

Introduction
Personalized medicine has been a goal for many decades. The advancement of scientific tools has brought us closer to achieving this goal—in particular, single cell RNA sequencing may hold the answers to many questions about cardiac development and disease. Single cell RNA sequencing (scRNA-seq) allows one to understand the gene expression in each cell of the body rather than an average expression of a heterogenous population of cells within a tissue. Knowledge of gene expression of single cells within a tissue has contributed to the understanding of how each cell type contributes to the anatomic structures of the heart. In this review, we will discuss how scRNA-seq has been utilized in the field of cardiovascular research to understand cardiogenesis.
History of scRNA-seq
The Surani lab, at the Gurdon Institute, recognized limitations in the field transcriptomics while conducting research in early embryonic development.2 They noted that in certain cases, only a small quantity of tissue was available, thus, they developed single cell RNA sequencing analysis in 2009. The group demonstrated that the number of transcripts detected by traditional methods such as microarray assays and quantitative PCR were much lower than scRNA-seq. Single cell sequencing detected 75% more genes than traditional methods. This seminal paper allowed for many more contributions and advances in the technical aspects of single cell RNA sequencing analysis. While this first study utilized manual pipetting to isolate single cells, the field has made more advances which we will review.
Technical Aspects (Work flow)
The first step in scRNA-seq includes sample preparation—an important step to ensure the integrity of single cells (Figure 1). Protocols to ensure maximal single cell count and minimal cell death must be determined. Different tissue requires different amount and duration of exposure to enzymes (i.e. a protease and collagenase) for cell isolation. Cell count of isolated viable cells should be >10,000 cells and thus, a protocol that ensures adequate cell count is important. Important factors to consider include the biological characteristics of the tissue and size of the cell when preparing samples.
Figure 1:
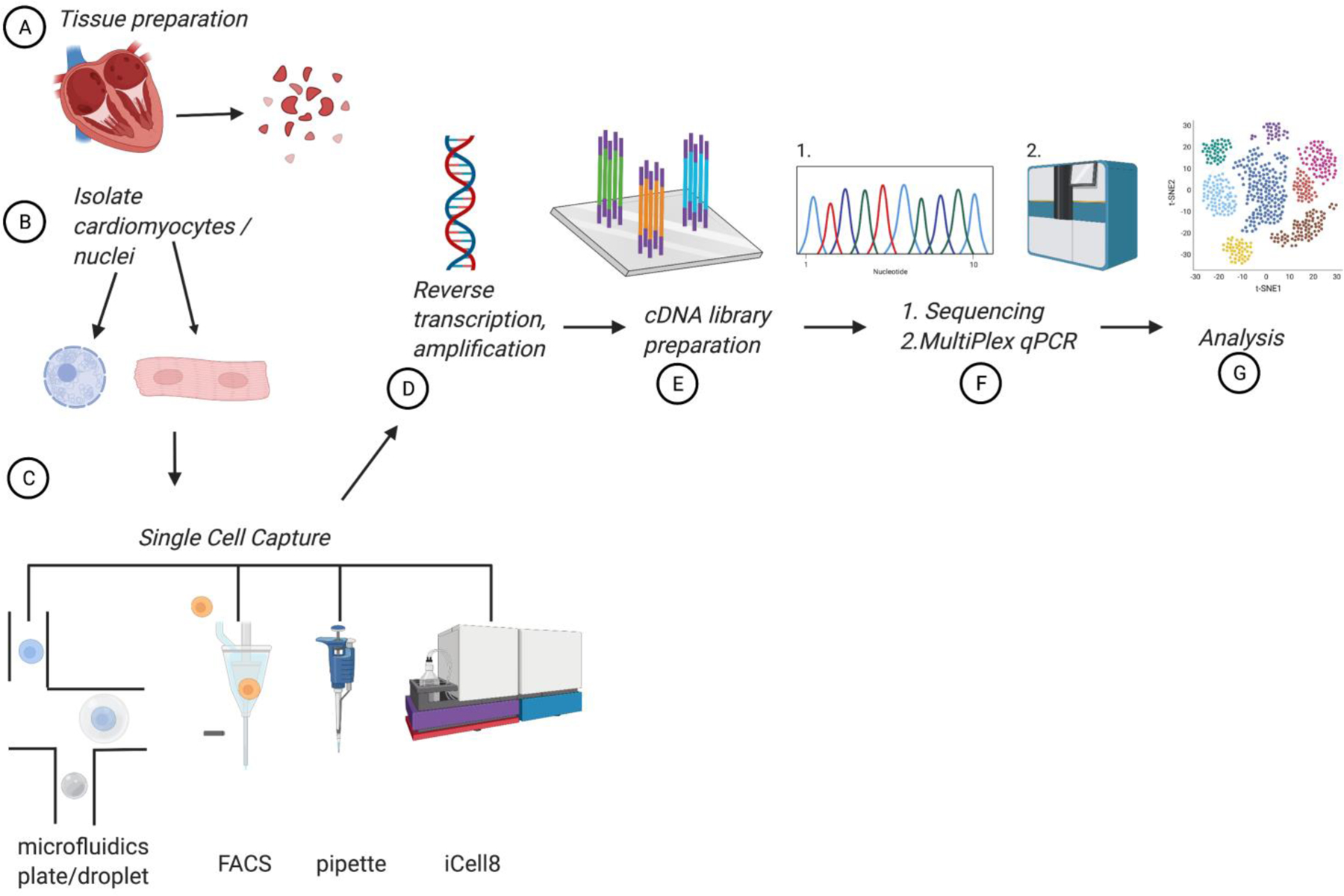
Steps to Single cell RNA sequencing. (A)Dissociation of the cardiac tissue using optimal digestion protocol. (B) Isolate whole cell cardiomyocyte or nuclei. (C) Single cell capture via various methods of single cell capture. (D) Reverse transcription of sc-RNA and PCR amplification. (E) cDNA library preparation. (F) (1) Next generation sequencing or (2) Multiplex qPCR generates readout. (G) Alignment, quality control and analysis.
In addition to various protocols required for different organs in the body, isolation of cardiomyocytes may also differ based on the characteristics of the age and origin of cardiac tissue. In comparison to the murine heart, the human heart is more fibrous which makes it more challenging to isolate cardiomyocytes without damaging the cells.2 Pimpalwar et al obtained a high yield of scRNA-seq data by isolating nuclei from cardiomyocytes frozen human hearts rather than whole cells. Bergmann et al isolated murine cardiomyocyte nuclei by density sedimentation, immunolabeling with antibodies against pericentriolar material 1 (PCM-1) and subsequent flow cytometry-based cell sorting.3,4 Dissection and homogenization of cardiac tissue is followed by lysis in order to free the nuclei from the cardiomyocyte cytoplasm. Following immunostaining against Nkx2.5 and PCM-1, the cardiomyocyte nuclei were then purified using flow cytometry. Although the content of RNA transcripts is lower from nuclei, single nuclei RNA analysis has been utilized by many research groups including the most comprehensive compendiums of gene expression from cells of the human heart that was recently published.5,6
The advantage of utilizing nuclei over whole adult cardiomyocyte is that it allows for obtaining information from a greater number of cells—thereby increasing cell count. However, it is important to note that the content of nuclear RNA is not only different but the yield of RNA transcripts is different from the nuclei compared to the cytoplasm.7 Bakken et al demonstrated that more transcripts are detected in whole cells (~11,000 genes) when compared to nuclei (~7,000) when processing cortical tissue.8
Upon isolation, single cells may need to be further purified according to specific biomarkers. One method of cell separation is Fluorescence-Activated Cell Sorting (FACS), which uses scatter and fluorescent signals to sort cells into 96 or 384 well plates. However, the nozzle size in FACS requires smaller cells (less than 50 μm in diameter) and thus, would provide a robust number of viable fetal cardiomyocytes and other smaller cells such as fibroblasts, however adult CM would be damaged in the process.9 A single CM is ~100 μm in size across the diameter and thus, requires a larger nozzle. To address the size, Gladka et al used a larger nozzle size (130 μm) with FACS in order to obtain single cells from adult murine tissue to conduct RNA sequencing.10 Similarly, Kannan et al isolated cardiomyocytes using large particle PACS (LP-FACS), which uses a channel size of 500 μm, and demonstrated an enriched population of viable cardiomyocytes with maximal RNA quality.11 In addition, Gladka et al also tested various enzymes to obtain an optimal number of viable cells with intact RNA, and found the use of Liberase for 15 minutes in 37 degrees Celcius water bath yielded optimal number of adult cardiomyocytes with intact RNA. Other enzymes used include trypsin and collagenase for cell isolation.10 Our lab is currently using trypsin and collagenase A and B to isolate whole human adult cardiomyocytes. To ensure data quality, high yield and intact RNA, investigators must each determine an optimal protocol for his/her tissue of interest.
The second step following single cell capture is the lysis of each cell and creating first complementary DNA (cDNA) via reverse transcriptase followed by a second copy of cDNA and polymerase chain reactions (PCR) amplification. During early days, there were two major single cell capture platforms - microfluid plate and microfluid droplet. Each of these methods offer advantages and disadvantages and have been reviewed previously.12,13 The microfluid plate system uses parallel microfluid channel, separates and isolates cells. Within the channels, cell lysis, reverse transcriptase, and multiplexing (pooling of a large number of cDNA libraries and sequenced simultaneously) occur, thus, the advantage of this system is that it allows for capturing of full-length RNA in each cell. The disadvantage of the microfluidic plate platform is time, cost, and limited number of cells captured per run resulting in ~100 to 1,000 cells per study.12,13
The microfluid droplet method is less time consuming and more cost effective on a per cell basis.14 In this system, each cell is captured in a droplet, typically in an oil emulsion that also contains the necessary reagents for cell barcoding and reverse transcription into 1st strand cDNA. These reagents may be incorporated in the form of a thermo-sensitive gel bead. Cells of various sizes can be captured using this system.12,13 One major distinction between the commonly used droplet-based system and the plate-based system is the 3’- or 5’-biased sequencing such that each cell only account for 10,000 to 100,000 reads thus providing significant cost saving per cell.12 The disadvantage of the droplet-based system includes a lower overall transcript recovery rate when compared to other methods.14
High-throughput single-cell RNA-seq methods assign limited unique molecular identifier (UMI) counts as gene expression values to single cells from shallow sequence reads, however limited gene counts are detected. Sasagawa et al developed a high-throughput single-cell RNA-seq method, Quartz-Seq2, to detect more genes.15 The Quartz-Seq2 methods has 5 steps with improvements in the reaction steps make it possible to effectively convert initial reads to UMI counts, at a rate of 30–50%, and detect more genes. The 5 steps include (1) single-cell collection using a cell sorter, (2) cell barcoding, (3) pooling of cell-barcoded cDNA, (4) whole-transcript amplification with improved efficiency of poly(A) tagging by 3.6-fold, and (5) library preparation for deep sequencing. In addition to Quartz-Seq2, other methods have also been developed to produce high quality data. The first full-length total RNA-sequencing method for single cells, RamDA-seq, (random displacement amplification sequencing) was developed by Hayashi et al. Their method consisted of combining reverse transcriptase-RamDA (RT-RamDA) and not-so-random primers (NSRs).16 RT-RamDA provides global cDNA amplification directly from RNA during reverse transcription, which benefits reverse transcription efficiency, simplifies the procedure, and decontaminates genomic DNA. NSRs enables random priming while preventing cDNA synthesis from rRNAs. Hayashi et al demonstrated that RamDA-seq has high sensitivity to non-poly(A) and full-length coverage for long transcripts exceeding 10 kb.
After reverse transcription and amplification, cDNA libraries are prepared, next generation sequencing generates readouts which are aligned to a reference genome. After alignment, quality control of the data ensures that readouts consist of single, viable cells. During quality control, cells that have physically stuck together called ‘doublets’ or ‘double enriched clusters’ and non-viable cells are not excluded from the dataset.12 Cell clusters are then assigned cell type identities based on cell markers, mitochondrial gene percentage and total gene count.12,13 For example, cardiomyocytes have high mitochondrial gene content (56–86%) compared to other cells (5–20%).8 Single cell RNA sequencing also allows identification of rare cell population in tissue.17 The data generated is then displayed utilizing non-linear reduction algorithms such as t-distributed stochastic neighboring embedding (t-SNE) and uniform manifold approximation and projection (UMAP). These methods consist of an algorithm that first finds similar data points and then arranges the points in a 2D plot such that the similar cells remain closer. Becht et al compared t-SNE with UMAP utilizing hematopoietic cells and determined that while t-SNE efficiently reveals local data structure and identifies distinct cell populations, it has slow computation time and is unable to meaningfully represent very large databases.18 UMAP has a shorter run time, preserves more of the global structure, and preserves continuity of cell subsets (i.e. does not separate CD4+ T cells from other T cell subsets), whereas t-SNE expands low dense areas and tends to ignore global relationships. Our lab has utilized t-SNE in our prior work and we are currently utilizing UMAP given its advantages. Paik et al has described comparison of clustering methods for scRNA-seq, thus, we will briefly state that there are several ways of clustering cells such as k-means and Louvain methods.12 Of note, principal component analysis (PCA), a linear statistical technique that reduces the number of experimental variables to a minimum amount, has been used in the past by researchers for analysis, however, it has high dropout and noise among data.19
One popular software for scRNA-seq data processing is Seurat version 3.0, an R package designed for quality control and analysis. Other R or Python packages for quality control, visualization and analysis include Monocle, Scanpy, Scater, and SINCERA.13 New algorithms and updates will enable researchers to visualize data in improved ways such as the newly updated Seurat version 4, which includes the ability to rapidly map query datasets to references.20 A recent benchmarking study of sc-RNA sequencing by Mereu et al provided insight into the differences in protocol performance.21 This group performed a study comparing 13 commonly used scRNA-seq and single-nucleus RNA-seq protocols applied to a heterogeneous reference sample resource consisting of human peripheral blood and mouse colon tissue. They defined strengths and weakness of key features of scRNA and snRNA analysis thereby providing guidance for scientists when designing studies. For example, they found significant differences among protocols in converting RNA molecules into sequencing libraries. Varying library complexities affected the protocol’s power to quantify gene expression levels and to identify cell-type markers. Thus, it is important to optimize protocols when performing single cell or single nuclei RNA sequencing studies.
Application scRNA-seq to Cardiac Development
Single cell RNA sequencing has revealed important information regarding cardiogenesis and cardiomyocyte maturation (Figure 2). Understanding cardiogenesis will provide insight into the origins of congenital heart disease which affects approximately 40,000 babies born each year according to the Center for Disease Control (CDC).22 Using scRNA-seq Delaughter et al and Li et al describe the consequences of Nkx2.5 deletion during fetal development and noted the requirement of this gene for establishing ventricular identity and cardiomyocyte maturation.23,24 Li et al identified 18 distinct subpopulations based on cell type and anatomical location and confirms the existence of gene expression gradients within embryonic trabecular and compact ventricular myocardium.24 Xiong et al further determined that Nkx2.5 regulates spatial expression of Cxcr2 and Cxcr4, chemokine receptors, that direct cardiac progenitor migration in the second heart field.25 Mutations in Nkx2.5 have been found to contribute to atrial septal defects, ventricular septal defects, Tetralogy of Fallot, conotruncal malformations (i.e truncus arterious), and hypoplastic left heart syndrome in children.26
Figure 2:
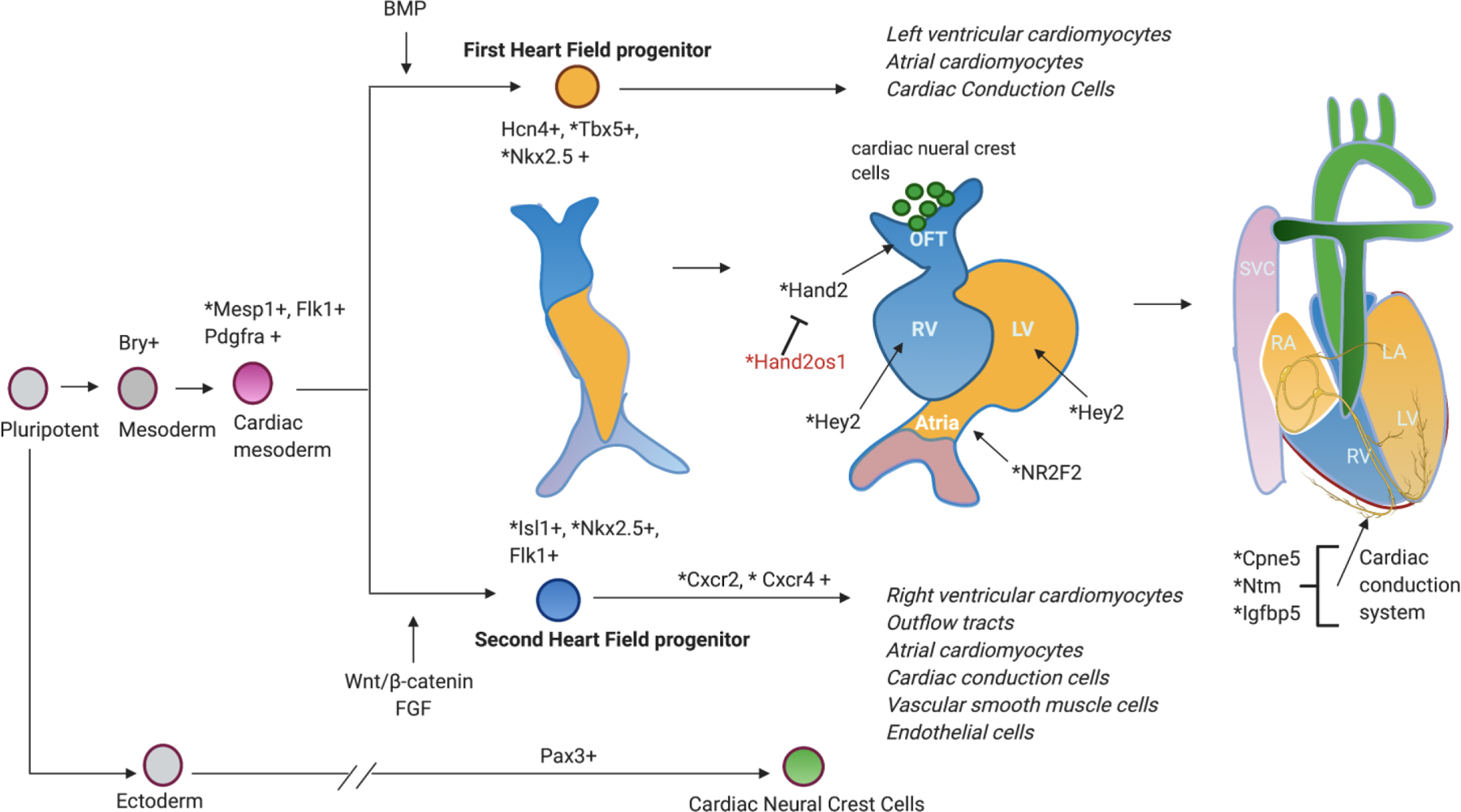
scRNA sequencing contribution to elucidation of cardiac genes role in heart development. Genes with *asterisk are discussed in this review. (Red) Non-coding RNA.
Early work by Bondue et al uncovered Mesp1 as an important driver of cardiovascular cell fate when the group over expressed Mesp1 during ESC differentiation which resulted in promotion and acceleration of cardiovascular cell fate.27,28 Lescroart et al utilized scRNA-seq to identifying distinct populations of Mesp1 cardiovascular progenitor cells and conclude that Mesp1 was required for exiting the pluripotent state and induction of cardiovascular gene expression.29 During organogenesis, the endocardial cushion of the heart consist of cells expressing endothelial-to-mesenchymal transition (EndMT) markers consistent with their endocardial cell origin and progression towards valve formation (Dong et al).30
Single cell RNA sequencing has also provided us better understanding about the genes that contribute to anatomical and electrical conduction of the developing heart with greater resolution compared to traditional models.1 Expression and lineage-tracing experiments have determined expression of Hand1 and Hand2 in developing epicardium, myocardium, and endocardium.31 Early study demonstrated that Hand2 knock out was embryonically lethal at the looping heart tube stage.32 A network based computational method predicted that Hand2 also marks outflow tract cells.30 Recently, using scRNA-seq approach, de Soysa et al profiled 36,000 cells from the cardiogenic region of mouse embryos and found that Hand2-null murine hearts show evidence of outflow tract myocardium specification defect.33 Interestingly, this led to improper differentiation and migration of right ventricular cardiomyocytes. Han et al combined murine models and scRNA-seq of 3,469 Hand2os1 knock out cells from murine embryos as well as 2,563 wild type cells to determine that Hand2 is regulated by non-coding RNA Hand2os1, thereby determining the mechanism by which the right ventricle fails to form into its anatomic structure.34
The anatomical landscape of the heart is of utmost importance during cardiac surgery to repair congenital heart lesions and in particular, the cells of the conduction system, AV valves and outflow tracts. Using scRNA-seq to determine the transcriptomic profile of the cells of the conduction system (Goodyer et al 2019),1 we found genes that are specific to each component (sinoatrial node, atrial-ventricular node, Purkinje fibers) of the cardiac conduction system. Investigation of these genes has provided insights into potential markers for improving the diagnostics, therapeutics, and interventions for cardiac arrhythmias and heart block. Other investigators have also utilized scRNA-seq to understand aortic and mitral valve development in postnatal mice and cardiac outflow tract development, respectively.35,36 As the outflow tract (OFT) wall changes from a myocardial to an arterial phenotype during cardiac development, several cell types are involved and many transcriptional changes occur. Liu et al performed single-cell transcriptomic sequencing of 55,611 mouse OFT cells from three successive developmental stages corresponding to the early, middle, and late stages of OFT remodeling and septation.32 The group identified cells that contribute to the development of the OFT and determine that the vascular smooth muscle cells transition to myocardial cells during OFT development. Transcriptional diversity of cells in the cardiac valve have been largely unknown until recently. Hullin et al found recently that melanocytes, immune, and endothelial cells are present in developing valves into the postnatal period and that interstitial cell clusters were different at various stages of cardiac valve development.36 These studies demonstrate that scRNA-seq has greatly improved our ability to detect rare cell populations contributing to cardiogenesis.
Most recently, large scale single cell RNA sequencing of human fetal tissue was published. Cao et al conducted scRNA-seq on 28 human fetuses ranging from 72 to 129 days in gestation, comprising a total of 4 million cells.5 They identified 667 cell types which they annotated based on cross matching with murine cell atlases, which they noted was straightforward despite species and developmental stage variability. They extracted nuclei from cryopreserved tissue with the exception of renal and digestive tissue in which they conducted scRNA-seq in whole cell and subjected 4 million single cells to UMAP visualization and Louvain clustering using Monocle 3. The group identified 16 main cell types in the fetal heart. The 16 cell types include cells which are known to be found in the heart such as endocardial cells, cardiomyocytes, smooth muscle cells, vascular endothelial cells, epicardial fat cells, stromal cells and lymphatic endothelial cells, and hematopoietic cell lineages including erythroblasts, myeloid cells, megakaryocytes, myeloid cells and lymphoid cells. However, they also noted Schwann cells, and three types of cells identified by markers called (14) SATB2_LRRC7 cells, (15) ELF3_AGBL2 positive cells, and (16) CLC_IL5RA positive cells. The LRRC7 gene codes for a protein, Densin-180, which is expressed primarily in the brain and is involved in synaptic regions of neurons.37 SATB2 gene codes for DNA-binding protein that specifically binds nuclear matrix attachment regions and is involved in chromatin remodeling and transcription regulation.38 It has been found to be a neuronal cell marker as well as markers of colorectal, bone and soft tissue cancers, and important in developmental of cortical neurons.38,39,40 ELF3, epithelial specific ets transcription factor, is involved in epithelial differentiation.41 Diseases associated with ABGL2 include cardiofaciocutaneous syndrome 2.42 The findings of these rare cells within fetal heart demonstrates the power of scRNA-seq and may warrant further investigation on the role that they play in the development of the heart. Most recently, Tyser et al combined single-cell RNA sequencing with high-resolution volume imaging and time-lapse microscopy to identify a population of progenitor cells which they termed as the juxta-cardiac field (JCF), representing the earliest known progenitors of the epicardium.43 This unique population of cells is located rostral to the cardiac crescent, at the confluence of the embryonic and extraembryonic mesoderm. Although the loss of function of the JCF marker, Mab21l2, leads to early embryonic lethality owing to a reduction in myocardium and proepicardium it is unclear whether the JCF is a pool of unipotent progenitors of cardiomyocytes and proepicardium or contains bipotent cells capable of giving rise to both.43 Single cell RNA sequencing allows discovery of new population of cells in the developing heart, thereby providing greater understanding of the role of specific cells in cardiogenesis.
Beyond cardiac development, scRNA-seq has provided insights into how the transcriptome of specific cells change to trigger pathology in cardiac disease in children. Hu et al analyzed the transcriptome of 15,083 nuclei from postnatal development of mitochondrial cardiomyopathy and found upregulation of Gdf15, a growth factor that is known to increase during inflammatory states.44,45Ablation of cardiac neural crest cells in an animal model results in failure of septation of the outflow, however, the molecular mechanisms were previously unknown.46 Bulk and scRNA-seq profiling of neural crest cells combined with reprogramming trunk neural crest cells to cardiac crest fate in vivo demonstrated that the transcriptional sub-circuit of Tgif1, Ets1, and Sox8 was critical for cardiac neural crest and heart development (Gandhi et al 2020).46
One of the many advantages of scRNA-seq involves the ability to determine the transcriptome of very few cells within small sample sizes. The AV nodal tissue is ~3–5 mm in adult heart and is much smaller in a fetus, however, Suryawanshi et al recently conducted scRNA-seq on a human fetus with congenital heart block along with three healthy fetuses and confirmed that an interferon-rich environment contributes to the pathophysiology of congenital heart block.47 Furthermore, the group identified a set of interferon-stimulated genes (TXNIIP, IFITM3, ISG15, IF116) expressed in all cell types in a patient with congenital hear block, which potentially contribute to the pathogenesis of congenital heart block via extracellular matrix deposition.47 The application of scRNA-seq will pave the way for new discovery and insight into determining mechanism and treatment for cardiac disease in the pediatric population. Beyond congenital heart diseases, scRNA-seq has been utilized to understand pathology such as heart failure and hypertrophy. Nomura et al conducted transverse aortic constriction (TAC) in murine models and found that the transcriptomic profiles changed drastically after TAC operation, and returned to the baseline state slowly thereafter.48
Cellular Interactomes, Spatial Transcriptomes, Trajectory Analysis
Cardiac maturation involves orchestration of multiple cells and a cellular environment. scRNA-seq can be utilized to understand interactions between cells. By integrating scRNA-seq data of murine hearts at multiple stages, Wang et al, constructed cellular interactomes and regulatory signal networks.49 The group demonstrated fibroblast subtypes switch from neonatal to adult thereby driving cardiomyocyte maturation. Single cell RNA sequencing has been utilized to investigate the role of fibroblasts in heart failure and immune cells,50,51 as well as ligand-receptor analysis between cardiac cell types in healthy and disease states in hopes of developing prognostic tools or guiding therapies.52
In addition to understanding cellular interactomes, spatial information is important to understand in the developing heart. Spatial transcriptomics allows visualization and quantitative analysis of the transcriptome with spatial resolution in individual tissue sections.53 By positioning histological sections on arrayed reverse transcription primers with unique positional barcodes, Stahl et al demonstrated RNA-sequencing data with maintained two-dimensional positional information from the mouse brain and human breast cancer.54 Asp et al combined scRNA-seq data of human embryonic cardiac cells, RNA sequencing data of spatial transcriptomics, and in situ sequencing data to generate a 3D gene expression atlas of the developing heart.55 In situ sequencing allows linkage between sequencing information and its location.56 Slide-seq is another approach that can be used to measures genome-wide expression at 10 micron resolution to provide spatially resolved gene expression data.57 Rodriques et al utilized slide-seq to localize cell types within the murine cerebellum and hippocampus, characterized spatial gene expression patterns in the Purkinje layer of mouse cerebellum, and defined the temporal evolution of cell-type-specific responses in a mouse model of traumatic brain injury.57 This method may be applied to other tissues such as the heart—particularly, during development in order to determine the cells that contribute to normal development as well as pathology.
Trajectory inference interprets a single cell as a snapshot of a continuous process and places cells along a continuous path that represents the evolution of the process by analyzing transcriptional changes between neighboring cells. There are several inference methods with various model paths including Monocle (simple linear),Wanderlust (bifurcating), Slingshot (bifurcating), and PAGA (multifurcating).7 Velocyto, another R package, distinguishes unspliced and spliced mRNAs.7
Single Cell RNA Sequencing Profiling of Human Induced Pluripotent Stem Cells
The use of human induced pluripotent stem cells (hiPSCs) have contributed to immense knowledge in the field of cardiac development and it is known that hiPSCs have fetal-like characteristics. Single cell RNA-seq of 43,168 hiPSCs during cardiac differentiation was performed and demonstrated that hypertrophic signaling is not sufficiently activated during monolayer-based cardiac differentiation, thereby preventing expression of HOPX and its activation of downstream genes governing the late stages of CM maturation.7,9 Other scRNA-seq studies have identified temporal and spatial gene expression changes and their key factors such as ISL1, NR2F2, TBX5, and HEY2 during cardiac differentiation. Given that single ventricle physiology results from many forms of congenital heart disease and failure of the single ventricle has placed pediatric and adult congenital patients at the greatest risk for mortality and morbidity, the use of hiPSC to understand intrinsic mechanisms of failure is warranted. We recently performed scRNA-seq on day 30 hiPSC-CMs from control and patient with HLHS and demonstrated that the HLHS iPSC-CMs had reduced mitochondrial respiration and oxidative metabolism.58 Similarly, Lam et al conducted scRNA-seq analysis on hiPSC lines from 3 patients with PA/IVS and healthy subjects and demonstrated that PA/IVS lines show attenuated maturation and development as well as diminished expression of cardiac contractile genes.59
Chromatin Accessibility at Single Cell Resolution
Probing of chromatin accessibility profiles of tens of thousands of cells simultaneously have been developed in order to better understand the factors that influence gene expression.9 Domcke et al 2020 created a human atlas of fetal chromatin accessibility by devising a three-level combinatorial indexing assay and applied it to 53 samples representing 15 organs, profiling ~800,000 single cells.60 They utilized single cell ATAC sequencing (scATAC-seq) to determine which transcription factors are involved in generating and maintaining diversity of cell types from an invariant genome. They used a linear regression model to ask which transcription factor motifs found in the accessible sites of each cell best explain its cell type affiliation (i.e. MEF2B for cardiomyocytes). Simultaneous profiling of histone modifications and gene expression in single cells was recently developed by Zhu et al utilizing murine brain tissue.61 This method allows integrative analysis of the resulting maps to identify distinct groups of genes subject to divergent epigenetic regulatory mechanisms. Compared to scATAC-seq, this method can reveal the functional states of cis regulatory elements to provide mechanistic insights of regulatory programs for each cell type from heterogeneous cellular environments.
Combining Genome Wide Association Studies & Organ Atlases
Investigators from the Tabula Muris Consortium performed scRNA-seq profiling of over 100,000 cells from 20 different adult murine organs and tissue to create an atlas of the adult mouse thereby opening the possibility of finding unique gene expression domains for every gene of interest.62 It is possible that by combining scRNA-seq data for murine cardiac tissue and scRNA-seq data from adult human heart tissue, one may be able to determine which cells and tissues express disease-associated genes of interest from genome wide association studies (GWAS).5,6 Although GWAS informs us about the genes associated with disease processes, these studies cannot tell us the specific mechanism nor the cells responsible for the development of disease. Analysis of the expression domain of GWAS candidates using scRNA-seq of human tissue may help to uncover novel mechanisms of disease pathogenesis.
Summary and Future Perspective
The development of scRNA-seq technology has provided significant insights into the key regulators of cardiogenesis and the pathophysiological mechanisms involved in congenital heart disease. The generation of mouse and human heart atlases to comprehensively profile as many cell types as possible, including rare cell populations, should provide extremely valuable gene expression databases for basic and translational investigators to identify new mechanisms of disease. By incorporating information derived from scRNA-seq databases with those obtained for perturbational studies, one would anticipate a major acceleration in our ability to develop personalized, optimal, and safer treatments for patients. With the greater incorporation of scRNA-seq technology in the future, we believe we would be one step closer to attaining the ultimate goal of personalized medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Goodyer WR, Beyersdorf BM, Paik DT, Tian L, Li G, Buikema JW, Chirikian O, Choi S, Venkatraman S, Adams EL, Tessier-Lavigne M, Wu JC, Wu SM. Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution. Circ Res. 2019August2;125(4):379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pimpalwar N, Czuba T, Smith ML, Nilsson J, Gidlöf O, Smith JG. Methods for isolation and transcriptional profiling of individual cells from the human heart. Heliyon. 2020December29;6(12):e05810. doi: 10.1016/j.heliyon.2020.e05810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergmann O, et al. (2009). “Evidence for cardiomyocyte renewal in humans.” Science 324(5923):98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann O and Jovinge S (2012). “Isolation of cardiomyocyte nuclei from post-mortem tissue.” J Vis Exp(65). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Litviňuková M, Talavera-López C, Maatz H et al. Cells of the adult human heart. Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tucker NR, et al. (2020). “Transcriptional and Cellular Diversity of the Human Heart.” Circulation 142(5):466–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao J, et al. (2020). “A human cell atlas of fetal gene expression.” Science 370(6518). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakken TE, et al. (2018). “Single-nucleus and single-cell transcriptomes compared in matched cortical cell types.” PLoS One 13(12): e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada S and Nomura S (2020). “Review of Single-Cell RNA Sequencing in the Heart.” Int J Mol Sci 21(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gladka MM, et al. (2018). “Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation.” Circulation 138(2): 166–180. [DOI] [PubMed] [Google Scholar]
- 11.Kannan S, et al. (2019). “Large Particle Fluorescence-Activated Cell Sorting Enables High-Quality Single-Cell RNA Sequencing and Functional Analysis of Adult Cardiomyocytes.” Circ Res 125(5): 567–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paik DT, et al. (2020). “Single-cell RNA sequencing in cardiovascular development, disease and medicine.” Nat Rev Cardiol 17(8): 457–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kashima Y, Sakamoto Y, Kaneko K et al. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med 52, 1419–1427 (2020). 10.1038/s12276-020-00499-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papalexi E, Satija R Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 18, 35–45 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Sasagawa Y, et al. (2018). “Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads.” Genome Biol 19(1): 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi T, et al. (2018). “Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs.” Nat Commun 9(1): 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jindal A, Gupta P, Jayadeva et al. Discovery of rare cells from voluminous single cell expression data. Nat Commun 9, 4719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Jolliffe IT and Cadima J (2016). “Principal component analysis: a review and recent developments.” Philos Trans A Math Phys Eng Sci 374(2065): 20150202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.“Beta release of Seurat 4.0.” Seurat, https://satijalab.org/seurat/ [Google Scholar]
- 21.Mereu E, et al. (2020). “Benchmarking single-cell RNA-sequencing protocols for cell atlas projects.” Nat Biotechnol 38(6): 747–755. [DOI] [PubMed] [Google Scholar]
- 22.Center for Disease Control and Prevention (December 2020). Data and Statistics on Congenital Heart Defects. National Center on Birth Defects and Developmental Disabilities. https://www.cdc.gov/ncbddd/heartdefects/data.html [Google Scholar]
- 23.DeLaughter DM; Bick AG;Wakimoto H; McKean D; Gorham JM; Kathiriya IS; Hinson JT; Homsy J;Gray J; Pu W; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. CDC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li G; Xu A; Sim S; Priest JR; Tian X; Khan T; Quertermous T; Zhou B; Tsao PS; Quake SR; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong H; Luo Y; Yue Y; Zhang J; Ai S; Li X;Wang X; Zhang YL;Wei Y; Li HH; et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circ. Res 2019,125, 398–410. [DOI] [PubMed] [Google Scholar]
- 26.Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {600584}: {Date last edited 10/30/2019}: World Wide Web URL: https://www.omim.org/entry/600584 [Google Scholar]
- 27.Bondue A Lapouge G Paulissen C Semeraro C Iacovino Kyba M Blanpain C Cell Stem Cell. 2008; 3 (this issue): 69–84 [DOI] [PubMed] [Google Scholar]
- 28.Wu SM (2008). “Mesp1 at the heart of mesoderm lineage specification.” Cell Stem Cell 3(1): 1–2. [DOI] [PubMed] [Google Scholar]
- 29.Lescroart F; Wang X; Lin X; Swedlund B; Gargouri S; Sànchez-Dànes A; Moignard V; Dubois C;Paulissen C; Kinston S; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong J; Hu Y; Fan X; Wu X; Mao Y; Hu B; Guo H; Wen L; Tang F Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 2018, 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hand Factors in Cardiac Development. George Rajani M. and Firulli Anthony B.. [DOI] [PMC free article] [PubMed]
- 32.Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, Olson EN. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat Genet. 1997June;16(2):154–60. doi: 10.1038/ng0697-154.Erratum in: Nat Genet 1997 Aug;16(4):410. [DOI] [PubMed] [Google Scholar]
- 33.de Soysa TY, Ranade SS, Okawa S, Ravichandran S, Huang Y, Salunga HT, Schricker A, Del Sol A, Gifford CA, Srivastava D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature. 2019August;572(7767):120–124. doi: 10.1038/s41586-019-1414-x.Epub 2019 Jul 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han X; Zhang J; Liu Y; Fan X; Ai S; Luo Y; Li X; Jin H; Luo S; Zheng H; et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development 2019, 146, dev176198. [DOI] [PubMed] [Google Scholar]
- 35.Liu X; Chen W; Li W; Li Y; Priest JR; Zhou B;Wang J; Zhou Z Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019, 28, 1346–1361. [DOI] [PubMed] [Google Scholar]
- 36.Hulin A; Hortells L; Gomez-Stallons MV; O’Donnell A; Chetal K; Adam M; Lancellotti P; Oury C; Potter SS; Salomonis N; et al. Maturation of heart valve cell populations during postnatal remodeling. Development 2019, 146, dev173047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.“LRRC7 Gene- Undiagnosed Diseases Network (UDN).” UDN, 2 January. 2020, undiagnosed.hms.harvard.edu/genes/lrrc7/. [Google Scholar]
- 38.Britanova O, et al. (2008). “Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex.” Neuron 57(3): 378–392. [DOI] [PubMed] [Google Scholar]
- 39.Zhang YJ, Chen JW, He XS, Zhang HZ, Ling YH, Wen JH, Deng WH, Li P, Yun JP, Xie D, Cai MY. SATB2 is a Promising Biomarker for Identifying a Colorectal Origin for Liver Metastatic Adenocarcinomas. EBioMedicine. 2018February;28:62–69. doi: 10.1016/j.ebiom.2018.01.001.Epub 2018 Jan 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creytens D and Ferdinande L (2015). “SATB2 is a novel marker of osteoblastic differentiation in bone and soft tissue tumours: comment on Conner et al. (2013).” Histopathology 67(2): 272–273. [DOI] [PubMed] [Google Scholar]
- 41.Brembeck FH, et al. (2000). “Dual function of the epithelial specific ets transcription factor, ELF3, in modulating differentiation.” Oncogene 19(15): 1941–1949. [DOI] [PubMed] [Google Scholar]
- 42.AGBL2 Gene (Protein Coding).” GeneCards, www.genecards.org/cgi-bin/carddisp.pl?gene=AGBL2. [Google Scholar]
- 43.Tyser RCV, et al. (2021). “Characterization of a common progenitor pool of the epicardium and myocardium.” Science 371(6533). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu P; Liu J; Zhao J; Wilkins BJ; Lupino K; Wu H; Pei L Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuca SA, et al. (2017). “The Relationship between Metabolic Parameters, Cardiac Parameters and MIC-1/GDF15 in Obese Children.” Exp Clin Endocrinol Diabetes 125(2): 86–90. [DOI] [PubMed] [Google Scholar]
- 46.Gandhi S, Ezin M, Bronner ME. Reprogramming Axial Level Identity to Rescue Neural-Crest-Related Congenital Heart Defects. Dev Cell. 2020May4;53(3):300–315.e4. doi: 10.1016/j.devcel.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suryawanshi H; Clancy R; Morozov P; Halushka MK; Buyon JP; Tuschl T Cell atlas of the fetal human heart and implications for autoimmune-mediated congenital heart block. Cardiovasc. Res 2020, 116, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nomura S, et al. (2018). “Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure.” Nat Commun 9(1): 4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Yao F, Wang L, Li Z, Ren Z, Li D, Zhang M, Han L, Wang SQ, Zhou B, Wang L. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat Commun. 2020May22;11(1):2585. doi: 10.1038/s41467-020-16204-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krstevski C, Cohen CD, Dona MSI, Pinto AR. New perspectives of the cardiac cellular landscape: mapping cellular mediators of cardiac fibrosis using single-cell transcriptomics. Biochem Soc Trans. 2020December1:BST20191255. doi: 10.1042/BST20191255. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 51.Gupta RK, Kuznicki J. Biological and Medical Importance of Cellular Heterogeneity Deciphered by Single-Cell RNA Sequencing. Cells. 2020July22;9(8):1751. doi: 10.3390/cells9081751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vidal R, et al. (2019). “Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart.” JCI Insight 4(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satija R, Farrell JA, Gennert D, Schier AF and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg Å, Pontén F, Costea PI, Sahlén P, Mulder J, Bergmann O, Lundeberg J, Frisén J. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016July1;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 55.Asp M; Giacomello S; Larsson L; Wu C; Fürth D; Qian X; Wärdell E; Custodio J; Reimegård J;Salmén F; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660 [DOI] [PubMed] [Google Scholar]
- 56.Ke R, Mignardi M, Pacureanu A et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods 10, 857–860 (2013). 10.1038/nmeth.2563 [DOI] [PubMed] [Google Scholar]
- 57.Rodriques SG, et al. (2019). “Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution.” Science 363(6434): 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paige SL, et al. (2020). “Patient-Specific Induced Pluripotent Stem Cells Implicate Intrinsic Impaired Contractility in Hypoplastic Left Heart Syndrome.” Circulation 142(16): 1605–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lam YY, Keung W, Chan CH, Geng L, Wong N, Brenière-Letuffe D, Li RA, Cheung YF. Single-Cell Transcriptomics of Engineered Cardiac Tissues From Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals Abnormal Developmental Trajectory and Intrinsic Contractile Defects in Hypoplastic Right Heart Syndrome. J Am Heart Assoc. 2020October20;9(20):e016528. doi: 10.1161/JAHA.120.016528.Epub 2020 Oct 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Domcke S, et al. (2020). “A human cell atlas of fetal chromatin accessibility.” Science 370(6518). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu C, et al. (2021). “Joint profiling of histone modifications and transcriptome in single cells from mouse brain.” Nat Methods 18(3): 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The Tabula Muris Consortium., Overall coordination., Schaum N et al. Single-cell transcriptomics of mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Figures 1 and 2 were created with Biorender.com with permission from vendor as outline in the user service agreement.