

Summary
Fibroblasts display extensive transcriptional heterogeneity, yet functional annotation and characterization of their heterocellular relationships remains incomplete. Using mass cytometry, we chart the stromal composition of 18 murine tissues and 5 spontaneous tumor models, with an emphasis on mesenchymal phenotypes. This analysis reveals extensive stromal heterogeneity across tissues and tumors, and identifies coordinated relationships between mesenchymal and immune cell subsets in pancreatic ductal adenocarcinoma. Expression of CD105 demarks two stable and functionally distinct pancreatic fibroblast lineages, which are also identified in murine and human healthy tissues and tumors. Whereas CD105-positive pancreatic fibroblasts are permissive for tumor growth in vivo, CD105-negative fibroblasts are highly tumor suppressive. This restrictive effect is entirely dependent on functional adaptive immunity. Collectively, these results reveal two functionally distinct pancreatic fibroblast lineages and highlight the importance of mesenchymal and immune cell interactions in restricting tumor growth.
Keywords: tumor microenvironment, cancer-associated fibroblast lineages, pancreatic cancer, CyTOF, tumor-restrictive fibroblasts, mass cytometry, CAF, CD105, Eng
Graphical abstract
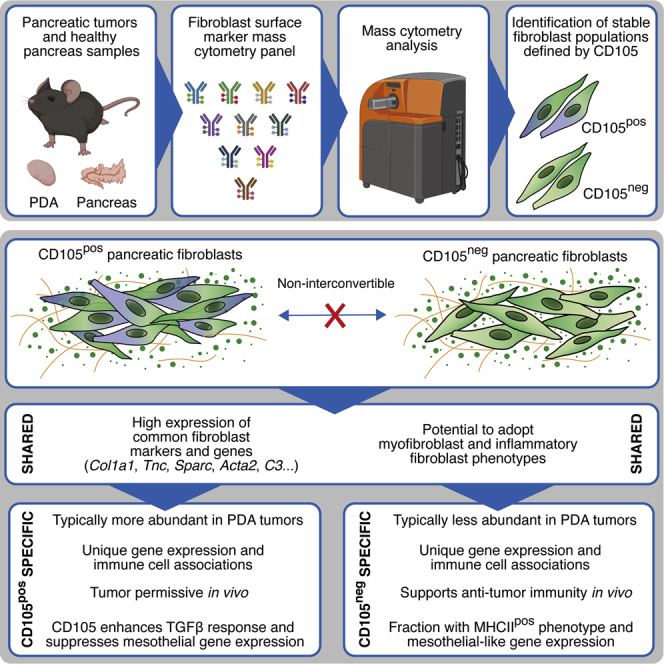
Highlights
-
•
Mass cytometry analysis of mesenchymal stroma in murine normal and tumor tissue
-
•
Mesenchymal heterogeneity is a feature of human and murine tissues and tumors
-
•
CD105 expression distinguishes two pancreatic fibroblast lineages
-
•
CD105neg pancreatic fibroblasts support anti-tumor immunity to control tumor growth
Hutton et al. use mass cytometry to chart stromal cells and describe mesenchymal states and lineages in pancreatic ductal adenocarcinoma. CD105 (Eng) expression distinguishes two pancreatic fibroblast lineages with distinct functions. CD105pos fibroblasts are tumor permissive, whereas CD105neg fibroblasts suppress tumor growth in a manner dependent on adaptive immunity.
Introduction
Stromal fibroblasts are critical to normal tissue homeostasis but are functionally subverted in fibrotic, inflammatory, and neoplastic disease (Dakin et al., 2018; Driskell and Watt, 2015; Sahai et al., 2020). Coerced fibroblasts, and their heterocellular interactions, have therefore become attractive therapeutic targets in multiple disease indications (Dakin et al., 2018; Sahai et al., 2020). In particular, cancer-associated fibroblasts (CAFs) have been ascribed pleiotropic pro-tumorigenic functions, such as extracellular matrix remodeling and tissue stiffening, escape from immune surveillance, and promotion of therapeutic resistance (Feig et al., 2013; Hirata et al., 2015; Sahai et al., 2020). However, genetic and pharmacological ablation of fibroblasts in preclinical mouse models reduces survival, and a clinical trial broadly targeting fibroblasts in pancreatic cancer patients was terminated due to disease acceleration (NCT01130142) (Catenacci et al., 2015; Kim et al., 2014; Özdemir et al., 2014; Rhim et al., 2014). Consequently, functionally opposing fibroblast populations have been hypothesized to co-exist in the tumor microenvironment (TME).
Cellular diversity arises from a combination of irreversible differentiation hierarchies (lineages) and distinct but plastic polarizations (states) (Croft et al., 2019; Janes, 2016; Tirosh et al., 2016; Wohlfahrt et al., 2019). For example, distinct lineages of spatially organized dermal fibroblasts arise during embryonic development and have discrete functions in adult skin homeostasis and wound repair (Driskell et al., 2013; Rinkevich et al., 2015). However, whether functionally distinct fibroblast lineages exist in other mammalian tissues is not known. Moreover, fibroblasts have the capacity to adopt at least two phenotypically distinct states, with myofibroblastic or inflammatory characteristics (Biffi et al., 2019; Kuppe et al., 2020; Öhlund et al., 2017). Determining whether distinct fibroblast lineages and phenotypes are associated with specific pathologies is necessary for the efficient application of stromal-targeting therapies (Helms et al., 2020; Sahai et al., 2020).
Genetically engineered mouse models (GEMMs) have been instrumental in interrogating the TME. The Pdx1-Cre;KrasLSL-G12D/+;Trp53LSL-R172H/+ (KPC) model of pancreatic ductal adenocarcinoma (PDA) recapitulates several aspects of the human disease, including genetic instability, therapeutic resistance, and an extensive desmoplastic microenvironment (Halbrook et al., 2019; Hingorani et al., 2005; Steele et al., 2016). Targeting specific pro-tumorigenic functions of fibroblasts in KPC tumors improves response to chemotherapy and sensitizes to immune checkpoint blockade (ICB) (Feig et al., 2013; Jiang et al., 2016; Miller et al., 2015; Shi et al., 2019). Similarly, targeting suppressive immune subsets also sensitizes to ICB and simultaneously alters desmoplasia, underscoring how mesenchymal and immune cell interactions balance pro- and anti-tumorigenic properties of the TME (Candido et al., 2018; Steele et al., 2016).
To chart mesenchymal and immune cell phenotypes in neoplastic disease, we immunophenotyped 14 million cells from 39 tumor samples, across 5 autochthonous murine models by mass cytometry (MC). In contrast to most CAF markers, CD105 demarks two discrete fibroblast populations in most normal and tumor-bearing tissues. The abundance of CD105pos and CD105neg CAFs correlate with distinct immune cell populations in PDA tumors, and diverge in their response to regulatory signals in the microenvironment. CD105pos pancreatic fibroblasts are permissive for tumor growth in vivo. In contrast, CD105neg fibroblasts potently restrict tumor growth, in a manner dependent on functional adaptive immunity and type 1 conventional dendritic cells (cDC1s).
Results
Single-cell immunophenotyping of mesenchymal stromal cells
A practical barrier for characterizing fibroblast functions is a lack of robust cell surface markers for live cell isolation. We therefore assembled an MC antibody panel, emphasizing mesenchymal cell surface markers, for subsequent purification and characterization (Table S1). Established immune and epithelial cell lineage markers were included to aid annotation of non-mesenchymal lineages (Bendall and Nolan, 2012; Bendall et al., 2011). To ensure that bona fide mesenchymal cell populations were distinguishable from immune, endothelial, and tumor cells, including tumor cells having undergone epithelial to mesenchymal transition, we analyzed tumors from Pdx1-Cre;KrasLSL-G12D/+;Trp53LSL-R172H/+;Rosa26LSL-tdRFP/LSL-tdRFP (RFPpos KPC) mice. High-dimensional phenotypes were visualized using UMAP projections, demonstrating tumor cell (RFPpos PCKhigh EpCAMpos) segregation from immune cells (CD45pos), endothelial cells (ECs) (CD31pos), and non-transformed mesenchymal stromal cells (RFPneg CD45neg CD31neg CD90pos), even when RFP was omitted from clustering (Figures S1A and S1B) (Becht et al., 2018; Van Gassen et al., 2015).
Phenotypic and compositional heterogeneity of pancreatic cancer-associated mesenchymal cells
To quantitatively annotate the composition of mesenchymal stromal cells in PDA, we analyzed 5 million cells from 19 tumors collected from KPC mice (Figures 1A–1D and S1C). Mesenchymal stromal cells constituted 12.6% ± 5.0% (mean ± standard deviation), CD45pos immune cells 39.3% ± 14.7%, and tumor cells 47.8% ± 17.7% of all viable single cells.
Figure 1.

Phenotypic and compositional heterogeneity of pancreatic cancer-associated mesenchymal cells
(A) UMAP projection of single mesenchymal stromal cells from n = 19 tumors, with color-coded FlowSOM clusters (1–20). Total of 5 × 105 cell displayed.
(B) Stacked bar graph displaying relative abundance of KPC PDA mesenchymal stromal subclusters. Color coded as in (A) and separated into major mesenchymal groups.
(C) Heatmap of marker median mass intensities (MMIs) displayed as Z scores. Each FlowSOM cluster was grouped by unsupervised hierarchical clustering based on marker MMIs. Cell-type annotations based on canonical markers are listed.
(D) UMAP projection from (A) displaying overlaid signal intensity of selected phenotypic markers.
(E) Whisker plot with relative frequency of CD105pos and CD105neg CAFs displayed as mean ± SD. n = 19 KPC tumors.
(F–J) Relative frequency of S-phase (F), apoptotic (G), αSMApos (H), MHCIIpos (I) and CD74pos (J) CAFs within total CD105pos and CD105neg CAFs. Paired populations from the same tumor samples are linked.
(K) Spearman correlation coefficients of all pairwise mesenchymal stroma cluster frequencies. CD105neg (orange) and CD105pos (green) CAF subsets highlighted.
Data are compared using paired t tests (E–J) or Spearman correlation adjusted for multiple testing using Benjamini-Hochberg correction (K). ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Immune and tumor cells were excluded and the remaining cells were clustered using FlowSOM and visualized by UMAP projection (Figures 1A–1D and S1D). The mesenchymal subset composition varied extensively within and between tumors (Figures 1A–1D; Table S1). CD31pos ECs comprised seven clusters, including three blood EC phenotypes (S-3, 4, and 5), one lymphatic EC cluster (S-11), and three other minor clusters (S-7, 17, and 18). Pericytes (S-15) form a single uniform and discrete cluster (Figures 1A–1D). Blood ECs (S-3, 4, and 5), form a continuum of phenotypes with graded abundance of MCAM, ITGβ3, and ITGα5 (Figures 1A–1D). Comparing EC subset abundances with macroscopic tumor features revealed an inverse relationship between the major blood EC cluster, S-4, and tumor weight (Figure S1E), suggesting that larger PDA tumors are not only poorly perfused due to vessel collapse, but also display insufficient vascularization (Olive et al., 2009; Provenzano et al., 2012).
The remaining clusters were designated as CAFs (8.2% ± 3.5% of all viable cells). PDPN, CD90, DES, and CD63 were abundant on most CAFs; however, these markers cannot be used in isolation to confidently identify all CAFs (Figures 1A–1D). Most markers, including αSMA, PDGFRα/β, MCAM, ICAM1, VCAM1, ITGα5, CD34, and CD73, displayed graded expression in several CAF clusters, revealing a spectrum of phenotypic states (Figures 1A–1D). For example, αSMA and PDGFRα displayed an inverse relationship across CAF subsets with αSMAhigh clusters (S-19 and 20) and αSMAlow/PDGFRαhigh clusters (S-6, 9, and 12) corresponding to myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs), respectively (Biffi et al., 2019; Elyada et al., 2019). The dipeptidylpeptidase CD26, which demarks a distinct fibroblast lineage in the skin, displayed graded expression in PDA tumors, indicative of phenotypic states rather than a defined lineage (Driskell et al., 2013; Rinkevich et al., 2015). In contrast, the transforming growth factor β receptor (TGF-βR) co-receptor, CD105, clearly separated two distinct CAF populations in all 19 tumor samples analyzed (Figures 1A–1D). The CD105pos:CD105neg CAF ratio varied widely between different PDA tumors, where CD105pos CAFs typically were more abundant (∼7:3 ratio) (Figure 1E). However, CD105neg CAFs were notably abundant in a minority of tumors. Moreover, CD105neg CAFs were more proliferative in tumors, but did not display any differences in apoptotic rate (Figures 1F and 1G). Most markers, including αSMA and PDGFRα, displayed graded expression in both CD105pos and CD105neg CAFs, indicating that both populations can acquire myCAF and iCAF characteristics (Figures 1C and 1D) (Biffi et al., 2019; Kuppe et al., 2020; Öhlund et al., 2017). The extent of myofibroblast polarization for both CD105pos and CD105neg CAFs was highly variable (31.4%–92.3% of all CAFs) and exhibited remarkable co-variation between CAFs from the same tumor, indicative of highly coordinated regulation of the myCAF phenotype within each tumor (Figures 1H and S1F). Conversely, the laminin binding ITGα6, the lipopolysaccharide co-receptor, CD14, and several proteins involved in major histocompatibility complex class II (MHCII) antigen presentation (MHCII and CD74) were almost exclusive to CD105neg CAFs (S-9 and 12), indicating that the majority of the recently described antigen-presenting CAF (apCAFs) fall within the CD105neg CAF subset (Figures 1A–1D, 1I, and 1J) (Elyada et al., 2019). Re-analysis of available single-cell transcriptomic (scRNA-seq) data (Elyada et al., 2019) confirmed that differential Eng (CD105) expression separates two CAF populations, with myCAF and iCAF signature gene expression in both Engpos and Engneg clusters and apCAF gene expression restricted to the Engneg cluster (Figure S1G). Finally, correlation analysis of the relative abundances between mesenchymal subsets revealed distinct coordinated relationships within, but not between, most CD105pos or CD105neg subsets, suggesting that each population responds distinctly to regulatory signals within the TME (Figure 1K).
Co-regulated CAF and immune subsets within the PDA tumor microenvironment
In addition to fibrotic expansion, developing PDA is characterized by a co-evolving tumor-permissive inflammation (Clark et al., 2007; Collins et al., 2012). To reveal phenotypic relationships between mesenchymal and immune cell populations in PDA, we used MC to annotate and quantify CD45pos CD3εneg (myeloid, natural killer, and B cell [MNB]) and CD45pos CD3εpos (T cell) subsets (Figures S2A–S2H) in tumors that had already been annotated for mesenchymal stromal composition.
All major immune subsets were identified and quantified (Figures S2A–S2H; Table S2) (Bendall et al., 2011; Spitzer et al., 2017). Monocytes (MNB-6, 8, 12) and macrophages (MNB-11, 14–20) were notably abundant and phenotypically heterogeneous, with graded expression of T cell inhibitory checkpoint ligands and chemotactic receptors (Figures S2A–S2D) (Di Mitri et al., 2019). In contrast, CD45pos CD3εpos T cells constituted only 4.0% ± 3.9% of all viable cells. CD4pos T cells were predominantly FOXP3pos T regulatory cells (Figures S2E–S2H). The majority of all CD8pos T cells (75.7% ± 23.4%) in these tumors were PD-1neg CD39neg bystanders, where only three minor CD8pos T cell subsets (T-3, 4, and 6) expressed markers indicative of T cell receptor engagement (Figures S2E–S2H) (Simoni et al., 2018). T-4 (PD-1high CD39high CD38high) resemble the “terminally exhausted” phenotype, with a lack of GZMB expression, high EOMES, and reduced effector function (Simoni et al., 2018; Thommen et al., 2018). T-3 (PD-1int CD39pos CD38neg GZMBpos CTLA-4pos 4-1BBpos T-BETpos) is phenotypically consistent with an active but not terminally exhausted phenotype and has been associated with improved capacity for expansion and tumor control (Leun et al., 2020; Philip et al., 2017). T-6 is the only CD8pos PD-1pos subset to express the transcription factor TCF-1, associated with stem/progenitor-like functions and high expansion potential during immunotherapy (Leun et al., 2020; Yost et al., 2019).
Annotation of the relative subset abundance, the proliferating cell fraction (%Ki67pos IdUpos cells), and apoptotic cell fraction (%CC3pos cells) revealed extensive variability of all stromal subsets between tumors (Figures 2A, 2B, and S3A; Table S2). For example, T cell subsets display striking variation in proliferation rates, where the CD8pos T cell subset, T-3, was highly proliferative only in some tumors (mean 33.8% ± 20.4%) (Figure 2B). Notably, three of the most proliferative mesenchymal subsets were all CD105neg CAFs, including the MHCIIpos CD74pos S-9 and S-12.
Figure 2.

Co-regulated CAF and immune subsets within the PDA tumor microenvironment
(A and B) Relative frequency within parental population (A) and proliferative fraction (Ki67pos IdUpos) (B) of annotated subsets. Data displayed as mean ± SD.
(C) Model of association between mesenchymal subset abundance and immune cell proliferation.
(D) Matrix of Spearman correlation coefficients of all pairwise mesenchymal subset frequencies and immune cell proliferation. CD105neg (orange) and CD105pos (green) CAF subsets highlighted.
(E–G) Spearman correlation analysis of S-9 (E, F) and S-19 (G) relative frequency with proliferative fraction of T-3 (E), T-19 (F), and T-10 (G) (top). ρ = Spearman correlation coefficient, 90% confidence intervals displayed. PDA tumors split into high (n = 7) or low (n = 8) fractions of S-9 (E and F) and S-19 (G) with proliferative fraction of T-3 (E), T-19 (F), and T-10 (G) displayed as mean ± SD (bottom).
(H) Model of positive (red) and negative (blue) correlations of CD105pos and CD105neg CAF subset abundance and proliferation of selected immune subsets.
Samples were compared using unpaired t tests (E–G) (bottom) or Spearman correlation adjusted for multiple testing using Benjamini-Hochberg correction (D, E–G) (top). ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figures S2 and S3 and Table S2.
To find potential heterocellular relationships, we leveraged the inherent variability between these spontaneous tumors and correlated the abundance, proliferation, and apoptotic fractions of stromal subsets in a pairwise manner (Figures S3B and S3C; Table S2) (Chevrier et al., 2017; Jackson et al., 2020). The abundance of specific mesenchymal subsets correlated with the proliferation rate of several immune subsets, reflecting possible directional interactions (Figures 2C–2G). For example, the abundance of mesenchymal subset S-9 (CD105neg MHCIIpos CD74pos) was positively correlated with the proliferation of several T cell subsets, including the antigen-experienced CD4 T cell subset (T-19) and the CD8pos CD39neg T-10 subset (Figures 2E–2G). Moreover, S-9 was the only mesenchymal subset positively associated with increased proliferation of the antigen experienced, but not terminally exhausted, T-3 subset (Figures 2D and 2E). In contrast, the CD105pos αSMAhigh CAF subsets (S-19 and 20) were anti-correlated with the proliferation of T-19 and 10 (Figures 2D and 2G). Markedly, some CD105pos and CD105neg mesenchymal subsets displayed opposing relationships with several immune subsets, suggestive of contrasting immune-modulatory effects (Figures 2D and 2H).
CD105 expression discriminates two distinct CAF populations in murine and human PDA
To determine whether CD105pos and CD105neg CAFs were also present in human PDA, we co-stained human resected samples for pan-cytokeratin (PCK) to mark epithelial cells, VIM or PDPN to mark CAFs and CD105. Both CD105pos and CD105neg CAFs were clearly identified and regionally distributed in the stroma, demonstrating that these CAF populations are preserved in human disease (Figures 3A and 3B; Table S3).
Figure 3.

CD105 expression discriminates two distinct CAF populations in murine and human PDA
(A and B) Immunohistochemistry (IHC) of human PDA tumor samples stained for pan-cytokeratin (PCK) (green), CD105 (yellow), DAPI (blue) and vimentin (VIM) (A) or podoplanin (PDPN) (B) (purple). Insert is magnified with arrows annotating vessels (right) (B). Representative images of n = 15 tumor samples. Scale bar = 500 μm..
(C and D) Fluorescence-activated cell sorting plots (C) and in vitro cultures (D) of CD105pos and CD105neg CAFs. Representative of n = 6 independent experiments. Scale bar, 150 μm.
(E–J) RNA-seq expression analysis of paired CD105pos (n = 6) and CD105neg (n = 6) PDA CAFs. Isolations from the same tumor sample are linked. Gene expression calculated as transcripts per kilobase million (TPM). Displaying Eng (the gene encoding CD105) (E), canonical fibroblast genes (F), canonical pericyte genes (G), myCAF- and iCAF-associated genes (H), and genes associated with fibroblast heterogeneity in other studies (I–J).
(K) Principal-component (PC) analysis of differentially expressed genes between CD105pos (n = 6, yellow) and CD105neg (n = 6, purple) PDA CAFs. DEGs determined using DEseq2 as >2 fold-change and Benjamini-Hochberg adjusted p < 0.05. Paired CAFs from the same tumor are linked.
(L) Ingenuity Pathway Analysis of CD105pos (Yellow) and CD105neg (purple) CAF DEGs, displaying upstream activators.
(M) Heatmap of expression levels of all 1007 CAF DEGs, displayed as row Z scores. Example DEGs are highlighted.
(N–P) CD105pos PDA CAF DEGs (N), CD105neg PDA CAF DEGs (O), and CD105neg CAF DEGs associated with mesothelial cell identity (P). Gene expression calculated as TPM. Isolations from the same tumor sample are linked.
Samples are compared using paired t tests (D–I and M–O). ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To establish if CD105pos and CD105neg CAFs are phenotypically distinct, we used the MC data to design a fluorescence-activated cell sorting (FACS) gating strategy and collected paired CD105pos and CD105neg CAFs from six KPC PDA tumors for gene expression analysis (Figures 3C–3P and S4A). In agreement with the MC analysis (Figure 1F), the relative abundance of CD105pos and CD105neg CAFs varied extensively between samples, and plated cells exhibited mesenchymal morphology (Figures 3D and S4A). Genes associated with general fibroblast identity, such as Col1a1, Col1a2, Vim, Pdpn, and Dcn, were expressed at equal levels in both the CD105pos and CD105neg populations and neither population had significantly different expression of the pericyte-associated gene Rgs5, however, CD105pos CAFs have higher Cspg4 expression (Figures 3E–3G). Genes associated with myCAF and iCAF identity as well as genes previously reported to define heterogeneous fibroblast populations, such as S100a4 (FSP1), Dpp4 (CD26), Dlk1, En1, Lrcc15, C5ar2 (GPR77), Mme (CD10), Sfrp1, Cxcl12, and Lif, were all equally expressed between CD105pos and CD105neg CAFs (Figures 3H and 3I) (Dominguez et al., 2020; Driskell et al., 2013; Feig et al., 2013; Lichtenberger et al., 2016; Rinkevich et al., 2015; Su et al., 2018). Expression of Fap and Ly6c1, which have previously been used for isolation of CAF populations, were enriched in CD105pos CAFs (Elyada et al., 2019; Feig et al., 2013). However, CD105neg CAFs variably express some level of these genes in vivo (Figure 3J).
Principal-component (PC) analysis of differentially expressed genes (DEGs) confirmed the major variance across samples (PC1) was related to CD105 status, suggesting that consistent differences in CD105pos and CD105neg CAF gene expression are conserved across tumors (Figure 3K). Ingenuity Pathway Analysis (IPA) highlighted several differentially engaged upstream regulators and pathways, with TGF-β signaling enriched in CD105pos CAFs, and LTBR, tumor necrosis factor alpha (TNF-α), nuclear factor κB (NF-κB), interleukin-6 (IL-6), JAK2, and STING1 signaling enriched in CD105neg CAFs (Figures 3L and S4B). A large number of genes encoding secreted products with known functional relevance in the TME were differentially expressed (Figures 3M–3O). For example, Postn, Cxcl14, and Igfbp5 were increased in CD105pos CAFs, and Cxcl2, Gas1, Bmp2, and Nos2 were elevated in CD105neg CAFs, which was also confirmed by re-analysis of available KPC scRNA-seq data (Figures 3M–3O, S4C, and S4D) (Elyada et al., 2019). Notably, single-cell Eng mRNA levels appear lower in iCAF-polarized CD105pos cells, which makes accurate annotation of CD105 status by single-cell mRNA levels alone challenging (Figures S4C and S1G). As expected, genes involved in MHCII antigen presentation were predominantly expressed in CD105neg CAFs, confirming an overlap between apCAFs and CD105neg CAFs (Figures 3O and S4D). Moreover, CD105neg CAFs express higher levels of several genes associated with mesothelial cell identity, including Wt1, Msln, Krt8/18, Upk3b, and Ezr, although the expression was non-uniform and restricted to a sub-fraction of Engneg cells (Figures 3P and S4D). Analysis of scRNA-seq data from human PDA tumors and normal adjacent tissue confirmed the presence of distinct ENGpos and ENGneg populations with expected distribution of CD105pos and CD105neg signature genes (Figures S4E–S4G) (Steele et al., 2020). Notably, myCAF and iCAF signature genes were also expressed across both ENGpos and ENGneg clusters in human PDA (Figure S4G). Collectively, this demonstrates the presence of CD105pos and CD105neg CAFs in human PDA and highlights their potential to differentially respond to and modify the inflammatory TME.
Phenotypic plasticity of mesenchymal marker expression
We hypothesized that lineage-restricted fibroblast subsets would be defined by distinct and stable marker expression, whereas graded marker expression likely reflects cellular plasticity (Figures 1A–1D). Since CAFs are tumor educated, we reasoned that fibroblasts from the normal tissue would reveal intrinsic differences in fibroblast hierarchies and therefore examined whether CD105 expression is stable or dynamically regulated in naive fibroblasts. Pancreatic fibroblasts (PaFs) were expanded from healthy tissue, revealing CD105pos and CD105neg PaF populations, which could be purified by FACS and cultured (Figure S5A). PaFs remained CD105pos or CD105neg after extended passaging and were able to generate stable cell lines (Figure 4A). Moreover, CD105 remained differentially expressed after treatment with tumor cell-conditioned medium, by direct tumor cell co-culture, or following extended culture with fibroblast-modulating signals TGF-β1, IL-1α, and interferon gamma (IFN-γ) (Figures 4B, 4C, and S5B). CD105 expression also distinguished two separate and stable populations in isolated human PaFs, and demarked two distinct fibroblast populations in non-tumor-bearing tissue adjacent to PDA (Figures 4D and S5C). Interestingly, CD105pos and CD105neg PaFs were discretely localized in the inflamed pancreas, with CD105pos PaFs observed in the intra-acinar regions of the pancreas and the CD105neg PaFs in the inter-acinar regions (Figure S5C). Finally, scRNA-seq analysis of in-vitro-expanded primary murine PaFs 7 days after isolation, confirmed that Eng expression defines the two major cell clusters, with expected expression of signature genes (Figures S5A, S5D–S5K). Eng transcripts were incompletely detected by scRNA-seq in clusters that have robust CD105 protein expression by flow cytometry (Figures S5A and S5I). Clustering was further divided by proliferation-associated genes (Figure S5K), indicating that differential Eng/CD105 expression captured the major source of heterogeneity in PaFs. Thus, CD105 is a key cell surface discriminator of two distinct human and murine PaF lineages.
Figure 4.

Phenotypic plasticity of mesenchymal marker expression
(A) Flow cytometry analysis of PDPN and CD105 in purified and in-vitro-cultured CD105pos and CD105neg pancreatic fibroblasts (PaFs) after 1 and 7 weeks. Plots are representative of n = 4 experiments. Relative frequencies shown in relevant quadrants.
(B) Normalized Eng mRNA expression in purified CD105pos (n = 4) and CD105neg PaFs (n = 4) treated with control (top) or KPC PDA conditioned medium (bottom). Data displayed as mean ± SD.
(C) Representative flow cytometry analysis (n = 4) of CD105 on GFPposCD105pos and GFPposCD105neg PaFs in mono- or co-culture with RFPpos KPC PDA tumor cells.
(D) Representative flow cytometry analysis (n = 3) of CD105 in isolated CD105pos and CD105neg human PaFs after >3 weeks of in vitro culture.
(E and F) MC analysis of primary PaFs treated with the indicated ligands for 3 days. Representative plots displaying relative frequencies of CD105pos and CD105neg PaFs.
(G and H) Heatmap of median marker intensity (MMI) displayed as column Z scores for each phenotypic marker on CD105pos (G) and CD105neg (H) PaFs after 3 days of treatment as indicated. Boxplots show MMI with upper and lower boundary of the interquartile range and whiskers denoting maximum and minimum values minus outliers, across all conditions.
(I and J) Representative flow cytometry analysis (n = 3) of CD105pos (I) and CD105neg (J) PaFs with IFN-γ, IFN-γ + KPC PDA conditioned medium, or IFN-γ + TGF-β1 treatment.
Samples are compared using unpaired t tests (B) (top and bottom). ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To subsequently determine how individual stimulations regulate marker expression in an unbiased, but experimentally controlled manner, we treated freshly isolated PaFs with 17 individual fibroblast-modulating signals for 72 h and analyzed marker expression by MC (Figures 4E–4H; Table S4). Importantly, distinct PaF populations remained clearly separated by bimodal CD105 expression across all stimulations (Figures 4E and 4F). Moreover, the relative abundance of CD105pos and CD105neg PaFs remained consistent across most of the 17 treatments, except for TGF-β1, which increased the fraction of CD105pos PaFs, and TNF-α and IFN-γ, which increased the relative abundance CD105neg PaFs (Figures 4E and 4F).
Hierarchical clustering of normalized median marker intensities revealed a diverse range of responses across both CD105pos and CD105neg populations, illustrating a high degree of phenotypic plasticity in both PaF populations (Figures 4G and 4H). Compared with the MC analysis of KPC tumors (Figures 1A–1D), markers with both broad and graded expression were dynamically regulated by individual signals, indicating that fibroblast surface marker abundance, in most cases, reflects dynamic changes in the local signaling environment. Some signals, such as TNF-α, IL-1α, and IL-1β, decrease αSMA and MCAM and increase PDGFRα and VCAM1 levels in both CD105pos and CD105neg populations, as expected. However, several other stimulations differentially regulate marker levels in CD105pos and CD105neg PaFs (Figures 4G and 4H). IFN-γ treatment increases MHCII, CD74, and CD80 in both CD105pos and CD105neg PaFs, which was verified using orthogonal flow cytometry (Figures 4I and 4J). In addition, this interferon-induced MHCIIpos fibroblast phenotype is inhibited in both CD105pos and CD105neg PaFs by simultaneous treatment with TGF-β1 or tumor cell-conditioned medium, suggesting that local signal integration shapes fibroblast phenotypes in vivo (Figures 4I and 4J). Finally, direct co-cultures of PaFs and pancreatic cancer cells induced a unique fibroblast marker signature, with elevated proliferation and expression of CD86 and CD90 (Figures 4G and 4H). This supports the notion that heterocellular interactions impose unique fibroblast phenotypes (Tape et al., 2016; Wei et al., 2020). Together, these results demonstrate that CD105 expression remains restricted and stable in isolated PaFs and therefore denote fibroblast lineages, whereas other tested markers are dynamically regulated and reflect fibroblast phenotypic plasticity.
Differential signaling engagement of CD105pos and CD105neg PaFs
To determine whether CD105pos and CD105neg PaFs also exhibit differences in their engagement of signaling networks, we analyzed selected signaling nodes in stimulated PaFs by MC (Figure 5A; Table S5). Strikingly, CD105pos and CD105neg PaFs exhibited distinct signaling responses even under controlled in vitro conditions. For example, IL-1α and IL-1β, engage NF-κB signaling more prominently in CD105pos PaFs; and leukemia inhibitory factor (LIF), as well as IL-6, which both engage the common gp130 co-receptor, have distinct effects on signaling across PaF populations, with greater STAT3 phosphorylation in CD105pos PaFs. This suggests that the two populations are intrinsically constrained in their signaling response.
Figure 5.

Differential signaling engagement of CD105pos and CD105neg PaFs
(A) MC analysis of CD105pos and CD105neg PaFs signaling. Data are displayed as median mass intensities (MMI) and column Z scores. Specific phosphorylation sites are annotated in brackets.
(B–D) RNA-seq analysis of CD105pos and CD105neg PaFs stimulated as displayed for 6 h (n = 3). DEGs were identified using DEseq2 with Benjamini-Hochberg adjusted p < 0.05. Data are displayed as Venn diagrams (top), with example genes listed (below). Unique DEGs of CD105pos PaFs in red, CD105neg PaFs in blue, and shared in purple. Numbers of significant DEGs are displayed in parenthesis.
(E and F) Expression of myCAF (E) and iCAF (F) genes from CD105pos and CD105neg PaFs stimulated with TGF-β1 or IL-1α (n = 4) for 3 days. Eng expression is also shown. Data displayed as mean ± SD.
Samples were compared using unpaired t tests (E and F). ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We subsequently compared early gene expression changes after short 6 h TGF-β1, IL-1α, or IFN-γ treatment (Figures 5B–5D; Table S5) to determine whether observed differences in cellular signaling response is reflected in gene expression. This analysis highlighted a selective engagement of early transcriptional networks, where CD105pos PaFs exhibited a significantly increased number of DEGs after stimulation with both TGF-β1 and IL-1α (Figures 5B and 5C). Although CD105 protein has no reported signaling capacity by itself, the receptor has been demonstrated to modulate the affinity of TGF-β family ligands to the TGF-βR signaling complex (Valluru et al., 2011). To test the role of CD105 in mediating TGF-β signaling, we used CRISPR-Cas-9 to delete Eng in CD105pos PaFs (Figure S6A). Loss of CD105 dampened the early transcriptional response to TGF-β1 compared with parental CD105pos PaFs, with a reduction from 151 DEGs in parental CD105pos PaFs to 76 DEGs in CD105KO PaFs (Figure 5B). In addition, the key TGF-β signaling mediator, Smad3 is more highly expressed in CD105pos PaFs (Figure S6B). Curiously, the expression of several mesothelial-associated genes (e.g., Msln and Upk3b) are increased in CD105pos PaFs when CD105 is deleted, demonstrating that CD105 itself suppresses a mesothelial-associated gene program in fibroblasts (Figure S6C). Moreover, both of the IL-1 receptors, Il1r1 and Il1rp, as well as the key signaling mediator, Myd88, were more highly expressed in CD105pos PaFs, which may underlie increased IL-1 sensitivity at this early time point (Figure S6B). Furthermore, Il6st (Gp130) is more abundant in CD105pos PaFs, providing a possible explanation for the increased sensitivity to LIF and IL-6 (Figures 5A and S6B). The response to IFN-γ was more equal and the majority of IFN-γ early DEGs (234 genes) were shared between the CD105pos and CD105neg PaFs, including well established IFN-γ response genes, such as Irf1. However, both populations have a number of unique DEGs, indicative of a largely similar “core” IFN-γ response, with some population-specific differences.
As the MC and ex vivo gene expression analysis indicated that both CD105pos and CD105neg CAFs adopt myCAF and iCAF phenotypes in vivo (Figures 1C, 1D, and 3H), we treated CD105pos and CD105neg PaFs with TGF-β1 or IL-1α for an extended period (72 h) and analyzed gene expression by qPCR (Figures 5E and 5F). In agreement with the MC analysis, prolonged stimulation induced myCAF and iCAF phenotypes in both CD105pos and CD105neg PaFs. Differential Eng expression was retained across all conditions although CD105pos PaFs that are iCAF-polarized have reduced Eng mRNA levels (Figures 5E and 5F). Thus, CD105pos and CD105neg PaFs appear to have different sensitivity to TGF-β1 and IL-1α activation at early time points (which may be more relevant when ligand abundance is limited) but both populations have the capacity to adopt both myCAF and iCAF phenotypes under extended stimulation (which may be more reflective of the extended activation that occurs in tumors).
CD105neg fibroblasts restrict tumor growth in vivo
To determine whether CD105pos and CD105neg fibroblasts differentially influence tumor growth in vivo we established a subcutaneous co-injection model, reasoning that possible confounding effects from resident PaFs would be bypassed in this simplified model. Indeed, tumors formed by injecting an established KPC PDA tumor cell line alone exhibit low-level infiltration of host fibroblasts at early time points (Figure S7A). Moreover, when KPC PDA tumor cells were mixed with an equal number of GFP-labeled PaFs and injected subcutaneously, GFPpos PaFs were retained after transplantation and exhibit stable differential CD105 expression (Figures 6A, S7B, and S7C).
Figure 6.

CD105neg fibroblasts restrict tumor growth in vivo
(A) Flow cytometry analysis of co-implanted GFPpos CD105pos or CD105neg PaFs 7 days after co-injection.
(B) Tumor growth of subcutaneous injection of 105 PDA tumor cells or co-transplantation with 105 CD105pos or CD105neg PaFs in syngeneic B6 mice. n = 5 mice per condition. Data are representative of n = 4 separate experiments. For the combined condition a 1:1 mixture of CD105pos:CD105neg PaFs was used and the total number of PaFs kept constant.
(C) Kaplan-Meier analysis of tumors exceeding a threshold volume of 400 mm3 (n = 4 independent studies, in total n = 14–22 mice per condition).
(D–F) As for (B) but with NOD-scid.Il2rg−/− (n = 4 to 5 per condition) (D) B6.Rag1−/− (n = 6 per condition) (E), and B6.Batf3−/− (n = 8 to 9 per condition) (F) mice.
(G) As for (A) but with CD105neg PaFs disrupted for H2Ab1, Cd74, and Cd80 expression. Non-targeting gRNA transfected CD105neg PaFs were used as control.
(H–J) Bulk RNA-seq analysis of co-injected PDA tumor cells with CD105pos (orange) and CD105neg (purple) PaFs at day 10. Heatmap of differentially expressed genes displayed as row Z scores (H), Ingenuity Pathway Analysis of differentially activated pathways (I), and upstream regulators (J).
Data are displayed as mean tumor volumes ± standard error of the mean (SEM) (B–G). Conditions were compared using two-way ANOVA (B and D–G) and log rank test (C). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Compared with control PDA tumor cells, co-injected CD105pos PaFs did not significantly influence tumor volume at endpoint (Figure 6B). However, co-injection of CD105neg PaFs dramatically restricts tumor growth and improves survival (Figures 6B and 6C), with 40% of mice exhibiting complete macroscopic and histological tumor regressions. Mixing CD105pos and CD105neg fibroblasts at a 1:1 ratio, but maintaining a constant total number of fibroblasts, also suppressed tumor growth; however, no full regressions were observed (Figure 6B). These findings are consistent across multiple independent studies and are reproduced with a second PDA tumor cell line and PaF lines with no GFP expression (Figures S7D–S7F). Thus, these results demonstrate that CD105pos PaFs are permissive to tumor growth, whereas CD105neg fibroblasts are highly tumor restrictive and that, in 1:1 mixtures of fibroblasts, the suppressive effect is dominant.
Co-injection of PDA tumor cells with CD105pos or CD105neg PaFs in NOD-scid.Il2rg−/− (deficient in innate and adaptive immune functions) or in Rag1−/− mice (deficient in mature T and B cells), did not affect tumor growth compared with mono-transplanted PDA tumor cells (Figures 6D and 6E). In addition, the restrictive effect of CD105neg on tumor growth was highly blunted in Baft3−/− animals, which lack cDC1s (Figure 6F). Thus, the growth-suppressive effect of CD105neg PaFs in vivo entirely depends on functional adaptive immunity, with a major contribution from cDC1s.
Since CD105neg CAFs are almost unique in expression of MHCII antigen presentation machinery in vivo (Figures 1D, 1I, 1J, and 3O), and can be induced to express these in vitro (Figures 4G–4J), we sought to explore whether MHCII, CD74, and CD80 were required for the in vivo tumor-suppressive effect of CD105neg PaFs. We disrupted the expression of these genes in CD105neg PaFs using CRISPR-Cas-9 (Figures S7G and S7H) and investigated the ensuing effect in vivo (Figure 6G). However, co-implanted CD105neg PaFs retained their restrictive capacity across all conditions. Thus, fibroblast MHCII antigen presentation is not required for the tumor-suppressive effect. To determine whether CD105 expression actively represses the tumor-restrictive phenotype, we co-implanted PDA tumor cells and CD105pos PaFs disrupted for CD105 expression (CD105KO) (Figure S6A). Injected CD105KO fibroblasts did not differentially affect tumor growth compared with non-targeting gRNA-transfected CD105pos PaFs (Figure S7I). Thus, CD105 is a useful marker of distinct fibroblast lineages but does not functionally contribute to the divergent tumor growth in vivo.
To investigate how co-injected CD105pos and CD105neg PaFs modulate the tumor microenvironment, we isolated developing tumors of similar size 10 days after injection and analyzed bulk gene expression profiles (Figures 6H–6J; Table S6). Consistent with loss of tumor suppression in animals with adaptive immunity deficiencies (Figures 6D–6F), we observed divergent engagement of immune-suppressive and immune-stimulatory transcriptional programs in CD105pos and CD105neg co-implanted tumors (Figures 6H–6J). DEG analysis highlighted increased expression of transcripts in CD105neg co-injected tumors that are associated with T cell infiltration (Cd3d, Lck, Zap70, Il2rb, Cd96), effector CD8 T cells/Th1 CD4 T cells/innate lymphocytes (Tbx21), T cell memory precursor differentiation (Il7r), cDC1 (Batf3), and general DC (Itgax) infiltration, and antigen presentation (H2-DMb1, H2-DMa, Cd74) (Figure 6H). Furthermore, IPA revealed engagement of pathways and upstream regulators with established roles in productive anti-tumor immune responses, such as DC maturation, T cell activation, IFN-γ signaling, and innate lymphoid signaling, in CD105neg PaF co-transplants (Figures 6I–6J) and MCPcounter analysis indicated an enrichment for DCs and CD8 T cells (Figure S7J) (Becht et al., 2016). Thus, these data support a role for CD105neg PaFs in establishing a tumor-suppressive inflammatory reaction.
CD105pos and CD105neg fibroblasts are identified in normal and tumor-bearing tissues
To expand the analysis of stromal fibroblasts beyond PDA and the pancreas, we analyzed low passage, primary fibroblast cultures from 18 normal tissues by MC, revealing both CD105pos and CD105neg fibroblast populations from most healthy murine tissues examined (Figures 7A and S8A; Table S7). Notably, the ratio of CD105pos and CD105neg fibroblasts varied across tissues, which may reflect inter-tissue heterogeneity or differential sensitivity to in vitro expansion. Fibroblast isolations, which initially appeared homogeneous (such as isolations from the liver and lung), contained both CD105pos and CD105neg fibroblasts when analyzed at earlier time points (Figures S8A and S8B), suggesting that fibroblast heterogeneity can be rapidly lost by in vitro culture. Consistent with the observation that CD105 expression is restricted in PaFs, FACS-purified CD105pos and CD105neg fibroblasts from the liver and lung also retained differential CD105 expression in vitro (Figure S8C). Furthermore, gene expression analysis comparing CD105pos and CD105neg pancreatic and liver fibroblasts confirmed CD105 status as the major source of variation (Figure S8D), highlighting DEGs associated with CD105 status (Figures S8E and S8F).
Figure 7.

CD105pos and CD105neg fibroblasts are identified in normal and tumor-bearing tissues
(A) MC analysis of in-vitro-expanded primary fibroblasts. Plots show PDPN and CD105 levels. LIN, EpCAM CD31 CD45.
(B) UMAP projection of CAFs from KPC pancreatic (n = 4), KPN colorectal (n = 5), MMTV-PyMT mammary (n = 4), KP lung (n = 4), and BRAFV600E melanoma (n = 3) GEMMs. FlowSOM clusters are color coded. Total of 5 × 105 cell displayed.
(C) Stacked bar graphs of GEMM CAF (GCAF) clusters displayed as a fraction of total CAFs. FlowSOM colors based on (B).
(D) Heatmap of marker median mass intensities (MMIs) displayed as Z scores. Each GCAF FlowSOM cluster is grouped by unsupervised hierarchical clustering based on marker MMIs. Cell-type annotations based on canonical phenotypic markers are listed. Tumor type/s that the GCAF clusters predominantly arise from are listed.
(E) UMAP projection from (B) displaying overlaid signal intensity of CD105 with annotated tumor types.
(F) UMAP projection from (B) displaying overlaid signal intensity of example markers. The tumor types of origin are highlighted: Pa, pancreatic; Co, colorectal; Ma, mammary; Lu, lung; Me, melanoma.
(G–I) Representative IHC analysis of human colorectal (n = 9), breast (n = 8), and lung adenocarcinoma (n = 6) tumor samples stained for pan-cytokeratin (PCK) (green), vimentin (VIM) (purple), CD105 (yellow), and DAPI (blue). Scale bar, 500 μm.
We subsequently analyzed CAFs in 20 tumors from 5 autochthonous GEMMs (Figures 7B–7F and S8G–S8J). FlowSOM clustering and UMAP visualization highlighted the presence of distinct CAF subsets across all tumors (Figures 7B–7F and S8G–S8J). Interestingly, while lung (KP), mammary (MMTV-PyMT), and melanoma (BRAFV600E) CAFs clustered according to their tissue of origin, several CAF clusters from pancreatic (KPC) and colorectal (KPN) tumors overlapped considerably, indicating phenotypic conservation of the CAFs between these two tissues (Figures 7B–7F). Few markers were broadly expressed (such as VIM, COLIV, CD63, and ITGαV) although none were expressed in all CAFs (Figures 7D, 7F, and S8G). Moreover, individual markers exhibited clear tissue-specific variation in vivo (Figures 7D and 7F). As expected, pancreatic tumors (KPC) contained mixtures of CD105pos and CD105neg CAFs, with CD105pos CAFs favored. In contrast, colorectal (KPN) and mammary (MMTV-PyMT) tumors contained more abundant CD105neg CAFs, whereas lung (KP) and melanoma (BRAFV600E) tumors were dominated by CD105pos CAFs (Figure 7E). This observation was supported by immunohistochemistry in FPPE human tumor samples (Figures 7G–7I and S8K–S8M; Table S7). Thus, CD105pos and CD105neg CAFs are present in normal and tumor-bearing mammalian tissue.
Discussion
Fibroblast lineages play distinct roles in development, homeostasis, and wound repair of the skin, and specific fibroblast states are increasingly recognized as regulators of immune cell function in inflammatory disease and cancer (Dominguez et al., 2020; Driskell et al., 2013; Koliaraki et al., 2020; Rinkevich et al., 2015). CD105 is well established as an abundant and robust marker for all EC subtypes, pericytes, and mesenchymal stem cells, and has furthermore been noted in the stroma of human prostate and colorectal tumors and in human healthy breast tissue (Kato et al., 2018; Lv et al., 2014; Morsing et al., 2016; Paauwe et al., 2018; Pittenger et al., 2019). The data presented here demonstrate that CD105 demarks two pancreatic fibroblast lineages with distinct tumor-permissive and restrictive functions: CD105pos fibroblasts are tumor permissive, whereas CD105neg fibroblasts restrict tumor growth in a manner that is dependent on functional adaptive immunity.
Within both CD105pos and CD105neg fibroblast populations, environmentally regulated signals further diversify the fibroblast repertoire. For example, CD105pos and CD105neg populations respond differently to fibroblast-modulating signals, such as LIF, IL-1, and TGF-β, and apCAF and mesothelial cell markers (MHCII and CD74) are predominantly expressed in CD105neg CAFs. Nonetheless, both populations express myCAF and iCAF markers in vivo, and isolated CD105pos and CD105neg PaFs can be induced to express myCAF and iCAF signature genes in vitro. Notably, CD105pos fibroblasts are transcriptionally more responsive to TGF-β1, and Eng expression is decreased in CD105pos iCAFs. This is in agreement with scRNA-seq data, which demonstrated expression of both myCAF and iCAF signature genes in both Engpos and Engneg subsets. However, within the Engpos subset, Eng expression correlates with the expression of established myCAF genes.
The detection of mesothelial cell transcripts (Wt1, Msln, Krt8/18, Upk3b) in a fraction of CD105neg PDA CAFs indicate a developmental relationship between CD105neg CAFs and the mesothelium. Indeed, mesothelial cells can adopt fibroblastic characteristics under TGF-β exposure (Namvar et al., 2018), and fibroblasts and smooth muscle cells in the lung and other trunk organs derive, in a sonic hedgehog-dependent process, from mesothelial precursors during tissue development (Cano et al., 2013; Dixit et al., 2013; Koopmans and Rinkevich, 2018; Rinkevich et al., 2012; Wilm, 2005). While mesothelial-like cells have been observed at the invasive edge of colorectal tumors (Gordillo et al., 2020), this cell type remains an otherwise understudied source of fibroblasts in tumors. The data presented here indicate that fibroblasts that are developmentally related to the mesothelium have intrinsically distinct functions. Future lineage-tracing studies will be needed to accurately determine whether CD105pos and CD105neg fibroblasts and CAFs arise from common or distinct differentiation hierarchies during tissue development.
The notable inter-tumoral heterogeneity of CAF subsets observed in PDA, combined with low preservation of CAF subsets across tumors in different organs, underscores the importance of determining the functional roles of CAF subsets across different tissues and tumors. For example, while CD26 defines fibroblasts lineages in the skin (Driskell et al., 2013; Rinkevich et al., 2015), it is dynamically regulated in PaFs. Moreover, although CD105pos and CD105neg fibroblasts isolated from the pancreas and liver show stable CD105 expression and exhibit conserved gene expression patterns, many genes remain expressed in a tissue-specific manner.
Single-cell technologies, such as MC, have been instrumental to define cellular subsets in the TME (Bendall et al., 2011; Chevrier et al., 2017). Whereas the abundance and phenotype within cell populations can be readily compared across tumors and tissues, tumor and stromal cells may exhibit differences in their liberation and details of local tissue structure is lost. Thus, future in situ studies are needed to establish regional differences in the cellular neighborhood of CD105pos and CD105neg CAFs.
Determining how inherent tumor-restrictive effects of naive CD105neg PaFs are bypassed as tumors develop, and whether anti-tumor immunity may also be regulated by CD105neg CAFs in established tumors is important. Indeed, the balance between CD105pos and CD105neg fibroblasts could be a key determinant of the local immune environment in PDA and may be exploited therapeutically. However, the function of CD105pos CAFs needs further characterization. Specifically, mono-injections of the PDA tumor cell lines used in this study grew aggressively and therefore only minimally depended on stromal-supportive signals, suggesting that tumor promoting effects from co-injected fibroblasts may not be fully captured with this model. Moreover, while CD105pos and CD105neg fibroblast populations are identified in multiple tumors and normal tissues, and also appear preserved in human tissue, further studies are needed to determine whether the tumor-permissive and restrictive functions of CD105pos and CD105neg fibroblasts are broadly conserved.
We envisage that the presented phenotypic atlas will accelerate much needed functional studies of the mesenchyme to improve our understanding of shared fibroblast features across different tissues to thereby enhance the application of stromal-targeting therapies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Fc block clone 2.4G2 | BD Biosciences | 558636 |
| Anti-mouse CD44 clone IM7 | Biolegend | 103002 |
| Anti-mouse EpCAM clone G8.8 | Biolegend | 118202 |
| Anti-mouse CD86 clone GL-1 | Biolegend | 105002 |
| Anti-mouse MCAM 141Pr clone ME-9F1 | Fluidigm | 3141016B |
| Anti-mouse ITGA5 clone 5H10-27(MFR5) | Biolegend | 103801 |
| Anti-mouse CD81 clone Eat-2 | Biolegend | 104902 |
| Anti-mouse CD87 clone 109801 | Thermo Fisher | MA5-23853 |
| Anti-PE 145Nd clone PE001 | Fluidigm | 3145006B |
| Anti-mouse ITGAV clone RMV-7 | Biolegend | 104102 |
| Anti-mouse ITGA2 clone Hma2 | Biolegend | 103501 |
| Anti-mouse PDGFRA 148Nd clone APA5 | Fluidigm | 3148018B |
| Anti-mouse PDPN clone 8.1.1 | Biolegend | 127402 |
| Anti-mouse CD24 (150Nd) clone M1/69 | Fluidigm | 3150009B |
| Anti-mouse PDGFRB (151Eu) clone APB5 | Fluidigm | 3151017B |
| Anti-mouse ICAM1 clone YN1/1.7.4 | Biolegend | 116102 |
| Anti-mouse CD63 clone NVG-2 | Biolegend | 143902 |
| Anti-mouse CD73 clone TY/11.8 | Biolegend | 127202 |
| Anti-FITC 160Gd clone FIT-22 | Fluidigm | 3160011B |
| Anti-mouse ITGB3 clone Cc9.G2 (HMB3-1) | Biolegend | 104302 |
| Anti-mouse CD34 clone MEC14.7 | Biolegend | 119302 |
| Anti-mouse ITGA6 clone GoH3 | Biolegend | 313602 |
| Anti-Biotin 165Ho clone 1D4-C5 | Fluidigm | 3165012B |
| Anti-mouse CD14 clone Sa14-2 | Biolegend | 123302 |
| Anti-mouse CD74 clone In1/CD74 | Biolegend | 151002 |
| Anti-mouse CD80 clone 16-10A1 | Biolegend | 104702 |
| Anti-mouse CD31 clone MEC13.3 | Biolegend | 102502 |
| Anti-mouse CD38 171Yb clone 90 | Fluidigm | 3171007B |
| Anti-mouse ITGB1 clone HMB1-1 | Biolegend | 102202 |
| Anti-mouse VCAM1 clone 429 (MVCAM.A) | Biolegend | 105702 |
| Anti-mouse CD45 175Lu clone 30-F11 | Fluidigm | 3175010B |
| Anti-APC 176Yb clone APC003 | Fluidigm | 3176007B |
| Anti-mouse MHCI clone 28-14-8 | Biolegend | 114502 |
| Anti-mouse MHCII 209Bi clone M5/114.15.2 | Fluidigm | 3209006B |
| Anti-mouse cytokeratin-7 clone RCK105 | Abcam | Ab9021 |
| Anti-mouse pan-cytokeratin clone C-11 | Biolegend | 628602 |
| Anti-mouse VIM 154Sm clone D21H3 | Fluidigm | 3154014A |
| Anti-mouse RFP clone 8E5.G7 | Rockland Inc | 200-301-379 |
| Anti-mouse DES clone Y66 | Abcam | ab271829 |
| Anti-mouse aSMA clone 1A4 | Abcam | ab240654 |
| Anti-human/mouse cleaved caspase-3 (CC3) clone D3E9 | Cell Signaling Technology | 9579 |
| Anti-human/mouse Ki67 clone So1A15 | Thermo Fisher | 14-5698-82 |
| Anti-mouse collagen-4 pAb | Abcam | ab6586 |
| Anti-mouse CD64 151Eu clone X54-5/7.1 | Fluidigm | 3151012B |
| Anti-mouse CD16/32 clone 93 | Biolegend | 101302 |
| Anti-mouse CD11b clone M1/70 | Biolegend | 101202 |
| Anti-mouse PDCA-1 clone 927 | Biolegend | 127002 |
| Anti-mouse CD68 clone FA-11 | Biolegend | 137002 |
| Anti-mouse Ly6G 141Pr clone 1A8 | Fluidigm | 3141008B |
| Anti-mouse Siglec-F clone E50-2440 | BD Biosciences | 552125 |
| Anti-mouse PD-L1 clone 10F.9G2 | Biolegend | 124302 |
| Anti-mouse F4/80 146Nd clone BM8 | Fluidigm | 3146008B |
| Anti-mouse CD3e clone 17A2 | BD Biosciences | 555273 |
| Anti-mouse CD19 149Sm clone 6D5 | Fluidigm | 3149002B |
| Anti-mouse CD1d clone 1B1 | Biolegend | 123502 |
| Anti-mouse CD11c clone N418 | Biolegend | 117302 |
| Anti-mouse XCR1 clone ZET | Biolegend | 148202 |
| Anti-mouse TCRb clone H57-597 | Biolegend | 109202 |
| Anti-mouse CD45 clone 30-F11 | Biolegend | 103102 |
| Anti-mouse CX3CR1 clone SA011F11 | Biolegend | 149002 |
| Anti-mouse CXCR2 clone SA044G4 | Biolegend | 149302 |
| Anti-mouse CSF1R clone AFS98 | Biolegend | 135502 |
| Anti-mouse CD40 clone HM40-3 | Biolegend | 102902 |
| Anti-mouse CD103 clone 2E7 | Biolegend | 121402 |
| Anti-mouse PD-L2 clone TY25 | Biolegend | 107202 |
| Anti-mouse VISTA clone MIH63 | Biolegend | 150202 |
| Anti-mouse SIRPa clone P84 | Biolegend | 144002 |
| Anti-mouse IL-4Ra clone I015F8 | Biolegend | 144802 |
| Anti-mouse CD206 169Tm clone C086C2 | Fluidigm | 3169021B |
| Anti-mouse CD49b 170Er clone HMa2 | Fluidigm | 3170008B |
| Anti-mouse CD80 171Yb clone 16-10A1 | Fluidigm | 3171008B |
| Anti-mouse CD86 172Yb clone GL1 | Fluidigm | 3172016B |
| Anti-mouse CD101 clone Moushi101 | Biolegend | Custom order |
| Anti-mouse NKp46 clone 29A1.4 | Biolegend | 137602 |
| Anti-mouse CD38 175Lu clone 90 | Fluidigm | 3175014B |
| Anti-mouse Ly-6C clone HK1.4 | Biolegend | 128002 |
| Anti-mouse CD24 clone M1/69 | Biolegend | 101802 |
| Anti-mouse Galectin-9 clone 9M1-3 | Thermo Fisher | 16-9116-85 |
| Anti-mouse iNOS 161Dy clone CXNFT | Fluidigm | 3161011B |
| Anti-mouse CXCR3 clone CXCR3-173 | Biolegend | 126502 |
| Anti-mouse GITR 143Nd clone DTA1 | Fluidigm | 3143019B |
| Anti-mouse CD69 145Nd clone H1.2F3 | Fluidigm | 3145005B |
| Anti-mouse TIGIT clone 1G9 | Biolegend | 142102 |
| Anti-mouse 4-1BB clone 17B5 | Biolegend | 106107 |
| Anti-mouse CD27 150Nd clone LG.3A10 | Fluidigm | 3150017B |
| Anti-mouse LAG3 clone C9B7W | Biolegend | 125202 |
| Anti-mouse CD8a 153Eu clone 53-6.7 | Fluidigm | 3153012B |
| Anti-mouse CTLA4 154Sm clone UC10-4B9 | Fluidigm | 3154008B |
| Anti-mouse CD4 clone RM4-5 | Biolegend | 100506 |
| Anti-mouse PD-1 159Tb clone 29F.1A12 | Fluidigm | 3159024B |
| Anti-mouse CD62L 160Gd clone MEL-14 | Fluidigm | 3160008B |
| Anti-mouse TIM3 162Dy clone RMT3-23 | Fluidigm | 3162029B |
| Anti-mouse CD49b clone HMa2 | Biolegend | 103501 |
| Anti-mouse OX40 clone OX-86 | Thermo Fisher | 14-1341-82 |
| Anti-mouse KLRG1 clone 2F1 | BD Biosciences | 562190 |
| Anti-mouse ICOS 168Er clone C398.4A | Fluidigm | 3168024B |
| Anti-mouse CD39 clone 24DMS1 | Thermo Fisher | 14-0391-82 |
| Anti-mouse SLAM clone TC15-12F12.2 | Biolegend | 115902 |
| Anti-mouse CD25 clone PC61 | Biolegend | 102002 |
| Anti-mouse CD127 174Yb clone A7R34 | Fluidigm | 3174013B |
| Anti-mouse TCRgd clone UC7-13D5 | Biolegend | 107502 |
| Anti-mouse GATA3 clone L50-823 | BD Biosciences | 558686 |
| Anti-mouse GZMB clone GB11 | Thermo Fisher | MA1-80734 |
| Anti-mouse TCF1 clone C63D9 | Cell Signaling Technology | 2203 |
| Anti-mouse EOMES clone Dan11mag | Thermo Fisher | 14-4875-82 |
| Anti-mouse TBET 161Dy clone 4B10 | Fluidigm | 3160010B |
| Anti-mouse FOXP3 165Ho clone FJK-16s | Fluidigm | 3165024A |
| Anti-RFP clone 8E5.G7 | Rockland Inc | 200-301-379 |
| Anti-human/mouse pMARPKAPK2 [T334] clone 27B7 | Cell Signaling Technology | 3007 |
| Anti-human/mouse pTAK1 [S412] | Cell Signaling Technology | 9339 |
| Anti-human/mouse pAMPKa [T172] clone 40H9 | Cell Signaling Technology | 2535 |
| Anti-human/mouse pPLCg2 [Y759] 144Nd clone K86-689.37 | Fluidigm | 3144015A |
| Anti-human/mouse pFAK [S910] clone K73-480 | BD Biosciences | Custom order |
| Anti-human/mouse pp90RSK [S380] clone D5D8 | Cell Signaling Technology | 12032 |
| Anti-human/mouse B-catenin 147Sm clone D10A8 | Fluidigm | 3147005A |
| Anti-human/mouse pSTAT4 [Y693] 148Nd clone 38/p-Stat4 | Fluidigm | 3148006A |
| Anti-human/mouse p4EBP1 [T37/T46] 149Sm clone 236B4 | Fluidigm | 3149005A |
| Anti-human/mouse pSTAT5 [Y694] 150Nd clone 47/Stat5 | Fluidigm | 3150005A |
| Anti-human/mouse pGSK3B [S9] clone D85E12 | Cell Signaling Technology | 5558 |
| Anti-human/mouse pAKT [S473] 152Eu clone D9E | Fluidigm | 3152005A |
| Anti-human/mouse pSTAT1 [Y701] 153Eu clone 58D6 | Fluidigm | 3153003A |
| Anti-human/mouse pSMAD1/5/9 [S463]/465]/[S463/465]/[S465/467] clone D5B10 | Cell Signaling Technology | 13820 |
| Anti-human/mouse p70S6K [T389] clone 1A5 | Cell Signaling Technology | 9206 |
| Anti-human/mouse pp38 [T180/182] 156Gd clone D3F9 | Fluidigm | 3156002A |
| Anti-human/mouse pSTAT3 158Gd clone Y705 | Fluidigm | 3158005A |
| Anti-human/mouse pMEK1/2 [S221] clone 166F8 | Cell Signaling Technology | 2338 |
| Anti-human/mouse pAKT [T308] clone D25E6 | Cell Signaling Technology | 13038 |
| Anti-human/mouse pSRC [Y418] clone SC1T2M3 | Thermo Fisher | 12-9034-82 |
| Anti-human/mouse pMKK3/6 [S189]/[S207] clone D8E9 | Cell Signaling Technology | 12280 |
| Anti-human/mouse cyclinB1 clone V152 | Cell Signaling Technology | 4135 |
| Anti-human/mouse IkBa 164Dy clone L35A5 | Fluidigm | 3164004A |
| Anti-human/mouse pCREB [S133] 165Ho clone 87G3 | Fluidigm | 3165009A |
| Anti-human/mouse pJAK2 [Y1007/1008] clone E132 | Abcam | ab219728 |
| Anti-human/mouse pERK1/2 [T202]/[Y204] 167Er clone D1314.4E | Fluidigm | 3167005A |
| Anti-human/mouse pIKKa/b [S176/180] clone 16A6 | Cell Signaling Technology | 2697 |
| Anti-human/mouse pSMAD2/3 [S465/467]/[S423/425] clone D27F4 | Cell Signaling Technology | 8828 |
| Anti-human/mouse pNFkBp65 [S536] clone 92H1 | Cell Signaling Technology | 3033 |
| Anti-human/mouse pMKK4 [S257] clone C36C11 | Cell Signaling Technology | 4514 |
| Anti-human/mouse pRelB [S552] clone D41B9 | Cell Signaling Technology | 5025 |
| Anti-human/mouse pPDK1 [S241] clone J66-653.44.22 | BD Biosciences | 558395 |
| Anti-human/mouse pRB [S807/S811] clone J112-906 | BD Biosciences | 558389 |
| Anti-human/mouse pS6 [S235/S236] (175Lu) clone N7-548 | Fluidigm | 3175009A |
| Anti-human/mouse pHH3 [S28] clone HTA28 | Biolegend | 641002 |
| Anti-GFP clone FM264C | Biolegend | 338002 |
| Anti-mouse CD90 APC clone G7 | Abcam | ab25322 |
| Anti-mouse CD105 Biotin clone MJ7/18 | Biolegend | 120404 |
| Anti-mouse ITGA1 PE clone HMa1 | Biolegend | 142604 |
| Anti-mouse CD26 FITC clone H194-112 | Biolegend | 137806 |
| Anti-mouse EpCAM FITC clone G8.8 | Biolegend | 118208 |
| Anti-mouse CD45 FITC clone 30-F11 | Biolegend | 103108 |
| Anti-mouse CD31 FITC clone MED13.3 | Biolegend | 102506 |
| Anti-mouse PDPN APC clone 8.1.1 | Biolegend | 127410 |
| Anti-mouse PDPN PE-Cy7 clone 8.1.1 | Biolegend | 127412 |
| Anti-mouse CD90 PE clone G7 | Abcam | ab24904 |
| Anti-mouse CD105 BV421 clone MJ7/18 | BD Biosciences | 562760 |
| Anti-mouse CD105 PE clone MJ7/18 | Biolegend | 120408 |
| Anti-mouse CD105 PE-Cy7 clone MJ7/18 | Biolegend | 120410 |
| Anti-mouse CD105 APC clone MJ7/18 | Biolegend | 120414 |
| Anti-mouse CD74 AF647 clone In1/CD74 | Biolegend | 151004 |
| Anti-mouse MHCII PE-Cy7 clone M5/114.15.2 | Biolegend | 107630 |
| Anti-mouse MHCII BV421 clone M5/114.15.2 | Biolegend | 107632 |
| Anti-mouse CD90 AF746 clone 5E10 | Biolegend | 328116 |
| Anti-mouse CD105 PE clone 43A3 | Biolegend | 323206 |
| Anti-human pan-Cytokeratinp pAb | Abcam | ab9377 |
| Anti-human PDPN clone D2-40 | Agilent Dako | M361901-2 |
| Anti-human CD105 clone 3A9 | CST | 14606 |
| Anti-human VIM clone D21H3 | CST | 5741 |
| Anti-mouse aSMA clone 1A4 | Sigma Aldrich | A5228 |
| Anti-GFP pAb | Abcam | ab13970 |
| Goat anti-chicken IgG pAb | Abcam | ab207998 |
| Bacterial and Virus Strains | ||
| NEB 5-Alpha Competent E. coli | New England BioLabs Inc. | C2987I |
| Biological samples | ||
| Human FFPE pancreatic ductal adenocarcinoma tumors | Manchester Cancer Research Centre (MCRC) Biobank | See Table S3 |
| Human FFPE colorectal adenocarcinoma tumors | Manchester Cancer Research Centre (MCRC) Biobank | See Table S7 |
| Human FFPE lung adenocarcioma tumors | Manchester Cancer Research Centre (MCRC) Biobank | See Table S7 |
| Human FFPE mammary invasive ductal carcinoma tumors | Manchester Cancer Research Centre (MCRC) Biobank | See Table S7 |
| Chemicals, peptides, and recombinant proteins | ||
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) | Thermo Fisher | 77720 |
| Yttrium chloride | Sigma Aldrich | 204919 |
| Lanthanum chloride | Sigma Aldrich | 203521 |
| 194-Pt monoisotopic cisplatin | Fluidigm | 201194 |
| 195-Pt monoisotopic cisplatin | BuyIsotope | Custom order |
| 196-Pt monoisotopic cisplatin | BuyIsotope | Custom order |
| 198-Pt monoisotopic cisplatin | Fluidigm | 201198 |
| 157Gd isotopically enriched gadolinium chloride | Trace Sciences | Quote |
| 105Pd isotopically enriched palladium nitrate | Trace Sciences | Quote |
| 106Pd isotopically enriched palladium nitrate | Trace Sciences | Quote |
| 108Pd isotopically enriched palladium nitrate | Trace Sciences | Quote |
| 110Pd isotopically enriched palladium nitrate | Trace Sciences | Quote |
| 113In isotopically enriched indium chloride | Trace Sciences | Quote |
| 115In isotopically enriched indium chloride | Trace Sciences | Quote |
| 5-iodo-2’-deoxyuridine (IdU) | Sigma Aldrich | 17125 |
| Sodium hydroxide (NaOH) | Sigma Aldrich | 757527 |
| Ammonium Acetate (NH4CH3CO2) | Sigma Aldrich | 372331 |
| Heparin Sodium Salt | Sigma Aldrich | H3393 |
| DMSO | Sigma Aldrich | D2650 |
| Sodium Azide | Sigma Aldrich | S8032 |
| Collagenase Type IV | Thermo Fisher | 17104019 |
| DNase1 | Sigma Aldrich | 10104159001 |
| Hyaluronidase | Sigma Aldrich | H3757 |
| Dispase II | Thermo Fisher | 17105041 |
| FOXP3 Fixation/Permeabilization Kit | Thermo Fisher | 00-5523-00 |
| 16% Paraformaldehyde (PFA) | Thermo Fisher | 28908 |
| EQ Four Element Calibration Beads | Fluidigm | 201078 |
| Cell-ID 125 uM Iridium Intercalator | Fluidigm | 201192A |
| 3% hydrogen peroxide | VWR | 23614.291P |
| Casein | Vector | SP5020 |
| Tris Buffer Saline with Tween 20 (TBST) | VWR | J77500.K8 |
| HyClone Antibiotic/Antimycotic | Fisher Scientific | 11536481 |
| Accutase Cell Detachment Solution | Sigma Aldrich | A6964 |
| Epitope Retrieval Solution 1 (ER1) | Leica Microsystems | AR9961 |
| Research Detection System 2 | Leica Microsystems | DS9777 |
| Bond Antibody Diluent | Leica Microsystems | AR9352 |
| EnVision HRP | Agilent | K4001/4003 |
| Premixed TSA520 Reagent | Perkin Elmer | FP1487001KT |
| Premixed TSA570 Reagent | Perkin Elmer | FP1488001KT |
| Premixed TSA650 Reagent | Perkin Elmer | FP1496001KT |
| 4′,6-diamidino-2-phenylindole (DAPI) | Thermo Fisher | 62248 |
| ProLong Gold Antifade Mountant | Thermo Fisher | P36930 |
| RBS Lysis Buffer | Biolegend | 420301 |
| 0.5 M EDTA | Thermo Fisher | 15575020 |
| Live/Dead Fixable Near-IR Dead Cell Stain Kit | Thermo Fisher | L10119 |
| Primocin | InvivoGen | ant-pm-1 |
| DMEM with glucose and L-glutamine | Thermo Fisher | 41966052 |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | A3294 |
| Fetal Bovine Albumin (FBS) | Thermo Fisher | 10270106 |
| Lipofectamine2000 | Thermo Fisher | 11668019 |
| Optimem Reduced Serum Media | Thermo Fisher | 31985070 |
| Polybrene | Sigma Aldrich | 107689 |
| Puromycin | Sigma Aldrich | P8833 |
| Polyethylenimine (PEI) | Sigma Aldrich | 764647 |
| Universal Mouse Reference RNA | Thermo Fisher | QS0640 |
| Reverse Transcription Buffer | Thermo Fisher | 18067017 |
| Mg2Cl2 | Thermo Fisher | R0971 |
| dNTP Mix | Thermo Fisher | R0191 |
| DL-Dithiothreitol (DTT) | Sigma Aldrich | 43815 |
| RNAse Inhibitor | Thermo Fisher | N8080119 |
| Random Hexamers | Thermo Fisher | N8080127 |
| Multiscribe Reverse Transciptase | Thermo Fisher | 4311235 |
| TaqMan Pre-Amp Master Mix | Applied Biosystems | 4391128 |
| RNAse-free water | Thermo Fisher | 10977035 |
| Assay Loading Reagent | Fluidigm | 85000736 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | 4304437 |
| GE Sample Loading Reagent | Fluidigm | 85000746 |
| Hygromycin B | Thermo Fisher | 10687010 |
| 1,4,7,10-tetraazacyclododecane-1,4,7-tris-acetic acid-10-maleimidoethylacetamide (mDOTA) | Macrocyclics | B-272 |
| Methanol | Fisher Scientific | 10767665 |
| Bis(2,2′-bipyridine)-4′-methyl-4-carboxybipyridine-ruthenium N-succinimidyl ester-bis(hexafluorophosphate) (ASCQ_Ru) | Sigma Aldrich | 96631 |
| Sodium Bicarbonate (NaHCO3) | Sigma Aldrich | 31437 |
| Phosphate-Buffered Saline (for in vivo) | Thermo Fisher | 10010056 |
| Growth Factor Reduced Matrigel | Corning | 356231 |
| Neutral Buffered Formalin | Genta Medical | BIB10L |
| Low pH Target Retrieval Buffer Ph6 | Agilent | S236984 |
| Vectastain Elite ABC HRP Kit | Vector | PK-6100 |
| 3,3'-diaminobenzidine (DAB) | Agilent | K3467 |
| Shandon Gill Haematoxylin | Thermo Fisher | 6765005 |
| Primary Cell P3 Nucleofector solution | Lonza | V4XP-3032 |
| Electroporation Enhancer Solution | Integrated DNA Technologies | Alt-R Cas9 Electroporation Enhancer, 2 nmol |
| PBS-based Antibody Stabilization Buffer | Candor Biosciences | 13150 |
| Maxpar water (for mass cytometry) | Fluidigm | 201069 |
| Phosphate-Buffered Saline (PBS) (for mass cytometry) | Fisher Scientific | 10091403 |
| Recombinant Cas-9 | Integrated DNA Technologies | 1081059 |
| Recombinant mouse TGFB1 | RnD Systems | 7666-MB-005 |
| Recombinant rat PDGF-BB | RnD Systems | 520-BB-050 |
| Recombinant mouse FGF2 | RnD Systems | 3139-FB-025 |
| Recombinant human/mouse Activin-A | RnD Systems | 338-AC-010 |
| Recombinant mouse BMP2 | RnD Systems | 355-BM-010 |
| Recombinant mouse BMP4 | RnD Systems | 5020-BP-010 |
| Recombinant mouse BMP9 | RnD Systems | 5566-BP-010 |
| Recombinant mouse MIF | Biolegend | 599504 |
| Recombinant mouse IFNg | PeproTech | 315-05 |
| Recombinant mouse TNFa | Peprotech | 315-01A |
| Recombinant mouse IL1a | RnD Systems | 400-ML-005 |
| Recombinant mouse IL1b | PeproTech | 211-11B |
| Recombinant mouse IL4 | PeproTech | AF-214-14 |
| Recombinant mouse IL13 | RnD Systems | 413-ML-005 |
| Recombinant mouse IL22 | RnD Systems | 582-ML-010 |
| Recombinant mouse LIF | PeproTech | 250-02 |
| Recombinant mouse BMP7 | RnD Systems | 5666-BP-010 |
| Recombinant mouse BMP10 | RnD Systems | 6038-BP-025 |
| Recombinant mouse IL6 | PeproTech | 216-16 |
| Critical commercial assays | ||
| MaxPar X8 Antibody Conjugation Kits (various metals) | Fluidigm | Mutiple e.g. 141Pr 201141A |
| Cell-ID 20-plex Pd Barcoding Kit | Fluidigm | 201060 |
| TruSeq Small RNA Library Kit | Illumina Inc. | 200-0012 |
| Agilent SureSelect Strand Specific RNA Library Prep Kit for Illumina Sequencing | Agilent | G9691B |
| Kapa Library Quantification Kit for Illumina Sequencing Platforms | Kapa Biosystems Inc. | KK4835 |
| Illumina HighSeq 500 High Output 1x75 bp Kit | Illumina Inc. | 200-24906 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| QIAGEN Plasmid Midi Kit | QIAGEN | 12145X4 |
| 96x96 Dynamic Array Chip | Fluidigm | |
| Deposited data | ||
| Mouse Pancreatic Tumor scRNA-seq dataset GSE129455 | Elyada et al. | GEO accession: GSE129455 |
| Human Pancreatic Tumor scRNA-seq dataset | Steele et al. | GEO accession: GSE155698 |
| Mouse in vitro Pancreatic Fibroblast (PaF) scRNA-seq raw data. | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse PDA CAF bulk RNA-seq dataset | This paper | GEO accession: GSE156985 |
| Mouse in vitro Pancreatic Fibroblast (PaF) bulk RNA-seq datset (various stimulations) | This paper | GEO accession: GSE157391 |
| Mouse in vitro Pancreatic Fibroblast (PaF) and Liver Fibroblast (LiF) bulk RNA-seq datset | This paper | GEO accession: GSE176057 |
| Mouse subcut CD105pos and CD105neg PaF co-transplant day 10 bulk RNA-seq dataset | This paper | GEO accession: GSE176056 |
| Mouse PDA mass cytometry dataset - Mesenchymal Stroma (S) panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse PDA mass cytometry dataset - Myeloid/NK/B (MNB) cell panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse PDA mass cytometry dataset - T cell panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse in vitro Pancreatic Fibroblast (PaF) mass cytometry dataset - Mesenchymal Stroma (S) panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse in vitro Pancreatic Fibroblast (PaF) mass cytometry dataset - Cell Signalling panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse multi-organ in vitro primary fibroblast mass cytometry dataset - Mesenchymal Stroma (S) panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Mouse multi-GEMM mass cytometry dataset - Mesenchymal Stroma (S) panel. Raw data | This paper | https://doi.org/10.5281/zenodo.4584773 |
| Experimental models: Cell lines | ||
| Mouse PDA B6KPC-TB32043 (PDA#1) | Gift from Dr. Kris Frese, CRUK MI, UK | |
| Mouse PDA B6KPC-TB32047 (PDA#2) | Gift from Dr. Kris Frese, CRUK MI, UK | |
| B6 CD105+ Pancreatic Fibroblasts (PaFs) | This paper | |
| B6 CD105- Pancreatic Firoblasts (PaFs) | This paper | |
| Phoenix Cells | ATCC (Pear et al., 1993) | Thermo Fisher |
| HEK293FT | Thermo Fisher | R70007 |
| Human Primary Pancreatic Fibroblasts (hPaFs) | Generon | H-6201 |
| B6 primary mouse embryonic fibroblasts (MEFs) | Generon | C57-6028 |
| Experimental models: Organisms/strains | ||
| Pdx1-Cre; KrasLSL-G12D/+;Trp53LSL-R172H/+(KPC) | Hingorani et al. (2005) | |
| Pdx1-Cre; KrasLSL-G12D/+;Trp53LSL-R172H/+; Rosa26LSL-tdRFP/LSL-tdRFP(RFP KPC) | Luche et al. (2007) | |
| C57BL/6JOIaHsd (B6) | Envigo | |
| NOD-scid.Il2rg-/- (NSG) | Charles River | |
| B6.Rag1-/- (RAG1) | Mombaerts et al. (1992) | |
| B6.Batf3-/-(BATF3) | Hildner et al. (2008) | |
| B6.Villin::CreER; KrasLSL-G12D/+; Trp53fl/fl; Rosa26Notch1icd/(KPN) | Jackstadt et al. (2019) | |
| MMTV-PyMT (MMTV-PyMT) | Guy et al. (1992) | |
| B6.KrasLSL-G12D/+; Trp53fl/fl (KP) | Jackson et al. (2001), Marino et al. (2000) | |
| B6.Tyr::CreER; BrafLSL-V600E/+ (BRAFV600E) | Dhomen et al. (2009) | |
| Oligonucleotides | ||
| RT-QPCR primer for Actb: forward, 5’-aaggccaaccgtgaaaagat-3’ | Sigma Aldrich | |
| RT- QPCR primer for Actb: reverse, 5’-gtggtacgaccagaggcatac-3’ | Sigma Aldrich | |
| RT- QPCR primer for Gapdh: forward, 5’-gggttcctataaatacggactgc-3’ | Sigma Aldrich | |
| RT-QPCR primer for Gapdh: reverse, 5’-ccattttgtctacgggacga-3’ | Sigma Aldrich | |
| RT-QPCR primer for Pgk1: forward, 5’-tacctgctggctggatgg-3’ | Sigma Aldrich | |
| RT-QPCR primer for Pgk1: reverse, 5’-cacagcctcggcatatttct-3’ | Sigma Aldrich | |
| RT-QPCR primer for Ppia: forward, 5’-gccaccctccctaactgc-3’ | Sigma Aldrich | |
| RT-QPCR primer for Ppia: reverse, 5’-gcgggctcctactagatggt-3’ | Sigma Aldrich | |
| RT-QPCR primer for Tbp: forward, 5’-ggcggtttggctaggttt-3’ | Sigma Aldrich | |
| RT-QPCR primer for Tbp: reverse, 5’-gggttatcttcacacaccatga-3’ | Sigma Aldrich | |
| RT-QPCR primer for Tubb4a: forward, 5’-gacctatcatggggacagtga-3’ | Sigma Aldrich | |
| RT-QPCR primer for Tubb4a: reverse, 5’-cggctctgggaacatagttt-3’ | Sigma Aldrich | |
| RT-QPRC primer for Acta2: forward, 5’-ctctcttccagccatctttcat-3’ | Sigma Aldrich | |
| RT-QPCR primer for Acta2: reverse, 5’-tataggtggtttcgtggatgc-3’ | Sigma Aldrich | |
| RT-QPCR primer for Col1a1: forward, 5’-caggcaagcctggtgaac-3’ | Sigma Aldrich | |
| RT-QPCR primer for Col1a1: reverse, 5’-aacctctctcgcctcttgc-3’ | Sigma Aldrich | |
| RT-QPCR primer for Ctgf: forward, 5’-tgacctggaggaaaacattaaga-3’ | Sigma Aldrich | |
| RT-QPCR primer for Ctgf: reverse, 5’-agccctgtatgtcttcacactg-3’ | Sigma Aldrich | |
| RT-QPCR primer for Fndc1: forward, 5’-tggtcctcaaggaacaaagtg-3’ | Sigma Aldrich | |
| RT-QPCR primer for Fndc1: reverse, 5’-ttctgcattcaacaccaagc-3’ | Sigma Aldrich | |
| RT-QPCR primer for Il6: forward, 5’-gctaccaaactggatataatcagga-3’ | Sigma Aldrich | |
| RT-QPCR primer for Il6: reverse, 5’-ccaggtagctatggtactccagaa-3’ | Sigma Aldrich | |
| RT-QPCR primer for Cxcl1: forward, 5’-gactccagccacactccaac-3’ | Sigma Aldrich | |
| RT-QPCR primer for Cxcl1: reverse, 5’-tgacagcgcagctcattg-3’ | Sigma Aldrich | |
| RT-QPCR primer for Ccl2: forward, 5’-catccacgtgttggctca-3’ | Sigma Aldrich | |
| RT-QPCR primer for Ccl2: reverse, 5’-gatcatcttgctggtgaatgagt-3’ | Sigma Aldrich | |
| RT-QPCR primer for Csf3: forward, 5’-ccaccttggacttgcttcag-3’ | Sigma Aldrich | |
| RT-QPCR primer for Csf3: reverse, 5’-ccacccctaggttttccatc-3’ | Sigma Aldrich | |
| sgRNA Scrambled non-targeting: 1, 5’-gcacuaccagagcuaacuca-3’ | Synthego | |
| sgRNA Scrambled non-targeting: 2, 5’-guacgucgguauaacuccuc-3’ | Synthego | |
| sgRNA Eng: 1, 5’-cucuuuc ugcgagaccugcu-3’ |
Synthego | |
| sgRNA Eng: 2, 5’-cggcugugaucuacagccug-3’ | Synthego | |
| sgRNA Eng: 3, 5’-ucaccc cuugugggguccac-3’ |
Synthego | |
| sgRNA H2Ab1: 1, 5’-ucucau ccacacagcuuauu-3’ |
Synthego | |
| sgRNA H2Ab1: 2, 5’-gaacc agcgcacuuugaucu-3’ |
Synthego | |
| sgRNA H2Ab1: 3, 5’-ugagg gccucuguccuggac-3’ |
Synthego | |
| sgRNA Cd74: 1, 5’-auuuc ggaagcuucaugcga-3’ |
Synthego | |
| sgRNA Cd74: 2, 5’-uuacuu ccuguaccagcaac-3’ |
Synthego | |
| sgRNA Cd74: 3, 5’-ugagg gccucuguccuggac-3’ |
Synthego | |
| sgRNA Cd80: 1, 5’-ggaca uggaaacuugaggag-3’ |
Synthego | |
| sgRNA Cd80: 2, 5’-cgucuu ucacaagugucuuc-3’ |
Synthego | |
| sgRNA Cd80: 3, 5’-uaagcucg cugggguuuuga-3’ |
Synthego | |
| Recombinant DNA | ||
| pBABE-puro SV40 LT plasmid | Addgene | 13970 |
| pCL-Eco plasmid | Addgene | 12371 |
| SFFV-eGFP plasmid | Harris et al. (2012) | |
| SFFV-mCherry plasmid | Harris et al. (2012) | |
| pCMV delta R8.2 packaging plasmid | Addgene | 12263 |
| pMD2.G envelope plasmid | Addgene | 12259 |
| pCSII-IRES2-hygro hTERT plasmid | Gift from Dr. Fernando Calvo, Institute of Cancer Research (ICR), UK | |
| Software and algorithms | ||
| FCS Normalization Tool | Fluidigm | |
| FCS Normalization Tool | Zunder et al. (2015) | |
| www.cytobank.org | Beckman Coulter | |
| Cytofkit2 | https://github.com/JinmiaoChenLab/cytofkit2 | |
| FlowSOM (Cytofkit2 implemnation) | Van Gassen et al. (2015) | |
| UMAP (Cytofkit2 implementation) | Becht et al. (2018) | |
| Qu-Path (v0.2.0-m9) | Bankhead et al. (2017) | |
| FASTQC tool (version 0.11.3) | https://github.com/s-andrews/FastQC | |
| STAR aligner (version 2.5.1b) | https://github.com/alexdobin/STAR | |
| Rsubread (version 1.28.1) | https://bioconductor.org/packages/release/bioc/html/Rsubread.html | |
| DESeq2 (version 3.10) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| Ingenuity Pathway Analysis (Dec 2020). Qiagen Digital Insights | https://digitalinsights.qiagen.com | |
| Seurat (version 3.1.5) | https://github.com/satijalab/seurat | |
| BioMark Real-Time PCR Analysis Software | Fluidigm | |
| GraphPad Prism software (version 7) | GraphPad Software Inc | |
| ComplexHeatmap | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html | |
| flowCore | https://www.bioconductor.org/packages/release/bioc/html/flowCore.html | |
| FlowJo (version 10.6.2) | BD LifeSciences | |
| Corrplot | https://github.com/taiyun/corrplot | |
| MCPcounter | https://github.com/ebecht/MCPcounter | |
| Other | ||
| GentleMACS Octo Dissociator | Miltenyi Biotech | |
| Helios Mass Cytometer | Fluidigm | |
| Super Sampler | Victorian Airship & Scientific Apparatus LLC | |
| BOND RX automated platform | Leica Microsystems | |
| VS120 microscope | Olympus Lifescience | |
| SCN400 | Leica microsystems | |
| BD FACS AriaIII | BD Biosciences | |
| BD LSRFortessa | BD Biosciences | |
| Vevo 3100 Imaging System | Fujifilm VisualSonics | |
| NextSeq 500 Sequencer | Illumina Inc. | |
| 4D-Nucleofector Core Unit | Lonza | |
| Nucleocuvette kit/strip | Lonza | V4XP-3032 |
| Nanodrop One/One Spectrophotometer | Thermo Fisher | |
| Luna Cell Counter | Logosbio | |
| Alpha 2-4 Benchtop Lyopholiser | MartinChrist Freeze Dryers | |
| C-tubes | Miltenyi Biotech | 130-096-334 |
| 50 kDa Microfilters | Merck Millipore | UFC505096 |
| 3 kDa Microfilters | Merck Millipore | UFC500324 |
| Braun Omnican 50 Insulin Syringe/Needles | VWR | 9151117 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Claus Jørgensen (claus.jorgensen@cruk.manchester.ac.uk)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Bulk RNA-seq data are avaliable at NCBI under GEO accession numbers GSE129455, GSE155698, GSE156985, GSE157391, GSE176056 and GSE176057. Mass cytometry and scRNA-seq data is available on Zenodo at https://doi.org/10.5281/zenodo.4584773.
This paper does not report original code
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
Experimental models
Animal models
Pdx1-Cre; KrasLSL-G12D/+; Trp53LSL-R172H/+ (KPC) mice (Hingorani et al., 2005); ‘RFP KPC’ mice were generated by crossing KPC and Rosa26LSL-tdRFP/LSL-tdRFP mice (Luche et al., 2007); B6.Villin::CreER; KrasLSL-G12D/+; Trp53fl/fl; Rosa26Notch1icd/+ (KPN) mice (Jackstadt et al., 2019); B6.KrasLSL-G12D/+; Trp53fl/fl (‘KP’) mice were generated by crossing B6.KrasLSL-G12D/+ mice (Jackson et al., 2001) and B6.Trp53fl/fl mice (Marino et al., 2000); B6.Tyr::CreER; BrafLSL-V600E/+ (BRAFV600E) mice (Dhomen et al., 2009) were generated by crossing B6.Tyr::CreER (Mercer et al., 2005) and B6.BrafLSL-V600E/+ mice (Yajima et al., 2006); MMTV-PyMT mice (Guy et al., 1992); B6.Rag1-/- mice (Mombaerts et al., 1992) and B6.Batf3-/- mice (Hildner et al., 2008). Animals were bred and maintained under pathogen-free conditions at University of Manchester and CRUK Beatson Institute (CRUK-BI), University of Glasgow. Female C57BL/6 (B6) mice were purchased from Envigo and used at ages specified for each experiment. Female NOD-scid.Il2rg-/- (NSG) mice were purchased from Charles River and used at 14 weeks of age. All animal experiments were performed under a UK Home Office License and in accordance with the ‘Animal (Scientific Procedures) Act of 1986’ under Project License Number 70/8745 and 70/8375 subject to review by the Animal Welfare and Ethical Review Body of Cancer Research UK Manchester Institute, University of Manchester (CRUK MI) and the University of Glasgow (UoG). Experiments are reported in accordance with Animal Research: Reporting of In Vivo Experiments (ARRIVE) 2.0 guidelines.
Human tissue samples
Formalin fixed paraffin embedded (FFPE) human pancreatic ductal adenocarcinoma, colorectal adenocarcinoma, mammary invasive ductal carcinoma and lung adenocarcinoma tumor samples were obtained with informed patient consent by the Manchester Cancer Research Centre (MCRC) Biobank in accordance with the Human Tissue Act 2004 (13_RIMA_04). The MCRC Biobank (ethics code: 18/NW/0092) is licensed by the Human Tissue Authority (license number: 30004) and is ethically approved as a research tissue bank by the South Manchester Research Ethics Committee (Ref: 07/H1003/161+5). The role of the MCRC Biobank is to distribute samples. For more information see www.mcrc.manchester.ac.uk/Biobank/Ethics-and-Licensing.
Cell lines and culture
To limit culture-induced phenotypic changes, all cells were used within one month of thawing and the same frozen batch used for all experiments when possible. All cell lines were regularly tested for mycoplasma infection. Primary pancreatic fibroblasts (PaFs) were expanded in vitro from the pancreas of 8-week-old female B6 mice, were never allowed to become confluent and were detached for splitting using Accutase Dissociation Solution. The murine PDA cancer cell lines used in this study (designated as ‘PDA’ and ‘PDA#2’ in the manuscript) are BL6KPC-TB32043 and BL6KPC-TB32047 and on a B6 background and were a kind gift from Dr. Kris Frese at CRUK MI. All cells were cultured in Cell Culture Media 10% (CCM(10)), consisting of 10% v/v FBS and 1% v/v HyClone Antibiotic/Antimycotic in DMEM with glucose and L-glutamine.
Methods details
Mass cytometry antibody conjugation
Supplier, clone and heavy-metal isotope tag of each mass cytometry antibody used in this study are listed in the Supplemental Information Table. Where possible targets were placed in higher sensitivity channels with minimal spill over from more abundant channels. The dedicated panel builder at dvssciences.com was used to estimate isotope and oxide spill-over and guide channel selection. Particular antibody clones were selected based on widespread use and extensive evidence of specific staining in the literature or from our own flow cytometry analysis. Where indicated antibodies were purchased pre-conjugated (Fluidigm). All other antibodies were purchased in carrier protein-free format and labelled with the indicated heavy-metal tag using Maxpar X8 Antibody Conjugation Kits (Fluidigm) (Han et al., 2018). 200 μg of each antibody was washed twice with 400 μL R buffer (Fluidigm) in a 50 kDa Microfilter (Merck Millipore, UFC505096) by centrifuging at 12,000 g at room temperature (RT) for 6 minutes. Antibodies were partially reduced using 200 μL of a 4 mM solution of tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Thermo Fisher, 77720) in R-buffer. After 25 min of TCEP reduction, antibodies were washed twice with C-buffer (Fluidigm). In parallel to antibody reduction, metal chelation was performed by adding 10ul of 50 μM lanthanide metal solutions to two equivalents of Maxpar X8 chelating polymer (Fluidigm) in 190 μL of L-buffer (Fluidigm) and incubating for 1 h at RT. The metal loaded polymers were washed once with L-buffer then once with C-buffer in 3 kDa Microfilters (Merck Millipore, UFC500324), by centrifuging at 12,000 g for 20 min. The metal loaded polymer in C-buffer was added to the partially reduced antibody and incubated at 37°C for 1.5 h. Conjugated antibodies were washed six times with W-buffer (Fluidigm), suspended in 100 μL W-buffer, vortexed and left for 5 min at RT before being reverse centrifuged into a fresh 1.6 mL collection tube. Protein content was assessed using a Nanodrop One/One Spectrophotometer (Thermo Fisher) and then 300 μL of PBS-based Antibody Stabilization Buffer (Candor Biosciences, 13150) containing 0.6 mg/mL sodium azide (Sigma Aldrich, S8032) was added and the conjugated antibodies stored at 4°C. To generate cisplatin conjugates, 200 μg of antibody was reduced as described above and incubated with 200 μL of 400 μM monoisotopic cisplatin (BuyIsotope, custom order) in C-buffer at 37°C for 1.5 h and washed and stored as for the polymer/lanthanide conjugates. For any antibody that showed low final protein content (<40% recovery), the process was repeated but with a 10 min TCEP reduction. If significant degradation was still observed, an alternative antibody clone was tested or the target was not included in the panel. Antibodies were titrated in panels by staining samples of known positive and negative controls. See Supplemental Information Table for Panels.
Tumor disaggregations
Buffers and reagents used in tissue processing and cell staining were checked for heavy-metal ion contamination, particularly barium contamination, and buffers were made up in non-glass containers that had not been detergent washed. 5-iodo-2’-deoxyuridine (IdU) (Sigma Aldrich, 17125) was solubilized overnight at RT under mixing, in a minimally basic solution of 0.01 M sodium hydroxide (NaOH) (Sigma Aldrich, 757527) in water, to 10 mg/mL concentration and filtered through a 0.22 μm pore mesh. To label cells in S-phase for mass cytometry studies, mice were injected intraperitoneally with 200 μL of IdU solution 2 h before the mouse was culled by Schedule 1 method and tissues collected. Tumor samples were quickly transferred into ice-cold phosphate-buffered saline (PBS) (Fisher Scientific, 10091403) on ice. All non-tumor tissue that was attached to the outer edge of the denser tumor core was removed, the surface of the tumor samples was carefully dried with sterile paper and tumor weight recorded. Samples were washed once with ice-cold RPMI media and minced with disposable scalpels in 2 mL of disaggregation buffer (DB), consisting of 2 mg/mL Collagenase Type IV (Thermo Fisher, 17104019), 1 mg/mL DNase1 (Sigma Aldrich, 10104159001) and 0.5 mg/mL Hyaluronidase (Sigma Aldrich, H3757) in RPMI. Tumor pieces less than 3 mm in length were transferred to a C-tube (Miltenyi Biotech, 130-096-334) and a further 3 mL of DB added. If tumors were >600 mg, the disaggregation was carried out in two C-tubes, each with a total of 5 mL DB. The C-tube was placed in a GentleMACS Octo Dissociator (Miltenyi Biotech), heating blocks fitted and tumors disaggregated using the automated 37C_m_TDK1 program. Once complete, the C-tube was centrifuged at 100 g for 2 min, to ensure the contents were gathered at the bottom of the tube. The sample was diluted with a further 5 mL of fresh and warmed DB, mixed well by pipetting and filtered into a 50 mL tube through a 70 μm strainer, which was then washed with 10 mL ice-cold RPMI to quench the digestion. The single cell suspension was pelleted at 300 g for 6 min and used for mass or flow cytometry staining.
Mass cytometry live/dead and extracellular staining
All cell pelleting was conducted using a swinging bucket centrifuge, with the braking speed reduced to avoid disruption to the cell pellet. Aspirations were done carefully and always left at least 50 μL of void volume above the pellet. Live cells were spun at 300 g for 6 min and fixed cells spun at 1000 g for 6 min. Particular care was taken during PBS-only washes to ensure that cells had pelleted completely. The disaggregated tumor cell pellet was resuspended in 300 μL of ice-cold PBS, vortexed well and 300 μL of 1 μM 198Pt monoisotopic cisplatin (Fluidigm, 201198) in PBS added, followed by vortexing. After exactly 1 min incubation, the staining was quenched with 20 mL of CSM-E (Cell Staining Buffer – Extracellular) consisting of 5 mg/ml Bovine Serum Albumin (BSA) (Sigma Aldrich, A3294), 0.5% v/v Fetal Bovine Albumin (FBS) (Thermo Fisher, 10270106) and 0.2 mg/mL DNAse1 in PBS. The cells were resuspended and counted using a Luna Cell Counter (Logosbio) on fluorescence mode. The propidium iodide and acridine orange staining allows for improved cell counting of disaggregated tissues compared with trypan blue-based methods. 3x106 cells were aliquoted into a 5 mL polypropylene FACS tube, washed with 3 mL CSM-E and pelleted. 20 μL of 100 U/mL heparin sodium salt (Sigma Aldrich, H3393) solution in PBS and 1 μL Fc block (BD Biosciences, 558636) was added. The contents were mixed by gentle rocking but no vortexing and incubated on ice for 5 min. A master mix of fluorophore-conjugated antibodies (see Supplemental Information Table) in 50 μL CSM-E was added, mixed by gentle rocking and incubated on ice in the dark. After 20 min the mixture was vortexed. After a total incubation of 45 min, the cells were washed once with 4 mL of CSM-E. A master mix of extracellular targeting, metal-conjugated antibodies (see Supplemental Information Table) in 50 μL of CSM-E was added, mixed by gentle rocking and incubated on ice in the dark. After 20 min the mixture was vortexed. After a total incubation of 45 min, the cells were washed twice with 4 mL of CSM-E. The cell pellet was resuspended in 100 μL of PBS and vortexed and 1 mL of FOXP3 Fixation/Permeabilization Kit (Thermo Fisher, 00-5523-00), 1x FOXP3 Fixation Buffer added, followed by thorough vortexing. After 30 min incubation at RT, 2 mL of 1x FOXP3 Permeabilization Buffer was added and the cells pelleted. The cell pellet was resuspended in 1 mL of 10% v/v DMSO (Sigma Aldrich, D2650) in CSM-I (Cell Staining Buffer – Intracellular), consisting of 5 mg/ml BSA and 0.2 mg/ml sodium azide in PBS, vortexed and frozen at -80°C. For staining the sample with the Myeloid/NK/B cell (MNB) panel, no extracellular Fc block was used. Instead, the cells were incubated with heparin solution for 5 min, followed by metal-conjugated anti-CD64 antibody for 10 min on ice, followed by metal-conjugated anti-CD16/32 antibody for 5 min on ice, before adding the remaining master mix of extracellular antibodies. This ensured strong metal labelling of Fc-receptors, which contributed to accurate sub-setting of the mononuclear phagocyte lineage.
Mass cytometry barcoding and intracellular staining
Once the samples for an entire study had been collected, all the frozen aliquots were allowed to thaw at RT and washed once with 4 mL PBS. Each of the cell pellets were resuspended in a unique barcoding aliquot from the Cell-ID 20-plex Pd Barcoding Kit (Fluidigm, 201060) in 1 mL of cold PBS, vortexed and incubated at RT for 15 min. After the incubation, the mixtures were diluted in 3 mL of CSM-I, pelleted and washed once more with 4 mL CSM-I. Each of the cell pellets from the samples to be included in the study were resuspended in 200 μL of 1x FOXP3 Permeabilization Buffer, pooled into a 5 mL polypropylene FACS tube and pelleted. For each sample included in the pooled sample, 10 μL of 100 U/mL heparin sodium salt in PBS and 0.5 μL of Fc block was added and the sample mixed by gently rocking. After incubating for 5 min at RT in the dark, a master mix of intracellular targeting, metal-conjugated antibodies (see Supplemental Information Table) in CSM-I was added. For each sample included in the pooled sample, one equivalent of antibody and 25 μL of CSM-I was used and scaled up as required. The sample was mixed by gentle rocking and incubated on ice in the dark. After 20 min the mixture was vortexed. After a total incubation of 45 min the cells were washed twice with 4 mL of CSM-I. The cell pellet was resuspended in 1 mL of PBS and vortexed well. For every individual sample included in the pooled sample, a minimum of 500 μL of 4% Paraformaldehyde (PFA) (Thermo Fisher, 28908) in PBS was added to ensure complete fixation, using larger tubes as needed. If during sample acquisition, the heavy-metal markers are seen to ‘streak’, this is an indication the cells were not sufficiently fixed at this stage. The sample was vortexed and stored overnight at 4°C in the dark.
Mass cytometry DNA staining and acquisition
On the day of acquisition, 0.5 μL of 125 μM of Cell-ID Iridium Intercalator (Fluidigm, 201192A) was added per individual sample included and vortexed well. After 1 h of incubation at RT the cells were washed once with PBS and aliquoting to allow staggered acquisition. Typically, a pooled sample containing >15 individual samples was stored as x4 cell pellets, each prepared just before acquisition. Each cell pellet was washed twice with water and resuspended at a concentration of 1x106 cells/mL in 15% EQ Four Element Calibration Beads (Fluidigm, 201078) in water, filtered twice through 70 μm Filcons (BD Biosciences, 340633) and acquired on a Helios Mass Cytometer (Fluidigm), using a Super Sampler (Victorian Airship & Scientific Apparatus LLC) to improve the consistency of sample delivery. The sample was acquired at a maximum of 500 events/s and sample lines and nebulizers were replaced each time an additional 5x106 events had been recorded.
Mass cytometry data processing
FCS files were normalized for signal-drift using the built-in Helios normalization tool (Fluidigm) and individual sample events deconvoluted using either the debarcoder (Fluidigm) or a stand-alone debarcoder (Zunder et al., 2015) with a Mahalanobis distance of 10 and 15 respectively and a minimum barcode separation of 0.26 for both. FCS files were uploaded to the cloud-based cytometry platform Cytobank (www.cytobank.org, Beckmann Coulter) and checked for consistent signal across the entire acquisition period, as well as clean and correct barcode deconvolution. Live cell events were selected based on DNA-191Ir positivity and cisplatin-198Pt negativity. Because samples were stained with cisplatin separately, this gating step was conducted using sample tailored gating. 191Ir+ debris and cell doublets and aggregates were removed based on event length. If possible, target cells were selected by manual biaxial gating: MNB cell events were selected as CD45+CD3ε- and T cell events selected as CD45+CD3ε+. Target cells were exported as FCS files and uploaded to the Cytofkit2 package (version 2.0.1). Cells were clustered using FlowSOM (Van Gassen et al., 2015) and visualized using UMAP projections and expression overlays (Becht et al., 2018), exporting cell data with annotated clusters for further downstream analysis. For target cells that consist of cell populations that are difficult to separate cleanly from non-target cells by simple biaxial gating, such as tumor mesenchymal stromal populations, an initial analysis of high-dimensional clustering and visualization was carried out which allowed use of the full dataset to cluster and annotate events. Clusters of target cells were exported and then re-uploaded for further analysis. Three KPC tumors could not be weighed (mouse #16,17 and 18) and one mouse did not receive an IdU injection (mouse #16), so this data is not present in the respective analysis. KPC mouse #19 was only stained using the mesenchymal stroma (S) antibody panel and not the MNB and T cell panels, which gave n=18 KPC PDA tumors in which each sample was stained with all three antibody panels.
Mouse organ disaggregations
Primary fibroblast/fibroblast-like cells were expanded from the following mouse organs: pancreas, colon, small intestine, mammary tissue, shaved back skin, stomach, mesentery adipose tissue, spleen, thymus, lungs, liver, kidneys, bladder, esophagus and heart. The entire hind legs were collected and bone marrow processed separately (see below). Unless otherwise stated, tissues were isolated from female 8-week-old B6 mice. The prostate was isolated from 8-week-old male B6 mice. The number of organs required for successful fibroblast expansion from each tissue is detailed in the Supplemental Information Table. All tissues were transferred to ice-cold sterile PBS on ice. The stomach, small intestine and colon were flushed clear with PBS. Some tissues were processed manually and others were disaggregated using a GentleMACS dissociator (See Supplemental Information Table). Previous experiments had indicated which method yielded the most fibroblasts from each tissue (for a full list of methods used, see Supplemental Information Table). Each tissue was transferred to a 10 cm cell culture dish and washed once with ice-cold RPMI. For some specific tissues, DB was supplemented with 0.5 mg/mL Dispase II (Thermo Fisher, 17105041) to aid disaggregation (noted in Supplemental Information Table). For manual disaggregation, 3 mL of RT DB was added and the tissue minced using disposable scalpels, with a further 17 mL of DB added once pieces were below 3 mm and incubated at 37°C for 25 min. The cells, tissue fragments and buffer were then transferred to a centrifuge tube and the contents allowed to settle for ∼10 s. The settled tissue pieces were transferred to a separate centrifuge and repeatedly mixed to break up the fragments. The contents of both centrifuge tubes were combined and quenched with 20 mL of cell culture media (CCM), consisting of 20% v/v FBS, 1% v/v HyClone Antibiotic/Antimycotic (Fisher Scientific, 11536481) and 0.2% v/v Primocin (InvivoGen, ant-pm-1) in DMEM with glucose and L-glutamine (Thermo Fisher, 41966052). Cells and remaining tissue fragments were centrifuged at 300 g for 6 min and resuspended in 40 mL CCM and transferred to a 225 cm2 culture flask. Methods for GentleMACS tissue disaggregations were specific for each tissue (listed in Supplemental Information Table). Once the program was complete the C-tube was centrifuged at 100 g for 2 min, to ensure contents were gathered at the bottom of the tube. The sample was diluted with a further 15 mL of fresh DB, mixed by pipetting and quenched with 20 mL CCM. Cells and remaining tissue fragments were centrifuged at 300 g for 6 min and resuspended in 40 mL CCM and plated into a 225 cm2 flask. For isolation of bone marrow stromal cells, muscle was removed from each pair of intact tibias and fibias, the ends of the bones cut with a disposable scalpel and the bone marrow flushed out with 5 mL DB using a needle and syringe. The combined bone marrow extracts were vortexed to break up clumps, quenched with 20 mL CCM, pelleted, resuspended in 40 mL CCM and transferred to a 225 cm2 culture flask. Frozen B6 primary mouse embryonic fibroblasts (MEFs) (Generon, C57-6028) were thawed and resuspended in 40 mL CCM and transferred to a 225 cm2 culture flask. All primary cell cultures were grown in an incubator at 37°C with 5% CO2, humidified air. Media was carefully replaced at 24 h and 48 h, taking care not to dislodge attached tissue fragments. Primary fibroblast isolations were used when the cells reached ∼50% confluence, which varied between 6-15 d (Supplemental Information Table). For isolations to be analyzed by mass cytometry, cells were lifted by aspirating media, washing with sterile PBS (Thermo Fisher, 10010023) and incubating with 10 mL of Accutase Cell Detachment Solution (Sigma Aldrich, A6964) at 37°C for 10 mins. The dissociation buffer was quenched with 30 mL CCM and the cells allowed to settle in the same flask (without washing out the dissociation buffer). This step removes many non-fibroblast cell types that do not survive detachment well. The following day 40 μL of 10 mM IdU solution in 0.2 M NaOH/water was added directly to the media, mixed by swirling and the cells incubated at 37°C for 20 min. Media was aspirated, cells lifted with 10 mL Accutase Cell Detachment Solution, diluted with 20 mL PBS and centrifuged at 300 g for 6 min at 4°C. The cell pellet was resuspended in 100 μL PBS, vortexed and 100 μL of 1 μM 198Pt cisplatin in PBS added, followed by vortexing. After exactly 1 min incubation, the staining was quenched with 10 mL of CSM-I, cells pelleted, resuspended in 2 mL CSM-I, counted and 3x106 cells aliquoted into a 5 mL polypropylene FACS tube. The remaining staining, acquisition and analysis steps were as described above for the ex vivo analysis.
Mass cytometry cell signaling panel generation
Supplier, clone and heavy-metal isotope tag of each antibody used in the cell signaling mass cytometry analysis are listed in the Supplemental Information Table. Where indicated antibodies were purchased pre-conjugated (Fluidigm). All other antibodies were labelled with the indicated metal tag using the Maxpar X8 antibody conjugation kit (Fluidigm), as described above. Cell signaling antibodies were titrated in panels against in vitro cell lines stimulated with recombinant cytokines and growth factors. Antibody clones were prioritized based on extensive prior validation (Kumar et al., 2020; Lun et al., 2017, 2019; Rapsomaniki et al., 2018) or confirmation of expected signal node activation during the antibody titration step. A custom 6-choose-3 barcode scheme using enriched isotopes of 105Pd, 106Pd, 108Pd, 110Pd, 113In and 115In (Trace Sciences) was generated using established methodology (Zunder et al., 2015). Stocks of 10 mM palladium/indium salt solutions in L-buffer were diluted 1:10 in 20 mM ammonium acetate (NH4CH3CO2) (Sigma Aldrich, 372331). 127 μL of these 1 mM palladium/indium nitrate solutions were added to 2 mg of 1,4,7,10-tetraazacyclododecane-1,4,7-tris-acetic acid-10-maleimidoethylacetamide (mDOTA) (Macrocyclics, B-272) in a 1.5 mL polypropylene tube giving a 2:1 ratio of chelator:metal. Volumes were adjusted based on the accurate weight of mDOTA. After 1 min of vortexing the tube was snap frozen in dry ice/ethanol bath and stored at -80°C. Tubes were opened and lyophilized overnight in a cooled Alpha 2-4 Benchtop Lyopholizer (MartinChrist Freeze Dryers), working quickly to ensure the contents did not thaw before being desiccated. The resulting powder was dissolved to 10 mM in dry DMSO (Sigma Aldrich, D2650) and an aliquot diluted 5000x to give a 2 μM working stock. This was titrated against PFA fixed and methanol permeabilized in vitro cells, to mimic final assay conditions as closely as possible. Once an optimal dilution for each of the six barcodes was found 1:1:1 mixtures were generated in a 6-choose-3 barcode scheme (Zunder et al., 2015) and each of the 20 possible combinations was titrated to ensure optimal staining, before being aliquoted and stored at -20°C.
Mass cytometry signaling analysis
1.5x106 mCherry+ CD105+ pancreatic fibroblasts and 1.5x106 GFP+ CD105- pancreatic fibroblasts were combined in 30 mL of reduced-serum cell culture media (CCM(0.5)), consisting of 0.5% v/v dialyzed FBS (dFBS) (Thermo Fisher, 26400044) and 1% v/v HyClone Antibiotic/Antimycotic in DMEM with glucose and L-glutamine, and plated into a 225 cm2 cell culture flask. After 24 h, 40 μL of 10 mM IdU solution in 0.2 M NaOH/water was added directly to the media, mixed well and the cells incubated at 37°C for a further 20 min. The media was aspirated and replaced with 28 mL of warmed CCM(0.5) containing cytokine or growth factors, as detailed in the Supplemental Information Table. The conditions consisted of x1 no stimulation control and x19 recombinant cytokine or growth factor stimulations. After exactly 5 min of stimulation at 37°C, 4 mL of 16% PFA was added to the media, to give a final PFA concentration of 2%, and immediately swirled over the cells to fix. After 30 min of fixation, the media was aspirated, and the attached cells washed with PBS, CSM-I, and then PBS. 15 mL of Accutase Cell Detachment Solution was added and the flasks incubated at 37°C for 15 min. Because buffer-mediated detachment is less efficient for fixed cells, a cell scraper was used to further detach the cells from the flask and collected in a centrifuge tube and diluted with 20 mL PBS. Cells were centrifuged at 1000 g for 6 min with reduced braking, aspirated and resuspended in 2 mL PBS, vortexed and permeabilized by slowly adding 5 mL of -20°C methanol (Fisher Scientific, 10767665) with vortexing, followed by incubation at -20°C for 20 min. The methanol-permeabilized cells were diluted with 10 mL PBS and then a further 10 mL CSM-I and pelleted. Cells were resuspended in PBS, counted and a maximum of 3x106 cells aliquoted into separate 5 mL polypropylene FACS tubes. After washing with 4mL of PBS and resuspending the cell pellets in void volume, one aliquot of each unique 6-choose-3 barcode dissolved in 1mL of ice-cold PBS was added to each sample and vortexed. Once a 30 min incubation at RT was complete, the cells were washed twice with 4 mL CSM-I, pooled into a single 5 mL polypropylene FACS tube in CSM-I and pelleted. The 20-sample pooled cell pellet was resuspended in the void volume and 200 μL of 100 U/mL heparin sodium salt solution and 10 μL of Fc block added. After 5 min at RT a master mix containing 20 equivalents of each antibody from the cell signaling panel (Supplemental Information Table) in 500 μL of CSM-I was added and vortexed. After staining for 2h at RT with regular vortexing, the sample was washed three times with 4 mL CSM-I and resuspended in 1 mL PBS, transferred to a larger centrifuge tube and fixed in 10 mL of 4% PFA in PBS. The sample was vortexed and stored at 4°C in the dark overnight. After the overnight fixation, the PFA/PBS was washed out with PBS and the cells incubated in 1mL of 100 ug/mL bis(2,2′-bipyridine)-4′-methyl-4-carboxybipyridine-ruthenium N-succinimidylester-bis(hexafluorophosphate) (ASCQ_Ru) (Sigma Aldrich, 96631) in 0.1 M sodium bicarbonate (NaHCO3) (Sigma Aldrich, 31437) solution for 1h at RT, before continuing with the PBS and water washes and acquisition, as described above.
Multiplexed immunofluorescence
Multiplexed Tyramide Signal Amplification (TSA) immunofluorescence staining was performed using the BOND RX automated platform (Leica Microsystems). 4um sections of FFPE tumors were cut and mounted on charged slides. Dewaxing and heat induced epitope retrieval of slides was automated on the Bond RX, using Epitope Retrieval Solution 1 (ER1) (Leica Microsystems, AR9961) for 20 min at 100°C. Using the Research Detection System 2 (Leica Microsystems, DS9777), endogenous peroxidase was blocked using 3% v/v hydrogen peroxide (VWR, 23622.260) in Tris Buffer Saline with Tween 20 (TBST) (VWR, J77500.K8) for 10 min and the slides further blocked with 10% w/v casein (Vector, SP5020) in TBST. Antibody application, detection and TSA amplification was conducted in three sequential rounds following the same general procedure: incubation with the primary antibody in Bond Antibody Diluent (Leica Microsystems, AR9352) for 30 min, followed by detection using EnVision HRP (Agilent, K4001/4003) for 30 min, followed by a specific premixed TSA reagent (Perkin Elmer) at 1/200 for 30 min. Antibody sequence and TSA-fluorophore selection were optimized to reduce non-specific staining and tyramide binding site competition. The first staining round used mouse anti-human CD105 antibody (CST clone 3A9) at 1/200 and TSA570 (FP1488001KT). The second round used rabbit anti-human pan-CK antibody (Abcam ab9377) at 1/200 and TSA520 (FP1487001KT). The third round used mouse anti-human podoplanin antibody (Dako cloneD2/40) at 1/100 or anti-human VIM antibody (CST clone D21H3) at 1/500 and TSA650 (FP1496001KT). Following labelling with TSA, antibodies were removed using a heat stripping step in ER1 for 10 min at 100°C. This was not applied following application of the third antibody. Finally, nuclei were counterstained with 0.33 ug/ml 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher, 62248) for 15 min and coverslipped with ProLong Gold Antifade Mountant (Thermo Fisher, P36930). Slides were scanned using a VS120 microscope (Olympus Lifescience) at 20x and analyzed using QuPath (v0.2.0-m9) (Bankhead et al., 2017).
FACS and flow cytometry
To isolate CD105+/- CAFs directly from PDA tumors, single cell suspensions were prepared as described above for analysis by mass cytometry. Red blood cells (RBCs) were lysed using 5 mL of ice-cold 1x RBS Lysis Buffer (Biolegend, 420301) for 2 min on ice. The lysis was quenched with 20 mL FACS buffer (FB), consisting of 2% v/v FBS and 2 mM ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher, 15575020) in PBS and pelleted by centrifugation at 300 g for 6 min with reduced braking. Cells were counted using a Luna Cell Counter on fluorescence mode, washed once with 20 mL PBS and stained with Live/Dead Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher, L10119), using 0.25 μL of reagent in 0.5 mL of ice-cold PBS per 1x106 cells. After 20 min on ice, the staining was quenched with 20 mL FB and cells pelleted. 0.25 μL of Fc block per 1x106 cells was added to the void volume and cells gently mixed. After 5 min on ice, a master mix containing anti-EpCAM-FITC, anti-CD45-FITC, anti-CD31-FITC, anti-PDPN-APC (all Biolegend), anti-CD90-PE (Abcam) and anti-CD105-BV421 (BD Biosciences) was added at 0.25 μL of each antibody in 20 μL FB per 1x106 cells. Cells were vortexed, stained on ice in the dark for 45 min, washed once with 20 mL FB, resuspended to 5x106 cells/mL, filtered through 70 μm Filcons into 5 mL polypropylene FACS tubes and sorted on a BD FACS AriaIII (BD Biosciences) using the gating strategy described in the manuscript. FACS sorted CD105+ and CD105- fibroblasts in CCM were centrifuged, aspirated and cells lysed in RLT buffer (QIAGEN) and RNA isolated using a RNeasy Micro Kit (QIAGEN, 74004), according to the manufacturer’s instructions. For flow cytometry analysis or FACS of in vitro cells (e.g. isolation and surface marker analysis of CD105+/- pancreatic fibroblasts), a similar protocol was used without the RBC lysis and dead cell staining steps, and alternative fluorophore conjugates were applied. For a full list of antibodies used for flow cytometry/FACS see Supplemental Information Table. For flow cytometry/FCAS analysis all samples were analyzed on a BD LSRFortessa (BD Biosciences). Flow cytometry/FACS plots were generated in Cytobank.
Bulk RNA sequencing and analysis
RNA was isolated from FACS purified cells or whole tumor lysates at timepoints indicated in the manuscript. Indexed PolyA libraries were prepared using 50 ng of total input RNA and 16 cycles of amplification with the Agilent SureSelect Strand Specific RNA Library Prep Kit for Illumina Sequencing (Agilent, G9691B). Libraries were quantified by qPCR using a Kapa Library Quantification Kit for Illumina Sequencing Platforms (Kapa Biosystems Inc., KK4835). Paired-end 75 base-pair sequencing was carried out by clustering 1.9-2.0 pM of the pooled libraries on a NextSeq 500 Sequencer (Illumina Inc.) Pre-alignment quality control was performed using the FASTQC tool (version 0.11.3). Raw sequencing reads were aligned to the mouse reference genome GRCm38/mm10 using STAR aligner (version 2.5.1b) and gene annotation was taken from Ensembl build 92. Read counts were determined by using the featureCounts function from the Bioconductor package Rsubread (version 1.28.1). For analysis of pancreatic fibroblast transcriptional response to recombinant protein stimulations, a similar protocol was applied but only single-end reads were measured. Differential gene expression analysis was performed using the Bioconductor package DESeq2 (Love et al., 2014). For the ex vivo KPC CAF analysis, a gene was called as significantly differentially expressed if its abundance changed more than 2-fold between populations of interest, with a Benjamini-Hochberg(BH)-adjusted p-value <0.05. For the in vitro pancreatic fibroblast stimulation analysis, batch effect correction was performed using DESeq2 as recommended by DESeq2 workflow guidelines. A gene was considered as differentially expressed if BH-adjusted p-value <0.05 between stimulation and baseline conditions. No fold change cut-off was applied. For single gene expression comparisons, values were calculated either as TPM or scaled/normalized expression values directly from the DEseq2 analysis. For TGFB and IL1 receptor and signaling mediator gene expression comparisons, read counts from the baseline and stimulation conditions were combined for comparison of gene expression between CD105+ and CD105- fibroblasts (these genes were not significantly differentially expressed between baseline and stimulation samples). Ingenuity Pathway Analysis (Qiagen) was conducted according to manufacturer's recommendations. MCPcounter analysis was performed by converting gene names to the human annotation and using standard paramters (Becht et al., 2016).
scRNA-seq re-analysis
Single-cell mRNA sequencing (scRNA-seq) data containing mouse PDA mesenchymal cells was obtained from a publicly available dataset (Elyada et al., 2019), available at Gene Expression Omnibus (GEO) under the accession number GSE129455. Pre-processing steps, analysis and visualizations were done using the R package Seurat (Butler et al., 2018). The downloaded data is already normalized by log-normalization. The built-in function ScaleData was implemented to centralize expression of each gene, to shift the mean values to 0 and scale the variance from -1 to 1. Then according to best practices, we allocated 2000 of the most variable genes using the built-in function FindVariableFeatures. Based on the obtained variable genes, principal component analysis (PCA) was implemented by the function RunPCA. We estimated 50 principal components for each cell and, after performing an elbow test (function ElbowPlot) and JaskStraw estimation (functions JackStraw, ScoreJackStraw and JackStrawPlot), selected the top 20 principal components for further analysis. For cell clustering, Seurat's graph-based k-Nearest Neighbors (kNN) approach was implemented (functions FindNeighbors and FindClusters). Dimension 20 and resolution 0.5 were used as clustering parameters. UMAP method was used for data visualization (function RunUMAP). Scatter plot visualizations were done by Seurat's functions DimPlot and FeaturePlot. A similar workflow was used to analyze the Steele et al. human pancreas and PDA dataset (Steele et al., 2020) Fibroblast clusters were enriched as outlined in Figure S3E
scRNA-seq of in vitro pancreatic fibroblasts
scRNA-seq on in vitro pancreatic fibroblasts was performed at Hubrecht single Cell Genomics according to an adapted version of the SORT-seq protocol (Muraro et al., 2016) with primers as previously described (Van den Brink et al., 2017). The pancreas from 3 8-week-old female B6 mice was disaggregated and the fibroblasts expanded in vitro as described above. After 7 d, cells were lifted and individual single cells were plated into 3 separate 384-well plates containing 384 primers and Mineral oil. A sperate flow cytometry experiment showed the majority of cells at this time point were EpCAM-CD45-CD31-PDPN+ fibroblasts. After sorting, plates were centrifuged at 300 g for 3 min and stored at -80° C. For amplification, cells were heat-lysed at 65° C followed by cDNA synthesis using the CEL-Seq2 protocol (Hashimshony et al., 2016) and robotic liquid handling platforms. After second strand cDNA synthesis, the barcoded material was pooled into libraries of 384 cells and amplified using in vitro transcription. Following amplification, the rest of the CEL-Seq2 protocol was followed for preparation of the amplified cDNA library, using TruSeq Small RNA Library Kits (Illumina, 2000012). The DNA library was paired-end sequenced on a Nextseq 500 Sequencer (Illumina) using an Illumina High Output 1x75 bp Kit (Illumina Inc., 20024906). During sequencing, Read 1 was assigned 26 bp and was used for identification of the Illumina library barcode, cell barcode and Unique Molecular Identifiers (UMIs). Read 2 was assigned 60 bp and used to map to the reference transcriptome of mm10 with Burrows-Wheeler Aligner (BWA) (Anders and Huber, 2010). Data was indexed using Samtools (version 1.9) and aggregated using the UMI-tools package (version 1.0.1). Single-cell transcriptomics analysis was done using the R package Seurat (Butler et al., 2018), as above.
In vitro fibroblast isolation and culture
Primary pancreatic fibroblasts (PaFs) were expanded in vitro from the pancreas of 8-week-old female B6 mice, as described above. At d7, cells were FACS sorted into LIN(CD45/CD31/EpCAM)-PDPN+CD90+CD105+ and LIN-PDPN+CD90+CD105- populations. Notably, Ficoll density gradient isolation of pancreatic fibroblasts from healthy murine pancreas, using standard methods, also yielded mixed CD105pos and CD105neg fibroblast populations. For immortalization, cells were transduced with SV40 LT (Addgene, 13970). After selection, cells were expanded, purity checked by flow cytometry and frozen stocks made and used for downstream functional assays. GFP and mCherry expressing target cells were generated using a second-generation lentiviral system (Harris et al., 2012) (Addgene 12263 and Addgene, 12259). No puromycin selection was carried out and GFP/mCherry expressing cells were isolated by FACS. For cancer cell and fibroblast co-culture experiments 2x106 mCherry+ PDA cells and 2x10ˆ6 GFP+ PaFs were plated in 20 mL CCM(10) in a 225 cm2 flask and analyzed after 48 h. To analyze the primary PaF surface marker changes under recombinant protein stimulation, primary PaFs were expanded in vitro as described, and incubated in 75 cm2 flasks with recombinant proteins in CCM(10) (at concentrations listed in the Supplemental Information Table) for 3 d. Cells were analyzed by mass cytometry as described above. To analyze the transcriptomic responses of CD105+ and CD105- PaFs to recombinant protein stimulation, 105 CD105+ and CD105- PaFs were plated into 6 well plates in 2 mL CCM(0.5). The following day the media was replaced with CCM(0.5) containing recombinant cytokines or growth factors (see Supplemental Information Table) and after 6 h, cells were aspirated, washed with ice-cold PBS and lysed using RLT buffer, fully detached with a cell scraper and RNA isolated using RNeasy Mini Kit (QIAGEN, 74104), according to the manufacturer’s instructions. Human pancreatic fibroblasts (hPaFs) (Generon, H-6201) were cultured in CCM, CD105+ and CD105- cells separated by FACS and cell lines generated using the lentiviral system described above with a pCSII-IRES2-hygro plasmid containing an hTERT expression inset, which was a kind gift from Dr. Farnando Calvo at the Institute of Cancer Research London, followed by 50 ug/mL hygromycin B (Thermo Fisher, 10687010) selection for 7 days.
BioMark HD multiplex qPCR
Assay primers and probes were designed using the Roche Universal Probe Library Assay Design Centre Tool (https://lifescience.roche.com/en_gb/brands/universal-probe-library.html). Where possible, primers were selected to span different exons to minimize amplification of genomic DNA. See Supplemental Information Table for primer sequences and TaqMan probe numbers. New primers and probes were validated by qPCR using Universal Mouse Reference RNA (Thermo Fisher, QS0640). cDNA was synthesized from 500 ng of RNA in a 50 μL reaction mixture of 1x Reverse Transcription Buffer (Thermo Fisher, 18067017), 1.75 mM Mg2Cl2 (Thermo Fisher, R0971), 2 mM dNTP Mix (Thermo Fisher, R0191), 5mM DL-Dithiothreitol (DTT) (Sigma Aldrich, 43815), 100 U/mL RNAse Inhibitor (Thermo Fisher, N8080119), 2.5 μM Random Hexamers (Thermo Fisher, N8080127) and 2500 U/mL Multiscribe Reverse Transciptase (Thermo Fisher, 4311235). Reverse transcription was carried out at 25°C for 10 min, 37°C for 60 min, 95°C for 5 min and 4°C indefinitely before being stored at -20°C. A pre-amplification of 2.5 μL of the cDNA mixture was conducted in 10 μL of 1x TaqMan Pre-Amp Master Mix (Applied Biosystems, 4391128) and a pool of all assay-specific primers at 5 nM (see Supplemental Information Table), by temperature cycling at 95°C for 10 min for 1 cycle, 95°C for 15 s and 60°C for 4 min for 14 cycles and 4 degree indefinitely until being diluted with 40 μL RNAse-free water (Thermo Fisher, 10977035) and storage at -20°C. Assay mixes for the qPCR reactions were made using 8 μM of each primer and 1 μM of the appropriate hydrolysis probe in 1x Assay Loading Reagent (Fluidigm, 85000736). Sample mixes were made by diluting the amplified cDNA 1:1 in TaqMan Universal PCR Master Mix (Applied Biosystems, 4304437) and GE Sample Loading Reagent (Fluidigm, 85000746). Samples and assays were carefully loaded on a 96x96 Dynamic Array Chip (Fluidigm, BMK-M-96.96) and analyzed according to manufacturer’s instructions using standard settings, auto-exposure settings and with ROX as the passive reference dye. Raw qPCR data was analyzed using the BioMark Real-Time PCR Analysis Software (Fluidigm). Assay dependent thresholds were used to calculate cycle threshold (Ct) values and relative expression calculated as: relative expression = 2-ΔCt, where: ΔCt = (Ct value gene A) – (Geometric mean (Ct values house-keeping genes)). A combination of house-keeping genes (Actb, Gapdh, Pgk1, Ppia, Tbp, Tubb4a) was used for normalization to mitigate potential confounding issues caused by differential housekeeping-gene expression between cell lines.
Genetically engineered annimal models
KPC colonies on mixed backgrounds were bred in-house in individually ventilated cages, under pathogen-free conditions at CRUK Beatson Institute (CRUK-BI) and maintained in conventional caging with environmental enrichment, access to standard chow and water ad libitum. Genotyping was performed by Transnetyx (Cordoba, TN, USA). Mice of both sexes were monitored 3 times weekly and when a diagnosis of pancreatic cancer was made by abdominal palpation, tumor growth was monitored by ultrasound imaging (Fujifilm VisualSonics, Vevo 3100 preclinical imaging system). Mice were culled by Schedule 1 method, as per institutional guidelines, when exhibiting moderate symptoms of PDA, such as swollen abdomen, loss of body conditioning resembling cachexia or reduced mobility. RFP KPC colonies on a mixed background were maintained under pathogen-free conditions at the UoM and monitored as described above. B6.Rag1-/- mice and B6.Batf3-/- mice were maintained at the UoM and both sexes used at >12 weeks of age. KPN mice of both sexes at 6-12 weeks age were injected intraperitoneally with a single dose of 2 mg tamoxifen (Sigma Aldrich, T5648) and primary colorectal tumors collected at clinical end point, defined as animal weight loss and/or hunching and/or cachexia. Female MMTV-PyMT mice were monitored for tumor growth by caliper measurement and tumors collected when total tumor volume was >900 mm3, typically across multiple foci. KP mice of both sexes at 8-14 weeks of age were anaesthetized using isoflurane and intranasally administered with 50 μL of 1x106 PFU replication-deficient Cre-expressing adenovirus, as per standard protocols (Meuwissen et al., 2001) and monitored for tumor formation by computerized tomography scans. Resulting lung tumors were collected 16 weeks after adenoviral induction. Female BRAFV600E mice, 8-12-weeks old, had 1 mg freshly prepared tamoxifen in ethanol applied to their shaven back. 4 weeks after transgene induction, the back skin was UV irradiated with a UV6 lamp (UV280-380 nm) every week for 4 weeks. Once tumors were visible, tumor volume was measured weekly and collected at a minimum volume of 500 mm3.
Subcutaneous co-transplant model
The majority of subcutaneous co-transplant studies in this study used syngeneic female B6 mice of 14 weeks of age. Where indicated female 14 week old NSG mice were used. Both male and female Rag1-/- and Batf3-/- mice of mixed ages >12 weeks old were used where indicated and sex/age matched across cohorts. During optimization, KPC PDA cells on a B6 background were injected in 100 μL PBS (Thermo Fisher, 10010056) but this fails to retain fibroblasts within the growing tumor (see manuscript Figure S6). For all subsequent transplants Growth Factor Reduced Matrigel (Corning, 356231) was used. Where possible, a single Matrigel lot was used for experiments to minimize the impact of lot-to-lot variation. Cancer cells (BL6KPC-TB32043 or BL6KPC-TB32047 from B6 fully backcrossed KPC mice) and fibroblasts were lifted 20-24 h before the day of injection using Accutase Cell Detachment Solution and 6x106 and 3x106 cells plated, respectively into 225 cm2 flasks. Cancer cells were cultured in CCM(10) and fibroblasts in CCM. One the day of injection, cells were lifted again using Accutase Cell Detachment Solution, washed twice with ice-cold PBS and counted in triplicate using a Luna Cell Counter on bright-field mode. The required number of cancer cells and fibroblasts were combined in 5 mL polypropylene FACS tubes, washed once more with PBS and carefully and fully aspirated. Ice-cold Matrigel were added using pre-cooled pipette tips to obtain a final concentration of each cell type of 5000 cells/uL and gently mixed on ice. Braun Omnican 50 Insulin Syringe/Needles (VWR, 9151117) were used to accurately measure 20 μL of cell/Matrigel mix with no dead-volume, which was injected subcutaneously into the right flank of the mouse (therefore giving 105 cancer cells and 105 fibroblasts). 20 μL injection volume was found to be the optimal balance between generating plugs with no necrosis and consistent injection volume. Tumor width and length were measured (blinded to the study) by calipers and tumor volume (V) calculated as V = (2xwidth)xlength)/2. Study end point was V>900 mm3, if the mouse lost >10% body weight or if a mouse’s health showed any other signs of deterioration e.g. loss of activity, altered breathing, behavioral changes. For gene expression analysis, subcutenous tumors were collected at day 10, lysed in 500 uL Trizol utilizing a TissueLyzer (QIAGEN) and RNA isolated using DirectZol kits (Zymo) following manufacturers recommendations.
Immunohistochemistry of subcutaneous tumors
Subcutaneous co-transplant tumors were collected at 7 and 30 d after implantation. Large tumors were cut in half. Samples were fixed for 24h in 10% v/v Neutral Buffered Formalin (Genta Medical, BIB10L), processed and paraffin embedded (Leica Microsystems). 4um cut sections were mounted onto charged glass slides and stained manually. Slides were dewaxed by x3 5 min xylene washes and rehydrated in 100%, 90% then 70% ethanol for 1 min each. Heat induced epitope retrieval was conducted using a Biocare Declocker at 110°C for 15 min and allowed to cool for 15 min using Low pH Target Retrieval Buffer Ph6 (Agilent, S236984). Slides were cooled in running water for 2 min and endogenous peroxidases blocked using 3% v/v hydrogen peroxide in TBST for 10 min. Following further washing in TBST, slides were blocked with 10% w/v casein in TBST for 20 min. Staining was conducted using chicken anti-GFP antibody (Abcam, ab13970) at 1/500 in TBST for 1 h at RT, followed by x2 5 min TBST washes. Detection of the primary antibody used a biotinylated goat anti-chicken IgG antibody (Abcam, ab207998) at 1/200 in TBST for 30 min at RT, followed by Vectastain Elite ABC HRP Kit (Vector, PK-6100) for 30 mins at RT, x2 5 min TBST washes and 3,3'-diaminobenzidine (DAB) (Agilent, K3467) for 5 min. Finally, nuclei were counterstained with 1x Shandon Gill Haematoxylin (Thermo Fisher, 6765005) and then dehydrated and coverslips applied before being scanned using an SCN400 (Leica microsystems) and analyzed using QuPath.
CRISPR-Cas-9 gene editing
For in vitro fibroblasts, we found nucleofection-based CRISPR-Cas-9 methods to be far superior to other methods to generate efficient gene knockouts. For each target gene, three separate gRNAs were designed (Synthego ‘Mulit-Guide’ platform), such that their spatial distribution favored large (>50bp) genomic deletions rather that small indels, resulting in improved knockout efficiency and consistency. gRNAs were synthesized and chemically modified to improve stability and reduce intracellular immune responses. Ribonucleoprotein (RNP) complexes were formed by diluting 2 μL of 100 μM of multi-gRNA (Synthego) in Tris-EDTA (TE) (Synthego) and 1 μL of 20 μM recombinant Cas-9 (Integrated DNA Technologies (IDT), 1081059) in RNase-free PBS in 12 μL Primary Cell P3 Nucleofector solution (Lonza, V4XP-3032) and incubating at RT for 20 min. To 2.5x105 cells in 5 μL Nucelofector solution, 0.8 μL of Electroporation Enhancer Solution (IDT, Alt-R Cas9 Electroporation Enhancer, 2 nmol) was added, followed by 15 μL of the RNP solution and mixed by pipetting. This was transferred to a well of a 20 μL Nucleocuvette Strip (Lonza, V4XP-3032) and transfected using a 4D-Nucleofector Core Unit (Lonza), using the CM-137 program. Cells were allowed to rest for 3 min before being plated in CCM and cultured as normal. Transfection with a pool of x2 separate non-targeting (NT) gRNAs (Synthego) was used to generate control cells. Gene knockout was confirmed in a split of the cells at the protein level by flow cytometry after 7 d, using IFNγ stimulation to induce expression of H2Ab1 and Cd74. Surface MHCII expression was measured and CD74 expression was measured by intracellular staining (CD74 is predominantly located in the ER/endosomes), using the FOXP3 Fixation/ Permeabilization kit. Cells were not purified further as gene knockout was consistently >95% for all targeted genes.
Quantification and statistical analysis
Plotting and statistical tests were performed in Prism (version 7, GraphPad Software Inc.) or R Statistical Software. For RNA-seq data visualization, differentially expressed genes (DEGs) from DESeq2 were scaled by library size using the function estimateSizeFactors, the data transformed by the function normTransform and the obtained expression values used for visualization. Heatmap plots were drawn using the R package ComplexHeatmap. Heatmap visualization of CyTOF data was achieved by first processing the data using the R package flowCore (Hahne et al., 2009). According to best practices, data was transformed by hyperbolic arc-sine with cofactor = 5 by the function asinh. The z-score was calculated by the function scale and heatmaps drawn using the R package ComplexHeatmap. Principle Component Analysis (PCA) plots were generated using the pcaplot function. For the abundance/phenotype cross-cluster correlation analysis, the number of cells in each FlowSOM cluster as a percentage of the total number of gated cells from each sample was used as the abundance input data. To calculate the fraction of proliferating and dying cells in each cluster, FCS files containing all target cells, including FlowSOM cluster annotation were exported from Cytofkit2 and uploaded to FlowJo (version 10.6.2, BD Life Sciences). S-phase cells were defined as cells with both Ki67 mass intensity signal ≥ 20 and IdU mass intensity signal ≥ 20. Dying cells were defined as cells with cleaved caspase-3 (CC3) mass intensity signal ≥ 8. The abundance of each FlowSOM cluster and the percentage of S-phase and dying cells within each FlowSOM cluster for each sample was exported and used as the phenotypic input data for the cross-correlation analysis. Since all antibody panels were measured on each of the 18/19 PDA samples, the abundance, proliferation and apoptosis data for each of the 20 FlowSOM clusters from each of the three panels (60 total clusters) was concatenated into one data frame for these 18 samples. Correlation analysis was performed on selected abundance, proliferation and apoptosis comparisons (see manuscript for specific comparisons), using Spearman correlation measurement with permutation testing adjusted for multiple testing using Benjamini-Hochberg correction. The correlation results were visualized by the R package corrplot.
Acknowledgments
This work was supported by Cancer Research UK Institute Awards C5759/A27412 (to C.J., A.M., R.M., C.S., and S.Z.), A17196 and A21139 (to O.J.S.), and A29996 (to J.P.M.), Experimental Medicine Programme Award (A25236 to C.J., O.J.S., and J.P.M.) and European Research Council Consolidator Award (ERC-2017-COG 772577 to C.J.).
A.L. received funding from ASCO Conquer Cancer Foundation Young Investigator Award and The Christie Charity. The authors would like to acknowledge colleagues at CRUK Manchester Institute Systems Oncology Team for valuable input, CRUK Manchester Institute core facilities, in particular flow cytometry, molecular biology, and visualization irradiation analysis, as well as the Biological Services Unit at the CRUK Beatson Institute. We would also like to thank William C. Hahn, Didier Trono, Inder Verma, Fernando Calvo, and Tim Somervaille for kindly sharing plasmids. Graphical abstract was created with images from BioRender.com.
Author contributions
C.H. planned and conducted the experiments, analyzed the data, and wrote the manuscript. A.B.-G., F.H., and E.H. planned and conducted the experiments and analyzed the data. A.B. analyzed the data. A.K. conducted computational analysis. X.Z. planned and conducted the experiments. A.B., S. Karim, S. Kemp, D.W., J.K., and V.P.-H. provided technical support, conducted experiments, and provided reagents. F.H., N.S., and S. Karim, S. Kemp conducted the experiments and analyzed the data. A.L., J.V., M.P.d.M., R.-F.J., O.J.S., F.L., C.S., M.M., A.M., L.C., and R.M. provided reagents and experimental advice. S.Z. provided reagents and oversight. J.P.M. conducted the experiments, and provided reagents and oversight. C.J. conceived the project, analyzed the data, provided oversight, and wrote the manuscript.
Declaration of interests
O.S. receives funding from Novartis, AstraZeneca, RedEx and Cancer Research Technology. C.J. receives funding from AstraZeneca. R.M. is an expert witness for Pfizer and, as a former employee of the Institute of Cancer Research (ICR) in London, may benefit financially from commercialized programs. C.S. and F.L. are former employees of the ICR in London and may benefit financially from commercialized programs. The other authors declare no competing interests.
Published: July 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2021.06.017.
Supplemental information
References
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P., Loughrey M.B., Fernandez J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E., Giraldo N.A., Lacroix L., Buttard B., Elarouci N., Petitprez F., Selves J., Laurent-Puig P., Sautès-Fridman C., Fridman W.H. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Bendall S.C., Nolan G.P. From single cells to deep phenotypes in cancer. Nat. Biotechnol. 2012;30:639–647. doi: 10.1038/nbt.2283. [DOI] [PubMed] [Google Scholar]
- Bendall S.C., Simonds E.F., Qiu P., Amir E.-A.D., Krutzik P.O., Finck R., Bruggner R.V., Melamed R., Trejo A., Ornatsky O.I. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G., Oni T.E., Spielman B., Hao Y., Elyada E., Park Y., Preall J., Tuveson D.A. IL1-Induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301. doi: 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido J.B., Morton J.P., Bailey P., Campbell A.D., Karim S.A., Jamieson T., Lapienyte L., Gopinathan A., Clark W., McGhee E.J. CSF1R+ macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. CellReports. 2018;23:1448–1460. doi: 10.1016/j.celrep.2018.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano E., Carmona R., Muñoz-Chápuli R. Wt1-expressing progenitors contribute to multiple tissues in the developing lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013;305:L322–L332. doi: 10.1152/ajplung.00424.2012. [DOI] [PubMed] [Google Scholar]
- Catenacci D.V.T., Junttila M.R., Karrison T., Bahary N., Horiba M.N., Nattam S.R., Marsh R., Wallace J., Kozloff M., Rajdev L. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015;33:4284–4292. doi: 10.1200/JCO.2015.62.8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S., Levine J.H., Zanotelli V.R.T., Silina K., Schulz D., Bacac M., Ries C.H., Ailles L., Jewett M.A.S., Moch H. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169:736–738.e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C.E., Hingorani S.R., Mick R., Combs C., Tuveson D.A., Vonderheide R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- Collins M.A., Bednar F., Zhang Y., Brisset J.-C., Galbán S., Galbán C.J., Rakshit S., Flannagan K.S., Adsay N.V., Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft A.P., Campos J., Jansen K., Turner J.D., Marshall J., Attar M., Savary L., Wehmeyer C., Naylor A.J., Kemble S. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570:246–251. doi: 10.1038/s41586-019-1263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakin S.G., Coles M., Sherlock J.P., Powrie F., Carr A.J., Buckley C.D. Pathogenic stromal cells as therapeutic targets in joint inflammation. Nat. Rev. Rheumatol. 2018;14:1–13. doi: 10.1038/s41584-018-0112-7. [DOI] [PubMed] [Google Scholar]
- Dhomen N., Reis-Filho J.S., da Rocha Dias S., Hayward R., Savage K., Delmas V., Larue L., Pritchard C., Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Di Mitri D., Mirenda M., Vasilevska J., Calcinotto A., Delaleu N., Revandkar A., Gil V., Boysen G., Losa M., Mosole S. Re-education of tumor-associated macrophages by CXCR2 blockade drives senescence and tumor inhibition in advanced prostate cancer. Cell Rep. 2019;28:2156–2168.e5. doi: 10.1016/j.celrep.2019.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R., Ai X., Fine A. Derivation of lung mesenchymal lineages from the fetal mesothelium requires hedgehog signaling for mesothelial cell entry. Development. 2013;140:4398–4406. doi: 10.1242/dev.098079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C.X., Muller S., Keerthivasan S., Koeppen H., Hung J., Gierke S., Breart B., Foreman O., Bainbridge T.W., Castiglioni A. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020;10:232–253. doi: 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- Driskell R.R., Watt F.M. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 2015;25:92–99. doi: 10.1016/j.tcb.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Driskell R.R., Lichtenberger B.M., Hoste E., Kretzschmar K., Simons Ben D., Charalambous M., Ferron S.R., Herault Y., Pavlovic G., Ferguson-Smith A.C. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504:277–281. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E., Bolisetty M., Laise P., Flynn W.F., Courtois E.T., Burkhart R.A., Teinor J.A., Belleau P., Biffi G., Lucito M.S. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C., Jones J.O., Kraman M., Wells R.J.B., Deonarine A., Chan D.S., Connell C.M., Roberts E.W., Zhao Q., Caballero O.L. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordillo C.H., Sandoval P., Muñoz-Hernández P., Pascual-Antón L., López-Cabrera M., Jiménez-Heffernan J.A. Mesothelial-to-mesenchymal transition contributes to the generation of carcinoma-associated fibroblasts in locally advanced primary colorectal carcinomas. Cancers. 2020;12:499. doi: 10.3390/cancers12020499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy C.T., Cardiff R.D., Muller W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F., LeMeur N., Brinkman R.R., Ellis B., Haaland P., Sarkar D., Spidlen J., Strain E., Gentleman R. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics. 2009;10:106. doi: 10.1186/1471-2105-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrook C.J., Pontious C., Kovalenko I., Lapienyte L., Dreyer S., Lee H.-J., Thurston G., Zhang Y., Lazarus J., Sajjakulnukit P. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29:1390–1399.e1396. doi: 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G., Spitzer M.H., Bendall S.C., Fantl W.J., Nolan G.P. Metal-isotope-tagged monoclonal antibodies for high-dimensional mass cytometry. Nat. Protoc. 2018;13:2121–2148. doi: 10.1038/s41596-018-0016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris W.J., Huang X., Lynch J.T., Spencer G.J., Hitchin J.R., Li Y., Ciceri F., Blaser J.G., Greystoke B.F., Jordan A.M. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K.J., Rozenblatt-Rosen O. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms E., Onate M.K., Sherman M.H. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 2020;10:1–10. doi: 10.1158/2159-8290.CD-19-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K., Edelson B.T., Purtha W.E., Diamond M., Matsushita H., Kohyama M., Calderon B., Schraml B.U., Unanue E.R., Diamond M.S. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hirata E., Girotti M.R., Viros A., Hooper S., Spencer-Dene B., Matsuda M., Larkin J., Marais R., Sahai E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015;27:574–588. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson E.L., Willis N., Mercer K., Bronson R.T., Crowley D., Montoya R., Jacks T., Tuveson D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson H.W., Fischer J.R., Zanotelli V.R.T., Ali H.R., Mechera R., Soysal S.D., Moch H., Muenst S., Varga Z., Weber W.P. The single-cell pathology landscape of breast cancer. Nature. 2020;177:1–25. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- Jackstadt R., van Hooff S.R., Leach J.D., Cortes-Lavaud X., Lohuis J.O., Ridgway R.A., Wouters V.M., Roper J., Kendall T.J., Roxburgh C.S. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell. 2019;36:319–336 e317. doi: 10.1016/j.ccell.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K.A. Single-cell states versus single-cell atlases—two classes of heterogeneity that differ in meaning and method. Curr. Opin. Biotechnol. 2016;39:120–125. doi: 10.1016/j.copbio.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Hegde S., Knolhoff B.L., Zhu Y., Herndon J.M., Meyer M.A., Nywening T.M., Hawkins W.G., Shapiro I.M., Weaver D.T. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016;22:1–13. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Placencio-Hickok V.R., Madhav A., Haldar S., Tripathi M., Billet S., Mishra R., Smith B., Rohena-Rivera K., Agarwal P. Heterogeneous cancer-associated fibroblast population potentiates neuroendocrine differentiation and castrate resistance in a CD105-dependent manner. Oncogene. 2018;303:1–15. doi: 10.1038/s41388-018-0461-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.J., Sahai V., Abel E.V., Griffith K.A., Greenson J.K., Takebe N., Khan G.N., Blau J.L., Craig R., Balis U.G. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014;20:5937–5945. doi: 10.1158/1078-0432.CCR-14-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliaraki V., Prados A., Armaka M., Kollias G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 2020;21:974–982. doi: 10.1038/s41590-020-0741-2. [DOI] [PubMed] [Google Scholar]
- Koopmans T., Rinkevich Y. Mesothelial to mesenchyme transition as a major developmental and pathological player in trunk organs and their cavities. Commun. Biol. 2018;1:170. doi: 10.1038/s42003-018-0180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Lun X.K., Bodenmiller B., Rodriguez Martinez M., Koeppl H. Stabilized reconstruction of signaling networks from single-cell cue-response data. Sci. Rep. 2020;10:1233. doi: 10.1038/s41598-019-56444-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppe C., Ibrahim M.M., Kranz J., Zhang X., Ziegler S., n J.P.-P.X., Jansen J., Reimer K.C., Smith J.R., Dobie R. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2020;589:1–42. doi: 10.1038/s41586-020-2941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leun A.M., Thommen D.S., Schumacher T.N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer. 2020;77:1–15. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenberger B.M., Mastrogiannaki M., Watt F.M. Epidermal β-catenin activation remodels the dermis via paracrine signalling to distinct fibroblast lineages. Nat. Commun. 2016;7:10537. doi: 10.1038/ncomms10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luche H., Weber O., Nageswara Rao T., Blum C., Fehling H.J. Faithful activation of an extra-bright red fluorescent protein in "knock-in" Cre-reporter mice ideally suited for lineage tracing studies. Eur. J. Immunol. 2007;37:43–53. doi: 10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- Lun X.K., Szklarczyk D., Gabor A., Dobberstein N., Zanotelli V.R.T., Saez-Rodriguez J., von Mering C., Bodenmiller B. Analysis of the human kinome and phosphatome by mass cytometry reveals overexpression-induced effects on cancer-related signaling. Mol. Cell. 2019;74:1086–1102 e1085. doi: 10.1016/j.molcel.2019.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun X.K., Zanotelli V.R., Wade J.D., Schapiro D., Tognetti M., Dobberstein N., Bodenmiller B. Influence of node abundance on signaling network state and dynamics analyzed by mass cytometry. Nat. Biotechnol. 2017;35:164–172. doi: 10.1038/nbt.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv F.-J., Tuan R.S., Cheung K.M.C., Leung V.Y.L. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408–1419. doi: 10.1002/stem.1681. [DOI] [PubMed] [Google Scholar]
- Marino S., Vooijs M., van Der Gulden H., Jonkers J., Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Mercer K., Giblett S., Green S., Lloyd D., DaRocha Dias S., Plumb M., Marais R., Pritchard C. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 2005;65:11493–11500. doi: 10.1158/0008-5472.CAN-05-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R., Linn S.C., van der Valk M., Mooi W.J., Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001;20:6551–6558. doi: 10.1038/sj.onc.1204837. [DOI] [PubMed] [Google Scholar]
- Miller B.W., Morton J.P., Pinese M., Saturno G., Jamieson N.B., McGhee E., Timpson P., Leach J., McGarry L., Shanks E. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P., Iacomini J., Johnson R.S., Herrup K., Tonegawa S., Papaioannou V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Morsing M., Klitgaard M.C., Jafari A., Villadsen R., Kassem M., Petersen O.W., Rønnov-Jessen L. Evidence of two distinct functionally specialized fibroblast lineages in breast stroma. Breast Cancer Res. 2016;18:108–119. doi: 10.1186/s13058-016-0769-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro M.J., Dharmadhikari G., Grun D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M.A., Carlotti F., de Koning E.J. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 2016;3:385–394 e383. doi: 10.1016/j.cels.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namvar S., Woolf A.S., Zeef L.A., Wilm T., Wilm B., Herrick S.E. Functional molecules in mesothelial-to-mesenchymal transition revealed by transcriptome analyses. J. Pathol. 2018;245:491–501. doi: 10.1002/path.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K.P., Jacobetz M.A., Davidson C.J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M.A., Caldwell M.E., Allard D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.-C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:1–16. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paauwe M., Schoonderwoerd M.J.A., Helderman R.F.C.P., Harryvan T.J., Groenewoud A., van Pelt G.W., Bor R., Hemmer D.M., Versteeg H.H., Snaar-Jagalska B.E. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clin. Cancer Res. 2018;24:6331–6344. doi: 10.1158/1078-0432.CCR-18-0329. [DOI] [PubMed] [Google Scholar]
- Philip M., Fairchild L., Sun L., Horste E.L., Camara S., Shakiba M., Scott A.C., Viale A., Lauer P., Merghoub T. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger M.F., Discher D.E., Péault B.M., Phinney D.G., Hare J.M., Caplan A.I. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen. Med. 2019;4:1–15. doi: 10.1038/s41536-019-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P.P., Cuevas C., Chang A.E., Goel V.K., Hoff Von D.D., Hingorani S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapsomaniki M.A., Lun X.K., Woerner S., Laumanns M., Bodenmiller B., Martinez M.R. CellCycleTRACER accounts for cell cycle and volume in mass cytometry data. Nat. Commun. 2018;9:632. doi: 10.1038/s41467-018-03005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim A.D., Oberstein P.E., Thomas D.H., Mirek E.T., Palermo C.F., Sastra S.A., Dekleva E.N., Saunders T., Becerra C.P., Tattersall I.W. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich Y., Walmsley G.G., Hu M.S., Maan Z.N., Newman A.M., Drukker M., Januszyk M., Krampitz G.W., Gurtner G.C., Lorenz H.P. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348:aaa2151. doi: 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich Y., Mori T., Sahoo D., Xu P.-X., Bermingham J.R., Weissman I.L. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat. Cell Biol. 2012;14:1251–1260. doi: 10.1038/ncb2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E., Astsaturov I., Cukierman E., DeNardo D.G., Egeblad M., Evans R.M., Fearon D., Greten F.R., Hingorani S.R., Hunter T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 2020;6:1–13. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Gao W., Lytle N.K., Huang P., Yuan X., Dann A.M., Ridinger-Saison M., DelGiorno K.E., Antal C.E., Liang G. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature. 2019;569:1–27. doi: 10.1038/s41586-019-1130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.-L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- Spitzer M.H., Carmi Y., Reticker-Flynn N.E., Kwek S.S., Madhireddy D., Martins M.M., Gherardini P.F., Prestwood T.R., Chabon J., Bendall S.C. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168:487–502.e15. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele C.W., Karim S.A., Leach J.D.G., Bailey P., Upstill-Goddard R., Rishi L., Foth M., Bryson S., McDaid K., Wilson Z. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29:832–845. doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele N.G., Carpenter E.S., Kemp S.B., Sirihorachai V.R., The S., Delrosario L., Lazarus J., Amir E.-a.D., Gunchick V., Espinoza C. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat. Cancer. 2020;1:1097–1112. doi: 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S., Chen J., Yao H., Liu J., Yu S., Lao L., Wang M., Luo M., Xing Y., Chen F. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 2018;172:1–33. doi: 10.1016/j.cell.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Tape C.J., Ling S., Dimitriadi M., McMahon K.M., Worboys J.D., Leong H.S., Norrie I.C., Miller C.J., Poulogiannis G., Lauffenburger D.A. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell. 2016;165:910–920. doi: 10.1016/j.cell.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen D.S., Koelzer V.H., Herzig P., Roller A., Trefny M., Dimeloe S., Kiialainen A., Hanhart J., Schill C., Hess C. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018;24:1–17. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valluru M., Staton C.A., Reed M.W.R., Brown N.J. Transforming growth factor-β and endoglin signaling orchestrate wound healing. Front. Physiol. 2011;2:89. doi: 10.3389/fphys.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Brink S.C., Sage F., Vertesy A., Spanjaard B., Peterson-Maduro J., Baron C.S., Robin C., van Oudenaarden A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods. 2017;14:935–936. doi: 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- Wei K., Korsunsky I., Marshall J.L., Gao A., Watts G.F.M., Major T., Croft A.P., Watts J., Blazar P.E., Lange J.K. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020;582:259–264. doi: 10.1038/s41586-020-2222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm B. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development. 2005;132:5317–5328. doi: 10.1242/dev.02141. [DOI] [PubMed] [Google Scholar]
- Wohlfahrt T., Rauber S., Uebe S., Luber M., Soare A., Ekici A., Weber S., Matei A.-E., Chen C.-W., Maier C. PU.1 controls fibroblast polarization and tissue fibrosis. Nature. 2019;566:1–27. doi: 10.1038/s41586-019-0896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima I., Belloir E., Bourgeois Y., Kumasaka M., Delmas V., Larue L. Spatiotemporal gene control by the Cre-ERT2 system in melanocytes. Genesis. 2006;44:34–43. doi: 10.1002/gene.20182. [DOI] [PubMed] [Google Scholar]
- Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019;25:1–33. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunder E.R., Finck R., Behbehani G.K., Amir el A.D., Krishnaswamy S., Gonzalez V.D., Lorang C.G., Bjornson Z., Spitzer M.H., Bodenmiller B. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015;10:316–333. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-seq data are avaliable at NCBI under GEO accession numbers GSE129455, GSE155698, GSE156985, GSE157391, GSE176056 and GSE176057. Mass cytometry and scRNA-seq data is available on Zenodo at https://doi.org/10.5281/zenodo.4584773.
This paper does not report original code
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.