

ABSTRACT
Malaria-associated acute respiratory distress syndrome (MA-ARDS) is a severe complication of malaria that occurs despite effective antimalarial treatment. Currently, noninvasive imaging procedures such as chest X-rays are used to assess edema in established MA-ARDS, but earlier detection methods are needed to reduce morbidity and mortality. The early stages of MA-ARDS are characterized by the infiltration of leukocytes, in particular monocytes/macrophages; thus, monitoring of immune infiltrates may provide a useful indicator of early pathology. In this study, Plasmodium berghei ANKA-infected C57BL/6 mice, a rodent model of MA-ARDS, were longitudinally imaged using the 18-kDa translocator protein (TSPO) imaging agent [18F]FEPPA as a marker of macrophage accumulation during the development of pathology and in response to combined artesunate and chloroquine diphosphate (ART+CQ) therapy. [18F]FEPPA uptake was compared to blood parasitemia levels and to levels of pulmonary immune cell infiltrates by using flow cytometry. Infected animals showed rapid increases in lung retention of [18F]FEPPA, correlating well with increases in blood parasitemia and pulmonary accumulation of interstitial inflammatory macrophages and major histocompatibility complex class II (MHC-II)-positive alveolar macrophages. Treatment with ART+CQ abrogated this increase in parasitemia and significantly reduced both lung uptake of [18F]FEPPA and levels of macrophage infiltrates. We conclude that retention of [18F]FEPPA in the lungs is well correlated with changes in blood parasitemia and levels of lung-associated macrophages during disease progression and in response to ART+CQ therapy. With further development, TSPO biomarkers may have the potential to accurately assess the early onset of MA-ARDS.
KEYWORDS: malaria, PET, lung, PbA, macrophages
INTRODUCTION
Malaria is a life-threatening disease that affects many tropical and subtropical areas of the world (1). Severe malaria, caused mostly by Plasmodium falciparum, is triggered by hyperinflammation, characterized by the activation of inflammatory cells and the overproduction of type 1 proinflammatory mediators (2), affecting both the central nervous system and the lungs (3). Malaria-associated acute respiratory distress syndrome (MA-ARDS) affects many adults infected with Plasmodium falciparum and can be life-threatening (4). MA-ARDS is characterized by alveolar inflammation, alveolar capillary membrane damage, and pulmonary edema and can develop either at initial presentation or after the initiation of treatment, when the parasitemia is falling and the patient is improving. Patients presenting with acute-onset dyspnea can rapidly progress to respiratory failure. Imaging procedures such as chest X-ray, computed tomography (CT), and magnetic resonance (MR) are currently gold-standard techniques in MA-ARDS identification and are fundamental for the diagnosis and analysis of lung function during MA-ARDS development (4). While these procedures provide useful anatomical information on lung structure and associated edema, they do not provide information about disease activity, such as the accumulation and activation of inflammatory cells. Postmortem lung histology shows that MA-ARDS is associated with the accumulation of leukocytes (monocytes, neutrophils, and macrophages) and with pulmonary edema (4–6). Monitoring of these immune infiltrates in the lung may aid in the earlier identification and treatment of MA-ARDS. However, assessment of this initial immune cell infiltrate is complex, and lung biopsy is invasive and does not represent the total pulmonary inflammatory burden. Thus, there is a pressing need to develop noninvasive ways to measure lung immune infiltrates. Positron emission tomography (PET) imaging with [18F]fluorodeoxyglucose ([18F]FDG) has been used clinically to assess lung inflammation in pneumonia and bronchiectasis but is a marker of glucose consumption, providing information about cell and tissue metabolism, not an effective marker for leukocyte accumulation or activity (7–12). PET radioligands have been developed which target the 18-kDa translocator protein (TSPO), a five-transmembrane-domain protein located on the outer membranes of mitochondria and overexpressed in activated macrophages. TSPO expression has been used to assess the activation of macrophages in numerous brain and lung inflammatory pathologies (13–17), and macrophages have been shown to be essential in the host response against malarial infection (18). Severe malaria (SM) models have been developed using different parasite–mouse strain combinations to decipher the pathogenic mechanisms underlying cerebral malaria (CM) and MA-ARDS. The most widely used model is Plasmodium berghei ANKA infection in C57BL/6 mice, which exhibits neuropathology and lung pathology with many similarities to human CM and MA-ARDS pathologies (19).
In the current study, we have utilized the TSPO imaging agent [18F]FEPPA{N-(2-(2-[18F]fluoroethoxy)benzyl)-N-(4-phenoxypyridin-3-yl)acetamide} to investigate the development of MA-ARDS pathology in C57BL/6 mice infected with P. berghei ANKA and their response to combined artesunate and chloroquine diphosphate (ART+CQ) therapy. Our results were paired with comparative analysis of lung-infiltrating immune cell subsets by use of flow cytometry.
RESULTS
Assessment of acute lung pathology and antimalarial treatment efficacy by using [18F]FEPPA PET imaging.
Using an established rodent model of malaria infection, where >80% of P. berghei ANKA-infected C57BL/6 mice developed a an immune-mediated lethal disease targeting principally the brain and the lung during the first week of infection, we aimed to investigate the ability of [18F]FEPPA PET imaging, a noninvasive imaging technique, to accurately monitor the in vivo development of malaria-associated lung pathology and the effectiveness of the antimalarial treatment response. Peripheral parasitemia in the P. berghei ANKA-infected group increased over time, reaching a maximum at 7 days postinfection (7 dpi), whereas it dropped significantly in the antimalarial-treated group. On a side note, 10 days after the cessation of antimalarial treatment, two mice (of the 10 tested over two experiments) showed signs of recrudescent parasites in the blood (Fig. 1B). [18F]FEPPA PET imaging was done on the P. berghei ANKA-infected group at 5, 6, and 7 dpi, and the group treated with combined artesunate and chloroquine drugs was imaged 48 h after the antimalarial treatment (day 7 post–P. berghei ANKA infection) and 3 and 10 days after the cessation of treatment (days 14 and 21 post–P. berghei ANKA infection, respectively).
FIG 1.
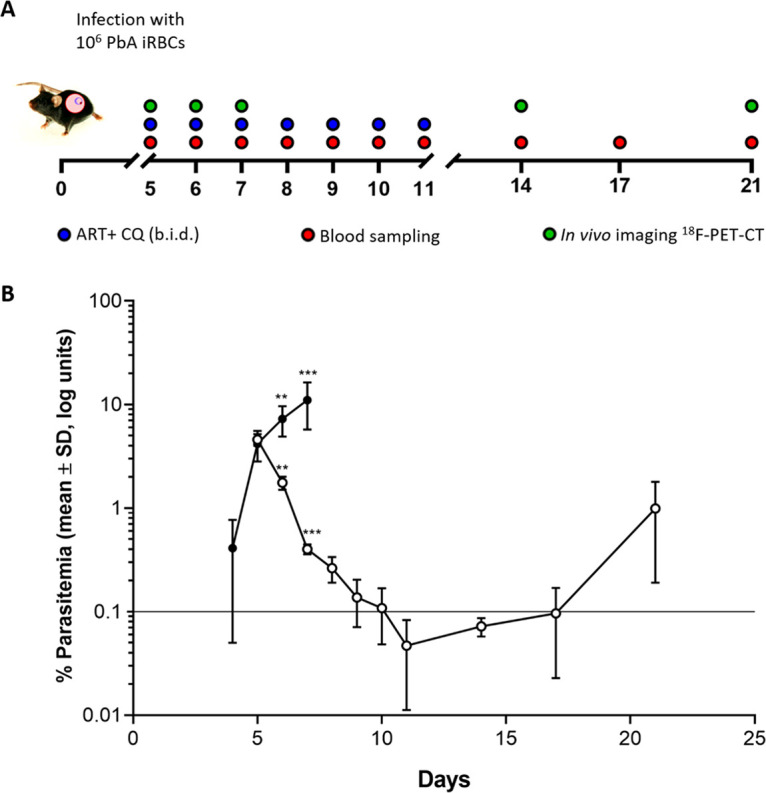
(A) Timeline showing drug-dosing regimen. C57BL/6 mice were infected with 106 P. berghei ANKA organisms on day 0. Animals were treated with combined artesunate (30 mg/kg) and chloroquine diphosphate (120 mg/kg) therapy or saline vehicle on days 5 to 11 inclusive (i.p., twice a day [b.i.d.]). Blood sampling was performed daily from days 5 to 11 and subsequently on days 14, 17, and 21 for parasite quantification. In vivo imaging was performed at 5, 6, and 7 dpi for the untreated group (n = 6) and at 7, 14, and 21 dpi for the treated group (n = 6). (B) Parasitemia levels in P. berghei ANKA-infected mice that were left untreated (n = 10) (filled circles) or treated with ART+CQ (n = 10) (open circles). Asterisks indicate significant differences (**, P < 0.01; ***, P < 0.001; ****, P < 0.001) between the log-transformed parasitemia levels in the two groups as determined by an unpaired Student t test. Data are shown as the mean log percentages of parasitemia ± SD.
In vivo [18F]FEPPA PET imaging showed a great contrast between the lungs of untreated P. berghei ANKA-infected C57BL/6 mice and those of mice in the antimalarial-treated group (Fig. 2). Lung uptake of [18F]FEPPA increased over the course of P. berghei ANKA infection (9.3% ± 1.6% of the injected dose [ID]/g at day 5; 16.2% ± 2.8% of ID/g at day 6), reaching a maximum at 7 dpi (29.0% ± 9.03% of ID/g) (Fig. 3A). Likewise, lung uptake of [18F]FEPPA was significantly decreased after 2 days of treatment with ART+CQ (day 7) (19.2% ± 4.4% of ID/g; P < 0.01). This reduction in uptake was further evident 3 days after the cessation of the full course of treatment (day 14) (9.1% ± 2.2% of ID/g; P < 0.001), when uptake decreased to levels similar to those in naive lungs (9.9% ± 0.9% of ID/g), levels that were maintained up to the end of the study on day 21 postinfection (10 days after the cessation of treatment) for the majority of animals (9.3% ± 4.1% of ID/g; P < 0.001) (Fig. 3B). Two animals showed increased lung uptake on day 21 but not enough to significantly change the group assessment. Previous studies have demonstrated that lowered parasitemia is associated with protection from vascular leakage (20). Here, we show that lung uptake of [18F]FEPPA correlates well with blood parasitemia across both arms of the study (Pearson’s r2 = 0.908) (Fig. 3C).
FIG 2.

Representative standardized PET-CT maximum-intensity projection images for the assessment of lung uptake of [18F]FEPPA in mice on days 5, 6, and 7 after P. berghei ANKA infection and after 2 days of combined ART+CQ treatment. Yellow arrows indicate lung uptake, and white arrows indicate kidney clearance.
FIG 3.
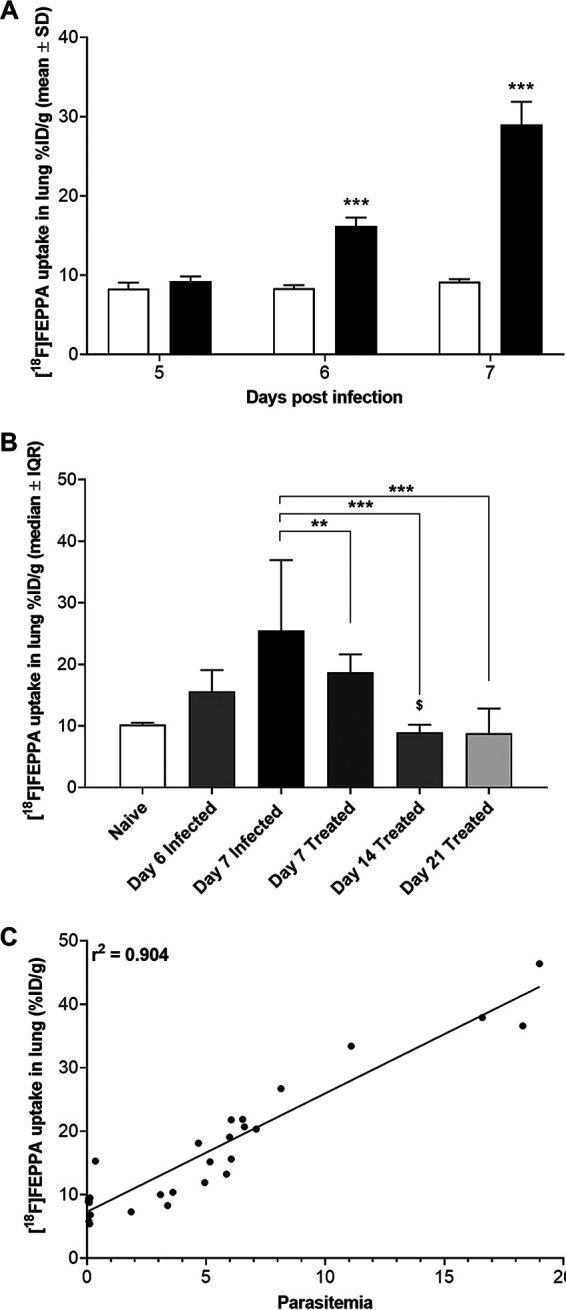
In vivo assessment of lung uptake of [18F]FEPPA from PET-CT defined volumes of interest (VOI) from groups of P. berghei ANKA-infected C57BL/6 mice (n = 6). (A) Lung uptake of [18F]FEPPA increased at 6 dpi (filled bar), reaching the maximum at 7 dpi. Asterisks indicate significant differences (***, P < 0.001) from background results for naive mice (open bar) by an unpaired Student t test. Data are shown as mean percentages of the ID per gram ± SD. (B) Treatment with combined ART+CQ led to significant decreases in [18F]FEPPA uptake in the lung 2 days after the initiation of treatment (day 7) (dark shaded bar) compared to uptake in untreated P. berghei ANKA-infected animals (filled bar). [18F]FEPPA uptake levels similar to background levels in naive mice (open bar) were observed 3 days (day 14) (medium shaded bar) and 10 days (day 21) (light shaded bar) after the cessation of treatment. Symbols indicate significant differences by one-way ANOVA (Kruskal-Wallis test with a Bonferroni posttest) in multiple comparisons against data for untreated (*, P < 0.05; **, P < 0.01; ***, P < 0.001) and treated ($, P < 0.05) P. berghei ANKA-infected animals on day 7. Data are shown as median percentages of ID per gram ± interquartile range (IQR). (C) Correlation of lung uptake of [18F]FEPPA with parasitemia over the time course studied (Pearson’s r2 = 0.904).
[18F]FEPPA uptake is associated with increased lung infiltration of monocytes/macrophages.
Analysis of postmortem lung histological sections revealed the accumulation of monocytes, macrophages, and other cell types sequestered in the lungs in malaria-infected patients or animals who presented respiratory distress (21–25).
Flow cytometry analysis of myeloid cells present in the lungs of infected untreated and treated mice showed significant accumulation of interstitial inflammatory macrophages (Fig. 4A) and major histocompatibility complex class II (MHC-II)-positive alveolar macrophages (Fig. 4B) at 6 and 7 dpi, but not monocyte-derived macrophages and MHC-II-negative alveolar macrophages, compared to levels in naive mice. Treatment with combined ART+CQ successfully abrogated the accumulation of both cell types within 2 days of the initiation of treatment, and this decrease was maintained up to 10 days after the cessation of treatment (Fig. 4A and B).
FIG 4.
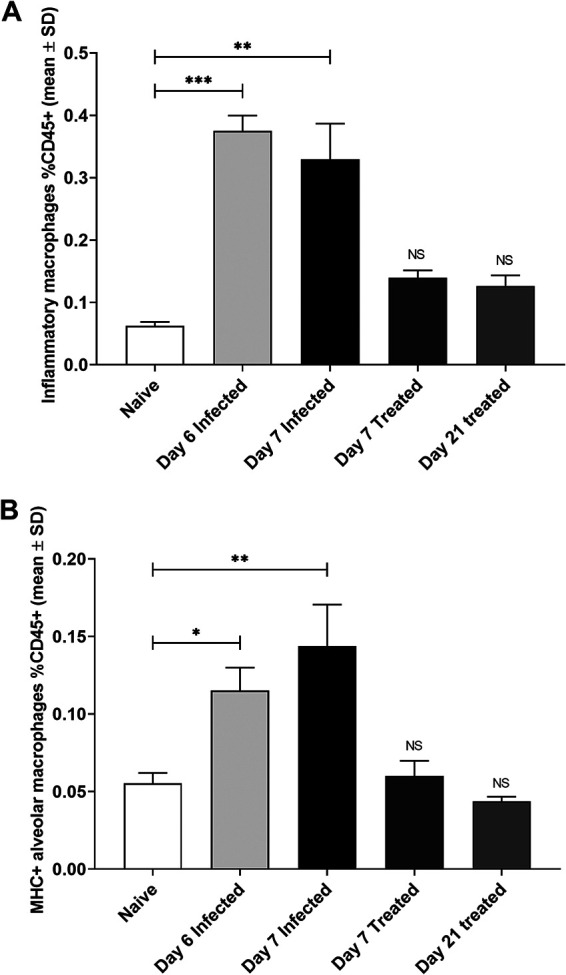
Assessment of myeloid cells in the lungs of infected mice by flow cytometry. (A) Levels of inflammatory macrophages increased significantly after P. berghei ANKA infection at 6 dpi (light shaded bar) and 7 dpi (filled bar) over background levels in naive animals (open bar). Treatment with combined ART+CQ led to significant decreases in levels of inflammatory macrophages 2 days after the initiation of treatment (day 7) (dark shaded bar) and 10 days after the cessation of treatment (day 21) (medium shaded bar) (n = 5). Asterisks indicate significant differences (**, P < 0.01; ***, P < 0.001) by one-way ANOVA. Data are shown as mean percentages of CD45+ cells ± SD. NS, no significant differences from results for naive animals. (B) Levels of MHC-positive alveolar macrophages increased significantly at 6 dpi (light shaded bar) and 7 dpi (filled bar) over background levels in naive animals (open bar).Treatment with combined ART+CQ led to significant decreases in MHC-positive alveolar macrophages 2 days after the initiation of treatment (day 7) (dark shaded bar) and 10 days after the cessation of treatment (day 21) (medium shaded bar) (n = 6). Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by one-way ANOVA. Data are shown as mean percentages of CD45+ cells ± SD. NS, no significant differences from results for naive animals.
DISCUSSION
The pathology of MA-ARDS is still not well understood, in contrast to the neurological symptoms in CM; it is difficult to assess lung pathology, and the initial symptoms of MA-ARDS can be easily missed. Typically, radiography is used to assess the development of pulmonary edema, but this does not differentiate the onset of MA-ARDS from other causes of pulmonary edema, such as cardiac failure or pneumonia. The initial phase of MA-ARDS is known as the exudative phase and is caused by the sequestration of parasitized red blood cells (pRBCs) in lung microvessels. These pRBCs bind to CD36 receptors expressed on lung endothelial cells, initiating and amplifying local immune responses (26). Many immune cells, including CD8+ T cells, monocytes, and macrophages, have been shown to be important in the early pathology of MA-ARDS (20, 22, 27, 28). Noninvasive imaging of these immune infiltrates may provide a way to detect this initial exudative phase, which could be used to complement the current clinical diagnostic measures of MA-ARDS (chest X-ray, CT, or MR), allowing for the earlier provision of interventional therapy. Recently, there has been an explosion in research aimed at developing noninvasive imaging techniques to assess infiltrating immune cells, focusing on infiltrates associated with immune responses in cancer (29–32). The current study has focused on the involvement of macrophages, since they have been shown to be critical early in the pathology of MA-ARDS, recognizing pathogen-associated molecular patterns and triggering innate immunity and host defenses. Macrophages have also been reported to participate in the entire pathogenesis of MA-ARDS and subsequent response to treatment, including the modulation of inflammatory responses and the repair of damaged lung tissues (33). The P. berghei ANKA mouse model is also a model for experimental CM (ECM), although MA-ARDS and ECM have different pathologies (20). As observed in MA-ARDS, monocytes/macrophages also accumulate in the brain; however, they have no role in ECM pathology, since mice deficient in monocytes/macrophages or depleted of these cells by drug treatment still develop ECM (34). For both MA-ARDS and ECM, the end phase of the pathogenic process is the rupture of the capillary barriers, and in both pathologies, this is mediated by pathogenic parasite-specific CD8 T cells (20, 35, 36). However, further experiments are needed to define the pathogenic pathways for the two pathologies, potentially utilizing murine models with a longer duration of pathology.
In this MA-ARDS model, longitudinal imaging with [18F]FEPPA correlated well with parasitemia, demonstrating a peak uptake in the lungs at 7 dpi, when pulmonary vascular leakage is known to occur; however, significant increases in lung uptake were also observed at 6 dpi, when pulmonary vascular leakage is still low (20). Interstitial inflammatory macrophages and lung-resident alveolar macrophages expressing MHC-II followed a similar trajectory, with significant increases observed early on day 6 postinfection and peaking at 7 dpi. Interstitial inflammatory macrophages are phagocytic and are associated with an earlier response, while alveolar macrophages are known as a second line of defense against pathogens. In response to an infection, alveolar macrophages go through a remodeling process, altering their surface marker phenotype, with surface expression of MHC-II found to be increased only upon infection (37). These data suggest that pulmonary retention of [18F]FEPPA is associated with a macrophage-driven inflammatory response and that significant increases in lung retention may be observable prior to vascular leakage. Macrophage imaging may also aid in the assessment of therapy efficacy. Numerous treatments have been assessed for CM, consisting mainly of antimalarial drugs that reduce the parasite load or corticosteroid treatment (38). In the current study, ART+CQ treatment rapidly reduced parasitemia to background levels, which were sustained in the majority of animals up to 10 days after the cessation of treatment. Likewise, ART+CQ treatment reduced levels of lung-associated interstitial inflammatory macrophages and lung-resident alveolar macrophages expressing MHC-II. However, while parasitemia decreased substantially within 2 days of treatment, [18F]FEPPA uptake in the lungs reached background levels only after the full course of treatment. Importantly, lung uptake was very sensitive to changes in parasitemia levels; animals that displayed recrudescent parasitemia on day 21 also showed enhanced [18F]FEPPA uptake, suggesting that lung expression of TSPO could potentially act as an early marker of recrudescence. While P. berghei ANKA infection in C57BL/6 mice exhibits many similarities to human CM and MA-ARDS (19), care should be taken in translating these findings, because the clinical course of malaria is complex, and animal models do not fully recapitulate the extent of clinical pathology.
Conclusions.
The current study is the first to assess the ability of TSPO radiotracers to track malaria pathology and the response to treatment in a murine model of MA-ARDS. The imaging data show that [18F]FEPPA is well correlated with changes in parasitemia levels and provides useful information on the pathology of malaria-infected lungs during disease progression and in response to therapy. The data presented suggest that further development of TSPO biomarkers may have potential for accurate assessment of the early onset of MA-ARDS.
MATERIALS AND METHODS
Animal procedures.
Animal procedures were carried out in accordance with the Institutional Animal Care and Use Committee Singapore (IACUC no. 181314). Male C57BL/6 mice aged 5 to 7 weeks were purchased from In Vivos Singapore, were housed under the same specific-pathogen-free conditions on a 12-h light-dark cycle, and had free access to food and water.
Parasite and infection parameters.
The transgenic Plasmodium berghei ANKA (231cl1) line expressing luciferase and green fluorescent protein (GFP) under the control of the ef1-α promoter was provided by Christian Engwerda (QIMR, Brisbane, Australia) (39). Animals were infected as described previously (20). Briefly, all mice were infected intraperitoneally (i.p.) with 106 parasitized red blood cells (pRBCs; prepared in 100 μl Alsever’s buffer). Parasitemia was monitored by flow cytometry (LSR II; Becton, Dickinson) daily from 5 to 11 days postinfection (dpi) and subsequently at 14, 17, and 21 dpi using an anti-CD45 monoclonal antibody (MAb) coupled to allophycocyanin (APC) (clone 30F11.1; Miltenyi), 5 μg/ml Hoechst 33342 (Sigma), and 8 μM dihydroethidium (Sigma) dyes (40).
Drug treatment.
Infected mice were treated with 30 mg artesunate (Guilin No. 2 Pharmaceutical Factory, China)/kg of body weight and 120 mg chloroquine diphosphate (Sigma-Aldrich)/kg of body weight at 5 dpi for 7 days. Artesunate (dissolved in 0.9% NaCl) and chloroquine (dissolved in distilled water and then filtered) were administered intraperitoneally twice a day (b.i.d.) (one dose in the morning and one in the afternoon, 8 h apart) (41).
Radiochemistry.
A no-carrier-added aqueous [18F]fluoride ion was produced via the [18O(p,n)18F] nuclear reaction (GE PETtrace 860 cyclotron). The compounds 2-(2-((N-(4-phenoxypyridin-3-yl)acetamido)methyl)phenoxy)ethyl 4-methylbenzenesulfonate (FEPPA precursor; catalog no. 1654) and N-(2-(2-fluoroethoxy)benzyl)-N-(4-phenoxypyridin-3-yl) acetamide (FEPPA reference standard; catalog no. 1655) were purchased from ABX GmbH, Germany. Acetonitrile (99.8%), anhydrous potassium carbonate (99.99%), 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (Kryptofix_222; 98%), phosphoric acid (85 wt% in H2O [99.99%]), and sodium bicarbonate (99.7%) were procured from Sigma-Aldrich Pte. Ltd. High-performance liquid chromatography (HPLC)-grade acetonitrile and Millex GV filters (pore size, 0.22 μm) were purchased from Merck Pte. Ltd., and 0.9% saline solution was purchased from Braun Medical Industries and was used as supplied. All reactions were carried out in closed Thermo Scientific conical reaction vials (capacity, 5 ml). Sep-Pak Accell Plus QMA Carbonate Plus Light cartridges (46 mg) were purchased from Waters Corporation and were preconditioned with 10 ml deionized water (Elga Purelab flex 3 system; water purity, 18.2 MΩ.cm) before use. Radiochemical isolation and purification were conducted with a semipreparative radio-HPLC system comprising two Knauer Smartline 1050 pumps, a manual injection valve (6-port/3-channel), a SmartMix 100 solvent mixer, a Smartline UV detector 2520, and a Flow-Count radio-HPLC NaI detection system. Quality-control analytical radio-HPLC was performed on an ultrafast liquid chromatography (UFLC) Shimadzu HPLC system equipped with a dual-wavelength UV detector and a NaI/photomultiplier tube (PMT)-radio detector (Flow-RAM; LabLogic). Radioactivity measurements were made with a CRC-55tPET dose calibrator (Capintec, USA).
PET-CT imaging.
The P. berghei ANKA-infected group was imaged on days 5, 6, and 7 postinfection, and the treated group on days 7 (2 days after treatment initiation), 14, and 21 (3 and 10 days after treatment cessation, respectively) using a Siemens Inveon PET-CT and a Mediso nanoScan 3T magnetic resonance imaging (MRI) system (Fig. 1A). Briefly, animals were injected with [18F]FEPPA (∼10 MBq per animal) via the lateral tail vein and were maintained heated and anesthetized throughout the procedure. Static PET acquisitions were performed at 60 to 80 min postinjection, and CT and T2 weighted fast-spin echo MR scans were used to delineate the areas of interest. Animals were monitored for maintenance of body temperature and respiration rate during imaging studies using the physiological monitoring systems. Postanalysis of reconstructed calibrated images was performed with Fiji and Amide software (version 10.3; SourceForge). Uptake of radioactivity was determined by the placement of a volume of interest (VOI) as delineated by CT or MR imaging.
Lung tissue immune-cell isolation.
Mice were anesthetized with ketamine (150 mg/kg)-xylazine (10 mg/kg) for 10 min, followed by intracardiac perfusion of phosphate-buffered saline (PBS). The lungs were removed, placed in complete RPMI medium (Life Technologies) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and cut into small pieces. Subsequently, each lung was digested with 50 mg/ml collagenase type 4 (Worthington, USA) and 10 mg/ml DNase I (Roche, Switzerland) for 1 h at 37°C. Cells were then homogenized using a 19½-G syringe, passed through a 70-μm cell strainer, and treated with cold ammonium-chloride-potassium (ACK) lysis buffer. The cells were washed, counted with a hemocytometer, stained with various antibodies conjugated with fluorochromes, and fixed in 1% paraformaldehyde (PFA).
Myeloid cell staining.
Cells isolated from PBS-perfused lungs were first stained with a Live/Dead Aqua stain kit (Life Technologies, USA) and then incubated with 50 μl blocking buffer (containing 1% rat and mouse serum [Sigma-Aldrich] in fluorescence-activated cell sorter [FACS] buffer [0.5% bovine serum albumin {BSA}, 2 mM EDTA in PBS]). The fluorochrome- or biotin-conjugated monoclonal antibodies specific for the mouse myeloid markers used are indicated in Table S1 in the supplemental material. All antibodies were purchased from either BD Biosciences, e-Biosciences, or BioLegend. The myeloid cells were acquired using a Fortessa cytometer (Becton, Dickinson), and the data were subsequently analyzed using FlowJo software, v.10.0 (TreeStar). Analysis was carried out by gating on singlets and Aqua− CD45+ cells (Fig. S1). The myeloid cells analyzed were monocyte-derived macrophages (MDM) (CD45+ MHC-II+ CD11c– CD11b+ CD64+), interstitial macrophages (referred to as interstitial inflammatory macrophages after infection) (CD45+ MHC-II– CD11c+ CD11b+ Ly6C– F480+), and alveolar macrophages (CD45+ MHC-II– CD11c+ F480+ or CD45+ MHC-II+ SSChigh F480+).
Statistical analysis.
Parasitemia values were assessed for normal distribution (Shapiro–Wilk test; P, 0.8215; W, 0.9668), log-transformed, and compared using an unpaired t test. All other normally distributed data were analyzed using one-way analysis of variance (ANOVA), and nonnormally distributed data were analyzed using a Kruskal-Wallis test with a Bonferroni posttest using GraphPad Prism, version 8.0.0 (GraphPad Software). A P value of <0.05 was considered statistically significant. Data are expressed as means ± standard deviations (SD) unless otherwise indicated.
ACKNOWLEDGMENTS
This project was funded by administered by the Agency for Science, Technology, and Research (A*STAR). The content is solely the responsibility of the authors and does not necessarily represent the official views of A*STAR.
We gratefully acknowledge the cyclotron radiochemistry team at the Clinical Imaging Research Centre (CIRC) for the provision of [18F]fluoride.
We declare that no competing interest exists.
J.L.G., C.C., and L.R. conceived and designed the experiments. J.L.G., S.V.H., P.X.H., S.Y.T.N., Z.W.C., B.R., and P.C. performed the experiments. J.L.G., S.V.H., P.X.H., S.Y.T.N., and Z.W.C. analyzed the data. Radiochemistry was performed by P.W.T., H.A.R., J.R.T., and E.G.R., J.L.G. and C.C. wrote the paper. S.V.H., C.C., L.R., and E.G.R. revised the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Julian L. Goggi, Email: Julian_goggi@sbic.a-star.edu.sg.
De’Broski R. Herbert, University of Pennsylvania
REFERENCES
- 1.WHO Global Malaria Programme. 30November2020. World malaria report 2020. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Schofield L, Grau GE. 2005. Immunological processes in malaria pathogenesis. Nat Rev Immunol 5:722–735. 10.1038/nri1686. [DOI] [PubMed] [Google Scholar]
- 3.Renia L, Howland SW, Claser C, Gruner AC, Suwanarusk R, Teo TH, Russell B, Ng LFP. 2012. Cerebral malaria: mysteries at the blood-brain barrier. Virulence 3:193–201. 10.4161/viru.19013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor WRJ, Hanson J, Turner GDH, White NJ, Dondorp AM. 2012. Respiratory manifestations of malaria. Chest 142:492–505. 10.1378/chest.11-2655. [DOI] [PubMed] [Google Scholar]
- 5.Asiedu DK, Sherman CB. 2000. Adult respiratory distress syndrome complicating Plasmodium falciparum malaria. Heart Lung 29:294–297. 10.1067/mhl.2000.106724. [DOI] [PubMed] [Google Scholar]
- 6.Eryüksel E, Gün D, Odabaşi Z, Karakurt S, Celikel T. 2009. Acute respiratory distress syndrome in Plasmodium falciparum malaria. Southeast Asian J Trop Med Public Health 40:1179–1182. [PubMed] [Google Scholar]
- 7.Sugiyama M, Ikeda E, Kawai S, Higuchi T, Zhang H, Khan N, Tomiyoshi K, Inoue T, Yamaguchi H, Katakura K, Endo K, Suzuki M. 2004. Cerebral metabolic reduction in severe malaria: fluorodeoxyglucose-positron emission tomography imaging in a primate model of severe human malaria with cerebral involvement. Am J Trop Med Hyg 71:542–545. 10.4269/ajtmh.2004.71.542. [DOI] [PubMed] [Google Scholar]
- 8.Braune A, Hofheinz F, Bluth T, Kiss T, Wittenstein J, Scharffenberg M, Kotzerke J, Gama de Abreu M. 2019. Comparison of static and dynamic 18F-FDG PET/CT for quantification of pulmonary inflammation in acute lung injury. J Nucl Med 60:1629–1634. 10.2967/jnumed.119.226597. [DOI] [PubMed] [Google Scholar]
- 9.de Prost N, Tucci MR, Melo MF. 2010. Assessment of lung inflammation with 18F-FDG PET during acute lung injury. AJR Am J Roentgenol 195:292–300. 10.2214/AJR.10.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deuschl C, Ruber T, Ernst L, Fendler WP, Kirchner J, Monninghoff C, Herrmann K, Quesada CM, Forsting M, Elger CE, Umutlu L. 2020. 18F-FDG-PET/MRI in the diagnostic work-up of limbic encephalitis. PLoS One 15:e0227906. 10.1371/journal.pone.0227906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shan W, Liu X, Wang Q. 2019. Teaching NeuroImages: 18F-FDG-PET/SPM analysis in 3 different stages from a patient with LGI-1 autoimmune encephalitis. Neurology 93:e1917–e1918. 10.1212/WNL.0000000000008473. [DOI] [PubMed] [Google Scholar]
- 12.Vass LD, Lee S, Wilson FJ, Fisk M, Cheriyan J, Wilkinson I. 2019. Reproducibility of compartmental modelling of 18F-FDG PET/CT to evaluate lung inflammation. EJNMMI Phys 6:26. 10.1186/s40658-019-0265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatori A, Yui J, Yamasaki T, Xie L, Kumata K, Fujinaga M, Yoshida Y, Ogawa M, Nengaki N, Kawamura K, Fukumura T, Zhang MR. 2012. PET imaging of lung inflammation with [18F]FEDAC, a radioligand for translocator protein (18 kDa). PLoS One 7:e45065. 10.1371/journal.pone.0045065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colasanti A, Guo Q, Muhlert N, Giannetti P, Onega M, Newbould RD, Ciccarelli O, Rison S, Thomas C, Nicholas R, Muraro PA, Malik O, Owen DR, Piccini P, Gunn RN, Rabiner EA, Matthews PM. 2014. In vivo assessment of brain white matter inflammation in multiple sclerosis with 18F-PBR111 PET. J Nucl Med 55:1112–1118. 10.2967/jnumed.113.135129. [DOI] [PubMed] [Google Scholar]
- 15.Dedeurwaerdere S, Callaghan PD, Pham T, Rahardjo GL, Amhaoul H, Berghofer P, Quinlivan M, Mattner F, Loc’h C, Katsifis A, Gregoire MC. 2012. PET imaging of brain inflammation during early epileptogenesis in a rat model of temporal lobe epilepsy. EJNMMI Res 2:60. 10.1186/2191-219X-2-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Missault S, Anckaerts C, Blockx I, Deleye S, Van Dam D, Barriche N, De Pauw G, Aertgeerts S, Valkenburg F, De Deyn PP, Verhaeghe J, Wyffels L, Van der Linden A, Staelens S, Verhoye M, Dedeurwaerdere S. 2019. Neuroimaging of subacute brain inflammation and microstructural changes predicts long-term functional outcome after experimental traumatic brain injury. J Neurotrauma 36:768–788. 10.1089/neu.2018.5704. [DOI] [PubMed] [Google Scholar]
- 17.Vignal N, Cisternino S, Rizzo-Padoin N, San C, Hontonnou F, Gele T, Decleves X, Sarda-Mantel L, Hosten B. 2018. [18F]FEPPA a TSPO radioligand: optimized radiosynthesis and evaluation as a PET radiotracer for brain inflammation in a peripheral LPS-injected mouse model. Molecules 23:1375. 10.3390/molecules23061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakley MS, Chorazeczewski JK, Aleshnick M, Anantharaman V, Majam V, Chawla B, Myers TG, Su Q, Okoth WA, Takeda K, Akue A, KuKuruga M, Aravind L, Kumar S. 2018. TCRβ-expressing macrophages induced by a pathogenic murine malaria correlate with parasite burden and enhanced phagocytic activity. PLoS One 13:e0201043. 10.1371/journal.pone.0201043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engwerda C, Belnoue E, Gruner AC, Renia L. 2005. Experimental models of cerebral malaria. Curr Top Microbiol Immunol 297:103–143. [PubMed] [Google Scholar]
- 20.Claser C, Nguee SYT, Balachander A, Howland SW, Becht E, Gunasegaran B, Hartimath SV, Lee AWQ, Ho JTT, Ong CB, Newell EW, Goggi J, Ng LG, Renia L. 2019. Lung endothelial cell antigen cross-presentation to CD8+ T cells drives malaria-associated lung injury. Nat Commun 10:4241. 10.1038/s41467-019-12017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epiphanio S, Campos MG, Pamplona A, Carapau D, Pena AC, Ataide R, Monteiro CA, Felix N, Costa-Silva A, Marinho CR, Dias S, Mota MM. 2010. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog 6:e1000916. 10.1371/journal.ppat.1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagasse HA, Anidi IU, Craig JM, Limjunyawong N, Poupore AK, Mitzner W, Scott AL. 2016. Recruited monocytes modulate malaria-induced lung injury through CD36-mediated clearance of sequestered infected erythrocytes. J Leukoc Biol 99:659–671. 10.1189/jlb.4HI0315-130RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovegrove FE, Gharib SA, Pena-Castillo L, Patel SN, Ruzinski JT, Hughes TR, Liles WC, Kain KC. 2008. Parasite burden and CD36-mediated sequestration are determinants of acute lung injury in an experimental malaria model. PLoS Pathog 4:e1000068. 10.1371/journal.ppat.1000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corbett CE, Duarte MI, Lancellotti CL, Silva MA, Andrade Junior HF. 1989. Cytoadherence in human falciparum malaria as a cause of respiratory distress. J Trop Med Hyg 92:112–120. [PubMed] [Google Scholar]
- 25.Duarte MI, Corbett CE, Boulos M, Amato Neto V. 1985. Ultrastructure of the lung in falciparum malaria. Am J Trop Med Hyg 34:31–35. 10.4269/ajtmh.1985.34.31. [DOI] [PubMed] [Google Scholar]
- 26.Frevert U, Nacer A, Cabrera M, Movila A, Leberl M. 2014. Imaging Plasmodium immunobiology in the liver, brain, and lung. Parasitol Int 63:171–186. 10.1016/j.parint.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozarslan N, Robinson JF, Gaw SL. 2019. Circulating monocytes, tissue macrophages, and malaria. J Trop Med 2019:3720838. 10.1155/2019/3720838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niewold P, Cohen A, van Vreden C, Getts DR, Grau GE, King NJC. 2018. Experimental severe malaria is resolved by targeting newly-identified monocyte subsets using immune-modifying particles combined with artesunate. Commun Biol 1:227. 10.1038/s42003-018-0216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kristensen LK, Frohlich C, Christensen C, Melander MC, Poulsen TT, Galler GR, Lantto J, Horak ID, Kragh M, Nielsen CH, Kjaer A. 2019. CD4+ and CD8a+ PET imaging predicts response to novel PD-1 checkpoint inhibitor: studies of Sym021 in syngeneic mouse cancer models. Theranostics 9:8221–8238. 10.7150/thno.37513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C, Cragnolini JJ, Bierie B, Gostissa M, Gorman J, Grotenbreg GM, Bhan A, Weinberg RA, Ploegh HL. 2017. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med 214:2243–2255. 10.1084/jem.20161950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rashidian M, LaFleur MW, Verschoor VL, Dongre A, Zhang Y, Nguyen TH, Kolifrath S, Aref AR, Lau CJ, Paweletz CP, Bu X, Freeman GJ, Barrasa MI, Weinberg RA, Sharpe AH, Ploegh HL. 2019. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc Natl Acad Sci U S A 116:16971–16980. 10.1073/pnas.1905005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goggi JL, Tan YX, Hartimath SV, Jieu B, Hwang YY, Jiang L, Boominathan R, Cheng P, Yuen TY, Chin HX, Tang JR, Larbi A, Chacko AM, Renia L, Johannes C, Robins EG. 2020. Granzyme B PET imaging of immune checkpoint inhibitor combinations in colon cancer phenotypes. Mol Imaging Biol 22:1392–1402. 10.1007/s11307-020-01519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van den Steen PE, Deroost K, Deckers J, Van Herck E, Struyf S, Opdenakker G. 2013. Pathogenesis of malaria-associated acute respiratory distress syndrome. Trends Parasitol 29:346–358. 10.1016/j.pt.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 34.Claser C, Malleret B, Gun SY, Wong AY, Chang ZW, Teo P, See PC, Howland SW, Ginhoux F, Renia L. 2011. CD8+ T cells and IFN-γ mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS One 6:e18720. 10.1371/journal.pone.0018720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howland SW, Claser C, Poh CM, Gun SY, Renia L. 2015. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol 37:221–231. 10.1007/s00281-015-0476-6. [DOI] [PubMed] [Google Scholar]
- 36.Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, Renia L. 2013. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med 5:984–999. 10.1002/emmm.201202273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guillon A, Arafa EI, Barker KA, Belkina AC, Martin I, Shenoy AT, Wooten AK, Lyon De Ana C, Dai A, Labadorf A, Escalante JH, Dooms H, Blasco H, Traber KE, Jones MR, Quinton LJ, Mizgerd JP. 2020. Pneumonia recovery reprograms the alveolar macrophage pool. JCI Insight 5:e133042. 10.1172/jci.insight.133042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van den Steen PE, Geurts N, Deroost K, Van Aelst I, Verhenne S, Heremans H, Van Damme J, Opdenakker G. 2010. Immunopathology and dexamethasone therapy in a new model for malaria-associated acute respiratory distress syndrome. Am J Respir Crit Care Med 181:957–968. 10.1164/rccm.200905-0786OC. [DOI] [PubMed] [Google Scholar]
- 39.Franke-Fayard B, Waters AP, Janse CJ. 2006. Real-time in vivo imaging of transgenic bioluminescent blood stages of rodent malaria parasites in mice. Nat Protoc 1:476–485. 10.1038/nprot.2006.69. [DOI] [PubMed] [Google Scholar]
- 40.Malleret B, Claser C, Ong AS, Suwanarusk R, Sriprawat K, Howland SW, Russell B, Nosten F, Renia L. 2011. A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci Rep 1:118. 10.1038/srep00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Claser C, Chang ZW, Russell B, Renia L. 2017. Adaptive immunity is essential in preventing recrudescence of Plasmodium yoelii malaria parasites after artesunate treatment. Cell Microbiol 19:e12763. 10.1111/cmi.12763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download IAI.00024-21-s0001.pdf, PDF file, 0.4 MB (450.5KB, pdf)