

ABSTRACT
Vibrio cholerae is a Gram-negative bacterium that causes the enteric disease cholera. V. cholerae colonization of the human intestine is dependent on the expression of both virulence genes and environmental adaptation genes involved in antimicrobial resistance. The expression of virulence genes, including the genes encoding the main virulence factors cholera toxin (CT) and the toxin-coregulated pilus (TCP), are coordinately regulated by the ToxR regulon. Tripartite transport systems belonging to the ATP binding cassette, major facilitator, and resistance-nodulation-division families are critical for V. cholerae pathogenesis. Transport systems belonging to these families contribute to myriad phenotypes, including protein secretion, antimicrobial resistance, and virulence. TolC plays a central role in bacterial physiology by functioning as the outer membrane pore protein for tripartite transport systems. Consistent with this, V. cholerae tolC was previously found to be required for MARTX toxin secretion and antimicrobial resistance. Here, we investigated the contribution of TolC to V. cholerae virulence. We documented that tolC was required for CT and TCP production in O1 El Tor V. cholerae. This phenotype was linked to repression of the critical ToxR regulon transcription factor aphA. Decreased aphA transcription correlated with increased expression of the LysR-family transcription factor leuO. Deletion of leuO restored aphA expression, and CT and TCP production, in a tolC mutant. The collective results document that tolC is required for ToxR regulon expression and further suggest that tolC participates in an efflux-dependent feedback circuit to regulate virulence gene expression.
KEYWORDS: TolC, cholera, virulence, RND, efflux, TolC, Vibrio cholerae
INTRODUCTION
Vibrio cholerae is a Gram-negative human pathogen that causes the acute diarrheal disease cholera (1). Cholera affects millions of people each year, particularly people in areas with poor sanitation. V. cholerae is a common inhabitant of fresh and saltwater environments from which people acquire cholera through the ingestion of V. cholerae contaminated water or food (2–4). Following ingestion, V. cholerae activates the production of virulence factors in the small intestine that facilitate colonization and the development of a severe secretory diarrhea that is the hallmark of the disease cholera.
The production of virulence factors by V. cholerae is under the transcriptional control of a hierarchical regulatory system called the ToxR regulon (5). The masthead of this regulon is ToxR, which was the first gene identified in the regulon (6). ToxR is a membrane-associated transcriptional regulator that modulates virulence factor production in response to environmental cues in the host. ToxR functions in conjunction with ToxS, which is encoded downstream from toxR and functions to stabilize ToxR (7–9). ToxR contains three domains: a periplasmic sensing domain (PPD) that is connected to a cytoplasmic DNA-binding domain by a single transmembrane-spanning domain (7, 10–12). Environmental sensing by ToxR is thought to be mediated through its PPD, which interacts with chemical cues in the periplasm to affect the activity of the DNA-binding domain at target genes. In the presence of activating signals, ToxR binds to the toxT promoter with another membrane-associated transcriptional regulator, TcpP, resulting in activation of toxT transcription. Expression of tcpP is regulated by two environmentally responsive transcription factors, AphA and AphB (13, 14). Expression of aphA is regulated by quorum sensing (13), while aphB is transcriptionally regulated by OmpR and posttranscriptionally by oxygen (15, 16). Once ToxT is produced, it directly activates the expression of the genes responsible to produce the two primary V. cholerae virulence factors: the toxin coregulated pilus (TCP) and cholera toxin (CT). TCP is a type IV pilus that is essential for V. cholerae colonization of the small intestine, whereas CT is an enterotoxin that is responsible for production of secretory diarrhea.
TolC is an outer membrane protein that was first identified in Escherichia coli (17). TolC is the archetype of a large family of outer membrane proteins belonging to the outer membrane efflux protein (OEP) superfamily that are ubiquitous among Gram-negative bacteria (18, 19). TolC plays critical roles in cellular physiology by serving as the outer membrane pore protein for transport systems belonging to the ATP-binding cassette (ABC), major facilitator (MF), and resistance-nodulation-division (RND) families. Transporters belonging to these families contribute to a multitude of phenotypes in Gram-negative bacteria, including antimicrobial resistance, metal homeostasis, protein secretion, metabolism, and virulence (20). This is true in V. cholerae, where deletion of tolC prevented MARTX toxin secretion and rendered the mutant strain hypersensitive to multiple antibiotics and highly attenuated for colonization of the infant mouse small intestine (21). These observations suggested that V. cholerae tolC was essential for both virulence and antimicrobial resistance, but the mechanism by which tolC contributed to virulence was not explored.
In this study, we investigated the contribution of V. cholerae TolC in virulence factor production in O1 El Tor strain N16961. We found that tolC was required for robust expression of the V. cholerae ToxR virulence regulon. Mutation of tolC resulted in attenuated CT and TCP production. This defect was linked to the LeuO-dependent repression of aphA. The collective results revealed that TolC has pleiotropic contributions to V. cholerae virulence, being required for virulence gene expression, MARTX toxin secretion, and resistance to antimicrobial compounds that are present in the host.
RESULTS
V. choleraetolC is required for virulence factor production.
Previous studies showed that a V. cholerae tolC mutant was highly attenuated for colonization of the infant mouse small intestine (21). This phenotype was attributed to increased antimicrobial sensitivity, but the contribution of tolC to virulence factor production was not assessed. Therefore, we tested the effect of tolC deletion on CT and TCP production in V. cholerae O1 El Tor strain N16961. We cultured N16961 and an isogenic ΔtolC mutant under virulence-inducing conditions (i.e., AKI conditions) and quantified CT and TcpA production as described in Materials and Methods (22). TcpA is the pilin subunit of the TCP and is used as a marker for TCP production (23). The results showed that tolC deletion resulted in an ∼66% reduction in CT production relative to the wild type (WT), with a corresponding decrease in TcpA production in the tolC mutant (Fig. 1A). Complementation of the ΔtolC mutant with pBAD18-tolC revealed that ectopic expression of tolC partially complemented the ΔtolC mutant for both CT and TcpA production (Fig. 1B). It is unknown why we only observed partial complementation, but we suspect that it may be due to overexpression toxicity, as ectopic overexpression of tolC was detrimental to cell growth. From these results we concluded that tolC is required for high-level production of virulence factors in V. cholerae.
FIG 1.

TolC is required for CT and TCP production in V. cholerae. The indicated V. cholerae strains were cultured under AKI conditions for 18 h, when culture aliquots were then collected and assayed for CT and TCP production (inset) as described in Materials and Methods. (A) CT and TcpA production in WT and ΔtolC strains. (B) Complementation of CT and TcpA production (inset) in the ΔtolC mutant by ectopic tolC expression from the arabinose-regulated promoter in pBAD18. The CT results are the mean and standard deviation from three independent experiments. Statistical significance was determined using Student's t test (A) and by analysis of variance (ANOVA) with the Tukey-Kramer multiple-comparison test (B). The TcpA Western blot is representative of three independent experiments. *, P ≤ 0.05.
TolC is required for induction of the ToxR regulon.
Deletion of tolC resulted in diminished CT and TcpA production. As CT and TCP are under the control of the ToxR regulon, this finding suggested that the mutation of tolC impacted the expression of the ToxR regulon. To test this, we introduced luciferase-based transcriptional reporters for CT and TCP production (i.e., ctxA and tcpA) and the five major ToxR regulon regulatory genes (i.e., aphA, aphB, toxT, tcpP and toxR) into N16961 and an isogenic ΔtolC mutant (Table 1). The resulting strains were then cultured under virulence-inducing conditions for 5 h, when gene expression was quantified. Five hours was selected as the time point to measure gene expression to capture effects on both early- and late-expressed genes in the hierarchical ToxR regulon. The results showed that ctxA and tcpA expression were reduced in the tolC-negative background relative to the WT (Fig. 2). This is consistent with the CT enzyme-linked immunosorbent assay (ELISA) and TcpA Western blot results presented in Fig. 1 and suggests that attenuated CT and TcpA production in the ΔtolC mutant results from decreased transcription and not from posttranscriptional regulation or a defect in protein secretion.
TABLE 1.
Strains, plasmids, and oligonucleotide primers
| Strain, plasmid, or primer | Description | Reference or source |
|---|---|---|
| E. coli | ||
| EC100 | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ− rpsL (Smr) nupG pir+ | Epicentre |
| SM10 λpir | thi-1 thr leu tonA lacY supE recA::RP4-2-4-Tc::Mu Kmr (λ pirR6K) | Lab collection |
| V. cholerae | ||
| JB58 | 01 El Tor strain N16961, ΔlacZ Smr | 25 |
| JB150 | JB58 ΔtolC | 21 |
| JB485 (ΔRND) | JB58 ΔvexB ΔvexD ΔvexF ΔvexH ΔvexK ΔvexM | 34 |
| YW535 | JB58 ΔtolC ΔleuO | This study |
| YW576 | JB485 ΔtolC | This study |
| C6706 | 01 El Tor strain C6706, Smr | Lab collection |
| Plasmids | ||
| pBAD18 | Arabinose-inducible expression vector, Ampr | 61 |
| pBAD18-tolC | pBAD18 expressing tolC from V. cholerae strain N16961, Ampr | 21 |
| pWM91-ΔtolC | oriR6k allelic exchange vector used to construct in-frame deletion of tolC, mob-oriT sacB Ampr | 21 |
| pWM91-ΔleuO | oriR6k allelic exchange vector used to construct in-frame deletion of leuO, mob-oriT sacB Ampr | 24 |
| pBBR-lux | luxCDABE-based promoter fusion vector, Cmr | 62 |
| TB085 | JB58-pTB17 (toxT-lux), Ampr | 52 |
| TB091 | JB58-pTB20 (tcpP-lux), Ampr | 52 |
| TB092 | JB58-pTB22 (tcpA-lux), Ampr | 52 |
| TB093 | JB58-pTB23 (ctxA-lux), Ampr | 52 |
| TB096 | JB150-pTB17 (toxT-lux), Ampr | This study |
| TB097 | JB150-pTB22 (tcpA-lux), Ampr | This study |
| TB098 | JB150-pTB23 (ctxA-lux), Ampr | This study |
| TB099 | JB150-pTB19 (aphA-lux), Ampr | This study |
| TB100 | JB150-pTB20 (tcpP-lux), Ampr | This study |
| TB102 | JB58-pTB19 (aphA-lux), Ampr | 52 |
| TB103 | JB150-pTB21 (toxR-lux), Ampr | This study |
| TB104 | JB58-pTB21 (toxR-lux), Ampr | 52 |
| TB106 | JB58-pTB18 lux-based promoter probe vector, Ampr | 52 |
| TB107 | JB150-pTB18 lux-based promoter probe vector, Ampr | This study |
| TB126 | JB58-pTB25 (aphB-lux), Ampr | 52 |
| TB127 | JB150-pTB25 (aphB-lux), Ampr | This study |
| TB139 | JB150-pTB32 (leuO-lux), Ampr | This study |
| TB140 | JB58-pTB32 (leuO-lux), Ampr | 52 |
| YV551 | YW535-pTB22 (tcpA-lux), Ampr | This study |
| YV553 | YW535-pTB17 (toxT-lux), Ampr | This study |
| YV585 | YV576-pTB19 (aphA-lux), Ampr | This study |
| YV587 | YV576-pTB23 (ctxA-lux), Ampr | This study |
| Oligonucleotide primers | ||
| AmpR-F | GCCCGCCTGATGAATGCTCATCCGGGAATTCTGACGGATGGCCTTTTTGCGTTTCT | |
| AmpR-R | CTCACCGTCTTTCATTGCCATACGGGAATTCTACAGGGCGCGTAAATCAATCTAAAG | |
| 61T-F-SacI | ATGAGCTCGTTTGACAGCTTATCATCGGAGCTC | |
| 61T-R-BamHI | TTGGATCCGTCGGGATCGCTAGTTAGTTAGG | |
| VC0164-qRT-F | CTGCTGCGCGAACGTAGTAG | |
| VC0164-qRT-R | ACGTAAATGGCGTCTCAACCC | |
| VC0629-qRT-F | GCTGCCGATTAAAGTCGAAGG | |
| VC0629-qRT-R | ACAATGGCGGGTTTACCATCTA | |
| VC0914-qRT-F | TTGCTCGATCGGTTTAGCTTGA | |
| VC0914-qRT-R | GTTCTGGCTTGATGCGTACATAC | |
| VC1673-qRT-F | CTTTAGCATCACCGGATGACTCAT | |
| VC1673-qRT-R | AACCGCATCCACGTTACAAC | |
| VC1757-qRT-F | GCTGATGCGCTATAACGGTCAG | |
| VC1757-qRT-R | CGCCGGTGACATTCGAGATA | |
| VCA0638-qRT-F | TGCCGTACAGTGGGCTATCC | |
| VCA0638-qRT-R | ACCCGCATGGATGATTACATCG |
FIG 2.

Effect of tolC on the expression of genes in the ToxR virulence regulon. V. cholerae N16961 (blue bars) and an isogenic ΔtolC mutant (blue bars) bearing the indicated ToxR regulon luciferase-based reporters were cultured under AKI conditions for 5 h when gene expression was quantified as relative light units (RLU) divided by the optical density at 600 nm. The results represent the average of RLU/OD and standard deviation from three independent experiments. Statistical significance was determined using Student's t test. *, P ≤ 0.05 relative to the WT.
We next assessed the expression of the genes that are upstream of ctxA and tcpA in the ToxR regulon. This revealed that expression of toxT, the direct regulator of ctxA and tcpA, was reduced in the tolC mutant (Fig. 2). Expression of toxT is under the control of ToxR and TcpP. While toxR transcription was unchanged in the tolC mutant (Fig. 2), the expression of tcpP was significantly reduced in the ΔtolC strain compared to the WT. The production of TcpP is regulated by AphA and AphB, which function synergistically at the tcpP promoter to activate its expression. The results here showed that the expression level of aphA, but not aphB, was reduced in the ΔtolC mutant (Fig. 2). These findings suggest that the attenuated CT and TCP production in the ΔtolC mutant results from decreased aphA transcription.
Deletion of tolC activates leuO transcription to repress virulence.
The expression of aphA is negatively regulated by quorum sensing and the LysR family transcriptional regulator LeuO (13, 24). However, the fact that N16961 is quorum sensing negative (25) strongly suggested that quorum sensing was not involved in virulence repression in the ΔtolC mutant. Therefore, we examined the effect of tolC deletion on leuO expression by introducing a leuO-lux transcriptional reporter (pTB32) into WT and ΔtolC strains. The resulting strains were cultured under virulence-inducing conditions for 5 h, when leuO expression was quantified. The results revealed a >2-fold increase in leuO expression in the ΔtolC mutant relative to the WT (Fig. 3A). As LeuO directly binds the aphA promoter to repress its transcription, these results suggest that LeuO was responsible for aphA repression and attenuated virulence factor production in the ΔtolC mutant. If this was true, we hypothesized that deletion of leuO in the ΔtolC background should restore the expression of aphA, ctxA, and tcpA in the ΔtolC mutant background. We therefore generated a ΔtolC ΔleuO double mutant and cultured WT, ΔtolC, and ΔtolC ΔleuO mutants bearing transcriptional reporters for aphA, ctxA, and tcpA under virulence-inducing conditions for 5 h before quantifying reporter expression. The results showed that aphA, ctxA, and tcpA expression was attenuated in the ΔtolC mutant and that deletion of leuO in the ΔtolC background restored WT-level expression of all three genes (Fig. 3B). The expression of aphA increased in the ΔtolC ΔleuO mutant relative to the WT, which is consistent with previous studies showing that LeuO directly bound to the aphA promoter to repress its expression (12). Consistent with the reporter data, deletion of leuO in the ΔtolC background restored both CT and TcpA production to WT levels (Fig. 3C). Taken together, these results supported the conclusion that tolC deletion resulted in increased leuO expression and that leuO was responsible for virulence attenuation in the ΔtolC mutant.
FIG 3.
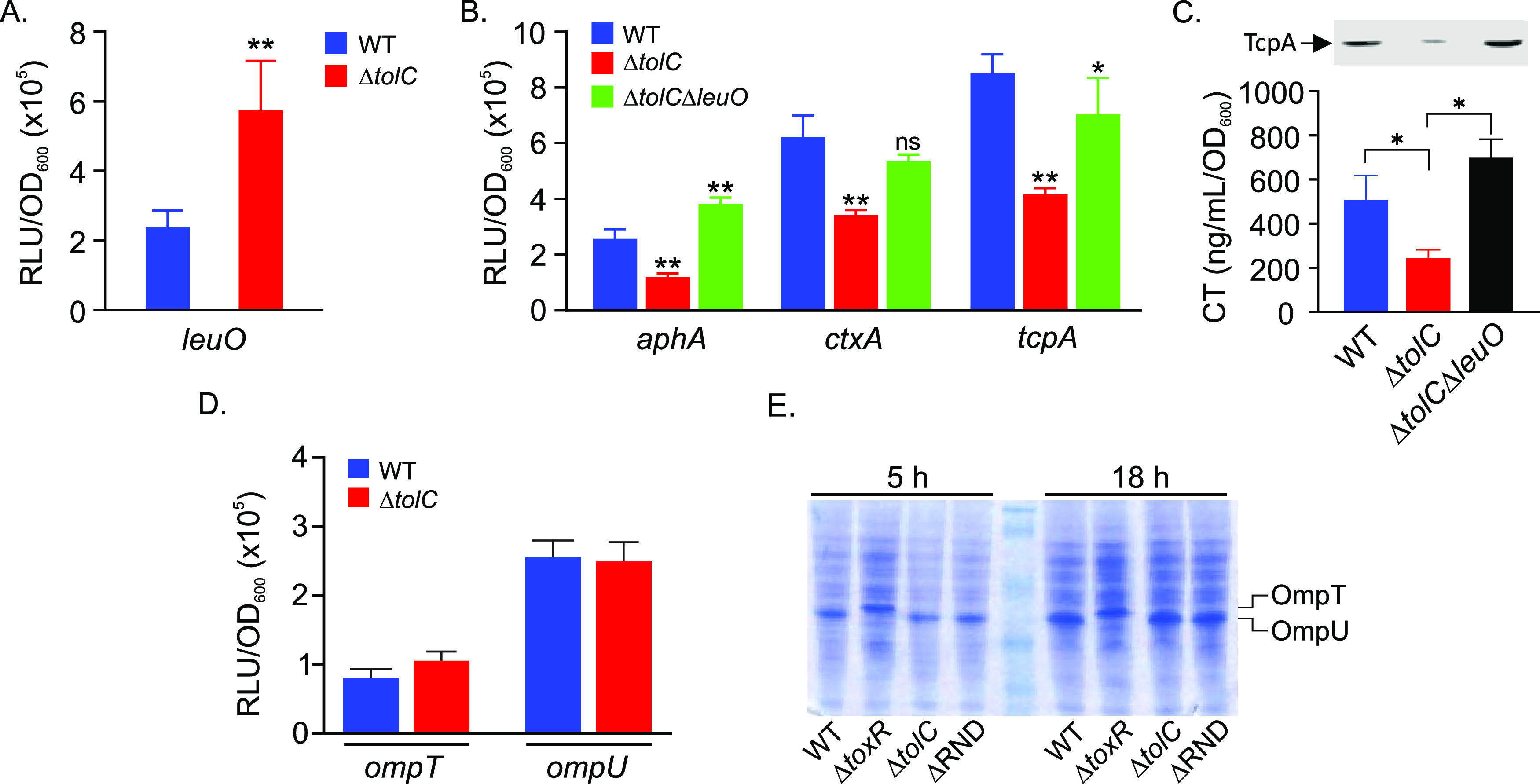
TolC-dependent virulence repression is mediated by LeuO. V. cholerae N16961 strain JB58 and isogenic ΔtolC and ΔtolC ΔleuO mutants bearing lux-based transcriptional reporters for leuO, aphA, ctxA, tcpA, ompT, and ompU were cultured under AKI conditions for 5 h when gene expression was quantified as relative light units (RLU) divided by the optical density at 600 nm. (A) Effect of tolC mutation on leuO expression in WT and ΔtolC strains. (B) Expression of aphA, ctxA, and tcpA in WT, ΔtolC, and ΔtolC ΔleuO strains. (C) CT and TcpA production (inset) in the indicated strains following overnight growth under AKI conditions. (D) Effect of tolC deletion on ompT and ompU expression. (E) SDS-PAGE gel stained with Coomassie blue. Whole-cell lysates of the indicated V. cholerae strains were prepared following growth under AKI conditions for 5 h or 18 h and resolved by SDS-PAGE. The OmpT and OmpU protein bands are indicated on the right. The results represent the average RLU/OD and standard deviation from three independent experiments. Statistical significance was determined using Student's t test (A) and a one-way ANOVA with Dunnett’s post hoc test (B and C). *, P < 0.01 relative to WT. **, P < 0.001; ns, not significant.
ToxR reciprocally regulates the expression of the ompT and ompU porins independently of TcpP (26–28), with ToxR activating ompU and repressing ompT during growth in rich medium (29, 30). ToxR-dependent induction of ompU was reported to be activated in response to bile salts and certain amino acids, with the latter resulting from increased toxRS expression (31–33). As our data suggested that toxRS transcription was not affected in the ΔtolC mutant, we tested whether tolC deletion affected the expression of porin transcription or production. We first introduced ompT and ompU transcriptional reporters into WT and ΔtolC strains. We then cultured the resulting strains under AKI conditions for 5 h before quantifying gene expression. The results showed that there was no significant difference in ompU or ompT expression in the ΔtolC mutant relative to the WT (Fig. 3D). We next examined OmpT and OmpU production using SDS-PAGE of whole-cell lysates from 5-h and 18-h cultures of the ΔtolC and ΔRND mutants. As ToxR activates ompU transcription and represses ompT transcription in rich media, we included the WT and the ΔtoxRS mutant to discriminate between the OmpT and OmpU protein bands on the SDS-PAGE gel. The results showed that OmpU was the major protein in WT lysates, whereas OmpT was the major protein in the ΔtoxRS mutant at both time points, which is consistent with previous reports (Fig. 3E). Porin production in the ΔtolC and ΔRND mutants was indistinguishable from the WT at both time points. These results indicated that neither tolC mutation nor impaired RND-mediated efflux affected ToxR-dependent porin regulation under the tested conditions. Thus, it appears that the tolC mutant phenocopies the RND efflux null mutant in activating leuO transcription without affecting porin expression.
TolC is required for robust aphA expression in an ex vivo intestine colonization model.
We sought to assess whether tolC affected aphA expression in vivo, but the tolC mutant exhibits a >500-fold increase in susceptibility to bile acids and other detergents relative to the WT, which precludes its ability to colonize infant mice (21, 34). We therefore tested the effect of tolC on aphA expression using an ex vivo small-intestine colonization model as previously described (35). To accomplish this, WT V. cholerae and isogenic ΔtolC and ΔtolC ΔleuO mutants bearing empty vector (pTB18) or aphA-lux (pTB19) were spotted onto the luminal surface of ∼1.0-cm-square segments of fresh porcine small intestine. The inoculated intestinal segments were then overlaid with LB broth and incubated at 37°C under anaerobic conditions. After a 5-h incubation, aphA expression on the surface of the intestinal segments was captured as emanating luminescence using an IVIS Lumina X5 imaging system (Fig. 4A), and luminescence production was quantified using the Living Image software (Fig. 4B). The results revealed the induction of aphA in the intestine segments inoculated with WT V. cholerae (Fig. 4). The level of aphA expression decreased by approximately 2.7-fold in the ΔtolC mutant relative to the WT, while deletion of leuO in the ΔtolC background (i.e., ΔtolC ΔleuO mutant) restored aphA expression to a level that was slightly greater than that of the WT (Fig. 4). There was low luminescence production in the empty vector control (i.e., WT-pTB18) and uninoculated control (Fig. 4). As there is no difference in the growth kinetics between these strains in vitro (not shown), these results suggest that tolC is required for robust aphA expression during V. cholerae colonization of the intestinal epithelium. These results are also consistent with the reporter studies described above (Fig. 3) and suggest that this tolC-dependent regulatory circuit is relevant in vivo.
FIG 4.

TolC affects aphA expression in an ex vivo small-intestine colonization model. The indicated V. cholerae N16961 strains bearing pTB18 (empty vector) or pTB19 (aphA-lux) were inoculated onto the surface of fresh porcine small intestine as described in Materials and Methods. The inoculated intestine segments were then incubated at 37°C in the absence of oxygen. After 5 h, the intestinal segments were imaged for luminescence production on an IVIS imaging system (A) with luminescence production being quantified using the Living Image software (B).
Deletion of tolC affects the expression of RND-family transporters.
The RND efflux systems are ubiquitous tripartite transporters in Gram-negative bacteria that contribute to a multitude of phenotypes, including antimicrobial resistance and virulence (36). In V. cholerae, TolC is believed to function as the outer membrane pore protein for all six of the V. cholerae RND efflux systems (18, 21). As the RND efflux systems are regulated by a feedback mechanism in response to their efflux substrates and tolC is required for their function, we tested if mutation of tolC affected the expression of the V. cholerae RND efflux systems. We therefore cultured WT and ΔtolC strains under AKI conditions for 5 h, when cell pellets were used to isolate total RNA. We then used quantitative reverse transcription-PCR (qRT-PCR) to assess the expression of each of the six V. cholerae RND efflux pump genes (i.e., vexB, vexD, vexF, vexH, vexK, and vexM). The results showed that the expression of vexB, vexD, and vexH increased in the ΔtolC mutant relative to the WT, with vexB and vexD being upregulated 5.2- and 4.7-fold (Fig. 5A), respectively. The increased expression of vexH was more moderate (∼1.5-fold). In contrast, the expression level of the vexF, vexK, and vexM efflux pump genes decreased by ∼2-fold relative to the WT. A similar feedback response among the vexB, vexD, and vexH RND efflux systems was noted in transcriptomics experiments comparing an RND-negative V. cholerae strain to the WT (12), which is consistent with the RND systems being regulated by a feedback mechanism. Taken together, these results provide additional support for the conclusion that tolC serves as the outer membrane pore for the V. cholerae RND efflux systems (21, 34).
FIG 5.
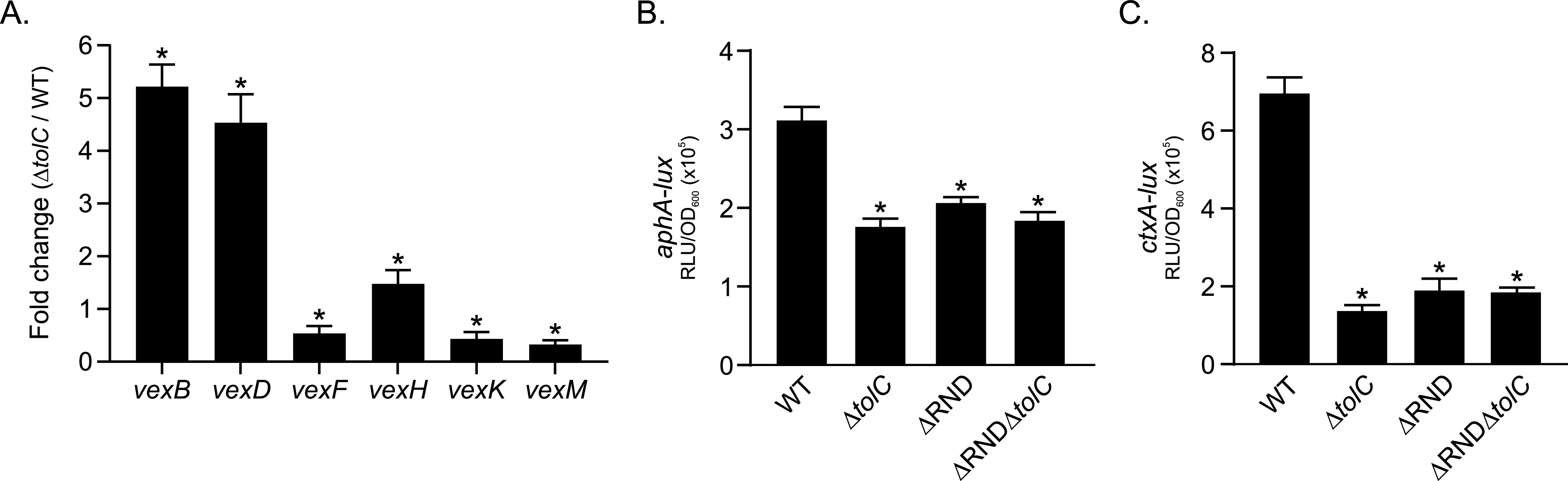
Effect of tolC on the expression of the V. cholerae RND efflux systems. (A) The expression level of the genes encoding the six V. cholerae RND-family pump proteins in the ΔtolC mutant relative to the WT. V. cholerae WT and ΔtolC strains were cultured under AKI conditions for 5 h, and total RNA was isolated and used to assess the expression level of the indicated genes by qRT-PCR as described in Materials and Methods. The results are the means and standard deviations from three independent experiments. Statistical significance was determined by the t test relative to a hypothetical fold change of 1.0. *, P ≤ 0.05. (B and C) Expression of aphA and ctxA in WT, ΔtolC, ΔRND, and ΔRND ΔtolC strains following growth under AKI conditions for 5 h. The indicated strains bearing aphA-lux (B) or ctxA-lux (C) reporters were cultured under AKI conditions for 5 h, when reporter gene expression was quantified as relative light units (RLU) and normalized by the optical density at 600 nm. The results are the means and standard deviations from three independent experiments. Statistical significance was determined using ANOVA with the Tukey-Kramer multiple-comparison test. *, P ≤ 0.05 relative to the WT.
To further confirm that the ΔtolC strain phenocopied the RND efflux-deficient mutant, we deleted tolC in RND efflux pump-deficient strain JB485 (i.e., ΔRND) and then quantified aphA and ctxA expression in WT, ΔtolC, ΔRND, and ΔRND ΔtolC strains following growth under virulence-inducing conditions. Like the leuO and porin results described above, the data showed that the ΔtolC mutant phenocopied ΔRND for both aphA and ctxA expression. Furthermore, deletion of tolC in ΔRND (i.e., the ΔRND ΔtolC mutant) did not impact the expression of either aphA or ctxA relative to that of the ΔRND or ΔtolC mutants (Fig. 5B and C). The fact that the ΔRND ΔtolC mutant phenocopied the ΔRND or ΔtolC mutant suggests that these mutants impact virulence via the same mechanism. From these results we concluded that virulence attenuation in the ΔtolC strain very likely resulted from the functional inactivation of RND-mediated efflux due to the loss of the RND system’s requisite outer membrane pore protein (i.e., TolC).
DISCUSSION
Here, we investigated the function of TolC in V. cholerae virulence factor production. We documented that deletion of tolC in O1 El Tor strain N16961 resulted in attenuated production of the critical virulence factors CT and TCP. Decreased CT and TCP production correlated with the activation of a negative regulatory circuit that resulted in increased expression of the LysR-family transcription factor leuO. LeuO was linked to repression of aphA transcription, which led to downregulation of the genes that are downstream of aphA in the ToxR regulon (Fig. 6). Confirmation of this regulatory circuit was provided by complementation studies and results showing that deletion of leuO in the tolC null background restored both aphA expression and virulence factor production. Collectively these results expand the function of TolC in V. cholerae biology and suggest that TolC has pleiotropic contributions to virulence.
FIG 6.

Working model for the function of TolC in V. cholerae virulence factor production. TolC functions as the outer membrane pore protein for the V. cholerae RND family efflux systems. In the absence of TolC, the RND family transporters no longer function and metabolites that are normally removed from the cell by the RND transporters accumulate intracellularly. The accumulated metabolites then interact with periplasmic sensors, including ToxR, to activate leuO transcription. LeuO is a repressor that binds to the aphA promoter to downregulate the ToxR regulon, leading to attenuated CT and TCP production. The RND efflux system image depicts the E. coli AcrAB-TolC system and is derived from PDB entry 5V5S (60).
TolC functions as the outer membrane pore for multiple classes of transporters, including ABC, MF, and RND transporters (19). Thus, it is not surprising that TolC has been implicated in the virulence of multiple human pathogens, including E. coli (37, 38), Salmonella (39), Haemophilus (40), Francisella (41, 42), Legionella (43), and Brucella (44). Consistent with its function as the outer membrane pore for multiple different transport systems, the contribution of TolC to pathogenesis is often pleiotropic. For example, TolC has frequently been linked to virulence in enteric pathogens, including in V. cholerae (21), due to its contribution to antimicrobial resistance. In addition, TolC is required for the secretion of virulence factors by type I secretion systems, including hemolysin (45) and enterotoxin (46) in E. coli and MARTX toxin in V. cholerae (21). However, studies linking TolC to the expression of virulence genes have been limited to Salmonella (47) and V. cholerae (described further below). However, the fact that many TolC-dependent transporters have been linked to virulence gene expression suggests that tolC has more extensive contributions to virulence gene expression than are currently appreciated (12, 36, 48–51).
The presented studies document the requirement for tolC in V. cholerae virulence factor production, but the mechanism linking tolC to leuO induction remains unclear. Recent studies defined a metabolic feedback regulatory loop in RND efflux-impaired V. cholerae (12, 52). These studies indicated that metabolites, which were substrates for the RND efflux systems, accumulated intracellularly in the absence of RND-mediated efflux. The accumulating metabolites then activated periplasmic sensors to affect transcriptional responses. This included leuO-dependent virulence repression via aphA, as observed here. Based on this, we propose that virulence repression in the ΔtolC mutant also resulted from the intracellular accumulation of metabolites that led to increased leuO transcription and the subsequent downregulation of the ToxR regulon (Fig. 6). The fact that deletion of leuO in the ΔtolC strain restored virulence factor production (Fig. 3C) and that a ΔtolC mutant phenocopied an RND efflux null mutant (Fig. 5B and C) provide support for this model. The chemical stimuli that were responsible for activating leuO in the RND impaired strains was not determined, but several metabolites that have been shown to activate leuO transcription are substrates of the RND efflux systems (e.g., indole, bile salts, cyclic dipeptides, and products of intermediate and central metabolism) and, thus, could contribute to the phenotype observed here (11, 24, 53). The fact that the V. cholerae ΔtolC strain phenocopies the antimicrobial susceptibility profile of an isogenic RND efflux-negative mutant (21, 34), combined with the feedback effects of tolC deletion on RND efflux system expression (Fig. 5), provide additional support for this model. Whether similar feedback regulatory mechanisms occur in other pathogens where tolC has been linked to virulence gene expression remains to be investigated.
There is also evidence to suggest that TolC functions in metabolite feedback regulation in V. cholerae strains belonging to the classical biotype. Classical biotype strains are believed to be responsible for pandemic cholera prior to 1961, when the seventh pandemic began with the emergence of the El Tor biotype (1, 54). El Tor biotype strains have since displaced the classical biotype in causing epidemic and pandemic disease. The two biotypes are differentiated by multiple phenotypes, including the conditions required for ToxR regulon activation. El Tor strains require AKI growth conditions to activate the ToxR regulon, whereas classical strains express the ToxR regulon in LB broth, pH 6.8, at 30°C and in minimal medium supplemented with asparagine, arginine, glutamic acid, and serine (NRES). Interestingly, deletion of tolC in a classical V. cholerae strain resulted in increased toxT expression under noninducing conditions (i.e., minimal medium lacking NRES) (55). The mechanism responsible for increased toxT transcription was not determined but was attributed to feedback regulation through ToxR or TcpP in response to the intracellular accumulation of efflux-dependent metabolic by-products. These results, while contrary to the regulatory effects of tolC mutation observed in El Tor biotypes, further support the conclusion that tolC affects gene expression via metabolite feedback processes that are likely linked to TolC’s function as the outer membrane pore protein for multiple transport systems.
While the data presented here document the effects of tolC on virulence gene expression, we note that TolC also affects other aspects of V. cholerae virulence. For example, previous studies have documented that V. cholerae tolC is required for secretion of MARTX toxin. MARTX toxin has been shown to target neutrophils to prevent clearance of V. cholerae in the gut (21, 56). Whether tolC is involved in the secretion of other proteins and whether tolC can affect virulence by other posttranscriptional mechanisms is unclear. We noted that deletion of tolC in V. cholerae O1 El Tor strain C6706 did not affect the production of the hemagglutinin (HA)/protease (data not shown), suggesting that it does not contribute to HA protease secretion (57).
In summary, the results presented here expand the function of TolC in V. cholerae pathogenesis and confirm that TolC makes pleiotropic contributions to V. cholerae pathogenesis. Given the conserved requirement for TolC in pathogenesis and antimicrobial resistance among Gram-negative pathogens, TolC is a viable target for the development of novel therapeutics that could inhibit virulence and/or potentiate antibiotics.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
V. cholerae O1 El Tor strain N16961 was acquired from our laboratory storage. N16961 JB58 (ΔlacZ Smr) and JB58ΔtolC strains were previously described (21). JB58 was used as the wild-type strain in this study. All lux reporter strains were constructed in our laboratory. E. coli strain EC100λpir (Epicentre, Madison, WI, USA) was used as the host strain for DNA cloning experiments. V. cholerae and E. coli strains were routinely grown in lysogeny broth (LB) medium or on LB agar plates at 37°C. In vitro virulence gene-inducing conditions for V. cholerae (i.e., AKI conditions) were performed by diluting overnight LB medium cultured strains (10−4) into 150- by 15-mm borosilicate glass test tubes containing 10 ml of AKI broth (4 g Difco yeast extract, 15 g Bacto peptone, and 5 g NaCl per liter, pH 7.4). This was followed by static incubation for 4 h at 37°C or until the optical density at 600 nm was greater than 0.08. Thereafter the cultures were transferred to 125-ml Erlenmeyer flasks and then incubated for 1 h or overnight at 37°C with shaking. Carbenicillin and streptomycin were added to the growth medium at 100 μg/ml when needed. Strains bearing the pBAD18 expression vector were cultured with the addition of 0.1% l-arabinose to induce expression from the arabinose-regulated promoter when necessary.
Chemicals and reagents.
Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Carbenicillin and streptomycin stock solutions were made in ultrapure water, filter sterilized, aliquoted, and stored at −20°C until needed. Oligonucleotides were designed based on the V. cholerae N16961 and C6706 genomes (25, 58) and purchased from Integrated DNA Technologies (Coralville, IA, USA), and enzymes for cloning were purchased from New England Biolabs (Beverly, MA, USA) (25).
Strain construction.
Strains, plasmids, and oligonucleotide primers are listed in Table 1. The construction of the ΔtolC mutant of V. cholerae strain N16961 has been previously reported (21). The ΔtolC ΔleuO and ΔRND ΔtolC strains were generated by allelic exchange using the pWM91ΔleuO and pWM91ΔtolC suicide vectors as previously described (21, 24). The construction of the lux-based reporter plasmids for the ToxR regulon genes in pTB18 has been described (52). Reporter plasmids were introduced into the V. cholerae strains by electroporation or conjugation from E. coli strain SM10λpir.
Virulence factor production.
Virulence factor production was assessed by quantifying cholera toxin using a GM1 enzyme-linked immunosorbent assay (ELISA) in the indicated strains following overnight growth under AKI conditions in AKI medium as previously described (59). Purified cholera toxin was used as a standard for quantitation. TcpA production was assessed by Western blotting using polyclonal antibodies to TcpA as previously described (24).
Porin production.
Whole-cell lysates were prepared from the indicated V. cholerae strains following growth under AKI conditions for 5 or 18 h. Culture aliquots were collected from the test strains, normalized by the optical density (OD) at 600 nm before being solubilized in sample buffer. The whole-cell lysates were then separated by SDS-PAGE on a 12% polyacrylamide gel and stained with Coomassie blue for visualization. WT and ΔtoxRS strains were included as controls to demark the OmpU and OmpT bands, respectively.
Transcriptional reporter assays.
V. cholerae N16961 strain JB58 and its isogenic ΔtolC mutant containing the indicated lux reporter plasmids were cultured under AKI growth conditions in AKI medium. At the indicated time points, culture aliquots (200 μl) were collected in triplicate and transferred to the wells of a 96-well white microtiter plate with a clear bottom. Luminescence production and the optical density at 600 nm were determined on a BioTek Synergy HT plate reader. The results were reported as the mean ± standard deviation number of relative luminescence units (RLU) divided by the optical density from three independent biological replicates.
Ex vivo virulence model.
The ex vivo virulence gene induction model was performed essentially as described previously (35). Briefly, fresh porcine small intestines were procured from the University of Pittsburgh Division of Animal Resources. The intestines were derived from discarded tissues of freshly euthanized animals. The intestines were splayed open, and 1-cm-square sections of the intestine were excised. The intestinal sections were placed into a petri dish, and 20 μl of V. cholerae inoculum was placed on the lumen side (faced up). The inoculum was prepared as follows. Overnight cultures grown in LB medium were diluted 1:100 into 3 ml of fresh LB medium and incubated with shaking at 37°C until they reached an optical density of ∼1.0, and then a 1-ml aliquot was collected from each culture, washed in an equal amount of minimum essential medium (MEM) twice before being resuspended in 500 μl of MEM to generate the inoculum. After inoculation, the covered petri dishes were incubated at 37°C in a sealed anaerobic jar containing a Mitsubishi AnaeroPack-Anaero to generate anaerobic conditions. After 4 to 5 h, the petri dishes were imaged using an IVIS Lumina X5 imaging system (Perkin Elmer), and luminescence production on the captured images was quantified using the Living Image software (Perkin Elmer).
Quantitative real-time PCR.
Total RNA was isolated from cultures of V. cholerae strains after 5 h of growth under AKI conditions in AKI medium using TRIzol as described by the manufacturer’s directions. cDNA was generated with the total RNA using Superscript III RT (Invitrogen). The expression level of specific genes was quantified by amplifying about 25 ng cDNA with 0.5 μM primers using SYBR green PCR mix (Fisher Scientific) in a Step One Plus real-time PCR system machine (Applied Biosystems). DNA gyrase (gyrA) was used as an internal control. Changes in gene expression were calculated by the 2−ΔΔCT method and are presented as means ± standard errors from three biological replicates, with each biological replicate generated from three technical replicates.
Statistical methods.
Statistical analyses of the data were performed using GraphPad Prism software (GraphPad Software). The specific statistical method used for each data set is described in the figure legend.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Allergy and Infectious Disease of the National Institutes of Health (NIH) under award R01AI132460. J.A.B. and D.E.K. were supported by NIH training grants T32AI138954 and T32AI049820, respectively. The IVIS Lumina S5 imaging system is a shared resource supported by NIH grant S10OD025011.
The content is solely the responsibility of the authors.
Contributor Information
James E. Bina, Email: JBina@pitt.edu.
Igor E. Brodsky, University of Pennsylvania
REFERENCES
- 1.Kaper JB, MorrisJG, Jr, Levine MM. 1995. Cholera. Clin Microbiol Rev 8:48–86. 10.1128/CMR.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakraborty S, Nair GB, Shinoda S. 1997. Pathogenic vibrios in the natural aquatic environment. Rev Environ Health 12:63–80. 10.1515/reveh.1997.12.2.63. [DOI] [PubMed] [Google Scholar]
- 3.Bina RF, Bina JE, Weng Y. 2020. Genome Sequence of Vibrio cholerae strain RFB16, isolated from North Park Lake in Allegheny County. Microbiol Resour Announc 9:e00111-20. 10.1128/MRA.00111-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daboul J, Weghorst L, DeAngelis C, Plecha SC, Saul-McBeth J, Matson JS. 2020. Characterization of Vibrio cholerae isolates from freshwater sources in northwest Ohio. PLoS One 15:e0238438. 10.1371/journal.pone.0238438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Childers BM, Klose KE. 2007. Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol 2:335–344. 10.2217/17460913.2.3.335. [DOI] [PubMed] [Google Scholar]
- 6.Waldor MK, Mekalanos JJ. 1994. ToxR regulates virulence gene expression in non-O1 strains of Vibrio cholerae that cause epidemic cholera. Infect Immun 62:72–78. 10.1128/iai.62.1.72-78.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Midgett CR, Almagro-Moreno S, Pellegrini M, Taylor RK, Skorupski K, Kull FJ. 2017. Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Mol Microbiol 105:258–272. 10.1111/mmi.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller VL, DiRita VJ, Mekalanos JJ. 1989. Identification of toxS, a regulatory gene whose product enhances toxR-mediated activation of the cholera toxin promoter. J Bacteriol 171:1288–1293. 10.1128/jb.171.3.1288-1293.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almagro-Moreno S, Root MZ, Taylor RK. 2015. Role of ToxS in the proteolytic cascade of virulence regulator ToxR in Vibrio cholerae. Mol Microbiol 98:963–976. 10.1111/mmi.13170. [DOI] [PubMed] [Google Scholar]
- 10.Lembke M, Hofler T, Walter AN, Tutz S, Fengler V, Schild S, Reidl J. 2020. Host stimuli and operator binding sites controlling protein interactions between virulence master regulator ToxR and ToxS in Vibrio cholerae. Mol Microbiol 114:262–278. 10.1111/mmi.14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ante VM, Bina XR, Howard MF, Sayeed S, Taylor DL, Bina JE. 2015. Vibrio cholerae leuO transcription is positively regulated by ToxR and contributes to bile resistance. J Bacteriol 197:3499–3510. 10.1128/JB.00419-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bina XR, Howard MF, Taylor-Mulneix DL, Ante VM, Kunkle DE, Bina JE. 2018. The Vibrio cholerae RND efflux systems impact virulence factor production and adaptive responses via periplasmic sensor proteins. PLoS Pathog 14:e1006804. 10.1371/journal.ppat.1006804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovacikova G, Skorupski K. 2002. Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol Microbiol 46:1135–1147. 10.1046/j.1365-2958.2002.03229.x. [DOI] [PubMed] [Google Scholar]
- 14.Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol 181:4250–4256. 10.1128/JB.181.14.4250-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. 2011. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci USA 108:810–815. 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunkle DE, Bina XR, Bina JE. 2020. Vibrio cholerae OmpR contributes to virulence repression and fitness at alkaline pH. Infect Immun 88:e00141-20. 10.1128/IAI.00141-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morona R, Reeves P. 1981. Molecular cloning of the tolC locus of Escherichia coli K-12 with the use of transposon Tn10. Mol Gen Genet 184:430–433. 10.1007/BF00352517. [DOI] [PubMed] [Google Scholar]
- 18.Paulsen IT, Park JH, Choi PS, SaierMH, Jr.. 1997. A family of gram-negative bacterial outer membrane factors that function in the export of proteins, carbohydrates, drugs and heavy metals from gram-negative bacteria. FEMS Microbiol Lett 156:1–8. 10.1016/S0378-1097(97)00379-0. [DOI] [PubMed] [Google Scholar]
- 19.Zgurskaya HI, Krishnamoorthy G, Ntreh A, Lu S. 2011. Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front Microbiol 2:189. 10.3389/fmicb.2011.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alcalde-Rico M, Hernando-Amado S, Blanco P, Martinez JL. 2016. Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front Microbiol 7:1483. 10.3389/fmicb.2016.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun 69:4681–4685. 10.1128/IAI.69.7.4681-4685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwanaga M, Yamamoto K. 1985. New medium for the production of cholera toxin by Vibrio cholerae O1 biotype El Tor. J Clin Microbiol 22:405–408. 10.1128/jcm.22.3.405-408.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faast R, Ogierman MA, Stroeher UH, Manning PA. 1989. Nucleotide sequence of the structural gene, tcpA, for a major pilin subunit of Vibrio cholerae. Gene 85:227–231. 10.1016/0378-1119(89)90486-1. [DOI] [PubMed] [Google Scholar]
- 24.Bina XR, Taylor DL, Vikram A, Ante VM, Bina JE. 2013. Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). mBio 4:e00366-13. 10.1128/mBio.00366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakrabarti SR, Chaudhuri K, Sen K, Das J. 1996. Porins of Vibrio cholerae: purification and characterization of OmpU. J Bacteriol 178:524–530. 10.1128/jb.178.2.524-530.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sperandio V, Bailey C, Giron JA, DiRita VJ, Silveira WD, Vettore AL, Kaper JB. 1996. Cloning and characterization of the gene encoding the OmpU outer membrane protein of Vibrio cholerae. Infect Immun 64:5406–5409. 10.1128/iai.64.12.5406-5409.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crawford JA, Kaper JB, DiRita VJ. 1998. Analysis of ToxR-dependent transcription activation of ompU, the gene encoding a major envelope protein in Vibrio cholerae. Mol Microbiol 29:235–246. 10.1046/j.1365-2958.1998.00925.x. [DOI] [PubMed] [Google Scholar]
- 30.Li CC, Crawford JA, DiRita VJ, Kaper JB. 2000. Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol Microbiol 35:189–203. 10.1046/j.1365-2958.2000.01699.x. [DOI] [PubMed] [Google Scholar]
- 31.Provenzano D, Schuhmacher DA, Barker JL, Klose KE. 2000. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect Immun 68:1491–1497. 10.1128/IAI.68.3.1491-1497.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mey AR, Butz HA, Payne SM. 2015. Vibrio cholerae CsrA regulates ToxR levels in response to amino acids and is essential for virulence. mBio 6:e01064-15. 10.1128/mBio.01064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mey AR, Craig SA, Payne SM. 2012. Effects of amino acid supplementation on porin expression and ToxR levels in Vibrio cholerae. Infect Immun 80:518–528. 10.1128/IAI.05851-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76:3595–3605. 10.1128/IAI.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci USA 110:2348–2353. 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colclough AL, Alav I, Whittle EE, Pugh HL, Darby EM, Legood SW, McNeil HE, Blair JM. 2020. RND efflux pumps in Gram-negative bacteria; regulation, structure and role in antibiotic resistance. Future Microbiol 15:143–157. 10.2217/fmb-2019-0235. [DOI] [PubMed] [Google Scholar]
- 37.Imuta N, Nishi J, Tokuda K, Fujiyama R, Manago K, Iwashita M, Sarantuya J, Kawano Y. 2008. The Escherichia coli efflux pump TolC promotes aggregation of enteroaggregative E. coli 042. Infect Immun 76:1247–1256. 10.1128/IAI.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mu X, Gao R, Xiao W, Gao Q, Cao C, Xu H, Gao S, Liu X. 2020. EntE, EntS and TolC synergistically contributed to the pathogenesis of APEC strain E058. Microb Pathog 141:103990. 10.1016/j.micpath.2020.103990. [DOI] [PubMed] [Google Scholar]
- 39.Stone BJ, Miller VL. 1995. Salmonella enteritidis has a homologue of tolC that is required for virulence in BALB/c mice. Mol Microbiol 17:701–712. 10.1111/j.1365-2958.1995.mmi_17040701.x. [DOI] [PubMed] [Google Scholar]
- 40.Trepod CM, Mott JE. 2004. Identification of the Haemophilus influenzae tolC gene by susceptibility profiles of insertionally inactivated efflux pump mutants. Antimicrob Agents Chemother 48:1416–1418. 10.1128/AAC.48.4.1416-1418.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gil H, Platz GJ, Forestal CA, Monfett M, Bakshi CS, Sellati TJ, Furie MB, Benach JL, Thanassi DG. 2006. Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proc Natl Acad Sci USA 103:12897–12902. 10.1073/pnas.0602582103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kopping EJ, Doyle CR, Sampath V, Thanassi DG. 2019. Contributions of TolC orthologs to Francisella tularensis Schu S4 multidrug resistance, modulation of host cell responses, and virulence. Infect Immun 87:e00823-18. 10.1128/IAI.00823-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferhat M, Atlan D, Vianney A, Lazzaroni JC, Doublet P, Gilbert C. 2009. The TolC protein of Legionella pneumophila plays a major role in multi-drug resistance and the early steps of host invasion. PLoS One 4:e7732. 10.1371/journal.pone.0007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Posadas DM, Martin FA, Sabio y Garcia JV, Spera JM, Delpino MV, Baldi P, Campos E, Cravero SL, Zorreguieta A. 2007. The TolC homologue of Brucella suis is involved in resistance to antimicrobial compounds and virulence. Infect Immun 75:379–389. 10.1128/IAI.01349-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vakharia H, German GJ, Misra R. 2001. Isolation and characterization of Escherichia coli tolC mutants defective in secreting enzymatically active alpha-hemolysin. J Bacteriol 183:6908–6916. 10.1128/JB.183.23.6908-6916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamanaka H, Nomura T, Fujii Y, Okamoto K. 1998. Need for TolC, an Escherichia coli outer membrane protein, in the secretion of heat-stable enterotoxin I across the outer membrane. Microb Pathog 25:111–120. 10.1006/mpat.1998.0211. [DOI] [PubMed] [Google Scholar]
- 47.Webber MA, Bailey AM, Blair JM, Morgan E, Stevens MP, Hinton JC, Ivens A, Wain J, Piddock LJ. 2009. The global consequence of disruption of the AcrAB-TolC efflux pump in Salmonella enterica includes reduced expression of SPI-1 and other attributes required to infect the host. J Bacteriol 191:4276–4285. 10.1128/JB.00363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buckner MM, Blair JM, La Ragione RM, Newcombe J, Dwyer DJ, Ivens A, Piddock LJ. 2016. Beyond antimicrobial resistance: evidence for a distinct role of the AcrD efflux pump in Salmonella biology. mBio 7:e01916-16. 10.1128/mBio.01916-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binns AN, Zhao J. 2020. The MexE/MexF/AmeC Efflux Pump of Agrobacterium tumefaciens and Its Role in Ti Plasmid Virulence Gene Expression. J Bacteriol 202:e00609-19. 10.1128/JB.00609-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Varela M, Corral J, Aranda J, Barbe J. 2019. Roles of efflux pumps from different superfamilies in the surface-associated motility and virulence of Acinetobacter baumannii ATCC 17978. Antimicrob Agents Chemother 63:e02190-18. 10.1128/AAC.02190-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alvarez-Ortega C, Olivares J, Martinez JL. 2013. RND multidrug efflux pumps: what are they good for? Front Microbiol 4:7. 10.3389/fmicb.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weng Y, Bina TF, Bina XR, Bina JE. 2021. ToxR mediates the antivirulence activity of phenyl-arginine-β-naphthylamide to attenuate Vibrio cholerae virulence. Infect Immun 89:e00147-21. 10.1128/IAI.00147-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Howard MF, Bina XR, Bina JE. 2019. Indole inhibits ToxR regulon expression in Vibrio cholerae. Infect Immun 87:e00776-18. 10.1128/IAI.00776-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sack DA, Sack RB, Nair GB, Siddique AK. 2004. Cholera. Lancet 363:223–233. 10.1016/s0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 55.Minato Y, Siefken RL, Hase CC. 2011. TolC affects virulence gene expression in Vibrio cholerae. J Bacteriol 193:5850–5852. 10.1128/JB.05222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woida PJ, Satchell KJF. 2020. The Vibrio cholerae MARTX toxin silences the inflammatory response to cytoskeletal damage before inducing actin cytoskeleton collapse. Sci Signal 13:aaw9447. 10.1126/scisignal.aaw9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benitez JA, Silva AJ. 2016. Vibrio cholerae hemagglutinin(HA)/protease: an extracellular metalloprotease with multiple pathogenic activities. Toxicon 115:55–62. 10.1016/j.toxicon.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weng Y, Bina XR, Bina JE. 2021. Complete genome sequence of Vibrio cholerae O1 El Tor strain C6706. Microbiol Resour Announc 10:e01301-20. 10.1128/MRA.01301-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bina XR, Bina JE. 2010. The cyclic dipeptide cyclo(Phe-Pro) inhibits cholera toxin and toxin-coregulated pilus production in O1 El Tor Vibrio cholerae. J Bacteriol 192:3829–3832. 10.1128/JB.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, Chiu W, Luisi BF, Du D. 2017. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. Elife 6:e24905. 10.7554/eLife.24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hammer BK, Bassler BL. 2007. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc Natl Acad Sci USA 104:11145–11149. 10.1073/pnas.0703860104. [DOI] [PMC free article] [PubMed] [Google Scholar]