

Abstract
Objective
Human adult articular cartilage (AC) has little capacity for repair, and joint surface injuries often result in osteoarthritis (OA), characterised by loss of matrix, hypertrophy and chondrocyte apoptosis. Inflammation mediated by interleukin (IL)-6 family cytokines has been identified as a critical driver of proarthritic changes in mouse and human joints, resulting in a feed-forward process driving expression of matrix degrading enzymes and IL-6 itself. Here we show that signalling through glycoprotein 130 (gp130), the common receptor for IL-6 family cytokines, can have both context-specific and cytokine-specific effects on articular chondrocytes and that a small molecule gp130 modulator can bias signalling towards anti-inflammatory and antidegenerative outputs.
Methods
High throughput screening of 170 000 compounds identified a small molecule gp130 modulator termed regulator of cartilage growth and differentiation (RCGD 423) that promotes atypical homodimeric signalling in the absence of cytokine ligands, driving transient increases in MYC and pSTAT3 while suppressing oncostatin M- and IL-6-mediated activation of ERK and NF-κB via direct competition for gp130 occupancy.
Results
This small molecule increased proliferation while reducing apoptosis and hypertrophic responses in adult chondrocytes in vitro. In a rat partial meniscectomy model, RCGD 423 greatly reduced chondrocyte hypertrophy, loss and degeneration while increasing chondrocyte proliferation beyond that observed in response to injury. Moreover, RCGD 423 improved cartilage healing in a rat full-thickness osteochondral defect model, increasing proliferation of mesenchymal cells in the defect and also inhibiting breakdown of cartilage matrix in de novo generated cartilage.
Conclusion
These results identify a novel strategy for AC remediation via small molecule-mediated modulation of gp130 signalling.
INTRODUCTION
Articular cartilage (AC) is an avascular, specialised tissue found in diarthrodial joints and acts as a substrate to enable fluid motion of joint surfaces. Adult AC is comprised of mostly extracellular matrix (ECM) and water, with chondrocytes constituting only 2%–5% of total tissue volume.1 Cartilaginous ECM consists mostly of collagens, with collagen II being the most abundant, and proteoglycans including aggrecan. Osteoarthritis (OA) is a degenerative joint disease whose hallmarks include degradation of ECM by proteases including matrix metalloproteinases (MMPs) and members of the disintegrin-like and a metalloproteinase with thrombospondin motif (ADAMTS) family, expression of developmental hypertrophy genes, apoptosis and localised compensatory proliferation termed chondrocyte cloning (reviewed in reference 22). During homoeostasis, articular chondrocytes do not undergo hypertrophy; however, in some pathological conditions, changes mimicking developmental hypertrophy can occur and drive OA.3,4 In addition to expression of matrix-degrading enzymes, chondrocytes upregulate collagen 10 (COL10A1), RUNX2 and alkaline phosphatase while downregulating articular chondrocyte genes including COL2A1, lubricin (PRG4) and SOX9.2 Eventually, AC undergoes calcification and chondrocytes are lost to apoptosis. Although the regenerative potential of mature AC is minimal, chondrocytes closest to injured regions on the joint surface in the superficial zone have been shown to proliferate;2,5,6 however, the frequency of cells that can divide and deposit large amounts of matrix is low and insufficient to enact repair.
The pathogenesis of OA often begins from an injury to AC, which establishes chronic, low-grade inflammation mediated by interleukin-6/glycoprotein 130 (IL-6/gp130) and other factors that promote hypertrophy, matrix degradation and eventual destruction of cartilage (reviewed in reference 77). The IL-6 family of cytokines share a common co-receptor, IL-6RST (gp130; signal transducer (ST)), and includes IL-6, leukaemia inhibitory factor (LIF), oncostatin M (OSM) and others.8 Signalling downstream of these cytokines involves activation of proteins including MAPKs, JAK/STAT proteins, AKT and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB);9 both activation of MAPKs (ERK1/2) and NF-κB have been linked to hypertrophy during OA.10,11 OSM promotes matrix loss and disease progression.12,13 IL-6 suppresses chondrocyte proliferation,14 promotes mineralisation in AC,15 downregulates matrix proteins16 and increases expression of proteases.17,18 Moreover, blockade of IL-6 in mouse models of OA is chondroprotective.17,18 Importantly, higher serum levels of IL-6 have been correlated with the development of OA in humans,19 and an antibody against IL-6R is currently in phase III trials for treatment of hand OA (NCT02477059). The downstream mediator of IL-6 signalling STAT3 has been demonstrated to have pleiotropic effects during chondrogenesis and in articular chondrocytes. During chondrogenic differentiation of multipotent mesenchymal stem cells, IL-6/STAT3 promote chondrocyte commitment and matrix production.20 Similarly, loss of STAT3 during limb formation results in increased hypertrophy, premature ossification and decreased SOX9 expression.21 This could potentially be regulated in part by the STAT3 target gene Myc, which both promotes proliferation and inhibits hypertrophy in developing chondrocytes.22 In contrast, inhibition of STAT3 downstream of exogenous IL-6 is chondroprotective, reducing the severity of OA-like pathology in a mouse model.17 These data implicate IL-6 family cytokine signalling as a major regulator of articular chondrocyte biology with potentially context-specific effects.
Here we define the molecular and functional outcomes downstream of gp130 signalling in articular chondrocytes and unveil a small molecule that selectively shifts the output of this pathway to achieve disease-modifying activity in two rat models of cartilage pathology.
RESULTS
Identification of RCGD 423 as a small molecule inhibitor of hypertrophy
Based on the literature defining the relationship between IL-6 family cytokines, hypertrophy and catabolism,12,13,23–26 we hypothesised that a small molecule regulating this pathway could prevent cartilage loss. To identify compounds that could inhibit hypertrophy, we isolated total limb cells from Col10a1-mCherry mice27 and cultured them in the presence of bone morphogenetic protein (BMP)-4, a driver of developmental chondrocyte hypertrophy;28 170 000 compounds were assayed for their ability to decrease the mCherry signal (figure 1A). Seventy-five were chosen for follow-up (figure 1B), and additional vetting based on reproducibility and magnitude of Col10a1 inhibition led to the selection of regulator of cartilage growth and differentiation (RCGD) 423 for continued characterisation (figure 1C). The ability of RCGD 423 to inhibit hypertrophy in human cells was confirmed by assessing RUNX2 and COL 10 levels in healthy and osteoarthritic articular chondrocytes following incubation with the compound (figure 1D); these results demonstrated that RCGD 423 could attenuate hypertrophy in vitro.
Figure 1.
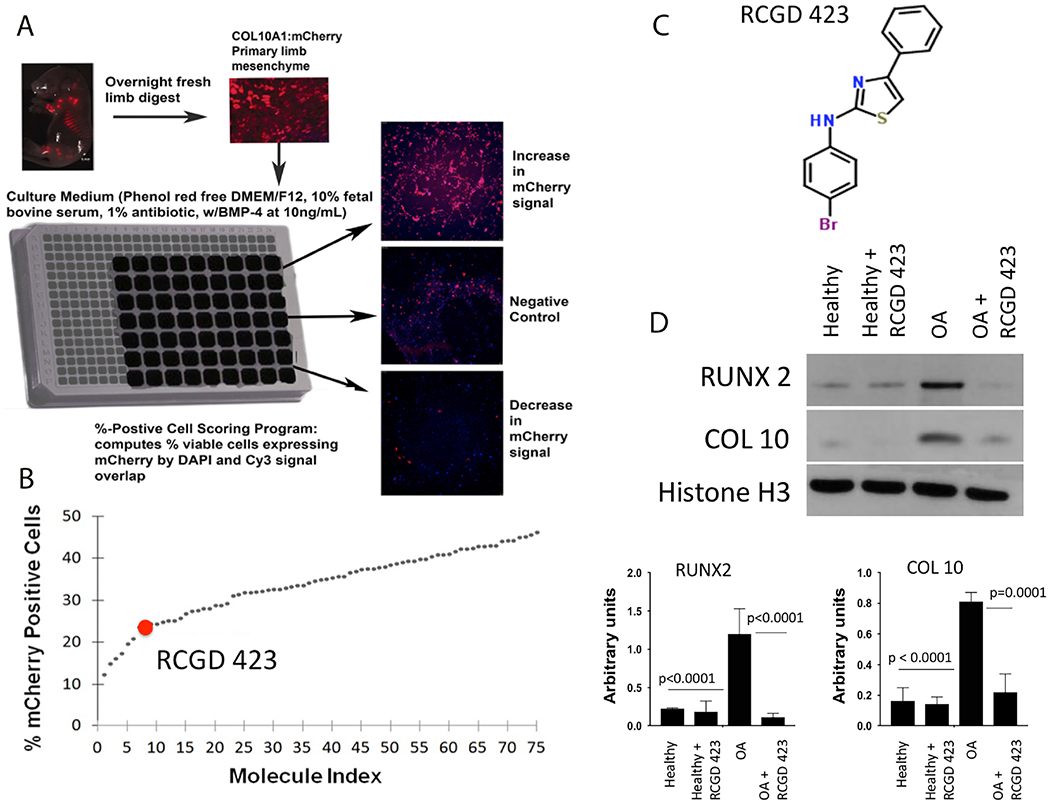
Small molecule screen to identify regulators of cartilage hypertrophy and differentiation. (A) Schematic representation of the high throughput screen performed to identify putative small molecule regulators of chondrocyte differentiation state. Limb mesenchymal cells were isolated from E13.5 mouse embryos carrying a Col10a1-mCherry transgene. Compounds were considered positive hits if they reduced mCherry signal after induction with the prodifferentiation factor BMP-4. (B) Quantitation of top 75 positive hits. (C) Structure of regulator of cartilage growth and differentiation (RCGD) 423. (D) RCGD 423 decreases levels of RUNX2 and COL 10 protein in articular chondrocytes from osteoarthritic donors (n=3). DMEM, Dulbecco’s Modified Eagle Media; OA, osteoarthritis.
RCGD 423 elicits different signalling in chondrocytes than IL-6 family cytokines and can inhibit their catabolic effects
Cartilage degeneration is a feed-forward process, as micro-environmental stresses including inflammation can interact with chondrocyte hypertrophy and loss to promote structural damage.29 In turn, these alterations promote matrix loss via MMPs (collagenases) and aggrecanases (ADAMTS4/5; figure 2A). To elucidate the mechanisms of action of IL-6 family cytokines, RCGD 423 and known proinflammatory cytokines in the promotion of degeneration, we stimulated pig articular chondrocytes and quantitated the levels of activated downstream signalling proteins (Figure 2B,C) as well as assessed the expression levels of MMP13, ADAMTS4 and ADAMTS5 (Figure 2D). Treatment with tumor necrosis factor (TNF)-α, a classic proinflammatory cytokine, resulted in strong activation of ERK1/2 and AKT, culminating in increased pNF-κB and upregulation of all catabolic enzymes. Within the IL-6 family cytokines, OSM acted in a similar fashion to TNF-α, while both LIF and IL-6 stimulated the activation of ERK1/2 and AKT to varying degrees and resulted in low-level upregulation of matrix proteases. In parallel with this catabolic signalling, LIF and IL-6 also increased levels of pSTAT3 and MYC, suggesting that both cytokines may have both procatabolic and antihypertrophic effects. RCGD 423 elicited a unique signalling profile, driving strong increases in pSTAT3 and MYC levels without activating AKT or NF-κB, resulting in a signalling milieu downstream of gp130 similar to actively proliferating and anabolic fetal chondrocytes (online supplementary figure 1A–C); concordantly, no upregulation of MMP13, ADAMTS4 and ADAMTS5 occurred. In fetal cartilage both STAT3 and MYC signalling are critically important for chondrocyte survival and proliferation, and activation of these pathways appears to be primarily driven by LIF (online supplementary figure 1C–E). We then assessed whether RCGD 423 could inhibit the catabolic effects of these cytokines. Incubation of human articular chondrocytes with IL-6, OSM and RCGD 423 decreased levels of pNF-κB and pERK1/2 when RCGD 423 was included (figure 2E) and reduced catabolic gene expression (online supplementary figure 2A). Moreover, this effect was not limited to chondrocytes, as RCGD 423 reduced both IL-6- and OSM-induced increases in pNF-κB and pERK 1/2 in human synoviocytes and peripheral blood mononuclear cells (PBMCs; online supplementary figure 2B,C). To assess if RCGD 423 could prevent matrix loss stimulated by IL-6 or OSM, we incubated explants of pig AC with either cytokine and the compound and measured neoepitopes of collagen and aggrecan released into the media;30 inclusion of RCGD 423 strongly reduced the production of cleavage products of both proteins (figure 2F). Together, these data demonstrate that IL-6 family cytokines have developmental stage-specific effects and elicit varying degrees of procatabolic, prohypertrophic responses in adult chondrocytes which can be inhibited by RCGD 423.
Figure 2.
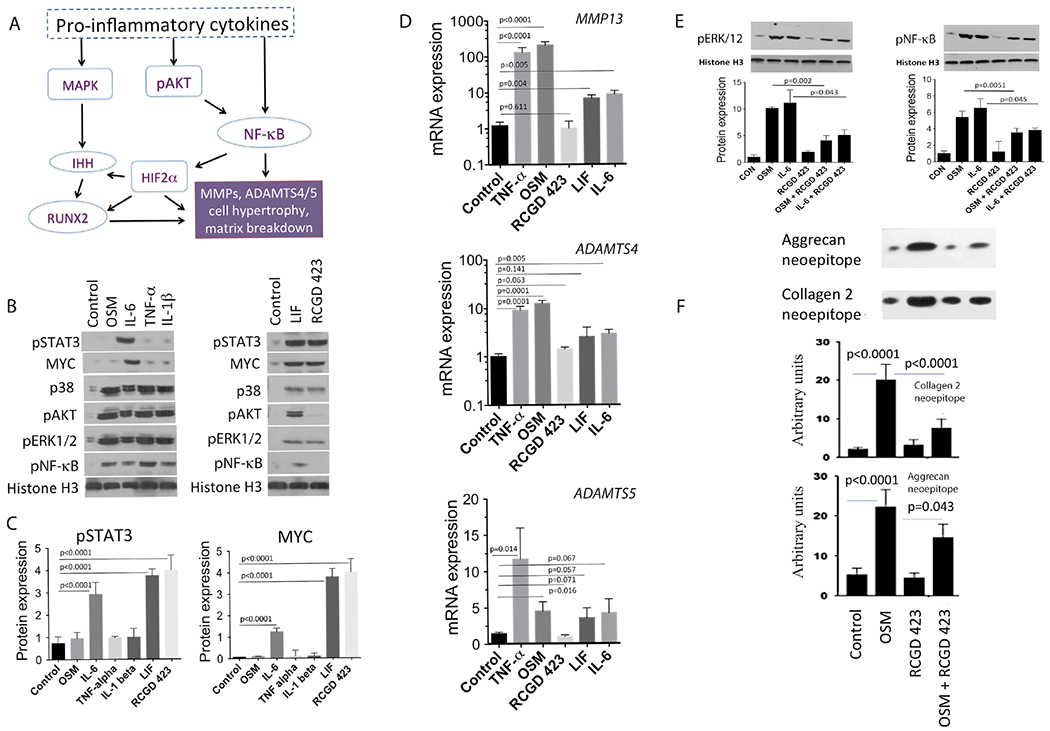
IL-6 family cytokines induce proinflammatory, catabolic signalling that can be directly competed by regulator of cartilage growth and differentiation (RCGD) 423. (A) Schematic of the proinflammatory signalling pathways that cause cell hypertrophy and matrix degradation in chondrocytes. (B) Adult pig articular chondrocytes were cultured for 24 hours with the indicated cytokines or RCGD 423 and the levels of pSTAT3 and MYC were quantified (C; n=3) with respect to histone H3. Representative data for other proteins in MAPK (p38 and pERK1/2), AKT (phospho-AKT; pAKT) or NF-κB (pNF-κB) are also shown. (D) Transcription of catabolic genes was determined via qPCR in adult pig articular chondrocytes treated with oncostatin M (OSM), IL-6, TNF-α, leukaemia inhibitory factor (LIF) or RCGD 423. Data are represented at mean±SD. (E) Adult human chondrocytes were incubated with the indicated cytokines in the presence or absence of RCGD 423 and the levels of downstream proteins quantitated with respect to histone H3. (F) Pig articular cartilage explants were incubated with OSM, RCGD 423 or both; levels of cleaved aggrecan and collagen epitopes in the supernatant are normalised to the wet weight of the explant. For all panels, n=3.
RCGD 423 stimulates proliferation and prevents apoptosis in adult articular chondrocytes
To assess the functional effects of RCGD 423 on adult human articular chondrocytes, we first validated that RCGD 423 significantly increased pSTAT3 and MYC (figure 3A) and in a dose-dependent and time-dependent manner (figure 3B). RCGD 423 also increased the proliferative potential and decreased apoptosis in articular chondrocytes (figure 3C,D) as well as increased proliferation in pig AC explants (figure 3E). We next evaluated the effects of RCGD 423 at the transcriptional level on human adult articular chondrocytes (n=7). As expected, the variability between these samples was high, so we focused our analysis on genes that increased more than 1.5-fold in four of seven replicates. GO analysis of these 1244 enriched genes revealed categories related to cell cycle, secretion and migration (figure 3F), while untreated cells were enriched for categories related to IL-6-mediated inflammation (online supplementary figure 3A); we confirmed RCGD 423 upregulated three of these genes with roles in cartilage biology (HAS331, FGF1832 and CDK633; online supplementary figure 3B). We hypothesised that RCGD 423 may promote proliferation through MYC and therefore focused on the 31 genes in the M phase Gene Ontology (GO) category enriched in drug treated cells. We compared these with MYC targets defined by ChIP-Seq,34 which gave a statistically significant overlap (P=0.0062; hypergeometric test). We then incubated pig articular chondrocytes with RCGD 423 and inhibitors of either STAT3 or MYC to directly assess the roles of these proteins in promoting proliferation. Blockade of STAT3 or MYC reduced proliferation downstream of RCGD 423 (figure 3G). Together, these data demonstrate that RCGD 423 stimulates increases in adult chondrocyte proliferation and survival and suggest that pSTAT3 and MYC may mediate these effects.
Figure 3.
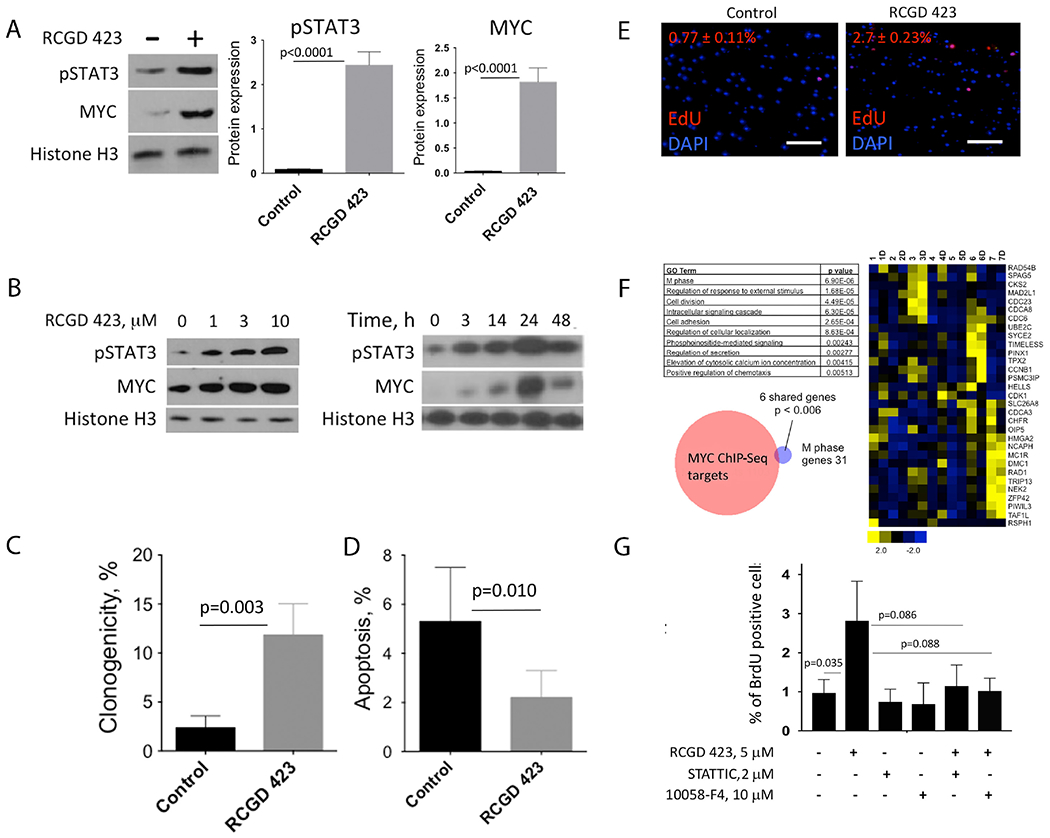
Regulator of cartilage growth and differentiation (RCGD) 423 inhibits apoptosis and promotes proliferation via induction of pSTAT3 and MYC in adult human chondrocytes. (A) Adult human articular chondrocytes were incubated with or without RCGD 423 and levels of MYC and pSTAT3 were quantified relative to histone H3 after 24 hours. (B) Increases in pSTAT3 and MYC proteins occurred in both a dose-dependent and time-dependent fashion following stimulation with RCGD 423. (C) Single human adult articular chondrocytes were cultured for 5 weeks with or without stimulation with RCGD 423 and assessed for colony formation. (D) Adult human articular chondrocytes were incubated with or without RCGD 423 in Mebiol hydrogel for 24 hours and then apoptotic cells were quantitated via flow cytometry for annexin V. (E) Proliferation in explants of adult pig articular cartilage in the absence or presence of RCGD 423 as shown by EdU incorporation. Scale bars represent 25 μm. P<0.0001. n=5. (F) Seven independent samples of human adult articular chondrocytes were cultured with or without RCGD 423 and then subjected to RNA-Seq. Genes that were significantly enriched in four of seven drug-treated samples (‘D’) when compared with their untreated controls were analysed using GO. Selected categories and their respective P values are shown. Heat map depicting the 31 genes in the ‘M phase’ GO category. Relative expression for all 14 matched samples are shown. Venn diagram depicting the overlap of the 31 genes from the ‘M phase’ GO category and a gene set defined by Zeller et al34 comprised of direct MYC target genes. (G) Adult human articular chondrocytes were cultured with RCGD 423 and inhibitors of either STAT3 (STATTIC) or MYC (10058-F4) for 48 hours; 5-bromo-2′-deoxyuridine (BrdU) incorporation was measured by flow cytometry. Unless noted, n=3. DAPI, 4′-6-diamidino-2-phenylindole.
RCGD 423 is a direct modulator of gp130 and acts via promoting homodimerisation
Given the molecular and functional effects of RCGD 423, we hypothesised that it may act within the gp130 signalling cascade. To evaluate this, we incubated pig articular chondrocytes with inhibitors of various proteins in this pathway in the presence of either LIF, a canonical activator of gp130, or RCGD 423 (figure 4A,B) and measured levels of pSTAT3 and MYC. Inhibitors of both JAKs and gp130 (SC144)35 greatly reduced pSTAT3 and MYC activation induced by both LIF and RCGD 423, while the compound induced gp130 activation in a dose-dependent manner (figure 4C), suggesting that RCGD 423 may directly interact with gp130 to induce signalling in the absence of ligand.
Figure 4.

Regulator of cartilage growth and differentiation (RCGD) 423 binds to domain 2 of glycoprotein 130 (gp130) and induces stable homodimerisation, thereby competing out IL-6 family cytokine receptors. (A) Levels of MYC and pSTAT3 protein after 24 hours in adult pig articular chondrocytes in the presence or absence of leukaemia inhibitory factor (LIF) and JAK or gp130 inhibitors. Histone H3 was used as a loading control. (B) Adult pig articular chondrocytes were cultured in the presence of the indicated combinations of RCGD 423, JAK and/or gp130 inhibitors and the levels of MYC and pSTAT3 proteins were quantitated relative to histone H3 after 24 hours. (C) Phosphorylation of gp130 (Tyr905) occurred in a dose-dependent manner after stimulation with RCGD 423. (D) Predicted binding site of RCGD 423 in the extracellular domain of gp130. The structure of the indicated gp130 domains is shown in ribbon diagram representation (left) as well as with electrostatic potential (blue, positive charge; red, negative charge; white, neutral) mapped onto the molecular surface (right). RCGD 423 and gp130 residues within 4Å surrounding it are shown in stick representation in the expanded views. Carbon atoms are shown in green for gp130 and pink for RCGD 423. Oxygen and nitrogen atoms are in red and blue, respectively; the bromine atom in RCGD 423 is shown in red, while the sulfur atom is in yellow. The electrostatic potential surfaces are drawn at ±3 kT/e. D1, domain 1; FNIII, fibronectin type-III. (E) Protein was isolated from cultures of Ba/F3 cells 24 hours after transfection with either full-length gp130 or gp130 lacking domain 2 (ΔD2). (F) BaF3 cells were transfected with the indicated variants of gp130. Three independent clones were used for transfection. (G) Schematic representation of experimental design to assess RCGD 423 interaction with gp130. Protein complexes were immunoprecipitated with an anti-Flag antibody; western blotting was performed with an anti-Myc-tag antibody. Representative results are shown from four independent experiments. (H) Pig articular chondrocytes were transfected with wild type (WT) gp130 or gp130 lacking four amino acids (S187–Y190; ΔS) and the levels of indicated proteins quantified by western blot. (I) Adult human chondrocytes were transfected with gp130-Flag and then incubated with IL-6, RCGD 423 or both; an anti-Flag antibody was used to immunoprecipitate gp130-associated proteins. Western blots for IL-6R and gp130-Flag were used to quantitate gp130/IL-6R interactions and normalise immunoprecipitated protein levels, respectively. Adult human chondrocytes transfected with gp130-Flag were incubated with various concentrations of RCGD 423 in the presence of oncostatin M (OSM) or IL-6; levels of immunoprecipitated (J) oncostatin M receptor (OSMR) or (K) IL-6R were used to calculate the dissociation constant (Kd) for each complex. For all panels, n=3.
To gain better understanding of how RCGD 423 could interact with gp130, we modelled its binding to gp130 using the Swissdock and Gold programmes; this revealed a potential high affinity binding site in domain 2 of the gp130 extracellular region (figure 4D). Notably, deletion of four residues proposed to interact with the drug (K151–R154) has been shown to occur in hepatocellular lesions, and overexpression of this gp130 mutant induced constitutive gp130/STAT3 signalling,36 while deletion of adjacent residues promoted unbridled gp130/STAT3 activation due to induction of stable homodimerisation of gp130. Based on these findings, we hypothesised that RCGD 423 may increase pSTAT3 by binding to domain 2 of gp130 and stabilising homodimers. To address this, we first transfected gp130−/− Ba/F3 cells (figure 4E)37 with plasmids encoding either full-length gp130 (WTgp130) or gp130 lacking domain 2 (gp130ΔD2) and cultured them with or without RCGD 423; in gp130ΔD2 cells, the compound did not induce increases in pSTAT3 and MYC (figure 4E,F). We then transfected cells with both Flag-tagged and Myc-tagged gp130 (figure 4G) and cultured them with or without RCGD 423. Cellular extracts were immunoprecipitated with an anti-Flag antibody and then subjected to western blot using an anti-Myc-tag antibody. Only in cells treated with RCGD 423 could we detect complexes containing both forms of gp130, demonstrating that RCGD 423 induced homodimers of gp130. Finally, to demonstrate that gp130 homodimerisation increases levels of pSTAT3 and MYC, we transfected pig articular chondrocytes with a mutant gp130 plasmid shown to form homodimers (ΔS36) and performed western blots (figure 4H).
To confirm that gp130 mediates the effects of RCGD 423 in vivo, we co-injected the compound into rat knee joints with either the gp130 inhibitor SC144 or a gp130 domain 2-specific blocking antibody;38 both abolished the effects of RCGD 423 (online supplementary figure 4A,B). These data demonstrate RCGD 423 acts via gp130 and support the hypothesis that the compound could inhibit IL-6 family cytokines by sequestering gp130 into homodimeric complexes. To directly address receptor competition, we transfected cells with gp130-Flag and treated them with IL-6, RCGD 423 or both and quantitated gp130/IL-6R interactions; inclusion of RCGD 423 dramatically reduced levels of this complex (figure 4I). Determination of the dissociation constant (Kd), a measure of the affinity between gp130 and its binding partners OSMR and IL-6R, in the presence of RCGD 423 and either OSM or IL-6 demonstrated a strong ability of the compound to interfere with ligand-mediated gp130 heterodimerisation (figure 4J,K). Overall, these data indicate that RCGD 423 promotes the formation of ligand-independent homodimers of gp130 that can prevent heterodimerisation with IL-6 family cytokine receptors. Although direct physical interaction of RCGD 423 with gp130 has not been experimentally demonstrated, the homodimer formation required the integrity of domain 2, which, based on in silico studies, is predicted to bind gp130.
RCGD 423 prevents AC degeneration in vivo
Based on these data, we hypothesised that RCGD 423 could promote cartilage retention following injury by inhibiting inflammation and hypertrophy and inducing proliferation. We first defined the half maximal effective concentration (EC50) of RCGD 423 on human articular chondrocytes by quantifying phosphorylation of gp130 and STAT3 as well as MYC protein levels; these results demonstrated an EC50 in the range of 4.5–7.2 μM (online supplementary figure 5). To address whether RCGD 423 could ameliorate cartilage degeneration, we adopted a rat partial meniscectomy model,39 in which 30%–50% of the meniscus is removed (online supplementary figure 7A). As a delivery vehicle, we employed US Food and Drug Administration-approved poly(lactic-co-glycolic) acid (PLGA) microspheres loaded with RCGD 423 (online supplementary figure 6A). Direct evaluation of offloading kinetics of RCGD 423 from PLGA microspheres showed that 20 μg of the compound could sustain a 0.1–0.3 μM concentration for at least 12 days (online supplementary figure 6B) in vitro, which would be fivefold to sevenfold higher in the context of a 150–200 μL rat joint.40 Based on these data, we designed a 6-week experiment in which operated animals would receive an intra-articular injection of 4 μg RCGD 423 loaded onto microspheres at the time of surgery and another 3 weeks later. This dosing strategy was projected to produce an average intra-articular concentration of ~0.3 μM RCGD 423, as this concentration consistently resulted in increased gp130 pathway activity in vitro (figure 4 and data not shown). Micro CT verified loss of cut meniscus (online supplementary figure 7B). We then used the Osteoarthritis Research Society International (OARSI) histological scoring system41 to quantify the extent of cartilage damage in all animals. Tissue sections were stained with Safranin O to detect proteoglycans (figure 5A); these results demonstrated dramatic loss of cartilage on the tibial plateau in control animals, while in RCGD 423-treated animals there was little to no cartilage degradation, structural damage or generation of osteophytes (figure 5A). Control animals showed increased levels of cartilage-degrading proteins and markers of hypertrophy, while these were mostly absent in RCGD 423-treated rats (figure 5B). To evaluate proliferation, animals were injected with RCGD 423 once a week and administered 5-ethynyl-2′-deoxyuridine (EdU) for 4 days after each injection; animals were sacrificed at 3 weeks and 6 weeks after meniscectomy (online supplementary figure 7C). These results demonstrated RCGD 423 enhanced the minimal chondrocyte proliferation occurring soon after surgery; later, the ability of the compound to induce proliferation was limited (figure 5C). Together, these data indicate that RCGD 423 can prevent cartilage degeneration in vivo, associated with decreasing levels of catabolic enzymes and an early proliferative response.
Figure 5.

Regulator of cartilage growth and differentiation (RCGD) 423 prevents articular cartilage degeneration in vivo. (A) Histological staining and quantitative assessment of cartilage degradation and changes in joint morphology of rat knee joints 6 weeks after partial meniscectomy surgery. RCGD 423-loaded or empty microspheres were injected intra-articularly at the time of surgery and 3 weeks later (online supplementary figure 7). Safranin O delineates proteoglycans; arrow indicates a representative osteophyte. Sham=joint capsule exposure but no meniscectomy. Scale bars represent 100 μm, n=8. (B) Sections of RCGD 423-treated or control joints were stained for matrix degrading enzymes and markers of hypertrophy. Representative images are shown; scale bars=25 μm. n=4. (C) Partial meniscectomies were performed on rats and RCGD 423 or saline injected intra-articularly immediately and at weeks 1 and 2 or at weeks 3, 4 and 5 (online supplementary figure 7); EdU was injected intraperitoneally each day for 4 days after each articular injection. EdU+ cells in articular cartilage were scored in four animals for each condition and time point; histone H3 was used to stain nuclei. Scale bars=25 μm.
RCGD 423 promotes cartilage repair in vivo
We then evaluated the ability of RCGD 423 to promote cartilage repair in a rat osteochondral defect model (online supplementary figure 8), in which new cartilage is generated by both synovial and bone marrow stromal cells.42,43 These defects spontaneously heal in 4 weeks, and cartilage repair was assessed at this time point;44 these results demonstrated a highly significant improvement in cartilage resurfacing in the presence of RCGD 423 (figure 6A). This result was further verified using traditional markers of chondrocyte identity (SOX9, COL 2) or hypertrophy (RUNX2, COL 10; figure 6B). In vitro experiments demonstrated the compound significantly decreased matrix degradation in cartilage pellets generated using pig synovial stromal cells (online supplementary figure 8A) in the presence of OSM, consistent with effects of RCGD 423 on cartilage explants exposed to either IL-6 or OSM. We hypothesised that enhanced repair may also be due to proliferation induced by RCGD 423. Two weeks after injury, EdU incorporation by cells in the defect was significantly increased in RCGD 423-treated rats; this effect was temporary, as at 4 weeks and 6 weeks when defects were fully repaired no difference was observed (figure 6C). Taken together, these data define the gp130 modulator RCGD 423 as an agent that can potentially improve cartilage healing following full-thickness injury by reducing catabolism and enhancing the proliferative response.
Figure 6.

Regulator of cartilage growth and differentiation (RCGD) 423 promotes cartilage repair following osteochondral injury. (A) Full-thickness osteochondral defects were created in the patellar grooves of rats. Saline (vehicle) or RCGD 423 were injected intra-articularly at the time of surgery and weekly afterwards. Animals were sacrificed at the indicated time points for Safranin O staining and histological scoring (4 weeks; n=8). Scale bars=100 μm. (B) Markers of chondrocyte identity, hypertrophy and fibrosis were assessed on the cells present in the defects of RCGD 423-treated and saline-treated animals. n=4. Scale bars=25 μm. (C) Osteochondral defects and treatments were conducted as in (A); EdU was injected intraperitoneally for 4 days after each treatment (online supplementary figure 8). EdU+ cells in the defects were scored in four animals for each condition and time point; histone H3 was used to stain nuclei. Scale bars=25 μm.
DISCUSSION
Hypertrophy driven by IL-6 family cytokines has emerged as one driver of OA. Here we show that small molecule-mediated modulation of gp130 signalling can bias output downstream of this pleiotropic pathway against hypertrophic, procatabolic effects and promote chondrocyte proliferation in vivo. We identified RCGD 423 based on its ability to inhibit developmental hypertrophy in mouse limb mesenchymal cells, and these effects were conserved in preventing disease-based hypertrophy in chondrocytes from human patients with OA. Molecularly, RCGD 423 promotes formation of active homodimers signalling primarily via pSTAT3/MYC; this mechanism of action can actively compete against IL-6 family cytokine-mediated heterodimerisation, thereby inhibiting the hypertrophic and catabolic effects of this pathway mediated by ERK1/2 and NF-κB. Importantly, gp130 signal modulation occurs in chondrocytes, synoviocytes and PBMCs, thus providing a means to combat the proinflammatory, procatabolic milieu found in destabilised and full-thickness injured rat joints. Together, our results provide additional insight into IL-6 family cytokine signalling in chondrocytes and nominate gp130 signal modulation as a potential therapeutic strategy for OA.
The function of IL-6 family members in cartilage biology and pathogenesis has been the focus of much study (reviewed in reference 4545). IL-6, OSM and LIF have been shown to promote OA, either through acting as proinflammatory cytokines or directly regulating matrix destruction.13,25,26 Consequently, all members of the IL-6 family are often considered to be detrimental to chondrocyte biology. However, IL-6 has been shown to be chondroprotective in ageing mice,46 in agreement with our data that during human development and when output of gp130 is modulated this pathway can be beneficial. Our data show that individual cytokines activate MAPK and NF-κB differently and result in varied transcriptional responses of catabolic genes (figure 2). Moreover, OSM elicits minimal upregulation of pSTAT3 or MYC, while IL-6 and LIF both increase levels of these proteins. These data suggest that there is great diversity in response to IL-6 family members in chondrocytes and that individual cytokines (eg, IL-6 and LIF) can have both positive (induction of proliferation) and negative (increases in NF-κB and catabolic gene expression) effects. RCGD 423 represents an interesting new facet of this story, as it uncouples some of the effects downstream of gp130 signalling, such as MAPK/NF-κB activation and catabolic gene expression, from STAT3/MYC activation. This bimodal mechanism of action will need to be evaluated in the context of more advanced cartilage injury to assess the ability of RCGD 423 to reverse pre-existing damage. Moreover, the compound can compete against procatabolic signalling mediated by both IL-6 and OSM, presenting an advantage over existing anti-IL-6/IL-6R therapies.
Although proliferation of adult articular chondrocytes is minimal (figure 5), we found moderate upregulation of proliferation induced by RCGD 423 both in vitro and in vivo. In vivo, EdU+ cells were found in both the most superficial as well as adjacent layers of cartilage, suggesting that a surprising number of resident chondrocytes are capable of responding to activation signals present in the injured joint. Chondrocyte cloning has been well documented in the past, but this is typically associated with later stages of disease progression;2 RCGD 423 significantly increased rates of EdU incorporation, and it will be critical in future work to determine the molecular identity of these cells as well as to address the relative contributions of proliferation versus anticatabolic effects to the reduction in cartilage degeneration supported by RCGD 423. Moreover, given the decrease in proliferation elicited by the compound at 6 weeks postinjury, it will be important to ascertain if cells capable of responding are limited in the number of divisions they can undergo as this would suggest that other cell types such as synovium43 may need to be harnessed for cartilage repair or that a therapeutic window exists for potential intervention.
LIF and other members of the IL-6 family have been shown to mediate proliferation and regeneration in a variety of cellular contexts. IL-6 and OSM have both been shown to be important for the regenerative response in the liver, acting upstream of STAT3 to promote proliferation.47 As we demonstrate here, levels of pSTAT3 and MYC are high in rapidly growing fetal tissues; temporary and controlled upregulation by a more MYC-inducing RCGD 423 analogue, recently synthesised by our group, may be beneficial for tissue repair (online supplementary figure 9) as was recently shown by Flores et al48 in the context of acceleration of the hair cycle. Intriguingly, Ocampo et al49 recently demonstrated that repeated, transient increases in systemic MYC protein improved regeneration and did not result in tumorigenesis. These results suggest that both intra-articular, as well as systemic administration of RCGD 423 may represent potential strategies to promote tissue repair; however, close monitoring of proliferation in all tissues will be critical. Our work indicates that gp130 can function as a node whose modulation may tip the balance in pathological conditions away from tissue degeneration and towards repair. In summary, we have identified a novel small molecule modulator of gp130 signalling that demonstrates prominent disease-modifying activity in two rat models of cartilage injury or/and degeneration. Optimised analogues of this compound may represent attractive therapeutic candidates for patients with both degenerative and inflammatory forms of arthritis.
Supplementary Material
Acknowledgements
The authors thank Ms Felicia Codrea and Ms Jessica Scholes of UCLA for their help with sample analysis and sorting. The authors also thank Dr Xinmin Li from UCLA CMC for his help with sequencing experiments and Dr Kenneth Dorshkind (UCLA) for critical reading of the manuscript, as well as Dr Zucman E Rossi for sharing reagents.
Funding
This work is supported by NIH grant K01AR061415, Department of Defense grant W81XWH-13-1-0465, California Institute for Regenerative Medicine grant RB5-07230, a USC Stevens Technology Advancement Grants award and a Wright Foundation award (all to DE) as well as a postdoctoral scholarship from Fundação de Amparoà Pesquisa do Estado de São Paulo E FAPESP (grant# 2015/08952E6) to CEF.
Footnotes
Additional material is published online only. To view please visit the journal online (http://dx.doi.org/10.1136/annrheumdis-2017-212037).
Correction notice This article has been corrected since it published Online First. The affiliation for Mohammad Parvez Alam has been corrected.
Competing interests DE, BVH and VJ are inventors onPCT/US16/20126. DE and BVH are cofounders and shareholdersof CarthroniX.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Primer sequences are available upon request; please contact the corresponding authors.
REFERENCES
- 1.Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health 2009;1:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage 2012;20:223–32. [DOI] [PubMed] [Google Scholar]
- 3.Saito T, Fukai A, Mabuchi A, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med 2010;16:678–86. [DOI] [PubMed] [Google Scholar]
- 4.Yang S, Kim J, Ryu JH, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med 2010;16:687–93. [DOI] [PubMed] [Google Scholar]
- 5.Yasuhara R, Ohta Y, Yuasa T, et al. Roles of β-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab Invest 2011;91:1739–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dowthwaite GP, Bishop JC, Redman SN, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci 2004;117(Pt 6):889–97. [DOI] [PubMed] [Google Scholar]
- 7.Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis 2013;5:77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheller J, Chalaris A, Schmidt-Arras D, et al. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011;1813:878–88. [DOI] [PubMed] [Google Scholar]
- 9.Ogura H, Murakami M, Okuyama Y, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008;29:628–36. [DOI] [PubMed] [Google Scholar]
- 10.B. Marcu K, Otero M, Olivotto E, et al. Goldring MB: NF-κB Signaling: Multiple angles to target OA. Current drug targets 2010;11:599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasadam I, van Gennip S, Friis T, et al. ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum 2010;62:1349–60. [DOI] [PubMed] [Google Scholar]
- 12.Beekhuizen M, van Osch GJ, Bot AG, et al. Inhibition of oncostatin M in osteoarthritic synovial fluid enhances GAG production in osteoarthritic cartilage repair. Eur Cell Mater 2013;26:80–90. [DOI] [PubMed] [Google Scholar]
- 13.Hui W, Rowan AD, Richards CD, et al. Oncostatin M in combination with tumor necrosis factor alpha induces cartilage damage and matrix metalloproteinase expression in vitro and in vivo. Arthritis Rheum 2003;48:3404–18. [DOI] [PubMed] [Google Scholar]
- 14.Jikko A, Wakisaka T, Iwamoto M, et al. Effects of interleukin-6 on proliferation and proteoglycan metabolism in articular chondrocyte cultures. Cell Biol Int 1998;22:615–21. [DOI] [PubMed] [Google Scholar]
- 15.Nasi S, So A, Combes C, et al. Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann Rheum Dis 2016;75:1372–9. [DOI] [PubMed] [Google Scholar]
- 16.Legendre F, Dudhia J, Pujol JP, et al. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression. J Biol Chem 2003;278:2903–12. [DOI] [PubMed] [Google Scholar]
- 17.Latourte A, Cherifi C, Maillet J, et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis 2017;76:748–55. [DOI] [PubMed] [Google Scholar]
- 18.Ryu JH, Yang S, Shin Y, et al. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum 2011;63:2732–43. [DOI] [PubMed] [Google Scholar]
- 19.Livshits G, Zhai G, Hart DJ, et al. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: The Chingford Study. Arthritis Rheum 2009;60:2037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo M, Yamaoka K, Sakata K, et al. Contribution of the Interleukin-6/STAT-3 Signaling Pathway to Chondrogenic Differentiation of Human Mesenchymal Stem Cells. Arthritis Rheumatol 2015;67:1250–60. [DOI] [PubMed] [Google Scholar]
- 21.Hall MD, Murray CA, Valdez MJ, et al. Mesoderm-specific Stat3 deletion affects expression of Sox9 yielding Sox9-dependent phenotypes. PLoS Genet 2017;13:e1006610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwamoto M, Yagami K, Lu Valle P, et al. Expression and role of c-myc in chondrocytes undergoing endochondral ossification. J Biol Chem 1993;268:9645–52. [PubMed] [Google Scholar]
- 23.Moran EM, Mullan R, McCormick J, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies. Arthritis Res Ther 2009;11:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotz M, Moats T, Villiger PM. Leukemia inhibitory factor is expressed in cartilage and synovium and can contribute to the pathogenesis of arthritis. J Clin Invest 1992;90:888–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henrotin YE, De Groote DD, Labasse AH, et al. Effects of exogenous IL-1 beta, TNF alpha, IL-6, IL-8 and LIF on cytokine production by human articular chondrocytes. Osteoarthritis Cartilage 1996;4:163–73. [DOI] [PubMed] [Google Scholar]
- 26.Hui W, Bell M, Carroll G. Soluble glycoprotein 130 (gp130) attenuates OSM- and LIF-induced cartilage proteoglycan catabolism. Cytokine 2000;12:151–5. [DOI] [PubMed] [Google Scholar]
- 27.Maye P, Fu Y, Butler DL, et al. Generation and characterization of Col10a1-mcherry reporter mice. Genesis 2011;49:410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L, Bluguermann C, Kyupelyan L, et al. Human developmental chondrogenesis as a basis for engineering chondrocytes from pluripotent stem cells. Stem Cell Reports 2013;1:575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunziker EB. Articular cartilage repair: basic science and clinical progress. A review of the current status and prospects. Osteoarthritis Cartilage 2002;10:432–63. [DOI] [PubMed] [Google Scholar]
- 30.Janusz MJ, Little CB, King LE, et al. Detection of aggrecanase- and MMP-generated catabolic neoepitopes in the rat iodoacetate model of cartilage degeneration. Osteoarthritis Cartilage 2004;12:720–8. [DOI] [PubMed] [Google Scholar]
- 31.Hiscock DR, Caterson B, Flannery CR. Expression of hyaluronan synthases in articular cartilage. Osteoarthritis Cartilage 2000;8:120–6. [DOI] [PubMed] [Google Scholar]
- 32.Mori Y, Saito T, Chang SH, S-h C, et al. Identification of fibroblast growth factor-18 as a molecule to protect adult articular cartilage by gene expression profiling. J Biol Chem 2014;289:10192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito K, Maruyama Z, Sakai A, et al. Overexpression of Cdk6 and Ccnd1 in chondrocytes inhibited chondrocyte maturation and caused p53-dependent apoptosis without enhancing proliferation. Oncogene 2014;33:1862–71. [DOI] [PubMed] [Google Scholar]
- 34.Zeller KI, Jegga AG, Aronow BJ, et al. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 2003;4:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu S, Grande F, Garofalo A, et al. Discovery of a novel orally active small-molecule gp130 inhibitor for the treatment of ovarian cancer. Mol Cancer Ther 2013;12:937–49. [DOI] [PubMed] [Google Scholar]
- 36.Rebouissou S, Amessou M, Couchy G, et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009;457:200–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stuhlmann-Laeisz C, Lang S, Chalaris A, et al. Forced dimerization of gp130 leads to constitutive STAT3 activation, cytokine-independent growth, and blockade of differentiation of embryonic stem cells. Mol Biol Cell 2006;17:2986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chevalier S, Fourcin M, Robledo O, et al. Interleukin-6 family of cytokines induced activation of different functional sites expressed by gp130 transducing protein. J Biol Chem 1996;271:14764–72. [DOI] [PubMed] [Google Scholar]
- 39.Lozoya KA, Flores JB. A novel rat osteoarthrosis model to assess apoptosis and matrix degradation. Pathol Res Pract 2000;196:729–45. [PubMed] [Google Scholar]
- 40.Dowd E, McQueen DS, Chessell IP, et al. P2X receptor-mediated excitation of nociceptive afferents in the normal and arthritic rat knee joint. Br J Pharmacol 1998;125:341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerwin N, Bendele AM, Glasson S, et al. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rat. Osteoarthritis Cartilage 2010;18(Suppl 3):S24–34. [DOI] [PubMed] [Google Scholar]
- 42.Eltawil NM, De Bari C, Achan P, et al. A novel in vivo murine model of cartilage regeneration. Age and strain-dependent outcome after joint surface injury. Osteoarthritis Cartilage 2009;17:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roelofs AJ, Zupan J, Riemen AHK, et al. Joint morphogenetic cells in the adult mammalian synovium. Nat Commun 2017;8:15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Driscoll SW, Keeley FW, Salter RB. Durability of regenerated articular cartilage produced by free autogenous periosteal grafts in major full-thickness defects in joint surfaces under the influence of continuous passive motion. A follow-up report at one year. J Bone Joint Surg Am 1988;70:595–606. [PubMed] [Google Scholar]
- 45.Kapoor M, Martel-Pelletier J, Lajeunesse D, et al. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 2011;7:33–42. [DOI] [PubMed] [Google Scholar]
- 46.de Hooge AS, van de Loo FA, Bennink MB, et al. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage 2005;13:66–73. [DOI] [PubMed] [Google Scholar]
- 47.Li W, Liang X, Kellendonk C, et al. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem 2002;277:28411–7. [DOI] [PubMed] [Google Scholar]
- 48.Flores A, Schell J, Krall AS, et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat Cell Biol 2017;19:1017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ocampo A, Reddy P, Martinez-Redondo P, et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016;167:1719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.