

Abstract
Type 2 diabetes mellitus (T2DM) associated non-alcoholic fatty liver disease (NAFLD) is the fourth-leading cause of death. Hyperglycemia induces various complications, including nephropathy, cirrhosis and eventually hepatocellular carcinoma (HCC). There are several etiological factors leading to liver disease development, which involve insulin resistance and oxidative stress. Free fatty acid (FFA) accumulation in the liver exerts oxidative and endoplasmic reticulum (ER) stresses. Hepatocyte injury induces release of inflammatory cytokines from Kupffer cells (KCs), which are responsible for activating hepatic stellate cells (HSCs). In this review, we will discuss various molecular targets for treating chronic liver diseases, including homeostasis of FFA, lipid metabolism, and decrease in hepatocyte apoptosis, role of growth factors, and regulation of epithelial-to-mesenchymal transition (EMT) and HSC activation. This review will also critically assess different strategies to enhance drug delivery to different cell types. Targeting nanocarriers to specific liver cell types have the potential to increase efficacy and suppress off-target effects.
Keywords: Liver fibrosis, diabetes, NAFLD, inflammation, cirrhosis, hepatocellular carcinoma of microRNA in n
Graphical abstract
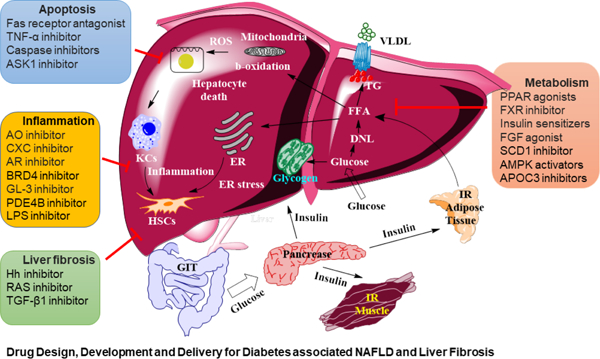
INTRODUCTION
Cirrhosis is a growing major health problem in the world. In the United States, the number of adults diagnosed with liver disease was 4.5 million and the number of deaths due to these complications was 44,358 in 2020 [1]. Around 50% of cirrhosis cases arise from non-alcoholic fatty liver disease (NAFLD) and excessive alcohol consumption [2]. Moreover, about 20% of individuals with cirrhosis progress to hepatocellular carcinoma (HCC), which is the third leading cause of cancer related deaths [3]. Obesity and its metabolic consequences are considered the major contributors to fat accumulation in the hepatocytes and are termed hepatic steatosis [4]. Diabetes and liver fibrosis are interrelated and promote each other. Under normal conditions, insulin increases the glucose transporter (GLUT) receptor-mediated blood glucose uptake into hepatocytes, muscles, and adipocytes. In hepatic steatosis, the glucose uptake capacity of hepatocytes is compromised, and blood glucose levels stay high despite enough insulin. Therefore, obesity in the presence of hepatic steatosis also induces insulin resistance and type 2 diabetes mellitus (T2DM) [5]. Excessive fat induces oxidative stress in the liver and causes hepatocyte damage, resulting in the activation of Kuffer cells (KCs) and the overproduction of inflammatory cytokines. Fibrosis is developed in response to tissue injury and is accompanied by extracellular matrix (ECM) accumulation which is secreted mainly by hepatic stellate cells (HSCs) [6]. Conversely, Long-standing diabetes predisposes patients to severe micro-and macrovascular complications in multiple organ systems. These include renal fibrogenesis, diabetic retinopathy, and cardiovascular complications such as hypertension, stroke, and coronary artery disease [7]. Therefore, understanding the pathogenesis of diabetes-associated with liver fibrosis is a timely endeavor as this will lend support to prevent and treat diabetes and hepatic complications.
Lipotoxicity and inflammatory reactions promote NAFLD progression to non-alcoholic steatohepatitis (NASH). Before the establishment of NASH, there are three major intermediate events: (i) lipid metabolism, including de novo lipid synthesis (DNL) and β-oxidation, (ii) inflammation, and (iii) hepatocellular apoptosis [8]. At some stages of non-alcoholic liver injury, fat accumulation in the liver plays a protective role. However, subsequent initiation of inflammatory reactions triggered by second insults which may lead to NASH progression [9]. To prevent it, several pharmacological agents have been evaluated, including insulin sensitizers, anti-apoptotic, anti-inflammatory and antifibrotic agents. However, current therapeutic approaches have shown a lack of efficacy or an unfavorable safety profile, making NASH an untreatable disease.
High levels of free fatty acids (FFA) in hepatocytes promote mitochondrial β-oxidation. Overburden of this metabolic pathway results in an imbalanced fatty acid metabolism and can cause mitochondrial dysfunction increasing oxidative stress and steatosis [10]. Formation of reactive oxygen species (ROS) induces lipid peroxidation, which can alter the mitochondrial DNA. Also, ROS bind to mitochondrial proteins and inhibit the electron transfer respiratory chain. Therefore, NASH patients display mitochondrial lesions and reduced activity of respiratory chain complexes [11]. Liver inflammation is fueled by excess production of inflammatory cytokines, including TNF-α, interleukin (IL)-1 and IL-6 by KCs that induce the progression of NAFLD to NASH. Hepatic inflammation activates HSCs, which undergo proliferation and enter the contractility phase. Moreover, their myofibroblast forms loose vitamin A content and start secreting excess collagen, thereby promoting excessive ECM deposition in the liver [6].
Although NASH is one of the most common liver diseases, there is still no effective therapeutic approaches due to complex etiology, silent disease features, lack of sensitive therapeutic evaluation methods, and inefficient drug delivery systems. Efforts are being made to address the issues related to lipid homeostasis, hepatic inflammation, and hepatocyte damage. Some of the newly discovered compounds with diverse chemical structures and mechanisms of action show a decline in fibrosis progression and decrease liver damage score in preclinical studies as well as in initial phase of clinical trials. However, this interesting field is still nascent and requires more intense drug discovery efforts to bear fruit with regulatory agency approvals. This review critically discusses novel discoveries in the settings of NAFLD treatment. Since some of these drugs may have adverse effects to other organs, an effective drug delivery system is needed to enhance their therapeutic effects and reduce toxic side-effects. Therefore, we focus on the development of nanocarriers for improved delivery of different therapeutic agents. Further, site-specific delivery of these anti-diabetic and anti-fibrotic drugs can be achieved by decorating the nanocarrier surface with different targeting ligands. This review will increase the awareness and understanding of critical challenges in the field not only to drug delivery researchers studying drug delivery but also to those working in drug discovery for liver fibrosis.
2. ROLE OF DIABETES AND METABOLIC DYSFUNCTION ON LIVER FIBROSIS
NAFLD is a metabolic disease caused by dysregulated glucose and lipid metabolism. NAFLD has a strong association with T2DM, as more than 90% of obese patients with T2DM also have NAFLD [12]. Obesity and high caloric diet contribute to the development of insulin resistance, which promotes hepatic lipid accumulation, lipotoxicity, liver injury, and inflammation. NAFLD is closely associated with insulin resistance and its prevalence among patients with T2DM is 55.5%, which is twice the general population’s prevalence without T2DM. The prevalence of NASH and liver fibrosis among patients with both T2DM and NAFLD is 67.3% and 17.02%, respectively. Moreover, the mortality rate among patients with T2DM and NAFLD is 585 per 10,000 people, significantly higher than other chronic liver diseases [13].
Many pathways are involved in insulin resistance in T2DM, and they have close association with NAFLD. Therefore, there is a great interest to discuss the link between lipid accumulation in the liver and lipid-induced hepatic insulin resistance. After a meal, high glucose in the blood is sensed by β-cells of the pancreas, which in turn results in insulin secretion. Insulin receptors are mainly found on muscles, adipose, and hepatic cells. Insulin binding to the insulin receptor tyrosine kinase (IRTK) promotes phosphorylation of insulin receptor substrates (IRS) and IRS2 [14]. Phosphorylation of IRS2 on tyrosine residue brings conformational changes and generates binding sites for phosphatidylinositol-3-OH kinase (PI3K). The binding of PI3K to IRS2 converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which in turn phosphorylates and activates PIP3 dependent kinase 1 (PDK1). Phosphorylated PDK1 and activates protein kinase B, commonly known as AKT. Activation of AKT brings two significant changes in the cells: inhibition of protein AS160 thereby resulting in translocation of glucose transport receptor 4 (GLUT4) to the cell membrane. GLUT4 mediates insulin-stimulated glucose uptake by skeletal muscle, heart, and adipose tissues [15]. However, in the liver, glucose is transported by GLUT2, irrespective of insulin resistance. Hyperglycemia in T2DM directly causes high hepatic uptake of glucose, and initiation of glycogen synthesis by inhibiting glycogen synthase kinase 3 beta (GSK3β), a natural inhibitor of glycogen synthase (GS) [16]. In the liver, glucose is either used for glycolysis, stored in the cells as glycogen, or feeds into de novo lipid (DNL) synthesis. High hepatic glucose level also causes hepatotoxicity, in part due to increased glucose metabolism. DNL synthesis in the liver further aggravates the elevated levels of FFA and TG. Both FFA and glucose are oxidized through TCA cycle during ATP synthesis in the mitochondria. The leaky electron transport chains lead to ROS production, which is the source of inflammation, cytotoxicity, and fibrosis. AKT activation also inhibits gluconeogenesis by decreasing phosphorylation and nuclear exclusion of the fork-head box protein FOXO1 [17].
The development of NAFLD is strongly associated with hepatic lipid content, which is affected by the balance between lipid uptake, DNL, and lipid export mechanisms. Hepatic lipid uptake is a function of substrate availability and transport into the hepatocytes. Adipose tissue lipolysis generates an excess of fatty acid in the circulation that promotes NAFLD and hepatic insulin resistance. Mouse models with liver or muscle-specific overexpression of lipoprotein lipase (LPL) have been shown to promote lipid accumulation and insulin resistance in these organs [18]. Further, DNL is threefold higher in patients with NAFLD than normal, representing a key feature of fatty livers. Fasted patients with obesity and NAFLD account for ~26% of hepatocellular triglycerides (TGs). Knockdown of the genes encoding the acetyl CoA carboxylases ACC1 and ACC2, which are crucial in the regulation of DNL and lipid oxidation, reduced liver triacylglycerol and protected mice from lipid-induced hepatic insulin resistance. DNL can be stimulated by both insulin via sterol regulatory element-binding protein 1c (SREBP‑1c) and glucose via carbohydrate response element-binding protein (ChREBP) [19]. Finally, the hepatocellular FFA pool can be further increased by impaired export of VLDL cholesterol in insulin-resistant patients with NASH. Upon meal ingestion, humans with insulin resistance exhibit reduced muscle glycogen synthesis, doubling of both liver TG levels and hepatic DNL. These observations indicate that muscle insulin resistance shifts post-prandial energy storage from muscle glycogen to hepatic lipid storage [20].
Phospholipases metabolize TGs present in high concentration in the liver cells to produce to diacylglycerol (DAG) as an intermediate product, which is a key mediator of lipid-induced hepatic insulin resistance. DAG increased concentration in the human liver is correlated well with hepatic insulin resistance [21]. In an HFD rat model, the hepatic glycogen synthetic and gluconeogenesis inhibitory capacity of insulin is diminished due to increased hepatic DAG content. DAG increases translocation of protein kinase-Cε (PKCε) isoform to the cell membrane. To date, PKCε has been shown to be involved in diverse cellular pathways including tumor-promoting effects of phorbol esters, in protection against cardiac ischemia-reperfusion injury, in regulating cell–cell junctions, in immune cell activation and in channel regulation [22]. In context to insulin resistance, PKCε phosphorylates the IRS2 protein on serine residues and thus, inhibits the insulin signaling cascade. Therefore, hepatic DAG content is a direct predictor of hepatic insulin resistance in obese humans.
Hepatic lipids undergo oxidation mainly in mitochondria. Mitochondrial FFA entry is controlled via carnitine O‑palmitoyltransferase 1 (CPT1), which is inhibited by insulin, malonyl-CoA, and fatty acyl CoA. CPT1 activates peroxisome proliferator-activated recep-tor alpha (PPARα), thereby stimulating β-oxidation of fatty acids [23]. Of note, reduced muscle mitochondrial function positively relates to liver fat content. Patients with T2DM also exhibit reductions in hepatocellular ATP concentrations and ATP synthase flux. Hepatic ATP synthesis correlates directly well with both peripheral and hepatic insulin sensitivity but inversely with body fat content. Obese individuals with or without steatosis have up to fivefold higher maximal mitochondrial respiration rates than lean individuals [24]. This excessive lipid overloading impairs antioxidant capacity and accelerate oxidative stress with mitochondrial leakage, resulting in NASH and aggravated insulin resistance.
Inflammation pathway also interferes with insulin signaling pathway. Adipose tissue has emerged as a major site of inflammation in obesity-related disorders. The proinflammatory cytokine levels of adipose tissues are significantly higher in the obese population [25]. Increased concentration of proinflammatory cytokines including IL-1β, IL-6, and TNFα and an increase in macrophage as well as invariant natural killer T (iNKT) numbers are common in adipose tissue of obese individuals. Whereas some cytokines such as IL-37, IL-10, IL-5, and interferon (IFN) -γ are characteristics of lean person’s adipose tissue [26]. Endoplasmic reticulum (ER) dysfunction is another commonly observed phenomenon in NASH [27, 28]. Abnormal protein folding within the ER creates stress stimuli, it starts unfolded protein response (UPR) through activation of 3 pathways involving 1 α (IRE1α), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 α (ATF6α) [29]. ER stress triggers inflammation during adipose tissue expansion and liver steatosis. Further, by virtue of excessive insulin production, apoptotic pancreatic β cells recruit monocytes to both local and neighboring tissues. Besides, the elevated level of FFA directly activates the proinflammatory response. TNF-α activates its receptor and leads to insulin resistance via phosphorylation of IRS1, resulting in increased lipolysis and the fatty acid influx into the liver, which exacerbates NAFLD [30]. Triggering of TNF-α receptor induces downstream inflammatory pathway via NF-κB signaling. NF-κB activation depends on the inhibitor of nuclear factor-κB kinase (IKK), which inhibits the activation of target genes. Inhibition of IKK with siRNA reversed insulin resistance in animal models [31].
2.1. Genetic risk factors
Although insulin resistance and NAFLD coexist and share common pathophysiological factors, each patient is unique in terms of his/her age, ethnicity, culture, and genetics that potentially affect treatment outcomes [32]. An inherent clarity in DNA sequencing has enabled us to interpret the patient’s disposition to disease and analyze treatment options. Certain genetic variations including derived from genes involved in lipid biology, including Patatin-like phospholipase domain-containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), glucokinase regulator (GCKR), membrane-bound O-acyltransferase domain-containing protein 7 (MBOAT7), and 17β-hydroxysteroid dehydrogenase type 13 (HSD17B13) are known to influence the progression of NAFLD, NASH and cirrhosis [33]. Further, aberrant DNA methylation also promotes NAFLD. In humans, the single nucleotide polymorphism (SNP) rs56225452, putatively representing a gain-of-function mutation in the fatty acid transporter protein 5 FATP5 promoter, was associated with insulin resistance and NAFLD [34]. Mice lacking FATP5 are protected from diet induced NAFLD and hepatic insulin resistance [35]. Apolipoprotein C3 (APOC3), which inhibits lipoprotein lipase (LPL) and hepatic lipase (HL), is associated with hypertriglyceridemia when mutated. As a result, the livers of individuals with APOC3 variants take up high amount of lipid from chylomicrons, remnants of lipoprotein particles, predisposing them to NAFLD hepatic insulin resistance [36].
Such variation needs to be evaluated in combination with clinical and environmental factors to personalize either drug choice or drug dose in individual patients [37]. The immense appreciation is to identify some of these critical “drivers” involved and pinpointing novel potential candidate targets for therapy.
2.2. Epigenetic risk factors
Lifestyle, epigenetic gene modification, and other preexisting conditions may affect energy homeostasis and contribute to the progression of T2D as well as NAFLD [38]. Every single step involved in the gene expression process has several controlling transcriptional or post-transcriptional factors. Although transcriptional control of gene is deciding in its expression, about 60% of all protein-coding genes are regulated post-transcriptionally. Transcription attenuation, alternative splicing, and targeting of messenger RNA by microRNAs (miRNAs) are some key regulation processes. miRNAs are 21–23 nt endogenous noncoding RNA molecules which can bind with mRNA based on “seed” sequence, therebyresulting in its transcription repression or degradation [6]. miRNA does not require complete complementarity to target mRNA, and single miRNA regulate a set of mRNAs, and a single mRNA can be targeted by several miRNAs. The differential expression of miRNAs has been linked to development of NAFLD and related disease [39]. Therefore, based on their expression profile, different diagnosis as well treatment strategies were developed and now are in early phase clinical trials. Furthermore, miRNA-based therapeutic targeting may enable individualized therapeutic management for liver fibrosis patients, paving the way for precision medicine.
The local or systemic inflammation by various immune cells also fuel in the development of NAFLD progression. Cytokines, and inflammatory molecules notably IL-1β, IL-17A, IL-6, TNF-α, and LPS activate multiple proinflammatory cascades that in unison promote liver injury. TNF-α released by KCs as well as generated oxidative stress mediate liver injury in NAFLD. KCs get activated in response to hepatocyte insult and gut-derived endotoxins (i.e., LPS), leading to excessive inflammatory cytokine production [40]. Moreover, hepatocytes can also secrete TNF-α in response to an increased supply of FFA by adipose tissue macrophages of obese individuals with T2DM. Thus, environmental risk factors influence the severity, levels of oxidative stress, the magnitude of the immune reactions, and the eventual liver fibrosis progression.
Age can alter the oxidative stress, blood flow and mitochondrial capacity and immune responses of the body. The probability of liver fibrosis progression for men in age between 60–70 is estimated 300 times higher than for a man of 20 −40 years [41]. Liver fibrosis progression rate in female gender is 10 times less than males, irrespective of age. Although more investigation is needed, but researchers have reported that estrogen is one of the critical factors responsible for this difference. Estrogen can block some profibrotic cytokines and inhibit HSC activation, collagen synthesis, thus reducing CCl4-induced hepatic fibrosis in rats. Estradiol (E2) is a potent endogenous antioxidant and diminish hepatic steatosis in animal models [42].
Based on genetic, environmental, and epigenetic factors, patients with similar pathological conditions may respond differently to the same treatments. Therefore, it emphasizes the rationale behind individual variability and provides the right treatment options for all patients. Interestingly, several studies conclude that patients’ signature genes are predictive of clinical outcomes and overall survival rates. These genetics-based predictions with clinical factors may help for predictions and design anti-fibrotic therapies.
3. MOLECULAR TARGETS OF LIVER INJURY, NAFLD, AND LIVER FIBROSIS
Approaches currently being investigated for the treatment of NAFLD/NASH and liver fibrosis treatment include, but are not restricted to, modulation of energy intake or increasing extra-hepatic energy expenditure, diminishing excessive lipid deposition in the liver, reducing hepatocyte apoptosis, decreasing the inflammatory responses in the liver, and decreasing ECM proteins deposition after injury in the liver. Among these, the best therapies maybe those that address the most proximal causes of energy overload. Such approaches are useful in treating NASH and addressing the other components of the metabolic syndrome. In this section, we present the most promising targets for treatment of different stages of liver disease.
3.1. Molecules affecting hepatic metabolism
3.1.1. Peroxisome proliferator-activated receptor activators
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptor superfamily, which is comprised of PPAR-α, PPAR-γ, and PPAR-β/δ. Each PPAR has a specific expression pattern in adipose tissues, liver, skeletal muscle, heart. PPARs regulate lipid and glucose metabolism and play a key role in liver energy homeostasis and regulating adipogenesis [43]. PPARs function as heterodimers in association with retinoid X receptor (RXR). Upon ligand-mediated activation, the coactivator complex binds to its specific DNA sequence called peroxisome proliferators response elements (PPREs) present in the promoter regions of target genes. In the absence of ligands, these heterodimers are associated with co-repressor complex, including silencing mediator of retinoid and thyroid (SMRT) hormone receptors, and nuclear receptor corepressor (NCoR), which blocks transcription [44]. The natural ligands of PPARs mainly include lipid-derived substrates such as unsaturated fatty acids, eicosanoids, oxidized LDL, VLDL, and linoleic acid derivatives. In contrast, synthetic molecules such as fibric acid derivatives (fibrates) and thiazolidinediones (also called glitazones) are their pharmacological agonists.
Drugs that can reduce the fatty acid burden of the liver could be ideal for NASH treatment. Fat could be reduced by either blocking its synthesis in the liver or by improving the insulin sensitivity of adipose tissues to prevent inappropriate peripheral lipolysis. PPAR-α gene negatively regulates the hepatic lipid uptake by modulating fatty acid transport, esterifying FFA, and increasing mitochondrial FFA oxidation [45]. APOC3 protein is found on circulating lipoproteins, including high density lipoprotein (HDL), low - density lipoprotein (LDL), and triglyceride -rich lipoproteins (TRLs). There is a PPAR binding site in the proximal promoter of the APOC3 gene, and therefore, fibrates suppress its expression. The circulatory lipid-lowering activity of PPAR agonists is by repression of hepatic APOC3 expression, which leads to increased lipoprotein catabolism. PPAR-α agonists are classes of compounds name fibrates, which reduce blood TG levels and inc rease HDL-cholesterol without significantly affecting LDL-cholesterol [46]. Also, PPAR-α activation inhibits inflammatory genes induced by NF-kB and decreases the expression of acute-phase inflammation response genes [47]. Although several PPAR-α agonists have been developed, only a handful of them could reach the clinics successfully (Figure 1).
Figure 1. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease (NAFLD).
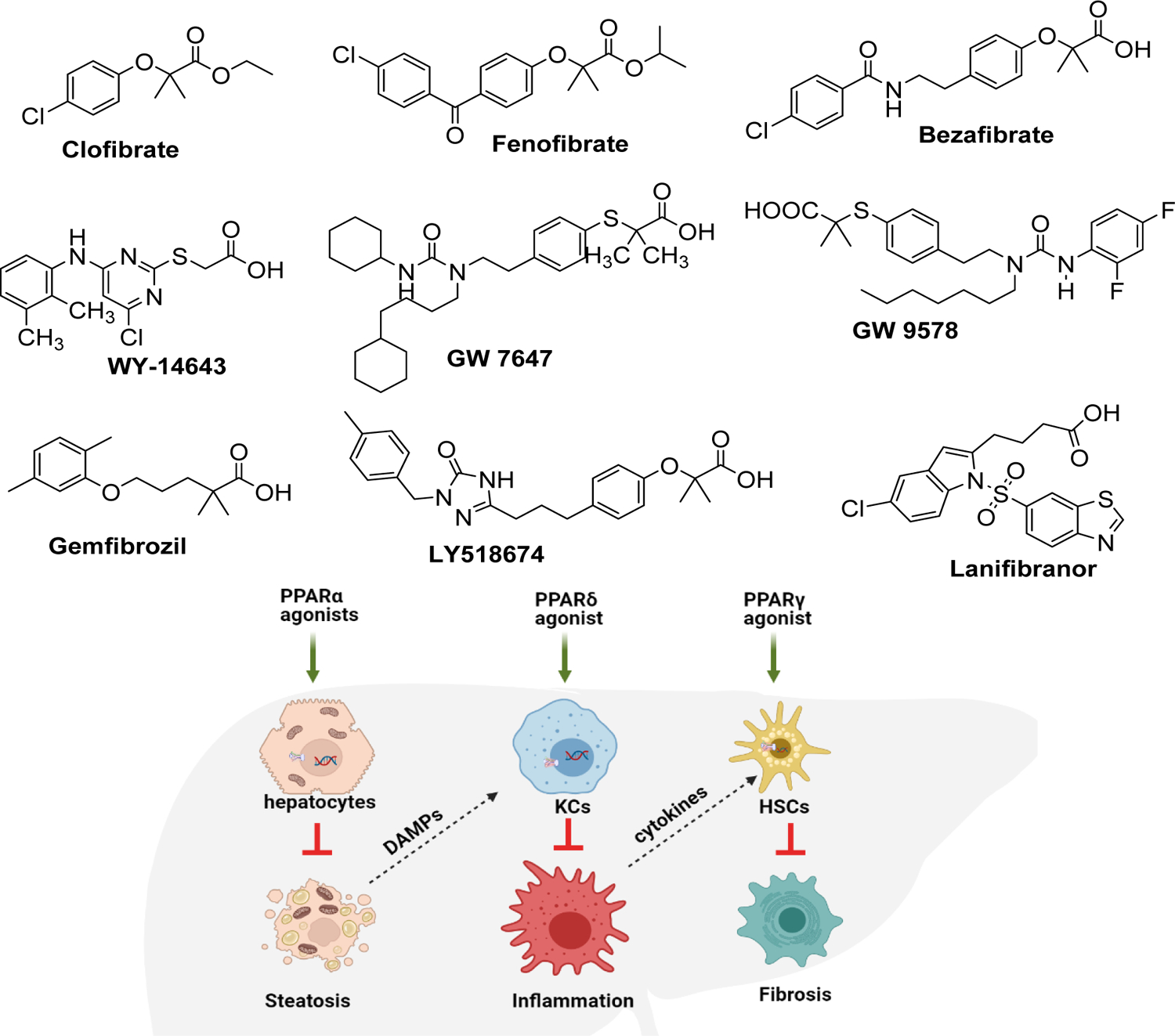
Various synthetic PPAR agonists are being evaluated for the treatment of NAFLD. PPAR-α activation leads to fatty acid β-oxidation in the mitochondria. The products can be later converted into ketone bodies (β-hydroxybutyrate or acetoacetate) or can be incorporated into TCA cycle as Acetyl CoA for further oxidation. PPAR- β/δ activation induces FOXO-1 transcription, which reduces hepatic gluconeogenesis and glucose uptake processes. Further, PPAR-β/δ activation suppresses inflammation by reducing IL-1β, IL-6, and NF-κB . Activation of PPAR-γ is linked to decreased hepatocyte lipogenesis and keeps HSCs in quiescent state. Dual or pan agonists targeting two or more of PPARs such as Lanifibranor has shown promising results in pre-clinical and clinical studies.
Fibrates are amphipathic compounds whose structural features include three components: a carboxylic acid head, an aromatic ring, and a lipophilic cyclic tail. Carboxylate interacts with the upper part of the PPAR-α binding pocket containing residues S280 (H3), Y314 (H5), H440 (H11), and Y464 (H12) to form hydrogen-bonding. Among these, Y464 binding is essential for maintaining the active protein conformation [48].
Clofibrate is a lipid-lowering agent used for controlling high cholesterol and TG levels in the blood. However, it causes significant toxicity in almost every organ system, especially in the liver and kidney. Different substitutions of carboxylic acid depict the potency and subtype selectivity. A series of experiments determined that the molecule’s high lipophilicity is required for optimal anti-dyslipidemia effects of fibrates. For instance, the substitution of the carboxyl moiety with 1,3,4-oxadiazole (1), phenyl mercapto triazole (2), and pyrazolone (3) increased their hypolipidemic activity. Clofibrate and fenofibrate are ester forms of the active moiety clofibric acid metabolized in vivo [49].
Several studies using fibrates such as fenofibrate and gemfibrozil have shown improvement in NAFLD patients’ biochemical and histological parameters [50]. However, most of these molecules are weak PPAR-α agonists and induce significant hepatotoxicity, with limited overall clinical efficacy. Gemfibrozil was produced by introducing a spacer between the carboxylic head and the aromatic ring. Further, dimethyl substitution on the 2,5 position of the aromatic ring showed significantly improved activity [51]. The ureido-thioisobutyric acid analog GW9578 with 7 carbon chain on the nitrogen increase PPAR-α selectivity by 300-fold on mice and 20-fold on the human receptors.
Further in vivo study in cholesterol/cholic acid-fed rodents showed the lipid, serum APOC3 and TLDL cholesterol-lowering effect at 500 times less dose of GW9578 than fenofibrate [52]. The compound WY14643 was created to interact with H12 in the PPAR-α ligand-binding domain (LBD). Unexpectedly, WY14643 occupies a different position in the ligand binding and thus provides a distinct ligand recognition mode than other fibrates. To improve physicochemical properties and PPAR-α selectivity (~200-fold), molecule GW7647 was discovered [53]. Similar to GW9578, molecule GW7647 has been shown to bind not only the receptor but also transactivate PPAR-α receptor [54]. LY-518674 has a disubstituted triazolone tethered by a two-carbon atom alkyl linker to the fibrate moiety. It shows a 10,000 more selectivity to PPAR-α compared to fenofibrate [55].
Fibrates alone are not very sufficient to produce desired pharmacological effects, or to show hepatotoxicity. Therefore, their pharmacophores have also been combined with other structures to obtain hybrid molecules with synergistic activity two molecules. For example, antioxidant and hepatoprotective effects of molecule 5-(4-methoxyphenyl-3H-1,2-dithiole-3-thione (ADT) was combined with fenofibrate [56]. Further, fenofibrate with the combination of pentoxifylline was evaluated in a patient with NAFLD. Although pentoxifylline did not improve Fenofibrate’s lipid homeostasis, it reduced the hepatic fibrosis as measured by levels of hyaluronic acid, transforming growth factor-beta 1 (TGF-β1), insulin resistance, and liver stiffness. Another study compared the therapeutic effects of fenofibrate and pioglitazone on a small number of NAFLD patients. The study showed significant improvement in liver enzyme profile, blood pressure profile, and BMI after treatment [57].
PPAR-γ protein exists in two isoforms: PPAR-γ1 and PPAR-γ2. The latter poses higher transcriptional activity compared to PPAR-γ1. Several PPAR-γ agonists effectively improve the histological condition of the liver from NASH mice [58]. PPAR-γ activation causes insulin sensitization and enhances glucose metabolism [59]. However, PPAR-γ activation often leads to several side effects, such as congestive heart failure, peripheral edema, bone fractures, and weight gain, therefore restrict their clinical applications [60]. Upon ligand binding, PPAR-γ forms a heterodimer with RXR-α and regulates transcription of target genes mainly in adipose tissues. PPAR-γ agonists are in clinical use for the treatment of T2D and regulates glucose, lipids, and protein metabolism [61]. However, phosphorylation of PPAR-γ at Ser273 is known to deminish its transcription activity and linked to obesity. Genes such as cyclin-dependent kinase 5 (CDK5) and growth differentiation factor 3 (GDF-3), are known to phosphorylated PPAR-γ, and their agonism has been demonstrated to have potent anti-diabetic effects [62].
PPAR-γ consists of 13 α helices and four β-sheets with typical helix H-3 to C terminus and one extra small helix H-2′. Helices H-3, H-7, H-10, and H-12, along with the β-sheets arranged in antiparallel orientation, constitute a large-ligand binding pocket of PPAR-γ [63]. PPAR-γ agonists binding switch the H12 α-helix and forms part of the ligand-dependent activation domain, AF-2, and closes on the ligand-binding site. The active form then binds to several co-activator proteins and control the transcription of target genes. Polyunsaturated fatty acids like linolenic acid, eicosapentaenoic acid, and 9-hydroxy-10, 12-octadecadienoic acid (9-HODE) are the natural agonists of these receptors [64]. Several synthetic thiazolidinediones PPAR-γ agonists are approved for T2DM. These family members share a standard methoxy-phenylmethyl-1,3-thiazolidine-2,4-dione backbone structure having an acidic head, an aromatic or aliphatic linker, and a hydrophobic tail. Their interaction with the binding domain can be adjusted by different substitutions on these chromophores. Ciglitazone was the first thiazolidinedione molecule discovered of this class, while troglitazone (CS-045) was the first approved drug with antidiabetic and anti-inflammatory activity [65].
Troglitazone possesses α-tocopherol moiety, which is known to inhibit lipid peroxidation. Troglitazone was withdrawn from the clinics due to its severe hepatotoxicity induced by its mitochondrial dysfunction activity. It was found that the hydroxyl group is involved in the toxic effects, and therefore compounds with hydroxyl groups protected by benzyl groups were designed by Reddy et al [66]. Rosiglitazone (BRL-49653) and pioglitazone are the other two approved molecules of the same category. Most of these molecules have a U-shaped configuration that binds with the Y-shaped active site by forming hydrogen-bonding with H323, H449, and Y473 amino acid residues [67]. Although rosiglitazone decreases insulin resistance and has anti-inflammatory effects, it does not reduce mouse NAFLD because activation of PPAR-γ also promotes SREBP-1c mediated lipogenesis, increases hepatic TGs and maintain the progression of NAFLD [68]. A promising strategy to decrease the toxic effects induced by full PPAR-γ agonists is to use partial agonists with different binding properties than full agonists. Farglitazar mostly shows hydrophobic interaction in sub-pocket near the H-12 helix via benzophenone, inhibits HSC activation, diminishes collagen secretion, and TNF-α reduction [69]. However, for patients with fibrosis of stages 2–4, treatment for 2 weeks with farglitazar did not alleviate fibrosis [70].
PPAR-δ regulates metabolism in the liver and other peripheral tissues. PPAR-δ overexpression leads to a decrease in insulin resistance, increases fatty acid oxidation, and decreases hepatic gluconeogenesis [71]. Most importantly, PPAR-δ exerts anti-inflammatory activities in macrophages and KCs [72]. PPAR-δ agonists have been shown to reduce liver fat, plasma lipids, increase insulin sensitivity, and decrease γ-glutamyltransferase (GGT) [73]. 15-Hydroxyeicosatetraenoic acid (15-HETE) is a natural compound that activates PPAR-β/δ receptors [74].
Simultaneous activation of two or more PPARs has been shown to be advantageous over individual activation. Many adverse side effects of single PPAR activation, such as fluid accumulation and weight gain, and increased frequency of congestive heart failure, are often associated with PPAR-γ agonists, such as rosiglitazone and pioglitazone. Dual PPAR agonists target two isotypes of PPARs, and work like hybrid molecules, and often have better efficacy than single inhibitors. Elafibranor (E)-2-(2,6-dimethyl-4-(3-(4-(methylthio)phenyl)-3-oxoprop-1-en-1-yl)phenoxy)-2-methylpropanoic acid) also known as GFT505, is a dual PPAR-α/δ agonist that has demonstrated efficacy in preclinical models of NAFLD, NASH and liver fibrosis [75]. Elafibranor shows hepatoprotection by reducing steatosis, inflammation, and fibrosis. It also improves insulin sensitivity, glucose homeostasis, and lipid metabolism in prediabetic and T2DM patients. Oral administration of elafibranor at the dose of 120 mg/day for a year showed benefits in NASH without progression to liver fibrosis and cirrhosis. In this study, elafibranor also improved their cardiometabolic risk profile [76]. However, there was a rise in serum creatinine in elafibranor treated group, but it was reversible. Further, elafibranor was failed to show the predefined primary endpoint of NASH resolution without worsening of fibrosis tested under the phase 3 trial in 1,070 patients (RESOLVE-IT Phase 3).
PPAR-β/δ are expressed mostly in skeletal muscle, adipocytes, macrophages, lungs, brain, and skin. In the liver, these receptors are expressed in hepatocytes, liver sinusoidal endothelial cells (LSECs), KCs, and HSCs [77]. PPAR-β/δ receptors regulate genes related to lipoprotein and glucose metabolism and genes related to inflammatory pathways [78]. Specifically, PPAR-β/δ increases monounsaturated fatty acids (MUFA) production in hepatocytes by upregulating stearoyl-CoA desaturase 1 (SCD1), a process that avoids lipotoxicity and protects against NAFLD [79]. Further, PPAR-β/δ promotes phosphatidylcholine (PC) 18:0/18:1 production in the liver, which activates PPAR-α and stimulates FA catabolism [80]. PPAR-β/δ also enhances adiponectin receptor 2 (AdipoR2) expression in the liver, leading to enhanced activity of 5’ adenosine monophosphate-activated protein kinase (AMPK) and suppression of lipogenesis and glycogen synthesis [81]. Similarly, PPAR-β/δ stimulates the insulin-induced gene-1 (INSIG1), suppressing SREBP1c levels, and lowers lipogenesis [82]. PPAR-β/δ activation reduces TNF-α or interferon-gamma (IFN-γ) expression in KCs, thus suppresses inflammation. Inversely, PPAR-β activation by small molecule L165041 is known to enhance HSC proliferation in case of acute and chronic liver inflammation [83].
Various PPAR-β/δ agonists have been developed recently, including saturated and unsaturated fatty acids (GW 501516), Carbaprostacyclin (GW 610742), VLDLs (GW 0742X, L-165041, retinoic acid). GW501516 is a selective PPAR-δ receptor agonist with high binding affinity (Ki = 1 nM). Binding of GW501516 to PPAR-δ stimulate PPAR-γ coactivator 1 alpha (PGC-1α). Treatment with GW501516 increases FA metabolism in skeletal muscle and provides protection against diet-induced obesity and T2DM in preclinical studies. Further, GW501516 was found to increase HDL and lowered VLDL in non-human primates [84]. Increased PGC-1α induces the transcription of nuclear respiratory factor 1 (NRF1) and NRF2, leading to increased expression of mitochondrial transcription factor A (mtTFA) as well as other nuclear-encoded mitochondria subunits of the electron transport chain complex such as β-ATP synthase, cytochrome c, and cytochrome c oxidase IV [85, 86]. mtTFA after translocating to the mitochondria stimulates mitochondrial biogenesis as manifested by stimulation of mitochondrial DNA replication and mitochondria gene expression [87, 88]. Prostacyclin (also called prostaglandin I2 or PGI2) activates carnitine palmitoyltransferase-1 (CPT-1) expression through PPAR-β/δ; the former plays an important role in mitochondrial fatty acid β-oxidation (FAO), which is s the major pathway of fatty acid degradation and is essential for maintaining energy homeostasis.
Dual PPAR-α and PPAR-γ agonists are also being evaluated for fatty liver complications [89]. Muraglitazar and tesaglitazar were tested in phase 3 clinical trial. However, due to the greater incidence of edema and heart failure, their further development was discontinued. Another drug Saroglitazar was significantly decreased plasma TGs and fasting plasma glucose level and increased HDL cholesterol level compared to the placebo. However, its further development was discontinued due to various toxicological reasons or a risk-benefit assessment. Lanifibranor (IVA337), a panPPAR agonist combines the pharmacological effects and control different components of the disease. Lanifibranor has shown clear beneficial effects in a preclinical model of liver fibrosis and portal hypertension [43]. A NATIVE study (EudraCT: 2016–001979-70, NCT: NCT03008070) assessed the safety and efficacy of a 24-week treatment with lanifibranor at the doses of 800 and 1200 mg/day in adult non-cirrhotic NASH patients [90]. The primary composite endpoint of patients having both NASH resolution and fibrosis improvement endpoint was met with statistical significance.
3.1.2. Farnesoid X receptor activators
Bile acid receptor (BAR), commonly known as Farnesoid X receptor (FXR), is expressed at high levels in the liver and intestine. FXR is responsible for the homeostasis of cholesterol metabolism and bile acid production. FXR contains both ligand and DNA binding domains. Ligand binding activates FXR and promotes its binding to DNA binding elements called FXR response elements (FXREs) and RNA polymerase II to modulate transcriptional activity. FXR itself does not bind to the putative bile acid response element (BARE) in its promoter region but induces expression of the typical nuclear receptor small heterodimer partner (SHP or NR0B2) [91]. SHP interacts with two other nuclear receptors that transactivate CYP7A1 expression via BARE regions, such as hepatic nuclear factor 4 (HNF4 or NR2A1) and liver receptor homolog-1 (LRH-1 or NR5A2). SHP repression of CYP7A1 gene transcription occurs by promoting the dissociation of coactivators linked to HNF4 and LRH-1 as well as by histone deacetylation of the promoter [92]. FXR activation lowers blood glucose, FFA, TGs, and total cholesterol. FXR activation also increases intestinal bile acid-binding protein (I-BABP) that facilitates bile acid transport from the intestines across enterocytes and portal circulation [93].
These functions make FXR a promising target to treat metabolic and chronic liver diseases. Various functions of FXR and its regulated pathways are shown in Figure 2. Most importantly, FXR by increasing FGF-19 secretion into the small intestine regulates carbohydrate and lipid metabolism, improves hyperinsulinemia, hepatic steatosis, and insulin sensitivity in preclinical models [94]. FGF-19 signals through fibroblast growth factor receptor 4 (FGFR4) cell surface receptor tyrosine kinase and suppresses CYP7A1, which results in decreased bile acid synthesis. All-trans retinoic acid (atRA) activates bile acid nuclear receptor FXR/NR1H4, resulting in downregulation of CYP7A1. It is the rate-limiting enzyme in bile acid biosynthesis. AtRA, in combination with ursodeoxycholic acid (UDCA) has been shown to reduce cholesterol significantly in the liver and improve cholestasis conditions [95]. A limited clinical trial of atRA in patients with primary sclerosing cholangitis (PSC) supports this mechanism in humans [96].
Figure 2. Farnesoid X Receptor (FXR) agonists in non-alcoholic hepatitis.
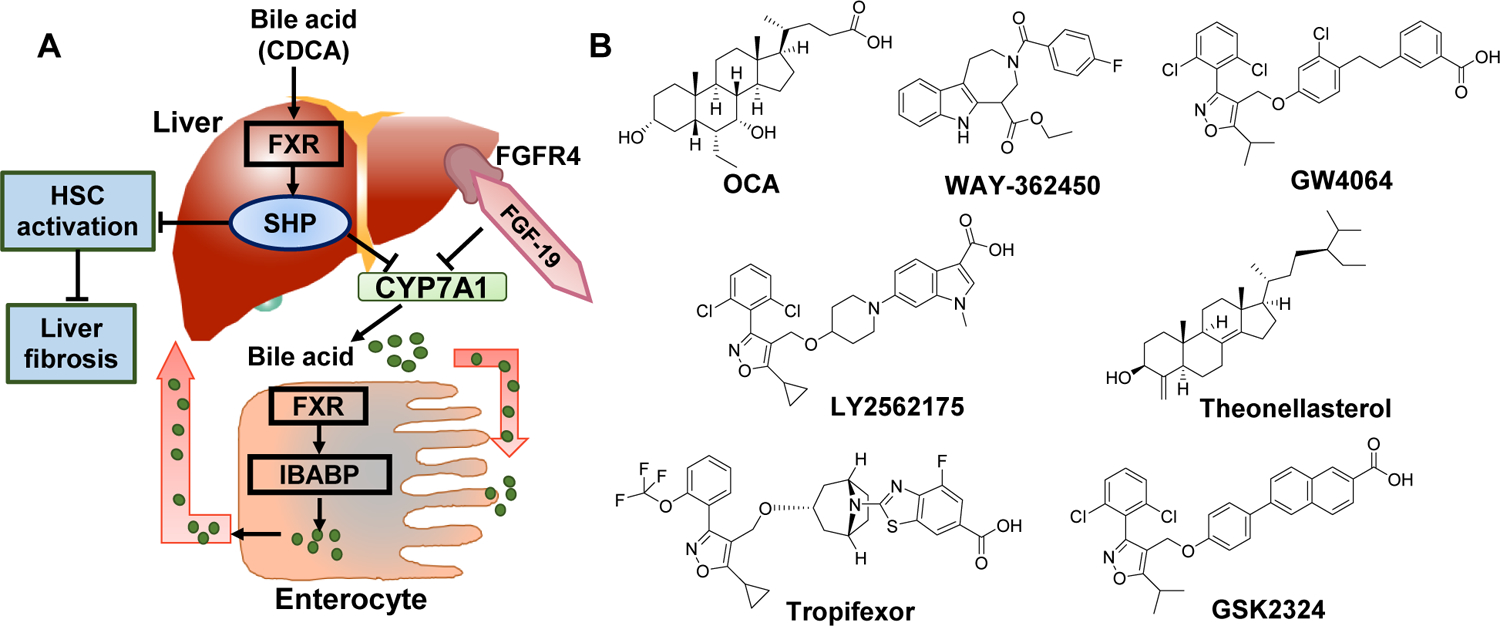
A) The role of FXRs in bile homeostasis and liver disease. B) Various FXR binding agents. CYP7A1 activity affects the overall rate of bile acid synthesis. Hepatic FXR activation reduces CYP7A1 mRNA expression by activating small heterodimer partner (SHP). FXR signaling plays an essential role in the control of hepatic de novo lipogenesis via SHP- mediated inhibition of SREBP-1c. SHP is also expressed in HSCs, and FXR ligands inhibit their activation and collagen synthesis. In enterocytes, FXR through Ileal bile acid-binding protein (IBABP), induces intestinal hormone fibroblast growth factor 19 (FGF19), which activates FGF receptor 4 (FGFR4) signaling in hepatocytes to inhibit CYP7A1 via triggering the extracellular stress-activated receptor kinase 1/2 (ERK1/2) pathway. Obeticholic acid (OCA), a semi-synthetic bile acid analogue of 6α-ethyl-chenodeoxycholic acid (6-ECDCA), is used as a medication for treatment of primary biliary cholangitis. In REGENERATE trial, OCA improved fibrosis in NASH patients.
Loss of function mutations in FXR encoding NR1H4 (low FXR expression) or mutations of FXR target genes ABCB11 (encoding BSEP) or ABCB4 encoding MDR3 lead to a severe form of cholestasis [97]. FXR also promotes liver regeneration by aiding hepatocyte growth after liver injury. The FXR-SHP regulatory cascade also inhibits HSC activation and thus promotes the resolution of liver fibrosis. Therefore, several FXR agonists have been evaluated for NASH and related diseases. Several metabolic intermediates, the mevalonic pathway precursor farnesol derivative, farnesyl pyrophosphate is FXR agonists. Chenodeoxycholic acid (CDCA) and conjugated CDCA species are potent natural agonists [98]. Obeticholic acid (OCA) is the first approved FXR agonist to treat (PBC). OCA is a 6α-ethyl derivative of CDCA and 100-fold higher FXR activation potency than its parent molecule. CDCA increases mRNA expression of bile acid efflux transporters, including the bile salt export pump (BSEP), multidrug resistance-associated protein 2 (MRP2), and organic solute transporter (OSTα/β), in hepatocytes [99]. OCA inhibits alkaline phosphatase (ALP) levels, total bilirubin levels (TBIL), and other liver disease markers in PBC patients. OCA at the dose of 25 mg significantly improved fibrosis and key components of NASH in patients in a phase 3 clinical trial (NCT02548351) [100]. However, OCA has steroidal bile acid-like chemical structure with low aqueous solubility and bioavailability. OCA and its metabolites do not follow the enterohepatic circulation resulting in a reduced PK/PD profile. Most noteworthy, OCA shows Takeda-G-protein-receptor-5 (TGR5) agonistic properties, with dose-dependent side effects such as pruritus [101]. Furthermore, OCA also decreases the conversion of cholesterol to bile acids, which results in increased serum levels of total and LDL cholesterol and decreased HDL, thus elevates the risk of cardiovascular disease.
To overcome these limitations, several non-steroidal FXR agonists have been synthesized. Small molecule GW4064 is a potent and selective non-steroidal FXR agonist. It is 100 times more potent than CDCA. However, its oral bioavailability is ≤ 10%, with a half-life of 3.5 h [102]. Therefore, there is a need to improve the PK properties of FXR agonists to be potentially used in the treatment of NAFLD. Efforts have been made to improve the structure of GW4064. When the stilbene moiety of GW4064 was replaced with a fused biaryl ring system such as isoquinoline in GSK2324 (EC50 = 50 nM, 102% efficacy) improved potency in vitro. However, this compound lacked the necessary physicochemical properties and PK profiles to progress into development [103]. Recently, in another study the central phenyl ring was replaced by a piperidine ring exemplified by LY2562175 (EC50 = 193 nM, 41% efficacy) with partial activity.
Another potent FXR agonist WAY-362450 (Turofexorate), which is 3-(3,4-difluorobenzoyl)-1,1-dimethyl-1,2,3,6-tetrahydro-azepino[4,5-b]indole-5-carboxylate. It was with acceptable oral bioavailability of 38% and a half-life of 25 h. In a preclinical study, WAY-362450 has been shown to significantly lower serum TG and total cholesterol levels and inhibit atherosclerotic aortic formation [104]. Tropifexor (LJN452) has a bicyclic [3.2.1] tropane linker ring, which was proved to the key component for its high potency [97].
Conversely, FXR activation is not always beneficial and may be detrimental in obstructive cholestasis patients. FXR activation suppresses the constitutive androstane receptor (CAR) to activate MRP4 promoter. MRP4 has a protective mechanism in obstructive cholestasis, and thus FXR activation may even aggravate the liver injury by inhibiting by CAR [105]. When FXR activity was antagonized by theonellasterol (a 4-methylene-24-hysteroid isolated from the marine sponge; Theonella swinhoei), led to increase in MRP4 expression in the liver, which protected against liver injury in cholestasis. CAR, in association with pregnane X receptor (PXR), regulate different target genes in cholestasis. Based on these observations, it is perceptible that a combination of FXR, PXR, and CAR agonists could be applied for optimum activity and reduced potential side effects of FXR activation alone. Besides, alternative/basolateral overflow, along with the renal excretion of bile acids, may be an attractive pathway for reducing the bile acid accumulation in cholestasis.
3.1.3. Insulin sensitizers
Glucose is converted into pyruvate through glycolysis in the cytoplasm. The pyruvate is transported in the mitochondria for oxidation. This process is accomplished by the transmembrane protein mitochondrial pyruvate carrier (MPC), which is composed of two significant subunits known as MPC1 and MPC2, which are the primary connection between non-oxidative and oxidative metabolism. After reaching in to mitochondria, pyruvate is either carboxylated to form oxaloacetate, which can be used for gluconeogenesis or converted to acetyl-coenzyme A (acetyl-CoA) and one molecule of carbon dioxide by the pyruvate dehydrogenase complex (PDC). After conversion to acetyl-CoA, it is either used of the tricarboxylic acid (TCA) cycle to produce energy or may be used to produce fatty acids, glucose, or amino acids.
Insulin sensitizers are designed to slow down pyruvate’s entry into mitochondria to prevent its conversion to acetyl-CoA [106]. Inhibition of either MCP1 or MCP2 results in decrease hepatocyte damage induced by HFD. MPC1 and MPC2 are located on the inner membrane, which are obligate components of an apparent complex that facilitates inhibitor sensitive pyruvate transport. A partial PPAR-γ agonist pioglitazone (MSDC-0160) has the inhibitory activity of MPC1 and MPC2 [107]. Interestingly, MSDC-0160 showed only a 50% reduction in circulating adiponectin (a marker of the amount of contribution of white adipose tissue) and only a 50% decrease in hematocrit (a marker for PPAR-γ expansion of plasma volume). These results show that PPAR-γ agonism is not needed for the insulin-sensitizing effects of MSDC-0160.
Further, MSDC-0602 was synthesized to retain the inhibitory activity of MPC1 while sparing PPAR-y binding activity. In PPARγ−/− mice, MSDC-0602 showed hepatocyte protection showing insulin-sensitizing effects without PPAR-γ activation. These effects were independent of PPAR-γ expression levels [108]. Further, MSDC-0602 and Liraglutide improved insulinemia and fatty liver disease in mice, alone and with liraglutide [109]. Recently, a Phase 2a clinical trial (12-month, Emminence) in NASH patients with or without T2D has been completed with MSDC-0602. The study outcomes showed that MSDC-0602K was well-tolerated improving liver enzyme levels, liver histology, and markers of glycemic control, in NASH patients. However, MSDC-0602K did not demonstrate statistically significant effects on primary and secondary liver histology endpoints [NCT02784444] [110]. Off note, MSDC-0160 and MSDC-0602 are being evaluated for the treatment of T2DM and show reduced side effects associated with PPAR-γ.
Dipeptidyl peptidase 4 (DPP4) is a membrane-associated peptidase with the primary function to deactivate a variety of bioactive peptides such as glucagon-like peptide-1 (GLP-1). GLP-1 released by L cells of the intestine regulates blood glucose by stimulating insulin release. GLP-1 binding to its receptors induces fatty acid oxidation pathways in hepatocytes and increases insulin sensitivity. The serum DPP4 levels are higher in NASH patients than controls, resulting in decreased GLP-1 activity [111]. Inactivation of GLP-1 causes glucose intolerance, T2DM, and hepatic steatosis. Several GLP-1 analogs are being developed and evaluated for treating T2DM.
A peptide named exendin-4 (brand name exenatide) was the first GLP-1 analog discovered from Gila monster venom with 53% amino acid similarity to GLP-1 [112]. The N-terminus of exendin-4 has a glycine in place of alanine and, therefore, resistance to DPP4 and its secondary and tertiary structures prevent its hydrolysis, thus have a much longer half-life and blood-glucose-lowering effect. Downside, Extendin-4 treatment results in several side effects, including medullary thyroid cancer, angioedema, pancreatitis, and kidney injury. Liraglutide is a synthetic analog of GLP-1 peptide with two modifications: a substitution of Arginine for Lysine at position 34 and an attachment of a C-16 FFA derivative glutamyl spacer to Lysine 26. The FFA chain can promote albumin binding of liraglutide and therefore decrease body clearance. Liraglutide was approved by the FDA in 2014 for treating T2DM [113]. In some patients, including those with T2DM and NAFLD, liraglutide, while improved serum liver enzyme levels but did not improve the overall disease [114].
There has been a lot of effort done in extending the half-life and decreasing the side effects of GLP-1 analogs, several of these new GLP-1 receptor agonists currently under clinical development. Exenatide is a FDA approved poly(D,L-lactic-co-glycolic acid) (PLGA) based microsphere formulation of synthetic extendin-4 peptide for treating T2DM [115]. Exenatide reduces the blood glucose level and provides an extended-release profile to reduce dosing frequency [116]. LY2189265 is a GLP-1 analog-Fc fusion protein, and LY2428757 is a PEGylated GLP-1 analog with once a week dose. CJC-1134-PC is a recombinant human serum albumin-exendin-4 conjugate that has a half-life of approximately eight days. Albiglutide is an albumin GLP-1 complex; both can be administered once weekly [117]. NN9535 is another long-acting GLP-1 analog designed using protein acylation technology for once-a-week injection. Taspoglutide and AVE0010 are two other GLP-1R agonists currently undergoing clinical trials. Semaglutide oral formulation of a GLP-1 agonist 2 doses, 7 mg and 14 mg are recently approved by the FDA. It is a co-formulation of semaglutide and sodium N-[8-(2-hydroxy benzoyl amino]caprylate (SNAC) [118]. Peptides are not stable in the gastrointestinal tract and their absorption is a challenge. Semaglutide is noncovalently associated with medium-chain length fatty acid-based system SNAC as an oral absorption enhancer to prevent enzyme degradation and facilitate absorption.
The other mechanism to increase GLP-1 level is by inhibiting dipeptidyl peptidase 4 (DPP4) enzyme. Several DPP4 inhibitors such as sitagliptin, saxagliptin, linagliptin, and alogliptin are approved by the FDA [119]. Some other DPP4 inhibitor molecules are also approved by regulatory agencies. For instance, vildagliptin was approved by the European Union, gemigliptin and evogliptin by South Korea, anagliptin, omarigliptin, trelagliptin, and teneligliptin by Japan, and gosogliptin (by Russia. A detailed review has been published to highlight various DPP4 inhibitors used for the treatment of T2DM, their benefits, and risks [120].
3.1.4. Fibroblast growth factor (FGF) signaling promoters
Fibroblast growth factors (FGF) are a family of cell signaling proteins. Specifically, FGF21 increases energy expenditure under specific stimuli. FGF21 is synthesized in multiple organs and can act on multiple target tissues in either a paracrine or an endocrine fashion [121]. The mechanism of FGF21 action signaling is complex that involves several FGF receptors (FGFRs) as well as an obligate coreceptor, Klotho-β (KLB). Co-expression of a given FGFR and KLB confer the tissue specificity of FGF signaling. FGF21 is predominantly released from hepatocytes and increased fatty acid oxidation as well as TCA cycle flux by inducing PGC-1α expression in the liver [122]. FGF21 increases glucose uptake by increasing GLUT1 expression [123]. Furthermore, FGF21 decreases serum free fatty acids by decreasing lipolytic gene expression in white adipose tissue. The ketogenic diet, which is a high-fat and low-carbohydrate diet, leads to increased energy consumption, improved glucose homeostasis, and increased expression of genes in the fatty acid oxidation pathway (Figure 3) [124]. FGF21 is an endocrine regulator of the ketotic state and FGF21 expression is induced by both KD and fasting. Hypothalamic neuropeptides, such as neuropeptide Y (NPY), agouti-related peptide (AgRP), are considered in inverse correlation with energy expenditures, while proopiomelanocortin (POMC) is in direct correlation with it. However, a ketogenic diet was shown to increase NPY and AgRP and decrease pro-opiomelanocortin (POMC), along with an increase in energy expenditure; It was against the general paradigm. FGF21 was found to upregulate PGC-1α and explained these inconsistent results with the ketogenic diet. Mitochondria controls cellular energy homeostasis, and their dysfunction leads to cell death and may be responsible for organ failure. FGF21 is regarded as a biomarker of mitochondrial dysfunction in several diseases [125].
Figure 3. Glucose-lowering effects of fibroblast growth factors (FGFs).
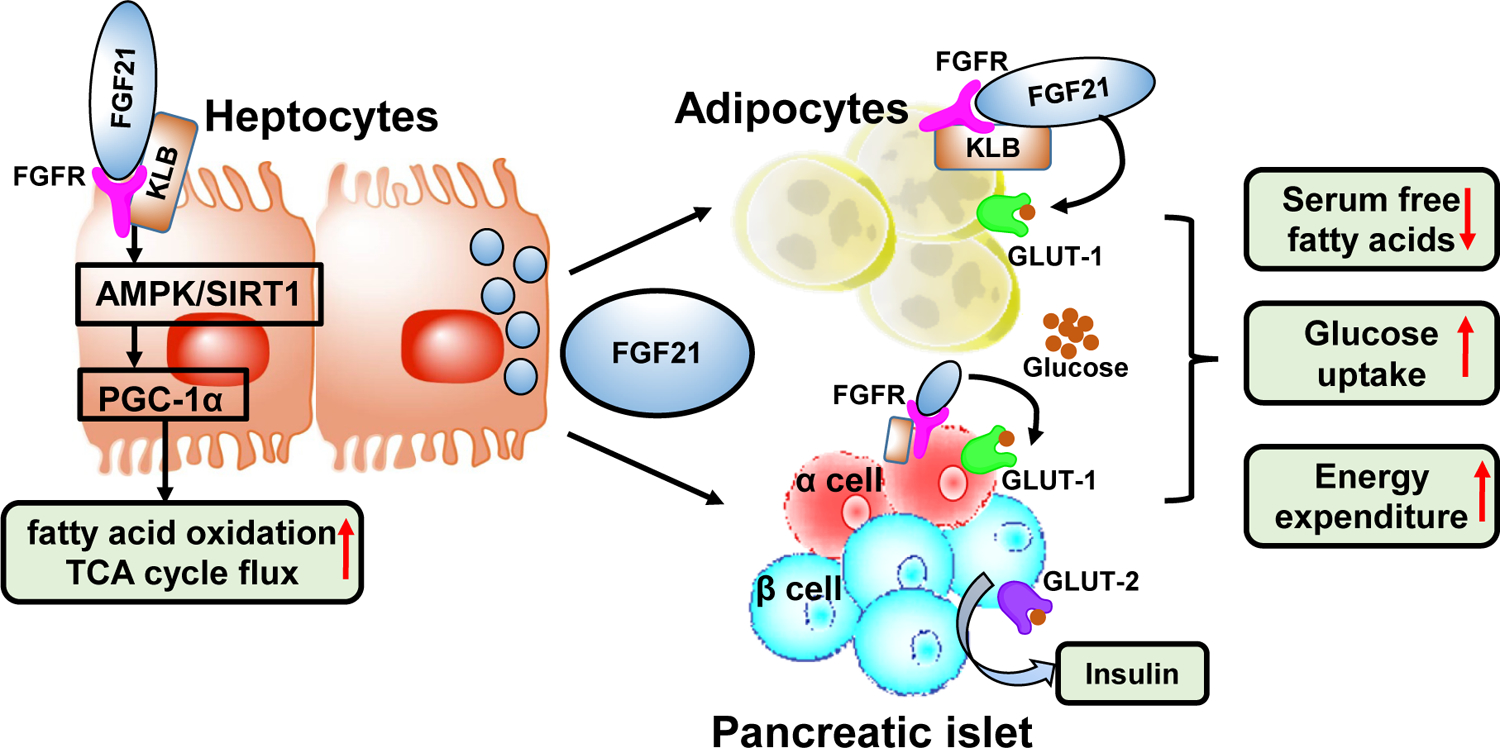
FGF21 is secreted by hepatocytes and stimulates glucose uptake in adipocytes while increases insulin secretions by pancreas. FGF21 requires the cofactor, b-Klotho (KLB) for binding its receptor FGFR and activation of the receptor auto-phosphorylation and signaling. In hepatocytes, via SIRT1 and AMPK dependent mechanism, FGF21 induces transcription coactivator, PGC-1α, which increases the mitochondrial activity and enhances oxidative capacity. In white adipose tissue, FGF21 induces the expression of glucose transporter, GLUT1, but prolonged exposure to FGF21 enhances lipolysis as well as increases thermogenesis. FGF21 induces insulin gene expression in islet cells, and this treatment alleviates β-cell dysfunction. A novel FGF21 version, LY2405319 with reduced tumorigenic potential and improved biophysical properties was investigated in Phase 1 clinical trial.
FGF21 and PPAR-β/δ regulate VLDL receptor expression in the liver. PPAR-β/δ-null mice and PPAR-β/δ−/− hepatocytes show higher VLDL receptor expression. On the other hand, FGF21 neutralizing antibody induces TG accumulation in PPAR-β/δ-null mice [126]. Further, increased VLDL receptor levels with reduced PPAR-β/δ mRNA levels are evident from the liver biopsies of NAFLD patients [127]. These findings were essential to understand the VLDL receptor levels controlling mechanism by PPARβ/δ and FGF21 in NASH and suggest novel treatment opportunities. In preclinical studies, the administration of FGF21 reversed hepatic steatosis, decreased obesity, and insulin resistance. Therefore, FGF21 peptide is being developed as a novel therapy for NASH treatment [128]. However, its clinical applications were halted by its short in vivo half-life and immunogenic activity.
Several approaches have been applied to improve recombinant human rhFGF21 in vivo properties. PEGylation of FGF21 at the N-terminal residue with 20 kDa mPEG-butyraldehyde (BMS-986036) is the most successful strategy till now, which improved its biostability and efficacy [129, 130]. Other strategies used for increased stability include introducing an additional disulfide bond through L118C and A134C mutations resulted in a novel FGF21 variant LY2405319. Further, the deletion of four N-terminal amino acids, His-Pro-Ile-Pro (HPIP), prevented FGF21 proteolytic cleavage. For large-scale homogenous protein production in yeast Pichia pastoris, a S167A mutation was introduced to eliminate O-linked glycosylation. Altogether these efforts of FGF21 re-engineering led to significant improvement in its biopharmaceutical properties [131].
FGF19 is another member of the FGF family, a gastrointestinal hormone responsible for bile acid synthesis. FGF19 stimulates glycogen synthesis and gluconeogenesis, thus regulating the pathology of NASH [132]. Circulating FGF19 concentration is reduced in NASH patients, suggesting that its modulation could benefit bile acid-related metabolic disorders. However, the therapeutic potential of FGF19 has been hindered by its hepatocarcinogenicity. Its expression is elevated in HCC patients.[133] FGF19 stimulates tumor progression by activating the signal transducer and activator of the transcription 3 (STAT3) pathway [134]. A recombinant non-tumorigenic variant of FGF19 protein, NGM282, was recently developed. NGM282 variant (also known as M70) has 5-amino acid deletion (P24–S28) coupled with the substitution of three amino acids (A30S, G31S, and H33L) at the active site. These modifications enabled NGM282 to retain CYP7A1 repressing ability while dropping its STAT3 activation properties. NGM282 showed hepatocarcinogenesis blocking properties. Treatment with NGM282 in NASH animal models showed a robust reduction in ALT and AST levels and had a definite improvement in all histological features associated with NASH. More importantly, NGM282 was well tolerated in healthy volunteers and was associated with reduction in serum concentrations of 7α-hydroxy-4-cholesten-3-one, a biomarker of hepatic CYP7A1 activity [135, 136].
3.1.5. Stearoyl coenzyme A desaturase 1 (SCD1) inhibitors
SCD1 is a key enzyme in fatty acid metabolism and catalyzes delta 9 monounsaturated palmitic and stearic acids. SCD1 activity results in the synthesis of monounsaturated fatty acids (MUFAs), the major FA of TGs, cholesteryl esters, and membrane phospholipids. SCD1 deficiency in mice has been demonstrated to reduce lipid synthesis and increase mitochondrial FA β-oxidation and insulin sensitivity [137]. Accordingly, the inhibition of SCD1 results in several beneficial effects, including reduction of liver fat, protection against insulin resistance, and protection against obesity. Several synthetic SCD1 inhibitors, including aramchol, CVT-12012, GSK1940029, MF-438, MK-8245, and SW203668 are being evaluated for their efficacy in preclinical as well as clinical studies (Figure 4). Aramchol is a conjugate of arachidic acid and cholic acid, which is in Phase 3 clinical trial for NAFLD. By inhibiting SCD1 activity, Aramchol was found to prevent and reduce NAFLD, and improve cholesterol metabolism, and even dissolve cholesterol gallstones in some cases. In a preclinical study, Aramchol treatment in MCD induced NASH mouse model downregulated SCD1, which led to decrease in hepatic FAs and TGs, ameliorated inflammation, and reversed fibrosis. Aramchol improved β-oxidation by increasing the flux through the trans-sulfuration pathway, leading to a rise in glutathione (GSH) [138]. In addition, Aramchol activates cholesterol efflux by stimulating the adenosine triphosphate–binding cassette transporter A1 (ABCA1), a pan-cellular cholesterol export pump, and has shown to have an anti-atherogenic effect in animal studies [139]. Aramchol once a day dose of 600 mg was tested in patients with biopsy-proven NASH who were obese and pre-diabetic or T2DM. The trial enrolled 240 patients with advanced NASH, with more than 60% having fibrosis in stages 2 and 3 [140].
Figure 4. The role of stearoyl-CoA desaturase 1 (SCD1) in the development of fatty liver diseases.
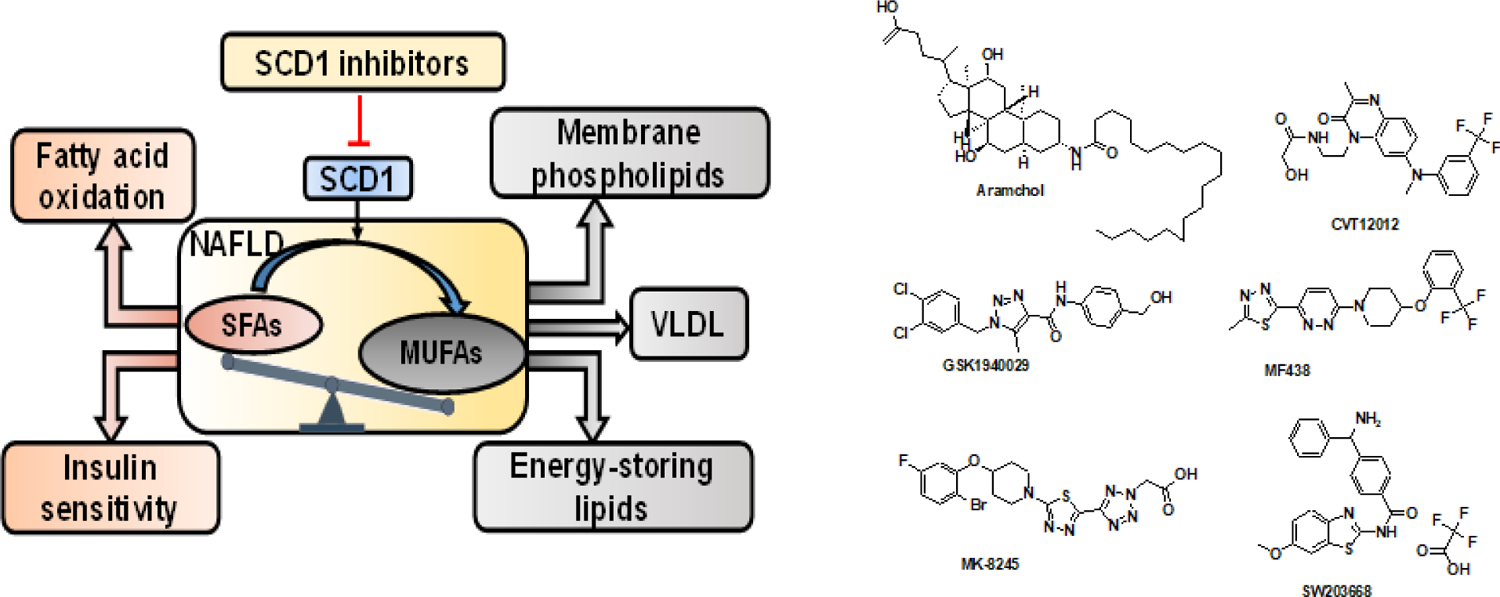
SCD1 converts saturated fatty acids (SFAs) to monounsaturated fatty acids (MUFAs). SCD1 maintains a balance between SFAs and MUFAs in the cells. SFAs could be used for β-oxidation and energy production by mitochondria. MUFAs products of SCD1 may be further elongated or incorporated into a variety of complex lipids including triglycerides (TGs), phospholipids, and other lipid species or exported through VLDL mechanism. Excess TG accumulation in the cells leads to steatosis and onset of NAFLD. SCD1 inhibition produces a significant enrichment of SFAs and promotes fatty acid oxidation and increases insulin sensitivity. SCD1 inhibitor Aramchol was tested in Phase 3 clinical trial.
3.1.6. Adenosine monophosphate-activated protein kinase activators
Adenosine monophosphate-activated protein kinase (AMPK) is an intracellular energy sensor that is involved in many biological processes. The activation of AMPK is found suppressed in fibrosis progression. AMPK activation can improve NAFLD in three ways: by suppressing de novo lipogenesis, restoring of mitochondrial function, and increasing fatty acid oxidation [141]. Several reports confirm the beneficiary effects of AMPK pathway activation in metabolic liver disease. Recently, Yao et al. have used a herbal medicine Dioscin, a steroid saponin, to treat NAFLD. Dioscin enhanced the phosphorylation and activation of AMPK [142]. Woods et al. increased AMPK activity in mice by mutation of a residue in AMPK, making it a worse substrate for dephosphorylation. This mutation decreased lipogenesis and protected against hepatic steatosis in high fructose-fed mice [143]. In an interesting study, Boudaba et al. have shown that AMPK knockout mice displayed normal hepatic lipid homeostasis and were not prone to NAFLD. However, re-activation of AMPK in AMPK knockout mice by a small molecule direct AMPK activator, A769662, could decrease hepatic TG and cholesterol content and restore fatty acid oxidation [144]. PF-06409577 an AMPK activator has also been shown to inhibit de novo lipid and cholesterol synthesis pathways and causes a reduction in hepatic lipids and mRNA expression of markers of hepatic fibrosis [145]. These findings indicate AMPK as a potential target for the treatment of fatty liver disease. Therefore, in a randomized clinical trial, safety and lipid lowering efficacy of Dithiolethiones drug oltipraz was evaluated in patients with NAFLD. Dithiolethiones, are novel class of AMPK activators, which prevent insulin resistance through AMPK-dependent p70 ribosomal S6 kinase-1 (S6K1) inhibition (NCT02068339). Further, oltipraz was also shown to inhibit liver X receptors (LXRg) and SREBP-c and thus decreases the expression of lipogenic genes and decrease fatty acid synthesis [146]. Compared with the placebo group 24-week oltipraz treatment significantly reduced the liver fat content and BMI in patients with NAFLD. However, the treatment did not significantly change in insulin resistance, liver enzymes, lipids, and cytokines.
3.1.7. Apolipoprotein C-3 inhibitors
Apolipoproteins are TG-rich lipoproteins (TRL) that transport cholesterol and TGs through the blood. The fat absorbed from the intestine is carried to the liver by apolipo protein (APO) particles called chylomicrons [147]. On the other hand, excess fat in the liver is packaged and exported in apolipoprotein carrier particles called very-low-density lipoprotein (VLDL) [148]. APOC3 is expressed and secreted in the liver and functions as an inhibitor of hepatic LDL receptor. Consequently, the plasma residence time of TRLs and their remnants increases. Thus, reduction in hepatic APOC3 can enhance the clearance of VLDL from the liver. The molecule Gemcabene (6, 6’-oxybis [2, 2-dimethyl-4-hexanoic acid] monocalcium salt), also known as PD72953, is an inhibitor of APOC3 and lipid-lowering compound. Gemcabene also decrease the TG and cholesterol production, associated with reduced hepatic APOC3 mRNA levels. This may result in increased VLDL remnant uptake and consequently lower LDL-C levels. Some studies also indicate that it posess an anti-inflammatory profile, associated with a lowered expression of hs-CRP gene regulating mechanisms. Gemcabene is an orally bioavailable small molecule of a dialkyl ether dicarboxylic acid [6,6′-oxybis (2,2-dimethylhexanoic acid)]. It is metabolized via glucuronidation by uridine diphosphate glucuronosyltransferase (UGT) 2B7 (UDT2B7) [149].
In hypercholesterolemia patients, Gemcabene significantly lowers LDL-C, ApoB, C-reactive protein (CRP), TG, and increases high-density lipoprotein cholesterol (HDL-C). To date, gemcabene has been administered to 895 healthy subjects and patients and has been observed to be well tolerated at the doses up to 900 mg once a day for up to 12 weeks [150]. Gemcabene with gemfibrozil has been shown to cause a marked elevation and enlargement of HDL and plasma TG reduction; however, no effect on the amount and size of HDL was observed [151]. When combined with a statin, gemcabene reduces LDL-C levels by 17–21% more than placebo. The safety and efficacy of gemcabene in patients with homozygous FH is currently being evaluated in a phase 2 study. Familial partial lipodystrophy (FPL) is a rare genetic disorder due to an abnormal distribution of fatty tissue and various metabolic abnormalities, including NASH. Gemphire Therapeutics Inc. initiated a phase 2a clinical trial of Gemcabene in adults with FPL (NCT03508687).
3.2. Molecules affecting hepatocyte apoptosis
During NASH, hepatocyte apoptosis is correlated with the severity of inflammation and fibrosis [152]. Saturated fatty acids (SFAs), as well as free cholesterol, are the critical mediators of lipotoxicity resulting in apoptotic cell death. There are two main mechanisms of apoptosis: extrinsic and intrinsic pathways. The extrinsic pathway is initiated by triggering of death receptors, including Fas, TNFR1, and TNF-related apoptosis-inducing ligand (TRAIL) receptors. Binding of ligands to these receptors iniates intracellular cascades that activate death-inducing proteolytic enzymes, especially caspases. APO-1/Fas (CD95) -mediated apoptosis is one of the mechanisms for hepatocyte apoptosis. Intrinsic apoptotic pathway is initiated by damage of the intracellular organelles, such as mitochondria, lysosomal permeabilization, and ER stress, among others. Inhibition of hepatocyte apoptosis therefore is a viable approach to decrease liver injury and related inflammation [153].
3.2.1. Fas receptor and Fas ligand antagonists
Fas receptor (FasR) is a glycosylated type 2 transmembrane protein that belongs to the TNF family. Its binding to FasL (CD95L or CD178) induces receptor trimerization and formation of the death-inducing signaling complex (DISC) [154]. Fas expression upregulation is a common feature in the liver samples of NASH patient. NAFLD induced in mice fed with a high carbohydrate diet show increased sensitivity to Fas-induced apoptosis of hepatocytes. This model recapitulates many features of human NAFLD, including obesity, insulin resistance, hyperleptinemia, elevated serum FFA and hepatic steatosis. Incubation of human liver cells to FFA resulted in the upregulation of Fas expression and increased sensitivity to Fas-induced apoptosis [155]. c-Met, also called tyrosine-protein kinase Met or hepatocyte growth factor receptor (HGFR) prevents Fas activation in healthy liver tissues. This interaction of Fas and Met is abrogated in both human and experimental NAFLD, resulting in increased Fas–FasL complex formation and increased apoptosis. Some death receptors, through several intracellular cascades, mediate pro-inflammatory responses. For instance, activation of NF-κB by death receptors is proinflammatory [156]. Thus, Fas-activation on hepatocytes also results in HSC activation and fibrosis. Therefore, blocking Fas stimulation may prevent hepatic fibrosis by blocking inflammation, not only apoptosis. There is no fas receptor inhibitor at present. However, recently a Fas receptor antagonist peptide ONL1204 (HHIYLGAVNYIY), has been reported. ONL1204 has shown anti-inflammatory and neuroprotective effects in a microbead-induced mouse model of glaucoma [157]. Therapeutic efficacy of ONL1204 in NAFLD/NASH model need to be evaluated.
3.2.2. TNF-α and its receptor inhibitors
TNF-α is a type 2 transmembrane protein responsible for signal transduction carcinogenesis as well as in immune reactions. It is a well-studied proinflammatory cytokine produced mainly by activated macrophages [158]. TNF-α ligand can bind to any of its two receptors: TNF-R1 (p55 or CD120a) and TNF-R2 (p75 or CD120b). TNF-R1 can induce both pro and anti-apoptotic signaling. Upon ligand binding, TNF-R1 engages TNFR-associated protein with death domain (TRADD), receptor-interacting protein 1 (RIP1), and TNFR-associated factor 2 (TRAF2) to form a complex called “complex I.” This complex internalizes and binds to FADD, resulting in caspase 8 activation and cell death. TNF-α/TNF-R1 can also induce c-Jun N-terminal kinases (JNK) activation and promote cell death [159].
In contrast, TNF-R2 activation results in the recruitment of TRAF2 and stimulates the pro-survival cellular inhibitor of apoptosis 1 and 2 (cIAP1/2) proteins. Together with TRAF2, cIAP1/2 proteins degrade the TRADD-bound ubiquitinated RIP1. Multiple ubiquitinations of RIP1 and NF-κB essential modulator [NEMO, also called IκB kinase (IKK)γ], engage the kinase TAK1 NEMO-containing IKK complex. IKKβ in the IKK complex becomes phosphorylated and phosphorylates NF-κB inhibitor IκBα that is subsequently cleaved. Released NF-κB translocates into the nucleus and induces survival target gene expression [160]. Pentoxifylline, a TNF-α inhibitor, improved NASH in a randomized placebo-controlled trial involving patients when administered three times a day at the dose of 400 mg [161]. Pentoxifylline results in weight loss with improved liver histological in patients with NAFLD/NASH [162].
3.2.3. Caspase inhibitors
Since caspases target critical cellular components, caspase inhibitors have been applied in several clinical conditions, including liver fibrosis. Caspase inhibitors IDN-1965 and Z-Asp-CMK reduce several apoptotic SEC after reperfusion in normothermic and cold ischemia models. Another caspase inhibitor GS-9450 is an irreversible selective inhibitor of caspases 1, 8, and 9 and showed some promising results in NAFLD patients [163]. Hepatocyte apoptosis is also profibrogenic in animal models of cholestasis. HSCs, after exposure to apoptotic bodies, i.e., DNA from apoptotic hepatocytes, become fibrogenic by activation of reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2), phagocytic NADPH oxidase [164].
Caspases 3 and 7, along with higher expression of Fas receptors by hepatocytes, are well correlated with NASH progression [165]. Hence, pan-caspase inhibitors were designed to inhibit all caspases and inhibit cellular apoptosis and tested as therapeutic agents for NASH. Emricasan ((L-alaninamide, N-[2-(1,1- dimethylethyl) apephenyl]-2-oxoglycyl-N-[(1S)-1-(carboxymethyl)-2-oxo-3-(2,3,5,6-tetrafluorophenoxy)propyl]) is a small molecule that irreversibly inhibit activated caspases. In preclinical models of cytokine-induced liver disease, cholestasis, and NASH. Emricasan reduced apoptosis of hepatocytes as well as HSC activation, resulting in reduced inflammation and fibrosis. The drug has a good safety profile, as patients with hepatitis C or NASH treated with emricasan once, twice, or three a day at doses from 5–400 mg daily for up to 12 weeks were well-tolerated without significant adverse events [166]. Several other clinical trials evaluated emricasan in different liver diseases. A randomized, double-blind Phase 2b clinical trial ENCORE-LF, enrolled 217 patients with decompensated NASH. Patients were randomized and received 5 mg or 25 mg of emricasan or placebo twice a day for at least 48 weeks. The trial’s endpoint was event-free survival, defined as a composite of all-cause mortality, new decompensation events, or ≥4 points progression in the Model for End-stage Liver Disease (MELD) score. The primary analysis showed no statistically significant differences between the treatment and placebo arms. Another double-blinded Phase 2b clinical trial named ENCORE-PH was conducted in 263 NASH patients with compensated or early decompensated cirrhosis and severe portal hypertension confirmed by the hepatic venous pressure gradient (HVPG) of ≥12 mmHg at baseline. The patient received 5, 25, or 50 mg of emricasan or placebo twice a day for 24 weeks. The trial showied no statistically significant differences between the treatment and placebo arms.
Another pan-caspase inhibitor VX-166, was shown to reduce liver apoptosis, inflammation, and fibrosis in experimental models without decreasing ALT levels and oxidative stress markers. The beneficial effects of VX-166 on liver fibrosis were attributed to a reduction in the uptake of apoptotic bodies by HSCs and their decreased activation (Figure 5). However, the drug had a modest effect on animal models with established steatosis/steatohepatitis [167]. Nivocasan (GS-9450) is a caspase inhibitor that reduced ALT and levels of cytokeratin 18 (CK-18), a central intermediate fragment protein in the liver in patients with hepatitis C and NASH. The drug also had favorable PK profiles; however, the potential risk of carcinogenesis was a concern regarding GS-9450 and caspase inhibitors in general [168].
Figure 5. Hepatocyte apoptosis in NAFLD.
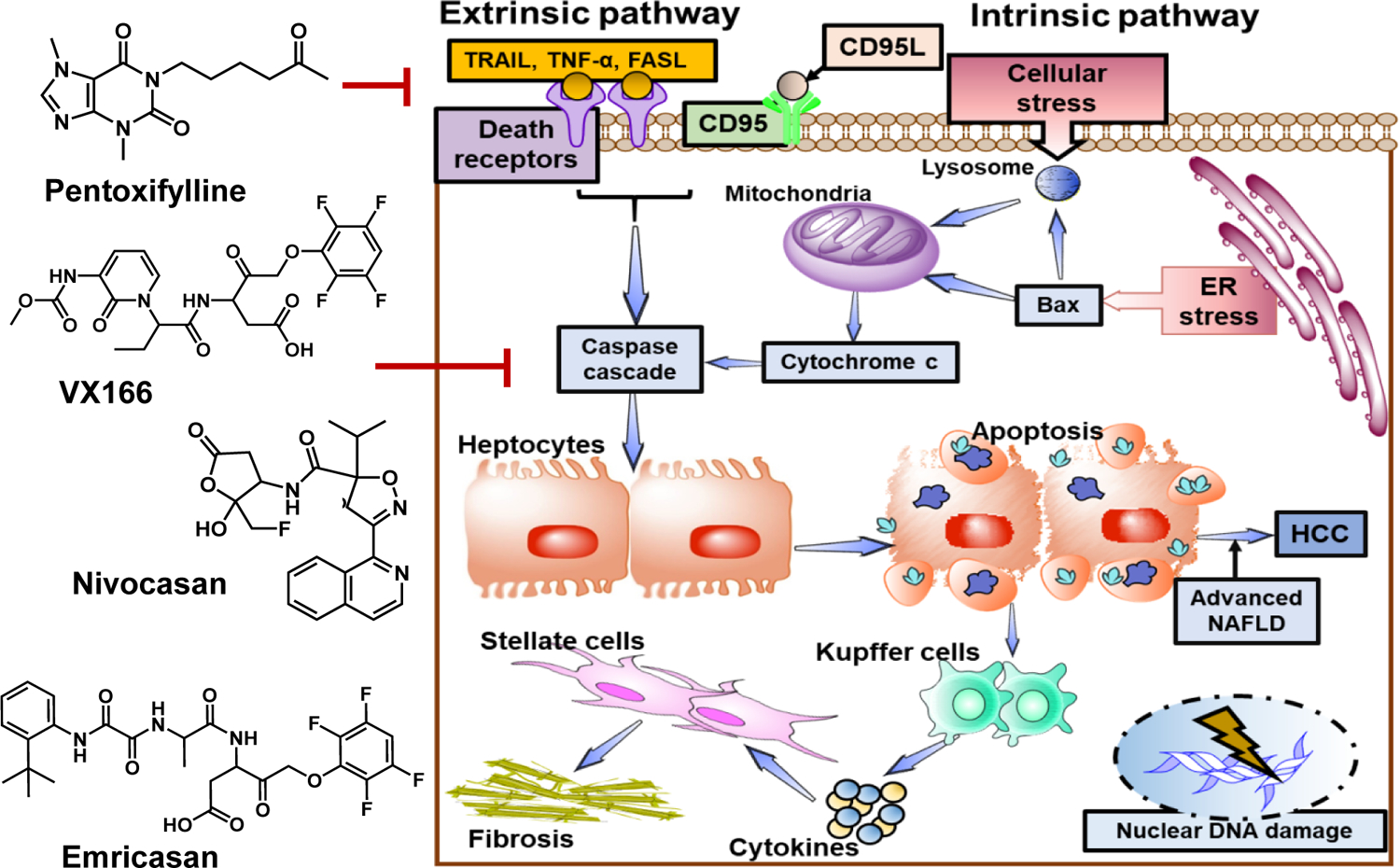
Lipids accumulation in hepatocytes makes them more susceptible to multiple secondary hits such as ROS and LPS that can lead to hepatocyte apoptosis and NASH progression. Apoptosis can occur by two main mechanisms: extrinsic and intrinsic pathways. In the extrinsic pathway, death receptors including Fas, TNFR1, and TNF-related apoptosis-inducing ligand (TRAIL) are activated. These receptors initiate intracellular cascades that activate death-inducing proteolytic enzymes, especially caspases. APO-1/Fas (CD95) -mediated apoptosis is one of the mechanisms for hepatocyte apoptosis. Intrinsic apoptotic pathway is initiated by damage of the intracellular organelles such as mitochondria, lysosomal permeabilization, ER stress, and nuclear DNA damage. Blocking apoptosis pathways may prevent hepatic fibrosis by reducing inflammation. Emricasan a pan-caspase inhibitor show antiapoptotic and anti-inflammatory effects. It was evaluated in a randomized, double-blind Phase 2b clinical trial in patients with decompensated NASH.
Several other apoptosis inhibitors like triacsin C, baicalin, and resveratrol have been evaluated in preclinical models of acute liver injury [169]. However, their efficacy and safety in chronic liver disease are not known. Inhibition of caspase activity might be ineffective in chronic liver injury, as caspase inhibition may promote premalignant cells and could potentially exacerbate the risk of cirrhosis and HCC. Another concern regarding the safety of caspase inhibitors is ALT overshoot after treatment withdrawal. The pan-caspase inhibition escape apoptosis of hepatocytes upon withdrawal, leading to massive apoptosis, which could result in acute liver failure [170].
3.2.4. Apoptosis signal-regulating kinase 1 inhibitor
Apoptosis signal-regulating kinase 1 (ASK1) is a member of the mitogen-activated protein kinase (MAPK) family, which executes intracellular signaling in response to various extrac ellular stimuli [171]. There are three leading MAPK families in this pathway: extracellular signal-regulating kinase (ERK) family, c-Jun N-terminal kinase (JNK) family, and p38 MAPK family. Each MAPK family is activated by distinct stimuli and changes the cell physiology. Other than stress, specific ligands such as death-associated protein 6 also known as DAXX, which is a Fas adapter protein, and TNFR-associated factor 2 also activaes ASK1 to induce apoptosis [172]. ASK1/JNK cascade phosphorylates and inactivates the antiapoptotic protein Bcl-2 [173]. In oxidative stress conditions of hepatocytes, ASK1 mediates phosphorylation of p38 MAPK leading to apoptosis and worsens fibrosis [174]. Therefore, inhibition of ASK1 could be an attractive approach to prevent hepatocyte death and treatment of NASH. In a preclinical study, small molecule ASK1 inhibitor selonsertib showed significant improvement in metabolism and NASH features such as hepatic steatosis, inflammation, and fibrosis. However, selonsertib, when tested in two randomized phase 3 trials for the anti-fibrotic efficacy in patients with NASH with bridging fibrosis (STELLAR-3 trial), and patients with compensated cirrhosis (STELLAR-4 trial), the drug failed to reach the primary efficacy endpoint of fibrosis.
Selonsertib has also been evaluated preclinically in combination with another drug for the treatment of fibrosis. Lysyl oxidase‐like molecule 2 (LOXL2) is an enzyme that catalyzes the crosslinking of collagen and elastin, leading to the stabilization of the ECM [175]. A humanized monoclonal antibody against LOXL2, simtuzumab, was tested for antifibrotic effect in the NASH model. However, in clinical studies of simtuzumab, monotherapy was not encouraging, and patients with F3 fibrosis (NCT01672866) and cirrhosis (NCT01672879) showed no benefits in reducing hepatic fibrosis. Further, selonsertib, in combination with simtuzumab, showed additive effects in a murine model of advanced fibrosis [176]. In another study, the combination of selonsertib and an acetylcoenzyme A carboxylase (ACC) inhibitor GS-9674 resulted in more significant anti-fibrotic activity than either agent alone in a rodent model of advanced fibrosis supported clinical evaluation of the combination. Selonsertib with GS-9674 and GS-0976 are currently being evaluated in a Phase 2 study in patients with NASH (NCT03449446).
3.3. Molecules affecting inflammatory pathways
NASH is characterized by extensive inflammation in the liver. Although lipotoxicity resulting from the build-up of proinflammatory lipids are the major cause, the inability of hepatocytes to cope with an increased metabolic load leads to ER stress, metabolic dysfunction, and production of ROS, which exacerbation of the hepatic inflammatory response [177]. Injured hepatocytes release DAMPs (damage-endogenous-associated molecular patterns) that activate proinflammatory signaling pathways via TLRs. Subsequent activation of KCs releases the proinflammatory cytokines and ligands. Several pathways including leukocyte extravasation signaling, chemokine signaling, production of nitric oxide, ROS, and Fcγ receptor-mediated phagocytosis in macrophages and monocytes are believed to play an important role in the inflammatory response in NASH. Inhibitors of these pathways thus represent an important tool for therapeutics development.
3.3.1. Amine oxidases inhibitors
Amine oxidase (AO) catalyzes the oxidation of endogenous amines such as histamine or dopamine. AO can be either flavin or copper dependent. Four amine oxidase copper (AOC) containing genes are known to code for different enzymes in humans. AOC-1 encodes for a diamine oxidase (DAO), possesses a histamine metabolism activity, and is predominantly found in the kidney, placenta, intestine, thymus, and seminal vesicles. AOC-2 encodes for amine oxidase, which metabolizes larger monoamines and is mainly found in the retina. AOC-3 is the best characterized AOC known as vascular adhesion protein-1 (VAP-1) and semicarbazide-sensitive amine oxidase (SSAO). AOC-3 is found in the adipocytes, smooth muscle cells, endothelial cells, liver, lung, aorta, and ileum. It is involved in leukocyte migration during inflammation and implicated in many human diseases.
AOC-3 protein has a membrane-bound distal site and a catalytic amine oxidase site. Both sites are critical for inhibiting leukocyte rolling, adhesion, and transmigration in response to inflammatory stimuli. Active AOC-3 catalyzes the oxidation of primary amines to aldehydes, releasing ammonia and hydrogen peroxide upon regeneration of the cofactor. At high concentrations, these compounds are cytotoxic and contribute to the pathogenesis of different vasculopathy and inflammation. Methylamine and aminoacetone are the endogenous substrates for SSAO/VAP-1. Active AOC-3 is upregulated in the liver and renal fibrosis and could be used as a biomarker of disease progression (Figure 6).
Figure 6. Various vascular adhesion protein (VAP-1) and semi carbazide-sensitive amine oxidase (SSAO) inhibitors for treating NAFLD and fibrosis.
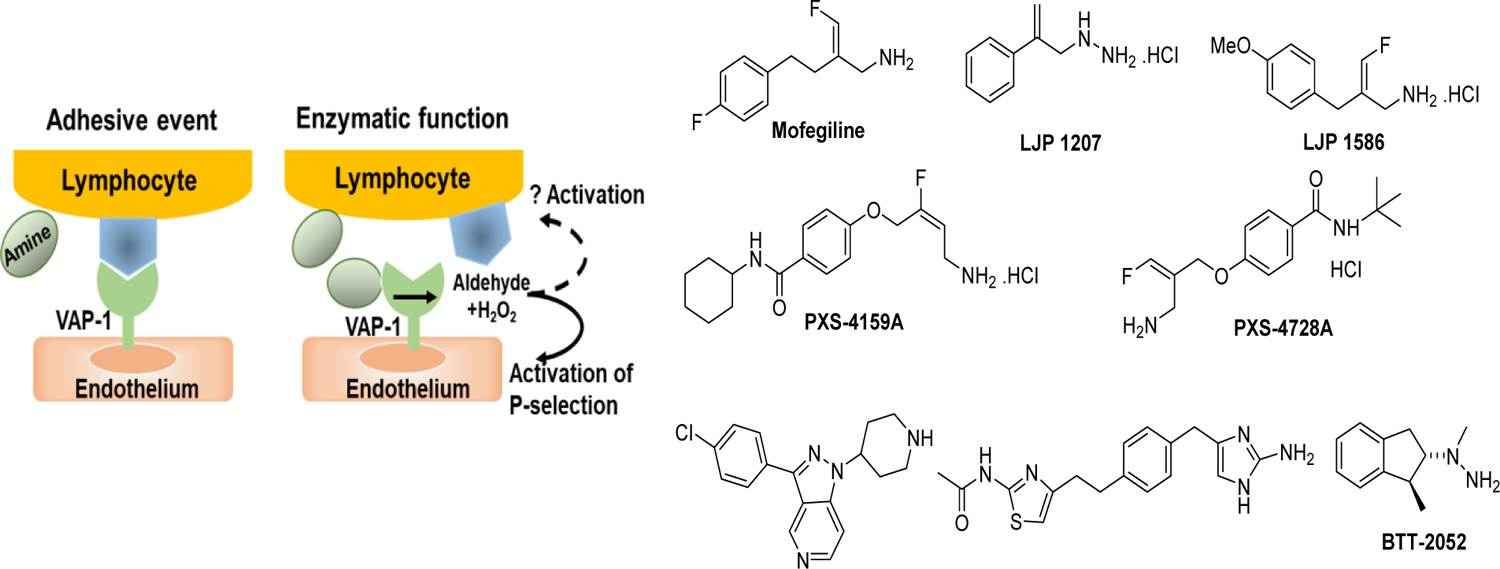
Neutrophil’s activation is a characteristic feature of inflammatory liver disease. Neutrophils can perform a series of functions, including degranulation, reactive oxygen species (ROS) generation, phagocytosis, and the formation of neutrophil extracellular traps (NETs)Upon injury, endothelial cells express P and E selectins, which serve as an anchoring ligands for neutrophils to adhere the endothelium layer. Amine oxidase (AO) are the enzymes that catalyze the oxidation of endogenous amines. One of the isoform AOC-3 also known as VAP-1 and SSAO catalyzes oxidation of primary amines of lymphocytes and enables them to transmigrate through the endothelial cell lining into the underlying parenchyma. In contrast, neutrophils can also inhibit liver injury by phagocytosing necrotic cellular debris and secrete hepatocyte growth factor (HGF) to induce hepatocyte regeneration. To inhibit neutrophil migration via their adhesion process, several small molecules are being developed. PXS-4728 was evaluated under a phase 2a study in patients with NASH. Although the treatment was well tolerated and showed inhibition of AOC3 activity in the plasma, compared to placebo, it was not developed further due to drug-drug interactions.
NASH is predominantly characterized by neutrophil infiltration and responsible for the non-specific immune response. In case of injury, neutrophils upregulate cell surface receptors such as CD44 and CD11b, which subsequently help their binding to endothelial cell surface adhesion molecules such as E-selectin, P-selectin, VAP-1/SSAO, and ICAM-1. This interaction enables neutrophils to transmigrate through the endothelial cell lining into the underlying parenchyma. Neutrophils release several inflammatory mediators and proteinases that contribute to progressive fibrosis and destruction of the liver parenchyma [178]. VAP-1 is a membrane-bound amine oxidase that promotes leukocyte infiltration to the liver. Hepatic VAP-1 expression is increases in patients with chronic liver disease including NAFLD. To inhibit VAP-1 activity, a binding peptide Siglec-9 was developed for imaging of inflammation and cancer [179]. This peptide binds to VAP-1-positive vessels in rheumatoid synovium, and the peptide conjugated with 68Ga-DOTA has recently successfully passed the phase 1 clinical trial. Several small molecules are being developed to inhibit neutrophil migration via their adhesion [178]. PXS-4728A is a small molecule inhibitor of VAP-1/SSAO enzymatic function.
However, the development of several of these molecules as therapeutics has been hampered by low selectivity, pharmacological differences across species, and poor PK properties. The crystal structure shows that SSAO/VAP-1 has an elongated binding pocket with the co-factor and copper sitting deep within the pocket and can accommodate more bulky groups. Mofegiline is a selective, irreversible of monoamine oxidase B (MAO-B) and semicarbazide-sensitive amine oxidase (SSAO). Mofegiline with a phenylethyl moiety tethered to the 2-position of 3-fluoroallylamine shows good interaction with the receptor. Mofegiline, when substitution at either the 3 or 4-positions of a benzene ring with hydrophilic polar group decreases its receptor binding. However, substituting secondary amides with larger lipophilic groups resulted in a potent and selective compound PXS-4159A (10 nM) [(E)-4-(4-amino-2-fluorobut-2-enyloxy)-N-cyclohexylbenzamide [180]. PXS-4159A showed a promising in vivo PK parameter. However, it suffered from a narrow therapeutic window in the mouse model because of low efficacy against rodent protein. A new allylamine-based inhibitor, PXS-4681A [(Z)-4-(2-(aminomethyl)-3-fluoroallyloxy)benzenesulfonamide], has been synthesized with ideal PK properties. PXS-4728 was initially developed for treating NASH and subsequently for diabetic retinopathy. The compound testing was discontinued primarily because of the risk of dose-dependent drug interactions. LJP1207, hydrazine derivative, displayed antiinflammatory activity but has the potential for toxicity upon prolonged administration [181]. Substitution of methoxy on benzene ring resulted in LJP 1586 [Z-3-fluoro-2-(4-methoxybenzyl)allylamine an orally active compound with reasonable inhibition of SSAO with IC50 of 4 nM. SzV-1287 [3-(3,4-diphenyl-1,3-oxazol-2-yl)propanal oxime] is another new molecule with VAP-1 inhibition and analgesic properties [182]. PXS-4728 was evaluated under a phase 2a study in patients with NASH. Although the treatment was well tolerated and showed inhibition of AOC3 activity in the plasma, compared to placebo, it was not developed further due to drug-drug interactions (NCT03166735) (Figure 6).
3.3.2. Chemokine signalling inhibitors
A chronic low-grade inflammatory response is directly connected to obesity, which increases the infiltration of macrophages in the adipose tissues. Macrophages can be either proinflammatory (M1) or anti-inflammatory (M2) [183]. In the fat tissues, an increase in M1 macrophages and a decrease in M2 macrophages is evident [184]. Specific chemokines and cytokines promote the M1 state. Chemokines stimulate the chemotaxis of target cells by activating mitogenic responses through specific G protein-coupled C-C chemokine receptors (CCRs). The chemokine monocyte chemoattractant protein (MCP)-1 and its receptor CCR2 increase the M1 population in fat tissue, as CCR2−/− mice show resistance to this switch [185].
Chemokine receptor-ligand interaction is crucial for the development of insulin resistance and hepatic steatosis in an HFD-induced obese mouse [186, 187]. CCR consists of seven transmembrane domains and amino and carboxyl termini.[188] Inhibition of CCR2 with propagermanium could prevent insulin resistance and steatosis in mutant diabetic as well as in wild-type mice [189, 190]. CCL2 can be released by KCs, stressed hepatocytes, stressed adipocytes, or activated HSCs. CCL2 mobilizes CCR2+ monocytes into the circulation and recruit them into the tissue.
Other than CCR2, genetic deletion or inhibition of CCR5 also results in reduced inflammation and fibrosis progression in the liver. CCR5 has widely expressed in the liver non-parenchymal cells and helps in recruiting lymphocytes in the liver. CCR5 activation recruits lymphocytes and stimulates the migration of HSCs to the injury site, which starts the accumulation of KCs [191]. CCR1 is also implicated in macrophage migration to the injured liver and the promotion of HSC activation. Although drugs designed to inhibit CCR1/2/5 is expected to dampen inflammation, and fibrogenic activation of HSCs, the high level of redundancy within the chemokine network makes this task a bit challenging. For example, CCR1 can not only interacts with CCL1 but additionally with CCL3/4/5,[192] CCR8 with CCL1 [193], CX3CR1 with CX3CL1 [194], and CXCR3-CXCL10 [195]. Thus, the effect of inhibition of one CC pathway is compensated by alternative CC. P-selectin glycoprotein ligand 1 (PSGL-1/CD162) and Mac-1 (CD11b/CD18) are the two attractive targets. Studies have shown that CXCR2 deficient mice or treated with CXCR2 antagonists; SCH527123, AZD8309, and SB-656933 inhibit lipopolysaccharides (LPS) induced inflammation due to neutrophils [196, 197].
CCR2 and CCR5 receptors share almost 72% sequence identity and are expressed on different cells in a complementary manner. Dual inhibition of these receptors, therefore, has been implemented for antiinflammatory therapy. The compound benzo[7]annulene (TAK-779) represents a potent, first non-peptide dual CCR5 and CCR2 receptor antagonists with IC50 = 1.4 nM, and 27 nM respectively (Figure 5) [198]. However, due to the quaternary ammonium group of TAK-779, its oral bioavailability was very low (~10%) and found irritant at the injection site [199]. The analog bearing polar sulfoxide instead of quaternary ammonium group TAK-652 (2) showed similar efficacy (IC50 = 3.1 nM for CCR5 and IC50 = 5.9 nM for CCR2 receptors) [200]. Structure-activity relationship (SAR) studies conclude that a compound with a reduced ring from a benzazocine to a benzazepine had similar CCR5 affinity while introducing an isobutyl side chainenzazocine ring increased CCR5 binding affinity. Cenicriviroc (CVC, TAK-652), is an orally active molecule with a mean half-life of ~40 h, and an excellent safety profile. In a recent clinical phase 2b study (Centaur), demonstrated an improvement in liver fibrosis at least one stage after a year of therapy. Oral administration of CVC at the dose of 150 mg once a day is being tested in a phase 3 trial, dubbed AURORA in NASH patients [201].
CCR inhibitors, in combination with other liver metabolism targeting drugs such as FXR agonist Tropifexor is being tested in a clinical trial (TANDEM). TANDEM (NCT03517540) has now progressed to Phase 2. Another nonsteroidal FXR agonist Cilofexor (GS-9674) demonstrated the biological activity and safety profile in healthy volunteers and strengthened its evaluation in patients with NASH and cholestatic liver disorders. Phase 2 studies with Cilofexor are ongoing in patients with NASH, primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC) [202]. Further, its combination with PPAR agonists or FGF-21 could be logical for treating steatohepatitis. [203].
3.3.3. Adenosine receptor agonists
Adenosine receptors (ARs) are G protein-coupled receptors (GPCRs) found in the human lung, liver, brain, aorta, and testis. ARs are involved in a variety of intracellular signaling pathways and physiological functions. ARs are a family of four subtypes; A1, A2A, A2B, and A3, where A1 and A3 ARs inhibit the adenylyl cyclase (AC) activity, whereas A2A and A2B increase the cAMP levels. Of these, A3AR and A2AR are of particular interest in cancer and fibrosis. A3AR is overexpressed on inflammatory cells and peripheral blood mononuclear cells (PBMCs) compared to healthy cells. The antiinflammatory effects of A3AR have been proven in preclinical models of inflammatory bowel disease (IBD), systemic toxemia, pulmonary inflammation, rheumatoid arthritis (RA), osteoarthritis, and liver inflammation.
The antiinflammatory activity of A3AR is through the deregulation of NF-kB signaling pathway, leading to inhibition of TNF-α, IL-6, IL-12, macrophage inflammatory proteins (MIPs)-1a, MIP-2 and receptor activator of NF-κB ligand (RANKL), resulting in apoptosis of inflammatory cells. Under stressed metabolic conditions, TNF-α upregulates protein kinase B (PKB)/AKT, which is an inhibitor of NF-kB light polypeptide gene enhancer in B cells (IkB) and IkB kinase (IKK). Upregulated NF-kB acts as an A3AR transcription factor. Further, transcription factors, such as c-Rel, MyoD, c-fos, GR, CREB, AP-1, GATA-1, C/EBP, c-Jun, and PU.1 are also known to bind the A3AR promoter region (Figure 8A). A3AR agonists administered in combination with chemotherapeutic agents to tumor-bearing mice prevented myelotoxic effects and prevented a decline in white blood cell (WBC) and neutrophil counts, resulting in full recovery of myeloid system parameters. This effect is because A3AR agonists induces the production of granulocyte colony-stimulating factor (G-CSF), which stimulates myeloid progenitor cell expansion in the bone marrow and increases the WBC and neutrophil counts in the peripheral blood. Additionally, A3AR agonist can protect against mitochondrial damage and helps preserve ATP production in immune cells. Caffeine, the major component of coffee, has AR antagonist properties and is found to significantly diminish hepatic injury and chronic liver disease [204].
Figure 8. Adenosine receptor (AR) agonists for treatment of NAFLD.
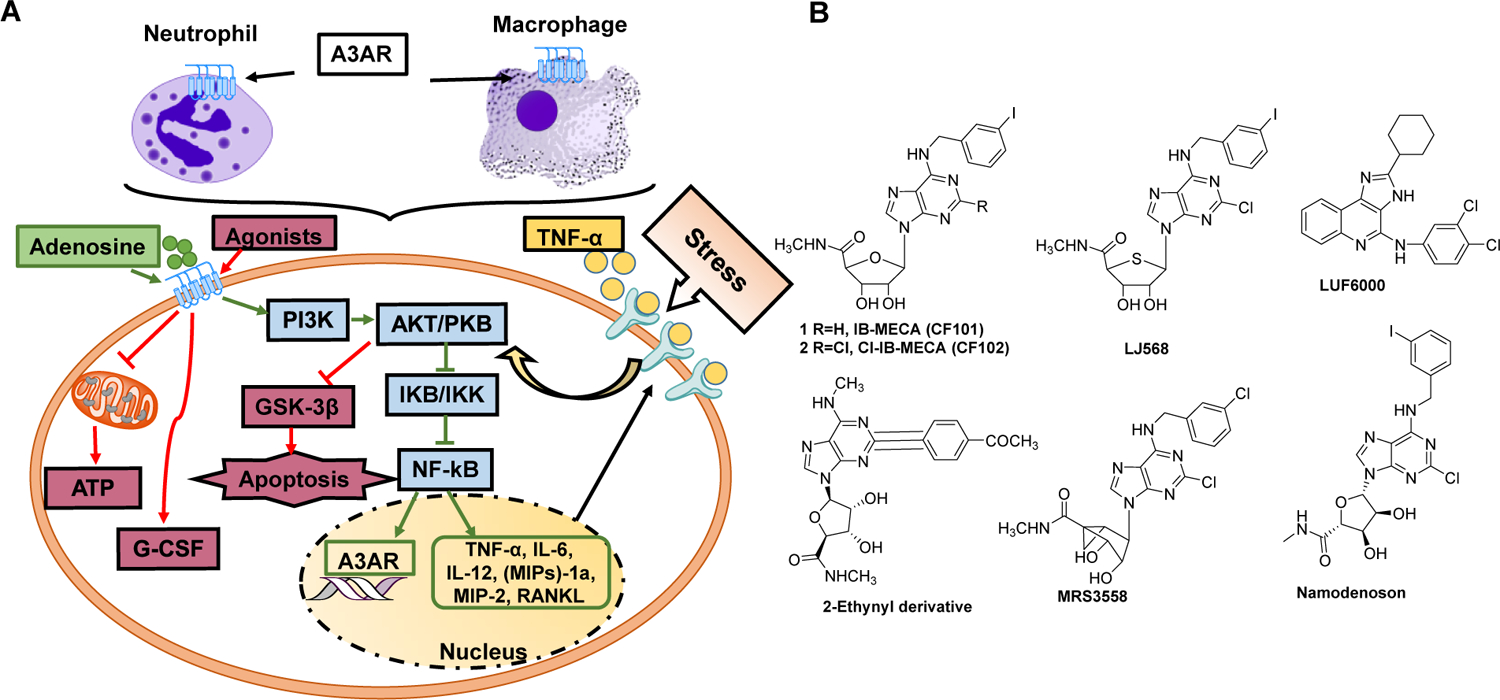
A) The role of ARs in inflammatory responses in NAFLD. ARs are G protein-coupled receptors (GPCRs) that regulate adenylyl cyclase (AC) activity. A3AR stimulation triggers increase of AC activity and cAMP production. cAMP activates protein kinase B (PKB), which is a negative regulator of inflammatory NF-kB pathway and glycogen synthase kinase 3 beta (GSK-3β) mediated cell apoptosis. B) There are various AR inhibitors. Namodenoson, a potent and selective A3AR agonist was evaluated in Phase 2 trials for the treatment of NAFLD and NASH. The drug successfully met the endpoints with 60 patients and the optimal dosage. The Phase IIb NASH trial of namodenoson is currently underway.
Although A3AR is an important target in drug development, several major problems complicate the development of AR agonists in general: (1) AR expression is ubiquitous in the body and may result in diverse side effects. (2) The low density of ARs in the target can reduce its desired treatment. (3) In general, nucleoside derivatives possess low efficacy (the ability to induce the required conformational change of the receptor), and their binding to receptors exert only a partial agonistic effect. (4) GPCRs are insoluble in environments lacking phospholipids. Thus, obtaining their 3D structural configuration is difficult by using standard structure determination techniques such as X-ray and NMR studies. (5) GPCRs are dynamic proteins, and ligand binding can induce a spectrum of conformations changes, and each confirmation is associated with its own signaling profile. Thus, the effect of various adenosine substitutions on affinity and relative maximal efficacy is a comparison by determining the concentration of activating the second messenger to a known full agonist [205].
A3AR agonists have been modified at the N6 and C2 positions of adenine as well as the ribose moiety and using a combination of these substitutions to optimize their interaction with A3AR. The binding of a nucleoside to A3AR and its activation of the receptor are separate processes and represent a distinct structural requirement (Figure 8B). With a small group such as methyl at the N6 position, affinity to hA3AR is reduced by some groups at C-2 such as trifluoromethyl, amino, aminomethyl, and N-methyl-carboxamidine (NMPC) and enhanced by another group such as cyano. With a larger N6-substituent, the 2-cyano substitution did not enhance the affinity but instead resulted in a reduction in the ability to activate the receptor.[206] There is no correlation between the affinity of a given nucleoside derivative in binding to A3AR and its ability to fully vs. partially activate the receptor. For instance, N6-benzyl and certain 2-position substituents on the adenine moiety reduce the relative efficacy at A3AR. 2-Chloro alone does not reduce A3AR efficacy, but in combination with a substituted N6-benzyl moiety, it leads to a further reduction. IB-MECA has an N6-iodobenzyl group, and a 5′-methylcarboxamido group, whereas in Cl-IB-MECA a 2-chloro group was introduced makes it more selective than IB-MECA. IB-MECA was tested in various conditions, which included RA, dry eye syndrome, and psoriasis. Oral administration of IB-MECA was safe and well-tolerated and resulted in improvement in some symptoms of diseases in various clinical trials [207]. Substituted on MECA structure bearing a methyl group in the N6-position and 2-pyridinyl)ethynyl substituent showed a remarkable selectivity and 7- to 8-fold more potency than Cl-IBMECA [208].
Modifications of the ribose moiety specifically on 5′ region is a prerequisite for A3AR activation. Conformational studies of the ribose moiety and its equivalents indicate that the ring oxygen is not required, and that the North (N) ring conformation is preferred in binding to A3AR. One means of locking the conformation of the ribose-like ring is through the use of bicyclo[3.1.0]hexane ring system, which assumes an (N)-envelope conformation. Based on these observations, CF502 (MRS3558, N-methanocarba-5′-uronamide derivatives was reported to be highly potent [209]. Further, LJ529 (2-chloro-N6-methyl-4’-thioadenosine-5’-methyluronamide) with the 4’-thionucleosides was found to be a potent compound [210]. A flexible 5′ -uronamide moiety is particularly well suited to maintaining efficacy and even overcomes the reduction of efficacy induced by various adenine substituents at the N6 and C2 positions. CP-532,903 [N6-(2,5-Dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] with an amine on 4’thioadenosine showed high potency to A3AR [211]. One family of selective A3AR antagonists comprises the derivatives of 1.4-dihydropiridines, which are known as inhibitors of L-type Ca2+ channels. After various modifications, including the introduction of a 6-phenyl group, these molecules bind with high affinity and selectivity for the human A3AR.
Several synthetic AR antagonist molecules have been developed, which are being tested in preclinical or clinical studies. Namodenoson is a synthetic adenosine derivative that binds with high selectivity to A3AR in pathological liver cells, inducing robust anti-inflammatory and anti-cancer effects. Namodenoson was tested in Phase 2 (60 patients) double-blind, placebo-controlled, dose-finding efficacy, and safety study enrolled 60 patients with NAFLD with or without NASH. The study shows that treatment with Namodenoson twice daily with 12.5 mg (n=21) or 25 mg (n=19) achieved its efficacy endpoints while continuing to demonstrate a good safety profile (www.can-fite.com).
Another critical member of this family is A2AR, which is activated upon liver injury and fibrosis. A2AR activation enhances HSC activation, and A2AR deficient mice are protected from CCl4 or thioacetamide-induced liver fibrosis. Further, A2AR helps in the hypoxic preconditioning of hepatocytes through the PI3K/PKB/AKT pathway and increases angiogenesis post-liver injury. A2AR inhibits proinflammatory cytokine formation in monocytes and macrophages acting via adenosine. Mice with macrophages deficient in A2AR display elevated and prolonged production of proinflammatory cytokines, including TNF-α and IFN-γ, in response to challenge with lipopolysaccharide (LPS) [212].
3.3.4. Bromodomain containing protein inhibitors
Bromodomain (BRD) containing proteins are transcriptional enhancers from the bromodomain and extra terminal domain (BET) family. The BET family of proteins including BRD2, BRD3, BRD4, and BRDT (germ cell-specific) contain N terminal tandem bromodomains, a structural feature that enables to bind acetylated histones in the promoter of genes and regulate their expression. BET proteins regulate the transcription of genes needed for normal development, maintenance of oncogenic gene expression, and physiological response to injury and infection. BET proteins contain first and second bromodomains (BD1 and BD2), each with a different role in the activity. BD1 domain is mainly involved in the maintenance of gene expression, including for cancer, whereas BD2 domain predominantly affects the rapid induction of genes such as inflammatory and autoimmune responses. After inflammatory stimuli such as cytokine (including IFN-y, Th17, IL-6) stimulation, the BET proteins are specifically recruited to target the gene prom oter region to induce gene expression. Specific BD2 inhibition has been shown to dampen inflammation in preclinical arthritis, psoriasis, and hepatitis. There are mainly two types of BETi under development. First, the type of molecules interacts with BET protein binding domains and inhibit their interaction with acetylated histones, including JQ1, OTX-015, I-BET762, BY27, RVX-208, GSK340, and ABBV-744. The second type of inhibitors are proteolysis-targeting chimeras (PROTACS) molecules, which binds to the target protein and recruits the E3 ligase complex and degrades BET protein after ubiquitination. These include dBET1, ARV-771, QCA570, BETd-260, and A1874 [213]. These PROTAC molecules usually consist of three parts: a target protein-ligand, an E3 ubiquitinated ligase ligand, and a ligation module. Among all these BET inhibitors (BETi), the selective BD1 (RVX 201, PLX51107) and BD2 (ABBV744) binding molecules are also undergoing clinical investigation.
BET activity is directly related to the gene expression signatures of steatosis, liver fibrosis, and patient-derived NASH [214]. BET promotes interferon-related proinflammatory pathways by upregulating expression of genes such as CXCL10 and STAT1. It has been shown that chemokine CXCL10 induces inflammation (TNF-α, IL-1β, and MCP-1) and regulates lipogenesis (SREBP-1c) and oxidative stress (CYP2E1 and C/EBPβ) in NASH. STAT1 upregulation has been shown to increase hepatocyte apoptosis and increase disease severity [215].
Transcriptomic profiling of diethylnitrosamine (DEN)/choline-deficient, L-amino-acid-defined HFD (CDAHFD) mouse livers show gene expression profile like patients with NASH. Treatment with JQ1 significantly reduced liver fibrosis measured by collagen deposition and expression of genes mediating fibrogenesis (ACTA2, TGFβ1 and TIMP1) as well as liver inflammation (CCL2 and TNF-α expression).
Among all BET proteins, research has focused mainly on the coactivator effects of BRD4 transcriptional functions because of its requirement to maintain oncogenic transcriptional programs. BRD4 binding to histone H3 lysine 27 acetylation (H3K27ac) is an epigenetic mark for active enhancers for several profibrotic transcription factors such as ETS1 and SRF SMAD3, and NF-κB. Gene ontology (GO) analyses of BRD4 putative target genes include the major profibrotic pathways such as focal adhesion, ECM-receptor interaction, integrin signaling, smooth muscle contraction, platelet-derived growth factor-beta (PDGF) signaling, NF-κB signaling, and JNK/MAPK signaling. BRD4 inhibitor JQ1 reverted the transcriptional liver programming of NASH, including the EMT, inflammation, IL2/STAT5, oxidative phosphorylation, bile acid, and fatty acid xenobiotic metabolism, and allograft rejection pathways. BRD4 inhibition mainly (~60%) reverted expression of H3K27ac-altered genes. JQ1 treatment did not affect BRD4 levels; instead, significantly reduced its binding on SOX9, EPCAM, KRT7, and NF-κB2 promoter [216]. Lipid accumulation in the liver is controlled by the coordinated action of several genes, including CYP8B1, dihydroxyacetone kinases (DAK), and perilipin 5 (PLIN5). Cyp8b1 gene is involved in the regulation of cholesterol metabolism by generating cholic acid. The DAK gene product converts dihydroxyacetone to dihydroxyacetone phosphate in the glycolytic system. PLIN5 is found in the periphery of lipid droplets and facilitate TG synthesis. BRD4 enhances the transcription of these three genes in fructose-force-fed mice compared to glucose-force-fed mice. BRD4 binding to the promoters of CYP8B1, DAK, and PLIN5 genes by JQ1 were found to suppress their expression [217].
BRD4 acts as a mitogenic regulator in HSC proliferation and promotes COL1A1 expression in response to TGF-β1 signaling. JQ1, a small molecule BRD4 inhibitor as a treatment for CCl4 induced liver fibrosis. JQ1 treatment in vitro suppressed the induction of key marker genes, including COL1A1, COL1A2, and α-SMA. Besides, early treatment with JQ1 could dramatically inhibit HSC activation and reverse or at least prevent further progression of liver fibrosis as indicated by reduced collagen and profibrotic markers. These findings strongly support that BRD4 is a therapeutic target for liver fibrosis [218]. Yamada et al. reported induction of genes related to lipid accumulation in the livers of mice force-fed with fructose [217]. Systemic administration of JQ1 inhibited histone acetylation and inhibited lipid accumulation in the liver.
Further, Jung et al. determined the role of BRD4 on FXR function in bile acid regulation and whether the beneficial effects of OCA are reduced by inhibiting BRD4 in cholestatic mice [219]. Inhibition of BRD4 disrupted bile acid homeostasis in mice, and FXR-mediated regulation of bile acid-related genes was BRD4-dependent. JQ1 or OCA treatment ameliorated hepatotoxicity, inflammation, and fibrosis in cholestatic mice but was antagonistic in combination. It was found that FXR mediates bile acid metabolism through the small heterodimer partner (SHP), which is a key regulator of bie acid. BRD4 epigenetically coactivates the induction of SHP. Inhibition of BRD4 with JQ1 abolished OCA-induced anti-inflammatory and anti-fibrotic effects.
3.3.5. Galectin-3 receptor inhibitors
Oxidation of excessive accumulated fat results in advanced lipoxidation end-product (ALE) and deposition of advanced glycation end-products (AGEs) in the liver. Receptor-mediated endocytosis of ALE triggers several signaling pathways leading to chronic low-grade inflammation and contribute to NASH. Also, the scavenging function of KCs is impaired, and result in the deposition of ALEs in various extra-hepatic organs, including vessels and increase the risk of NAFLD related cardiovascular risk [220]. FA metabolites induce ER stress and increased production of reactive oxygen species (ROS) by mitochondrial β-oxidation. When ER capacity is overwhelmed by an excess of fat, ROS are produced by peroxisomal β-oxidation and ER ω-oxidation. In positive feedback, ROS trigger κB kinase-β/NF-κB/TNF-α cycle and initiates and maintains a T helper 1 (Th1)- and macrophage 1 (M1)-mediated inflammation.
There was significant increase in the plasma levels of ALE/AGE in patients with cirrhosis, which correlates well with the disease severity [221]. ALEs/AGEs are selectively recognized by scavenger receptors (SRs), TLRs, and AGE receptors present in different liver cell types. For instance, macrophages express SR-A and SR-A/CD36 receptors for ALE endocytosis. Hepatocytes predominantly express the receptor for AGE (RAGE), whereas galectin-3 is overexpressed in KCs and SLECs. Levels of both RAGE and galectin-3 receptors increase with the extent of liver damage. RAGE is upregulated during HSC activation, and its blockade reduces experimental hepatic fibrosis. Galectin-3 promotes an M2 macrophage state in mice, which is associated with increased fibrosis of the liver, lung, kidney, heart, and vascular system [222, 223]. While deletion of galectin-3 gene blocks TGF-β–mediated HSC activation and ECM production. However, some studies have shown that mice with galectin-3 disruption develop NAFLD/spontaneously with aging, thus warrant further investigation to define its exact role.
Galectin-3 plays an important role in the uptake of ALE/AGE by LSECs, and its ablation resulted in reduced accumulation within the liver and increased serum levels of these end-products compounds [220]. Galectin-3 also modulates VEGF/VEGFR-2 signaling pathway in endothelial cells in response to hypoxia, and its deficiency attenuates inflammatory angiogenesis in vivo [224, 225]. Several galactin-3 inhibitors are being tested in preclinical and clinical setup for antifibrotic effect in different organ fibrosis. Two approaches have been taken to develop drugs that bind galactin-3: large polysaccharides that contain galactose (GR-MD-02, belapectin; Galectin Therapeutics), and modified disaccharides (TD139; Galecto Biotech). GR-MD-02 is a modified, naturally occurring galactoarabino-rhamnogalacturonan polysaccharide (50 kDa), that contains side chains of 1,4-β-D-galactose (Gal) and 1,5-α-L-arabinose (Ara) galactose as a drug that binds to galactin-3.[226] TD139 is disaccharide 1,1′-sulfanediyl-bis-{3-deoxy-3-[4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl]-β-d-galactopyranoside} molecule. Both molecules are not well absorbed orally and must be given parenterally, although TD139 could also be delivered by inhalation for idiopathic pulmonary fibrosis (IPF). TD139 has a higher affinity for the carbohydrate recognition domain of galactin-3, while per drug molecule of GR-MD-02 has ~5 molecules of galactin-3 binding domains. Regardless of these crucial differences, both molecules show a similar effect in a pulmonary fibrosis mouse model [227].
Recently, a phase 2b randomized trial of galentin inhibitor belapectin (GR-MD-02) a) was completed in patients with NASH, cirrhosis, and portal hypertension [223]. Patients with NASH, cirrhosis, and portal hypertension (hepatic venous pressure gradient [HVPG] ≥ 6 mm Hg) received biweekly infusions of 2 mg/kg, 8 mg/kg belapectin, or placebo for 52 weeks. The primary and secondary endpoints were changed in HVPG (Δ HVPG), and changes in liver histology at the end of 52-week period compared with baseline. Belapectin was safe in the patient when administered at the dose of 2mg/kg. However, there was no significant difference in ΔHVPG between belapectin groups at both doses and placebo groups. Further, belapectin treatment did not show any significant effect on fibrosis or NAFLD liver. In patients without esophageal varices at baseline, 2 mg/kg belapectin reduced HVPG at 52 weeks compared to baseline, and reduced development of new varices. The company plans to advance belapectin into phase III testing.
A synthetic low-molecular weight carbohydrate-based compound lactulosyl-L-leucine (Lac-L-Leu) has been shown to have galactin-3 antagonizing properties [228]. Further, Lac-L-Leu synergistically augmented paclitaxel’s efficacy in human cancer cells and inhibited clonogenic survival, and induced apoptosis in metastatic cells. Lac-L-Leu/paclitaxel combination was functionally linked with increased mitochondrial damage and caused a reversal and eradication of the advanced metastatic disease in 56% of experimental animals.
3.3.6. Phosphodiesterase inhibitors
Cyclic nucleotide monophosphates (guanosine; cGMP and adenosine; cAMP) are the second messengers of the cells and regulate metabolic hemostasis and inflammation. cAMP plays a pharmacological role in inflammation, cognition, lipogenesis, proliferation, apoptosis, and differentiation. Cellular cAMP levels are tightly controlled by a balance between inducing substances such as epinephrine and glucagon and degradation of cAMP into AMP by phosphodiesterases (PDEs). The superfamily of PDE enzymes comprises 11 isotypes (PDE1–PDE11), and each type differing in structure, substrate specificity, inhibitor selectivity, and differentially expressed in cells and tissues. cAMP specific PDE4 is overexpressed in immunocytes, including T cells, monocytes, macrophages, neutrophils, dendritic cells, and eosinophils. In the case of inflammation, the intracellular cAMP levels are found low; thus, inhibition of PDE4 is one of the best strategies to modulate the inflammatory responses because it bypasses the complex antigen receptor-specific immunoregulatory mechanisms [229].
Increased cAMP level leads to the release of a catalytic subunit from the regulatory subunit and exhibits the activation of protein kinase A (PKA), resulting in phosphorylation of downstream protein targets. Cyclic nucleotide modulators, including PKA, cyclic nucleotide-gated ion channels, and exchange factors (Epac1/2) are directly activated by cAMP and contribute to the formation of cAMP signalosomes. PKA regulates gene expression by mediating a transcription factor, cAMP response element-binding (CREB) protein, activating transcription factor 1 (ATF-1), and cAMP-responsive element modulator (CREM) and recruit CREB binding protein (CBP) or the homologous protein p300, leading to the reduction of inflammatory cytokines and increase in anti-inflammatory cytokines. Notably, the activity can be stimulated upon the phosphorylation of p65 on Ser276 by PKA, and PKA activation could regulate the transcriptional activity of NF-κB by modulating its interaction with CBP and p300 without IκBα degradation or NF-κB DNA binding activity, which results in the downregulation of inflammatory responses. Additionally, PKA activation could interfere with B-cell lymphoma 6 protein (Bcl-6)-mediated synthesis of proinflammatory cytokines and proliferation of immune cells. Increase in cAMP levels showed multiple effects on cell proliferation, differentiation, immune responses, increased glucose production, lipogenesis inhibition, and improved oxidative stress [230].
PDE4 is a downstream component of β-adrenoceptor, and N-methyl-D-aspartic acid (NMDA) receptor mediated signaling pathway. Receptors for activated C kinase 1 (RACK1) and Akinase-anchoring proteins (AKAPs) and proteins that contain SH3 domains act as the interacting proteins that affect the intracellular localization and function of PDE4. PDE4 inhibition modulates both innate and adaptive responses. For instance, PDE4 inhibition showed regulatory activities in macrophages, neutrophils, monocytes, and dendritic cells. Besides, PDE4 inhibition showed excellent effects on T cell receptor (TCR)-induced activation of T cells, resulting decreased cytokine and chemokine secretion from T helper 1 (Th1), Th2, and Th17 cells. Selective PDE4 inhibitors such as roflumilast, apremilast, and crisaborole have been developed and used in asthma, chronic obstructive pulmonary disease (COPD), psoriasis, atopic dermatitis (AD), inflammatory bowel diseases (IBD), rheumatic arthritis (RA), lupus, and neuroinflammation. However, most of these inhibitors are nonspecific and cause nausea, emesis, gastrointestinal effects, and other adverse effects which largely impeded the clinical application.
ASP9831 is a new PDE4 inhibitor with antiinflammatory properties mainly directed at activated macrophages and KCs. Upon stimulation by lipopolysaccharide (LPS), ASP9831 inhibits TNF-α production in the nanomolar range from both human and rat peripheral blood mononuclear cells (PBMCs). Further, ASP9831 showed a significant reduction in ALT activity in 3 distinct acute hepatitis models: D-galactosamine and D-galactosamine plus LPS in rats and concanavalin A in mice. In MCD induced NASH, ASP9831 administration at 1 and 3 mg/kg reduced 40% to 50% in ALT levels and improved histologic necroinflammation. Based on these data, ASP9831 was evaluated in Phase 1 and 2a clinical trials for anti-inflammatory and antifibrotic potential in patients with NASH [231]. Unfortunately, no significant change was observed for liver injury biomarkers and in the biomarker adiponectin, cytokeratin 18, and TNF-α between ASP9831 and placebo groups. Interestingly, while >50% inhibition of TNF-α expression was observed in healthy individuals treated with 100 or 200 mg ASP9831 daily but not in patients with NASH, possibly reducing TNF-α signaling through cAMP induction could trigger compensatory inflammatory pathways, resulting in TNF-α upregulation [168].
3.3.7. Anti-LPS hyperimmune bovine colostrum
Metabolic imbalance often leads to insulin resistance and inflammation which is characterized by immune cells such as CD4+ T lymphocytes infiltrate into the inflamed tissues. Regulatory T cells (Tregs, CD4+ Foxp3+) play an essential role in maintaining peripheral tolerance, preventing autoimmune diseases, and limiting chronic inflammatory diseases by preventing autoreactive T cells [232]. Accordingly, it is not surprising that a reduction of Tregs is observed in the fat of insulin-resistant models of obesity. Tregs induction can ameliorate the effect on the leptin-deficient (ob/ob) mouse model of T2DM associated with an increase in the number of splenic CD4+ CD25+, CD4+ CD25+ Foxp3+, and CD3+ NK1.1+ regulatory lymphocytes [233].
Bovine colostrum (BC) is the milk of lactating mammals that is secreted during the first 72 hours following birth. Colostrum delivers bioactive proteins that possess a wide range of biological activities that promote the normal development of innate immune system and maturation. Immunoglobulins (IgG) are the primary immune components of the acquired immune system presented in colostrum. The immunological activity of bovine IgG in milk from cows immunized against human pathogens is like that of IgG in human milk. For example, skimed milk from cows immunized with a polyvalent human gut bacterial vaccine was shown to reduce elevated blood cholesterol and TG concentrations significantly. IMM-124E is an anti-LPS hyperimmune BC. Treatment with IMM124-E showed a beneficial effect on the lipid profile and liver injury of treated patients. One month of oral treatment of 124-E affected Tregs, which is accompanied by an increase in serum IL-6 and a small increase in adiponectin levels. IMM124-E induces NKT cells and provides an ameliorating effect in ob/ob mice. The BC also neutralizes LPS and reduces the influx of LPS from the gut to inhibit enteropathogenic endotoxemia.
Oral administration of Imm124-E in patients with metabolic syndrome was safe and alleviated insulin resistance, liver damage, and hyperlipidemia [234]. These beneficial effects were also accompanied by increased serum levels of GLP-1, adiponectin, and Tregs. Colostrum contains a high amount of insulin-like growth factor (IGF-1), stimulating glucose utilization to alleviate diabetic symptoms. IMM-124E is tested orally three times a day in a phase II trial in patients with any stage biopsy-proven NASH by Immuron Ltd., (IMRN). The primary endpoint is the change in liver fat content confirmed by MRI and the change in ALT. In addition to the adult NASH study, IMM-124E is also evaluated in a phase II study in children with pediatric NAFLD.
3.4. Molecules affecting extracellular matrix/remodeling and HSC activation
Activated KCs in the inflammatory liver release profibrogenic cytokines to induce quiescient HSCs into activated myofibroblast phenotype. Activated HSCs produce high amount of extracellular matrix (ECM) leading to progressive fibrosis [235]. ECM is rich in fibrillary and nonfibrillar collagen and other proteins such as elastin, laminin, and fibronectin; hyaluronan, aggrecan, fibromodulin, decorin, biglycan, glypicans, and syndecans. ECM represents a complex network which meant to restore the architecture damaged tissue and provide the mechanical stability [236]. However, prolong ECM deposition increases its crosslinking that renders it more resistant to degradation, and often result in incomplete reversibility of advanced fibrosis. ECM contributes to typical complications such as portal hypertension and loss of functional liver mass.
Although HSC activation is a central process in all type of liver injuries, recent single-cell RNA sequencing study showed involvement of other cells type such as mesothelia/portal fibroblasts, vascular smooth muscle cells, and scar-associated mesenchymal cells in profibrogenic response [237]. ECM deposition is associated with dysregulation of ECM remodeling proteins, i.e., increased expression of TIMPs and inefficient removal of excess fibrillary collagen by metalloproteases (MMPs). Targeting mechanisms resulting in dysregulation of critical molecular pathways in activated HSCs or MFs represent a promising therapeutic approach. Several molecular pathways including hedgehog (Hh), renin-angiotensin, and TGF-β1 are known to participate in HSC activation and ECM secretion. Yet, there is a long road ahead to translate these molecular mechanisms for therapies in human.
3.4.1. Hedgehog pathway inhibitors
Sonic hedgehog (Shh) ligand is secreted by stressed hepatocytes, which leads to several events related to lipid metabolism and NASH development. Hedgehog (Hh) pathway includes three morphogen proteins; sonic Hh (Shh), Indian Hh (Ihh), and Desert Hh (Dhh), which regulate the developmental events in the embryo and wound healing process in adult tissue. In vertebrates, Hh ligand binding to its receptor ‘Patched’ evokes its inhibitory effects on the Smoothened (SMO) receptor and subsequently leads to GLI translocation into the nucleus. GLI is a transcription factor family consists of GLI1, GLI2, and GLI3. GLI2 and GLI3 are the primary effectors to induce Hh signaling, whereas GLI1 is their direct target and functions to amplify the transcriptional response. Moreover, GLI2 is an Hh dependent transcriptional activator, and GLI3 is a repressor of Hh signaling [238].
Hh pathway shows various effects related to glucose metabolism, insulin secretion, lipid generation, and adipocyte differentiation. For instance, SHH inhibits adipocyte differentiation by diverting preadipocytes away from adipogenesis and inducing white adipose tissue development. Hh signaling elicits these effects by inducing anti-adipogenic transcription factors such as Gata2 [239]. While the Hh-GLI2 axis drives lipogenesis in adipocytes, leading to increased obesity in adult mice. Further, postnatal constitutive activation of Hh pathway in adipocytes by either mutated SMO (SmoM2) or by overexpression of GLI2 (ΔNGli2) suppresses both white adipose tissue (WAT) and brown adipose tissue (BAT) accumulation in mice on a high fat diet (HFD). Adipocyte de novo formation (hyperplasia) and hypertrophy contribute to fat accumulation in response to a HFD. The Hh signaling through GLI2 induces Wnt expression, which subsequently inhibits adipocyte differentiation and the conversion of glucose to lipids [238]. In β-cells, elevated Hh signaling leads to impaired β-cell function and insulin secretion, resulting in glucose intolerance in transgenic mice. The mature β-cell exclude precursor cell markers Hes1 and Sox9, while Hh signaling stimulates their expression and impair the functionality of these cells. Sustained high Hh levels in β-cell erodes their identity and eventually led to undifferentiated pancreatic tumors [240]. As discussed before, PPAR-γ is a master regulator of glucose homeostasis, and its depletion induces insulin resistance. Recently it was shown that Shh by ERK-dependent pathway decreases the stability of PPAR-γ in subcutaneous adipose tissue and result in insulin resistance [241]. ERK phosphorylation induced by Shh increased PPARγ phosphorylation at Ser-112 and decreased its protein levels via E3 ubiquitin protein ligase neural precursor cells expressed developmentally downregulated protein 4 (NEDD4–1) dependent ubiquitination.
The Hh pathway modulates the regeneration and repair responses, inflammation and accelerates NASH progression to liver fibrosis. Shh stimulates HSCs and portal fibroblasts (PF), thereby promote fibrosis responses. Lipotoxicity in these cells results in c-Jun N-terminal kinase (JNK) activation, subsequently induces Shh expression. Shh prevents cell death and continuously being secreted persistently despite lipotoxic insults [242]. Inhibition of Hh signaling in the liver may lead to deleting ballooned hepatocytes and reduced downstream events, including HSC actication, inflammatory reactions, and even HCC development. Osteopontin (OPN) is an extracellular matrix protein, which is one of the Hh target genes and plays pathological roles in several liver diseases. Osteopontin (OPN) is secreted by macrophages, neutrophils, and HSCs in the liver [243]. Although the hepatocyte-specific role of Shh signaling is not fully understood, in one study, it was found that mice with liver‐specific knockout of Smoothened (SMO LKO) reduced macrophage activation and inhibited proinflammatory cytokine expression [244]. Further, inhibition of SMO with Vismodegib or Sonidegib moderately reduced serum levels of TG and cholesterols, improved glucose tolerance, and significantly reduced the expression of SREBP1 and SCD1 in the liver [244]. Hh pathway inhibition decreased murine macrophage marker F4/80‐positive cells and mRNA levels of TNF‐α, IL‐1β, MCP1, and IL‐6. Activated macrophages (M1 phenotype) produce pro-inflammatory cytokines, whereas alternatively activated macrophages (M2) possess the ability to attenuate inflammation. In the case of the body’s higher energy balance, macrophages in the adipose tissue tend to polarize to the M1 type responsible for insulin resistance. It was found that adipocytes communicate with macrophages mainly by exosomes caring Shh. In T2DM patients, serum exosomes are the elevated proportion of Shh-positive. Shh increases the PI3K phosphorylation in macrophages, essential for their survival and activation [245].
Yes-associated protein 1 (YAP1) is a stem cell-associated transcription coactivator in the Hippo pathway responsible for controlling adult liver size [246]. Very few cells express YAP, including localizing along hepatic sinusoids and within small ductular-appearing cells immediately adjacent to portal tracts in healthy adult liver. In cholestatic liver disease, nuclear YAP positive ductular cells accumulate periportal. NAFLD OPN and YAP expressions are co-localized in NAFLD patient samepls, suggesting that YAP positive cells promote fibrosis in NASH [247]. Hippo-YAP/TAZ signaling is activated by cell shape, cell–cell interactions, and ECM stiffness. HSCs and myofibroblasts also express high YAP/TAZ levels upon activation.
Moreover, inhibition of YAP diminished fibrogenesis in CCl4 treated mice. Martin et al. have further found that YAP is a critical mediator able to perpetuate integrin β1 in profibroblast phenotype. Integrin β1 has been extensively studied and is required to produce ECM and myofibroblast (MF) proliferation. Inhibition of YAP could attenuate liver fibrosis in vivo by mediating integrin β1 [248]. The Ihh ligand is a TAZ/TEAD target, and increased YAP/TAZ in hepatocytes during NAFLD progression leads to its secretion, which then acts on HSCs to promote the expression of pro-fibrotic genes [249].
Hh signaling activates quiescent HSCs into ECM-secreting MFs, contributing to increased vascular resistance, thereby promoting portal hypertension. Upregulation of Hh ligands also leads to the transformation of cholangiocytes and hepatocytes into MFs through EMT. Furthermore, the fibrotic liver has matrix deposition in the portal tracts or in perisinusoidal spaces, resulting in liver sinusoidal endothelial cell (LSEC) capillarization (loss of fenestrae) and poor extravasation to liver fibrogenic cells. Hh inhibition prevents LSEC capillarization in chronic and acute liver injuries, thus increasing the liver’s drug accumulation. Furthermore, differentiated LSEC prevents HSC activation and causes a reversal of activated myofibroblastic HSCs to quiescence. Because of the Hh pathway’s deep involvement in inflammation, liver fibrosis, and cancers, its inhibition has provided an attractive approach. While most Hh pathway inhibitors target SMO, some small molecules targeting the signaling cascade downstream of SMO were also developed. The details of small molecules’ discovery as Hh inhibitors from the diverse chemical background with their extensive structure-activity relationship (SAR) have been reviewed recently by Bariwal et al [250].
Our lab co-delivered Vismodegib and Rosiglitazone, a PPAR-γ agonist, for liver fibrosis treatment. Our study showed that Hh ligands are highly upregulated in common bile duct ligation (CBDL) induced liver fibrotic mice and their inhibition by Vismodegib. Delivery of Hh inhibitor alone reduced liver injury and attenuated fibrosis in CBDL mice as indicated by injury markers and fibrotic gene expression. Co-delivery of Vismodegib and Rosiglitazone showed a synergic effect on fibrosis treatment, improving both the Hh pathway and PPAR-γ are potent targets for treating liver fibrosis [251]. Further, our group has also synthesized vismodegib analog MDB5 to improve its efficacy in treating liver fibrosis. Delivery of MDB5 loaded NPs to CBDL mice significantly inhibited the Hh pathway. MDB5 loaded NPs also decreased serum AST and ALT levels, revered hepatic pathological damage, reduced collagen accumulation, and prevented EMT. More importantly, MDB5 showed higher efficiency compared with Vismodegib [252].
3.4.2. Renin-angiotensin system antagonists
Renin-angiotensin system (RAS) is classically considered a hormonal cascade, which regulates cardiovascular, renal, and adrenal functions by media hydrolytic balance and blood pressure through angiotensin II actions. Angiotensin-converting enzyme (ACE) converts angiotensin I into angiotensin II (Ang II). The increased ACE/ACE II ratio leads to vasoconstriction. Fibrosis progression rate (FPR) is commonly associated with arterial hypertension. Therefore, the RAS system is directly associated with the disease and favors steatosis, inflammation, and fibrogenesis via HSC activation [253].
Ang II could exacerbate liver fibrosis by stimulating TGF-β1 via Ang II receptor type 1 (AT1), while its interaction with angiotensin II receptor type 2 (AT2) has an anti-fibrogenic effect. It is reasonable that ACE inhibitors and AT1 receptor antagonists could serve fibrosis treatment targets [254]. Josson et al. demonstrated that early administration of captopril reduced collagen deposition, α-SMA production, TGF-β1 level, and increased ECM degradation enzyme matrix metalloproteinase (MMP)-2 and MMP-9 activity as compared to CBDL rats [255].
There are several pathways by which angiotensin-receptor inhibitors may be beneficial for NAFLD treatment [256]. RAS may also promote insulin resistance and modulate cytokine and adipokine production. Angiotensin II receptor blockers (ARB) may benefit from insulin resistance by selective stimulation of PPAR-γ. RAS inhibition can be achieved by ACE-inhibitors (ACEI) or by ARB. ACEI and ARB are also widely prescribed for reno-protection in individuals with diabetes. In crosssectional studies, RAS inhibition protected patients with hypertension from severe fibrosis and NAFLD, and was associated with reduced liver stiffness in patients with chronic kidney disease [257]. Significant improvements in the systolic blood pressure, AST levels, ALT, and liver fibrosis-related biomarkers were seen when 7 hypertensive patients with NASH were treated with 50 mg losartan daily for 48 weeks. Improvements in necroinflammation, ballooning, and fibrosis were seen in some patients, but there was no change in steatosis. Angiotensin II receptor blocker, telmisartan, has the unique qualities of a PPAR-γ modulator. In a blinded pilot study, 54 patients with biopsy-proven NASH and mild-moderate hypertension were randomly assigned to either telmisartan (20 mg/d) or valsartan (80 mg/d). Both medications improved transaminase levels and insulin resistance, but this improvement was more profound in the telmisartan group, showing a significant decrease in NASH activity score and fibrosis. In a study using rats on a high-fat, high-carbohydrate diet, it was found that telmisartan, but not valsartan, promoted increases in caloric expenditure and protected against diet-induced weight gain. Telmisartan reduced visceral fat accumulation, decreased adipocyte size, and reduced hepatic TG levels to a much greater extent than valsartan.
In contrast, a recent 12-month, randomized, open-label study in 137 patients with NASH showed no additional benefit on liver histology with combination therapy with rosiglitazone and losartan (50 mg/d) compared to rosiglitazone alone. Thus, data from human studies are limited and contradictory. A double-blind, randomized controlled trial of 50 mg Losartan once a day versus placebo for 96 weeks in patients with histological evidence of NASH has been conducted. The trial under-recruited due to the widespread use of ACEI and ARBs in the study population, and as a result, the study was unable to determine whether losartan has anti-fibrotic effects in the liver [258].
3.4.3. Transforming growth factor-beta 1 (TGF-β1) inhibitors
Liver injury results in a rapid induction of transforming TGF-β1, which involves in extensive signaling pathways in metabolism and fibrosis. TGF-β1 is also a negative regulator of hepatocytes and an inducer of parenchymal cell apoptosis. It exerts multiple functions, including both profibrogenic and anti-inflammatory effects. Okuno et al. used a serine protease inhibitor, camostat mesylate (CMM), as a TGF-β1 inhibitor to significantly reduce HSC activation, α-SMA and collagen production in rats with hepatic fibrosis induced by porcine serum [259]. A liver histological study showed suppressed fibrosis with CMM treatment, supporting TGF-β1 inhibitor as chronic fibrosis treatment. Several pathogenetic mechanisms result in excess ECM deposition (Figure 9), where TGF-β1 is one such crucial cytokine mediating fibrosis response.
Figure 9. Role of transforming growth factor beta 1 (TGF-β1) in progression of liver fibrosis.
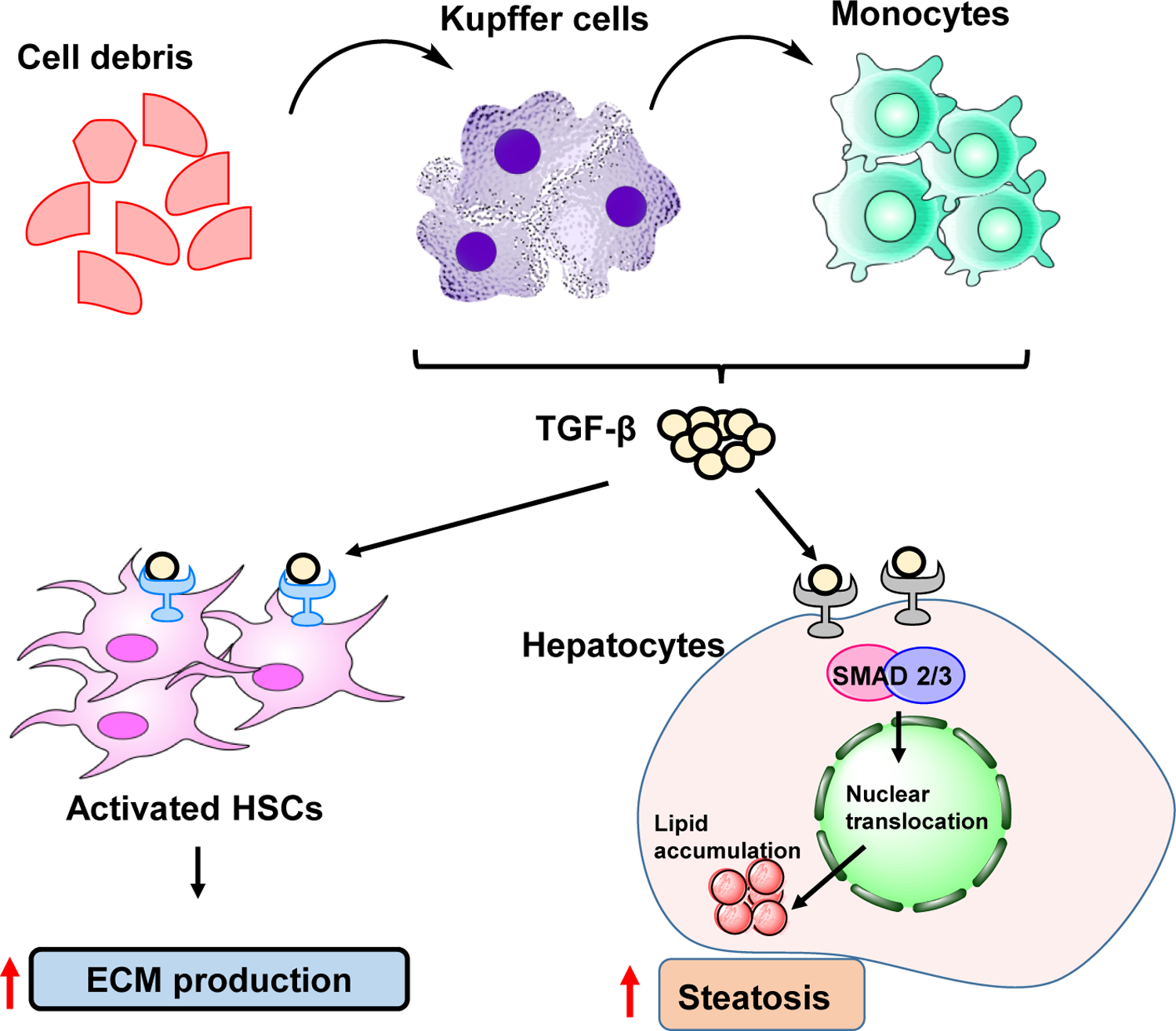
Kupffer cells (KCs), monocytes, and injured hepatocytes release TGF-β, the main cytokine in fibrosis, which causes the activation of HSCs and more accumulation of ECM protein in the liver. In the hepatocytes, TGF-β promote accumulation of lipids and promote steatosis.
Forkhead O transcription factors (FOXOs) are the downstream mediator of insulin signaling and their activity is modulated by AKT and other kinases. After meal, insulin stimulated AKT phosphorylate FOXOs and decrease their nuclear localization, while in fasting FOXOs are less phosphorylated and actively promote gluconeogenesis and lipolysis and inhibit glycolysis and lipogenesis. FOXOs also regulate hepatic lipid metabolism by modulating multiple pathways including inhibition of lipogenesis through suppression of SREBP-1c and glucokinase. FOXOs also modulate sirtuin (SIRT) enzymes, espatially SIRT1 and SIRT6. Both SIRT1 and SIRT6 inhibit lipogenesis and promote fatty acid oxidation in the liver. FOXOs control SIRT activity by increasing the expression of rate-limiting enzyme in the NAD biosynthesis nicotinamide phosphoribosyltransferase (NAMPT). FOXOs also promote hepatic lipolysis by upregulation of adipose triglyceride lipase (ATGL) that catalyzes the first step of lipolysis and downregulation of the G0/S1 switch 2 gene (G0S2), an inhibitor of ATGL. In addition, FOXOs also increase breakdown of lipid droplets through activation of the autophagy pathway as autophagy related 14 (ATG14) has been shown to be a direct target of FOXOs. Deletion of hepatic FOXO1/3/4 has been shown to markedly aggravate NAFLD phenotype by increasing hepatic inflammation and fibrosis, especially on the HFC diet. FOXO3 is a negative regulator or TGF-β1 pathway [260]. FOXO3 upregulate SMAD7 protein expression and therefore decrease liver fibrosis.
Within last decade, various pharmaceutical companies stepped up for NASH therapy development. Therefore, many drugs being actively pursued in various stages of for clinical trials. Here, we review some of the most promising targets for NASH, as well as describe compounds being developed against these targets in the clinic (Table 1).
Table 1.
Molecular targets of liver injury, NAFLD and fibrosis.
| Types | Targets | Active agents | Advantages | Clinical trial status or pre-clinical study | Ref. |
|---|---|---|---|---|---|
| Insulin-sensitizers | PPAR agonist | Pioglitazone | Decrease in blood glucose level | Phase 4 completed /NCT00994682 | Ahn et al. |
| Lanifibranor | Reduces hepatic glucogenesis, lipogenesis, and steatosis | Phase 2b/NCT03008070 | Sven et al. | ||
| FXR agonist | Obeticholic acid | Regulates the metabolic homeostasis of bile acids | Phase 3 completed/NCT02548351 | Younissi et al. | |
| GLP-1 agonist | Liraglutide | GLP-1 receptor antagonist for NAFLD/NASH prevention/treatment | Phase 2 completed/NCT01237119 | Mantovani et al. | |
| FGFR agonist | Pegbelfermin | Regulates lipid and glucose metabolism | Phase 2 active, not recruiting/NCT03486899 | Charles et al. | |
| SCD1 inhibitor | Aramchol | Decreases the synthesis and increases β oxidation of fatty acids | Phase 2 and 3 completed/NCT02279524 | Safadi et al. | |
| AMPK agonist | PF-06409577 | Reduces de novo lipogenesis to slow the development of NAFLD | Evaluated in rodent and primate models | Esquejo et al. | |
| APOC3 inhibitor | Gemcabene | Reduces hepatic de novo triglyceride and cholesterol synthesis | Phase 2 completed in FPLD adults/NCT03508687 | Oniciu et al. | |
| Anti-apoptosis | Pan-caspase inhibitor | Emricasan | Reduce cell death in liver injury due to apoptosis | Phase 2b NASH/NCT02960204 | Tsao et al. |
| ASK1 inhibitor | Selonsertib | Combination with Simtuzumab for NASH and stage 2–3 fibrosis | Phase 3 terminated/NCT03053050 | Loomba et al. | |
| TNF-α inhibitor | Pentoxifylline | A nonspecific PDE inhibitor upregulates cAMP and reduces TNF-α | Phase 2 completed/NCT00590161 | Du et al. | |
| Anti-inflammation | Amine oxidase inhibitor | BI 1,467,335 | Good oral bioavailability and nanomolar potency | Phase 2 completed/NCT03166735 | Schilter et al. |
| CCR2 and CCR5 antagonist | Cenicriviroc | Inhibits macrophage accumulation in the liver and ameliorates fibrosis | Phase 2 terminated/NCT03059446 | Anstee et al. | |
| Adenosine receptor agonist | Namodenoson | Inhibits c-AMP, PKA, PI3K and p-Akt and protects from liver damage | Phase 2 Not yet recruiting/NCT04697810 | Fishman et al. | |
| BRD4 inhibitor | JQ1 | Enhanced sensitivity of BRD4 and block of HSC activation | Evaluated in rodent and primate preclinical models | Ding et al. | |
| Galectin-3 antagonist | GR-MD-02 | Reverse liver fibrosis/cirrhosis and reduces portal hypertension | Phase 2 completed/NCT02421094 | Chalasani et al. | |
| PDE3/4 inhibitor | Roflumilast | Normalizes hydroxyproline, TGF-β1 and NF-κB levels | Phase 2 terminated/NCT01703260 | Essam et al. | |
| Anti-LPS antibody | Imm124-E | Exerts an immunomodulatory effect and alleviates target Organ damage and lipid profile | Phase 2 completed/NCT02316717 | Mizrahi et al. | |
| Anti-fibrosis | Hedgehog inhibitor | MDB5 | Better inhibition of Hedgehog downstream GLI target genes than GDC-0449 | Evaluated in rodent models | Kumar et al. |
| Angiotensin II receptor blocker | Losartan | Reduces PAI-1 production and improves insulin sensitivity | Phase 2 completed in pediatric NAFLD/NCT03467217 Phase 2 completed/NCT01913470 Phase 3 completed in NASH/NCT01051219 |
Vos et al. | |
| TGF-β1 inhibitor | Vactosertib | Inhibits TGF-β1 activation | Evaluated in rodent and primate preclinical models | Kim et al. |
4.0. DRUG DELIVERY AND TARGETING FOR TREATING LIVER FIBROSIS
Liver fibrosis disrupts the normal architecture, leads to scar fomation and perturbs the liver functions, especially in the end-stage of cirrhosis. Due to multiple factors, most of the preclinical findings in liver fibrosis are not being translated to humans. Therefore, there is an urgent need of clinically effective treatment. One major obstacle is the inability of conventional therapy to deliver the adequate concentration of therapeutic agents into the fibrotic liver. Drug delivey approach has the potential to some of the issues. Here, we discuss some of the common challenges and drug delivery systems being explored in treating fibrosis.
4.1. Barriers to liver fibrosis therapy
Despite significant improvement in our understanding of liver fibrogenesis, there is no standard treatment. Due to excess deposition of ECM proteins in the fibrotic liver, perfusion is less, resulting in decreased uptake of the antifibrotic drug by fibrogenic cells, consequently suboptimal efficacy. LSECs constitute liver sinusoids, with discontinuous fenestrae. In adults, LSECs express embryological as well as endothelial cell markers due to their complex origin. In a normal liver, differentiated LSECs maintain HSCs in their quiescent state. LSECs also regulate sinusoidal blood flow and thus maintain portal pressure low. LSECs allow molecules such as metabolites, plasma proteins, drug molecules, and small chylomicron, viruses (<200 nm), and exosomes to pass throw into the space of Disse. Following liver injury, de-differentiation of LSECs occurs called capillarization and result in HSC activation and macrophages [261]. After capillarization, fenestra disappears, which further inhibits the hepatic uptake of anti-fibrotic agents. ECM deposition in the space of Disse decreases the exchange of molecules [262]. Further, delivery of anti-fibrotic agents to specific cell types such as effector HSCs, and KCs in the liver is a challenge. HSCs constitute a small portion of the liver mass, and targeting these cells is difficult. Activated KCs that abide with HSCs can take up a substantial amount of delivery carriers and dilute the therapeutic response.
4.2. Drug delivery systems used for liver fibrosis
The conventional anti-fibrotic treatments fail in clinical settings because of non-specific drug disposition. The nanomedicine is a promising tool for loading the therapeutic agents for both acute and chronic liver diseases in animal models. Nanocarriers can achieve liver-specific anti-fibrotic drug delivery using with or without active targeting to the fibrotic region. Without drug targeting decorations, most drugs from nanocarriers are taken up by hepatocytes with the facilitation of different transporters on the sinusoidal site of hepatocytes, which may lead to unexpected adverse effects and cytotoxicity.
Depending on disease types and cell of interest, the approach for drug delivery may vary. Each cell type has specific receptors and a preference for uptake drug carriers which could be utilized to target using nenocarriers. For instance, NPs with a hydrophobic surface are more rapidly removed from circulation by KCs. Further, KCs and LSECs recognize NPs based on deposited LDL, human serum albumin (HSA), and negatively charged NPs by scavenger receptors. At the same time, hepatocytes uptake more positively charged particles. Several HSC-specific targeting carriers have also been designed and explored in the past. One difficulty in targeting HSCs is their number is only 10–15% of the liver cells’ total population. In liver fibrosis, HSCs have been the primary target for drug delivery using nanomedicine. To improve the pharmacological effects of antifibrotic drugs, various receptor types on activated HSCs have been identified, and nanocarriers decorated with targeting ligands were applied in the literature (Table 2, Figure 10).
Table 2.
Drug delivery system for liver fibrosis therapy.
| Strategy | Drug | Particle size | Targeting ligand | Polymer/Formulation | Reference |
|---|---|---|---|---|---|
| Hepatocyte targeting | Oxymatrine | – | RGD peptide | Liposomes | Chai et al. |
| siRNA against apoB | <200 nm | – | PEI-pullulan/siRNA complex | Kang et al. | |
| Quercetin | – | Galactosylate | Liposomes | Mandal et al. | |
| HSC targeting | siRNA against gp46 | 153.9 nm | Vitamin A | Liposomes | Sato et al. |
| Vismodegib | 80 nm | cRGDyK | Lipoid S100 and mPEG2000-DSPE/Liposome | Li et al. | |
| Antisense oligodeoxynucleotide against collagen I | 115 nm | Retinol | PEI/Complex of PEI/ASO | Zhang et al. | |
| Silibinin and siCOL1α1 | 151 nm | Vitamin A | PLGA-PSPE-PEG/Micelles | Qiao et al. | |
| siRNA against TGF-β1 | – | Galactosylate | Galactosylated PEG | Zhu et al. | |
| IFN-γ | <100 nm | Cyclic peptide | mPEG2000-DSPE/Liposome | Li et al. | |
| Silibinin | 40 nm | Hyaluronic acid (HA) | Lipoid E80/DOCA-Na/Micelles | Li et al. | |
| – | – | PDGFR binding peptide (C * SRNLIDC * ) | Peptide-modified albumin | Beljaars et al. | |
| Hepatocyte growth factor (HGF) | 50–100 nm | – | HGF incorporated chitosan nanoparticle | Pulavendran et al. | |
| siRNA against TGF-β1 | 100 nm | Hyaluronic acid (HA) | siRNA/(PEI-SS)-g-HA complex targeting HSC | Park et al. | |
| – | – | Nerve growth factor peptide (NGFp) | Ad serotype 5-derived wild-type vector | Reetz et al. | |
| Kupffer cell targeting | siRNA against migration inhibitory factor | 139.1 nm | Glucan | BG34–10-Re-I/siRNA nanoparticle | Zhang et al. |
| 350.5 nm | Phospholipid serine | Ac-Dex nanoparticle | Wang et al. |
Figure 10. Drug delivery systems used for treating liver fibrosis.
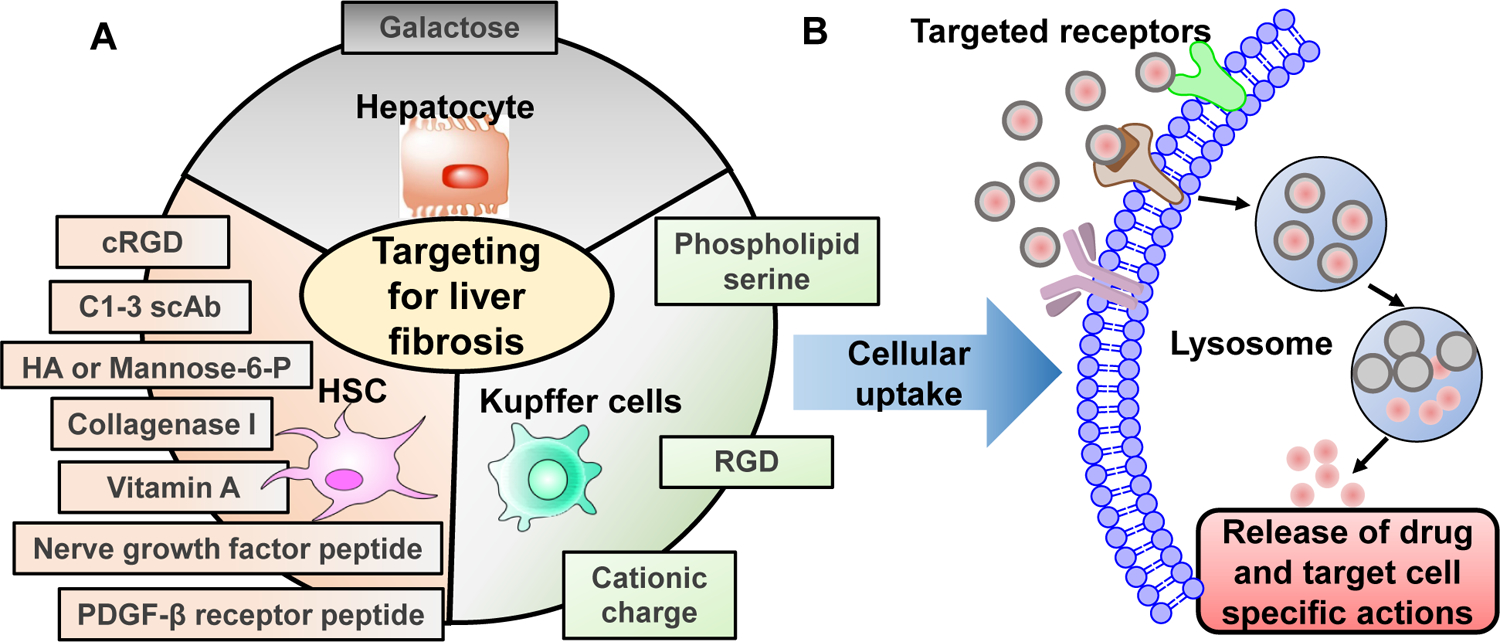
Cell-specific receptors could be used for targeted delivery of nanomedicine for fibrosis/cirrhosis. (B) Ligand decoration on nanocarries facilitate receptor binding and uptake of compounds resulting in target cell-specific actions.
4.2.1. Targeting ECM to enhance delivery to the liver cells.
Due to the excessive fibrosis, collagen deposited in the space of Disse upon hepatic injury hinders the delivery of nanoformulations. Due to silent nature of disease, liver fibrosis is often diagnosed in late stage at which ECM is already prominent in the hepatic fibrosis tissue. ECM is abundant in condensed fiber network which blocks NP activity and significantly reduces NP diffusion efficiency. In addition, ECM compresses the blood and lymphatic vessels, resulting in high interstitial fluid pressure which prevents NP convection and NP transport being dependent only on diffusion. Therefore, thaere is a critical need to improve the deep penetration capabilities of NPs under such poor perfusion conditions.
It has been reported that nanocarriers bearing a biochemical matrix degradation agent such as collagenase, hyaluronidase, MMPs, and bromelain could disrupt ECM to improve NP transport efficiency. However, protein delivery in vivo is a difficult task hindered by their rapid degradation, immunogenic effects, and accumulation at the non target site. To solve some of these issues, Fan et al. developed micelles using poly-(lactic-co-glycolic)-b-poly (ethylene glycol) (PLGA-PEG) decorated with collagenase I and retinol (CRM). Collagenase I enhanced the penetration of micelles, and retinol increased HSC specificity in the liver. The efficacy and uptake of collagenase I and retinol decorated micelles were investigated and in human HSCs (LX-2 cells) in the presence of an excessive collagen I barrier. The in vivo biodistribution, antifibrotic activity and toxicity profile of these polymeric micelles loaded with tyrosine kinase inhibitor were analyzed. Drug loaded CRM showed optimal antifibrotic activity in CCl4 induced liver fibrosis model. Micelles primorily showed liver accumulation and efficient HSC targeting without exerting any acute or chronic toxicity [263]. Similarly, Zinger at el. reported collagenase loaded liposomes called “collagozome” to disrupt collagen in CCL4 induced liver fibrosis [264]. Liposomes were composed of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC): cholesterol: 1,2-distearoyl-sn-glycero-3-phosphorylethanolamine (DSPE)-PEG2000. Liposomes predominantly accumulated in the liver. When injected in CCl4 induced liver fibrotic mice, collagozomes decreased collagen levels up to 35–49% compared to empty liposomes and decreased the severity of the disease.
There is an inverse correlation exist between collagen-I degrading MMPs and liver fibrosis progression. Liver fibrosis is characterized by increased tissue inhitors of metalloprotease (TIMP) and consequently reduced MMP levels. Recently, Geervliet et al. reported MMP-1 decorated polymersomes (MMPsomes) for the treatment of liver fibrosis. MMPsomes are polymeric vesicles with defined aqueous compartment mimicking cell- and virus-dimensions. MMPsomes enhanced relative MMP-1/TIMP-1 expression which then efficiently degraded collagen I and thereby ameliorated liver fibrosis in CCl4-induced early liver fibrogenesis mouse model [265].
Stem bromelain is a cysteine protease derived from pineapple which is orally administered for its anti-inflammatory and anticoagulant properties. Parodi et al. developed mesoporous silica nanoparticles (MSN) decorated with proteolytic bromelain. This surface modification showed increased particle uptake in endothelial, macrophage, and an increased ability to digest and diffuse in tumor ECM in vitro and in vivo [266]. HA is an anionic, nonsulfated glycosaminoglycan, composed of D-glucuronic acid and N-acetyl-D-glucosamine, which is a major ECM component. In fact, serum hyaluronan and hyaluronidase are early markers of toxic liver injury [267]. Strategies for depleting HA via decreasing its synthesis or degrading coild beneifit in liver fibrosis. Hyaluronidase-1 and hyaluronidase-2 are the two enzymes majorly responsible for HA catabolism. 4-methylumbelliferone (4MU) is a naturally occurring coumarin, which decrease HA deposition via the depletion of cellular UDP-glucuronic acid dehydrogenase (UDP-GlucUA) and downregulation of hyaluronan synthase 2 (HAS2) and HAS3 gene expression. HAS2 expression is upregulated by TGF-β1 through Wilms tumor 1 in HSCs and secreted HA promotes fibrogenic phenotypes of HSCs through activation of CD44, TLR4, and Notch1 signaling. Yang et al. showed 4-MU treatment suppressed liver fibrosis by decreasing HA content, Notch1 mRNA expression, HSC activation, and macrophage infiltration [268]. Further, Andreichenko et al. used 4MU in CCl4-induced liver fibrosis in mice [269]. 4MU treatment alleviated liver fibrosis by reducing hyaluronan deposition and downregulating follistatin like 1 (FSTL1) expression. Further, 4MU suppressed HSC activation and altered macrophage localization. Yang et al. used 4-MU for treatment of mouse model of choline-deficient L-amino acid-defined diet (CDAA)-induced NASH. 4-MU effectively ameliorated NASH-associated steatosis, hepatocyte injury, inflammation, and fibrotic response [270]. Rajan et al. prepared hyaluronidase core-5-fluorouracil-loaded chitosan-polyethylene glycol-gelatin polymer nanocomposites (CS-HYL-5-FU). These nanocomposites were in the size range between 300 – 580 nm with a clear, smooth surface and fine morphology, drug release, and cytotoxicity [271]. Although these nanoparticles can be potential carriers for targeted and controlled drug delivery, but their potency has not been checked in a suitable in-vivo model.
Hepatocyte targeting
Hepatocyte targeting is little challenging, as these cells do not express specific surface receptors which can be used for drug targeting. Hepatocytes express an extracellular glycoprotein called the asialoglycoprotein receptor (ASGPR). The expression level of asialoglycoprotein receptor (ASGPR) is liver fibrosis stage dependent and could be utilized for precise treatment of liver fibrosis. Galactose is the specific ligand of ASGPR. Therefore, delivery systems modified with galactose have been explored to target hepatocytes and improve delivery efficiency. Mandal et al. used galactose-modified liposomes loading quercetin (QC) to combat arsenic-induced hepatic fibrogenesis [272]. Galactosylated liposomes containing quercetin (QC) could improve drug delivery to the the hepatocytes and protect the liver from fibrosis. Further, Zhang et al. used ASGPR targeting tracer for in vivo SPECT/CT imaging and quantification of fibrosis severity for the chronic liver disease [273]. Kang et al. designed pullulan-containing PEI/siRNA complexes for enhanced delivery into mice liver through the tail vein injection [274]. Pullulan is a polysaccharide consisting of three α−1, 4-linked glucose polymers with different α−1, 6-glucosidic linkages and has high affinity for the ASGPR.
Hepatocytes show dynamin-dependent macropinocytosis which could be used as a strategy for improving hepatocyte targeting in liver diseases. Nanocomplexes mimicking natural lipoproteins termed lipopeptide nanoparticles (LPNP) could efficiently target and hepatocytes in vivo. Dong et al. prepared lysine conjugated lipid cKK-E12 based LPNP for siRNA against factor VII (a blood clotting factor) in mice liver and siRNA targeting TTR in nonhuman primates [275]. Further, siRNA cell uptake and gene silencing efficiency by cKK-E12 was significantly enhanced by addition of apolipoprotein (ApoE) proteins into LPNPs.
4.2.2. HSC targeting
HSCs are the proper targets for antifibrotic agents. The challenge of targeting HSCs is mainly due to its low percentage of 5% to 8% in total liver cells [276]. HSCs express retinol-binding protein receptor (RBPR). Vitamin A (VA) is the major ligand for these receptors [277]. Therefore, VA-targeted delivery carriers could be utilized for targeted drug delivery systems. Liposomes encapsulating siRNA against human heat shock protein 47 (siHSP47) were decorated with vitamin A (VA-lip-siHSP47). HSP47 is an ER protein that is transiently associated with procollagen and is involved in collagen maturation and secretion under normal conditions [278]. Deletion of HSP47 causes ER stress-mediated apoptosis of HSCs [279]. VA-liposomes contacting siHSP47 showed regression of fibrosis in dinitrosamine (DMN), CCl4, and CBDL induced liver fibrosis, animal models. The therapeutic effect was surmised to because of inhibition of collagen secretion by HSP47 and concomitant degradation of predeposited collagen by collagenase activity [200].
In another study, polymeric nanoparticles (NPs) encapsulating nitric oxide (NO) donor molecules S-nitrosoglutathione and decorated with vitamin A were designed. In the in-vitro experiment, Vitamin A conjugated nanoparticles showed a significantly higher uptake by HSCs than KCs, LSECs or NPs. Further, higher accumulated in the liver of CBDL rats when compared to normal rat due to a larger number of HSCs in former animals. These vitamin A coated NPs release nitric oxide (NO) in liver cells, resulted in inhibition of collagen α1 (COL1α1) and α-SMA. Moreover, NO-releasing NPs targeted with vitamin A not only attenuate endothelin-1 (ET-1), which elicits HSC contraction but also alleviates hemodynamic disorders in CBL-induced portal hypertension evidenced by decreasing portal pressure (≈20%) and unchanging mean arterial pressure [280].
Surface modification of NPs with biological ligands has limited clinical success because of the layering of serum proteins on active ligands in the blood circulation. Control of the layering proteins on the vehicle surfaces can avoid this masking effect. The corona formation can be controlled for recruiting selective proteins to its component and utilize these proteins for tissue/cell-targeting purposes. In a study, Zhang et al. prepared a retinol-conjugated polyetherimide (RcP) NPs capable of selectively recruit the retinol-binding protein 4 (RBP4) on its corona. RBP4 was found to bind retinol and direct the antisense oligonucleotide (ASO)-laden RcP carrier to HSCs. After injection into mice, most ASO molecules were accumulated in HSCs. However, naked ASO was found mainly in the endothelial cells and macrophages. ASO-laden RcP particles in two different mouse models of liver fibrosis induced by CCl4 and CBDL effectively suppressed the expression of type I collagen and consequently ameliorated hepatic fibrosis [281]. VA conjugated with low molecular weight polyethylenimine (PEI) was used to target NPs to the HSCs. PEI possesses a positive charge, which formed complexes with nucleotide (RcP). This NP system actively recruited plasma proteins such as RBP4 to form corona on the surface.
Qiao et al. reported micellar formulation of poly (lactide-co-glycolide)-polyspermine-poly (ethylene glycol)-vitamin A (PLGA-PSPE-PEG-VA), for co-delivery of silibinin and siCOL1α1 to suppress collagen production synergistically. PLGA-PSPE-PEG-VA is an amphiphilic copolymer that self-assembled into micelles at low concentrations. Drug loaded micelles showed very low cytotoxicity and hemolytic activity in vitro and were well tolerated in vivo. Of note, micelles effectively accumulated in fibrotic livers and specifically targeted activated HSCs, resulting in decreased α1 production and ameliorated liver fibrosis compared with silibinin loaded micelles or siCOL1α1 loaded non targeted micelles [282].
HSCs reside in the space of Disse and procude excess collagen, whose deposition in the liver hinders drug delivery to activated HSCs. Nanocarriers with proteolytic enzymes on their surface can degrade ECM and facilitate drug delivery to the fibrotic liver. Monomeric type I collagen fiber consists of two extended α1 chains and one α2 chain, twisted into a triple helix and resistant to most protease except collagenase. Fan et al. developed polymeric micelles based on poly (lactic-co-glycolic)-b-poly(ethylene glycol)-maleimide (PLGA-PEG-Mal) co-decorated with collagenase I and retinol (termed CRM) for liver fibrosis therapy [263]. Collagenase I decoration facilitated CRM to permeation acriss fibrotic ECM, and the retinol decoration was specifically recognized by HSCs. The DiR (red fluorescent dye) loaded CRM exhibited excellent accumulation in the liver of normal and 4 or 8 wk CCl4 treated liver fibrosis, mouse models. Further, in mice injected with DiI dye loaded CRM showed fluorescence merged with the green fluorescence of HSCs to generate strong yellow signals indicating that most of the dye was in activated HSCs.
Integrin receptors mediate cellular adhesion and reaction to the ECM and regulate cell growth, survival/apoptosis, differentiation, and migration. Among the integrin family, integrin αvβ3 interacts with various ECM proteins like vitronectin, fibrinogen, and fibronectin via arginine-glycine-aspartate (RGD) motif. Integrin αvβ3 is found upregulated on activated HSCs compared to quotient and promotes later survival and proliferation [283]. RGD sequence has a binding affinity with integrin αvβ3and can be used to target HSCs in the fibrotic liver [284]. A cyclic peptide containing RGD (cRGD) recognizing collagen type VI receptor on HSCs was conjugated to sterically stable liposomes (SSLs) for delivering Interferon-alpha1b (IFN-α1b). In CBDL rats, a biodistribution study showed 10-fold more accumulation of cRGD-SSLs in HSCs isolated from CBDL rats than unlabeled SSLs. cRGD-SSL encapsulating INF-α1b exhibited significantly reduced the extent of liver fibrosis compared with CBDL control rats or CBDL rats treated with SSLs encapsulating IFN-α1b [285]. In a separate study, Li et al. decorated dendrimer nanoprobes with cyclic arginine-glycine-aspartic acid pentapeptide (cRGDyK) to track HSCs. An MRI modality was used to visualize hepatic Den-RGD deposition in a thioacetamide-induced mouse model of liver fibrosis. Nanoprobes conjugated with cRGDyK bind specifically to activated HSCs expressing αvß3 receptors. A T1-weighed MR signal MRI study showed that Den-RGD tracer deposition increased in parallel with the severity of liver fibrosis [286].
Oxymatrine (OM) is an alkaloid extracted from the medicinal plant Sophora alopecuraides L. OM exhibits an antifibrotic effect through acting against lipid peroxidation and inhibiting the TGF-β1 signaling pathway. To facilitate OM binding to HSCs, RGD liposomes containing OM were developed and tested in rats with CCl4-induced hepatic fibrosis. They demonstrated that OM-RGD liposomes were bound explicitly to HSCs and enhanced OM’s liver-protective effect compared with OM-liposome group [287]. Li et al. encapsulated hepatocyte growth factor (HGF) in sterically stabilized liposomes (SSL) to protect from in vivo degradation. Further, SSL was decorated with cRGD peptides for targeting. Injection of RGD–SSL–HGF induced more significant remission of liver cirrhosis than the injection of SSL–HGF, HGF alone, HGF plus RGD–SSL, and saline [288]. Further, Vismodegib loaded cRGDyK liposomes effectively inhibited the Hh pathway and prevented the activation of HSCs in vitro, and alleviated liver fibrosis in vivo [289]. Pulavendran et al. incorporated HGF into chitosan nanoparticles (CNP) by ionic gelation method. Cirrhotic mice either with hematopoietic stem cells (HSC) or mesenchymal stem cells (MSCs) were treated with HGF-CNP or saline control. HGF-CNP enhanced the differentiation of stem cells into hepatocytes and reversed the hepatic fibrosis [290].
Activated HSCs also express mannose 6-phosphate/insulin-like growth factor II (M6P/IGF-II) receptors. Beljaars et al. conjugated M6P on albumin at different proportions and showed that M6P modified albumin accumulated in liver tissue, specifically on HSCs [291]. We also used M6P decorated methoxy poly (ethylene glycol)-block-poly (2-methyl-2-carboxyl-propylene carbonate)-graft-dodecanol (PEG-PCD) polymeric micelles for vismodegib delivery to HSCs [292]. Micelles with different M6P ligand densities (10, 20, and 30% w/w) were administered to normal and liver fibrotic mice, and biodistribution of vismodegib was determined at 30 and 120 min post systemic administration. There was a significant increase in vismodegib accumulation in the liver when M6P-conjugated mixed micelles were injected. Micelles decorated with 30% M6P-ligand density showed the highest drug concentration in the liver. Of note, HSCs were isolated after injection and accounted for 14.19% of vismodegib for M6P-targeted micelles in fibrotic mice compared to 5.62% of non-targeted micelles.
Further, liposomes encapsulating inactivated hemagglutinating virus of Japan (HVJ) containing a plasmid DNA holding luciferase as reporter gene were decorated with albumin-M6P showed efficient delivery to the fibrotic liver [293]. Zhu et al. used heterobifunctional PEG (ortho-pyridyl) disulfide-poly (ethylene glycol) – N-hydroxysuccinimidyl ester (OPSS-PEG-NHS) as a backbone to conjugate M6P at its NHS end and 3’-sense strand of siRNA at its OPSS end via a disulfide bond. After transfection, the level of TGF-β1 protein in the cell medium was found 40% decreased compared to siTGF-β1 measured by ELISA [294].
Dilinoleoylphosphatidylcholine (DLPC) has been shown to reduce HSC proliferation, α-SMA expression, and collagen synthesis by HSCs [295]. Further, it reduces TNF-α production by KCs [296]. Taking advantage of it, Adrian et al. developed liposomes incorporating DLPC into the lipid bilayer. Further, the surface of DLPC-containing liposomes was modified with M6P-HSA. The antifibrotic effects of DLPC-containing liposomes were evaluated in a CBDL rat model of liver fibrosis. DLPC-containing liposomes displayed hepatoprotective effects in livers of CBDL rats [297]. Rho kinase pathway is involved in activation, migration, and contraction of HSCs. However, the Rho pathway is expressed in the whole body, and its generalized inhibition may result in unwanted effects. Therefore, Beuge et al. conjugated Rho kinase inhibitor to M6P-HSA carrier for HSC specific delivery. M6P-HSA conjugated Y27632 (Y27-conjugate) decreased protein expression of phosphorylated myosin light chain 2 (pMLC2) and vinculin, downstream of Rho-kinase; the Y27-conjugate showed high specificity and did not show activity in rat aortas. In CCl4-induced liver fibrosis, Y27-conjugate, but not with a free drug, reduced pMLC2 expression in livers 24 h after injection, and significantly reduced collagen deposition [298].
However, M6P targeting approach has a significant limitation. In a study, M6P-FITC-dextran-loaded particles were incubated with different liver cell types. Surprisingly, particles were taken up not only by HSCs but also by ECs and hepatocytes and effectively by KCs. Therefore, M6P directed drug delivery strategy is suitable not specifically to HSCs, and both M6P/IGF-II and scavenger receptors can detect and bind to M6P-HSA subunit. Activated HSCs also overexpress CD44 receptors. Hyaluronic acid (HA) and chondroitin sulfate (CS) is the natural ligand of CD44 receptors. Among these, CS has a higher affinity than HA to CD44 receptors. Recently, a self-assembled micelle system based on chondroitin sulfate−deoxycholic acid conjugate (CS−DOCA), phospholipid, and sodium deoxycholate were reported [299]. CS decorated micelles loaded with doxorubicin (DOX), and retinoic acid (RA) were evaluated for therapeutic efficacy in a CCl4 induced liver fibrosis model in rats. Study results showed that FITC-labeled CS micelles endocytosis by HSCs via a CD44 receptor-mediated internalization pathway and selectively accumulated in the Golgi apparatus. CCl4 induced fibrotic rats, when treated with DOX+RA-CS micelles showed a significantly reduced number of highly proliferative HSCs collagen fibers deposition, inflammatory lesions, and hydroxyproline content in the liver tissues. Further, silibinin-loaded hyaluronic acid (SLB-HA) micelles were designed for the treatment of hepatic fibrosis [300].
PDGF-β is secreted by both resident and infiltrating cells of the liver and causes extensive proliferation, migration, and contraction of primarily HSCs and myofibroblasts. HSCs positively express PDGF-β receptors. Hence, a cyclic peptide (pPB) that mimics the binding site of PDGF-β to its endogenous receptor was developed for their targeting. Arginine and isoleucine residues and their adjacent amino acids represent PDGF-β receptor binding moieties in pPB peptide CSRNLIDC (C denotes the cyclizing cysteine residues) [301]. Multiple (10) pPB moieties coupled to human serum albumin yielded a highly HSC-specific carrier macromolecule pPB-HAS. In the CBDL rat’s liver, pPB-HSA highly accumulated in PDGF-β receptor-expressing HSCs in contrast to unmodified HSA (P<0.001) [302]. Further, Li et al. developed SSLs modified with cyclic peptides (pPB) with a specific affinity for PDGFR-β, encapsulated IFN-γ (pPB-SSL-IFN-γ). In healthy rats, pPB-SSL-IFN-γ showed higher accumulation in the livers and had a longer half-life than free IFN-γ. Furthermore, thioacetamide-induced liver fibrotic rats, FITC-labeled pPB-SSL were predominantly localized in activated HSCs accompanied by enhanced anti-fibrotic effect of PB-SSL-IFN-γ treatment [303].
HA as discussed earlier is a polysaccharide capable of specific delivery to the liver tissues with HA receptors. A liver-specific, low molecular weight polyethyleneimine (PEI) grafted hyaluronic acid [(PEI-SS)-g-HA] conjugates were designed for the delivery of TGF- β siRNA. siRNA/(PEI-SS)-g-HA complex showed 80% gene silencing in-vitro in the presence of 10 vol% serum and 50% silencing in the presence of 50 vol% serum in B16F1 melanoma cells and activated HSCs. Furthermore, the complex showed an antifibrotic effect on cirrhosis with a significantly reduced nodule formation, collagen content, and HSC number [304]. Activated HSCs, overexpress death receptor 5 on the cell surface, making them more susceptible to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induced apoptosis than normal HSCs. Therefore, HA–TRAIL conjugate was developed for targeting HSC to resolve liver fibrosis. Infra red (IR) dye-labeled HA–TRAIL conjugate showed target-specificity in vivo. Surprisingly, HA–TRAIL conjugate was detected for more than 4 days after a single intravenous injection into rats. Further antifibrotic effect of HA–TRAIL conjugate was confirmed in an N-nitrosodimethylamine-induced liver fibrosis rat models [305].
Synaptophysin (SYN) is a major transmembrane glycoprotein of small (30–80 nm) electron-translucent (SET) vesicles containing neurotransmitters. Expression of SYN has been demonstrated in quiescent as well as activated human and rat HSCs [306]. A recombinant human monoclonal single-chain antibody (C1–3 scAb) against a conserved peptide sequence present in an extracellular domain of synaptophysin was therefore generated. C1–3 scAb was conjugated to an HSC apoptosis-inducing compound gliotoxin (GT). C1–3-GT conjugates significantly reduced the numbers of liver myofibroblasts present in CCl4 induced liver fibrosis model and resolved fibrosis without affecting the number of KCs [307].
HSCs also express p75 NT receptor (p75NTR), which is a part of the TNF superfamily. The structure of p75NTR features a cysteine repeat motif, which acts as a ligand-binding domain and a cytoplasmic portion called the death domain. Natural ligands of 75NTR are the NTs with nerve growth factor (NGF)as a prototype and BDNF, NT-3, and NT-4. Binding of NGF to p75NTR induces apoptosis of activated HSCs and can selectively target and eliminate HSCs. Therefore, Reetz et al. developed a peptide (NGFP) derived from the NGF and conjugated it to the adenoviral (Adv) vector surface via the PEG linker. NGFp coupled. Adv resulted in a reduced hepatocellular tropism and an enhanced adenoviral-mediated gene transfer to HSCs in CBDL mice model [308, 309].
4.2.3. KCs targeting
KCs are closely associated with the hepatic scar, which can act on HSCs to induce a pro-fibrotic phenotype because of a rich source of inflammatory cytokines. Macrophages can produce and activate several regulatory factors, such as PDGF, which is a potent stimulator of myofibroblast proliferation; TGF-β, which acts to increase the production of ECM and TIMP-1 by myofibroblasts; IL-1β and TNF-α, which are proinflammatory cytokines; and the number of chemokines, which can induce further inflammatory cell recruitment to perpetuate the proinflammatory profibrotic stimulus. Therefore, KCs are considered as potential targets in combatting fibrosis. The success of targeting liver macrophages involves the use of different nanoparticle types with spherical shape, positive charge, or a diameter >200 nm to allow them to be preferentially taken up by the KCs and appropriate ligands to increase accumulation in the specific phenotype of KCs to avoid unexpected adverse effects.
Drugs can be delivered to macrophages by passive and active targeting. NPs are easily identified and swallowed by macrophages as a foreign object. Specifically after intravenous injection, NPs with positive surface charge tend to adsorb on the plasma proteins and are easily recognized by macrophages [310]. However, NPs modified by PEG on the surface has been shown to weaken this ability. Besides macrophages can precisely identify the signal exposed from apoptotic cells, such as phospholipid serine (PS). Therefore, NPs modified by PS can be identified as apoptotic cells by macrophages and enhance the macrophages-targeting ability. Wang et al. demonstrated that PS-coated NPs have the ability to efficiently target macrophages [311]. They recently designed PS-modified nanostructured lipid carriers that prolonged the drug’s retention time, enhanced its bioavailability and delivery efficiency to the liver, resulting in reduced liver fibrosis in vivo. In addition, Kim et al. also developed a glucan-based siRNA carrier system BG34–10-Re-I/siRNA for macrophage-targeted siRNA delivery. Therefore, macrophage targeting delivery systems may have a promising future for the treatment of liver fibrosis.
5. Concluding remarks and future prospectives
Patients who are obese or have T2DM are at an increased risk of developing NASH and if not controlled progresses to cirrhosis and eventually to HCC. The high prevalence of T2DM and obesity is expected to make NASH one of the most common causes of advanced liver diseases. Further, NASH is associate with a higher risk of death from cirrhosis, HCC and cardiovascular diseases. Given a vast numberof drugs acurrently being investigated, the arsenal of medications will be available for treating NASH is likely to significantly expand in the coming years. The ideal treatment of NASH should improve liver histology and cardiovascular outcomes and have good tolerability and an excellent safety profile. Diabetic NASH patients could be preferentially treated with novel antidiabetic drugs such as glucagon-like peptide-1 receptor agonists (GLP-1RAs) and sodium-glucose cotransporter 2 (SGLT2) inhibitors. Reversal of steatohepatitis with at least no worsening of fibrosis has been considered as a primary endpoint of clinical trials. However, no treatment to date has shown histological improvement of NASH in greater than 50% of patients and resolution rates of fibrosis are remarkably low. NASH is a heterogeneous disease and likely has different underlying contributing factors in different patients. This may reflect ineffectiveness of therapies, inability to overcome continuing caloric excess, or the heterogeneity in pathway activation between subjects.
Because insulin resistance is central to the pathogenesis of NASH, many new NASH drugs have insulin sensitization as one of their primary modes of action; however, other drug classes such as anti-inflammatory and antifibrotic are also emerging. There is an urgent need of developing antifibrotic agents for NASH treatment, as fibrosis significantly complicate and induce progression to end-stage disease. However, it should be noted that therapies based simply on pure antifibrotic effects do not address the drivers of disease progression such as cell stress, apoptosis and inflammation. It is also imperative to treat other components of the metabolic syndrome, including hypertension, dyslipidemia and insulin resistance or diabetes. It is possible that no one-medication-fits-all strategy will be successful and future research should focus on combination therapies. In addition, longer-term studies are essential to establish safety and the effects of other metabolic comorbidities.
Various approaches have been developed for drug delivery to fibrotic livers. Nanocarriers show great potential for selective drug delivery to targeting cells. Apart from physical entrapment into NPs or micelles, drugs can also be conjugated to polymer backbone via ester, amide, or pH-sensitive bonds is another way to improve drug loading and plasma circulation. A large number of formulations have been prepared till date for liver targeting by polymeric nanocarriers as targeting ligands. Nevertheless, liver cell specific delivery needs sophisticated synthesis and involves a lot of engineering tools, which creates many opportunities for liver fibrosis therapy but also needs complex synthesis and may introduce heterogeneity to the final products. That may be the reason that very few nanomedicines are approved or tested in the clinical trials for liver fibrosis. Apart from small molecules, gene therapy and immunotherapy are being evaluated for liver fibrosis treatment.
However, several nanocarriers have failed to deliver therapeutic agents efficiently due to the fibrotic liver due to the closure of the sinusoidal gaps and excessive collagen deposition.
In conclusion, this review discussed various molecular targets and strategies to overcome biological barriers to the effective treatment of NAFLD and liver fibrosis. Several nanocarriers have the potential to increase efficacy and decrease off-target effects by enhancing drug delivery and targeting to specific liver cell types.
Figure 7. Chemokines in the progress of NAFLD.
Chemokines mediate the liver inflammation by controlling the migration of various hepatic and immune cells. The C-C chemokine receptor types 2 (CCR2) and 5 (CCR5) and their respective ligands (CCL2 and CCL3–5) are involved in the development of NAFLD and NASH. Therefore, CCR2 and CCR5 have been established as promising therapeutic targets for NASH. TAK-779 was the first non peptide CCR5 receptor antagonist. To improve the oral bioavailability, cenicriviroc (CVC) was developed for dual CCR2 and CCR5 inhibition. CVC demonstrated anti-fibrotic effects in the Phase 2b clinical study in adults with NASH and liver fibrosis.
Acknowledgements
The Faculty Startup fund University of Nebraska Medical Center are duly acknowledged for providing financial support for this work. RIM is supported by the National Institutes of Health (R01NS116037).
Abbreviations
- ALE
Advanced lipoxidation end-product
- ALT
Alanine aminotransferase
- AMPK
Adenosine monophosphate-activated protein kinase
- ApoB
Apolipoprotein B
- AR
Adenosine receptor
- ASK
Apoptosis signal-regulating kinase
- AST
Aspartate transaminase
- ATF6
Activating transcription factor 6
- AUC
Area under the curve
- BC
Bovine colostrum
- BIL
Bilirubin
- CAR
Constitutive androstane receptor
- CBDL
Common bile duct ligation
- CCl4
Carbon tetrachloride
- CCR
Chemokine receptor
- CDCA
Chenodeoxycholic acid
- CTGF
Connective tissue growth factor
- E2
Estradiol
- ECM
Extracellular matrix
- EMT
Epithelial-to-mesenchymal transition
- ER
Endoplasmic reticulum
- ERK
Extracellular signal-regulating kinase
- ET-1
Endothelin-1
- EV
Extracellular vesicles
- FAH
Fumarylacetoacetate hydrolase
- FAK
Focal adhesion kinase
- FFA
Free fatty acids
- FGF
Fibroblast growth factor
- FXR
Farnesoid X receptor
- GPCR
G protein-coupled receptor
- GSDs
Glycogen storage disease
- GSH
glutathione
- HA
Hyaluronic acid
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- Hh
Hedgehog
- HSCs
Hepatic stellate cells
- HSP47
Heat shock protein 47
- IFN-1β
Interferon 1β
- IGF1
Insulin-like growth factor
- IL-6
Interlukin-6
- JNK
c-jun N-terminal protein kinase
- IKK
Inhibitor of nuclear factor-κB kinase
- KC
KCs
- LAL
Lysosomal acid lipase
- LDL
Low-density lipoprotein
- LNP
Lipid nanoparticles
- LPS
Lipopolysaccharides
- LSEC
Liver sinusoidal endothelial cell
- MAA
Malondialdehyde-acetaldehyde
- miRNAs
microRNAs
- M6P
Mannose 6 phosphate
- M6P-PEG-PCD
M6P--methyl-2-carboxyl-propylene carbonate)-graft-dodecanol
- MCD
Methionine and choline deficient
- MSCs
Mesenchymal stem cells
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- NF-κB
Nuclear factor-κB
- NPs
Nanoparticles
- OM
Oxymatrine
- PAM
Protospacer adjacent motif
- PBMC
Peripheral blood mononuclear cell
- PD
Pharmacodynamics
- PDE4
Phosphodiesterase 4
- PDGF-β
Platelet-derived growth factor beta
- PH
Portal hypertension
- PK
Pharmacokinetics
- PPAR
Peroxisome proliferator-activated receptor
- RBP
Retinol binding protein 4
- RGD
Arginine-Glycine-Aspartate
- ROS
Reactive oxygen species
- RXR
Retinoid X receptor
- SCD1
Stearoyl-coenzyme A desaturase 1
- SFA
Saturated fatty acids
- sgRNA
Single guide RNA
- SNP
Single nucleotide polymorphism
- SPECT
Single-photon emission computed tomography
- SREBPs
Sterol regulatory element binding proteins
- SSL
Sterically stabilized liposomes
- T2DM
Type 2 diabetes mellitus
- TGs
Triglycerides
- TGF-β
Transforming growth factor
- TIMP1
Tissue inhibitor of metalloproteinase 1
- TNF-α
Tumor necrosis factor alpha
- TRAF2
TNFR-associated factor 2
- TRAIL
Tumor necrosis factor-related apoptosis-inducing ligand
- UPR
Unfolded protein response
- VLDL
Very-low-density lipoprotein
- WD
Wilson’s Disease
- ZFNs
Zinc finger nucleases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest
References
- [1].Centers for Disease Control and Prevention, Chronic Liver Disease and Cirrhosis, CDC 2021 (2021) .
- [2].Mitra S, De A, Chowdhury A, Epidemiology of non-alcoholic and alcoholic fatty liver diseases, Transl. Gastroenterol. Hepatol 5 (2020) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, Xu S, Zhang H, Cui G, Chen X, Sun R, Wen H, Lerut JP, Kan Q, Li L, Zheng S, Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma, Gut 68 (2019) 1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Makri E, Goulas A, Polyzos SA, Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease, Arch. Med. Res 52 (2021) 25–37. [DOI] [PubMed] [Google Scholar]
- [5].Samuel VT, Shulman GI, The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux, J. Clin. Invest 126 (2016) 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kumar V, Mahato RI, Delivery and targeting of miRNAs for treating liver fibrosis, Pharm. Res 32 (2015) 341–361. [DOI] [PubMed] [Google Scholar]
- [7].Cole JB, Florez JC, Genetics of diabetes mellitus and diabetes complications, Nat. Rev. Nephrol 16 (2020) 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Su Q, Kumar V, Sud N, Mahato RI, MicroRNAs in the pathogenesis and treatment of progressive liver injury in NAFLD and liver fibrosis, Adv. Drug Deliv. Rev 129 (2018) 54–63. [DOI] [PubMed] [Google Scholar]
- [9].Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M, Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008, Clin. Gastroenterol. Hepatol 9 (2011) 524–530.e1; quiz e60. [DOI] [PubMed] [Google Scholar]
- [10].Reddy JK, Hashimoto T, Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system, Annu. Rev. Nutr 21 (2001) 193–230. [DOI] [PubMed] [Google Scholar]
- [11].Dornas W, Schuppan D, Mitochondrial oxidative injury: a key player in nonalcoholic fatty liver disease, Am. J. Physiol. Gastrointest. Liver Physiol 319 (2020) G400–G411. [DOI] [PubMed] [Google Scholar]
- [12].Tolman KG, Fonseca V, Dalpiaz A, Tan MH, Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease, Diabetes Care 30 (2007) 734–743. [DOI] [PubMed] [Google Scholar]
- [13].Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, Qiu Y, Burns L, Afendy A, Nader F, The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis, J. Hepatol 71 (2019) 793–801. [DOI] [PubMed] [Google Scholar]
- [14].Copps KD, White MF, Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2, Diabetologia 55 (2012) 2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chadt A, Al-Hasani H, Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease, Pflugers Arch 472 (2020) 1273–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wagman AS, Johnson KW, Bussiere DE, Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes, Curr. Pharm. Des 10 (2004) 1105–1137. [DOI] [PubMed] [Google Scholar]
- [17].Smith GC, Turner N, FOX01 Is the Headline Akt Regulating Hepatic Glucose Metabolism, Endocrinology 158 (2017) 2436–2438. [DOI] [PubMed] [Google Scholar]
- [18].Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, Lutz EP, Kako Y, Velez-Carrasco W, Goldberg IJ, Breslow JL, Shulman GI, Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 7522–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ, Mechanisms of NAFLD development and therapeutic strategies, Nat. Med 24 (2018) 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Geng Y, Faber KN, de Meijer VE, Blokzijl H, Moshage H, How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int 15 (2021) 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Eichmann TO, Lass A, DAG tales: the multiple faces of diacylglycerol--stereochemistry, metabolism, and signaling, Cell Mol. Life Sci 72 (2015) 3931–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gassaway BM, Petersen MC, Surovtseva YV, Barber KW, Sheetz JB, Aerni HR, Merkel JS, Samuel VT, Shulman GI, Rinehart J, PKCepsilon contributes to lipid-induced insulin resistance through cross talk with p70S6K and through previously unknown regulators of insulin signaling, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E8996–E9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, Luo J, Heinrich B, Yu SJ, Zhang Q, Wilson A, Shi ZD, Swenson R, Greten TF, Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development, Cell. Death Dis 9 (2018) 620–018–0687–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hodson L, Gunn PJ, The regulation of hepatic fatty acid synthesis and partitioning: the effect of nutritional state, Nat. Rev. Endocrinol 15 (2019) 689–700. [DOI] [PubMed] [Google Scholar]
- [25].Ballak DB, Li S, Cavalli G, Stahl JL, Tengesdal IW, van Diepen JA, Kluck V, Swartzwelter B, Azam T, Tack CJ, Stienstra R, Mandrup-Poulsen T, Seals DR, Dinarello CA, Interleukin-37 treatment of mice with metabolic syndrome improves insulin sensitivity and reduces pro-inflammatory cytokine production in adipose tissue, J. Biol. Chem 293 (2018) 14224–14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pirola L, Ferraz JC, Role of pro- and anti-inflammatory phenomena in the physiopathology of type 2 diabetes and obesity, World J. Biol. Chem 8 (2017) 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tilg H, Moschen AR, Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis, Hepatology 52 (2010) 1836–1846. [DOI] [PubMed] [Google Scholar]
- [28].Dara L, Ji C, Kaplowitz N, The contribution of endoplasmic reticulum stress to liver diseases, Hepatology 53 (2011) 1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Maiers JL, Malhi H, Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis, Semin. Liver Dis 39 (2019) 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ishizuka K, Usui I, Kanatani Y, Bukhari A, He J, Fujisaka S, Yamazaki Y, Suzuki H, Hiratani K, Ishiki M, Iwata M, Urakaze M, Haruta T, Kobayashi M, Chronic tumor necrosis factor-alpha treatment causes insulin resistance via insulin receptor substrate-1 serine phosphorylation and suppressor of cytokine signaling-3 induction in 3T3-L1 adipocytes, Endocrinology 148 (2007) 2994–3003. [DOI] [PubMed] [Google Scholar]
- [31].Austin RL, Rune A, Bouzakri K, Zierath JR, Krook A, siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle, Diabetes 57 (2008) 2066–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].National Academies of Sciences, Engineering, and Medicine, Health and Medicine Division, Board on Health Sciences Policy, Roundtable on Genomics and Precision Health, Forum on Drug Discovery, Development, and Translation, (2017) .
- [33].Romeo S, Sanyal A, Valenti L, Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease, Cell. Metab 31 (2020) 35–45. [DOI] [PubMed] [Google Scholar]
- [34].Auinger A, Valenti L, Pfeuffer M, Helwig U, Herrmann J, Fracanzani AL, Dongiovanni P, Fargion S, Schrezenmeir J, Rubin D, A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis, Horm. Metab. Res 42 (2010) 854–859. [DOI] [PubMed] [Google Scholar]
- [35].Doege H, Baillie RA, Ortegon AM, Tsang B, Wu Q, Punreddy S, Hirsch D, Watson N, Gimeno RE, Stahl A, Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis, Gastroenterology 130 (2006) 1245–1258. [DOI] [PubMed] [Google Scholar]
- [36].Xu QY, Li H, Cao HX, Pan Q, Fan JG, APOC3 rs2070667 Associates with Serum Triglyceride Profile and Hepatic Inflammation in Nonalcoholic Fatty Liver Disease, Biomed. Res. Int 2020 (2020) 8869674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pirmohamed M, Pharmacogenetics for the prescriber, Medicine 44 (2016) 412–415. [Google Scholar]
- [38].Houghton D, Stewart CJ, Day CP, Trenell M, Gut Microbiota and Lifestyle Interventions in NAFLD, Int. J. Mol. Sci 17 (2016) 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lopez-Pastor AR, Infante-Menendez J, Escribano O, Gomez-Hernandez A, miRNA Dysregulation in the Development of Non-Alcoholic Fatty Liver Disease and the Related Disorders Type 2 Diabetes Mellitus and Cardiovascular Disease, Front. Med. (Lausanne) 7 (2020) 527059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Oates JR, McKell MC, Moreno-Fernandez ME, Damen MSMA, Deepe GS Jr, Qualls JE, Divanovic S, Macrophage Function in the Pathogenesis of Non-alcoholic Fatty Liver Disease: The Mac Attack, Front. Immunol 10 (2019) 2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Deuffic S, Buffat L, Poynard T, Valleron AJ, Modeling the hepatitis C virus epidemic in France, Hepatology 29 (1999) 1596–1601. [DOI] [PubMed] [Google Scholar]
- [42].Bissell DM, Sex and hepatic fibrosis, Hepatology 29 (1999) 988–989. [DOI] [PubMed] [Google Scholar]
- [43].Boyer-Diaz Z, Aristu-Zabalza P, Andres-Rozas M, Robert C, Ortega-Ribera M, Fernandez-Iglesias A, Broqua P, Junien JL, Wettstein G, Bosch J, Gracia-Sancho J, Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease, J. Hepatol (2020) . [DOI] [PubMed]
- [44].Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S, The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases, J. Adv. Pharm. Technol. Res 2 (2011) 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fruchart JC, Duriez P, Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism, Drugs Today (Barc) 42 (2006) 39–64. [DOI] [PubMed] [Google Scholar]
- [46].Lalloyer F, Staels B, Fibrates, glitazones, and peroxisome proliferator-activated receptors, Arterioscler. Thromb. Vasc. Biol 30 (2010) 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, Van Hul W, Mertens I, Hubens G, Van Marck E, Michielsen P, Van Gaal L, Staels B, PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis, J. Hepatol 63 (2015) 164–173. [DOI] [PubMed] [Google Scholar]
- [48].Bernardes A, Souza PC, Muniz JR, Ricci CG, Ayers SD, Parekh NM, Godoy AS, Trivella DB, Reinach P, Webb P, Skaf MS, Polikarpov I, Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: a new mode of ligand recognition and receptor stabilization, J. Mol. Biol 425 (2013) 2878–2893. [DOI] [PubMed] [Google Scholar]
- [49].Dobordzhginidze LM, Gratsianskii NA, Fibrates: mechanism of action, effect on levels of lipids and lipoproteins, risk of coronary events. Part 1. Clofibrate, gemfibrosil, besafibrate, Kardiologiia 44 (2004) 96–103. [PubMed] [Google Scholar]
- [50].Choudhary NS, Kumar N, Duseja A, Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease, J. Clin. Exp. Hepatol 9 (2019) 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Creger PL, Moersch GW, Neuklis WA, Structure/activity relationship of gemfibrozil (CI-719) and related compounds, Proc. R. Soc. Med 69Suppl 2 (1976) 3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brown PJ, Stuart LW, Hurley KP, Lewis MC, Winegar DA, Wilson JG, Wilkison WO, Ittoop OR, Willson TM, Identification of a subtype selective human PPARalpha agonist through parallel-array synthesis, Bioorg. Med. Chem. Lett 11 (2001) 1225–1227. [DOI] [PubMed] [Google Scholar]
- [53].Brown PJ, Plunket KD, Moore LB, Lewis MC, Wilson JG, Sundseth SS, Koble CS, Wu Z, Chapman JM, Lehmann JM, Kliewer SA, Willson TM, A ureido-thioisobutyric acid (GW9578) is a subtype-selective PPARalpha agonist with potent lipid-lowering activity, J. Med. Chem 42 (1999) 3785–3788. [DOI] [PubMed] [Google Scholar]
- [54].Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM, The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR), Toxicol. Sci 90 (2006) 269–295. [DOI] [PubMed] [Google Scholar]
- [55].Xu Y, Mayhugh D, Saeed A, Wang X, Thompson RC, Dominianni SJ, Kauffman RF, Singh J, Bean JS, Bensch WR, Barr RJ, Osborne J, Montrose-Rafizadeh C, Zink RW, Yumibe NP, Huang N, Luffer-Atlas D, Rungta D, Maise DE, Mantlo NB, Design and synthesis of a potent and selective triazolone-based peroxisome proliferator-activated receptor alpha agonist, J. Med. Chem 46 (2003) 5121–5124. [DOI] [PubMed] [Google Scholar]
- [56].Chen ZZ, Xie YD, Shao LH, Wang QT, Xu YH, Bian XL, 5-(4-Hydroxyphenyl)-3H-1,2-dithiole-3-thione-based fibrates as potential hypolipidemic and hepatoprotective agents, Bioorg. Med. Chem. Lett 28 (2018) 3787–3792. [DOI] [PubMed] [Google Scholar]
- [57].Yaghoubi M, Jafari S, Sajedi B, Gohari S, Akbarieh S, Heydari AH, Jameshoorani M, Comparison of fenofibrate and pioglitazone effects on patients with nonalcoholic fatty liver disease, Eur. J. Gastroenterol. Hepatol 29 (2017) 1385–1388. [DOI] [PubMed] [Google Scholar]
- [58].Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, Ranvir R, Kadam S, Patel H, Swain P, Roy SS, Das N, Karmakar E, Wahli W, Patel PR, Dual PPARalpha/gamma agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models, Liver Int 38 (2018) 1084–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, Hong SH, Castro GL, Yin YQ, Nelson MC, Hsiao G, Greaves DR, Downes M, Yu RT, Olefsky JM, Evans RM, PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 22504–22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hall JA, Ramachandran D, Roh HC, DiSpirito JR, Belchior T, Zushin PH, Palmer C, Hong S, Mina AI, Liu B, Deng Z, Aryal P, Jacobs C, Tenen D, Brown CW, Charles JF, Shulman GI, Kahn BB, Tsai LTY, Rosen ED, Spiegelman BM, Banks AS, Obesity-Linked PPARgamma S273 Phosphorylation Promotes Insulin Resistance through Growth Differentiation Factor 3, Cell. Metab 32 (2020) 665–675.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Arnold SV, Inzucchi SE, Echouffo-Tcheugui JB, Tang F, Lam CSP, Sperling LS, Kosiborod M, Understanding Contemporary Use of Thiazolidinediones, Circ. Heart Fail 12 (2019) e005855. [DOI] [PubMed] [Google Scholar]
- [62].Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM, Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5, Nature 466 (2010) 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bland RD, Clarke TL, Harden LB, Rapid infusion of sodium bicarbonate and albumin into high-risk premature infants soon after birth: a controlled, prospective trial, Am. J. Obstet. Gynecol 124 (1976) 263–267. [DOI] [PubMed] [Google Scholar]
- [64].Wang L, Waltenberger B, Pferschy-Wenzig EM, Blunder M, Liu X, Malainer C, Blazevic T, Schwaiger S, Rollinger JM, Heiss EH, Schuster D, Kopp B, Bauer R, Stuppner H, Dirsch VM, Atanasov AG, Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARgamma): a review, Biochem. Pharmacol 92 (2014) 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yasmin S, Capone F, Laghezza A, Piaz FD, Loiodice F, Vijayan V, Devadasan V, Mondal SK, Atli O, Baysal M, Pattnaik AK, Jayaprakash V, Lavecchia A, Novel Benzylidene Thiazolidinedione Derivatives as Partial PPARgamma Agonists and their Antidiabetic Effects on Type 2 Diabetes, Sci. Rep 7 (2017) 14453–017–14776–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Reddy KA, Lohray BB, Bhushan V, Reddy AS, Rao Mamidi NV, Reddy PP, Saibaba V, Reddy NJ, Suryaprakash A, Misra P, Vikramadithyan RK, Rajagopalan R, Novel antidiabetic and hypolipidemic agents. 5. Hydroxyl versus benzyloxy containing chroman derivatives, J. Med. Chem 42 (1999) 3265–3278. [DOI] [PubMed] [Google Scholar]
- [67].Jang JY, Bae H, Lee YJ, Choi YI, Kim HJ, Park SB, Suh SW, Kim SW, Han BW, Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARgamma, Sci. Rep 8 (2018) 31–017–18274–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lima KG, Schneider Levorse VG, Rosa Garcia MC, de Souza Basso B, Pasqualotto Costa B, Antunes GL, Luft C, Haute GV, Leal Xavier L, Donadio MVF, Rodrigues de Oliveira J, Octyl gallate induces hepatic steatosis in HepG2 cells through the regulation of SREBP-1c and PPAR-gamma gene expression, EXCLI J 19 (2020) 962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C, Casini A, Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro, Gastroenterology 122 (2002) 1924–1940. [DOI] [PubMed] [Google Scholar]
- [70].McHutchison J, Goodman Z, Patel K, Makhlouf H, Rodriguez-Torres M, Shiffman M, Rockey D, Husa P, Chuang WL, Levine R, Jonas M, Theodore D, Brigandi R, Webster A, Schultz M, Watson H, Stancil B, Gardner S, Farglitizar Study Investigators, Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection, Gastroenterology 138 (2010) 1365–73, 1373.e1–2. [DOI] [PubMed] [Google Scholar]
- [71].Bojic LA, Huff MW, Peroxisome proliferator-activated receptor delta: a multifaceted metabolic player, Curr. Opin. Lipidol 24 (2013) 171–177. [DOI] [PubMed] [Google Scholar]
- [72].Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A, Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance, Cell. Metab 7 (2008) 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu Y, Colby JK, Zuo X, Jaoude J, Wei D, Shureiqi I, The Role of PPAR-delta in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor, Int. J. Mol. Sci 19 (2018) 10.3390/ijms19113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Naruhn S, Meissner W, Adhikary T, Kaddatz K, Klein T, Watzer B, Muller-Brusselbach S, Muller R, 15-Hydroxyeicosatetraenoic Acid is a Preferential Peroxisome Proliferator-Activated Receptor Beta/delta Agonist, Mol. Pharmacol 77 (2010) 171–184. [DOI] [PubMed] [Google Scholar]
- [75].Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, Baron M, Lucas A, Tailleux A, Hum DW, Ratziu V, Cariou B, Hanf R, Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis, Hepatology 58 (2013) 1941–1952. [DOI] [PubMed] [Google Scholar]
- [76].Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero-Gomez M, Boursier J, Abdelmalek M, Caldwell S, Drenth J, Anstee QM, Hum D, Hanf R, Roudot A, Megnien S, Staels B, Sanyal A, GOLDEN-505 Investigator Study Group, Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening, Gastroenterology 150 (2016) 1147–1159.e5. [DOI] [PubMed] [Google Scholar]
- [77].Hoekstra M, Kruijt JK, Van Eck M, Van Berkel TJ, Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells, J. Biol. Chem 278 (2003) 25448–25453. [DOI] [PubMed] [Google Scholar]
- [78].Sanderson LM, Boekschoten MV, Desvergne B, Muller M, Kersten S, Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver, Physiol. Genomics 41 (2010) 42–52. [DOI] [PubMed] [Google Scholar]
- [79].Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, Lonardo A, Carulli N, Loria P, Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes, J. Gastroenterol. Hepatol 24 (2009) 830–840. [DOI] [PubMed] [Google Scholar]
- [80].Liu S, Brown JD, Stanya KJ, Homan E, Leidl M, Inouye K, Bhargava P, Gangl MR, Dai L, Hatano B, Hotamisligil GS, Saghatelian A, Plutzky J, Lee CH, A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use, Nature 502 (2013) 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Horike N, Sakoda H, Kushiyama A, Ono H, Fujishiro M, Kamata H, Nishiyama K, Uchijima Y, Kurihara Y, Kurihara H, Asano T, AMP-activated protein kinase activation increases phosphorylation of glycogen synthase kinase 3beta and thereby reduces cAMP-responsive element transcriptional activity and phosphoenolpyruvate carboxykinase C gene expression in the liver, J. Biol. Chem 283 (2008) 33902–33910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Qin X, Xie X, Fan Y, Tian J, Guan Y, Wang X, Zhu Y, Wang N, Peroxisome proliferator-activated receptor-delta induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice, Hepatology 48 (2008) 432–441. [DOI] [PubMed] [Google Scholar]
- [83].Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J, Heirman C, Quartier E, Schuit F, Wahli W, Geerts A, Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells, Gastroenterology 124 (2003) 184–201. [DOI] [PubMed] [Google Scholar]
- [84].Ronnelid J, Huang YH, Norrlander T, Rogberg S, Nilsson B, Gustafsson R, Klareskog L, Short-term kinetics of the humoral anti-C1q response in SLE using the ELISPOT method: fast decline in production in response to steroids, Scand. J. Immunol 40 (1994) 243–250. [DOI] [PubMed] [Google Scholar]
- [85].Scarpulla RC, Nuclear activators and coactivators in mammalian mitochondrial biogenesis, Biochim. Biophys. Acta 1576 (2002) 1–14. [DOI] [PubMed] [Google Scholar]
- [86].Scarpulla RC, Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells, Gene 286 (2002) 81–89. [DOI] [PubMed] [Google Scholar]
- [87].Garesse R, Vallejo CG, Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes, Gene 263 (2001) 1–16. [DOI] [PubMed] [Google Scholar]
- [88].Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA, Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice, Nat. Genet 18 (1998) 231–236. [DOI] [PubMed] [Google Scholar]
- [89].Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS, Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence, Int. J. Mol. Sci 20 (2019) 10.3390/ijms20205055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sven MF, Pierre B, Manal FA, Quentin MA, Elisabetta B, Vlad R, Philippe HM, Bruno S, Jean-Louis J, Pierre B, Jean-Louis A, A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study, Contemp. Clin. Trials 98 (2020) 106170. [DOI] [PubMed] [Google Scholar]
- [91].Chiang JY, Kimmel R, Weinberger C, Stroup D, Farnesoid X receptor responds to bile acids and represses cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription, J. Biol. Chem 275 (2000) 10918–10924. [DOI] [PubMed] [Google Scholar]
- [92].Kemper JK, Kim H, Miao J, Bhalla S, Bae Y, Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP, Mol. Cell. Biol 24 (2004) 7707–7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gong YZ, Everett ET, Schwartz DA, Norris JS, Wilson FA, Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol, Proc. Natl. Acad. Sci. U. S. A 91 (1994) 4741–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Jiang L, Zhang H, Xiao D, Wei H, Chen Y, Farnesoid X receptor (FXR): Structures and ligands, Comput. Struct. Biotechnol. J 19 (2021) 2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].He H, Mennone A, Boyer JL, Cai SY, Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells, Hepatology 53 (2011) 548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Assis DN, Abdelghany O, Cai SY, Gossard AA, Eaton JE, Keach JC, Deng Y, Setchell KD, Ciarleglio M, Lindor KD, Boyer JL, Combination Therapy of All-Trans Retinoic Acid With Ursodeoxycholic Acid in Patients With Primary Sclerosing Cholangitis: A Human Pilot Study, J. Clin. Gastroenterol 51 (2017) e11–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, Mutnick D, Bursulaya B, Schmeits J, Wu X, Bao D, Zoll J, Kim Y, Groessl T, McNamara P, Seidel HM, Molteni V, Liu B, Phimister A, Joseph SB, Laffitte B, Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH), J. Med. Chem 60 (2017) 9960–9973. [DOI] [PubMed] [Google Scholar]
- [98].Merk D, Sreeramulu S, Kudlinzki D, Saxena K, Linhard V, Gande SL, Hiller F, Lamers C, Nilsson E, Aagaard A, Wissler L, Dekker N, Bamberg K, Schubert-Zsilavecz M, Schwalbe H, Molecular tuning of farnesoid X receptor partial agonism, Nat. Commun 10 (2019) 2915–019–10853–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Guo C, LaCerte C, Edwards JE, Brouwer KR, Brouwer KLR, Farnesoid X Receptor Agonists Obeticholic Acid and Chenodeoxycholic Acid Increase Bile Acid Efflux in Sandwich-Cultured Human Hepatocytes: Functional Evidence and Mechanisms, J. Pharmacol. Exp. Ther 365 (2018) 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, Bedossa P, Geier A, Beckebaum S, Newsome PN, Sheridan D, Sheikh MY, Trotter J, Knapple W, Lawitz E, Abdelmalek MF, Kowdley KV, Montano-Loza AJ, Boursier J, Mathurin P, Bugianesi E, Mazzella G, Olveira A, Cortez-Pinto H, Graupera I, Orr D, Gluud LL, Dufour JF, Shapiro D, Campagna J, Zaru L, MacConell L, Shringarpure R, Harrison S, Sanyal AJ, REGENERATE Study Investigators, Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial, Lancet 394 (2019) 2184–2196. [DOI] [PubMed] [Google Scholar]
- [101].Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, Cevikbas F, Steinhoff M, Nassini R, Materazzi S, Guerrero-Alba R, Valdez-Morales E, Cottrell GS, Schoonjans K, Geppetti P, Vanner SJ, Bunnett NW, Corvera CU, The TGR5 receptor mediates bile acid-induced itch and analgesia, J. Clin. Invest 123 (2013) 1513–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Chiang PC, Thompson DC, Ghosh S, Heitmeier MR, A formulation-enabled preclinical efficacy assessment of a farnesoid X receptor agonist, GW4064, in hamsters and cynomolgus monkeys, J. Pharm. Sci 100 (2011) 4722–4733. [DOI] [PubMed] [Google Scholar]
- [103].Bass JY, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Mills WY, Navas F 3rd, Parks DJ, Smalley TL Jr, Spearing PK, Todd D, Williams SP, Wisely GB, Conformationally constrained farnesoid X receptor (FXR) agonists: heteroaryl replacements of the naphthalene, Bioorg. Med. Chem. Lett 21 (2011) 1206–1213. [DOI] [PubMed] [Google Scholar]
- [104].Flatt B, Martin R, Wang TL, Mahaney P, Murphy B, Gu XH, Foster P, Li J, Pircher P, Petrowski M, Schulman I, Westin S, Wrobel J, Yan G, Bischoff E, Daige C, Mohan R, Discovery of XL335 (WAY-362450), a highly potent, selective, and orally active agonist of the farnesoid X receptor (FXR), J. Med. Chem 52 (2009) 904–907. [DOI] [PubMed] [Google Scholar]
- [105].Renga B, Migliorati M, Mencarelli A, Cipriani S, D’Amore C, Distrutti E, Fiorucci S, Farnesoid X receptor suppresses constitutive androstane receptor activity at the multidrug resistance protein-4 promoter, Biochim. Biophys. Acta 1809 (2011) 157–165. [DOI] [PubMed] [Google Scholar]
- [106].Colca JR, The TZD insulin sensitizer clue provides a new route into diabetes drug discovery, Expert Opin. Drug Discov 10 (2015) 1259–1270. [DOI] [PubMed] [Google Scholar]
- [107].Colca JR, VanderLugt JT, Adams WJ, Shashlo A, McDonald WG, Liang J, Zhou R, Orloff DG, Clinical proof-of-concept study with MSDC-0160, a prototype mTOT-modulating insulin sensitizer, Clin. Pharmacol. Ther 93 (2013) 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, McDonald WG, Colca JR, Kletzien RF, Finck BN, Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor gamma-sparing thiazolidinedione, J. Biol. Chem 287 (2012) 23537–23548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kamm DR, Pyles KD, Sharpe MC, Healy LN, Colca JR, McCommis KS, Novel insulin sensitizer MSDC-0602K improves insulinemia and fatty liver disease in mice, alone and in combination with liraglutide, J. Biol. Chem (2021) 100807. [DOI] [PMC free article] [PubMed]
- [110].Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, Lee BH, Loomba R, Cusi K, Kolterman O, Cotter G, Dittrich HC, Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study, J. Hepatol 72 (2020) 613–626. [DOI] [PubMed] [Google Scholar]
- [111].Sagara M, Iijima T, Kase M, Kato K, Sakurai S, Tomaru T, Jojima T, Usui I, Aso Y, Serum levels of soluble dipeptidyl peptidase-4 in type 2 diabetes are associated with severity of liver fibrosis evaluated by transient elastography (FibroScan) and the FAST (FibroScan-AST) score, a novel index of non-alcoholic steatohepatitis with significant fibrosis, J. Diabetes Complications 35 (2021) 107885. [DOI] [PubMed] [Google Scholar]
- [112].Yap MKK, Misuan N, Exendin-4 from Heloderma suspectum venom: From discovery to its latest application as type II diabetes combatant, Basic Clin. Pharmacol. Toxicol 124 (2019) 513–527. [DOI] [PubMed] [Google Scholar]
- [113].Crane J, McGowan B, The GLP-1 agonist, liraglutide, as a pharmacotherapy for obesity, Ther. Adv. Chronic Dis 7 (2016) 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Mantovani A, Byrne CD, Scorletti E, Mantzoros CS, Targher G, Efficacy and safety of anti-hyperglycaemic drugs in patients with non-alcoholic fatty liver disease with or without diabetes: An updated systematic review of randomized controlled trials, Diabetes Metab 46 (2020) 427–441. [DOI] [PubMed] [Google Scholar]
- [115].DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P, Encapsulation of exenatide in poly-(D,L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes, Diabetes Technol. Ther 13 (2011) 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Best JH, Boye KS, Rubin RR, Cao D, Kim TH, Peyrot M, Improved treatment satisfaction and weight-related quality of life with exenatide once weekly or twice daily, Diabet. Med 26 (2009) 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yu M, Benjamin MM, Srinivasan S, Morin EE, Shishatskaya EI, Schwendeman SP, Schwendeman A, Battle of GLP-1 delivery technologies, Adv. Drug Deliv. Rev 130 (2018) 113–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, McGowan BM, Rosenstock J, Tran MTD, Wadden TA, Wharton S, Yokote K, Zeuthen N, Kushner RF, STEP 1 Study Group, Once-Weekly Semaglutide in Adults with Overweight or Obesity, N. Engl. J. Med 384 (2021) 989. [DOI] [PubMed] [Google Scholar]
- [119].Dipeptidyl Peptidase-4 Inhibitors, in: Anonymous LiverTox: Clinical and Research Information on Drug-Induced Liver Injury, Bethesda (MD), 2012, . [PubMed] [Google Scholar]
- [120].Deacon CF, Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus, Nat. Rev. Endocrinol 16 (2020) 642–653. [DOI] [PubMed] [Google Scholar]
- [121].Degirolamo C, Sabba C, Moschetta A, Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23, Nat. Rev. Drug Discov 15 (2016) 51–69. [DOI] [PubMed] [Google Scholar]
- [122].Chau MD, Gao J, Yang Q, Wu Z, Gromada J, Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 12553–12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB, FGF-21 as a novel metabolic regulator, J. Clin. Invest 115 (2005) 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, Furukawa N, Marino FE, Liu FF, Kahn BB, Libermann TA, Maratos-Flier E, A high-fat, ketogenic diet induces a unique metabolic state in mice, Am. J. Physiol. Endocrinol. Metab 292 (2007) E1724–39. [DOI] [PubMed] [Google Scholar]
- [125].Ruiz-Margain A, Pohlmann A, Ryan P, Schierwagen R, Chi-Cervera LA, Jansen C, Mendez-Guerrero O, Flores-Garcia NC, Lehmann J, Torre A, Macias-Rodriguez RU, Trebicka J, Fibroblast growth factor 21 is an early predictor of acute-on-chronic liver failure in critically ill patients with cirrhosis, Liver Transpl 24 (2018) 595–605. [DOI] [PubMed] [Google Scholar]
- [126].Zarei M, Barroso E, Palomer X, Dai J, Rada P, Quesada-Lopez T, Escola-Gil JC, Cedo L, Zali MR, Molaei M, Dabiri R, Vazquez S, Pujol E, Valverde AM, Villarroya F, Liu Y, Wahli W, Vazquez-Carrera M, Hepatic regulation of VLDL receptor by PPARbeta/delta and FGF21 modulates non-alcoholic fatty liver disease, Mol. Metab 8 (2018) 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, Endo H, Takahashi H, Inamori M, Kobayashi N, Kirikoshi H, Kubota K, Saito S, Nakajima A, Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis, Hepatology 50 (2009) 772–780. [DOI] [PubMed] [Google Scholar]
- [128].Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, Itoh N, Wang Y, Bornstein SR, Xu A, Li X, Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice, Cell. Metab 17 (2013) 779–789. [DOI] [PubMed] [Google Scholar]
- [129].Huang Z, Wang H, Lu M, Sun C, Wu X, Tan Y, Ye C, Zhu G, Wang X, Cai L, Li X, A better anti-diabetic recombinant human fibroblast growth factor 21 (rhFGF21) modified with polyethylene glycol, PLoS One 6 (2011) e20669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Charles ED, Neuschwander-Tetri BA, Pablo Frias J, Kundu S, Luo Y, Tirucherai GS, Christian R, Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study, Obesity (Silver Spring) 27 (2019) 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kharitonenkov A, Beals JM, Micanovic R, Strifler BA, Rathnachalam R, Wroblewski VJ, Li S, Koester A, Ford AM, Coskun T, Dunbar JD, Cheng CC, Frye CC, Bumol TF, Moller DE, Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319, PLoS One 8 (2013) e58575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Henriksson E, Andersen B, FGF19 and FGF21 for the Treatment of NASH-Two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 From Mice to Human, Front. Endocrinol. (Lausanne) 11 (2020) 601349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Gadaleta RM, Scialpi N, Peres C, Cariello M, Ko B, Luo J, Porru E, Roda A, Sabba C, Moschetta A, Suppression of Hepatic Bile Acid Synthesis by a non-tumorigenic FGF19 analogue Protects Mice from Fibrosis and Hepatocarcinogenesis, Sci. Rep 8 (2018) 17210–018-35496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Zhou M, Wang X, Phung V, Lindhout DA, Mondal K, Hsu JY, Yang H, Humphrey M, Ding X, Arora T, Learned RM, DePaoli AM, Tian H, Ling L, Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19, Cancer Res 74 (2014) 3306–3316. [DOI] [PubMed] [Google Scholar]
- [135].Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, Kugelmas M, Bashir MR, Jaros MJ, Ling L, Rossi SJ, DePaoli AM, Loomba R, NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial, Lancet 391 (2018) 1174–1185. [DOI] [PubMed] [Google Scholar]
- [136].Harrison SA, Rossi SJ, Paredes AH, Trotter JF, Bashir MR, Guy CD, Banerjee R, Jaros MJ, Owers S, Baxter BA, Ling L, DePaoli AM, NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients With Nonalcoholic Steatohepatitis, Hepatology 71 (2020) 1198–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lounis MA, Escoula Q, Veillette C, Bergeron KF, Ntambi JM, Mounier C, SCD1 deficiency protects mice against ethanol-induced liver injury, Biochim. Biophys. Acta 1861 (2016) 1662–1670. [DOI] [PubMed] [Google Scholar]
- [138].Iruarrizaga-Lejarreta M, Varela-Rey M, Fernandez-Ramos D, Martinez-Arranz I, Delgado TC, Simon J, Juan VG, delaCruz-Villar L, Azkargorta M, Lavin JL, Mayo R, Van Liempd SM, Aurrekoetxea I, Buque X, Cave DD, Pena A, Rodriguez-Cuesta J, Aransay AM, Elortza F, Falcon-Perez JM, Aspichueta P, Hayardeny L, Noureddin M, Sanyal AJ, Alonso C, Anguita J, Martinez-Chantar ML, Lu SC, Mato JM, Role of Aramchol in steatohepatitis and fibrosis in mice, Hepatol. Commun 1 (2017) 911–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Goldiner I, van der Velde AE, Vandenberghe KE, van Wijland MA, Halpern Z, Gilat T, Konikoff FM, Veldman RJ, Groen AK, ABCA1-dependent but apoA-I-independent cholesterol efflux mediated by fatty acid-bile acid conjugates (FABACs), Biochem. J 396 (2006) 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, Oren R, FLORA Group, The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease, Clin. Gastroenterol. Hepatol 12 (2014) 2085–91.e1. [DOI] [PubMed] [Google Scholar]
- [141].Garcia D, Hellberg K, Chaix A, Wallace M, Herzig S, Badur MG, Lin T, Shokhirev MN, Pinto AFM, Ross DS, Saghatelian A, Panda S, Dow LE, Metallo CM, Shaw RJ, Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD, Cell. Rep 26 (2019) 192–208.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Yao H, Tao X, Xu L, Qi Y, Yin L, Han X, Xu Y, Zheng L, Peng J, Dioscin alleviates non-alcoholic fatty liver disease through adjusting lipid metabolism via SIRT1/AMPK signaling pathway, Pharmacol. Res 131 (2018) 51–60. [DOI] [PubMed] [Google Scholar]
- [143].Woods A, Azzout-Marniche D, Foretz M, Stein SC, Lemarchand P, Ferre P, Foufelle F, Carling D, Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase, Mol. Cell. Biol 20 (2000) 6704–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M, AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development, EBioMedicine 28 (2018) 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Esquejo RM, Salatto CT, Delmore J, Albuquerque B, Reyes A, Shi Y, Moccia R, Cokorinos E, Peloquin M, Monetti M, Barricklow J, Bollinger E, Smith BK, Day EA, Nguyen C, Geoghegan KF, Kreeger JM, Opsahl A, Ward J, Kalgutkar AS, Tess D, Butler L, Shirai N, Osborne TF, Steinberg GR, Birnbaum MJ, Cameron KO, Miller RA, Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models, EBioMedicine 31 (2018) 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Hwahng SH, Ki SH, Bae EJ, Kim HE, Kim SG, Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones, Hepatology 49 (2009) 1913–1925. [DOI] [PubMed] [Google Scholar]
- [147].Ko CW, Qu J, Black DD, Tso P, Regulation of intestinal lipid metabolism: current concepts and relevance to disease, Nat. Rev. Gastroenterol. Hepatol 17 (2020) 169–183. [DOI] [PubMed] [Google Scholar]
- [148].Oniciu DC, Hashiguchi T, Shibazaki Y, Bisgaier CL, Gemcabene downregulates inflammatory, lipid-altering and cell-signaling genes in the STAM model of NASH, PLoS One 13 (2018) e0194568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sirtori CR, Yamashita S, Greco MF, Corsini A, Watts GF, Ruscica M, Recent advances in synthetic pharmacotherapies for dyslipidaemias, Eur. J. Prev. Cardiol 27 (2020) 1576–1596. [DOI] [PubMed] [Google Scholar]
- [150].Stein E, Bays H, Koren M, Bakker-Arkema R, Bisgaier C, Efficacy and safety of gemcabene as add-on to stable statin therapy in hypercholesterolemic patients, J. Clin. Lipidol 10 (2016) 1212–1222. [DOI] [PubMed] [Google Scholar]
- [151].Bisgaier CL, Essenburg AD, Barnett BC, Auerbach BJ, Haubenwallner S, Leff T, White AD, Creger P, Pape ME, Rea TJ, Newton RS, A novel compound that elevates high density lipoprotein and activates the peroxisome proliferator activated receptor, J. Lipid Res 39 (1998) 17–30. [PubMed] [Google Scholar]
- [152].Hirsova P, Bohm F, Dohnalkova E, Nozickova B, Heikenwalder M, Gores GJ, Weber A, Hepatocyte apoptosis is tumor promoting in murine nonalcoholic steatohepatitis, Cell. Death Dis 11 (2020) 80–020–2283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Akazawa Y, Nakao K, To die or not to die: death signaling in nonalcoholic fatty liver disease, J. Gastroenterol 53 (2018) 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Akazawa Y, Gores GJ, Death receptor-mediated liver injury, Semin. Liver Dis 27 (2007) 327–338. [DOI] [PubMed] [Google Scholar]
- [155].Zou C, Ma J, Wang X, Guo L, Zhu Z, Stoops J, Eaker AE, Johnson CJ, Strom S, Michalopoulos GK, DeFrances MC, Zarnegar R, Lack of Fas antagonism by Met in human fatty liver disease, Nat. Med 13 (2007) 1078–1085. [DOI] [PubMed] [Google Scholar]
- [156].Cubero FJ, Woitok MM, Zoubek ME, de Bruin A, Hatting M, Trautwein C, Disruption of the FasL/Fas axis protects against inflammation-derived tumorigenesis in chronic liver disease, Cell. Death Dis 10 (2019) 115–019–1391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Krishnan A, Kocab AJ, Zacks DN, Marshak-Rothstein A, Gregory-Ksander M, A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma, J. Neuroinflammation 16 (2019) 184–019–1576–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Agbanoma G, Li C, Ennis D, Palfreeman AC, Williams LM, Brennan FM, Production of TNF-alpha in macrophages activated by T cells, compared with lipopolysaccharide, uses distinct IL-10-dependent regulatory mechanism, J. Immunol 188 (2012) 1307–1317. [DOI] [PubMed] [Google Scholar]
- [159].Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, Teufel A, Itzel T, Longerich T, Maier O, Fischer R, Kontermann RE, Pfizenmaier K, Schulze-Osthoff K, Bantel H, TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice, Cell. Death Dis 11 (2020) 212–020–2411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Sheng Y, Li F, Qin Z, TNF Receptor 2 Makes Tumor Necrosis Factor a Friend of Tumors, Front. Immunol 9 (2018) 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Zein CO, Yerian LM, Gogate P, Lopez R, Kirwan JP, Feldstein AE, McCullough AJ, Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial, Hepatology 54 (2011) 1610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Du J, Ma YY, Yu CH, Li YM, Effects of pentoxifylline on nonalcoholic fatty liver disease: a meta-analysis, World J. Gastroenterol 20 (2014) 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Bouziana SD, Tziomalos K, Inhibition of apoptosis in the management of nonalcoholic fatty liver disease, World J. Gastrointest. Pharmacol. Ther 4 (2013) 4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Alkhouri N, Carter-Kent C, Feldstein AE, Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications, Expert Rev. Gastroenterol. Hepatol 5 (2011) 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H, Kaneko T, Fujisawa M, Higuchi T, Nakamura H, Matsumoto N, Yamagami H, Ogawa M, Imazu H, Kuroda K, Moriyama M, Apoptosis and non-alcoholic fatty liver diseases, World J. Gastroenterol 24 (2018) 2661–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Garcia-Tsao G, Bosch J, Kayali Z, Harrison SA, Abdelmalek MF, Lawitz E, Satapathy SK, Ghabril M, Shiffman ML, Younes ZH, Thuluvath PJ, Berzigotti A, Albillos A, Robinson JM, Hagerty DT, Chan JL, Sanyal AJ, IDN-6556–14 Investigators(double dagger), Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension, J. Hepatol 72 (2020) 885–895. [DOI] [PubMed] [Google Scholar]
- [167].Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, Omenetti A, Jung Y, Teaberry V, Choi SS, Guy CD, Pollard J, Charlton P, Diehl AM, Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis, Hepatology 50 (2009) 1421–1430. [DOI] [PubMed] [Google Scholar]
- [168].Ratziu V, Sheikh MY, Sanyal AJ, Lim JK, Conjeevaram H, Chalasani N, Abdelmalek M, Bakken A, Renou C, Palmer M, Levine RA, Bhandari BR, Cornpropst M, Liang W, King B, Mondou E, Rousseau FS, McHutchison J, Chojkier M, A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis, Hepatology 55 (2012) 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Zhong X, Liu H, Baicalin attenuates diet induced nonalcoholic steatohepatitis by inhibiting inflammation and oxidative stress via suppressing JNK signaling pathways, Biomed. Pharmacother 98 (2018) 111–117. [DOI] [PubMed] [Google Scholar]
- [170].Pockros PJ, Schiff ER, Shiffman ML, McHutchison JG, Gish RG, Afdhal NH, Makhviladze M, Huyghe M, Hecht D, Oltersdorf T, Shapiro DA, Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C, Hepatology 46 (2007) 324–329. [DOI] [PubMed] [Google Scholar]
- [171].Sayama K, Hanakawa Y, Shirakata Y, Yamasaki K, Sawada Y, Sun L, Yamanishi K, Ichijo H, Hashimoto K, Apoptosis signal-regulating kinase 1 (ASK1) is an intracellular inducer of keratinocyte differentiation, J. Biol. Chem 276 (2001) 999–1004. [DOI] [PubMed] [Google Scholar]
- [172].Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D, Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx, Science 281 (1998) 1860–1863. [DOI] [PubMed] [Google Scholar]
- [173].Yamamoto K, Ichijo H, Korsmeyer SJ, BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M, Mol. Cell. Biol 19 (1999) 8469–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Yamamoto E, Dong YF, Kataoka K, Yamashita T, Tokutomi Y, Matsuba S, Ichijo H, Ogawa H, Kim-Mitsuyama S, Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition, Hypertension 52 (2008) 573–580. [DOI] [PubMed] [Google Scholar]
- [175].Haase VH, Hypoxia-inducible factor signaling in the development of kidney fibrosis, Fibrogenesis Tissue Repair 5 (2012) S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, Diehl AM, Djedjos CS, Han L, Myers RP, Subramanian GM, McHutchison JG, Goodman ZD, Afdhal NH, Charlton MR, GS-US-384–1497 Investigators, The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial, Hepatology 67 (2018) 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Morrison MC, Kleemann R, van Koppen A, Hanemaaijer R, Verschuren L, Key Inflammatory Processes in Human NASH Are Reflected in Ldlr(−/−).Leiden Mice: A Translational Gene Profiling Study, Front. Physiol 9 (2018) 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Schilter HC, Collison A, Russo RC, Foot JS, Yow TT, Vieira AT, Tavares LD, Mattes J, Teixeira MM, Jarolimek W, Effects of an anti-inflammatory VAP-1/SSAO inhibitor, PXS-4728A, on pulmonary neutrophil migration, Respir. Res 16 (2015) 42–015–0200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Virtanen H, Autio A, Siitonen R, Liljenback H, Saanijoki T, Lankinen P, Makila J, Kakela M, Teuho J, Savisto N, Jaakkola K, Jalkanen S, Roivainen A, 68Ga-DOTA-Siglec-9--a new imaging tool to detect synovitis, Arthritis Res. Ther 17 (2015) 308–015–0826–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Foot JS, Deodhar M, Turner CI, Yin P, van Dam EM, Silva DG, Olivieri A, Holt A, McDonald IA, The discovery and development of selective 3-fluoro-4-aryloxyallylamine inhibitors of the amine oxidase activity of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 (SSAO/VAP-1), Bioorg. Med. Chem. Lett 22 (2012) 3935–3940. [DOI] [PubMed] [Google Scholar]
- [181].O’Rourke AM, Wang EY, Miller A, Podar EM, Scheyhing K, Huang L, Kessler C, Gao H, Ton-Nu HT, Macdonald MT, Jones DS, Linnik MD, Anti-inflammatory effects of LJP 1586 [Z-3-fluoro-2-(4-methoxybenzyl)allylamine hydrochloride], an amine-based inhibitor of semicarbazide-sensitive amine oxidase activity, J. Pharmacol. Exp. Ther 324 (2008) 867–875. [DOI] [PubMed] [Google Scholar]
- [182].Horvath A, Menghis A, Botz B, Borbely E, Kemeny A, Tekus V, Csepregi JZ, Mocsai A, Juhasz T, Zakany R, Bogdan D, Matyus P, Keeble J, Pinter E, Helyes Z, Analgesic and Anti-Inflammatory Effects of the Novel Semicarbazide-Sensitive Amine-Oxidase Inhibitor SzV-1287 in Chronic Arthritis Models of the Mouse, Sci. Rep 7 (2017) 39863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A, Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance, Nature 447 (2007) 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M, The chemokine system in diverse forms of macrophage activation and polarization, Trends Immunol 25 (2004) 677–686. [DOI] [PubMed] [Google Scholar]
- [185].Ito A, Suganami T, Yamauchi A, Degawa-Yamauchi M, Tanaka M, Kouyama R, Kobayashi Y, Nitta N, Yasuda K, Hirata Y, Kuziel WA, Takeya M, Kanegasaki S, Kamei Y, Ogawa Y, Role of CC chemokine receptor 2 in bone marrow cells in the recruitment of macrophages into obese adipose tissue, J. Biol. Chem 283 (2008) 35715–35723. [DOI] [PubMed] [Google Scholar]
- [186].Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M, MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity, J. Clin. Invest 116 (2006) 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW Jr, CCR2 modulates inflammatory and metabolic effects of high-fat feeding, J. Clin. Invest 116 (2006) 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Barmania F, Pepper MS, C-C chemokine receptor type five (CCR5): An emerging target for the control of HIV infection, Appl. Transl. Genom 2 (2013) 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Tamura Y, Sugimoto M, Murayama T, Minami M, Nishikaze Y, Ariyasu H, Akamizu T, Kita T, Yokode M, Arai H, C-C chemokine receptor 2 inhibitor improves diet-induced development of insulin resistance and hepatic steatosis in mice, J. Atheroscler. Thromb 17 (2010) 219–228. [DOI] [PubMed] [Google Scholar]
- [190].Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, Ariyasu H, Akamizu T, Kita T, Yokode M, Arai H, Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice, Arterioscler. Thromb. Vasc. Biol 28 (2008) 2195–2201. [DOI] [PubMed] [Google Scholar]
- [191].Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, Schwabe RF, CCR1 and CCR5 promote hepatic fibrosis in mice, J. Clin. Invest 119 (2009) 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Heinrichs D, Berres ML, Nellen A, Fischer P, Scholten D, Trautwein C, Wasmuth HE, Sahin H, The chemokine CCL3 promotes experimental liver fibrosis in mice, PLoS One 8 (2013) e66106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Tacke F, Functional role of intrahepatic monocyte subsets for the progression of liver inflammation and liver fibrosis in vivo, Fibrogenesis Tissue Repair 5 (2012) S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Aoyama T, Inokuchi S, Brenner DA, Seki E, CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice, Hepatology 52 (2010) 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Groom JR, Luster AD, CXCR3 ligands: redundant, collaborative and antagonistic functions, Immunol. Cell Biol 89 (2011) 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Goncalves AS, Appelberg R, The involvement of the chemokine receptor CXCR2 in neutrophil recruitment in LPS-induced inflammation and in Mycobacterium avium infection, Scand. J. Immunol 55 (2002) 585–591. [DOI] [PubMed] [Google Scholar]
- [197].Holz O, Khalilieh S, Ludwig-Sengpiel A, Watz H, Stryszak P, Soni P, Tsai M, Sadeh J, Magnussen H, SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced neutrophilia in healthy subjects, Eur. Respir. J 35 (2010) 564–570. [DOI] [PubMed] [Google Scholar]
- [198].Shiraishi M, Aramaki Y, Seto M, Imoto H, Nishikawa Y, Kanzaki N, Okamoto M, Sawada H, Nishimura O, Baba M, Fujino M, Discovery of novel, potent, and selective small-molecule CCR5 antagonists as anti-HIV-1 agents: synthesis and biological evaluation of anilide derivatives with a quaternary ammonium moiety, J. Med. Chem 43 (2000) 2049–2063. [DOI] [PubMed] [Google Scholar]
- [199].Junker A, Kokornaczyk AK, Zweemer AJ, Frehland B, Schepmann D, Yamaguchi J, Itami K, Faust A, Hermann S, Wagner S, Schafers M, Koch M, Weiss C, Heitman LH, Kopka K, Wunsch B, Synthesis, binding affinity and structure-activity relationships of novel, selective and dual targeting CCR2 and CCR5 receptor antagonists, Org. Biomol. Chem 13 (2015) 2407–2422. [DOI] [PubMed] [Google Scholar]
- [200].Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, Takimoto R, Takada K, Miyanishi K, Matsunaga T, Takayama T, Niitsu Y, Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone, Nat. Biotechnol 26 (2008) 431–442. [DOI] [PubMed] [Google Scholar]
- [201].Anstee QM, Neuschwander-Tetri BA, Wong VW, Abdelmalek MF, Younossi ZM, Yuan J, Pecoraro ML, Seyedkazemi S, Fischer L, Bedossa P, Goodman Z, Alkhouri N, Tacke F, Sanyal A, Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design, Contemp. Clin. Trials 89 (2020) 105922. [DOI] [PubMed] [Google Scholar]
- [202].Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, Kayali Z, Wong VW, Greenbloom S, Jayakumar S, Shiffman ML, Freilich B, Lawitz EJ, Gane EJ, Harting E, Xu J, Billin AN, Chung C, Djedjos CS, Subramanian GM, Myers RP, Middleton MS, Rinella M, Noureddin M, Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial, Hepatology 72 (2020) 58–71. [DOI] [PubMed] [Google Scholar]
- [203].Yu D, Cai SY, Mennone A, Vig P, Boyer JL, Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents, Liver Int 38 (2018) 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, Olman MA, Pritchard MT, Nagy LE, Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis, PLoS One 8 (2013) e69114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Kimand SK, Jacobson KA, Three-dimensional quantitative structure-activity relationship of nucleosides acting at the A3 adenosine receptor: analysis of binding and relative efficacy, J. Chem. Inf. Model 47 (2007) 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Ohno M, Gao ZG, Van Rompaey P, Tchilibon S, Kim SK, Harris BA, Gross AS, Duong HT, Van Calenbergh S, Jacobson KA, Modulation of adenosine receptor affinity and intrinsic efficacy in adenine nucleosides substituted at the 2-position, Bioorg. Med. Chem 12 (2004) 2995–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Koscso B, Csoka B, Pacher P, Hasko G, Investigational A(3) adenosine receptor targeting agents, Expert Opin. Investig. Drugs 20 (2011) 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Volpini R, Buccioni M, Dal Ben D, Lambertucci C, Lammi C, Marucci G, Ramadori AT, Klotz KN, Cristalli G, Synthesis and biological evaluation of 2-alkynyl-N6-methyl-5’-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A3 receptor, J. Med. Chem 52 (2009) 7897–7900. [DOI] [PubMed] [Google Scholar]
- [209].Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA, (N)-methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists, J. Med. Chem 48 (2005) 1745–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, Gao ZG, Lu C, Duong HT, Gunaga P, Lee SK, Jin DZ, Chun MW, Moon HR, Structure-activity relationships of 2-chloro-N6-substituted-4’-thioadenosine-5’-uronamides as highly potent and selective agonists at the human A3 adenosine receptor, J. Med. Chem 49 (2006) 273–281. [DOI] [PubMed] [Google Scholar]
- [211].Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, Knight DR, Novel N6-substituted adenosine 5’-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury, Am. J. Physiol. Heart Circ. Physiol 285 (2003) H2780–7. [DOI] [PubMed] [Google Scholar]
- [212].Koroskenyi K, Kiss B, Szondy Z, Adenosine A2A receptor signaling attenuates LPS-induced pro-inflammatory cytokine formation of mouse macrophages by inducing the expression of DUSP1, Biochim. Biophys. Acta 1863 (2016) 1461–1471. [DOI] [PubMed] [Google Scholar]
- [213].Jiang F, Wei Q, Li H, Li H, Cui Y, Ma Y, Chen H, Cao P, Lu T, Chen Y, Discovery of novel small molecule induced selective degradation of the bromodomain and extra-terminal (BET) bromodomain protein BRD4 and BRD2 with cellular potencies, Bioorg. Med. Chem 28 (2020) 115181. [DOI] [PubMed] [Google Scholar]
- [214].Middleton SA, Rajpal N, Cutler L, Mander P, Rioja I, Prinjha RK, Rajpal D, Agarwal P, Kumar V, BET Inhibition Improves NASH and Liver Fibrosis, Sci. Rep 8 (2018) 17257–018–35653–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Zhang X, Shen J, Man K, Chu ES, Yau TO, Sung JC, Go MY, Deng J, Lu L, Wong VW, Sung JJ, Farrell G, Yu J, CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis, J. Hepatol 61 (2014) 1365–1375. [DOI] [PubMed] [Google Scholar]
- [216].Juhling F, Hamdane N, Crouchet E, Li S, El Saghire H, Mukherji A, Fujiwara N, Oudot MA, Thumann C, Saviano A, Roca Suarez AA, Goto K, Masia R, Sojoodi M, Arora G, Aikata H, Ono A, Tabrizian P, Schwartz M, Polyak SJ, Davidson I, Schmidl C, Bock C, Schuster C, Chayama K, Pessaux P, Tanabe KK, Hoshida Y, Zeisel MB, Duong FH, Fuchs BC, Baumert TF, Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma, Gut 70 (2021) 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Yamada A, Honma K, Mochizuki K, Goda T, BRD4 regulates fructose-inducible lipid accumulation-related genes in the mouse liver, Metabolism 65 (2016) 1478–1488. [DOI] [PubMed] [Google Scholar]
- [218].Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M, He M, Liddle C, Downes M, Evans RM, BRD4 is a novel therapeutic target for liver fibrosis, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 15713–15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Jung H, Chen J, Hu X, Sun H, Wu SY, Chiang CM, Kemper B, Chen LF, Kemper J, BRD4 inhibition and FXR activation, individually beneficial in cholestasis, are antagonistic in combination, JCI Insight (2020) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Iacobini C, Menini S, Ricci C, Blasetti Fantauzzi C, Scipioni A, Salvi L, Cordone S, Delucchi F, Serino M, Federici M, Pricci F, Pugliese G, Galectin-3 ablation protects mice from diet-induced NASH: a major scavenging role for galectin-3 in liver, J. Hepatol 54 (2011) 975–983. [DOI] [PubMed] [Google Scholar]
- [221].Sebekova K, Kupcova V, Schinzel R, Heidland A, Markedly elevated levels of plasma advanced glycation end products in patients with liver cirrhosis - amelioration by liver transplantation, J. Hepatol 36 (2002) 66–71. [DOI] [PubMed] [Google Scholar]
- [222].MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T, Regulation of alternative macrophage activation by galectin-3, J. Immunol 180 (2008) 2650–2658. [DOI] [PubMed] [Google Scholar]
- [223].Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, Noureddin M, Pyko M, Shiffman M, Sanyal A, Allgood A, Shlevin H, Horton R, Zomer E, Irish W, Goodman Z, Harrison SA, Traber PG, Belapectin (GR-MD-02) Study Investigators, Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension, Gastroenterology 158 (2020) 1334–1345.e5. [DOI] [PubMed] [Google Scholar]
- [224].Markowska AI, Jefferies KC, Panjwani N, Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells, J. Biol. Chem 286 (2011) 29913–29921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Chen WS, Cao Z, Leffler H, Nilsson UJ, Panjwani N, Galectin-3 Inhibition by a Small-Molecule Inhibitor Reduces Both Pathological Corneal Neovascularization and Fibrosis, Invest. Ophthalmol. Vis. Sci 58 (2017) 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Traber PG, Chou H, Zomer E, Hong F, Klyosov A, Fiel MI, Friedman SL, Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease, PLoS One 8 (2013) e75361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Traber PG, Zomer E, Therapy of experimental NASH and fibrosis with galectin inhibitors, PLoS One 8 (2013) e83481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Glinsky VV, Kiriakova G, Glinskii OV, Mossine VV, Mawhinney TP, Turk JR, Glinskii AB, Huxley VH, Price JE, Glinsky GV, Synthetic galectin-3 inhibitor increases metastatic cancer cell sensitivity to taxol-induced apoptosis in vitro and in vivo, Neoplasia 11 (2009) 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Kumar N, Goldminz AM, Kim N, Gottlieb AB, Phosphodiesterase 4-targeted treatments for autoimmune diseases, BMC Med 11 (2013) 96–7015–11–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Essam RM, Ahmed LA, Abdelsalam RM, El-Khatib AS, Phosphodiestrase-1 and 4 inhibitors ameliorate liver fibrosis in rats: Modulation of cAMP/CREB/TLR4 inflammatory and fibrogenic pathways, Life Sci 222 (2019) 245–254. [DOI] [PubMed] [Google Scholar]
- [231].Ratziu V, Bedossa P, Francque SM, Larrey D, Aithal GP, Serfaty L, Voiculescu M, Preotescu L, Nevens F, De Ledinghen V, Kirchner GI, Trunecka P, Ryder SD, Day CP, Takeda J, Traudtner K, Lack of efficacy of an inhibitor of PDE4 in phase 1 and 2 trials of patients with nonalcoholic steatohepatitis, Clin. Gastroenterol. Hepatol 12 (2014) 1724–30.e5. [DOI] [PubMed] [Google Scholar]
- [232].Asano M, Toda M, Sakaguchi N, Sakaguchi S, Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation, J. Exp. Med 184 (1996) 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D, Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters, Nat. Med 15 (2009) 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Mizrahi M, Shabat Y, Ben Ya’acov A, Lalazar G, Adar T, Wong V, Muller B, Rawlin G, Ilan Y, Alleviation of insulin resistance and liver damage by oral administration of Imm124-E is mediated by increased Tregs and associated with increased serum GLP-1 and adiponectin: results of a phase I/II clinical trial in NASH, J. Inflamm. Res 5 (2012) 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Kumar V, Mondal G, Dutta R, Mahato RI, Co-delivery of small molecule hedgehog inhibitor and miRNA for treating liver fibrosis, Biomaterials 76 (2016) 144–156. [DOI] [PubMed] [Google Scholar]
- [236].Friedman SL, Extracellular matrix, in: Dufour P-CJ-F (Ed.), Signaling pathways in liver diseases (3rd ed.), 2015, pp. 85–96.
- [237].Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC, Resolving the fibrotic niche of human liver cirrhosis at single-cell level, Nature 575 (2019) 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Shi Y, Long F, Hedgehog signaling via Gli2 prevents obesity induced by high-fat diet in adult mice, eLife 6 (2017) e31649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM, Hedgehog signaling plays a conserved role in inhibiting fat formation, Cell. Metab 3 (2006) 25–34. [DOI] [PubMed] [Google Scholar]
- [240].Landsman L, Parent A, Hebrok M, Elevated Hedgehog/Gli signaling causes beta-cell dedifferentiation in mice, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 17010–17015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Yao Q, Liu J, Xiao L, Wang N, Sonic hedgehog signaling instigates high-fat diet-induced insulin resistance by targeting PPARgamma stability, J. Biol. Chem 294 (2019) 3284–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Hirsova P, Gores GJ, Ballooned hepatocytes, undead cells, sonic hedgehog, and vitamin E: therapeutic implications for nonalcoholic steatohepatitis, Hepatology 61 (2015) 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, Mi Z, Pereira TA, Zdanowicz M, Malladi P, Chen Y, Moylan C, Jung Y, Bhattacharya SD, Teaberry V, Omenetti A, Abdelmalek MF, Guy CD, Adams DH, Kuo PC, Michelotti GA, Whitington PF, Diehl AM, Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis, Hepatology 53 (2011) 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Kwon H, Song K, Han C, Chen W, Wang Y, Dash S, Lim K, Wu T, Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease, Hepatology 63 (2016) 1155–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Song M, Han L, Chen FF, Wang D, Wang F, Zhang L, Wang ZH, Zhong M, Tang MX, Zhang W, Adipocyte-Derived Exosomes Carrying Sonic Hedgehog Mediate M1 Macrophage Polarization-Induced Insulin Resistance via Ptch and PI3K Pathways, Cell. Physiol. Biochem 48 (2018) 1416–1432. [DOI] [PubMed] [Google Scholar]
- [246].Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR, YAP1 increases organ size and expands undifferentiated progenitor cells, Curr. Biol 17 (2007) 2054–2060. [DOI] [PubMed] [Google Scholar]
- [247].Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, Diehl AM, Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease, J. Hepatol 63 (2015) 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Moon H, Cho K, Shin S, Kim DY, Han KH, Ro SW, High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway, Int. J. Mol. Sci 20 (2019) 10.3390/ijms20030581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, Tabas I, Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis, Cell. Metab 24 (2016) 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Bariwal J, Kumar V, Dong Y, Mahato RI, Design of Hedgehog pathway inhibitors for cancer treatment, Med. Res. Rev 39 (2019) 1137–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Kumar V, Mundra V, Mahato RI, Nanomedicines of Hedgehog inhibitor and PPAR-gamma agonist for treating liver fibrosis, Pharm. Res 31 (2014) 1158–1169. [DOI] [PubMed] [Google Scholar]
- [252].Kumar V, Dong Y, Kumar V, Almawash S, Mahato RI, The use of micelles to deliver potential hedgehog pathway inhibitor for the treatment of liver fibrosis, Theranostics 9 (2019) 7537–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Park JG, Mok JS, Han YI, Park TS, Kang KW, Choi CS, Park HD, Park J, Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan, Sci. Rep 9 (2019) 4003–019–40322–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Haghbin H, Gangwani MK, Ravi SJK, Perisetti A, Aziz M, Goyal H, Nawras A, Sodeman T, Nonalcoholic fatty liver disease and atrial fibrillation: possible pathophysiological links and therapeutic interventions, Ann. Gastroenterol 33 (2020) 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Jonsson JR, Clouston AD, Ando Y, Kelemen LI, Horn MJ, Adamson MD, Purdie DM, Powell EE, Angiotensin-converting enzyme inhibition attenuates the progression of rat hepatic fibrosis, Gastroenterology 121 (2001) 148–155. [DOI] [PubMed] [Google Scholar]
- [256].Vos MB, Jin R, Konomi JV, Cleeton R, Cruz J, Karpen S, Rodriguez DS, Frediani JK, McCracken C, Welsh J, A randomized, controlled, crossover pilot study of losartan for pediatric nonalcoholic fatty liver disease, Pilot Feasibility Stud 4 (2018) 109–018–0306–4. eCollection 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Pelusi S, Petta S, Rosso C, Borroni V, Fracanzani AL, Dongiovanni P, Craxi A, Bugianesi E, Fargion S, Valenti L, Renin-Angiotensin System Inhibitors, Type 2 Diabetes and Fibrosis Progression: An Observational Study in Patients with Nonalcoholic Fatty Liver Disease, PLoS One 11 (2016) e0163069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].McPherson S, Wilkinson N, Tiniakos D, Wilkinson J, Burt AD, McColl E, Stocken DD, Steen N, Barnes J, Goudie N, Stewart S, Bury Y, Mann D, Anstee QM, Day CP, A randomised controlled trial of losartan as an anti-fibrotic agent in non-alcoholic steatohepatitis, PLoS One 12 (2017) e0175717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Okuno M, Akita K, Moriwaki H, Kawada N, Ikeda K, Kaneda K, Suzuki Y, Kojima S, Prevention of rat hepatic fibrosis by the protease inhibitor, camostat mesilate, via reduced generation of active TGF-beta, Gastroenterology 120 (2001) 1784–1800. [DOI] [PubMed] [Google Scholar]
- [260].Kato M, Yuan H, Xu ZG, Lanting L, Li SL, Wang M, Hu MC, Reddy MA, Natarajan R, Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease, J. Am. Soc. Nephrol 17 (2006) 3325–3335. [DOI] [PubMed] [Google Scholar]
- [261].Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, Tamaki K, Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression, Lab. Invest 95 (2015) 1130–1144. [DOI] [PubMed] [Google Scholar]
- [262].Varin F, Huet PM, Hepatic microcirculation in the perfused cirrhotic rat liver, J. Clin. Invest 76 (1985) 1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Fan QQ, Zhang CL, Qiao JB, Cui PF, Xing L, Oh YK, Jiang HL, Extracellular matrix-penetrating nanodrill micelles for liver fibrosis therapy, Biomaterials 230 (2020) 119616. [DOI] [PubMed] [Google Scholar]
- [264].Zinger A, Koren L, Adir O, Poley M, Alyan M, Yaari Z, Noor N, Krinsky N, Simon A, Gibori H, Krayem M, Mumblat Y, Kasten S, Ofir S, Fridman E, Milman N, Lubtow MM, Liba L, Shklover J, Shainsky-Roitman J, Binenbaum Y, Hershkovitz D, Gil Z, Dvir T, Luxenhofer R, Satchi-Fainaro R, Schroeder A, Collagenase Nanoparticles Enhance the Penetration of Drugs into Pancreatic Tumors, ACS Nano 13 (2019) 11008–11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Geervliet E, Moreno S, Baiamonte L, Booijink R, Boye S, Wang P, Voit B, Lederer A, Appelhans D, Bansal R, Matrix metalloproteinase-1 decorated polymersomes, a surface-active extracellular matrix therapeutic, potentiates collagen degradation and attenuates early liver fibrosis, J. Control. Release 332 (2021) 594–607. [DOI] [PubMed] [Google Scholar]
- [266].Parodi A, Haddix SG, Taghipour N, Scaria S, Taraballi F, Cevenini A, Yazdi IK, Corbo C, Palomba R, Khaled SZ, Martinez JO, Brown BS, Isenhart L, Tasciotti E, Bromelain surface modification increases the diffusion of silica nanoparticles in the tumor extracellular matrix, ACS Nano 8 (2014) 9874–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].George J, Stern R, Serum hyaluronan and hyaluronidase: very early markers of toxic liver injury, Clin. Chim. Acta 348 (2004) 189–197. [DOI] [PubMed] [Google Scholar]
- [268].Yang YM, Noureddin M, Liu C, Ohashi K, Kim SY, Ramnath D, Powell EE, Sweet MJ, Roh YS, Hsin IF, Deng N, Liu Z, Liang J, Mena E, Shouhed D, Schwabe RF, Jiang D, Lu SC, Noble PW, Seki E, Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis, Sci. Transl. Med 11 (2019) 10.1126/scitranslmed.aat9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Andreichenko IN, Tsitrina AA, Fokin AV, Gabdulkhakova AI, Maltsev DI, Perelman GS, Bulgakova EV, Kulikov AM, Mikaelyan AS, Kotelevtsev YV, 4-methylumbelliferone Prevents Liver Fibrosis by Affecting Hyaluronan Deposition, FSTL1 Expression and Cell Localization, Int. J. Mol. Sci 20 (2019) 10.3390/ijms20246301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Yang YM, Wang Z, Matsuda M, Seki E, Inhibition of hyaluronan synthesis by 4-methylumbelliferone ameliorates non-alcoholic steatohepatitis in choline-deficient L-amino acid-defined diet-induced murine model, Arch. Pharm. Res 44 (2021) 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Rajan M, Raj V, Al-Arfaj AA, Murugan AM, Hyaluronidase enzyme core-5fluorouracil-loaded chitosan-PEG-gelatin polymer nanocomposites as targeted and controlled drug delivery vehicles, Int. J. Pharm 453 (2013) 514–522. [DOI] [PubMed] [Google Scholar]
- [272].Mandal AK, Das S, Basu MK, Chakrabarti RN, Das N, Hepatoprotective activity of liposomal flavonoid against arsenite-induced liver fibrosis, J. Pharmacol. Exp. Ther 320 (2007) 994–1001. [DOI] [PubMed] [Google Scholar]
- [273].Zhang D, Guo Z, Zhang P, Li Y, Su X, You L, Gao M, Liu C, Wu H, Zhang X, Simplified quantification method for in vivo SPECT/CT imaging of asialoglycoprotein receptor with (99m)Tc-p(VLA-co-VNI) to assess and stage hepatic fibrosis in mice, Sci. Rep 6 (2016) 25377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Kang JH, Tachibana Y, Kamata W, Mahara A, Harada-Shiba M, Yamaoka T, Liver-targeted siRNA delivery by polyethylenimine (PEI)-pullulan carrier, Bioorg. Med. Chem 18 (2010) 3946–3950. [DOI] [PubMed] [Google Scholar]
- [275].Dong Y, Love KT, Dorkin JR, Sirirungruang S, Zhang Y, Chen D, Bogorad RL, Yin H, Chen Y, Vegas AJ, Alabi CA, Sahay G, Olejnik KT, Wang W, Schroeder A, Lytton-Jean AK, Siegwart DJ, Akinc A, Barnes C, Barros SA, Carioto M, Fitzgerald K, Hettinger J, Kumar V, Novobrantseva TI, Qin J, Querbes W, Koteliansky V, Langer R, Anderson DG, Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 3955–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Chen Z, Jain A, Liu H, Zhao Z, Cheng K, Targeted Drug Delivery to Hepatic Stellate Cells for the Treatment of Liver Fibrosis, J. Pharmacol. Exp. Ther 370 (2019) 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H, A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A, Science 315 (2007) 820–825. [DOI] [PubMed] [Google Scholar]
- [278].Nagata K, Hsp47: a collagen-specific molecular chaperone, Trends Biochem. Sci 21 (1996) 22–26. [DOI] [PubMed] [Google Scholar]
- [279].Kawasaki K, Ushioda R, Ito S, Ikeda K, Masago Y, Nagata K, Deletion of the collagen-specific molecular chaperone Hsp47 causes endoplasmic reticulum stress-mediated apoptosis of hepatic stellate cells, J. Biol. Chem 290 (2015) 3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Duong HT, Dong Z, Su L, Boyer C, George J, Davis TP, Wang J, The use of nanoparticles to deliver nitric oxide to hepatic stellate cells for treating liver fibrosis and portal hypertension, Small 11 (2015) 2291–2304. [DOI] [PubMed] [Google Scholar]
- [281].Zhang Z, Wang C, Zha Y, Hu W, Gao Z, Zang Y, Chen J, Zhang J, Dong L, Corona-directed nucleic acid delivery into hepatic stellate cells for liver fibrosis therapy, ACS Nano 9 (2015) 2405–2419. [DOI] [PubMed] [Google Scholar]
- [282].Qiao JB, Fan QQ, Xing L, Cui PF, He YJ, Zhu JC, Wang L, Pang T, Oh YK, Zhang C, Jiang HL, Vitamin A-decorated biocompatible micelles for chemogene therapy of liver fibrosis, J. Control. Release 283 (2018) 113–125. [DOI] [PubMed] [Google Scholar]
- [283].Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC, Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells, J. Biol. Chem 279 (2004) 23996–24006. [DOI] [PubMed] [Google Scholar]
- [284].Beljaars L, Molema G, Schuppan D, Geerts A, De Bleser PJ, Weert B, Meijer DK, Poelstra K, Successful targeting to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor, J. Biol. Chem 275 (2000) 12743–12751. [DOI] [PubMed] [Google Scholar]
- [285].Du SL, Pan H, Lu WY, Wang J, Wu J, Wang JY, Cyclic Arg-Gly-Asp peptide-labeled liposomes for targeting drug therapy of hepatic fibrosis in rats, J. Pharmacol. Exp. Ther 322 (2007) 560–568. [DOI] [PubMed] [Google Scholar]
- [286].Li F, Yan H, Wang J, Li C, Wu J, Wu S, Rao S, Gao X, Jin Q, Non-invasively differentiating extent of liver fibrosis by visualizing hepatic integrin alphavbeta3 expression with an MRI modality in mice, Biomaterials 102 (2016) 162–174. [DOI] [PubMed] [Google Scholar]
- [287].Chai NL, Fu Q, Shi H, Cai CH, Wan J, Xu SP, Wu BY, Oxymatrine liposome attenuates hepatic fibrosis via targeting hepatic stellate cells, World J. Gastroenterol 18 (2012) 4199–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Li F, Sun JY, Wang JY, Du SL, Lu WY, Liu M, Xie C, Shi JY, Effect of hepatocyte growth factor encapsulated in targeted liposomes on liver cirrhosis, J. Control. Release 131 (2008) 77–82. [DOI] [PubMed] [Google Scholar]
- [289].Li Y, Pu S, Liu Q, Li R, Zhang J, Wu T, Chen L, Li H, Yang X, Zou M, Xiao J, Xie W, He J, An integrin-based nanoparticle that targets activated hepatic stellate cells and alleviates liver fibrosis, J. Control. Release 303 (2019) 77–90. [DOI] [PubMed] [Google Scholar]
- [290].Pulavendran S, Rose C, Mandal AB, Hepatocyte growth factor incorporated chitosan nanoparticles augment the differentiation of stem cell into hepatocytes for the recovery of liver cirrhosis in mice, J. Nanobiotechnology 9 (2011) 15–3155–9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Beljaars L, Molema G, Weert B, Bonnema H, Olinga P, Groothuis GM, Meijer DK, Poelstra K, Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells, Hepatology 29 (1999) 1486–1493. [DOI] [PubMed] [Google Scholar]
- [292].Dutta R, Kumar V, Peng Y, Evande RE, Grem JL, Mahato RI, Pharmacokinetics and Biodistribution of GDC-0449 Loaded Micelles in Normal and Liver Fibrotic Mice, Pharm. Res 34 (2017) 564–578. [DOI] [PubMed] [Google Scholar]
- [293].Adrian JE, Kamps JA, Poelstra K, Scherphof GL, Meijer DK, Kaneda Y, Delivery of viral vectors to hepatic stellate cells in fibrotic livers using HVJ envelopes fused with targeted liposomes, J. Drug Target 15 (2007) 75–82. [DOI] [PubMed] [Google Scholar]
- [294].Zhu L, Mahato RI, Targeted delivery of siRNA to hepatocytes and hepatic stellate cells by bioconjugation, Bioconjug. Chem 21 (2010) 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Poniachik J, Baraona E, Zhao J, Lieber CS, Dilinoleoylphosphatidylcholine decreases hepatic stellate cell activation, J. Lab. Clin. Med 133 (1999) 342–348. [DOI] [PubMed] [Google Scholar]
- [296].Cao Q, Mak KM, Lieber CS, Dilinoleoylphosphatidylcholine decreases LPS-induced TNF-alpha generation in Kupffer cells of ethanol-fed rats: respective roles of MAPKs and NF-kappaB, Biochem. Biophys. Res. Commun 294 (2002) 849–853. [DOI] [PubMed] [Google Scholar]
- [297].Adrian JE, Poelstra K, Scherphof GL, Meijer DK, van Loenen-Weemaes AM, Reker-Smit C, Morselt HW, Zwiers P, Kamps JA, Effects of a new bioactive lipid-based drug carrier on cultured hepatic stellate cells and liver fibrosis in bile duct-ligated rats, J. Pharmacol. Exp. Ther 321 (2007) 536–543. [DOI] [PubMed] [Google Scholar]
- [298].van Beuge MM, Prakash J, Lacombe M, Gosens R, Post E, Reker-Smit C, Beljaars L, Poelstra K, Reduction of fibrogenesis by selective delivery of a Rho kinase inhibitor to hepatic stellate cells in mice, J. Pharmacol. Exp. Ther 337 (2011) 628–635. [DOI] [PubMed] [Google Scholar]
- [299].Luo J, Zhang P, Zhao T, Jia M, Yin P, Li W, Zhang ZR, Fu Y, Gong T, Golgi Apparatus-Targeted Chondroitin-Modified Nanomicelles Suppress Hepatic Stellate Cell Activation for the Management of Liver Fibrosis, ACS Nano 13 (2019) 3910–3923. [DOI] [PubMed] [Google Scholar]
- [300].Li W, Zhou C, Fu Y, Chen T, Liu X, Zhang Z, Gong T, Targeted delivery of hyaluronic acid nanomicelles to hepatic stellate cells in hepatic fibrosis rats, Acta Pharm. Sin. B 10 (2020) 693–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Clements JM, Bawden LJ, Bloxidge RE, Catlin G, Cook AL, Craig S, Drummond AH, Edwards RM, Fallon A, Green DR, Two PDGF-B chain residues, arginine 27 and isoleucine 30, mediate receptor binding and activation, EMBO J 10 (1991) 4113–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Beljaars L, Weert B, Geerts A, Meijer DK, Poelstra K, The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue, Biochem. Pharmacol 66 (2003) 1307–1317. [DOI] [PubMed] [Google Scholar]
- [303].Li F, Li QH, Wang JY, Zhan CY, Xie C, Lu WY, Effects of interferon-gamma liposomes targeted to platelet-derived growth factor receptor-beta on hepatic fibrosis in rats, J. Control. Release 159 (2012) 261–270. [DOI] [PubMed] [Google Scholar]
- [304].Park K, Hong SW, Hur W, Lee MY, Yang JA, Kim SW, Yoon SK, Hahn SK, Target specific systemic delivery of TGF-beta siRNA/(PEI-SS)-g-HA complex for the treatment of liver cirrhosis, Biomaterials 32 (2011) 4951–4958. [DOI] [PubMed] [Google Scholar]
- [305].Yang JA, Kong WH, Sung DK, Kim H, Kim TH, Lee KC, Hahn SK, Hyaluronic acid-tumor necrosis factor-related apoptosis-inducing ligand conjugate for targeted treatment of liver fibrosis, Acta Biomater 12 (2015) 174–182. [DOI] [PubMed] [Google Scholar]
- [306].Cassiman D, van Pelt J, De Vos R, Van Lommel F, Desmet V, Yap SH, Roskams T, Synaptophysin: A novel marker for human and rat hepatic stellate cells, Am. J. Pathol 155 (1999) 1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Douglass A, Wallace K, Parr R, Park J, Durward E, Broadbent I, Barelle C, Porter AJ, Wright MC, Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury, J. Hepatol 49 (2008) 88–98. [DOI] [PubMed] [Google Scholar]
- [308].Reetz J, Genz B, Meier C, Kowtharapu BS, Timm F, Vollmar B, Herchenroder O, Abshagen K, Putzer BM, Development of Adenoviral Delivery Systems to Target Hepatic Stellate Cells In Vivo, PLoS One 8 (2013) e67091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Schon HT, Bartneck M, Borkham-Kamphorst E, Nattermann J, Lammers T, Tacke F, Weiskirchen R, Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis, Front. Pharmacol 7 (2016) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW, Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination, J. Control. Release 240 (2016) 332–348. [DOI] [PubMed] [Google Scholar]
- [311].Enhancement of macrophage uptake via phosphatidylserine-coated acetalated dextran nanoparticles, - Journal of Drug Delivery Science and Technology - 57. [Google Scholar]