

Abstract
The number of viral vector-based gene therapies (GTx) continues to grow with two products (Zolgensma® and Luxturna®) approved in the USA as of March 2021. To date, the most commonly used vectors are adeno-associated virus-based (AAV). The pre-existing humoral immunity against AAV (anti-AAV antibodies) has been well described and is expected as a consequence of prior AAV exposure. Anti-AAV antibodies may present an immune barrier to successful AAV transduction and hence negatively impact clinical efficacy and may also result in adverse events (AEs) due to the formation of large immune complexes. Patients may be screened for the presence of anti-AAV antibodies, including neutralizing (NAb) and total binding antibodies (TAb) prior to treatment with the GTx. Recommendations for the development and validation of anti-AAV NAb detection methods have been presented elsewhere. This manuscript covers considerations related to anti-AAV TAb-detecting protocols, including the advantages of the use of TAb methods, selection of assay controls and reagents, and parameters critical to monitoring assay performance. This manuscript was authored by a group of scientists involved in GTx development representing eleven organizations. It is our intent to provide recommendations and guidance to industry sponsors, academic laboratories, and regulatory agencies working on AAV-based GTx viral vector modalities with the goal of achieving a more consistent approach to anti-AAV TAb assessment.
Graphical abstract
KEY WORDS: adenovirus associated virus, gene therapy, immunogenicity, total anti-GTx antibody
INTRODUCTION
Gene therapies (GTx) were first used in clinical studies in the 1980s in an ex vivo setting where the genetic modification of patient cells could occur under carefully controlled laboratory conditions. As the field of GTx matured, approaches to in vivo gene replacement and gene modification evolved. These approaches generally utilize the administration of viruses, with adeno-associated viruses (AAV) proving to be effective vectors for the delivery of genetic information. Administration of viral vectors for in vivo gene therapy and the ability of these vectors to deliver and express an exogenous gene may be impacted by a number of patient-dependent factors, including the immune response.
Anti-AAV vector antibodies may be either pre-existing or treatment-induced. Pre-existing anti-AAV vector antibodies commonly arise as a consequence of natural, non-pathogenic AAV infection with their prevalence varying with different AAV serotypes, geographic region, and age of the population. Additionally, the titer and avidity of anti-AAV antibodies may vary over time in any subject. Further, it is expected that most, if not all, patients without detectable pre-existing anti-AAV antibodies will seroconvert to antibody positive following systemic administration of AAV gene therapy vectors. This could present a constraint for GTx that experience a decrease in product activity over time since they may preclude re-administration of the GTx (1–4). Upon GTx administration, the levels of treatment-induced antibodies in non-human primate (NHP) studies were significantly elevated as compared to the antibody levels at baseline (5). Similarly, in patients administered Zolgensma® by intravenous infusion who were required to have anti-AAV9 antibody titers ≤1:50 prior to treatment, the post-treatment anti-AAV9 antibody titers reached at least 1:102,400 in every case, with most titers exceeding 1:819,200 (6). Other non-antibody-based factors found in circulation may act as transduction inhibitors and could be detected in the neutralizing antibody (NAb) assay, more appropriately described as a transduction inhibition (TI) assay (7–9).
In this manuscript, the authors focus on the review of topics related to the detection and evaluation of total antibody (TAb) response against AAV vectors. Other types of immune response, including cellular immunity against AAV and humoral or cellular anti-transgene protein responses, remain outside of the scope of this publication.
Important considerations related to anti-AAV TAb-detecting assays are reviewed in this manuscript, including benefits of detecting TAb response and selection of assay critical reagents and parameters. Authors intend to provide recommendations and guidance to industrial sponsors, academic laboratories, and regulatory scientists working on AAV-based GTx viral vector modalities with the goal to reach consistency in anti-AAV TAb assessment.
TOTAL ANTIBODY ASSAYS AS AN ALTERNATIVE TO NEUTRALIZING ANTIBODY ASSAYS
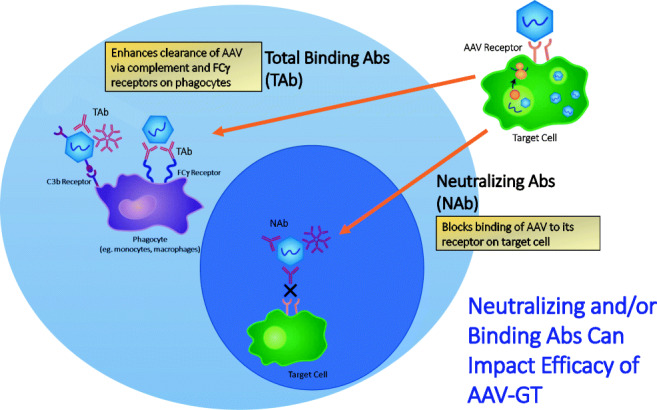
The term TAb refers to all antibodies that bind to a specific antigen, irrespective of isotype, epitope, or neutralizing ability. Detection of anti-AAV TAbs therefore facilitates a comprehensive assessment of various modes of potential impact that pre-existing or treatment-induced AAV capsid-specific antibodies may have on gene therapy treatment outcomes. The most direct impact is capsid neutralization, either by blocking epitopes on the capsid that play a role in receptor-mediated uptake into target cells or by hindering intracellular processes that lead to uncoating of the capsid (10–12). TAbs in this group may be further characterized in a cell-based transduction inhibition assay to determine the magnitude of the inhibitory effects. While some degree of correlation between anti-AAV TAb and NAb titers has been reported (13), the prevalence of TAbs is generally considered to be higher since a moderate fraction (~20%) of antibody-positive samples contain low to undetectable NAb titers assuming comparable assay sensitivities (5,14).
While the latest generation of cell-based NAb assays may have improved sensitivity, emerging evidence suggests that this increased in vitro sensitivity may come at the expense of in vivo relevance (13). A recent report describing the improved sensitivity of a cell-based NAb assay evaluating the pre-treatment plasma of hemophilia B patients receiving an AAV5-FIX gene therapy showed no relationship between the presence of low-titer pre-treatment anti-AAV5 NAbs and therapeutic efficacy (13). Though depletion of IgG did reduce reported NAb titers, AAV5-specific IgG could only be detected in pre-treatment serum samples using an ELISA method from patients with higher NAb titers, suggesting the detection of non-antibody-based inhibitory factors in the NAb assay. Similarly, non-clinical data from cynomolgus monkeys treated with an AAV5-FVIII GTx construct demonstrated diminished expression of FVIII only in animals with detectable anti-AAV5 antibodies (5). Animals were also tested for the presence of TI in a cell-based method that is similar to a NAb protocol. The terms TI and NAb have been applied interchangeably. Animals that tested positive in the NAb assay but had no detectable anti-AAV5 antibody titers all had normal levels of FVIII expression. Similar to the clinical data described above, there was a trend toward animals with higher NAb titers also having detectable anti-AAV5 antibodies. Overall, these data suggest that due to improved sensitivity and absence of specificity, the latest generation of cell-based NAb assays may be detecting low levels of non-antibody plasma factors that limit the transduction efficiency in vitro but may have limited clinical relevance. Conversely, there have been examples of NAb and TAb assay discordance where antibody is detected in the absence of a neutralizing signal. In one study reporting on characterization of the TI and TAb assay, 5 of 100 (5%) healthy donors and 2 of 24 (8%) of hemophilia A patients met this criterion with positive TAb titers in the absence of neutralization in the TI assay (14). This data highlights one potential risk in enrolling subjects solely based on the TAb assay as there are subjects with anti-AAV binding antibodies that are non-neutralizing and, though meeting an exclusion criterion for pre-existing immunity, may be predicted to respond to gene therapy. However, though these antibodies may be non-neutralizing for uptake of AAV by a cell line in vitro, they may alter the biodistribution in vivo and/or enhance clearance through Fc-receptor-mediated uptake into cells of the reticuloendothelial system. Though this redirection may inhibit or enhance full transduction of target tissue, one study noted increased uptake of vector genomes into the liver in the presence of binding, non-neutralizing antibody, while no increase in transgene expression was detected (10). Reduced efficacy is not the only concern with respect to pre-existing anti-AAV antibodies. Of further consideration is that these subjects may be at greater risk for safety-related adverse events and infusion-related reactions emanating from AAV antibody-mediated immune complex formation and complement activation (15). These potential safety concerns exist regardless of whether the pre-existing antibodies are neutralizing or not.
Opsonization is another mechanism by which anti-AAV TAbs may impact AAV GTx efficacy. Antibodies bound to the capsid can increase Fcγ-receptor-mediated uptake of the GTx vector by immune cells, such as dendritic cells (DCs) and macrophages, which could lead to an altered biodistribution of the vector, increased clearance of the AAV, resulting in a lower distribution to a target tissue, and/or inflammation (16). Finally, mechanism of anti-AAV TAbs impact may be based on ability to form large immune complexes that may trigger the classical C1q-dependent complement pathway, which could result in further opsonization, inflammation and attraction of other immune cells mediated by C3a and C5a proteins, and assembly of a membrane attack complex (17).
Currently, single administration is the primary approach of delivering AAV GTx therapeutics administered systemically. For a single administration AAV GTx, pre-existing antibodies, rather than treatment-induced or treatment-boosted antibodies, may impact the efficacy of the AAV GTx due to the rapid partitioning of the AAV into the cellular compartment of blood and tissues and lymphoid organs, such as spleen and lymph nodes (10,18). As a consequence, it is likely that the vast majority of the vector dose will have been cleared from the plasma compartment prior to the advent of treatment-induced or treatment-boosted antibody responses that could reduce efficacy. In accordance, anti-AAV antibody responses arising in response to dose administration were shown to not impact safety or efficacy (4). Relevance of anti-AAV antibodies found in blood compartment to the GTx modalities administered into immune-privileged tissues, for example intravitreally (IVT), is briefly discussed in the “ASSAY MATRIX SELECTION” section.
The presence of anti-AAV TAbs could be used as a biomarker of previous exposure to AAV. Such previous encounters between the virus and the host may have formed a lasting immune memory in the B cell, the T cell, or both compartments of adaptive immunity. While the presence of these memory cells may not directly influence the fate of a gene therapy vector during dose administration, they could still modulate how the pre-sensitized gene therapy recipient responds to the presence of vector-transduced target cells in the long run, thus potentially impacting durability of the treatment. This, currently theoretical, concern may be more applicable to certain tissues than others and is reliant on the duration of expression of AAV peptides on the cell surface. For example, it is known that most, if not all, AAVs will distribute to the liver. A cellular response to the AAV may lead to lysis of hepatocytes which may be evident by increases in serum transaminases (19). If the target cell type for efficacy is the hepatocyte, this may also lead to an attenuated efficacy. If the target cell type is not the hepatocyte, e.g., the target cell type is within the brain, the impact on efficacy may be limited since antibodies in blood are unlikely to distribute to the brain tissue and interact with the transduced cells. More studies are needed to fully understand the clinical implications of anti-AAV TAbs as a predictive immune biomarker for treatment success.
NATURE OF THE TAb ASSAY ANALYTES
Anti-AAV TAbs can be defined as immunoglobulins that bind to the capsid of an AAV vector. Assays detecting TAbs should be capable of assessing the presence of all anti-AAV immunoglobulin isotypes found in circulation, including the IgG and IgM subclasses. Detection of other classes of anti-AAV immunoglobulins is rarely considered critical, except potentially for the IgE class. Regulatory expectations for assays detecting the presence of specific IgE class antibodies are relatively unique and have been defined elsewhere (20). In general, it should be expected that TAb assays at minimum detect the presence of IgM and IgG antibody classes specific to the viral capsid as the main analytes for the test. Selection of the assay format is likely to impact the ability of the method to detect any given immunoglobulin isotype of anti-viral vector antibodies. In a typical TAb method, the choice of assay format therefore should be driven by the need to detect multiple or a single, specific isotype. From a practical point of view, it is preferred to use a single assay that can detect multiple or all anti-AAV antibody isotypes. Subsequently, a separate method designed to detect a specific subclass of anti-AAV antibodies, for example, IgG or IgM, may be added if a detailed characterization of the immune response is required.
ANALYTICAL METHODOLOGIES APPLIED FOR TAb DETECTION
With respect to platform, format, and design, anti-AAV TAb assays are very similar to conventional anti-drug antibody (ADA) assays. The main difference lies in the requirement for the use of the therapeutic drug as the capture antigen and anti-drug-specific antibodies as the positive control in the traditional ADA assays. This is in contrast to anti-AAV TAb assays where commercial viral vectors of the same serotype for plate coating and commercial anti-vector antibodies as positive control may be acceptable. However, comparable antigenicity between GTx and commercial vectors may sometimes be a questionable assumption. Depending on the manufacturing cell line, purification process, and the achieved ratio of empty vs. full capsids, antigenic epitopes and/or binding strength may differ between different vector preparations (21,22). This should at least be considered when choosing the target antigen for anti-AAV capsid TAb assays, and some bridging data may be required to demonstrate the commercial vector as a suitable surrogate antigen for the GTx. It should also be noted that the supply of the therapeutic GTx may be limited at the early stages of development. Considering that typical TAb assays require a large amount of vector regardless of whether they use the classical ELISA format or other immunoassay platforms, this may lead to the diversion of the GTx material from patient treatment to TAb assays. Consequently, TAb assays may use commercially available AAV capsids which usually are empty capsids or empty capsids that are harvested during the purification of the GTx. Although the potential, as yet unproven, differences between the binding of anti-AAV antibodies to the GTx intended for clinical use versus empty capsids are acknowledged, sponsors may choose to take a pragmatic approach (e.g., using commercially available empty capsids rather than striving for perfection by using the GTx product for clinical trials). It should also be noted that by using empty capsid as a reagent, lot-to-lot comparative evaluation should be considered (23). It may be advantageous to create a platform assay that can be used across multiple GTx programs of the same AAV capsid serotype, thereby reducing the cost of drug development.
The potential ADA assay interference from residual drug in post-treatment study incurred samples, commonly referred to as ADA assay drug tolerance, is often considered and addressed for common protein-based biotherapeutics (20). Fast clearance of the GTx from the blood compartment suggests limited, if any, impact from the residual GTx therapeutic on the performance of the TAb assays. Additionally, no significant level of circulating AAV, including GTx therapeutic, is expected to be present prior to treatment, i.e., at the point of collection of pre-treatment samples. Therefore, the value of the assessment of the drug tolerance may be limited and could be determined based on specific needs of a program.
Antigen-Capture and Bridging Formats of TAb Protocols
Anti-GTx TAb assays could be designed with either antigen-capture or bridging formats. Figure 1 presents various adaptations of assay format and platform for an anti-AAV TAb assay. In the antigen-capture format, the AAV capsid is coated directly onto the ELISA or electrochemiluminescence (ECL) microtiter plate. Following a blocking step, samples are loaded onto the assay plate to allow for the binding of TAbs to the coated capsids. Subsequently, the plate is washed to remove the unbound material, and a detection agent is added. The detection system utilized in this step could be species-specific enzyme-conjugated (e.g., horseradish peroxidase (HRP)-tagged) (24–29), or ruthenylated anti-Ig antibodies depending on the ELISA versus ECL platform (Figure 1a and b, respectively). Several groups apply multiple isotype-specific labeled detection reagents (13,30–32). An indirect ELISA approach was employed where AAV capsids were bound to the ECL plate, incubated with the test sample, and detected by ruthenium-labeled protein A, G, or L (5). If the method is intended for a specific class of anti-AAV antibodies, isotype-specific secondary antibodies may be used for detection. Alternatively, HRP-conjugated or ruthenylated protein A, G, or A/G may substitute for secondary detection antibodies (Figure 1c). Where enzyme-conjugated detection systems are used in the ELISA platform, the enzyme substrate is added as the last step for colorimetric detection, and the assay signal is monitored in absorbance units following the addition of stop reagent. If a ruthenylated detection system is utilized, the assay signal is measured in luminescence units after the addition of the read buffer. An antigen-capture format is recommended when information about isotypic composition of TAb response is needed. The assay can be re-developed with relative ease to detect IgG, IgM, or other isotypes through a substitution of isotype-specific secondary detection antibodies.
Figure 1.

Total anti-AAV antibody assay formats. a Antigen-capture format on ELISA platform: capture phase, viral capsid, and detection phase, horse radish peroxidase (HRP)-conjugated secondary detection antibody. b Antigen-capture format on ECL platform: capture phase, viral capsid, and detection phase, ruthenylated secondary detection antibody. c Antigen-capture format on ELISA platform: capture phase, viral capsid, and detection phase, HRP-conjugated protein A, G, or A/G. d Bridging format on ELISA platform: capture phase, viral capsid, and detection phase, biotinylated viral capsid in combination with streptavidin-HRP. e Bridging format on ECL platform using streptavidin coated ECL plates: capture phase, biotin-labeled viral capsid, and detection phase, ruthenium-labeled viral capsid. f A different iteration of bridging ECL assay in e where unlabeled viral capsid is coated onto bare ECL plates
Anti-GTx TAb assays may also be developed in bridging format on both ELISA and ECL platforms. In this assay format on the ECL platform, AAV capsids bound to ECL plate are bridged to ruthenium-labeled AAV capsids through anti-capsid antibodies present in the test sample (14). The advantage of the bridging format is that it is agnostic to antibody isotypes and detects all antibodies that are at least bivalent (the only exception being monovalent, bispecific IgG4 antibodies). This makes the bridging format a powerful tool for initial screening assays. For ELISA bridging assays, unlabeled viral capsid is coated onto standard immunoassay microtiter plates, and biotinylated capsid in combination with streptavidin-HRP is used for detection (Figure 1d). On the ECL platform, biotinylated and ruthenylated AAV capsids may be used as capture and detector, respectively, using standard or high bind streptavidin-coated plates (Figure 1e). An ECL-based bridging assay may also be designed using unlabeled capsid for coating and ruthenylated capsid as the detection reagent (Figure 1f). A summary of the characteristics of the antigen-capture versus bridging assay formats is presented in Table I.
Table I.
Comparison of the Characteristics of the Antigen-Capture and Bridging Assay Formats with Respect to Application, Design, and Methodology
| Assay parameter | Antigen-capture format | Bridging format |
|---|---|---|
| Capture reagent | AAV capsid | AAV capsid |
| Detector reagent | Labeled secondary antibody | Labeled AAV capsid |
| Applicable platforms | ELISA, ECL | ELISA, ECL |
| Isotype specificity detection | Feasible | Not feasible |
| Species specificity | Feasible | Not feasible |
| Sensitivity | Comparable to other formats | Comparable to other formats |
| Ease of development | Comparable to other formats | Comparable to other formats |
| Homogeneous protocol | Feasible | Feasible |
| Heterogeneous protocol | Feasible | Feasible |
Homogeneous vs. Heterogeneous Protocol
Irrespective of platform or format, a homogeneous (co-incubation) or a heterogeneous (stepwise) protocol may be followed to develop anti-AAV TAb assays. For a homogeneous antigen-capture format, test sample and the secondary detection antibody are added simultaneously onto the plate. For a homogeneous bridging TAb assay where unlabeled viral capsid is coated onto the plate, test sample and the labeled capsid are co-incubated. A bridging homogeneous protocol based on ECL bridging assay involves co-incubation of samples with biotinylated and ruthenylated viral capsids which are used as the capture and the detection reagents. Alternatively, incubations may be conducted sequentially forming a heterogeneous version of the method. Selection of homogeneous versus heterogeneous methods should be based on the performance characteristics of the assay and should be investigated during method development. There are no pre-determined guidelines as to which protocol better suits a particular TAb assay. Depending on the assay and the properties of its critical reagents, either a homogeneous or a heterogeneous method may be selected based on whether the method would lead to improved assay selectivity, antigen tolerance, or specificity.
Confirmatory Assay
Most anti-AAV TAb screening assays described above may be followed up or accompanied by a confirmatory assay. This additional step is performed to ensure the specificity of the signal obtained while screening the samples. A common confirmatory assay format consists of signal depletion due to competition with an excess amount of exogenously added, unlabeled AAV capsid. The unlabeled capsids compete with the labelled capture and/or detection capsid reagents for the limited amount of anti-AAV antibodies contained in a test sample, thus leading to a reduction in screening signal which may be normalized and expressed, for example, as percent depletion of assay signal in comparison to the signal generated during the corresponding screening assay. Alternative approaches include immunoglobulin removal from the test sample using generic immunoadsorption resins (22,33), or specific anti-AAV immunoglobulin removal using customized AAV capsid-conjugated resins (25).
TAb ASSAY CRITICAL REAGENT CONSIDERATIONS
While several formats of TAb assays are possible, all will require the use of AAV capsid-derived reagents. Such viral capsid-related critical reagents are applied as a capture-only or as capture and detection reagents, depending on the chosen format for the TAb assay. Unlabeled viral capsid-related reagents may also be applied during confirmatory assay step. Capsid reagents can be the GTx capsid itself or a suitable surrogate, e.g., an empty (also referred as light) version of the GTx capsid or a representative protein the GTx capsid, while keeping in mind that specific manufacturing process details may impact capsid configuration. Yet another alternative is to use a reporter gene carrying version of the GTx vector with the same AAV serotype, which is also often a preferred reagent in cell-based NAb assays designed to detect neutralizing anti-AAV antibodies.
Since the main goal of the TAb assay is to detect antibodies specific to the broad variety of epitopes present on the surface of the viral capsid, the use of the intact viral capsid reagent is recommended. Peptide or protein fragment-based reagent may not represent all epitopes required and, therefore, limit the ability of the assay to detect all anti-capsid antibodies. Additionally, some of the epitopes found on an isolated capsid proteins or fragments thereof may not be exposed or accessible on the native capsid surface. Use of these reagents as target antigen in TAb assays may result in the inadvertent detection of irrelevant antibodies.
It is debatable whether the use of an empty capsid vs. a capsid carrying the transgene or a reporter gene has an impact on the ability to detect anti-AAV capsid antibodies. On one hand, some evidence exists that capsids carrying a payload may have different affinity of binding to antibodies tested (22). It is, however, not clear at the moment whether this reported difference in binding abilities expands to all anti-AAV capsid antibodies found in vivo. On the other hand, GTx preparations may still contain empty capsid in addition to the transgene carrying vectors, and because the reported difference in the binding affinities is relatively limited, it is considered that the use of empty capsid as a target antigen may be appropriate.
Further, the use of a reporter gene carrying vector as target antigen could allow for an alignment with the NAb assay which can be viewed as a plus, but only if a NAb (TI) assay is used. Specificity of a reporter gene carrying vector-based TAb method against transgene carrying GTx may be confirmed during assay development phase. This can be done by conducting a competition test where transgene carrying GTx vector is added to a PC-spiked sample resulting in reduction of the assay signal. If assay specificity is confirmed, the data could form a rational argument in favor of using a surrogate reporter gene vector in both TAb and NAb assays if a sponsor uses both assays. It should be noted that there are no regulatory requirements to utilize both TAb and NAb assays in support of GTx modalities. Some sponsors use only either Tab assay, or Nab assay, but very few sponsors use both.
Some of the commonly used AAV serotypes are commercially available and can be used in relevant TAb assays for the assessment of the antibody response to a GTx. One should ensure that the viral vector used to construct the GTx has no additional mutations and is identical to the wild-type version. Another consideration is related to any contaminants or co-purified components of the virus expression system. If cell lines used to produce GTx vector or commercial reagent are different, it is possible that co-purified components of the final assay reagent will also vary. The resulting assay may detect antibodies that are specific to not only the capsid epitopes but also to any other immunogenic components found in the assay capture or detector reagent. However, this concern is not unique to GTxs and applies to all biologic drugs. In addition to wild-type AAV vectors, lab-engineered serotypes, that contain modifications to the capsid protein sequence, have been actively introduced (34). Caution should be applied when using commercially available TAb assays when detecting immune response against lab-engineered serotype vectors.
Similar to protein-based biotherapeutics, production process modifications may introduce changes in the GTx vector product composition. A significant change in the nature and concentration of contaminants or co-purified components could potentially result in inaccurate results from the TAb. However, at this point in time, this concern is theoretical in nature rather than a proven one. Ideally, the viral capsid reagent should be produced using a process identical or very similar to that used to generate clinically applied GTx vector material. One may consider a comparative analysis of analytical specifications available for the assay capsid reagents and the clinically applied GTx vector material, and if they are different, one may consider bridging studies to confirm comparable antigenicity. It should be noted that comparability assessments for immunoassays are fraught with challenges, and it may not be feasible to truly show comparability. For example, the data from titration assays are often reported as the reciprocal of the dilution required to produce a signal that is above a certain value (cut point). Consequently, small changes in the OD may cause a result to be reported as a different value. Such inherent variability makes comparison between assays a challenging endeavor, a point that has been acknowledged by health authorities in package inserts.
POSITIVE CONTROL (PC) PARAMETERS, TYPES, AND LEVELS
Multiple serotype-specific anti-AAV capsid monoclonal and polyclonal antibodies are commercially available and can be applied in TAb assays. Proprietary antibodies may be required if GTx vectors are unique, and there is a desire to develop antibody reagent using GTx as the immunogen (35). Antibodies with various binding affinities and different specificities would be used as positive controls for the TAb assay. It is practical and recommended by the authors to apply commercial reagents wherever possible to facilitate cross-product data comparison.
Serotype specificity may be achieved by applying monoclonal positive control reagents. The question regarding the need for strict serotype specificity of the PC reagent is debatable. While strict PC antibody specificity against viral serotype can be desired in some cases, a broader approach based on a given cross-reactive antibody reagent with appropriate target specificity and binding affinity may also be applied. The latter allows application of commercial reagents and is more practical. Serotype-specific or cross-reacting PC reagent may be used to characterize some of the assay performance parameters, including assay sensitivity, precision, selectivity, and/or robustness. Other parameters should be defined based on analysis of the negative control or naive individual matrix samples. These include assay specificity, assay matrix interference, and cut point. It is critical to understand that serotype specificity demonstrated by using serotype-specific PC reagent does not guarantee strict serotype specificity of the antibody response detected in study samples, where polyclonal, cross-reactive antibody responses are expected (4). Thus, a cross-reacting PC with broad specificity may be preferred but not required.
Affinity of the PC reagent together with the TAb assay design defines whether the method will have acceptable assay sensitivity, commonly expected to be at or below 100 ng/mL of PC antibody in undiluted matrix. When using a bridging format of detection, higher affinity of the interaction may be required to achieve acceptable assay sensitivity. Due to the multivalent nature of IgM isotype antibodies, high avidity of binding can also ensure that relatively low affinity IgM response can still be detected. Sufficiently characterized PC antibody reagent will ensure acceptable assay performance, including assay sensitivity. However, no specific PC antibody affinity requirements are proposed here, and the need for affinity assessment should be defined on a case-by-case basis.
Similar to ADA detecting methods applied for other biotherapeutic modalities, no true standard reference material can be available for anti-AAV TAb-detecting protocols. The PC antibody reagent is instead used as a suitability control and only serves the purpose of demonstrating that the assay performance remains acceptable and similar to that observed during the initial assay validation phase. Although not commonly viewed as critical, the human origin of the PC reagent may become important based on the assay setup. For example, in the direct binding assay format, in order to use a non-human origin PC antibody, one would need to confirm that the anti-human immunoglobulin detection reagent has sufficient cross-reactivity with the PC reagent or alternatively to create a separate detection protocol of the PC reagent alone. Such complexity can be avoided by using a human PC antibody reagent, although obtaining a human antibody specific to the viral vector serotype of interest may be challenging.
As generally recommended for other assays designed to detect antibody responses to biotherapeutics, each method should include negative and positive control samples in order to monitor the assay performance (20). The PC antibody is commonly evaluated at low (LPC) and high (HPC) concentrations. The mid-concentration PC sample may be used although this should be regarded as optional. Alternatively, the PC sample may be tested at multiple dilutions in a titration test.
NEGATIVE CONTROL (NC) SELECTION
Negative matrix (i.e., biological material without AAV-binding antibodies) plays several important roles in GTx TAb assays. At a minimum, the negative matrix is used as a control to monitor assay performance and as a diluent for the assay PC controls. For screening and confirmatory assay tiers (if used), negative matrix control may be further used to normalize signal and adjust for inter-plate variation. For titer assays, a negative matrix may be used as a diluent to ensure consistent matrix concentration at each dilution.
Sufficient amounts of negative matrix should be prepared and stored to support method development, validation, and sample testing. Consequently, a pooled matrix from several individuals is typically required. For many AAV vectors used in generating GTx, a significant proportion of treatment-naïve donors are expected to have vector cross-reacting antibodies (36–38). High prevalence of pre-existing antibodies greatly complicates preparation of a pooled negative control material. Depending on availability of matrices and the frequency of individuals with capsid-binding antibodies, different strategies may be applied.
If relatively few individuals have antibodies, individuals may be screened, and anti-AAV antibody-negative samples are pooled to prepare matrix sample serving as the negative control. While samples with high positivity will likely stand out, samples with lower amounts of reactive antibodies may be difficult to discern; therefore, a confirmatory step is recommended in order to accurately identify and exclude antibody-positive samples for the negative control preparation. Consequently, it may be necessary to estimate screening and confirmatory cut points as part of method development.
In situations where large numbers of individuals have antibodies, screening to remove positive individuals may not be feasible to generate the required volume of matrix, and immunodepletion may be preferred. Non-specific immunodepletion may be performed through use of reagents that bind multiple antibody isotypes (e.g., Protein A, Protein G, Protein L) (14,25), whereas specific immunodepletion may be performed by using an affinity column to selectively remove antibodies reactive to the AAV capsid of interest (22). Because non-specific immunodepletion may alter assay background, one should demonstrate that the signal of the negative control is not too far below the assay cut point (20); otherwise non-specific immunodepletion may not be a suitable approach to generate a negative matrix. Specific immunodepletion, which could be performed to deplete serotype-specific antibodies (22), should use the same capsid that will be used for the TAb assay. This specific approach is unlikely to alter assay background because most immunoglobulins will remain in the sample following immunodepletion.
Both specific and non-specific immunodepletion protocols have the potential to produce negative control matrix that will generate a significantly reduced assay signal. This may lead to an artificially inflated rate of reported anti-AAV TAb prevalence and titer in study samples. If immunodepletion approach is selected, the NC produced signal should be carefully compared to the assay cut point value.
Negative matrix should match the sample type(s) that will be evaluated in the assay (e.g., species, specimen type, anticoagulant). Some matrices (e.g., CSF, aqueous humor) may not be available in sufficient quantities, potentially requiring the use of a surrogate matrix. Suitability of a surrogate matrix should be demonstrated, and any impact may be mitigated by including a limited number of matrix matching controls (39).
SAMPLE TESTING STRATEGY
Serum (or other matrices such as plasma, CSF, aqueous humor, vitreous humor, or saliva) may be analyzed using a classical (screening, confirmatory, titration) testing paradigm (40–42). The intent of the screening and confirmatory tests is to reduce the number of samples that are subsequently tested in the titration assay since the titration assay is resource intensive and is of low throughput. However, there can be a strong rationale for only analyzing the sample in a titration assay. For example, for patient selection, a titration result may be required, and the turnaround time for the sample analysis may be rate-limiting for the treatment of the patient. In this case, solely analyzing the sample in the titration assay may be the best approach. Post-treatment, most if not all, patients will develop an anti-AAV antibody response. Therefore, the use of a screening and confirmatory assay will only add time and cost to the sample analysis. Therefore, some sponsors do not conduct screening and confirmatory assays and only perform the titration assay.
Classical tiered-based approach to TAb testing may be considered in case anti-GTx serotype antibody specificity and cross-reactivity with other AAV serotypes needs to be understood. Clinical trials for GTx often consist of a small number of patients and hence a low number of samples; therefore, screening and confirmation tests may be done in parallel. A comparison of pre- and post-dose sample titers may be necessary irrespective of the cut point assessment approach. An increase in post-dose versus pre-dose titer signifies treatment emergent anti-AAV capsid immune responses.
Reporting TAb assay results in titer units calculated as a reciprocal of sample dilution is recommended. Consistent reporting in titer units will facilitate the ability to compare among studies and programs. It has to be pointed out that assay to assay differences prevent direct comparison of results.
ASSAY MATRIX SELECTION
The sample matrix that is used for the analysis of anti-AAV antibodies will depend on many factors and may differ between, and within, programs depending upon the questions that are being asked. Serum is the matrix that is most often used to detect presence of anti-AAV antibodies, regardless of the route of administration for gene therapy. Measurement of the titers of pre-existing anti-AAV antibodies can be important for mitigating the impact of these antibodies on efficacy of IV administered gene therapies, or the acute safety of intravenous (IV), intrathecal (IT), or cisterna magna (ICM) administered gene therapies. Serum is also a relevant matrix for measuring treatment-induced or treatment-boosted anti-AAV antibody titers which may potentially be associated with certain systemic adverse events such as cytotoxic immune response (43).
In addition to serum, plasma is also often used for anti-AAV antibody detection. These two matrices are top choices due to relative ease of sample collection. Although plasma is used less frequently, at times it may be the only option. In pediatric clinical trials, blood volume often limits the number of analytes that can be measured. It may not be possible to collect contemporaneous samples to assess cellular responses (as measured by an ELISpot assay) and humoral responses (anti-AAV antibodies). Therefore, it may be necessary to collect whole blood that is used to prepare PBMCs and plasma as a byproduct that can be used for the measurement of anti-AAV antibodies. However, it is acknowledged that it is not possible to collect the large volume (e.g., 8–10 mL) of blood required for the cellular response at multiple timepoints, if it is feasible to collect such samples at all. Consequently, the assessment of anti-AAV antibodies within a clinical trial may require the analysis of both plasma and serum samples. Another example where plasma samples are preferred are GTx clinical trials in the area of coagulation disorders, such as hemophilia A and B; collection of serum samples may be difficult due to the inability to form proper blood clots in the absence of any medical treatment.
Application of serum as the assay matrix for the detection of anti-AAV antibodies may not be the fully relevant, depending on route of administration (e.g., intrathecal, intravitreal, subretinal), and detection of anti-AAV antibodies in other matrices could be of relevance. For example, for intravitreally administered gene therapies, the measurement of anti-AAV antibodies in vitreous humor or aqueous humor may be of importance. However, the collection of vitreous fluid can only easily be performed immediately prior to the intravitreal (IVT) administration. This limits the utility of the collected sample since it cannot be used for patient selection. However, the collection of aqueous humor can be made repeatedly and may offer hope as a surrogate matrix for the vitreous. Similarly, cerebrospinal fluid (CSF) is often collected prior to IT administration with the same limitations that exist for the collection of vitreous. It has been documented that anti-AAV antibody titers are lower in CSF and vitreous than in the corresponding serum samples, and it is likely that anti-AAV antibodies do not exist in the subretinal space, which is an artificial space created by the act of injecting the gene therapy (44,45). Collection of CSF samples post-dose remains more feasible than the collection of vitreous post-dose samples. It will be, therefore, important to understand the relationship between anti-AAV titers in rare matrices and proposed surrogate matrix, such as serum.
One matrix that may be of utility for anti-AAV antibody testing but has rarely been used to date is saliva. It is possible that saliva may be used as a surrogate matrix for serum, which would be more convenient for the patient and also allow for more frequent sampling. More data are needed to investigate the presence of anti-AAV TAbs in saliva and the correlation with serum or plasma antibody levels.
KEY TAb ASSAY VALIDATION PARAMETERS
Assay Sensitivity
Sensitivity parameter for the assays designed to detect total anti-capsid antibodies can be assessed similar to the approach applied for ADA detection methods (40). Current FDA guidance, describing regulatory expectations for anti-biotherapeutic immunogenicity detection protocols, (20) suggests that a typical ADA assay should be developed with a minimal target sensitivity of 100 ng/mL. It has been broadly demonstrated that sensitivity of ADA assays commonly meets and exceeds such proposed minimal requirements (46). Because the sensitivity of either ADA or anti-AAV TAb assay is expected to be defined based on the performance of the assay-positive control, low affinity PC reagents may present a challenge in achieving the required value. In these cases, assay conditions, including nature of the PC used, should be re-evaluated. The PC reagent is not expected to represent actual anti-AAV capsid antibodies in patient samples, and the exact correlation between the ability of the assay to detect PC antibodies at a given concentration and clinically observed safety or efficacy signals remains unclear. Thus, assays with greater than 100 ng/mL sensitivity in detecting PC may be still capable of detecting clinically relevant TAbs. The ultimate desired clinically relevant TAb assay sensitivity may also depend on several other factors, including antibody epitope specificity, binding affinity, isotype composition, effective concentration in patients’ blood, and more.
Serotype Specificity of Detected TAb
It has been shown that there is a prevalence of anti-AAV immunity in the general population (37). Anti-AAV antibodies have also been shown to be highly cross-reactive across AAV serotypes (37,47). Treatment-naïve donors assessed for anti-AAV binding antibodies were found TAb positive for AAV serotypes 1, 2, 5, 6, and 9 with the highest sero-prevalence being against AAV2 reported (48,49). The degree of cross-reactivity varied depending on the AAV serotype and was proposed to be contingent on the degree of homology between AAV sequence (50). The high degree of cross-reactivity of anti-AAV TAb activity may be distinguished from a lower level of cross-reactivity of neutralizing antibody responses (51–55). High complexity and multi-step process of viral transduction is likely to require a high specificity of neutralizing antibody binding which should be distinguished from a simple binding event which is detected in the case of the TAb assay. The actual level of cross-reactivity may be dependent on the analytical methodology and antibody titers. High degree of alignment of anti-AAV5- or anti-AAV8-neutralizing activity was reported for samples with high anti-AAV2 NAb titers (47). In general, anti-AAV antibody response to various serotypes, including age-dependent rise of the anti-AAV immunity, has been associated with environmental exposure to highly prevalent AAV2 serotype (47,56).
Recently, the sero-prevalence of pre-existing anti-AAV9 antibodies was assessed in patients enrolled in clinical trials of onasemnogene abeparvovec (intravenous and intrathecal administration) and managed access programs (intravenous). Through December 31, 2019, 196 patients and 155 biologic mothers were screened for anti-AAV9 antibodies using a TAb assay. Of these, 15 patients (7.7%) and 23 biologic mothers (14.8%) had titers >1:50 on their initial screening tests. Eleven patients (5.6%) had elevated titers on their final screening tests (57).
The need to demonstrate whether the anti-AAV antibodies detected in the TAb assay are specific for the GTx serotype of interest may be important under some circumstances, for example, if GTx of different serotypes are available for the treatment of the disease. This could be achieved by performing confirmatory test. Presence of serotype-specific antibodies should then lead to signal depletion compared to the untreated sample. This is not to say that the TAb assay should be designed to achieve exclusive serotype specificity since such assays would not detect antibodies that bind to both the serotype of interest and other related serotypes. It may not be technically feasible to develop serotype-specific assays due to the high homology of capsid sequences across various AAVs. Additionally, the reason for detecting anti-AAV antibodies in clinical trials is to understand the impact of all antibodies that bind to the serotype of the GTx, not to understand the impact of antibodies that bind only to the serotype of the GTx. However, the cross-reactivity of the antibody response may be of great importance when the patient may be subsequently treated with another vector-based drug, either as another GTx or as a vaccine. For example, following recent development of vaccines against SARS-COV2 virus, sponsors may consider whether the antibodies generated by the investigated GTx can bind to viral serotype used by vaccine product, e.g., Ad26 (58,59).
Factors other than immunoglobulins can play a role in the overall effect on the degree of vector transduction inhibition in NAb assays. Depending on the assay setup, TAb protocols can report capsid-specific interaction (bridging assay formats, Figure 1d, e, and f) or presence of anti-capsid-specific IgG/IgM or other isotype immunoglobulins (direct binding assay formats, Figure 1a, b, and c). The need to determine whether the binding is immunoglobulin-based may require an additional characterization of the TAb activity or a need for a sample pre-treatment step, e.g., isolated immunoglobulin fraction. Any additional characterization of the immune response should be value-driven since it will be costly and time-consuming and require a larger sample volume. Only antibody characterization approaches that can add significant value to the interpretation of the study data should be considered.
Precision
Assay precision is defined based on the observed variability of the assay raw signal or assay signal-to-noise ratio (S/N). Typically, assay precision is calculated based on results obtained in the intra- and inter-assay experiments. In these experiments, samples are tested in two or more replicates. The inter-assay evaluation most commonly includes multiple (3 or more) runs conducted by several (2 or more) analysts on different days. This approach is to ensure that sufficient data are collected to appropriately assess assay performance.
Typical ligand-binding assay precision acceptance criteria for both intra- and inter-assay performance (e.g., CV% less than 20%) are applicable for anti-AAV TAb protocols (40).
Selectivity and Matrix Interference
In the assay selectivity test, the influence of naturally occurring matrix components in individual donor samples is evaluated. To assess selectivity, positive control samples at the LPC concentration are evaluated to confirm the assay ability to detect PC at the low concentration levels under interference of various matrix components. PC reagent is spiked into several, typically 10, individual TAb-negative donor samples to be evaluated in the assay. At least 80% of LPC level-spiked samples should score TAb positive. Unspiked individual samples are also tested with the expectation that at least 80% of samples should score TAb negative. Ideally, selectivity should be performed using both healthy and disease-state samples although healthy donor samples may be applied if disease-state samples are difficult to procure.
Additional evaluation of assay performance in samples containing varying amounts of known and potentially interfering matrix components should be considered. This includes the assessment of the impact of lipids (lipemic samples), hemoglobin (hemolytic samples), and bilirubin (icteric samples) (20). Such components may have limited or no impact on assay performance, and tests can be conducted during assay development phase only. Other matrix components that are expected to be present in samples at the time of collection may be considered and tested, including various co-medications and anti-coagulants (e.g., heparin). The overall criteria for the acceptance of the test should be similar to the criteria applied for the selectivity test.
Assay Cut Point
TAb detection methods are semi-quantitative and employ a threshold value, referred to as assay cut point, that determines whether a sample contains detectable levels of anti-AAV antibodies and thus is reported as positive. A multistep tier-based approach, commonly used to evaluate ADA against protein-based biotherapeutics (20,40,60), is suggested for the TAb protocols. Individual cut point values are assigned to screening and confirmatory methods. Such cut points are method specific and identified during assay qualification or validation phase, prior to clinical sample testing. Both screening and confirmatory assay cut points are determined based on statistical analysis of data obtained for a large number of drug naïve individual study relevant samples (20,40). For example, assay data are collected for 50 or more individual samples obtained from treatment-naïve subjects that are tested in 3 to 6 runs. The resulting data set is analyzed for the presence of statistical and biological outliers which are removed. Typically, in the next step, the 95th (screening test) or 99th (confirmatory test) percentile of the data distribution is identified by an appropriate statistical approach. The methodology has been described in detail elsewhere (20,40,41).
Similarly, application of a statistically defined cut point value is proposed for anti-AAV TAb assays. One of the practical challenges and potential limitations of this approach is the high prevalence of pre-existing anti-AAV capsid antibodies in treatment-naïve subjects. Selection of a large pool (50 or more) of individual samples that do not demonstrate detectable presence of anti-capsid TAb may, therefore, be a concern. The distribution of sample-generated assay responses is often continuous, not allowing for a clean separation between positive and negative sample population as it has been observed in other cases (42). One possible solution is to identify true anti-AAV TAb-positive samples in a confirmatory test. Unlabeled GTx-spiked samples that produce 50% or greater assay signal reduction may be removed from the cut point data analysis as biological outliers containing pre-existing anti-AAV TAbs. The proposed 50% cut-off is based on the assumption that such a high impact on assay response suggests a clear indication of a true anti-capsid-binding biological component in the sample. Data set produced by samples with less than 50% of signal reduction can be applied to calculate screening and confirmatory cut point values for the anti-AAV TAb method. It is clear that an analysis of a large number of samples will be required to finally yield at least 50 TAb-negative individual samples needed for the statistical calculation of assay cut points.
VALUE OF ANTI-AAV TAb ISOTYPE CHARACTERIZATION
In either the T-cell-dependent or T-cell-independent pathways of the immune response, anti-capsid antibody development can be expected within the days and weeks after GTx exposure. Initially, there will be development of a low affinity, typically IgM isotype immunoglobulins, response. Maturation of the B-cell response can further result in immunoglobulin class switching, increase in binding affinity, and potentially in a modulation of the binding epitopes. Resulting anti-capsid antibody pools consist of a collection of various immunoglobulin isotypes, including various IgG subclasses, although development of other isotypes is also possible. Understanding of the exact isotype composition of anti-capsid immune response may be important as it could potentially allow for a more detailed explanation of post-dose toxicities or facilitate the patient selection process prior to the treatment. Detection of IgEs specific to a biotherapeutic presents a separate case, as this class of immunoglobulins has been linked to various hypersensitivity responses, including a type I response. However, hypersensitivity reactions to AAV GTx have been rarely, if at all, observed to date. The significance of understanding the presence of other classes and subclasses of immunoglobulins is less clear at this point (61).
The pre-existing anti-AAV antibodies will predominantly be the IgG isotype (49). Prevalence and isotype composition of pre-existing anti-AAV8 antibodies was investigated in cohorts of healthy donors and patients with hemophilia B from different geographical regions (38). It was shown that majority of anti-AAV8-positive samples contained IgG (71%) or a mix of IgG and IgM (12%) isotypes. A more sensitive TAb assay demonstrated a very strong correlation between IgG isotype of anti-AAV8 TAb and NAb responses, suggesting connection between mature antibody response and anti-AAV8-neutralizing activity (38). While detection of IgG4 subclass antibodies may indicate a highly mature response, this specific subclass has been associated with reduced effector function as compared to other IgG subclasses, for example, IgG1 (62,63). The ability of anti-capsid antibodies to interact with and activate complement cascade components to bind the Fc-gamma receptors may be reflected in the clinical safety observations. For example, IgM, IgG1, and IgG3 immunoglobulins have high capability to activate complement, as opposed to the IgG2 and IgG4 subclasses (64–67). Both affinity and avidity of antibodies usually correlate with immunoglobulin class and subclass. IgM immunoglobulins are typically low affinity but may present high avidity due to the repetitive nature of the capsid protein structure that could allow multiple Fab arms on the pentameric IgM molecule to engage with the target antigen, barring any steric constraints. Binding affinity of the IgG isotype antibodies is typically higher due to somatic hypermutation that occurs concurrently with class switching in germinal centers (65,68). It is possible that presence of IgM isotype may help to explain complement related hypersensitivity reaction or that the presence of IgG4 subclass may facilitate with understanding of the maturity of immune response, particularly pre-dose. Although a significant amount of data is available regarding potential implications of IgM and IgG subclasses development, we are still learning about whether and how specific isotype information can benefit treatment decisions related to a given GTx product.
At the present time, various methodologies are available to assess the isotype composition of anti-AAV capsid immune response. These include LBA protocols allowing to detect a particular isotype, as well as multiplexing methods that can detect multiple isotypes in each sample in one run (69). Other emerging methodologies have been discussed, including liquid chromatography-mass spectrometry (LCMS)-based protocols (70).
VALUE AND CHALLENGE OF STANDARDIZING TAb ASSAYS ACROSS THE INDUSTRY
Since GTx treatment decisions can bear substantial implications for the course of the disease in patients, it may be helpful to harmonize how anti-AAV TAb assays are performed across the industry. This could lead to a higher degree of transparency, reliability, and comparability for a key methodology that is used to predict treatment success of gene therapies. As more experience is gained with regard to the predictability of clinical outcomes based on anti-AAV antibody status and titer, the standardization of antibody measurements would also enable a better comparison between different clinical trials. Clinical lessons learned about pre-existing antibody titers in AAV5 gene therapy may even be informative for other AAV serotype gene therapies, if the same route of administration is used, such that anti-AAV antibodies would be expected to pose similar barriers to successful dosing.
Without assay standardization, however, a comparison of anti-AAV antibody titers across different TAb assays is highly difficult and oftentimes nearly, if not completely, impossible, due to differences in assay sensitivity, reagents, cut points, sample dilution schemes, and the different mathematical calculations for antibody titer determination. Currently, measurements of anti-AAV antibodies are not standardized and thus method and clinical trial specific. This restricts our ability to reach broader conclusions about the impact of pre-existing immunity on AAV gene therapy outcomes, thus decelerating progress made by the field as whole. However, sponsors with a large portfolio of GTx of the same serotype will be able to use the same TAb assay across their portfolio and apply learnings across programs. Standards in assessing pre-existing immunity would also help leveling the ground for competing companies in the same disease area, as it would remove the opportunity to use inadequate anti-AAV TAb assays, or less restrictive screening criteria in order to identify a larger patient population as eligible for treatment.
The fundamental challenge for standardizing the assessment of pre-existing anti-AAV immunity using TAb assays is to seek industry and health authority agreement that this type of assay is clinically meaningful to measure anti-AAV antibody levels. In contrast to cell-based NAb assays, TAb assays detect the binding of antibodies to the AAV capsid, leaving out of consideration any potential functional consequences of this interaction. As stated earlier in this manuscript, while a good correlation between TAb and NAb titers, in particular for higher antibody levels, is typically expected, presence of non-neutralizing anti-AAV antibodies is possible and would require an orthogonal assessment in both TAb and NAb assays (5,14,71).
Standardization for quantitative bioanalytical assays has been achieved in the past by establishing global calibration standards, for example, the WHO International Standards for coagulation factor levels in human plasma (72). In contrast to quantitative assays, however, anti-AAV TAb assays do not have an objective calibration standard that fully corresponds to the measured analyte. This problem stems from the heterogeneous nature of the analyte, a polyclonal mixture of AAV-specific antibodies of various isotypes that are highly diverse in composition across human individuals. Therefore, anti-AAV TAb assays are not usually performed by absolute quantification using interpolation against a concentration curve of standard material but rather by performing relative quantification using sample dilution to determine antibody titers. Reporting of absolute anti-AAV antibody matrix concentrations in reference to a global calibration standard (i.e., as international antibody units per mL) would thus be a very difficult approach.
Other challenges for the standardization of anti-AAV TAb assays arise from the use of different assay technologies and formats, with varying sensitivities and diverse reagents across the pharmaceutical industry. In addition, multiple statistical approaches exist to establish cut points used for discriminating positive from negative samples and to determine the antibody titer in a positive sample using the results from serial sample dilutions (endpoint titer vs. interpolated titer). Given that at least some of these differences are due to internal choices made during assay development and validation, they may be amenable to a certain degree of standardization, as detailed below.
An important milestone for standardization of anti-AAV TAb assays would be their consistent use for testing of pre-existing AAV immunity in patients across all clinical gene therapy trials. The TAb assay may even be run concurrently to an otherwise preferred cell-based NAb assay, which is used by some sponsors for enrollment decisions, but additionally performing a TAb assay would constitute an important step toward generating data that can be consistently cross-referenced with clinical outcomes in each trial. Ideally, TAb data would encompass patients found to be both eligible and ineligible for a particular AAV gene therapy in question.
To enable further standardization of anti-AAV TAb assays, harmonizing the use of certain critical assay reagents, such as the AAV capsid material that serves as the target antigen, could be considered. Commercially available AAV reference standards exist for some serotypes (e.g., ATCC: VR-1616, VR-1816) and could be established for additional serotypes if desired. As discussed above, this approach would require a careful evaluation to ensure comparable antigenicity between the AAV reference material and the AAV vector under development, given the potentially diverging manufacturing conditions and the different sequence and length of the vector genomes.
It is difficult to gauge the potential value of using AAV reference materials as target antigens in anti-AAV TAb assays since these assays do not quantify AAV capsids but rather determine the magnitude of antibody responses directed against them. It would also be counter-productive to standardize the exact amount of AAV capsid utilized in the assay to be combined with a certain volume of test sample, because this parameter is frequently optimized during assay development to achieve the desired sensitivity for antibody detection.
Instead of standardizing assay reagents, formats, and technology platforms, it appears more promising to seek industry and health authority alignment for mandatory method performance characteristics and for regulated procedures associated with data analysis. For example, assessing the minimally required dilution (MRD) of a test sample that is needed to avoid matrix interference, or achieving sufficient sensitivity and specificity for anti-AAV antibody detection, is of utmost importance for generating reliable results. Moreover, statistical procedures that lead to cut point establishment could be harmonized, similar to those used for ADA assays (20), as described earlier in the manuscript. One needs to remember that FDA guidance on immunogenicity assessment, issued in 2019, specifically excludes application for the gene therapy modalities. As random collections of donor samples frequently contain pre-existing AAV antibodies, harmonized and transparent procedures are needed to identify and exclude baseline-positive samples, as well as statistical outliers from cut point analyses.
Finally, antibody titer results can be reported by TAb assays either as the reciprocal of the highest sample dilution that was still tested positive (endpoint method), or as the reciprocal of the sample dilution interpolated at the titer cut point, as this would be the highest sample dilution theoretically expected to still test positive (interpolated titer). The latter approach tends to report higher but potentially more accurate titers, in particular if relatively large dilution steps (greater than 1:3 dilutions) are used for the test samples. Thus, harmonized guidelines could be beneficial to direct which titer dilution scheme (1:2, 1:3, or 1:5) and associated titer reporting algorithms should be used. In summary, many anti-AAV TAb method performance parameters and data analysis procedures could be amenable to industry-wide standardization, if all stakeholders (industry, regulators, diagnostic companies, patient advocates) choose to seek alignment.
It is worth noting that the above efforts to standardize anti-AAV TAb assays would still be unlikely to achieve full data comparability. Numerical antibody titers reported by TAb assays heavily depend on assay sensitivity: a more sensitive assay will report a higher antibody titer for the same sample than a less sensitive assay, since more sample dilutions are required until the sample no longer tests positive. PC reagent antibodies can be monoclonal or polyclonal, highly purified and crude serum, but in any case, they usually differ between different sponsors. It is, therefore, currently not possible to ensure comparable sensitivity between anti-AAV TAb assays that use different PC antibodies, even if the same concentration (ng per mL) was validated as the limit of detection (LOD). Hence, the current assessments of sensitivity performed during method validation are insufficient to enable a direct comparison of antibody titer results.
A similar shortcoming had previously been recognized by the ADA community when comparing results from TAb assays that detect anti-EPO antibodies (73). The solution implemented in this case was the establishment of a global and publicly available antibody reference panel, consisting of a set of highly purified, monoclonal human anti-EPO antibodies (i.e., WHO positive control anti-EPO reference panel).
With regard to AAV gene therapy, monoclonal antibodies against AAV capsids could be isolated and purified, in order to test them under defined conditions in different anti-AAV TAb assays across various laboratories. For each anti-AAV antibody in the panel, an LOD could be determined, and titer results could be correlated with antibody levels expressed as mass concentration units. These data could then be compared between different sponsors to gain insights into assay sensitivities for various types of antibodies and how the reported titer results could potentially be translated. As the establishment of a global reference panel would be a time-consuming and resource-intensive endeavor, meaningful engagement of stakeholders will critically depend on the perceived value for sponsors, regulators, caregivers, and patients.
As a short-term and more cost-efficient alternative to establishing reference panels containing purified human monoclonal anti-AAV antibodies, a defined set of crude human plasma or serum samples containing polyclonal mixtures of anti-AAV antibodies could be distributed to individual laboratories to perform proficiency testing. Each laboratory would test the same set of samples and report back the obtained titer results. This would help benchmark relative sensitivity and magnitude of titer measurements for each TAb assay across sponsors and also reveal the overall degree of variation that currently exists between different laboratory tests. The disadvantage of this approach would be the finite quantity of the human samples used for cross-testing, thus restricting longitudinal assessment and trending of method performance.
It should be noted that in attempting to standardize TAb assays, we may be constraining our ability to provide the most appropriate bioanalytical support. For example, once a standardized assay format had been agreed upon, there would be no incentive to develop assays that may be an improvement upon the standardized assays. Defining a sample dilution scheme that must be followed by all sponsors would mean that a sponsor could not deviate from that scheme when it may be appropriate to use small dilution steps to characterize a low-titer response and larger dilution steps to characterize a high titer response. Additionally, standardization of reagents will increase the criticality of those reagents, making them highly vulnerable to supply chain disruptions.
APPLICATION OF THE TAb ASSAY DATA
Total anti-AAV antibody data can be used as a risk mitigation strategy and as a tool to determine the association of anti-AAV antibodies to the safety and/or efficacy of the gene therapy. As mentioned above, TAb data can be used to identify subjects for treatment with a gene therapy. An argument in favor of using TAb assays may be that any type of pre-existing anti-AAV antibodies, neutralizing and non-neutralizing, is a specific biomarker that indicates prior exposure to AAV. Anti-AAV antibodies may also accelerate the uptake and clearance of AAV vectors by other immune cells, thus compromising the efficacy of AAV gene therapy. More research is needed to better understand the immune correlates of successful gene therapy outcomes, and it appears that currently the broad detection capabilities of anti-AAV TAb assays offer a key advantage over other assays due to its ease of execution, quick turn-around time. In addition to the scientific reasons for using a TAb assay rather than a NAb assay, there are pragmatic reasons for this choice. TAb assays will be more robust than NAb assays, and their data quality consequently better suited for use as a companion diagnostic or a point-of-care assay than cell-based NAb assays.
Anti-AAV TAb methods have been used for patient stratification during clinical studies. The sample will be collected in a pre-defined timeframe (e.g., within 1–2 months prior to administration of the gene therapy). If the titer meets the pre-defined criterion for patient eligibility, then the patient may be dosed with the gene therapy contingent upon the patient meeting all other eligibility criteria. If the TAb titer does not meet the predefined criterion, another sample may be collected at later time, and the patient will be re-screened. If the titer does meet the acceptance criterion, the patient is eligible for treatment. If the titer does not meet the acceptance criterion, further rescreening may be performed, depending on the observed titer and the likelihood that the titer may decline to an acceptable level in a reasonable time frame. Plasmapheresis or immuno-adsorption-based anti-AAV antibody depletion and IgG-clearing patient pre-treatment strategies have been proposed to reduce the impact of pre-existing anti-AAV vector antibodies (22,25,74).
There is much debate about what represents a suitable eligibility criterion with respect to pre-existing antibody titer. Often the criterion is selected based on an abundance of caution and represents the lowest detectable titer (i.e., subjects are required to be seronegative). The eligibility criteria are not often based upon actual data that show a relationship between anti-AAV antibody titer and safety and/or efficacy variables. It should be noted that for some routes of administration (e.g., intrathecal, intravitreal, subretinal), serum circulating anti-AAV antibodies may have a different overall level of impact on the treatment. Therefore, the use of a stringent anti-AAV Ab titer threshold as an exclusion criterion for GTx administered by routes other than intravenous may unnecessarily exclude patients from receiving a beneficial life-changing therapy. It should be noted that although the initial clinical trial for Luxturna® used an exclusion criterion of 1:1000 for serum anti-AAV2 antibodies, no such exclusion criterion was used for subsequent studies nor is there a requirement included in the product label (75). Similarly, the impact of pre-existing serum anti-AAV antibodies on the safety of AAV gene therapies is likely to be far less for those gene therapies that are administered by non-intravenous routes compared to those administered intravenously. Although the systemic bioavailability of a gene therapy after intrathecal administration may be high, the systemic concentrations of the AAV gene therapy will be low and hence the safety concerns due to the presence of pre-existing anti-AAV antibodies in serum is likely to be lower. Consequently, it may be feasible to administer AAV gene therapies by the intrathecal route to patients with titers of pre-existing antibodies that are greater than the threshold antibody titers used for patient eligibility in intravenously administered gene therapies. However, more research is required to determine how high the threshold titer would be for patient stratification during AAV gene therapies by the intrathecal route.
For the treatment of newborn patients, monitoring of the maternal serum anti-AAV antibody titers may be advantageous. The immune repertoire of the newborn will reflect that of the mother as a consequence of placental transfer of IgGs, and it will take several months for the mother’s immunity to wane in the infant and for the infant to develop their own anti-AAV antibody repertoire based upon environmental exposure. By screening the mother for the presence of anti-AAV antibodies, it is possible to provide some guidance as to whether the newborn may be eligible for treatment with an intravenously administered AAV gene therapy or whether the treatment may need to be delayed until the infant’s passive anti-AAV antibody titers have waned. More research is required to determine the relationship between maternal and newborn serum anti-AAV antibody titers and the time required for the maternally transferred antibodies to decline.
REGULATORY CONSIDERATIONS
Screening prior to enrollment in a clinical study may be a useful strategy to identify patients who are most likely to benefit from investigational GTx. The regulatory considerations for GTx TAb and NAb assays during clinical studies have not been fully defined. However, a qualified test may suffice for phase I/II studies, while registration studies may require a validated test.
The assays used to identify patients for treatment with marketed gene therapies in the USA will require a companion diagnostic assay (CDx) or laboratory-developed test (LDT). In the USA, regulation of LDTs falls under the Clinical Laboratory Improvement Amendments (CLIA) program which regulates laboratories that perform testing on patient specimens in order to ensure accurate and reliable test results (76). However, debates continue regarding US FDA’s assertion that they regulate LDTs under “enforcement discretion.”
Regardless of the regulatory oversight, for patient inclusion/exclusion in clinical trials intended for treatment with an investigational gene therapy, anti-AAV antibody assays may need to be run in a facility regulated under the CLIA regulations using personnel trained to perform testing under these regulations for US patients. This requirement would not apply to natural history studies (i.e., studies in which no therapeutic agent is administered) or samples collected at the time of dosing or post-treatment since the data are not used for patient selection or the determining how to treat the patient. Industry experience suggests that this may not always be necessary if principles of good clinical practice (GCP) and general regulatory recommendations for validated immunogenicity assays are followed. However, if the data collected is intended to support a submission to US FDA for subsequent approval as a CDx, FDA may expect that the pre-screening testing be performed using facilities and personnel regulated under CLIA. This is an area of great debate in the industry and is a topic that needs clear guidance from the FDA.
It should also be noted that CLIA does not apply globally as it is governed by the US regulations, and thus assay validations should be deemed suitable for the specific geographic region where the patient samples are derived from. Attention should be paid to where the samples are collected and analyzed, including specific country, state, or other appropriate jurisdictions. During clinical development, these assays may be run in a single lab as a laboratory-developed test (LDT); however, upon marketing of the therapeutic product, it may be preferable to have a US FDA-approved or EU CE-marked companion diagnostic test (CDx) in place to perform the assays.
GTx clinical studies may form the basis for a CDx requirement if prospective patients with detectable pre-existing antibodies are excluded. The term “companion diagnostic” is defined by the FDA (https://www.fda.gov/medical-devices/vitro-diagnostics/companion-diagnostics). Recent FDA guidance documents, covering Human Gene Therapy for Rare Diseases (77) and Human Gene Therapy for Hemophilia (78), describe considerations for the study population where pre-existing antibodies to the GTx product may block delivery of the therapeutic vector genome, pose a potential risk to safety, and /or limit therapeutic potential. In such cases where patients are excluded on the basis of pre-existing antibodies, both guidance documents state that the sponsor should strongly consider contemporaneous development of a companion diagnostic. Submission of the premarket application (PMA) for companion diagnostic and submission of BLA/MMA for a gene therapy product should be coordinated to support contemporaneous marketing authorizations.
As an alternative to developing a CDx test, patient screening may be accomplished with LDTs. Some current examples from marketed GTx products include Zolgensma® and Luxturna®. Clinical trials of Zolgensma® used a laboratory-developed ELISA assay where anti-AAV9 titers above 50 were not studied. Patients receiving the marketed GTx are recommended to have an anti-AAV9 Ab titer of ≤50 prior to infusion. For Luxturna®, indicated for treatment of Leber congenital amaurosis and administered via subretinal injection, pre-existing immunity to AAV2 was assessed during clinical development but did not appear to affect treatment safety or efficacy, and therefore this assessment was not requested prior to therapeutic administration during the post-approval phase. Detailed discussion of CDx and LDT is beyond the scope of this manuscript. Both tests are reviewed by the FDA and should meet expected targets for method performance parameters. The extent of method validation necessary for assays detecting pre-existing antibodies should match the stage of product development, and the level of validation should be “fit-for-purpose” or appropriate for the intended purpose of the study.
SUMMARY AND CONCLUSIONS
The expected immune response against GTx is likely multi-factorial and may be as complex as the therapeutic itself. GTxs consist of multiple components that may be affected by the immune response and, therefore, may require multiple assays to fully understand patient immune response to the therapeutic. The goal of this paper is to guide the immunogenicity assessment of these products, resulting in the delivery of relevant data that, in context with safety and efficacy measures, will allow for the delivery of beneficial life-changing therapies to the patient population.
Pre-existing or treatment-induced anti-AAV antibody immunity has been broadly reported (29,37). The prevalence of pre-existing anti-AAV antibodies is highly dependent on a number of factors, including AAV serotype, geographical region, and age of the patient (37). Individual characteristics of the anti-AAV response are variable and may include both TAb and NAb. Parameters and expectations for cell-based neutralizing anti-AAV antibody detection assays have been discussed elsewhere (79). Here, we review various considerations relevant to the detection of the TAb anti-AAV antibody response, including applicable analytical protocols, assay format, positive and negative control selection, sample matrix selection, sample testing strategies, key assay validation parameters, value and challenges of method standardization, and regulatory considerations.
Total antibody detecting methods are a valuable alternative to the NAb detection protocols. TAb methods are typically LBA based, relatively easy to establish, and robust in production. TAb protocols may also offer a significant cost benefit when compared to complex cell-based NAb methods. While common NAb methods can also be detecting immunoglobulin-based transduction inhibitors, a TAb protocol can be designed as antibody specific to identify the total binding antibodies (both neutralizing and non-neutralizing) that can impact post-administration AAV vector concentrations through clearance, e.g., opsonization, but do not possess ability to directly neutralize cell transduction. More patients are expected to be identified as TAb positive than NAb positive due to typically higher TAb titers and overall expected higher TAb assay sensitivity. It remains to be determined whether and at what level the TAb positivity can be directly correlated with an impact on GTx treatment. The actual answer is likely to be product and treatment specific.
Analytical platforms and assay validation parameters relevant to the TAb-detecting protocols are generally similar to those that have been broadly accepted for ADA detection methods (20,40). The similarity between ADA and anti-AAV TAb protocols includes possible application of a tier-based approach to sample analysis where initial screening and confirmatory tests are followed by the final titer determination test. Considering that treatment decision is typically based on reported patient TAb titer, direct titer determination with no prior screening and confirmatory analysis may be applied. Based on the typical assay formats and platforms used, TAb protocols are expected to detect majority of the anti-AAV antibody isotypes with the IgM and IgG classes to be the main analytes for the test.
Harmonization of anti-AAV antibody assays presents an opportunity to combine immunogenicity data obtained in various studies, improve level of reliability for a critical methodology, and overall advance our ability to predict success of GTx-based treatment. The semi-quantitative nature of TAb assays due to the lack of a true calibration standard, use of various analytical platforms and assay formats, and diversity of reagents are examples of challenges currently standing in the way of standardization of TAb protocols. Some of these are based on the decisions made during assay development, e.g., selection of certain critical reagents and use of commercial AAV serotype reference standards where appropriate. Even more relevant is to apply an industry wide approach to evaluation of assay performance and data reporting. Strong examples of the latter are agreements on a statistical methodology to calculate assay cut point value and reporting assay results as reciprocal of the sample dilution value. A set of standardized antibody reagents or pooled matrix samples may be established and distributed to analytical labs to perform proficiency testing in order to establish comparable baseline for the assay sensitivity and performance.
Overall, TAb-detecting protocols present an attractive alternative to often complex and variable NAb methods. TAb assays may detect pre-existing anti-AAV antibodies where potentially less sensitive NAb methods would not and therefore warn about prior exposure to the virus. TAb assays also offer significant advantages, e.g., ease of use, relatively straightforward to establish and validate, high cost efficiency, and improved assay turnaround time. More needs to be done to understand the connection between TAb assay results and possible impacts on safety and efficacy in the clinical studies.
Acknowledgements
We want to thank all members of the AAPS Immunogenicity of GTx team, including Michele Fiscella, Shan He, and many others for providing critical suggestions during development of this manuscript. We also want to thank AAPS for general support and providing a general platform that enabled these discussions.
Abbreviations
- GTx
Gene therapy
- AAV
Adeno-associated virus
- NHP
Non-human primate
- CDx
Companion diagnostic test
- FDA
US Food and Drug Administration
- BLA
Biological license application
- LDT
Laboratory-developed test
- ELISA
Enzyme-linked immunosorbent assay
- NAb
Neutralizing antibody
- TAb
Total antibody
- TI
Transduction inhibition
- DC
Dendritic cells
- MAC
Membrane attack complex
- HRP
Horseradish peroxidase
- ADA
Anti-drug antibody
- ECL
Electrochemiluminescence
- CV
Coefficient of variance
- MRD
Minimum required dilution
- LOD
Limit of detection
- EPO
Erythropoietin
- WHO
World Health Organization
Author Contribution
B.G, M.A, G.B, T.H., M.H., V.J., B.L., J.M., M.M., R.N., M.O., K.R., C.V., and B.W contributed equally in writing of the review
Declarations
Conflict of Interest
The authors are employed by and receive compensation from companies that are involved in development of gene therapy modality therapeutics and are listed on the title page of the manuscript. The authors have no other relevant affiliations or financial involvements with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Disclaimer
The views and conclusion presented in this paper are those of the authors and do not necessarily reflect the representative affiliation or company’s position on the subject.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.George LA, Ragni MV, Rasko JEJ, Raffini LJ, Samelson-Jones BJ, Ozelo M, Hazbon M, Runowski AR, Wellman JA, Wachtel K, Chen Y, Anguela XM, Kuranda K, Mingozzi F, High KA. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Molecular Therapy : the Journal of the American Society of Gene Therapy. 2020;28(9):2073–2082. doi: 10.1016/j.ymthe.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D'Avola D, López-Franco E, Sangro B, Pañeda A, Grossios N, Gil-Farina I, et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J Hepatol. 2016;65(4):776–783. doi: 10.1016/j.jhep.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Miesbach W, Meijer K, Coppens M, Kampmann P, Klamroth R, Schutgens R, Tangelder M, Castaman G, Schwäble J, Bonig H, Seifried E, Cattaneo F, Meyer C, Leebeek FWG. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long BR, Veron P, Kuranda K, Hardet R, Mitchell N, Hayes GM, Wong WY, Lau K, Li M, Hock MB, Zoog SJ, Vettermann C, Mingozzi F, Schweighardt B. Early phase clinical immunogenicity of valoctocogene roxaparvovec, an AAV5-mediated gene therapy for hemophilia A. Molecular Therapy : the Journal of the American Society of Gene Therapy. 2021;29(2):597–610. doi: 10.1016/j.ymthe.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long BR, Sandza K, Holcomb J, Crockett L, Hayes GM, Arens J, Fonck C, Tsuruda LS, Schweighardt B, O’Neill CA, Zoog S, Vettermann C. The impact of pre-existing immunity on the non-clinical pharmacodynamics of AAV5-based gene therapy. Mol Ther Methods Clin Dev. 2019;13:440–452. doi: 10.1016/j.omtm.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.AveXis Inc. ZOLGENSMA. Highlights of prescribing information. https://www.fda.gov/media/126109/download (2019). Accessed 24 March 2021.
- 7.Berry G, Murlidharan G, Asokan A. 30. Modulation of intracellular calcium enhances AAV transduction in the CNS. Mol Ther. 2016;24:S14. doi: 10.1016/S1525-0016(16)32839-8. [DOI] [Google Scholar]
- 8.Hirosue S, Senn K, Clément N, Nonnenmacher M, Gigout L, Linden RM, Weber T. Effect of inhibition of dynein function and microtubule-altering drugs on AAV2 transduction. Virology. 2007;367(1):10–18. doi: 10.1016/j.virol.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virella-Lowell I, Poirier A, Chesnut KA, Brantly M, Flotte TR. Inhibition of recombinant adeno-associated virus (rAAV) transduction by bronchial secretions from cystic fibrosis patients. Gene Ther. 2000;7(20):1783–1789. doi: 10.1038/sj.gt.3301268. [DOI] [PubMed] [Google Scholar]
- 10.Fitzpatrick Z, Leborgne C, Barbon E, Masat E, Ronzitti G, van Wittenberghe L, Vignaud A, Collaud F, Charles S, Simon Sola M, Jouen F, Boyer O, Mingozzi F. Influence of pre-existing anti-capsid neutralizing and binding antibodies on AAV vector transduction. Mol Ther Methods Clin Dev. 2018;9:119–129. doi: 10.1016/j.omtm.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harbison CE, Weichert WS, Gurda BL, Chiorini JA, Agbandje-McKenna M, Parrish CR. Examining the cross-reactivity and neutralization mechanisms of a panel of mAbs against adeno-associated virus serotypes 1 and 5. J Gen Virol. 2012;93(Pt 2):347–355. doi: 10.1099/vir.0.035113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wobus CE, Hügle-Dörr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74(19):9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majowicz A, Nijmeijer B, Lampen MH, Spronck L, de Haan M, Petry H, van Deventer SJ, Meyer C, Tangelder M, Ferreira V. Therapeutic hFIX activity achieved after single AAV5-hFIX treatment in hemophilia B patients and NHPs with pre-existing anti-AAV5 NABs. Mol Ther Methods Clin Dev. 2019;14:27–36. doi: 10.1016/j.omtm.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falese L, Sandza K, Yates B, Triffault S, Gangar S, Long B, Tsuruda L, Carter B, Vettermann C, Zoog SJ, Fong S. Strategy to detect pre-existing immunity to AAV gene therapy. Gene Ther. 2017;24(12):768–778. doi: 10.1038/gt.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaiss AK, Cotter MJ, White LR, Clark SA, Wong NCW, Holers VM, Bartlett JS, Muruve DA. Complement is an essential component of the immune response to adeno-associated virus vectors. J Virol. 2008;82(6):2727–2740. doi: 10.1128/JVI.01990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng YS, Agbandje-McKenna M. Mapping the AAV capsid host antibody response toward the development of second generation gene delivery vectors. Front Immunol. 2014;5:9. doi: 10.3389/fimmu.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers GL, Martino AT, Aslanidi GV, Jayandharan GR, Srivastava A, Herzog RW. Innate immune responses to AAV vectors. Front Microbiol. 2011;2:194. doi: 10.3389/fmicb.2011.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meadows AS, Pineda RJ, Goodchild L, Bobo TA, Fu H. Threshold for pre-existing antibody levels limiting transduction efficiency of systemic rAAV9 gene delivery: relevance for translation. Mol Ther Methods Clin Dev. 2019;13:453–462. doi: 10.1016/j.omtm.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.FDA: Immunogenicity testing of therapeutic protein products —developing and validating assays for anti-drug antibody detection. Guidance for Industry. https://www.fda.gov/media/119788/download (2019). Accessed July 2019.
- 21.Rumachik NG, Malaker SA, Poweleit N, Maynard LH, Adams CM, Leib RD, Cirolia G, Thomas D, Stamnes S, Holt K, Sinn P, May AP, Paulk NK. Methods matter: standard production platforms for recombinant aav produce chemically and functionally distinct vectors. Mol Ther Methods Clin Dev. 2020;18:98–118. doi: 10.1016/j.omtm.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertin B, Veron P, Leborgne C, Deschamps JY, Moullec S, Fromes Y, Collaud F, Boutin S, Latournerie V, van Wittenberghe L, Delache B, le Grand R, Dereuddre-Bosquet N, Benveniste O, Moullier P, Masurier C, Merten O, Mingozzi F. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci Rep. 2020;10(1):864. doi: 10.1038/s41598-020-57893-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King LE, Farley E, Imazato M, Keefe J, Khan M, Ma M, Pihl KS, Sriraman P. Ligand binding assay critical reagents and their stability: recommendations and best practices from the Global Bioanalysis Consortium Harmonization Team. AAPS J. 2014;16(3):504–515. doi: 10.1208/s12248-014-9583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira V, Twisk J, Kwikkers K, Aronica E, Brisson D, Methot J, Petry H, Gaudet D. Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPL(S447X)) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum Gene Ther. 2014;25(3):180–188. doi: 10.1089/hum.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salas D, Kwikkers KL, Zabaleta N, Bazo A, Petry H, van Deventer SJ, Aseguinolaza GG, Ferreira V. Immunoadsorption enables successful rAAV5-mediated repeated hepatic gene delivery in nonhuman primates. Blood Adv. 2019;3(17):2632–2641. doi: 10.1182/bloodadvances.2019000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corti M, Liberati C, Smith BK, Lawson LA, Tuna IS, Conlon TJ, Coleman KE, Islam S, Herzog RW, Fuller DD, Collins SW, Byrne BJ. Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by Pompe disease. Hum Gene Ther Clin Dev. 2017;28(4):208–218. doi: 10.1089/humc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zygmunt DA, Crowe KE, Flanigan KM, Martin PT. Comparison of serum rAAV serotype-specific antibodies in patients with Duchenne muscular dystrophy, Becker muscular dystrophy, inclusion body myositis, or GNE myopathy. Hum Gene Ther. 2017;28(9):737–746. doi: 10.1089/hum.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavita U, Dai Y, Salvador L, Miller W, Adam LP, Levesque PC, Zhang YJ, Ji QC, Pillutla RC. Development of a chemiluminescent ELISA method for the detection of total anti-adeno associated virus serotype 9 (AAV9) antibodies. Hum Gene Ther Methods. 2018;29(6):237–250. doi: 10.1089/hgtb.2018.131. [DOI] [PubMed] [Google Scholar]
- 29.Leborgne C, Latournerie V, Boutin S, Desgue D, Quéré A, Pignot E, Collaud F, Charles S, Simon Sola M, Masat E, Jouen F, Boyer O, Masurier C, Mingozzi F, Veron P. Prevalence and long-term monitoring of humoral immunity against adeno-associated virus in Duchenne muscular dystrophy patients. Cell Immunol. 2019;342:103780. doi: 10.1016/j.cellimm.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Chirmule N, Propert K, Magosin S, Qian Y, Qian R, Wilson J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999;6(9):1574–1583. doi: 10.1038/sj.gt.3300994. [DOI] [PubMed] [Google Scholar]
- 31.Murphy SL, Li H, Mingozzi F, Sabatino DE, Hui DJ, Edmonson SA, High KA. Diverse IgG subclass responses to adeno-associated virus infection and vector administration. J Med Virol. 2009;81(1):65–74. doi: 10.1002/jmv.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calcedo R, Chichester JA, Wilson JM. Assessment of humoral, innate, and T-cell immune responses to adeno-associated virus vectors. Human Gene Ther Methods. 2018;29(2):86–95. doi: 10.1089/hgtb.2018.038. [DOI] [PubMed] [Google Scholar]
- 33.Sanchez S, Barger T, Zhou L, Hale M, Mytych D, Gupta S, Swanson SJ, Civoli F. Strategy to confirm the presence of anti-erythropoietin neutralizing antibodies in human serum. J Pharm Biomed Anal. 2011;55(5):1265–1274. doi: 10.1016/j.jpba.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 34.Lugin ML, Lee RT, Kwon YJ. Synthetically engineered adeno-associated virus for efficient, safe, and versatile gene therapy applications. ACS Nano. 2020;14(11):14262–14283. doi: 10.1021/acsnano.0c03850. [DOI] [PubMed] [Google Scholar]
- 35.Tseng YS, Vliet KV, Rao L, McKenna R, Byrne BJ, Asokan A, Agbandje-McKenna M. Generation and characterization of anti-adeno-associated virus serotype 8 (AAV8) and anti-AAV9 monoclonal antibodies. J Virol Methods. 2016;236:105–110. doi: 10.1016/j.jviromet.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199(3):381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 38.Kruzik A, Fetahagic D, Hartlieb B, Dorn S, Koppensteiner H, Horling FM, Scheiflinger F, Reipert BM, de la Rosa M. Prevalence of anti-adeno-associated virus immune responses in international cohorts of healthy donors. Mol Ther Methods Clin Dev. 2019;14:126–133. doi: 10.1016/j.omtm.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azadeh M, Sondag P, Wang Y, Raines M, Sailstad J. Quality controls in ligand binding assays: recommendations and best practices for preparation, qualification, maintenance of lot to lot consistency, and prevention of assay drift. AAPS J. 2019;21(5):89. doi: 10.1208/s12248-019-0354-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, Fiscella M, Gorovits B, Kirschner S, Moxness M, Parish T, Quarmby V, Smith H, Smith W, Zuckerman LA, Koren E. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–1281. doi: 10.1016/j.jpba.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Devanarayan V, Smith WC, Brunelle RL, Seger ME, Krug K, Bowsher RR. Recommendations for systematic statistical computation of immunogenicity cut points. AAPS J. 2017;19(5):1487–1498. doi: 10.1208/s12248-017-0107-3. [DOI] [PubMed] [Google Scholar]
- 42.Kumar SC, DelCarpini JA, Qu Q, Kane M, Gorovits B. Mitigation of pre-existing antibodies to a biotherapeutic in non-clinical species when establishing anti-drug antibody assay cutpoint. AAPS J. 2017;19(1):313–319. doi: 10.1208/s12248-016-0011-2. [DOI] [PubMed] [Google Scholar]
- 43.Samelson-Jones BJ, Finn JD, Favaro P, Wright JF, Arruda VR. Timing of intensive immunosuppression impacts risk of transgene antibodies after AAV gene therapy in nonhuman primates. Mol Ther Methods Clin Dev. 2020;17:1129–1138. doi: 10.1016/j.omtm.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee S, Kang IK, Kim JH, Jung BK, Park K, Chang H, et al. Relationship between neutralizing antibodies against adeno-associated virus in the vitreous and serum: effects on retinal gene therapy. Transl Vis Sci Technol. 2019;8(2):14. doi: 10.1167/tvst.8.2.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anna Majowicz AV, Floris van Waes, Bozena Bohuslavova, Zdenka Ellederova, Jan Motlik, Joseph Higgins, Sander van Deventer, Pavlina Konstantinova, Valerie Ferreira: Pre-existing anti-adeno-associated virus (AAV) serotype 5 neutralizing antibodies (NABs) titers in minipig serum do not reflect levels of antiAAV5 NABs titers in their cerebrospinal fluid (CSF). http://uniqure.com/2019%20CHDI%20-%20Pre-existing%20AAV5%20Neutralizing%20Antibodies%20Titers%20in%20Minipig%20Serum%20Do%20Not%20Reflect%20levels%20of%20ANTI-AAV5%20NABs%20Titers%20in%20Their%20CSF.pdf (2019). Accessed 6 April 2021.
- 46.Song S, Yang L, Trepicchio WL, Wyant T. Understanding the supersensitive anti-drug antibody assay: unexpected high anti-drug antibody incidence and its clinical relevance. J Immunol Res. 2016;2016:3072586–3072588. doi: 10.1155/2016/3072586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li C, Narkbunnam N, Samulski RJ, Asokan A, Hu G, Jacobson LJ, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012;19(3):288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- 48.Tobiasch E, Burguete T, Klein-Bauernschmitt P, Heilbronn R, Schlehofer JR. Discrimination between different types of human adeno-associated viruses in clinical samples by PCR. J Virol Methods. 1998;71(1):17–25. doi: 10.1016/s0166-0934(97)00198-5. [DOI] [PubMed] [Google Scholar]
- 49.Erles K, Sebökovà P, Schlehofer JR. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV) J Med Virol. 1999;59(3):406–411. doi: 10.1002/(sici)1096-9071(199911)59:3<406::aid-jmv22>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 50.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5(3):285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 51.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(18):11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peden CS, Burger C, Muzyczka N, Mandel RJ. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J Virol. 2004;78(12):6344–6359. doi: 10.1128/jvi.78.12.6344-6359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grimm D, Kay MA. From virus evolution to vector revolution: use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr Gene Ther. 2003;3(4):281–304. doi: 10.2174/1566523034578285. [DOI] [PubMed] [Google Scholar]
- 54.Rivière C, Danos O, Douar AM. Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther. 2006;13(17):1300–1308. doi: 10.1038/sj.gt.3302766. [DOI] [PubMed] [Google Scholar]
- 55.Zaiss AK, Muruve DA. Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 2008;15(11):808–816. doi: 10.1038/gt.2008.54. [DOI] [PubMed] [Google Scholar]
- 56.Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Pogoda JM, Provost R, Guerrero J, Hajjar RJ, Zsebo KM. Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther. 2016;23(3):313–319. doi: 10.1038/gt.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Day JW, Finkel RS, Mercuri E, Swoboda KJ, Menier M, van Olden R, Tauscher-Wisniewski S, Mendell JR. Adeno-associated virus serotype 9 antibodies in patients screened for treatment with onasemnogene abeparvovec. Mol TherMethods Clin Dev. 2021;21:76–82. doi: 10.1016/j.omtm.2021.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mercado NB, Zahn R, Wegmann F, Loos C, Chandrashekar A, Yu J, Liu J, Peter L, McMahan K, Tostanoski LH, He X, Martinez DR, Rutten L, Bos R, van Manen D, Vellinga J, Custers J, Langedijk JP, Kwaks T, Bakkers MJG, Zuijdgeest D, Rosendahl Huber SK, Atyeo C, Fischinger S, Burke JS, Feldman J, Hauser BM, Caradonna TM, Bondzie EA, Dagotto G, Gebre MS, Hoffman E, Jacob-Dolan C, Kirilova M, Li Z, Lin Z, Mahrokhian SH, Maxfield LF, Nampanya F, Nityanandam R, Nkolola JP, Patel S, Ventura JD, Verrington K, Wan H, Pessaint L, van Ry A, Blade K, Strasbaugh A, Cabus M, Brown R, Cook A, Zouantchangadou S, Teow E, Andersen H, Lewis MG, Cai Y, Chen B, Schmidt AG, Reeves RK, Baric RS, Lauffenburger DA, Alter G, Stoffels P, Mammen M, van Hoof J, Schuitemaker H, Barouch DH. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature. 2020;586(7830):583–588. doi: 10.1038/s41586-020-2607-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stephenson KE, Le Gars M, Sadoff J, de Groot AM, Heerwegh D, Truyers C, et al. Immunogenicity of the Ad26.COV2.S vaccine for COVID-19. JAMA : the Journal of the American Medical Association. 2021. 10.1001/jama.2021.3645. [DOI] [PMC free article] [PubMed]
- 60.Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, Barrett YC, Devanarayan V, Gorovits B, Gupta S, Parish T, Quarmby V, Moxness M, Swanson SJ, Taniguchi G, Zuckerman LA, Stebbins CC, Mire-Sluis A. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333(1-2):1–9. doi: 10.1016/j.jim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 61.Gorovits B. Current considerations for immunoglobulin isotype characterization of antibody response against biotherapeutics. AAPS J. 2020;22(6):144. doi: 10.1208/s12248-020-00530-4. [DOI] [PubMed] [Google Scholar]
- 62.Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39(4):469–477. doi: 10.1111/j.1365-2222.2009.03207.x. [DOI] [PubMed] [Google Scholar]
- 63.van der Neut KM, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317(5844):1554–1557. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 64.Vultaggio A, Matucci A, Nencini F, Pratesi S, Parronchi P, Rossi O, Romagnani S, Maggi E. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy. 2010;65(5):657–661. doi: 10.1111/j.1398-9995.2009.02280.x. [DOI] [PubMed] [Google Scholar]
- 65.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bindon CI, Hale G, Brüggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. 1988;168(1):127–142. doi: 10.1084/jem.168.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343(1):227–235. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang JK, Alt FW, Yeap L-S. Related mechanisms of antibody somatic hypermutation and class switch recombination. Microbiol Spectr. 2015;3(1):MDNA3-2014. 10.1128/microbiolspec.MDNA3-0037-2014. [DOI] [PMC free article] [PubMed]
- 69.Partridge MA, Purushothama S, Elango C, Lu Y. Emerging technologies and generic assays for the detection of anti-drug antibodies. J Immunol Res. 2016;2016:6262383–6262386. doi: 10.1155/2016/6262383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen LZ, Roos D, Philip E. Development of immunocapture-LC/MS assay for simultaneous ADA isotyping and semiquantitation. J Immunol Res. 2016;2016:7682472–7682414. doi: 10.1155/2016/7682472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daniel HDJ, Kumar S, Kannangai R, Lakshmi KM, Agbandje-McKenna M, Coleman KE, et al. Prevalence of AAV3 capsid binding and neutralizing antibodies in healthy and individuals with hemophilia B from India. Hum Gene Ther. 2020;32:451–457. doi: 10.1089/hum.2020.258. [DOI] [PubMed] [Google Scholar]
- 72.Hubbard A.R. JS: Calibration of the proposed WHO 1st international standard for blood coagulation factor V in human plasma. https://www.who.int/bloodproducts/publications/05-2007%20BCF%20V%20in%20plasma.pdf (2005). Accessed 24 Mar 2021.
- 73.Mytych DT, Barger TE, King C, Grauer S, Haldankar R, Hsu E, Wu MM, Shiwalkar M, Sanchez S, Kuck A, Civoli F, Sun J, Swanson SJ. Development and characterization of a human antibody reference panel against erythropoietin suitable for the standardization of ESA immunogenicity testing. J Immunol Methods. 2012;382(1-2):129–141. doi: 10.1016/j.jim.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 74.Leborgne C, Barbon E, Alexander JM, Hanby H, Delignat S, Cohen DM, Collaud F, Muraleetharan S, Lupo D, Silverberg J, Huang K, van Wittengerghe L, Marolleau B, Miranda A, Fabiano A, Daventure V, Beck H, Anguela XM, Ronzitti G, Armour SM, Lacroix-Desmazes S, Mingozzi F. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med. 2020;26(7):1096–1101. doi: 10.1038/s41591-020-0911-7. [DOI] [PubMed] [Google Scholar]
- 75.Spark T: LUXTURNA Package Insert. https://www.fda.gov/media/109906/download (2017). Accessed 2019.
- 76.CMS: Centers for Medicare & Medicaid Services. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA (2021). Accessed 29 June 2021.
- 77.U.S. Food and Drug Administration. Human gene therapy for rare diseases. https://www.fda.gov/media/113807/download (2020). Accessed 3 April 2021.
- 78.Administration USFaD: U.S. Food and Drug Administration. Human gene therapy for hemophilia. https://www.fda.gov/media/113799/download (2020). Accessed 3 April 2021.
- 79.Gorovits B, Fiscella M, Havert M, Koren E, Long B, Milton M, Purushothama S. Recommendations for the development of cell-based anti-viral vector neutralizing antibody assays. AAPS J. 2020;22(2):24. doi: 10.1208/s12248-019-0403-1. [DOI] [PubMed] [Google Scholar]