

Abstract
White matter stroke (WMS) occurs as small infarcts in deep penetrating blood vessels in the brain and affects the regions of the brain that carry connections, termed the subcortical white matter. WMS progresses over years and has devastating clinical consequences. Unlike large grey matter strokes, WMS disrupts the axonal architecture of the brain and depletes astrocytes, oligodendrocyte lineage cells, axons and myelinating cells, resulting in abnormalities of gait and executive function. An astrocytic cell-based therapy is positioned as a strong therapeutic candidate after WMS. In this study we report, the reliable generation of a novel stem cell-based therapeutic product, glial enriched progenitors (GEPs) derived from human induced pluripotent stem cells (hiPSCs). By transient treatment of hiPSC derived neural progenitors (hiPSC-NPCs) with the small molecule deferoxamine, a prolyl hydroxylase inhibitor, for three days hiPSC-NPCs become permanently biased towards an astrocytic fate, producing hiPSC-GEPs. In preparation for clinical application, we have developed qualification assays to ensure identity, safety, purity, and viability of the cells prior to manufacture. Using tailored q-RT-PCR-based assays, we have demonstrated the lack of pluripotency in our final therapeutic candidate cells (hiPSC-GEPs) and we have identified the unique genetic profile of hiPSC-GEPs that is clearly distinct from the parent lines, hiPSCs and iPSC-NPCs. After completion of the viability assay, we have stablished the therapeutic window of use for hiPSC-GEPs in future clinical applications (7 h). Lastly, we were able to reliably and consistently produce a safe therapeutic final product negative for contamination by any human or murine viral pathogens, selected bacteria, common laboratory mycoplasmas, growth of any aerobes, anaerobes, yeast, or fungi and 100 times less endotoxin levels than the maximum acceptable value. This study demonstrates the reliable and safe generation of patient derived hiPSC-GEPs that are clinically ready as a cell-based therapeutic approach for WMS.
Keywords: hiPSC, Astrocytes, White matter, Stroke, Manufacturing, Assays
1. Introduction
The ability to generate human induced pluripotent stem cells (hiPSCs) from fibroblasts has created a vast potential in the field of regenerative medicine. This was contingent on the advent of cell reprogramming via exogenous expression of transcription factors (Takahashi and Yamanaka, 2006). The use of hiPSCs has expanded research in disease modeling and tissue differentiation (Robinton and Daley, 2012). Controlled differentiation into a target cell type can provide insight to mechanisms of disease and repair and offer potential for medical application. Using human stem cell-based therapies to target diseases and injury of the central nervous system (CNS) has been of immense interest due to limitations of endogenous repair in the human brain (Goldman, 2016).
White matter stroke (WMS) constitutes 30% of all stroke subtypes. WMS occurs as small infarcts in deep penetrating blood vessels in the brain and affects the regions of the brain that carry connections, termed the subcortical white matter. WMS disrupts the axonal architecture of the brain and depletes astrocytes, oligodendrocyte lineage cells, axons and myelinating cells (Rosenzweig and Carmichael, 2015; Sozmen et al., 2012, 2009; Llorente et al., 2021). WMS progresses over years and has devastating clinical consequences (Vernooij et al., 2007; Knopman et al., 2011). These infarcts produce hemiparesis with incomplete recovery and accumulate to produce gait abnormalities, verbal processing deficits and difficulties in executive functioning that present as vascular dementia (Marin and Carmichael, 2018). Currently, there are no therapeutic approaches for this disease.
Prior studies have shown recovery of neurological functions in stroke models following stem/progenitor cell transplantation (Bliss et al., 2007; Chu et al., 2004; Jeong et al., 2003; Kelly et al., 2004; Kokaia et al., 2018; Llorente et al., 2021). Therefore, a cell-based therapy that can replace lost glia and induce structural repair in WMS is of great promise. hiPSC derived neural progenitor cells (hiPSC-NPCs) have the potential to differentiate into neurons and glia. However, these cells have a bias towards a neuronal lineage (Elkabetz et al., 2008). To address this obstacle, we have previously described a protocol that biases hiPSC-NPCs towards an astrocytic fate via modulation of oxygen tension in culture (Xie et al., 2014; Xie and Lowry, 2018; Llorente et al., 2021). This process can be mimicked by a brief exposure to the prolyl hydroxylase inhibitor deferoxamine (DFX). DFX is added to hiPSC-NPC cultures for a 3-day period to produce this astrocyte-biased differentiation process (Llorente et al., 2021). This approach allows for rapid and efficient production of astrocytes derived from an hiPSC source (hiPSC-GEPs).
hiPSC-GEPs possess many advantages over current cell-based therapies in development. First, hiPSC-GEPs can theoretically be produced in a manner that is at least 4 times faster than protocols which produce similar cell types (Kokaia et al., 2018; Wang et al., 2013; Llorente et al., 2021). Second, beyond this increase in speed of differentiation, DFX confers a permanent ‘switch’ in cell fate that should eliminate issues with lineage restriction that have previously hampered safe use in clinical practice (Goldman, 2016; Llorente et al., 2021). Third, the use of specific cells (hiPSC-GEPs) as opposed to more general neural or progenitor cells currently used in trials for CNS repair (mesenchymal stromal cells, fetal neural progenitors and ES/iPSC-derived neural progenitors) will likely confer a decrease in off-target effects and an increase in repair capacity (Wang et al., 2016; Steinbeck and Studer, 2015). Despite these enticing prospects, hiPSC-GEPs have yet to be shown to be a consistent, scalable, and a safe therapeutic candidate. Previous work on cell-based therapies that target the eye, CNS, and bone marrow has paved the way for effectively evaluating the clinical prospects for novel hiPSC-derived cell lines (Steinbeck and Studer, 2015; Hazim et al., 2017; Karumbayaram et al., 2012; Park et al., 2015).
This paper reports, reliable generation of the novel stem cell-based therapeutic product hiPSC-GEP that is permanently biased towards an astrocytic fate. We perform in vitro characterization of the hiPSC-GEPs and define a clear transcriptomic profile specific to the cells’ lineage. Additionally, we have developed the safety, purity, and viability assays of the final therapeutic product in preparation for possible clinical application. The ability to differentiate patient derived hiPSCs into glial enriched progenitors provides a strong candidate for a cell-based therapy to target white matter stroke and other neurodegenerative diseases.
2. Materials and methods
2.1. Derivation of fibroblasts
Fibroblast derivation and hiPSC generation were performed in an established good manufacturing practice (GMP) facility at the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA – California Institute for Regenerative Medicine Shared Research Laboratories. hiPSC-NPC and hiPSC-GEP differentiation was performed in a laboratory within the UCLA Department of Molecular, Cell, and Developmental Biology.
Incubation with 1 mg/mL AOF Collagenase A (LS00415; Worthington Biochemical) for 1 h was used to dissociate fibroblasts from 1 mm2 skin biopsies. Isolated cells were plated onto CellStart (A1014201; Gibco) coated plates in FibroGRO medium (SCM037; EMD Millipore). Confluent cells were then dissociated and passaged using TrypLE Select (12563-011; Invitrogen). Purity of these cells was analyzed by determining the fraction of cells expressing fibroblast markers and only cells stable in culture for 5 or more passages continued through the workflow.
2.2. Generation and maintenance of hiPSCs
Stable fibroblasts were transfected with the Venezuelan Equine Encephalitis (VEE) virus RNA Replicon RNA system from EMD Millipore (SCR550). This system enables the reprogramming of fibroblasts to ‘xeno-free’ hiPSCs via use of a non-integrating synthetic polycistronic RNA replicon expressing OCT4, SOX2, GLIS1, and KLF4. Selection of successfully transfected fibroblasts was performed with puromycin and B18R (GF156; EMD Millipore) for 9–11 days. Upon removal of B18R, and subsequent elimination of the RNA replicon, cells were passaged onto Matrigel (Corning) coated plates. Following 3–4 weeks of maintenance, selected hiPSC colonies were re-plated and maintained on feeder-free Matrigel coated plates with a 1:1 formulation of TeSR2 (Stem Cell Technologies) and NutriStem medium (Stemgent). A total of four hiPSC lines, each from a unique donor, were successfully generated through this process and subsequently used in this study.
Once generated, hiPSCs were cultured as described previously in accordance with UCLA Embryonic Stem Cell Research Oversight committee (Awe et al., 2013). Feeder-free hiPSC lines were maintained with mTeSR1 (Stem Cell Technologies) on Matrigel and passaged mechanically using ReLeSR (Stem Cell Technologies).
2.3. Differentiation of hiPS-NPCs and hiPS-GEPs
We previously described a protocol for the accelerated generation of lineage-restricted glial enriched progenitors from a population of hiPSCs via modulation of apparent oxygen tension (Xie et al., 2014; Xie and Lowry, 2018; Llorente et al., 2021). Rosettes were generated by mechanically passaging large, sparse hiPSC colonies onto Matrigel coated plates in mTeSR1 for 4 days. Media was changed daily. mTeSR1 was then replaced by Rosette Media + 2i containing Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 with N2 and B27 supplements (Invitrogen), 20 ng/mL basic fibroblast growth factor (FGF) (R&D Systems), 1 uM retinoic acid (RA; Sigma), 1 uM Purmorphamine (Stem Cell Technologies), 2 uM SB431542 (Stem Cell Technologies), and 0.05 uM LDN193189 (Stem Cell Technologies). This media was changed twice daily. Following 6–10 days of growth, rosettes were picked using tweezers and plated onto Matrigel coated plates in hiPSC-NPC medium containing DMEM/F12, N2 and B27, 20 ng/mL basic FGF, and 50 ng/mL EGF (GIBCO). hiPSC-NPC media was changed every other day. hiPSC-NPCs in this media were twice expanded and homogenized using TrypLE to achieve a uniform population of twice-passaged hiPSC-NPCs (P2 hiPSC-NPCs). To permanently bias hiPSC-NPCs towards an astrocytic fate (hiPSC-GEPs) deferoxamine (Cayman Chemicals) was administered to the hiPSC-NPCs as follows: Day1: 50 uM and Days 2 and 3: 100 uM. As this bias in fate is permanent, generated hiPSC-GEPs (P2 hiPSC-GEPs) were then maintained in hiPSC-NPC media.
2.4. Karyotype test
Karyotyping by G-banding on all four hiPSC lines, as well as their differentiated hiPSC-GEP counterpart, was performed by Cell Line Genetics. PluriTest, an assessment of pluripotency, was conducted by Life Technologies on representative cells from all produced populations (3 cell types × 4 donors).
2.5. Pluripotency test
To determine the pluripotency of our final product (hiPSC-GEPs), the transcriptome of all samples was analyzed and processed in the PluriTest™ algorithm to generate a Pluripotency and Novelty Score.
The Pluripotency Score is based on many samples (pluripotent, somatic, and tissues) in the stem cell model matrix which consists of an extensive reference set of > 450 cell/tissue types, including 223 hESC lines, 41 hiPSC lines, somatic cells, and tissues. Samples with positive pluripotency values are more similar to the pluripotent samples in the model matrix than to all other classes of samples in the matrix. The Novelty Score is based on well-characterized PSCs in the stem cell model matrix. A low novelty score indicates that the tested sample can be well reconstructed based on existing data from other well-characterized hiPSC and hESC lines.
Cells were prepared according to the PureLink™ RNA Mini Kit and quantified using a NanoDrop™. Purified RNA samples were prepared according to the DNA-free™ Kit to remove potential contaminating genomic DNA. Finally, 100 ng total RNA was used to prepare the GeneChip® for the PluriTest™. Samples were then analyzed using the PluriTest™ algorithm that integrates gene expression data to authenticate pluripotency status.
2.6. Commercial assessment of pathogenic contamination
For safety assessment, all four hiPSC-GEP lines at clinically predicted concentrations were tested for presence of endotoxin via Limulus Amebocyte Lysate challenge (Kinetic Chromogenic Microplate Testing; Charles River), bacteriostatic/fungistatic properties (VRL Eurofins), and microbial growth within 14 days of inoculation into liquid media (USP < 71>; VRL Eurofins). The four hiPSC-GEP lines were also tested for human viral pathogens (h-IMPACT I; IDEXX BioAnalytics), murine viral pathogens (IMPACT VIII; IDEXX BioAnalytics), and mycoplasma (h-IMPACT I and IMPACT VIII; IDEXX BioAnalytics) via proprietary PCR.
2.7. Immunohistochemistry and confocal microscopy
Immunofluorescent staining was performed as described (Patterson et al., 2014). P2 hiPSC-NPCs were thawed onto 24 well flat-bottomed Bio-One SensoPlates (Greiner). After reaching confluency, cells were fixed with 4% (w/v) paraformaldehyde (Electron Microscopy Sciences) in PBS. Antibodies used include the following mouse HuNu (Abcam), goat anti-Sox2 (Abcam), rabbit anti-Pax6 (Biolegend), and DAPI (Thermo Fisher).
High-resolution confocal images in Z-stacks were acquired (Nikon C2). Images were taken at 20x magnification with the same parameters for scanning across all wells. NIS-Elements JOBS acquisition and analysis designer software performed 3D analysis of cell number with the same threshold for each channel for all wells. The percentage of SOX2 and PAX6 positive neural progenitors was calculated with a positive staining area from each marker and normalized based on cell number (DAPI staining) via JOBS analysis. P-value was calculated with Student’s t-test.
2.8. Quantitative real-time PCR (q-RT-PCR)
Samples of messenger ribonucleic acid (mRNA) for quantitative real-time polymerase chain reaction (q-RT-PCR) were obtained as previously described (Anuncibay-Soto et al., 2014). Briefly, total RNA was extracted using the Zymo Research Quick-RNA Microprep Kit. The high-capacity complementary deoxyribonucleic acid (cDNA) reverse transcription kit (Applied Biosystems, Foster City, CA, USA) was used to perform reverse transcription following the manufacturer’s instructions. Reactions were carried out for 10 min at 25 °C, 2 h at 37 °C and heated to 85 °C for 5 s to end the reaction.
Real-time PCR was carried out on a Roche LightCycler 480 Instrument II. A LightCycler® 480 SYBR Green I Master Mix (LifeScience, Roche) was used, and the following settings were programmed: 1 cycle of 30 s at 95 °C, 40 cycles of 10 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C, 1 cycle of 5 s at 95 °C, 1 min at 65 °C, and continuous at 97 °C, and 1 cycle of 30 s at 40 °C. Cycle thresholds (Ct) for the different genes were selected immediately above the baseline and within the linear range on the log scale. Each reaction (10 μL) was made using 5 nM of each primer (Table 1), 1 μL cDNA aliquot, 5 μL of SYBR® Green PCR Master Mix and 3.4 μL of H2O2 molecular grade water.
Table 1.
Primers used in quantitative real-time PCR studies.
| Gene Name | Sequence |
|---|---|
| TFRC | TCT GGA TAA AGC GGT TCT TGG |
| CCC AGT TGC TGT CCT GAT ATA G | |
| ADM | CTG CCC AGA CCC TTA TTC G |
| GTT CAT GCT CTG GCG GTA G | |
| PDK1 | CAA GAG TTG CCT GTC AGA CTG |
| CAA GAA GCT CCT GAA GAC TCT G | |
| RAB20 | CGC TCC TAC AAC ATC TCC ATC |
| TCC TCC AGC TCC ACC AG | |
| STC2 | GAG AAC AAC TCT TGT GAG ATT CG |
| CGT CTT TGA TGA ATG ACT TGC C | |
| SLC16A3 | ACA AGT TCT CCA GTG CCA TT |
| GTA GAC GTG GGT CGC ATC | |
| PLOD2 | GAG CAT TGA GTC CTG ATG GAT A |
| CTG ATC GGA GTG TCT TTC CTT | |
| GAPDH | ACA TCG CTC AGA CAC CAT G |
| TGT AGT TGA GGT CAA TGA AGG G | |
| OCT4 | TGT GTC TAT CTA CTG TGT CCC A |
| GTT GGA GGG AAG GTG AAG TTC | |
| NANOG | CCT TCT GCG TCA CAC CAT T |
| AAC TCT CCA ACA TCC TGA ACC |
Increases in fluorescence of SYBR® Green during the amplification process were analyzed with Sequence Detector software (Roche). Fold changes for the different comparisons were expressed as (2 − ΔCt), where ΔCt = Cttarget − CtGAPDH (Livak and Schmittgen, 2001). Ct values correspond °C to the cycle number at which the fluorescence signal crossed the designated threshold. Experiments were performed in accordance with the Minimum Information for Publication of Quantitative Real-time PCR Experiments (MIQE) guidelines (Bustin et al., 2009).
2.9. RNA-Sequencing
4 months post-DFX treatment total RNA was isolated from hiPSC-NPCS and hiPCS-GEPs using a RNeasy Mini Kit (QIAGEN). Library preparation was performed using TruSeq Standard RNA LT Kit (Illumina) following the standard total RNA sample preparation protocol. The sequencing reactions were run on HiSeq 2000 as single-end 100 bp.
2.10. RNA-Sequencing analysis
Tophat was used to align reads to the hg19 genome assembly using the default settings in Galaxy (http://galaxy.hoffman2.idre.ucla.edu). Multimappers, unmapped reads, and low-quality alignments were excluded from analysis. Counts obtained using featureCounts with Gencode annotations were analyzed with the R package edgeR, which uses a negative binomial generalized log-linear model. An unadjusted p-value threshold of 0.05 was imposed for the likelihood ratio test to select differentially expressed genes.
3. Results
3.1. Experimental design
A total of four hiPSC lines, each from a unique donor, have been successfully generated and differentiated into hiPSC-NCPS and hiPSC-GEPs on this study (3 cell types X 4 different patients). Furthermore, we have tested 5 hiPSC-NPCs and hiPSC-GEPs independent batches from each donor and found no significant differences between batches in any of the qualification assays described in this study (data not shown).
3.2. Reliable generation of hiPSC-NPCs and hiPSC-GEPs
The protocol for the rapid production of hiPSC-NPCs from hiPSCs through a rosette intermediate is well-established (Chambers et al., 2009). However, protocols for the generation of a lineage restricted glial enriched progenitor are less well defined and quite lengthy (~200 days) (Bliss et al., 2007). Recently, we reported a method to accelerate this process through modulation of the apparent oxygen tension in culture via small molecules such as deferoxamine (Xie et al., 2014; Xie and Lowry, 2018). First, we curated a protocol combining the established method for the production of hiPSC-NPCs and the generated a homogenous population of P2 hiPSC-NPCs from each of four hiPSC lines within 30 days. This finding is confirmed via staining for SOX2 and PAX6, established markers of neural progenitors (Fig. 1B). Quantification of this staining revealed that all putative neural progenitor populations achieved the previously reported threshold of 60% SOX2/PAX6 expression (Fig. 1C).
Fig. 1.

Generation of hiPSC-GEPs. (A) ~ 35-day workflow to produce hiPSC-GEPs from patient derived iPSCs. (B) Confocal staining of hiPSC-NPCs from all four donors. Cells are stained with the nuclear stain DAPI, the neural progenitor markers SOX2 and PAX6, and the human lineage marker HuNu. (C) Quantification of cells expressing PAX6/SOX2. All populations expressed > 60% confirming their hiPSC-NPC identity.
Following confirmation of hiPSC-NPC identity, we treated a subset of each population with a 3-day course of DFX (Day 1: 50 uM, Days 2 and 3: 100 uM). In doing so, we successfully generated hiPSC-GEPs from all four donors within a total of 35 days.
To determine cell specificity of the newly developed hiPSC-GEPs we have performed a transcriptional analysis 4 months after DFX treatment. This approach confirms the fate commitment of the cells using detailed gene expression data, and also at this long-term time point, also tests the permanency of this fate commitment. The transcriptional analysis showed that astrocyte specific genes were significantly induced in hiPSC-GEPs, such as S100A10 and CD44, while neuron, oligodendrocyte, or oligodendrocyte progenitor cell (OPC)-related genes are suppressed (Fig. 2). These results show that hiPSC-GEPs retained a long-term astrocyte differentiation bias even after brief treatment in vitro with DFX.
Fig. 2.
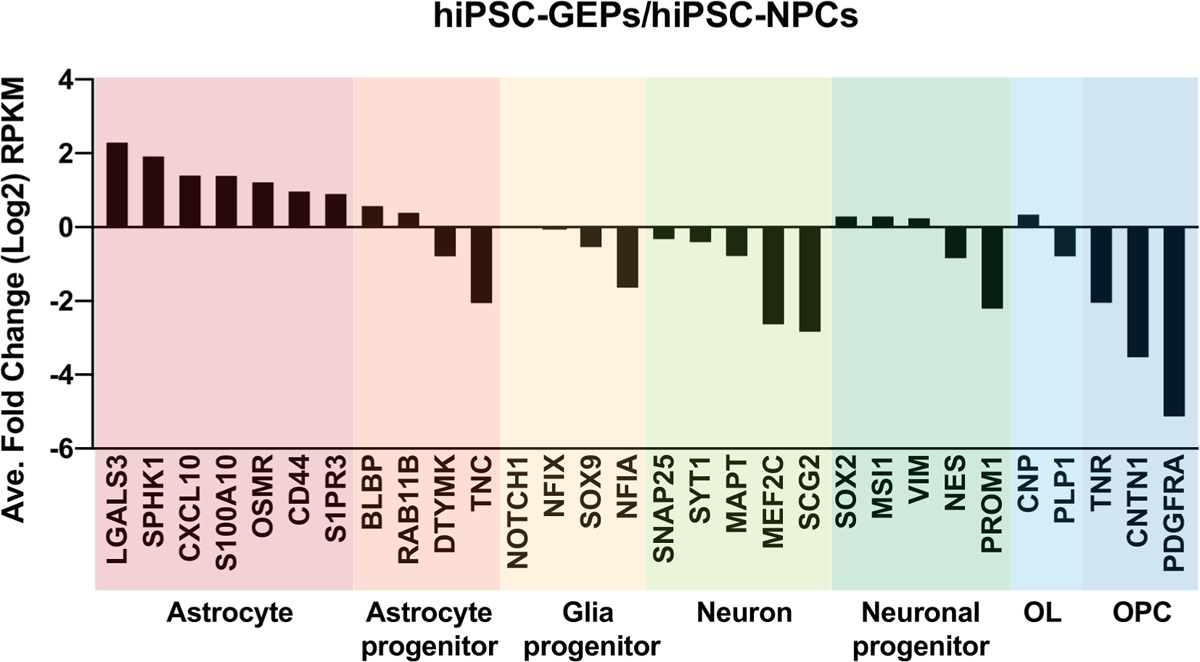
hiPSC-GEPs transcriptional profile. Panel shows relative fold change (hiPSC-GEP versus hiPSC-NPC) of cell type specific genes in log scale by RNA-seq (RPKM).
As hiPSC-GEPs are a terminal cell type and putative therapeutic candidate, karyotyping by G-banding was performed on all four populations of cells, every batch of cells analyzed presented a normal karyotype (Fig. 3A; Cell Line Genetics). Confirmation of lineage restriction of hiPSC-NPCs and hiPSC-GEPs was analyzed and processed in the PluriTest™ algorithm to generate a Pluripotency and Novelty Score (Fig. 3B; Life Technologies). As observed on Fig. 2B, 4 representative batches of hiPS presented the highest pluripotency score values and cluster tightly together. However, the 4 representative batches of hiPSC-NPCs and hiPSC-GEPs analyzed, lost their pluripotency (presented low pluripotency score values) and cluster closely to the non-iPSC control sample, which indicates that the final product is free of pluripotent contamination.
Fig. 3.
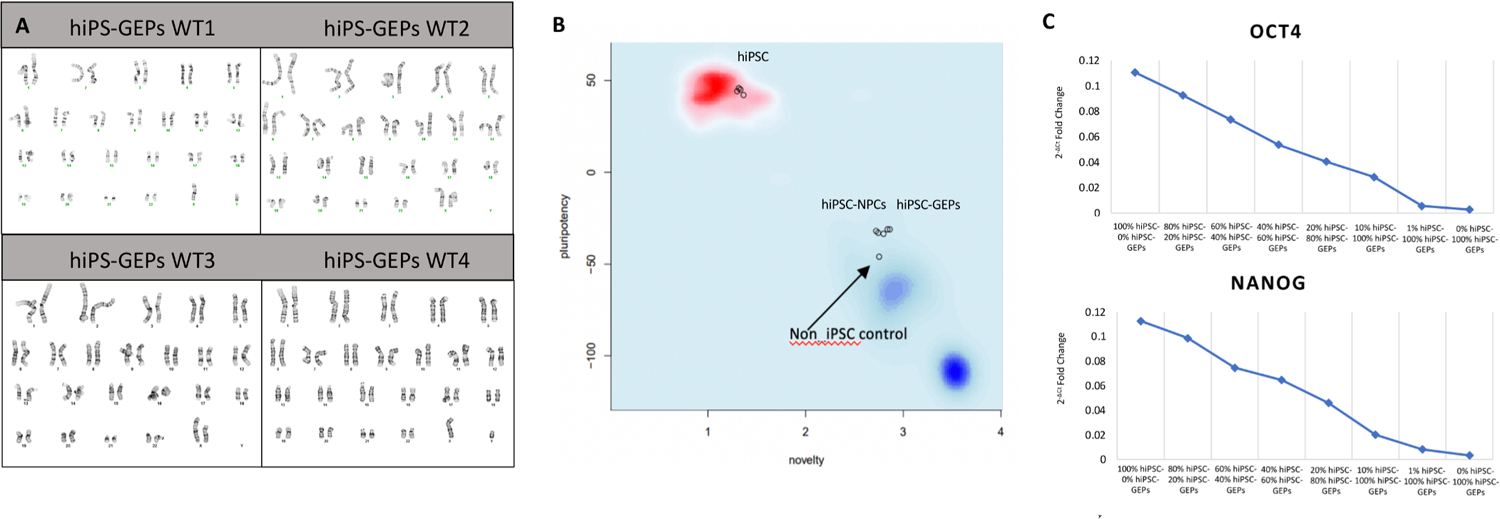
Karyotype and purity assay. (A) G-Banding karyotype of hiPSC-GEPs from all four donors performed by Cell Line genetics. (B) Assessment of pluripotency of hiPSCs, hiPSC-NPCs, and hiPSC-GEPs performed by Life Technologies.
3.3. Purity assay
We have developed a q-RT-PCR-based assay for quantifying undifferentiated pluripotent contamination in the final hiPSC-GEP population. This assessment is important to avoid teratogenicity of a potential cell-based therapy. The q-RT-PCR-based assay was aimed to determine the presence of contamination from the initial (hiPS). This assay can be further used to establish acceptable limits of pluripotent contamination prior to transplant.
Presence of mRNA from pluripotent markers NANOG and OCT4 was used as a proxy for pluripotent contamination (Hazim et al., 2017; Barrett et al., 2014). To define the minimum pluripotent contamination that may be detected using this method, we spiked 1, 10, 20, 40, 60, 80 and 100% hiPSCs into hiPSC-GEPs. Total RNA was isolated from these mixed cells and a one step-q-RT-PCR test performed. GAPDH was used as the reference gene. We successfully detected NANOG and OCT4 expression in all mixtures spiked with hiPSCs. This indicates that the q-RT-PCR-based assay is able to detect at least 1% of residual pluripotent contamination in a population of hiPSC-GEPs (Fig. 3C).
3.4. Identity assay
We have developed a q-RT-PCR-based assay to assess the composition of the population containing the intended therapeutic candidate (hiPSC-GEPs). The main goal of the identity assay is to validate an identity profile that is unique to hiPSC-GEPs and clearly distinct from both hiPSCs and hiPSC-NPCs, the two origin cell types prior to hiPSC-GEPs in the differentiation protocol. Based on this q-RT-PCR-based assay we have determined the unique identity profile of our intended therapeutic candidate (hiPSC-GEPs) as ADM/RAB20/PLOD2/STC2 + and STLC16A3/OCT4/NANOG- (Fig. 4A, B).
Fig. 4.
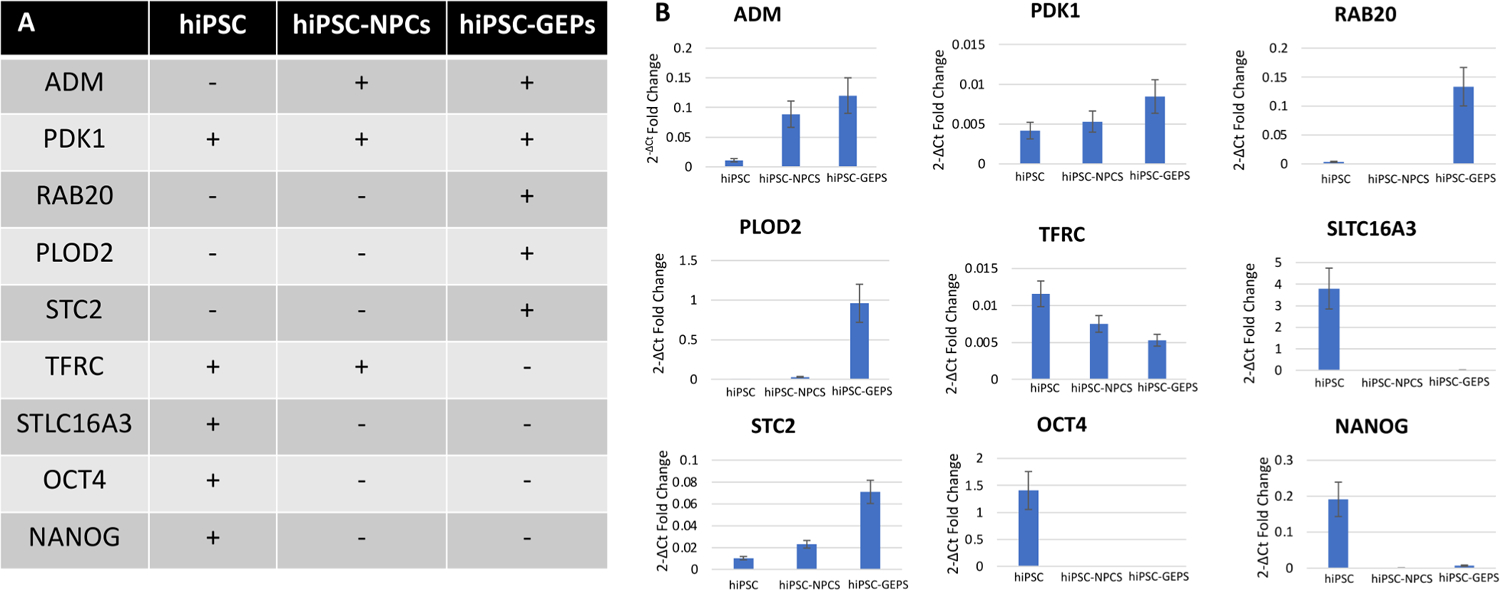
Identity assay. (A) Identity profiles of hiPSCs, hiPSC-NPCs, and hiPSC-GEPs. (B) Quantification of expression of differentially expressed genes between hiPSCs, hiPSC-NPCs, and hiPSC-GEPs as determined by q-RT-PCR. (C) Linearity quantification of expression of pluripotent markers (OCT4 and NANOG) between hiPSCs, hiPSC-NPCs, and hiPSC-GEPs as determined by q-RT-PCR.
To define the minimum pluripotent contamination (% hiPSCs) that may be detected using this method, we spiked serial concentrations of % hiPSCs into hiPSC-GEPs (Fig. S1). We were able to detect TFRC, ADM, PDK1, PLOD2, RAB20, STC2, SLC16A3 expression in all spiked mixtures, indicating that the q-RT-PCR-based assay is able to detect at least 1% of residual hiPSCs contamination in a population of hiPSC-GEPs (Fig. S1).
The identity markers used on this study (ADM/RAB20/PLOD2/STLC16A3/STC2/OCT4/NANOG) were selected based on an in vitro transcriptional analysis (RNA-seq) to determine the most upregulated and downregulated unique genes expressed by hiPSC, hiPSC-NPCS and hiPSC-GEPs.
3.5. Viability assay
In order to assess the clinical feasibility of implementing hiPSC-GEP transplants, we first assessed the stability of the three cell types in either their respective media or retinal pigment epithelium (RPE) buffer. RPE Buffer is a Food and Drug Administration (FDA) approved solution that was previously found to significantly extend the stability of RPE cells in suspension, therefore lengthening the window of time from cell collection to transplant (Hazim et al., 2017; Karumbayaram et al., 2012). Stability was calculated by suspending the cells at clinical concentration at 4 °C and manually counting cells at 0 to 72 h post-cell culture collection. Survival curves for all three cell types were nearly identical when suspended in their respective media or RPE buffer, which demonstrates that RPE buffer had no beneficial effect on the stability of our cells in suspension (Fig. 5A). Therefore, we decided that collecting cells in their appropriate media is the most ideal for experimentation and clinical application.
Fig. 5.
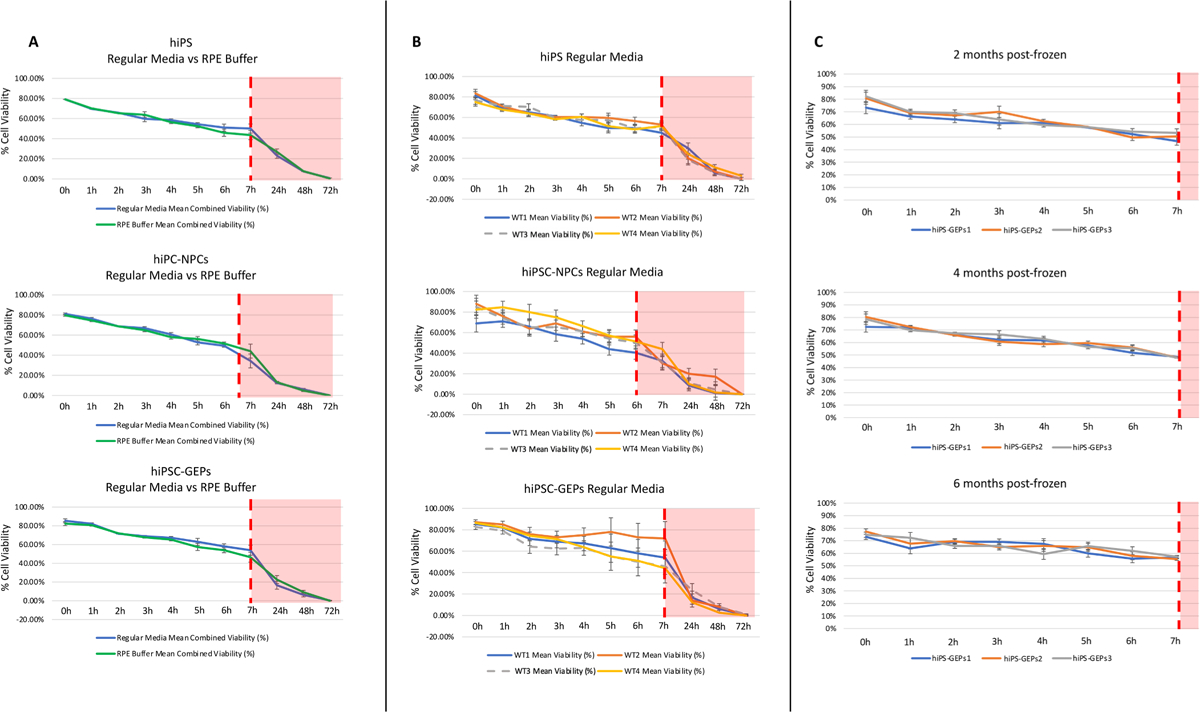
Viability Assay. (A) Viability quantification of all 12 populations in appropriate media or RPE buffer. (B) Comparison of the average viability of each cell type in appropriate media or RPE buffer. (C) Post-thaw viability of hiPSC-GEPs from all four donors after varying time in liquid nitrogen storage at − 180 °C.
To further explore the cells’ longevity in suspension, we assessed the stability of all three cell types from all four donors suspended in their appropriate media at clinical concentration at 4 °C (Fig. 5B). The % of hiPSCs and hiPSC-NPCs survival decreased from 80% to ~ 40% in the first 7 h post-collection (Fig. 5B). Interestingly, hiPSC-GEPs were observed to have the highest survival ratio compared with hiPSC and hiSPC-NPCs, the % of hiPSC-GEPs survival decreased from 80% to ~ 60% in the first 7 h post-collection (Fig. 5B). Similar work on cell-based transplants have established an acceptable survival ratio of 60–70% at transplantation (Hazim et al., 2017; Karumbayaram et al., 2012). We have established a 7-hour window from hiPSC-GEP cells preparation at the bench to transplant to the patient in the operating room with a cell viability between 60 and 70%.
In our clinical application we aim to perform hiPSC-GEPs transplants directly from liquid nitrogen storage. The same viability assessment as described above was therefore carried out on hiPSC-GEPs that had been thawed from 2, 4- and 6-months liquid nitrogen storage. hiPSC-GEPs were found to retain their same 7-hour survival curve independent of time spent in liquid nitrogen storage (up to 6 months; Fig. 5C). This provides promising evidence that hiPSC-GEPs could be transplanted directly from liquid nitrogen storage, which means that each cell lot does not need to be generated “fresh” for each transplant and enhances the efficiency of future clinical studies.
For scale up for Phase 1 and Phase 2 clinical trials, the estimated human dose is 10 million to 40 million cells per patient: based on the white matter stroke volume in the mouse (0.027 cm3) and dose (100,000 hiPS-GEP cells) and the range of white matter lesion volume in humans that is associated with neurological dysfunction (3 cm3 to 10 cm3) (Au et al., 2006; Wang et al., 2015). This target human cell number is consistent with other progenitor cell white matter transplant clinical trials (Gupta et al., 2012) Based on preliminary stroke clinical trials the estimated trial size is 20 patients (Phase 1) and 70 patients (Phase 2) (Kalladka et al., 2016; Steinberg et al., 2018, 2016; Gupta et al., 2019; Muir et al., 1999). This means an estimate of 9 billion to 36 billion cells. The expansion of hiPSC-GEPS production is: 1 million hiPSC (one vial) produces 3 wells (1mil cells/well) of P0 hiPSC-NPC. These are expanded to 12 wells (1 mil cells/well) at P1 hiPSC-NPC, then to 36 wells (1 mil cells/well) at P2 hiPSC-NPC, which then produces 36 million hiPSC-GEPs. A total of 100 vials of hiPSC (1 million hiPSC per vial) will be needed to generate the necessary amount of hiPSC-GEPs to complete phase 1 and phase 2 clinical trials (3.6×109 hiPSC-GEPs necessary for Phases 1 and 2 clinical trials). Together, the ability to produce a vast quantity of hiPSC-GEPs and directly transplant the cells from liquid nitrogen storage demonstrates the feasibility of hiPSC-GEPs in clinical application.
3.6. Safety assay
To demonstrate the safety of this terminal therapeutic candidate, all four lines of hiPSC-GEPs were put through several rigorous commercially available assays for the detection of contaminants.
For assessment of viral, mycoplasma, and select bacterial contamination, representative samples from all four hiPSC-GEP lines were sent to IDEXX BioAnalytics for H-IMPACT I and IMPACT VIII testing (IDEXX BioAnalytics). All samples tested negative by proprietary PCR for contamination by any human or murine viral pathogens, select bacteria, and common laboratory mycoplasmas (Fig. 6A).
Fig. 6.

Safety assay. (A) Clean assessment of viral, mycoplasmal, and bacterial contamination in four representative samples of hiPSC-GEPs as performed by IDEXX BioAnalytics (H-IMPACT I and IMPACT VIII). (B) Acceptably low concentration of endotoxin in 10 representative samples of hiPSC-GEPs as determined by Charles River Kinetic Chromogenic LAL Microplate Testing. The maximum accepted value is 35 EU/mL. (C) Clean assessment of microbial contamination in 12 representative samples of hiPSC-GEPs as performed by VRL Eurofins (USP < 71 >).
To determine the endotoxin contaminants in the final product, we sent 10 representative samples of hiPSC-GEPs to Charles River for Kinetic Chromogenic LAL Microplate Testing. Using the Endotoxin Limit calculation K/M, where K = 0.2 EU/70 kg (average patient weight)/1hr and M = 0.4 uL for a dose of 40×106 cells in 0.4 uL of saline (the maximum foreseeable dose use in humans), we calculated the maximum acceptable endotoxin concentration to be 0.035 EU/uL or 35 EU/mL. The maximum endotoxin concentration detected in any of our samples was 0.6857 EU/mL, almost 100 times less than the maximum acceptable value (Fig. 6B).
For further assessment of microbial contamination, all four lines of hiPSC-GEPs were sent in triplicate to VRL Eurofins. After determining the cells had no bacteriostatic/fungistatic properties that would interfere with further testing, aliquots form all 12 samples were directly inoculated into liquid media (USP < 71 > VRL Eurofins). Inoculation was performed in a controlled ISO certified environment. Following 14 days of incubation, all samples were found to be void of growth of any aerobes, anaerobes, yeast, or fungi (Fig. 6C).
4. Discussion
Due to the limited endogenous repair capacity of the brain, cell-based therapies are of interest in targeting neurodegenerative diseases. WMS results in demyelination of axonal tracts and depletion of astrocytes and oligodendrocytes. There is limited repair after a WMS occurs, as well as limited differentiation of OPCs to mature myelinating oligodendrocytes (Sozmen et al., 2019; Llorente et al., 2021). Studies have shown that after WMS, OPCs proliferate and move to the lesion site, however differentiation is blocked (Sozmen et al., 2019, 2016; Llorente et al., 2021). Astrocytes have been previously reported to play a key role in oligodendrocyte progenitor cell (OPC) proliferation and differentiation in CNS development (Clemente et al., 2013). In white matter injury, astrocytes were observed to support OPCs and promote maturation of oligodendrocytes, promoting gross repair and remyelination (Miyamoto et al., 2015; Jiang et al., 2016; Sorensen et al., 2008; Llorente et al., 2021). As a result of these findings, we hypothesize an astrocytic cell-based therapy would be appropriate after WMS.
Here we report reliable generation of the novel cell type hiPSC-derived GEPs. Through modulation of oxygen tension by treating hiPSC-NPC cultures with the small molecule deferoxamine (DFX) for 3 days, the cells are permanently biased towards an astrocytic fate (Xie et al., 2014; Llorente et al., 2021). The importance of the Hypoxia inducible factor (HIF) pathway in development has been previously reported (Lendahl et al., 2009; Simon and Keith, 2008). Hypoxia inducible factors (HIFs) regulate cellular responses in hypoxic conditions. When O2 is less than atmospheric concentration the regulatory prolyl hydroxylases (PHDs) are diminished. As a result, HIF1α is stabilized and target genes are activated. DFX mimics this process, stabilizing HIF in neural stem cells by chelating Fe2+, thus inhibiting PHDs from enzymatic activity (Xie et al., 2014; Xie and Lowry, 2018). HIF promotes astrocyte lineage differentiation and in low concentrations of oxygen facilitates differentiation into astrocytes (Xie et al., 2014; Xie and Lowry, 2018). Utilization of DFX allows for rapid and efficient production of hiPSC-GEPs that are clinically viable.
Generation of hiPSC-GEPs as described in this report has significant advantages. Previously described protocols of astrocyte derivation from human iPSCs and human ESCs are lengthy, typically ranging from 2 to 6 months (Krencik et al., 2011; Krencik and Zhang, 2011; Shaltouki et al., 2013; Dhara et al., 2008). Additionally, OPC’s that have been reported as potential therapeutic approaches to promote remyelination in the brain, also require long periods of time for differentiation. Protocols deriving OPCs require multiple steps and typically require 3 to 6 months in culture (Wang et al., 2013; Douvaras et al., 2014; Stacpoole et al., 2013; Sundberg et al., 2010). The preparation of hiPSC-GEPs requires only 35 days in culture from patient derived hiPSCs. Following addition of DFX into hiPSC-NPC cultures for 3 days, the cell type of interest is produced and ready to use. Moreover, deferoxamine is an FDA approved iron-chelating agent to treat acute iron poisoning for decades. Therefore, its safety for clinical applications has been well established. The hiPSC-GEPs have been cultured under GMP-compatible conditions, congruent with the goal of developing a cell-based therapy for clinical trial. We characterized hiPSC-GEPs by q-RT-PCR-based transcriptomic assays and confirmed their novel identity as unique from hiPSCs and hiPSC-NPCs. We successfully report the key genetic markers ADM, RAB20, PLOD2 and STC2 that the hiPSC-GEPs are positive for and STLC16A3 is negative. These makers are specific to hiPSC-GEPs and are necessary to create a genetic profile essential for clinical use. In addition, qualification assays have been performed such as stability, purity, safety and sterility. Viability assays established the time frame during which hiPSC-GEPs could be collected and prepared for transplantation in future clinical application as 6–7 h. Further assessment of hiPSC-GEP viability suggested the ability to both produce sufficient quantities of cells for human treatment as well as the ability to transplant the cells directly from liquid nitrogen. Purity assays were completed to evaluate any presence of contaminating pluripotent cells and distinguish the hiPSC-GEPs as a pure population. The safety assays for endotoxin, murine viruses, human viruses, mycoplasma, and microbes were performed to ensure that cultures are contaminant free. These assays are imperative in establishing a cell therapy product. The protocol we described here produces a viable cell product that is ready for clinical manufacturing.
These results confirm hiPSC-GEPs as a viable cell product for therapy after WMS. Other glial and myelin disorders occurring in the brain and spinal cord may benefit from this stem cell-based therapy. These disorders include multiple sclerosis (MS), cerebral palsy, periventricular leukomalacia (PVL) and pediatric leukodystrophies (Goldman, 2016). Disorders resulting in demyelination would be appropriate targets of a glial progenitor cell replacement therapy that gives rise to astrocytes (Fox et al., 2014; Goldman et al., 2012; Franklin and French-Constant, 2008). Like white matter stroke, in chronic multiple sclerosis lesions, remyelination is halted due to blockage of OPC differentiation (Kuhlmann et al., 2008; Clemente et al., 2011). An astrocytic therapy that can promote OPC differentiation to mature oligodendrocytes, inducing remyelination and repair could be of interest for MS patients. Other diseases that are characterized by neuronal degeneration such as Alzheimer’s disease (AD), Amyotrophic Lateral Sclerosis (ALS), Parkinson’s Disease (PD) and Huntington’s disease (HD) are of interest as well. Many studies have displayed the role of astrocytes in neuroprotection and restoration in these diseases (Bronzuoli et al., 2017; Pehar et al., 2017; Vargas and Johnson, 2010). Astrocytic cell-based therapies have been shown to slow disease progression and restore neurologic function in various animal models (Lepore et al., 2008; Gowing et al., 2014; Thomsen et al., 2018; Giralt et al., 2010; Llorente et al., 2021). In addition to neurodegenerative diseases, injuries to the CNS such as traumatic brain injury and spinal cord injury may benefit from hiPSC-GEP transplants. Transplanting astrocytes or astrocyte precursors in SCI has been shown to facilitate axonal growth by forming bridges across lesions in the spinal cord and encourage repair (Hasegawa et al., 2005; Filous et al., 2010; Jin et al., 2011). Due to the ability of the hiPSC-GEPs to differentiate into astrocytes, this cell-based therapy has the potential to target a multitude of neurodegenerative diseases and injuries. We have placed emphasis on characterizing and qualifying the hiPSC-GEPs here for suitable transplantation as therapy for humans after WMS and potentially other neurodegenerative diseases.
5. Conclusion
In summary, we have demonstrated reliable generation of a novel stem cell-based therapeutic, hiPSC-GEPs derived from patient fibroblasts. By addition of the small molecule deferoxamine to hiPSC-NPC cultures for a 3-day period hiPSC-GEPs are rapidly and efficiently produced. We have demonstrated that these progenitor cells are permanently biased to an astrocytic fate, that prove to be advantageous as a cell-based therapy after WMS. The protocol described here generates a safe and viable cell product ready for clinical manufacturing.
Supplementary Material
Acknowledgement
We thank the UCLA Neuroscience genomic core.
Funding
This research was supported by Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, Paul G Allen Family Foundation, Allen Frontiers Group Eli and Edythe Broad Center for Regenerative Medicine, Rose Hills Scholar Award and CIRM DISC2-09018.
Abbreviations:
- WMS
White matter stroke
- GEPs
Glial enriched progenitors
- hiPSCs
human induced pluripotent stem cells
- hiPSC-NPCs
hiPSC derived neural progenitors
- hiPSC-GEPs
hiPSC derived glial enriched progenitors
- CNS
Central nervous system
- DFX
Deferoxamine
- GMP
Good manufacturing practice
- VEE
Venezuelan Equine Encephalitis
- DMEM
Dulbecco’s Modified Eagle’s Medium
- RA
Retinoic acid
- FGF
Fibroblast growth factor
- q-RT-PCR
Quantitative real time polymerase chain reaction
- mRNA
messenger ribonucleic acid
- RPE
retinal pigment epithelium
- FDA
Food and Drug administration
- OPCs
Oligodendrocyte progenitor cells
- PHDs
prolyl hydroxylases
- hESCs
human embryonic stem cells
- MS
Multiple sclerosis
- PVL
Periventricular leukomalacia
- AD
Alzheimer’s disease
- ALS
Amyotrophic Lateral Sclerosis
- PD
Parkinson’s Disease
- HD
Huntington’s disease
- SCI
Spinal cord injury
Footnotes
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: S Thomas Carmichael reports financial support was provided by Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. S Thomas Carmichael reports financial support was provided by California Institute of Regenerative Medicine. William Lowry reports financial support was provided by Paul G Allen Family Foundation. William Lowry reports financial support was provided by Allen Frontiers Group. William Lowry reports financial support was provided by Eli and Edythe Broad Center for Regenerative Medicine. S Thomas Carmichael has patent INDUCED PLURIPOTENT STEM CELL DERIVED GLIAL ENRICHED PROGENITOR CELLS FOR THE TREATMENT OF WHITE MATTER STROKE pending to University of California.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scr.2021.102458.
References
- Takahashi K, Yamanaka S, 2006. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126 (4), 663–676. [DOI] [PubMed] [Google Scholar]
- Robinton DA, Daley GQ, 2012. The promise of induced pluripotent stem cells in research and therapy. Nature 481 (7381), 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S, 2016. Stem and Progenitor Cell-Based Therapy of the Central Nervous System: Hopes, Hype, and Wishful Thinking. Cell Stem Cell 18 (2), 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig S, Carmichael ST, 2015. The Axon–glia Unit in White Matter Stroke: Mechanisms of Damage and Recovery. Brain Res. 1623, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozmen EG, Hinman JD, Carmichael ST, 2012. Models That Matter: White Matter Stroke Models. Neurotherapeutics 9 (2), 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozmen EG, Kolekar A, Havton LA, Carmichael ST, 2009. A White Matter Stroke Model in the Mouse: Axonal Damage, Progenitor Responses and MRI Correlates. J. Neurosci. Methods 180 (2), 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente IL, Xie Y, Mazzitelli JA, Hatanaka EA, Cinkornpumin J, Miller DR, Lin Y, Lowry WE, Carmichael ST, 2021. Patient-derived glial enriched progenitors repair functional deficits due to white matter stroke and vascular dementia in rodents. Sci. Transl. Med 13 (590), eaaz6747. 10.1126/scitranslmed.aaz6747. [DOI] [PubMed] [Google Scholar]
- Vernooij MW, Ikram MA, Tanghe HL, Vincent AJPE, Hofman A, Krestin GP, Niessen WJ, Breteler MMB, van der Lugt A, 2007. Incidental findings on brain MRI in the general population. N. Engl. J. Med 357 (18), 1821–1828. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Penman AD, Catellier DJ, Coker LH, Shibata DK, Sharrett AR, Mosley TH, 2011. Vascular risk factors and longitudinal changes on brain MRI: the ARIC study. Neurology 76 (22), 1879–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin MA, Carmichael ST, 2018. Stroke in CNS white matter: Models and mechanisms. Neurosci. Lett 684, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss T, Guzman R, Daadi M, Steinberg GK, 2007. Cell Transplantation Therapy for Stroke. Stroke 38 (2), 817–826. [DOI] [PubMed] [Google Scholar]
- Chu K, Kim M, Park K-I, Jeong S-W, Park H-K, Jung K-H, Lee S-T, Kang L, Lee K, Park D-K, Kim SU, Roh J-K, 2004. Human neural stem cells improve sensorimotor deficits in the adult rat brain with experimental focal ischemia. Brain Res. 1016 (2), 145–153. [DOI] [PubMed] [Google Scholar]
- Jeong S-W, Chu K, Jung K-H, Kim SU, Kim M, Roh J-K, 2003. Human neural stem cell transplantation promotes functional recovery in rats with experimental intracerebral hemorrhage. Stroke 34 (9), 2258–2263. [DOI] [PubMed] [Google Scholar]
- Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, Masel J, Yenari MA, Weissman IL, Uchida N, Palmer T, Steinberg GK, 2004. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc. Natl. Acad. Sci. U. S. A 101 (32), 11839–11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia Z, Llorente IL, Carmichael ST, 2018. Customized Brain Cells For Stroke Patients using Pluripotent Stem Cells. Stroke 49 (5), 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L, 2008. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22 (2), 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhang J, Lin Y, Gaeta X, Meng X, Wisidagama DRR, 20140. Defining the Role of Oxygen Tension in Human Neural Progenitor Fate. Stem Cell Rep. 3, 743–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Lowry WE, 2018. Manipulation of neural progenitor fate through the oxygen sensing pathway. Methods 133, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.u., Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, Goldman S, 2013. Human iPSC-Derived Oligodendrocyte Progenitor Cells Can Myelinate and Rescue a Mouse Model of Congenital Hypomyelination. Cell Stem Cell 12 (2), 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao Z, Rege SV, Wang M, Si G, Zhou Y.i., Wang S.u., Griffin JH, Goldman SA, Zlokovic BV, 2016. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat. Med 22 (9), 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck J, Studer L, 2015. Moving stem cells to the clinic: potential and limitations for brain repair. Neuron 86 (1), 187–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazim RA, Karumbayaram S, Jiang M, Dimashkie A, Lopes VS, Li D, Burgess BL, Vijayaraj P, Alva-Ornelas JA, Zack JA, Kohn DB, Gomperts BN, Pyle AD, Lowry WE, Williams DS, 2017. Differentiation of RPE cells from integration-free iPS cells and their cell biological characterization. Stem Cell Res. Ther 8, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karumbayaram S, Lee P, Azghadi SF, Cooper AR, Patterson M, Kohn DB, Pyle A, Clark A, Byrne J, Zack JA, Path K, Lowry WE, 2012. From skin biopsy to neurons through a pluripotent intermediate under good manufacturing practice protocols. Stem Cells Transl. Med 1, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B, Yoo KH, Kim C, 2015. Hematopoietic stem cell expansion and generation: the ways to make a breakthrough. Blood Res. 50 (4), 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awe JP, Lee PC, Ramathal C, Vega-Crespo A, Durruthy-Durruthy J, Cooper A, Karumbayaram S, Lowry WE, Clark AT, Zack JA, Sebastiano V, Kohn DB, Pyle AD, Martin MG, Lipshutz GS, Phelps PE, Pera RAR, Byrne JA, 2013. Generation and characterization of transgene-free human induced pluripotent stem cells and conversion to putative clinical-grade status. Stem Cell Res. Ther 4 (4), 87. 10.1186/scrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Gaeta X, Loo K, Edwards M, Smale S, Cinkornpumin J, Xie Y, Listgarten J, Azghadi S, Douglass SM, Pellegrini M, Lowry WE, 2014. let-7 miRNAs can act through notch to regulate human gliogenesis. Stem Cell Rep. 3 (5), 758–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anuncibay-Soto B, Pérez-Rodríguez D, Llorente IL, Regueiro-Purriños MM, Gonzalo-Orden JM, Fernández-López A, 2014. Global cerebral ischemia modifies the vascular adhesion molecules, inflammation and apoptosis after 48 hours of reperfusion in age-dependent way. Age 36 (5), 9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCt. Method, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT, 2009. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 55 (4), 611–622. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L, 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotech 27 (3), 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett R, Ornelas L, Yeager N, Mandefro B, Sahabian A, Lenaeus L, Targan SR, Svendsen CN, Sareen D, 2014. Reliable generation of induced pluripotent stem cells from human lymphoblastoid cell lines. Stem Cells Transl. Med 3 (12), 1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au R, Massaro JM, Wolf PA, Young ME, Beiser A, Seshadri S, D’Agostino RB, DeCarli C, 2006. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch. Neurol 63 (2), 246–250. [DOI] [PubMed] [Google Scholar]
- Wang R, Fratiglioni L, Laukka EJ, Loevden M, Kalpouzos G, Keller L, Graff C, Salami A, Backman L, Qiu C, 2015. Effects of vascular risk factors and APOE Œμ4 on white matter integrity and cognitive decline. Neurology 84 (11), 1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, Caverzasi E, Gaetano L, Mandelli ML, Ryan T, Perry R, Farrell J, Jeremy RJ, Stephen MU, Huhn SL, Barkovich AJ, Rowitch DH, 2012. Neural stem cell engraftment and myelination in the human brain. Sci. Transl. Med 4(155), ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalladka D, Sinden J, Pollock K, Haig C, McLean J, Smith W, McConnachie A, Santosh C, Bath PM, Dunn L, Muir KW, 2016. Human neural stem cells in patients with chronic ischaemic storke (PISCES): a phase 1, first-in-man- study. Lancet 388 (10046), 787–796. [DOI] [PubMed] [Google Scholar]
- Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Kim AS, Johnson JN, Bates D, Poggio G, Case C, McGrogan M, Yankee EW, Schwartz NE, 2018. Two-year safety and clinical outcomes in chronic ischemic stroke patients after implantation of modified bone marrow-derived mesenchymal stem cells (SB623): a phase 1/2a study. J. Neurosurg 23, 1–11. [DOI] [PubMed] [Google Scholar]
- Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB, Kim AS, Johnson JN, Bates D, King B, Case C, McGrogan M, Yankee EW, Schwartz NE, 2016. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: A phase 1/2a study. Stroke 47 (7), 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Henry RG, Kang S-M, Strober J, Lim DA, Ryan T, Perry R, Farrell J, Ulman M, Rajalingam R, Gage A, Huhn SL, Barkovich AJ, Rowitch DH, 2019. Long-Term Safety, Immunologic Response, and Imaging Outcomes following Neural Stem Cell Transplantation for Pelizaeus-Merzbacher Disease. Stem Cell Rep. 13 (2), 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW, Weir CJ, Alwan W, Squire IB, Lees KR, 1999. C-reactive protein and outcome after ischemic stroke. Stroke 30 (5), 981–985. [DOI] [PubMed] [Google Scholar]
- Sozmen EG, DiTullio DJ, Rosenzweig S, Hinman JD, Bridges SP, Marin MA, Kawaguchi R, Coppola G, Carmichael ST, 2019. White Matter Stroke Induces a Unique Oligo-Astrocyte Niche That Inhibits Recovery. J. Neurosci 39 (47), 9343–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozmen EG, Rosenzweig S, Llorente IL, DiTullio DJ, Machnicki M, Vinters HV, Havton LA, Giger RJ, Hinman JD, Carmichael ST, 2016. Nogo receptor blockade overcomes remyelination failure after white matter stroke and stimulates functional recovery in aged mice. Proc. Natl Acad. Sci. USA 113 (52), E8453–E8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente D, Ortega MC, Melero-Jerez C, de Castro F, 2013. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front. Cell Neurosci 7, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Shindo A, Liang AC, Maeda M, Egawa N, Itoh K, Lo EK, Lok J, Ihara M, Arai K, 2015. Astrocytes Promote Oligodendrogenesis after White Matter Damage via Brain-Derived Neurotrophic Factor. J. Neurosci 35 (41), 14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Chen C, Liu X-B, Pleasure D, Liu Y, Deng W, 2016. Human iPSC-Derived Immature Astroglia Promote Oligodendrogenesis by Increasing TIMP-1 Secretion. Cell Rep. 15 (6), 1303–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen A, Moffat K, Thomson C, Barnett SC, 2008. Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia 56 (7), 750–763. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Lee KL, Yang H, Poellinger L, 2009. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet 10 (12), 821–832. [DOI] [PubMed] [Google Scholar]
- Simon MC, Keith B, 2008. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol 9 (4), 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, Weick JP, Liu Y, Zhang Z-J, Zhang S-C, 2011. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat. Biotech 29 (6), 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krencik R, Zhang S-C, 2011. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat. Prot 6 (11), 1710–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltouki A, Peng J, Liu Q, Rao MS, Zeng X, 2013. Efficient Generation of Astrocytes from Human Pluripotent Stem Cells in Defined Conditions. Stem Cells 31 (5), 941–952. [DOI] [PubMed] [Google Scholar]
- Dhara SK, Hasneen K, Machacek DW, Boyd NL, Rao RR, Stice SL, 2008. Human neural progenitor cells derived from embryonic stem cells in feeder-free cultures. Differentiation 76 (5), 454–464. [DOI] [PubMed] [Google Scholar]
- Douvaras P, Wang J, Zimmer M, Hanchuk S, O’Bara M, Sadiq S, Sim F, Goldman J, Fossati V, 2014. Efficient Generation of Myelinating Oligodendrocytes from Primary Progressive Multiple Sclerosis Patients by Induced Pluripotent Stem Cells. Stem Cell Rep 3 (2), 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole SL, Spitzer S, Bilican B, Compston A, Karadottir R, Chandran S, Franklin RM, 2013. High Yields of Oligodendrocyte Lineage Cells from Human Embryonic Stem Cells at Physiological Oxygen Tensions for Evaluation of Translational Biology. Stem Cell Rep. 1 (5), 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg M, Skottman H, Suuronen R, Narkilahti S, 2010. Production and isolation of NG2+ oligodendrocyte precursors from human embryonic stem cells in defined serum-free medium. Stem Cell Res. 5 (2), 91–103. [DOI] [PubMed] [Google Scholar]
- Fox I, Daley GQ, Goldman SA, Huard J, Kamp TJ, Trucco MJ, 2014. Use of differentiated pluripotent stem cells in replacement therapy for treating disease. Science, 345, 1247391–1247391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SA, Nedergaard M, Windrem MS, 2012. Glial Progenitor Cell-Based Treatment and Modeling of Neurological Disease. Science 338 (6106), 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM, ffrench-Constant C, 2008. Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci 9 (11), 839–855. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Bruck W, 2008. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131 (7), 1749–1758. [DOI] [PubMed] [Google Scholar]
- Clemente D, Ortega MC, Arenzana FJ, de Castro F, 2011. FGF-2 and Anosmin-1 Are Selectively Expressed in Different Types of Multiple Sclerosis Lesions. J. Neurosci 31 (42), 14899–14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronzuoli M, Facchinetti R, Steardo L, Scuderi C, 2017. Astrocyte: An Innovative Approach for Alzheimer’s Disease Therapy. Curr. Pharm. Des 23 (33), 4979–4989. [DOI] [PubMed] [Google Scholar]
- Pehar M, Harlan B, Killoy K, Vargas M, 2017. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des 23 (33), 5010–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA, 2010. Astrogliosis in amyotrophic lateral sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics 7 (4), 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, Maragakis NJ, 2008. Focal transplantation–based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci 11 (11), 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowing G, Shelley B, Staggenborg K, Hurley A, Avalos P, Victoroff J, Latter J, Garcia L, Svendsen CN, 2014. Glial cell line-derived neurotrophic factor-secreting human neural progenitors show long-term survival, maturation into astrocytes, and no tumor formation following transplantation into the spinal cord of immunocompromised rats. NeuroReport 25, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen GM, Avalos P, Ma AA, Alkaslasi M, Cho N, Wyss L, Vit J-P, Godoy M, Suezaki P, Shelest O, Bankiewicz KS, Svendsen CN, 2018. Transplantation of Neural Progenitor Cells Expressing Glial Cell Line-Derived Neurotrophic Factor into the Motor Cortex as a Strategy to Treat Amyotrophic Lateral Sclerosis. Stem Cells 36 (7), 1122–1131. [DOI] [PubMed] [Google Scholar]
- Giralt A, Friedman HC, Caneda-Ferrón B, Urbán N, Moreno E, Rubio N, Blanco J, Peterson A, Canals JM, Alberch J, 2010. BDNF regulation under GFAP promoter provides engineered astrocytes as a new approach for long-term protection in Huntington’s disease. Gene Ther. 17 (10), 1294–1308. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Chang Y-W, Li H, Berlin Y, Ikeda O, Kane-Goldsmith N, Grumet M, 2005. Embryonic radial glia bridge spinal cord lesions and promote functional recovery following spinal cord injury. Exp. Neurol 193 (2), 394–410. [DOI] [PubMed] [Google Scholar]
- Filous AR, Miller JH, Coulson-Thomas YM, Horn KP, Alilain WJ, Silver J, 2010. Immature astrocytes promote CNS axonal regeneration when combined with chondroitinase ABC. Dev. Neurobiol 70 (12), 826–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Neuhuber B, Singh A, Bouyer J, Lepore A, Bonner J, Himes T, Campanelli JT, Fischer I, 2011. Transplantation of Human Glial Restricted Progenitors and Derived Astrocytes into a Contusion Model of Spinal Cord Injury. J. Neurotrauma 28 (4), 579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.