

Supplemental Digital Content is Available in the Text. Dual leucine zipper kinase inhibition is a promising therapeutic strategy against chemotherapy-induced neurotoxicities including chemotherapy-induced peripheral neuropathy and chemotherapy-induced cognitive impairments.
Keywords: DLK, Map3k12, Chemotherapy, Peripheral neuropathy, Cognitive impairment, Neuroimmune, Mitochondria
Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) and chemotherapy-induced cognitive impairments (CICI) are common, often severe neurotoxic side effects of cancer treatment that greatly reduce quality of life of cancer patients and survivors. Currently, there are no Food and Drug Administration-approved agents for the prevention or curative treatment of CIPN or CICI. The dual leucine zipper kinase (DLK) is a key mediator of axonal degeneration that is localized to axons and coordinates the neuronal response to injury. We developed a novel brain-penetrant DLK inhibitor, IACS′8287, which demonstrates potent and highly selective inhibition of DLK in vitro and in vivo. Coadministration of IACS′8287 with the platinum derivative cisplatin prevents mechanical allodynia, loss of intraepidermal nerve fibers in the hind paws, cognitive deficits, and impairments in brain connectivity in mice, all without interfering with the antitumor activity of cisplatin. The protective effects of IACS′8287 are associated with preservation of mitochondrial function in dorsal root ganglion neurons and in brain synaptosomes. In addition, RNA sequencing analysis of dorsal root ganglia reveals modulation of genes involved in neuronal activity and markers for immune cell infiltration by DLK inhibition. These data indicate that CIPN and CICI require DLK signaling in mice, and DLK inhibitors could become an attractive treatment in the clinic when coadministered with cisplatin, and potentially other chemotherapeutic agents, to prevent neurotoxicities as a result of cancer treatment.
1. Introduction
As advances in cancer therapies improve survival, increasing concerns have been raised around long-term side effects of cancer treatment. Chemotherapy-induced peripheral neuropathy (CIPN) and cognitive impairments (CICI) affect up to 75% of patients receiving chemotherapy and can last for months and even years after treatment completion.51,53 Common symptoms of CIPN include persistent shooting or burning pain in the absence of noxious stimuli, tingling, and loss of sensation, which typically present in a “stocking and glove” distribution.20 In CICI, patients experience deficits in memory, attention, information processing, and executive functioning.51 As there are no Food and Drug Administration-approved agents that prevent or treat these disorders, CIPN and CICI can greatly impair quality of life of patients and sometimes even lead to delay or discontinuation of cancer treatment.24
The dual leucine zipper kinase (DLK) is a neuronally enriched mitogen-activated kinase kinase kinase (MAP3K) that activates the c-Jun N-terminal kinase (JNK)/c-Jun pathway.13 In neurons, DLK's primary function is the detection of axonal injury. Axonal injury leads to activation of DLK and reduction of axonal NAD+ levels through induction of the NAD+ degrading enzyme SARM1 and concomitant degradation of the NAD+ synthesizing enzyme NMNAT2, which ultimately result in axon disintegration and neuronal death.17,47 Therefore, inhibiting DLK could provide therapeutic benefits for injuries or diseases that are characterized by axonal injury. Accordingly, genetic ablation or pharmacological inhibition of DLK are beneficial in mouse models of AD and ALS,31,49 and conditional knockout of DLK in sensory neurons prevented development of mechanical allodynia in mouse models of nerve injury.25,63
The exact mechanisms underlying CIPN are debated. Given that the longest sensory neurons are preferentially affected, disruption of axonal transport and mitochondrial function is likely involved, as axons are highly reliant on energetically demanding transport of cargo.20,41 This hypothesis suggests that active axon degeneration programs could be activated after chemotherapy. In line with this view, genetic deletion of Sarm1 or expression of a gain-of-function mutant form of Nmnat2 is protective in animal models of CIPN.15–17,58,61 As DLK participates in the same pathway, DLK inhibition could also block the neuronal injury response to chemotherapy and thereby prevent CIPN. The mechanisms underlying CICI are even less established, particularly given that many CICI-causing therapeutics have poor penetration into the central nervous system (CNS).11 Disruption of functional brain connectivity and white matter integrity occur after chemotherapy in humans,21 as well as in rodents.8 In addition, rodent models indicate a critical role for synaptic mitochondrial dysfunction and synaptic loss in the pathogenesis of CICI.7,40 Interestingly, DLK contributes to synaptic loss in neurodegenerative diseases,31,33 suggesting that inhibiting DLK may also prevent CICI.
In the current study, we investigated the potential of pharmacologically inhibiting DLK for prevention of CIPN and CICI induced by the platinum class chemotherapeutic cisplatin in mice. We used a novel compound, IACS′8287, which demonstrates selective inhibition of DLK kinase activity in vitro and in vivo. Our data indicate that DLK inhibition prevents cisplatin-induced CIPN and CICI and highlight a potential clinical application of DLK inhibitors.
2. Materials and methods
2.1. Human dual leucine zipper kinase–induced p-c-jun cell assay
HEK293 cells expressing doxycycline (DOX)-inducible hDLK-His protein were maintained in Dulbecco's Modified Eagle Medium media (Thermal Fisher Scientific, Waltham, MA) containing 10% fetal bovine serum and 1 µg/mL puromycin. DOX-inducible hDLK-His HEK293 cells were plated into 384-well plate (40,000 cells/20 µL/well) in maintenance medium with or without 1.5 μg/mL DOX. The cell plates were incubated at 37°C, 5% CO2 for 20 hours, and then treated with dimethyl sulfoxide (DMSO) or increasing concentrations of IACS′8287 for 5 hours. Cellular p-c-Jun levels were measured after cell lysis using the p-c-Jun (Ser63) cellular assay kit (Cisbio, Bedford, MA) following manufacturer instructions.
2.2. Human neurotrophic receptor tyrosine kinase 1 (TrkA) cell assay
HEK293 cells expressing the C-terminal HA-tagged human neurotrophic receptor tyrosine kinase 1 (TrkA) protein were maintained and further expanded in Dulbecco's Modified Eagle Medium media containing 10% fetal bovine serum and 200 µg/mL hygromycin. The TrkA HEK293 cells were plated into 96-well plate (50,000 cells/well) in the maintenance medium. The cells were incubated at 37°C, 5% CO2 for 20 hours, and treated with DMSO or increasing concentrations of IACS′8287 for 30 minutes. Cells were then stimulated with 15 ng/mL of human nerve growth factor (NGF) for 5 minutes and lysed for measurement of cellular phospho-TrkA levels using the phospho-TrkA DuoSet IC ELISA detection kit (R&D Systems, Minneapolis, MN) following manufacturer protocol.
2.3. Dual leucine zipper kinase–mediated neuroprotection in rat primary dorsal root ganglion sensory neurons
Rat embryonic dorsal root ganglia (DRG) were purchased from Lonza (Basel, Switzerland). The cells were dissociated by incubating in 0.25% Trypsin-EDTA (Gibco, Waltham, MA) for 20 minutes at 37°C, followed by gentle trituration with a fire-polished glass Pasteur pipette. Triturated cells were passed through a 70 µM nylon cell strainer and then pelleted down at 200 × g for 5 minutes at room temperature. Cells were then resuspended in NbActive1 media (BrainBits, New York, NY) supplemented with 100 ng/mL mouse NGF, 70 µM uridine, and 30 µM 5-fluoro-2-deoxyuridine (Sigma-Aldrich, St. Louis, MO). Cells were seeded in a 96-well plate precoated with 10 µg/mL laminin (Sigma-Aldrich). At 5 days in vitro, cells were treated with IACS′8287 followed by addition of 0.1 mg/mL NGF/proNGF neutralizing antibody 1 hour after compound treatment. Cells were incubated for 72 hours, and then stained with calcein AM and Hoechst 33342 (Sigma-Aldrich), and imaged on an Operetta High-Content Imaging Station (PerkinElmer, Waltham, MA). Thirteen 20X images/well were collected, the sum of the length (in microns) of all healthy neurites in each well was determined, and the data were normalized to values measured in control wells in which neurons were maintained in the presence of NGF and DMSO.
2.4. Binding affinity assay
The dissociation constant Kd was determined in the KINOMEscan KdELECT service at Eurofins DiscoveRx (Fremont, CA, https://www.discoverx.com/services/drug-discovery-development-services/kinase-profiling). The binding between IACS′8287 and a panel of kinases including DLK and 403 other kinases was determined using the KINOMEscan. In brief, binding of IACS′8287 to kinase active site will prevent kinase binding to the immobilized ligand, thereby reducing the amount of kinase captured as determined with quantitative polymerase chain reaction (qPCR). Conversely, binding to solid support of the kinases that are not bound by the IACS′8287 will not be affected. This assay does not require adenosine triphosphate (ATP) and thereby reports true thermodynamic interaction affinities.
2.5. Animals and drug administration
Male and female wild-type C57Bl/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were used at 10 to 12 weeks of age. All experimental procedures were consistent with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and the Ethical Issues of the International Association for the Study of Pain66 and were approved by the Institution for Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center. Experiments were performed and reported in compliance with the ARRIVE guidelines.28 For CIPN and CICI experiments, cisplatin was administered intraperitoneally (i.p.) at a dose of 2.3 mg/kg for a total of 10 doses as previously described.45 The DLK inhibitor IACS′8287 was formulated by dissolving the appropriate amount of compound with 0.5% methylcellulose (Sigma-Aldrich). The solution was vortexed and then sonicated at max power with no heat for 10 minutes, followed by homogenization with a Polytron homogenizer at half-maximal setting with a dispersing aggregate attachment for 5 minutes. The dosing solution was used immediately and placed on a rotating shaker between doses. The compound was administered via oral gavage at 30 mg/kg once daily starting 1 day before each cisplatin injection cycle and 1 hour before each cisplatin injections as preventive treatments.43–45 For pharmacokinetic (PK) and pharmacodynamic analysis, a single dose of IACS′8287 or vehicle was administered via oral gavage. For tumor volume studies, female BALB/c nude mice (6-8 week old, 18-22 g, Shanghai BK Laboratory Animal Co, Ltd, Shanghai, China) were used. After tumor establishment, cisplatin (4 mg/kg) or phosphate-buffered saline (PBS) was administered i.p. every 4 days for a total of 6 doses, whereas IACS′8287 (30 mg/kg) or vehicle was administered orally once daily for the entire 21 days of the study.
2.6. Tumor volume
The effect of IACS′8287 on the antitumor efficacy of cisplatin was determined in an NCI-H69 lung cancer xenograft model in female BALB/c nude mice. Tumors were established by subcutaneous injection of 10 × 106 NCI-H69 cells in 0.2 mL of PBS with matrigel (1:1) for tumor development. Tumor volume was measured every 3 to 4 days throughout the course of the 21-day experiment.
2.7. Pharmacokinetic analysis
Plasma and brainstem were collected 105 minutes after drug administration for PK analysis. Plasma samples were precipitated with acetonitrile containing an internal standard (5 ng/mL) and were diluted 1:1 with water before liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis. Brainstem samples were homogenized in 80:20 methanol:water. Supernatants from the homogenates were mixed with 1:1 acetonitrile:water solution containing the internal standard for LC-MS/MS analysis using the Acquity ultra performance liquid chromatography (UPLC) system coupled with a Xevo TQ-S triple quadrupole mass spectrometer.
2.8. Western blotting
Frozen cerebellar tissue or frozen DRG tissue was homogenized in radioimmunoprecipitation assay buffer (RIPA) buffer containing 2× Halt protease and phosphatase inhibitor cocktail (Invitrogen, Carlsbad, CA). 35 µg of total protein was then separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE) using NuPAGE 4 to 12% Bis-Tris Gels (Invitrogen) and 1× MES running buffer (Invitrogen). The proteins were transferred to nitrocellulose membranes (Invitrogen). Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE). Primary antibodies against DLK (75-355, 1:500, NeuroMab, Davis, CA), phospho-c-Jun (Ser63) (9261, 1:250, Cell Signaling Technology, Danvers, MA), c-Jun (9156, 1:250, Cell Signaling Technology, Danvers, MA), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (MAB374, 1:5000, Millipore, Burlington, MA) were used, followed by LI-COR secondary antibody (1:5000). The membranes were imaged using the LI-COR Odyssey Imager and analyzed with Image Studio 4.0 software.
2.9. KiNativ kinase profiling
16 male C57Bl/6 mice were randomly divided into 7 treatment groups: vehicle (n = 4); IACS′8287 1.88 mg/kg (n = 2); IACS′8287 3.75 mg/kg (n = 2); IACS′8287 7.5 mg/kg (n = 2); IACS′8287 15 mg/kg (n = 2); IACS′8287 30 mg/kg (n = 2); and IACS′8287 60 mg/kg (n = 2). 105 minutes after a single dose of either vehicle or IACS′8287, the animals were killed and frontal cortices were isolated and shipped frozen to ActivX Biosciences Inc. (La Jolla, CA) for KiNativ in situ kinase profiling (http://www.activxprobes.com/).
2.10. Von Frey test
Mechanical allodynia was measured with von Frey hairs and the “up and down” method (Stoelting, Wood Dale, IL) as described previously.6,30 C57Bl/6 mice were randomly divided into 4 experimental groups (PBS + vehicle, cisplatin + vehicle, PBS + IACS′8287, and cisplatin + IACS′8287) with n = 8 per group for males and n = 4 per group for females. The test was conducted on day 0 (before the start of drug treatment), day 8 (after 1 cycle of treatment), and days 17 and 18 (after 2 cycles of treatment).
2.11. Cognitive behavioral testing
Mice were randomly divided into 4 experimental groups (PBS + vehicle, cisplatin + vehicle, PBS + IACS′8287, and cisplatin + IACS′8287) with n = 12 per group. Cognitive behavioral testing was performed 1 week after completion of the treatment cycles. Y-maze,22 novel object/place recognition,2 and the puzzle box test3 were conducted sequentially as previously described.8,40
2.12. Resting-state functional magnetic resonance imaging
Four mice per group from the 4 experimental groups were used for resting-state functional magnetic resonance imaging (rsfMRI) testing 1 month after completion of the treatment cycles. In vivo rsfMRI data were obtained from mice with a 7 T Bruker BioSpec (Bruker BioSpin, Billerica, MA) scanner as previously described.26
2.13. RNA sequencing
Mice were randomly divided into 4 experimental groups (PBS + vehicle, cisplatin + vehicle, PBS + IACS′8287, and cisplatin + IACS′8287) with n = 4 per group. Mice were treated with 1 cycle of the DLK inhibitor IACS′8287 and cisplatin. L3-L6 DRG were collected 4 hours after the last injection of cisplatin. Total RNA was isolated with RNAeasy MinElute Cleanup Kit (Qiagen, Hilden, Germany). mRNA sequencing and data analysis were performed as previously described.43 Heatmap for log2 fold change of significantly altered genes was plotted using Morpheus (https://software.broadinstitute.org/morpheus/). Ingenuity Pathway Analysis (IPA; Qiagen Inc.) was used for further pathway analysis.
2.14. Immunohistochemistry
The glabrous skin of the hind paws were collected from mice 1 month after completion of the treatment cycles (n = 8), and then processed and stained for PGP-9.5 (ab108986, 1:500, Abcam) and collagen IV (1340-01, 1:100, Southern Biotech, Birmingham, AL) as described previously.45
2.15. Mitochondrial bioenergetics analysis
Mice were killed 1 month after completion of the treatment cycles. XF24 Flux Analyzer (Agilent Technologies Inc, Santa Clara, CA) was used for measurement of mitochondrial oxygen consumption rate. Dorsal root ganglia neurons from 6 mice per group39,42 or synaptosomes from 8 mice per group40 were isolated and tested as described previously.
2.16. Statistics
Data are expressed as mean ± SEM. Student t test, and one-way or two-way analysis of variance with Tukey post-hoc analysis (GraphPad, San Diego, CA) were performed as appropriate. The fMRI data were analyzed using nonparametric permutation testing. P < 0.05 was considered statistically significant for all analyses.
3. Results
3.1. Selectivity and potency of the novel compound IACS′8287 against dual leucine zipper kinase
To investigate the potential of targeting DLK for chemotherapy-induced neurotoxicities, we developed a novel DLK inhibitor IACS′8287,55 with optimized properties that allow us to investigate DLK function in mouse models. IACS′8287 showed high affinity for human recombinant DLK in an in vitro binding assay, demonstrating a Kd of 15.0 + 6.0 nM (n = 6). We then determined the binding of IACS′8287 to a panel of 403 kinases at 1 μM, a concentration approximately 100-fold higher than its Kd for DLK binding. IACS′8287 showed no significant binding against 399 kinases; modest binding activity was identified for leucine zipper kinase (LZK) (IC50 = 904 nM), TRKA (IC50 = 420 nM), and MEK5 (IC50 = 236 nM). These data indicate that IACS′8287 has good binding affinity and selectivity towards DLK.
Next, we examined the cellular activity of IACS′8287 using a DOX-inducible DLK-overexpressing HEK293 cell line. DOX induction led to activation of the JNK/c-Jun pathway and phosphorylation of c-Jun at Ser63 (p-c-Jun) (Fig. 1A). IACS′8287 dose-dependently reduced p-c-Jun (IC50 = 454.2 nM) (Fig. 1B); this potency decrease relative to the binding assay is explained by the high concentration of the competing endogenous ligand ATP in cells. To investigate neuroprotective efficacy, we used a rat embryonic DRG neuron model and found that IACS′8287 dose-dependently protected cultured DRG neurons from neurite retraction caused by NGF withdrawal (IC50 = 510 nM) (Figs. 1C and D). Because the selectivity data indicated that IACS′8287 has slight binding affinity for TRKA, which mediates NGF signaling and thus could confound data interpretation, we used a TRKA cellular kinase activity assay to assess this activity. No inhibition was found in the TRKA cellular assay for up to 10,000 nM of IACS′8287.
Figure 1.
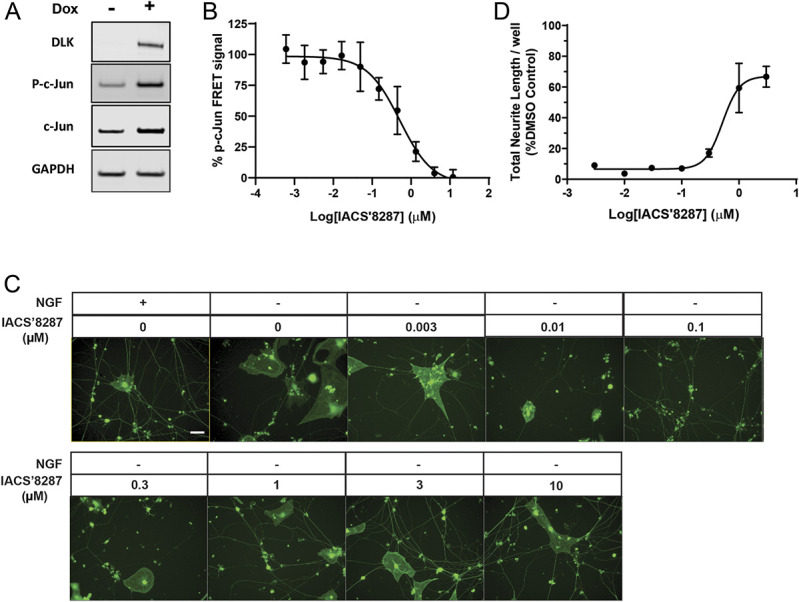
In vitro potency of the novel compound IACS′8287 against DLK. (A) Representative Western blots showing induction of DLK and p-c-Jun by doxycycline (DOX) in HEK293 cell line stably transfected with DOX-inducible DLK. GAPDH was used as loading control. (B) Dose–response curve for the DLK inhibitor IACS′8287 in attenuating p-c-Jun level. p-c-Jun signal was calculated and plotted as percent of the p-c-Jun signal measured with DMSO treatment (n = 6). (C) Representative images of DRG neurons cultured in the presence and absence of NGF, in the presence of IACS′8287 at different concentrations. Scale bar = 20 μm, magnification ×20. (D) Dose–response curve for the effect of IACS′8287 in the preservation of neurite integrity. Thirteen 20x images were collected per well, the sum of the length of all healthy neurites in each well was determined, and the data were normalized to control wells in which neurons were maintained in the presence of NGF and DMSO (n = 3). Results are expressed as mean ± SEM. DLK, dual leucine zipper kinase; DRG, dorsal root ganglion; NGF, nerve growth factor.
3.2. In vivo target engagement of IACS′8287
Next, we assessed the PK of IACS′8287 in mice. After a 10 mg/kg oral dose of IACS′8287, bioanalysis revealed that drug remained in plasma and brain in micromolar concentrations for up to 24 hours, with a brain/plasma ratio of 0.7. The plasma t1/2 of IACS′8287 was ∼3.5 hours, and the time to reach maximal plasma and brain concentration (tmax) was approximately 105 minutes (Fig. 2A; and Supplementary Fig. 1A, available at http://links.lww.com/PAIN/B326). With nonspecific protein binding for IACS′8287 in plasma and brain being measured in vitro as 83% and 87.9%, respectively, we estimated that sufficient unbound drug would be present to inhibit DLK in vivo. We have shown that constitutive expression of p-c-Jun is DLK-dependent in adult mouse cerebellum and is an accurate measure of CNS target engagement for DLK inhibitors.19 We thus measured p-c-Jun inhibition in cerebellum of mice that received a single administration of increasing doses of IACS′8287. IACS′8287 dose-dependently decreased p-c-Jun/c-Jun beginning at 3 mg/kg, whereas similar extent of p-c-Jun reduction (∼70%) was observed at 10, 30, and 100 mg/kg doses (Fig. 2B). We plotted plasma and brain drug concentrations against percent p-c-Jun/c-Jun reduction to determine the in vivo IC50 of IACS′8287, which was 1.29 µM and 1.43 µM for plasma (Supplementary Fig. 1B, available at http://links.lww.com/PAIN/B326) and brain (Fig. 2C), respectively. After accounting for protein binding, the IC50 for free plasma and brain concentrations were determined to be 0.218 µM and 0.172 µM, respectively (Supplementary Fig. 1C, D, available at http://links.lww.com/PAIN/B326). These potency values are within 3-fold of the potency in the HEK293 cellular assay with human DLK.
Figure 2.

In vivo potency and selectivity of the novel compound IACS′8287 against DLK. (A) Plasma and brain concentration of IACS′8287 at different time points after single oral administration of the compound at 10 mg/kg (n = 3). (B) Representative Western blot and quantification for p-c-Jun and c-Jun in cerebellar lysates from mice dosed with vehicle (n = 8) or IACS′8287 at 0.1 (n = 4), 0.3 (n = 8), 1 (n = 8), 3 (n = 4), 10 (n = 8), 30 (n = 4), or 100 (n = 4) mg/kg (one-way ANOVA, F (7, 40) = 37.4, P < 0.0001; ***P < 0.001). (C) Dose–response curve for IACS′8287 in reduction of p-c-Jun level in mouse cerebellum. Percent p-c-Jun/c-Jun for each mouse was plotted against the corresponding brain concentrations of IACS′8287. (D) Binding of IACS′8287 to DLK in mouse cerebellum in response to treatment of vehicle (n = 2) and different doses of the compound (n = 2). (E) Dose–response curve for IACS′8287 binding to DLK in mouse cerebellum. Percent DLK binding was plotted against the corresponding brain concentrations of IACS′8287. (F) Graphical representation of the kinases examined for kinome selectivity in mice dosed with IACS′8287 by ActivX KiNativ analysis at 30 mg/kg. Results are expressed as mean ± SEM. ANOVA, analysis of variance; DLK, dual leucine zipper kinase.
To assess selectivity in vivo, we used KiNativ in situ kinase profiling, a mass spectrometry-based technique that measures direct binding of compound to the kinome. Using this method, all kinases that are sufficiently expressed in a tissue and bind an ATP analog probe are captured and detected; there will be reduced capture and detection of a kinase if it is occupied by an ATP site-directed kinase inhibitor. Mice were given a single dose of IACS′8287 at 1.88, 3.75, 7.5, 15, 30, or 60 mg/kg, and brains were isolated at tmax for KiNativ in situ kinase profiling. IACS′8287 at doses of 15 mg/kg and higher abolished detection of DLK (>95% loss of signal), indicating that IACS′8287 fully occupied the ATP binding sites of DLK in vivo (Figs. 2D and E). Pharmacokinetic modeling indicated that a single oral dose of 30 mg/kg would cover the IC50 for 24 hours; thus, we examined the selectivity of IACS′8287 at this dose against 246 kinases detectable in brain. At tmax, IACS′8287 fully blocked detection of DLK (97%) and, to a lesser extent, LZK (40%). No significant competition (>35%) to the other 244 kinases was detected (Fig. 2F). Thus, IACS′8287 dosed orally at 30 mg/kg exhibited full target occupancy along with maximal p-c-Jun inhibition while maintaining selectivity against other brain kinases except for a less potent interaction with DLK's close homolog LZK.
3.3. Dual leucine zipper kinase inhibition protects against cisplatin-induced peripheral neuropathy in mice
To determine the effects of DLK inhibition on CIPN, we used a well-established mouse model, in which 2 cycles of cisplatin cause mechanical allodynia and loss of intraepidermal nerve fibers.45 Male C57Bl/6 mice were injected with 2.3 mg/kg of cisplatin for 5 days, followed by 5 days of rest, and then another cycle of cisplatin treatment for 5 days. IACS′8287 (30 mg/kg) was administered orally 1 day before the start of each cisplatin cycle and 1 hour before each cisplatin injection (Fig. 3A). This dose of IACS′8287 selectively inhibited DLK and exceeded the plasma IC50 for 24 hours (Fig. 2). To confirm that IACS′8287 blocks DLK signaling in the DRG, we collected whole DRG 4 hours after completion of the first treatment cycle (on day 5) for analysis of p-c-Jun in the DRG. One cycle of cisplatin treatment significantly, but modestly, induced p-c-Jun, whereas IACS′8287 treatment reduced p-c-Jun in PBS-treated animals and blocked cisplatin-mediated p-c-Jun induction (Fig. 3B), indicating target engagement in the DRG.
Figure 3.
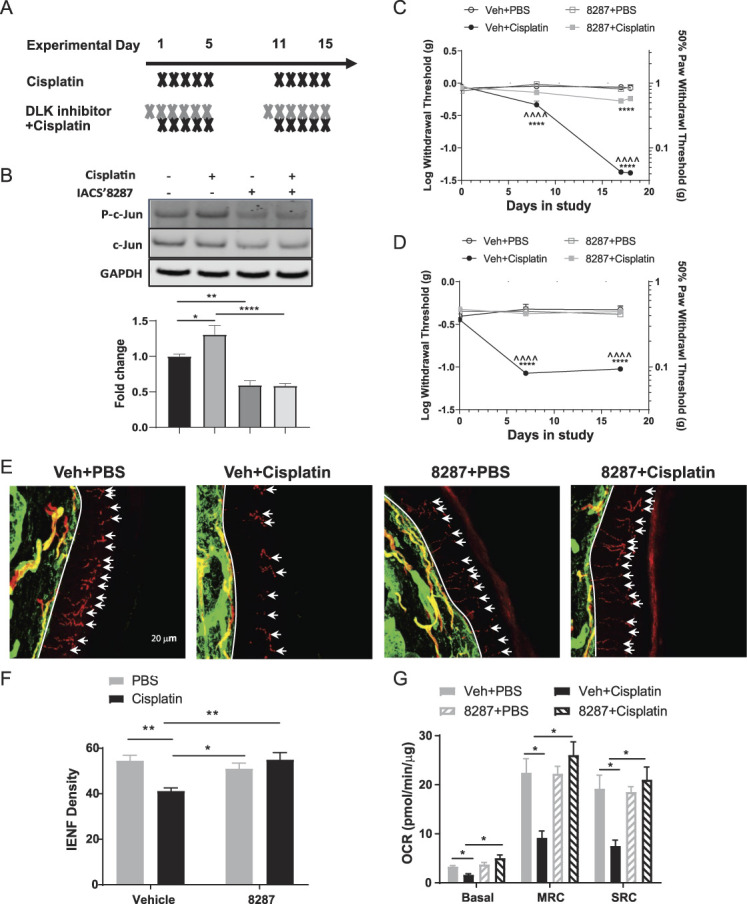
The DLK inhibitor IACS′8287 prevents cisplatin induced peripheral neuropathy. (A) Treatment schedule of IACS′8287 and cisplatin. IACS′8287 (30 mg/kg) or vehicle via oral gavage was given 1 day before start of each cisplatin cycle and 1 hour before each cisplatin or PBS i.p. injection for 2 treatment cycles. (B) Representative Western blot and quantification for p-c-Jun, c-Jun, and GAPDH in DRG from mice received one cycles of IACS′8287 and cisplatin treatment (n = 8, two-way ANOVA reveal significant main effect on interaction: F (1, 28) = 4.601, P = 0.0408, and significant IACS′8287 treatment effect: F (1, 28) = 58.55, P < 0.0001; *P < 0.05, **P < 0.01, ****P < 0.0001). Sensitivity to mechanical stimulation of the hind paws was monitored over time using the von Frey test. Coadministration of IACS′8287 prevented cisplatin-induced mechanical allodynia in (C) male mice (n = 8, statistical analysis was conducted using Log[withdrawal threshold (g)], two-way repeated-measures ANOVA reveal significant main effect on interaction: F (9, 84) = 105, P < 0.0001, significant treatment effect: F (3, 28) = 98.73, P < 0.0001, and significant time effect: F (3, 84) = 438.8, P < 0.0001; Tukey post hoc analysis: ***P < 0.001, ****P < 0.0001 as compared to Veh+PBS; ^^^^P < 0.0001 as compared to IACS′8287+Cisplatin) and (D) female mice (statistical analysis was conducted using Log[withdrawal threshold (g)], two-way repeated-measures ANOVA reveal significant main effect on interaction: F (6, 27) = 15.78, P < 0.0001, significant treatment effect: F (2, 9) = 13.72, P = 0.0018, and significant time effect: F (3, 27) = 89.31, P < 0.0001; Tukey post hoc analysis: ****P < 0.0001 as compared to Veh+PBS; ^^^^P < 0.0001 as compared to IACS′8287+Cisplatin). Paw biopsies were obtained from the hind paws of mice that received 2 cycles of cisplatin with or without the DLK inhibitor IACS′8287. Tissues were stained with antibodies against PGP9.5 (red) and collagen (green). (E) Representative images from each treatment group, scale bar = 20 μm, magnification ×40. (F) Quantification of IENF density expressed as the number of nerve fibers crossing the basement membrane/length of the basement membrane (mm) (n = 8, two-way ANOVA reveal significant main effect on interaction: F (1, 28) = 12.68, P = 0.0013; Tukey post hoc analysis: *P < 0.05, **P < 0.01). (E) Lumbar DRG neurons were isolated after completion of behavioral testing for mitochondrial bioenergetic analysis using the Seahorse XFe24 Flux Analyzer. Changes induced by cisplatin and IACS′8287 in (G) basal OCR, MRC, and SRC (n = 6, two-way ANOVA reveal significant main effect on interaction: F (1, 20) = 18.88, P = 0.0003 for basal OCR; F (1, 20) = 22.57, P = 0.0001 for MRC; F (1, 20) = 17.70, P = 0.0004 for SRC; Tukey post hoc analysis: * P < 0.05). Results are expressed as mean ± SEM. ANOVA, analysis of variance; DLK, dual leucine zipper kinase; DRG, dorsal root ganglion; IENF, intraepidermal nerve fibers; MRC, maximal respiratory capacity.
Von Frey measurements taken during and after cisplatin treatment demonstrated that coadministration of IACS′8287 prevented cisplatin-induced mechanical allodynia in both male and female mice (Figs. 3C and D). To determine whether the protective effects of IACS′8287 extend to prevention of axonal damage, we measured intraepidermal nerve fibers in the glabrous skin from distal extremities, which are commonly affected in CIPN.29 As expected, cisplatin reduced IENF density in the hind paws; coadministration of IACS′8287 prevented loss of these terminal nerve fibers (Figs. 3E and F). IACS′8287 seemed to specifically block neurotoxicity as it did not prevent cisplatin-induced body weight loss (data not shown).
3.4. Dual leucine zipper kinase inhibition prevents cisplatin-induced bioenergetic deficits in the dorsal root ganglion sensory neurons
Mitochondrial dysfunction has been suggested to underlie chemotherapy-induced mechanical allodynia.4,41 To determine if IACS′8287 prevented cisplatin-induced mitochondrial dysfunction, we measured mitochondrial bioenergetics in isolated DRG neurons. Two cycles of cisplatin treatment decreased basal respiration, maximal respiratory capacity, and spare respiratory capacity (SRC), indicating impaired mitochondrial oxidative phosphorylation. Importantly, these deficits were prevented by IACS′8287 coadministration (Fig. 3G). Thus, IACS′8287 preserves mitochondrial function in DRG neurons during cisplatin treatment.
3.5. RNA sequencing analysis reveals that dual leucine zipper kinase inhibition regulates neurological function and immune responses in dorsal root ganglia
To better understand the mechanism by which DLK inhibition prevents CIPN, we collected whole DRG 4 hours after completion of the first treatment cycle (Fig. 3A, day 0-5) for RNAseq. Consistent with DLK activity being low in uninjured DRG, administration of IACS′8287 alone changed the expression of only 9 transcripts (padj < 0.05), including Sh2d3c, Dusp1, and Dusp8, which encode JNK signaling proteins that are consistently downregulated by genetic deletion or pharmacological inhibition of DLK.19,62 By contrast, administration of IACS′8287 to cisplatin-treated mice changed the expression of 1689 genes, suggesting a robust induction of DLK signaling activity in DRG during cisplatin treatment. Consistent with its profound effect on allodynia and axonal integrity, cisplatin alone changed the expression of 1337 genes compared with controls (Fig. 4A). Among these differentially expressed genes, 582 were significantly altered both by cisplatin alone and by IACS′8287 plus cisplatin (Fig. 4A). The heatmap of log2 fold changes of the genes significantly altered by cisplatin alone and/or IACS′8287 plus cisplatin indicates that most genes show opposite direction of change between these conditions (Fig. 4B), as confirmed by correlation analysis (R = −0.65, P < 0.0001) (Fig. 4C), suggesting DLK inhibition counteracts many of the transcriptional effects of cisplatin.
Figure 4.
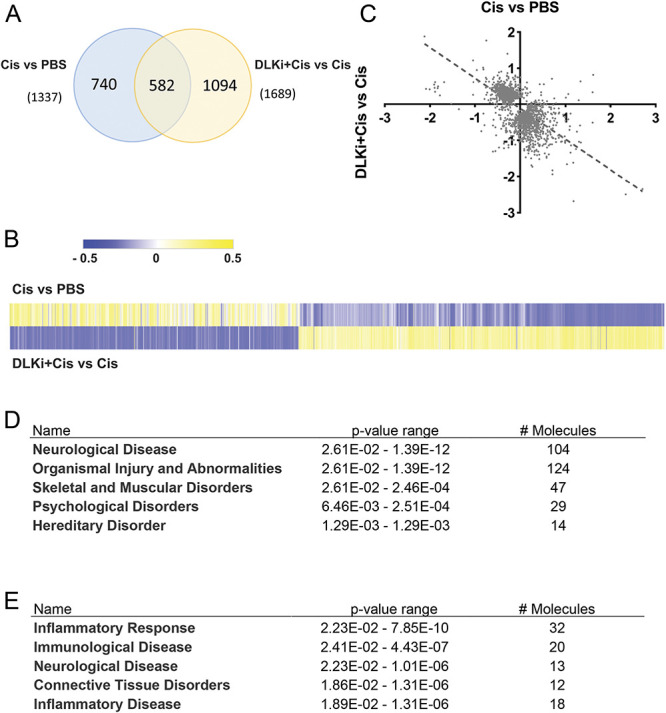
The DLK inhibitor IACS′8287 prevents transcriptome changes induced by cisplatin in the DRG. (A) Differentially expressed genes (DEGs) (padj<0.05) induced by cisplatin in comparison to PBS treatment and by IACS′8287 in cisplatin-treated mice. Venn diagram analysis shows that 582 genes were significantly altered in both comparisons. 740 genes were significantly altered only by cisplatin alone, whereas 1094 genes were significantly altered only by IACS′8287 in cisplatin-treated mice as compared to mice treated with cisplatin alone. (B) Heat map for the 2416 genes in (A) plotted using log2 fold changes induced by cisplatin and by IACS′8287 in cisplatin-treated mice. (C) Correlation plots for the log2 fold change of the 2416 genes induced by cisplatin and by IACS′8287 in cisplatin-treated mice. (D) Ingenuity pathway analysis of the 504 genes downregulated by cisplatin and upregulated by coadministration of IACS′8287. (E) Ingenuity pathway analysis of the 72 genes upregulated by cisplatin and downregulated by coadministration of IACS′8287. Results are expressed as mean ± SEM. DLK, dual leucine zipper kinase; DRG, dorsal root ganglion.
Of the 582 genes that were significantly altered by both cisplatin alone and by IACS′8287 in cisplatin-treated mice, 504 genes were downregulated by cisplatin and upregulated by IACS′8287 coadministration, whereas 72 genes were upregulated by cisplatin and downregulated when IACS′8287 was coadministered. Ingenuity pathway analysis indicates that genes that were downregulated by cisplatin and upregulated by IACS′8287 largely mapped to neurological disease and organismal injury and abnormalities (Fig. 4D). Conversely, genes that were upregulated by cisplatin and downregulated by IACS′8287 primarily mapped to inflammatory response pathways (Fig. 4E). Manual interrogation of the data revealed that expression of the axonal maintenance factor Nmnat2 was downregulated by cisplatin, whereas IACS′8287 normalized this downregulation. Furthermore, many genes implicated in pain response were altered by cisplatin, and these changes were prevented by IACS′8287 (Table 1). For example, although calpain 1127 was the top upregulated gene by cisplatin, it was significantly downregulated in cisplatin mice that received IACS′8287. Many transcripts encoding voltage-gated potassium channels (Kcnb1, Kcnc2, Kcnc3, Kcnc4, Kcnh2, Kcnk3, Kcnk12, Kcnn2, Kcnq2, and Kcns1) were decreased by cisplatin, whereas the alterations were mitigated in the presence of IACS′8287. The Nogo protein (Rtn4) and Nogo receptor (Rtn4r), the lack of which is implicated in neuropathic and inflammatory pain conditions,23,34 were also downregulated by cisplatin and upregulated by IACS′8287. Furthermore, in line with a role for immune cells in the development of neuropathic pain in CIPN,32 many transcripts encoding leukocyte markers such as Itgam, Treml2, Il18rap, Clec5a, Cd33, Sell (selectin L), Il17rb, and the cytokine Il34 were induced by cisplatin. Increased expression of these genes was also prevented by IACS′8287. Taken together, our data suggest that inhibition of DLK has global impact on cisplatin-induced transcriptomic changes in DRG, implicating effects on neurons and infiltrating immune cells.
Table 1.
Selection of genes through manual interrogation of the RNAseq data listed in alphabetical order.
| Symbol | Cis vs PBS | DLKi+Cis vs Cis | ||||
|---|---|---|---|---|---|---|
| log2 fold change | P | Padj | log2 fold change | P | Padj | |
| Capn11 | 2.71 | 0.000686 | 0.0123 | −2.349 | 0.00258 | 0.0307 |
| Cd33 | 0.484 | 0.000607 | 0.0111 | −0.744 | 0.0000699 | 0.00235 |
| Clec5a | 0.898 | 0.00112 | 0.0175 | −1.045 | 0.000135 | 0.00379 |
| Il17rb | 0.522 | 0.00147 | 0.0214 | −0.484 | 0.00325 | 0.0362 |
| Il18rap | 0.921 | 0.000015 | 0.000565 | −1.035 | 0.000336 | 0.00738 |
| Il34 | 0.274 | 0.000306 | 0.0065 | −0.34 | 0.000306 | 0.0069 |
| Itgam | 0.364 | 0.00378 | 0.0433 | −0.64 | 0.000407 | 0.00849 |
| Kcnb1 | −0.245 | 0.00389 | 0.0443 | 0.333 | 0.00176 | 0.0236 |
| Kcnc2 | −0.27 | 0.000212 | 0.00481 | 0.307 | 0.00000707 | 0.000438 |
| Kcnc3 | −0.491 | 1.9E-09 | 0.000000605 | 0.577 | 1.24E-09 | 0.000000613 |
| Kcnc4 | −0.314 | 0.00439 | 0.0483 | 0.344 | 0.00474 | 0.0471 |
| Kcnh2 | −0.377 | 0.000000299 | 0.0000283 | 0.29 | 0.00452 | 0.0457 |
| Kcnk12 | −0.386 | 0.00413 | 0.0462 | 0.398 | 0.00154 | 0.0217 |
| Kcnk3 | −0.723 | 0.00000555 | 0.000265 | 0.492 | 0.000563 | 0.0106 |
| Kcnn2 | −0.397 | 0.000069 | 0.00193 | 0.425 | 0.0000441 | 0.00167 |
| Kcnq2 | −0.231 | 0.000161 | 0.00381 | 0.392 | 0.0000252 | 0.00111 |
| Kcns1 | −0.31 | 0.000607 | 0.0111 | 0.361 | 0.00205 | 0.0264 |
| Kctd17 | −0.205 | 0.00091 | 0.015 | 0.174 | 0.00432 | 0.0442 |
| Nmnat2 | −0.177 | 0.0065 | 0.0495 | 0.256 | 0.00473 | 0.0471 |
| Rtn4 | −0.219 | 0.00073 | 0.0129 | 0.306 | 0.000971 | 0.0156 |
| Rtn4r | −0.353 | 0.000376 | 0.00764 | 0.47 | 0.00021 | 0.00522 |
| Sell | 0.572 | 0.00255 | 0.0322 | −0.824 | 0.000174 | 0.00457 |
| Treml2 | 0.991 | 0.0000884 | 0.00233 | −1.074 | 0.0000697 | 0.00235 |
3.6. Dual leucine zipper kinase inhibition protects against cisplatin-induced cognitive impairments in mice
We next used a battery of functional assays to assess whether DLK inhibition also protects against CICI. Mice underwent the treatment cycles for cisplatin and IACS′8287 (Fig. 3A); cognitive testing started a week after treatment completion. In the Y-maze spontaneous alternation test for spatial working memory, cisplatin induced a significant decrease in the alternation rate, and this deficit was prevented by IACS′8287 (Fig. 5A). Coadministration of IACS′8287 also prevented impaired performance in the novel object and place recognition test (NOPRT) for spatial memory (Fig. 5B). In the puzzle box test, which is a problem-solving task to study executive functioning, the time for mice to get through different obstacles and reach the goal box is evaluated. All mice entered the goal box at the same rate without obstacles, indicating that cisplatin did not affect motivation or the ability to execute the task. However, when a difficult obstacle was present (cardboard plug, trials 8-11), cisplatin-treated mice required significantly more time to enter the goal box. The increase in time was not due to lack of motivation, as cisplatin-treated mice spent significant amount of time exploring the tunnel.8 Coadministration of IACS′8287 normalized the time taken to enter the goal box in the difficult trials, indicating preservation of executive functioning (Fig. 5C).
Figure 5.
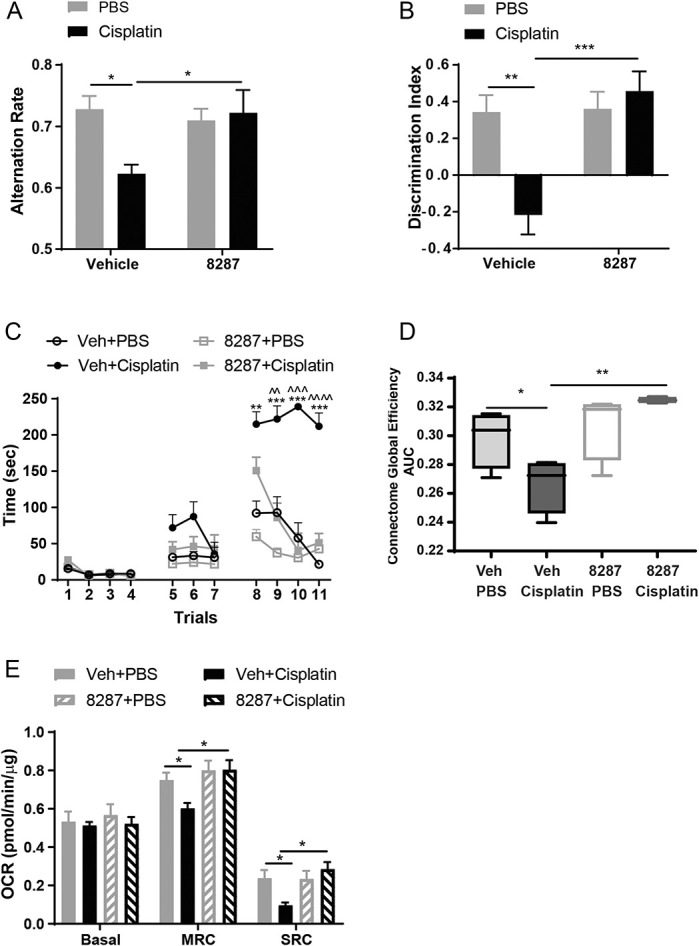
The DLK inhibitor IACS′8287 prevents cisplatin-induced cognitive impairments. Behavioral tests including Y-maze, NOPRT, and the puzzle box tests were started 1 week after the last administration of IACS′8287 and cisplatin. (A) IACS′8287 prevented cisplatin-induced spatial working memory deficits in the Y-maze test. The percentage of perfect alternations (alternation rate) was calculated (n = 12, two-way ANOVA reveals significant main effect on interaction: F (1, 44) = 5.398, P = 0.0251; Tukey post hoc analysis: *P < 0.05). (B) IACS′8287 prevented cisplatin-induced spatial memory deficits in NOPRT. The discrimination index was used as a measure for the preference for the novel object (n = 12, two-way ANOVA reveals significant main effect on interaction: F (1, 44) = 9.598, P = 0.034; Tukey post hoc analysis: **P < 0.01, ***P < 0.0001). (C) IACS′8287 prevented cisplatin-induced impairments in executive functioning tested in the puzzle box test. Trials 1 to 4, the underpass was unobstructed; trials 5 to 7, the underpass was filled with corncob bedding; trials 8 to 11, the underpass was covered by the cardboard plug. The time it took for the mice to enter the goal box was used as an index for executive functioning. (n = 12, two-way ANOVA reveals significant main effect on interaction: F (30, 402) = 12.08, P < 0.0001; Tukey post hoc analysis: **P < 0.01, ***P < 0.001 vs Veh+PBS, ^^P < 0.01, ^^^P < 0.001, ^^^^P < 0.0001 vs IACS′8287+cisplatin). (D) fMRI analysis of connectome global efficiency. IACS′8287 prevented cisplatin-induced disruption of functional connectome global efficiency (n = 4, nonparametric permutation testing, *P < 0.05, **P < 0.01). (E) Oxygen consumption rates were analyzed in isolated synaptosomes using the Seahorse XFe24 Flux Analyzer. Changes induced by cisplatin and IACS′8287 in basal OCR, MRC, and SRC (n = 8, two-way ANOVA reveals significant main effect on interaction: F (1, 28) = 11, P = 0.0025 for MRC; F (1, 28) = 8.933, P = 0.0058 for SRC; Tukey post hoc analysis: *P < 0.05). Results are expressed as mean ± SEM. ANOVA, analysis of variance; DLK, dual leucine zipper kinase; fMRI, resting state functional magnetic resonance imaging; MRC, maximal respiratory capacity.
3.7. Dual leucine zipper kinase inhibition prevents cisplatin-induced deficits in resting-state functional magnetic resonance imaging functional connectivity
Functional magnetic resonance imaging has been used to uncover the neural basis for the cognitive deficits reported by patients with CICI52 and thus provides an independent means of measuring brain function that's translatable to humans. We have shown that cisplatin impairs brain functional connectivity in mice.8 To determine if DLK inhibition also impacts brain functional connectivity, we measured rsfMRI in anesthetized mice after completion of behavioral testing. As previously observed, cisplatin reduced global coupling efficiency between functionally related brain regions. Mice coadministered IACS′8287 with cisplatin did not show this deficit, which is in line with the protective effects of IACS′8287 against cognitive impairments (Fig. 5D).
3.8. Dual leucine zipper kinase inhibition prevents cisplatin-induced synaptosomal bioenergetic deficits
Synaptic mitochondrial dysfunction is a key player in cisplatin-induced cognitive impairments.8 Thus, we tested if DLK inhibition impacted cisplatin-induced deficits in synaptosomal mitochondrial bioenergetics. Analysis of mitochondrial function in synaptosomes demonstrated that cisplatin-treated mice showed reduced maximal respiratory capacity and spare respiratory capacity. Coadministration of IACS′8287 preserved synaptosomal mitochondrial bioenergetics (Fig. 5E).
3.9. Inhibition of dual leucine zipper kinase does not interfere with the antitumor activity of chemotherapeutics
To be safely used in patients undergoing chemotherapy, IACS′8287 cannot promote tumor growth, interfere with the antitumor efficacy, or increase the overall toxicity of chemotherapeutics. To the best of our knowledge, DLK has not been reported as a tumor suppressor, and examination of public databases indicated that DLK is neither highly induced nor commonly deleted or mutated in a wide range of tumor samples, indicating low risk of DLK inhibition on tumor progression.48 Nevertheless, we examined the effect of IACS′8287 in the NCI-H69 human lung cancer xenograft model, a model featuring aggressive tumor growth that responds well to cisplatin. Mice were inoculated subcutaneously with tumor cells and treated with cisplatin in the presence or absence of IACS′8287 after tumor establishment. NCI-H69 tumor xenografts grew substantially over the study course in control mice. Tumor growth was not affected by IACS′8287 treatment alone. Cisplatin treatment significantly reduced tumor growth, and the efficacy of cisplatin was not affected by IACS′8287 coadministration (Fig. 6A). IACS′8287 was well tolerated and did not affect overall health or body weight (Fig. 6B).
Figure 6.
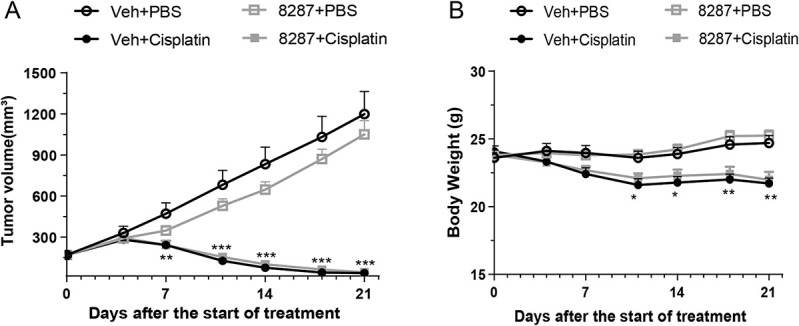
The DLK inhibitor IACS′8287 does not interfere with the antitumor activity of cisplatin. Tumor volume (A) and body weight (B) were measured every 3 to 4 days across the 21-day study period. In PBS-treated mice, NCI-H69 tumor xenografts grew substantially over the course of the study. Cisplatin treatment substantially reduced tumor volume and body weight. IACS′8287 treatment did not have any impact on tumor volume or body weight change in either PBS or cisplatin treated mice (n = 12, two-way ANOVA reveals significant main effect on interaction: F (18, 264) = 44.08, P < 0.0001 for tumor volume; F (18, 264) = 14.01, P < 0.0001 for body weight; Tukey post hoc analysis: *P < 0.05, **P < 0.01, ***P < 0.001 vs Veh+PBS group). Results are expressed as mean ± SEM. ANOVA, analysis of variance; DLK, dual leucine zipper kinase.
4. Discussion
Activation of the MAPK-JNK pathway has long been implicated in neuronal injury signaling and the development and maintenance of neuropathic pain.14 JNK-targeting strategies, including small molecule ATP-competitive inhibitors and peptide inhibitors, have been explored for treating neurodegenerative diseases and chronic pain.14,64 However, despite the appeal of targeting the JNK pathway, JNK inhibitors are limited by the lack of selectivity against the many JNK substrates, leading to severe mechanism-based toxicity.46 Dual leucine zipper kinase, as a neuronally enriched kinase upstream in the MAPK-JNK pathway, mediates a core mechanism for neuronal injury responses, and therefore is an attractive drug target for neuronal injury and neurodegenerative conditions.54 Dual leucine zipper kinase activates JNK through activating its upstream kinases MKK4/7, and it has been shown to selectively regulate JNK-based stress response pathway to mediate axon degeneration and neuronal apoptosis without interfering with other aspects of JNK signaling.18 Moreover, given that mice with somatic DLK knockout seem healthy,50 pharmacological inhibition of DLK is expected to be well tolerated. We developed a novel DLK inhibitor with optimized drug-like properties and investigated the potential of this compound for prevention of CIPN and CICI in mice. Our results demonstrate that DLK is an essential component in the molecular pathway leading to neurotoxic effects of chemotherapy, as DLK inhibition prevented signs of cisplatin-induced abnormalities in the nervous system, including mechanical allodynia, IENF loss, cognitive impairments, and disruption of brain functional connectivity. The protective effects are not explained by drug-drug interaction that blocks the bioactivity of cisplatin, as coadministration of our DLK inhibitor did not prevent cisplatin-induced weight loss or interfere with its antitumor efficacy. Our conclusion regarding a key role for DLK in the development of CIPN and CICI is based on studies with IACS′8287, which is highly selective with no measurable binding to over 200 other kinases expressed in the brain other than LZK (Fig. 2D). However, it remains possible that partial inhibition of LZK or another non-DLK protein contributes to the efficacy of IACS′8287. Future studies with targeted deletion of DLK will further delineate the role of DLK in CIPN and CICI.
IACS′8287 is readily brain-penetrant and prevents deficits associated with cisplatin treatment in the CNS. Chemotherapy-induced cognitive impairments shares pathological features with neurodegenerative diseases such as AD, including memory deficits, loss of synapses, white matter damage, changes in brain functional connectivity, and mitochondrial dysfunction.7–9,21,40,59 Dual leucine zipper kinase inhibition and genetic ablation are effective in mouse models of AD and ALS,31,49 and our results extend those findings to CICI. In model systems, cisplatin induces synaptic loss,1,7,40 demyelination,8 loss of neurogenesis,9,36 and tau pathology.7,40 It will be interesting to determine whether DLK contributes to these aspects of CICI in future studies.
Consistent with previous reports,18 we confirmed that DLK is required for NGF depletion-induced neurite retraction in vitro. In addition, we showed that DLK is involved in IENF loss in vivo. These findings are consistent with the role of DLK in the axonal response to various stress conditions including axotomy and vincristine treatment in vitro.47,60 Target engagement of IACS′8287 in the peripheral nervous system was demonstrated by the reduction of p-c-Jun levels in the DRG. We only observed a small induction of p-c-Jun by cisplatin in the DRG, suggesting that cisplatin induces p-c-Jun in a subpopulation of cells in the DRG. This observation is consistent with our RNAseq data revealing no apparent regulation of c-Jun target genes by cisplatin and raises the intriguing possibility that c-Jun-independent but DLK-dependent mechanisms also mediate cisplatin-induced neurotoxicity. It has been shown previously that pharmacological inhibition of DLK elevates NMNAT2 abundance and protects axons from injury-induced degeneration.57 Furthermore, a gain-of-function mutant form of Nmnat2 was protective against CIPN in mice.61 Our RNAseq analysis revealed that Nmnat2 transcript was reduced by cisplatin, and this reduction was prevented by IACS′8287. Therefore, modulation of NMNAT2 likely contributes to the protective effects of DLK inhibition.
IACS′8287 prevented cisplatin-induced neuronal mitochondrial dysfunction, which suggests that DLK triggers mitochondrial dysfunction after cisplatin treatment. However, the mechanisms linking DLK to mitochondrial dysfunction are unclear. Previous RNAseq analysis of DRG from mice treated with 2 cycles of cisplatin showed that mitochondrial dysfunction and oxidative phosphorylation were the top canonical pathways regulated by cisplatin.43 However, the RNAseq data here, from mice treated with one cycle of cisplatin, showed no such changes in response to cisplatin. Therefore, mitochondrial dysfunction may not be detectable at transcriptional level in DRG until the second cycle of cisplatin treatment. Given the critical role of NAD+ in mitochondrial energy metabolism,10 DLK-mediated regulation of the NAD+ synthesizing enzyme NMNAT2 could play a role here. Interestingly, 2 recent studies showed that mitochondrial dysfunction led to decreased synthesis of NMNAT2 in DRG neurons37 and acceleration of DLK-mediated NMNAT2 degradation.56 Therefore, DLK might be involved in a feed-forward cycle that drives cisplatin-induced neuronal pathology through modulating mitochondrial function and regulating NMNAT2 level.
Previous studies described a role for DLK in vincristine neurotoxicity in vitro15,47; we now show that DLK contributes to neuropathic pain induced by chemotherapy in a sex-independent manner. RNAseq analysis of DRG revealed that many genes implicated in pain response were altered by cisplatin and these changes were dampened or prevented by IACS′8287. Among them were genes encoding voltage-gated potassium channels, which impact pain signaling by regulating electrophysiological activity of sensory neurons.12 Of note, one of the potassium channel genes reported in the current study, Kcnq2, is downregulated in sensory neurons in a model of oxaliplatin-induced neuropathic pain as well.35 Thus, normalization of the expression of these potassium channels could contribute to the effects of DLK inhibition on cisplatin-induced mechanical allodynia. Calpains, the calcium-dependent cysteine proteases, have been implicated in allodynia.27 We found here that calpain 11 was increased by cisplatin treatment and this increase was prevented by IACS′8287. The genes encoding Nogo protein and Nogo receptor were downregulated by cisplatin and upregulated by IACS′8287. As local administration of Nogo proteins rescues neuropathic pain and inflammatory pain,23,34 regulation of this pathway could also contribute to the protective effect of DLK inhibition.
Interestingly, genes involved in the immune response account for a large number of transcripts coregulated by cisplatin and IACS′8287. Infiltration of leukocytes into the peripheral nervous system is associated with development of neuropathic pain in CIPN.32 For example, increased macrophage infiltration into DRG and sciatic nerve was observed in preclinical models of paclitaxel- or oxaliplatin-induced neuropathy.5,38,65 Consistently, we observed upregulation of monocyte-/macrophage-specific genes Itgam and Treml2 in DRG of cisplatin-treated mice. In addition, cell surface markers of leukocytes such as Il18rap, Clec5a, Cd33, selectin L, Il17rb, Il34, and many others were upregulated. Interestingly, DLK inhibition prevented these inductions. These data are consistent with recent studies in nerve injury models where DLK was implicated in regulation of immune cell infiltration by promoting neuronal secretion of CCL2 and CSF1 in response to injury.25 Although we did not capture these specific changes in the current RNAseq analysis, the DLK-dependent increase in immune cell markers in our study are consistent with a role for DLK in the communication between neurons and the immune system. Future studies that determine whether DLK mediates a specific communication between injured neurons with immune cells will be of interest.
In summary, our findings provide evidence that DLK inhibitors are promising therapeutics for the prevention of CIPN and CICI, 2 frequent and debilitating side effects of cancer treatment.
Conflict of interest statement
The effect of IACS-8287 was discovered by MD Anderson and licensed to Magnolia Neurosciences Corporation. MD Anderson has equity in Magnolia Tejas Corporation.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B326.
Acknowledgements
The work was supported by generous philanthropic donations to the Neurodegeneration Consortium, grant 20160903 from the Alzheimer Drug Discovery Foundation (Ray), and grants CA208371 (C.J.H. and A.K.), NS073939 (A.K. and C.J.H.), and CA227064 (A.K. and C.J.H.) from the National Institutes of Health. The RNA sequencing work was done with technical support from The University of Texas MD Anderson Cancer Center's Sequencing and Microarray Facility, supported by The University of Texas MD Anderson Cancer Center Support Grant NIH CA016672.
Author contributions: Participated in research design: J. Ma, S. Goodwani, P.J. Acton, V. Buggia-Prevot, M.J. Soth, P. Jones, W.J. Ray, A. Kavelaars, and C.J. Heijnen. Conducted experiments: J. Ma, S. Goodwani, P.J. Acton, S.R. Kesler, I. Jamal, I.D. Mahant, Z. Liu, F. Mseeh, B.L. Roth, C. Chakroborty, Q. Wu, Y. Jiang, and K. Le. Performed data analysis: J. Ma, S. Goodwani, I. Jamal, S.R. Kesler, Z. Liu, F. Mseeh, B.L. Roth, C. Chakraborty, B. Peng, Q. Wu, and Y. Jiang. Wrote or contributed to the writing of the manuscript: J. Ma, S. Goodwani, P.J. Acton, V. Buggia-Prevot, Ray, A. Kavelaars, and C.J. Heijnen.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
Contributor Information
Jiacheng Ma, Email: jma5@mdanderson.org.
Sunil Goodwani, Email: sggoodwani@mdanderson.org.
Paul J. Acton, Email: pjacton@mdanderson.org.
Virginie Buggia-Prevot, Email: virginie.buggiaprevot@gmail.com.
Shelli R. Kesler, Email: srkesler@austin.utexas.edu.
Imran Jamal, Email: IJamal@mdanderson.org.
Iteeben D. Mahant, Email: idmahant@mdanderson.org.
Zhen Liu, Email: zliu8@mdanderson.org.
Faika Mseeh, Email: Fmseeh@mdanderson.org.
Bruce L. Roth, Email: rothbr@msn.com.
Chaitali Chakraborty, Email: chaitali19@gmail.com.
Bo Peng, Email: bo.peng@bcm.edu.
Qi Wu, Email: QWu6@mdanderson.org.
Yongying Jiang, Email: yjiang4@mdanderson.org.
Kang Le, Email: kle3@mdanderson.org.
Michael J. Soth, Email: mjsoth@mdanderson.org.
Philip Jones, Email: pjones3@mdanderson.org.
Annemieke Kavelaars, Email: akavelaars@mdanderson.org.
William J. Ray, Email: wjray@mdanderson.org.
References
- [1].Andres AL, Gong X, Di K, Bota DA. Low-doses of cisplatin injure hippocampal synapses: a mechanism for “chemo” brain? Exp Neurol 2014;255:137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process 2012;13:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ben Abdallah NM, Fuss J, Trusel M, Galsworthy MJ, Bobsin K, Colacicco G, Deacon RM, Riva MA, Kellendonk C, Sprengel R, Lipp HP, Gass P. The puzzle box as a simple and efficient behavioral test for exploring impairments of general cognition and executive functions in mouse models of schizophrenia. Exp Neurol 2011;227:42–52. [DOI] [PubMed] [Google Scholar]
- [4].Bennett GJ, Doyle T, Salvemini D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol 2014;10:326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boyette-Davis J, Dougherty PM. Protection against oxaliplatin-induced mechanical hyperalgesia and intraepidermal nerve fiber loss by minocycline. Exp Neurol 2011;229:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- [7].Chiang ACA, Huo X, Kavelaars A, Heijnen CJ. Chemotherapy accelerates age-related development of tauopathy and results in loss of synaptic integrity and cognitive impairment. Brain Behav Immun 2019;79:319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chiu GS, Boukelmoune N, Chiang ACA, Peng B, Rao V, Kingsley C, Liu HL, Kavelaars A, Kesler SR, Heijnen CJ. Nasal administration of mesenchymal stem cells restores cisplatin-induced cognitive impairment and brain damage in mice. Oncotarget 2018;9:35581–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chiu GS, Maj MA, Rizvi S, Dantzer R, Vichaya EG, Laumet G, Kavelaars A, Heijnen CJ. Pifithrin-mu prevents cisplatin-induced chemobrain by preserving neuronal mitochondrial function. Cancer Res 2017;77:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, Zhang Z, Migaud ME, Rabinowitz JD, Baur JA. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 2018;7:e33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res 2007;13:1663–74. [DOI] [PubMed] [Google Scholar]
- [12].Du X, Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol 2013;11:621–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fan G, Merritt SE, Kortenjann M, Shaw PE, Holzman LB. Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J Biol Chem 1996;271:24788–93. [DOI] [PubMed] [Google Scholar]
- [14].Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neurosci Lett 2008;437:180–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Hoke A, Milbrandt J, DiAntonio A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 2019;4:e129920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016;139:3092–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon self-destruction: New links among SARM1, MAPKs, and NAD+ metabolism. Neuron 2016;89:449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol 2011;194:751–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Goodwani S, Fernandez C, Acton PJ, Buggia-Prevot V, McReynolds ML, Ma J, Hu CH, Hamby ME, Jiang Y, Le K, Soth MJ, Jones P, Ray WJ. Dual leucine zipper kinase is constitutively active in the adult mouse brain and has both stress-induced and homeostatic functions. Int J Mol Sci 2020;21:4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Grisold W, Cavaletti G, Windebank AJ. Peripheral neuropathies from chemotherapeutics and targeted agents: diagnosis, treatment, and prevention. Neuro Oncol 2012;14(suppl 4):iv45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hosseini SM, Kesler SR. Multivariate pattern analysis of FMRI in breast cancer survivors and healthy women. J Int Neuropsychol Soc 2014;20:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, BrentClark H, Carlson G. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 1995;15:1203–18. [DOI] [PubMed] [Google Scholar]
- [23].Hu F, Liu HC, Su DQ, Chen HJ, Chan SO, Wang Y, Wang J. Nogo-A promotes inflammatory heat hyperalgesia by maintaining TRPV-1 function in the rat dorsal root ganglion neuron. FASEB J 2019;33:668–82. [DOI] [PubMed] [Google Scholar]
- [24].Hu S, Huang KM, Adams EJ, Loprinzi CL, Lustberg MB. Recent developments of novel pharmacologic therapeutics for prevention of chemotherapy-induced peripheral neuropathy. Clin Cancer Res 2019;25:6295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hu Z, Deng N, Liu K, Zeng W. DLK mediates the neuronal intrinsic immune response and regulates glial reaction and neuropathic pain. Exp Neurol 2019;322:113056. [DOI] [PubMed] [Google Scholar]
- [26].Kesler SR, Acton P, Rao V, Ray WJ. Functional and structural connectome properties in the 5XFAD transgenic mouse model of Alzheimer's disease. Netw Neurosci 2018;2:241–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kharatmal SB, Singh JN, Sharma SS. Calpain inhibitor, MDL 28170 confer electrophysiological, nociceptive and biochemical improvement in diabetic neuropathy. Neuropharmacology 2015;97:113–21. [DOI] [PubMed] [Google Scholar]
- [28].Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother 2010;1:94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Koskinen MJ, Kautio AL, Haanpaa ML, Haapasalo HK, Kellokumpu-Lehtinen PL, Saarto T, Hietaharju AJ. Intraepidermal nerve fibre density in cancer patients receiving adjuvant chemotherapy. Anticancer Res 2011;31:4413–16. [PubMed] [Google Scholar]
- [30].Krukowski K, Ma J, Golonzhka O, Laumet GO, Gutti T, van Duzer JH, Mazitschek R, Jarpe MB, Heijnen CJ, Kavelaars A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. PAIN 2017;158:1126–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, Gogineni A, Sengupta Ghosh A, Jiang Z, Lee SH, Maloney J, Gandham VD, Pozniak CD, Wang B, Lee S, Siu M, Patel S, Modrusan Z, Liu X, Rudhard Y, Baca M, Gustafson A, Kaminker J, Carano RAD, Huang EJ, Foreman O, Weimer R, Scearce-Levie K, Lewcock JW. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci Transl Med 2017;9:eaag0394. [DOI] [PubMed] [Google Scholar]
- [32].Lees JG, Makker PG, Tonkin RS, Abdulla M, Park SB, Goldstein D, Moalem-Taylor G. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur J Cancer 2017;73:22–9. [DOI] [PubMed] [Google Scholar]
- [33].Li J, Zhang YV, Asghari Adib E, Stanchev DT, Xiong X, Klinedinst S, Soppina P, Jahn TR, Hume RI, Rasse TM, Collins CA. Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104. Elife 2017;6:e24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li L, Qin H, Shi W, Gao G. Local Nogo-66 administration reduces neuropathic pain after sciatic nerve transection in rat. Neurosci Lett 2007;424:145–8. [DOI] [PubMed] [Google Scholar]
- [35].Ling J, Erol F, Viatchenko-Karpinski V, Kanda H, Gu JG. Orofacial neuropathic pain induced by oxaliplatin: downregulation of KCNQ2 channels in V2 trigeminal ganglion neurons and treatment by the KCNQ2 channel potentiator retigabine. Mol Pain 2017;13:1744806917724715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lomeli N, Di K, Czerniawski J, Guzowski JF, Bota DA. Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radic Biol Med 2017;102:274–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Loreto A, Hill CS, Hewitt VL, Orsomando G, Angeletti C, Gilley J, Lucci C, Sanchez-Martinez A, Whitworth AJ, Conforti L, Dajas-Bailador F, Coleman MP. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol Dis 2020;134:104678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Luo X, Huh Y, Bang S, He Q, Zhang L, Matsuda M, Ji RR. Macrophage toll-like receptor 9 contributes to chemotherapy-induced neuropathic pain in male mice. J Neurosci 2019;39:6848–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BS, Dobrowsky RT. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J Pharmacol Exp Ther 2014;348:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ma J, Huo X, Jarpe MB, Kavelaars A, Heijnen CJ. Pharmacological inhibition of HDAC6 reverses cognitive impairment and tau pathology as a result of cisplatin treatment. Acta Neuropathol Commun 2018;6:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ma J, Kavelaars A, Dougherty PM, Heijnen CJ. Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: targeting the source. Cancer 2018;124:2289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ma J, Pan P, Anyika M, Blagg BS, Dobrowsky RT. Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chem Neurosci 2015;6:1637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ma J, Trinh RT, Mahant ID, Peng B, Matthias P, Heijnen CJ, Kavelaars A. Cell-specific role of histone deacetylase 6 in chemotherapy-induced mechanical allodynia and loss of intraepidermal nerve fibers. Pain 2019;160:2877–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Maj MA, Ma J, Krukowski KN, Kavelaars A, Heijnen CJ. Inhibition of mitochondrial p53 accumulation by PFT-mu prevents cisplatin-induced peripheral neuropathy. Front Mol Neurosci 2017;10:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mao-Ying QL, Kavelaars A, Krukowski K, Huo XJ, Zhou W, Price TJ, Cleeland C, Heijnen CJ. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS One 2014;9:e100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Messoussi A, Feneyrolles C, Bros A, Deroide A, Dayde-Cazals B, Cheve G, Van Hijfte N, Fauvel B, Bougrin K, Yasri A. Recent progress in the design, study, and development of c-Jun N-terminal kinase inhibitors as anticancer agents. Chem Biol 2014;21:1433–43. [DOI] [PubMed] [Google Scholar]
- [47].Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci 2009;12:387–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park SJ, Yoon BH, Kim SK, Kim SY. GENT2: an updated gene expression database for normal and tumor tissues. BMC Med Genomics 2019;12(suppl 5):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Patel S, Meilandt WJ, Erickson RI, Chen J, Deshmukh G, Estrada AA, Fuji RN, Gibbons P, Gustafson A, Harris SF, Imperio J, Liu W, Liu X, Liu Y, Lyssikatos JP, Ma C, Yin J, Lewcock JW, Siu M. Selective inhibitors of dual leucine zipper kinase (DLK, MAP3K12) with activity in a model of alzheimer's disease. J Med Chem 2017;60:8083–102. [DOI] [PubMed] [Google Scholar]
- [50].Pozniak CD, Sengupta Ghosh A, Gogineni A, Hanson JE, Lee SH, Larson JL, Solanoy H, Bustos D, Li H, Ngu H, Jubb AM, Ayalon G, Wu J, Scearce-Levie K, Zhou Q, Weimer RM, Kirkpatrick DS, Lewcock JW. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J Exp Med 2013;210:2553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sales MVC, Suemoto CK, Apolinario D, Serrao V, Andrade CS, Conceicao DM, Amaro E, Jr, de Melo BAR, Riechelmann RP. Effects of adjuvant chemotherapy on cognitive function of patients with early-stage colorectal cancer. Clin Colorectal Cancer 2019;18:19–27. [DOI] [PubMed] [Google Scholar]
- [52].Scherling CS, Smith A. Opening up the window into “chemobrain”: a neuroimaging review. Sensors (Basel) 2013;13:3169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shah A, Hoffman EM, Mauermann ML, Loprinzi CL, Windebank AJ, Klein CJ, Staff NP. Incidence and disease burden of chemotherapy-induced peripheral neuropathy in a population-based cohort. J Neurol Neurosurg Psychiatry 2018;89:636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Siu M, Sengupta Ghosh A, Lewcock JW. Dual leucine zipper kinase inhibitors for the treatment of neurodegeneration. J Med Chem 2018;61:8078–87. [DOI] [PubMed] [Google Scholar]
- [55].Soth MJL G, Le K, Cross J, Jones P. Bicyclo[1.1.1]pentane inhibitors of dual leucine zipper (DLK) kinase for the treatment of disease, 2018. [Google Scholar]
- [56].Summers DW, Frey E, Walker LJ, Milbrandt J, DiAntonio A. DLK activation synergizes with mitochondrial dysfunction to downregulate axon survival factors and promote SARM1-dependent axon degeneration. Mol Neurobiol 2020;57:1146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Summers DW, Milbrandt J, DiAntonio A. Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc Natl Acad Sci U S A 2018;115:E8746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Turkiew E, Falconer D, Reed N, Hoke A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J Peripher Nerv Syst 2017;22:162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Walczak P, Janowski M. Chemobrain as a product of growing success in chemotherapy - focus on glia as both a victim and a cure. Neuropsychiatry 2019;9:2207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife 2017;6:e22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang MS, Davis AA, Culver DG, Glass JD. WldS mice are resistant to paclitaxel (taxol) neuropathy. Ann Neurol 2002;52:442–7. [DOI] [PubMed] [Google Scholar]
- [62].Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A 2013;110:4039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wlaschin JJ, Gluski JM, Nguyen E, Silberberg H, Thompson JH, Chesler AT, Le Pichon CE. Dual leucine zipper kinase is required for mechanical allodynia and microgliosis after nerve injury. Elife 2018;7:e33910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yarza R, Vela S, Solas M, Ramirez MJ. c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for alzheimer's disease. Front Pharmacol 2015;6:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, Dougherty PM. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain 2016;17:775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. PAIN 1983;16:109–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B326.