to 35.25; P = 0.0009; participants = 54; studies = 1; I2 = 0%), but not at four weeks (random‐effects OR 1.24, 95% CI 0.06 to 26.93; P = 0.89; participants = 99; studies = 1; I2 = 0%) (Analysis 2.21). Nausea/vomiting was more likely to occur on the day of infusion in participants receiving ketamine over midazolam (random‐effects OR 3.62, 95% CI 1.13 to 11.58; P = 0.03; participants = 152; studies = 2; I2 = 0%), but this did not continue at one week (random‐effects OR 2.57, 95% CI 0.78 to 8.52; P = 0.12; participants = 126; studies = 2; I2 = 0%) or four weeks (random‐effects OR 7.12, 95% CI 0.40 to 125.66; P = 0.18; participants = 99; studies = 1; I2 = 0%) (Analysis 2.32). Sleepiness/drowsiness was also more likely with ketamine over midazolam on the day of infusion (random‐effects OR 0.21, 95% CI 0.07 to 0.66; P = 0.008; participants = 80; studies = 1; I2 = 0%), but not at one week (random‐effects OR 2.57, 95% CI 0.43 to 15.41; P = 0.30; participants = 54; studies = 1; I2 = 100%) (Analysis 2.44).
2.5. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 5: AE Blurred vision
2.14. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 14: AE Dizziness
2.18. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 18: AE General malaise
2.21. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 21: AE Increased blood pressure or heart rate
2.32. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 32: AE Nausea/vomiting
2.44. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 44: AE Sleepiness/drowsiness
No differences between ketamine and midazolam were found for any other adverse event outcomes (Analysis 2.2; Analysis 2.3; Analysis 2.4; Analysis 2.6; Analysis 2.7; Analysis 2.8; Analysis 2.9; Analysis 2.10; Analysis 2.11; Analysis 2.12; Analysis 2.13; Analysis 2.15; Analysis 2.16; Analysis 2.17; Analysis 2.19; Analysis 2.20; Analysis 2.22; Analysis 2.23; Analysis 2.24; Analysis 2.25; Analysis 2.26; Analysis 2.27; Analysis 2.28; Analysis 2.29; Analysis 2.30; Analysis 2.31; Analysis 2.33; Analysis 2.34; Analysis 2.35; Analysis 2.36; Analysis 2.37; Analysis 2.38; Analysis 2.39; Analysis 2.40; Analysis 2.41; Analysis 2.42; Analysis 2.43; Analysis 2.44; Analysis 2.45; Analysis 2.46; Analysis 2.47; Analysis 2.48; Analysis 2.49; Analysis 2.50; Analysis 2.51; Analysis 2.52).
2.2. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 2: AE Abnormal dreams
2.3. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 3: AE Agitation/anxiety
2.4. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 4: AE Back pain
2.6. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 6: AE Chest pain
2.7. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 7: AE Chills
2.8. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 8: AE Constipation
2.9. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 9: AE Decreased energy
2.10. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 10: AE Decreased libido
2.11. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 11: AE Depression
2.12. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 12: AE Diarrhea
2.13. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 13: AE Difficulty swallowing
2.15. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 15: AE Dry mouth
2.16. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 16: AE Dry skin
2.17. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 17: AE Fatigue
2.19. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 19: AE Insomnia
2.20. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 20: AE Headache
2.22. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 22: AE Increase in systolic blood pressure and heart rate
2.23. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 23: AE Increased perspiration
2.24. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 24: AE Indigestion
2.25. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 25: AE Insomnia
2.26. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 26: AE Irritability
2.27. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 27: AE Itching
2.28. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 28: AE Loss of consciousness
2.29. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 29: AE Memory problems
2.30. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 30: AE Muscle/bone/joint pain
2.31. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 31: AE Nasal congestion
2.33. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 33: AE Numbness
2.34. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 34: AE Pain in extremities
2.35. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 35: AE Palpitations
2.36. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 36: AE Poor concentration
2.37. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 37: AE Poor co‐ordination
2.38. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 38: AE Poor quality sleep
2.39. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 39: AE Rash
2.40. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 40: AE Reduced duration of sleep
2.41. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 41: AE Restlessness
2.42. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 42: AE Sensory disturbance
2.43. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 43: AE Sexual dysfunction
2.45. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 45: AE Stomach or abdominal discomfort
2.46. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 46: AE Suicide attempt
2.47. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 47: AE Suicidal ideas
2.48. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 48: AE Tachycardia
2.49. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 49: AE Tinnitus
2.50. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 50: AE Tooth Abscess
2.51. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 51: AE Tremor
2.52. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 52: AE Urination issues
2.3 Efficacy: number of participants who achieve remission
Effect sizes favoured ketamine over midazolam, however wide confidence intervals affected the certainty of these results at all time points: at 24 hours (random‐effects OR 2.21, 95% CI 0.67 to 7.32; P = 0.19; participants = 122; studies = 2; I2 = 40%), low‐certainty evidence, at 72 hours (random‐effects OR 1.73, 95% CI 0.74 to 4.04; P = 0.20; participants = 118; studies = 2; I2 = 0%), at one week (random‐effects OR 1.86, 95% CI 0.80 to 4.32; P = 0.15; participants = 126; studies = 2; I2 = 0%), at two weeks (random‐effects OR 2.29, 95% CI 0.76 to 6.92; P = 0.14; participants = 53; studies = 1; I2 = 0%), at four weeks (random‐effects OR 0.50, 95% CI 0.01 to 19.56; P = 0.71; participants = 5; studies = 1; I2 = 0%), and at three months (Analysis 2.53).
2.53. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 53: Remission rate
2.4 Change scores on depression scale from baseline
We found evidence that ketamine was more effective than midazolam at reducing depression scores at 24 hours (random‐effects SMD ‐0.49, 95% CI ‐0.87 to ‐0.10; P = 0.01; participants = 297; studies = 4; I2 = 56%), very low‐certainty evidence, at 72 hours (random‐effects SMD ‐0.39, 95% CI ‐0.70 to ‐0.08; P = 0.01; participants = 207; studies = 3; I2 = 0%), at one week (random‐effects SMD ‐0.38, 95% CI ‐0.69 to ‐0.08; P = 0.01; participants = 212; studies = 3; I2 = 0%), and at four weeks (random‐effects SMD ‐0.57, 95% CI ‐1.10 to ‐0.04; P = 0.03; participants = 86; studies = 1; I2 = 0%) (Analysis 2.54). However there was no effect at two weeks (random‐effects SMD ‐0.37, 95% CI ‐0.84 to 0.10; P = 0.12; participants = 137; studies = 2; I2 = 36%).
2.54. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 54: Depression rating scale score
2.5 Suicidality
Ketamine was more effective than midazolam in reducing suicidal ideation (random‐effects MD ‐1.32, 95% CI ‐2.52 to ‐0.12; P = 0.03; participants = 57; studies = 1; I2 = 0%) (Analysis 2.55).
2.55. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 55: Suicidal ideation composite
2.6 Cognition
No data were available for this outcome.
2.7 Quality of life
No data were available for this outcome.
2.8 Cost to healthcare services
No data were available for this outcome.
2.9 Acceptability: total dropouts
We found no difference between ketamine and midazolam in terms of participants who dropped out due to any cause (random‐effects OR 0.33, 95% CI 0.05 to 2.09; P = 0.24; 1 study, 72 participants), low‐certainty evidence (Analysis 2.56).
2.56. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 56: Acceptability
3. Ketamine versus thiopental
Two studies contributed to this comparison (Jagtiani 2014; Yoosefi 2014), providing data for 72 hours, two weeks and four weeks. In both studies, ketamine or thiopental was used as an anaesthetic agent for patients undergoing ECT treatment.
3.1 Efficacy: number of participants who respond to treatment
There was no evidence that ketamine was more effective than thiopental in helping participants to achieve response at 72 hours (random‐effects OR 2.64, 95% CI 0.10 to 69.88; P = 0.56; 1 study, 31 participants) or at four weeks (random‐effects OR 0.81, 95% CI 0.05 to 14.28; P = 0.89; 1 study, 31 participants) (Analysis 3.1). There were no participants who met response at two weeks in either the ketamine or thiopental group (1 study, 31 participants).
3.1. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 1: Response rate
3.2 Adverse events
Participants receiving thiopental reported more increased secretions than those receiving ketamine (random‐effects OR 3.86, 95% CI 0.93 to 16.05; P = 0.06; participants = 60; studies = 1; I2 = 0%) (Analysis 3.7).
3.7. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 7: AE Increased secretions
There was no evidence of any difference in the occurrence of any other adverse events Analysis 3.6; Analysis 3.2; Analysis 3.4; Analysis 3.8; Analysis 3.5; Analysis 3.3.
3.6. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 6: AE Heart Rate Rise
3.2. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 2: AE Blood Pressure Rise
3.4. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 4: AE Emergence reactions
3.8. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 8: AE Nausea/vomiting
3.5. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 5: AE Headache
3.3. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 3: AE Delirium
3.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
3.4 Change scores on depression scale from baseline
We found some evidence that ketamine was more effective than thiopental in improving depression rating scale scores at 72 hours (random‐effects MD ‐3.87, 95% CI ‐6.08 to ‐1.66; P = 0.0006; 1 study, 29 participants), at one week (random‐effects MD ‐6.96, 95% CI ‐9.82 to ‐4.10; P < 0.00001; participants = 60; studies = 1; I2 = 0%); and two weeks (random‐effects MD ‐3.46, 95% CI ‐4.88 to ‐2.04; P < 0.00001; participants = 89; studies = 2; I2 = 97%) (Analysis 3.9). No difference was found between ketamine and thiopental at four weeks (random‐effects MD ‐0.22, 95% CI ‐2.64 to 2.20; P = 0.86; 1 study, 29 participants). However, these findings were based on a very small number of participants.
3.9. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 9: Depression rating scale score
3.5 Suicidality
No data were available for this outcome.
3.6 Cognition
No data were available for this outcome.
3.7 Quality of life
No data were available for this outcome.
3.8 Cost to healthcare services
No data were available for this outcome.
3.9 Acceptability: total dropouts
We found no difference between ketamine and thiopental in terms of participants who dropped out due to any cause (random‐effects OR 4.68, 95% CI 0.21 to 105.89; P = 0.33; 1 study, 31 participants) (Analysis 3.10).
3.10. Analysis.

Comparison 3: Ketamine versus Thiopental, Outcome 10: Acceptability
4. Ketamine versus methohexital
One study contributed to this comparison (Carspecken 2018), providing depression rating scale score data for 72 hours.
4.1 Efficacy: number of participants who respond to treatment
No data were available for this outcome.
4.2 Adverse events
No data were available for this outcome.
4.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
4.4 Change scores on depression scale from baseline
There was no difference in depression rating scale scores between ketamine and methohexital at 72 hours (random‐effects MD ‐0.80, 95% CI ‐4.45 to 2.85; P = 0.67; participants = 50; studies = 1; I2 = 0%) (Analysis 4.1).
4.1. Analysis.

Comparison 4: Ketamine versus Methohexital, Outcome 1: Depression rating scale score
4.5 Suicidality
No data were available for this outcome.
4.6 Cognition
No data were available for this outcome.
4.7 Quality of life
No data were available for this outcome.
4.8 Cost to healthcare services
No data were available for this outcome.
4.9 Acceptability: total dropouts
No data were available for this outcome.
5. Ketamine versus propofol
One study contributed to this comparison (Fernie 2017), providing depression rating scale score data at two weeks and three months.
5.1 Efficacy: number of participants who respond to treatment
No data were available for this outcome.
5.2 Adverse events
No data were available for this outcome.
5.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
5.4 Change scores on depression scale from baseline
No evidence was found of a difference in depression rating scale scores between ketamine and propofol at two weeks (random‐effects MD 3.67, 95% CI ‐0.84 to 8.18; P = 0.11; participants = 31; studies = 1, or at three months (random‐effects MD 2.00, 95% CI ‐4.93 to 8.93; P = 0.57; participants = 26; studies = 1), based on the small number of participants included in this analysis (Analysis 5.1).
5.1. Analysis.

Comparison 5: Ketamine versus Propofol, Outcome 1: Depression rating scale score
5.5 Suicidality
No data were available for this outcome.
5.6 Cognition
No data were available for this outcome.
5.7 Quality of life
No data were available for this outcome.
5.8 Cost to healthcare services
No data were available for this outcome.
5.9 Acceptability: total dropouts
No data were available for this outcome.
6. Ketamine versus remifentanil hydrochloride
One study contributed to this comparison (Sumner 2020), providing depression rating scale score data at 24 hours, one week and two weeks.
6.1 Efficacy: number of participants who respond to treatment
No data were available for this outcome.
6.2 Adverse events
No data were available for this outcome.
6.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
6.4 Change scores on depression scale from baseline
There was some evidence that ketamine improved depression rating scale scores over remifentanil hydrochloride at 24 hours (random‐effects MD ‐7.74, 95% CI ‐14.03 to ‐1.45; P = 0.02; participants = 30; studies = 1; I2 = 0%), and at one week (random‐effects MD ‐7.54, 95% CI ‐14.13 to ‐0.95; P = 0.02; participants = 30; studies = 1; I2 = 0%). Results from one small study found no difference at two weeks (random‐effects MD ‐1.00, 95% CI ‐6.98 to 4.98; P = 0.74; participants = 30; studies = 1; I2 = 0%) (Analysis 6.1).
6.1. Analysis.

Comparison 6: Ketamine versus Remifentanil hydrochloride, Outcome 1: Depression rating scale score
6.5 Suicidality
No data were available for this outcome.
6.6 Cognition
No data were available for this outcome.
6.7 Quality of life
No data were available for this outcome.
6.8 Cost to healthcare services
No data were available for this outcome.
6.9 Acceptability: total dropouts
No data were available for this outcome.
7. Ketamine versus esketamine
One study contributed to this comparison (Correia‐Melo 2020), providing response efficacy data at 24 hours, 72 hours, and one week.
7.1 Efficacy: number of participants who respond to treatment
There was no difference in response between ketamine and esketamine at 24 hours (random‐effects OR 1.07, 95% CI 0.40 to 2.89; P = 0.89; participants = 63; studies = 1; I2 = 0%), at 72 hours (random‐effects OR 1.56, 95% CI 0.58 to 4.22; P = 0.38; participants = 63; studies = 1; I2 = 100%), and at one week (random‐effects OR 2.34, 95% CI 0.85 to 6.45; P = 0.10; participants = 63; studies = 1; I2 = 0%) (Analysis 7.1).
7.1. Analysis.

Comparison 7: Ketamine versus Esketamine, Outcome 1: Response Rate
7.2 Adverse events
No data were available for this outcome.
7.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
7.4 Change scores on depression scale from baseline
No data were available for this outcome.
7.5 Suicidality
No data were available for this outcome.
7.6 Cognition
We found no difference in cognition scores between ketamine and esketamine during infusion (random‐effects MD 3.30, 95% CI ‐4.70 to 11.30; P = 0.42; participants = 63; studies = 1; I2 = 0%) (Analysis 7.2).
7.2. Analysis.

Comparison 7: Ketamine versus Esketamine, Outcome 2: Cognition
7.7 Quality of life
No data were available for this outcome.
7.8 Cost to healthcare services
No data were available for this outcome.
7.9 Acceptability: total dropouts
No data were available for this outcome.
C. Ketamine versus ECT
8. Ketamine versus ECT
One study contributed to this comparison (Ghasemi 2013), providing data for 24 hours, 72 hours, one week and two weeks.
8.1 Efficacy: number of participants who respond to treatment
We found very limited evidence that ketamine was more effective than ECT in terms of response at 24 hours (random‐effects OR 28.00, 95% CI 2.07 to 379.25; P = 0.01; 1 study, 18 participants) and 72 hours (random‐effects OR 12.25, 95% CI 1.33 to 113.06; P = 0.03; 1 study, 18 participants) (Analysis 8.1). Results from one very small study found no difference in response efficacy between ketamine and ECT at one week (random‐effects OR 3.35, 95% CI 0.12 to 93.83; P = 0.48; 1 study, 18 participants) and at two weeks (random‐effects OR 3.35, 95% CI 0.12 to 93.83; P = 0.48; 1 study, 18 participants).
8.1. Analysis.

Comparison 8: Ketamine versus ECT, Outcome 1: Response rate
8.2 Adverse events
We found no difference between ketamine and ECT in terms of adverse events. However, the only adverse events reported in this study were blood pressure and heart rate.
8.3 Efficacy: number of participants who achieve remission
Results from one small study found no difference in terms of remission between ECT and ketamine at any time point; at 24 hours (random‐effects OR 3.35, 95% CI 0.12 to 93.83; P = 0.48; 1 study, 18 participants); at 72 hours (random‐effects OR 3.35, 95% CI 0.12 to 93.83; P = 0.48; 1 study, 18 participants); at one week (random‐effects OR 10.23, 95% CI 0.45 to 233.23; P = 0.14; 1 study, 18 participants); and at two weeks (random‐effects OR 4.00, 95% CI 0.33 to 48.66; P = 0.28; 1 study, 18 participants) (Analysis 8.3).
8.3. Analysis.

Comparison 8: Ketamine versus ECT, Outcome 3: Remission rate
8.4 Change scores on depression scale from baseline
We found some evidence that ketamine may be more effective than ECT at 24 hours (random‐effects MD ‐8.90, 95% CI ‐11.72 to ‐6.08; P < 0.00001; participants = 18; studies = 1; I2 = 0%), at 72 hours (random‐effects MD ‐9.00, 95% CI ‐14.24 to ‐3.76; P = 0.0008; 1 study, 18 participants) and at one week (random‐effects MD ‐6.66, 95% CI ‐11.20 to ‐2.12; P = 0.004; 1 study, 18 participants). No difference in effect of ketamine compared with ECT at two weeks, although this may be impacted by the small sample included in this analysis (random‐effects MD ‐4.45, 95% CI ‐9.01 to 0.11; P = 0.06; 1 study, 18 participants) (Analysis 8.4).
8.4. Analysis.

Comparison 8: Ketamine versus ECT, Outcome 4: Depression rating scale score
8.5 Suicidality
No data were available for this outcome.
8.6 Cognition
No data were available for this outcome.
8.7 Quality of life
No data were available for this outcome.
8.8 Cost to healthcare services
No data were available for this outcome.
8.9 Acceptability: total dropouts
There were no patients who dropped out of the trial in either the ketamine or ECT group in Ghasemi 2013.
D. Other glutamate receptor modulators versus placebo
9. Esketamine versus placebo
Nine studies contributed to this comparison (Canuso 2018; Daly 2018; Fedgchin 2019; Fu 2020; Ionescu 2020; Jarventausta 2013; Ochs‐Ross 2020; Popova 2019; Singh 2016 b). Data ere available for 24 hours, 72 hours, one week, two weeks, four weeks, and three months. Table 3
9.1 Efficacy: number of participants who respond to treatment
Esketamine was more efficacious than placebo in terms of response at 24 hours (random‐effects OR 2.11, 95% CI 1.20 to 3.68; P = 0.009; participants = 1071; studies = 5; I2 = 50%), low‐certainty evidence, at one week (random‐effects OR 1.60, 95% CI 1.09 to 2.34; P = 0.02; participants = 1115; studies = 6; I2 = 20%), at two weeks (random‐effects OR 1.57, 95% CI 1.09 to 2.28; P = 0.02; participants = 451; studies = 2; I2 = 0%), and at four weeks (random‐effects OR 1.84, 95% CI 1.44 to 2.37; P < 0.00001; participants = 1117; studies = 5; I2 = 0%) (Analysis 9.1). There was no difference found at 72 hours (random‐effects OR 1.34, 95% CI 0.92 to 1.96; P = 0.13; participants = 451; studies = 2; I2 = 0%). Figure 6.
9.1. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 1: Response rate
6.

Forest plot of comparison: 9 Esketamine versus placebo, outcome: 9.1 Response rate.
9.2 Adverse events
Participants assigned to receive esketamine reported more changes in blood pressure over esketamine (random‐effects OR 2.67, 95% CI 1.52 to 4.70; P = 0.0007; participants = 933; studies = 4; I2 = 0%) (Analysis 9.5). There was an increase in constipation in those receiving esketamine over placebo (random‐effects OR 4.07, 95% CI 1.60 to 10.39; P = 0.003; participants = 452; studies = 2; I2 = 0%) (Analysis 9.6). Dissociative symptoms were also increased for esketamine over placebo (random‐effects OR 8.76, 95% CI 5.19 to 14.77; P < 0.00001; participants = 933; studies = 4; I2 = 0%) (Analysis 9.11), as was dizziness (random‐effects OR 3.67, 95% CI 2.54 to 5.31; P < 0.00001; participants = 933; studies = 4; I2 = 0%) (Analysis 9.12), and dizziness postural (random‐effects OR 4.70, 95% CI 1.06 to 20.80; P = 0.04; participants = 569; studies = 2; I2 = 7%) (Analysis 9.13). Participants receiving esketamine were found to be more likely to feel drunk (random‐effects OR 7.58, 95% CI 1.37 to 41.77; P = 0.02; participants = 571; studies = 2; I2 = 0%) (Analysis 9.17), and experience nausea/vomiting (random‐effects OR 3.24, 95% CI 1.84 to 5.72; P < 0.0001; participants = 933; studies = 4; I2 = 60%) (Analysis 9.26). Paresthesia/neuropathy exacerbation was also increased in participants receiving esketamine compared with placebo (random‐effects OR 3.51, 95% CI 1.62 to 7.62; P = 0.001; participants = 708; studies = 3; I2 = 18%) (Analysis 9.27), as was sensory disturbance (random‐effects OR 7.25, 95% CI 3.55 to 14.78; P < 0.00001; participants = 796; studies = 3; I2 = 0%) (Analysis 9.30). There was a difference in favour of placebo over esketamine for sedation (random‐effects OR 5.31, 95% CI 2.18 to 12.94; P = 0.0002; participants = 796; studies = 3; I2 = 0%) (Analysis 9.31) and sleepiness/drowsiness (random‐effects OR 2.11, 95% CI 1.39 to 3.21; P = 0.0005; participants = 796; studies = 3; I2 = 0%) (Analysis 9.32). We also found that esketamine increased vertigo (random‐effects OR 12.25, 95% CI 4.09 to 36.67; P < 0.00001; participants = 796; studies = 3; I2 = 0%) (Analysis 9.39) and blurred vision (random‐effects OR 3.02, 95% CI 1.37 to 6.66; P = 0.006; participants = 796; studies = 3; I2 = 13%) compared with placebo (Analysis 9.40).
9.5. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 5: AE Change in blood pressure
9.6. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 6: AE Constipation
9.11. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 11: AE Dissociative symptoms
9.12. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 12: AE Dizziness
9.13. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 13: AE Dizziness postural
9.17. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 17: AE Feeling drunk
9.26. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 26: AE Nausea/vomiting
9.27. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 27: AE Paresthesia/neuropathy exacerbation
9.30. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 30: AE Sensory disturbance
9.31. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 31: AE Sedation
9.32. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 32: AE Sleepiness/drowsiness
9.39. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 39: AE Vertigo
9.40. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 40: AE Vision blurred
We found no differences between esketamine and placebo for any other adverse events (Analysis 9.2; Analysis 9.3; Analysis 9.4; Analysis 9.7; Analysis 9.8; Analysis 9.9; Analysis 9.10; Analysis 9.14; Analysis 9.15; Analysis 9.16; Analysis 9.18; Analysis 9.19; Analysis 9.20; Analysis 9.21; Analysis 9.22; Analysis 9.23; Analysis 9.24; Analysis 9.25; Analysis 9.28; Analysis 9.29; Analysis 9.33; Analysis 9.34; Analysis 9.35; Analysis 9.36; Analysis 9.37; Analysis 9.38).
9.2. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 2: AE Aggression
9.3. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 3: AE Agitation/anxiety
9.4. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 4: AE Arrhythmia
9.7. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 7: AE Depersonalisation/derealization
9.8. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 8: AE Depression
9.9. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 9: AE Diabetic ketoacidosis
9.10. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 10: AE Diarrhoea
9.14. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 14: AE Double vision
9.15. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 15: AE Euphoria
9.16. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 16: AE Fatigue
9.18. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 18: AE Headache
9.19. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 19: AE Hypertransaminasemia
9.20. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 20: AE Increased sweating
9.21. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 21: AE Infections and Infestations
9.22. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 22: AE Insomnia
9.23. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 23: AE Lethargy
9.24. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 24: AE Mental impairment
9.25. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 25: AE Nasal discomfort
9.28. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 28: AE Pericardial effusion
9.29. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 29: AE Pneumothorax
9.33. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 33: AE Sore throat
9.34. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 34: AE Suicide attempt
9.35. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 35: AE Suicidal ideas
9.36. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 36: AE Taste perversion
9.37. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 37: AE Tremor
9.38. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 38: AE Urination issues
9.3 Efficacy: number of participants who achieve remission
Participants assigned esketamine treatment achieved remission more than those receiving placebo at 24 hours (random‐effects OR 2.74, 95% CI 1.71 to 4.40; P < 0.0001; participants = 894; studies = 5; I2 = 0%), moderate‐certainty evidence, at two weeks (random‐effects OR 1.52, 95% CI 1.07 to 2.16; P = 0.02; participants = 832; studies = 4; I2 = 0%), and at four weeks (random‐effects OR 1.57, 95% CI 1.18 to 2.10; P = 0.002; participants = 957; studies = 5; I2 = 0%) (Analysis 9.41). There were no differences found at 72 hours (random‐effects OR 1.55, 95% CI 0.91 to 2.64; P = 0.11; participants = 517; studies = 3; I2 = 24%) or at one week (random‐effects OR 1.54, 95% CI 0.88 to 2.69; P = 0.13; participants = 948; studies = 6; I2 = 30%).
9.41. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 41: Remission rate
9.4 Change scores on depression scale from baseline
Esketamine reduced depression rating scale scores over placebo at 24 hours (random‐effects SMD ‐0.31, 95% CI ‐0.45 to ‐0.17; P < 0.0001; participants = 824; studies = 4; I2 = 0%), moderate‐certainty evidence, at 72 hours (random‐effects SMD ‐0.30, 95% CI ‐0.50 to ‐0.11; P = 0.002; participants = 517; studies = 3; I2 = 14%), at one week (random‐effects SMD ‐0.23, 95% CI ‐0.37 to ‐0.10; P = 0.0008; participants = 884), at two weeks (random‐effects SMD ‐0.21, 95% CI ‐0.34 to ‐0.07; P = 0.003; participants = 857; studies = 4; I2 = 0%), and at four weeks (random‐effects SMD ‐0.27, 95% CI ‐0.39 to ‐0.16; P < 0.00001; participants = 1182; studies = 6; I2 = 0%) (Analysis 9.42). However, there was no difference at three months (random‐effects SMD ‐0.12, 95% CI ‐0.75 to 0.52; P = 0.72; participants = 38; studies = 1; I2 = 0%).
9.42. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 42: Depression rating scale score
9.5 Suicidality
There were no differences in suicidal ideation scores at any time point: at 24 hours (random‐effects MD ‐0.15, 95% CI ‐0.44 to 0.15; P = 0.33; participants = 450; studies = 2; I2 = 0%), at 72 hours (random‐effects MD ‐0.20, 95% CI ‐0.49 to 0.08; P = 0.16; participants = 451; studies = 2; I2 = 0%), at one week (random‐effects MD 0.01, 95% CI ‐0.10 to 0.13; P = 0.83; participants = 660; studies = 3; I2 = 0%), at two weeks (random‐effects MD ‐0.10, 95% CI ‐0.22 to 0.02; P = 0.10; participants = 659; studies = 3; I2 = 0%), and at four weeks (random‐effects MD ‐0.04, 95% CI ‐0.12 to 0.05; P = 0.40; participants = 647; studies = 3; I2 = 0%) (Analysis 9.43).
9.43. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 43: Suicidal ideation composite
9.6 Cognition
No data were available for this outcome.
9.7 Quality of life
No data were available for this outcome.
9.8 Cost to healthcare services
No data were available for this outcome.
9.9 Acceptability: total dropouts
We found no difference between esketamine and placebo in terms of participants who dropped out due to any cause (random‐effects OR 1.58, 95% CI 0.92 to 2.73; P = 0.10; participants = 773; studies = 5; I2 = 8%) moderate‐certainty evidence (Analysis 9.44).
9.44. Analysis.

Comparison 9: Esketamine versus placebo, Outcome 44: Acceptability
10. Memantine versus placebo
Four studies contributed to this comparison, including one new study (Amidfar 2016; Omranifard 2014; Smith 2013; Zarate 2006b), providing data at one week, two weeks, four weeks and three months.
10.1 Efficacy: number of participants who respond to treatment
We found no evidence that memantine was more effective than placebo in response at any time point; at one week (random‐effects OR 1.07, 95% CI 0.06 to 18.82; P = 0.96; participants = 63; studies = 2; I2 = 0%) ; two weeks (random‐effects OR 0.31, 95% CI 0.01 to 8.28; P = 0.49; participants = 32; studies = 1; I2 = 0%); four weeks (random‐effects OR 1.22, 95% CI 0.25 to 5.89; P = 0.81; participants = 185; studies = 4; I2 = 57%); and at three months (random‐effects OR 0.48, 95% CI 0.18 to 1.24; P = 0.13; participants = 123; studies = 3; I2 = 0%) (Analysis 10.1).
10.1. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 1: Response rate
10.2 Adverse events
We found no difference between memantine and placebo for any adverse events (Analysis 10.2; Analysis 10.3; Analysis 10.4; Analysis 10.5; Analysis 10.6; Analysis 10.7; Analysis 10.8; Analysis 10.9; Analysis 10.10; Analysis 10.11; Analysis 10.12; Analysis 10.13; Analysis 10.14; Analysis 10.15; Analysis 10.16; Analysis 10.17; Analysis 10.18; Analysis 10.19; Analysis 10.20; Analysis 10.21; Analysis 10.22; Analysis 10.23; Analysis 10.24; Analysis 10.25; Analysis 10.26; Analysis 10.27; Analysis 10.28; Analysis 10.29; Analysis 10.30; Analysis 10.31; Analysis 10.32; Analysis 10.33; Analysis 10.34; Analysis 10.35; Analysis 10.36; Analysis 10.37; Analysis 10.38; Analysis 10.39; Analysis 10.40; Analysis 10.41; Analysis 10.42; Analysis 10.43; Analysis 10.44; Analysis 10.45; Analysis 10.46; Analysis 10.47; Analysis 10.48; Analysis 10.49; Analysis 10.50; Analysis 10.51; Analysis 10.52; Analysis 10.53; Analysis 10.54; Analysis 10.55; Analysis 10.56; Analysis 10.57; Analysis 10.58; Analysis 10.59).
10.2. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 2: AE Abdominal Pain
10.3. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 3: AE Active suicidal ideation
10.4. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 4: AE Agitation/anxiety
10.5. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 5: AE Appetite increase
10.6. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 6: AE Back pain
10.7. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 7: AE Balance or gait problems
10.8. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 8: AE Carbohydrate craving
10.9. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 9: AE Chest pain
10.10. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 10: AE Chills
10.11. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 11: AE Clammy hands
10.12. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 12: AE Confusion/decreased mental clarity
10.13. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 13: AE Conjunctival swelling
10.14. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 14: AE Constipation
10.15. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 15: AE Decreased appetite
10.16. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 16: AE Delusions
10.17. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 17: AE Diaphoresis
10.18. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 18: AE Difficulty breathing
10.19. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 19: AE Dissociative symptoms
10.20. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 20: AE Dizziness
10.21. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 21: AE Dry mouth
10.22. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 22: AE Dyskinesia
10.23. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 23: AE Dyspepsia
10.24. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 24: AE Ear pain/jaw pain
10.25. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 25: AE Emotional lability
10.26. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 26: AE Eye photosensitivity
10.27. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 27: AE Facial twitching
10.28. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 28: AE Falls
10.29. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 29: AE Fatigue
10.30. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 30: AE Feeling flushed/hot
10.31. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 31: AE Generalised aches
10.32. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 32: AE Head pressure/ear pressure
10.33. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 33: AE Headache
10.34. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 34: AE Heart palpitations
10.35. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 35: AE Hypomania/mania
10.36. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 36: AE Increased menstrual pain
10.37. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 37: AE Insomnia
10.38. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 38: AE Internal sensation of speed or rapid thoughts
10.39. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 39: AE Irritability
10.40. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 40: AE Leg weakness
10.41. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 41: AE Nausea
10.42. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 42: AE Nightmares
10.43. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 43: AE Paresthesia/neuropathy exacerbation
10.44. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 44: AE Passive suicidal ideation
10.45. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 45: AE Perceived weight gain
10.46. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 46: AE Perceived weight loss
10.47. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 47: AE Pruritus
10.48. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 48: AE Rash
10.49. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 49: AE Sedation
10.50. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 50: AE Skin lesion
10.51. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 51: AE Sleepiness/drowsiness
10.52. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 52: AE Sleepwalking
10.53. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 53: AE Sore throat
10.54. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 54: AE Taste perversion
10.55. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 55: AE Tinnitus
10.56. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 56: AE Upper respiratory infection symptoms
10.57. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 57: AE Vomiting
10.58. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 58: AE Worsened acne
10.59. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 59: AE Worsened sleep apnoea
10.3 Efficacy: number of participants who achieve remission
We found no evidence that memantine was more effective than placebo in remission at any time point; at one week (random‐effects OR ‐0.11, 95% CI ‐1.10 to 0.89; P = 0.26; participants = 59; studies = 2; I2 = 72%); at four weeks (random‐effects OR 1.39, 95% CI 0.46 to 4.26; P = 0.56; participants = 185; studies = 4; I2 = 0%); or at three months (random‐effects OR 0.76, 95% CI 0.15 to 3.77; P = 0.74; participants = 123; studies = 3; I2 = 0%) (Analysis 10.60).
10.60. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 60: Remission rate
10.4 Change scores on depression scale from baseline
There was no difference in depression rating scale score changes between memantine and placebo at any of the time points; at one week (random‐effects SMD ‐0.11, 95% CI ‐1.10 to 0.89; P = 0.84; participants = 59; studies = 2; I2 = 72%); at two weeks (random‐effects SMD ‐0.09, 95% CI ‐0.83 to 0.65; P = 0.81; participants = 28; studies = 1; I2 = 100%); at four weeks (random‐effects SMD 0.11, 95% CI ‐0.26 to 0.48; P = 0.56; participants = 112; studies = 3; I2 = 0%); and at three months (random‐effects SMD 0.23, 95% CI ‐0.14 to 0.61; P = 0.22; participants = 110; studies = 3; I2 = 0%) (Analysis 10.61).
10.61. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 61: Depression scale rating score
10.5 Suicidality
No data were available for this outcome.
10.6 Cognition
No data were available for this outcome.
10.7 Quality of life
We found no difference between memantine and placebo in quality of life at four weeks (random‐effects MD ‐0.70, 95% CI ‐5.04 to 3.64; P = 0.75; 1 study, 57 participants) and at three months (random‐effects MD ‐1.21, 95% CI ‐5.78 to 3.36; P = 0.60; 1 study, 57 participants) (Analysis 10.62).
10.62. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 62: Quality of life
10.8 Cost to healthcare services
No data were available for this outcome.
10.9 Acceptability: total dropouts
We found no difference between memantine and placebo in terms of participants who dropped out due to any cause (random‐effects OR 0.78, 95% CI 0.23 to 2.66; P = 0.69; I2 = 0%; 3 studies, 123 participants) (Analysis 10.63), nor due to side effects (random‐effects OR 0.68, 95% CI 0.10 to 4.47; P = 0.68; I2= 0%; 2 studies, 63 participants) (Analysis 10.64).
10.63. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 63: Acceptability
10.64. Analysis.

Comparison 10: Memantine versus Placebo, Outcome 64: Acceptability ‐ adverse events
11. Lanicemine (AZD6765) versus placebo
Two studies contributed to this comparison, including one new study (Zarate 2013; Sanacora 2017), providing outcome data at 24 hours, 72 hours, one week and four weeks.
11.1 Efficacy: number of participants who respond to treatment
There was no evidence that lanicemine was more effective than placebo in response at any time point; at 24 hours (random‐effects OR 7.74, 95% CI 0.35 to 170.10; P = 0.19; 1 study, 22 participants); at 72 hours (random‐effects OR 2.74, 95% CI 0.10 to 74.87; P = 0.55; 1 study, 22 participants); at one week (random‐effects OR 2.74, 95% CI 0.10 to 74.87; P = 0.55; 1 study, 22 participants); or at four weeks (random‐effects OR 1.03, 95% CI 0.63 to 1.69; P = 0.92; participants = 298; studies = 1; I2 = 0%) (Analysis 11.1).
11.1. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 1: Response rate
11.2 Adverse events
Lanicemine was found to increase the incidence of dizziness over placebo (random‐effects OR 5.02, 95% CI 2.46 to 10.26; P < 0.00001; participants = 301; studies = 1; I2 = 0%) (Analysis 11.6).
11.6. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 6: AE Dizziness
There were no other differences between lanicemine and placebo for adverse events (Analysis 11.2; Analysis 11.3; Analysis 11.4; Analysis 11.5; Analysis 11.7; Analysis 11.8; Analysis 11.9; Analysis 11.10; Analysis 11.11; Analysis 11.12; Analysis 11.13; Analysis 11.14; Analysis 11.15; Analysis 11.16).
11.2. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 2: AE Agitation/anxiety
11.3. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 3: AE Back pain
11.4. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 4: AE Blood Pressure Rise
11.5. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 5: AE Dissociative symptoms
11.7. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 7: AE Dry mouth
11.8. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 8: AE Feeling drunk
11.9. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 9: AE Insomnia
11.10. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 10: AE Muscle/bone/joint pain
11.11. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 11: AE Nausea
11.12. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 12: AE Rash
11.13. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 13: AE Sedation
11.14. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 14: AE Upper respiratory infection symptoms
11.15. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 15: AE Vomiting
11.16. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 16: AE Weight gain
11.3 Efficacy: number of participants who achieve remission
We found no evidence that lanicemine was more effective than placebo in remission at 24 hours (random‐effects OR 5.00, 95% CI 0.21 to 117.21; P = 0.32; 1 study, 22 participants), 72 hours (random‐effects OR 2.74, 95% CI 0.10 to 74.87; P = 0.55; 1 study, 22 participants); at one week (random‐effects OR 2.74, 95% CI 0.10 to 74.87; P = 0.55; 1 study, 22 participants); or at four weeks (random‐effects OR 1.38, 95% CI 0.75 to 2.52; P = 0.30; participants = 298; studies = 1; I2 = 0%) (Analysis 11.17).
11.17. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 17: Remission rate
11.4 Change scores on depression scale from baseline
There was no difference between lanicemine and placebo at any time point. The effect of lanicemine compared with placebo at 24 hours was (random‐effects MD ‐8.65, 95% CI ‐17.81 to 0.51; P = 0.06; 1 study, 22 participants), at 72 hours was (random‐effects MD ‐6.27, 95% CI ‐13.93 to 1.39; P = 0.11; 1 study, 21 participants), at one week was (random‐effects MD ‐6.55, 95% CI ‐14.07 to 0.97; P = 0.09; 1 study, 21 participants), at four weeks was (random‐effects MD ‐0.11, 95% CI ‐1.42 to 1.20; P = 0.87; participants = 298; studies = 1; I2 = 0%), and at three months was (random‐effects MD 0.51, 95% CI ‐1.05 to 2.07; P = 0.52; participants = 298; studies = 1; I2 = 0%) (Analysis 11.18).
11.18. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 18: Depression rating scale score
11.5 Suicidality
No data were available for this outcome.
11.6 Cognition
No data were available for this outcome.
11.7 Quality of life
No data were available for this outcome.
11.8 Cost to healthcare services
No data were available for this outcome.
11.9 Acceptability: total dropouts
We found no difference between lanicemine and placebo in terms of participants who dropped out due to any cause (random‐effects OR 2.74, 95% CI 0.10 to 74.87; P = 0.55, participants = 22, studies = 1) (Analysis 11.19).
11.19. Analysis.

Comparison 11: Lanicemine (AZD6765) versus Placebo, Outcome 19: Acceptability
12. Org26576 versus placebo
Two studies contributed to this comparison (Nations 2012 (part I); Nations 2012 (part II)), providing data at 24 hours, 72 hours, one week, two weeks and four weeks.
12.1 Efficacy: number of participants who respond to treatment
There was no evidence that Org26576 was more effective than placebo in achieving response at any time point; at 24 hours (random‐effects OR 0.81, 95% CI 0.09 to 7.13; P = 0.85; I2 = 0%; 2 studies, 54 participants); at 72 hours (random‐effects OR 0.80, 95% CI 0.16 to 3.90; P = 0.78; I2 = 0%; 2 studies, 54 participants); at one week (random‐effects OR 1.40, 95% CI 0.31 to 6.28; P = 0.63; I2 = 0%; 2 studies, 54 participants); at two weeks (random‐effects OR 2.24, 95% CI 0.61 to 8.22; P = 0.22; I2 = 0%; 2 studies, 54 participants), and at four weeks (random‐effects OR 0.82, 95% CI 0.18 to 3.74; P = 0.80; 1 study, 30 participants) (Analysis 12.1).
12.1. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 1: Response rate
12.2 Adverse events
Org26576 resulted in an increased number of reports of nausea over placebo (random‐effects OR 4.50, 95% CI 1.05 to 19.28; P = 0.04; 2 studies, 54 participants) (Analysis 12.13).
12.13. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 13: AE Nausea
We found no other differences in adverse events for Org26576 and placebo (Analysis 12.2; Analysis 12.3; Analysis 12.4; Analysis 12.5; Analysis 12.6; Analysis 12.7; Analysis 12.8; Analysis 12.9; Analysis 12.10; Analysis 12.11; Analysis 12.12; Analysis 12.13; Analysis 12.14; Analysis 12.15; Analysis 12.16; Analysis 12.17; Analysis 12.18; Analysis 12.19; Analysis 12.20).
12.2. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 2: AE Abnormal dreams
12.3. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 3: AE Back pain
12.4. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 4: AE Disturbance in attention
12.5. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 5: AE Dizziness
12.6. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 6: AE Fatigue
12.7. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 7: AE Feeling drunk
12.8. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 8: AE Headache
12.9. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 9: AE Insomnia
12.10. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 10: AE Irritability
12.11. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 11: AE Muscle twitching
12.12. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 12: AE Nasal congestion
12.14. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 14: AE Palpitations
12.15. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 15: AE Post‐lumbar puncture syndrome
12.16. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 16: AE Rash
12.17. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 17: AE Sedation
12.18. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 18: AE Sensory disturbance
12.19. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 19: AE Sleepiness/drowsiness
12.20. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 20: AE Total
12.3 Efficacy: number of participants who achieve remission
We found that there were no participants who met remission at 24 hours in either study, and no participants who met remission at 72 hours in Nations 2012 (part II). There was no evidence that Org26576 was more effective than placebo in remission rates at all other time points; at 72 hours (random‐effects OR 0.47, 95% CI 0.03 to 8.60; P = 0.61; 1 study, 24 participants); at one week (random‐effects OR 1.52, 95% CI 0.21 to 11.06; P = 0.68; I2 = 0%; 2 studies, 54 participants); at two weeks (random‐effects OR 2.29, 95% CI 0.43 to 12.15; P = 0.33; I2 = 0%; 2 studies, 54 participants) and at four weeks (random‐effects OR 0.64, 95% CI 0.13 to 3.14; P = 0.59; 1 study, 30 participants) (Analysis 12.21).
12.21. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 21: Remission rate
12.4 Change scores on depression scale from baseline
We found no evidence that Org26576 was more effective than placebo at any time point; at 24 hours (random‐effects MD ‐0.51, 95% CI ‐4.14 to 3.13; P = 0.78; I2 = 0%; 2 studies, 54 participants); at 72 hours (random‐effects MD ‐0.88, 95% CI ‐4.67 to 2.91; P = 0.65; I2 = 0%; 2 studies, 54 participants); at one week (random‐effects MD ‐1.43, 95% CI ‐5.31 to 2.44; P = 0.47; I2= 0%; 2 studies, 54 participants); at two weeks (random‐effects MD ‐2.61, 95% CI ‐7.32 to 2.09; P = 0.28; I2 = 0%; 2 studies, 54 participants) and at four weeks (random‐effects MD ‐1.25, 95% CI ‐8.14 to 5.64; P = 0.72; 1 study, 30 participants) (Analysis 12.22).
12.22. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 22: Depression rating scale score
12.5 Suicidality
No data were available for this outcome.
12.6 Cognition
No data were available for this outcome.
12.7 Quality of life
No data were available for this outcome.
12.8 Cost to healthcare services
No data were available for this outcome.
12.9 Acceptability: dropouts due to adverse effects
We found no difference between Org26576 and placebo in terms of participants who dropped out due to side effects (random‐effects OR 0.50, 95% CI 0.05 to 5.17; P = 0.56; I2 = 0%; 2 studies, 54 participants) (Analysis 12.23). No information about all‐cause dropouts was reported.
12.23. Analysis.

Comparison 12: Org 26576 versus Placebo, Outcome 23: Acceptability ‐ adverse events
13. Riluzole versus placebo
Two studies contributed to this comparison, including one new study (Ibrahim 2012a; Salardini 2016), providing data at 24 hours, 72 hours, one week, two weeks and four weeks.
13.1 Efficacy: number of participants who respond to treatment
There was no evidence that riluzole was more effective than placebo in increasing response rates at any other time point; at 24 hours (random‐effects OR 1.23, 95% CI 0.35 to 4.36; P = 0.75; participants = 42; studies = 1; I2 = 0%); at 72 hours (random‐effects OR 2.62, 95% CI 0.64 to 10.61; P = 0.18; participants = 42; studies = 1; I2 = 0%); or at two weeks (random‐effects OR 1.41, 95% CI 0.27 to 7.26; P = 0.68; participants = 102; studies = 2; I2 = 0%), or at four weeks (random‐effects OR 1.57, 95% CI 0.09 to 28.00; P = 0.76; participants = 102; studies = 2; I2 = 80%) (Analysis 13.1).
13.1. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 1: Response rate
13.2 Adverse events
We found no difference between riluzole and placebo in terms of adverse events (Analysis 13.2; Analysis 13.3; Analysis 13.4; Analysis 13.5; Analysis 13.6; Analysis 13.7; Analysis 13.8; Analysis 13.9; Analysis 13.10; Analysis 13.11; Analysis 13.12; Analysis 13.13; Analysis 13.14; Analysis 13.15; Analysis 13.16; Analysis 13.17; Analysis 13.18; Analysis 13.19; Analysis 13.20; Analysis 13.21; Analysis 13.22; Analysis 13.23; Analysis 13.24; Analysis 13.25; Analysis 13.26; Analysis 13.27; Analysis 13.28; Analysis 13.29; Analysis 13.30; Analysis 13.31; Analysis 13.32; Analysis 13.33; Analysis 13.34; Analysis 13.35; Analysis 13.36; Analysis 13.37; Analysis 13.38; Analysis 13.39; Analysis 13.40; Analysis 13.41; Analysis 13.42; Analysis 13.43; Analysis 13.44; Analysis 13.45; Analysis 13.46; Analysis 13.47; Analysis 13.48; Analysis 13.49; Analysis 13.50; Analysis 13.51).
13.2. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 2: AE Abdominal Pain
13.3. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 3: AE Appetite decrease
13.4. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 4: AE Appetite increase
13.5. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 5: AE Agitation/anxiety
13.6. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 6: AE Blurred vision
13.7. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 7: AE Chest pain
13.8. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 8: AE Concentration difficulty
13.9. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 9: AE Confusion
13.10. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 10: AE Constipation
13.11. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 11: AE Coughing
13.12. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 12: AE Cramps
13.13. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 13: AE Decreased appetite
13.14. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 14: AE Decreased motor activity
13.15. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 15: AE Decreased libido
13.16. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 16: AE Dental problems
13.17. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 17: AE Depression
13.18. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 18: AE Dermatologic/skin irritation/lesions
13.19. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 19: AE Diarrhoea
13.20. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 20: AE Dizziness
13.21. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 21: AE Dry mouth
13.22. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 22: AE Eye irritation
13.23. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 23: AE Flatulence
13.24. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 24: AE Flu/upper respiratory infection
13.25. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 25: AE Genital discomfort
13.26. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 26: AE Gum problems
13.27. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 27: AE Headache
13.28. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 28: AE Increased libido
13.29. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 29: AE Increased thirst
13.30. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 30: AE Insomnia
13.31. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 31: AE Irritability
13.32. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 32: AE Memory problems
13.33. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 33: AE Mouth ulcer
13.34. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 34: AE Muscle/bone/joint pain
13.35. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 35: AE Nasal congestion
13.36. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 36: AE Nausea
13.37. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 37: AE Oedema
13.38. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 38: AE Sexual dysfunction
13.39. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 39: AE Shortness of breath
13.40. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 40: AE Sleepiness/drowsiness
13.41. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 41: AE Sore throat
13.42. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 42: AE Sore tongue
13.43. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 43: AE Stomach or abdominal discomfort
13.44. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 44: AE Suicidal ideas
13.45. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 45: AE Sweating
13.46. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 46: AE Tachycardia
13.47. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 47: AE Tinnitus
13.48. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 48: AE Tiredness/fatigue
13.49. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 49: AE Urination problems
13.50. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 50: AE Weight gain
13.51. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 51: AE Weight loss
13.3 Efficacy: number of participants who achieve remission
There was no evidence that riluzole was more effective than placebo in remission at any time point; at 24 hours (random‐effects OR 0.71, 95% CI 0.14 to 3.64; P = 0.68; 1 study, 42 participants); at 72 hours (random‐effects OR 1.33, 95% CI 0.30 to 5.84; P = 0.71; 1 study, 42 participants); at one week (random‐effects OR 1.00, 95% CI 0.18 to 5.63; P = 1.00; 1 study, 42 participants); at two weeks (random‐effects OR 1.00, 95% CI 0.13 to 7.85; P = 1.00; 1 study, 42 participants) and at four weeks (random‐effects (OR 1.19, 95% CI 0.12 to 12.13; P = 0.88; participants = 102; studies = 2; I2 = 65%) (Analysis 13.52).
13.52. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 52: Remission rate
13.4 Change scores on depression scale from baseline
There was no evidence that riluzole was more effective than placebo at any time point; at 24 hours (random‐effects SMD ‐0.26, 95% CI ‐0.87 to 0.35; P 0.40; participants = 42; studies = 1; I2 = 0%); at 72 hours (random‐effects SMD ‐0.25, 95% CI ‐0.86 to 0.37; P = 0.43; participants = 41; studies = 1; I2 = 0%); one week (random‐effects SMD ‐0.06, 95% CI ‐0.70 to 0.58; P = 0.85; participants = 38; studies = 1; I2 = 0%); at two weeks (random‐effects SMD ‐0.36, 95% CI ‐1.20 to 0.47; P = 0.39; participants = 97; studies = 2; I2 = 75%); and at four weeks (random‐effects SMD ‐0.18, 95% CI ‐1.19 to 0.84; P = 0.73; participants = 87; studies = 2; I2 = 79%) (Analysis 13.53).
13.53. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 53: Depression rating scale score
13.5 Suicidaility
No data were available for this outcome.
13.6 Cognition
No data were available for this outcome.
13.7 Quality of life
No data were available for this outcome.
13.8 Cost to healthcare services
No data were available for this outcome.
13.9 Acceptability: total dropouts
We found no difference between riluzole and placebo in terms of participants who dropped out due to any cause (fixed‐effects OR 0.81, 95% CI 0.23 to 2.88; P = 0.75; 1 study, 42 participants) (Analysis 13.54).
13.54. Analysis.

Comparison 13: Riluzole versus Placebo, Outcome 54: Acceptability
14. Atomoxetine versus placebo
One study contributed to this comparison (Michelson 2007), providing outcome data only at three months.
14.1 Efficacy: number of participants who respond to treatment
We found no evidence that atomoxetine was more effective than placebo in response at three months (OR 1.25, 95% CI 0.63 to 2.47; P = 0.52; 1 study, 146 participants) (Analysis 14.1).
14.1. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 1: Response rate
14.2 Adverse events
Atomoxetine treatment was associated with higher incidence of constipation over placebo (OR 17.06, 95% CI 0.96 to 304.51; P = 0.05; 1 study, 146 participants; Analysis 14.3), dry mouth (OR 20.86, 95% CI 2.68 to 162.03; P = 0.004; 1 study, 146 participants ‐ Analysis 14.7) and insomnia (OR 9.13, 95% CI 1.11 to 74.95; P = 0.04; 1 study, 146 participants; Analysis 14.12).
14.3. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 3: AE Constipation
14.7. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 7: AE Dry mouth
14.12. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 12: AE Insomnia
14.3 Efficacy: number of participants who achieve remission
We found no evidence that atomoxetine was more effective than placebo in remission at three months (OR 1.34, 95% CI 0.67 to 2.67; P = 0.41; 1 study, 146 participants) (Analysis 14.16).
14.16. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 16: Remission rate
14.4 Change scores on depression scale from baseline
There was no difference between atomoxetine and placebo at three months (MD ‐1.60, 95% CI ‐3.88 to 0.68; P = 0.17; 1 study, 141 participants) (Analysis 14.17).
14.17. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 17: Depression rating scale score
14.5 Suicidality
No data were available for this outcome.
14.6 Cognition
No data were available for this outcome.
14.7 Quality of life
No data were available for this outcome.
14.8 Cost to healthcare services
No data were available for this outcome.
14.9 Acceptability: total dropouts and dropouts due to adverse effects
We found no difference between atomoxetine and placebo in terms of participants who dropped out due to any cause (OR 1.03, 95% CI 0.44 to 2.41; P = 0.94; 1 study; 146 participants; Analysis 14.18), nor in terms of participants who dropped out due to side effects (OR 1.88, 95% CI 0.53 to 6.74; P = 0.33; 1 study; 146 participants; Analysis 14.19).
14.18. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 18: Acceptability
14.19. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 19: Acceptability ‐ adverse events
15. Basimglurant versus placebo
One study contributed to this comparison (Quiroz 2016), providing data at four weeks.
15.1 Efficacy: number of participants who respond to treatment
There was no evidence that basimglurant was more effective than placebo at four weeks (OR 0.98, 95% CI 0.62 to 1.55; P = 0.92; participants = 332; studies = 1; I2 = 0%) (Analysis 15.1).
15.1. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 1: Response rate
15.2 Adverse events
Participants assigned to treatment with basimglurant reported more dizziness over placebo (OR 2.77, 95% CI 1.12 to 6.86; P = 0.03; participants = 332; studies = 1; I2 = 0%) (Analysis 15.2).However those receiving basimglurant had lower incidence of nasopharyngitis than those receiving placebo (OR 0.06, 95% CI 0.01 to 0.46; P = 0.007; participants = 332; studies = 1; I2 = 0%) (Analysis 15.7).
15.2. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 2: AE Dizziness
15.7. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 7: AE Nasopharyngitis
15.3 Efficacy: number of participants who achieve remission
We found no evidence of any difference between basimglurant and placebo for remission rates at four weeks (OR 1.05, 95% CI 0.64 to 1.73; P = 0.84; participants = 332; studies = 1; I2 = 0%) (Analysis 15.9).
15.9. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 9: Remission rate
15.4 Change scores on depression scale from baseline
There was no evidence of a difference between basimglurant and placebo for depression rating scale scores at four weeks (MD ‐0.39, 95% CI ‐1.66 to 0.88; P = 0.55; participants = 332; studies = 1; I2 = 0%) (Analysis 15.10).
15.10. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 10: Depression rating scale score
15.5 Suicidality
No data were available for this outcome.
15.6 Cognition
No data were available for this outcome.
15.7 Quality of life
No data were available for this outcome.
15.8 Cost to healthcare services
No data were available for this outcome.
15.9 Acceptability: total dropouts
No data were available for this outcome.
16. Citicoline versus placebo
One study contributed to this comparison (Roohi‐Azizi 2017), providing data at four weeks.
16.1 Efficacy: number of participants who respond to treatment
There was no evidence that citicoline was more effective than placebo at four weeks (OR 4.47, 95% CI 0.83 to 24.19; P = 0.08; participants = 50; studies = 1; I2 = 0%) (Analysis 16.1).
16.1. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 1: Response rate
16.2 Adverse events
There were no differences in adverse events between citicoline and placebo (Analysis 16.2; Analysis 16.3; Analysis 16.4; Analysis 16.5; Analysis 16.6; Analysis 16.7; Analysis 16.8; Analysis 16.9; Analysis 16.10).
16.2. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 2: AE Abdominal Pain
16.3. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 3: AE Appetite decrease
16.4. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 4: AE Appetite increase
16.5. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 5: AE Dizziness
16.6. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 6: AE Diarrhoea
16.7. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 7: AE Headache
16.8. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 8: AE Insomnia
16.9. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 9: AE Nausea
16.10. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 10: AE Sedation
16.3 Efficacy: number of participants who achieve remission
Citicoline was more efficacious in achieving remission than placebo at four weeks (OR 3.27, 95% CI 1.01 to 10.62; P = 0.05; participants = 50; studies = 1; I2 = 0%) (Analysis 16.11)
16.11. Analysis.

Comparison 16: Citicoline versus Placebo, Outcome 11: Remission rate
16.4 Change scores on depression scale from baseline
No data were available for this outcome.
16.5 Suicidality
No data were available for this outcome.
16.6 Cognition
No data were available for this outcome.
16.7 Quality of life
No data were available for this outcome.
16.8 Cost to healthcare services
No data were available for this outcome.
16.9 Acceptability: total dropouts
No data were available for this outcome.
17. CP‐101,606 versus placebo
One study contributed to this comparison (Preskorn 2008), providing continuous outcome data at 24 hours, one week and two weeks.
17.1 Efficacy: number of participants who respond to treatment
No data were available for this outcome
17.2 Adverse events
We found no difference between CP‐101, 606 and placebo in terms of adverse events (1 study, 30 participants) (Analysis 17.1; Analysis 17.2).
17.1. Analysis.

Comparison 17: CP‐101,606 versus Placebo, Outcome 1: AE Change in blood pressure
17.2. Analysis.

Comparison 17: CP‐101,606 versus Placebo, Outcome 2: AE Dissociative reaction
17.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
17.4 Change scores on depression scale from baseline
We found no difference between CP‐101,606 and placebo at 24 hours (MD ‐0.40, 95% CI ‐6.75 to 5.95; P = 0.90; 1 study, 30 participants) (Analysis 17.3). CP‐101,606 was more effective in reducing depression rating scale scores at one week over placebo (MD ‐7.10, 95% CI ‐13.42 to ‐0.78; P = 0.03; 1 study, 26 participants). At two weeks we observed no difference between CP‐101,606 and placebo (MD ‐2.90, 95% CI ‐12.06 to 6.26; P = 0.53; 1 study, 20 participants).
17.3. Analysis.

Comparison 17: CP‐101,606 versus Placebo, Outcome 3: Depression rating scale score
17.5 Suicidality
No data were available for this outcome.
17.6 Cognition
No data were available for this outcome.
17.7 Quality of life
No data were available for this outcome.
17.8 Cost to healthcare services
No data were available for this outcome.
17.9 Acceptability: total dropouts and dropouts due to adverse effects
We found no difference between CP‐101,606 and placebo in terms of participants who dropped out due to any cause (OR 0.29, 95% CI 0.06 to 1.45; P = 0.13; 1 study, 30 participants) (Analysis 17.4). No participants dropped out due to side effects.
17.4. Analysis.

Comparison 17: CP‐101,606 versus Placebo, Outcome 4: Acceptability
18. D‐cycloserine versus placebo
One study contributed to this comparison (Heresco‐Levy 2013), providing data only at two and four weeks.
18.1 Efficacy: number of participants who respond to treatment
We found no difference between D‐cycloserine and placebo in terms of response at two weeks (OR 5.33, 95% CI 0.51 to 56.24; P = 0.16; 1 study, 26 participants), or at four weeks (OR 3.44, 95% CI 0.53 to 22.43; P = 0.20; 1 study, 26 participants) (Analysis 18.1).
18.1. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 1: Response rate
18.2 Adverse events
We found no difference between D‐cycloserine and placebo in terms of any adverse event (Analysis 18.2; Analysis 18.3; Analysis 18.4; Analysis 18.5; Analysis 18.6; Analysis 18.7; Analysis 18.8; Analysis 18.9; Analysis 18.10; Analysis 18.11; Analysis 18.12; Analysis 18.13; Analysis 18.14).
18.2. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 2: AE Agitation/anxiety
18.3. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 3: AE Concentration difficulties
18.4. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 4: AE Constipation
18.5. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 5: AE Failing memory
18.6. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 6: AE Headache
18.7. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 7: AE Increased dream activity
18.8. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 8: AE Increased duration of sleep
18.9. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 9: AE Increased sexual desire
18.10. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 10: AE Increased tendency to sweat
18.11. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 11: AE Nausea/vomiting
18.12. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 12: AE Palpitation/tachycardia
18.13. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 13: AE Sleepiness/drowsiness
18.14. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 14: AE Urination issues
18.3 Efficacy: number of participants who achieve remission
There were no participants who met remission in either the D‐cycloserine or placebo group at two weeks and there was no difference in terms of remission between D‐cycloserine and placebo at four weeks (OR 9.00, 95% CI 0.42 to 194.07; P = 0.16; 1 study, 26 participants) (Analysis 18.15).
18.15. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 15: Remission rate
18.4 Change scores on depression scale from baseline
There was no evidence of a difference between D‐cycloserine and placebo at two weeks (MD ‐5.00, 95% CI ‐11.08 to 1.08; P = 0.11; 1 study, 25 participants), or at four weeks (MD ‐7.00, 95% CI ‐14.53 to 0.53; P = 0.07; 1 study, 23 participants) (Analysis 18.16).
18.16. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 16: Depression rating scale score
18.5 Suicidality
No data were available for this outcome.
18.6 Cognition
No data were available for this outcome.
18.7 Quality of life
No data were available for this outcome.
18.8 Cost to healthcare services
No data were available for this outcome.
18.9 Acceptability: total dropouts
We found no difference between D‐cycloserine and placebo in terms of participants who dropped out due to any cause (OR 3.60, 95% CI 0.32 to 40.23; P = 0.30; 1 study, 26 participants) (Analysis 18.17).
18.17. Analysis.

Comparison 18: D‐cycloserine versus Placebo, Outcome 17: Acceptability
19. Decoglurant versus placebo
One study contributed to this comparison (Umbricht 2020), providing data only at four weeks.
19.1 Efficacy: number of participants who respond to treatment
Participants assigned to treatment with decoglurant were more likely to respond to treatment than those receiving a placebo at four weeks (OR 2.04, 95% CI 1.23 to 3.38; P = 0.006; participants = 309; studies = 1; I2 = 0%) (Analysis 19.1).
19.1. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 1: Response Rate
19.2 Adverse events
Decoglurant was associated with more reports of dizziness than placebo (OR 2.01, 95% CI 1.03 to 3.94; P = 0.04; participants = 357; studies = 1; I2 = 0%) (Analysis 19.3).
19.3. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 3: AE Dizziness
There were no other differences in adverse events between decoglurant and placebo (Analysis 19.2; Analysis 19.4; Analysis 19.5; Analysis 19.6; Analysis 19.7).
19.2. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 2: AE Diarrhea
19.4. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 4: AE Headache
19.5. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 5: AE Nausea
19.6. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 6: AE Sleepiness/drowsiness
19.7. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 7: AE Vomiting
19.3 Efficacy: number of participants who achieve remission
We found no evidence of any differences between decoglurant and placebo for remission rates at four weeks (OR 1.60, 95% CI 0.95 to 2.69; P = 0.08; participants = 309; studies = 1; I2 = 0%) (Analysis 19.8).
19.8. Analysis.

Comparison 19: Decoglurant (mGlu2/3) versus placebo, Outcome 8: Remission rate
19.4 Change scores on depression scale from baseline
No data were available for this outcome.
19.5 Suicidality
No data were available for this outcome.
19.6 Cognition
No data were available for this outcome.
19.7 Quality of life
No data were available for this outcome.
19.8 Cost to healthcare services
No data were available for this outcome.
19.9 Acceptability: total dropouts
No data were available for this outcome.
20. MK‐0657 versus placebo
Only one cross‐over study (five patients overall) contributed to this comparison (Ibrahim 2012b), providing data at 24 hours, 72 hours, one week and two weeks.
20.1 Efficacy: number of participants who respond to treatment
There were no responders in the MK‐0657 or placebo group in any of the time points measured: 24 hours; 72 hours; one week and two weeks.
20.2 Adverse events
No data were available for this outcome.
20.3 Efficacy: number of participants who achieve remission
No patients met remission in the MK‐0657 or placebo group at any of the time points measures: 24 hours; 72 hours; one week and two weeks.
20.4 Change scores on depression scale from baseline
We found no difference between MK‐0657 and placebo at 24 hours (MD 4.17, 95% CI ‐7.21 to 15.55; P = 0.47; 1 study, 5 participants), 72 hours (MD ‐2.83, 95% CI ‐14.21 to 8.55; P = 0.63; 1 study, 5 participants), one week (MD 0.67, 95% CI ‐13.16 to 14.50; P = 0.92; 1 study, 5 participants) and at two weeks (MD 3.50, 95% CI ‐8.67 to 15.67; P = 0.57; 1 study, 5 participants) (Analysis 20.1).
20.1. Analysis.

Comparison 20: MK‐0657 versus Placebo, Outcome 1: Depression rating scale score
20.5 Suicidality
No data were available for this outcome.
20.6 Cognition
No data were available for this outcome.
20.7 Quality of life
No data were available for this outcome.
20.8 Cost to healthcare services
No data were available for this outcome.
20.9 Acceptability: total dropouts
No patients dropped out of the trial in either the MK‐0657 or placebo group.
21. N‐acetylcysteine versus placebo
One study contributed to this comparison (Berk 2014), providing data at two weeks, four weeks and three months.
21.1 Efficacy: number of participants who respond to treatment
There was no evidence that N‐acetylcysteine was more effective than placebo in response at any time point; at two weeks (OR 0.77, 95% CI 0.38 to 1.55; P = 0.46; 1 study, 269 participants); at four weeks (OR 0.91, 95% CI 0.52 to 1.61; P = 0.75; 1 study, 269 participants) and at three months (OR 1.38, 95% CI 0.83 to 2.30; P = 0.21; 1 study, 269 participants) (Analysis 21.1).
21.1. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 1: Response rate
21.2 Adverse events
Participants receiving N‐acetylcysteine were more likely to report gastrointestinal problems than those receiving placebo (OR 2.26, 95% CI 1.27 to 4.02; P = 0.006; 1 study, 169 participants) (Analysis 21.3).
21.3. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 3: AE Gastrointestinal problems
No other differences in adverse events were found (Analysis 21.2; Analysis 21.4; Analysis 21.5).
21.2. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 2: AE Back pain
21.4. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 4: AE Joint pain
21.5. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 5: AE Muscle spasms
21.3 Efficacy: number of participants who achieve remission
We found no difference in remission between N‐acetylcysteine and placebo at two weeks (OR 0.48, 95% CI 0.12 to 1.98; P = 0.31; 1 study, 269 participants) or at three months (OR 1.45, 95% CI 0.79 to 2.68; P = 0.23; 1 study, 269 participants) (Analysis 21.6). However, N‐acetylcysteine produced higher remission rates over placebo at four weeks (OR 0.41, 95% CI 0.17 to 0.97; P = 0.04; 1 study, 269 participants).
21.6. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 6: Remission rate
21.4 Change scores on depression scale from baseline
We found no difference between N‐acetylcysteine and placebo at two weeks (MD ‐0.40, 95% CI ‐2.06 to 1.26; P = 0.64; 1 study, 252 participants), at four weeks (MD ‐1.20, 95% CI ‐3.28 to 0.88; P = 0.26; 1 study, 252 participants) and at three months (MD ‐1.50, 95% CI ‐4.14 to 1.14; P = 0.26; 1 study, 207 participants) (Analysis 21.7).
21.7. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 7: Depression rating scale score
21.5 Suicidality
No data were available for this outcome.
21.6 Cognition
No data were available for this outcome.
21.7 Quality of life
We found no difference between N‐acetylcysteine and placebo at three months (MD ‐0.10, 95% CI ‐2.74 to 2.54; P = 0.94; 1 study, 207 participants) (Analysis 21.8).
21.8. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 8: Quality of life
21.8 Cost to healthcare services
No data were available for this outcome.
21.9 Acceptability: total dropouts and dropouts due to adverse events
We found no difference between N‐acetylcysteine and placebo in terms of participants who dropped out due to any cause (OR 0.67, 95% CI 0.36 to 1.24; P = 0.20; 1 study, 269 participants; Analysis 21.9), nor due to side effects (OR 0.49, 95% CI 0.04 to 5.50; P = 0.57; 1 study, 269 participants; Analysis 21.10).
21.9. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 9: Acceptability
21.10. Analysis.

Comparison 21: N‐acetylcysteine versus Placebo, Outcome 10: Acceptability ‐ adverse events
E. Other glutamate receptor modulators versus other pharmacologically active agents
22. Sarcosine versus citalopram
One study contributed to this comparison (Huang 2013), providing data only at two and four weeks.
22.1 Efficacy: number of participants who respond to treatment
We found no difference in terms of response between sarcosine and citalopram at two weeks (OR 8.14, 95% CI 0.88 to 75.48; P = 0.06; 1 study, 40 participants). A difference in favour of sarcosine was found at four weeks (OR 6.93, 95% CI 1.53 to 31.38; P = 0.01; 1 study, 40 participants) (Analysis 22.1).
22.1. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 1: Response rate
22.2 Adverse events
A higher number of participants receiving sarcosine treatment experienced adverse events over citalopram (OR 0.04, 95% CI 0.00 to 0.68; P = 0.03; 1 study, 40 participants) (Analysis 22.2).
22.2. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 2: AE Total
22.3 Efficacy: number of participants who achieve remission
We found no difference in terms of remission between sarcosine and citalopram at two weeks (OR 14.55, 95% CI 0.75 to 283.37; P = 0.08; 1 study, 40 participants). Sarcosine treatment resulted in more frequent remission over citalopram at four weeks (OR 27.88, 95% CI 1.48 to 526.12; P = 0.03; 1 study, 40 participants) (Analysis 22.20).
22.20. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 20: Remission rate
22.4 Change scores on depression scale from baseline
We found evidence that sarcosine was more effective than citalopram at two weeks (MD ‐5.50, 95% CI ‐10.12 to ‐0.88; P = 0.02; 1 study, 40 participants). The effect of sarcosine compared with citalopram at four weeks was MD ‐4.00, 95% CI ‐8.30 to 0.30; P = 0.07; 1 study, 31 participants (Analysis 22.21).
22.21. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 21: Depression rating scale score
22.5 Suicidality
No data were available for this outcome.
22.6 Cognition
No data were available for this outcome.
22.7 Quality of life
No data were available for this outcome.
22.8 Cost to healthcare services
No data were available for this outcome.
22.9 Acceptability: total dropouts and dropouts due to adverse events
We found no difference between sarcosine and citalopram in terms of participants who dropped out due to any cause (OR 0.52, 95% CI 0.14 to 1.92; P = 0.33; 1 study, 40 participants) (Analysis 22.22). In Huang 2013 no participants dropped out due to side effects.
22.22. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 22: Acceptability
Subgroup analyses
Two comparisons had enough data to complete the pre‐planned subgroup analyses (ketamine versus placebo and esketamine versus placebo).
23. Ketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting)
Four studies contributed to this subgroup analysis (Arabzadeh 2018; Hu 2016; Ionescu 2018; Su 2017), providing data at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
23.1 Efficacy: number of participants who respond to treatment
Ketamine treatment in outpatient settings resulted in increased response efficacy at 72 hours over placebo (random‐effects OR 33.46, 95% CI 1.65 to 677.83; P = 0.02; participants = 27; studies = 1; I2 = 0%), one week (random‐effects OR 33.46, 95% CI 1.65 to 677.83; P = 0.02; participants = 27; studies = 1; I2 = 0%), and two weeks (random‐effects OR 20.80, 95% CI 2.04 to 211.79; P = 0.01; participants = 27; studies = 1; I2 = 0%) (Analysis 23.1). There was no difference at 24 hours (random‐effects OR 18.76, 95% CI 0.92 to 383.10; P = 0.06; participants = 27; studies = 1; I2 = 0%), four weeks (random‐effects OR 2.00, 95% CI 0.68 to 5.85; P = 0.21; participants = 132; studies = 3; I2 = 41%), or three months (random‐effects OR 3.95, 95% CI 0.16 to 97.23; P = 0.40; participants = 47; studies = 2; I2 = 80%).
23.1. Analysis.

Comparison 23: Ketamine versus Placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 1: Response rate
23.2 Efficacy: number of participants who achieve remission
We found no differences in terms of remission between ketamine and placebo in outpatient treatment settings at any time points: at 24 hours (random‐effects OR 3.48, 95% CI 0.13 to 93.30; P = 0.46; participants = 27; studies = 1; I2 = 0%), at 72 hours (random‐effects OR 6.30, 95% CI 0.27 to 144.70; P = 0.25; participants = 27; studies = 1; I2 = 0%), at one week (random‐effects OR 6.30, 95% CI 0.27 to 144.70; P = 0.25; participants = 27; studies = 1; I2 = 0%), at two weeks (random‐effects OR 3.90, 95% CI 0.35 to 43.36; P = 0.27; participants = 27; studies = 1; I2 = 0%), at four weeks (random‐effects OR 2.19, 95% CI 0.85 to 5.66; P = 0.11; participants = 132; studies = 3; I2 = 0%), and at three months (random‐effects OR 1.09, 95% CI 0.45 to 2.67; P = 0.85; participants = 90; studies = 2; I2 = 0%) (Analysis 23.2).
23.2. Analysis.

Comparison 23: Ketamine versus Placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 2: Remission rate
23.3 Change scores on depression scale from baseline
We found evidence that ketamine was more effective at reducing depression rating scale scores than placebo in outpatient treatment settings at two weeks (random‐effects SMD ‐0.73, 95% CI ‐1.31 to ‐0.15; P = 0.01; participants = 126; studies = 3; I2 = 50%) and at four weeks (random‐effects SMD ‐0.68, 95% CI ‐1.07 to ‐0.29; P = 0.0006; participants = 107; studies = 2; I2 = 0%) (Analysis 23.3). There were no differences observed between ketamine and placebo at 24 hours (random‐effects SMD ‐0.47, 95% CI ‐1.11 to 0.18; P = 0.16; participants = 75; studies = 2; I2 = 42%), at 72 hours (random‐effects SMD ‐0.28, 95% CI ‐1.18 to 0.62; P = 0.54; participants = 94; studies = 3; I2 = 75%), or at one week (random‐effects SMD ‐0.52, 95% CI ‐1.69 to 0.65; P = 0.38; participants = 45; studies = 2; I2 = 72%).
23.3. Analysis.

Comparison 23: Ketamine versus Placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 3: Depression rating scale score
23.4 Suicidality
There were no differences in suicidality between ketamine and placebo in outpatient settings at any time point: at 24 hours (random‐effects MD 0.02, 95% CI ‐0.78 to 0.82; P = 0.96; participants = 48; studies = 1; I2 = 0%); at 72 hours (random‐effects MD 0.34, 95% CI ‐0.25 to 0.93; P = 0.26; participants = 68; studies = 2; I2 = 10%), at one week (random‐effects MD ‐0.30, 95% CI ‐1.56 to 0.96; P = 0.64; participants = 19; studies = 1; I2 = 0%), at two weeks (random‐effects MD ‐0.20, 95% CI ‐1.46 to 1.06; P = 0.76; participants = 19; studies = 1; I2 = 0%) (Analysis 23.4).
23.4. Analysis.

Comparison 23: Ketamine versus Placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 4: Suicidal ideation composite
24. Ketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting)
Three studies contributed to this subgroup analysis (Loo 2012; Sos 2013; Zarate 2006a), providing data at 24 hours, 72 hours, one week, and two weeks.
24.1 Efficacy: number of participants who respond to treatment
Ketamine was more efficacious than placebo in achieving response in inpatient treatment settings at 24 hours (random‐effects OR 15.11, 95% CI 1.97 to 115.92; P = 0.009; participants = 48; studies = 2; I2 = 10%) and 72 hours (random‐effects OR 14.00, 95% CI 2.07 to 94.75; P = 0.007; participants = 48; studies = 2; I2 = 0%) (Analysis 24.1). There was no difference in response between ketamine and placebo at one week (random‐effects OR 3.41, 95% CI 0.95 to 12.27; P = 0.06; participants = 99; studies = 3; I2 = 21%) and two weeks (random‐effects OR 0.93, 95% CI 0.31 to 2.83; P = 0.90; participants = 51; studies = 1; I2 = 0%).
24.1. Analysis.

Comparison 24: Ketamine versus Placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 1: Response rate
24.2 Efficacy: number of participants who achieve remission
Ketamine had higher efficacy for remission over placebo when administered in an inpatient setting at 24 hours (random‐effects OR 6.60, 95% CI 0.96 to 45.09; P = 0.05; participants = 48; studies = 2; I2 = 0%), at 72 hours (random‐effects OR 7.88, 95% CI 1.17 to 53.21; P = 0.03; participants = 48; studies = 2; I2 = 0%), and at one week (random‐effects OR 7.24, 95% CI 1.70 to 30.81; P = 0.007; participants = 99; studies = 3; I2 = 0%) (Analysis 24.2). There was no difference in remission at two weeks (random‐effects OR 0.95, 95% CI 0.28 to 3.24; P = 0.93; participants = 51; studies = 1; I2 = 100%).
24.2. Analysis.

Comparison 24: Ketamine versus Placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 2: Remission rate
24.3 Change scores on depression scale from baseline
There was evidence suggesting a decrease in depression rating scale scores for ketamine over placebo in inpatient treatment settings at 24 hours (random‐effects SMD ‐1.63, 95% CI ‐2.86 to ‐0.39; P = 0.010; participants = 46; studies = 2; I2 = 62%) , at 72 hours (random‐effects SMD ‐1.21, 95% CI ‐1.87 to ‐0.55; P = 0.0003; participants = 46; studies = 2; I2 = 0%) , and at one week (random‐effects SMD ‐0.75, 95% CI ‐1.19 to ‐0.31; P = 0.0008; participants = 91; studies = 3; I2 = 0%) (Analysis 24.3). There was no difference in change on depression rating scale scores between ketamine and placebo at two weeks (random‐effects SMD ‐0.10, 95% CI ‐0.68 to 0.48; P = 0.74; participants = 46; studies = 1; I2 = 0%).
24.3. Analysis.

Comparison 24: Ketamine versus Placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 3: Depression rating scale score
24.4 Suicidality
No data were available for this outcome.
25. Esketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting)
Three studies contributed to this subgroup analysis (Daly 2018; Fedgchin 2019; Popova 2019), contributing data at 24 hours, one week, two weeks, four week, and three months.
25.1 Efficacy: number of participants who respond to treatment
We found a difference in response rates favouring esketamine over placebo in outpatient treatment settings at 24 hours (random‐effects OR 4.33, 95% CI 1.08 to 17.31; P = 0.04; participants = 620; studies = 3; I2 = 66%), one week (random‐effects OR 2.73, 95% CI 1.41 to 5.28; P = 0.003; participants = 632; studies = 3; I2 = 0%), and four weeks (random‐effects OR 1.92, 95% CI 1.34 to 2.75; P = 0.0004; participants = 543; studies = 2; I2 = 0%) (Analysis 25.1).
25.1. Analysis.

Comparison 25: Esketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 1: Response rate
25.2 Efficacy: number of participants who achieve remission
There was an increased likelihood of remission in those receiving esketamine in outpatient treatment settings over placebo at 24 hours (random‐effects OR 6.51, 95% CI 1.93 to 21.92; P = 0.002; participants = 377; studies = 2; I2 = 0%), one week (random‐effects OR 7.76, 95% CI 1.75 to 34.48; P = 0.007; participants = 399; studies = 2; I2 = 0%), and at two weeks (random‐effects OR 2.30, 95% CI 1.02 to 5.17; P = 0.04; participants = 315; studies = 1; I2 = 0%) (Analysis 25.2). We found no difference between esketamine and placebo in outpatient settings at four weeks (random‐effects OR 1.39, 95% CI 0.84 to 2.30; P = 0.20; participants = 317; studies = 1; I2 = 0%).
25.2. Analysis.

Comparison 25: Esketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 2: Remission rate
25.3 Change scores on depression scale from baseline
We found a decrease in depression scores from baseline in participants allocated to esketamine over placebo in outpatient treatment settings at 24 hours (random‐effects SMD ‐0.25, 95% CI ‐0.49 to ‐0.01; P = 0.04; participants = 310; studies = 1; I2 = 100%), at one week (random‐effects SMD ‐0.28, 95% CI ‐0.51 to ‐0.06; P = 0.01; participants = 340; studies = 1; I2 = 0%), at two weeks (random‐effects SMD ‐0.32, 95% CI ‐0.54 to ‐0.09; P = 0.006; participants = 340; studies = 1; I2 = 0%), and at four weeks (random‐effects SMD ‐0.28, 95% CI ‐0.45 to ‐0.10; P = 0.002; participants = 213; studies = 2; I2 = 0%) (Analysis 25.3). No difference was found at three months (random‐effects SMD ‐0.12, 95% CI ‐0.75 to 0.52; P = 0.72; participants = 38; studies = 1; I2 = 0%).
25.3. Analysis.

Comparison 25: Esketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 3: Depression rating scale score
25.4 Suicidality
There were no differences in suicidality between esketamine and placebo in outpatient treatment settings at any time point: at one week (random‐effects MD 0.05, 95% CI ‐0.08 to 0.18; P = 0.43; participants = 209; studies = 1; I2 = 0%), at two weeks (random‐effects MD ‐0.07, 95% CI ‐0.21 to 0.07; P = 0.31; participants = 208; studies = 1; I2 = 0%), or at four weeks (random‐effects MD ‐0.02, 95% CI ‐0.11 to 0.07; P = 0.65; participants = 196; studies = 1; I2 = 0%) (Analysis 25.4).
25.4. Analysis.

Comparison 25: Esketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting), Outcome 4: Suicidal ideation composite
26. Esketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting)
Two studies contributed data to this subgroup analysis (Canuso 2018; Fu 2020), providing data at 24 hours, 72 hours, one week, two weeks, and four weeks.
26.1 Efficacy: number of participants who respond to treatment
Esketamine had higher response efficacy over placebo in inpatient treatment settings at two weeks (random‐effects OR 1.71, 95% CI 1.01 to 2.91; P = 0.05; participants = 224; studies = 1; I2 = 0%) and four weeks (random‐effects OR 1.85, 95% CI 1.09 to 3.14; P = 0.02; participants = 224; studies = 1; I2 = 100%) only (Analysis 26.1). No difference was found between esketamine and placebo at 24 hours (random‐effects OR 1.40, 95% CI 0.79 to 2.49; P = 0.25; participants = 224; studies = 1; I2 = 0%), at 72 hours (random‐effects OR 1.52, 95% CI 0.88 to 2.62; P = 0.13; participants = 224; studies = 1; I2 = 0%), or at one week (random‐effects OR 1.29, 95% CI 0.76 to 2.18; P = 0.35; participants = 224; studies = 1; I2 = 0%).
26.1. Analysis.

Comparison 26: Esketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 1: Response rate
26.2 Efficacy: number of participants who achieve remission
Esketamine had increased remission rates over placebo in inpatient treatment settings at 24 hours only (random‐effects OR 2.25, 95% CI 1.13 to 4.49; P = 0.02; participants = 290; studies = 2; I2 = 0%) (Analysis 26.2). There was no difference at any other time point: at 72 hours (random‐effects OR 1.61, 95% CI 0.59 to 4.34; P = 0.35; participants = 290; studies = 2; I2 = 54%), at one week (random‐effects OR 1.22, 95% CI 0.70 to 2.13; P = 0.49; participants = 290; studies = 2; I2 = 0%), at two weeks (random‐effects OR 1.39, 95% CI 0.83 to 2.32; P = 0.21; participants = 290; studies = 2; I2 = 0%), or at four weeks (random‐effects OR 1.48, 95% CI 0.91 to 2.42; P = 0.11; participants = 290; studies = 2; I2 = 0%).
26.2. Analysis.

Comparison 26: Esketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 2: Remission rate
26.3 Change scores on depression scale from baseline
We found that esketamine decreased depression rating scale scores over placebo in inpatient treatment settings at 24 hours (random‐effects SMD ‐0.35, 95% CI ‐0.58 to ‐0.12; P = 0.003; participants = 290; studies = 2; I2 = 0%), at 72 hours (random‐effects SMD ‐0.41, 95% CI ‐0.64 to ‐0.17; P = 0.0006; participants = 290; studies = 2; I2 = 0%), and at four weeks (random‐effects SMD ‐0.25, 95% CI ‐0.48 to ‐0.02; P = 0.03; participants = 290; studies = 2; I2 = 0%) (Analysis 26.3). No difference was found between esketamine and placebo at one week (random‐effects SMD ‐0.20, 95% CI ‐0.43 to 0.04; P = 0.10; participants = 290; studies = 2; I2 = 0%) or at two weeks (random‐effects SMD ‐0.14, 95% CI ‐0.37 to 0.09; P = 0.23; participants = 290; studies = 2; I2 = 0%).
26.3. Analysis.

Comparison 26: Esketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 3: Depression rating scale score
26.4 Suicidality
We found no difference in suicidality between esketamine and placebo at any time point: at 24 hours (random‐effects MD ‐0.20, 95% CI ‐0.63 to 0.23; P = 0.36; participants = 224; studies = 1; I2 = 0%), at 72 hours (random‐effects MD ‐0.30, 95% CI ‐0.69 to 0.09; P = 0.14; participants = 224; studies = 1; I2 = 0%), at one week (random‐effects MD ‐0.20, 95% CI ‐0.56 to 0.16; P = 0.28; participants = 224; studies = 1; I2 = 0%), at two weeks (random‐effects MD ‐0.10, 95% CI ‐0.43 to 0.23; P = 0.55; participants = 224; studies = 1; I2 = 0%), and at four weeks (random‐effects MD ‐0.20, 95% CI ‐0.53 to 0.13; P = 0.25; participants = 224; studies = 1; I2 = 0%; Analysis 26.4).
26.4. Analysis.

Comparison 26: Esketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting), Outcome 4: Suicidal ideation composite
27. Esketamine versus placebo (pre‐planned subgroup analysis: excluding elderly populations >65 years)
Eight studies contributed to this subgroup analysis (Canuso 2018; Daly 2018; Fedgchin 2019; Fu 2020; Ionescu 2020; Jarventausta 2013; Popova 2019; Singh 2016 b), with one study excluded as it recruited solely older adults aged 65 and over (Ochs‐Ross 2020). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
27.1 Efficacy: number of participants who respond to treatment
Participants receiving esketamine (excluding elderly populations) were more likely to achieve response than those receiving placebo at 24 hours (random‐effects OR 2.11, 95% CI 1.20 to 3.68; P = 0.009; participants = 1071; studies = 5; I2 = 50%), at one week, (random‐effects OR 1.64, 95% CI 1.05 to 2.54; P = 0.03; participants = 1083; studies = 5; I2 = 35%), at two weeks (random‐effects OR 1.57, 95% CI 1.09 to 2.28; P = 0.02; participants = 451; studies = 2; I2 = 0%), and at four weeks (random‐effects OR 1.81, 95% CI 1.40 to 2.34; P < 0.00001; participants = 994; studies = 4; I2 = 0%) (Analysis 27.1). However there was no difference found between esketamine and placebo at 72 hours (random‐effects OR 1.34, 95% CI 0.92 to 1.96; P = 0.13; participants = 451; studies = 2; I2 = 0%).
27.1. Analysis.

Comparison 27: Esketamine versus placebo (pre‐planned subgroup analysis: excluding elderly populations >65 years), Outcome 1: Response rate
27.2 Efficacy: number of participants who achieve remission
Esketamine treatment led to higher remission rates over placebo excluding elderly populations at 24 hours (random‐effects OR 2.74, 95% CI 1.71 to 4.40; P < 0.0001; participants = 894; studies = 5; I2 = 0%), two weeks (random‐effects OR 1.52, 95% CI 1.07 to 2.16; P = 0.02; participants = 832; studies = 4; I2 = 0%), and at four weeks (random‐effects OR 1.51, 95% CI 1.12 to 2.04; P = 0.006; participants = 834; studies = 4; I2 = 0%) (Analysis 27.2). No difference was found at 72 hours (random‐effects OR 1.55, 95% CI 0.91 to 2.64; P = 0.11; participants = 517; studies = 3; I2 = 24%), or at one week (random‐effects OR 1.62, 95% CI 0.91 to 2.89; P = 0.10; participants = 916; studies = 5; I2 = 36%).
27.2. Analysis.

Comparison 27: Esketamine versus placebo (pre‐planned subgroup analysis: excluding elderly populations >65 years), Outcome 2: Remission rate
27.3 Change scores on depression scale from baseline
A decrease in depression rating scale scores was found in esketamine over placebo when excluding elderly populations at 24 hours (random‐effects SMD ‐0.31, 95% CI ‐0.45 to ‐0.17; P < 0.0001; participants = 824; studies = 4; I2 = 0%), at 72 hours (random‐effects SMD ‐0.30, 95% CI ‐0.50 to ‐0.11; P = 0.002; participants = 517; studies = 3; I2 = 14%), at one week (random‐effects SMD ‐0.24, 95% CI ‐0.37 to ‐0.10; P = 0.0007; participants = 857; studies = 4; I2 = 0%), at two weeks (random‐effects SMD ‐0.21, 95% CI ‐0.34 to ‐0.07; P = 0.003; participants = 857; studies = 4; I2 = 0%), and at four weeks (random‐effects SMD ‐0.27, 95% CI ‐0.40 to ‐0.15; P < 0.0001; participants = 1059; studies = 5; I2 = 0%) (Analysis 27.3). No difference was found between esketamine and placebo was found at three months (random‐effects SMD ‐0.12, 95% CI ‐0.75 to 0.52; P = 0.72; participants = 38; studies = 1; I2 = 0%).
27.3. Analysis.

Comparison 27: Esketamine versus placebo (pre‐planned subgroup analysis: excluding elderly populations >65 years), Outcome 3: Depression rating scale score
27.4 Suicidality
We found no differences in suicidality between esketamine and placebo in the excluding elderly populations subgroup at any time point: at 24 hours (random‐effects MD ‐0.15, 95% CI ‐0.44 to 0.15; P = 0.33; participants = 450; studies = 2; I2 = 0%), at 72 hours (random‐effects MD ‐0.20, 95% CI ‐0.49 to 0.08; P = 0.16; participants = 451; studies = 2; I2 = 0%), at one week (random‐effects MD 0.01, 95% CI ‐0.10 to 0.13; P = 83; participants = 660; studies = 3; I2 = 0%), at two weeks (random‐effects MD ‐0.10, 95% CI ‐0.22 to 0.02; P = 0.10; participants = 659; studies = 3; I2 = 0%), or at four weeks (random‐effects MD ‐0.04, 95% CI ‐0.12 to 0.05; P = 0.40; participants = 647; studies = 3; I2 = 0%) (Analysis 27.4).
27.4. Analysis.

Comparison 27: Esketamine versus placebo (pre‐planned subgroup analysis: excluding elderly populations >65 years), Outcome 4: Suicidal ideation composite
Sensitivity analyses
Two comparisons had enough data to complete sensitivity analyses (ketamine versus placebo and esketamine versus placebo).
28. Ketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features)
Eleven studies contributed to this sensitivity analysis (Arabzadeh 2018; Chen 2017; Chen 2018; Hu 2016; Ionescu 2018; Li 2016; Singh 2016 a; Sos 2013; Su 2017; Tiger 2020; Zarate 2006a). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
28.1 Efficacy: number of participants who respond to treatment
Ketamine had higher efficacy in terms of response over placebo in studies excluding participants with bipolar disorder or psychotic features at 24 hours (random‐effects OR 4.33, 95% CI 1.47 to 12.80; P = 0.008; participants = 177; studies = 6; I2 = 28%), at 72 hours (random‐effects OR 17.99, 95% CI 3.58 to 90.34; P = 0.0004; participants = 75; studies = 3; I2 = 0%), at one week (random‐effects OR 14.32, 95% CI 2.90 to 70.64; P = 0.001; participants = 75; studies = 3; I2 = 0%), at two weeks (random‐effects OR 15.73, 95% CI 4.71 to 52.51; P < 0.00001; participants = 85; studies = 2; I2 = 0%) (Analysis 28.1). No difference was found between ketamine and placebo at four weeks (random‐effects OR 2.00, 95% CI 0.68 to 5.85; participants = 132; P = 0.21; studies = 3; I2 = 41%), and at three months (random‐effects OR 3.95, 95% CI 0.16 to 97.23; P = 0.40; participants = 47; studies = 2; I2 = 80%).
28.1. Analysis.

Comparison 28: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 1: Response rate
28.2 Efficacy: number of participants who achieve remission
Participants assigned to ketamine treatment achieved remission at a higher rather than those assigned to placebo treatment in studies that did not include participants with bipolar disorder or psychotic features at 24 hours (random‐effects OR 5.60, 95% CI 1.07 to 29.46; P = 0.04; participants = 75; studies = 3; I2 = 0%), very low‐certainty evidence, at 72 hours (random‐effects OR 7.42, 95% CI 1.45 to 37.89; P = 0.02; participants = 75; studies = 3; I2 = 0%), at one week (random‐effects OR 9.02, 95% CI 1.80 to 45.31; P = 0.008; participants = 75; studies = 3; I2 = 0%), and at two weeks (random‐effects OR 7.50, 95% CI 1.51 to 37.22; P = 0.01; participants = 85; studies = 2; I2 = 0%) (Analysis 28.2). No difference was found at four weeks (random‐effects OR 2.19, 95% CI 0.85 to 5.66; P = 0.11; participants = 132; studies = 3; I2 = 0%) or three months (random‐effects OR 1.29, 95% CI 0.14 to 11.54; P = 0.82; participants = 20; studies = 1; I2 = 0%).
28.2. Analysis.

Comparison 28: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 2: Remission rate
28.3 Change scores on depression scale from baseline
We found a reduction in depression rating scale scores in ketamine over placebo for studies that excluded participants with bipolar disorder or psychotic features at all time points: at 24 hours (random‐effects SMD ‐0.88, 95% CI ‐1.31 to ‐0.46; P < 0.0001; participants = 223; studies = 7; I2 = 50%), at 72 hours (random‐effects SMD ‐0.63, 95% CI ‐1.29 to 0.04; P = 0.06; participants = 140; studies = 5; I2 = 69%), at one week (random‐effects SMD ‐0.76, 95% CI ‐1.34 to ‐0.19; P = 0.010; participants = 90; studies = 4; I2 = 40%), at two weeks (random‐effects SMD ‐0.73, 95% CI ‐1.31 to ‐0.15; P = 0.01; participants = 126; studies = 3; I2 = 50%), and at four weeks (random‐effects SMD ‐0.68, 95% CI ‐1.07 to ‐0.29; P = 0.0006; participants = 107; studies = 2; I2 = 0%) (Analysis 28.3).
28.3. Analysis.

Comparison 28: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 3: Depression rating scale score
28.4 Suicidality
No differences in suicidality were found between ketamine and placebo in studies excluding participants with bipolar or psychotic features at any time point: at 24 hours (random‐effects MD 0.02, 95% CI ‐0.78 to 0.82; P = 0.96; participants = 48; studies = 1; I2 = 0%), at 72 hours (random‐effects MD 0.34, 95% CI ‐0.25 to 0.93; P = 0.26; participants = 68; studies = 2; I2 = 10%), at one week (random‐effects MD ‐0.30, 95% CI ‐1.56 to 0.96; P = 0.64 participants = 19; studies = 1; I2 = 0%), or at two weeks (random‐effects MD ‐0.20, 95% CI ‐1.46 to 1.06; P = 0.76; participants = 19; studies = 1; I2 = 0%) (Analysis 28.4).
28.4. Analysis.

Comparison 28: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 4: Suicidal ideation composite
29. Ketamine versus placebo (pre‐planned sensitivity analysis: excluding treatment resistant populations)
Four studies contributed to this sensitivity analysis (Arabzadeh 2018; Berman 2000; Sos 2013; Tiger 2020). Data were provided at 24 hours, 72 hours, one week, two weeks, and four weeks.
29.1 Efficacy: number of participants who respond to treatment
Ketamine produced higher response rates over placebo in studies excluding treatment resistant populations was found at 72 hours (random‐effects OR 15.32, 95% CI 1.58 to 148.09; P = 0.02; participants = 38; studies = 2; I2 = 0%), one week (random‐effects OR 10.29, 95% CI 0.97 to 108.81; P = 0.05; participants = 30; studies = 1; I2 = 0%), and at four weeks (random‐effects OR 4.31, 95% CI 1.48 to 12.56; P = 0.007; participants = 81; studies = 1; I2 = 0%) (Analysis 29.1). No differences were found at 24 hours (random‐effects OR 2.31, 95% CI 0.65 to 8.14; P = 0.19; participants = 68; studies = 3; I2 = 0%).
29.1. Analysis.

Comparison 29: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 1: Response rate
29.2 Efficacy: number of participants who achieve remission
Remission was achieved more frequently in participants receiving ketamine over placebo in studies excluding treatment resistant populations was found at one week (random‐effects OR 10.29, 95% CI 0.97 to 108.81; P = 0.05; participants = 30; studies = 1; I2 = 0%) (Analysis 29.2). However, no differences were found at any other time point: at 24 hours (random‐effects OR 6.75, 95% CI 0.61 to 75.27; P = 0.12; participants = 30; studies = 1; I2 = 0%), at 72 hours (random‐effects OR 5.63, 95% CI 0.77 to 40.99; P = 0.09; participants = 38; studies = 2; I2 = 0%), or at four weeks (random‐effects OR 1.59, 95% CI 0.51 to 4.98; P = 0.42; participants = 81; studies = 1; I2 = 0%).
29.2. Analysis.

Comparison 29: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 2: Remission rate
29.3 Change scores on depression scale from baseline
We found a decrease in depression scores in participants allocated to receive ketamine over placebo within studies that excluded treatment resistant patients at all time points: at 24 hours (random‐effects SMD ‐1.06, 95% CI ‐1.61 to ‐0.52; P = 0.0001; participants = 66; studies = 3; I2 = 0%), at 72 hours (random‐effects SMD ‐1.20, 95% CI ‐1.96 to ‐0.44; P = 0.002; participants = 36; studies = 2; I2 = 0%), at one week (random‐effects SMD ‐1.19, 95% CI ‐1.97 to ‐0.42; P = 0.003; participants = 35; studies = 2; I2 = 0%), at two weeks (random‐effects SMD ‐0.90, 95% CI ‐1.36 to ‐0.45; P = 0.0001; participants = 81; studies = 1; I2 = 0%), and at four weeks (random‐effects SMD ‐0.77, 95% CI ‐1.22 to ‐0.31; P = 0.0009; participants = 81; studies = 1; I2 = 0%) (Analysis 29.3).
29.3. Analysis.

Comparison 29: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 3: Depression rating scale score
29.4 Suicidality
No data were available for this outcome.
30. Ketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%)
Eleven studies contributed to this sensitivity analysis (Arabzadeh 2018; Berman 2000; Chen 2017; Chen 2018; Hu 2016; Li 2016; Loo 2012; Sos 2013; Su 2017; Tiger 2020; Zarate 2006a). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
30.1 Efficacy: number of participants who respond to treatment
Ketamine was more efficacious than placebo in creating response when excluding trials with a dropout rate greater than 20% at 24 hours (random‐effects OR 3.94, 95% CI 1.54 to 10.10; P = 0.004; participants = 185; studies = 7; I2 = 14%), at 72 hours (random‐effects OR 15.84, 95% CI 3.68 to 68.12; P = 0.0002; participants = 83; studies = 4; I2 = 0%), at one week (random‐effects OR 5.69, 95% CI 1.34 to 24.11; P = 0.02; participants = 126; studies = 4; I2 = 39%), and at three months (random‐effects OR 20.00, 95% CI 2.77 to 144.31; P = 0.003; participants = 27; studies = 1; I2 = 100%) (Analysis 30.1). No difference was found at two weeks (random‐effects OR 3.72, 95% CI 0.17 to 79.32; P = 0.40; participants = 78; studies = 2; I2 = 83%) or at four weeks (random‐effects OR 2.00, 95% CI 0.68 to 5.85; P = 0.21; participants = 132; studies = 3; I2 = 41%).
30.1. Analysis.

Comparison 30: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 1: Response rate
30.2 Efficacy: number of participants who achieve remission
Ketamine treatment was associated with higher remission rates over placebo when trials with a dropout rate greater than 20% were excluded at 24 hours (random‐effects OR 5.60, 95% CI 1.07 to 29.46; P 0.04; participants = 75; studies = 3; I2 = 0%), at 72 hours (random‐effects OR 6.60, 95% CI 1.51 to 28.92; P = 0.01; participants = 83; studies = 4; I2 = 0%), and at one week (OR 7.06, 95% CI 1.90 to 26.31; P = 0.004; participants = 126; studies = 4; I2 = 0%) (Analysis 30.2). No difference was found at two weeks (random‐effects OR 1.30, 95% CI 0.41 to 4.12; P = 0.66; participants = 78; studies = 2; I2 = 5%) or at four weeks (random‐effects OR 2.60, 95% CI 0.60 to 11.33; P = 0.20; participants = 108; studies = 2; I2 = 35%).
30.2. Analysis.

Comparison 30: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 2: Remission rate
30.3 Change scores on depression scale from baseline
A decrease in depression rating scale scores was found for ketamine over placebo when excluding trials with a dropout rate greater than 20% at all time points: at 24 hours (random‐effects SMD ‐0.87, 95% CI ‐1.26 to ‐0.48; P < 0.0001; participants = 231; studies = 8; I2 = 41%), very low‐certainty evidence, at 72 hours (random‐effects SMD ‐0.86, 95% CI ‐1.24 to ‐0.48; P < 0.00001; participants = 128; studies = 5; I2 = 0%), at one week (random‐effects SMD ‐0.85, 95% CI ‐1.23 to ‐0.47; P < 0.0001; participants = 124; studies = 5; I2 = 0%), at two weeks (random‐effects SMD ‐0.68, 95% CI ‐1.29 to ‐0.08; P = 0.03; participants = 153; studies = 3; I2 = 66%), and at four weeks (random‐effects SMD ‐0.68, 95% CI ‐1.07 to ‐0.29; P = 0.0006; participants = 107; studies = 2; I2 = 0%) (Analysis 30.3).
30.3. Analysis.

Comparison 30: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 3: Depression rating scale score
30.4 Suicidality
No differences were found for suicidality between ketamine and placebo when trials with a dropout rate greater than 20% were excluded at 24 hours (random‐effects MD 0.02, 95% CI ‐0.78 to 0.82; P = 0.96; participants = 48; studies = 1; I2 = 0%) or at 72 hours (random‐effects MD 0.09, 95% CI ‐0.63 to 0.81; P = 0.81; participants = 48; studies = 1; I2 = 0%) (Analysis 30.4).
30.4. Analysis.

Comparison 30: Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 4: Suicidal ideation composite
31. Ketamine versus placebo (post‐hoc sensitivity analysis: excluding multiple doses)
Eight studies contributed to this sensitivity analysis (Berman 2000; Chen 2018; Hu 2016; Li 2016; Sos 2013; Su 2017; Tiger 2020; Zarate 2006a). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
31.1 Efficacy: number of participants who respond to treatment
Ketamine treatment resulted in larger numbers of participants responding to treatment over placebo when excluding studies that administered multiple doses of study drugs at 24 hours (random‐effects OR 3.94, 95% CI 1.54 to 10.10; P 0.004; participants = 185; studies = 7; I2 = 14%), at 72 hours (random‐effects OR 15.84, 95% CI 3.68 to 68.12; P = 0.0002; participants = 83; studies = 4; I2 = 0%), at one week (random‐effects OR 14.32, 95% CI 2.90 to 70.64; P = 0.001; participants = 75; studies = 3; I2 = 0%), at two weeks (random‐effects OR 20.80, 95% CI 2.04 to 211.79; P = 0.01; participants = 27; studies = 1; I2 = 0%), and at three months (random‐effects OR 20.00, 95% CI 2.77 to 144.31; P = 0.003; participants = 27; studies = 1; I2 = 100%) (Analysis 31.1). No difference was found between ketamine and placebo at four weeks (random‐effects OR 1.60, 95% CI 0.35 to 7.40; P = 0.55; participants = 27; studies = 1; I2 = 100%).
31.1. Analysis.

Comparison 31: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding multiple doses), Outcome 1: Response rate
31.2 Efficacy: number of participants who achieve remission
More participants receiving ketamine achieved remission than those receiving placebo in single dose administration studies at 24 hours (random‐effects OR 5.60, 95% CI 1.07 to 29.46; P 0.04; participants = 75; studies = 3; I2 = 0%), at 72 hours (random‐effects OR 6.60, 95% CI 1.51 to 28.92; P = 0.01; participants = 83; studies = 4; I2 = 0%), at one week (random‐effects OR 9.02, 95% CI 1.80 to 45.31; P = 0.008; participants = 75; studies = 3; I2 = 0%), and at four weeks (random‐effects OR 8.12, 95% CI 0.80 to 82.73; P = 0.08; participants = 27; studies = 1; I2 = 0%) (Analysis 31.2). No difference in remission was found at two weeks (random‐effects OR 1.30, 95% CI 0.41 to 4.12; P = 0.66; participants = 78; studies = 2; I2 = 5%)
31.2. Analysis.

Comparison 31: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding multiple doses), Outcome 2: Remission rate
31.3 Change scores on depression scale from baseline
A decrease in depression rating scale scores was found in participants randomised to receive ketamine over placebo when excluding multiple dose trials at 24 hours (random‐effects SMD ‐0.87, 95% CI ‐1.26 to ‐0.48; P < 0.0001; participants = 231; studies = 8; I2 = 41%), at 72 hours (random‐effects SMD ‐0.68, 95% CI ‐1.28 to ‐0.07; P = 0.03; participants = 148; studies = 6; I2 = 62%), at one week (random‐effects SMD ‐1.07, 95% CI ‐1.57 to ‐0.58; P < 0.0001; participants = 78; studies = 4; I2 = 0%), and at two weeks (random‐effects SMD ‐1.14, 95% CI ‐1.98 to ‐0.30; P = 0.08; participants = 26; studies = 1; I2 = 0%) (Analysis 31.3). No difference was found at four weeks (random‐effects SMD ‐0.43, 95% CI ‐1.21 to 0.35; P = 0.28; participants = 26; studies = 1; I2 = 0%).
31.3. Analysis.

Comparison 31: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding multiple doses), Outcome 3: Depression rating scale score
31.4 Suicidality
No differences in suicidality were found between ketamine and placebo in single dose trials at 24 hours (MD 0.02, 95% CI ‐0.78 to 0.82; P = 0.96; participants = 48; studies = 1; I2 = 0%) or at 72 hours (MD 0.09, 95% CI ‐0.63 to 0.81; P = 0.81; participants = 48; studies = 1; I2 = 0%) (Analysis 31.4).
31.4. Analysis.

Comparison 31: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding multiple doses), Outcome 4: Suicidal ideation composite
32. Ketamine versus placebo (post‐hoc sensitivity analysis: excluding add‐on ECT studies)
Eleven studies contributed to this sensitivity analysis (Arabzadeh 2018; Berman 2000; Chen 2018; Hu 2016; Ionescu 2018; Li 2016; Singh 2016 a; Sos 2013; Su 2017; Tiger 2020; Zarate 2006a). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
32.1 Efficacy: number of participants who respond to treatment
Ketamine was more efficacious than placebo for response when excluding add‐on ECT studies at 24 hours (random‐effects OR 3.94, 95% CI 1.54 to 10.10; P = 0.004; participants = 185; studies = 7; I2 = 14%), at 72 hours (random‐effects OR 15.84, 95% CI 3.68 to 68.12; P = 0.0002; participants = 83; studies = 4; I2 = 0%), at one week (random‐effects OR 14.32, 95% CI 2.90 to 70.64; P = 0.001; participants = 75; studies = 3; I2 = 0%), and at two weeks (random‐effects OR 15.73, 95% CI 4.71 to 52.51; P < 0.00001; participants = 85; studies = 2; I2 = 0%) (Analysis 32.1). No difference was found between ketamine and placebo at four weeks (random‐effects OR 2.00, 95% CI 0.68 to 5.85; P = 0.21; participants = 132; studies = 3; I2 = 41%) or at three months (random‐effects OR 3.95, 95% CI 0.16 to 97.23; P = 0.40; participants = 47; studies = 2; I2 = 80%).
32.1. Analysis.

Comparison 32: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding add‐on ECT studies), Outcome 1: Response rate
32.2 Efficacy: number of participants who achieve remission
Remission rates were increased in participants assigned to receive ketamine over placebo in studies administering study medications without ECT at 24 hours (random‐effects OR 5.60, 95% CI 1.07 to 29.46; P = 0.04; participants = 75; studies = 3; I2 = 0%), at 72 hours (random‐effects OR 6.60, 95% CI 1.51 to 28.92; P = 0.01; participants = 83; studies = 4; I2 = 0%), at one week (random‐effects OR 9.02, 95% CI 1.80 to 45.31; P = 0.008; participants = 75; studies = 3; I2 = 0%), and at two weeks (random‐effects OR 7.50, 95% CI 1.51 to 37.22; P = 0.01; participants = 85; studies = 2; I2 = 0%) (Analysis 32.2). No difference was found at four weeks (random‐effects OR 2.19. 95% CI 0.85 to 5.66, participants‐ 132, studies = 3; I2 = 0%), or at three months (random‐effects OR 1.29, 95% CI 0.14 to 11.54; P = 0.82; participants = 20; studies = 1; I2 = 0%).
32.2. Analysis.

Comparison 32: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding add‐on ECT studies), Outcome 2: Remission rate
32.3 Change scores on depression scale from baseline
We found a decrease in depression rating scale scores in those allocated ketamine over placebo when excluding add‐on ECT trials at all time points: at 24 hours (random‐effects SMD ‐0.87, 95% CI ‐1.26 to ‐0.48; P < 0.0001; participants = 231; studies = 8; I2 = 41%), at 72 hours (random‐effects SMD ‐0.68, 95% CI ‐1.28 to ‐0.07; P = 0.03; participants = 148; studies = 6; I2 = 62%), at one week (random‐effects SMD ‐0.80, 95% CI ‐1.31 to ‐0.30; P = 0.002; participants = 97; studies = 5; I2 = 24%), at two weeks (random‐effects SMD ‐0.73, 95% CI ‐1.31 to ‐0.15; P = 0.01; participants = 126; studies = 3; I2 = 50%), and at four weeks (random‐effects SMD ‐0.68, 95% CI ‐1.07 to ‐0.29; P = 0.0006; participants = 107; studies = 2; I2 = 0%) (Analysis 32.3).
32.3. Analysis.

Comparison 32: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding add‐on ECT studies), Outcome 3: Depression rating scale score
32.4 Suicidality
No differences were found in suicidality between ketamine and placebo excluding trials using add‐on ECT at any time point: at 24 hours (random‐effects MD 0.02, 95% CI ‐0.78 to 0.82; P = 0.96; participants = 48; studies = 1; I2 = 0%), at 72 hours (random‐effects MD 0.34, 95% CI ‐0.25 to 0.93; P = 0.26; participants = 68; studies = 2; I2 = 10%), at one week (random‐effects MD ‐0.30, 95% CI ‐1.56 to 0.96; P = 0.64; participants = 19; studies = 1; I2 = 0%), and at two weeks (random‐effects MD ‐0.20, 95% CI ‐1.46 to 1.06; P = 0.76; participants = 19; studies = 1; I2 = 0%) (Analysis 32.4).
32.4. Analysis.

Comparison 32: Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding add‐on ECT studies), Outcome 4: Suicidal ideation composite
33. Esketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features)
Eight studies contributed to this sensitivity analysis (Canuso 2018; Daly 2018; Fedgchin 2019; Fu 2020; Ionescu 2020; Ochs‐Ross 2020; Popova 2019; Singh 2016 b). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
33.1 Efficacy: number of participants who respond to treatment
Esketamine treatment increased response rates compared with placebo at 24 hours when excluding studies that included participants with bipolar disorder or psychotic features (random‐effects OR 2.11, 95% CI 1.20 to 3.68; P = 0.009; participants = 1071; studies = 5; I2 = 50%), at one week (random‐effects OR 1.64, 95% CI 1.05 to 2.54; P = 0.03; participants = 1083; studies = 5; I2 = 35%), at two weeks (random‐effects OR 1.57, 95% CI 1.09 to 2.28; P = 0.02; participants = 451; studies = 2; I2 = 0%), and at four weeks (random‐effects OR 1.84, 95% CI 1.44 to 2.37; P<0.00001; participants = 1117; studies = 5; I2 = 0%) (Analysis 33.1). No difference was found between esketamine and placebo at 72 hours (random‐effects OR 1.34, 95% CI 0.92 to 1.96; P = 0.13; P < 0.13; participants = 451; studies = 2; I2 = 0%).
33.1. Analysis.

Comparison 33: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 1: Response rate
33.2 Efficacy: number of participants who achieve remission
Remission was higher in participants receiving esketamine over placebo in studies that did not include participants with bipolar disorder or psychotic features at 24 hours (random‐effects OR 2.74, 95% CI 1.71 to 4.40; P < 0.0001; participants = 894; studies = 5; I2 = 0%), at two weeks (random‐effects OR 1.52, 95% CI 1.07 to 2.16; P = 0.02; participants = 832; studies = 4; I2 = 0%), and at four weeks (random‐effects OR 1.57, 95% CI 1.18 to 2.10; P = 0.002; participants = 957; studies = 5; I2 = 0%) (Analysis 33.2). No difference was found at 72 hours (random‐effects OR 1.55, 95% CI 0.91 to 2.64; P = 0.11; participants = 517; studies = 3; I2 = 24%), at one week (random‐effects OR 1.62, 95% CI 0.91 to 2.89; P = 0.10; participants = 916; studies = 5; I2 = 36%).
33.2. Analysis.

Comparison 33: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 2: Remission rate
33.3 Change scores on depression scale from baseline
Esketamine was associated with a decrease in depression rating scale scores over placebo when studies including participants with bipolar disorder or psychotic features were excluded at 24 hours (random‐effects SMD ‐0.31, 95% CI ‐0.45 to ‐0.17; P < 0.0001; participants = 824; studies = 4; I2 = 0%), at 72 hours (random‐effects SMD ‐0.30, 95% CI ‐0.50 to ‐0.11; P = 0.002; participants = 517; studies = 3; I2 = 14%), at one week (random‐effects SMD ‐0.24, 95% CI ‐0.37 to ‐0.10; P = 0.0007; participants = 857; studies = 4; I2 = 0%), at two weeks (random‐effects SMD ‐0.21, 95% CI ‐0.34 to ‐0.07; P = 0.003; participants = 857; studies = 4; I2 = 0%), and at four weeks (random‐effects SMD ‐0.27, 95% CI ‐0.39 to ‐0.16; P < 0.00001; participants = 1182; studies = 6; I2 = 0%) (Analysis 33.3). No difference between esketamine and placebo was found at three months (random‐effects SMD ‐0.12, 95% CI ‐0.75 to 0.52; P = 0.72; participants = 38; studies = 1; I2 = 0%).
33.3. Analysis.

Comparison 33: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 3: Depression rating scale score
33.4 Suicidality
No differences in suicidality were found between esketamine and placebo when excluding studies that included participants with bipolar disorder or psychotic features at any time points: at 24 hours (random‐effects MD ‐0.15, 95% CI ‐0.44 to 0.15; P = 0.33; participants = 450; studies = 2; I2 = 0%), at 72 hours (random‐effects MD ‐0.20, 95% CI ‐0.49 to 0.08; P = 0.16; participants = 451; studies = 2; I2 = 0%), at one week (random‐effects MD 0.01, 95% CI ‐0.10 to 0.13; P = 0.83; participants = 660; studies = 3; I2 = 0%), at two weeks (random‐effects MD ‐0.10, 95% CI ‐0.22 to 0.02; P = 0.10; participants = 659; studies = 3; I2 = 0%), and at four weeks (random‐effects MD ‐0.04, 95% CI ‐0.12 to 0.05; P = 0.40; participants = 647; studies = 3; I2 = 0%) (Analysis 33.4).
33.4. Analysis.

Comparison 33: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features), Outcome 4: Suicidal ideation composite
34. Esketamine versus placebo (pre‐planned sensitivity analysis: excluding treatment resistant populations)
Three studies contributed to this sensitivity analysis (Canuso 2018; Fu 2020; Ionescu 2020). Data were provided at 24 hours, 72 hours, one week, two weeks, and four weeks.
34.1 Efficacy: number of participants who respond to treatment
Esketamine had higher response efficacy over placebo when excluding treatment resistant populations at 24 hours (random‐effects OR 1.55, 95% CI 1.03 to 2.33; P = 0.03; participants = 451; studies = 2; I2 = 0%), two weeks (random‐effects OR 1.57, 95% CI 1.09 to 2.28; P = 0.02; participants = 451; studies = 2; I2 = 0%), and at four weeks (random‐effects OR 1.70, 95% CI 1.17 to 2.46; P = 0.006; participants = 451; studies = 2; I2 = 0%) (Analysis 34.1). There was no difference found between esketamine and placebo at 72 hours (random‐effects OR 1.34, 95% CI 0.92 to 1.96; P = 0.13; participants = 451; studies = 2; I2 = 0%), and at one week (random‐effects OR 1.23, 95% CI 0.85 to 1.78; P = 0.28; participants = 451; studies = 2; I2 = 0%).
34.1. Analysis.

Comparison 34: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 1: Response rate
34.2 Efficacy: number of participants who achieve remission
Remission rates were greater in participants receiving esketamine over placebo when excluding treatment resistant populations at 24 hours (random‐effects OR 2.35, 95% CI 1.40 to 3.92; P = 0.001; participants = 517; studies = 3; I2 = 0%), and at four weeks (random‐effects OR 1.58, 95% CI 1.10 to 2.29; P = 0.01; participants = 517; studies = 3; I2 = 0%) (Analysis 34.2). No difference was found at 72 hours (random‐effects OR 1.55, 95% CI 0.91 to 2.64; P = 0.11; participants = 517; studies = 3; I2 = 24%), at one week (random‐effects OR 1.31, 95% CI 0.86 to 2.01; P = 0.21; participants = 517; studies = 3; I2 = 0%), or at two weeks (random‐effects OR 1.38, 95% CI 0.93 to 2.04; P = 0.11; participants = 517; studies = 3; I2 = 0%).
34.2. Analysis.

Comparison 34: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 2: Remission rate
34.3 Change scores on depression scale from baseline
There was a decrease in depression rating scale scores found in esketamine over placebo when treatment resistant populations were excluded at 24 hours (random‐effects SMD ‐0.34, 95% CI ‐0.52 to ‐0.17; P = 0.0001; participants = 514; studies = 3; I2 = 0%), at 72 hours (random‐effects SMD ‐0.30, 95% CI ‐0.50 to ‐0.11; P = 0.002; participants = 517; studies = 3; I2 = 14%), at one week (random‐effects SMD ‐0.21, 95% CI ‐0.38 to ‐0.04; P = 0.02; participants = 517; studies = 3; I2 = 0%), and at four weeks (random‐effects SMD ‐0.27, 95% CI ‐0.44 to ‐0.10; P = 0.002; participants = 517; studies = 3; I2 = 0%) (Analysis 34.3). No difference was found between esketamine and placebo at two weeks (random‐effects SMD ‐0.14, 95% CI ‐0.32 to 0.03; P = 0.10; participants = 517; studies = 3; I2 = 0%).
34.3. Analysis.

Comparison 34: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 3: Depression rating scale score
34.4 Suicidality
No differences in suicidality were found between esketamine and placebo when excluding treatment resistant populations at any time point: at 24 hours (random‐effects MD ‐0.15, 95% CI ‐0.44 to 0.15; P = 0.33; participants = 450; studies = 2; I2 = 0%), at 72 hours (random‐effects MD ‐0.20, 95% CI ‐0.49 to 0.08; P = 0.16; participants = 451; studies = 2; I2 = 0%), at one week (random‐effects MD ‐0.15, 95% CI ‐0.41 to 0.11; P = 0.26; participants = 451; studies = 2; I2 = 0%), at two weeks (random‐effects MD ‐0.19, 95% CI ‐0.43 to 0.05; P = 0.12; participants = 451; studies = 2; I2 = 0%), and at four weeks (random‐effects MD ‐0.15, 95% CI ‐0.39 to 0.09; P = 0.22; participants = 451; studies = 2; I2 = 0%) (Analysis 34.4).
34.4. Analysis.

Comparison 34: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding TRD populations), Outcome 4: Suicidal ideation composite
35. Esketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%)
Eight studies contributed to this sensitivity analysis (Daly 2018; Fedgchin 2019; Fu 2020; Ionescu 2020; Jarventausta 2013; Ochs‐Ross 2020; Popova 2019; Singh 2016 b). Data were provided at 24 hours, 72 hours, one week, two weeks, four weeks, and three months.
35.1 Efficacy: number of participants who respond to treatment
Esketamine administration increased response rates over placebo when excluding trials with a dropout rate greater than 20% at 24 hours (random‐effects OR 2.11, 95% CI 1.20 to 3.68; P = 0.009; participants = 1071; studies = 5; I2 = 50%), at one week (random‐effects OR 1.60, 95% CI 1.09 to 2.34; P = 0.02; participants = 1115; studies = 6; I2 = 20%), at two weeks (random‐effects OR 1.57, 95% CI 1.09 to 2.28; P = 0.02; participants = 451; studies = 2; I2 = 0%), and at four weeks (random‐effects OR 1.84, 95% CI 1.44 to 2.37; P < 0.00001; participants = 1117; studies = 5; I2 = 0%) (Analysis 35.1). No differences in response were found between esketamine and placebo at 72 hours (random‐effects OR 1.34, 95% CI 0.92 to 1.96; P = 0.13; participants = 451; studies = 2; I2 = 0%).
35.1. Analysis.

Comparison 35: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 1: Response rate
35.2 Efficacy: number of participants who achieve remission
Esketamine was associated with higher remission numbers of participants achieving remission over placebo in trials with a dropout rate less than 20% at 24 hours (random‐effects OR 2.88, 95% CI 1.72 to 4.81; P < 0.0001; participants = 828; studies = 4; I2 = 0%), at two weeks (random‐effects OR 1.51, 95% CI 1.04 to 2.19; P = 0.03; participants = 766; studies = 3; I2 = 0%), and at four weeks (random‐effects OR 1.60, 95% CI 1.19 to 2.16; P = 0.002; participants = 891; studies = 4; I2 = 0%) (Analysis 35.2). No differences were found at 72 hours (random‐effects OR 1.76, 95% CI 0.97 to 3.18; P = 0.06; participants = 451; studies = 2; I2 = 31%), or at one week (random‐effects OR 1.79, 95% CI 0.93 to 3.42; P = 0.08; participants = 882; studies = 5; I2 = 33%)
35.2. Analysis.

Comparison 35: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 2: Remission rate
35.3 Change scores on depression scale from baseline
We found a decrease in depression rating scale scores in esketamine over placebo when excluding trials with a dropout rate greater than 20% at 24 hours (random‐effects SMD ‐0.29, 95% CI ‐0.43 to ‐0.14; P = 0.0001; participants = 758; studies = 3; I2 = 0%), at 72 hours (random‐effects SMD ‐0.26, 95% CI ‐0.45 to ‐0.07; P = 0.007; participants = 451; studies = 2; I2 = 4%), at one week (random‐effects SMD ‐0.23, 95% CI ‐0.37 to ‐0.09; P = 0.001; participants = 818; studies = 4; I2 = 0%), at two weeks (random‐effects SMD ‐0.21, 95% CI ‐0.36 to ‐0.07; P = 0.004; participants = 791; studies = 3; I2 = 0%), and at four weeks (random‐effects SMD ‐0.27, 95% CI ‐0.39 to ‐0.15; P < 0.0001; participants = 1116; studies = 5; I2 = 0%) (Analysis 35.3). No differences were found at three months (random‐effects SMD ‐0.12, 95% CI ‐0.75 to 0.52; P = 0.72; participants = 38; studies = 1; I2 = 0%).
35.3. Analysis.

Comparison 35: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 3: Depression rating scale score
35.4 Suicidality
No differences in suicidality were found between esketamine and placebo when excluding trials with a dropout rate greater than 20% at any time point: at 24 hours (random‐effects MD ‐0.15, 95% CI ‐0.44 to 0.15; P = 0.33; participants = 450; studies = 2; I2 = 0%), at 72 hours (random‐effects MD ‐0.20, 95% CI ‐0.49 to 0.08; P = 0.16; participants = 451; studies = 2; I2 = 0%), at one week (random‐effects MD 0.01, 95% CI ‐0.10 to 0.13; P = 0.83; participants = 660; studies = 3; I2 = 0%), at two weeks (random‐effects MD ‐0.10, 95% CI ‐0.22 to 0.02; P = 0.10; participants = 659; studies = 3; I2 = 0%), and at four weeks (random‐effects MD ‐0.04, 95% CI ‐0.12 to 0.05; P = 0.40; participants = 647; studies = 3; I2 = 0%) (Analysis 35.4).
35.4. Analysis.

Comparison 35: Esketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%, Outcome 4: Suicidal ideation composite
Discussion
Summary of main results
This updated systematic review assessed the efficacy and acceptability of ketamine and other glutamate receptor modulators for the treatment of unipolar depression. We identified a total of 64 randomised controlled trials, involving 5299 participants and 16 glutamate receptor modulators (of which data were available for 15). It is important to note that the included studies in the present review had, on average, small to very small sample sizes, and furthermore there was a lack of data available on important pre‐defined outcomes, including side effects.
Efficacy: Ketamine versus placebo
Overall, we found potential evidence of efficacy of ketamine over placebo up to one week, in terms of response, remission and change in depressive symptoms. Not all the included studies reported data at all the time points prespecified in this review, so we cannot rule out the possibility that results would have been different if all studies contributed to all outcomes at every time point. The updated review included new studies with longer‐term data, with three studies providing data for up to three months (Anderson 2017; Hu 2016; Ionescu 2018), which found a lack of efficacy of ketamine over placebo at this time point for response and remission. This indicates that although ketamine may be an effective treatment compared to placebo in the short term, this effect could be lost after treatment ends. No differences were found in suicidality at any time point, suggesting that ketamine is not an effective treatment for suicidality over placebo. However, this was based on only two studies.
We were able to conduct a number of subgroup analyses for the ketamine versus placebo comparison. Inpatients appeared to have a faster response to ketamine than outpatients, with higher response rates at 24 hours over placebo, and both inpatients and outpatients observing higher response with ketamine at 72 hours. This seemed to be sustained for longer in outpatients (at one week and two weeks), which was not seen in inpatients. We found no evidence of a difference in remission between ketamine and placebo in outpatients, although this was based on extremely small participant numbers. Ketamine was likely more effective than placebo at 24 hours, 72 hours, and one week for inpatients. For depression scores on rating scales there were decreases for inpatients administered ketamine over placebo at 24 hours, 72 hours, and one week, whereas depression scores for outpatients only saw decreases for ketamine over placebo at two weeks and four weeks. Limited data identified no differences in suicidality at any time point for outpatients, and no data were available for inpatients. These subgroup analyses suggest that the effects of ketamine may be experienced differently depending on the setting in which the drug is administered. However, this finding could also reflect differences in the clinical characteristics of the patients treated in inpatient versus outpatient settings. Additionally, the sample sizes in these analyses are too small to be conclusive.
We consistently found possible increased response and remission rates favouring ketamine over placebo up to one week in all sensitivity analyses. Depression rating scale scores may have been decreased for those administered ketamine over placebo for up to two weeks in all sensitivity analyses, and up to four weeks in all but one analysis (excluding multiple doses). No differences were found in all sensitivity analyses about suicidality, but data were limited.
Efficacy: ketamine versus other pharmacologically active agents and: electroconvulsive therapy (ECT)
Ketamine appeared to be more effective than midazolam in the short term. Only one study provided data at three months for response and remission (Gálvez 2018), with ketamine not being more effective than midazolam for either outcome. This is in line with other evidence supporting the notion that ketamine can be an effective drug in treating depressive symptoms in the short term, but this effect is not sustained when treatment ends. Results from ketamine versus thiopental, remifentanil hydrochloride and ECT comparisons also supported this conclusion, although very limited data were available. Comparisons with methohexital and propofol showed no differences. However, data were only available for one study in each comparison and only for depression rating scale score outcomes. Also these comparisons occurred in the context of ECT treatment which complicates interpretation.
Efficacy: esketamine
There was also evidence of esketamine being efficacious over placebo at 24 hours, two weeks, and four weeks for response, remission, and depression rating scale scores. Evidence was mixed at other time points. Only one study provided data at three months, and only for depression rating scale scores, showing uncertain evidence of no difference between esketamine and placebo (Fedgchin 2019). This suggests short‐term effectiveness of esketamine for treatment of depression, but further studies exploring long‐term outcomes are needed. There were no differences in suicidality between esketamine over placebo at any time point.
Several subgroup and sensitivity analyses were able to be conducted for the esketamine versus placebo comparison. In the subgroup analyses we found that outpatients may have a very quick increased response to esketamine, whilst inpatients could take longer to respond. No differences in suicidality between esketamine and placebo were found in any subgroups.
Sensitivity analyses for esketamine versus placebo did not materially change the results from the primary analysis. No differences were found between esketamine and placebo at any time point for suicidality, suggesting that esketamine is not effective in reducing suicidal ideation.
When compared head‐to‐head, one study showed no difference in depression rating scale scores between ketamine and esketamine at any time point (Correia‐Melo 2020). However, data were only provided up to one week, and minimal data were available for other outcomes in this study.
Adverse events
In terms of adverse events, ketamine increased the incidence of agitation/anxiety, confusion, and dissociative symptoms over placebo. When compared to midazolam, ketamine was more likely to be associated with blurred vision, dizziness, general malaise, increased blood pressure or heart rate, nausea/vomiting, sleepiness/drowsiness. However, the majority of these effects were short‐lived on the day of infusion, and were not observed at one to seven days post‐infusion. Esketamine was more likely than placebo to be associated with change in blood pressure, constipation, dissociative symptoms, dizziness, dizziness postural, feeling drunk, nausea/vomiting, paresthesia/neuropathy exacerbation, sensory disturbance, sedation, sleepiness/drowsiness, vertigo, and vision blurred.
Efficacy: other glutamate receptor modulators
We found very limited evidence for the antidepressant efficacy of the remaining 13 glutamate receptor modulators, with the only effects found being citicoline over placebo remission rate at four weeks, decoglurant over placebo response at four weeks, and response rate of sarcosine over citalopram at two weeks.
Overall completeness and applicability of evidence
The overall completeness of the evidence was found to be limited. We set out nine outcomes in the protocol and found that many trials did not provide data on all of these outcomes. The primary outcomes of response rate and adverse events were not reported in all studies, but response was imputed where possible. However, adverse events data were not accessible in some trials. We found no data, or very limited data, on certain prespecified outcomes, namely: suicidality, cognition, quality of life, costs to healthcare services and dropouts due to lack of efficacy. The great majority of the included trials in this review were placebo‐controlled, with only 16 studies comparing a glutamate receptor modulator with an active comparison. This limits the completeness of the comparative evidence (Cipriani 2020; Naci 2020). The literature search for this review identified a large number of ongoing studies. These trials could contribute key data and of course will be included in future updates of the review.
In terms of applicability of evidence, all participants in the included studies of the present review met standardised diagnostic criteria of a depressive episode according to the DSM‐IV, DSM‐IV‐TR, or DSM‐5. However, there was variation in regards to the severity of depression in the recruited participants, with severities including moderate, severe and treatment resistant, which may impact the intervention effect (Deeks 2021). There was also disparity among the studies in terms of how the interventions were administered. In many of the trials, participants received concomitant psychotropic medications (for example, continuing the pre‐existing antidepressant), some received concomitant ECT, and the length and route of administration also varied.
Quality of the evidence
It was difficult to judge the overall quality of the retrieved evidence, as the most important items of the risk of bias tool (random sequence generation, allocation concealment and blinding of outcome assessment) were deemed ''unclear'' in many studies. This might be due to problems in the reporting of the included studies; however this could potentially bias our results and limit the reliability of the findings of the review.
The vast majority of studies in this review included small sample sizes (50 out of the 64 studies had an overall study sample size below 100). This is not unusual in psychiatry; however, in certain comparisons there were too few participants to be able to draw any meaningful conclusion. For example, in the MK‐0657 versus placebo comparison, there were only five participants in total.
An important factor to take into consideration is the bias that may have occurred in blinding procedures. Given the profile of ketamine and its psychotomimetic side effects, participants and personnel, particularly in comparisons with inactive placebo, would probably not have remained unaware of treatment arm allocation, despite attempts to blind them. Of the included studies assessing the efficacy of ketamine, most did not test the blinding or provide any information relating to whether the intended blinding was effective. This should be considered a major limitation, which is likely to result in a biased assessment of the intervention effect.
The certainty of evidence according to GRADE (Atkins 2004) was very low to moderate in the comparison between ketamine and placebo (Table 1), and also for ketamine versus midazolam (Table 2). The GRADE certainty of evidence the comparison between esketamine and placebo was very low to high (Table 3).
Potential biases in the review process
We identified 64 studies, but we could only include 54 of them in the meta‐analysis due to the unavailability of data despite contacting the authors. We cannot rule out that this may have had an impact on the pooled results, as these 10 studies may have contributed additional important data (Mavridis 2014).
We could not evaluate study publication bias or outcome reporting bias due to the small number of included studies in each comparison and due to the unavailability of the study protocols. Although we searched extensively for relevant trials, it is possible that unpublished trials remain unknown to us.
In order to generate as much data as possible, according to the review protocol we imputed response and remission rates, as we did previously in other Cochrane Reviews (Furukawa 2005; Magni 2013; Watanabe 2011). In addition, in one case we imputed standard deviations (SDs) (Yoosefi 2014) using a validated method (Furukawa 2006). Imputation of SDs might affect the results, by widening the confidence interval and reducing its weight in the analysis, underestimating the overall treatment effect (Aitken 2019). Due to limited data in the ketamine versus thiopental comparison, however, Yoosefi 2014 was frequently the only study contributing to outcomes, so a reduction in weighting would not significantly impact our results and interpretation.
In this review, we used the following definition of treatment‐resistant definition: inadequate response to at least two antidepressants. This definition can limit the interpretation of our findings, as it enables inclusion of patients who have merely failed treatment with two selective serotonin reuptake inhibitors and no other classes of antidepressants or psychotherapy. Some definitions of treatment‐resistant depression demand that a trial of an antidepressant can only be regarded as adequate if the drug is take for a minimum duration and dose. Other definitions will count a trial of an antidepressant which was not tolerated. A majority of surveyed clinicians indicated that they would define treatment resistant depression as an adequate trial of two to three antidepressant medications plus a 10‐ to 12‐week trial of evidence‐based psychotherapy (Brown 2019). While recognising that this definition may limit generalisability of this review to real‐world clinical practice (Turner 2019), it is also used by the majority of depression treatment studies, so a pragmatic decision was made to use it.
There is also some discussion in the scientific literature about the classification of drugs (Malhi 2019). Unlike ketamine and memantine which have well‐characterised primary effects on glutamate receptors, some of the treatments included in this review (for instance, atomoxetine) have an action on glutamate receptors, but have other pharmacological properties that are thought more important in their mode of action. However, these complex pharmacological properties are not always recognised in drug descriptions. For example, a publication on neuroscience‐based drug nomenclature described atomoxetine as a norepinephrine reuptake inhibitor only, without mention of its action on glutamate receptors (Zohar 2014). However, in the present review we wished to adopt a more pragmatic approach to better inform clinical practice. We therefore decided to run a very comprehensive search of the scientific literature, and compared individual drugs in order to avoid the clustering of different interventions into non‐homogeneous drug classes.
Agreements and disagreements with other studies or reviews
In line with findings of previous reviews (Caddy 2015; McGirr 2015; Naughton 2014; Marcantoni 2020; Memon 2020), the present review demonstrated a rapid onset of antidepressant effect for ketamine, which lasts up to one week. However, the majority of these reviews have not adopted the same strict inclusion criteria as in the present review. Most previously conducted reviews have included data from non‐randomised studies and from both phases of cross‐over trials (Marcantoni 2020), which may have overestimated the efficacy of ketamine due to selection bias and carry‐over effect. In order to be clinically informative and rely only on the most robust results, in the present review we only included data from the first phase of cross‐over trials and included only double blind or single blind randomised controlled trials |9RCTs).
We also found a rapid onset of antidepressant effect for esketamine, which was consistent with a systematic review by Zheng 2020. Zheng concluded that an antidepressant effect lasted for at least 28 days; however we were unable to find an effect at 72 hours and one week. This may be because Zheng only compared data between baseline and endpoint (which ranged between 8 to 28 days), whilst we examined data for all available time points.
Of further note is that previous reviews included studies of patients diagnosed with unipolar and bipolar depression in the same analyses (Caddy 2014; Coyle 2015; Marcantoni 2020). By contrast, in this review we have focused specifically on unipolar depression because of the specific clinical features that differentiate this disorder from bipolar depression.
Authors' conclusions
Implications for practice.
The present review has provided evidence for a short‐term antidepressant effect of ketamine compared with placebo and midazolam, and esketamine compared with placebo in adults with unipolar depression. Our confidence in the findings of the review is limited by the risk of performance bias and low number of trials contributing data to the meta‐analysis for each comparison (Cipriani 2019). The majority of comparisons contain only one study, with the largest body of evidence included in a single analysis being only from eight studies. There was no robust evidence for the use of other glutamate receptor modulators in depression.
This review continues to support the rapid antidepressant effect of ketamine, and also found a rapid antidepressant effect of esketamine, an important finding given the typical delayed onset of action of traditional antidepressants, especially since many of these studies recruited patients with severe or treatment‐resistant depression. There was no evidence for a longer‐term effect of acute ketamine administration after one week, and whilst continued esketamine was effective over placebo at four weeks there was no effectiveness at 72 hours or one week. More studies with longer‐term outcomes are needed to confirm these results.
The majority of studies included in this review which compared ketamine with placebo, administered the ketamine intravenously, with only one trial administering the drug orally. The difficulties of administering via intravenous infusion in a clinical setting (i.e. equipment, time, staff requirements), create limitations to the accessibility of this treatment. Most esketamine trials studied intranasal administration, which may be a promising alternative in improving applications to clinical practice.
The present review provides additional information about the side‐effect profile of ketamine and esketamine, with additional data on a multitude of adverse events associated with this drug. Given these identified side effects, and the adverse effects that have been linked to ketamine abuse, such as cognitive impairments and bladder dysfunction, it is important that both the short and long‐term side effects are carefully considered for clinical application.
Whilst this updated review has demonstrated some promising evidence for the use of ketamine as an antidepressant, it is clear that there are still challenges for clinical application that require careful consideration and further research.
Implications for research.
Thecertainty of the evidence in the present review was assessed as very low to high quality according to GRADE (Atkins 2004). There were very few trials included in each comparison, and furthermore, the numbers of participants included in each trial were small (Jones 2019). It is therefore apparent that in order to draw robust conclusions about ketamine's antidepressant effects, there is a need for studies to be conducted that are of high methodological standard and that assess important outcomes such as cognition and adverse events, as well as efficacy.
Most trials included in the present meta‐analysis examining the efficacy of ketamine used intravenous administration, for which there are practical limitations as outlined above. Additional routes of administration should be explored in further randomised controlled trials (RCTS) to understand whether different modes of administration can be effective in order to improve accessibility.
There were limited data regarding long‐term effects of ketamine and esketamine, but the available evidence suggested that neither drug has sustained anti‐depressant effects after the end of treatment or prevents relapse at three months. Therefore further research could examine the use and efficacy of the delivery of psychotherapy (such as cognitive behavioral therapy (CBT)) or other medications following administration of glutamate receptor modulators (Zhou 2020).
Only six RCTs were identified that compared a psychoactive drug to ketamine, five in which midazolam efficacy was compared with ketamine, and one comparing ketamine and esketamine. This highlights the need for future research to focus on conducting RCTs which compare antidepressants with other psychoactive drugs (Cipriani 2010; Koesters 2013; Watanabe 2008). Furthermore, given the inadequate blinding methods that could potentially occur when comparing ketamine versus placebo (given its side‐effect profile), conducting more head‐to‐head comparisons that use an active control drug with similar effects to ketamine are needed.
In the present review the majority of studies provided patients with concomitant medication alongside the trial, demonstrating heterogeneity in the methodology (Efthimiou 2019). The role that concomitant medication may play in ketamine's antidepressant effect is currently unclear, and thus future research should explore and clarify this.
What's new
| Date | Event | Description |
|---|---|---|
| 17 November 2021 | Amended | Typo in the abstract corrected. |
History
Protocol first published: Issue 4, 2015 Review first published: Issue 9, 2015
| Date | Event | Description |
|---|---|---|
| 13 September 2021 | Amended | Typo corrected in the search methods and Appendix 3. |
| 9 September 2021 | New citation required but conclusions have not changed | Review has been updated: Ketamine and esketamine may be more efficacious than placebo at 24 hours, however evidence is of low or very low quality. |
| 9 September 2021 | New search has been performed | Thirty‐nine new trials identified. |
Acknowledgements
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Common Mental Disorders (CCMD) Group. Andrea Cipriani is supported by the National Institute for Health Research (NIHR) Oxford Cognitive Health Clinical Research Facility, by an NIHR Research Professorship (grant RP‐2017‐08‐ST2‐006), by the NIHR Oxford and Thames Valley Applied Research Collaboration and by the NIHR Oxford Health Biomedical Research Centre (grant BRC‐1215‐20005). The views expressed are those of the authors and not necessarily those of the UK National Health Service, the NIHR, or the UK Department of Health. KH is an NIHR Senior Investigator (Emeritus). Rupert McShane is supported by the NIHR Oxford Health Biomedical Research Centre.
We would like to thank the following authors for their help in retrieving or providing additional information on included or excluded studies: Hamid Afshar, Michael Berk, Robert Berman, Margarida Bretas‐Bastos, Sue Cotton, Olivia Dean, Mehdi Ghasemi, Charles Green, Michael Grunebaum, Uriel Heresco‐Levy, Kaija Jarventausta, Daniel Javitt, Olli Kampman, John Krystal, Steven Lippmann, Colleen Loo, David Luckenbaugh, Sanjay Mathew, Kari Nations, Tomas Novak, Georgios Paslakis, Vanina Popova, Lisa Roach, Tarek Shams, Paulo Shiroma, Eric Smith, Peter Sos, Rachael Sumner, Mikael Tiger, Jari Tiihonen, Christine Ulbricht, Elena Velasco Diez, Janet Williams and Carlos Zarate
The authors and the CCMD Editorial Team are also grateful to the following peer reviewers for their time and comments: Simon Davies, Nuala Livingstone and Jean Sellar‐Edmunds. They would also like to thank Cochrane Copy Edit Support for the team's help.
Disclaimer: The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, the National Health Service (NHS), or the Department of Health and Social Care.
Appendices
Appendix 1. Search strategies (2015‐2020)
Ovid MEDLINE databases
Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to July 28 2020> [Date limited 2015 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 depression/ 2 depressive disorder/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ 3 *Mood Disorders/ or *Affective Symptoms/ 4 "bipolar and related disorders"/ or bipolar disorder/ 5 (depression or depressive? or MDD or dysthymi*).ti,ab,kf. 6 depressed.ti. or (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).ab,kf. 7 (affective disorder* or affective spectrum disorder* or affective state* or affective symptom* or mixed state* or mood disorder*).ti,ab,kf. 8 or/1‐7 9 Amantadine/ or Memantine/ 10 Atomoxetine Hydrochloride/ 11 Acetylcysteine/tu 12 Cycloserine/ 13 Dextromethorphan/ 14 *Excitatory Amino Acid Antagonists/tu 15 ((glutamate* or glutamin* or glutathione* or glycin*) adj2 (modulat* or inhibit* or system?)).ti,ab,kf,hw. 16 Ketamine/ 17 N‐Methylaspartate/ 18 Quinolines/tu 19 Riluzole/ 20 Sarcosine/ 21 Tramadol/ 22 *receptors, glutamate/ or *receptors, ionotropic glutamate/ or *receptors, ampa/ or *receptors, kainic acid/ or *receptors, n‐methyl‐d‐aspartate/ 23 receptors, glutamate/de, ai or receptors, ionotropic glutamate/de, ai or receptors, ampa/de, ai or receptors, kainic acid/de, ai or receptors, n‐methyl‐d‐aspartate/de, ag, ai 24 Glycine Plasma Membrane Transport Proteins/ai 25 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or (GLYX 13 or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or AZD 6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or ETS 6103 or viotra) or ampa or cerc 301 or cerc301 or d‐serin* or GluN2B or mGlu* or N acetyl cysteine* or N acetylcysteine or N methyl D aspartate or NMDA? or nrx 1074 or nrx1074 or kainite or NR2B or sarcosin* or NAC).ti,ab,kf. 26 (Org 26576 or Org26576 or CP‐101,606 or CP101606).ti,ab,kf. 27 Cytidine.ti,ab,kf,hw. 28 or/9‐27 29 controlled clinical trial.pt. 30 randomized controlled trial.pt. 31 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kf. 32 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or crossover or cross‐over or design* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,kf. 33 placebo*.ab,ti,kf. 34 trial.ab,ti,kf. 35 groups.ab. 36 (control* and (trial or study or group*) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kf,hw. 37 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,kf. 38 random allocation/ or single‐blind method/ or double‐blind method/ 39 or/29‐38 40 exp animals/ not humans.sh. 41 39 not 40 42 8 and 28 and 41 43 (2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dt,ep,ez. 44 42 and 43 45 8 and (26 or 27) and 41 46 44 or 45 *************************** Ovid Embase <1980 to 2020 Week 30> [Date limited 2015 onwards] Search Strategy: 1 *depression/ or depression/dt or agitated depression/ or atypical depression/ or chronic depression/ or depressive psychosis/ or endogenous depression/ or involutional depression/ or late life depression/ or major depression/ or masked depression/ or "mixed anxiety and depression"/ or reactive depression/ or recurrent brief depression/ or treatment resistant depression/ 2 bipolar disorder/ or bipolar depression/ or bipolar i disorder/ or bipolar ii disorder/ or "mixed mania and depression"/ 3 mood disorder/ or major affective disorder/ or minor affective disorder/ 4 (depression or depressive? or MDD or TRD or dysthymi*).ti,ab,kw. 5 depressed.ti. or (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).ab,kw. 6 (affective spectrum disorder* or affective state* or mixed state*).ti,ab,kw. 7 or/1‐6 8 amantadine/ 9 memantine/ 10 atomoxetine/ 11 acetylcysteine/ 12 cycloserine/ 13 dextromethorphan/ 14 *glutamic acid/ 15 (glutamate* adj2 (modulat* or inhibit* or system?)).ti,ab,kw. 16 ketamine/ 17 Esketamine/ or Norketamine/ 18 n methyl dextro aspartic acid/ 19 *n methyl dextro aspartic acid receptor/ 20 quinoline/ 21 riluzole/ 22 Sarcosine/ 23 Tramadol/ 24 AZD 6765/ or "mk 0657"/ 25 *n methyl dextro aspartic acid receptor blocking agent/ or n methyl dextro aspartic acid receptor stimulating agent/ 26 *excitatory amino acid receptor/ or *glutamate receptor/ or exp *ionotropic receptor antagonist/ 27 AMPA receptor positive allosteric modulator/ 28 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or (GLYX 13 or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or AZD 6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or ETS 6103 or viotra) or ampa or cerc 301 or cerc301 or d‐serin* or GluN2B or mGlu* or N acetyl cysteine* or N acetylcysteine or N methyl D aspartate or NMDA? or nrx 1074 or nrx1074 or kainite or NR2B or sarcosin* or NAC).ti,ab,kw. 29 (Org 26576 or Org26576 or CP‐101,606 or CP101606).ti,ab,kw,hw. 30 Cytidine.ti,ab,kw,hw. 31 or/8‐30 32 randomized controlled trial/ 33 randomization.de. 34 controlled clinical trial/ and (Disease Management or Drug Therapy or Prevention or Rehabilitation or Therapy).fs. 35 *clinical trial/ 36 placebo.de. 37 placebo.ti,ab. 38 trial.ti. 39 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kw. 40 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,kw. 41 ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).mp. 42 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kw,hw. 43 or/32‐42 44 ((animal or nonhuman) not (human and (animal or nonhuman))).de. 45 43 not 44 46 7 and 31 and 45 47 (2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dc,dp 48 46 and 47 49 7 and (29 or 30) and 45 50 48 or 49 51 (review.ab. and review.pt.) not trial.ti. 52 50 not 51 *************************** Ovid PsycINFO <1806 to July Week 3> [Date limited 2015 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 major depression/ or anaclitic depression/ or dysthymic disorder/ or endogenous depression/ or late life depression/ or reactive depression/ or recurrent depression/ or treatment resistant depression/ 2 exp "Depression (Emotion)"/ or Atypical Depression/ 3 bipolar disorder/ 4 *Affective Disorders/ 5 (depression or depressive? or MDD or TRD or dysthymi*).ti,ab,id. 6 depressed.ti. or (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).ab,id. 7 (affective disorder* or affective spectrum disorder* or affective state* or affective symptom* or mixed state* or mood disorder*).ti,ab,id. 8 or/1‐7 9 amantadine/ 10 atomoxetine/ 11 glutamate receptors/ or glutamic acid/ 12 ketamine/ 13 n‐methyl‐d‐aspartate/ 14 tramadol/ 15 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or (GLYX 13 or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or AZD 6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or ETS 6103 or viotra) or ampa or cerc 301 or cerc301 or d‐serin* or GluN2B or mGlu* or N acetyl cysteine* or N acetylcysteine or N methyl D aspartate or NMDA? or nrx 1074 or nrx1074 or kainite or NR2B or sarcosin* or NAC).ti,ab,id,hw. 16 (Org 26576 or Org26576 or CP‐101,606 or CP101606).ti,ab,id. 17 Cytidine.ti,ab,id. 18 or/9‐17 19 clinical trials.sh. 20 (randomi#ed or randomi#ation or randomi#ing).ti,ab,id. 21 (RCT or at random or (random* adj3 (assign* or allocat* or control* or crossover or cross‐over or design* or divide* or division or number))).ti,ab,id. 22 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,id,hw. 23 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,id. 24 trial.ti. 25 placebo.ti,ab,id,hw. 26 treatment outcome.md. 27 treatment effectiveness evaluation.sh. 28 mental health program evaluation.sh. 29 or/19‐28 30 8 and 18 and 29 31 (2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,an. 32 30 and 31 33 8 and (16 or 17) and 29 34 32 or 33 *************************** Ovid XSearch: Esketamine MEDLINE, Embase, PsycINFO (all years, searched 30 July 2020) Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 esketamine.mp. 2 (depression or depressive? or MDD or dysthymi*).mp. 3 depressed.ti. 4 (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).mp. 5 2 or 3 or 4 6 1 and 5 7 (randomi#ed or randomi#ation or randomi#ing or (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or crossover or cross‐over or design* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*)))).ti,ab,kf,kw,id. 8 (placebo* or trial).ab,ti,kf,kw,id. or groups.ab. 9 (control* and (trial or study or group*) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kf,kw,id,hw. 10 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,kf,kw,id. 11 (controlled clinical trial or randomized controlled trial).pt. 12 random allocation/ or single‐blind method/ or double‐blind method/ 13 randomized controlled trial/ 14 randomization.de. 15 controlled clinical trial/ and (Disease Management or Drug Therapy or Prevention or Rehabilitation or Therapy).fs. 16 *clinical trial/ 17 placebo.de. 18 treatment outcome.md. or treatment effectiveness evaluation.sh. or mental health program evaluation.sh. 19 or/7‐18 20 6 and 19 21 remove duplicates from 20
*************************** Cochrane Central Register of Controlled Trials (CENTRAL) [All Years to Issue 7, 2020] IDSearch #1 MeSH descriptor: [Depression] this term only #2 MeSH descriptor: [Depressive Disorder] this term only #3 MeSH descriptor: [Depressive Disorder, Major] this term only #4 MeSH descriptor: [Depressive Disorder, Treatment‐Resistant] this term only #5 MeSH descriptor: [Dysthymic Disorder] this term only #6 MeSH descriptor: [Mood Disorders] this term only #7 MeSH descriptor: [Affective Symptoms] this term only #8 MeSH descriptor: [Bipolar and Related Disorders] explode all trees #9 (depress* or MDD or TRD or dysthymi*):ti,ab,kw (Word variations have been searched) #10 "affective disorder*" or "affective spectrum disorder*" or "affective state*" or "affective symptom*" or "mixed state*" or "mood disorder*":ti,ab,kw (Word variations have been searched) #11 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 #12 MeSH descriptor: [Adamantane] explode all trees #13 MeSH descriptor: [Atomoxetine Hydrochloride] this term only #14 MeSH descriptor: [Acetylcysteine] this term only #15 MeSH descriptor: [Cycloserine] this term only #16 MeSH descriptor: [Dextromethorphan] this term only #17 MeSH descriptor: [Excitatory Amino Acid Antagonists] explode all trees #18 ((glutamate* or glutamin* or glutathione* or glycin*) near/2 (modulat* or inhibit* or system*)):ti,ab,kw (Word variations have been searched) #19 MeSH descriptor: [Ketamine] this term only #20 MeSH descriptor: [N‐Methylaspartate] this term only #21 MeSH descriptor: [Quinolines] this term only #22 MeSH descriptor: [Riluzole] this term only #23 MeSH descriptor: [Sarcosine] this term only #24 MeSH descriptor: [N‐substituted Glycines] this term only #25 MeSH descriptor: [Tramadol] this term only #26 MeSH descriptor: [Receptors, Glutamate] explode all trees #27 MeSH descriptor: [Glycine Plasma Membrane Transport Proteins] this term only #28 MeSH descriptor: [Glutamate Plasma Membrane Transport Proteins] explode all trees #29 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or ("GLYX 13" or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or "AZD 6765") or esketamine or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or "ETS 6103" or viotra) or ampa or "cerc 301" or cerc301 or d‐serin* or GluN2B or mGlu* or "acetyl cysteine*" or acetylcysteine or "N methyl D aspartate" or NMDA* or "nrx 1074" or nrx1074 or kainite or NR2B or sarcosin* or NAC):ti,ab,kw (Word variations have been searched) #30 "Org 26576" or Org26576 or CP‐101,606 or CP101606:ti,ab,kw (Word variations have been searched) #31 Cytidine:ti,ab,kw (Word variations have been searched) #32 MeSH descriptor: [Cytidine] this term only #33 #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 #34 #11 and #33 #35 MeSH descriptor: [Anesthesia and Analgesia] explode all trees #36 sedation or anesthe* or anaesthe*:ti (Word variations have been searched) #37 ((respiratory or respiration or myocardial) next depression) or (depressed blood pressure):ti,ab,kw (Word variations have been searched) #38 (depression next (co or si)):kw (Word variations have been searched) #39 analgesi*:ti (Word variations have been searched) #40 #34 not (#35 or #36 or #37 or #38 or #39) #41 SR‐DEPRESSN or HS‐DEPRESSN #42 #40 not #41 *************************** Trial Registers WHO International Clinical Trials Registry Platform 1. (depression AND acetylcysteine OR depression AND amantadine OR depression AND atomoxetine OR depression AND AZD6765 OR depression AND AZD 6765 OR depression AND cerc 301 OR depression AND cerc301 OR depression AND cycloserine OR depression AND d‐cycloserine OR depression AND dextromethorphan OR depression AND d‐serine OR depression AND ETS6103 OR depression AND ETS 6103 OR depression AND esketamine OR depression AND ketamine OR depression AND ketalar OR depression AND ketaject OR depression AND ketanest OR depression AND kainite OR depression AND lanicemine OR depression AND memantine OR depression AND norketamine OR depression AND MK 0657 OR depression AND MK0657 OR depression AND nrx 1074 OR depression AND nrx1074 OR depression AND N‐acetyl‐cysteinene OR depression AND N‐acetylcysteine OR depression AND N‐methyl‐D‐aspartate OR depression AND NMDA OR depression AND quinoline OR depression AND rapastinel OR depression AND rellidep OR depression AND GLYX 13 OR depression AND GLYX13 OR depression AND riluzole OR depression AND sarcosine OR depression AND Tramadol OR depression AND viotra) 2. (depression AND glutamic acid OR depression AND glutamatergic OR depression AND glutamate AND modulation OR depression AND ampa OR depression AND GluN2B OR depression AND mGlu* or depression AND NR2B) 3. (depressive AND acetylcysteine OR depressive AND amantadine OR depressive AND atomoxetine OR depressive AND AZD6765 OR depressive AND AZD 6765 OR depressive AND cerc 301 OR depressive AND cerc301 OR depressive AND cycloserine OR depressive AND d‐cycloserine OR depressive AND dextromethorphan OR depressive AND d‐serine OR depressive AND ETS6103 OR depressive AND ETS 6103 OR depressive AND esketamine OR depressive AND ketamine OR depressive AND ketalar OR depressive AND ketaject OR depressive AND ketanest OR depressive AND kainite OR depressive AND lanicemine OR depressive AND memantine OR depressive AND norketamine OR depressive AND MK 0657 OR depressive AND MK0657 OR depressive AND nrx 1074 OR depressive AND nrx1074 OR depressive AND N‐acetyl‐cysteinene OR depressive AND N‐acetylcysteine OR depressive AND N‐methyl‐D‐aspartate OR depressive AND NMDA OR depressive AND quinoline OR depressive AND rapastinel OR depressive AND rellidep OR depressive AND GLYX 13 OR depressive AND GLYX13 OR depressive AND riluzole OR depressive AND sarcosine OR depressive AND Tramadol OR depressive AND viotra) 4. (depressive AND glutamic acid OR depressive AND glutamatergic OR depressive AND glutamate AND modulation OR depressive AND ampa OR depressive AND GluN2B OR depressive AND mGlu* or depressive AND NR2B) 5. (bipolar AND acetylcysteine OR bipolar AND amantadine OR bipolar AND atomoxetine OR bipolar AND AZD6765 OR bipolar AND AZD 6765 OR bipolar AND cerc 301 OR bipolar AND cerc301 OR bipolar AND cycloserine OR bipolar AND d‐cycloserine OR bipolar AND dextromethorphan OR bipolar AND d‐serine OR bipolar AND ETS6103 OR bipolar AND ETS 6103 OR bipolar AND esketamine OR bipolar AND ketamine OR bipolar AND ketalar OR bipolar AND ketaject OR bipolar AND ketanest OR bipolar AND kainite OR bipolar AND lanicemine OR bipolar AND memantine OR bipolar AND norketamine OR bipolar AND MK 0657 OR bipolar AND MK0657 OR bipolar AND nrx 1074 OR bipolar AND nrx1074 OR bipolar AND N‐acetyl‐cysteinene OR bipolar AND N‐acetylcysteine OR bipolar AND N‐methyl‐D‐aspartate OR bipolar AND NMDA OR bipolar AND quinoline OR bipolar AND rapastinel OR bipolar AND rellidep OR bipolar AND GLYX 13 OR bipolar AND GLYX13 OR bipolar AND riluzole OR bipolar AND sarcosine OR bipolar AND Tramadol OR bipolar AND viotra) 6. (bipolar AND glutamic acid OR bipolar AND glutamatergic OR bipolar AND glutamate AND modulation OR bipolar AND ampa OR bipolar AND GluN2B OR bipolar AND mGlu* or bipolar AND NR2B) 7. or/1‐6 ClinicalTrials.gov depression OR depressive OR MDD OR bipolar AND acetylcysteine OR amantadine OR atomoxetine OR AZD6765 OR AZD 6765 OR cerc 301 OR cerc301 OR cycloserine OR d‐cycloserine OR dextromethorphan OR d‐serine OR ETS6103 OR ETS 6103 OR esketamine OR ketamine OR ketalar OR ketaject OR ketanest OR kainite OR lanicemine OR memantine OR norketamine OR MK 0657 OR MK0657 OR nrx 1074 OR nrx1074 OR N‐acetyl‐cysteinene OR N‐acetylcysteine OR N‐methyl‐D‐aspartate OR NMDA OR quinoline OR rapastinel OR rellidep OR GLYX 13 OR GLYX13 OR riluzole OR sarcosine OR Tramadol OR viotra OR glutamic acid OR glutamatergic OR glutamate modulation OR ampa OR GluN2B OR mGlu OR NR2B ************************************************************************************************
Appendix 2. Searches to 2015 c/o Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR
The information specialist with CCMD searched their specialised register (all years to 9 Jan 2015) using the following terms.
#1. (depress* or dysthymi* or "affective disorder*" or “affective spectrum disorder*” or “affective state*” or "affective symptom*" or "mixed state*" or "mood disorder*" or MDD or unipolar or bipolar):ti,ab,kw,ky,emt,mh,mc #2. (amantadin* or atomoxetin* or *cycloserin* or dextromethorphan or "GLYX 13" or "MK 0657" or (ketamin* or Ketalar or Ketaject or Ketanest) or (lanicemin* or AZD6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or viotra) or ampa or “cerc 301” or “d serin*” or glun2b or glutamate or glutamin* or glutamatergic or glutathione* or glycin* or mglu* or "N acetyl cysteine*" or “N methyl D aspartate” or nmda or “nrx 1074” or kainite or nr2b or sarcosin* or NAC):ti,ab,kw,ky,emt,mh,mc #3. (#1 and #2)
[Key to field codes: ti:title; ab:abstract; kw:keywords: ky:additional keywords; emt:EMTREE headings; mh:MeSH headings; mc:MeSH checkwords]
Details of the CCMDCTR
The Cochrane Common Mental Disorders Group (CCMD) maintains two archived clinical trials registers at its editorial base in York, UK: a references register and a studies‐based register. The CCMDCTR‐References Register contains over 40,000 reports of RCTs in depression, anxiety and neurosis. Approximately 50% of these references have been tagged to individual coded trials. The coded trials are held in the CCMDCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary; (please contact the CCMD Information Specialists for further details). Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950 to 2016), Embase (1974 to 2016) and PsycINFO (1967 to 2016); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trial registers via the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCMD's generic search strategies (used to identify RCTs) can be found on the Group's website, (cmd.cochrane.org/specialised-register), with an example of the core MEDLINE search (used to inform the register) listed below. The Group’s Specialised Register has fallen out‐of‐date with the Editorial Group’s move from Bristol to York in the summer of 2016.
Core search strategy used to inform the Cochrane Common Mental Disorders Group's Specialised Register: OVID MEDLINE (to June 2016)
A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/ 2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf. 3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.) 4. (1 and 2 and 3) Records were screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs were tagged to the appropriate study record. Similar weekly search alerts were also conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
Appendix 3. Adverse events search
Ovid MEDLINE databases
Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to July 28 2020> [Date limited 2014 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2 (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3 (side effect* or treatment emergent or undesirable effect*).ti,ab. 4 (suicid* or death*).mp. 5 (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6 ae.fs. 7 to.fs. 8 or/1‐7 9 (atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 10 amantadine/ae, po, to 11 Ketamine/ae, po, to 12 Dextromethorphan/ae, po, to 13 Memantine/ae, po, to 14 Riluzole/ae, po, to 15 Cycloserine/ae, po, to 16 Quinidine/ae, po, to 17 Tramadol/ae, po, to 18 or/10‐17 19 (amantadine or Ketamine or Dextromethorphan or Memantine or Riluzole or Cycloserine or Quinidine or Tramadol).ti,sh. 20 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 21 (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,ab,sh. 22 exp animals/ not humans.sh. 23 exp Anesthesia/ 24 ((8 and 9 and 21) or ((18 or (19 and 20)) and 21)) not (22 or 23) 25 (2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dt,ep,ez. 26 24 and 25 27 (Org 26576 or Org26576 or CP‐101,606 or CP101606).mp. 28 Quinolinic Acid/ae, to [Adverse Effects, Toxicity] 29 Sarcosine/ae, to [Adverse Effects, Toxicity] 30 Cytidine/ae, to [Adverse Effects, Toxicity] 31 (cytidine or sarcosine or quinolinic acid).ti,sh. 32 8 and 27 33 ((21 and 28) or 29 or 30) not (22 or 23) 34 ((31 and 20) or (31 and 8 and 21)) not (22 or 23) 35 26 or 32 or 33 or 34 ***************************
Ovid Embase <1980 to 2020 Week 30> [Date limited 2014 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2 (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3 (side effect* or treatment emergent or undesirable effect*).ti,ab. 4 (suicid* or death*).mp. 5 (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6 ae.fs. 7 to.fs. 8 or/1‐7 9 ("GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 10 (Org 26576 or Org26576 or CP‐101,606 or CP101606).mp. 11 *Ketamine/ae, to 12 *Atomoxetine/ae, to 13 *amantadine/ae, to 14 *Dextromethorphan/ae, to 15 *Memantine/ae, to 16 Riluzole/ae, to 17 *Cycloserine/ae, to 18 *Quinidine/ae, to 19 *Tramadol/ae, to 20 cytidine/ae, to or quinolinic acid/ae, to or sarcosine/ae, to 21 or/11‐20 22 (amantadine or atomoxetine or Ketamine or Dextromethorphan or Memantine or Riluzole or Cycloserine or Quinidine or Tramadol).ti,sh. 23 (cytidine or sarcosine or quinolinic acid).ti,sh. 24 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 25 (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,sh. 26 ((animals or nonhuman) not (humans and (animals or nonhuman))).sh. 27 exp *anesthesiological procedure/ 28 (8 and (9 or 10)) not (26 or 27) 29 (((or/11‐19) and 25) or (22 and 24 and 25)) not (26 or 27) 30 (2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dc,dp. 31 29 and 30 32 ((20 and 25) or (23 and 24)) not (26 or 27) 33 28 or 31 or 32 *************************** OvidPsycINFO <1806 to July 2020 Week 3> [Date limited 2014 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or safety or side effect* or contraindication* or toxicity).ti,id,sh,tm. 2 (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks or toxicity).ti,id,ab. 3 (side effect* or treatment emergent or undesirable effect*).ti,id,ab. 4 (suicid* or death*).ti,ab,id,sh,tm. 5 (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,id,sh,tm. 6 or/1‐5 7 (Ketamin* or Ketaject or Ketalar or Ketanest or Ketaset or Ketalean or Vetalar or amantadin* or atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,ab,id,sh. 8 N‐Methyl‐D‐Aspartate/ 9 or/7‐8 10 (animal not ((human or inpatient or outpatient) and animal)).po. 11 (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,id,sh,tm,ab. 12 (6 and 9 and 11) not 10 13 (2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,an. 14 12 and 13 15 (Org 26576 or Org26576 or CP‐101,606 or CP101606 or cytidine or sarcosine or quinolinic acid).ti,ab,id,sh 16 (15 and 6) not 10 17 14 or 16 ***************************
Adverse effects of ketamine and other glutamate receptor modulators (OVID databases to 11‐Nov‐2014) (Version 1)
OVID MEDLINE was searched using the following terms: 1. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2. (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3. (side effect* or treatment emergent or undesirable effect*).ti,ab. 4. (suicid* or death*).mp. 5. (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6. ae.fs. [Floating Subheading: Adverse Effects ‐ MEDLINE] 7. to.fs. [Floating Subheading: Toxicity – MEDLINE] 8. ct.fs. [Floating Subheading: Contraindications ‐ MEDLINE] 9. or/1‐8 10. (atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 11. *Amantadine/ae,to 12. *Cycloserine/ae,to 13. *Dextromethorphan/ae,to 14. *Ketamine/ae,to 15. *Memantine/ae,to 16. *Quinidine/ae,to 17. Riluzole/ae,to 18. *Tramadol/ae,to 19. or/11‐18 20. (amantadine or ketamine or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,sh. 21. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 22. (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,ab,sh. 23. exp animals/ not humans.sh. 24. exp *anesthesia 25. ((9 and 10 and 22) or ((19 or (20 and 21)) and 22)) not (23 or 24)
OVID EMBASE was searched using the following terms: 1. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2. (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3. (side effect* or treatment emergent or undesirable effect*).ti,ab. 4. (suicid* or death*).mp. 5. (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6. ae.fs. [Floating Subheading: Adverse Drug Reaction ‐ EMBASE] 7. to.fs. [Floating Subheading: Drug Toxicity – EMBASE] 8. or/1‐7 9. ("GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 10. *Amantadine/ae,to 11. *Atomoxetine/ae,to 12. *Cycloserine/ae,to 13. *Dextromethorphan/ae,to 14. *Ketamine/ae,to 15. *Memantine/ae,to 16. *Quinidine/ae,to 17. Riluzole/ae,to 18. *Tramadol/ae,to 19. or/10‐18 20. (amantadine or atomoxetine or ketamine or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,sh. 21. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 22. (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,sh. 23. ((animal*1 or nonhuman) not (human*1 and (animal*1 or nonhuman))).sh. 24. exp *anesthesiological procedure/ 25. ((8 and 9 and 22) or ((19 or (20 and 21)) and 22)) not (23 or 24)
OVID PsycINFO was searched using a more sensitive set of terms: 1. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or safety or side effect* or contraindication* or toxicity).ti,id,sh,tm. 2. (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks or toxicity).ti,id,ab. 3. (side effect* or treatment emergent or undesirable effect*).ti,id,ab. 4. (suicid* or death*).ti,ab,id,sh,tm. 5. (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,id,sh,tm. 6. or/1‐5 7. (ketamin* or ketaject or ketalar or ketanest or ketaset or ketalean or vetalar or amantadin* or atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,ab,id,sh. 8. N‐Methyl‐D‐Aspartate/ or Tramadol/ 9. or/7‐8 10. (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,ab,id,sh,tm. 11. (animal not ((human or inpatient or outpatient) and animal)).po. 12. (6 and 9 and 10) not 11
Data and analyses
Comparison 1. Ketamine versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Response rate | 12 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1.1 at 24 hours | 7 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 3.94 [1.54, 10.10] |
| 1.1.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 15.84 [3.68, 68.12] |
| 1.1.3 at 1 week | 5 | 196 | Odds Ratio (M‐H, Random, 95% CI) | 3.76 [0.98, 14.42] |
| 1.1.4 at 2 weeks | 4 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 2.92 [0.48, 17.78] |
| 1.1.5 at 4 weeks | 4 | 202 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [0.50, 3.77] |
| 1.1.6 at 3 months | 3 | 117 | Odds Ratio (M‐H, Random, 95% CI) | 1.95 [0.24, 15.69] |
| 1.2 AE Abdominal Pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.3 AE Agitation/anxiety | 3 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 3.44 [1.07, 11.04] |
| 1.4 AE Blurred vision | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.5 AE Change in blood pressure | 2 | 157 | Odds Ratio (M‐H, Random, 95% CI) | 3.23 [0.49, 21.31] |
| 1.6 AE Confusion | 2 | 76 | Odds Ratio (M‐H, Random, 95% CI) | 3.76 [1.13, 12.47] |
| 1.7 AE Dissociative symptoms | 3 | 145 | Odds Ratio (M‐H, Random, 95% CI) | 7.72 [1.31, 45.51] |
| 1.8 AE Dizziness | 3 | 196 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.52, 5.81] |
| 1.9 AE Emotional blunting | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.10 AE Euphoria | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.11 AE Hallucinations | 3 | 203 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.45, 10.78] |
| 1.12 AE Headache | 2 | 194 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.62, 2.45] |
| 1.13 AE Infections and Infestations | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.14 AE Loss of Appetite | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.15 AE Mania/hypomania | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.16 AE Musculoskeletal and connective tissue disorders | 2 | 137 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.07, 26.35] |
| 1.17 AE Nausea/vomiting | 5 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 1.83 [0.81, 4.13] |
| 1.18 AE Nervousness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.19 AE Nervous system disorders | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.20 AE Palpitations | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.21 AE Psychiatric disorders | 2 | 137 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.30, 2.93] |
| 1.22 AE Restlessness | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.23 AE Skin and subcutaneous tissue disorders | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.24 AE Suicidal Ideas | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.25 AE Tremor | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.26 Remission rate | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.26.1 at 24 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 5.60 [1.07, 29.46] |
| 1.26.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 6.60 [1.51, 28.92] |
| 1.26.3 at 1 week | 5 | 196 | Odds Ratio (M‐H, Random, 95% CI) | 4.64 [1.37, 15.68] |
| 1.26.4 at 2 weeks | 4 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 1.67 [0.38, 7.27] |
| 1.26.5 at 4 weeks | 4 | 202 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.54, 3.95] |
| 1.26.6 at 3 months | 2 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 1.09 [0.45, 2.67] |
| 1.27 Depression rating scale score | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.27.1 at 24 hours | 8 | 231 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.48] |
| 1.27.2 at 72 hours | 6 | 148 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.28, ‐0.07] |
| 1.27.3 at 1 week | 6 | 143 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.72 [‐1.10, ‐0.33] |
| 1.27.4 at 2 weeks | 5 | 236 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.90, 0.04] |
| 1.27.5 at 4 weeks | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.07, ‐0.29] |
| 1.28 Suicidal ideation composite | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.28.1 at 24 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.78, 0.82] |
| 1.28.2 at 72 hours | 2 | 68 | Mean Difference (IV, Random, 95% CI) | 0.34 [‐0.25, 0.93] |
| 1.28.3 at 1 week | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.56, 0.96] |
| 1.28.4 at 2 weeks | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.46, 1.06] |
| 1.29 Cognition scores | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.29.1 Immediate‐term Memory | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 0.80 [0.12, 1.48] |
| 1.29.2 Short‐term Memory | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 6.90 [5.01, 8.79] |
| 1.29.3 Long‐term Memory | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 4.50 [2.79, 6.21] |
| 1.30 Quality of Life | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.30.1 EQ‐5D‐3L INDEX at 2 weeks | 1 | 64 | Mean Difference (IV, Random, 95% CI) | 0.11 [‐0.05, 0.27] |
| 1.31 Acceptability | 6 | 201 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.19, 8.28] |
Comparison 2. Ketamine versus Midazolam.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Response rate | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1.1 at 24 hours | 4 | 296 | Odds Ratio (M‐H, Random, 95% CI) | 2.48 [1.00, 6.18] |
| 2.1.2 at 72 hours | 3 | 218 | Odds Ratio (M‐H, Random, 95% CI) | 2.20 [0.92, 5.28] |
| 2.1.3 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 3.11 [1.38, 7.04] |
| 2.1.4 at 2 weeks | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 4.89 [1.49, 16.10] |
| 2.1.5 at 4 weeks | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.01, 19.56] |
| 2.1.6 at 3 months | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 3.00 [0.08, 115.34] |
| 2.2 AE Abnormal dreams | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.2.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.3 AE Agitation/anxiety | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.3.1 Infusion day | 1 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 1.99 [0.69, 5.75] |
| 2.3.2 at 1 week | 3 | 278 | Odds Ratio (M‐H, Random, 95% CI) | 1.32 [0.69, 2.50] |
| 2.3.3 48‐72 hours after last treatment | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.01, 19.56] |
| 2.4 AE Back pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.4.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.5 AE Blurred vision | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.5.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 8.52 [1.80, 40.39] |
| 2.5.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.23, 4.70] |
| 2.6 AE Chest pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.2 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.7 AE Chills | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.7.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.8 AE Constipation | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.8.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 2.80 [0.13, 60.66] |
| 2.8.2 at 1 week | 3 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 1.98 [0.42, 9.27] |
| 2.9 AE Decreased energy | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.9.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.30, 5.47] |
| 2.9.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.44, 3.13] |
| 2.10 AE Decreased libido | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.10.1 48‐72 hours after last treatment | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.11 AE Depression | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.11.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.12 AE Diarrhea | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.12.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.09, 12.37] |
| 2.12.2 at 1 week | 2 | 152 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.18, 3.83] |
| 2.12.3 at 4 weeks | 1 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.06, 26.93] |
| 2.13 AE Difficulty swallowing | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.13.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.14 AE Dizziness | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.14.1 Infusion day | 2 | 224 | Odds Ratio (M‐H, Random, 95% CI) | 3.42 [1.44, 8.14] |
| 2.14.2 at 1 week | 4 | 283 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.43, 2.56] |
| 2.15 AE Dry mouth | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.15.1 Infusion day | 2 | 152 | Odds Ratio (M‐H, Random, 95% CI) | 2.10 [0.66, 6.70] |
| 2.15.2 at 1 week | 3 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.22, 2.91] |
| 2.15.3 48‐72 hours after last treatment | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 3.00 [0.08, 115.34] |
| 2.16 AE Dry skin | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.16.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.16.2 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.17 AE Fatigue | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.17.1 Infusion day | 2 | 152 | Odds Ratio (M‐H, Random, 95% CI) | 0.29 [0.00, 22.84] |
| 2.17.2 at 1 week | 4 | 211 | Odds Ratio (M‐H, Random, 95% CI) | 1.42 [0.62, 3.27] |
| 2.18 AE General malaise | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.18.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 0.18 [0.04, 0.75] |
| 2.18.2 at 1 week | 3 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 1.90 [0.32, 11.44] |
| 2.19 AE Insomnia | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.19.1 at 1 week | 1 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 2.57 [0.43, 15.41] |
| 2.19.2 at 4 weeks | 1 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.06, 26.93] |
| 2.20 AE Headache | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.20.1 Infusion day | 2 | 152 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.19, 4.56] |
| 2.20.2 at 1 week | 3 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.32, 2.29] |
| 2.20.3 at 4 weeks | 1 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 5.18 [0.29, 93.01] |
| 2.21 AE Increased blood pressure or heart rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.21.1 at 1 week | 1 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 9.37 [2.49, 35.25] |
| 2.21.2 at 4 weeks | 1 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.06, 26.93] |
| 2.22 AE Increase in systolic blood pressure and heart rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.22.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.23 AE Increased perspiration | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.23.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 2.86 [0.32, 25.91] |
| 2.23.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.48 [0.23, 9.58] |
| 2.24 AE Indigestion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.24.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.25 AE Insomnia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.25.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.26 AE Irritability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.26.1 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.27 AE Itching | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.27.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.09, 12.37] |
| 2.27.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 3.54 [0.58, 21.59] |
| 2.27.3 48‐72 hours after last treatment | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 8.33 [0.22, 320.38] |
| 2.28 AE Loss of consciousness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.28.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.29 AE Memory problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.29.1 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.30 AE Muscle/bone/joint pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.30.1 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.31 AE Nasal congestion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.31.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.32 AE Nausea/vomiting | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.32.1 Infusion day | 2 | 152 | Odds Ratio (M‐H, Random, 95% CI) | 3.62 [1.13, 11.58] |
| 2.32.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 2.57 [0.78, 8.52] |
| 2.32.3 at 4 weeks | 1 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 7.12 [0.40, 125.66] |
| 2.33 AE Numbness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.33.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.34 AE Pain in extremities | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.34.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.35 AE Palpitations | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.35.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.35.2 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.36 AE Poor concentration | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.36.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 3.94 [0.81, 19.27] |
| 2.36.2 at 1 week | 3 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [0.18, 12.31] |
| 2.37 AE Poor co‐ordination | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.37.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.37.2 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.38 AE Poor quality sleep | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.38.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.39 AE Rash | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.39.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.39.2 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.40 AE Reduced duration of sleep | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.40.1 48‐72 hours after last treatment | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.01, 19.56] |
| 2.40.2 at 1 week | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.22] |
| 2.41 AE Restlessness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.41.1 48‐72 hours after last treatment | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 3.00 [0.08, 115.34] |
| 2.41.2 at 1 week | 1 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 6.28 [0.29, 137.16] |
| 2.42 AE Sensory disturbance | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.42.1 Infusion day | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.42.2 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.43 AE Sexual dysfunction | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.43.1 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.44 AE Sleepiness/drowsiness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.44.1 Infusion day | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.21 [0.07, 0.66] |
| 2.44.2 at 1 week | 1 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 2.57 [0.43, 15.41] |
| 2.45 AE Stomach or abdominal discomfort | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.45.1 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.46 AE Suicide attempt | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.46.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.47 AE Suicidal ideas | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.47.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.48 AE Tachycardia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.48.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.49 AE Tinnitus | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.49.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.09, 12.37] |
| 2.49.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.26, 3.73] |
| 2.49.3 48‐72 hours after last treatment | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [0.05, 78.25] |
| 2.50 AE Tooth Abscess | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.50.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.51 AE Tremor | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.51.1 Infusion day | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 7.99 [0.43, 147.87] |
| 2.51.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.14 [0.23, 5.74] |
| 2.52 AE Urination issues | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.52.1 Infusion day | 1 | 216 | Odds Ratio (M‐H, Random, 95% CI) | Not estimable |
| 2.52.2 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.08, 63.59] |
| 2.53 Remission rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.53.1 at 24 hours | 2 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 2.21 [0.67, 7.32] |
| 2.53.2 at 72 hours | 2 | 118 | Odds Ratio (M‐H, Random, 95% CI) | 1.73 [0.74, 4.04] |
| 2.53.3 at 1 week | 2 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.86 [0.80, 4.32] |
| 2.53.4 at 2 weeks | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 2.29 [0.76, 6.92] |
| 2.53.5 at 4 weeks | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.01, 19.56] |
| 2.53.6 at 3 months | 1 | 5 | Odds Ratio (M‐H, Random, 95% CI) | Not estimable |
| 2.54 Depression rating scale score | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.54.1 at 24 hours | 4 | 297 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐0.87, ‐0.10] |
| 2.54.2 at 72 hours | 3 | 207 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.70, ‐0.08] |
| 2.54.3 at 1 week | 3 | 212 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.69, ‐0.08] |
| 2.54.4 at 2 weeks | 2 | 137 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.84, 0.10] |
| 2.54.5 at 4 weeks | 1 | 86 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.57 [‐1.10, ‐0.04] |
| 2.55 Suicidal ideation composite | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐1.32 [‐2.52, ‐0.12] |
| 2.56 Acceptability | 1 | 72 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.05, 2.09] |
Comparison 3. Ketamine versus Thiopental.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1.1 at 3 days | 1 | 31 | Odds Ratio (M‐H, Random, 95% CI) | 2.64 [0.10, 69.88] |
| 3.1.2 at 4 weeks | 1 | 31 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.05, 14.28] |
| 3.2 AE Blood Pressure Rise | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.3 AE Delirium | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.4 AE Emergence reactions | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.5 AE Headache | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.6 AE Heart Rate Rise | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.7 AE Increased secretions | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.8 AE Nausea/vomiting | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.9 Depression rating scale score | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.9.1 at 72 hours | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐3.87 [‐6.08, ‐1.66] |
| 3.9.2 at 1 week | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐6.96 [‐9.82, ‐4.10] |
| 3.9.3 at 2 weeks | 2 | 89 | Mean Difference (IV, Random, 95% CI) | ‐4.84 [‐13.42, 3.73] |
| 3.9.4 at 4 weeks | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐2.64, 2.20] |
| 3.10 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 4. Ketamine versus Methohexital.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4.1.1 At 72 hours | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 5. Ketamine versus Propofol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1.1 At 2 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1.2 At 3 months | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 6. Ketamine versus Remifentanil hydrochloride.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1.1 at 24 hours | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐7.74 [‐14.03, ‐1.45] |
| 6.1.2 at 1 week | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐7.54 [‐14.13, ‐0.95] |
| 6.1.3 at 2 weeks | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐6.98, 4.98] |
Comparison 7. Ketamine versus Esketamine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Response Rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1.1 at 24 hours | 1 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.40, 2.89] |
| 7.1.2 at 72 hours | 1 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 1.56 [0.58, 4.22] |
| 7.1.3 at 1 week | 1 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 2.34 [0.85, 6.45] |
| 7.2 Cognition | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.2.1 CADSS scores during infusion | 1 | 63 | Mean Difference (IV, Random, 95% CI) | 3.30 [‐4.70, 11.30] |
Comparison 8. Ketamine versus ECT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.1 at 24 hours | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.2 at 72 hours | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.3 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.4 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2 AE Increase in systolic blood pressure and heart rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.3 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.3.1 at 24 hours | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.3.2 at 72 hours | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.3.3 at 1 week | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.3.4 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.4 Depression rating scale score | 1 | 72 | Mean Difference (IV, Random, 95% CI) | ‐2.98 [‐7.07, 1.12] |
| 8.4.1 at 24 hours | 1 | 18 | Mean Difference (IV, Random, 95% CI) | ‐8.90 [‐11.72, ‐6.08] |
| 8.4.2 at 72 hours | 1 | 18 | Mean Difference (IV, Random, 95% CI) | ‐3.40 [‐5.99, ‐0.81] |
| 8.4.3 at 1 week | 1 | 18 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐3.45, 1.45] |
| 8.4.4 at 2 weeks | 1 | 18 | Mean Difference (IV, Random, 95% CI) | 1.20 [‐1.20, 3.60] |
8.2. Analysis.

Comparison 8: Ketamine versus ECT, Outcome 2: AE Increase in systolic blood pressure and heart rate
Comparison 9. Esketamine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Response rate | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1.1 at 24 hours | 5 | 1071 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [1.20, 3.68] |
| 9.1.2 at 72 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.92, 1.96] |
| 9.1.3 at 1 week | 6 | 1115 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [1.09, 2.34] |
| 9.1.4 at 2 weeks | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.09, 2.28] |
| 9.1.5 at 4 weeks | 5 | 1117 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [1.44, 2.37] |
| 9.2 AE Aggression | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.3 AE Agitation/anxiety | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.46, 2.42] |
| 9.4 AE Arrhythmia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.5 AE Change in blood pressure | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 2.67 [1.52, 4.70] |
| 9.6 AE Constipation | 2 | 452 | Odds Ratio (M‐H, Random, 95% CI) | 4.07 [1.60, 10.39] |
| 9.7 AE Depersonalisation/derealization | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.8 AE Depression | 2 | 452 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.08, 5.05] |
| 9.9 AE Diabetic ketoacidosis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.10 AE Diarrhoea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.11 AE Dissociative symptoms | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 8.76 [5.19, 14.77] |
| 9.12 AE Dizziness | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 3.67 [2.54, 5.31] |
| 9.13 AE Dizziness postural | 2 | 569 | Odds Ratio (M‐H, Random, 95% CI) | 4.70 [1.06, 20.80] |
| 9.14 AE Double vision | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.15 AE Euphoria | 2 | 571 | Odds Ratio (M‐H, Random, 95% CI) | 5.27 [0.94, 29.64] |
| 9.16 AE Fatigue | 2 | 481 | Odds Ratio (M‐H, Random, 95% CI) | 1.90 [0.89, 4.04] |
| 9.17 AE Feeling drunk | 2 | 571 | Odds Ratio (M‐H, Random, 95% CI) | 7.58 [1.37, 41.77] |
| 9.18 AE Headache | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.80, 1.74] |
| 9.19 AE Hypertransaminasemia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.20 AE Increased sweating | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.21 AE Infections and Infestations | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.22 AE Insomnia | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.53, 1.42] |
| 9.23 AE Lethargy | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.24 AE Mental impairment | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.25 AE Nasal discomfort | 2 | 571 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.42, 1.68] |
| 9.26 AE Nausea/vomiting | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 3.24 [1.84, 5.72] |
| 9.27 AE Paresthesia/neuropathy exacerbation | 3 | 708 | Odds Ratio (M‐H, Random, 95% CI) | 3.51 [1.62, 7.62] |
| 9.28 AE Pericardial effusion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.29 AE Pneumothorax | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.30 AE Sensory disturbance | 3 | 796 | Odds Ratio (M‐H, Random, 95% CI) | 7.25 [3.55, 14.78] |
| 9.31 AE Sedation | 3 | 796 | Odds Ratio (M‐H, Random, 95% CI) | 5.31 [2.18, 12.94] |
| 9.32 AE Sleepiness/drowsiness | 3 | 796 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [1.39, 3.21] |
| 9.33 AE Sore throat | 2 | 571 | Odds Ratio (M‐H, Random, 95% CI) | 1.65 [0.70, 3.87] |
| 9.34 AE Suicide attempt | 2 | 452 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.24, 4.02] |
| 9.35 AE Suicidal ideas | 3 | 796 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.38, 1.26] |
| 9.36 AE Taste perversion | 4 | 933 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.95, 2.04] |
| 9.37 AE Tremor | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.38 AE Urination issues | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.39 AE Vertigo | 3 | 796 | Odds Ratio (M‐H, Random, 95% CI) | 12.25 [4.09, 36.67] |
| 9.40 AE Vision blurred | 3 | 796 | Odds Ratio (M‐H, Random, 95% CI) | 3.02 [1.37, 6.66] |
| 9.41 Remission rate | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.41.1 at 24 hours | 5 | 894 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [1.71, 4.40] |
| 9.41.2 at 72 hours | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.91, 2.64] |
| 9.41.3 at 1 week | 6 | 948 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.88, 2.69] |
| 9.41.4 at 2 weeks | 4 | 832 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [1.07, 2.16] |
| 9.41.5 at 4 weeks | 5 | 957 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.18, 2.10] |
| 9.42 Depression rating scale score | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.42.1 at 24 hours | 4 | 824 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.45, ‐0.17] |
| 9.42.2 at 72 hours | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.11] |
| 9.42.3 at 1 week | 5 | 884 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.37, ‐0.10] |
| 9.42.4 at 2 weeks | 4 | 857 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.34, ‐0.07] |
| 9.42.5 at 4 weeks | 6 | 1182 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.39, ‐0.16] |
| 9.42.6 at 3 months | 1 | 38 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.75, 0.52] |
| 9.43 Suicidal ideation composite | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.43.1 at 24 hours | 2 | 450 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.44, 0.15] |
| 9.43.2 at 72 hours | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.49, 0.08] |
| 9.43.3 at 1 week | 3 | 660 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.10, 0.13] |
| 9.43.4 at 2 weeks | 3 | 659 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.22, 0.02] |
| 9.43.5 at 4 weeks | 3 | 647 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.12, 0.05] |
| 9.44 Acceptability | 5 | 773 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.92, 2.73] |
Comparison 10. Memantine versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Response rate | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1.1 at 1 week | 2 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.06, 18.82] |
| 10.1.2 at 2 weeks | 1 | 32 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 8.28] |
| 10.1.3 at 4 weeks | 4 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.25, 5.89] |
| 10.1.4 at 3 months | 3 | 123 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.18, 1.24] |
| 10.2 AE Abdominal Pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.3 AE Active suicidal ideation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.4 AE Agitation/anxiety | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.5 AE Appetite increase | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.6 AE Back pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.7 AE Balance or gait problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.8 AE Carbohydrate craving | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.9 AE Chest pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.10 AE Chills | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.11 AE Clammy hands | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.12 AE Confusion/decreased mental clarity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.13 AE Conjunctival swelling | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.14 AE Constipation | 2 | 88 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.20, 3.29] |
| 10.15 AE Decreased appetite | 2 | 93 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.24, 10.10] |
| 10.16 AE Delusions | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.17 AE Diaphoresis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.18 AE Difficulty breathing | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.19 AE Dissociative symptoms | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.20 AE Dizziness | 3 | 181 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.33, 2.13] |
| 10.21 AE Dry mouth | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.22 AE Dyskinesia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.23 AE Dyspepsia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.24 AE Ear pain/jaw pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.25 AE Emotional lability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.26 AE Eye photosensitivity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.27 AE Facial twitching | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.28 AE Falls | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.29 AE Fatigue | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.30 AE Feeling flushed/hot | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.31 AE Generalised aches | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.32 AE Head pressure/ear pressure | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.33 AE Headache | 3 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.56, 3.72] |
| 10.34 AE Heart palpitations | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.35 AE Hypomania/mania | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.36 AE Increased menstrual pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.37 AE Insomnia | 2 | 93 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.19, 2.58] |
| 10.38 AE Internal sensation of speed or rapid thoughts | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.39 AE Irritability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.40 AE Leg weakness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.41 AE Nausea | 3 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.17, 3.38] |
| 10.42 AE Nightmares | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.43 AE Paresthesia/neuropathy exacerbation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.44 AE Passive suicidal ideation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.45 AE Perceived weight gain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.46 AE Perceived weight loss | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.47 AE Pruritus | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.48 AE Rash | 3 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.25, 8.55] |
| 10.49 AE Sedation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.50 AE Skin lesion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.51 AE Sleepiness/drowsiness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.52 AE Sleepwalking | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.53 AE Sore throat | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.54 AE Taste perversion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.55 AE Tinnitus | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.56 AE Upper respiratory infection symptoms | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.57 AE Vomiting | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.58 AE Worsened acne | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.59 AE Worsened sleep apnoea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.60 Remission rate | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.60.1 at 1 week | 2 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 6.11 [0.27, 138.45] |
| 10.60.2 at 4 weeks | 4 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.46, 4.26] |
| 10.60.3 at 3 months | 3 | 123 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.15, 3.77] |
| 10.61 Depression scale rating score | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.61.1 at 1 week | 2 | 59 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐1.10, 0.89] |
| 10.61.2 at 2 weeks | 1 | 28 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.83, 0.65] |
| 10.61.3 at 4 weeks | 3 | 112 | Std. Mean Difference (IV, Random, 95% CI) | 0.11 [‐0.26, 0.48] |
| 10.61.4 at 3 months | 3 | 110 | Std. Mean Difference (IV, Random, 95% CI) | 0.23 [‐0.14, 0.61] |
| 10.62 Quality of life | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.62.1 at 4 weeks | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐5.04, 3.64] |
| 10.62.2 at 3 months | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐1.21 [‐5.78, 3.36] |
| 10.63 Acceptability | 3 | 123 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.23, 2.66] |
| 10.64 Acceptability ‐ adverse events | 2 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.10, 4.47] |
Comparison 11. Lanicemine (AZD6765) versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Response rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1.1 at 24 hours | 1 | 22 | Odds Ratio (M‐H, Random, 95% CI) | 7.74 [0.35, 170.10] |
| 11.1.2 at 72 hours | 1 | 22 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [0.10, 74.87] |
| 11.1.3 at 1 week | 1 | 22 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [0.10, 74.87] |
| 11.1.4 at 4 weeks | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.63, 1.69] |
| 11.2 AE Agitation/anxiety | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.3 AE Back pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.4 AE Blood Pressure Rise | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.5 AE Dissociative symptoms | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.6 AE Dizziness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.7 AE Dry mouth | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.8 AE Feeling drunk | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.9 AE Insomnia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.10 AE Muscle/bone/joint pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.11 AE Nausea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.12 AE Rash | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.13 AE Sedation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.14 AE Upper respiratory infection symptoms | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.15 AE Vomiting | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.16 AE Weight gain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.17 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.17.1 at 24 hours | 1 | 22 | Odds Ratio (M‐H, Random, 95% CI) | 5.00 [0.21, 117.21] |
| 11.17.2 at 72 hours | 1 | 22 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [0.10, 74.87] |
| 11.17.3 at 1 week | 1 | 22 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [0.10, 74.87] |
| 11.17.4 at 4 weeks | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.75, 2.52] |
| 11.18 Depression rating scale score | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.18.1 at 24 hours | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐8.65 [‐17.81, 0.51] |
| 11.18.2 at 72 hours | 1 | 21 | Mean Difference (IV, Random, 95% CI) | ‐6.27 [‐13.93, 1.39] |
| 11.18.3 at 1 week | 1 | 21 | Mean Difference (IV, Random, 95% CI) | ‐6.55 [‐14.07, 0.97] |
| 11.18.4 at 4 weeks | 1 | 298 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐1.42, 1.20] |
| 11.18.5 at 3 months | 1 | 298 | Mean Difference (IV, Random, 95% CI) | 0.51 [‐1.05, 2.07] |
| 11.19 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 12. Org 26576 versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Response rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1.1 at 24 hours | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.09, 7.13] |
| 12.1.2 at 72 hours | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.16, 3.90] |
| 12.1.3 at 1 week | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [0.31, 6.28] |
| 12.1.4 at 2 weeks | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 2.24 [0.61, 8.22] |
| 12.1.5 at 4 weeks | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.18, 3.74] |
| 12.2 AE Abnormal dreams | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.3 AE Back pain | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.4 AE Disturbance in attention | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.5 AE Dizziness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.6 AE Fatigue | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.7 AE Feeling drunk | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.8 AE Headache | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.9 AE Insomnia | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.10 AE Irritability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.11 AE Muscle twitching | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.12 AE Nasal congestion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.13 AE Nausea | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.14 AE Palpitations | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.15 AE Post‐lumbar puncture syndrome | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.16 AE Rash | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.17 AE Sedation | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.18 AE Sensory disturbance | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.19 AE Sleepiness/drowsiness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.20 AE Total | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.21 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.21.1 at 72 hours | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.03, 8.60] |
| 12.21.2 at 1 week | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [0.21, 11.06] |
| 12.21.3 at 2 weeks | 2 | 54 | Odds Ratio (M‐H, Random, 95% CI) | 2.29 [0.43, 12.15] |
| 12.21.4 at 4 weeks | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.13, 3.14] |
| 12.22 Depression rating scale score | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12.22.1 at 24 hours | 2 | 54 | Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐4.14, 3.13] |
| 12.22.2 at 72 hours | 2 | 54 | Mean Difference (IV, Random, 95% CI) | ‐0.88 [‐4.67, 2.91] |
| 12.22.3 at 1 week | 2 | 54 | Mean Difference (IV, Random, 95% CI) | ‐1.43 [‐5.31, 2.44] |
| 12.22.4 at 2 weeks | 2 | 54 | Mean Difference (IV, Random, 95% CI) | ‐2.61 [‐7.32, 2.09] |
| 12.22.5 at 4 weeks | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐1.25 [‐8.14, 5.64] |
| 12.23 Acceptability ‐ adverse events | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 13. Riluzole versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Response rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1.1 at 24 hours | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.35, 4.36] |
| 13.1.2 at 72 hours | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 2.62 [0.64, 10.61] |
| 13.1.3 at 1 week | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 2.40 [0.51, 11.26] |
| 13.1.4 at 2 weeks | 2 | 102 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.27, 7.26] |
| 13.1.5 at 4 weeks | 2 | 102 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.09, 28.00] |
| 13.2 AE Abdominal Pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.3 AE Appetite decrease | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.4 AE Appetite increase | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.5 AE Agitation/anxiety | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.6 AE Blurred vision | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.7 AE Chest pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.8 AE Concentration difficulty | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.9 AE Confusion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.10 AE Constipation | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.11 AE Coughing | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.12 AE Cramps | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.13 AE Decreased appetite | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.14 AE Decreased motor activity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.15 AE Decreased libido | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.16 AE Dental problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.17 AE Depression | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.18 AE Dermatologic/skin irritation/lesions | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.19 AE Diarrhoea | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.20 AE Dizziness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.21 AE Dry mouth | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.22 AE Eye irritation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.23 AE Flatulence | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.24 AE Flu/upper respiratory infection | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.25 AE Genital discomfort | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.26 AE Gum problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.27 AE Headache | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.28 AE Increased libido | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.29 AE Increased thirst | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.30 AE Insomnia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.31 AE Irritability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.32 AE Memory problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.33 AE Mouth ulcer | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.34 AE Muscle/bone/joint pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.35 AE Nasal congestion | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.36 AE Nausea | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.37 AE Oedema | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.38 AE Sexual dysfunction | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.39 AE Shortness of breath | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.40 AE Sleepiness/drowsiness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.41 AE Sore throat | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.42 AE Sore tongue | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.43 AE Stomach or abdominal discomfort | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.44 AE Suicidal ideas | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.45 AE Sweating | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.46 AE Tachycardia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.47 AE Tinnitus | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.48 AE Tiredness/fatigue | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.49 AE Urination problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.50 AE Weight gain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.51 AE Weight loss | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.52 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.52.1 at 24 hours | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.14, 3.64] |
| 13.52.2 at 72 hours | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 1.33 [0.30, 5.84] |
| 13.52.3 at 1 week | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 1.00 [0.18, 5.63] |
| 13.52.4 at 2 weeks | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 1.00 [0.13, 7.85] |
| 13.52.5 at 4 weeks | 2 | 102 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.12, 12.13] |
| 13.53 Depression rating scale score | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13.53.1 at 24 hours | 1 | 42 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.87, 0.35] |
| 13.53.2 at 72 hours | 1 | 41 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.86, 0.37] |
| 13.53.3 at 1 week | 1 | 38 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.70, 0.58] |
| 13.53.4 at 2 weeks | 2 | 97 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐1.20, 0.47] |
| 13.53.5 at 4 weeks | 2 | 87 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐1.19, 0.84] |
| 13.54 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 14. Atomoxetine versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.1 at 3 months | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.2 AE Agitation/anxiety | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.3 AE Constipation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.4 AE Depressed mood | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.5 AE Diarrhea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.6 AE Dizziness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.7 AE Dry mouth | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.8 AE Fatigue | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.9 AE Flatulence | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.10 AE Headache | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.11 AE Increased sweating | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.12 AE Insomnia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.13 AE Nasopharyngitis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.14 AE Nausea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.15 AE Tremor | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.16 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.16.1 at 3 months | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.17 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 14.17.1 at 3 months | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 14.18 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.19 Acceptability ‐ adverse events | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.20 Acceptability ‐ lack of efficacy | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
14.2. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 2: AE Agitation/anxiety
14.4. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 4: AE Depressed mood
14.5. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 5: AE Diarrhea
14.6. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 6: AE Dizziness
14.8. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 8: AE Fatigue
14.9. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 9: AE Flatulence
14.10. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 10: AE Headache
14.11. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 11: AE Increased sweating
14.13. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 13: AE Nasopharyngitis
14.14. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 14: AE Nausea
14.15. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 15: AE Tremor
14.20. Analysis.

Comparison 14: Atomoxetine versus Placebo, Outcome 20: Acceptability ‐ lack of efficacy
Comparison 15. Basimglurant versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.2 AE Dizziness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3 AE Dry mouth | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.4 AE Fatigue | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.5 AE Headache | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.6 AE Insomnia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.7 AE Nasopharyngitis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.8 AE Nausea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.9 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.9.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.10 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 15.10.1 at 4 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
15.3. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 3: AE Dry mouth
15.4. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 4: AE Fatigue
15.5. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 5: AE Headache
15.6. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 6: AE Insomnia
15.8. Analysis.

Comparison 15: Basimglurant versus Placebo, Outcome 8: AE Nausea
Comparison 16. Citicoline versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.1.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.2 AE Abdominal Pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.3 AE Appetite decrease | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.4 AE Appetite increase | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.5 AE Dizziness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.6 AE Diarrhoea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.7 AE Headache | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.8 AE Insomnia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.9 AE Nausea | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.10 AE Sedation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.11 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.11.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 17. CP‐101,606 versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 AE Change in blood pressure | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.2 AE Dissociative reaction | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.3 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 17.3.1 at 24 hours | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 17.3.2 at 1 week | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 17.3.3 at 2 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 17.4 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 18. D‐cycloserine versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.1.1 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.1.2 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2 AE Agitation/anxiety | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.3 AE Concentration difficulties | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.4 AE Constipation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.5 AE Failing memory | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.6 AE Headache | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.7 AE Increased dream activity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.8 AE Increased duration of sleep | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.9 AE Increased sexual desire | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.10 AE Increased tendency to sweat | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.11 AE Nausea/vomiting | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.12 AE Palpitation/tachycardia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.13 AE Sleepiness/drowsiness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.14 AE Urination issues | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.15 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.15.1 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.16 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 18.16.1 at 2 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 18.16.2 at 4 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 18.17 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 19. Decoglurant (mGlu2/3) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 Response Rate | 1 | 309 | Odds Ratio (M‐H, Random, 95% CI) | 2.04 [1.23, 3.38] |
| 19.1.1 at 4 weeks | 1 | 309 | Odds Ratio (M‐H, Random, 95% CI) | 2.04 [1.23, 3.38] |
| 19.2 AE Diarrhea | 1 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.42, 1.99] |
| 19.3 AE Dizziness | 1 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 2.01 [1.03, 3.94] |
| 19.4 AE Headache | 1 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.61, 1.70] |
| 19.5 AE Nausea | 1 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.74, 2.59] |
| 19.6 AE Sleepiness/drowsiness | 1 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 4.78 [0.61, 37.26] |
| 19.7 AE Vomiting | 1 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [0.79, 5.65] |
| 19.8 Remission rate | 1 | 309 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.95, 2.69] |
| 19.8.1 at 4 weeks | 1 | 309 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.95, 2.69] |
Comparison 20. MK‐0657 versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 20.1 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20.1.1 at 24 hours | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20.1.2 at 72 hours | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20.1.3 at 1 week | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20.1.4 at 2 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 21. N‐acetylcysteine versus Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 21.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.1 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.2 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.3 at 3 months | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.2 AE Back pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.3 AE Gastrointestinal problems | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.4 AE Joint pain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.5 AE Muscle spasms | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.6 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.6.1 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.6.2 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.6.3 at 3 months | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.7 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 21.7.1 at 2 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 21.7.2 at 4 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 21.7.3 at 3 months | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 21.8 Quality of life | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 21.8.1 at 3 months | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 21.9 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.10 Acceptability ‐ adverse events | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 22. Sarcosine versus Citalopram.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 22.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.1.1 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.1.2 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.2 AE Total | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.3 AE Agitation/anxiety | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.4 AE Asthenia/increased fatigability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.5 AE Constipation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.6 AE Concentration difficulties | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.7 AE Depression | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.8 AE Dizziness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.9 AE Dystonia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.10 AE Headache/migraine | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.11 AE Increased dream activity | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.12 AE Increased duration of sleep | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.13 AE Nausea/vomiting | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.14 AE Palpitations/tachycardia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.15 AE Reduced duration of sleep | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.16 AE Sleepiness/drowsiness | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.17 AE Tremor | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.18 AE Weight gain | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.19 AE Weight loss | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.20 Remission rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.20.1 at 2 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.20.2 at 4 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.21 Depression rating scale score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 22.21.1 at 2 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 22.21.2 at 4 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 22.22 Acceptability | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
22.3. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 3: AE Agitation/anxiety
22.4. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 4: AE Asthenia/increased fatigability
22.5. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 5: AE Constipation
22.6. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 6: AE Concentration difficulties
22.7. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 7: AE Depression
22.8. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 8: AE Dizziness
22.9. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 9: AE Dystonia
22.10. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 10: AE Headache/migraine
22.11. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 11: AE Increased dream activity
22.12. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 12: AE Increased duration of sleep
22.13. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 13: AE Nausea/vomiting
22.14. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 14: AE Palpitations/tachycardia
22.15. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 15: AE Reduced duration of sleep
22.16. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 16: AE Sleepiness/drowsiness
22.17. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 17: AE Tremor
22.18. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 18: AE Weight gain
22.19. Analysis.

Comparison 22: Sarcosine versus Citalopram, Outcome 19: AE Weight loss
Comparison 23. Ketamine versus Placebo (pre‐planned subgroup analysis: outpatient treatment setting).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 23.1 Response rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 23.1.1 at 24 hours | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 18.76 [0.92, 383.10] |
| 23.1.2 at 72 hours | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 33.46 [1.65, 677.83] |
| 23.1.3 at 1 week | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 33.46 [1.65, 677.83] |
| 23.1.4 at 2 weeks | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 20.80 [2.04, 211.79] |
| 23.1.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [0.68, 5.85] |
| 23.1.6 at 3 months | 2 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 3.95 [0.16, 97.23] |
| 23.2 Remission rate | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 23.2.1 at 24 hours | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 3.48 [0.13, 93.30] |
| 23.2.2 at 72 hours | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 6.30 [0.27, 144.70] |
| 23.2.3 at 1 week | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 6.30 [0.27, 144.70] |
| 23.2.4 at 2 weeks | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 3.90 [0.35, 43.36] |
| 23.2.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.85, 5.66] |
| 23.2.6 at 3 months | 2 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 1.09 [0.45, 2.67] |
| 23.3 Depression rating scale score | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 23.3.1 at 24 hours | 2 | 75 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐1.11, 0.18] |
| 23.3.2 at 72 hours | 3 | 94 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐1.18, 0.62] |
| 23.3.3 at 1 week | 2 | 45 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.52 [‐1.69, 0.65] |
| 23.3.4 at 2 weeks | 3 | 126 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.73 [‐1.31, ‐0.15] |
| 23.3.5 at 4 weeks | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.07, ‐0.29] |
| 23.4 Suicidal ideation composite | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 23.4.1 at 24 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.78, 0.82] |
| 23.4.2 at 72 hours | 2 | 68 | Mean Difference (IV, Random, 95% CI) | 0.34 [‐0.25, 0.93] |
| 23.4.3 at 1 week | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.56, 0.96] |
| 23.4.4 at 2 weeks | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.46, 1.06] |
Comparison 24. Ketamine versus Placebo (pre‐planned subgroup analysis: inpatient treatment setting).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 24.1 Response rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 24.1.1 at 24 hours | 2 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 15.11 [1.97, 115.92] |
| 24.1.2 at 72 hours | 2 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 14.00 [2.07, 94.75] |
| 24.1.3 at 1 week | 3 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 3.41 [0.95, 12.27] |
| 24.1.4 at 2 weeks | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.31, 2.83] |
| 24.2 Remission rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 24.2.1 at 24 hours | 2 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 6.60 [0.96, 45.09] |
| 24.2.2 at 72 hours | 2 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 7.88 [1.17, 53.21] |
| 24.2.3 at 1 week | 3 | 99 | Odds Ratio (M‐H, Random, 95% CI) | 7.24 [1.70, 30.81] |
| 24.2.4 at 2 weeks | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.28, 3.24] |
| 24.3 Depression rating scale score | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 24.3.1 at 24 hours | 2 | 46 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.63 [‐2.86, ‐0.39] |
| 24.3.2 at 72 hours | 2 | 46 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.21 [‐1.87, ‐0.55] |
| 24.3.3 at 1 week | 3 | 91 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.19, ‐0.31] |
| 24.3.4 at 2 weeks | 1 | 46 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.68, 0.48] |
Comparison 25. Esketamine versus placebo (pre‐planned subgroup analysis: outpatient treatment setting).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 25.1 Response rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 25.1.1 at 24 hours | 3 | 620 | Odds Ratio (M‐H, Random, 95% CI) | 4.33 [1.08, 17.31] |
| 25.1.2 at 1 week | 3 | 632 | Odds Ratio (M‐H, Random, 95% CI) | 2.73 [1.41, 5.28] |
| 25.1.3 at 4 weeks | 2 | 543 | Odds Ratio (M‐H, Random, 95% CI) | 1.92 [1.34, 2.75] |
| 25.2 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 25.2.1 at 24 hours | 2 | 377 | Odds Ratio (M‐H, Random, 95% CI) | 6.51 [1.93, 21.92] |
| 25.2.2 at 1 week | 2 | 399 | Odds Ratio (M‐H, Random, 95% CI) | 7.76 [1.75, 34.48] |
| 25.2.3 at 2 weeks | 1 | 315 | Odds Ratio (M‐H, Random, 95% CI) | 2.30 [1.02, 5.17] |
| 25.2.4 at 4 weeks | 1 | 317 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.84, 2.30] |
| 25.3 Depression rating scale score | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 25.3.1 at 24 hours | 1 | 310 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.49, ‐0.01] |
| 25.3.2 at 1 week | 1 | 340 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.51, ‐0.06] |
| 25.3.3 at 2 weeks | 1 | 340 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.54, ‐0.09] |
| 25.3.4 at 4 weeks | 2 | 542 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.45, ‐0.10] |
| 25.3.5 at 3 months | 1 | 38 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.75, 0.52] |
| 25.4 Suicidal ideation composite | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 25.4.1 at 1 week | 1 | 209 | Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.08, 0.18] |
| 25.4.2 at 2 weeks | 1 | 208 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.21, 0.07] |
| 25.4.3 at 4 weeks | 1 | 196 | Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.11, 0.07] |
Comparison 26. Esketamine versus placebo (pre‐planned subgroup analysis: inpatient treatment setting).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 26.1 Response rate | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 26.1.1 at 24 hours | 1 | 224 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [0.79, 2.49] |
| 26.1.2 at 72 hours | 1 | 224 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [0.88, 2.62] |
| 26.1.3 at 1 week | 1 | 224 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.76, 2.18] |
| 26.1.4 at 2 weeks | 1 | 224 | Odds Ratio (M‐H, Random, 95% CI) | 1.71 [1.01, 2.91] |
| 26.1.5 at 4 weeks | 1 | 224 | Odds Ratio (M‐H, Random, 95% CI) | 1.85 [1.09, 3.14] |
| 26.2 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 26.2.1 at 24 hours | 2 | 290 | Odds Ratio (M‐H, Random, 95% CI) | 2.25 [1.13, 4.49] |
| 26.2.2 at 72 hours | 2 | 290 | Odds Ratio (M‐H, Random, 95% CI) | 1.61 [0.59, 4.34] |
| 26.2.3 at 1 week | 2 | 290 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.70, 2.13] |
| 26.2.4 at 2 weeks | 2 | 290 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.83, 2.32] |
| 26.2.5 at 4 weeks | 2 | 290 | Odds Ratio (M‐H, Random, 95% CI) | 1.48 [0.91, 2.42] |
| 26.3 Depression rating scale score | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 26.3.1 at 24 hours | 2 | 290 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.58, ‐0.12] |
| 26.3.2 at 72 hours | 2 | 290 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.41 [‐0.64, ‐0.17] |
| 26.3.3 at 1 week | 2 | 290 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.43, 0.04] |
| 26.3.4 at 2 weeks | 2 | 290 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.37, 0.09] |
| 26.3.5 at 4 weeks | 2 | 290 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.48, ‐0.02] |
| 26.4 Suicidal ideation composite | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 26.4.1 at 24 hours | 1 | 224 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.63, 0.23] |
| 26.4.2 at 72 hours | 1 | 224 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.69, 0.09] |
| 26.4.3 at 1 week | 1 | 224 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.56, 0.16] |
| 26.4.4 at 2 weeks | 1 | 224 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.43, 0.23] |
| 26.4.5 at 4 weeks | 1 | 224 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.53, 0.13] |
Comparison 27. Esketamine versus placebo (pre‐planned subgroup analysis: excluding elderly populations >65 years).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 27.1 Response rate | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 27.1.1 at 24 hours | 5 | 1071 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [1.20, 3.68] |
| 27.1.2 at 72 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.92, 1.96] |
| 27.1.3 at 1 week | 5 | 1083 | Odds Ratio (M‐H, Random, 95% CI) | 1.64 [1.05, 2.54] |
| 27.1.4 at 2 weeks | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.09, 2.28] |
| 27.1.5 at 4 weeks | 4 | 994 | Odds Ratio (M‐H, Random, 95% CI) | 1.81 [1.40, 2.34] |
| 27.2 Remission rate | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 27.2.1 at 24 hours | 5 | 894 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [1.71, 4.40] |
| 27.2.2 at 72 hours | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.91, 2.64] |
| 27.2.3 at 1 week | 5 | 916 | Odds Ratio (M‐H, Random, 95% CI) | 1.62 [0.91, 2.89] |
| 27.2.4 at 2 weeks | 4 | 832 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [1.07, 2.16] |
| 27.2.5 at 4 weeks | 4 | 834 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [1.12, 2.04] |
| 27.3 Depression rating scale score | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 27.3.1 at 24 hours | 4 | 824 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.45, ‐0.17] |
| 27.3.2 at 72 hours | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.11] |
| 27.3.3 at 1 week | 4 | 857 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.37, ‐0.10] |
| 27.3.4 at 2 weeks | 4 | 857 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.34, ‐0.07] |
| 27.3.5 at 4 weeks | 5 | 1059 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.40, ‐0.15] |
| 27.3.6 at 3 months | 1 | 38 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.75, 0.52] |
| 27.4 Suicidal ideation composite | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 27.4.1 at 24 hours | 2 | 450 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.44, 0.15] |
| 27.4.2 at 72 hours | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.49, 0.08] |
| 27.4.3 at 1 week | 3 | 660 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.10, 0.13] |
| 27.4.4 at 2 weeks | 3 | 659 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.22, 0.02] |
| 27.4.5 at 4 weeks | 3 | 647 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.12, 0.05] |
Comparison 28. Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 28.1 Response rate | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 28.1.1 at 24 hours | 6 | 177 | Odds Ratio (M‐H, Random, 95% CI) | 4.33 [1.47, 12.80] |
| 28.1.2 at 72 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 17.99 [3.58, 90.34] |
| 28.1.3 at 1 week | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 14.32 [2.90, 70.64] |
| 28.1.4 at 2 weeks | 2 | 85 | Odds Ratio (M‐H, Random, 95% CI) | 15.73 [4.71, 52.51] |
| 28.1.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [0.68, 5.85] |
| 28.1.6 at 3 months | 2 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 3.95 [0.16, 97.23] |
| 28.2 Remission rate | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 28.2.1 at 24 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 5.60 [1.07, 29.46] |
| 28.2.2 at 72 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 7.42 [1.45, 37.89] |
| 28.2.3 at 1 week | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 9.02 [1.80, 45.31] |
| 28.2.4 at 2 weeks | 2 | 85 | Odds Ratio (M‐H, Random, 95% CI) | 7.50 [1.51, 37.22] |
| 28.2.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.85, 5.66] |
| 28.2.6 at 3 months | 1 | 20 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.14, 11.54] |
| 28.3 Depression rating scale score | 9 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 28.3.1 at 24 hours | 7 | 223 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.88 [‐1.31, ‐0.46] |
| 28.3.2 at 72 hours | 5 | 140 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.63 [‐1.29, 0.04] |
| 28.3.3 at 1 week | 4 | 90 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.76 [‐1.34, ‐0.19] |
| 28.3.4 at 2 weeks | 3 | 126 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.73 [‐1.31, ‐0.15] |
| 28.3.5 at 4 weeks | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.07, ‐0.29] |
| 28.4 Suicidal ideation composite | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 28.4.1 at 24 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.78, 0.82] |
| 28.4.2 at 72 hours | 2 | 68 | Mean Difference (IV, Random, 95% CI) | 0.34 [‐0.25, 0.93] |
| 28.4.3 at 1 week | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.56, 0.96] |
| 28.4.4 at 2 weeks | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.46, 1.06] |
Comparison 29. Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding TRD populations).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 29.1 Response rate | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 29.1.1 at 24 hours | 3 | 68 | Odds Ratio (M‐H, Random, 95% CI) | 2.31 [0.65, 8.14] |
| 29.1.2 at 72 hours | 2 | 38 | Odds Ratio (M‐H, Random, 95% CI) | 15.32 [1.58, 148.09] |
| 29.1.3 at 1 week | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 10.29 [0.97, 108.81] |
| 29.1.4 at 4 weeks | 1 | 81 | Odds Ratio (M‐H, Random, 95% CI) | 4.31 [1.48, 12.56] |
| 29.2 Remission rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 29.2.1 at 24 hours | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 6.75 [0.61, 75.27] |
| 29.2.2 at 72 hours | 2 | 38 | Odds Ratio (M‐H, Random, 95% CI) | 5.63 [0.77, 40.99] |
| 29.2.3 at 1 week | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 10.29 [0.97, 108.81] |
| 29.2.4 at 4 weeks | 1 | 81 | Odds Ratio (M‐H, Random, 95% CI) | 1.59 [0.51, 4.98] |
| 29.3 Depression rating scale score | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 29.3.1 at 24 hours | 3 | 66 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.06 [‐1.61, ‐0.52] |
| 29.3.2 at 72 hours | 2 | 36 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.20 [‐1.96, ‐0.44] |
| 29.3.3 at 1 week | 2 | 35 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.19 [‐1.97, ‐0.42] |
| 29.3.4 at 2 weeks | 1 | 81 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.36, ‐0.45] |
| 29.3.5 at 4 weeks | 1 | 81 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.77 [‐1.22, ‐0.31] |
Comparison 30. Ketamine versus Placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 30.1 Response rate | 10 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 30.1.1 at 24 hours | 7 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 3.94 [1.54, 10.10] |
| 30.1.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 15.84 [3.68, 68.12] |
| 30.1.3 at 1 week | 4 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 5.69 [1.34, 24.11] |
| 30.1.4 at 2 weeks | 2 | 78 | Odds Ratio (M‐H, Random, 95% CI) | 3.72 [0.17, 79.32] |
| 30.1.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [0.68, 5.85] |
| 30.1.6 at 3 months | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 20.00 [2.77, 144.31] |
| 30.2 Remission rate | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 30.2.1 at 24 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 5.60 [1.07, 29.46] |
| 30.2.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 6.60 [1.51, 28.92] |
| 30.2.3 at 1 week | 4 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 7.06 [1.90, 26.31] |
| 30.2.4 at 2 weeks | 2 | 78 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.41, 4.12] |
| 30.2.5 at 4 weeks | 2 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 2.60 [0.60, 11.33] |
| 30.3 Depression rating scale score | 10 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 30.3.1 at 24 hours | 8 | 231 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.48] |
| 30.3.2 at 72 hours | 5 | 128 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.86 [‐1.24, ‐0.48] |
| 30.3.3 at 1 week | 5 | 124 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.85 [‐1.23, ‐0.47] |
| 30.3.4 at 2 weeks | 3 | 153 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.29, ‐0.08] |
| 30.3.5 at 4 weeks | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.07, ‐0.29] |
| 30.4 Suicidal ideation composite | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 30.4.1 at 24 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.78, 0.82] |
| 30.4.2 at 72 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.63, 0.81] |
Comparison 31. Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding multiple doses).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 31.1 Response rate | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 31.1.1 at 24 hours | 7 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 3.94 [1.54, 10.10] |
| 31.1.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 15.84 [3.68, 68.12] |
| 31.1.3 at 1 week | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 14.32 [2.90, 70.64] |
| 31.1.4 at 2 weeks | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 20.80 [2.04, 211.79] |
| 31.1.5 at 4 weeks | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.35, 7.40] |
| 31.1.6 at 3 months | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 20.00 [2.77, 144.31] |
| 31.2 Remission rate | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 31.2.1 at 24 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 5.60 [1.07, 29.46] |
| 31.2.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 6.60 [1.51, 28.92] |
| 31.2.3 at 1 week | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 9.02 [1.80, 45.31] |
| 31.2.4 at 2 weeks | 2 | 78 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.41, 4.12] |
| 31.2.5 at 4 weeks | 1 | 27 | Odds Ratio (M‐H, Random, 95% CI) | 8.12 [0.80, 82.73] |
| 31.3 Depression rating scale score | 9 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 31.3.1 at 24 hours | 8 | 231 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.48] |
| 31.3.2 at 72 hours | 6 | 148 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.28, ‐0.07] |
| 31.3.3 at 1 week | 4 | 78 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.07 [‐1.57, ‐0.58] |
| 31.3.4 at 2 weeks | 1 | 26 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.14 [‐1.98, ‐0.30] |
| 31.3.5 at 4 weeks | 1 | 26 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐1.21, 0.35] |
| 31.4 Suicidal ideation composite | 1 | 96 | Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.48, 0.59] |
| 31.4.1 at 24 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.78, 0.82] |
| 31.4.2 at 72 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.63, 0.81] |
Comparison 32. Ketamine versus Placebo (post‐hoc sensitivity analysis: excluding add‐on ECT studies).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 32.1 Response rate | 10 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 32.1.1 at 24 hours | 7 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 3.94 [1.54, 10.10] |
| 32.1.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 15.84 [3.68, 68.12] |
| 32.1.3 at 1 week | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 14.32 [2.90, 70.64] |
| 32.1.4 at 2 weeks | 2 | 85 | Odds Ratio (M‐H, Random, 95% CI) | 15.73 [4.71, 52.51] |
| 32.1.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [0.68, 5.85] |
| 32.1.6 at 3 months | 2 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 3.95 [0.16, 97.23] |
| 32.2 Remission rate | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 32.2.1 at 24 hours | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 5.60 [1.07, 29.46] |
| 32.2.2 at 72 hours | 4 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 6.60 [1.51, 28.92] |
| 32.2.3 at 1 week | 3 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 9.02 [1.80, 45.31] |
| 32.2.4 at 2 weeks | 2 | 85 | Odds Ratio (M‐H, Random, 95% CI) | 7.50 [1.51, 37.22] |
| 32.2.5 at 4 weeks | 3 | 132 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.85, 5.66] |
| 32.2.6 at 3 months | 1 | 20 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.14, 11.54] |
| 32.3 Depression rating scale score | 10 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 32.3.1 at 24 hours | 8 | 231 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.48] |
| 32.3.2 at 72 hours | 6 | 148 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.28, ‐0.07] |
| 32.3.3 at 1 week | 5 | 97 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.80 [‐1.31, ‐0.30] |
| 32.3.4 at 2 weeks | 3 | 126 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.73 [‐1.31, ‐0.15] |
| 32.3.5 at 4 weeks | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.68 [‐1.07, ‐0.29] |
| 32.4 Suicidal ideation composite | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 32.4.1 at 24 hours | 1 | 48 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.78, 0.82] |
| 32.4.2 at 72 hours | 2 | 68 | Mean Difference (IV, Random, 95% CI) | 0.34 [‐0.25, 0.93] |
| 32.4.3 at 1 week | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.56, 0.96] |
| 32.4.4 at 2 weeks | 1 | 19 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.46, 1.06] |
Comparison 33. Esketamine versus placebo (pre‐planned sensitivity analysis: excluding studies that included participants with bipolar disorder or psychotic features).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 33.1 Response rate | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 33.1.1 at 24 hours | 5 | 1071 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [1.20, 3.68] |
| 33.1.2 at 72 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.92, 1.96] |
| 33.1.3 at 1 week | 5 | 1083 | Odds Ratio (M‐H, Random, 95% CI) | 1.64 [1.05, 2.54] |
| 33.1.4 at 2 weeks | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.09, 2.28] |
| 33.1.5 at 4 weeks | 5 | 1117 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [1.44, 2.37] |
| 33.2 Remission rate | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 33.2.1 at 24 hours | 5 | 894 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [1.71, 4.40] |
| 33.2.2 at 72 hours | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.91, 2.64] |
| 33.2.3 at 1 week | 5 | 916 | Odds Ratio (M‐H, Random, 95% CI) | 1.62 [0.91, 2.89] |
| 33.2.4 at 2 weeks | 4 | 832 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [1.07, 2.16] |
| 33.2.5 at 4 weeks | 5 | 957 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.18, 2.10] |
| 33.3 Depression rating scale score | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 33.3.1 at 24 hours | 4 | 824 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.45, ‐0.17] |
| 33.3.2 at 72 hours | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.11] |
| 33.3.3 at 1 week | 4 | 857 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.37, ‐0.10] |
| 33.3.4 at 2 weeks | 4 | 857 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.34, ‐0.07] |
| 33.3.5 at 4 weeks | 6 | 1182 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.39, ‐0.16] |
| 33.3.6 at 3 months | 1 | 38 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.75, 0.52] |
| 33.4 Suicidal ideation composite | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 33.4.1 at 24 hours | 2 | 450 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.44, 0.15] |
| 33.4.2 at 72 hours | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.49, 0.08] |
| 33.4.3 at 1 week | 3 | 660 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.10, 0.13] |
| 33.4.4 at 2 weeks | 3 | 659 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.22, 0.02] |
| 33.4.5 at 4 weeks | 3 | 647 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.12, 0.05] |
Comparison 34. Esketamine versus placebo (pre‐planned sensitivity analysis: excluding TRD populations).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 34.1 Response rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 34.1.1 at 24 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [1.03, 2.33] |
| 34.1.2 at 72 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.92, 1.96] |
| 34.1.3 at 1 week | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.85, 1.78] |
| 34.1.4 at 2 weeks | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.09, 2.28] |
| 34.1.5 at 4 weeks | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [1.17, 2.46] |
| 34.2 Remission rate | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 34.2.1 at 24 hours | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 2.35 [1.40, 3.92] |
| 34.2.2 at 72 hours | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.91, 2.64] |
| 34.2.3 at 1 week | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [0.86, 2.01] |
| 34.2.4 at 2 weeks | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.93, 2.04] |
| 34.2.5 at 4 weeks | 3 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [1.10, 2.29] |
| 34.3 Depression rating scale score | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 34.3.1 at 24 hours | 3 | 514 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐0.52, ‐0.17] |
| 34.3.2 at 72 hours | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.50, ‐0.11] |
| 34.3.3 at 1 week | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.38, ‐0.04] |
| 34.3.4 at 2 weeks | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.32, 0.03] |
| 34.3.5 at 4 weeks | 3 | 517 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.44, ‐0.10] |
| 34.4 Suicidal ideation composite | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 34.4.1 at 24 hours | 2 | 450 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.44, 0.15] |
| 34.4.2 at 72 hours | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.49, 0.08] |
| 34.4.3 at 1 week | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.41, 0.11] |
| 34.4.4 at 2 weeks | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.43, 0.05] |
| 34.4.5 at 4 weeks | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.39, 0.09] |
Comparison 35. Esketamine versus placebo (pre‐planned sensitivity analysis: excluding trials with a dropout rate greater than 20%.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 35.1 Response rate | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 35.1.1 at 24 hours | 5 | 1071 | Odds Ratio (M‐H, Random, 95% CI) | 2.11 [1.20, 3.68] |
| 35.1.2 at 72 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.92, 1.96] |
| 35.1.3 at 1 week | 6 | 1115 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [1.09, 2.34] |
| 35.1.4 at 2 weeks | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.09, 2.28] |
| 35.1.5 at 4 weeks | 5 | 1117 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [1.44, 2.37] |
| 35.2 Remission rate | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 35.2.1 at 24 hours | 4 | 828 | Odds Ratio (M‐H, Random, 95% CI) | 2.88 [1.72, 4.81] |
| 35.2.2 at 72 hours | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [0.97, 3.18] |
| 35.2.3 at 1 week | 5 | 882 | Odds Ratio (M‐H, Random, 95% CI) | 1.79 [0.93, 3.42] |
| 35.2.4 at 2 weeks | 3 | 766 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [1.04, 2.19] |
| 35.2.5 at 4 weeks | 4 | 891 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [1.19, 2.16] |
| 35.3 Depression rating scale score | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 35.3.1 at 24 hours | 3 | 758 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.43, ‐0.14] |
| 35.3.2 at 72 hours | 2 | 451 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.45, ‐0.07] |
| 35.3.3 at 1 week | 4 | 818 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.37, ‐0.09] |
| 35.3.4 at 2 weeks | 3 | 791 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.36, ‐0.07] |
| 35.3.5 at 4 weeks | 5 | 1116 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.39, ‐0.15] |
| 35.3.6 at 3 months | 1 | 38 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.75, 0.52] |
| 35.4 Suicidal ideation composite | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 35.4.1 at 24 hours | 2 | 450 | Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.44, 0.15] |
| 35.4.2 at 72 hours | 2 | 451 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.49, 0.08] |
| 35.4.3 at 1 week | 3 | 660 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.10, 0.13] |
| 35.4.4 at 2 weeks | 3 | 659 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.22, 0.02] |
| 35.4.5 at 4 weeks | 3 | 647 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.12, 0.05] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Abbasinazari 2015.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode
N: 40 Age: memantine 5 mg/day group M = 36.5 (SD = 12.0); placebo group M = 41.6 (SD = 11.2) Sex: memantine 5 mg/day group 65% female; placebo group 50% female Baseline depression severity: not reported. |
|
| Interventions | Given 5 mg/day of memantine or placebo beginning the day before the first session of ECT until the fourth session of ECT Memantine 5 mg/day (N = 20) Placebo (N = 20) Concomitant treatment :not stated |
|
| Outcomes | Modified Mental State Examination (MMSE) | |
| Notes | Not included in analysis due to study not using depression rating scales (assessed cognition scores only) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A sequence was computer‐generated to randomly assign patients to two groups in a 1:1 ratio. This sequence was generated in blocks of 4, 8 and 12 using the 'blockrand' extension of the R Project software package." |
| Allocation concealment (selection bias) | Low risk | Quote: "Knowledge of this sequence was available only to a nurse not involved in volunteer recruitment. This nurse allocated patients to either the placebo group or the memantine group by flipping a coin." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study described as double‐blind. Quote: "A strategy of numbered boxes was used for sequence concealment". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study described as double‐blind. Quote: "A strategy of numbered boxes was used for sequence concealment". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts (zero) |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Average HAM‐D scores are missing |
| Other bias | Low risk | No other potential sources of bias identified |
Amidfar 2016.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group trial | |
| Participants |
Diagnosis: DSM‐IV moderate‐to‐severe major depressive episode
N: 66 randomised, 62 completed the study Age: memantine 20 mg/day group M = 34.19 (SD = 7.06); placebo group M = 33.58 (SD = 7.40) Sex: memantine 20 mg/day group 29% female; placebo group 41% female Baseline depression severity: memantine 20 mg/day group HDRS M = 24.29 (SD = 1.79); placebo group HDRS M = 23.93 (SD = 3.38) |
|
| Interventions | All patients, regardless of their assigned group, received 100 mg/day sertraline for the first week and 200 mg/day for the subsequent 5 weeks. Patients randomly received either memantine 20 mg/day or placebo for 6 weeks. Memantine was administered half‐dose (10 mg/day) for the first week of the trial. Memantine 20 mg/day group (N = 33), placebo group (N = 33). Concomitant treatment: no ‐ patients who had any history of antidepressant use in the last 1 month were excluded from the trial. |
|
| Outcomes | HDRS Response rates (≥ 50% reduction in the HDRS score) Remission rates (HDRS score ≤ 7) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "An independent party randomized study subjects by the permuted randomization block method using a computerized random number generator (allocation ratio 1:1) to recieve either memantine or placebo in addition to their standard treatment." |
| Allocation concealment (selection bias) | Low risk | Quote: "Allocation concealment was done using sequentially numbered, sealed, opaque and stapled envelopes. An aluminium foil was put inside envelopes to make the content of the envelopes impermeable to intense light." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The patients and the physician who referred the patient, the raters and the statistician were all blinded to treatment allocation". No further details given. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The patients and the physician who referred the patient, the raters and the statistician were all blinded to treatment allocation". No further details given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Exclusion, withdrawal and drop out rates at each time point are recorded |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other potential sources of bias identified |
Anderson 2017.
| Study characteristics | ||
| Methods | Multicentre, double‐blind, randomised, parallel‐group, superiority trial | |
| Participants |
Diagnosis: DSM‐IV moderate‐to‐severe unipolar or bipolar depressive episode
N: 79 randomised, 70 included in modified intention‐to‐treat analysis Age: ketamine 0.5 mg/kg group M = 52.5 (SD = 11.9); placebo saline group M = 56.4 (SD = 12.4) Sex: ketamine 0.5 mg/kg group 67% female; placebo saline group 60% female Baseline depression severity: ketamine 0.5 mg/kg group MADRS score M = 31.8 (SD = 7.4); placebo saline group MADRS score M = 35.2 (SD = 8.4) |
|
| Interventions | Ketamine (0.5 mg/kg) or saline was given as a slow intravenous bolus directly before anaesthetic induction at each treatment. ECT treatments were administered twice weekly. Ketamine 0.5mg/kg group (N = 33) Placebo saline group (N = 37) Concomitant treatment: yes, oral psychotropic medication was continued by the treating clinical team during the trial |
|
| Outcomes | MADRS EQ‐5D‐3L Response rates (≥50% decrease in MADRS score from baseline) Remission rates (MADRS score ≤ 10) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned (1:1) to receive either ketamine or saline as anaesthetic for their ECT treatment. Randomisation was done by the Christie Hospital Clinical Trials Co‐ordination Unit (CTU) by use of permuted block randomisation, which varied randomly between four and eight, and was stratified by the NHS Trust." |
| Allocation concealment (selection bias) | Low risk | "Both patients and assessment and ECT treatment teams were masked to treatment allocation, although the anaesthetists administering the study medication were not. The anaesthetist broke the seal away from the psychiatric ECT team, drew up the trial medication into a syringe, and disposed of the ampoule without revealing which drug was being administered. Researchers responsible for outcome assessment did not attend ECT sessions. To assess success of masking, patients and assessors were invited to complete a questionnaire to indicate suspected treatment group after four ECTs and at the end of the ECT course, and the results were sent directly to the CTU in a sealed envelope for data entry.” |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding was tested and deemed effective. Quote: "Masking of treatment allocation was successful as assessed by questionnaire; patient guesses were correct in 28 (48%) of 58 patients who completed the questionnaire at mid‐ECT and 30 (56%) of 54 patient guesses at the end of treatment, while 35 (56%) of 63 assessor guesses at mid‐ECT and 28 (51%) of 55 assessor guesses at the end of treatment were correct, which did not differ significantly from chance." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessor blinding was assessed to be effective. Quote: "Masking of treatment allocation was successful as assessed by questionnaire... 35 (56%) of 63 assessor guesses at mid‐ECT and 28 (51%) of 55 assessor guesses at the end of treatment were correct, which did not differ significantly from chance." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Exclusion, withdrawal and drop out rates at each time point are recorded |
| Selective reporting (reporting bias) | Low risk | Protocol available and data for all time points reported. |
| Other bias | Low risk | No other potential sources of bias identified |
Arabzadeh 2018.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, double‐blind, controlled trial | |
| Participants |
Diagnosis: DSM‐5 major depressive disorder
N: 90 randomised, 81 completed the study Age: ketamine 50 mg/day group M = 34.31 (SD = 6.73); placebo group M = 33.72 (SD = 8.34) Sex: ketamine 50 mg/day group 36.6% female; placebo group 40.0% female Baseline depression severity: ketamine 50 mg/day group HDRS score M = 24.17 (SD = 2.31); placebo group HDRS score M = 24.62 (SD = 3.52) |
|
| Interventions | All patients received setraline (150 mg a day). As an adjuvant, they recieved either 50 mg/day ketamine or placebo. Patients were followed for 6 weeks. Ketamine 50 mg/day group (N = 41) ‐ prescribed as 25 mg twice daily. Placebo group (N = 40) Concomitant treatment: No, patients were excluded from the study if they had received antidepressant drugs within the previous month. |
|
| Outcomes | HDRS Side effects Response rates (≥ 50% reduction in HDRS score at the termination of the trial) Remission rates (HDRS scors ≤ 7 at the termination of the trial). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization of patients to either ketamine or placebo groups was done by a computerized random number generator (allocation ratio 1/1)." |
| Allocation concealment (selection bias) | Low risk | Quote: "Allocation was concealed using successively numbered, opaque, and sealed envelopes". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The patients, the physician, and the statistician were all blind to allocation. The placebo and ketamine capsules were identical in shape, size, color, texture, and odor." No further details given. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The patients, the physician, and the statistician were all blind to allocation. The placebo and ketamine capsules were identical in shape, size, color, texture, and odor." No further details given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Exclusion and withdrawal rates at each time point are recorded |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other potential sources of bias identified |
Berk 2014.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; MADRS score ≥ 18
N: 269 Age:N‐Acetylcysteine group M = 29.9 (SD = 13.0); placebo group M = 50.5 (SD = 12.5) Sex:N‐Acetylcysteine group 84% female; placebo group 75% female Baseline depression severity:N‐Acetylcysteine group MADRS = 27.7 (SD = 5.8); placebo group MADRS = 28.1 (SD = 5.8) |
|
| Interventions | 12 weeks of treatment N‐Acetylcysteine (N = 135), 2 x 500 mg capsules twice daily Placebo (N =134), 2 capsules twice daily dusted in N‐Acetylcysteine to ensure similar odour Concomitant treatment: yes, patients continued existing treatments for depression |
|
| Outcomes | Montgomery Asberg Rating Scale ) (MADRS) Response rate (≥ 50% reduction in MADRS scores) Remission rate (MADRS score ≤ 7) Clinical Global Impression‐Improvement and Clinical Global Impression‐Severity HARS Global Assessment of Functioning SOFAS SLICE‐LIFE LIFE‐RIFT Q‐LES‐Q Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given on randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''To facilitate double‐blinding, the trial medications (both N‐acetylcysteine and placebo) were dispensed in identical numbers and capsule formulations in sealed containers by the trial pharmacist. Furthermore, to mask the distinct smell of the N‐acetylcysteine preparation, the placebo capsules were dusted with a tiny amount of N‐acetylcysteine so that all capsules had a similar odour'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study described as double‐blind, trial pharmacist dispensed medications but unlikely to have conducted outcome assessments, however this is unclear |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts, state exclusion of patients from analysis based on no post‐baseline data |
| Selective reporting (reporting bias) | Unclear risk | Protocol available, unclear if all prespecified outcomes reported in published report |
| Other bias | Low risk | No other potential sources of bias identified |
Berman 2000.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode (1 bipolar disorder, depressed)
N: 9 Age: M = 37 (SD = 10.0) Sex: 56% female Baseline depression severity: ketamine group HRSD = 33.0 (SD = 6.7); placebo group HRSD = 26.9 (SD = 5.8) |
|
| Interventions | 1 single IV infusion Ketamine (N = 4) 0.5 mg/kg infused over 40 minutes Placebo (N = 3) saline solution infused over 40 minutes Concomitant treatment: no, patients had a 2‐week drug‐free period before commencing treatment |
|
| Outcomes | HRSD Response rate (≥50% reduction in HRSD scores) BDI VAS BPRS |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given on randomisation procedure beyond quote: ''four participants were randomly assigned'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details given on blinding beyond quote: '''in a randomised, double‐blinded manner'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given on blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2/9 participants withdrew to institute antidepressant treatment |
| Selective reporting (reporting bias) | High risk | Scores not reported for each measured time point (only baseline and final) and BDI scores missing |
| Other bias | Low risk | No other potential sources of bias identified |
Canuso 2018.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled study | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder, confirmed by MINI N: 68 Age: M = 35.8 (SD = 13.03) Sex: 62.5% female Baseline depression severity: esketamine group MADRS M = 38.5 (SD = 6.17); placebo group MADRS M = 38.8 (SD = 7.02) |
|
| Interventions | 4 weeks treatment phase of intranasal esketamine (84 mg) or placebo administered twice weekly Concomitant treatment: Yes, all patients received standard‐of‐care antidepressants alongside the study drug |
|
| Outcomes | MADRS Beck Scale for Suicide Ideation CGI‐S Adverse events CADSS |
|
| Notes | Authors kindly provided additional data for remission rates according to this review's definition and MADRS mean scores. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomization was balanced using randomly permuted blocks” |
| Allocation concealment (selection bias) | Low risk | Quote: ““A computerized system was used for randomization; investigators were not provided with the randomization codes.” |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: ““A computerized system was used for randomization; investigators were not provided with the randomization codes.” Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study described as double‐blind, however no information on whether the effectiveness of the blind was tested. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Participant flowchart included (p2) |
| Selective reporting (reporting bias) | Low risk | Protocol available online, expected outcomes reported. |
| Other bias | High risk | Study funded by Janssen. Authors are associated with and receive payments from pharmaceutical companies |
Carspecken 2018.
| Study characteristics | ||
| Methods | Double‐blinded randomised clinical trial | |
| Participants | Veterans scheduled for an index course of ECT for treatment of a major depressive episode associated with treatment resistant major depressive disorder Diagnosis: DSM‐V major depressive disorder N: 52 Age:kKetamine group M = 50 (SD = 12); methohexital M = 47 (SD 12) Sex: ketamine 26% female; Methohexital 11% female Baseline depression severity: ketamine group PHQ‐9 M = 21.1 (SD = 3.9); Methohexital group PHQ‐9 M = 21.5 (SD = 3.6) |
|
| Interventions | Intravenous racemic ketamine (1‐2 mg/kg) or methohexital (1‐2 mg/kg), in addition to ketorolac 30 mg for induction of general anesthesia. Participants in the ketamine arm received 1 mg to 2 mg of IV midazolam to mitigate post‐procedural psychedelic effects. Concomitant treatment: yes. Quote: "Outpatient benzodiazepines were discontinued 48 hours before ECT, whereas all other psychiatric medications were continued during the study." |
|
| Outcomes | HAM‐D PHQ‐9 MOCA |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “"Patient randomization was performed using permuted block randomization to ensure number of subjects assigned to each treatment arm was balanced" |
| Allocation concealment (selection bias) | Low risk | Quote: “"Patient randomization was performed using permuted block randomization to ensure number of subjects assigned to each treatment arm was balanced" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “"Subjects and depression score raters were blinded to study drug intervention. Because of feasibility issues, anesthesiologists and psychiatrists were not blinded during ECT sessions." Effectiveness of blinding not assessed. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Effectiveness of blinding not assessed. Unclear if cognitive measured were blinded as they were completed by psychiatrists who were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study CONSORT flow chart included. |
| Selective reporting (reporting bias) | Low risk | Study protocol available, outcomes reported as expected. |
| Other bias | Low risk | None identified. |
Chen 2017.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, double‐blind trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive episode
N: 132 randomised, 127 completed the study Age: ketamine 0.3 mg/kg group M = 40.94 (SD = 15.41); placebo saline group M = 37.44 (SD = 14.16) Sex: ketamine 0.3 mg/kg group 66.7% female; placebo saline group 64.1% female Baseline depression severity: ketamine 0.3mg/kg group HAM‐D score M = 38.05 (SD = 3.23); placebo saline group HAM‐D score M = 37.43 (SD = 2.64) |
|
| Interventions | Patients assigned to the study group were anesthetised using 1.5 mg/kg propofol and 0.3 mg/kg ketamine delivered intravenously. Patients assigned to the control group were anesthestised using 1.5 mg/kg propofol and normal saline. The ECT was administered 3 times a week (12 sessions of ECT in total). Ketamine 0.3 mg/kg group (N = 63) Placebo saline group (N = 64) Concomitant treatment: Not reported |
|
| Outcomes | Wechsler Memory Scale‐Chinese Revision (WMS‐RC) MMSE HAM‐D Remission rates (sustained HAM‐D score <10 after 2 consecutive ECTs) 4‐item positie symptom subscale of the Brief Psychatric Rating Scale (BPRS+) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "All recruited participants were randomly assigned into 1 of 2 groups: the control group and the study group. Half the patients were placed in each group using a computer‐generated random number table." No further details given. |
| Allocation concealment (selection bias) | Low risk | Quote: "The patients, treatment teams, and the outcome assessors were all blinded to the intervention allocation." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding stated but not tested. Quote: "The anesthesiologists, patients, and outcome raters were all blind to the group assignments and the anesthetic regimen." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding stated but not tested. Ketamine and normal saline were both prepared before each ECT treatment, quote: "by a specialized nurse who was blind to the study design". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Exclusion and withdrawal rates at each time point are recorded |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other potential sources of bias identified |
Chen 2018.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive episode
N: 24 Age: ketamine 0.5m g/kg group M = 51.1 (SD = 13.6); ketamine 0.2 mg/kg group M = 49.8 (SD = 11.1); placebo group saline M = 46.3 (SD = 8.1) Sex: ketamine 0.5 mg/kg group 100% female; ketamine 0.2 mg/kg group 62.5% female; placebo group saline 62.5% female Baseline depression severity: ketamine 0.5 mg/kg group HDRS‐17 score M = 24.00 (SD = 1.93); ketamine 0.2 mg/kg group HDRS‐17 score M = 27.13 (SD = 3.23); placebo group saline HDRS‐17 score M = 24.63 (SD = 4.63) |
|
| Interventions | 1 single IV infusion over 40 minutes: Ketamine 0.5 mg/kg (N = 8) Ketamine 0.2 mg/kg (N = 8) Placebo saline (N = 8) Concomitant treatment: yes, at least 2‐week concomitant stable antidepressant treatment |
|
| Outcomes | HDRS‐17 Response rates (≥50% on HDRS‐17) F‐FDG‐PET scan |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "we followed the same protocol as in our previous study (Li et al., 2016)". Quote (Li et al., 2016): "patients were randomized in a 1:1:1/A:B:C ratio to each of the respective experiment groups". |
| Allocation concealment (selection bias) | Low risk | Quote: "we followed the same protocol as in our previous study (Li et al., 2016)". Quote (Li et al., 2016): "An independent research nurse who was not involved in the study was responsible for the random allocation and was not allowed to release any information to others, including the study nurse who applied the intravenous injection." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "we followed the same protocol as in our previous study (Li et al., 2016)". Quote (Li et al., 2016): The entire study procedure was double‐blinded, and participants and staff were all blind to the treatment assignment. Blinding stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''double blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts (zero) |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other potential sources of bias identified |
Correia‐Melo 2020.
| Study characteristics | ||
| Methods | Randomised, double‐blind, active‐controlled, bicentre, noninferiority clinical trial, with two parallel groups. | |
| Participants |
Diagnosis: DSM‐IV MDD diagnosis N: 63 Age: esketamine M = 45.5 (SD=14.5); Ketamine = 48.7 (SD=15.1) Sex: esketamine = 55.8% female (N = 19); ketamine = 70.3% female (N = 19). Baseline depression severity: MADRS score esketamine M = 3.1 (SD = 9.3); MADRS score ketamine M = 32.9 (SD = 5.3) |
|
| Interventions | Participants were randomised on a 1:1 ratio into two groups of either ketamine (Clortamina®, BioChimico, 10 mL ampoules, 50 mg/mL and dose: 0.5 mg/kg) or esketamine (Ketamin®, Cristália, 2 mL ampoules, 50 mg/mL and dose: 0.25 mg/kg). Both drugs were diluted in 100ml saline and administered intravenously over 40 minutes. | |
| Outcomes | MADRS Response Rate Adverse events CADSS |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Participants were randomized on a 1:1 ratio into two groups of either ketamine (Clortamina®, BioChimico, 10 mL ampoules, 50 mg/mL and dose: 0.5 mg/kg) or esketamine (Ketamin®, Cristália, 2 mL ampoules, 50 mg/mL and dose: 0.25 mg/kg, through an electronic randomization platform (http://www.randomizer.org) (Urbaniak and Plous, 2013)." |
| Allocation concealment (selection bias) | Low risk | Quote: "Only the single investigator, who was responsible for both centres’ randomization and allocation processes, and the nurse responsible for drug preparation, were aware of the drug being infused." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding stated but not tested |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All assessments were conducted by investigators blind to treatment allocation.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers included same as numbers in outcome data. |
| Selective reporting (reporting bias) | Low risk | Protocol expected outcomes are reported as planned |
| Other bias | Low risk | None identified. |
Daly 2018.
| Study characteristics | ||
| Methods | Double‐blind, doubly randomised, delayed‐start, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder
N: 67 randomised Age: M = 44.7 (SD = 10.0) Sex: 56.72% female Baseline depression severity: placebo group MADRS score M = 29.3 (SD = 5.79); esketamine 28 mg group MADRS score M = 31.3 (SD = 7.09); esketamine 56 mg group MADRS score M = 34.9 (SD = 6.13); esketamine 84mg group MADRS score M = 30.4 (SD = 4.67) |
|
| Interventions | Participants were randomised 3:1:1:1 to receive intranasal placebo, esketamine 28 mg, esketamine 56 mg, or esketamine 84mg twice weekly for 1 week. Participants still experiencing moderate‐severe symptoms were then re‐randomised for 1 further week. Concomitant treament: yes, the antidepressant that participants had been receiving immediately before study entry was continued unchanged. |
|
| Outcomes | MADRS QIDS‐SR Remission rates (MADRS total score ≤10) Response rates (≥50% improvement from baseline from total MADRS score) |
|
| Notes | Only data from the first week of treatment used since participants were re‐randomised. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "At the beginning of the double‐blind period 1, eligible participants were randomized (3:1:1:1) to intranasal placebo or esketamine 28, 56, or 84mg, twice weekly based on 2 computer‐generated randomization schedules (periods 1 and 2). Randomization was balanced by using randomly permuted blocks and stratified by study center. At the end of period 1, those randomized placebo who had moderate to severe symptoms...were ranomized (1:1:1:1) to intranasal esketamine 28, 56, or 84mg or placebo twice weekly." |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details given on blinding beyond quote, "double‐blind, doubly randomized study". Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given on blinding beyond quote, "double‐blind, doubly randomized study". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Exclusion and withdrawal rates at each time point are recorded |
| Selective reporting (reporting bias) | High risk | Protocol unavailable. Additional material quotes another study arm (esketamine 14 mg) which is not reported in this study. |
| Other bias | High risk | This study was funded by Janssen Research & Development, LLC. Drs Daly, Singh, Fedgchin, Cooper, Lim, Van Nueten, Manji, and Drevets are employees of Janssen Research & Development, LLC and hold company stock/stock options. Dr Manji holds a patent, which is assigned to Icahn School of Medicine at Mount Sinai, Yale University, and the National Institutes of Health; no financial benefit was received from this patent. |
Downey 2016.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐groups trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode
N: 60 (randomised) Age: lanicemine M = 26.7; ketamine M = 27.1; Placebo M = 25.7 Sex: lanicemine group 60% female; ketamine 61.9% female; Placebo group 57.9% female Baseline depression severity: lanicemine group BDI score M = 34.5 (SD = 9.3); Ketamine group BDI score M = 30.94 (SD = 7.1); Placebo group BDI score M = 25.72 (SD = 7.67) |
|
| Interventions | Participants received a constant intravenous infusion of lanicemine (100 mg total dose), ketamine (0.5 mg/kg total dose) or placebo (0.9% saline) each made up to 40 mL volume and infused for 60 minutes. Concomitant treatment: quote: "Participants were drug‐free but not drug naive" |
|
| Outcomes | BDI MADRS CADSS BOLD phMRI signal in the sgACC |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "participants were randomized". No further details given on randomisation procedure. |
| Allocation concealment (selection bias) | Low risk | Quote: "The research pharmacies at each site held separate randomization codes and dispensed the infusions on the day of the experiment labelled with a code number". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The research pharmacies at each site held separate randomization codes and dispensed the infusions on the day of the experiment labelled with a code number". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information given. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study reports dropouts (zero). |
| Selective reporting (reporting bias) | Unclear risk | No information given. |
| Other bias | High risk | Potential bias due to funding by pharmaceutical company. Quote: "This study was wholly sponsored by AstraZeneca". |
Fava 2018.
| Study characteristics | ||
| Methods | Double‐blind placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder, confirmed by the Structured Clinical Interview for DSM‐IV‐Patient Edition (SCID‐I/P) N: 99 Age: ketamine 0.1 mg/kg group M = 43.1 (SD = 11.9); ketamine 0.2 mg/kg group M = 45.5 (SD = 14.6); ketamine 0.5 mg/kg group M = 48.6 (SD = 12.9); ketamine 1.0 mg/kg group M = 47.4 (SD = 10.1); midazolam group M = 45.6 (SD = 13.8) Sex: ketamine 0.1 mg/kg group 55.6% female; ketamine 0.2 mg/kg group 45% female; ketamine 0.5 mg/kg group 50% female; ketamine 1.0 mg/kg group 40% female; midazolam group 57.9% female Baseline depression severity: ketamine 0.1 mg/kg group HAM‐D‐6 M = 12.6 (SD = 1.8); ketamine 0.2 mg/kg group HAM‐D‐6 M = 12.8 (SD = 2.5); ketamine 0.5 mg/kg group HAM‐D‐6 M = 12.6 (SD = 1.5); ketamine 1.0 mg/kg group HAM‐D‐6 13.1 (SD 2.3); midazolam group HAM‐D‐6 M = 13.1 (SD = 2.3) |
|
| Interventions | Five arms in a 1:1:1:1:1 fashion: a single intravenous dose of ketamine 0.1 mg/kg (n = 18), a single dose of ketamine 0.2 mg/kg (n = 20), a single dose of ketamine 0.5 mg/kg (n = 22), a single dose of ketamine 1.0 mg/kg (n = 20), and a single dose of midazolam 0.045 mg/kg (active placebo) (n = 19). Concomitant medications: patients on exclusionary concomitant psychotropic medications (e.g. opioids, tramadol, valproic acid, lamotrigine, carbamazepine, barbiturates, eszopiclone, stimulants, NMDA receptor antagonists such as memantine) were included only if they had been free of the exclusionary medication post‐taper for five half lives within the maximum screening period (28 days). |
|
| Outcomes | HAM‐D‐6 MADRS CGI‐S CGI‐I SDQ PAS |
|
| Notes | Authors kindly provided additional data which has been used in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomly assigned to one of five 40 min infusion arms in a 1:1:1:1:1 fashion. Prior to randomization, patients were grouped by body mass index (BMI) (group I: BMI ≤ 30; group II: BMI > 30), and were block randomized into each arm of the study, with the mg/kg ratio being maintained across all BMIs." |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomly assigned to one of five 40 min infusion arms in a 1:1:1:1:1 fashion. Prior to randomization, patients were grouped by body mass index (BMI) (group I: BMI ≤ 30; group II: BMI > 30), and were block randomized into each arm of the study, with the mg/kg ratio being maintained across all BMIs." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding tested and largely uneffective: quote: "Regarding unblinding, both clinicians’ and participants’ guesses of treatment assignment were significantly related to actual treatment group (p < 0.01 for both), where both groups were able to correctly guess assignment to ketamine for the 0.5 mg/kg (100% and 77% guessed correctly by clinicians and participants, respectively) and the 1.0 mg/kg (95% correctly guessed by both) ketamine doses, but not for the 0.1 mg/kg (50%, 56%, respectively) and 0.2 mg/kg doses (55%, 45%, respectively). Assignment to placebo was guessed correctly 42% by clinicians and 37% by participants." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding tested and largely uneffective, which may have impacted upon outcome assessments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | CONSORT diagram published (p3) |
| Selective reporting (reporting bias) | Low risk | Protocol available online, outcomes reported as expected. |
| Other bias | High risk | Extensive links with pharmaceutical industry creates potential for bias. |
Fedgchin 2019.
| Study characteristics | ||
| Methods | Phase 3 randomised, double‐blind, active‐controlled multicentre trial | |
| Participants |
Diagnosis: DSM‐5 single episode or recurrent major depressive disorder without psychotic features
N: 346 randomised (342 included in analysis) Age: esketamine 56 mg = 46.4 (SD=11.8); esketamine 84 mg = 45.7 (SD = 11.10); Placebo = 46.8 (SD=11.36). Sex: esketamine 56 mg group 70.4% female ; esketamine 84 mg 69.3% female; Placebo = 71.7% female. Baseline depression severity: esketamine 56 mg group MADRS 37.4 (SD = 4.76); esketamine 84 mg group MADRS 37.8 (SD = 5.58); Placebo group MADRS 37.5 (SD = 6.16). |
|
| Interventions | Participants were randomised to receive the following intranasal treatments twice weekly for four weeks: Esketamine 56 mg Esketamine 84 mg Placebo nasal spray (with bittering agent to simulate the taste of esketamine solution. Participants also initiated one of the following oral antidepressants to take alongside the randomised treatment: Duloxetine Escitalopram Sertraline Venlafaxine Extended Release (XR) Concomitant treatment: Yes |
|
| Outcomes | MADRS CADSS CGADR CGI‐S PHQ‐9 Response (≥50% improvement on MADRS from baseline) Remission (MADRS total score ≤12) |
|
| Notes | Authors kindly provided additional data for remission rates according to this review's definition and MADRS mean scores | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were randomised to interventions in a 1:1:1 sequence. |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was balanced by using randomly permuted blocks and stratified by country and class of oral antidepressant" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | MADRS assessments were completed by independent, blinded, remote raters, however not tested. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Higher rates of withdrawal and AEs in treatment group |
| Selective reporting (reporting bias) | Low risk | Trial registered (NCT02417064) outcomes reported as expected. |
| Other bias | High risk | Authors are associated with and receive payments from pharmaceutical companies. |
Fernie 2017.
| Study characteristics | ||
| Methods | Double‐blind, parallel‐design, randomised controlled trial | |
| Participants |
Diagnosis: major depression
N: 40 Age: ketamine group M = 51.76 (SD = 9.97); propofol group M = 49.88 (SD = 12.53). Sex: ketamine group 55% female ; propofol group 55% female. Baseline depression severity: ketamine group HRSD M = 27.19 (SD = 6.47); Propofol group HRSD M = 24.79 (SD = 8.50). |
|
| Interventions | Patients receiving ECT on an informal basis were randomised to either ketamine (up to 2 mg/kg) or propofol (up to 2.5 mg/kg) intravenous (bolus) as an anesthetic before ECT. Bilateral ECT was performed twice a week until their consultant psychiatrist decided to end their treatment or treatment was stopped for medical reasons. | |
| Outcomes | HRSD MADRS CANTAB SRM |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was performed over the internet or telephone using an independent randomisation service (CHaRT, University of Aberdeen) by the principal investigator (I.C.R.) or ECT nurses and recorded in medical notes as drug A or B". |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomisation was performed over the internet or telephone using an independent randomisation service (CHaRT, University of Aberdeen) by the principal investigator (I.C.R.) or ECT nurses and recorded in medical notes as drug A or B". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients, their treating medical teams and all researchers making assessments were masked to the anaesthetic allocation". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients, their treating medical teams and all researchers making assessments were masked to the anaesthetic allocation". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High rate of withdrawal (14 patients). Five of these patients withdrew prior to commencement of ECT and before post‐ECT followups. Four of these were allocated to receive ketamine. |
| Selective reporting (reporting bias) | High risk | Protocol registration available online, however changes to planned analysis had to be made due to high withdrawal rate. |
| Other bias | Low risk | None identified. |
Fu 2020.
| Study characteristics | ||
| Methods | Phase 3 double‐ blind, randomised, placebo‐controlled, multi‐centre study | |
| Participants |
Diagnosis: DSM‐5 Major depressive disorder and active suicidal ideation with intent
N: 226 Age: esketamine group M = 40.8 (SD = 13.17); placebo group M = 37.9 (SD = 12.54). Sex: esketamine group 68.8% female ; placebo group 66.1% female. Baseline depression severity: esketamine group MADRS M = 41.3 (SD = 5.87); Placebo group MADRS M = 41.0 (SD = 6.29). |
|
| Interventions | Patients were randomised to receive 84 mg esketamine nasal spray or matching placebo nasal spray administered twice weekly for 4 weeks. Concomitant medications: standard‐of‐care oral antidepressant(s) treatment was initiated or optimised for participants in both groups at the time of randomisation. |
|
| Outcomes | MADRS CGI‐SS‐r Remission Response Adverse events |
|
| Notes | Authors kindly provided additional data for remission rates according to this review's definition, MADRS and CGI‐SS‐r scores. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Eligible patients were randomized (1:1), based on a computer‐generated randomization scheduled”. |
| Allocation concealment (selection bias) | Low risk | Quote:“Eligible patients were randomized (1:1), based on a computer‐generated randomization scheduled...Randomization was balanced using randomly permuted blocks and stratified by study center and type of standard‐of‐care antidepressant”. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not tested. Likely unblinding of participants due to known dissociative effects of esketamine. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Participant attrition reported in supplementary figure 2. Withdrawals explained and balanced across groups. |
| Selective reporting (reporting bias) | Low risk | Trial registration available (NCT03039192). Outcomes reported as planned |
| Other bias | High risk | Funded by pharmaceutical company whom authors are employed by. |
Gálvez 2018.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled pilot study | |
| Participants |
Diagnosis: DSM‐5 unipolar major depressive episode (1 bipolar disorder, depressed)
N: 5 Mean age:ketamine 100 mg group M = 58.7; active control (midazolam 4.5 mg) group M = 50 Sex: ketamine 100 mg group 66.7% female; active control (midazolam 4.5 mg) group 0% female Baseline depression severity: unclear |
|
| Interventions | Participants self‐administered intranasal sprays containing either ketamine or an active control (three times a week for 2 weeks, then weekly for 2 weeks). All treatment sessions were observed, supervised and documented by research staff. Ketamine 100 mg group (N = 3) Active control (midazolam 4.5 mg) group (N = 2) Concomitant treatments: yes, however no changes in medication dosage were permitted for 4 weeks prior to the study entry or during the study. |
|
| Outcomes | CADSS PRISE CogState computerised‐battery MADRS HAM‐A CGI‐S SF‐12 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was conducted using permuted blocks via a computer‐generated block randomisation sequence." |
| Allocation concealment (selection bias) | Unclear risk | No more information beyond, quote: "Treatment allocation was sequential". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study is reported to be "double‐blind". No more information beyond, "All personnel involved in the study (treaters, raters, study psychiatrists) were blinded to the randomisation sequence, with the exception of the trial statistician and the trial pharmacist." Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No more information beyond, quote: "All personnel involved in the study (treaters, raters, study psychiatrists) were blinded to the randomisation sequence, with the exception of the trial statistician and the trial pharmacist." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts and explains reason for early termination of the study. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Low risk | No other potential sources of bias identified |
Ghasemi 2013.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode (1 bipolar disorder, depressed)
N: 18 Age: ketamine group M = 35.22 (SD = 13.63); ECT group M =40 (SD = 16.41) Sex: ketamine group 56% female; ECT group 56% female Baseline depression severity: ketamine group HRSD = 30.22 (SD = 5.78); ECT group HRSD = 35.88 (SD = 6.47) |
|
| Interventions | 1 week of treatment Ketamine (N = 9) 0.5 mg/kg over 45 minutes, 1 IV infusion every 48 hours ECT (N = 9) 3 bilateral ECT sessions every 48 hours. During each ECT procedure, patients were administered 0.5 mg atropine followed by 2–3 mg/kg thiopental intravenously(IV); succinylcholine (0.5 mg/kg) was administered as a muscle relaxant after the induction of anaesthesia Concomitant treatment: yes, patients continued existing treatments for depression |
|
| Outcomes | BDI HRSD Response rates (≥ 50% reduction in HRSD scores) Adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given on randomisation procedure beyond ''randomly assigned'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Treatment team members, including physicians and psychologists conducting the rating scales, were blinded to the treatment group except for the anesthesiologist'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''Treatment team members, including physicians and psychologists conducting the rating scales, were blinded to the treatment group except for the anesthesiologist'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts (zero) |
| Selective reporting (reporting bias) | Low risk | Protocol unavailable, but means and SDs of all measures specified in methods section reported at all time points |
| Other bias | Low risk | No other potential sources of bias identified |
Grunebaum 2018.
| Study characteristics | ||
| Methods | Randomised, double‐blind, controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode
N: 80 Age: ketamine 0.5 mg/kg group M = 38.4 (SD = 13.2); midazolam 0.02 mg/kg group M = 40.7 (SD = 13.1) Sex: ketamine 0.5 mg/kg group 55% female; midazolam 0.02 mg/kg group 65% female Baseline depression severity: ketamine 0.5 mg/kg group HAM‐D score M = 22.2 (SD = 4.6); midazolam 0.02 mg/kg group HAM‐D score M = 22.6 (SD = 3.9) |
|
| Interventions | Participants were given intravenous racemic ketamine hydrochloride at 0.5 mg/kg or midazolam at 0.02 mg/kg in 100 mL normal saline infused over 40 minutes. Ketamine 0.5 mg/kg group (N = 40) Midazolam 0.02 mg/kg group (N = 40) Concomitant treatment: yes, participants were allowed to continue on stable dosages of current psychiatric medications, except that benzodiazepines could not be taken within 24 hours before the infusion. |
|
| Outcomes | HAM‐D SSI BDI Profile of Mood States (POMS) CADSS BPRS |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A permuted, blocked design was used, with 1:1 assignment between treatments and block size randomized between 4 and 6 with equal probability. Randomization was stratified on two baseline factors" |
| Allocation concealment (selection bias) | Unclear risk | No information given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No further information given beyond, quote: "Patients and study personnel were blinded to treatment." Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further information given beyond, quote:"Patients and study personnel were blinded to treatment." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal and drop out rates reported. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Low risk | No other potential sources of bias identified |
Heresco‐Levy 2006.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; ≥ 4 weeks of treatment with an antidepressant drug; score ≥ 18 on HRSD‐21
N: 22 Age: M = 56.9 (SD = 12.4) Sex: 45% female Baseline depression severity: HRSD = 27.2 (SD = 5.2) |
|
| Interventions | 6 weeks of treatment D‐cycloserine (N = 9) 250 mg orally per day Placebo (N= 13) in identical capsules to D‐cycloserine Concomitant treatment: y es, patients continued on existing psychotropic medications |
|
| Outcomes | HRSD HAMA Zung Self Rating Depression Scale PANSS UKU Side Effects Rating Scale | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''subjects were randomly allocated, without blocking, stratification or other restrictions'' no further details given |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Clinical and research staff, patients and their families were unaware of and could not determine the study drug assignment by appearance or otherwise'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''double blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available, unclear if all prespecified outcomes reported in published report |
| Other bias | Low risk | No other potential sources of bias identified |
Heresco‐Levy 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; insufficient therapeutic response during the current episode, defined as a ≥ 20 score on the HRSD‐21 despite two or more adequate antidepressant medication trials
N: 26 Age: M = 53.0 (SD = 10.2) Sex: 62% female Baseline depression severity: D‐cycloserine group HRSD = 25.1 (SD = 5.6); placebo group HRSD = 27.2 (SD = 4.9) |
|
| Interventions | 6 weeks of treatment D‐cycloserine (N = 13) gradually titrated: 250 mg (one capsule)/d for 3 days ‐> 500 mg (two capsules)/d for 18 days ‐> 750 mg (three capsules)/day for 1 week ‐> and 1000 mg (four capsules)/d for the last 2 weeks Placebo (N = 13) in identical capsules to D‐cycloserine and according to same dose escalation schedule Concomitant treatment: yes, patients continued on existing psychotropic medications |
|
| Outcomes | HRSD
Response rate (≥ 50% reduction in HRSD scores) Remission rate (HRSD score ≤ 7) HAMA CGI BDI Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''randomly allocated using a block size of four'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Clinical and research staff, patients and their families were unaware of and could not determine the study drug assignment by appearance or otherwise'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''double blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable, but data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Hu 2016.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder
N: 30 randomised (27 completed the trial) Age: escitalopram and placebo group M = 41.0 (SD = 11.1); escitalopram andIV ketamine group M = 36.7 (SD = 14.0) Sex: escitalopram and placebo group 71.4% female; escitalopram and IV ketamine group 53.8% female Baseline depression severity: escitalopram and placebo group MADRS score M = 32.3 (SD = 6.5); escitalopram and IV ketamine group MADRS score M = 36.5 (SD = 7.8) |
|
| Interventions | Patients meeting entry criteria entered a 2‐week wash‐out phase of previously taken psychtropic medications (fluoxetine = 4 weeks). Patients were then randomised to 4 weeks of fixed‐dose escitalopram10 mg/day plus a single saline solution infusion (placebo) or fixed dose escitalopram 10 mg/day plus a single sub‐anaesthetic dose of i.v. ketamine hydrochloride administered over 40minutes. Escitalopram and placebo group (N = 14) Escitalopram andIV ketamine group (N = 13) Concomitant treatments: yes, other medications not affecting the central nervous system were allowed. |
|
| Outcomes | MADRS Response rates (≥50% reduction from the baseline MADRS) Remitter rates (MADRS total score ≤10) QIDS‐SR BPRS YMRS CADSS |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No further information beyond, "patients were randomized according to a table of random numbers". |
| Allocation concealment (selection bias) | Unclear risk | No information given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information given beyond, "The anesthesiologist was blind to the group membership of patients". No direct information regarding blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Two raters with >5 years of clinical experience and blind to the study protocol and treatment assignments indepently assessed patients" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study reports dropouts. Dropout last assessment carried forward. |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | Funded by University. No conflict of interests. |
Huang 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score ≥ 18 score on HRSD‐17
N: 40 Age: sarcosine group M = 37.2 (SD = 11.3); citalopram group M = 35.7 (SD = 9.5) Sex: sarcosine group 30% female; citalopram group 45% female Baseline depression severity: Sarcosine group HRSD = 23.7 (SD = 5.8); citalopram group HRSD = 24.5 (SD = 5.3) |
|
| Interventions | 6 weeks of treatment. The dose was initiated at 1 capsule per day in the first 2 weeks, and titrated to 1 capsuletwice daily in weeks 3–4, and 1 capsule in the morning and 2 capsules before sleep in weeks 5–6 if clinically indicated Sarcosine (N = 20) 500 mg capsules Citalopram (N = 20) 20 mg capsules Concomitant treatment: no, patients had to be at drug‐free for over 3 months to enter trial |
|
| Outcomes | HRSD
GAF
Remission (score < 7 on HRSD)
Response rate (≥ 50% reduction in HRSD scores) CGI‐S Adverse events |
|
| Notes | No other potential sources of bias identified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Patients were randomly assigned in blocks of six subjects to receive citalopram or sarcosine in a 1:1 ratio'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''Medication was provided in coded containers'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Patients, caregivers, and investigators (except the investigational pharmacist) were all masked to the assignment. Medication was provided in coded containers'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''investigators...were all masked to the assignment'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout rate at each time point is recorded |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Ibrahim 2012a.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score ≥ 22 score on MADRS; an inadequate response to at least two therapeutic trials of an antidepressant
N: 42 Age: riluzole group M = 47.2 (SD = 13.3); placebo group M = 47.2 (SD = 13.0) Sex: riluzole group 38% female; placebo group 38% female Baseline depression severity: riluzole group MADRS = 32.7 (SD = 3.7); placebo group MADRS = 32.7 (SD = 5.7) |
|
| Interventions | 4 weeks of treatment All patients first received a single open‐label IV infusion of 0.5 mg/kg of ketamine hydrochloride over 40 minutes, then were randomised to riluzole or placebo Riluzole (N = 21) dose titrated; initiated and maintained at 100 mg/day (50 mg twice daily). Dose could be flexibly increased in increments of 50 mg to a maximum of 200 mg/day Dose escalations continued on a weekly basis until treatment‐limiting side effects or completion of the study Placbeo (N = 21) daily capsules Concomitant treatment: No, patients had a 2‐week drug‐free period before commencing treatment |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) HRSD BDI VAS HAMA BPRS CADSS YMRS SSI Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given on randomisation procedure other than quote: ''patients were randomised'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''All study investigators, staff, and patients were blind to riluzole or placebo assignment'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''All study investigators, staff, and patients were blind to riluzole or placebo assignment'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given on blinding of outcome assessment other than 'quote:'double blind'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | n reported alongside means at each time point in data provided by author through correspondence |
| Selective reporting (reporting bias) | High risk | Protocol unavailable and only one of 8 secondary measures reported |
| Other bias | Low risk | No other potential sources of bias identified |
Ibrahim 2012b.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score ≥22 score on MADRS; an inadequate response to at least two therapeutic trials of an antidepressant
N: 5 Age: not stated Sex: not stated Baseline depression severity: not stated |
|
| Interventions | 12 days treatment MK‐0657 (N = 3) 1 mg capsules, 8 daily in the morning. Dose titrated; initial dose 4 mg/day, increased every 4 days by 2 mg/day until treatment‐limiting side effects or completion of the study. 8 mg/day was treatment target for all participants unless tolerability issues ensued Placebo (N = 2) capsules, same dose escalation as MK‐0657 Concomitant treatment: no, patients had a 2‐week drug‐free period before commencing treatment |
|
| Outcomes | MADRS Response rates (≥ 50% reduction in MADRS scores) Remission rates (< 10 on MADRS) HRSD BDI VAS HAMA BPRS CADSS YMRS SHAPS |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given on randomisation procedure other than quote:''randomised in a double‐blind manner'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment procedure |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''a double‐blind dummy design was used throughout the study'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given on blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Requested data reports dropout rates alongside each time point |
| Selective reporting (reporting bias) | Unclear risk | No results tables available in original publication. Data requested was provided when the author was contacted |
| Other bias | Low risk | No other potential sources of bias identified |
Ionescu 2018.
| Study characteristics | ||
| Methods | Randomised, double blind, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode
N: 26 Age: ketamine 45.5 (SD = 13.6); placebo 45.3 (SD = 11.7) Sex: ketamine 54% female; placebo 23% female Baseline depression severity: Ketamine HDRS = 31.6 (SD=5.2); Placebo 26.3 (SD=4.8) |
|
| Interventions | Six 45 minute intravenous infusions over three weeks (two infusions per week): Ketamine 0.5mg/kg (N=13) Placebo (saline) (N=13) Concomitant treatment: Participants were maintained on their stable outpatient medication regime prior to the start of the study and during infusion. |
|
| Outcomes | HDRS C‐SSRS SI score C‐SSRS SI intensity rating CADSS Response (≥50% improvement on HDRS) Remission (HDRS score ≤7) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Group allocation was completed by a computer generated randomization algorithm". |
| Allocation concealment (selection bias) | Low risk | Quote: "Group allocation was completed by a computer generated randomization algorithm". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "All clinicians, patients, and raters were blind to the randomization assignments". Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All clinicians, patients, and raters were blind to the randomization assignments...A study doctor administered assessments of depression and SI". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for screen failures and withdrawals were provided. |
| Selective reporting (reporting bias) | Unclear risk | Paper states "This study is registered on the ClinicalTrials.gov Registry with (http://www.clinicaltrials.gov; NCT01582945)", however this registration is for a previous open‐label study. |
| Other bias | High risk | Study funded by and authors employed by pharmaceutical company. |
Ionescu 2020.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled, multicentre study | |
| Participants |
Diagnosis: DSM‐5 major depressive disorder with suicidal ideation
N: 230 Age: esketamine M = 40.2 (SD=12.72); placebo M = 41.4 (SD = 13.43) Sex: esketamine 60.5% female; placebo 59.3% female Baseline depression severity: esketamine group MADRS M = 39.5 (SD = 5.19); placebo group MADRS M = 39.9 (SD = 5.76) |
|
| Interventions | Participants were randomised to receive either 84 mg esketamine nasal spray or matching placebo nasal spray twice weekly for 4 weeks. Concomitant medications: all participants received standard‐of‐care oral antidepressant(s) initiated or optimised at randomisation. |
|
| Outcomes | MADRS CGI‐SS‐r Response Remission Adverse events |
|
| Notes | Authors kindly provided additional data for remission rates according to this review's definition, MADRS and CGI‐SS‐r scores. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were randomized (1:1) to 84 mg esketamine nasal spray or matching placebo nasal spray according to a computer‐generated schedule". |
| Allocation concealment (selection bias) | Low risk | Quote: "Eligible patients were randomized (1:1) to 84 mg esketamine nasal spray or matching placebo nasal spray according to a computer‐generated schedule". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Different raters were used for efficacy and safety ratings, however it is likely patients were unblinded by dissociative side effects of esketamine. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Flow diagram of participants throughout study included. Participant withdrawal rate reported, similar between groups. |
| Selective reporting (reporting bias) | Low risk | Trial registered (NCT03097133), outcomes reported as expected. |
| Other bias | High risk | Study funded by and authors employed by pharmaceutical company. |
Jagtiani 2014.
| Study characteristics | ||
| Methods | Randomised, double‐blind, controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive episode
N: 60 Age: MECT and thiopentone 2.5mg/kg = 34.97 (SD = 8.17); MECT and ketamine 1mg/kg = 35.37 (SD = 8.97) Sex: MECT and thiopentone 2.5mg/kg group 43.3% female; MECT and ketamine 1mg/kg group 56.7% female Baseline depression severity: MECT and thiopentone 2.5mg/kg group HAMD17 = 32.00 (SD = 7.60); MECT and ketamine 1mg/kg group HAMD17 = 29.53 (SD = 4.56) |
|
| Interventions | MECT was given three times a week: MECT and thiopentone 2.5 mg/kg (N = 30) MECT and ketamine 1 mg/kg (N = 30) Concomitant treatment: yes, succinylcholine (0.5 mg/kg) was given intravenously as muscle relaxant after induction of anaesthesia. The participants in both groups received antidepressant medications randomly as decided by the treating psychiatrists who were not concerned with the study. |
|
| Outcomes | HAMD17 BDI MMSE Side effects |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patient’s group was decided by picking up a chit randomly from the container by the nurse on ECT duty." |
| Allocation concealment (selection bias) | Low risk | Quote: "Each patient was allotted randomly to either group ‘A’ or ‘B’ by the nursing assistant, who was not involved in the study, for deciding the choice of anaesthetic drug" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The researchers (AJ and HK), and the patients were blind to the choice anaesthetic drug done by anaesthetist (NM)." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The researchers (AJ and HK) and the patients were blind to the choice anesthetic drug done by anesthetist (NM). The choice of anesthetic drug was revealed only after the data analysis was complete." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts (zero) “None of the patients withdrew from the study” Participant flow diagram provided for numbers enrolled, excluded and refused to participate. [Figure 1]. |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable No protocol found – trial registration number not in paper. |
| Other bias | Low risk | No other potential sources of bias identified. |
Jarventausta 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV severe or psychotic major depressive disorder; an inadequate response to at least two therapeutic trials of an antidepressant Diagnosis of psychotic depression: S‐ketamine group = 31.3%; saline group = 31.3% N: 32 Age: S‐ketamine group M = 48.8 (range = 23‐81); saline group M = 53.7 (range 24‐81) Sex: S‐ketamine group 50% female; saline group 68.7% female Baseline depression severity: S‐ketamine group MADRS = 36.9 (range = 31‐50); saline group MADRS = 37.3 (range = 27‐49) |
|
| Interventions | S‐ketamine (N = 16) 0.4 mg/kg as a bolus Saline (N = 16) Following S‐ketamine or saline, all patients received an initial bolus of 0.5 mg/kg propofol, then given using a dose‐titration (mean dose = 99.5 mg/kg) Patients received ECT sessions three times a week until they reached remission (MADRS score ≤ 7) or no further symptom reduction was achieved during the last 2 ECT sessions Concomitant treatment: Yes, patients remained on existing psychotropic medication |
|
| Outcomes | MADRS Response rates (≥ 50% reduction in MADRS scores) Remission rates (≤ 7 on MADRS) BDI |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given on randomisation procedure, simply ''randomized'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment procedure |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details given on blinding of participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''all the rating were done blindly to S‐ketamine by experienced nurses'' |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Requested data to authors (waiting for a reply) |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Requested data to authors (waiting for a reply) |
| Other bias | Low risk | No other potential sources of bias identified |
Kuşçu 2015.
| Study characteristics | ||
| Methods | Randomised control trial | |
| Participants |
Diagnosis: DSM‐IV major depression
N: 58 Age: thiopental 4 mg/kg M = 42.7 (SD = 15.8); ketamine 1 mg/kg M = 44.8 (SD = 11.6); ketamine 1 mg/kg and thiopental 4 mg/kg M = 38.6 (SD = 6.8) Sex: thiopental 4 mg/kg group 45% female; ketamine 1 mg/kg group 47.4% female; ketamine 1 mg/kg and thiopental 4 mg/kg group 55.6% female Baseline depression severity: thiopental 4 mg/kg group HDRS score M = 17.6 (SD = 4.9); ketamine 1mg/kg group HDRS score M = 20.0 (SD = 4.0); ketamine 1 mg/kg and thiopental 4 mg/kg group HDRS score M = 19.7 (SD = 4.3) |
|
| Interventions | All patients received 8 session of ECT as bilateral, bitemporal, 3 times a week. Depending on randomised group assignment, patients were intravenously administered thiopental (4 mg/kg), ketamine (1 mg/kg), or thiopental (4 mg/kg) and ketamine (1 mg/kg) prior to ECT. | |
| Outcomes | HDRS Response rates (50% decrease in HDRS scores) HAM‐A Systolic artery blood pressure Diastolic artery blood pressure Heart rate (HR) Peripheral oxygen sauration (SpO2) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly divided into three groups according to the anaesthesia used." No more information given. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The patients were randomly divided into three groups according to the anaesthesia used." No more information given. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. Quote: "information about the method to be applied was given to the patients and their family." Quote: "ECT operation was conducted by a psychiatrist who did not know which anaesthesia method was applied". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not tested. Quote: "ECT operation was conducted by a psychiatrist who did not know which anaesthesia method was applied". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts (N = 3) |
| Selective reporting (reporting bias) | High risk | Protocol unavailable. Number of participants differs across tables with no explanation. |
| Other bias | Low risk | No other potential sources of bias identified |
Lapidus 2014.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; IDS‐C score ≥ 30
N: 20 Age: M = 48.0 (SD = 12.8) Sex: 50% female Baseline depression severity: IDS‐C = 42.7 (SD = 8.5) |
|
| Interventions | 1 single intranasal infusion Ketamine (N = 10) 50 mg Placebo (N = 10) Study drug or placebo provided in identical syringes containing clear solutions of either 100 mg/mL ketamine in.9% saline or saline alone. 5 intranasal applications of solution separated by 5 minutes Concomitant treatment: Yes, patients continued on existing psychotropic medications |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) QIDS‐SR HAMA BPRS CADSS YMRS SAFTEE Adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''The order of treatment periods was randomly assigned by the research pharmacy using permuted blocks of size four'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''The order of treatment periods was randomly assigned by the research pharmacy using permuted blocks of size four'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''All study investigators, anesthesiologists, and raters were blind to treatment assignment. Study drug or placebo was provided in identical syringes, containing clear solutions of either 100 mg/mL ketamine in.9% saline or saline alone'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''Raters were blind to treatment assignment'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Li 2016.
| Study characteristics | ||
| Methods | Double‐blind, randomised, controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR Major Depressive Disorder
N: 48 Age: ketamine 0.2 mg/kg group M = 44.4 (SD = 10.8); ketamine 0.5 mg/kg group M = 43.3 (SD = 11.9); saline group M = 49.9 (SD = 8.1) Sex: ketamine 0.2 mg/kg group 68.75% female ; ketamine 0.5 mg/kg group 68.75% female; saline group 81.25% female Baseline depression severity: ketamine 0.2 mg/kg group HDRS‐17 M = 20.9 (SD = 5.6); ketamine 0.5 mg/kg group HDRS‐17 M = 22.6 (SD = 5.8); saline group HDRS‐17 M = 22.8 (SD = 3.9) |
|
| Interventions | Patients were randomised to receive an intravenous infusion of either ketamine 0.2 mg/kg, ketamine 0.5 mg/kg, or normal saline placebo. | |
| Outcomes | HDRS‐17 MRI F‐FDG PET scans BPRS (positive symptoms subscale) Response (≥50% reduction in HDRS‐17 score from baseline) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomized in a 1:1:1/A:B:C ratio" |
| Allocation concealment (selection bias) | Low risk | Quote: "An independent research nurse who was not involved in the study was responsible for the random allocation and was not allowed to release any information to others, including the study nurse who applied the intravenous injection". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The entire study procedure was double‐blinded, and participants and staff were all blind to the treatment assignment". Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals reported ‐ all participants completed the entire study (no dropouts) |
| Selective reporting (reporting bias) | High risk | Trial registration available online (UMIN000016985). Outcomes differ from register, HDRS‐17 used in trial instead of MADRS. |
| Other bias | Low risk | None identified. |
Loo 2012.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode (9 bipolar disorder, depressed)
N: 46 Age: ketamine group = 45.2 (SD = 15.6); placebo group = 41.4 (SD = 12.0) Sex: ketamine group 50% female; placebo group 71% female Baseline depression severity: ketamine group MADRS = 32.1 (SD = 4.5); placebo group MADRS = 32.7 (SD = 7.9) |
|
| Interventions | All patients first received ECT 3 times a week. After induction of unconsciousness with IV thiopentone (3‐5mg/kg), patients randomly assigned to ketamine or placebo Ketamine (N = 26) 0.5 mg/kg IV Placebo (N = 25) saline The same procedure was followed for all ECT treatment sessions. A mean dose of 40.2 mg/kg (SD = 8.0) was delivered in the ketamine group Concomitant treatment: yes, 19 patients remained on medications during ECT and 3 commenced new medication in 2 weeks prior to beginning ECT |
|
| Outcomes | MADRS
Response rate (not defined) Remission rate (not defined) Adverse events Neuropsychological outcomes |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Participants were randomly assigned (by computer‐generated random number sequence)'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''Participants and clinical staff other than the ECT anaesthetist were masked to treatment condition'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Participants and clinical staff other than the ECT anaesthetist were masked to treatment condition... The integrity of blinding was not formally assessed but there were no indications that participants or raters were aware of the treatment assignment (as reported during ratings interviews), apart from one participant who was withdrawn because she was inadvertently unblinded." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given on blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not all participants followed up at 1 week post‐ECT. Dropout rates provided in published article do not match up with those provided by author |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable, but all outcomes fully reported |
| Other bias | Low risk | No other potential sources of bias identified |
Michelson 2007.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; HRSD‐17 score ≥ 18
N: 146 Age: atomoxetine group = 44.0 (SD = 12.3); placebo group = 45.5 (SD = 13.8) Sex: aAtomoxetine group 65% female; placebo group 66% female Baseline depression severity: atomoxetine group HRSD = 23.4 (SD = 3.5); placebo group MADRS = 23.1 (SD = 4.3) |
|
| Interventions | All patients first received 8 weeks of treatment with sertraline, initiated at 100 mg/day and increased in 50 mg increments to a maximum of 200 mg/day, based on efficacy and tolerability. Patients with a score > 4 on the Maier and Phillip core mood severity scale (MPS) of the HRSD were randomly assigned to 8 weeks of sertaline combined with atomoxetine or placebo Atomoxetine (N = 72) initiated at 40 mg/day, could be increased in 40 mg increments to a maximum of 120 mg/day, based on efficacy and tolerability Placebo (N = 74) The dose of sertraline during randomised treatment was fixed at 150 mg/day or 100 mg/day for patients who were unable to tolerate 150 mg/day during initial sertraline treatment Concomitant treatment: Unclear |
|
| Outcomes | Maier and Phillip core mood severity scale (MPS) of the HRSD HRSD CGI‐S Remission rate (MPS score ≤ 4 and no single item >1) Non response rate (< 30% reduction in MPS) Partial response rate (> 30% reduction in MPS but MPS > 4) Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No details given on random sequence generation, other than quote: ''were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No details given on random sequence generation, other than quote ''were randomly assigned under double‐blind conditions'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | To minimise rating bias, investigators and patients were blind to the symptom severity threshold for randomisation. To preserve this blinding, patients who met the response criteria (MPS ≤ 4 and no single item > 1) after the initial 8 weeks continued sertraline monotherapy in the randomised phase but were not included in the analyses of results from the randomised phase of the trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given on blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Responders data are per individual and only out of 26 and 28 rather than 72 and 74. Seems unclear |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other potential sources of bias identified |
Murrough 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; an inadequate response to at least three therapeutic trials of an antidepressant according to the criteria of the Antidepressant Treatment History Form; IDS‐C score ≥ 32
N: 73 Age: ketamine group M = 46.9 (SD = 12.8); midazolam group M = 42.7 (SD = 11.6) Sex: ketamine group 55% female; midazolam group 44% female Baseline depression severity: ketamine group MADRS = 32.6 (SD = 6.1); midazolam group MADRS = 31.1 (SD = 5.6) |
|
| Interventions | 1 single IV infusion Ketamine (N = 48) 0.5 mg/kg infused over 40 minutes Midazolam (N = 25) 0.045 mg/kg infused over 40 minutes Concomitant treatment: no, patients were free of other psychotropic medications with the exception of a stable dose of nonbenzodiazepine hypnotic |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) QIDS‐SR CGI‐I and CGI‐S PRISE CADSS BPRS Adverse events |
|
| Notes |
Secondary publication:Price 2014 Consistent with above characteristics, except for the following details: N: 57 Age: Ketamine group M = 48.6 (SD = 11.4); midazolam group M = 43.8 (SD = 10.9) Sex: Ketamine group 56% female; midazolam group 48% female Baseline depression severity: Ketamine group MADRS = 33.3 (SD = 5.6); midazolam group MADRS = 32.4 (SD = 4.8) Outcomes: BSS SI composite (sum of z‐scores on BSS, MADRS‐SI and QIDS‐SI) IAT‐ Death IAT‐ Escape |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Randomly assigned on a 2:1 ratio'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''The study research pharmacist prepared sealed envelopes that contained the drug identity..'' |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: ''...all other study personnel, including investigators, anesthesiologists, raters, patients, and data analysts, were masked to treatment assignment''. Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''Trained raters, who were not involved in the infusion‐day procedures and who were unaware of treatment group assignment and infusion‐related side effects, performed clinical assessments for the primary outcome at 24 hours and subsequent evaluations'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Nations 2012 (part I).
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score ≥ 9 but ≤ 20 on QIDS‐C
N: 24 Age: Org 26576 group M = 36.7 (SD = 10.3); placebo group M = 34.1 (SD = 12.7) Sex: Org 26576 group 31.2% female; placebo group 12.5% female Baseline depression severity: Org 26576 group QIDS‐C = 14.0 (SD = 2.8); placebo group QIDS‐C = 15.8 (SD = 2.0) |
|
| Interventions | 10‐16 days treatment, depending on titration schedule Org 26576 (N = 16) 100 mg to 600 mg twice daily with titration steps every 3 days. 4 sequential cohorts of 6 patients, cohorts A, B and C started at progressively higher doses, all rising to 600 mg twice daily; cohort D evaluated CSF pharmacokinetics at the lower end of the dosing range Placebo (N = 8) Indistinguishable capsules containing placebo, 50 mg or 100 mg Org 26576 Concomitant treatment: no, patients were taking no other psychotropic medication throughout study |
|
| Outcomes | MADRS
CGI‐S
CGI‐I
Response rate (≥ 50% reduction in MADRS scores)
Remission rate (MADRS score ≤ 10) Cognitive functioning and social acuity Neuroendocrine parameters and BDNF EEG Bioanalysis |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Eligible patients were randomized to receive Org 26576 or placebo via a centrally‐generated randomization list'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''Org 26576 and placebo were prepared as indistinguishable capsules. An unblinded on‐site pharmacist was responsible for preparing study medication according to the randomization list and for dispensing blinded medication to clinical staff for patient administration'' |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on blinding given, other than ''double‐blind'' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information on outcome assessment blinding given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Endpoint data uses LOCF, unsure when dropout occurred |
| Selective reporting (reporting bias) | Unclear risk | Data for all time points reported |
| Other bias | Low risk | No other potential sources of bias identified |
Nations 2012 (part II).
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score ≥ 9 but ≤ 20 on QIDS‐C
N: 30 Age: Org 26576 100 mg group M = 38.3 (SD = 14.4); Org 26576 400 mg group M = 35.6 (SD = 12.8); placebo group M = 31.1 (SD = 6.5) Sex: Org 26576 100 mg group 40% female; Org 26576 400 mg group 40% female; placebo group 50% female Baseline depression severity: Org 26576 100 mg group QIDS‐C = 15.0 (SD = 2.5); Org 26576 400 mg group QIDS‐C = 15.3 (SD = 1.1); placebo group QIDS‐C = 17.0 (SD = 1.9) |
|
| Interventions | 28 days treatment Org 26576 100 mg twice daily (N = 10) Org 26576 400 mg wice daily (N = 10) Placebo (N = 10) Indistinguishable capsules containing placebo, 50 or 100 mg Org 26576 Concomitant treatment: no, patients were taking no other psychotropic medication throughout study |
|
| Outcomes | MADRS
CGI‐S
CGI‐I
Response rate (≥ 50% reduction in MADRS scores)
Remission rate (MADRS score ≤ 10) Cognitive functioning and social acuity Neuroendocrine parameters and BDNF EEG Bioanalysis |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''Patients in Part II were randomized to receive Org 26576 100 mg BID, Org 26576 400 mg BID, or placebo in a 1:1:1 ratio'' |
| Allocation concealment (selection bias) | Unclear risk | No information on allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on blinding provided, other than 'double‐blind' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information on blinding of outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Endpoint data uses LOCF, unsure when dropout occurred |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data for all time points reported |
| Other bias | Low risk | No other potential sources of bias identified |
Ochs‐Ross 2020.
| Study characteristics | ||
| Methods | Randomised, double‐blind, active controlled, multicentre trial | |
| Participants |
Diagnosis: DSM‐5 single episode or recurrent major depressive disorder without psychotic features
N: 138 randomised (137 included in analysis) Age: esketamine = 70.6 (SD = 4.79); placebo = 69.4 (SD = 4.15). Sex: esketamine group 62.5% female; placebo = 61.5% female. Baseline depression severity: esketamine group MADRS 35.5 (SD = 5.91); Placebo group MADRS 34.8 (SD = 6.44). |
|
| Interventions | Patients were randomised to receive either flexibly dosed esketamine (28 mg, 56 mg, or 84 mg) plus a newly initiated antidepressant, or a placebo plus newly initiated antidepressant for four weeks. Concomitant treatment: Yes |
|
| Outcomes | MADRS CGI‐S Response (≥ 50% improvement on MADRS from baseline) Remission (MADRS total score ≤12) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were randomized in a 1:1 ratio to one of two treatments: ESK+AD (N=72) or AD+PBO (N=66). Randomization was stratified by country and class of oral AD (SNRI or SSRI)". |
| Allocation concealment (selection bias) | Low risk | Quote: "A computer‐generated randomization schedule was used to randomize patients (1:1)!". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "MADRS scores were obtained remotely by telephone by independent rates using a structured clinical interview guide (SIGMA) for additional (triple) blinding in this study. The raters had no knowledge of the patient's response to treatment including adverse or dissociative effects". Effectiveness of patient or personnel blinding not tested. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers of withdrawn participants and reasons for withdrawal listed. |
| Selective reporting (reporting bias) | Low risk | Trial registered (NCT02422186), outcomes reported as planned. |
| Other bias | High risk | Authors are associated with and receive payments from pharmaceutical companies. |
Omranifard 2014.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score > 17 on HRSD‐24
N: 60 Age: memantine group M = 67.5 (SD = 5.4); placebo group M = 68.9 (SD = 6.1) Sex: memantine group 61% female; placebo group 59% female Baseline depression severity: memantine group HRSD = 29.8 (SD = 7.0); placebo group HRSD = 28.8 (SD = 7.2) |
|
| Interventions | 8 weeks treatment Memantine (N = 28) dose titrated; 5 mg/day for first week, 5 mg twice daily for second week, 5 mg in the morning and 10 mg in evening for third week, 10 mg twice daily for fourth week. Target dose of 20 mg/day continued until eighth week Placebo (N = 29) one dose daily for first week, twice daily thereafter. Capsules same shape and taste as memantine Concomitant treatment: yes, all patients received citalopram 10 mg/daily initiation dose for first week and continued with 20 mg/daily for the rest of the study |
|
| Outcomes | HRSD
Response rate (≥ 50% reduction in HRSD scores) Remission rate (score < 7 on HRSD) Geriatric depression scale (GDS‐15) WHO‐QOL MMSE |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Selected patients were randomized into two groups: Intervention and placebo groups using permuted block design sampling with size of two for each block. Two consecutive patients were randomly allocated to the treatment and control groups in each step till to complete the sample size'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''Both the examiner and the patients were unaware of the component of the drugs and they used the drugs in the name of A and B. A questionnaire was filled secretly and every patient received a code for trial'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Both the examiner and the patients were unaware of the component of the drugs and they used the drugs in the name of A and B. A questionnaire was filled secretly and every patient received a code for trial'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''A questionnaire was filled secretly and every patient received a code for trial'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropout figures |
| Selective reporting (reporting bias) | High risk | Not all secondary outcome measures reported |
| Other bias | Low risk | No other potential sources of bias identified |
Popova 2019.
| Study characteristics | ||
| Methods | Randomised, double‐blind trial | |
| Participants |
Diagnosis: DSM‐5 single episode (≥ 2 years) or recurrent major depressive disorder without psychotic features confirmed by MINI; score ≥ 34 on the IDS‐C; treatment‐resistant depression (nonresponse to at least two antidepressants in the current episode).
N: 227 Age: esketamine group M = 44.9 (SD = 12.5); placebo group M = 46.4 (SD = 11.14) Sex: esketamine group 65.8% female; placebo group 57.8% female Baseline depression severity: esketamine group MADRS M = 37.0 (SD = 5.69); placebo group MADRS M = 37.3 (SD = 5.66) |
|
| Interventions | Participants were randomised to receive esketamine (56 mg or 84 mg) + oral antidepressant, or oral antidepressant + placebo (N = 109) twice weekly for 4 weeks. Esketamine or placebo was administered intranasally and combined with a newly initiated open‐label oral antidepressant administered daily. Concomitant treatment: All participants were assigned to a new open‐label oral antidepressant administered daily. |
|
| Outcomes | MADRS Response Remission C‐SSRS Adverse events |
|
| Notes | Authors kindly provided additional C‐SSRS data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “A computer‐generated randomization schedule was used to randomly assign eligible patients in a 1:1 ratio”. |
| Allocation concealment (selection bias) | Low risk | Quote: “Randomization was balanced by using randomly permuted blocks and was stratified by country and by class of oral antidepressant (serotonin‐norepinephrine reuptake inhibitor [SNRI] or selective serotonin reuptake inhibitor [SSRS]" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "To maintain blinding, a bittering agent was added to the intranasal placebo to simulate the taste of the esketamine solution, and three devices were administered to all patients at all sessions; in the esketamine plus antidepressant arm". "Because esketamine exhibits transient dissociative effects that are difficult to blind, possibly biasing the site staff supervising the dosing, all MADRS assessments (used for the primary endpoint, the first key secondary endpoint, and calculation of response and remission rates) were performed by independent, remote (by telephone) raters who were blind to the protocol details, including study visit, the patient’s clinical status, and side effects during the trial." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "While eventual efficacy bias was mitigated by using independent remote MADRS raters, it is possible that the specific adverse event profile of esketamine affected the blind for the study patients. Although patients were not specifically asked whether they believed they had received drug or placebo, it is noteworthy that the dissociation ratings (CADSS scores) increased in the control group who received placebo nasal spray, providing evidence of adequate blinding". But there was no assessment of whether blinding was effective or not and known side effects of ketamine may have unblinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Participant flow diagram reports attrition rates (figure S1 in online supplementary data) |
| Selective reporting (reporting bias) | Low risk | Trial registration available online (NCT02418585), outcomes reported as expected. |
| Other bias | High risk | Authors are employees of Janssen Research and Development, who supported the study financially. |
Preskorn 2008.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; score ≥ 18 on HRSD‐17; not benefited from at least 1 adequate trial of a SSRI
N: 30 Age: Ranged from 21‐54 Sex: 73% female Baseline depression severity: not stated |
|
| Interventions | All patients first received 6 weeks of treatment with paroxetine, starting at 20 mg/day and escalated to 40 mg/day within 1 week, and with a single‐blind IV placebo infusion after the third week. Patients who did not respond to paroxetine (< 20% improvement in HRSD score) were randomised to receive 1 single IV infusion of CP‐101, 606 or placebo, whilst continuing on paroxetine for 4 weeks CP‐101, 606 (N = 15) first 7 patients received 0.75 mg/kg per hour for 1.5 hours, followed by 0.15 mg/kg for 6.5 hours. Due to adverse events experienced by these patients, infusion duration was reduced to 1.5 hours at 0.5 mg/kg per hour Placebo (N = 15) duration of placebo infusions matched that of CP‐101, 606 Concomitant treatment: yes, paroxetine as described above |
|
| Outcomes | MADRS
HRSD Response rate (≥ 50% reduction in HRSD scores) Remission rate (score < 7 on HRSD) Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information given on random sequence generation method, other than 'randomly assigned' |
| Allocation concealment (selection bias) | Low risk | Quote: "randomly assigned in a double‐blind fashion....All infusions (periods 1 and 2) were identical in terms of volume infused and duration" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "randomly assigned in a double‐blind fashion....All infusions (periods 1 and 2) were identical in terms of volume infused and duration.Neither the infusion staff nor the patients discussed with the efficacy rating staff how the patients did during the infusions" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Neither the infusion staff nor the patients discussed with the efficacy rating staff how the patients did during the infusions. The efficacy rating staff were on the unit only for the ratings, leaving before the infusion started and returning 24 hours later for the day 2 ratings |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout data reported fully alongside means |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Some outcomes not reported so author was contacted |
| Other bias | Low risk | No other potential sources of bias identified |
Preskorn 2015.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder without psychotic features N: 120 randomised, 116 received treatment Age: GLYX‐13 1mg/kg M = 41.7 (SD = 8.1); GLYX‐13 5 mg/kg M = 44.0 (SD = 11.6); GLYX‐13 10 mg/kg M = 44.2 (SD = 11.5); GLYX‐13 30 mg/kg M = 47.3 (SD = 6.9); placebo M = 44.5 (SD = 12.5) Sex: GLYX‐13 1mg/kg female 56%; GLYX‐13 5 mg/kg female 60%; GLYX‐13 10 mg/kg female 64.7%; GLYX‐13 30 mg/kg female 47.6%; placebo female 63.6% Baseline depression severity: GLYX‐13 1 mg/kg HAM‐D17 = 26.10; GLYX‐13 5 mg/kg HAM‐D17 = 25.20; GLYX‐13 10 mg/kg HAM‐D17 = 25.10; GLYX‐13 30 mg/kg HAM‐D17 = 24.60; placebo HAM‐D17 = 26.10 |
|
| Interventions | A single intravenous dose of GLYX‐13 (1 mg, 5, 10 mg, or 30mg/kg) or placebo was administered. GLYX‐13 1 mg/kg (N = 25) GLYX‐13 5 mg/kg (N = 20) GLYX‐13 10 mg/kg (N = 17) GLYX‐13 30 mg/kg (N = 21) Placebo (N = 33) Concomitant treatments: No, 14‐day wash‐out period of anti‐depressant medication completed prior to study entry and no new anti‐depressant drug could be recevied after randomisation. |
|
| Outcomes | HAM‐D17 Bech‐6 Brief Psychiatric Rating Scale positive symptoms subscale Columbia Suicide Severity Rating Scale Response (50% improvement from baseline HAM‐D17 score) Remission (HAM‐D17 score <10) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were allocated to treatment groups in a block of 8 randomization sequences generated by a statistician not otherwise associated with the study and assigned sequentially using an interactive webbased randomization assignment system". |
| Allocation concealment (selection bias) | Low risk | Quote: "Subjects were allocated to treatment groups in a block of 8 randomization sequences generated by a statistician not otherwise associated with the study and assigned sequentially using an interactive webbased randomization assignment system". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not tested. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers of withdrawn participants and reasons for withdrawal listed. |
| Selective reporting (reporting bias) | Low risk | Protocol available online, outcomes reported in paper. |
| Other bias | High risk | Potential for bias due to funding source. Quote: "Funding for this study was provided in its entirety by Naurex, Inc, 1801 Maple Avenue, Suite 4300, Evanston IL 60201, which owns patents, patent applications, and commercialization rights to GLYX‐13". |
Quiroz 2016.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled, multi‐centre trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder N: 333 Age: basimglurant 0.5 mg group M = 45.8 (SD = 10.8); basimglurant 1.5 mg group M = 47.0 (SD = 17.1); placebo group M = 47.1 (SD = 11.3) Sex: basimglurant 0.5bmg group 62.5% female; basimglurant 1.5bmg group 69.4% female; placebo group 63.3% female Baseline depression severity: basimglurant 0.5bmg group MADRS = 31.1 (SD = 3.9); basimglurant 1.5bmg group MADRS = 31.3 (SD = 4.6); placebo group MADRS = 31.1 (SD = 4.7) |
|
| Interventions | Patients were randomised to receive 6‐weeks of treatment with 0.5 mg basimglurant, 1.5 mg basimglurant, or placebo, orally once daily. This was adjunctive to their ongoing antidepressant medication therapy. | |
| Outcomes | MADRS QIDS‐SR16 CGI‐S SDS (items 2‐3) Q‐LES‐Q‐SF PGI‐I Response (≥50% reduction in MADRS total score from baseline) Remission (MADRS total score ≤10) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients randomised in a 1:1:1 ratio. |
| Allocation concealment (selection bias) | Low risk | Patients randomised in a 1:1:1 ratio. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not tested. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Diagram of patient numbers and reasons for withdrawal included in figure 1 (page 678). |
| Selective reporting (reporting bias) | Low risk | Trial registration available online (NCT01437657). All outcomes reported. |
| Other bias | High risk | Potential funding bias. Quote: "This study was sponsored by F. Hoffmann‐La Roche Ltd." |
Roohi‐Azizi 2017.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder N: 54 Age: citicoline group M = 36.0 (SD = 8.11); placebo group M = 35.8 (SD = 11.10) Sex: citicoline group 64% female; placebo group 76% female Baseline depression severity: citicoline group HDRS score M = 24.76 (SD = 4.40); placebo group HDRS score M = 24.40 (SD = 4.24) |
|
| Interventions | Patients were randomised to receive either citicoline (100 mg) or placebo every 12 hours for 6 weeks. Both groups also received citalopram (20 mg/day first week; 40 mg/day subsequent 5 weeks). | |
| Outcomes | HDRS Early improvement ((≥20% reduction in HRDS score within the first 2 weeks) Response to treatment (≥50% reduction in the HDRS score) Remission rate (HDRS score ≤ 7) Time needed to respond to treatment Adverse effects |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was conducted by using a computerized random number generator (blocks of 4, allocation ratio of 1:1)." |
| Allocation concealment (selection bias) | Low risk | Quote: "An independent party who was not involved elsewhere in the study was responsible for the generation of randomization codes. Concealment of allocation was performed using sequentially numbered, sealed, opaque, and stapled envelopes. Separate individuals were responsible for randomization and treatment allocation, as well as rating." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. Quote: "The participants, research investigators, raters, and the statistician were all blinded to treatment allocation". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not tested. Quote: "The participants, research investigators, raters, and the statistician were all blinded to treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Diagram of patient numbers and reasons for withdrawal included in figure 1 (page 2). Patient withdrawals evenly spread across groups. |
| Selective reporting (reporting bias) | Low risk | Trial registration available online (IRCT201502191556N74). All outcomes reported. |
| Other bias | Low risk | None identified. |
Salardini 2016.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder N: 64 Age: riluzole group M = 34.56 (SD = 7.23); placebo group M = 33.23 (SD = 7.25) Sex: riluzole group 26.7% female; placebo group 36.7% female Baseline depression severity: riluzole group HDRS score M = 24.43 (SD = 2.14); placebo group HDRS score M = 23.63 (SD = 3.61) |
|
| Interventions | Participants were randomised to receive either 50 mg riluzole bi‐daily or placebo for six weeks. All patients received 20 mg/day citalopram for the first week and 40 mg/day for the subsequent 5 weeks. Participants were not allowed to undergo any behavioral intervention therapy or use any psychotropic drugs or undergo ECT during the course of the trial. | |
| Outcomes | HDRS Adverse events Time needed to respond to treatment Response (≥50% reduction in the HDRS score Remission (HDRS score ≤7) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed by the permuted randomization block method using a computerized random number generator by an independent party (allocation ratio 1:1, blocks of four)" |
| Allocation concealment (selection bias) | Low risk | Quote: "Allocation concealment was performed using sequentially numbered, sealed, opaque, and stapled envelopes. An aluminum foil inside the envelope rendered the content of envelope impermeable to intense light. Riluzole and placebo tablets were identical in their size, shape, color, texture and odor. The patients, the nurses, the physician who referred the patient, the investigator, and the raters were all blinded to treatment allocation" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients and the raters guessed wrongly about the allocated treatment in more than 50% of allocations" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The patients and the raters guessed wrongly about the allocated treatment in more than 50% of allocations" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Flow diagram of patients in the study and reasons for trial discontinuation included in figure 1 (page 27). |
| Selective reporting (reporting bias) | Low risk | Trial registration available online (IRCT201307181556N54), all outcomes reported as planned. |
| Other bias | Low risk | None identified |
Salehi 2015.
| Study characteristics | ||
| Methods | Blind, randomised clinical trial | |
| Participants |
Diagnosis: DSM‐IV‐TR drug‐resistant major depression N: 160 Age: Not stated Sex: ketamine group 53.8% female; thiopental group 53.8% female Baseline depression severity: ketamine group HDRS‐17 score M = 29.82 (SD = 7.3); placebo group HDRS‐17 score M = 28.86 (SD = 7.6) |
|
| Interventions | Patients were randomised to receive either ketamine 0.8 mg/kg or sodium thopental 1‐1.5m g/kg intravenously as anesthetic before ECT for 30‐90 seconds 3 times per week. | |
| Outcomes | HDRS‐17 Recovery time Post‐anesthesia complications |
|
| Notes | Title says double‐blind, but text says single‐blind. Not enough information given to verify. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation methods were used to randomly allocate participants. |
| Allocation concealment (selection bias) | Unclear risk | No details given |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. Unclear whether single or double blind (contradictory information in title of article and text). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No details about how outcome assessments were conducted. Unclear if assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information available |
| Selective reporting (reporting bias) | Low risk | Trial registration available online (IRCT2015013012642N13). All outcomes reported |
| Other bias | Low risk | None identified |
Sanacora 2014 (a).
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; at least two adequate lifetime antidepressant trials that failed; HRSD‐17 score ≥ 20 N: 34 Age: lanicemine group M = 47.6 (SE = 3.0); placebo group M = 43.9 (SE = 3.0) Sex: lanicemine group 56% female; placebo group 61% female Baseline depression severity: lanicemine group HAM‐D = 25.9 (SD = 1.1); placebo group MADRS = 25.4 (SD = 0.8) |
|
| Interventions | 1 single IV infusion Lanicemine (AZD6765) (N = 16) 100 mg infused over 60 minutes Placebo (N = 18) 0.9% saline infused over 60 minutes Concomitant treatment: Unclear |
|
| Outcomes | MADRS
VAS
BPRS
CogState Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''double‐blind randomized study'' no further details given |
| Allocation concealment (selection bias) | Unclear risk | Quote: ''double‐blind randomized study'' no further details given |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: ''double‐blind'' no further details given |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''double‐blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts not reported |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Sanacora 2014 (b).
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; history of poor response (defined as treatment failure on two or more antidepressants after exposure at adequate doses or maximum tolerated doses for ≥ 4 weeks); HRSD‐17 score ≥ 26; CGI‐S score ≥ 5; QIDS‐SR score ≥ 21. However, to improve recruitment criteria reduced to HRSD‐17 score ≥ 20; CGI‐S score ≥ 4; QIDS‐SR score ≥ 16 N: 152 Age: lanicemine 100 mg group M = 45.9 (SD = 10.0); lanicemine 150 mg group M = 46.6 (SD = 9.4); placebo group M = 44.4 (SD = 10.1) Sex: lanicemine 100 mg group 71% female; lanicemine 150 mg group 61% female; placebo group 61% female Baseline depression severity: lanicemine 100 mg group MADRS = 33.3 (SD = 5.6); lanicemine 150 mg group MADRS = 34.1 (SD= 5.0); placebo group MADRS = 33.5 (SD = 4.5) |
|
| Interventions | 3 weeks treatment Lanicemine (AZD6765) (N = 51) 100 mg, 3 IV infusions per week Lanicemine (AZD6765) (N = 51) 150 mg, 3 IV infusions per week Placebo (N = 50) 150 mg, 3 IV infusions per week Concomitant treatment: Yes, treatment adjunct to ongoing psychotropics that included at least one antidepressant |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) Remission rate (MADRS score ≤ 10) HAMA HRSD QIDS‐SR CGI‐S CGI‐I Q‐LES‐Q CADSS Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''patients were randomised in a 1:1:1 ratio'' no further details given |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: ''double‐blind'' no further details given |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''double‐blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Sanacora 2017.
| Study characteristics | ||
| Methods | Randomised, parallel‐arm, double‐blind, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder N: 302 randomised, 301 went through to the study Age: lanicemine 50 mg group M = 47.7 (SD = 11.19); lanicemine group 100mg M = 47.5 (SD = 11.89); Saline group M = 49.5 (SD = 11.12) Sex: Lanicemine 50mg group 61.4% female; Lanicemine 100 mg group 69.3% female; placebo group 65% female Baseline depression severity: lanicemine 50 mg group MADRS score M = 36.55 (SD = 4.67); lanicemine 100 mg group MADRS score M = 36.02 (SD = 4,74); placebo group MADRS score M = 35.64 (SD = 4.84) |
|
| Interventions | Participants completed a washout phase of up to 6 weeks before being randomised to the treatment phase for 12 weeks. Patients were randomly allocated to receive intravenous infusion of lanicemine 50 mg, lanicemine 100 mg, or placebo (saline). | |
| Outcomes | MADRS
Response (≥50% reduction from baseline in MADRS total score) Remission (MADRS total score ≤10) CGI‐S CGI‐I QIDS‐SR‐16 Sheehan Disability Scale |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects entering the study drug treatment phase were randomized in balanced blocks equally (1:1:1 ratio), using a unique randomization code generated via Interactive Voice Response System". |
| Allocation concealment (selection bias) | Low risk | Quote: "Packaging and labeling of study medications could not be used by the investigators or subjects to determine randomization assignment". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The investigator, patient, and study staff were all blinded." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The investigator, patient, and study staff were all blinded." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal rates are distributed almost equally across the treatment groups. Reasons for withdrawal are detailed in figure 1 (page 846). |
| Selective reporting (reporting bias) | High risk | Trial registration available online (NCT01482221). Post‐treatment follow up phase changed from 8 weeks in protocol to 2 weeks due to difficulties retaining subjects. |
| Other bias | High risk | Potential bias due to funding by pharmaceutical company. Sponsored by AstraZeneca. |
Shams Alizadeh 2015.
| Study characteristics | ||
| Methods | Randomised, double‐blind trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder N: 44 Age: ketamine 0.3 mg/kg group M = 34.27 (SD =10.66); placebo saline 5 mL group M = 35.1 (SD=12.44) Sex: ketamine 0.3mg/kg group 72.7% female; placebo saline 5 mL group 65% female Baseline depression severity: ketamine 0.3 mg/kg group HRSD score M = 35.4 (SD=6.7); placebo saline 5 mL group HRSD score M = 36.44 (SD=7.17) |
|
| Interventions | The intervention group received 0.3 mg/kg of ketamine hydrochloride intravenously which was diluted with 5mL of saline. In the control group, ketamine was replaced with 5mL of Normal saline. After 30 seconds, both groups received 0.5 mg of atropine IV, prior to ECT. Ketamine group (N = 22) Control group (N = 22) Concomitant treatments:aAll participants received ECT. Yes, no changes were made in the type and doses of the already prescribed drugs. |
|
| Outcomes | HRSD Cognitive Performance Recovery Time |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No further information beyond, "The patients were allocated randomly using the block randomization method." |
| Allocation concealment (selection bias) | Unclear risk | No information given on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No further information beyond, "The patients were also blind to the received medication" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The HRSD scores, vital signs, and duration of reorientation were collected by an author who was blind to group assignment". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers of withdrawn participants and reasons for withdrawal reported. |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other potential sources of bias identified |
Shiroma 2020.
| Study characteristics | ||
| Methods | Randomised, double‐blind, active placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder N: 54 Age: ketamine group M = 54.4 (SD = 13.8); midazolam group M = 51.2 (SD = 12.5) Sex: ketamine group 12% female; midazolam group 17.2% female Baseline depression severity: Ketamine group MADRS M = 34.88 (SD=7.80); Midazolam group MADRS M = 33.82 (SD=5.02) |
|
| Interventions | Participants were randomised to receive 6 ketamine (0.5mmg/kg) infusions or 5 midazolam (0.045mmg/kg) plus 1 ketamine (0.5mmg/kg) infusion. They received six infusions over a 12‐day period. Concomitant medications: participants were allowed to continue concomitant psychiatric medication regimen on stable dosages for at least 6 weeks prior to study onset. |
|
| Outcomes | MADRS mean change Response (≥50% MADRS reduction from baseline) Adverse events |
|
| Notes | Authors provided additional data for 5 ketamine infusions vs. 5 midazolam infusions. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomization was conducted using permutated blocks of 4 and 1:1 assignment between treatments”. |
| Allocation concealment (selection bias) | Low risk | Quote: "Group assignments for each participant was concealed in sequentially numbered, sealed, opaque envelopes.” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding tested with limited effectiveness for both personnel and participants. Quote: "At the end of the last infusion (midazolam plus single ketamine versus six ketamine),10.7% among midazolam cases and 50.0% among ketamine cases guessed incorrectly about assigned treatment (X2 1df = 9.12; P = 0.002). Raters, who were different during infusions days from those rating antidepressant outcomes at 24 h., incorrectly guessed 20.8% of midazolam cases and 7.1% of repeated ketamine cases prior to the last infusion (X2 1df = 1.25; P = 0.26). After the last infusion, 6.9 and 26% raters were incorrect about treatment assignment among midazolam plus single ketamine and repeated ketamine cases, respectively (X2 1df = 3.62; P = 0.06)." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding tested with limited effectiveness for both personnel and participants. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Participant recruitment and withdrawals documented. No participants withdrew after baseline assessment. |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. |
| Other bias | Low risk | None identified. |
Singh 2016 a.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR major depressive disorder without psychotic features N: 68 Age: ketamine 2 weekly group M = 45.7 (SD = 9.6); placebo 2 weekly group M = 40.3 (SD = 11.8); ketamine 3 weekly group M = 43.3 (SD = 12.0); placebo 3 weekly group M = 46.1 (SD = 10.5) Sex: ketamine 2 weekly group 66.7% female; placebo 2 weekly group 75% female; ketamine 3 weekly group 70.6% female; placebo 3 weekly group 56.3% female Baseline depression severity: ketamine 2 weekly MADRS score M = 33.3 (SD = 4.9); placebo 2 weekly MADRS score M = 35.6 (SD = 3.8); Ketamine 3 weekly MADRS score M = 35.4 (SD = 5.3); Placebo 3 weekly MADRS score M = 36.8 (SD = 5.8) |
|
| Interventions | Patients were randomised to one of four treatment groups: intravenous ketamine (0.5 mg/kg) two or three times weekly or intravenous placebo (0.9% sodium chloride for injection) two or three times weekly, administered over 40 minutes. Study drugs were administered on days 1, 4, 8, 11, 15, 18, 22, and 25 for the twice weekly regimen and on days 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, 24, and 26 for the thrice‐weekly regimen. | |
| Outcomes | MADRS Early onset of clinical response (improvement ≥50% from baseline in MADRS score during week 1 that was maintained through day 15) Responders (number of patients with a ≥50% reduction from baseline in MADRS score) Remitters (number of patients with a MADRS score ≤10) Change in MADRS score from baseline through day 29 CGI‐S CGI‐I PGI‐S PGI‐C |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized in a 1:1:1:1 ratio... Randomization was based on a computer‐generated randomization scheme, balanced by the use of randomly permuted blocks and stratified by study center". |
| Allocation concealment (selection bias) | Low risk | Quote: "The investigators, patients, and all study staff were kept blind to assigned treatment at randomization. An unblinded pharmacist was accountable for study drug preparation to ensure the integrity of blinding." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The investigators, patients, and all study staff were kept blind to assigned treatment at randomization. An unblinded pharmacist was accountable for study drug preparation to ensure the integrity of blinding." Stated but not testd. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The investigators, patients, and all study staff were kept blind to assigned treatment at randomization. An unblinded pharmacist was accountable for study drug preparation to ensure the integrity of blinding." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Participant flow chart detailing all reasons for withdrawal included in figure 1 (page 818). |
| Selective reporting (reporting bias) | Low risk | Clinical trial registration available online (NCT01627782), all outcomes reported |
| Other bias | Low risk | None identified |
Singh 2016 b.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR recurrent major depressive disorder without psychotic features N: 30 Age: esketamine .20 mg/kg group M = 44.7 (SD = 13.38); esketamine.40 mg/kg group M = 41.8 (SD = 11.63); placebo group M = 42.7 (SD = 10.89) Sex: esketamine .20 mg/kg group 56% female; esketamine.40 mg/kg group 64% female; placebo group 60% female Baseline depression severity: esketamine .20 mg/kg group MADRS score M = 33.1 (SD = 3.55); esketamine.40 mg/kg group MADRS M = 33.7 (SD = 5.82); placebo group MADRS score M = 33.9 (SD = 4.15) |
|
| Interventions | On day 1, patients were randomsly assigned to receive an IV infusion of .20mmg/kg or.40mmg/kg esketamine or placebo (.9% saline solution) over 40 minutes. On day 4 (second dose), responders received the same treatment as day 1. For those on placebo, non‐responders were randomly assigned to .20mmg/kg or.40mmg/kg esketamine. Non‐responders who received 20mmg/kg or 40mmg/kg on day 1 received esketamine 40mmg/kg on day 4. |
|
| Outcomes | MADRS Response (reduction of >50% in the MADRS total score on days 2, 3, or 4) QIDS‐SR CGI‐S CGI‐I PGI‐S PGI‐C TEAEs Clinical laboratory tests 12‐lead ECG Vital signs Physical examinations C‐SSRS CADSS BPRS MGH‐CPFQ |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Central randomization was implemented based on a computer generated randomization schedule prepared by the sponsor before the study. The randomization was balanced by using randomly permuted blocks and was stratified by study center". |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Before dosing, the unblinded pharmacist at each study site contacted the randomization center and provided the required subject information. The randomization center assigned a randomization number to the subject and informed the unblinded pharmacist at the site about the assigned treatment" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "During the study, the subject was assessed by qualified trained site raters who were blinded to the subject’s treatment" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for participant withdrawal noted on page 426 |
| Selective reporting (reporting bias) | Low risk | Clinical trials registration available online (NCT01640080). All outcomes reported in paper. |
| Other bias | High risk | Potential bias due to funding being provided by a pharmaceutical company, Janssen Research & Development. |
Smith 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive episode; HRSD‐17 score ≥ 16 N: 31 Age: memantine group M = 54.80 (SD = 6.17); placebo group M = 49.75 (SD = 11.68) Sex: memantine group 53.33% female; placebo group 68.75% female Baseline depression severity: memantine group MADRS = 27.47 (SD = 8.32); placebo group MADRS = 27.38 (SD = 6.95) |
|
| Interventions | 8 weeks treatment Memantine (N = 15) flexibly dosed: all participants began on 5 mg/day and the dose was increased by 5 mg/day at weekly intervals as tolerated to a maximum dose of 20 mg/day. In the absence of limiting adverse effects, this dose was achieved approximately 22 days into the eight‐week (56‐day) trial Placebo (N = 16) Concomitant treatment: yes, patients received one of the following medications at the following stable dosages for the previous 25 days or more prior to study entry: mirtazapine (≥ 15 mg/day), fluoxetine, paroxetine, or citalopram (≥ 20 mg/day), paroxetine controlled‐release (≥ 25 mg/day), sertraline or desvenlafaxine extended‐release (≥ 50 mg/day), duloxetine (≥ 60 mg/day), fluvoxamine extended‐release (≥ 100 mg/day), venlafaxine or venlafaxine extended‐release (≥ 150 mg/day), fluvoxamine (≥ 200 mg/day), bupropion or bupropion sustained‐release (≥ 300 mg/day). Participants were not permitted to make antidepressant drug dosage changes during the 8‐week trial participation |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) Remission rate (MADRS score ≤ 12) QIDS‐SR HAMA SADS BSS Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Participants...were randomized...using a block randomization design by the University of Massachusetts Medical School Investigational Pharmacy. A printed list computer generated from the Website www.randomization.com was used'' |
| Allocation concealment (selection bias) | Low risk | Quote: ''Participants...were randomized...using a block randomization design by the University of Massachusetts Medical School Investigational Pharmacy. A printed list computer generated from the Website www.randomization.com was used'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''...with allocation concealed from participants, research staff and investigators....All patients, research staff and clinical investigators remained blinded throughout the trial, with the exception of an unintentional unblinding of 1 of the PIs (E.G.S.) for a single participant receiving placebo who had completed the trial'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''All patients, research staff and clinical investigators remained blinded throughout the trial..'' |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Sos 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; MADRS score ≥ 20 N: 30 Age: ketamine group M = 42.2 (SD = 15.1); placebo group M = 44.6 (SD = 10.9) Sex: ketamine group 54.55% female; placebo group 47.37% female Baseline depression severity: ketamine group MADRS = 20.4 (SD = 4.7); placebo group MADRS = 24.6 (SD = 4.8) |
|
| Interventions | 1 single IV infusion Ketamine (N = 11) loading dose of 0.27 mg/kg for the first 10 minutes, followed by a maintenance infusion of 0.27 mg/kg within 20 minutes Placebo (N = 19) 0.9% saline solution Concomitant treatment: yes, patients were on a stable dose of antidepressant medication for a minimum of three weeks prior to admission and remained on the same medications and dosages throughout the duration of the study |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) BPRS Plasma level of ketamine and nor‐ketamine during infusion |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Subjects were randomized by a flip of a coin (Armitage 1982)'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: ''double blind'' no further details given |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''double blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Su 2017.
| Study characteristics | ||
| Methods | Double‐blind, randomised, parallel‐group, placebo controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder N: 71 Age: ketamine 0.5 mg/kg group M = 48.5 (SD = 11.0); ketamine 0.2 mg/kg group M = 45.0 (SD = 12.3); placebo group M = 48.6 (SD = 8.2) Sex: ketamine 0.5 mg/kg group 87.5% female; ketamine 0.2 mg/kg 73.9% female; placebo group 62.5% female Baseline depression severity: ketamine 0.5 mg/kg group HAMD‐17 score M = 23.0 (SD = 4.9); ketamine 0.2 mg/kg group HAMD‐17 score M = 23.1 (SD = 4.8); placebo group HAMD‐17 score M = 23.3 (SD = 4.1) |
|
| Interventions | Patients were randomised to receive a 40‐minute intravenous infusion ketamine (0.2 mg/kg or 0.5 mg/kg) or saline. | |
| Outcomes | HAMD‐17 MADRS BDI Blood pressure Heart rate Digit pulse oximetry Response (≥ 50% reduction of HAMD‐17 score at any two daily HAMD measures during the period of 24 to 96 hours (days 2 to 5) after infusion. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details on randomisation procedures given |
| Allocation concealment (selection bias) | Unclear risk | No details on allocation concealment given |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details about blinding of outcome assessment given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Consort flow diagram with details of attrition included in figure 1 (page 2843) |
| Selective reporting (reporting bias) | Low risk | Trial registration available online (UMIN000016985). Outcomes all reported. |
| Other bias | Low risk | None identified |
Sumner 2020.
| Study characteristics | ||
| Methods | Randomised, double‐blind, active placebo‐ controlled cross‐over design. | |
| Participants |
Diagnosis: participants who met DSM‐IV criteria for major depressive disorder. N: 30 Age: 30.7 (8.85) Sex: 15 male (50%), 15 female (50%) Baseline depression severity: ketamine group MADRS M = 30.00 (SD = 5.21); Placebo MADRS M = 30.33 (SD = 4.65). |
|
| Interventions | Participants were randomised to receive one dose of racemic ketamine (0.25 mg/kg bolus, followed a 0.25 mg/kg/hour infusion for 45 minutes), or the active placebo remifentanil hydrochloride as a 9‐minute infusion using a target‐controlled infusion system to achieve 1.7 ng/mL plasma concentration using the Minto pharmacokinetic model. | |
| Outcomes | MADRS Response |
|
| Notes | Authors kindly provided additional data for the first half of the cross‐over. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Order counterbalanced. |
| Allocation concealment (selection bias) | Unclear risk | Order randomised as stated, but it is unclear how or by what method. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding measures were not effective & high risk of performance bias: quote: “most (88%) participants were able to correctly identify their ketamine session during a debrief. This is a known issue with psychoactive drug research and is not unique to current study." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | To maintain study blinding, the MADRS assessor was never present during the acute phase of drug effects and patients were explicitly instructed not to share their experiences during the treatment sessions with the assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No data missing. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Low risk | None identified. |
Tiger 2020.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled trial | |
| Participants |
Diagnosis: major depressive episode according to M.I.N.I., with MADRS ≥ 20 N: 30 Age: ketamine group M = 39.2 (SD = 11.7); placebo group M = 37.1 (SD = 11.1) Sex: ketamine group 40% female; placebo group 60% female Baseline depression severity: ketamine group MADRS M = 26.3 (SD = 6.58); placebo group MADRS M = 30.8 (SD = 4.92) |
|
| Interventions | Patients were randomised to receive either 0.5 mg/kg racemic ketamine diluted in 100 mL isotonic NaCl solution, given as an intravenous infusion over 40 minutes. Placebo treatment was an isotonic NaCl solution only. Concomitant medication: ongoing pharmacological treatment was washed out for a time corresponding to at least five half‐lives of each drug prior to beginning study treatment. |
|
| Outcomes | MADRS Response Adverse events (obtained from author) PET |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed with sealed opaque envelopes, each containing one of the study treatment allocations. Before the start of treatment, a nurse not involved in patient assessments opened a new envelope, assigning the patient to the randomized treatment allocation". |
| Allocation concealment (selection bias) | Low risk | Quote:"Randomization was performed with sealed opaque envelopes, each containing one of the study treatment allocations. Before the start of treatment, a nurse not involved in patient assessments opened a new envelope, assigning the patient to the randomized treatment allocation". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Patient blinding not effective: quote:"Patients who received active treatment in the double blinded phase were all convinced they received ketamine, while four out of ten placebo treated patients thought that they were actually given the active treatment". Unknown whether personnel blinding was effective. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Patients and personnel blinded, however this was not effective for patients, unclear for personnel. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals reported in article: quote:"No patient was discontinued after randomization". |
| Selective reporting (reporting bias) | Unclear risk | No protocol accesible. |
| Other bias | Low risk | None identified. |
Umbricht 2020.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, double‐blind phase 2 trial. | |
| Participants |
Diagnosis: DSM‐IV‐TR Major Depressive Disorder N: 357 randomised Age: Placebo group M = 46 (SD=11.2); decoglurant 5 mg group M = 46.9 (SD = 10.7); decoglurant 15 mg group M = 46.9 (SD = 10.9), decoglurant 30 mg group M = 44.5 (SD = 13.1) Sex: Placebo group 40.7% female; decoglurant 5 mg group 69.7% female; decoglurant 15 mg group 71.6% female; decoglurant 30 mg group 63.8% female Baseline depression severity: placebo group MADRS M = 30.9 (SD=5.9); decoglurant 5 mg group MADRS M= 30.5 (SD = 5.8); decoglurant 15 mg group MADRS M = 30.9 (SD=5.7); decoglurant 30 mg group MADRS M = 31.2 (SD = 7.4). |
|
| Interventions | Participants were randomised to receive decoglurant (5mg, 15 mg, 30 mg) or placebo once daily for 6 weeks in addition to existing permitted medications. | |
| Outcomes | MADRS Response Remission |
|
| Notes | Remission rate is not consistent with study definition. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation codes genereated and put into web‐based response system. |
| Allocation concealment (selection bias) | Low risk | Computer generated and stratified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Fully blinded centralised raters. Trial registration states participants blinded too. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinded raters assessed MADRS score. Not clear how medications were concealed from participant and personelle. MADRS scores assessed by the centralised raters tended to be smaller than those assessed by site raters, particularly in the placebo group (page 5). Personnel and participants blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for missing data are explained, and not related to outcome. CONSORT diagram depicts participant flow through study. Similar numbers withdrew from each group. ITT analysis. |
| Selective reporting (reporting bias) | Low risk | Outcomes reported according to protocol outcomes. Trial registration NCT01457677, all expected outcomes reported. |
| Other bias | High risk | Sponsor bias ‐ pharma company. |
Yoosefi 2014.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; HAM‐D score ≥ 18 N: 31 Age: ketamine group M = 40.87; thiopental group M = 47 Sex: ketamine group 46.67% female; thiopental group 50% female Baseline depression severity: ketamine group HAM‐D = 23.60; thiopental group HAM‐D = 22.86 |
|
| Interventions | ECT performed 3 times a week for 2 weeks, using a dose‐titration protocol. After recording baseline variables, patients received 0.5 mg of IV atropine. Patients then randomised to: Ketamine (N =17) 1 to 2 mg/kg Thiopental (N = 14) 2 to 3 mg/kg Succinylcholine 0.5 mg/kg administered after patients became unconscious. Concomitant treatment: unclear |
|
| Outcomes | HAM‐D Response rate (60% reduction in HAM‐D scores) MMSE |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''patients were randomized...based on a table of random numbers...'' |
| Allocation concealment (selection bias) | High risk | Quote: ''...patients were randomized by the research executive manager (one of the investigators)...'' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''The study patients, the anesthesiologist (primary investigator), and the rater of the scales were all blind to the intervention allocation concealment'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: ''...the rater of the scales were all blind to the intervention allocation concealment'' |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other potential sources of bias identified |
Zarate 2006a.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; HRSD‐21 ≥18; at least two adequate lifetime antidepressant trials that failed, as assessed by the Antidepressant Treatment History Form N: 18 Age: M = 46.7 (SD = 11.2) Sex: 66.7% female Baseline depression severity: ketamine group HRSD = 24.89; placebo group HRSD = 24.44 |
|
| Interventions | 1 single IV infusion Ketamine (N = 9) 0.5 mg/kg infused over 40 minutes Placebo (N = 9) 0.9% saline infused over 40 minutes Concomitant treatment: No, patients had a 2‐week drug‐free period before commencing treatment |
|
| Outcomes | HRSD Response rate (≥ 50% reduction in HRSD scores) Remission rate (HRSD score ≤ 7) BDI BPRS YMRS VAS |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Patients were randomly assigned to the order in which they received the 2 infusions via a random‐numbers chart'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''Double‐blind'' no further details given |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Zarate 2006b.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; MADRS score ≥ 22 N: 32 Age: memantine group M = 47.1 (SD = 12.3); placebo group M = 46.1 (SD = 9.4) Sex: memantine group 56% female; placebo group 44% female Baseline depression severity: memantine group MADRS = 30.23; placebo group MADRS = 31.73 |
|
| Interventions | 8 weeks of treatment Memantine (N = 16) gradually titrated: 5 mg/day and increased by 5 mg/week as tolerated up to a maximum of 20 mg/day Placebo (N =16) Concomitant treatment: zopidem 5 mg to 0 mg/day as needed for insomnia (no more than three times per week and not within 8 hours of ratings). No other psychotropic medication allowed |
|
| Outcomes | MADRS Response rate (not defined) CGI HAMA |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''Randomly assigned'' no further information given |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: ''Double‐blind'' no further information given |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: ''Double‐blind'' no further information given |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
Zarate 2013.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled cross‐over trial | |
| Participants |
Diagnosis: DSM‐IV major depressive disorder; MADRS score ≥ 22 N: 22 Age: M = 51.5 (SD = 10.1) Sex: 45% female Baseline depression severity: AZD6765 group MADRS = 31.75; placebo group MADRS = 36.80 |
|
| Interventions | One single IV infusion AZD6765 (N = 12) 150 mg infused over 60 minutes Placebo (N = 10) 0.9% saline infused over 60 minutes Concomitant treatment: no, patients were not allowed to receive any other psychotropic medications (including benzodiazepines) |
|
| Outcomes | MADRS Response rate (≥ 50% reduction in MADRS scores) Remission rate (MADRS score < 10) HRSD BDI VAS HAMA BPRS CADSS YMRS Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''Patients were assigned to receive the two infusions via a random numbers chart'' |
| Allocation concealment (selection bias) | Unclear risk | No details given on allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: ''Study solutions were supplied in identical 45 mL syringes containing either .9% of saline or 150 mg of AZD6765, which forms a clear solution when dissolved in.9% saline. All staff were blind to whether drug or placebo was being administered'' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: All staff were blind to whether drug or placebo was being administered....Patient ratings were performed by research nurses or psychologists who trained together to establish reliability'' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study reports dropouts |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable. Data reported matches methods |
| Other bias | Low risk | No other potential sources of bias identified |
AEs: adverse effects; BDI: Beck Depression Inventory; BDNF: brain‐derived neurotrophic factor; BPRS: Brief Psychiatric Rating Scale; BSS: Beck Scale for Suicide Ideation; CADSS: Clinician‐Administered Dissociative States Scale; CGI: Clinical Global Interview; CGI‐I: Clinical Global Impression – Global Improvement; CGI‐S: Clinical Global Impression – Severity; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition; DSM‐V: Diagnostic and Statistical Manual of Mental Disorders, 5th Edition; ECT: electroconvulsive therapy; GAF: Global Assessment of Functioning; GDS: Geriatric Depression Scale; HAMA: Hamilton Anxiety Rating Scale;: IDS‐C Inventory of Depressive Symptoms‐Clinician rated; HRSD: Hamilton Rating Scale for Depression; ITT: intention‐to‐treat; IV: intravenous; LIFE‐RIFT: Range of Impaired Functioning Tool; LOCF: last observation carried forward; MADRS: Montgomery‐Asberg Depression Rating Scale; MECT: Modified Electroconvulsive therapy; MINI: Mini International Neuropsychiatric Interview; MMSE: Mini Mental State Examination; MPS: Maier and Phillip core mood severity scale; NaCL: sodium chloride; PANSS: Positive and Negative Syndrome Scale; PRISE: Patient‐Rated Inventory of Side Effects; QIDS: Quick Inventory of Depressive Symptomatology; Q‐LES‐Q: Quality of Life Enjoyment and Satisfaction Questionnaire; SAFTEE: Systematic Assessment for Treatment Emergent Events; SD: standard deviation; SF‐12: Short Form; SHAPS: Snaith‐Hamilton Pleasure Scale; SLICE/LIFE: Streamlined Longitudinal Interview Clinical Evaluation from the Longitudinal Interval Follow‐up Evaluation; SOFAS: Social and Occupational Functioning Assessment Scale; SSI: Scale of Suicidal Ideation; UKU: UKU Side Effect Rating Scale; VAS: Visual Analogue Scale; VEGF: Vascular endothelial growth factor; WHO‐QOL: The World Health Organization Quality of Life; YMRS: Young Mania Rating Scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aftanas 2019 | Wrong population |
| Barzman 2005 | Wrong intervention |
| Bondolfi 2000 | Comorbidity |
| Burger 2016 | wrong diagnostic criteria |
| Chen 2020 | Wrong design |
| Erdil 2015 | wrong diagnostic criteria |
| Giese 2014 | Comorbidity |
| Huey 2005 | Comorbidity |
| Irwin 2010 | Comorbidity |
| Liebrenz 2009 | Comorbidity |
| O'Gorman 2019 | Wrong design |
| Park 2020 | Wrong design |
| Rasmussen 2014 | Comorbidity |
| Rosenblat 2019 | Wrong design |
| Sharma 2020 | Wrong population |
| Shiroma 2020 | Wrong design |
| Zhang 2018 | Comorbidity |
| Zhong 2016 | Comorbidity |
RCT ‐ randomised controlled trial
Characteristics of studies awaiting classification [ordered by study ID]
IRCT201104092266N2.
| Methods | In a single‐blind, randomised clinical trial, 40 major depressive cases who referred to ECT selecting with a simple randomisation method and will be assigned to 2 case and control groups (20 persons in each group). Control group receives 3 times/week bi‐temporal ECT until complete symptom remission by a psychiatrist and anaesthesiologist (as a routine of treatment with ECT program) who are not in the research team. Intervention group receives 3 times/ week slow infusion of 0.5 mg/kg ketamine until complete symptom remission with direct supervision of an anaesthesiologist. Severity of major depressive symptoms will be evaluated in both group using HRSD prior to intervention, prior to each intervention session, one week, 1, 2 and 3 months after final intervention session by a psychiatrist who is blind to interventions. Cognitive function evaluate using Adult Wechsler Memory Scale, both group will be evaluated periodically for other potential side effects by a psychiatric resident who is blind to interventions |
| Participants | IInclusion criteria: known case of major depressive disorder without psychotic feature based on DSM IV‐TR who referred to ECT by a psychiatrist; age between 20‐50 years; signing the consent form Exclusion criteria: history of psychosis or being psychotic; substance abuse during recent 3 months; breastfeeding mothers; having medical incapacitating disease such as: renal, hepatic, gastrointestinal, cardiovascular, endocrine, neurological and hematological disease based on history and clinical and paraclinical evaluations; history of seizure with unknown origin; uncontrolled hypo‐ or hyper‐thyroidism ; simultaneous other psychiatric interventions |
| Interventions | Intervention 1: Intervention group:receives 3 times/week slow infusion of 0.5 mg/kg ketamine until complete symptom remission with direct supervision of an anaesthesiologist Intervention 2: Control group: receives 3 times/week bi‐temporal ECT until complete symptom remission by a psychiatrist and anaesthesiologist |
| Outcomes | Primary outcome: severity of major depressive symptoms. Timepoint: prior to each intervention session, one week, 1, 2 and 3 months after intervention. Method of measurement: structured interview based on HRSD. Secondary outcome: cognitive Side effects. Time point: prior to intervention, one week and one month after intervention. Method of measurement: Adult Wechsler Memory Scale |
| Notes |
ISRCTN87057460.
| Methods | This is a three‐arm, randomised, parallel study designed to assess the inhibition of noradrenaline and 5‐HT uptake by venlafaxine, paroxetine and atomoxetine. Approximately 40 depressed patients will be randomised to one of three treatment groups with the goal of having at least 10 participants complete the study in each group. The investigators involved in the tyramine test or the collecting of biochemical data will be blind to the medications used by patients. This study will be conducted on an outpatient basis |
| Participants |
Inclusion criteria 1. Male or female patients between 18 and 65 years of age 2. Diagnosis of major depression according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) (American Psychiatry Association, 1994) using the Structured Clinical Interview for Depression (SCID) (Spitzer 1992) 3. Initial global score 18 on the 17‐item HRSD 4. Written informed consent signed by the participant Exclusion criteria 1. Evidence of significant physical illness contraindicating the use of venlafaxine, paroxetine or atomoxetine found on physical or in the laboratory data obtained during the first week of the study 2. Evidence of suicidality or severity of depression precluding safe participation in the study 3. Mental retardation (IQ lower than 80) rendering the response to investigators unreliable 4. Pregnancy, or absence of adequate contraceptive method in women with childbearing potential 5. Concurrent use of psychotropic medication such as antipsychotics, mood stabilisers or regular use of high doses of benzodiazepines 6. Lack of response or intolerance to optimal doses of paroxetine, venlafaxine or atomoxetine 7. Participation in another clinical trial within 30 days of entry into the current study Interventions |
| Interventions | Venlafaxine, paroxetine and atomoxetine |
| Outcomes | The primary objective of this study is to find evidence of a dose‐dependent inhibition of noradrenaline reuptake starting of venlafaxine at 150 mg/day. A secondary objective of this study is to show a lack of effect of paroxetine on noradrenaline reuptake at doses of up to 50 mg/day |
| Notes |
NCT01046630.
| Methods | This was a multicentre, double‐blind, placebo‐controlled, parallel group study in males and females with Major Depressive Disorder (MDD). The maximum study period is approximately 42 days (6 weeks) for individual participants. The study was divided into two parts. Participants in Part 1 were randomised 1:1 to ketamine or placebo; participants in Part 2 were randomised 2:2:5 to ketamine, placebo or AZD6765. Study procedures were the same in both Part 1 and Part 2 |
| Participants | Sixty‐four male and female participants between the ages of 18 and 45 years old with MDD were to be randomised to obtain 60 evaluable participants. Twenty‐four evaluable participants in Part 1 (12 per arm; ketamine and placebo) and 36 evaluable participants in Part 2 (20 AZD6765, 8 ketamine and 8 placebo) |
| Interventions | AZD6765: participants received a single infusion of AZD6765 100 mg (15 mg/mL, IV infusion). The infusion was of a final maximum volume of 40 mL given over 60 minutes Batch number(s): 10‐003824AZ and 10‐005001AZ Ketamine: participants received a single infusion of ketamine 0.5 mg/kg (10 mg/mL Injection) intravenously. The infusion was of a final maximum volume of 40 mL given over 60 minutes Batch number(s): 42800A and 09‐008003AZ |
| Outcomes | Primary: BOLD signal in the BA25 area |
| Notes |
NCT01482221.
| Methods | A Multicentre, randomised, double‐blind, parallel group, placebo‐controlled, phase IIb efficacy and safety study of adjunctive AZD6765 in patients with major depressive disorder (MDD) and a history of inadequate response to antidepressants |
| Participants | Male or female patients aged 18 to 70 years, inclusive. The patient must have a clinical diagnosis of major depressive disorder with a lifetime history of inadequate response to at least 3 antidepressants |
| Interventions | Arm 1: 50 mg (AZD6765 Solution for Infusion, 0.5 mg/mL) by IV infusion; Arm 2: 100 mg (AZD6765 Solution for Infusion, 1.0 mg/mL) by IV infusion; Arm 3: 0.9 sodium chloride [normal saline] solution for injection by IV infusion |
| Outcomes | Change from baseline to Week 6 in the Montgomery‐Asberg Depression Rating Scale (MADRS) total score. Time frame: will be scored at weeks 1 (baseline), 2, 3, 4, 5 and 6 |
| Notes |
NCT01627782.
| Methods | This is a double‐blind (patients and study personnel do not know the identity of the administered treatments), randomised (the drug is assigned by chance), placebo‐controlled (placebo is a substance that appears identical to the treatment and has no active ingredients), parallel arm study (each group of patients will be treated at the same time). The study will consist of a screening phase of up to 4 weeks, a 4‐week double‐blind treatment phase (Day 1 to Day 29), and a 3‐week post‐treatment (follow‐up) phase. In the double‐blind phase, patients will receive over 4 weeks either IV infusions of placebo (2 or 3 times weekly) or IV infusions of ketamine (2 or 3 times weekly). The total study duration for each patient will be a maximum of 13 weeks |
| Participants | Ages eligible for study: 18 to 64 years Genders eligible for study: both Accepts healthy volunteers: no Inclusion Criteria • Be medically stable on the basis of clinical laboratory tests performed at screening • Meet diagnostic criteria for recurrent major depressive disorder (MDD), without psychotic features • Have a history of inadequate response, i.e. treatment was not successful, to at least 1 antidepressant • Have an Inventory of Depressive Symptoms‐Clinician rated, 30 item (IDS‐C30) total score >= 40 at screening and predose at Day 1 • Inpatient or agreed to be admitted to the clinic on each dosing day Exclusion Criteria • Has uncontrolled hypertension • Has a history of, or current signs and symptoms of diseases, infections or conditions that in the opinion of the investigator, would make participation not be in the best interest (e.g. compromise the well‐being) of the patient or that could prevent, limit, or confound the protocol‐specified assessments • Has known allergies, hypersensitivity, or intolerance to ketamine or its excipients • Is unable to read and understand the consent forms and patient reported outcomes, complete study‐related procedures, and/or communicate with the study staff |
| Interventions | IV ketamine versus placebo |
| Outcomes | Primary outcome measures: The change from Day 1 (baseline) to Day 15 in depressive symptoms using the Montgomery Asberg Depression Rating Scale (MADRS) total score. Time frame: Day 1, Day 15 |
| Notes |
NCT03965871.
| Methods | Randomised, multiple dose, placebo‐controlled, double‐blind, multicentre study |
| Participants | 88 participants with bipolar depression |
| Interventions | Drug: esketamine DPI ‐ low dose Drug: esketamine DPI ‐ medium dose Drug: esketamine DPI ‐ high dose Drug: placebo DPI |
| Outcomes | MADRS Response Remission Time to relapse C‐SSRS BPRS YMRS Adverse events |
| Notes | Trial completed February 2021, no results available yet. |
NCT04019704.
| Methods | Randomised, double‐blind, placebo‐controlled study |
| Participants | 327 participants with depression |
| Interventions | Drug: AXS‐05 Drug: placebo |
| Outcomes | Adverse events MADRS |
| Notes | Trial completed December 2019, no results available yet. |
NCT04035798.
| Methods | Randomised double‐blind placebo‐controlled study |
| Participants | Estimated 120 participants with bipolar II disorder |
| Interventions | Memantine 5mg (1 capsule) per day for 12 weeks |
| Outcomes | Memory function Executive function Attention Processing speed Inflammatory status HDRS YMRS |
| Notes | Recruiting |
NCT04210804.
| Methods | Double‐blind placebo controlled study |
| Participants | 80 participants with bipolar depression |
| Interventions | Dietary Supplement: Smoothie 1g eicosapentaenoic acid (EPA) and 1g docoshaexanoic acid (DHA) |
| Outcomes | Episodes of depression or elation Psychometric measures of depression or elation Adverse effects Continuation rate Time to relapse of depression or elation |
| Notes | No results available |
BPRS: Brief Psychiatric Rating Scale; ; ECT: electroconvulsive therapyIDS‐C30: Inventory of Depressive Symptomatology, clinician rating (30 items); IV: intravenous; MADRS: Montgomery‐Asberg Depression Rating Scale; YMRS: Young Mania; Rating Scale; 5‐HT: 5‐hydroxytryptamine..
Characteristics of ongoing studies [ordered by study ID]
EUCTR2011‐001520‐37‐SE.
| Study name | Ketamine as an alternative to electroconvulsive therapy for treatment of major depressive disorder ‐ KETECT EUCTR2011‐001520‐37‐SE |
| Methods | To compare the antidepressant effect of subanaesthetic ketamine with ECT. |
| Participants | Principal inclusion criteria: ASA grade 1‐3 Age 18‐65 Major depressive episode according to DSM‐IV Montgomery Asberg Depression Rating Scale (MADRS) ≥ 20 Offered and accepted ECT Understands and speaks Swedish |
| Interventions | Ketamine versus saline |
| Outcomes | E.5.1 Primary end point(s): antidepressive effect E.5.1.1 time point(s) of evaluation of this endpoint: the assessment is made before treatment, 1 hour and 4‐5 hours after and the day after each treatment session. A major evaluation occurs after 6 treatments to determine continued participation in the study. Patients are monitored weekly for up to four weeks after treatment or at relapse |
| Starting date | Date of Competent Authority Decision: 2014‐02‐14 |
| Contact information | Ida.ellerstrom@gmail.com |
| Notes |
EUCTR2011‐005476‐41‐GB.
| Study name | Ketamine augmentation of ECT to improve outcomes in depression |
| Methods | Ketamine versus saline treatment will reduce ECT‐induced cognitive impairments as measured by being able to learn new verbal information (anterograde verbal memory), remember personal events from their past (autobiographical memory) and saying the names of objects fluently (verbal fluency) |
| Participants | Patient Inclusion criteria: 1. Male or female aged 18 years and above; 2. Current DSM‐IV diagnosis of a major depressive episode, moderate or severe as part of unipolar or bipolar disorder mood disorder diagnosed by the Mini International Neuropsychiatric Interview (MINI); 3. American Society of Anaesthesiologists (ASA) score (excluding mental health considerations in the scoring) of 1, 2 or stable 3, and judged as suitable to receive ketamine by an anaesthetist; 4.Verbal IQ ≥ 85, sufficiently fluent in English to validly complete neuropsychological testing; 5.Capacity to give informed consent; 6. Willing to undertake neuropsychological testing as part of the study Healthy control inclusion criteria: 1. Aged 18 years or more; 2. Currently psychiatrically well, confirmed through MINI interview and no current psychotropic medication; 3. In good physical health Patient exclusion criteria: 1. DSM‐IV diagnosis of a primary psychotic or schizoaffective disorder, current primary obsessive compulsive disorder or anorexia nervosa; 2. History of drug or alcohol dependence (DSM‐IV criteria) within the last year; 3. ECT in last 6 months (to avoid confounding the assessment of cognitive outcomes) or has previously received ECT in the current trial; 4. Known hypersensitivity or contraindication to ketamine or excipients in the injection, including significant cardiovascular disease, uncontrolled hypertension, glaucoma, cirrhosis or significant liver impairment; 5. Known hypersensitivity or contraindication to concomitant medications used for ECT: thiopentone (thiopental), propofol and suxamethonium or excipients in the injections; 6. Evidence of organic brain disease including dementia, neurological illness or injury, or medical illness which may significantly affect neuropsychological function; 7. Detained under the Mental Health Act (1983 as amended 2007) or unable to give informed consent; 8. Pregnancy, or at risk of pregnancy and not taking adequate contraception, breastfeeding; 9. Score ≤ 24 on the Mini Mental State Examination (MMSE); 10. In the subgroup receiving MRI based investigation (fMRI, MRS and ASL) contraindication to MRI (e.g. metal implants or foreign bodies such as from a surgical implant, accident or injury) Control exclusion Criteria: 1. Personal history of psychiatric disorder, as revealed by MINI interview; 2. First degree family history of major psychiatric illness requiring treatment; 3. Significant physical illness including organic brain disease, neurological illness or injury that could interfere with interpretation of results; 4. Psychotropic medication or other medication that could interfere with interpretation of results; 5. Score ≤ 24 on the Mini Mental State Examination (MMSE); 6. In the subgroup receiving MRI based investigation (fMRI, MRS and ASL) contraindication to MRI (e.g. metal implants or foreign bodies such as from a surgical implant, accident or injury) |
| Interventions | Ketamine versus saline |
| Outcomes | Change in memory between baseline and end of ECT course measured by: Hopkins Verbal Learning Test – Revised (HVLT‐R) Autobiographical Memory Interview ‐ short form (AMI‐SF) Controlled Oral Word Association Test (COWAT) |
| Starting date | 2012‐05‐18 |
| Contact information | ian.anderson@manchester.ac.uk |
| Notes |
IRCT201307181556N54.
| Study name | Riluzole as adjuvant therapy in the treatment of moderate to severe major depression: a double ‐ blind placebo‐controlled trial IRCT201307181556N54 |
| Methods | The purpose of the present investigation is to assess the efficacy of riluzole as an adjuvant agent in the treatment of major depression in a six‐week double‐blind, placebo controlled trial. 40 adult outpatients or inpatients who meet the DSM‐ IV‐TR criteria for major depression will participate in the trial. Patients who have a baseline HRSD score of at least 22 will be allocated into two groups. 20 patients will receive citalopram 40 mg/day plus riluzole 100 mg/day and 20 participants will receive citalopram 40 mg/day plus placebo. Patients were assessed by a psychiatrist at baseline and after 2, 4 and 6 weeks after the medication started. Depression severity will be assessed by HRSD which will be the primary outcome measure |
| Participants | 40 adult outpatients or inpatient who meet the DSM‐ IV‐TR criteria for major depression will participate in the trial. Patients who have a baseline HRSD score of at least 22 will be allocated into two groups |
| Interventions | 20 patients will receive citalopram 40 mg/day plus riluzole 100 mg/day and 20 participants will receive citalopram 40 mg/day plus placebo |
| Outcomes | Depression severity will be assessed by HRSD which will be the primary outcome measure |
| Starting date | 2013‐08‐23 |
| Contact information | Dr. Shahin Akhondzadeh, S.akhond@sina.tums.ac.ir |
| Notes |
NCT00988663.
| Study name | Memantine augmentation of electroconvulsive therapy in patients with major depression |
| Methods | Patients will be assigned randomly either to a treatment group or a placebo groups. All patients in both groups will be receiving standard ECT. The treatment group will receive memantine. All patients will be given a battery of cognitive tests and test of depression before ECT treatments start, after the 6th ECT treatment and after the completions of ECT. An analysis will be performed to see if memantine causes any impact on the response to ECT and prevents memory and cognitive impairment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Memantine versus placebo |
| Outcomes | Primary outcome measures: assessment of whether memantine protects memory and cognitive impairment caused by ECT. Time frame: 6 to 8 weeks |
| Starting date | November 2009 |
| Contact information | mailto:jerry‐lewis%40uiowa.edu?subject=NCT00988663, Memantine ECT trial, The Impact of Memantine on Electroconvulsive Therapy (ECT): Will it Improve Response and Protect Against Cognitive Problems? |
| Notes | NCT00988663 |
NCT01179009.
| Study name | A safe ketamine‐based therapy for treatment‐resistant depression |
| Methods | We will be test whether a 100‐hour ketamine infusion would be more effective than the standard 40‐minute ketamine infusion currently used in other TRD studies. We will randomise participants to one of 2 arms: (1) 100‐hour (+/‐ 4 hours) ketamine infusion plus clonidine for the entire infusion (2) 40‐minute ketamine infusion (plus clonidine) following a 100+/‐ hour saline infusion. All participants will receive clonidine, an alpha‐2 agonist, to minimise side effects of ketamine (namely, brief/mild psychotic and cognitive symptoms) A subset of 20 patients with TRD will receive a 100‐hour (+/‐ 4‐hours) ketamine infusion with head MRIs pre (2) and post (1) infusion. Little research has been done on the mechanism of ketamine's putative antidepressant action. There is now a consensus that, in early stages of the novel treatment development for depression, clinical studies should be paired with mechanistic studies (neuroimaging) to understand the underlying mechanism and validate this as a treatment target. Ketamine is thought to have an antidepressant effect by increasing synaptic connections and therefore increasing connectivity in critical cognitive/emotional circuits Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine 40 minutes versus 100 hour |
| Outcomes | Primary outcome measures: reduction in (1) Clinical Global Interview (CGI) scores and (2) Montgomery‐Asberg Depression Rating Scale (MADRS) scores. Time frame: approximately 5 years. [Designated as safety issue: yes]. Primary Aim 1: to evaluate the efficacy and tolerability of a single safener for the prevention of ketamine‐induced psychotomimetic effects in healthy humans. Primary Aim 2: to evaluate the effect of a standardised IV ketamine plus optimal safener combination treatment on change in the severity of depression in patients with TRD |
| Starting date | April 2012 |
| Contact information | mailto:schweigj%40psychiatry.wustl.edu?subject=NCT01179009, 10‐0000, Treatment Resistant Depression (Pilot) |
| Notes | NCT01179009 |
NCT01204918.
| Study name | Efficacy and tolerability of riluzole in treatment resistant depression |
| Methods | Allocation: randomised Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Group A inclusion/exclusion Inclusioc criteria
Exclusion criteria
Group B inclusion/exclusion Inclusion criteria
Exclusion criteria
|
| Interventions | Riluzole 100 mg versus placebo added to ongoing SSRI or SNRI antidepressant |
| Outcomes | Primary outcome measures: change in Montgomery and Asberg Depression Rating Scale (MADRS). Time frame: 8 weeks of therapy. [Designated as safety issue: No]. This 10‐item instrument is completed by the clinician by using a structured interview and defined anchor points, and aims to quantify the degree of depression over the past 7 days. The MADRS is a widely studied instrument for depression, and its reliability and validity are high. This instrument is administered at every study visit during the double‐blind RCT, and at the screening, and baseline |
| Starting date | June 2011 |
| Contact information | Gerard Sanacora, MD PhD; Yale University |
| Notes | NCT01204918 |
NCT01260649.
| Study name | N‐methyl‐D‐aspartate antagonist (ketamine) augmentation of electroconvulsive treatment for severe major depression |
| Methods | Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, investigator) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus placebo |
| Outcomes | Primary outcome measures: HRSD‐28. Time frame: one month. [ Designated as safety issue: No]. HRSD will be administered at every ECT treatment, and 7‐10 days after last ECT (approximately 1 month after baseline) |
| Starting date | November 2010 |
| Contact information | mailto:ccusin%40partners.org?subject=NCT01260649, 2010P001672, N‐methyl‐D‐aspartate Antagonist (Ketamine) Augmentation of Electroconvulsive Treatment for Severe Major Depression |
| Notes | NCT01260649 |
NCT01441505.
| Study name | A study of ketamine as an antidepressant |
| Methods | This clinical study consists of two phases. In Phase I, participants who satisfy inclusion criteria will receive ketamine at variable doses (0.1 mg/kg to 0.5 mg/kg) or a placebo (saline, or 0.01 mg/kg midazolam) once a week up to 6 weeks. If participants qualify for Phase II, they will receive repeated sessions of ketamine at variable doses over three weeks. During both phases, mood, psychiatric, and neuropsychological outcomes will be measured Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: cross‐over assignment Masking: double‐blind participant, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Drug: ketamine IV, IM, or SC will be administered in Phase I and II Drug: saline or midazolam (active placebo) saline, or midazolam 0.01 mg/kg will be administered in Phase III |
| Outcomes | Primary outcome measures: change from baseline on Depression Rating Scales. Time frame: before, 4 hours after, and 24 hours after ketamine session |
| Starting date | September 2011 |
| Contact information | mailto:TMSandDCS%40unsw.edu.au?subject=NCT01441505, HREC 10409, A Study of Ketamine as an Antidepressant |
| Notes | NCT01441505 |
NCT01557712.
| Study name | Estimate the efficiency of the association of an injection of ketamine and the venlafaxine in the severe major depressive disorder for six weeks (KETADEP) |
| Methods | The objective of this study is to evaluate the effectiveness of ketamine (infusion of 0.5 mg/kg) and venlafaxine compared to the use of venlafaxine alone in the treatment of major depression (MADRS score ≥ 20 ) to six weeks of treatment Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: single group assignment Masking: double‐blind (participant, caregiver, investigator) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | ketamine (infusion of 0.5 mg/kg) and venlafaxine compared to the use of venlafaxine alone |
| Outcomes | Primary outcome measures: depressive state. Time frame: 6 weeks. [Designated as safety issue: yes]. Assessment of depression by MADRS defining six weeks: the state of clinical response defined by a MADRS score less than 50% in MADRS score at baseline initial set.the state of clinical remission is defined by obtaining a MADRS score ≤ 7 |
| Starting date | February 2012 |
| Contact information | mailto:jholtzmann%40chu‐grenoble.fr?subject=NCT01557712, 1129, Estimate the Efficiency of the Association of an Injection of Ketamine and the Venlafaxine in the Severe Major Depressive Disorder for Six Weeks |
| Notes | NCT01557712 |
NCT01558063.
| Study name | The antidepressant action of ketamine: brain chemistry |
| Methods | Ketamine will be given in a dose of 0.0 (placebo), 0.1, 0.2, 0.3, 0.4, or 0.5 mg/kg. If a patient does not respond to ketamine after the first infusion, it may be because s/he received ketamine placebo or the dose of ketamine was too low. In that case, an optional second scan and infusion of active ketamine (0.5 mg/kg) will be offered. This second scan will occur no later than weeks after the first scan/infusion (as scheduling permits). There is no guarantee that the patient will respond to the second ketamine infusion. Patients enrolled in the study are eligible for up to 6 months treatment with their study psychiatrist after the ketamine infusion(s). During this time, patients will be responsible for the cost of the conventional antidepressants but all doctors' visits will be free of charge. Healthy volunteers: healthy controls will receive an infusion of ketamine at a single dose (0.5 mg/kg). Volunteers will only receive one MRI scan and infusion Allocation: randomised Endpoint classification: efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | ketamine 0.1 ‐ 0.5 mg/kg, saline IV |
| Outcomes | Primary outcome measures: ketamine dose‐response curve. Time frame: baseline and Day 1 (post‐ketamine). [Designated as safety issue: no]. The primary outcome is the dose‐response curve as it refers to ketamine inducing a dose‐dependent reduction in the HRSD‐24 scores of patients with major depressive disorder |
| Starting date | February 2012 |
| Contact information | mailto:scolaro%40nyspi.columbia.edu?subject=NCT01558063, 6460, Ketamine in the Treatment of Depression |
| Notes | NCT01558063 |
NCT01613820.
| Study name | Combination of anticholinergic and glutamatergic effects in treatment‐resistant major depressive disorder. A pilot study |
| Methods | We therefore plan to investigate the feasibility and efficacy of open‐label repeated intravenous administration of ketamine and scopolamine combined in this population of severely depressed, treatment‐resistant patients |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketaine + placebo, scopolamine + placebo, ketamine + scopolamine |
| Outcomes | Primary outcome measures: HRSD‐28. Time frame: up to 4 months. [Designated as safety issue: No]. Participants will be assessed with HRSD‐28 |
| Starting date | September 2015 |
| Contact information | Cristina Cusin, M.D., MGH Department of Psychiatry |
| Notes | NCT01613820 |
NCT01667926.
| Study name | Randomized, double‐blind ketamine augmentation in chronically suicidal, treatment‐resistant major depression |
| Methods | Ketamine infusions resulted in an acute reduction in global depression scores and in severity of suicidal ideation. The investigators therefore plan to investigate the feasibility and efficacy of repeated intravenous administration of ketamine in severely depressed, treatment resistant patients |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine/saline 6 infusions of ketamine over three weeks |
| Outcomes | Primary outcome measures: HRSD‐28. Time frame: up to 4 months. [Designated as safety issue: No]. Participants will be assessed with HAM‐D |
| Starting date | January 2013 |
| Contact information | Cristina Cusin, M.D., MGH Department of Psychiatry |
| Notes |
NCT01684163.
| Study name | Phase 2, double‐blind, placebo controlled, randomized withdrawal, parallel efficacy and safety study of GLYX‐13 in participants with inadequate/partial response to antidepressants during the current episode of major depressive disorder |
| Methods | GLYX‐13 is a NMDA receptor glycine site partial agonist being studied in participants with major depressive disorder (depression) who have responded inadequately to another antidepressant drug during the current episode. This trial will assess the effects of GLYX‐13 on depression when added to another antidepressant drug that the patient is already taking |
| Participants | Male and female participants. Aged 18 to 65 years. Meets Diagnostic and Statistical Manual, Fourth Edition, Text Revision (DSM‐IV‐TR) criteria for major depressive disorder (MDD). Current episode has lasted ≥ 8 weeks before. Screening with an inadequate response (< 50% reduction in the Antidepressant Treatment Response Questionnaire [ATRQ]) to all approved antidepressant agent(s) administered at an adequate dose and duration for the current episode. Taking no antidepressant agent currently or taking an SSRI or SNRI from among the following: SSRI: SSRIs: citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, paroxetine CR, sertraline; SNRIs: desvenlafaxine, duloxetine, venlafaxine, venlafaxine XR HRSD‐17 score ≥ 18 at screening. HRSD‐17 score ≥ 18 at predose baseline. Female participants of childbearing potential with a negative serum pregnancy test prior to entry into the study and who are practicing an adequate method of birth control (e.g. oral or parenteral contraceptives, intrauterine device, barrier, abstinence) and who do not plan to become pregnant during the course of the study. Female participants may be included without a negative serum pregnancy test if they are surgically sterile or at least 2 years post‐menopausal |
| Interventions | Experimental Arm 1: GLYX‐13, 5 mg/kg Experimental Arm 2: GLYX‐13, 10 mg/kg Comparator Arm 3: placebo saline injection |
| Outcomes | Primary Outcome Measures: Change in HRSD score. Secondary Outcome Measures: Clinical Global Impression of Change |
| Starting date | October 2012 |
| Contact information | Lee Bastin, RN Ed.D., Naurex, Inc |
| Notes |
NCT01700829.
| Study name | Ketamine versus midazolam: testing rapid relief of suicide risk in depression |
| Methods | Allocation: randomised
Endpoint classification: efficacy study
Intervention model: parallel assignment
Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) Patients currently taking psychiatric medications may continue them during the study. However, if a patient is taking a benzodiazepine (such as ativan, klonopin, or xanax), they will be able to take up to 2 mg per day of lorazepam during the week before the infusion, but none will be permitted in the 24 hours pre‐infusion. Also, zolpidem (ambien) will not be permitted in the 24 hours pre‐infusion. If a person chooses to participate, their dose of benzodiazepine may need to be reduced so that they can do without it during the 24 hours pre‐infusion Depressed participants are randomly assigned to receive a single dose of ketamine(0.5 mg/kg) or midazolam (0.02 mg/kg), which is given slowly, in a vein, over about 40 minutes. The study is "double‐blind," meaning patients and study staff will not know which medication is in the infusion If a patient does not respond to the first infusion, and s/he received midazolam, then s/he will be offered the option of a second infusion, this time with ketamine (0.5 mg/kg). S/he will then start treatment with a standard antidepressant, unless s/he is not already taking one. After the infusion(s), participants will have weekly research interviews for 6 weeks to monitor response. If a patient does have a sufficient infusion response, and s/he is not already taking an antidepressant, then s/he will receive 6 weeks antidepressant research treatment with Sertraline, Fluoxetine, Paroxetine, or Escitalopram, followed by open clinical treatment. However, if s/he is already taking an antidepressant, then s/he will receive open treatment. If s/he does not have a sufficient infusion response, then s/he will receive open treatment Participation in this study requires a brief inpatient stay, at no cost, at the New York State Psychiatric Institute (NYSPI) Eligible participants enrolled in this study will be offered medication management visits at no cost for a total of up to 6 months from the date of enrolment combining inpatient and outpatient treatment. Study medications (sertraline, fluoxetine, paroxetine, escitalopram, lorazepam, zolpidem) will be at no cost during the 6 months. The study will not provide other medications at no cost |
| Participants | Inclusion criteria
EXxclusion criteria
|
| Interventions | Ketamine versus midazolam |
| Outcomes | Primary outcome measures: reduction of suicidal ideation. Time frame: At 24 hours post‐infusion. [Designated as safety issue: No]. Reduction of suicidal ideation in depressed patients with moderate to severe suicidal thoughts from the pre‐infusion baseline to 24 hours after the infusion with ketamine or midazolam, a sedative not known to reduce suicidal ideation |
| Starting date | June 2012 |
| Contact information | mailto:marverj%40nyspi.columbia.edu?subject=NCT01700829, #6598, Ketamine in the Treatment of Suicidal Depression |
| Notes | NCT01700829 |
NCT01703039.
| Study name | Riluzole augmentation pilot in depression (RAPID) trial |
| Methods | Allocation: randomised Endpoint classification: efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants |
Exclusion criteria
Disallowed therapies include: other psychotropic medications, including antipsychotics, mood stabilisers, benzodiazepines, barbiturates, other sedative‐hypnotics, chronic opiates, or additional antidepressants, psychotherapy, electroconvulsive therapy, vagal nerve stimulations therapy, transcranial magnetic stimulation therapy, or phototherapy |
| Interventions | Sertraline + riluzole versus placebo |
| Outcomes | Primary outcome measures: mean change in HRSD score from baseline to endpoint at 8 weeks. Time frame: 8 weeks. [Designated as safety issue: yes] Proportion of patients experiencing an antidepressant response (> 50% reduction in HRSD) at endpoint of 8 weeks. Time frame: 8 weeks. [Designated as safety issue: no] Proportion of patients experiencing remission from depression (HRSD < 7) at endpoint of 8 weeks. Time frame: 8 weeks. [Designated as safety issue: no] |
| Starting date | January 2013 |
| Contact information | mailto:DWOLFE%40PARTNERS.ORG?subject=NCT01703039, 2012P001841, Riluzole Augmentation Pilot in Depression (RAPID) Trial |
| Notes | NCT01703039 |
NCT01868802.
| Study name | Ketamine for treatment‐resistant depression: a multicentric clinical trial in Mexican population |
| Methods | A randomised multicentric parallel arms study involving the use of ketamine for treatment‐resistant depression will be held at three national health provider clinics in the Mexican population Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus placebo |
| Outcomes | Primary outcome measures: changes in baseline HRSD Score. Time frame: 20 minutes before and 40 minutes after ketamine infusion. [Designated as safety issue: no]. The HRSD baseline score will be measured 20 minutes before ketamine infusion. After 40 minutes post‐infusion, a second HRSD score will be obtained |
| Starting date | September 2013 |
| Contact information | mailto:paul%40lamothe.com?subject=NCT01868802, ABC KET‐DRT‐01‐2013, Ketamine for Treatment‐resistant Depression: A Multicentric Clinical Trial in Mexican Population |
| Notes | NCT01868802 |
NCT01880593.
| Study name | Ketamine plus lithium as a novel pharmacotherapeutic strategy in treatment‐resistant depression |
| Methods | The purpose of this study is to test the antidepressant effect of ketamine when given repeatedly over a period of 1 week, as well as the use of Lithium as a relapse‐prevention strategy for patients with treatment‐resistant depression (TRD) who respond to an initial series of ketamine infusions. Ketamine is a Food and Drug Administration approved anaesthetic (a drug used to produce loss of consciousness before and during surgery). Ketamine is not approved for the treatment of major depressive disorder and is considered experimental in this study. An additional purpose of this study is to research the effects of ketamine on brain function. You may qualify to take part in this research study because you have been diagnosed with major depressive disorder (MDD) and have not responded to past treatments |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine + lithium versus placebo |
| Outcomes | Primary outcome measures: Montgomery Asberg Depression Rating Scale. Time frame: 2 weeks after last ketamine infusion |
| Starting date | July 2013 |
| Contact information | mailto:jaclyn.schwartz%40mssm.edu?subject=NCT01880593, GCO 13‐0365, Ketamine Plus Lithium in Treatment‐Resistant Depression |
| Notes | NCT01880593 |
NCT01881763.
| Study name | Comparing therapeutic efficacy and cognitive side effects of electroconvulsive therapy (ECT) using ketamine versus methohexital anesthesia |
| Methods | Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus methohexital |
| Outcomes | Primary outcome measures: Time to achieve remission. Time frame: days required to achieve remission (on average 3‐4 weeks). [Designated as safety issue: No]. Remission is defined as two consecutive HRSD‐24 scores < 10, and HRSD‐24 total score does not increase > 3 points on the second consecutive HRSD‐24, or remains < 6 at the last two consecutive treatments. HRSD‐24 scores are used to define remission |
| Starting date | June 2010 |
| Contact information | mailto:skaliora%40nshs.edu?subject=NCT01881763, 10‐127, Ketamine as an Augmentation Strategy for Electroconvulsive Therapy (ECT) in Depression |
| Notes | NCT01881763 |
NCT01882829.
| Study name | Targeting the NMDA glutamate receptor as novel antidepressant strategy: a pilot clinical trial of nuedexta in treatment‐resistant major depression |
| Methods | The current project aims to test the safety, tolerability and efficacy of nuedexta ‐ containing the NMDA antagonist Endpoint classification: safety/efficacy study Intervention model: single group assignment Masking: open‐label Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: nuedexta (dextromethorphan/quinidine) 45/10 mg every 12 hours x 8 weeks |
| Outcomes | Primary outcome measures: change in Montgomery‐Asberg Depression Rating Scale. Time frame: at baseline and visit 6 (week 10). [Designated as safety issue: No]. The Montgomery‐Asberg Depression Rating Scale is a 10‐item instrument used for the evaluation of depressive symptoms in adults and for the assessment of any changes to those symptoms. Each of the 10 items is rated on a scale of 0 to 6, with differing descriptors for each item. These individual item scores are added together to form a total score, which can range between 0 and 60 points. The MADRS is specifically designed to detect changes in depression severity in the context of a medication treatment trial and is the used as the primary outcome of the current study. Primary outcome is change in MADRS at Visit 6 (Week 10) |
| Starting date | July 2013 |
| Contact information | mailto:seharish.moughal%40mssm.edu?subject=NCT01882829, GCO 13‐0389, Nuedexta in Treatment‐Resistant Major Depression |
| Notes | NCT01882829 |
NCT01887990.
| Study name | Treatment of suicidal ideation with intravenous ketamine infusion |
| Methods | Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus saline |
| Outcomes | Primary outcome measures: suicidality. Time frame: 2 weeks. [Designated as safety issue: Yes]. Scales and questionnaires Beck Scale for Suicidal Ideation |
| Starting date | May 2012 |
| Contact information | mailto:rshelton%40uab.edu?subject=NCT01887990, F120307001, Treatment of Suicidal Ideation With Intravenous Ketamine Infusion |
| Notes | NCT01887990 |
NCT01902004.
| Study name | Treatment of geriatric depression with mild cognitive impairment: a double‐blind placebo‐controlled trial of namenda (memantine) augmentation of lexapro (escitalopram) in depressed patients at least 60 years of age |
| Methods | Double‐blind placebo‐controlled trial of namenda (memantine) as an augmentation to lexapro (escitalopram) in depressed older adults 60 years of age and older Enrolled participants will be provided with 10‐20 mg of escitalopram for 12 months, and concurrently randomly assigned to either memantine or placebo groups Allocation: randomised Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | escitalopram + placebo/memantine |
| Outcomes | Primary outcome measures: Change in HRSD scores. Time frame: measured at 6 months and 12 months |
| Starting date | October 2013 |
| Contact information | mailto:nstcyr%40mednet.ucla.edu?subject=NCT01902004, R‐01 MH097892, Brain Aging and Treatment Response in Geriatric Depression |
| Notes | NCT01902004 |
NCT01920555.
| Study name | Double‐blind, placebo‐controlled trial of ketamine therapy in treatment‐resistant depression (TRD) |
| Methods | The primary objective is to investigate whether all doses (0.1 mg/kg, 0.2 mg/kg, 0.5 mg/kg, and 1.0 mg/kg) of ketamine are superior to active placebo (midazolam 0.045 mg/kg) therapy in the acute treatment of patients with treatment resistant depression within 72 hours (Day 3), when added to ongoing and stable antidepressant therapy |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine 0.1, 0.2, 0.5, 1 mg/kg versus midazolam |
| Outcomes | Primary outcome measures: HRSD‐6. Time frame: past 24 hours. [Designated as safety issue: No]. This instrument is completed with a structured interview guide by the clinician based on his/her assessment of the patient's symptoms. This structured interview has been validated for use with time frames shorter than one week. The time frame for this scale is the past 24 hours |
| Starting date | December 2014 |
| Contact information | mailto:kgilardi%40partners.org?subject=NCT01920555, RAP‐003, Double‐Blind, Placebo‐Controlled Trial of Ketamine Therapy in Treatment‐Resistant Depression (TRD) |
| Notes | NCT01920555 |
NCT01935115.
| Study name | A prospective randomized double‐blinded control trial using ketamine or propofol anesthesia for electroconvulsive therapy: improving treatment‐resistant depression |
| Methods | Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | ketamine 0.75 mg/kg intravenously versus propofol 1 mg/kg |
| Outcomes | Primary outcome measures: The primary outcome is defined as the number of ECT treatments required to reach a 50% reduction in baseline MADRS (Montgomery‐Asberg Depression Scale) score. Time frame: after 8 treatments or completion of therapy for an expected average of 4 weeks |
| Starting date | September 2013 |
| Contact information | mailto:J_Gamble%40yahoo.com?subject=NCT01935115, UofSKetamine‐01, Comparing Ketamine and Propofol Anesthesia for Electroconvulsive Therapy |
| Notes | NCT01935115 |
NCT01944293.
| Study name | Ketamine versus midazolam in bipolar depression |
| Methods | Allocation: randomised Endpoint classification: efficacy study Intervention Model: parallel assignment Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus midazolam |
| Outcomes | Primary outcome measures: Reduction of suicidal ideation measured with the Beck Scale for Suicidal Ideation. Time frame: At 24 hours post‐infusion. [Designated as safety issue: No]. Reduction of suicidal ideation in bipolar disorder during a Major Depressive Episode (MDE), with moderate to severe suicidal thoughts, from the pre‐infusion baseline to 24 hours after the infusion with ketamine or midazolam, a sedative not known to reduce suicidal ideation |
| Starting date | September 2013 |
| Contact information | mailto:marverj%40nyspi.columbia.edu?subject=NCT01944293, #6785, Ketamine for Suicidality in Bipolar Depression |
| Notes | NCT01944293 |
NCT01945047.
| Study name | Phase 2 optimization of the antidepressant action of ketamine in treatment‐resistant depression and investigations on its mechanism of action |
| Methods | The phases of this study compare response to a single KET injection to 6 injections over 2 weeks. Next, KET responders are given 1 injection a week for 3 weeks of either KET or the sedative agent to determine if beneficial effects of KET are maintained, and to assess duration of its benefits after repeated administration |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus midazolam |
| Outcomes | Primary outcome measures: Efficacy of ketamine over midazolam in double‐blind study for efficacy of relief for Major Depressive Disorder. Time frame: 2 weeks. [Designated as safety issue: Yes]. Phase 1 double‐blind treatment with ketamine or midazolam then cross‐over. Will assess efficacy of each for relief of Major Depressive Symptoms through assessment using the HRSD‐17 |
| Starting date | May 2013 |
| Contact information | mailto:wendy.fusee%40theroyal.ca?subject=NCT01945047, REB2012023, Action of Ketamine in Treatment‐Resistant Depression |
| Notes | NCT01945047 |
NCT01957410.
| Study name | An open‐label and double‐blind study to investigate evoked potentials as markers of ketamine‐induced cortical plasticity in participants with major depressive disorder |
| Methods | To evaluate if somatosensory evoked potentials (SEPs) and motor evoked potentials (MEPs) obtained with electroencephalography (EEG) and electromyography (EMG) can be used to detect changes in cortical plasticity in responders to a single IV infusion of ketamine as compared to non‐responders Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus placebo |
| Outcomes | Primary outcome measures: Comparison of change from baseline to 4 hr post‐dose on Day 1 in somatosensory evoked potential amplitudes (SEPs) between ketamine responders and ketamine non‐responders. Time frame: baseline and 4 hours post‐dose on Day. [Designated as safety issue: No]. SEPs are the electrical signals generated by the nervous system in response to somatosensory stimuli ‐ typically through electrical stimulation of the median nerve. SEPs are read on the skull with electroencephalography (EEG). SEPs will be carried out as described by Cornwell and colleagues (Cornwell 2012) with the exception that instead of using magnetoencephalography SEPs will be recorded using a 64‐channel EEG system. Identical procedures will be employed prior to and after study drug administration |
| Starting date | February 2014 |
| Contact information | jnj.ct@sylogent.com |
| Notes | NCT01957410 |
NCT02012335.
| Study name | Ketamine use in electroconvulsive therapy: clinical, cognitives and neurotrophic outcomes |
| Methods | Allocation: randomised
Endpoint Classification: efficacy study
Intervention Model: parallel assignment
Masking: double‐blind (participant, investigator)
Primary Purpose: treatment This study will compare the clinical response to brief pulse ECT with infusion of ketamine 0.5 mg/kg versus brief pulse electroconvulsive therapy with infusion of placebo (saline) in major depression. We also will compare levels of cognitive impairment among these groups, compare levels of quality of life among these groups, compare levels of BDNF among these groups. We also will study if levels of 25‐hydroxyvitamin D are associated with cognitive impairment in participants undergoing ECT |
| Participants | Inclusion criteria ‐ Patients with unipolar and bipolar depression from The Psychiatric Unit of Hospital de Clinicas de Porto Alegre (diagnosis will be established by the Structured Clinical Interview for DSM‐IV Axis I) Exclusion criteria
|
| Interventions | Brief pulse ECT with 0.05 mg/kg ketamine versus saline infusion in each session |
| Outcomes | Primary outcome measures: HRSD‐17 |
| Starting date | January 2014 |
| Contact information | Federal University of Rio Grande do Sul / Hospital de Clínicas de Porto Alegre (HCPA), Porto Alegre, RS, Brazil, 90035‐003 |
| Notes | NCT02012335 |
NCT02014363.
| Study name | Safety and efficacy study comparing ETS6103 with amitriptyline in the treatment of major depressive disorder (MDD) (ETS6103‐003) |
| Methods | Double‐blind, non‐inferiority study to evaluate the antidepressant activity of ETS6103 compared with amitriptyline in the treatment of major depressive disorder (MDD) in patients who have an unsatisfactory response to selective serotonin re‐uptake inhibitors (SSRIs) |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | ETS6103 (Low, high dose), Amitriptyline |
| Outcomes | Primary outcome measures: The mean difference in baseline‐adjusted (Montgomery‐Asberg Depression Scale) MADRS score at the end of treatment. Time frame: 8 weeks. [Designated as safety issue: No]. The MADRS will be measured at every visit |
| Starting date | October 2013 |
| Contact information | mailto:alan%40cpsresearch.co.uk?subject=NCT02014363, ETS6103‐003, Safety and Efficacy Study Comparing ETS6103 With Amitriptyline in the Treatment of Major Depressive Disorder (MDD) |
| Notes |
NCT02037503.
| Study name | Effect of oral ketamine treatment on suicidal ideation and drug resistant major depression, a clinical and fMRI study |
| Methods | In a double‐blind, placebo‐controlled trial, patients admitted to the emergency department after a suicide attempt will be randomised into two groups: one will be given a daily subanaesthetic dose of oral ketamine, while the second group will receive a daily dose of placebo. Participants will be followed up for 21 days. Some of the participants will also undergo functional MRI scans before and after the first ketamine intake Allocation: randomised Endpoint classification: efficacy study Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria Suicidal ideation group:
For the depression group
Exclusion criteria for all groups
|
| Interventions | Ketamine versus saline |
| Outcomes | Primary outcome measures: Resolution of suicidal ideation. Time frame: within 3 weeks of enrolment |
| Starting date | January 2014 |
| Contact information | |
| Notes | NCT02037503 |
NCT02067793.
| Study name | Phase 2, randomized, double‐blind, multiple‐dose level, placebo controlled, single intravenous dose, parallel efficacy and safety study of NRX‐1074 in participants with major depressive disorder |
| Methods | The purpose of this study is to evaluate the efficacy and safety of NRX‐1074 following a single intravenous dose in participants with major depressive disorder |
| Participants | 18 Years to 65 Years Inclusion criteria Male and female participants Aged 18 to 65 years Meets Diagnostic and Statistical Manual, Fourth Edition, Text Revision (DSM‐IV‐TR) criteria for major depressive disorder (MDD) Current episode has lasted ≥ 8 weeks before Screening HRSD‐17 score ≥ 21 before beginning the washout of all current antidepressant agents and/or adjuvant agents HRSD‐17 score ≥ 21 at baseline (after 14 days of washout of current antidepressant agents) Based on both the investigator and Naurex medical monitor's clinical judgment, participants with eating disorders, obsessive compulsive disorder (OCD), panic disorder, post‐traumatic stress disorder (PTSD), and generalised anxiety disorders secondary to major depressive episodes are permitted |
| Interventions | NRX‐1074 1 mg, 5 mg, 10 mg, placebo |
| Outcomes | Primary outcome measures: To evaluate the mean change from baseline in HRSD‐17 score for each NRX‐1074 dose group versus the placebo group's mean change. Time frame: Day 1, Day 3, Day 7, Day 14. [Designated as safety issue: No] |
| Starting date | April 2014 |
| Contact information | Ronald M Burch, MD PhD |
| Notes | NCT02067793 |
NCT02106325.
| Study name | A randomized, double‐blinded controlled trial of an N‐Methyl D‐Aspartate antagonist as a rapidly‐acting antidepressant in depressed emergency department patients |
| Methods | Investigators will conduct a trial to evaluate the use of ketamine as an alternate treatment for people with major depressive disorder. This study plans to explore the potential that ketamine's rapid antidepressant action holds for improving outcomes in patients presenting to the Emergency Department with severe depression. Since this is a controlled trial we will use an IV of Ketamine or and equivalent volume of Diphenhydramine. Sixty participants will be randomly assigned to receive Ketamine (30 participants) or Benadryl (30 participants). Investigators will then compare measures of mood pre‐ and post‐infusion in the Emergency Department. to supplement self‐reported measures of depressive symptoms(e.g. mood), investigators will obtain objective measures of the biological aspects of Major Depressive Disorder. With the current evidence pertaining to our hypothesis, investigators hope to add to the growing scientific studies surrounding such a vulnerable population Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: single group assignment Masking: double‐blind (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Ketamine versus diphenhydramine |
| Outcomes | Primary outcome measures: Evaluate the effects of ketamine on depressive symptomatology. Time frame: 0 ‐ 16 weeks. [Designated as safety issue: No]. Montgomery‐Asberg Depressive Rating Scale |
| Starting date | December 2013 |
| Contact information | mailto:stephen.ross%40nyumc.org?subject=NCT02106325, S13‐00794, Ketamine as a Rapidly‐Acting Antidepressant in Depressed Emergency Department Patients |
| Notes | NCT02106325 |
NCT02139540.
| Study name | Nitrous oxide as treatment for major depression ‐ a pilot study |
| Methods | A pilot randomised placebo‐controlled double‐blind cross‐over study in which patients will receive up to 50% nitrous oxide in oxygen or up to 50% oxygen in air for a period of one hour in addition to standard medical therapy. Depression severity will be assessed by a blinded observer pre‐treatment, 30 minutes and 2 hours post‐treatment using the HRSD Allocation: randomised Endpoint classification: safety/efficacy study Intervention model: cross‐over assignment Masking: double‐blind (participant, caregiver, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Inclusion Criteria:
Exclusion criteria History of:
|
| Interventions | N2O/placebo |
| Outcomes | Primary outcome measures: Change in HRSD‐21. Time frame: baseline and 24 hours |
| Starting date | November 2012 |
| Contact information | Peter Nagele, MD, MSc; Washington University School of Medicine |
| Notes | NCT02139540 |
NCT02153502.
| Study name | A Phase 2, multicenter, randomized, double‐blind, placebo‐controlled study to assess the efficacy, safety, and tolerability of AVP‐786 (deuterium modified dextromethorphan hydrobromide/quinidine sulfate) as an adjunctive therapy in patients with major depressive disorder with an inadequate response to antidepressant treatment |
| Methods | The objectives of this 10‐week study are to evaluate the efficacy, safety, and tolerability of AVP 786 as an adjunctive therapy compared with placebo in patients with major depressive disorder (MDD) who have shown an inadequate response to standard antidepressant treatment. A secondary objective of this study is to assess the pharmacokinetics (PK) of AVP‐786 and potential correlations with pharmacodynamic effects |
| Participants |
|
| Interventions | AVP‐786 versus placebo |
| Outcomes | Primary outcome measures: Montgomery‐Ǻsberg Depression Rating Scale (MADRS) total score |
| Starting date | July 2014 |
| Contact information | mailto:ldoan%40avanir.com?subject=NCT02153502, 14‐AVP‐786‐201, Efficacy, Safety, and Tolerability Study of AVP‐786 as an Adjunctive Therapy in Patients With Major Depressive Disorder With an Inadequate Response to Antidepressant Treatment |
| Notes | NCT02153502 |
NCT02267980.
| Study name | Effect of the addition of ketamine to sevoflurane anesthesia in electroconvulsive therapy |
| Methods | Patients will randomly allocate, to either a sevoflurane‐ketamine (Group SK), sevoflurane‐saline (Group SS) receiving group. Mean arterial pressure (MAP) and heart rate (HR) will record prior to anaesthetic induction (T1); following anaesthetic induction (T2); and 0, 1, 3, and 10 min after the seizure has ended (T3, T4, T5, and T6, respectively). Motor and EEG seizure durations will be recorded |
| Participants | 18 Years to 70 Years Inclusion Criteria: major depressive patients |
| Interventions | Sevoflurane‐ketamine (Group SK) or sevoflurane‐saline (Group SS) |
| Outcomes | Primary outcome measures: seizure duration. Time frame: During electroconvulsive therapy (30 minutes). [Designated as safety issue: Yes]. The time from application of the ECT stimulus to the cessation of tonic‐clonic motor activity in the isolated arm |
| Starting date | July 2014 |
| Contact information | Contact: Feray Erdil, MD feray.erdil@inonu.edu.tr |
| Notes | NCT02267980 |
NCT02295787.
| Study name | Intranasal ketamine for late‐life depression and suicidal ideation |
| Methods | A single subanaesthetic dose (0.5 mg/kg, intravenous) of ketamine (an anti‐glutamatergic compound) rapidly (within 24 hours) and robustly decreases depression and suicidality in treatment‐resistant patients, and this effect is sustained for about one week. Intranasal administration has similar effects, good tolerability, and significantly easier administration. We propose to investigate intranasal ketamine as a novel anti‐suicidal treatment for patients with LLD and suicidal ideation. This study will be a randomised, placebo‐controlled, double‐blind trial; intranasal doses will be administered 50 mg every three days. We will enrol 30 patients with a diagnosis of LLD. All patients must be depressed, must endorse some level of suicidal ideation, and must not have a history of psychosis or substance misuse disorders The proposed study involves three phases: 1) 2‐week medication stabilisation period; 2) double‐blind treatment with intranasal ketamine versus placebo, with visits every three days for six total administrations; 3) prospective follow‐up for 3 months, with visits every two weeks. Clinical assessment scales to be administered at each visit will include the HRSD; primary outcome measure; Columbia‐Suicide Severity Rating Scale (C‐SSRS); Clinical Global Impression (CGI); and side‐effect measures. Self‐rated scales measuring depression, anxiety, level of functioning, and quality of life will also be administered. In accordance with NIMH research guidelines for patients at risk for suicide, and in collaboration with the IRB, a detailed safety plan has been developed for the management of worsening suicidality ‐ and has been implemented for other depression studies in our department. All adverse events will be recorded to determine tolerability. To analyse the efficacy data, we will apply a linear random mixed model for longitudinal data. The change from baseline to post‐baseline times will be the dependent variable, with time as the independent variable; response will be defined as ≥ 50% improvement from baseline on the HRSD. Kaplan‐Meier survival estimates will be used to analyse time‐to‐relapse If intranasal ketamine is found to be safe, effective, and well‐tolerated in patients with LLD and suicidal ideation, it can clinically impact outpatient psychiatric settings. From a research perspective, the discovery of a rapidly‐acting anti‐suicidal agent for treating LLD and suicidal ideation has the potential to: 1) decrease the morbidity and mortality that results from late‐life depression; 2) offer new insights into the neurobiology of depression in older age; and 3) pave the way for the development of other novel therapeutics and treatment targets |
| Participants | Ages eligible for study: 65 Years and older Genders eligible for study: Both Accepts healthy volunteers: No Inclusion criteria 1) be ≥ 65 years old, 2) provide written informed consent, 3) meet criteria for a primary psychiatric diagnosis of major depressive disorder according to the Structured Clinical Interview for DSM‐IV (SCID) and have a HRSD‐28 total score ≥ 20; depression may have started at any time point in their life, and certain comorbid diagnoses (e.g., anxiety disorders) will be allowed insofar as they are not the primary psychiatric diagnosis, 4) have a history of ≥ 2 failed medication trials during the current episode (per the Massachusetts General Hospital Antidepressant Treatment History Questionnaire; MGH ATRQ), 5) endorse suicidal ideation for more than 2 months, per the Columbia Suicide Severity Rating Scale (C‐SSRS) and have a HRSD‐28 suicide item score ≥ 1 (thoughts that life isn't worth living), 6) be on a stable antidepressant regimen for ≥ 14 days prior to Study Phase II, 7) maintain a treating psychiatrist who is in agreement with study participation, and 8) have a reliable chaperone accompany them home following the completion of the intranasal administration Exclusion criteria •Patients will be excluded if any of the following criteria are met: 1) delirium or dementia diagnosis, 2) unstable medical illness, 3) history of clinically significant cardiovascular disease or electrocardiogram (EKG) findings, or medical conditions that put the patient at risk for possible cardiac side effects, 4) history of multiple adverse drug reactions, 5) current/past history of psychotic disorders or homicidality, 6) active substance use disorders (except nicotine and caffeine) within the past six months, positive urine toxicology screen, or past history of ketamine abuse, 7) requirement of excluded medications (narcotics, barbiturates, theophylline, or St. John's Wort), or 8) concurrent or recent participation in other research studies |
| Interventions | Ketamine versus placebo |
| Outcomes | Primary outcome measures: HRSD |
| Starting date | October 2015 |
| Contact information | Dawn F Ionescu, MD, mailto:dionescu%40partners.org?subject=NCT02295787, 2014D006212, Intranasal Ketamine for Late‐Life Depression and Suicidal Ideation |
| Notes | NCT02295787 |
NCT02299440.
| Study name | Evaluation of the effects of ketamine in the acute phase of suicidal ideation: a multicenter randomized double‐blind trial |
| Methods | EXPERIMENTAL: Ketamine ‐ Patients randomised to this group will be treated via Ketamine infusion Intervention: Baseline evaluation Intervention: 1st perfusion of ketamine Intervention: Follow‐up between perfusions Intervention: 2nd perfusion of ketamine Intervention: Follow‐up after perfusions. Other: Baseline evaluation Before perfusions begin, each patient will have a baseline evaluation including the following: the Columbia Suicide Severity Rating Scale (CSSRS), the Beck Scale for Suicide Ideation (BSSI), a physical pain VAS (visual analogue scale), a mental pain VAS, the Clinical Global Impressions Scale (CGI‐S), Beck's Hopeless scale (BHS), the 16‐item Quick Inventory of Depressive Symptomatology (QIDS‐SR16), the Inventory of Depressive Symptomatology for the Clinician (IDS‐C30), the Patient Rated Inventory of Side Effects (PRISE), the Young Mania Rating Scale (YMRS), and the Brief Psychiatric Rating Scale (BPRS). Drug: 1st perfusion of ketamine A is performed: 0.5 mg/kg diluted in saline, administered over 40 minutes by intravenous (IV) pump and cardiorespiratory monitoring (Day 0). Other: Follow‐up between perfusions. Patients will be re‐evaluated with a selection of questionnaires at 40 minutes, 120 minutes, 4 hours, and 24 hours after the end of the first perfusion, and then again at 48 hours after the end of the first perfusion and right before the second perfusion. Drug: 2nd perfusion of ketamine A is performed: 0.5 mg/kg diluted in saline, administered over 40 minutes by intravenous (IV) pump and cardiorespiratory monitoring (Day 2). Other: Follow‐up after perfusions. Patients will be re‐evaluated with a selection of questionnaires at 40 minutes, 120 minutes, and 4 hours after the end of the second perfusion, and then again at Day 3, Day 4, Week 2, Week 4 and Week 6 COMPARATOR: Placebo/control (saline) |
| Participants | 18 Years and older Inclusion criteria: French speaking patients freely hospitalised for prevention of suicide and who have a medium or high suicide risk score according to a MINI structured interview. The patient is able to understand how the study is carried out and the tests performed. The patient is deemed capable of giving his/her informed consent. The patient has been correctly informed. The patient must have given his/her informed and signed consent. The patient must be insured or beneficiary of a health insurance plan. Presence of suicidal ideation according to the SSI score (score > 3). Negative pregnancy test for women of childbearing age |
| Interventions | Ketamine versus placebo |
| Outcomes | Columbia Suicide Severity Rating Scale (CSSRS), the Beck Scale for Suicide Ideation (BSSI), a physical pain VAS (visual analogue scale), a mental pain VAS, the Clinical Global Impressions Scale (CGI‐S), Beck's Hopeless scale (BHS), the 16‐item Quick Inventory of Depressive Symptomatology (QIDS‐SR16), the Inventory of Depressive Symptomatology for the Clinician (IDS‐C30), the Patient Rated Inventory of Side Effects (PRISE), the Young Mania Rating Scale (YMRS) and the Brief Psychiatric Rating Scale (BPRS) |
| Starting date | April 2015 |
| Contact information | Contact: Mocrane Abbar, MD |
| Notes | NCT02299440 |
NCT02305394.
| Study name | Effect of subanesthetic dose of ketamine combined with propofol on cognitive function in depressive patients undergoing electroconvulsive therapy ‐ a randomized control double‐blind clinical trial |
| Methods | Propofol 1.5 mg/kg and ketamine 0.3 mg/kg will be administered to participants separately by intravenous infusion.When patients become unconscious, succinylcholine 1 mg/kg (a muscle relaxant) will be administered intravenously. After 1 minute of succinylcholine infused, ECT will be performed with bitemporal electrode placement using a stimulus dose of 1.0 millisecond pulse width, 60 Hz frequency, 6.0 second stimulus duration, and 0.8‐A maximal stimulus intensity |
| Participants | 18 years to 65 years Inclusion Criteria: diagnosed with moderate or severe depression according to Diagnostic and Statistical Manual of Mental Disorders |
| Interventions | Arm 1: propofol 1.5 mg/kg and ketamine 0.3 mg/kg will be administered to participants separately by intravenous infusion Arm 2: propofol 1.5 mg/kg and normal saline [weight(kg)×0.3÷10] ml |
| Outcomes | Primary outcome measures: Mini‐Mental State examination score. Time frame: at 24 hours after the sixth ECT. [Designated as safety issue: No]. Mini‐Mental State examination score will be measured at 24 hours after the sixth ECT |
| Starting date | January 2015 |
| Contact information | Qibin Chen mailto:403497559%40qq.com?subject=NCT02305394, CYYYMZ‐006, Effect of Subanesthetic Dose of Ketamine Combined With Propofol on Cognitive Function in Depressive Patients Undergoing Electroconvulsive Therapy |
| Notes | NCT02305394 |
NCT04082858.
| Study name | Ketamine Interleaved With Electroconvulsive Therapy for Depression |
| Methods | Pragmatic, randomised, controlled, parallel group, pilot clinical trial |
| Participants |
|
| Interventions | Experimental: ketamine Participants will receive twice‐weekly infusions of ketamine at 0.05mg/kg for the duration of treatment with ECT. All infusions will be administered by a Consultant Anaesthetist. Active Comparator: Midazolam Participants will receive twice‐weekly infusions of midazolam at 0.045mg/kg for the duration of treatment with ECT. All infusions will be administered by a Consultant Anaesthetist. |
| Outcomes | Primary Outcome Measures :Montgomery Asberg Depression Rating Scale (MADRS) [ Time Frame: 18 weeks ] Secondary Outcome Measures : The Quick Inventory of Depressive Symptoms, self‐report version (QIDS‐SR16) [ Time Frame: 18 weeks ] The Clinician‐Administered Dissociative States Scale (CADSS) [ Time Frame: 6 weeks ] The Brief Psychiatric Rating Scale (BPRS) [ Time Frame: 6 weeks ] Young Mania Rating Scale (YMRS; mood item) [ Time Frame: 6 weeks ] The Patient‐Rated Inventory of Side Effects (PRISE) [ Time Frame: 6 weeks ] The Montreal Cognitive Assessment (MoCA) [ Time Frame: 18 weeks ] |
| Starting date | January 6, 2020 |
| Contact information | d.mcloughlin@tcd.ie |
| Notes | NCT04082858 |
NCT04116528.
| Study name | Opiate Suicide Study in Patients With Major Depression (AFSP) |
| Methods | RCT |
| Participants | Diagnosed with Major Depressive Disorder (MDD), single or recurrent, and currently experiencing a Major Depressive Episode (MDE) of at least eight weeks in duration, prior to screening, according to the criteria defined in the Diagnosis and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision(DSM‐IV‐TR™). |
| Interventions | Ketamine Every eligible participant will receive 0.5mg/kg IV given over 40 minutes Active Comparator: Buprenorphine or Placebo Buprenorphine or placebo once daily for 4 weeks |
| Outcomes | Beck Suicidal Ideation Scale total score opioid activity of ketamine as well as buprenorphine Serum prolactin level Pupillometry |
| Starting date | August 1, 2020 |
| Contact information | jhawk@stanford.edu |
| Notes | NCT04116528 |
NCT04234776.
| Study name | Intramuscular Ketamine Versus Aripiprazole and Escitalopram in the Treatment of Resistant Depression |
| Methods | RCT |
| Participants |
|
| Interventions | Experimental: Rapid‐acting antidepressant Particpants eligible to participate in the study will receive IM ketamine and will use 2 placebo tablets as randomized. Active Comparator: Comparator Participants eligible to participate in the study will receive IM saline and will use escitalopram 15 mg and aripiprazole 5 mg as randomised |
| Outcomes | CHANGE IN DEPRESSIVE SYMPTOMS ASSESSED WITH Montomery‐Åsberg Depression Rating Scale 3 times a week in once month(phase II) Once a week in six months (phase III) Once a week in once month (Phase IV) 46 secondary outcome measures |
| Starting date | April 3, 2018 |
| Contact information | Ricardo A Moreno, MD, PhD, Department and Institute of Psychiatry, University of Sao Paulo |
| Notes | NCT04234776 |
NCT04399070.
| Study name | The Effect of S‐ketamine for Patients Undergoing Electroconvulsive Therapy (ECT) (ECT) |
| Methods | RCT |
| Participants |
|
| Interventions | Placebo Comparator: Propofol group patients were treated with propofol 1 mg/kg and saline bolus infusion before ECT Active Comparator: Ketamine group patients were treated with propofol 1 mg/kg and ketamine 0.5 mg/kg bolus infusion before ECT Experimental: S‐ketamine group patients were treated with propofol 1 mg/kg and S‐ketamine 0.25 mg/kg bolus infusion before ECT |
| Outcomes |
|
| Starting date | August 1, 2020 |
| Contact information | qiuyan_mz@126.com |
| Notes | NCT04399070 |
NTR3753.
| Study name | ECT and memantine |
| Methods | DB RCT |
| Participants | DSM‐IV criteria for unipolar or bipolar depression and a clinical indication for electroconvulsive treatment (ECT) |
| Interventions |
|
| Outcomes | Scores on a standard cognitive test battery. This test is administered before, during and after ECT and at follow‐up. This test battery has been proven to be sensitive to cognitive side‐effects of depression and also for the ECT effects in depression |
| Starting date | 1‐dec‐2012 |
| Contact information | r.kok@parnassia.nl |
| Notes |
Phillips 2020.
| Study name | A randomized, crossover comparison of ketamine and electroconvulsive therapy for treatment of major depressive episodes: a Canadian biomarker integration network in depression (CAN‐BIND) study protocol. |
| Methods | RCT |
| Participants | 240 patients with major depressive disorder or bipolar disorder experiencing a MDE |
| Interventions | Randomised (1:1) to a course of ECT or racemic IV ketamine (0.5 mg/kg) administered 3 times/week for 3 or 4 weeks |
| Outcomes | The primary outcome measure is change in MADRS scores after randomised treatment as assessed by raters blind to treatment modality. |
| Starting date | Registered September 17, 2018. |
| Contact information | Jennifer.Phillips@theroyal.ca |
| Notes | Phillips 2020 |
ASA: American Society of Anaesthesiologists;BMI: body mass index;CGI: Clinical Global Impression scale;COPD: chronic obstructive pulmonary disease; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition; ECT: electroconvulsive therapy; HDRS: Hamilton Depression Rating Scale; IM: intramuscular; IV: intravenous; MADRS: Montgomery‐Asberg Depression Rating Scale; MDD: Major Depressive Disorder; MRI: magnetic resonance imaging; MMSE: Mini Mental State Examination; PI: principal investigator; RCT: randomised controlled trial; SC: subcutaneous; SCID: Structured Clinical Interview for Depression; ULN: upper limit of normal.
Differences between protocol and review
Considering a significant proportion of included studies recruited patients with treatment‐resistant depression, we decided to add ECT as one of the active comparisons. Due to the lack of data available on quality of life, it was decided to include any validated measure of the outcome.
We removed the third objective, (’to investigate the adverse effects of ketamine and other glutamate receptor modulators in unipolar major depressive disorder, including general prevalence of adverse effects, compared with placebo or other antidepressant agents’) in order to make it clearer that whilst we did do a search for adverse events data, in the end we only included data from RCTs.
In order to address the comments of the peer reviewers, we decided to use a different threshold for depression severity (25 rather than 27 on HRSD‐17), and changed the references accordingly.
Extra detail was added about the implementation of the random‐effects model (see Data synthesis). The protocol stated: "We will use a random‐effects model because it has the highest generalisability for empirical examination of summary effect measures in meta‐analyses (Furukawa 2002). We will routinely examine the robustness of this summary measure by calculating the fixed‐effect model and random‐effects model ORs. We will report material differences between the models.We will calculate the pooled MD or SMD as appropriate with corresponding 95% CI for continuous outcomes. We will also use the random‐effects model for continuous outcomes. However, we will also routinely perform fixed‐effect analyses to investigate the effect of the choice of method on the effect estimates. We will report material differences between the models."
Contributions of authors
AC, RMcS, and KH conceived the review. RD, AB, CH, RS, SH, and SS selected the studies, appraised their quality and extracted data. RD, AB, RS, SH and SS entered the data into RevMan and RD carried out the analyses. RD and AC drafted the manuscript and all other authors critically reviewed the text.
Sources of support
Internal sources
University of Oxford, UK
External sources
No sources of support provided
Declarations of interest
Rebecca Dean: none known.
Claudia Hurducas: none known.
Sarah Hollingsworth: none known.
Tahnee Marquardt: none known.
Stella Spyridi: none known.
Phil Cowen: PJC has a patent with the University of Oxford on the use of ebselen in treatment‐resistant depression
Keith Hawton: none known.
Rupert McShane: Rupert McShane runs NHS and self‐pay ketamine clinics for Oxford Health NHS Foundation Trust. Rupert has undertaken educational and scientific advisory board work for Janssen Pharmaceuticals to support educational and research activity, no funds are received personally. Janssen supported Rupert's attendance at the APA conference in New York in 2018. Rupert has undertaken scientific advisory board work for Sage pharmaceuticals, no funds are directly received.
Erick Turner: Erick Turner has no financial interest in esketamine, ketamine or competing treatments. Erick previously worked as a Medical Officer for the US Food and Drug Administration (FDA), charged with reviewing applications submitted by pharmaceutical companies and determining whether the evidence on drug efficacy and safety met the FDA’s criteria for US marketing approval. Erick is a former member of the Psychopharmacologic Drugs Advisory Committee which, together with the Drug Safety and Risk Management Advisory Committee, convened in Feb 2019, to advise the FDA on whether to approve esketamine, although Erick did not take part in that particular meeting due to matters related to the government shutdown.
Andrea Cipriani: Andrea Cipriani has received research and consultancy fees from INCiPiT (Italian Network for Paediatric Trials), CARIPLO Foundation and Angelini Pharma.
Edited (no change to conclusions)
References
References to studies included in this review
Abbasinazari 2015 {published data only}
- Abbasinazari M, Adib-Eshgh L, Rostami A, Beyraghi N, Dabir S, Jafari R. Memantine in the prevention or alleviation of electroconvulsive therapy induces cognitive disorders: a placebo controlled trial. Asian Journal of Psychiatry 2015;15:5-9. [DOI: 10.1016/j.ajp.2015.04.002] [DOI] [PubMed] [Google Scholar]
Amidfar 2016 {published data only}
- Amidfar M, Khiabany M, Kohi A, Salardini E, Arbabi M, Roohi Azizi M, et al. Effect of memantine combination therapy on symptoms in patients with moderate-to-severe depressive disorder: ramdomized, double-blind, placebo-controlled study. Journal of Clinical Pharmacy and Therapeutics 2016;42(1):44-50. [DOI] [PubMed] [Google Scholar]
Anderson 2017 {published data only}
- Anderson IM, Blamire A, Branton T, Clark R, Downey D, Dunn G, et al. Ketamine augmentation of electroconvulsive therapy to improve neuropsychological and clinical outcomes in depression (Ketamine-ECT): a multicentre, double-blind, randomised, parallel-group, superiority trial. Lancet Psychiatry 2017;4(5):365-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Arabzadeh 2018 {published data only}
- Arabzadeh S, Hakkikazazi E, Shahmansouri N, Tafakhori A, Ghajar A, Jafarinia M, et al. Does oral administration of ketamine accelerate response to treatment in major depressive disorder? Results of a double-blind controlled trial. Journal of Affective Disorders 2018;235:236-41. [DOI] [PubMed] [Google Scholar]
Berk 2014 {published and unpublished data}
- Berk M, Dean OM, Cotton SM, Jeavons S, Tanious M, Kohlmann K, et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. Journal of Clinical Psychiatry 2014;75:628-36. [DOI] [PubMed] [Google Scholar]
Berman 2000 {published and unpublished data}
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biological Psychiatry 2000;47:351-4. [DOI] [PubMed] [Google Scholar]
Canuso 2018 {published and unpublished data}
- Canuso CM, Singh JB, Fedgchin M, Alphs L, Lane R, Lim P, et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind randomised placebo-controlled study. American Journal of Psychiatry 2018;175(7):620630. [DOI: 10.1176/appi.ajp.2018.17060720] [DOI] [PubMed] [Google Scholar]
Carspecken 2018 {published data only}
- Carspecken CW, Borisovskaya A, Lan S-T, Heller K, Buchholz J, Ruskin D, et al. Ketamine anesthesia does not improve depression scores in electroconvulsive therapy: a randomised clinical trial. Journal of Neurosurgical Anesthesiology 2018;30(4):305-13. [DOI: 10.1097/ANA.0000000000000511] [DOI] [PubMed] [Google Scholar]
Chen 2017 {published data only}
- Chen Q, Dong J, Luo J, Ren L, Min S, Hao X, et al. Effects of low-dose ketamine on the antidepressant efficacy and suicidal ideations in patients undergoing electroconvulsive therapy. Journal of ECT 2020;36(1):25-30. [DOI: 10.1097/YCT.0000000000000636] [DOI] [PubMed] [Google Scholar]
- Chen Q, Min S, Hao X, Peng L, Meng H, Luo Q, et al. Effect of Low dose of ketamine on learning memory function in patients undergoing electroconvulsive therapy-a randomized, double-blind, controlled clinical study. Journal of ECT 2017;33(2):89-95. [DOI] [PubMed] [Google Scholar]
Chen 2018 {published data only}
- Chen MH, Li CT, Lin WC, Hong CJ, Tu PC, Bai YM, et al. Persistent antidepressant effect of low-dose ketamine and activation in the supplementary motor area and anterior cingulate cortex in treatment resistant depression: a randomized control study. Journal of Affective Disorders 2018;225:709-14. [DOI: 10.1016/j.jad.2017.09.008] [DOI] [PubMed] [Google Scholar]
Correia‐Melo 2020 {published data only (unpublished sought but not used)}
- Correia-Melo FS, Leala GC, Vieira F, Jesus-Nunes AP, Mello RP, et al. Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: a randomized, double blind, non-inferiority study. Journal of Affective Disorders 29 July 2019;264:527-34. [DOI] [PubMed] [Google Scholar]
Daly 2018 {published data only}
- Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry 2018;75(2):139-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Downey 2016 {published data only}
- Abdallah CG, Dutta A, Averill CL, McKie S, Akiki TJ, Averill LA, et al. Ketamine, but not the NMDAR antagonist lanicemine, increases prefrontal global connectivity in depressed patients. Chronic Stress 2018;2:1-9. [DOI: 10.1177/2470547018796102] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey D, Dutta A, McKie S, Dawson GR, Dourish CT, Craig K, et al. Comparing the actions of lanicemine and ketamine in depression: key role of the anterior cingulate. European Neuropsychopharmacology 2016;26(6):994-1003. [DOI: ] [DOI] [PubMed] [Google Scholar]
Fava 2018 {published and unpublished data}
- Fava M, Freeman MP, Flynn M, Judge H, Hoeppner BB, Cusin C, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Molecular Psychiatry Epub 2018 Oct 3. [DOI: 10.1038/s41380-018-0256-5] [DOI] [PMC free article] [PubMed]
- Fava M, Freeman PM, Flynn M, Judge H, Hoepnner BB, Cusin C, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenousketamine as adjunctive therapy in treatment-resistant depression(TRD). Molecular Psychiatry 2020;25(7):1592-603. [DOI: 10.1038/s41380-018-0256-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fedgchin 2019 {published and unpublished data}
- Fedgchin M, Trivedi M, Daly EJ, Melkote R, Lane L, Lim P, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressantin treatment-resistant depression: results of a randomized, double-blind, active-controlled study(TRANSFORM-1). International Journal of Neuropsychopharmacology 2019;22(10):616-30. [DOI: 10.1093/ijnp/pyz039] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedgchin M, Trivedi M, Daly EJ, Melkote R, Lane R, Lim P, et al. Randomized, double-blind study of fixed-dose intranasal esketamine plus oral antidepressant vs. active control in treatment-resistant depression. In: Poster presentation at the 9th Biennial Conference of the International Society for Affective Disorders and the Houston Mood Disorders Conference. September 2018.
Fernie 2017 {published data only}
- Fernie G, Currie J, Perrin JS, Stewart CA, Anderson V, Bennett DM, et al. Ketamine as the anaesthetic for electroconvulsive therapy: the KANECT randomised controlled trial. British Journal of Psychiatry June 2017;210(6):422-8. [DOI: 10.1192/bjp.bp.116.189134] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fu 2020 {published and unpublished data}
- Fu DJ, Ionescu DF, Li X, Lane R, Lim P, Sanacora G, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (Aspire I). Journal of Clinical Psychiatry 2020;81(3):e1-e9. [https://doi.org/10.4088/JCP.19m13191] [DOI] [PubMed] [Google Scholar]
Gálvez 2018 {published data only}
- Gálvez V, Huggins C, Glue P, Martin D, Somogyi AA, Alonzo A, et al. Repeated intranasal ketamine for treatment-resistant depression - the way to go? Results from a pilot randomised controlled trial. Journal of Psychopharmacology 2018;32(4):397-407. [DOI] [PubMed] [Google Scholar]
Ghasemi 2013 {published and unpublished data}
- Ghasemi M, Kazemi MH, Yoosefi A, Ghasemi A, Paragomi P, Amini H, et al. Rapid antidepressant effects of repeated doses of ketamine compared with electroconvulsive therapy in hospitalized patients with major depressive disorder. Psychiatry Research 2013;215:355-61. [DOI] [PubMed] [Google Scholar]
Grunebaum 2018 {published data only}
- Grunebaum MF, Galfalvy HC, Choo T-H, Keilp JG, Moitra VK, Parris MS, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. American Journal of Psychiatry 2018;175(4):327-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heresco‐Levy 2006 {published and unpublished data}
- Heresco-Levy U, Javitt DC, Gelfin Y, Gorelik E, Bar M, Blanaru M, et al. Controlled trial of D-cycloserine adjuvant therapy for treatment-resistant major depressive disorder. Journal of Affective Disorders 2006;93:239-43. [DOI] [PubMed] [Google Scholar]
Heresco‐Levy 2013 {published and unpublished data}
- Heresco-Levy U, Gelfin G, Bloch B, Levin R, Edelman S, Javitt DC, et al. A Randomized add-on trial of high-dose d-cycloserine for treatment-resistant depression. International Journal of Neuropsychopharmacology 2013;16:501-6. [DOI] [PubMed] [Google Scholar]
Hu 2016 {published data only}
- Hu YD, Xiang YT, Fang JX, Zu S, Sha S, Shi H, et al. Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: results from a ransomized, placebo-controlled 4-week study. Psychological Medicine 2016;46(3):623-35. [DOI] [PubMed] [Google Scholar]
Huang 2013 {published data only}
- Huang CC, Wei IH, Huang CL, Chen KT, Tsai MH, Tsai P, et al. Inhibition of glycine transporter-I as a novel mechanism for the treatment of depression. Biological Psychiatry 2013;74(10):734-41. [DOI] [PubMed] [Google Scholar]
Ibrahim 2012a {published and unpublished data}
- Ibrahim L, Diazgranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs Add-on riluzole: Results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology 2012;37:1526-33. [Riluzole] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ibrahim 2012b {published and unpublished data}
- Ibrahim L, Diazgranados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring J, et al. A randomized, placebo-controlled crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. Journal of Clinical Psychopharmacology 2012;32:551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ionescu 2018 {published data only}
- Ionescu DF, Bentley KH, Eikermann M, Taylor N, Akeju O, Swee MB, et al. Repeat-dose ketamine augmentation for treatment-resistant depression with chronic suicidal ideation: A randomized, double blind, placebo controlled trial. Journal of Affective Disorders September 2018;243:516-24. [DOI: ] [DOI] [PubMed] [Google Scholar]
Ionescu 2020 {published and unpublished data}
- Ionescu DF, Fu D-J, Qiu X, Lane R, Lim P, Kasper S, et al. Esketamine nasal spray for rapid reduction of depressive symptoms in patients with major depressive disorder who have active suicide ideation with intent: results of a phase 3, double-blind, randomized study (ASPIRE II). International Journal of Neuropsychopharmacology 2020;24(1):1-10. [DOI: 10.1093/ijnp/pyaa068] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jagtiani 2014 {published data only}
- Jagtiani A, Khurana H, Malhotra N, Gandhi R. efficacy of ketamine versus thiopentone-assisted modified electroconvulsive therapy in major depression. Journal of ECT September 2014;30(3):251-6. [DOI: 10.1097/YCT.0000000000000158] [DOI] [Google Scholar]
- Jagtiani A, Khurana H, Malhotra N. Comparison of efficacy of ketamine versus thiopentone‑assisted modified electroconvulsive therapy in major depression. Indian Journal of Psychiatry 2019;61(3):258-64.. [DOI: 10.4103/psychiatry.IndianJPsychiatry_386_18] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jarventausta 2013 {published and unpublished data}
- Jarventausta K, Chrapek W, Kampman O, Tuohimaa K, Bjorkqvist M, Hakkinen H, et al. Effects of S-ketamine as an anesthetic adjuvant to propofol on treatment response to electroconvulsive therapy in treatment-resistant depression: a randomized pilot study. Journal of ECT 2013;29:158-61. [DOI] [PubMed] [Google Scholar]
Kuşçu 2015 {published data only}
- Kuscu OO, Karacaer F, Biricik E, Gulec E, Tamam L, Gunes Y. Effect of ketamine, thiopental and ketamine-thiopental combination during electroconvulsive therapy for depression. Turkish Journal of Anaesthesiology and Reanimation 2015;43:313-7. [DOI: DOI: 10.5152/TJAR.2015.92668] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lapidus 2014 {published and unpublished data}
- Lapidus KA, Levitch CF, Perez AM, Brallier JW, Parides MK, Soleimani L, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biological Psychiatry 2014;76(12):970-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2016 {published data only}
- Li CT, Chen MH, Lin WC, Hong CJ, Yang BH, Liu RS, et al. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: a randomized controlled study. Human Brain Mapping January 2016;37:1080-90. [DOI: 10.1002/hbm.23085] [DOI] [PMC free article] [PubMed] [Google Scholar]
Loo 2012 {published and unpublished data}
- Loo CK, Katalinic N, Garfield JB, Sainsbury K, Hadzi-Pavlovic D, Mac-Pherson R. Neuropsychological and mood effects of ketamine in electroconvulsive therapy: a randomised controlled trial. Journal of Affective Disorders 2012;142:233-40. [DOI] [PubMed] [Google Scholar]
Michelson 2007 {published and unpublished data}
- Michelson D, Adler LA, Amsterdam JD, Dunner DL, Nierenberg AA, Reimherr FW, et al. Addition of atomoxetine for depression incompletely responsive to sertraline: a randomized, double-blind, placebo-controlled study. Journal of Clinical Psychiatry 2007;68:582-7. [DOI] [PubMed] [Google Scholar]
Murrough 2013 {published and unpublished data}
- Murrough JW, Burdick KE, Levitch CF, Perez AM, Brallier JW, Chang LC, et al. Neurocognitive effects of ketamine and association with antidepressant response in individuals with treatment-resistant depression: a randomized controlled trial. Neuropsychopharmacology. 2015;40(5):1084-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Iosifescu DV, Chang LC, Al JRK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. American Journal of Psychiatry 2013;170:1134-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RB, Iosifescu DV, Murrough JW, Chang LC, Al Jurdi RK, Iqbal SZ, et al. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment-resistant depression. Depression and Anxiety 2014;31:335-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nations 2012 (part I) {published and unpublished data}
- Nations KR, Dogterom P, Bursi R, Schipper J, Greenwald S, Zraket D, et al. Examination of Org 26576, an AMPA receptor positive allosteric modulator, in patients diagnosed with major depressive disorder: an exploratory, randomized, double-blind, placebo-controlled trial. Journal of Psychopharmacology 2012;26:1525-39. [DOI] [PubMed] [Google Scholar]
Nations 2012 (part II) {published and unpublished data}
- Nations KR, Dogterom P, Bursi R, Schipper J, Greenwald S, Zraket D, et al. Examination of Org 26576, an AMPA receptor positive allosteric modulator, in patients diagnosed with major depressive disorder: An exploratory, randomized, double-blind, placebo-controlled trial. Journal of psychopharmacology 2012;26:1525-39. [DOI] [PubMed] [Google Scholar]
Ochs‐Ross 2020 {published data only}
- Ochs-Ross R, Daly EJ, Lane R, Zhang Y, Lim P, Foster K, et al. Efficacy and safety of intranasal esketamine plus an oral antidepressant in elderly patients with treatment-resistant depression. In: Poster presentation at the 2018 Annual Meeting of the American Psychiatric Association. May 2018.
- Ochs-Ross R, Daly EKJ, Zhang Y, Lane R, Lim P, Randall LM et al. Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment-resistant depression - TRANSFORM-3. Am J of Geriatric Psychiatry 2020;28(2):121-141. [DOI: ] [DOI] [PubMed] [Google Scholar]
Omranifard 2014 {published and unpublished data}
- Omranifard V, Shirzadi E, Samandari S, Afshar H, Maracy MR. Memantine add on to citalopram in elderly patients with depression: A double-blind placebo-controlled study. Journal of Research in Medical Sciences 2014;19:525-30. [PMC free article] [PubMed] [Google Scholar]
Popova 2019 {published and unpublished data}
- Popova V, Daly EK, Madhukar T, Cooper K, Lane R, Lim P et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. American Journal of Psychiatry June 2019;176(6):428-38. [DOI] [PubMed] [Google Scholar]
Preskorn 2008 {published and unpublished data}
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. Journal of Clinical Psychopharmacology 2008;28:631-7. [DOI] [PubMed] [Google Scholar]
Preskorn 2015 {published data only}
- Preskorn S, Macaluso M, Mehra V, Zammit G, Moskal JR, Burch RM. Randomized proof of concept trial of GLYX-13, an N-Methyl-D-Aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. Journal of Psychiatric Practice March 2015;21(2):140-149. [DOI: 10.1097/01.pra.0000462606.17725.93] [DOI] [PubMed] [Google Scholar]
Quiroz 2016 {published data only}
- Quiroz JA, Tamburri P, Deptula D, Banken L, Ulrich B, Rabbia M, et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression. JAMA Psychiatry June 2016;73(7):675684. [DOI: 10.1001/jamapsychiatry.2016.0838] [DOI] [PubMed] [Google Scholar]
Roohi‐Azizi 2017 {published data only}
- Roohi-Azizi M, Arabzadeh S, Amidfar M, Salimi S, Zarindast MR, Ralaei A, et al. Citicoline combination therapy for major depressive disorder: A randomized, double-blind, placebo-controlled trial. Clinical Neuropharmacology January/February 2017;40(1):5. [DOI: 10.1097/WNF.0000000000000185] [DOI] [PubMed] [Google Scholar]
Salardini 2016 {published data only}
- Salardini E, Zeinoddini A, Mohammadinejad P, Khodaie-Ardakani M-R, Zahraei N, Zeinoddini A, et al. Riluzole combination therapy for moderate-to-severe major depressive disorder: A randomized, double-blind, placebo-controlled trial. Journal of Psychiatric Research 2016;75:24-30. [DOI: 10.1016/j.jpsychires.2016.01.003] [DOI] [PubMed] [Google Scholar]
Salehi 2015 {published data only}
- Salehi B, Mohammadbeigi A, Kamali AR, Taheri-Nejad MR, Moshiri I. Impact comparison of ketamine and sodium thiopental on anesthesia during electroconvulsive therapy in major depression patients with drug‑resistant; a double‑blind randomized clinical trial. Annals of Cardiac Anaesthesia 2015;18:486-90. [DOI: 10.4103/0971-9784.166444] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sanacora 2014 (a) {published and unpublished data}
- Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Molecular Psychiatry 2014;19(9):978-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sanacora 2014 (b) {published and unpublished data}
- Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Molecular Psychiatry 2014;19(9):978-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sanacora 2017 {published data only}
- Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, Schronen JP, et al. Adjunctive lanicemine (AZD6765) in oatients with major depressive disorder and history of inadequate response to antidepressants: randomized, placebo-controlled study. Neuropsychopharmacology 2017;42:844853. [DOI: 10.1038/npp.2016.224] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shams Alizadeh 2015 {published data only}
- Shams Alizadeh NS, Maroufi A, Nasseri K, Sadeghi Najafabadi SH, Mousavi Taghiabad A, Gharibi F, et al. Antidepressant effect of combined ketamine and electroconvulsive therapy on patients with major depressive disorder: a randomized trial. Iranian Journal of Psychiatry and Behavioural Sciences 2015;9(3):e1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shiroma 2020 {published and unpublished data}
- Shiroma PR, Thuas P, Wels J, Albott S, Erbes C, Tye S, et al. A randomized, double-blind, active placebo-controlled study of efficacy, safety and durability of repeated vs single subanesthetic ketamine for treatment-resistant depression. Translational Psychiatry 2020;10(1):206. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2016 a {published data only}
- Singh JB, Fedgchin M, Dally EJ, De Boer P, Cooper K, Lim P, et al. A Double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. American Journal of Psychiatry August 2016;173(8):816-26. [DOI: 10.1176/appi.ajp.2016.16010037] [DOI] [PubMed] [Google Scholar]
Singh 2016 b {published data only}
- Singh JB, Fedgchin M, Daly E, Xi L, Melman C, De Bruecker G, Tadic A, Sienaert P, Wiegand F, Manji H, Drevets WC, Van Nueten L. Intravenous esketamine in adult treatment-resistant depression: A double-blind, double-randomization, placebo-controlled study. Biological Psychiatry September 2016;80:424-431. [DOI: 10.1016/j.biopsych.2015.10.018] [DOI] [PubMed] [Google Scholar]
Smith 2013 {published and unpublished data}
- Smith EG, Deligiannidis KM, Ulbricht CM, Landolin CS, Patel JK, Rothschild AJ. Antidepressant augmentation using the N-methyl-D-aspartate antagonist memantine: A randomized, double-blind, placebo-controlled trial. Journal of Clinical Psychiatry Diseases of the Nervous System 2013;74:966-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sos 2013 {published and unpublished data}
- Sos P, Klirova M, Novak T, Kohutova B, Horacek J, Palenicek T. Relationship of ketamine's antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinology Letters 2013;34(4):287-93. [PubMed] [Google Scholar]
Su 2017 {published data only}
- Chen MH, Lin WC, Tu PC, Li CT, Bai YM, Tsai SJ, et al. Antidepressant and antisuicidal effects of ketamine on the functional connectivity of prefrontal cortex-related circuits in treatment-resistant depression: a double-blind, placebo-controlled, randomized, longitudinal resting fMRI study. Journal of Affected Disorders 2019;259:15-20. [CLINICAL TRIAL REGISTRATION: R000019001- UMIN000016985] [DOI: ] [DOI] [PubMed] [Google Scholar]
- Su T-P, Chen M-H, Li C-T, Lin W-C, Hong C-J, Gueorguieva R, et al. Dose-related effects of adjunctive ketamine in Taiwanese patients with treatment-resistant depression. Neuropsychopharmacology 2017;42(13):2482-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sumner 2020 {published and unpublished data}
- Sumner RL, McMillian R, Spriggs MJ, Campbell D, Malpas G, Maxwell E, Deng C, Hay J, Ponton R, Kirk IJ, Sundram F, Muthukumaraswamy SD. Ketamine Enhances Visual Sensory Evoked Potential Long-term Potentiation in Patients With Major Depressive Disorder. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging 2020;5(1):45-55. [DOI: 10.1016/j.bpsc.2019.07.002] [DOI] [PubMed] [Google Scholar]
Tiger 2020 {published and unpublished data}
- Tiger M, Veldman ER, Ekman C-J, Halldin C, Svenningsson P, Lundberg J. A randomized placebo-controlled PET study of ketamine's effect on serotonin1B receptor binding in patients with SSRI-resistant depression. Translational Psychiatry 2020;10(1):159. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Umbricht 2020 {published data only (unpublished sought but not used)}
- Umbricht D, Niggli M, Sanwald-Ducray P, Deptula D, Moore R, Grunbauer W, et al. Randomised, double-blind, placebo-controlled trial of the mglu2/3 negative allosteric modulator decoglurant in partially refractory major depressive disorder. Journal of Clinical Psychiatry 2020;81(4):18m12470. [DOI: 10.4088/JCP.18m12470] [DOI] [PubMed] [Google Scholar]
Yoosefi 2014 {published and unpublished data}
- Yoosefi A, Sepehri AS, Kargar M, Akhondzadeh S, Sadeghi M, Rafei A, et al. Comparing effects of ketamine and thiopental administration during electroconvulsive therapy in patients with major depressive disorder: a randomized, double-blind study. Journal of ECT 2014;30:15-21. [DOI] [PubMed] [Google Scholar]
Zarate 2006a {published and unpublished data}
- Zarate C, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry 2006;63:856-64. [DOI] [PubMed] [Google Scholar]
Zarate 2006b {published and unpublished data}
- Zarate C, Singh JB, Quiroz JA, De JG, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. American Journal of Psychiatry 2006;163:153-5. [DOI] [PubMed] [Google Scholar]
Zarate 2013 {published and unpublished data}
- Zarate C, Mathews D, Ibrahim L, Chaves JF, Marquardt C, Ukoh I, et al. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biological Psychiatry 2013;74:257-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Aftanas 2019 {published data only}
- Aftanas LI, Bazanova OM, Khabarov AN, Pustovoit SM, Brack IV. Placebo-controlled study of xenon effect on the emotions and frequency of the EEG alpha-oscillations [Russian]. Vestnik Rossiiskoi Akademii Meditsinskikh Nauk 2019;74(5):342-50. [Google Scholar]
Barzman 2005 {published data only}
- Barzman DH, DelBello MP, Kowatch RA, Gernert B, Fleck DE, Pathak S, et al. The effectiveness and tolerability of aripiprazole for pediatric bipolar disorders: A retrospective chart review. Journal of Child and Adolescent Psychopharmacology 2005;14:593-600. [DOI] [PubMed] [Google Scholar]
Bondolfi 2000 {published data only}
- Bondolfi G, Lissner C, Kosel M, Eap CB, Baumann P. Fluoxetine augmentation in citalopram non-responders: Pharmacokinetic and clinical consequences. International Journal of Neuropsychopharmacology 2000;3:55-60. [DOI] [PubMed] [Google Scholar]
Burger 2016 {published data only}
- Burger J, Capobianco M, Lovern R, Boche B, Ross E, Darracq MA, et al. A double-blinded, randomized, placebo-controlled sub-dissociative dose ketamine pilot study in the treatment of acute depression and suicidality in a military emergency department setting. Military Medicine 2016;181(10):1195-9. [DOI] [PubMed] [Google Scholar]
Chen 2020 {published data only}
- Chen MH, Lin WC, Wu HJ, Bai YM, Li CT, Tsai SJ, et al. Efficacy of low-dose ketamine infusion in anxious vs nonanxious depression: revisiting the adjunctive ketamine study of Taiwanese patients with treatment-resistant depression. CNS Spectrums 2020;26(4):1-6. [DOI: 10.1017/S1092852920001194] [DOI] [PubMed] [Google Scholar]
Erdil 2015 {published data only}
- Erdil F, Begeç Z, Kayhan GE, Yoloğlu S, Ersoy MÖ, Durmuş M. Effects of sevoflurane or ketamine on the QTc interval during electroconvulsive therapy. Journal of Anasthesia 2015;29(2):180-5. [DOI] [PubMed] [Google Scholar]
Giese 2014 {published data only}
- Giese M, Beck J, Brand S, Muheim F, Hemmeter U, Hatzinger M, et al. Fast BDNF serum level increase and diurnal BDNF oscillations are associated with therapeutic response after partial sleep deprivation. Journal of Psychiatric Research 2014;59:1-7. [DOI] [PubMed] [Google Scholar]
Huey 2005 {published data only}
- Huey ED, Dustin IH, Overman GP, Mirza N, Sunderland T. Development of subtle psychotic symptoms with memantine: A case report 4 [letter]. Journal of Clinical Psychiatry 2005;66:658-9. [DOI] [PubMed] [Google Scholar]
Irwin 2010 {published data only}
- Irwin SA, Iglewicz A. Oral ketamine for the rapid treatment of depression and anxiety in patients receiving hospice care. Journal of Palliative Medicine 2010;13:903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liebrenz 2009 {published data only}
- Liebrenz M, Stohler R, Borgeat A. Repeated intravenous ketamine therapy in a patient with treatment-resistant major depression. World Journal of Biological Psychiatry 2009;10:640-3. [DOI] [PubMed] [Google Scholar]
O'Gorman 2019 {published data only}
- O'Gorman C, Anderson A, Jacobson M, Jones A, Tabuteau H. P. 601 AXS-05 (dextromethorphan/bupropion), a novel, oral, investigational agent for major depressive disorder: Results of a randomized, double-blind, active-controlled, multi-center trial (ASCEND).. European Neuropsychopharmacology 2019;29:S410. [Google Scholar]
Park 2020 {published data only}
- Park LT, uckenbaugh SJ, Pennybaker MA, Hopkins ID, Denter MS, Lener B, et al. The effects of ketamine on typical and atypical depressive symptoms. Acta Psychiatrica Scandinavica 2020;142(5):394-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rasmussen 2014 {published data only}
- Rasmussen KG, Kung S, Lapid MI, Oesterle TS, Geske JR, Nuttall GA, et al. A randomized comparison of ketamine versus methohexital anesthesia in electroconvulsive therapy. Psychiatry Research 2014;215:362-5. [DOI] [PubMed] [Google Scholar]
Rosenblat 2019 {published data only}
- Rosenblat, JD. Potential differences in antidepressant effects of oral ketamine liquid suspension versus compounded capsules. British Journal of Psychiatry 2019;215(1):434. [DOI] [PubMed] [Google Scholar]
Sharma 2020 {published data only}
- Sharma, R K. Antidepressant effects of ketamine and ECT: a pilot comparison. Journal of Affective Disorders 2020;276:260-6. [DOI] [PubMed] [Google Scholar]
Shiroma 2020 {published data only}
- Shiroma PR. A randomized, double-blind, active placebo-controlled study of efficacy, safety, and durability of repeated vs single subanesthetic ketamine for treatment-resistant depression. Translational Psychiatry 2020;10(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2018 {published data only}
- Zhang M, Rosenheck R, Lin X, Li Q, Zhou Y, Xiao, et al. A randomized clinical trial of adjunctive ketamine anesthesia in electro-convulsive therapy for depression. Journal of Affective Disorders 2018;227:372-8. [DOI] [PubMed] [Google Scholar]
Zhong 2016 {published data only}
- Zhong X, He H, Zhang C, Wang Z, Jiang M, Li Q, et al. Mood and neuropsychological effects of different doses of ketamine in electroconvulsive therapy for treatment-resistant depression. Journal of Affective Disorders 2016;201:124-30. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
IRCT201104092266N2 {published data only}
- IRCT201104092266N2. Comparison of the effect of electroconvulsive therapy and intravenous infusion of ketamine on control and relapse of depressive symptoms in depressive cases who are candidates of electro convulsive therapy. http://www.irct.ir/searchresult.php?id=2266&number=2 (August 28th 2015).
ISRCTN87057460 {published data only}
- ISRCTN87057460. Effectiveness of the dual serotonin norepinephrine reuptake inhibitor venlafaxine in depressed patients. http://www.isrctn.com/ISRCTN87057460 (August 28th 2015).
NCT01046630 {published data only}
- NCT01046630. A Phase I, multi-centre, double-blind, placebo-controlled parallel group study to assess the pharmacoMRI effects of AZD6765 in male and female subjects fulfilling the criteria for major depressive disorder. http://clinicaltrials.gov/show/NCT01046630 (August 28th 2015).
NCT01482221 {published data only}
- NCT01482221. A multicenter, randomized, double-blind, parallel group, placebo-controlled, phase IIb efficacy and safety study of adjunctive AZD6765 in patients with major depressive disorder (MDD) and a history of inadequate response to antidepressants study D6702C00031. http://clinicaltrials.gov/show/NCT01482221 (August 28th 2015).
NCT01627782 {published data only}
- NCT01627782. A double-blind, randomized, placebo-controlled, parallel group, dose frequency study of ketamine in subjects with treatment-resistant depression. http://clinicaltrials.gov/show/NCT01627782 (August 28th 2015).
NCT03965871 {published data only}
NCT04019704 {published data only}
NCT04035798 {published data only}
NCT04210804 {published data only}
References to ongoing studies
EUCTR2011‐001520‐37‐SE {published data only}
- EUCTR2011-001520-37-SE. Ketamine as an alternative to electroconvulsive therapy for treatment of major depressive disorder. https://www.clinicaltrialsregister.eu/ctr-search/search?query=Vestibular+Disorders (August 28th 2015).
EUCTR2011‐005476‐41‐GB {published data only}
- EUCTR2011-005476-41-GB. Ketamine augmentation of ECT to improve outcomes in depression - Ketamine-ECT study. https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-005476-41/GB (August 28th 2015). [DOI] [PMC free article] [PubMed]
IRCT201307181556N54 {published data only}
- IRCT201307181556N54. Riluzole as adjuvant therapy in the treatment of moderate to severe major depression: a double - blind placebo-controlled trial. http://www.irct.ir/searchresult.php?keyword=&id=1556&number=54&prt=5237&total=10&m=1 (August 28th 2015).
NCT00988663 {published data only}
- NCT00988663. Memantine augmentation of electroconvulsive therapy in patients with major depression. http://clinicaltrials.gov/show/NCT00988663 (August 28th 2015).
NCT01179009 {published data only}
- NCT01179009. A safe ketamine-based therapy for treatment resistant depression. http://clinicaltrials.gov/show/NCT01179009 (August 28th 2015).
NCT01204918 {published data only}
- NCT01204918. Efficacy and tolerability of riluzole in treatment resistant depression. http://clinicaltrials.gov/show/NCT01204918 (August 28th 2015).
NCT01260649 {published data only}
- NCT01260649. N-methyl-D-aspartate antagonist (ketamine) augmentation of electroconvulsive treatment for severe major depression. http://clinicaltrials.gov/show/NCT01260649 (August 28th 2015).
NCT01441505 {published data only}
- NCT01441505. A study of ketamine as an antidepressant. http://clinicaltrials.gov/show/NCT01441505 (August 28th 2015).
NCT01557712 {published data only}
- NCT01557712. Estimate the efficiency of the association of an injection of ketamine and the venlafaxine in the severe major depressive disorder for six weeks (KETADEP). http://clinicaltrials.gov/show/NCT01557712 (August 28th 2015).
NCT01558063 {published data only}
- NCT01558063. The antidepressant action of ketamine: brain chemistry. http://clinicaltrials.gov/show/NCT01558063 (August 28th 2015).
NCT01613820 {published data only}
- NCT01613820. Combination of anticholinergic and glutamatergic effects in treatment-resistant major depressive disorder. A pilot study. http://clinicaltrials.gov/show/NCT01613820 (August 28th 2015).
NCT01667926 {published data only}
- NCT01667926. Randomized, double-blind ketamine augmentation in chronically suicidal, treatment-resistant major depression. http://clinicaltrials.gov/show/NCT01667926 (August 28th 2015).
NCT01684163 {published data only}
- NCT01684163. Phase 2, double-blind, placebo controlled, randomized withdrawal, parallel efficacy and safety study of GLYX-13 in subjects with inadequate/partial response to antidepressants during the current episode of major depressive disorder. http://clinicaltrials.gov/show/NCT01684163 (August 28th 2015).
NCT01700829 {published data only}
- NCT01700829. Ketamine vs. midazolam: testing rapid relief of suicide risk in depression. http://clinicaltrials.gov/show/NCT01700829 (August 28th 2015).
NCT01703039 {published data only}
- NCT01703039. Riluzole augmentation pilot in depression (RAPID) trial. http://clinicaltrials.gov/show/NCT01703039 (August 28th 2015).
NCT01868802 {published data only}
- NCT01868802. Ketamine for treatment-resistant depression: a multicentric clinical trial in Mexican population. http://clinicaltrials.gov/show/NCT01868802 (August 28th 2015).
NCT01880593 {published data only}
- NCT01880593. Ketamine plus lithium as a novel pharmacotherapeutic strategy in treatment-resistant depression. http://clinicaltrials.gov/show/NCT01880593 (August 28th 2015).
NCT01881763 {published data only}
- NCT01881763. Comparing therapeutic efficacy and cognitive side effects of electroconvulsive therapy (ECT) using ketamine versus methohexital anesthesia. http://clinicaltrials.gov/show/NCT01881763 (August 28th 2015).
NCT01882829 {published data only}
- NCT01882829. Targeting the NMDA glutamate receptor as novel antidepressant strategy: a pilot clinical trial of nuedexta in treatment-resistant major depression. http://clinicaltrials.gov/show/NCT01882829 (August 28th 2015).
NCT01887990 {published data only}
- NCT01887990. Treatment of suicidal ideation with intravenous ketamine infusion. http://clinicaltrials.gov/show/NCT01887990 (August 28th 2015).
NCT01902004 {published data only}
- NCT01902004. Treatment of geriatric depression with mild cognitive impairment: a double-blind placebo-controlled trial of namenda (memantine) augmentation of lexapro (escitalopram) in depressed patients at least 60 years of age. http://clinicaltrials.gov/show/NCT01902004 (August 28th 2015).
NCT01920555 {published data only}
- NCT01920555. Double-blind, placebo-controlled trial of ketamine therapy in treatment-resistant depression (TRD). http://clinicaltrials.gov/show/NCT01920555 (August 28th 2015).
NCT01935115 {published data only}
- NCT01935115. A prospective randomized double blinded control trial using ketamine or propofol anesthesia for electroconvulsive therapy: improving treatment-resistant depression. http://clinicaltrials.gov/show/NCT01935115 (August 28th 2015).
NCT01944293 {published data only}
- NCT01944293. Ketamine vs. midazolam in bipolar depression. http://clinicaltrials.gov/show/NCT01944293 (August 28th 2015).
NCT01945047 {published data only}
- NCT01945047. Phase 2 optimization of the antidepressant action of ketamine in treatment-resistant depression and investigations on its mechanism of action. http://clinicaltrials.gov/show/NCT01945047 (August 28th 2015).
NCT01957410 {published data only}
- NCT01957410. An open-label and double-blind study to investigate evoked potentials as markers of ketamine-induced cortical plasticity in subjects with major depressive disorder. http://clinicaltrials.gov/show/NCT01957410 (August 28th 2015).
NCT02012335 {published data only}
- NCT02012335. Ketamine use in electroconvulsive therapy: clinical, cognitives and neurotrophic outcomes. http://clinicaltrials.gov/show/NCT02012335 (August 28th 2015).
NCT02014363 {published data only}
- NCT02014363. Double blind, non-inferiority study to evaluate the antidepressant activity of ETS6103 compared with amitriptyline in the treatment of major depressive disorder (MDD) in patients who have an unsatisfactory response to selective serotonin re-uptake Inhibitors (SSRIs). http://clinicaltrials.gov/show/NCT02014363 (August 28th 2015).
NCT02037503 {published data only}
- NCT02037503. Effect of oral ketamine treatment on suicidal ideation and drug resistant major depression, a clinical and fMRI study. http://clinicaltrials.gov/show/NCT02037503 (August 28th 2015).
NCT02067793 {published data only}
- NCT02067793. Phase 2, randomized, double-blind, multiple-dose level, placebo controlled, single intravenous dose, parallel efficacy and safety study of NRX-1074 in subjects with major depressive disorder. http://clinicaltrials.gov/show/NCT02067793 (August 28th 2015).
NCT02106325 {published data only}
- NCT02106325. A randomized, double-blinded controlled trial of an N-methyl D-aspartate antagonist as a rapidly-acting antidepressant in depressed emergency department patients. http://clinicaltrials.gov/show/NCT02106325 (August 28th 2015).
NCT02139540 {published data only}
- NCT02139540. Nitrous oxide as treatment for major depression - a pilot study. http://clinicaltrials.gov/show/NCT02139540 (August 28th 2015).
NCT02153502 {published data only}
- NCT02153502. A phase 2, multicenter, randomized, double-blind, placebo-controlled study to assess the efficacy, safety, and tolerability of AVP-786 (deuterium modified dextromethorphan hydrobromide/quinidine sulfate) as an adjunctive therapy in patients with Major depressive disorder with an inadequate response to antidepressant treatment. http://clinicaltrials.gov/show/NCT02153502 (August 28th 2015).
NCT02267980 {published data only}
- NCT02267980. Effect of the addition of ketamine to sevoflurane anesthesia in electroconvulsive therapy. http://clinicaltrials.gov/show/NCT02267980 (August 28th 2015). [DOI] [PubMed]
NCT02295787 {published data only}
- NCT02295787. Intranasal ketamine for late-life depression and suicidal ideation. http://clinicaltrials.gov/show/NCT02295787 (August 28th 2015).
NCT02299440 {published data only}
- NCT02299440. Evaluation of the effects of ketamine in the acute phase of suicidal ideation: a multicenter randomized double-blind trial. http://clinicaltrials.gov/show/NCT02299440 (August 28th 2015).
NCT02305394 {published data only}
- NCT02305394. Effect of subanesthetic dose of ketamine combined with propofol on cognitive function in depressive patients undergoing electroconvulsive therapy - a randomized control double-blind clinical trial. http://clinicaltrials.gov/show/NCT02305394 (August 28th 2015).
NCT04082858 {published data only}
- NCT04082858. Ketamine interleaved with electroconvulsive therapy for depression. https://clinicaltrials.gov/show/NCT04082858. 2019.
NCT04116528 {published data only}
- Opiate Suicide Study in Patients With Major Depression (AFSP). Ongoing study. August 1, 2020. Contact author for more information.
NCT04234776 {published data only}
- NCT04234776. Intramuscular ketamine versus aripiprazole and escitalopram in the treatment of resistant depression. https://clinicaltrials.gov/show/NCT04234776. 2019.
NCT04399070 {published data only}
- NCT04399070. The Effect of S-ketamine for patients undergoing electroconvulsive therapy (ECT. https://clinicaltrials.gov/show/NCT04399070. 2020.
NTR3753 {published data only}
- NTR3753. ECT and Memantine. http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=3753 (August 28th 2015).
Phillips 2020 {published data only}
- Phillips. A randomized, crossover comparison of ketamine and electroconvulsive therapy for treatment of major depressive episodes: a Canadian biomarker integration network in depression (CAN-BIND) study protocol. BMC Psychiatry 2020;20(1):268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Abdallah 2018
- Abdallah C, Averil, LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Rapamycin, an Immunosuppressant and mTORC1 Inhibitor, triples the antidepressant response rate of ketamine at 2 weeks following treatment: a double-blind, placebo-controlled, cross-over, randomized clinical trial. bioRxiv 2018:p.500959.
Aitken 2019
- Aitken C, Mavridis D. Reasoning under uncertainty. Evidence-Based Mental Health 2019;22:44-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edition. Washington, D.C: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edition - Revised. Washington, D.C: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edition. Washington, D.C: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edition, Text Revision (DSM-IV-TR). Washington, DC: American Psychiatric Association, 2000. [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. Arlington (VA): American Psychiatric Association, 2013. [Google Scholar]
Arroll 2009
- Arroll B, Elley CR, Fishman T, Goodyear-Smith FA, Kenealy T, Blashki G, et al. Antidepressants versus placebo for depression in primary care. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD007954. [DOI: 10.1002/14651858.CD007954] [DOI] [PMC free article] [PubMed] [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al, GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bandelow 2006
- Bandelow B, Baldwin DS, Dolberg OT, Andersen HF, Stein DJ. What is the threshold for symptomatic response and remission for major depressive disorder, panic disorder, social anxiety disorder, and generalized anxiety disorder? Journal of Clinical Psychiatry 2006;67(9):1428–34. [DOI] [PubMed] [Google Scholar]
Birkenhager 1995
- Birkenhager TK, Moleman P, Nolen WA. Benzodiazepines for depression? A review of the literature. International Clinical Psychopharmacology 1995;10(3):181-95. [DOI] [PubMed] [Google Scholar]
Brookes 2001
- Brookes ST, Whitley E, Peters TJ, Mulheran PA, Egger M, Davey Smith G. Subgroup analyses in randomized controlled trials: quantifying the risks of false-positives and false-negatives. Health Technology Assessment 2001;5(33):1–56. [DOI] [PubMed] [Google Scholar]
Brookes 2004
- Brookes ST, Whitely E, Egger M, Smith GD, Mulheran PA, Peters TJ. Subgroup analyses in randomized trials: risks of subgroup-specific analyses; power and sample size for the interaction test. Journal of Clinical Epidemiology 2004;57(3):229-36. [DOI] [PubMed] [Google Scholar]
Brown 2019
- Brown S, Rittenbach K, Cheung S, McKean G, MacMaster FP, Clement F. Current and Common Definitions of Treatment-Resistant Depression: Findings from a Systematic Review and Qualitative Interviews.. Canadian Journal of Psychiatry 2019;64(6):380-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Caddy 2014
- Caddy C, Giaroli G, White TP, Shergill SS, Tracy DK. Ketamine as the prototype glutamatergic antidepressant: pharmacodynamic actions, and a systematic review and meta-analysis of efficacy. Therapeutic Advances in Psychopharmacology 2014;4(2):75-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2009
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, et al. Efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009;373(9665):746–58. [DOI] [PubMed] [Google Scholar]
Cipriani 2010
- Cipriani A, La Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 1. Art. No: CD006117. [DOI: 10.1002/14651858.CD006117.pub4] [DOI] [PubMed] [Google Scholar]
Cipriani 2012
- Cipriani A, Koesters M, Furukawa TA, Nosè M, Purgato M, Omori IM, et al. Duloxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2012, Issue 10. Art. No: CD006533. [DOI: 10.1002/14651858.CD006533.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2019
- Cipriani A, Tomlinson A. Providing the most appropriate care to our individual patients. Evidence-Based Mental Health 2019;22:1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2020
- Cipriani A, Ioannidis JP, Rothwell PM, Glasziou P, Li T, Hernandez AF, et al. Generating comparative evidence on new drugs and devices after approval. Lancet 2020;395(10228):998-1010. [DOI: ] [DOI] [PubMed] [Google Scholar]
Coppen 1967
- Coppen A. The biochemistry of affective disorders. British Journal of Psychiatry 1967;113:1237-64. [DOI] [PubMed] [Google Scholar]
Cotter 2001
- Cotter D, Mackay D, Landau S, Kerwin R Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder.. Archives of General Psychiatry 2001;58(6):545553. [DOI] [PubMed] [Google Scholar]
Coyle 2015
- Coyle CM, Laws KR. The use of ketamine as an antidepressant: a systematic review and meta-analysis. Human Psychopharmacology 2015;30:152-63. [DOI] [PubMed] [Google Scholar]
Dean 2021
- Dean RL, Marquardt T, Hurducas C, Spyridi S, Barnes A, Smith R, et al. Ketamine and other glutamate receptor modulators for depression in bipolar disorder in adults. Cochrane Database of Systematic Reviews (in press). [DOI] [PMC free article] [PubMed]
Deeks 2021
- Deeks JJ, Higgins JP, Altman DG (editors). Chapter 10: Analysing data and undertaking meta-analyses.. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editors(s). Cochrane Handbook for Systematic Reviews of Interventions. 6.2 edition. Cochrane, 2021. [Google Scholar]
De Leo 2014
- De Leo D, Too LS. Suicide and depression. In: Okpaku, SO, editors(s). Essentials of Global Mental Health. Cambridge: Cambridge University Press, 2014:367-73. [Google Scholar]
Diamond 2014
- Diamond PR, Farmery AD, Atkinson S, Haldar J, Williams N, Cowen PJ, et al. Ketamine infusions for treatment resistant depression: a series of 28 patients treated weekly or twice weekly in an ECT clinic. Journal of Psychopharmacology 2014;28(6):536-44. [DOI] [PubMed] [Google Scholar]
Donohue 2007
- Donohue JM, Pincus HA. Reducing the societal burden of depression: a review of economic costs, quality of care and effects of treatment. Pharmacoeconomics 2007;25:7-24. [DOI] [PubMed] [Google Scholar]
Dozois 2004
- Dozois D JA, Dobson KS. Depression. In: Antony M M, Barlow D H, editors(s). Handbook of Assessment and Treatment Planning for Psychological Disorders. New York: Guilford Press, 2004:259-99. [Google Scholar]
Efthimiou 2019
- Efthimiou O. Statistics in pills: meta-analysis of rare events. Evidence-Based Mental Health 2019;22:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315(7109):629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving crossover trials: methodological issues. International Journal of Epidemiology 2002;31(1):140–9. [DOI] [PubMed] [Google Scholar]
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26(1):57–63. [DOI] [PubMed] [Google Scholar]
Furukawa 2002
- Furukawa TA, Guyatt GH, Griffith LE. An empirical study of summary effect measures in meta-analyses. International Journal of Epidemiology 2002;31(1):72–6. [DOI] [PubMed] [Google Scholar]
Furukawa 2005
- Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta-analysis. International Clinical Psychopharmacology 2005;20(1):49–52. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta-analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7–10. [DOI] [PubMed] [Google Scholar]
Furukawa 2007a
- Furukawa TA, Watanabe N, Omori IM, Montori VM, Guyatt GH. Association between unreported outcomes and effect size estimates in Cochrane meta-analyses. JAMA 2007;297(5):468–70. [DOI] [PubMed] [Google Scholar]
Furukawa 2007b
- Furukawa TA, Akechi T, Azuma H, Okuyama T, Higuchi T. Evidence-based guidelines for interpretation of the Hamilton Rating Scale for Depression. Journal of Clinical Psychopharmacology 2007;27(5):531–4. [DOI] [PubMed] [Google Scholar]
Godfrey 2018
- odfrey KM, Gardner AC, Kwon S, Chea W, Muthukumaraswamy SD. Differences in excitatory and inhibitory neurotransmitter levels between depressed patients and healthy controls: a systematic review and meta-analysis. Journal of Psychiatric Research. 2018;105:33-44. [DOI] [PubMed] [Google Scholar]
Godlewska 2018
- Godlewska, BR, Masaki, C, Sharpley, AL, Cowen, PJ and Emir, UE. Brain glutamate in medication-free depressed patients: a proton MRS study at 7 Tesla. Psychological medicine. Psychological medicine 2018;48(10):1731-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Goh 2019
- Goh KK, Chen CH, Chiu YH, Lu ML. Lamotrigine augmentation in treatment-resistant unipolar depression: a comprehensive meta-analysis of efficacy and safety. Journal of Psychopharmacology 2019;33(6):700-13. [DOI: 10.1177/0269881119844199] [DOI] [PubMed] [Google Scholar]
GradePro GDT 2020
- GRADEpro GDT: GRADEpro Guideline Development Tool [Software]. Available from gradepro.org. 2020.
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville (MD): U.S. Department of Health and Human Services, 1976. [Google Scholar]
Guyatt 1998
- Guyatt GH, Juniper EF, Walter SD, Griffith LE, Goldstein RS. Interpreting treatment effects in randomised trials. BMJ 1998;316(7132):690–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hasler 2010a
- Hasler G. Pathophysiology of depression: do we have any solid evidence of interest to clinicians? World Psychiatry 2010;9:155-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hawton 2009
- Hawton K, Heeringen K. Suicide. Lancet 2009;373:1372-81. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011].
Higgins 2011d
- Higgins JP, Green S (editors). 7.7.3.8 Combining groups. In: Higgins JP, Green S, editors(s). Cochrane Handbook for Systematic Reviews of Interventions. 5.1.0 edition. Cochrane Collaboration, March 2011. [Google Scholar]
Higgins 2020
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al editors). Cochrane Handbook for Systematic Reviews of Interventions. Available from www.training.cochrane.org/handbook. 2020;Version 6.1.
Hirschfeld 2000
- Hirschfeld RM. History and evolution of the monoamine hypothesis of depression. Jourmal of Clinical Psychiatry 2000;61 Suppl 6:4-6. [PubMed] [Google Scholar]
Jones 2019
- Jones H, Cipriani A. Barriers and incentives to recruitment in mental health clinical trials. Evidence-Based Mental Health 2019;22:49-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Koesters 2013
- Koesters M, Guaiana G, Cipriani A, Becker T, Barbui C. Agomelatine efficacy and acceptability revisited: systematic review and meta-analysis of published and unpublished randomised trials. British Journal of Psychiatry 2013;203(3):179-87. [DOI] [PubMed] [Google Scholar]
Li 2010
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010;329:959-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lim 2018
- Lim GY, Tam WW, Lu Y, Ho CS, Zhang MW, Ho RC. Prevalence of depression in the community from 30 countries between 1994 and 2014. Scientific Reports 2018;8(1):2861. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Magni 2013
- Magni LR, Purgato M, Gastaldon C, Papola D, Furukawa TA, Cipriani A, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2013, Issue 7. Art. No: CD004185. [DOI: 10.1002/14651858.CD004185.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Malhi 2019
- Malhi G, Bell E, Outhred T. Getting irritable about irritability? Evidence-Based Mental Health 2019;22(3):93-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marcantoni 2020
- Marcantoni W, Akoumba BS, Wassef M, Mayrand J, Lai H, Richard-Devantoy S, et al. A systematic review and meta-analysis of the efficacy of intravenous ketamine infusion for treatment resistant depression: January 2009 – January 2019. Journal of Affective Disorders 2020;277:831-41. [DOI: 10.1016/j.jad.2020.09.007] [DOI] [PubMed] [Google Scholar]
Mavridis 2014
- Mavridis D, Chaimani A, Efthimiou O, Leucht S Salanti G. Addressing missing outcome data in meta-analysis. Evidence Based Mental Health 2014;17(3):85-9. [DOI] [PubMed] [Google Scholar]
McCloud 2015
- McCloud TL, Caddy C, Jochim J, Rendell JM, Diamond PR, Shuttleworth C, et al. Ketamine and other glutamate receptor modulators for depression in bipolar disorder in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. Art. No: CD011611. [DOI: 10.1002/14651858.CD011611.pub2] [DOI] [PubMed] [Google Scholar]
McGirr 2015
- McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychological Medicine 2015;45:693-704. [DOI] [PubMed] [Google Scholar]
Memon 2020
- Memon RI, Naveed S, Faquih AE, Fida A, Abbas N, Chaudry AM, et al. rffectiveness and safety of ketamine for unipolar depression: a systematic Review. Psychiatric Quarterly 2020;91:1147-92. [DOI: 10.1007/s11126-020-09830-6] [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. BMJ 2009;339:2535. [PMC free article] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382–9. [DOI] [PubMed] [Google Scholar]
Moriguchi 2019
- Moriguchi S, Takamiya A, Noda Y, Horita N, Wada M, Tsugawa S, et al. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies.. Molecular Psychiatry 2019;24(7):952-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muller 2003
- Muller MJ, Himmerich H, Kienzle B, Szegedi A. Differentiating moderate and severe depression using the Montgomery-Asberg depression rating scale (MADRS). Journal of Affective Disorders 2003;77(3):255–60. [DOI] [PubMed] [Google Scholar]
Naci 2020
- Naci H, Salcher-Konrad M, Kesselheim AS, Wieseler B, Rochaiz L, Redberg RF, et al. Generating comparative evidence on new drugs and devices before approval. Lancet 2020;395(10228):986-97. [DOI: ] [DOI] [PubMed] [Google Scholar]
Naughton 2014
- Naughton M, Clarke G, O'Leary OF, Cryan JF, Dinan TG. A review of ketamine in affective disorders: current evidence of clinical efficacy, limitations of use and pre-clinical evidence on proposed mechanisms of action. Journal of Affective Disorders 2014;156:24-35. [DOI] [PubMed] [Google Scholar]
NICE 2009
- National Institute for Health and Clinical Excellence. Depression in adults: The treatment and management of depression in adults (CG90). http://publications.nice.org.uk/depression-in-adults-cg90 2009.
Petty 1984
- Petty F, Sherman AD. Plasma GABA levels in psychiatric illness. Journal of Affective Disorders 1984;6:131-8. [DOI] [PubMed] [Google Scholar]
Price 2014
- Price RB, Iosifescu DV, Murrough JW, Chang LC, Al Jurdi RK, Iqbal SZ, et al. Effects of ketamine on explicit and implicit suicidal cognition: A randomized controlled trial in treatment-resistant depression. Depression and Anxiety 2014;31:335-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rapaport 2005
- Rapaport MH, Clary C, Fayyad R, Endicott J. Quality-of-life impairment in depressive and anxiety disorders. American Journal of Psychiatry 2005;162:1171-8. [DOI] [PubMed] [Google Scholar]
Revman 2020 [Computer program]
- The Cochrane Collaboration Review Manager. Version Version 5.4. The Cochrane Collaboration, 2020.
Ruhe 2007
- Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Molecular Psychiatry 2007;12:331-59. [DOI] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editors. GRADE handbook for grading quality of evidence and strength of recommendations.. The GRADE Working Group, 2013. [Google Scholar]
Solmi 2016
- Solmi M, Veronese N, Zaninotto L, Loos ML, Gao K, Schaffer A, et al. Lamotrigine compared to placebo and other agents with antidepressant activity in patients with unipolar and bipolar depression: a comprehensive meta-analysis of efficacy and safety outcomes in short-term trials. CNS Spectrums 2016;21(5):403-18. [DOI: 10.1017/S1092852916000523] [DOI] [PubMed] [Google Scholar]
Spitzer 1978
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives General Psychiatry 1978;35(6):773–82. [DOI] [PubMed] [Google Scholar]
Sterne 2000
- Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta-analysis: power of statistical tests and prevalence in the literature. Journal of Clinical Epidemiology 2000;53(11):1119–29. [DOI] [PubMed] [Google Scholar]
Suh 1997
- Suh T, Gallo JJ. Symptom profiles of depression among general medical service users compared with specialty mental health service users. Psychological Medicine 1997;27(5):1051–63. [DOI] [PubMed] [Google Scholar]
Turner 2019
- Turner, E. Esketamine for treatment-resistant depression: seven concerns about efficacy and FDA approval. Lancet Psychiatry 2019;6(12):977-9. [DOI: 10.1016/S2215-0366(19)30394-3] [DOI] [PubMed] [Google Scholar]
Valentine 2011
- Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Research 2011;191:122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Watanabe 2008
- Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, McGuire H, et al. Mirtazapine versus other antidepressants in the acute-phase treatment of adults with major depression: systematic review and meta-analysis. Journal of Clinical Psychiatry 2008;69(9):1404-15. [DOI] [PubMed] [Google Scholar]
Watanabe 2011
- Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, Churchill R, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2011, Issue 12. Art. No: CD006528. [DOI: 10.1002/14651858.CD006528.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization (WHO). The ICD-10 Classification of Mental and Behavioural Disorders. Geneva: WHO, 1992. [Google Scholar]
WHO 2008a
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems, 10th Revision (ICD-10). New York, NY: WHO, 2008. [Google Scholar]
WHO 2008b
- World Health Organization (WHO). The global burden of disease: 2004 update. Geneva: WHO, 2008. [Google Scholar]
WHO 2012
- World Health Organization (WHO). In: Sixty-fifth World Health Assembly: daily notes on proceedings. Geneva: World Health Organization, 2012.
Wilkinson 2019
- Wilkinson ST, and Sanacora G. A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems.. Drug Discovery Today 2019;24(2):606-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Williams 2018
- Williams NR, Heifets BD, Blasey C, Sudheimer K, Pannu J, Pankow H, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. American Journal of Psychiatry 2018;175(12):1205-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoon 2019
- Yoon G, Petrakis IL, Krystal JH. Association of combined naltrexone and ketamine with depressive symptoms in a case series of patients with depression and alcohol use disorder.. JAMA Psychiatry 2019;76(3):337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zheng 2019
- Zheng W, Cai DB, Zheng W, Sim K, Ungvari GS, Peng XJ, et al. Brexanolone for postpartum depression: a meta-analysis of randomized controlled studies.. Psychiatry Research 2019;279:83-9. [DOI] [PubMed] [Google Scholar]
Zheng 2020
- Zheng W, Cai DB, Xiang YQ, Zheng W, Jiang WL, Sim K, et al. Adjunctive intranasal esketamine for major depressive disorder: a systematic review of randomized double-blind controlled-placebo studies. Journal of Affective Disorders 2020;265:63-70. [DOI: 10.1016/j.jad.2020.01.002] [DOI] [PubMed] [Google Scholar]
Zhou 2020
- Zhou X, Teng T, Zhang Y, Del Giovane C, Furukawa TA, Weisz JR, et al. Comparative efficacy and acceptability of antidepressants, psychotherapies, and their combination for acute treatment of children and adolescents with depressive disorder: a systematic review and network meta-analysis. Lancet Psychiatry 2020;7(7):581-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zohar 2014
- Zohar J, Nutt DJ, Kupfer DJ, Moller HJ, Yamawaki S, Spedding M, et al. A proposal for an updated neuropsychopharmacological nomenclature. European Neuropsychopharmacology 2014;24(7):1005-14. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Amit 2015
- Amit BH, Caddy C, McCloud TL, Rendell JM, Hawton K, Diamond PR, , et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database of Systematic Reviews 2015, Issue 4. Art. No: CD011612. [DOI: 10.1002/14651858.CD011612] [DOI] [PubMed] [Google Scholar]
Caddy 2015
- Caddy C, Amit BH, McCloud TL, Rendell JM, Furukawa TA, McShane R, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. Art. No: CD011612. [DOI: 10.1002/14651858.CD011612.pub2] [DOI] [PubMed] [Google Scholar]