

Abstract
Leak currents, defined as voltage and time independent flows of ions across cell membranes, are central to cellular electrical excitability control. The K2P (KCNK) potassium channel class comprises an ion channel family that produces potassium leak currents that oppose excitation and stabilize the resting membrane potential in cells in the brain, cardiovascular system, immune system, and sensory organs. Due to their widespread tissue distribution, K2Ps contribute to many physiological and pathophysiological processes including anesthesia, pain, arrythmias, ischemia, hypertension, migraine, intraocular pressure regulation, and lung injury responses. Structural studies of six homomeric K2Ps have established the basic architecture of this channel family, revealed key moving parts involved in K2P function, uncovered the importance of asymmetric pinching and dilation motions in the K2P selectivity filter (SF) C-type gate, and defined two K2P structural classes based on the absence or presence of an intracellular gate. Further, a series of structures characterizing K2P:modulator interactions have revealed a striking polysite pharmacology housed within a relatively modestly sized (~70 kDa) channel. Binding sites for small molecules or lipids that control channel function are found at every layer of the channel structure, starting from its extracellular side through the portion that interacts with the membrane bilayer inner leaflet. This framework provides the basis for understanding how gating cues sensed by different channel parts control function and how small molecules and lipids modulate K2P activity. Such knowledge should catalyze development of new K2P modulators to probe function and treat a wide range of disorders.
Keywords: K2P channel, selectivity filter C-type gate, M2 and M4 helix transmembrane motions, polysite pharmacology
Introduction
The term ‘leak’ tends to have negative connotations, is often used as a pejorative, especially in politics, and is something best avoided by those worried about roofs, buckets, or other important barriers or containers. To the contrary, leak is not a bad thing for cells, especially those involved in producing the bioelectricity that runs our hearts, brains, muscles, and sensory organs. Indeed, background or ‘leak’ potassium currents, defined broadly as those lacking voltage or time dependency, have been recognized for >70 years as essential actors in affecting the electrical excitability of cells.1–3 Ion channel proteins responsible for this activity first became apparent ~25 years ago with the identification of the founding members of what is now known as the K2P (KCNK) family of potassium channels: K2P1.1 (TWIK-1),4 K2P2.1 (TREK-1),5 K2P3.1 (TASK-1),6 and K2P4.1(TRAAK)7 (Figure 1(a)).8,9 The subsequent study of K2Ps has revealed that these channels are much ‘more than just leak’.10 K2Ps are highly regulated by diverse factors including both chemical and physical gating cues9,11,12 and although their pharmacology remains underdeveloped, they present promising targets for new therapeutics aimed at pain, migraine, ischemia, depression, glaucoma, and lung injury. Structural studies over the past ~8 years have begun to reveal the core architecture responsible for K2P function, as well as a rich array of sites that can be targeted by small molecule modulators. Here, we focus on these key molecular advances and highlight some of the exciting new directions that are on the horizon for this very positive form of leak.
Figure 1.

K2P channel family relationships and structure. (a) K2P family dendrogram. Subfamilies and key characteristics are indicated. Asterisks indicate structurally characterized K2Ps. (b) K2P subunit diagram. Pore domains 1 and 2 (PD1 and PD2), transmembrane helices (M1–M4), pore helices (P1 and P2), selectivity filters (SF1 and SF2), and Cap domain are indicated. (c and d) Cartoon diagram of the K2P2.1 (TREK-1) structure (PDB:6CQ6).37 Chains are colored marine and orange. Potassium ions are purple. Grey lines indicate membrane. c, side view, (d) cytoplasmic view. Channel elements are labeled as in ‘b’. ‘EIP’ denotes the extracellular ion pathway.
The K2P family of channels belongs to the larger voltage-gated ion channel (VGIC) superfamily.13 There are 15 human K2P subunits8,9 comprising six subfamilies that are regulated by various stimuli (Figure 1(a)). The name ‘K2P’ derives from their unique subunit architecture. Each K2P subunit comprises two pore-forming domains, PD1 and PD2. Both of these domains contain two transmembrane helices (M1-M2 and M3-M4) that are bridged by a pore helix (P1 and P2) and selectivity filter (SF1 and SF2) (Figure 1(b)). K2P subunits dimerize to create a channel having an intrinsically heterotetrameric pore due to the fact that PD1 and PD2 are not identical. Unlike voltage-gated or inward rectifier channels from the VGIC superfamily, K2Ps conduct ions over the entire physiological voltage range, in line with their role as ‘leak’ channels. This leak current is not invariant and can be tuned by a remarkable array of chemical and physical gating cues that include pressure, temperature, extracellular and intracellular pH, lipids, phosphorylation, and agents such as volatile anesthetics and antidepressants.9,14
Studies of K2P function have uncovered that members of this family are important for diverse classes of physiological responses and pathological conditions such as action potential propagation,15,16 anesthetic responses,17,18 microglial surveillance,19 sleep duration,20 pain,21–23 arrhythmia,24 ischemia,17,25,26 cardiac fibrosis,27 depression,28 migraine,29 intraocular pressure regulation,30 pulmonary hypertension,31 and lung injury.32 Due to their unusual topology, diverse gating stimuli, and poor pharmacology, K2Ps have remained the most poorly understood potassium channel class.8,9,33–35 Nevertheless, the structures of six homomeric K2Ps are now known (Figure 1 (a))36–41 and reveal a conserved overall architecture that defines the K2P family (Figure 1(c) and (d)).
In addition to the need to answer basic questions about how K2Ps function and are regulated, the lack of good pharmacological tools to manipulate K2Ps remains a challenge for understanding and controlling their physiological functions.42,43 The demand to address this deficit has not gone unnoticed and a number of recent discoveries of a variety of K2P modulators are beginning to define new pharmacological tools directed at this channel family.23,37–38,44–49 Further, in spite of their modest size (~70 kDa), the first studies of K2P structural pharmacology have revealed a strikingly lush structural landscape for functional control by various types of modulators.37,40,50,51 This unexpectedly rich polysite pharmacology provides the means to dissect mechanisms of K2P function and is the starting point for new ventures to elaborate K2P pharmacology.
K2P structure: Similar to other potassium channels, but different
Common to other potassium channels, each of the K2P pore domains carries the potassium channel selectivity filter signature sequence (TxTTxGYGD),52 encompassing the strands that directly coordinate the permeant ions and the C-terminal end of the pore helix (Figure 2(a)). There are several notable deviations from this conserved sequence. In PD1, the first Thr of this sequence is always a hydrophobic residue and the final Asp is replaced with Asn, His, Tyr, or Met. In PD2, the ‘Y’ of the ‘GYG’ is either Phe or Leu. Despite these sequence differences, which may impact function, structures of various K2P members show that the fundamental architecture of what is essentially the active site of the channel is not different from other potassium channels (Figure 2(b)).36–41
Figure 2.

K2P pore region structure. (a) Sequence alignment of selectivity filter and loop regions of PD1 and PD2 for the indicated channels. P1, M2, P2, and M4 helices (blue), SF1 and SF2 (orange), SF1-M2 loop (red), and SF2-M4 loop (light blue) are indicated. Terminal residue of the selectivity filter is highlighted. (b) Superposition of the selectivity filters and permeant ions for: K2P1.1 (TWIK-1) (3UKM)36 (red), K2P2.1 (TREK-1) (6CQ6)37 (smudge), K2P2.1 (TREK1):ML335 (6CQ8)37 (deep salmon), K2P2.1 (TREK-1):ML402 (6CQ9),37 K2P3.1 (TASK-1) (6RV2) (orange),38 K2P3.1 (TASK-1):BAY1000493 (6RV3) (yellow orange),38 K2P3.1 (TASK-1):BAY2341237(6RV4) (olive),38 K2P4.1 (TRAAK) (3UM7)39 (aquamarine), (4I9W)56 (limon), (4WFE) (forest green),58 (4WFF) (white),58 (4WFG) (grey),58 (4WFH) (black),58 K2P4.1 (TRAAK) G124I (4RUE) (blue)57, K2P4.1 (TRAAK) W262S (4RUF) (lime green),57 K2P5.1 (TASK-2) pH 6.5 (6WLV) (deep olive),41 K2P5.1 (TASK-2) pH 8.5 (6WM0) (light teal),41 K2P10.1 (TREK-2) (4BW5)40 (pink), (4XDJ)40 (magenta), (4XDK)40 (purple). SF1, SF2 and ion binding positions, S1-S4, are indicated. Ions are shown as spheres and colored according to the parent structure. (c) K2P pore domains (PD1 and PD2) highlighting the inherent heterotetrameric nature of the pore and the domain-swapped positions of M1. (d) Superposition of K2P2.1 (TREK-1) (PDB:6CQ6)37 P1-SF1-M2 (orange) and P2-SF2-M4 (slate). SF1-M2 loop (red) and SF2-M4 loop (blue) and portions having a shared conformation (dark blue) are indicated. Residue labels indicate the SF1-M2 and SF2-M4 loop ends and structural divergence point (Pro150/Ala259). GenBank sequences in ‘a’ are: K2P2.1 (TREK-1), AAD47569.1; K2P10.1 (TREK-2), AAL95705.1; K2P4.1 (TRAAK), AAF64062.1; K2P3.1 (TASK-1), AAC51777.1; K2P9.1 (TASK-3), AAF63708.1; K2P15.1 (TASK-5), AAG33127.1; K2P5.1 (TASK-2), AAC79458.1; K2P16.1 (TALK-1), AAK49532.1; K2P17.1 (TALK-2), AAK49533.1; K2P1.1 (TWIK-1), AAB01688.1, K2P6.1 (TWIK-2), AAD22980.1; K2P12.1 (THIK-2), AAG32313.1; K2P13.1 (THIK-1), AAG32314.1; K2P18.1 (TRESK), BAC78527.1.
Most of the K2P structure is within the membrane (Figure 1(c)). PD1 and PD2 assemble to form the single pore in which M2 and M4 line the central cavity and M1 and M3 face the bilayer. The two transmembrane pairs, M1/M2 and M3/M4 interact closely with each other to form the supporting structure of the P1 and P2 pore helices and their attendant selectivity filters, respectively. The four SF elements are arranged along the channel central axis where they coordinate the permeant ions (Figure 2(b)). Because of their dimeric structure, the K2P pore is intrinsically heterotetrameric having two PD1s and two PD2s arranged around the central axis (Figure 2(c)). The linker connecting the SF to the inner helix of its pore domain has different lengths in PD1 versus PD2 (Figure 2(a) and (d)). The SF1-M2 linker has six residues, similar to most other potassium channels, whereas the SF2-M4 linker is more than twice that length having between 12–14 residues (Figure 2(a) and (d)). The one exception is K2P18.1 (TRESK) in which the SF2-M4 linker has the canonical six residue length. These differences have important consequences for the role of the SF in channel gating and point to the SF2-M4 loop as a unique functional feature of K2Ps.53
K2Ps have a distinctive extracellular structure termed the Cap domain that is formed by a pair of helices from each subunit.36,39 One of these helices is contiguous with the M1 transmembrane helix (Figure 1(c)). The Cap has a largely conserved disulfide bond at its peak54,55 and forms an archway over the mouth of the channel pore that creates a bifurcated pathway, termed the ‘Extracellular ion pathway’ (EIP),36,39 through which potassium ions exit the channel under physiological conditions (Figure 1(c)).
Structural studies have uncovered one other unusual aspect of the K2P pore domain architecture. In K2P2.1 (TREK-1),37,51,53 K2P3.1 (TASK-1),38 K2P4.1 (TRAAK),56–58 K2P5.1 (TASK-2),41 and K2P10.1 (TREK-2),40 the M1 segment is domain swapped between the subunits, an arrangement enabled by the interposition of the Cap domain helices between M1 and P1. The result is that the PD1 structural module is comprised of M1 from one subunit and M2, P1, and the selectivity filter of the other (Figure 2(c)). Domain swapping, which involves the formation of intertwined protein assemblies by exchange of identical structural elements, is observed in many classes of soluble and transmembrane proteins,59–62 although its functional relevance is often not clear.59,61,63 The only K2P structure in which this arrangement has not been reported is K2P1.1 (TWIK-1)36. Further, such pore domain intertwining has not been seen in other VGIC superfamily members. The predominance of domain-swapped pores in the K2P family raises questions about how such interlocked structures form during K2P biogenesis, how easily they are taken apart, and whether such interlinking affects K2P stability or function. Moreover, some K2P subunits can assemble as heterodimers.29,64–71 Although it is not known whether heterodimeric K2Ps also have this domain swap, the impact of associating M1 of one subunit with the M2/P1 elements of the pore domain from the heterodimer partner could result in functional differences between domain-swapped versus non-domain swapped versions if both are able to form. Biogenesis and folding of most channels, including K2Ps, is largely a black box. Unravelling how these domain swapped structures form, and their consequences for function and regulation is a key question for future studies.
K2P channel moving parts
In order to respond to signals, a protein must have some moving parts. The structures of K2Ps determined to date have revealed three moving elements that are tied to function. Two of these involve the two pore-lining helices, M2 and M4.40–41,56–58 The third encompasses the structure that forms and supports the selectivity filter C-type gate.37,40,41,53
Unlike most members of the VGIC superfamily, K2P gating is primarily controlled at the selectivity filter (SF) C-type gate.53,72–77 Structures from the TWIK36 and TREK subfamilies37,40,51,53,56–58 showed channels that lack an intracellular barrier and that have an unhindered path from the cytoplasm to the SF (Figure 3(a)). These observations are consistent with functional data indicating that these channels,72–75,78 as well as other K2Ps, such as K2P18.1 (TRESK),78 lack an intracellular gate. Prior functional studies suggested that some K2Ps may have an intracellular gate79–80 and two recent structures of representatives from the TASK38 and TALK41 subfamilies have provided the first structural examples of how such a gate forms. In K2P3.1 (TASK-1) a pair of short helical segments (~two turns) at the distal end of M4 kink to produce a barrier at the intracellular mouth of the channel termed the ‘X-gate’38 (Figure 3(b)). By contrast, the intracellular barrier of the pH 6.5 K2P5.1 (TASK-2) structure shows a pinching between the two M4 helices that appears mediated by a pair of lysine sidechains41 (Figure 3(b)). Thus, it appears based on the current available data that the K2Ps will break into two structural classes, those without an inner gate, such as the TREK subfamily and K2P1.1 (TWIK-1), and those that have some type of intracellular barrier like K2P3.1 (TASK-1)38 and K2P5.1 (TASK2).41 Understanding these class distinctions and whether there are additional variations to this classification will be important for delineating gating mechanisms and for developing subtype selective pharmacologies that can exploit these differences. Defining how the other K2P homodimers fit into these classes, and uncovering whether there are further variations on these themes, especially among heterodimeric K2Ps, is an important line of investigation for the near future.
Figure 3.

K2P structure comparison highlighting the M4 position and presence or absence of a lower gate. (a) K2Ps having only the SF gate: K2P1.1 (TWIK-1) (PDB:3UKM),36 K2P2.1 (TREK-1) (PDB:6CQ6),37 K2P4.1 (TRAAK) (PDB:4WFG),58 K2P4.1 (TRAAK) (PDB:4WFF),58 K2P4.1 W262S (PDB:4RUF),57 K2P4.1 (TRAAK) G124I (PDB:4RUE),57 K2P10.1 (TREK-2) (PDB:4BW5),40 K2P10.1 (TREK-2) (PDB:4XDJ).40 (b) K2Ps having both an SF gate and a Lower gate K2P3.1 (TASK-1) (PDB:6RV2)38 and K2P5.1 (TASK-2) pH 8.5 (PDB:6WM0).41 SF, M4, X-gate, Intracellular gate (IC gate) are indicated. ‘Up’ and ‘down’ indicate M4 position. Shading indicates view from the side (blue) and intracellular face (orange).
The ‘up’ and ‘down’ movements of M4
The most obvious moving part of K2Ps is the M4 helix. The position of this helix varies when compared across structures from different subtypes (Figure 3(a)) and in multiple structures of K2P4.1 (TRAAK)56–58 and K2P10.1 (TREK-2).40 The extremes of these M4 positions have been termed ‘up’ and ‘down’ (Figures 3(a) and 4(a) and (b)). In the ‘down’ conformation M4 is approximately straight, crosses the membrane at a ~45° angle and makes no contacts with the M2 helix of the opposite subunit. This conformation leaves a 5–10Å gap that faces the lipid bilayer. ‘Down’ structures have shown that hydrophobic entities (either detergents or lipids),36,56,58 as well as the inhibitor norfluoxetine (Figure 4(b)) can occupy a site formed by this gap that is just under the P2 helix, termed the ‘Fenestration site’. It has been proposed that lipid tails may reach through this gap to block ion permeation58; however, functional tests have not supported this hypothesis.74,81,82 In the ‘up’, conformation, M4 bends at its midpoint near a conserved glycine (G260 in human K2P4.1 (TRAAK)) towards the bilayer core by approximately 25°. This conformational change allows the C-terminal half of M4 to pack against M2, closes the fenestration (Figure 4(a) and (b)),and also changes the shape of the channel near the intracellular end of M4 that is thought to be the principal site of lipid modulation by phosphatidylinositol 4,5-bisphosphate (PIP2) in the TREK subfamily.37,57 Intriguingly, functional and molecular simulation studies have suggested that the hydrophobicity of particular M2 and M4 residues near the fenestration may affect function in some K2Ps by enabling transient dewetting of the central pore through a process termed ‘hydrophobic gating’.83–85 However, it remains unclear how this process might be regulated by the state of the M4 helix.
Figure 4.

K2P transmembrane moving parts. (a) Superposition of the P2-SF2-M4 portion for: K2P1.1 (TWIK-1) (3UKM)36 (red), K2P2.1 (TREK-1) (6CQ6)37 (smudge), K2P2.1 (TREK-1):ML335 (6CQ8)37 (deep salmon), K2P2.1 (TREK-1):ML402 (6CQ9),37 K2P3.1 (TASK-1) (6RV2) (orange),38 K2P3.1 (TASK-1):BAY1000493 (6RV3) (yellow orange),38 K2P3.1 (TASK-1):BAY2341237(6RV4) (olive),38 K2P4.1 (TRAAK) (3UM7)39 (aquamarine), (4I9W)56 (limon), (4WFE) (forest green),58 (4WFF) (white),58 (4WFG) (grey),58 (4WFH) (black),58 K2P4.1 (TRAAK) G124I (4RUE) (blue),57 K2P4.1 (TRAAK) W262S (4RUF) (lime green),57 K2P5.1 (TASK-2) pH 6.5 (6WLV) (deep olive),41 K2P5.1 (TASK-2) pH 8.5 (6WM0) (light teal),41 K2P10.1 (TREK-2) (4BW5)40 (pink), (4XDJ)40 (magenta), (4XDK)40 (purple). Select structures are indicated. Arrow indicates point of M4 bend. (b) View of the ‘up’ (left) and ‘down’ (right) M4 conformations in K2P10.1 (TREK-2). Fenestration site is occupied by norfluoxetine (space filling) and is indicated by the red circle. (c) Sequence comparisons of the M2 helix and elements that frame the Fenestration site. Residues that interact with the Fenestration site ligand are indicated by the black asterisks and are highlighted in magenta (cf. Figure 6). TREK subfamily ‘GVG’ sequence is indicated by the arrows. Green asterisk indicates the position of azo-isoflurane labeling.143 (d) Buckling at the conserved TREK family ‘GVG’ sequence in K2P4.1 (TRAAK). Dashed red oval highlights the site of the i,i + 4 to i,i + 3 hydrogen bond shift. Residue numbers are from human K2P4.1 (TRAAK) PDB:4RUE.57 Genbank sequences in ‘c’ are the same as those in Figure 2(a).
These up/down M4 motions are reminiscent of similar motions of the pore-lining helices of fourfold symmetric channels from the VGIC superfamily, but whereas in VGICs a similar motion towards an ‘up-like’ conformation of four pore-lining helices leads to an opening of the intracellular gate, this is not so for K2Ps. In both the ‘up’ and ‘down’ conformations some K2Ps have an unobstructed path for ions to traverse from the cytoplasm to the selectivity filter (Figure 3(a)). Consequently, assigning functional correlates to the ‘up’ and ‘down’ conformations has not been straightforward. Initial reports from studies of the mechanosensitive TREK subfamily had contradictory interpretations with one suggesting that the ‘up’ state58 represented the active state – reasoning that the expanded cross sectional area of the ‘up’ state would be favored by in-plane membrane tension, while another observed that the ‘down’ state resulted from gain-of-function (GOF) mutations.57 It has been noted that the mechanosensitive TREK K2P subfamily shows relatively small differences in cross sectional area between ‘up’ and ‘down’ states compared to analogous gating changes in other mechanosensitive channels (ex. ΔA < 5 nm2 in K2P10.1 (TREK-2) versus 20 nm2 in MscL and 120 nm2 in Piezo1)12. Nevertheless, molecular dynamics studies of K2P10.1 (TREK-2) have shown that lipid bilayer stretching favors the ‘up’ state, likely as a result of complex changes in the internal pressure of the membrane and protein-lipid packing alterations, in addition to protein cross-sectional area changes.82,86–87 To add to the complexity, functional experiments have shown that activation is not a simple dichotomy of an active ‘up’ state and inactive ‘down’ state, but in fact, both the ‘up’ and the ‘down’ states are active and are sensitive to additional regulation.81
Based on the established centrality of the SF gate and the observation that activators reduce the mobility of the C-type gate,37,53 it has been proposed that M4 movements are coupled to the dynamics of the selectivity filter C-type gate, providing a rationalization for how both extreme positions could result in active channels.37,53,87–88 This structural explanation for resolving the apparent paradox with an ‘everybody wins’ mechanism is attractive, even though the details of the energetic coupling between M4 and the SF gate remain incompletely understood. Intriguingly, a recent report describes a K2P10.1 (TREK-2) high activity mode that retains sensitivity to norfluoxetine, and thus, must be in a ‘down’ conformation.89 This result supports the idea that the ‘down’ state can be a high activity state as inferred from the studies of gain-of-function mutants57 and underscores that there remains much to be learned about how M4 motions in K2Ps influence activity.
Both K2P3.1 (TASK-1)38 and K2P5.1 (TASK-2)41 structures that show a closure at the intracellular mouth of the channel have a ‘down’ M4 position that includes additional structural rearrangements that block the pore (Figure 3(b)). It is clear from the physical barriers made by the X-gate and the intracellular pH regulated gate that some part of these elements must move to let both ions and blocker molecules pass, an idea supported by functional studies of K2P3.1 (TASK-1),38,79 K2P5.1 (TASK2),90 and the drosophila K2P KCNK0.80 In the case of K2P5.1 (TASK-2)41, the motions required to open the inner gate are modest, largely involving changes in the sidechains. The distal end of the X-gate has a region termed the ‘latch’ that interacts extensively with M1 and M2, and it has been proposed that disruption of these interactions would allow M4 to move towards and ‘up’ conformation as the gate opens.38 Interestingly, mutations in and around the latch structure in TASK subfamily members are linked to pulmonary hypertension and a developmental disorder known as Birk-Barel syndrome,91 further highlighting the importance of this element in channel function.92 Understanding the extent of such motions, as well as the stability of these inner gate structures is a key next step that will enrich our understanding of how M4 moves, how changes in its position affect K2P function, how its motions can be exploited to gain control of specific K2Ps, and the extent to which such M4 motions are present in other K2P family members.
Buckling motions of M2
The TREK subfamily represents a group of polymodal ion channels that are affected by both physical and chemical gating cues.12 Structural studies of two K2P4.1 (TRAAK) GOF mutants that impact responses to temperature and pressure activation72–73 showed that the M4 ‘down’ state resulted in a second, functionally relevant conformational change.57 The ‘down’ motion of M4 separates this helix from M2 of the neighboring subunit, but creates a new interface with M2 from its own subunit. These changes deform M2 in a way that leads to its buckling at a conserved glycine GXG motif found in the TREK subfamily57 (Figure 4(c) and (d)). Interestingly, the deformation includes an apparent backbone hydrogen bond shift from i, i + 4 (Leu161-Gly165) to i, i + 3 (Ala162, Gly165) in human K2P4.1 (TRAAK) (PDB: 4RUE)57 (Figure 4(d)), a low-energy rearrangement that has been associated with functionally relevant helix deformations in other membrane proteins.93 Mutation at this GVG motif glycine impacts function and supports the idea that M2 buckling is important for TREK subfamily activation. It is interesting that this glycine is also conserved in the TASK and TALK subfamilies (Figure 4(c)) and coincides with the center of a ~5° bend in M2. A better understanding of the potential roles of motions in M2 as well as other parts of the K2P transmembrane structure may uncover additional factors that influence K2P dynamics and function.
Pinching and dilation of the K2P the selectivity filter ‘C-type’ gate
Numerous studies have established the primacy of the selectivity filter (C-type) gate for K2P function.72–74,76,77,90,94–96 This form of gating occurs in various potassium channel classes and displays a hallmark sensitivity to external potassium due to its dependency on interactions between the permeant ions and selectivity filter.73,74,76,97–101 The interdependency on channel:permeant ion interactions for function can be thought of as a type of ‘substrate assisted catalysis’ wherein interactions with the permeant ion are essential for maintaining the integrity of the selectivity filter active conformation. Although structural studies of exemplar homotetrameric potassium channels have uncovered various types of selectivity filter rearrangements attributed to C-type gating,100,102–108 whether the essence of potassium channel C-type gating involves pinching,100,102–105 dilation101, or more subtle selectivity filter changes106–108 has remained unresolved. Furthermore, even though structures of different K2Ps brought to light changes in M2 and M4 conformations that impact activity36,37,39,40,56–58, these studies did not reveal selectivity filter conformational changes that could explain how K2P C-type gating occurs (Figure 2(b)). Notably, all were determined under conditions of high concentrations (150–200 mM) of permeant ions. Thus, the extent to which K2P C-type gating mechanisms are similar to those described for homotetrameric channels100,102–107 and whether the innate heterodimeric K2P selectivity filter architecture conferred unique properties to their C-type gates had remained unclear.
Recent crystal structures of K2P2.1 (TREK-1) determined under seven different potassium concentrations spanning 0–200 mM, alone and complexed with a small molecule C-type gate activator, ML335, together with molecular dynamics simulations demonstrated that K2P selectivity filters use both pinching and dilation mechanisms to control the C-type gate.53 These structures show that under low potassium concentrations (<50 mM [K+)) both selectivity filter elements that interact with the permeant ions, SF1 and SF2, undergo potassium-dependent structural rearrangements that are accompanied by the loss of the potassium ions at the S1 and S2 sites in the outer portion of the selectivity filter (Figure 5(a)). These conformational changes pinch SF1 at the extracellular mouth of the pore and dilate the exterior portion of SF2 and unfold the linker that connects SF2 to M4 (SF2-M4 loop) (Figure 5(a)). Hence, K2Ps use both types of mechanisms that have been proposed to control potassium channel selectivity filter C-type gates – pinching and dilation. Binding of the small molecule activator ML335 to a site behind the selectivity filter, the K2P modulator pocket, completely suppresses these conformational changes and the loss of potassium ions at low potassium concentrations (Figure 5(b) and (c)).53 These studies also identified an important functional role for the uniquely long SF2-M4 loop found in the K2P family. This channel element is stabilized by a conserved hydrogen bond network, the ‘M3 glutamate network’, centered on Glu234 from the M3 helix, unfolds when SF2 dilates, and connects the C-type gate with gating cues sensed by other parts of the channel. Disruption of the M3 glutamate network by design in K2P2.1 (TREK-1) and K2P3.1 (TASK-1)53 or by a K2P3.1 (TASK-1) mutation associated with pulmonary hypertension92 impairs function and establishes the key role of this network and the SF2-M4 loop in diverse K2Ps even for those, such as K2P3.1(TASK-1), that have intracellular gates. Together, these findings show that asymmetric order–disorder transitions enabled by the K2P heterodimeric architecture are at the heart of K2P gating mechanisms.
Figure 5.

Structural changes in the K2P selectivity filter C-type gate. (a) SF1 and SF2 structures in low [K+] showing the pinched and dilated conformations. (b) SF1 and SF2 structures in low [K+] showing the conductive conformation with ML335 (surface and sticks). (c) Structure of K2P2.1 (TREK-1) (solid) highlighting SF2 and the surrounding structure (wireframe), ML335 (black, space filling), and potassium ions (purple spheres). Orange arrows show the flow of K+ through the channel. Grey bars indicate the membrane. (d) Model for K2P gating at the K2P C-type gate. Grey lines indicate mobile elements. Only P1 and M4 are depicted for simplicity. Activation involves rigidification of the SF gate elements and increased ion flow. K+ ions are shown as purple spheres. ML335 is shown in space filling. In ‘a’ and ‘b’ ion positions and select SF residues are indicated.
Several other structural studies have also hinted at asymmetric motions in the K2P SF gate. In the K2P10.1 (TREK-2) ‘M4-down’ structures SF1, but not SF2, adopts a small inward pinch relative to its position in the ‘M4-up’ structure.40 A similar motion has been observed in SF1 of the pH 6.5 structure of K2P5.1 (TASK-2) relative to the pH 8.5 structure.41 In both cases, these changes distort the coordination environment of ions at the S0 and S1 positions of the selectivity filter and no ions were observed in those sites. These selectivity filter changes are not as extreme as those seen in K2P2.1 (TREK-1)53 and the degree to which they diminish ion permeation is not yet clear. Nevertheless, such local, asymmetric structural distortions of the selectivity filter further highlight the intrinsically heterodimeric character of the K2P architecture.
Taken together, these findings support a model in which under basal conditions the K2P C-type gate transits between an inactive state in which the K2P selectivity filter and its supporting architecture are dynamic and has a low potassium ion occupancy53,77 to a rigidified, conductive state in which most potassium binding sites are occupied (Figure 5(d)).37 Ion permeation requires limiting filter mobility which can be achieved by ligand binding to the K2P modulator pocket or by conformational changes transmitted through the M4 helix.37 These structural changes shift the conformational equilibrium of the channel to the active state, enable permeant ions to organize and stabilize the K2P conductive state37,50,77 and may also be induced by the action of modulators that act on other sites in the channel.50,109 Developing a better understanding of the types of SF changes that can control K2P function, how conformational changes in transmembrane elements affect the SF gate, and how diversification of intrinsically asymmetric K2P architecture through heterodimer formation augments C-type gate function remain key unsolved issues.
The polysite pharmacology of K2Ps
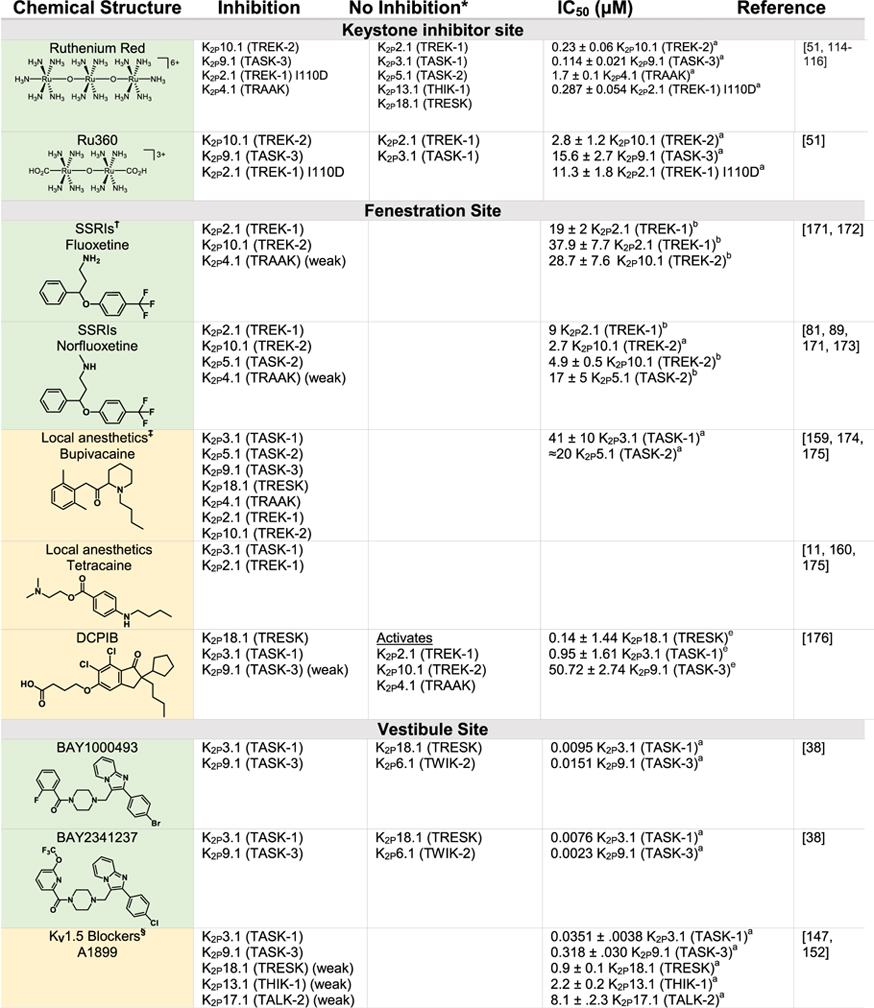
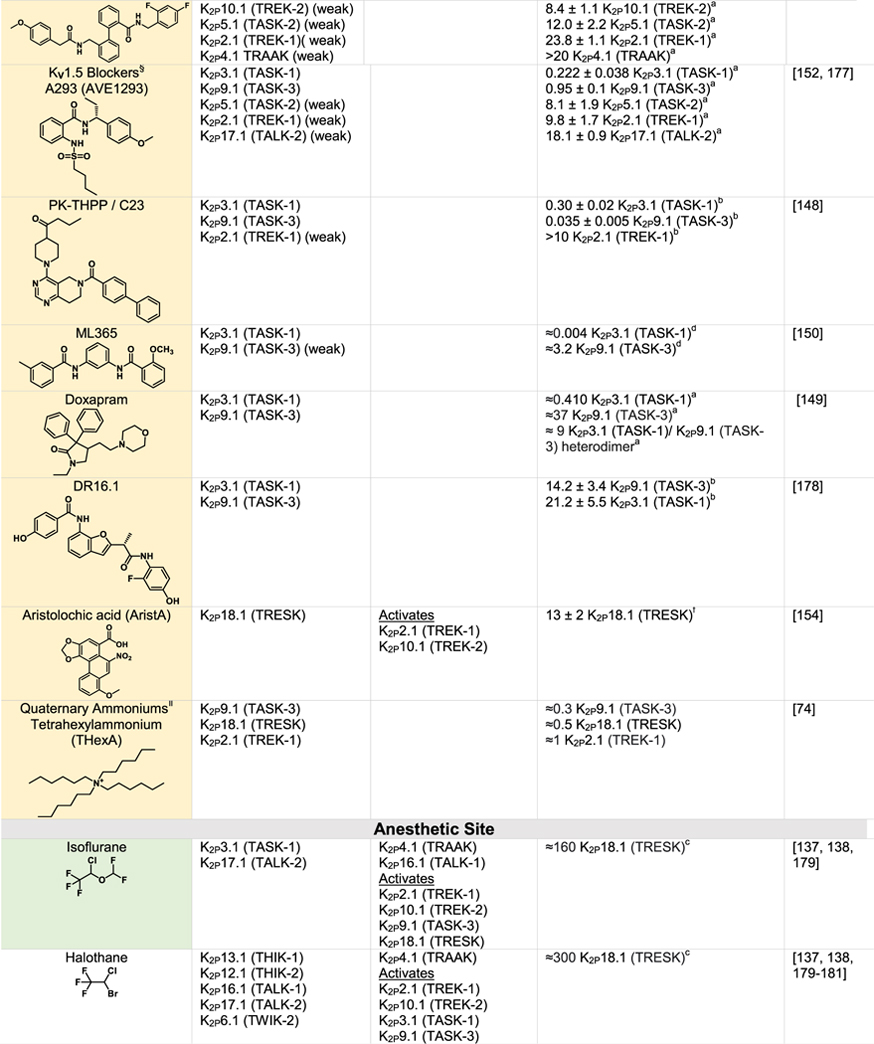
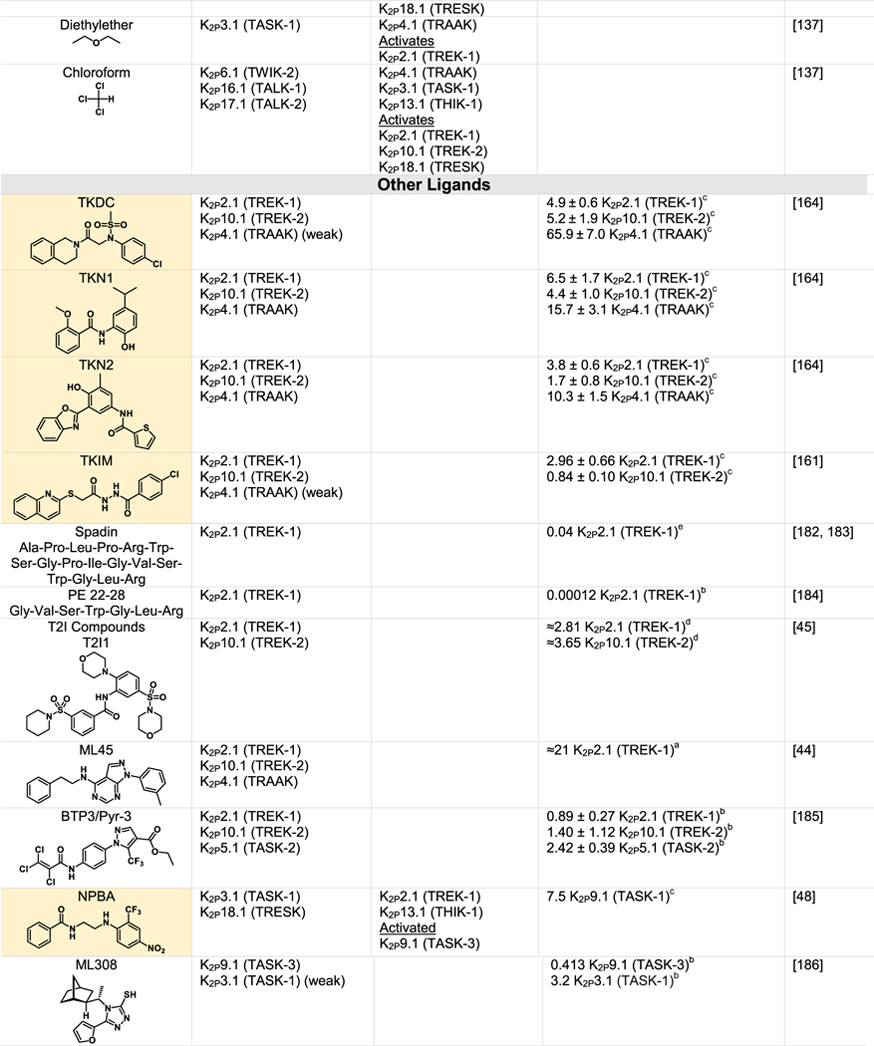
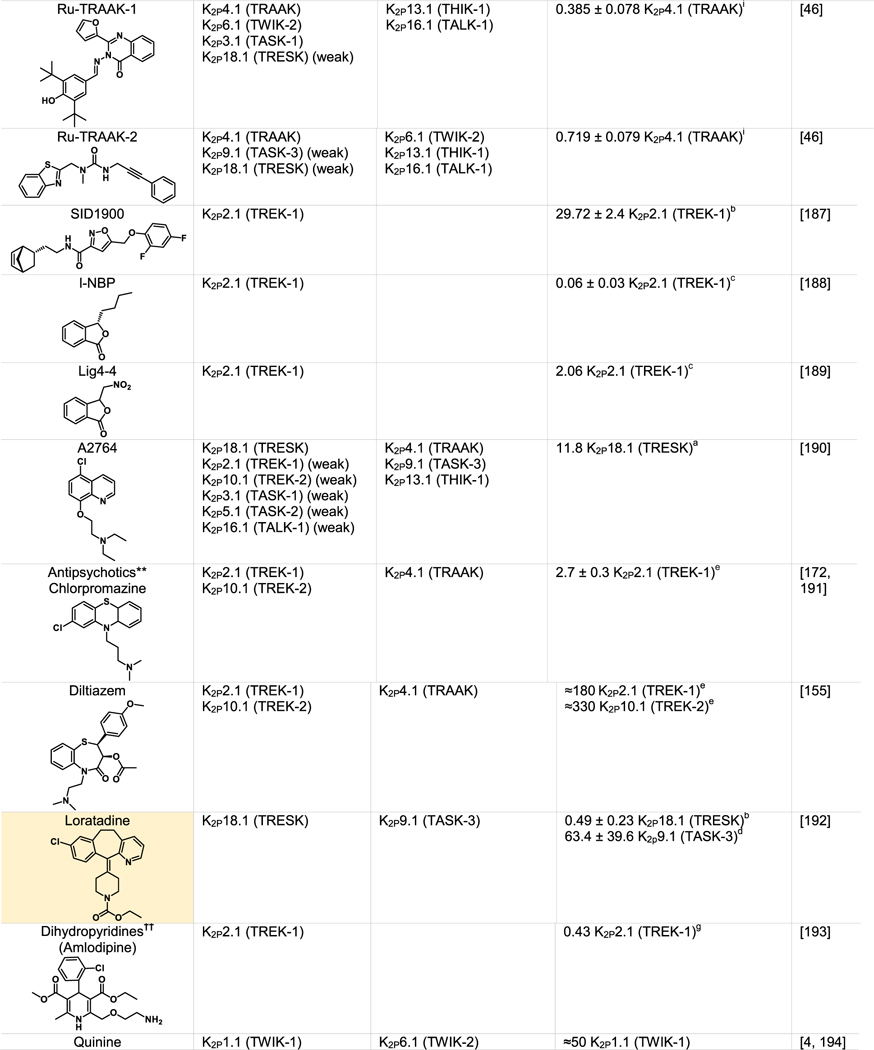
Although K2Ps have been largely viewed as pharmacological orphans, this perspective is changing rapidly due to both structural studies and the ongoing identification and characterization of new K2P modulators42,43,110 (Tables 1 and 2). Structural studies of K2Ps complexed with different types of modulators show a remarkably rich structural landscape for functional control, especially given the modest size of these channels (~70 kDa). Binding sites for modulators have been found at every layer of the protein, starting from its extracellular side through the portion that interacts with the membrane bilayer inner leaflet (Figure 6). This polysite pharmacology comprises five structurally defined binding sites for small molecules or lipids arranged from the outside to the inside: the Keystone inhibitor site,51 the K2P modulator pocket,37 the Vestibule site,38 the Fenestration site,40,50 and the Modulatory lipid site.37 Each offers a distinct structural environment and mechanism for controlling K2P function. As K2P modulators have been the subject of many recent reviews,42,43,110,111 we focus here on the current state of knowledge of the K2P structural pharmacology and try to provide context for some of the growing number of K2P modulators whose sites of action remain to be structurally defined.
Table 1.
|
|
|
|
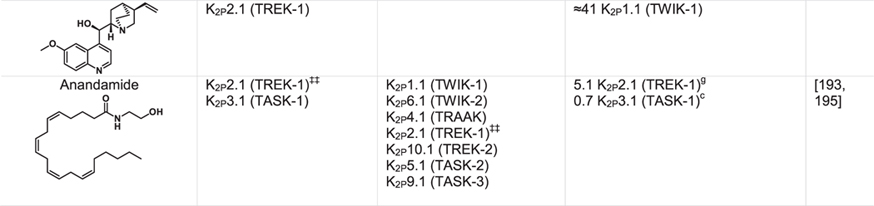
|
Green indicates structurally validated inhibitors. Yellow indicates inhibitors supported by mutagenesis or mutagenesis combined with simulation studies. Cells used to measure activity are indicated as follows:
Oocytes,
HEK cells,
CHO Cells,
Thallium flux assay,
COS-7 cells,
tsA201 cells, AZF cells,
hippocampal pyramidal neurons,
Lysosome flux assay.
Only K2Ps tested against the inhibitor are listed.
Other SSRIs such as citalopram and paroxetine inhibit K2Ps and likely bind to the fenestration site.
Other structurally similar local anesthetics such as lidocaine, ropivacaine, mepivacaine and etidocaine likely bind K2Ps similarly to bupivacaine.
Many KV1.5 blockers have been identified as being more potent antagonists for TASK channels. In addition to A1899 and A293, ICAGEN-4, MSD-D and A1899 derivatives have all been shown to potently inhibit TASK channels.
K2Ps are traditionally insensitive to quaternary ammoniums applied extracellularly, however are sensitive to intracellular application. Quaternary ammoniums with longer alkyl chains inhibit K2Ps more strongly than those with short alkyl chains.
Both typical antipsychotics and atypical antipsychotics (fluphenazine, haloperidol, loxapine, clozapine, etc.) have been shown to modulate K2P channels.
Other dihydropyridines such as niguldipine and nifedipine antagonize TREK channels.
Table 2.
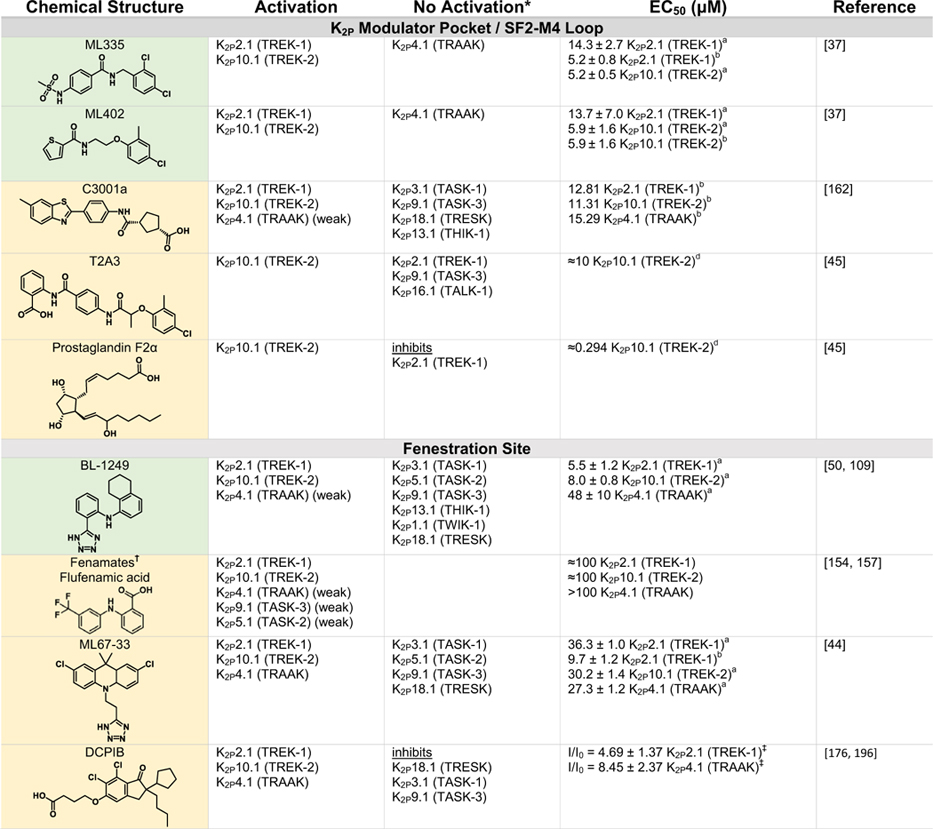
|
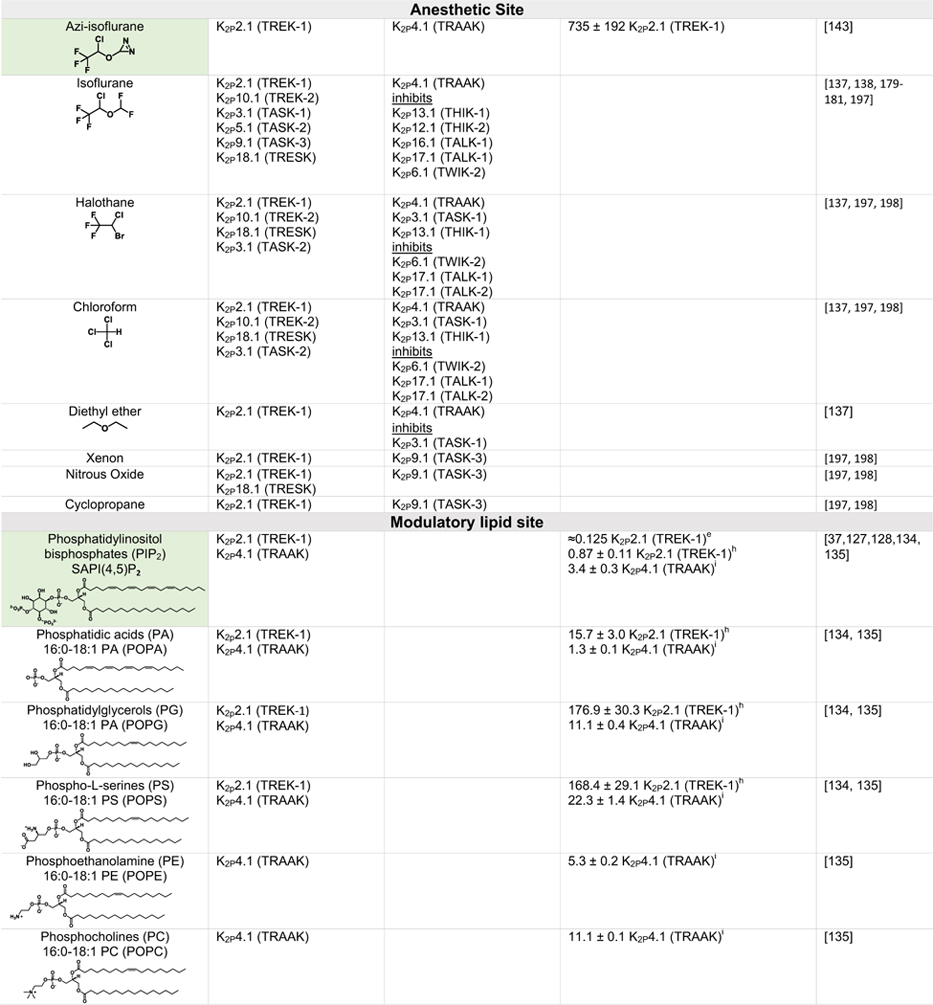
|
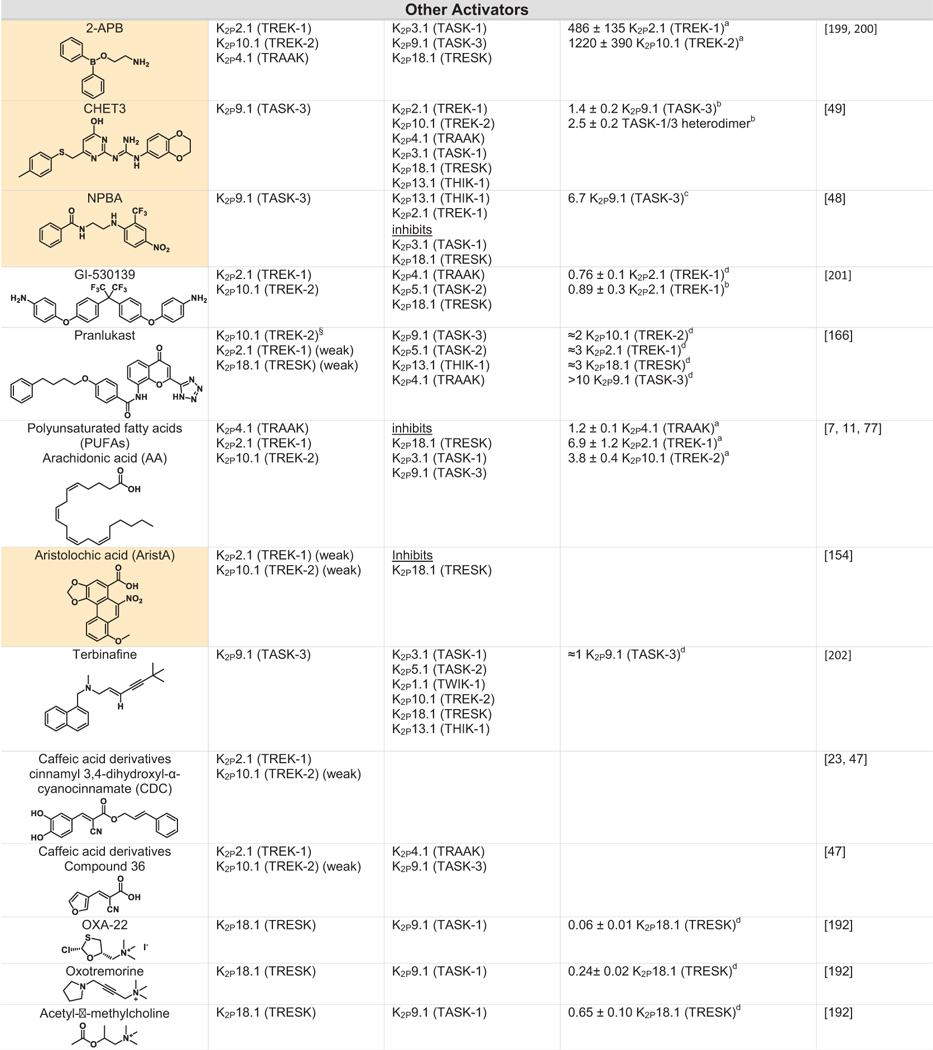
|
Green indicates structurally validated activators Yellow indicates activators supported by mutagenesis or mutagenesis combined with simulation studies. Cells used to measure activity are indicated as follows
Oocytes,
HEK cells,
CHO Cells,
Thallium flux assay,
COS cells,
tsA201 cells,
AZF cells,
fluorescent binding assay,
Kd values from native mass spectrometry with K2P4.1a (TRAAK) isoform.
Only K2Ps tested against the activator are listed (other K2Ps may not have been tested).
Other closely related fenamates like mefenamic acid and niflumic acid possess similar activity and selectivity to flufenamic acid.
I/I0 values at 10 lM DCPIB. EC50 values were not determined as Emax was not reached, even at very high DCPIB concentrations (100 lM).
While the EC50s for Pranlukast against the TREK family are nearly identical, the fold-activation observed for K2P10.1 (TREK-2) was much higher than for K2P2.1 (TREK-1) or K2P4.1 (TRAAK).
Figure 6.

Polysite model of K2P structural pharmacology. Central cartoon shows the locations of structurally defined K2P small molecule binding sites including the Keystone inhibitor site (magenta) (PDB: 6V3I),51 K2P modulator pocket (cyan) (PDB:6CQ8),37 Fenestration site (green) (PDB:4XDK),40 Modulatory lipid site (grey) (PDB:6W8C),53 and Vestibule site (orange) (PDB: 6RV3).38 Cap, Extracellular ion pathway (EIP), and ‘C-type’ SF gate are indicated. Potassium ions are shown (purple). Grey lines denote the lipid bilayer. Black boxes show the details of the individual sites. Grey boxes show modulator chemical structures. X-gate is not shown in the central cartoon.
The Keystone inhibitor site – ‘Finger in the dam’ polynuclear ruthenium amines
The trinuclear oxo-bridged ruthenium amine polycation ruthenium red (RuR) (Figure 6, Table 1)112 has many biological applications,113 including a ~50 year legacy as an inhibitor of diverse ion channels. RuR inhibits two TREK subfamily K2Ps, K2P4.1 (TRAAK)68,114 and K2P10.1 (TREK2),114 as well as K2P9.1 (TASK-3).115–117 Functional studies established that a negatively charged residue at the K2P Cap domain base comprises a key RuR sensitivity determinant in natively RuR sensitive channels.115–117 Further, mutational studies showed that placing a negatively charged residue at the Cap base is sufficient for rendering a non-RuR sensitive K2P responsive to RuR inhibition.51,114
Structural studies of an RuR-sensitive K2P2.1 (TREK-1) mutant, I110D,51 have recently revealed the mechanism by which this inhibition occurs (Figure 6). RuR inhibits K2Ps using a 1:1 stoichiometry matching prior functional studies68,114 that places one ruthenium amine moiety directly over the channel pore at the site of the S0 ion, while the remainder of the RuR molecule occupies one of the two branches of the EIP (Figure 6). The negatively charged residues that form the RuR-sensitivity determinant at the Cap domain base constitute the ‘Keystone inhibitor site’ and directly coordinate RuR through a multipronged interaction to several RuR elements (Figure 6). Further structural and functional investigation demonstrated that the dinuclear ruthenium amine, Ru360 (Table 1),118 an inhibitor of the mitochondrial calcium uniporter119–121 not previously known to affect potassium channels, also binds to the Keystone inhibitor site in a similar way to RuR, although with lower affinity, likely due to its decreased positive charge.51 Replacing the SF1 Asn147 at the SF external mouth with an acidic residue in conjunction with the I110D mutation yielded designer RuR super-responders having IC50s in the low nanomolar range.51 Because of the shared K2P pore architecture, combination of this protein engineering strategy with efforts to make novel polyruthenium amine derivatives122 could provide new means for exploring the function of any K2P subtype. Together, these studies define a ‘finger in the dam’ mechanism in which polynuclear ruthenium amines binding to the Keystone inhibitor site creates an electrostatic and physical barrier that prevents the flow of potassium ions through the selectivity filter. The ability of these relatively simple compounds to reach through the EIP to stop function raises the possibility that other classes of molecules could work using the same mechanism. In particular, biologics, such as nanobodies or peptide toxins, may be particularly suited to exploiting this unique feature of the EIP.
The K2P modulator pocket – A cryptic small molecule binding site for K2P control
Structures of K2P2.1 (TREK-1) complexed with two novel activators, ML335 (N-[(2,4-dichlorophe nyl)methyl]-4-(methanesulfonamido) benzamide) and ML402 (N-[2-(4-chloro-2-methylphenoxy)ethyl] thiophene-2-carboxamide), revealed the presence of an L-shaped pocket behind the selectivity filter in the P1-M4 interface, the K2P modulator pocket (Figure 6, Table 2). This small molecule binding site resides in an intersubunit interface involved in C-type gating72,73 and is unrelated to any previously known small molecule binding pocket in the VGIC superfamily.37 Both compounds bind in similar ways, act as molecular wedges that stabilize the P1-M4 interface, and directly activate the channel selectivity filter C-type gate.37,53 In the unliganded structure, P1-M4 interface interactions occlude the K2P modulator pocket. Small movements of a few residues are required for the compounds to bind, making this a cryptic site that relies on conformational change, similar to cryptic sites described for soluble proteins.123 The intersubunit interface that these two compounds stabilize is also the site of GOF mutations57,72,73 and highlights the general importance of intersubunit interfaces for channel control.
ML335 and ML402 show a strong subtype selectivity, activating K2P2.1 (TREK-1) and K2P10.1 (TREK-2) but not K2P4.1 (TRAAK).37 Selectivity originates from a single lysine residue on the N-terminal end of M4 that makes a cation-p interaction with the upper ring of the activators (Figure 6). The equivalent K2P4.1 (TRAAK) residue is a glutamine and exchanging K → Q in K2P2.1 (TREK1) and Q → K in K2P4.1 (TRAAK) at this position is sufficient for rendering the former insensitive to ML335 and ML402 activation and the later sensitive to activation by both compounds.37 The importance of a single amino acid difference in an otherwise conserved small molecule binding pocket underscores the potential for exploiting local differences and structural knowledge to develop subtype-selective K2P modulators.
There are no known natural ligands for the K2P modulator pocket, but given that P1-M4 interface is central for integrating gating cues that arise in other parts of the protein, particularly the C-terminal tail,37,53,72,73 it seems likely that endogenous compounds such as lipids, metabolites, signaling molecules, or regulatory proteins may target this site. Defining its roles in other K2P subfamilies and identifying natural effectors for this site remain important directions for future study.
The Fenestration site – A dual-action binding site
The M4 transmembrane helix is a key moving part of K2Ps and in the TREK subfamily this element conveys gating cues from temperature,57,72,73,81 pressure,73,81,82 and phosphorylation72 to the C-type gate. The M4 ‘down’ state creates an opening just below the P2 pore helix that faces the center of the membrane bilayer,36,39,57,58 the ‘Fenestration site’. Structural studies of K2P10.1 (TREK-2) have demonstrated that the K2P inhibitors fluoxetine and norfluoxetine bind here40 (Figure 6, Table 1). Remarkably, crystal structures of a K2P10.1 (TREK2) complex with a brominated version of an activator, BL-1249, although not defining density for the entire compound, strongly indicate that this molecule, and perhaps other activators, also bind to the Fenestration site created by the ‘down’ M4 position.50 Hence, the Fenestration site has the curious property of being a dual-action site where ligand binding can either stimulate or suppress channel activity.
How can the binding of a small molecule to the same site yield opposite functional outcomes of inhibition and activation? Clearly, the answer cannot be in stabilization of the M4 ‘down’ state over the ‘up’ state, as the binding of both inhibitors and activators to the Fenestration site requires an M4 ‘down’ conformation.40,50 Given the location of the Fenestration site just below the selectivity filter, one plausible mode of action would be for Fenestration site binders to affect the C-type gate conformational dynamics.89 Additionally, Fenestration site activators, such as BL-1249, may stabilize the filter by using a negatively charged moiety to recruit potassium ions to the aqueous cavity and thereby alter ion site occupancies in the selectivity filter.50 Notably, the Fenestration site is found throughout the VGIC superfamily and serves as the site of action for multiple types of activators of different classes of potassium channels.50 Understanding the relationship between Fenestration site occupation and its effects on the selectivity filter C-type gate, how permeant ion recruitment activates the SF gate, and the properties of activators and inhibitors that can inhabit this site is an important challenge for the future development of K2P modulators. Further, unlike the K2P modulator pocket which is composed of elements of the same subunit chain, the Fenestration site comprises elements from both chains of the K2P dimer (Figures 4(c) and 6). Hence, it has potential as a site for the discovery or design of compounds that exploit these differences to achieve selective modulation of homodimeric versus heterodimeric K2Ps.
The Vestibule site – X-gate traps
Potassium channels can be blocked by compounds such as quaternary amines that enter the central cavity and block ion flow through the filter.124–126 K2Ps share this property (Table 1) and it has been exploited to probe various features of the K2P inner pore.50,74 Recently, structures of K2P3.1 (TASK-1) complexed with two different inhibitors, BAY10000493 ((4-{[2-(4-bromophenyl) imidazo[1,2-a]pyridin-3-yl]methyl}piperazin-1-yl)(2-fluorophenyl)methanone) and BAY2341237 (4-{[2-(4-chlorophenyl)imidazo[1,2-a]pyridin-3-yl]methyl} piperazin-1-yl)[6-(trifluoromethoxy)pyridin2-yl]methanone) have revealed a novel mode in which these compounds are trapped in the intracellular vestibule by the closure of the cytoplasmic X-gate.38 Notably, neither compound interacts directly with the X-gate, but rather makes intimate contacts to the upper part of the vestibule (Figure 6). Nevertheless, the stability of the X-gate can affect how readily these compounds can be washed out of the channel.38 Both compounds also make many contacts to residues from each subunit, raising the possibility that it may be possible to identify derivatives that can distinguish homomeric from heteromeric channels. Understanding the interplay between compounds that bind to the vestibule site and the motions of the X-gate is an interesting new direction that will likely aid in the development of other compounds that can affect K2Ps at the vestibule site.
The Modulatory lipid site – PIP2 and the C-terminal tail
The signaling lipid PIP2 is an important modulatory lipid for TREK subfamily channels.127–129 The likely PIP2 site of action has been located in a series of K2P2.1 (TREK-1) structures.37,53 These show the presence of a phospholipid that copurified with the channel and that was bound to a groove formed by the M1/M2/M4 interface (Figure 6). The phosphoinositol headgroup contacts an electropositive patch on the C-terminal tail comprising five residues implicated in PIP2 modulation (Arg297, Lys301, Lys302, Lys304, and R311)127,128 (Figure 6). This region also contains two other regulatory sites, the Glu306 intracellular proton sensor130 and Ser300 inhibitory phosphorylation site.131 Notably, the PIP2-interacting residues are in a part of the M4 helix that is most affected by movements of between the ‘up’ and ‘down’ positions. Hence, it seems likely regulation of these channels by the modulatory lipid, intracellular pH sensor, and phosphorylation site are all tightly intertwined with M4 motions.132 Further study is needed to unravel questions about these interactions such as probing whether other activatory lipids such as phosphatidyl serine and phosphatidic acid127,133 compete with PIP2 at this site, whether this site can be targeted by small molecules, and to define how changes in this lower part of the channel impact the dynamics and function of the C-type gate. Further, the effects of PIP2 on K2P2.1 (TREK-1) are complex, involving activation and inhibition.128,134 Determining if the Modulatory lipid site has a role in both actions, whether there are additional lipid binding sites, and whether other activating lipids that bind TREK subfamily members134,135 (Table 2) affect this site or other parts of the channel is an important direction for future studies.
Members of the TASK and TALK subfamilies are also known to bind and be modulated by specific lipids129,136 and recent structures have begun to hint at the molecular details of these interactions. Structures of K2P3.1 (TASK-1) revealed a potential binding site for cholesterol in a site contacting M1, M2, M4, and key elements of the X-gate that is roughly spatially coincident with K2P2.1 (TREK-1) PIP2 site.38 Mutations at this site and depletion of cholesterol from bilayers in functional experiments modify K2P3.1 (TASK-1) activity, indicating a role for cholesterol or its analogues in stabilizing X-gate conformations. K2P5.1 (TASK-2) is known to be activated by a variety of phosphatidylinositol lipids that interact with its C-terminal tail.136 The K2P5.1 (TASK-2) structures show that a polybasic region suspected to be involved in these interactions exists at least in part as a membrane parallel helical extension of M4, similar to the electropositive PIP2 binding patch on M4 in K2P2.1 (TREK-1).41 While most of the molecular details of how lipids regulate K2Ps remain to be elucidated, it is intriguing that many key determinants for lipid modulation are at the distal end of the movable M4 helix, suggesting that they exploit its motion to convey gating commands to the C-type gate.72 Given that most of the K2P mass is solvated by the lipid bilayer, emerging approaches such as direct measure of K2P:lipid interactions by mass spectrometry135 and the ability to image K2Ps in lipid environments41 will have a key role in uncovering the variations on the use of the Modulatory lipid site and existence of other specific lipid binding sites housed with the K2P architecture.
General anesthetics – A site for gases in the pore
Various type of general anesthetics comprise one of the best studied K2P modulator classes.43 Volatile anesthetics such as halothane and isoflurane activate K2P2.1 (TREK-1), K2P9.1 (TREK-2), K2P3.1 (TASK-1), K2P9.1 (TASK-3) and K2P18.1 (TRESK) (Table 2).137,138 Although no structural information is yet available about where such molecules bind, a number of studies indicate that they have a specific binding site within the K2P inner cavity139–143. In particular, the recent use of an unbiased photoaffinity labeling approach utilizing the isoflurane derivative azi-isoflurane combined with mass spectrometry has identified a key glycine in M2 of K2P2.1 (TREK-1) that sits at the junction where M2, M3, and M4 converge143 (Figure 4(c), Table 2). This site is a hotspot for residues involved in responses to stretch, thermal responses, and the activator BL-1249.40,57,58,109 These findings set the stage for further studies of how anesthetics influence K2P function that will be important for resolving the ongoing debates about the relative contributions of direct binding modes versus proposed mechanisms of indirect modulation,144 defining whether other activators of the TREK and TRESK families, such as chloroform, nitrous oxide, and xenon (Table 2) target the same region, and defining why some anesthetics such as halothane activate K2P2.1 (TREK-1)137 but inhibit K2P13.1 (THIK-1) and K2P12.1 (THIK-2).145
K2P modulators - structural matchmaking
The desire to turn K2Ps into a channel class with good pharmacology has produced a growing list of activators and inhibitors (Tables 1 and 2).42,43,110,111 The mechanisms of action and binding sites for these modulators remain to be defined by direct methods such as X-ray crystallography, cryo-electron microscopy, or chemical crosslinking. Here, we recount some of these exciting molecules, particularly those where docking or mutagenesis studies have provided suggestions for potential binding sites and place them in the context of the defined structural pharmacology sites mentioned above.
Vestibule site modulators – An investigative TASK to be undertaken
Many potassium channels are blocked by compounds that act within the central cavity124–126,146 and functional studies have long indicated that K2Ps share this proclivity for block from the intracellular side74,142,147 (Table 1). Structural definition of the Vestibule site delimited by the X-gate in K2P3.1 (TASK-1)38 together with prior studies identifying the TASK subfamily central cavity as a hotspot for the action of a varied antagonists111 highlight the potential for modulators to act in the K2P inner cavity. Mutational and computational studies with antagonists A1899,147 A293,147 PK-THPP,148 doxapram,142,149 ML365150 and DR16.1151 indicate they share key interactions with hydrophobic residues directly below the selectivity filter and inhibit channel activity by directly blocking the channel pore (Table 1).142,147,152,153 While small differences between K2P3.1 (TASK-1) and K2P9.1 (TASK-3) lead to differences in the potency and selectivity of these blockers, the chemical structures of the inhibitors share a similar topology, incorporating aromatic rings, hydrogen bond acceptors and hydrophobic groups.111 Aristolochic acid (AristA), a compound identified in traditional medicinal plants, was found to activate K2P2.1 (TREK-1) and K2P10.1 (TREK-2) while having no effect on K2P4.1 (TRAAK) and causing inhibition of K2P18.1 (TRESK) (Table 2).154 Mutations at residues F145 and F352 in the presumptive K2P18.1 (TRESK) inner pore ablated the blocking effect of AristA,154 a result that suggests that AristA binds in the inner cavity. It would seem that exploiting differences between K2Ps that have an inner gate and those that do not might be a productive direction for developing subfamily specific compounds that act at the Vestibule site. Further, whether there are variations on the theme of the Vestibule site and whether multiple, distinct binding pockets exist within the K2P inner cavity remains to be discovered.
Fenestration site – Or very close
Nonsteroidal anti-inflammatory fenamates such as flufenamic acid (FFA) activate members of the TREK subfamily155–157 and K2P18.1 (TRESK).158 Based on the behavior of the related compound BL-124950,109 the Fenestration site seems like a strong candidate for their site of action (Table 2). It is thought that the negatively charged tetrazole moiety of BL-1249 creates a binding site for potassium ions in the central cavity and thereby stabilizes the selectivity filter C-type gate.50 Similar structural motifs can be found in FFA as well as other suspected Fenestration site activators ML67–3344,50 and DCPIB50 (Table 2), raising the possibility that these structurally diverse compounds activate K2Ps using this shared mechanism. Based on a combination of mutagenesis and molecular dynamics simulations, the local anesthetic bupivacaine has also been proposed to bind at the Fenestration site159 (Table 1). Other local anesthetics, such as lidocaine and ropivacaine, by virtue of their structural similarity to bupivacaine, seem likely to bind at the Fenestration site to inhibit these channels. The effect of local anesthetics may be more complicated, as in addition to direct action, they have been proposed to indirectly antagonize K2P2.1 (TREK-1) through inhibition of phospholipase D2 (PLD2).160 A highly selective K2P9.1 (TASK-3) agonist, CHET3 (Table 2), has also been predicted to bind underneath the selectivity filter between the M2 and M4 helices in an analogous position to BL-1249.49 The K2P2.1 (TREK-1) inhibitor, TKIM (Table 1) may also bind in the inner cavity.161 Defining the extent to which these and other modulators bind at the Fenestration site, the Vestibule site, or some variation of both, and whether conformational changes in the position of M4 are linked to action at such sites should provide new perspectives on the richness and druggabilty of what is clearly a hotspot of K2P modulation by small molecules.
K2P modulator pocket – Will it open for other modulators?.
No natural or synthetic activators of the K2P modulator pocket besides ML335 and ML402 are yet known. However, two small molecule screens have identified TREK subfamily activators that may act on the K2P modulator pocket or its surrounding elements. Mutagenesis and computational modelling suggest that C3001a acts at the K2P modulator pocket, although perhaps in a different manner than ML335 and ML402 (Table 2).162 T2A3 and the bioactive lipid 11-deoxy prostaglandin F2a were identified as highly specific K2P10.1 (TREK-2) activators through a high-throughput thallium flux screen (Table 2).45 Functional studies of K2P2.1 (TREK-1)/K2P10.1 (TREK-2) chimeras identified a short amino acid sequence in the SF2-M4 loop that governs the K2P10.1 (TREK-2) selectivity of both activators.45 The SF2-M4 loop is a key element of the K2P modulator pocket, is one of the few divergent portions of these two TREK subfamily channels (Figure 2(A)), and has an important role in C-type gating.53 Interestingly, the chemical structure of T2A3 is strikingly similar to ML335 and ML402. The main difference is the presence of an additional benzoate moiety, which is attached to part of the shared scaffold known as the ‘upper ring’.37 Hence, it seems likely that T2A3 would bind K2P10.1 (TREK-2) in a pose similar to ML335 and ML402 in which the T2A3 benzoate would protrude from the K2P modulator pocket and enable T2A3 to interact with the SF2-M4 loop in a way that capitalizes on the net charge difference between the K2P10.1 (TREK-2) and K2P2.1 (TREK-1) SF2-M4 loops (+1 versus −2 net charge at pH 7.0, respectively).163 These structural features could explain the selective activation of K2P10.1 (TREK-2) by T2A3 and its ability to inhibit K2P2.1 (TREK-1) at high concentrations. Further investigation into the pharmacological basis of how K2P modulator pocket binders can exploit differences among the SF2-M4 loops, which is one of the most diverse elements of this binding site (Figure 2(a)), could offer routes to more potent and selective K2P modulators.
How many more modulatory sites are packed into the K2P structure?.
The five structurally defined modulator sites within the compact K2P 70 kDa frame provide a surprisingly rich framework for modulators to influence K2P function. Intriguingly, there are suggestions that the count of sites where external agents can influence K2P function will not stop at five. For example, the small molecule TKDC, an inhibitor of the TREK subfamily (Table 1), has been proposed to act on a site on the external part of the Cap.164 2-APB, an activator of K2P2.1 (TREK-1) and K2P10.1 (TREK-2) has been proposed to have a binding site within thechannel C-terminal domain based on truncation studies165 (Table 2). Pranlukast, a leukotriene receptor antagonist, is a relatively selective K2P10.1 (TREK-2) activator (Table 2)166 that based on mutational studies does not act at either the K2P modulator pocket or the BL-1249 site, leaving open the possibility that other sites that mediate activation are present in the K2P framework. While the proposed modes of action of these compounds as well the many other inhibitors and activators (Tables 1 and 2) require structural validation, there is good reason to believe that the number of sites comprising of the polysite pharmacology of K2Ps will increase.
Perspectives.
The diversity of moving parts that affect K2P function and the many modulator binding sites housed within the compact K2P architecture underscores the structural complexity that undergirds channels known by the deceptively simple moniker of ‘leak potassium channels’. Understanding how the various K2P moving parts work together to control function and respond to natural gating cues remains a crucial challenge. Without question, structure determination efforts will continue to deepen our understanding of how gating cues impact the SF gate and inform the extent to which other K2Ps can be classified into those having an inner gate and those without such a feature. Promising lines of inquiry include obtaining structures of the remaining uncharacterized K2Ps homodimers, structures of K2Ps in lipid environments, additional structures of K2P:modulator complexes, especially for those whose binding sites are not yet clear (Tables 1 and 2), and structures of K2P heterodimers. Given the diverse structural sites where modulator binding can influence function, sorting out the synergies among the defined modulator binding sites and whether multiple modulators can work in concert to influence function is also a key next step. The question of the role of domain-swapping in function remains unsolved. Given how tightly intertwined the subunits in the domain-swapped configuration are, understanding how this feat of architectural gymnastics comes about during channel biogenesis and assembly and the extent to which such intertwining can be undone during the life cycle of a K2P channel remains an intriguing an unaddressed direction.
The importance of K2P heterodimer formation is becoming ever more appreciated,71 and a key open question is how heterodimer formation affects the structure and dynamics of K2P relative to the homomeric isoforms. As the Keystone inhibitor site, the Fenestration site, and the Vestibule site have contributions from each subunit, variations in the residues that comprise these sites has the potential to provide another layer of diversification in function and perhaps pharmacology between homodimer and heterodimer K2Ps. Further, in cells K2Ps are not lone actors but are part of larger assemblies of signaling and scaffolding proteins.167 The involvement of K2Ps in multiprotein assemblies is still understudied and steps to understand how the formation of K2Pcentered protein complexes affect function, structure, and pharmacology is a key direction for addressing the diverse roles of this channel family in physiologically relevant settings. Together, these recent advances in K2P structure and structural pharmacology, the ever growing appreciation of the importance of K2Ps in physiological and pathophysiological function,9,43,168–170 and the key K2P frontier questions outlined here show that these ‘leak channels’ are far more fascinating and important than their much maligned name might suggest.
Acknowledgements
We thank K. Brejc and M. Grabe for comments on the manuscript. This work was supported by NIH R01-MH093603 to D.L.M.
Footnotes
CRediT authorship contribution statement
Andrew M. Natale: Conceptualization, Visualization, Writing - original draft, Writing - review & editing. Parker E. Deal: Conceptualization, Visualization, Writing - original draft, Writing - review & editing. Daniel L. Minor: Conceptualization, Visualization, Writing - original draft, Writing - review & editing, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Hodgkin AL, Huxley AF, (1947). Potassium leakage from an active nerve fibre. J. Physiol, 106, 341–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodgkin AL, Huxley AF, (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol, 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodgkin AL, Katz B, (1949). The effect of sodium ions on the electrical activity of giant axon of the squid. J. Physiol, 108, 37–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, et al. , (1996). TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J., 15, 1004–1011. [PMC free article] [PubMed] [Google Scholar]
- 5.Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, et al. , (1996). Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J., 15, 6854–6862. [PMC free article] [PubMed] [Google Scholar]
- 6.Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M, (1997). TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J., 16, 5464–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, et al. , (1998). A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J., 17, 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enyedi P, Czirjak G, (2010). Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev, 90, 559–605. [DOI] [PubMed] [Google Scholar]
- 9.Feliciangeli S, Chatelain FC, Bichet D, Lesage F, (2015). The family of K2P channels: salient structural and functional properties. J. Physiol, 593, 2587–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renigunta V, Schlichthorl G, Daut J, (2015). Much more than a leak: structure and function of K(2)pchannels. Pflugers Arch., 467, 867–894. [DOI] [PubMed] [Google Scholar]
- 11.Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, et al. , (1998). A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J., 17, 4283–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douguet D, Honore E, (2019). Mammalian Mechanoelectrical Transduction: Structure and Function of Force-Gated Ion Channels. Cell, 179, 340–354. [DOI] [PubMed] [Google Scholar]
- 13.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA, (2005). Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev, 57, 387–395. [DOI] [PubMed] [Google Scholar]
- 14.Mathie A, Al-Moubarak E, Veale EL, (2010). Gating of two pore domain potassium channels. J. Physiol, 588, 3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanda H, Ling J, Tonomura S, Noguchi K, Matalon S, Gu JG, (2019). TREK-1 and TRAAK Are Principal K (+) Channels at the Nodes of Ranvier for Rapid Action Potential Conduction on Mammalian Myelinated Afferent Nerves. Neuron, 104, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brohawn SG, Wang W, Handler A, Campbell EB, Schwarz JR, MacKinnon R, (2019). The mechanosensitive ion channel TRAAK is localized to the mammalian node of Ranvier. eLife, 8, e50403. 10.7554/eLife.50403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, et al. , (2004). TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J., 23, 2684–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazarenko RM, Fortuna MG, Shi Y, Mulkey DK, Takakura AC, Moreira TS, et al. , (2010). Anesthetic activation of central respiratory chemoreceptor neurons involves inhibition of a THIK-1-like background K(+) current. J. Neurosci, 30, 9324–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madry C, Kyrargyri V, Arancibia-Carcamo IL, Jolivet R, Kohsaka S, Bryan RM, et al. , (2018). Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK1. Neuron, 97299–312 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida K, Shi S, Ukai-Tadenuma M, Fujishima H, Ohno RI, Ueda HR, (2018). Leak potassium channels regulate sleep duration. Proc. Natl. Acad. Sci. USA, 115, E9459–E9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, et al. , (2006). TREK-1, a K+ channel involved in polymodal pain perception. EMBO J., 25, 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devilliers M, Busserolles J, Lolignier S, Deval E, Pereira V, Alloui A, et al. , (2013). Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nature Commun., 4, 2941. [DOI] [PubMed] [Google Scholar]
- 23.Vivier D, Soussia IB, Rodrigues N, Lolignier S, Devilliers M, Chatelain FC, et al. , (2017). Development of the first Two-Pore Domain Potassium Channel TREK-1 (TWIK-Related K+ Channel 1)-selective agonist possessing in vivo anti-nociceptive activity. J. Med. Chem, 60, 1076–1088. [DOI] [PubMed] [Google Scholar]
- 24.Decher N, Ortiz-Bonnin B, Friedrich C, Schewe M, Kiper AK, Rinne S, et al. , (2017). Sodium permeable and “hypersensitive” TREK-1 channels cause ventricular tachycardia. EMBO Mol. Med, 9, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laigle C, Confort-Gouny S, Le Fur Y, Cozzone PJ, Viola A, (2012). Deletion of TRAAK potassium channel affects brain metabolism and protects against ischemia. PLoS ONE, 7, e53266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X, Liu Y, Chen X, Sun Q, Tang R, Wang W, et al. , (2013). Involvement of TREK-1 activity in astrocyte function and neuroprotection under simulated ischemia conditions. J Mol Neuroscience: MN, 49, 499–506. [DOI] [PubMed] [Google Scholar]
- 27.Abraham DM, Lee TE, Watson LJ, Mao L, Chandok G, Wang HG, et al. , (2018). The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Invest, 128, 4843–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thummler S, Peng XD, et al. , (2006). Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nature Neurosci., 9, 1134–1141. [DOI] [PubMed] [Google Scholar]
- 29.Royal P, Andres-Bilbe A, Avalos Prado P, Verkest C, Wdziekonski B, Schaub S, et al. , (2019). Migraine-Associated TRESK Mutations Increase Neuronal Excitability through Alternative Translation Initiation and Inhibition of TREK. Neuron, 101232–45 e6. [DOI] [PubMed] [Google Scholar]
- 30.Yarishkin O, Phuong TTT, Bretz CA, Olsen KW, Baumann JM, Lakk M, et al. , (2018). TREK-1 channels regulate pressure sensitivity and calcium signaling in trabecular meshwork cells. J. Gen. Physiol, 150, 1660–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambert M, Boet A, Rucker-Martin C, Mendes-Ferreira P, Capuano V, Hatem S, et al. , (2018). Loss of KCNK3 is a hallmark of RV hypertrophy/dysfunction associated with pulmonary hypertension. Cardiovasc. Res, 114, 880–893. [DOI] [PubMed] [Google Scholar]
- 32.Zyrianova T, Lopez B, Olcese R, Belperio J, Waters CM, Wong L, et al. , (2020). K2P2.1 (TREK-1) potassium channel activation protects against hyperoxia-induced lung injury. Sci. Rep, 10, 22011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayliss DA, Barrett PQ, (2008). Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol. Sci, 29, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honore E, (2007). The neuronal background K2P channels: focus on TREK1. Nature Rev. Neurosci, 8, 251–261. [DOI] [PubMed] [Google Scholar]
- 35.Lesage F, Barhanin J, (2011). Molecular physiology of pH-sensitive background K(2P) channels. Physiology, 26, 424–437. [DOI] [PubMed] [Google Scholar]
- 36.Miller AN, Long SB, (2012). Crystal structure of the human two-pore domain potassium channel K2P1. Science, 335, 432–436. [DOI] [PubMed] [Google Scholar]
- 37.Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, et al. , (2017). K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature, 547, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rödström KEJ, Kiper AK, Zhang W, Rinne S, Pike ACW, Goldstein M, et al. , (2020). A lower X-gate in TASK channels traps inhibitors within the vestibule. Nature, 582, 443–447. [DOI] [PubMed] [Google Scholar]
- 39.Brohawn SG, del Marmol J, MacKinnon R, (2012). Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science, 335, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong YY, Pike AC, Mackenzie A, McClenaghan C, Aryal P, Dong L, et al. , (2015). K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science, 347, 1256–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li BB, Rietmeijer RA, Brohawn SG, (2020). Structural basis for pH gating of the two-pore domain K (+) channel TASK2. Nature, 586, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sterbuleac D, (2019). Molecular determinants of chemical modulation of two-pore domain potassium channels. Chem. Biol. Drug Des, 94, 1596–1614. [DOI] [PubMed] [Google Scholar]
- 43.Mathie A, Veale EL, Cunningham KP, Holden RG, Wright PD, (2021). Two-pore Domain potassium channels as drug targets: anesthesia and beyond. Annu. Rev. Pharmacol. Toxicol, 61, 401–420. [DOI] [PubMed] [Google Scholar]
- 44.Bagriantsev SN, Ang KH, Gallardo-Godoy A, Clark KA, Arkin MR, Renslo AR, et al. , (2013). A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem. Biol, 8, 1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dadi PK, Vierra NC, Days EL, Dickerson M, Vinson PN, Weaver CD, et al. , (2017). Selective small molecule activators of TREK-2 channels stimulate DRG c-fiber nociceptor K2P currents and limit calcium influx. ACS Chem. Neurosci, 8, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su ZW, Brown EC, Wang WW, MacKinnon R, (2016). Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. P Natl Acad Sci USA, 113, 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodrigues N, Bennis K, Vivier D, Pereira V, Chapuy E, et al. , (2014). Synthesis and structure-activity relationship study of substituted caffeate esters as antinociceptive agents modulating the TREK-1 channel. Eur. J. Med. Chem, 75, 391–402. [DOI] [PubMed] [Google Scholar]
- 48.Tian F, Qiu Y, Lan X, Li M, Yang H, Gao Z, (2019). A small-molecule compound selectively activates K2P channel TASK-3 by acting at two distant clusters of residues. Mol. Pharmacol, 96, 26–35. [DOI] [PubMed] [Google Scholar]
- 49.Liao P, Qiu Y, Mo Y, Fu J, Song Z, Huang L, et al. ,(2019). Selective activation of TWIK-related acid-sensitive K(+) 3 subunit-containing channels is analgesic in rodent models. Sci. Transl. Med, 11, eaaw8434. 10.1126/scitranslmed.aaw8434. [DOI] [PubMed]
- 50.Schewe M, Sun H, Mert U, Mackenzie A, Pike ACW, Schulz F, et al. , (2019). A pharmacological master key mechanism that unlocks the selectivity filter gate in K (+) channels. Science, 363, 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pope L, Lolicato M, Minor DL, (2020). Polynuclear ruthenium amines inhibit K2P channels via a “Finger in the Dam” mechanism. Cell Chem. Biol, 27, 511–524.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heginbotham L, Lu Z, Abramson T, MacKinnon R, (1994). Mutations in the K+ channel signature sequence. Biophys. J, 66, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lolicato M, Natale AM, Abderemane-Ali F, Crottes D, Capponi S, Duman R, et al. , (2020). K2P channel C-type gating involves asymmetric selectivity filter orderdisorder transitions. Sci. Adv, 6, eabc9174. [DOI] [PMC free article] [PubMed]
- 54.Goldstein M, Rinne S, Kiper AK, Ramirez D, Netter MF, Bustos D, et al. , (2016). Functional mutagenesis screens reveal the ‘cap structure’ formation in disulfide-bridge free TASK channels. Sci. Rep, 6, 19492. 10.1038/srep19492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lesage F, Reyes R, Fink M, Duprat F, Guillemare E, Lazdunski M, (1996). Dimerization of TWIK-1 K+ channel subunits via a disulfide bridge. EMBO J., 15, 6400–6407. [PMC free article] [PubMed] [Google Scholar]
- 56.Brohawn SG, Campbell EB, MacKinnon R, (2013). Domain-swapped chain connectivity and gated membrane access in a Fab-mediated crystal of the human TRAAK K+ channel. Proc. Natl. Acad. Sci. USA, 110, 2129–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lolicato M, Riegelhaupt PM, Arrigoni C, Clark KA, Minor DL Jr., (2014). Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K(2P) channels. Neuron, 84, 1198–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brohawn SG, Campbell EB, MacKinnon R, (2014). Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature, 516, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wodak SJ, Malevanets A, MacKinnon SS, (2015). The landscape of intertwined associations in homooligomeric proteins. Biophys. J, 109, 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bennett MJ, Eisenberg D, (2004). The evolving role of 3D domain swapping in proteins. Structure, 12, 1339–1341. [DOI] [PubMed] [Google Scholar]
- 61.Rousseau F, Schymkowitz J, Itzhaki LS, (2012). Implications of 3D domain swapping for protein folding, misfolding and function. Adv. Exp. Med. Biol, 747, 137–152. [DOI] [PubMed] [Google Scholar]
- 62.Kimberlin CR, Meshcheriakova A, Palty R, Raveh A, Karbat I, Reuveny E, et al. , (2019). SARAF luminal domain structure reveals a novel domain-swapped betasandwich fold important for SOCE modulation. J. Mol. Biol, 431, 2869–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mackinnon SS, Malevanets A, Wodak SJ, (2013). Intertwined associations in structures of homooligomeric proteins. Structure, 21, 638–649. [DOI] [PubMed] [Google Scholar]
- 64.Blin S, Ben Soussia I, Kim EJ, Brau F, Kang D, Lesage F, et al. , (2016). Mixing and matching TREK/TRAAK subunits generate heterodimeric K2P channels with unique properties. Proc. Natl. Acad. Sci. USA, 113, 4200–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blin S, Chatelain FC, Feliciangeli S, Kang D, Lesage F, Bichet D, (2014). Tandem pore domain halothane-inhibited K+ channel subunits THIK1 and THIK2 assemble and form active channels. J. Biol. Chem, 289, 28202–28212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berg AP, Talley EM, Manger JP, Bayliss DA, (2004). Motoneurons express heteromeric TWIK-related acid-sensitive K+ (TASK) channels containing TASK-1 (KCNK3) and TASK-3 (KCNK9) subunits. J. Neurosci, 24, 6693–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kang D, Han J, Talley EM, Bayliss DA, Kim D, (2004). Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J. Physiol, 554, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Czirjak G, Enyedi P, (2002). Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem, 277, 5426–5432. [DOI] [PubMed] [Google Scholar]
- 69.Lengyel M, Czirjak G, Enyedi P, (2016). Formation of functional heterodimers by TREK-1 and TREK-2 two-pore domain potassium channel subunits. J. Biol. Chem, 291, 13649–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levitz J, Royal P, Comoglio Y, Wdziekonski B, Schaub S, Clemens DM, et al. , (2016). Heterodimerization within the TREK channel subfamily produces a diverse family of highly regulated potassium channels. Proc. Natl. Acad. Sci. USA, 113, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khoubza L, Chatelain FC, Feliciangeli S, Lesage F, Bichet D, (2021). Physiological roles of heteromerization: focus on the two-pore domain potassium channels. J. Physiol, 599, 1041–1055. [DOI] [PubMed] [Google Scholar]
- 72.Bagriantsev SN, Clark KA, Minor DL Jr., (2012). Metabolic and thermal stimuli control K(2P)2.1 (TREK-1) through modular sensory and gating domains. EMBO J., 31, 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bagriantsev SN, Peyronnet R, Clark KA, Honore E, Minor DL Jr., (2011). Multiple modalities converge on a common gate to control K2P channel function. EMBO J., 30, 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piechotta PL, Rapedius M, Stansfeld PJ, Bollepalli MK, Ehrlich G, Andres-Enguix I, et al. , (2011). The pore structure and gating mechanism of K2P channels. EMBO J., 30, 3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rapedius M, Schmidt MR, Sharma C, Stansfeld PJ, Sansom MS, Baukrowitz T, et al. , (2012). State-independent intracellular access of quaternary ammonium blockers to the pore of TREK-1. Channels (Austin)., 6, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cohen A, Ben-Abu Y, Hen S, Zilberberg N, (2008). A novel mechanism for human K2P2.1 channel gating. Facilitation of C-type gating by protonation of extracellular histidine residues. J. Biol. Chem, 283, 19448–19455. [DOI] [PubMed] [Google Scholar]
- 77.Schewe M, Nematian-Ardestani E, Sun H, Musinszki M, Cordeiro S, Bucci G, et al. , (2016). A Non-canonical Voltage-Sensing Mechanism Controls Gating in K2P K(+) Channels. Cell, 164, 937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lengyel M, Czirjak G, Enyedi P, (2018). TRESK background potassium channel is not gated at the helix bundle crossing near the cytoplasmic end of the pore. PLoS ONE, 13, e0197622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ashmole I, Vavoulis DV, Stansfeld PJ, Mehta PR, Feng JF, Sutcliffe MJ, et al. , (2009). The response of the tandem pore potassium channel TASK-3 (K(2P)9.1) to voltage: gating at the cytoplasmic mouth. J. Physiol, 587, 4769–4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ben-Abu Y, Zhou Y, Zilberberg N, Yifrach O, (2009). Inverse coupling in leak and voltage-activated K+ channel gates underlies distinct roles in electrical signaling. Nature Struct. Mol. Biol, 16, 71–79. [DOI] [PubMed] [Google Scholar]
- 81.McClenaghan C, Schewe M, Aryal P, Carpenter EP, Baukrowitz T, Tucker SJ, (2016). Polymodal activation of the TREK-2 K2P channel produces structurally distinct open states. J. Gen. Physiol, 147, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aryal P, Jarerattanachat V, Clausen MV, Schewe M, McClenaghan C, Argent L, et al. , (2017). Bilayer-Mediated Structural Transitions Control Mechanosensitivity of the TREK-2 K2P Channel. Structure., 25, 708–718.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aryal P, Sansom MS, Tucker SJ, (2015). Hydrophobic gating in ion channels. J. Mol. Biol, 427, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aryal P, Abd-Wahab F, Bucci G, Sansom MS, Tucker SJ, (2014). A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nature Commun., 5, 4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ben Soussia I, El Mouridi S, Kang D, Leclercq-Blondel A, Khoubza L, Tardy P, et al. , (2019). Mutation of a single residue promotes gating of vertebrate and invertebrate two-pore domain potassium channels. Nature Commun., 10, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clausen MV, Jarerattanachat V, Carpenter EP, Sansom MSP, Tucker SJ, (2017). Asymmetric mechanosensitivity in a eukaryotic ion channel. Proc. Natl. Acad. Sci. USA, 114, E8343–E8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brennecke JT, de Groot BL, (2018). Mechanism of Mechanosensitive Gating of the TREK-2 Potassium Channel. Biophys. J ., 114, 1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harrigan MP, McKiernan KA, Shanmugasundaram V, Denny RA, Pande VS, (2017). Markov modeling reveals novel intracellular modulation of the human TREK-2 selectivity filter. Sci. Rep, 7, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Proks P, Schewe M, Conrad LJ, Rao S, Rathje K, Rödström KEJ, et al. A Mechanistic Basis for Inhibition of TREK-2 K2P Channels by Norfluoxetine. bioRxiv. 2020; 10.1101/2020.10.29.360966. [DOI]
- 90.Niemeyer MI, Cid LP, Pena-Munzenmayer G, Sepulveda FV, (2010). Separate Gating Mechanisms Mediate the Regulation of K2P Potassium Channel TASK2 by Intra- and Extracellular pH. J. Biol. Chem, 285, 16467–16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barel O, Shalev SA, Ofir R, Cohen A, Zlotogora J, Shorer Z, et al. , (2008). Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am. J. Hum. Genet, 83, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, et al. , (2013). A novel channelopathy in pulmonary arterial hypertension. The New England journal of medicine., 369, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cao Z, Bowie JU, (2012). Shifting hydrogen bonds may produce flexible transmembrane helices. Proc. Natl. Acad. Sci. USA, 109, 8121–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nematian-Ardestani E, Abd-Wahab F, Chatelain FC, Sun H, Schewe M, Baukrowitz T, et al. , (2020). Selectivity filter instability dominates the low intrinsic activity of the TWIK-1 K2P K(+) channel. J. Biol. Chem, 295, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yuill KH, Stansfeld PJ, Ashmole I, Sutcliffe MJ, Stanfield PR, (2007). The selectivity, voltage-dependence and acid sensitivity of the tandem pore potassium channel TASK-1: contributions of the pore domains. Pflugers Arch., 455, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Niemeyer MI, Gonzalez-Nilo FD, Zuniga L, Gonzalez W, Cid LP, Sepulveda FV, (2007). Neutralization of a single arginine residue gates open a two-pore domain, alkali-activated K+ channel. Proc. Natl. Acad. Sci. USA, 104, 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW, (1993). Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels, 1, 61–71. [PubMed] [Google Scholar]
- 98.Baukrowitz T, Yellen G, (1995). Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron, 15, 951–960. [DOI] [PubMed] [Google Scholar]
- 99.Pardo LA, Heinemann SH, Terlau H, Ludewig U, Lorra C, Pongs O, et al. , (1992). Extracellular K+ specifically modulates a rat brain K+ channel. Proc. Natl. Acad. Sci. USA, 89, 2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cordero-Morales JF, Cuello LG, Zhao Y, Jogini V, Cortes DM, Roux B, et al. , (2006). Molecular determinants of gating at the potassium-channel selectivity filter. Nature Struct. Mol. Biol, 13, 311–318. [DOI] [PubMed] [Google Scholar]
- 101.Hoshi T, Armstrong CM, (2013). C-type inactivation of voltage-gated K+ channels: pore constriction or dilation?. J. Gen. Physiol, 141, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou Y, MacKinnon R, (2003). The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J. Mol. Biol, 333, 965–975. [DOI] [PubMed] [Google Scholar]
- 103.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R, (2001). Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature, 414, 43–48. [DOI] [PubMed] [Google Scholar]
- 104.Cuello LG, Cortes DM, Perozo E, (2017). The gating cycle of a K(+) channel at atomic resolution. eLife, 6, e28032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cuello LG, Jogini V, Cortes DM, Perozo E, (2010). Structural mechanism of C-type inactivation in K(+) channels. Nature, 466, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pau V, Zhou Y, Ramu Y, Xu Y, Lu Z, (2017). Crystal structure of an inactivated mutant mammalian voltage-gated K(+) channel. Nature Struct. Mol. Biol, 24, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang W, MacKinnon R, (2017). Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell, 169, 422–430.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Matthies D, Bae C, Toombes GE, Fox T, Bartesaghi A, Subramaniam S, et al. , (2018). Singleparticle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs. eLife, 7. [DOI] [PMC free article] [PubMed]
- 109.Pope L, Arrigoni C, Lou H, Bryant C, Gallardo-Godoy A, Renslo AR, et al. , (2018). Protein and chemical determinants of BL-1249 Action And Selectivity for K2P channels. ACS Chem. Neurosci, 9, 3153–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Decher N, Rinne S, Bedoya M, Gonzalez W, Kiper AK, (2021). Molecular pharmacology of K2P potassium channels. Cell. Physiol. Biochem, 55, 87–107. [DOI] [PubMed] [Google Scholar]
- 111.Bedoya M, Rinne S, Kiper AK, Decher N, Gonzalez W, Ramirez D, (2019). TASK channels pharmacology: new challenges in drug design. J. Med. Chem, 62, 10044–10058. [DOI] [PubMed] [Google Scholar]
- 112.Fletcher JM, Greenfield BF, Hardy CJ, Scargill D, Woodhead JL, (1961). Ruthenium Red. J. Chem. Soc,, 2000–2006.
- 113.Clarke MJ, (2002). Ruthenium metallopharmaceuticals. Coordin. Chem. Rev, 232, 69–93. [Google Scholar]
- 114.Braun G, Lengyel M, Enyedi P, Czirjak G, (2015). Differential sensitivity of TREK-1, TREK-2 and TRAAK background potassium channels to the polycationic dye ruthenium red. Br. J. Pharmacol, 172, 1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Musset B, Meuth SG, Liu GX, Derst C, Wegner S, Pape HC, et al. , (2006). Effects of divalent cations and spermine on the K+ channel TASK-3 and on the outward current in thalamic neurons. J. Physiol, 572, 639–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Czirjak G, Enyedi P, (2003). Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Mol. Pharmacol, 63, 646–652. [DOI] [PubMed] [Google Scholar]
- 117.Gonzalez W, Zuniga L, Cid LP, Arevalo B, Niemeyer MI, Sepulveda FV, (2013). An extracellular ion pathway plays a central role in the cooperative gating of a K(2P) K+ channel by extracellular pH. J. Biol. Chem, 288, 5984–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ying WL, Emerson J, Clarke MJ, Sanadi DR, (1991). Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry, 30, 4949–4952. [DOI] [PubMed] [Google Scholar]
- 119.Kirichok Y, Krapivinsky G, Clapham DE, (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature, 427, 360–364. [DOI] [PubMed] [Google Scholar]
- 120.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. , (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature, 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, et al. , (2016). Architecture of the mitochondrial calcium uniporter. Nature, 533, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Woods JJ, Wilson JJ, (2019). Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr. Opin. Chem. Biol, 55, 9–18. [DOI] [PubMed] [Google Scholar]
- 123.Hardy JA, Wells JA, (2004). Searching for new allosteric sites in enzymes. Curr. Opin. Struct. Biol, 14, 706–715. [DOI] [PubMed] [Google Scholar]
- 124.Lenaeus MJ, Vamvouka M, Focia PJ, Gross A, (2005). Structural basis of TEA blockade in a model potassium channel. Nature Struct. Mol. Biol, 12, 454–459. [DOI] [PubMed] [Google Scholar]
- 125.Yohannan S, Hu Y, Zhou Y, (2007). Crystallographic study of the tetrabutylammonium block to the KcsA K+ channel. J. Mol. Biol, 366, 806–814. [DOI] [PubMed] [Google Scholar]
- 126.Zhou M, Morais-Cabral JH, Mann S, MacKinnon R, (2001). Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature, 411, 657–661. [DOI] [PubMed] [Google Scholar]
- 127.Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honore E, (2005). A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J., 24, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chemin J, Patel AJ, Duprat F, Sachs F, Lazdunski M, Honore E, (2007). Up- and down-regulation of the mechano-gated K(2P) channel TREK-1 by PIP (2) and other membrane phospholipids. Pflugers Arch., 455, 97–103. [DOI] [PubMed] [Google Scholar]
- 129.Lopes CM, Rohacs T, Czirjak G, Balla T, Enyedi P, Logothetis DE, (2005). PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J. Physiol, 564, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Honore E, Maingret F, Lazdunski M, Patel AJ, (2002). An intracellular proton sensor commands lipid-and mechano-gating of the K(+) channel TREK-1. EMBO J., 21, 2968–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Murbartian J, Lei Q, Sando JJ, Bayliss DA, (2005). Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J. Biol. Chem, 280, 30175–30184. [DOI] [PubMed] [Google Scholar]
- 132.Soussia IB, Choveau FS, Blin S, Kim EJ, Feliciangeli S, Chatelain FC, et al. , (2018). Antagonistic effect of a cytoplasmic domain on the basal activity of polymodal potassium channels. Front. Mol. Neurosci, 11, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chemin J, Patel A, Duprat F, Zanzouri M, Lazdunski M, Honore E, (2005). Lysophosphatidic acid-operated K + channels. J. Biol. Chem, 280, 4415–4421. [DOI] [PubMed] [Google Scholar]
- 134.Cabanos C, Wang M, Han X, Hansen SB, (2017). A soluble fluorescent binding assay reveals PIP2 antagonism of TREK-1 channels. Cell Rep., 20, 1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schrecke S, Zhu Y, McCabe JW, Bartz M, Packianathan C, Zhao M, et al. , (2021). Selective regulation of human TRAAK channels by biologically active phospholipids. Nature Chem. Biol, 17, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Niemeyer MI, Cid LP, Paulais M, Teulon J, Sepulveda FV, (2017). Phosphatidylinositol (4,5)bisphosphate dynamically regulates the K2P background K(+) channel TASK-2. Sci. Rep, 7, 45407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Patel AJ, Honore E, Lesage F, Fink M, Romey G, Lazdunski M, (1999). Inhalational anesthetics activate two-pore-domain background K+ channels. Nature Neurosci., 2, 422–426. [DOI] [PubMed] [Google Scholar]
- 138.Liu C, Au JD, Zou HL, Cotten JF, Yost CS, (2004). Potent activation of the human tandem pore domain K channel TRESK with clinical concentrations of volatile anesthetics. Anesth. Analg, 99, 1715–1722. [DOI] [PubMed] [Google Scholar]
- 139.Luethy A, Boghosian JD, Srikantha R, Cotten JF, (2017). Halogenated ether, alcohol, and alkane anesthetics activate TASK-3 tandem pore potassium channels likely through a common mechanism. Mol. Pharmacol, 91, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Andres-Enguix I, Caley A, Yustos R, Schumacher MA, Spanu PD, Dickinson R, et al. , (2007). Determinants of the anesthetic sensitivity of two-pore domain acid-sensitive potassium channels: molecular cloning of an anesthetic-activated potassium channel from Lymnaea stagnalis. J. Biol. Chem, 282, 20977–20990. [DOI] [PubMed] [Google Scholar]
- 141.Conway KE, Cotten JF, (2012). Covalent modification of a volatile anesthetic regulatory site activates TASK-3 (KCNK9) tandem-pore potassium channels. Mol. Pharmacol, 81, 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chokshi RH, Larsen AT, Bhayana B, Cotten JF, (2015). Breathing stimulant compounds inhibit TASK-3 potassium channel function likely by binding at a common site in the channel pore. Mol. Pharmacol, 88, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wague A, Joseph TT, Woll KA, Bu W, Vaidya KA,Bhanu NV, et al. , (2020). Mechanistic insights into volatile anesthetic modulation of K2P channels. eLife, 2, e59839. 10.7554/eLife.59839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pavel MA, Petersen EN, Wang H, Lerner RA, Hansen SB, (2020). Studies on the mechanism of general anesthesia. Proc. Natl. Acad. Sci. USA, 117, 13757–13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, et al. , (2001). THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J. Biol. Chem, 276, 7302–7311. [DOI] [PubMed] [Google Scholar]
- 146.Asai T, Adachi N, Moriya T, Oki H, Maru T, Kawasaki M, et al. , (2021). Cryo-EM structure of K(+)bound hERG Channel Complexed With The Blocker astemizole. Structure, 29203–12 e4. [DOI] [PubMed] [Google Scholar]
- 147.Streit AK, Netter MF, Kempf F, Walecki M, Rinne S, Bollepalli MK, et al. , (2011). A specific two-pore domain potassium channel blocker defines the structure of the TASK-1 open pore. J. Biol. Chem, 286, 13977–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Coburn CA, Luo Y, Cui M, Wang J, Soll R, Dong J, et al. , (2012). Discovery of a pharmacologically active antagonist of the two-pore-domain potassium channel K2P9.1 (TASK-3). ChemMedChem, 7, 123–133. [DOI] [PubMed] [Google Scholar]
- 149.Cotten JF, Keshavaprasad B, Laster MJ, Eger EI 2nd, Yost CS, (2006). The ventilatory stimulant doxapram inhibits TASK tandem pore (K2P) potassium channel function but does not affect minimum alveolar anesthetic concentration. Anesth. Analg, 102, 779–785. [DOI] [PubMed] [Google Scholar]
- 150.Flaherty DP, Simpson DS, Miller M, Maki BE, Zou B, Shi J, et al. , (2014). Potent and selective inhibitors of the TASK-1 potassium channel through chemical optimization of a bis-amide scaffold. Bioorg. Med. Chem. Letters, 24, 3968–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ramirez D, Bedoya M, Kiper AK, Rinne S, Morales-Navarro S, Hernandez-Rodriguez EW, et al. , (2019). Structure/activity analysis of TASK-3 channel antagonists based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine. Int. J. Mol. Sci, 20, 2252. 10.3390/ijms20092252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kiper AK, Rinne S, Rolfes C, Ramirez D, Seebohm G, Netter MF, et al. , (2015). Kv1.5 blockers preferentially inhibit TASK-1 channels: TASK-1 as a target against atrial fibrillation and obstructive sleep apnea?. Pflugers Arch., 467, 1081–1090. [DOI] [PubMed] [Google Scholar]
- 153.O’Donohoe PB, Huskens N, Turner PJ, Pandit JJ, Buckler KJ, (2018). A1899, PK-THPP, ML365, and Doxapram inhibit endogenous TASK channels and excite calcium signaling in carotid body type-1 cells. Physiol. Rep, 6, 10.14814/phy2.13876e13876. [DOI] [PMC free article] [PubMed]
- 154.Veale EL, Mathie A, (2016). Aristolochic acid, a plant extract used in the treatment of pain and linked to Balkan endemic nephropathy, is a regulator of K2P channels. Br. J. Pharmacol, 173, 1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Takahira M, Sakurai M, Sakurada N, Sugiyama K, (2005). Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pflugers Arch., 451, 474–478. [DOI] [PubMed] [Google Scholar]
- 156.Tertyshnikova S, Knox RJ, Plym MJ, Thalody G, Griffin C, Neelands T, et al. , (2005). BL-1249 [(5,6,7,8tetrahydro-naphthalen-1-yl)-[2-(1H-tetrazol-5-yl)-phenyl]amine]: a putative potassium channel opener with bladder-relaxant properties. J. Pharmacol. Exp. Therap, 313, 250–259. [DOI] [PubMed] [Google Scholar]
- 157.Veale EL, Al-Moubarak E, Bajaria N, Omoto K, Cao L, Tucker SJ, et al. , (2014). Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol. Pharmacol, 85, 671–681. [DOI] [PubMed] [Google Scholar]
- 158.Monteillier A, Loucif A, Omoto K, Stevens EB, Lainez S, Saintot PP, et al. , (2016). Investigation of the structure activity relationship of flufenamic acid derivatives at the human TRESK channel K2P18.1. Bioorg. Med. Chem. Lett, 26, 4919–4924. [DOI] [PubMed] [Google Scholar]
- 159.Rinne S, Kiper AK, Vowinkel KS, Ramirez D, Schewe M, Bedoya M, et al. , (2019). The molecular basis for an allosteric inhibition of K(+)-flux gating in K2P channels. eLife, 8, e39476. 10.7554/eLife.39476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pavel MA, Chung HW, Petersen EN, Hansen SB, (2019). Polymodal mechanism for TWIK-related K+ channel inhibition by local anesthetic. Anesth. Analg, 129, 973–982. [DOI] [PubMed] [Google Scholar]
- 161.Ma Y, Luo Q, Fu J, Che Y, Guo F, Mei L, et al. , (2020). Discovery of an inhibitor for the TREK-1 channel targeting an intermediate transition state of channel gating. J. Med. Chem, 63, 10972–10983. [DOI] [PubMed] [Google Scholar]
- 162.Qiu Y, Huang L, Fu J, Han C, Fang J, Liao P, et al. , (2020). TREK channel family activator with a well-defined structure-activation relationship for pain and neurogenic inflammation. J. Med. Chem, 63, 3665–3677. [DOI] [PubMed] [Google Scholar]
- 163.Sandoz G, Douguet D, Chatelain F, Lazdunski M, Lesage F, (2009). Extracellular acidification exerts opposite actions on TREK1 and TREK2 potassium channels via a single conserved histidine residue. Proc. Natl. Acad. Sci. USA, 106, 14628–14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Luo Q, Chen L, Cheng X, Ma Y, Li X, Zhang B, et al. , (2017). An allosteric ligand-binding site in the extracellular cap of K2P channels. Nature Commun., 8, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhuo RG, Peng P, Liu XY, Yan HT, Xu JP, Zheng JQ, et al. , (2016). Intersubunit concerted cooperative and cis-type mechanisms modulate allosteric gating in two-pore-domain potassium channel TREK-2. Front. Cell. Neurosci, 10, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wright PD, McCoull D, Walsh Y, Large JM, Hadrys BW, Gaurilcikaite E, et al. , (2019). Pranlukast is a novel small molecule activator of the two-pore domain potassium channel TREK2. Biochem. Biophys. Res. Commun, 520, 35–40. [DOI] [PubMed] [Google Scholar]
- 167.Noel J, Sandoz G, Lesage F, (2011). Molecular regulations governing TREK and TRAAK channel functions. Channels (Austin)., 5, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee LM, Muntefering T, Budde T, Meuth SG, Ruck T, (2021). Pathophysiological Role of K2P channels in human diseases. Cell. Physiol. Biochem, 55, 65–86. [DOI] [PubMed] [Google Scholar]
- 169.Wiedmann F, Rinne S, Donner B, Decher N, Katus HA, Schmidt C, (2021). Mechanosensitive TREK-1 two-pore-domain potassium (K2P) channels in the cardiovascular system. Prog. Biophys. Mol. Biol, 159, 126–135. [DOI] [PubMed] [Google Scholar]
- 170.Djillani A, Mazella J, Heurteaux C, Borsotto M, (2019). Role of TREK-1 in health and disease, focus on the central nervous system. Front. Pharmacol, 10, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A, (2005). Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br. J. Pharmacol, 144, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim EJ, Lee DK, Hong SG, Han J, Kang D,(2017). Activation of TREK-1, but Not TREK-2, channel by mood stabilizers. Int. J. Mol. Sci, 18, 2460. 10.3390/ijms18112460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bustos D, Bedoya M, Ramirez D, Concha G, Zuniga L, Decher N, et al. , (2020). Elucidating the structural basis of the intracellular pH sensing mechanism of TASK2 K2P channels. Int. J. Mol. Sci, 21, 532. 10.3390/ijms21020532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Meadows HJ, Randall AD, (2001). Functional characterisation of human TASK-3, an acid-sensitive two-pore domain potassium channel. Neuropharmacology, 40, 551–559. [DOI] [PubMed] [Google Scholar]
- 175.Kindler CH, Yost CS, Gray AT, (1999). Local anesthetic inhibition of baseline potassium channels with two pore domains in tandem. Anesthesiology, 90, 1092–1102. [DOI] [PubMed] [Google Scholar]
- 176.Lv J, Liang Y, Zhang S, Lan Q, Xu Z, Wu X, et al. , (2019). DCPIB, an inhibitor of volume-regulated anion channels, distinctly modulates K2P channels. ACS Chem. Neurosci, 10, 2786–2793. [DOI] [PubMed] [Google Scholar]
- 177.Putzke C, Wemhoner K, Sachse FB, Rinne S, Schlichthorl G, Li XT, et al. , (2007). The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc. Res, 75, 59–68. [DOI] [PubMed] [Google Scholar]
- 178.Ramirez D, Concha G, Arevalo B, Prent-Penaloza L, Zuniga L, Kiper AK, et al. , (2019). Discovery of novel TASK-3 channel blockers using a pharmacophore-based virtual screening. Int. J. Mol. Sci, 20, 4014. 10.3390/ijms20164014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, Romey G, et al. , (2001). Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochem. Biophys. Res. Commun, 282, 249–256. [DOI] [PubMed] [Google Scholar]
- 180.Renigunta V, Zou X, Kling S, Schlichthorl G, Daut J, (2014). Breaking the silence: functional expression of the two-pore-domain potassium channel THIK-2. Pflugers Arch., 466, 1735–1745. [DOI] [PubMed] [Google Scholar]
- 181.Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honore E, (2000). TWIK-2, an inactivating 2P domain K+ channel. J. Biol. Chem, 275, 28722–28730. [DOI] [PubMed] [Google Scholar]
- 182.Mazella J, Petrault O, Lucas G, Deval E, Beraud-Dufour S, Gandin C, et al. , (2010). Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: a new concept in the antidepressant drug design. PLoS Biol., 8, e1000355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Moha Ou Maati H, Veyssiere J, Labbal F, Coppola T, Gandin C, Widmann C, et al. , (2012). Spadin as a new antidepressant: absence of TREK-1-related side effects. Neuropharmacology, 62, 278–288. [DOI] [PubMed] [Google Scholar]
- 184.Djillani A, Pietri M, Moreno S, Heurteaux C, Mazella J, Borsotto M, (2017). Shortened spadin analogs display better TREK-1 inhibition, in vivo stability and antidepressant activity. Front. Pharmacol, 8, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kim HJ, Woo J, Nam Y, Nam JH, Kim WK, (2016). Differential modulation of TWIK-related K(+) channel (TREK) and TWIK-related acid-sensitive K(+) channel 2 (TASK2) activity by pyrazole compounds. Eur. J. Pharmacol, 791, 686–695. [DOI] [PubMed] [Google Scholar]
- 186.Miller MR, Zou B, Shi J, Flaherty DP, Simpson DS, Yao T, et al. (2010) Development of a Selective Chemical Inhibitor for the Two-Pore Potassium Channel, KCNK9. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD). [PubMed] [Google Scholar]
- 187.Ye D, Li Y, Zhang X, Guo F, Geng L, Zhang Q, et al. , (2015). TREK1 channel blockade induces an antidepressant-like response synergizing with 5-HT1A receptor signaling. Eur. Neuropsychopharmacol, 25, 2426–2436. [DOI] [PubMed] [Google Scholar]
- 188.Ji XC, Zhao WH, Cao DX, Shi QQ, Wang XL, (2011). Novel neuroprotectant chiral 3-n-butylphthalide inhibits tandem-pore-domain potassium channel TREK-1. Acta Pharmacol. Sin, 32, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang W, Liu D, Xiao Q, Cai J, Feng N, Xu S, et al. , (2018). Lig4–4 selectively inhibits TREK-1 and plays potent neuroprotective roles in vitro and in rat MCAO model. Neurosci. Letters, 671, 93–98. [DOI] [PubMed] [Google Scholar]
- 190.Lengyel M, Erdelyi F, Pergel E, Balint-Polonka A, Dobolyi A, Bozsaki P, et al. , (2019). Chemically Modified Derivatives of the Activator Compound Cloxyquin Exert Inhibitory Effect on TRESK (K2P18.1) Background Potassium Channel. Mol. Pharmacol, 95, 652–660. [DOI] [PubMed] [Google Scholar]
- 191.Thummler S, Duprat F, Lazdunski M, (2007). Antipsychotics inhibit TREK but not TRAAK channels. Biochem. Biophys. Res. Commun, 354, 284–289. [DOI] [PubMed] [Google Scholar]
- 192.Bruner JK, Zou B, Zhang H, Zhang Y, Schmidt K, Li M, (2014). Identification of novel small molecule modulators of K2P18.1 two-pore potassium channel. Eur. J. Pharmacol, 740, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu H, Enyeart JA, Enyeart JJ, (2007). Potent inhibition of native TREK-1 K+ channels by selected dihydropyridine Ca2+ channel antagonists. J. Pharmacol. Exp. Therap, 323, 39–48. [DOI] [PubMed] [Google Scholar]
- 194.Chavez RA, Gray AT, Zhao BB, Kindler CH, Mazurek MJ, Mehta Y, et al. , (1999). TWIK-2, a new weak inward rectifying member of the tandem pore domain potassium channel family. J. Biol. Chem, 274, 7887–7892. [DOI] [PubMed] [Google Scholar]
- 195.Maingret F, Patel AJ, Lazdunski M, Honoré E, (2001). The endocannabinoid anandamide is a direct and selective blocker of the backgroun K+ channel TASK-1. EMBO J., 20, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Minieri L, Pivonkova H, Caprini M, Harantova L, Anderova M, Ferroni S, (2013). The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. Br. J. Pharmacol, 168, 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gray AT, Zhao BB, Kindler CH, Winegar BD, Mazurek MJ, Xu J, et al. , (2000). Volatile anesthetics activate the human tandem pore domain baseline K+ channel KCNK5. Anesthesiology, 92, 1722–1730. [DOI] [PubMed] [Google Scholar]
- 198.Gruss M, Bushell TJ, Bright DP, Lieb WR, Mathie A, Franks NP, (2004). Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol. Pharmacol, 65, 443–452. [DOI] [PubMed] [Google Scholar]
- 199.Zhuo RG, Peng P, Liu XY, Yan HT, Xu JP, Zheng JQ, et al. , (2016). Allosteric coupling between proximal C-terminus and selectivity filter is facilitated by the movement of transmembrane segment 4 in TREK-2 channel. Sci. Rep, 6, 21248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Beltran L, Beltran M, Aguado A, Gisselmann G, Hatt H, (2013). 2-Aminoethoxydiphenyl borate activates the mechanically gated human KCNK channels KCNK 2 (TREK-1), KCNK 4 (TRAAK), and KCNK 10 (TREK-2). Front. Pharmacol, 4, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Loucif AJC, Saintot PP, Liu J, Antonio BM, Zellmer SG, Yoger K, et al. , (2018). GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P) channel opener, reduces rat dorsal root ganglion neuron excitability. Br. J. Pharmacol, 175, 2272–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wright PD, Veale EL, McCoull D, Tickle DC, Large JM, Ococks E, et al. , (2017). Terbinafine is a novel and selective activator of the two-pore domain potassium channel TASK3. Biochem. Biophys. Res. Commun, 493, 444–450. [DOI] [PubMed] [Google Scholar]