

Abstract
Corona Virus Disease 2019 (COVID-19), referred to as ‘New Coronary Pneumonia’, is a type of acute infectious disease caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection. Mpro is one of the main targets for treating COVID-19. The current research on Mpro mainly focuses on the repurposing of old drugs, and there are only a few novel ligands that inhibit Mpro. In this research, we used computational free energy calculation to screen a compound library against Mpro, and discovered four novel compounds with the two best compounds (AG-690/13507628 and AG-690/13507724) having experimental measured IC50 of just under 3 μM and low cell toxicity. Detailed decomposition of the interactions between the inhibitors and Mpro reveals key interacting residues and interactions that determine the activity. The results from this study should provide a basis for further development of anti-SARS-CoV-2 drugs.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, main protease, inhibitory activity, virtual screening
Introduction
Coronavirus Disease (COVID-19), which is referred as ‘New Coronary Pneumonia’, is caused by the infection of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The main symptoms of COVID-19 include respiratory symptoms, fever, cough, and breathing difficulties (Struyf et al., 2020). In more severe cases, the infection can lead to pneumonia, severe acute respiratory syndrome, kidney failure, and even death (Wang et al., 2020). Since the outbreak of the pandemic in later 2019, the coronavirus has been rapidly discovered in the world because of its own pathogenic characteristic and infectious characteristic. The number of confirmed infections has surpassed that of SARS, and the number of deaths has continued to increase, causing serious social harm and economic losses. The World Health Organization announced that the outbreak of New Coronary Pneumonia has constituted a ‘Public Health Emergency of International Concern’ (“Coronavirus disease,” 2020). Therefore, the development of therapeutic drugs against the SARS-CoV-2 is a very urgent task.
The genome of SARS-CoV-2, which likes most of the Coronavirus genome, encodes two large polyproteins, namely PP1A and PP1AB (Thiel et al., 2003). These two polyproteins are cleaved by two proteases Mpro (3 C-like protease or 3CLpro) and PLpro (papain-like protease) which are encoded by the ORF 1a/b (Hilgenfeld, 2014). The main protease Mpro is crucial for virus replication and controlling of host cell response, which makes it important for the propagation of the virus. Therefore, it is considered as the one of the key targets in the development of anti-SARS-CoV-2 drugs.
Mpro is a dimer with monomer contains two domains: the N-terminal catalytic domain and the C-terminal domain (Lee et al., 2005). The sequences of Mpro in SARS-CoV and SARS-CoV-2 have a sequence identity of 96% (Gil et al., 2020). Around the active site, the conserved binding pocket of Mpro is composed of four subsites (S1’, S1, S2, and S4) well accommodating the substrate (Figure 1) (Xue et al., 2008). Moreover, the high-resolution crystal structure of SARS-CoV-2 Mpro in complex with the inhibitor N3 has been solved (Protein Data Bank ID: 7BQY) (Jin et al., 2020), which provides an excellent basis for structure-based drug design.
Figure 1.

The structure of SARS-CoV-2 Mpro. The coordinates are from PDB entry 7BQY, and the N3 inhibitor is shown in stick. The substrate-binding pocket is shown in the inset with subsites labeled.
Recently, a number of inhibitors against Mpro have been discovered. For example, Jin et al. reported that the IC50 of Ebselen, an anti-inflammatory drug (Jin et al., 2020), was 0.67 μM. In the study of Li et al., several old drugs like Dipyridamole, Candesarta, and Cilexetil were found to have a inhibitory effect in vitro (Li et al., 2020). Moreover, various inhibitors against SARS-CoV-1 Mpro and other antiviral drugs including Boceprevir, Nariaprevir, GC-376, MG-132, Calpain inhibitor II, and Calpain inhibitor XII have been proven effective against the Mpro of SARS-CoV2 in vitro (Ma et al., 2020). A number of novel inhibitors of Mpro have also been discovered by research groups. Dai et al. discovered several novel polypeptide analogues with IC50 of approximately 0.05 μM (Wenhao et al., 2020). The alpha-ketoamide inhibitors such as 13 b have good inhibitory activity with IC50 of 0.67 μM (Linlin Zhang et al., 2020). Small-molecule compounds such as N-substituted isatin have shown enzymatic activity at sub-micromolar level (Liu et al., 2020). An indole moiety inhibits for Mpro could block the infectivity of SARS-CoV-2 with EC50 values of 15 ± 4 and 4.2 ± 0.7 μM for GRL-1720 and 5 h (Hattori et al., 2021). Recently, Qiao et al. discovered six compounds of the Mpro inhibitors with nanomolar or low micromolar EC50 values, including MI-09 (0.86 μM), MI-12 (0.53 μM), MI-14 (0.66 μM), MI-28 (0.67 μM), MI-30 (0.54 μM), and MI-31 (0.83 μM) (Qiao et al., 2021). Zhang et al. obtained a series of non-covalent inhibitors by modifying Perampanel, and the IC50 of best molecule reached 18 nM (Zhang et al., 2021). Despite these exciting progresses, discovering novel Mpro inhibitors to enrich our arsenal against SARS-CoV-2 is still in urgent need.
In this study, we used virtual screening to identify Mpro inhibitors from the Specs compound library. Four active compounds with a novel scaffold and micromolar IC50 values were discovered. Furthermore, these compounds have multiple modifiable chemical sites, low cytotoxicity and potentials for further optimization.
Methods
Virtual screening
The virtual screening was performed using Glide (Friesner et al., 2004). The structure of SARS-CoV-2 Mpro (PDB ID:7BQY) (Jin et al., 2020) was used to generate the receptor grid for docking simulations. The center of the grid was determined using the inhibitor N3 in the structure with a grid box of 30 Å. The Van der Waals radius scaling factor was 1.0 and partial charge cutoff was 0.25. We used the Specs library (Delft, Netherlands: http://www.specs.net) the ligand library. The ligands were prepared with LigPrep (Friesner et al., 2004) at target pH 7.2 ± 0.2 with the OPLS3e force field (Harder et al., 2016).
Molecular dynamics simulations
We used the gaff force field to obtain parameters for the compounds (Wang et al., 2004). The charges for the all the ligands were calculated using the AM1-BCC method in antechamber of Amber18 (Salomon-Ferrer et al., 2013). Protonation of the protein-ligand complex was performed at pH 7.4. Molecular dynamics (MD) simulations were carried out using Amber18 with the ff14SB force field (Maier et al., 2015). We solvated the protein-ligand complex in a cubic box using TIP3P water, which extends 10.0 Å away from the solute. Cut-off of non-bonded interactions was set to 10 Å. We added counter-ions to neutralize the systems. Each system was minimized for 10,000 steps and then heated from 0 to 300 K in 300 ps with Langevin dynamics. Berendsen barostat was used to control the pressure at 1.0 atm. Production runs were 10-ns long in an NPT ensemble with a time step of 2 fs. The trajectories were recorded every 1 ps. We used the SHAKE algorithm to constrain the bonds involving hydrogen atoms (Andersen, 1983).
Binding free energy calculation
We used the alanine-scanning in combination with interaction entropy (ASIE) method to calculate the binding free energy (Yan et al., 2017). In this approach, it is assumed that the contribution of the mutated alanine to the binding free energy is negligible. Therefore, the contribution of a specific residue (Zhou et al., 2018) to the total binding free energy is given by the energy difference when the wildtype residue X is mutated to A (alanine):
| (1) |
The gas-phase binding free energy and the solvation component are evaluated by the following scheme:
| (2) |
| (3) |
The gas-phase component of the binding free energy is and which represent the energy between the ligand L and alanine and the wildtype residue X. The enthalpy and entropy of gas-phase energy were calculated with the standard molecular mechanics and the IE method (Ben-Shalom et al., 2017; Duan et al., 2016; Qiu et al., 2018; Song et al., 2018), respectively
| (4) |
| (5) |
where, and were the van der Waals and electrostatic interaction energies between the ligand and residue X and A, respectively. The exponential argument denotes and is the deviation from average. We evaluated the exponential average by discrete time averaging,
| (6) |
where N is the number of MD snapshots. Finally, Eq. (2) becomes
| (7) |
The MM/GBSA method (igb = 8 in Amber18) (Genheden & Ryde, 2015; Massova & Kollman, 2000; Onufriev et al., 2004) is used to calculate the solvation free energy by
| (8) |
where and are the electrostatic solvation free energy and the nonpolar solvation free energy, respectively. is given by an empirical solvent-accessible surface area (SASA) formula:
| (9) |
The γ and β values we used here are the standard values of 0.00542 kcal/(mol·Å2) and 0.92 kcal/mol. In our ASIE calculation, different dielectric constant of 1, 3, and 5 are used for nonpolar, polar, and charged residues (Hou et al., 2011; Petukh et al., 2015).
The total protein-ligand binding free energy can be approximated by the summation (Liu et al., 2018; Zhou et al., 2018),
| (10) |
where the summation is over residues that are within 5 Å of the ligand. A total of 100 snapshots from the last 5 ns of the MD trajectory were used to calculate the binding energy with the ASIE method.
Activity of the protein
The SARS-CoV-2 Mpro protein was purchased from Kactus Biosystems Co, Ltd (Figure S1). PBS-EDTA (4 × PE, pH 7.4, LEAGEN) with 4 mM DTT (TCI, CAS RN 3483-12-3) was diluted as the buffer solution. We used a FRET-based Mpro substrate of SARS-CoV-2 Dabcyl-KTSAVLQ/SGFRKME (Edans) (Go Top Peptide Biotech Co, Ltd). According to the preliminary experiment read by an imaging reader (BioTek, SYNERGY4), we set filters for excitation at 340 nm and emission at 535 nm. We set up six concentrations of the substrate at 100 μM, 50 μM, 25 μM, 12.5 μM, 6.25 μM, and 3.125 μM to measure the activity of the protein. The reaction progress was monitored for 30 min. In the first 20 min, the reaction kinetics were monitored every 90 s, and the data was used to calculate the initial velocity via linear regression in Prism 8 with the classic Michaelis–Menten equation.
Enzymatic assays
For the screening of inhibitors and IC50 measurements, 100 nM Mpro was incubated with the ligands at 30 °C for 60 min in reaction buffer, then the reaction was initiated by adding 5 µM FRET substrate and monitored for 1 h. The IC50 value was calculated by plotting the initial velocity against various concentrations of ligands using a dose-response curve in the Prism 8 software.
The compound concentration was 500 μM, 100 μM, 10 μM, 1 μM, 0.1 μM, and 0.01 μM for the measurement the IC50 values. The data from imaging reader was calculated with the equation below:
where stands for protease, drug, buffer and substrate; stands for buffer and substrate; stands for protease, DMSO, buffer and substrate.
Cytotoxicity test
Human Umbilical Vein Endothelial Cells (HUVEC) were seeded into a 96-well plate. After the cells adhere to the wall, the compounds were diluted to 100 μM and 500 μM for administration. Each concentration was tested with three replicates. CCK-8 reagent was added 72 h after the administration, and incubated for 2 h. Then the plate was read with an imaging reader (BioTek, SYNERGY4). The GraphPad Prism 8 software was used to calculate the cell activity inhibition plot for normal cells.
Results
Virtual screening
We used the complex structure of SARS-CoV-2 Mpro and the N3 inhibitor (PDB ID: 7BQY) (Jin et al., 2020) for in silico screening against the Specs library with Glide. The top 400 compounds from docking were selected for binding free energy calculation using the ASIE method. The top 200 compounds from ASIE were manually inspected, and 20 compounds were selected based on the calculated binding free energy from ASIE, 5 compounds were selected according to Glide XP score, and 5 were selected based on experience of medicinal chemistry. At last, 30 compounds were purchased from Specs (Table 1).
Table 1.
The Glide XP score of the 30 purchased compounds.
| Specs ID | Glide XP score | Specs ID | Glide XP score |
|---|---|---|---|
| AE-562/12222311 | –9.726 | AK-918/42028521 | –8.343 |
| AE-848/07789006 | –8.128 | AK-918/42293923 | –8.033 |
| AF-399/34897016 | –8.058 | AL-398/12677067 | –8.493 |
| AG-205/12085061 | –8.663 | AO-022/43235300 | –8.865 |
| AG-650/41069129 | –8.358 | AO-081/15245021 | –8.427 |
| AG-670/40968109 | –8.042 | AO-081/41756050 | –8.725 |
| AG-690/08355055 | –8.032 | AO-554/14700005 | –8.525 |
| AG-690/11763097 | –9.087 | AP-064/41684917 | –8.277 |
| AG-690/13507628 | –8.457 | AP-064/41684959 | –8.029 |
| AG-690/40753764 | –8.037 | AP-064/42646016 | –8.875 |
| AG-690/40753996 | –8.176 | AP-124/40904362 | –8.051 |
| AH-487/15274490 | –8.710 | AP-970/13521174 | –8.685 |
| AJ-030/14523202 | –8.317 | AP-970/42898518 | –8.898 |
| AJ-292/13489193 | –8.015 | AR-360/42760471 | –8.262 |
| AK-778/43464919 | –8.494 | AT-057/43469634 | –8.515 |
Biological activity of the compounds
We first tested the activity of the wildtype Mpro, and found that its Km (119.5 μM) and Vmax (55.61 nM/s) are close to the values reported in a previous study (Li et al., 2020) (Figure S2). The activity of the 30 compounds were then tested at two concentrations of 10 μM (Figure S3) and 100 μM. At 10 μM, one of the compounds, AG-690/13507628, was found to inhibit the protein activity by 70%, and its IC50 value was determined to be 9.457 μM (Figure S4a). The IC50 of the positive reference Ebselen was 0.786 μM in our assay (Figure S4b).
Figure 2.

The IC50 of the four active compounds (95% Confidence Interval). Each experiment was repeated three times.
Figure 3.

Compound AG-690/13507628, AG-690/13508164, and AG-690/13507724 have little cytotoxicity on HUVEC cells at 100 µM. HUVEC cells were treated with the indicated compounds for 72 h and their cytotoxicity was determined by CCK8 assay.
Figure 4.

The free energy contributions of the dominant residues of Mpro in binding to four compounds.
We further purchased 30 compounds that share a similar skeleton with AG-690/13507628 in Specs and performed additional screening at concentrations of 10 μM (Figure S5) and 50 μM. Six of the compounds (including AG-690/13507628) showed detectable activities. The IC50 values of four out of the six compounds were determined as AG-690/13507628 (2.806 μM), AG-690/13507724 (2.961 μM), AG-690/13507750 (19.19 μM), AG-690/13508164 (16.905 μM) (Figure 2; Table 2). The decreased IC50 of AG-690/13507628 from 9.457 μM to 2.806 μM in the second measurement is likely due to different experimental setup.
Figure 5.

The stacked bars graph for free energy contributions of the dominant residues of Mpro in binding to four compounds.
Table 2.
The structures and IC50 of the four active compounds.
| Compound | Specs ID | 2D-structure | IC50 (μM) |
|---|---|---|---|
| 1 | AG-690/13507628 |
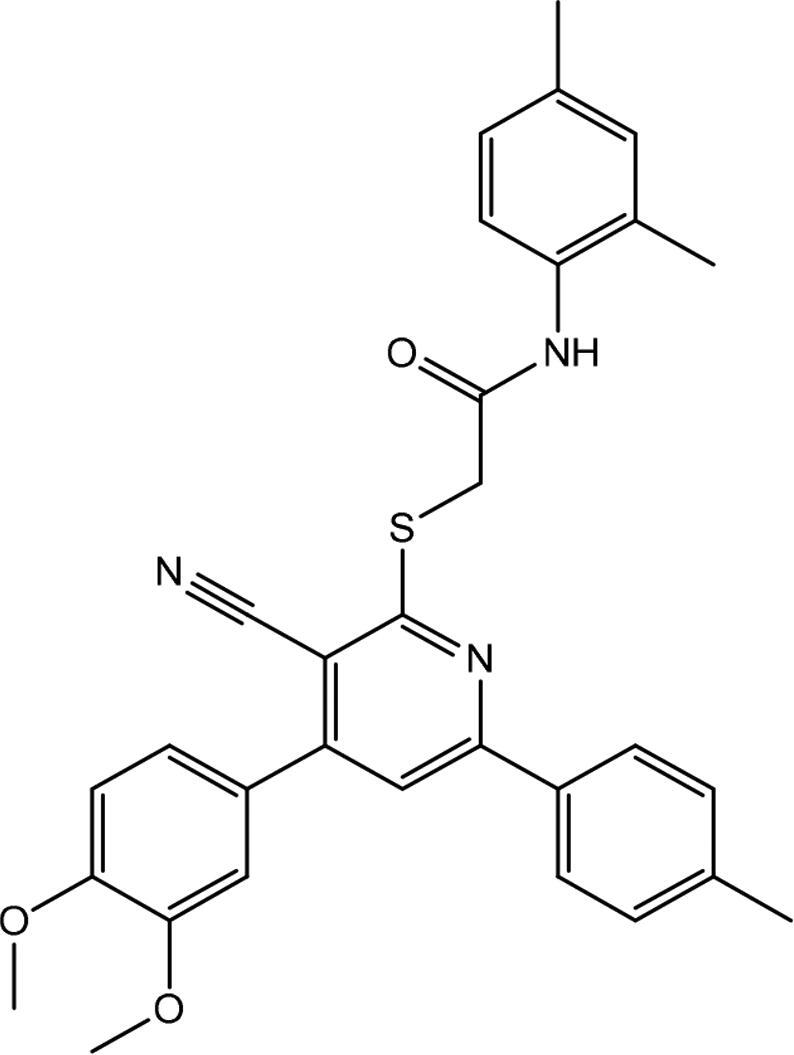
|
2.806 ± 0.642 |
| 2 | AG-690/13507724 |
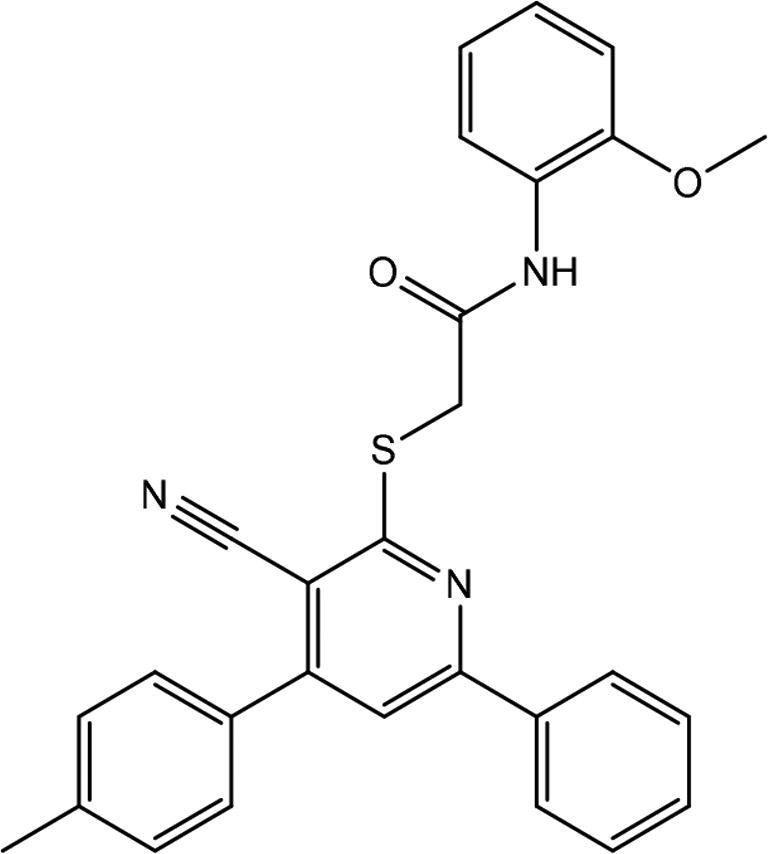
|
2.961 ± 0.636 |
| 3 | AG-690/13507757 |
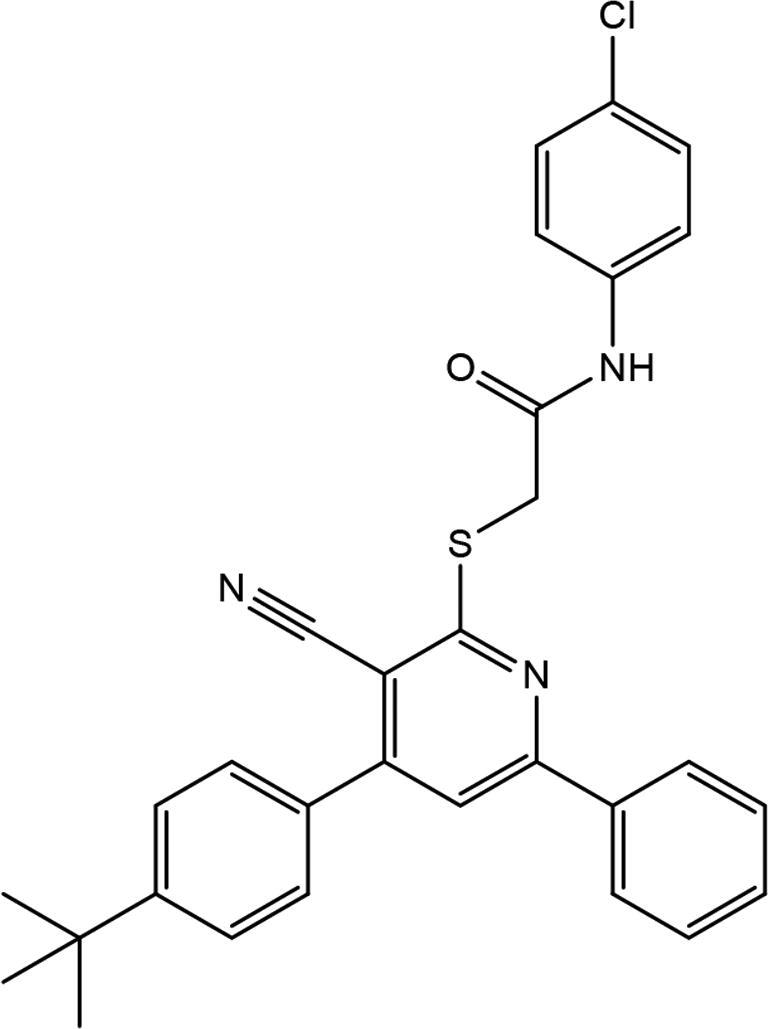
|
19.19 ± 1.94 |
| 4 | AG-690/13508164 |
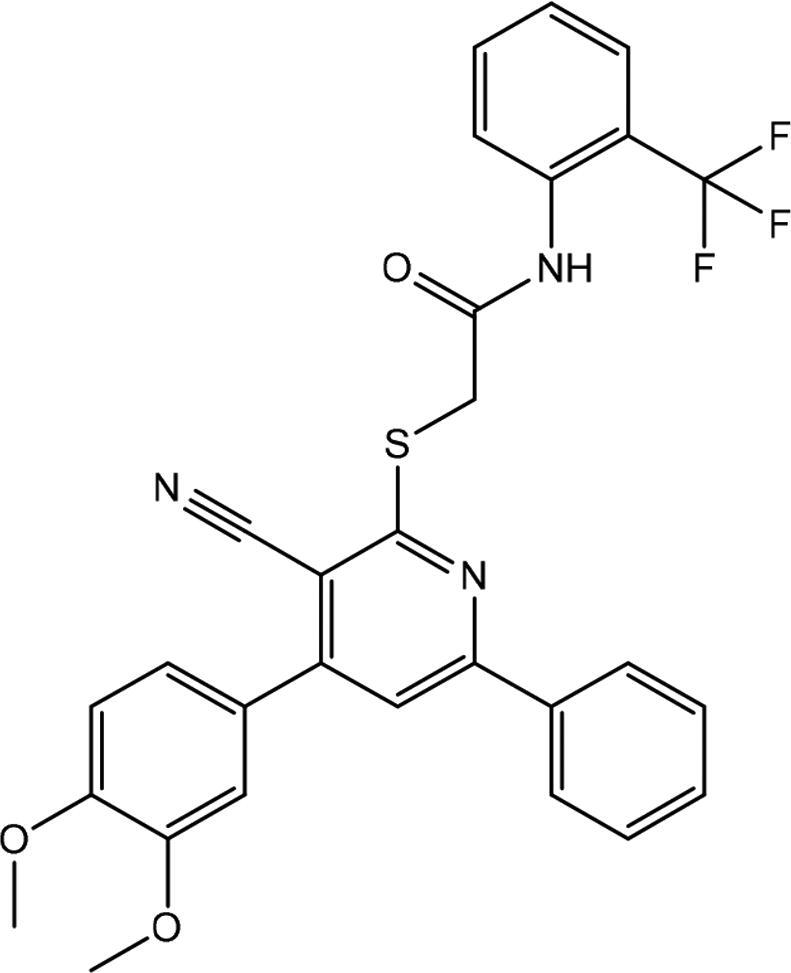
|
16.905 ± 2.795 |
Cytotoxicity
We next determined the cytotoxicity of the four active compounds (Figure 3). Due to different solubility of the compounds, the concentration of dimethyl sulfoxide (DMSO) and dimethylformamide (DMF) was 10‰ for AG-690/13507724 and 4‰ for the other three compounds. Compound AG-690/13507628, AG-690/13508164, and AG-690/13507724 have little effect on cell viability, but compound AG-690/13507757 has a greater inhibitory effect on cell growth. Considering that the IC50 of AG-690/13507628 and AG-690/13507724 are 2.806 μM and 2.961 μM, respectively, these two compounds may hold promise for further optimizations.
Computational alanine scanning analysis
We performed MD simulations for these four compounds and details of the simulations are described in the Method section. Each Mpro-compound complex was simulation for 100 ns with three independent replicas. The RMSDs of the compounds are between 2 to 4 Å in the simulations, suggesting they are overall stable (Figure S6). Our computational alanine scanning analysis shows that residues His41, Met49, Leu27 and Thr25 contribute dominantly to the binding as shown in Figures 4 and 5. These four residues are conservative in binding to all these four compounds, indicating similar binding mechanism.
Figure 6.

Interaction diagram of pocket residues (within 4 Å) with two best compounds AG-690/13507628 (a, c) and AG-690/13507724 (b, d). The figure was prepared with PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC) and LigPlot (Laskowski & Swindells, 2011).
Discussion
To gain insights of the interactions between the active compounds and Mpro, we used Autodock vina (Trott & Olson, 2010) to generate the complex structures of two compounds AG-690/13507628 and AG-690/13507724, whose complex structures was extracted from the last frame in the 100 ns trajectory, that have better activity than the other two (Figure 6). These two compounds form specific interactions with four key residues including His41, Met49, Leu27 and Thr25. The hydrophilic cyano group is exposed to the solvent, together with the amide group, form a lever-like structure so that the benzene ring connected to the 6th position of the pyridine ring is inserted stably into the deep cavity. The benzene ring fits into the groove between Met165 and His41, and form π-π stacking interaction with His41.
Decomposition of the binding free energy of AG-690/13507628 using ASIE reveals six hot-spot residues: His41, Met49, Met165, Leu27, Thr25 and Gln189 (Table 3), which is in agreement with the structural analysis above. Among these residues, His41 is the dominant contributing residue with binding energy contribution great than 1.5 kcal/mol, and the main interaction being the van der Waals energy.
Table 3.
Residue-specific binding free energy of compound AG-690/13507628 from ASIE.
| Residue | ΔΔEvdw | ΔΔEele | ΔΔGB | ΔΔNP | ΔΔH | –TΔΔS | ΔΔG |
|---|---|---|---|---|---|---|---|
| 189GLN | –3.81 | –1.35 | 1.60 | –0.28 | –3.84 | 1.03 | –2.81 |
| 41HIE | –2.50 | –0.97 | 1.08 | –0.10 | –2.49 | 0.64 | –1.84 |
| 49MET | –2.40 | –0.06 | 0.68 | –0.17 | –1.94 | 0.80 | –1.14 |
| 27LEU | –0.82 | –0.03 | –0.03 | –0.06 | –0.94 | 0.08 | –0.87 |
| 165MET | –1.26 | –0.42 | 0.57 | –0.07 | –1.18 | 0.62 | –0.56 |
| 25THR | –0.98 | –0.13 | 0.36 | –0.14 | –0.89 | 0.37 | –0.51 |
| Total | –18.30 | –4.31 | 6.62 | –1.09 | –17.09 | 5.52 | –11.57 |
Only residues with ΔΔG < –0.50 kcal/mol are listed and all value are in kcal/mol.
Furthermore, the activity data obtained in this study also allows side-by-side comparison of active and non-active compounds to reveal key determinants of binding strength. To this end, we first compare in detail two compounds AG-690/13507757 (IC50 = 19.19 μM) and AG-690/13507750 (non-active) that have high structural similarity but distinct activities (Figure 7a; Table S1). On the benzene ring attached to the amide, the chlorine group is closer to the hydroxyl group of Thr25 (2.5 Å) when it is in the para-position (AG-690/13507757), which facilitates the formation of hydrogen bonds with the hydroxyl group. In contrast, when the chlorine group is in the inter-position (AG-690/13507750), it is away from Thr25 (3.0 Å), therefore decrease the inhibitory activity.
Figure 7.

Comparison of active and non-active compounds. (a) AG-690/13507757 (active, yellow sticks) and AG-690/13507750 (non-active, green sticks). (b) AG-690/13507724 (active, pink sticks), AG-690/13507754 (non-active, blue sticks) and AG-690/13507732(nonactive, red sticks).
In AG-690/13507724 (IC50 = 2.961 μM), when the substituent adjacent to the benzene ring attached to the amide is methoxy, it is easy to have a stable polar interaction with the carboxamide of Asn142 (2.6 Å). However, when the substituent is a benzene ring (AG-690/13507754, non-active, Figure 7b; Table S2), it is prone to collide with the surrounding amino acids due to the large size of the benzene ring. In the non-active compound AG-690/13507732, the substituent is a trifluoromethyl group (Figure 7b; Table S1), which cannot interact strongly with Asn142 in a stable manner due to its increased distance with Asn142 (3.1 Å).
Conclusion
Although FDA has issued an emergency use authorization on vaccines against COVID-19, drugs for anti-SARS-Cov2 are still extremely important (Lau et al., 2020). In this study, we discovered four inhibitors of SARS-Cov2 Mpro. To assess the structural novelty of these inhibitors, we used Rdkit to calculate the Morgan fingerprint similarity of these four inhibitors with other known inhibitors, and the highest score was only 0.513 (Table S2). Three of these compounds have low toxicity and several modifiable sites for further optimization. For example, the three benzene rings are all easily modifiable sites and have a considerable modification space: the phenyl ring at the 6th position of the pyridine ring may be replaced with hydrophobic group to form hydrophobic interactions with residue Met165. We have additionally predicted the ADMET properties of the four compounds using ADMETlab 2.0 (Xiong et al., 2021) and included the data in Table S3. Our compounds show good absorption, distribution and Excretion properties, but less desirable LogS, LogD and LogP values, indicating directions of further optimization. These results may provide a basis for further development of anti-SARS-CoV-2 drugs.
Supplementary Material
Acknowledgment
We thank NYU Shanghai and the ECNU Multifunctional Platform for Innovation (001) for providing us computer time.
Funding Statement
This work was supported by the National Natural Science Foundation of China (Grant 91753103, 31700646, 21933010), and the Natural Science Foundation of Shanghai (Grant 19ZR1473600).
References
- Andersen, H. C. (1983). Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. Journal of Computational Physics, 52(1), 24–34. 10.1016/0021-9991(83)90014-1 [DOI] [Google Scholar]
- Ben-Shalom, I. Y., Pfeiffer-Marek, S., Baringhaus, K. H., & Gohlke, H. (2017). Efficient approximation of ligand rotational and translational entropy changes upon binding for use in MM-PBSA calculations. Journal of Chemical Information and Modeling, 57(2), 170–189. 10.1021/acs.jcim.6b00373 [DOI] [PubMed] [Google Scholar]
- Coronavirus disease (COVID-19) Situation Report – 161 . (2020). WHO. [Google Scholar]
- Duan, L., Liu, X., & Zhang, J. Z. (2016). Interaction entropy: A new paradigm for highly efficient and reliable computation of protein-ligand binding free energy. Journal of the American Chemical Society, 138(17), 5722–5728. 10.1021/jacs.6b02682 [DOI] [PubMed] [Google Scholar]
- Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., Repasky, M. P., Knoll, E. H., Shelley, M., Perry, J. K., Shaw, D. E., Francis, P., & Shenkin, P. S. (2004). Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry, 47(7), 1739–1749. 10.1021/jm0306430 [DOI] [PubMed] [Google Scholar]
- Genheden, S., & Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery, 10(5), 449–461. 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil, C., Ginex, T., Maestro, I., Nozal, V., Barrado-Gil, L., Cuesta-Geijo, M. A., Urquiza, J., Ramirez, D., Alonso, C., Campillo, N. E., & Martinez, A. (2020). COVID-19: Drug targets and potential treatments. Journal of Medicinal Chemistry, 63(21), 12359–12386. 10.1021/acs.jmedchem.0c00606 [DOI] [PubMed] [Google Scholar]
- Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., Wang, L., Lupyan, D., Dahlgren, M. K., Knight, J. L., Kaus, J. W., Cerutti, D. S., Krilov, G., Jorgensen, W. L., Abel, R., & Friesner, R. A. (2016). OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. Journal of Chemical Theory and Computation, 12(1), 281–296. 10.1021/acs.jctc.5b00864 [DOI] [PubMed] [Google Scholar]
- Hattori, S. I., Higashi-Kuwata, N., Hayashi, H., Allu, S. R., Raghavaiah, J., Bulut, H., Das, D., Anson, B. J., Lendy, E. K., Takamatsu, Y., Takamune, N., Kishimoto, N., Murayama, K., Hasegawa, K., Li, M., Davis, D. A., Kodama, E. N., Yarchoan, R., Wlodawer, A., … Mitsuya, H. (2021). A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nature Communications, 12(1), 668. 10.1038/s41467-021-20900-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld, R. (2014). From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. The FEBS Journal, 281(18), 4085–4096. 10.1111/febs.12936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, T., Wang, J., Li, Y., & Wang, W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of Chemical Information and Modeling, 51(1), 69–82. 10.1021/ci100275a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- Lau, S. Y., Wang, P., Mok, B. W., Zhang, A. J., Chu, H., Lee, A. C., Deng, S., Chen, P., Chan, K. H., Song, W., Chen, Z., To, K. K., Chan, J. F., Yuen, K. Y., & Chen, H. (2020). Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerging Microbes & Infections, 9(1), 837–842. 10.1080/22221751.2020.1756700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, T. W., Cherney, M. M., Huitema, C., Liu, J., James, K. E., Powers, J. C., Eltis, L. D., & James, M. N. (2005). Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. Journal of Molecular Biology, 353(5), 1137–1151. 10.1016/j.jmb.2005.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., Li, X., Huang, Y. Y., Wu, Y., Liu, R., Zhou, L., Lin, Y., Wu, D., Zhang, L., Liu, H., Xu, X., Yu, K., Zhang, Y., Cui, J., Zhan, C. G., Wang, X., & Luo, H. B. (2020). Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proceedings of National Academy of Sciences of the United States of America, 117, 27381–27387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linlin Zhang, D. L., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science,368(6489), 409-412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P., Liu, H., Sun, Q., Liang, H., Li, C., Deng, X., Liu, Y., & Lai, L. (2020). Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. European Journal of Medicinal Chemistry, 206, 112702. 10.1016/j.ejmech.2020.112702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., Peng, L., Zhou, Y., Zhang, Y., & Zhang, J. Z. H. (2018). Computational alanine scanning with interaction entropy for protein-ligand binding free energies. Journal of Chemical Theory and Computation, 14(3), 1772–1780. 10.1021/acs.jctc.7b01295 [DOI] [PubMed] [Google Scholar]
- Ma, C., Sacco, M. D., Hurst, B., Townsend, J. A., Hu, Y., Szeto, T., Zhang, X., Tarbet, B., Marty, M. T., Chen, Y., & Wang, J. (2020). Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Research, 30(8), 678–692. 10.1038/s41422-020-0356-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova, I., & Kollman, P. A. (2000). Combined molecular mechanical and continuμM solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspectives in Drug Discovery and Design, 18(1), 113–135. 10.1023/A:1008763014207 [DOI] [Google Scholar]
- Onufriev, A., Bashford, D., & Case, D. A. (2004). Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins, 55(2), 383–394. 10.1002/prot.20033 [DOI] [PubMed] [Google Scholar]
- Petukh, M., Li, M., & Alexov, E. (2015). Predicting binding free energy change caused by point mutations with knowledge-modified MM/PBSA method. PLoS Computational Biology, 11(7), e1004276. 10.1371/journal.pcbi.1004276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, J., Li, Y.-S., Zeng, R., Liu, F.-L., Luo, R.-H., Huang, C., Wang, Y.-F., Zhang, J., Quan, B., Shen, C., Mao, X., Liu, X., Sun, W., Yang, W., Ni, X., Wang, K., Xu, L., Duan, Z.-L., Zou, Q.-C., … Yang, S. (2021). SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science (New York, N.Y.), 371(6536), 1374–1378. 10.1126/science.abf1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, L. Q., Yan, Y. N., Sun, Z. X., Song, J. N., & Zhang, J. Z. H. (2018). Interaction entropy for computational alanine scanning in protein-protein binding. Wiley Interdisciplinary Reviews: Computational Molecular Science, 8(2), e1342. 10.1002/wcms.1342 [DOI] [Google Scholar]
- Salomon-Ferrer, R., Case, D. A., & Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science, 3(2), 198–210. 10.1002/wcms.1121 [DOI] [Google Scholar]
- Song, J., Qiu, L., & Zhang, J. Z. H. (2018). An efficient method for computing excess free energy of liquid. Science China Chemistry, 61(1), 135–140. 10.1007/s11426-017-9106-3 [DOI] [Google Scholar]
- Struyf, T., Deeks, J. J., Dinnes, J., Takwoingi, Y., Davenport, C., Leeflang, M. M., Spijker, R., Hooft, L., Emperador, D., Dittrich, S., Domen, J., Horn, S. R. A., Van den Bruel, A., & Cochrane, C.-D. T. A. G. (2020). Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19 disease. Cochrane Database Syst Rev, 7, CD013665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.0 , Schrödinger, LLC [Software].
- Thiel, V., Ivanov, K. A., Putics, Á., Hertzig, T., Schelle, B., Bayer, S., Weißbrich, B., Snijder, E. J., Rabenau, H., Doerr, H. W., Gorbalenya, A. E., & Ziebuhr, J. (2003). Mechanisms and enzymes involved in SARS coronavirus genome expression. The Journal of General Virology, 84(Pt 9), 2305–2315. 10.1099/vir.0.19424-0 [DOI] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D., Hu, B., Hu, C., Zhu, F., Liu, X., Zhang, J., Wang, B., Xiang, H., Cheng, Z., Xiong, Y., Zhao, Y., Li, Y., Wang, X., & Peng, Z. (2020). Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA, 323(11), 1061–1069. 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- Wenhao, D, Zhang, B., Jiang, X.-M., Su, H., Li, J., Zhao, Y., Xie, X., Jin, Z., Peng, J., Liu, F., Li, C., Li, Y., Bai, F., Wang, H., Cheng, X., Cen, X., Hu, S., Yang, X., Wang, J., … Liu, X. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science, 368(6497), 1331–1335. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, G., Wu, Z., Yi, J., Fu, L., Yang, Z., Hsieh, C., Yin, M., Zeng, X., Wu, C., Chen, X., Hou, T., & Cao, D. (2021). ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Research, 49(W1), W5–W14. https://doi.org/10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, X., Yu, H., Yang, H., Xue, F., Wu, Z., Shen, W., Li, J., Zhou, Z., Ding, Y., Zhao, Q., Zhang, X. C., Liao, M., Bartlam, M., & Rao, Z. (2008). Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. Journal of Virology, 82(5), 2515–2527. 10.1128/JVI.02114-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Y., Yang, M., Ji, C. G., & Zhang, J. Z. H. (2017). Interaction entropy for computational alanine scanning. Journal of Chemical Information and Modeling, 57(5), 1112–1122. 10.1021/acs.jcim.6b00734 [DOI] [PubMed] [Google Scholar]
- Zhang, C.-H., Stone, E. A., Deshmukh, M., Ippolito, J. A., Ghahremanpour, M. M., Tirado-Rives, J., Spasov, K. A., Zhang, S., Takeo, Y., Kudalkar, S. N., Liang, Z., Isaacs, F., Lindenbach, B., Miller, S. J., Anderson, K. S., & Jorgensen, W. L. (2021). Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Central Science, 7(3), 467–475. 10.1021/acscentsci.1c00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Liu, X., Zhang, Y., Peng, L., & Zhang, J. Z. H. (2018). Residue-specific free energy analysis in ligand bindings to JAK2. Molecular Physics, 116(19–20), 2633–2641. 10.1080/00268976.2018.1442596 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.