

Abstract
The mortality risk of coronavirus disease 2019 (COVID-19) patients has been linked to the cytokine storm caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Understanding the inflammatory responses shared between COVID-19 and other infectious diseases that feature cytokine storms may therefore help in developing improved therapeutic strategies. Here, we use integrative analysis of single-cell transcriptomes to characterize the inflammatory signatures of peripheral blood mononuclear cells from patients with COVID-19, sepsis, and HIV infection. We identify ten hyperinflammatory cell subtypes in which monocytes are the main contributors to the transcriptional differences in these infections. Monocytes from COVID-19 patients share hyperinflammatory signatures with HIV infection and immunosuppressive signatures with sepsis. Finally, we construct a “three-stage” model of heterogeneity among COVID-19 patients, related to the hyperinflammatory and immunosuppressive signatures in monocytes. Our study thus reveals cellular and molecular insights about inflammatory responses to SARS-CoV-2 infection and provides therapeutic guidance to improve treatments for subsets of COVID-19 patients.
Keywords: SARS-CoV-2, COVID-19, inflammation, immunosuppression
Graphical abstract
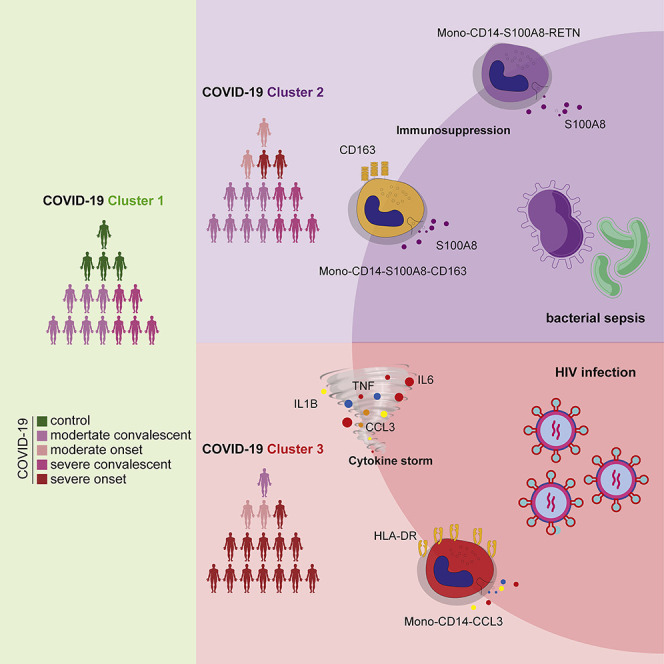
Liu et al. perform integrative analysis of single-cell transcriptomes to characterize the inflammatory signatures of peripheral blood mononuclear cells from patients with COVID-19, sepsis, and HIV infection. Analysis of heterogeneity among COVID-19 patients reveals a “three-stage” model, which is related to the hyperinflammatory and immunosuppressive signatures in monocytes.
Introduction
Currently, coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is still threatening public health in more than 200 countries. As of December 1, 2020, over 61.8 million people have been infected and more than 1.4 million deaths have been reported globally by the World Health Organization (WHO). Studies have shown that the elevated expression of proinflammatory cytokines, also known as a “cytokine storm,” is one of the main causes of death in COVID-19 (Del Valle et al., 2020; Giamarellos-Bourboulis et al., 2020; Pedersen and Ho, 2020; Zhou et al., 2020). Elevated levels of IL-10 and of proinflammatory cytokines, including interleukin (IL)-6, IL-1, and tumor necrosis factor (TNF)-α, have been associated with severe cases of COVID-19 (Chen et al., 2020; Huang et al., 2020; Mathew et al., 2020). Further studies have demonstrated that a subpopulation of monocytes with inflammatory states in the peripheral blood is a major source of these cytokines in severe COVID-19 patients (Guo et al., 2020; Ren et al., 2021).
Inflammation is closely related to the disease progression of infectious diseases (Hotchkiss et al., 2013b; Tay et al., 2020; Tien et al., 2010; Zhu et al., 2020). For example, during the period of acute HIV infection, a large number of cytokines and chemokines, such as interferon (IFN)-α, IFN-γ, IL-6, IL-10, TNF-α, and MCP-1, are induced, and their expression may enhance the early replication of the virus (Freeman et al., 2016; Muema et al., 2020; Stacey et al., 2009; Teigler et al., 2018). A recent study using single-cell RNA sequencing (RNA-seq) technology revealed the gene modules (GMs) that vary with time and cell subtypes after HIV infection, such as the inflammation-related modules driven by IL1B, TNF, and OSM (Kazer et al., 2020). Cytokine storms are one of the key mechanisms of lethality in COVID-19 (Hu et al., 2021).
Sepsis is a disease syndrome with high mortality; a comprehensive report estimated that sepsis is associated with one in five deaths worldwide (Rudd et al., 2020). Sepsis-induced systemic immune response disorder and dysregulated cytokine production play key roles in the development of multiple organ dysfunction (Bozza et al., 2007; Riedemann et al., 2003; Schulte et al., 2013). Recently, a single-cell study reported a subset of CD14+ monocytes (Reyes et al., 2020a) characteristic of sepsis. These monocytes were similar to myeloid-derived suppressor cells (MDSCs) and presented an immunosuppressive function that is dependent on the levels of IL-6 and IL-10 in the plasma (Reyes et al., 2020b). Importantly, the gene transcriptional signature of MDSC-like monocytes is also upregulated in severe COVID-19 patients (Schulte-Schrepping et al., 2020; Silvin et al., 2020). Integrative analysis enables the pooling of datasets produced across diseases and conditions, which may facilitate the discovery of cross-disease commonalities on a unified scale (Stuart and Satija, 2019). We integrated COVID-19 and sepsis peripheral blood mononuclear cells (PBMCs) transcriptomic profiles to explore whether COVID-19 patients have similar immunosuppressive cellular modalities as sepsis patients.
Previous studies suggested that the inflammatory response of COVID-19 patients was similar to that of sepsis and HIV infection (Collora et al., 2021; Remy et al., 2020); however, it is not clear which cell types may account for the commonalities of these responses. In addition, dysfunctions in the immune responses of COVID-19 patients have been independently characterized (Su et al., 2020; Wen et al., 2020; Wilk et al., 2020), and it is now clear that patients with different disease statuses and genetic backgrounds can have complex and varied molecular mechanisms that are associated with severe symptoms (Arunachalam et al., 2020; Pairo-Castineira et al., 2021; Su et al., 2020). There is still much to learn about the heterogeneity in COVID-19 patients, and investigations of dysregulated inflammatory responses in these patients are likely to yield insights that can improve clinical outcomes. Here, we conducted an integrative evaluation of a large-scale single-cell transcriptome analysis of PBMCs from patients with COVID-19, sepsis, and HIV infection. We identified the transcriptome characteristics shared between COVID-19 and sepsis or HIV infection. Our heterogeneity analysis revealed a “three-stage” model of COVID-19 and the underlying molecular factors and pathways, which provides potential guidance for precision treatment of COVID-19 patients.
Results
Integrated single-cell transcriptome atlas of PBMCs from patients with COVID-19, sepsis, and HIV infection
To investigate the peripheral immune responses elicited by different infectious diseases, we collected single-cell RNA-seq datasets of PBMCs from patients with COVID-19 (Ren et al., 2021), bacterial sepsis (Reyes et al., 2020a), and HIV-1 infection (Kazer et al., 2020) (Figure 1 A). Specifically, the COVID-19 single-cell datasets were generated at multiple institutions and hospitals (Ren et al., 2021) and included 67 samples taken from patients with an active disease (with severe disease, n = 48; with moderate disease, n = 19), 85 samples of taken from patients during the convalescent phase (from severe disease, n = 36; from moderate disease, n = 49), and healthy donors (n = 20). The patients with sepsis included two independent cohorts with five clinical entities, namely, urinary tract infection (UTI) with leukocytosis (Leuk-UTI, n = 10), UTI with mild or transient organ dysfunction (Int-URO, n = 7), UTI with clear or persistent organ dysfunction (URO, n = 10), and Bac-SEP (n = 4), indicating those in hospital wards or intensive care units (ICU-SEPs, n = 8) (Reyes et al., 2020a). Likewise, the HIV transcriptome dataset was composed of 28 historical HIV samples from 4 patients sampled at multiple time points: 0 weeks, 1 week, 2 weeks, 3 weeks, 4 weeks, 6 months, and 1 year after the first detectable viremia (Kazer et al., 2020) (Figure 1A; Table S1).
Figure 1.

Single-cell transcriptional landscape of COVID-19, sepsis, and HIV infection
(A) A schematic outline depicting the workflow for data collection from published literature and subsequent integrated analysis. Numbers indicate the number of samples of different cohorts (COVID-19, sepsis, HIV infection) and the number of single-cell transcriptomes analyzed.
(B) Top, uniform manifold approximation and projection (UMAP) embeddings of integrated single-cell transcriptomes of COVID-19, sepsis, HIV infection, and healthy donors’ (HDs’) samples. Cells are colored by cell subtypes, and dashed circles indicate the major cell types. Bottom, UMAP embeddings of different datasets illustrating no obvious batch effect in this integrated atlas.
(C) Violin plots of canonical markers (columns) for major cell types (rows).
(D) Dot plots of the proportion shifts of cell subtypes in COVID-19, sepsis, and HIV, compared to HDs. The circle color indicates enrichment (red) or depletion (blue), and the circle size indicates the values of log2(Disease/HD). Mono, monocytes; DCs, dendritic cells; Neu, neutrophils; Mega, megakaryocytes.
After removing cell doublets using Scrublet (Wolock et al., 2019) and filtering out low-quality cells, we applied Harmony (Korsunsky et al., 2019) to integrate the single-cell RNA-seq datasets from patients with COVID-19, sepsis, and HIV infection, analyzing a total of 1,080,252 single-cell transcriptomes. We then normalized the gene expression matrix and performed clustering analysis, which identified 28 cell clusters, and visualized the clusters using uniform manifold approximation and projection (UMAP). Cell types including T cells, natural killer (NK) cells, B cells, myeloid cells (monocytes, dendritic cells [DCs], neutrophils, and megakaryocytes), and MKI67+ proliferative lymphocytes were identified (Figures 1B, 1C, and S1A).
To further investigate the composition of cell subtypes of different lineages, we performed subclustering separately for the major immune cell types (B cells, NK cells, T cells, and myeloid cells) and identified 4 B cell clusters, 3 NK cell clusters, 11 myeloid cell clusters, and 9 T cell clusters based on known markers and differentially expressed genes (DEGs) (Figures 1B and S1B). In addition, we confirmed that the cells of each cluster showed remarkable consistency in cell identity with those previously annotated in the literature (Kazer et al., 2020; Ren et al., 2021; Reyes et al., 2020a) (Figure S1C), indicating that we obtained a reliable single-cell transcriptome atlas of PBMCs from patients with COVID-19, sepsis, and HIV infection.
We then investigated the divergence in the proportions of various cell types in these patients. The percentages of plasma cells and classical monocytes (CD14+ CD16– monocytes) were increased, while those of CD4+ T cells, CD8+ T cells, and NK cells were decreased, in COVID-19 patients relative to controls and particularly in patients in the severe onset state, which was consistent with previous reports (Schulte-Schrepping et al., 2020; Wen et al., 2020). The same trends in cell percentages were detected for the HIV infection and sepsis samples (Figures 1D and S2). Notably, nonclassical monocytes (CD14– CD16+ monocytes, Mono-CD16) and DCs were reduced in COVID-19 patients with severe onset status but increased in patients with sepsis and HIV infection (Figures 1D and S2). In contrast, the proportion of megakaryocytes, which was increased in the COVID-19 patients with severe onset status, was decreased in the patients with sepsis and HIV infection (Figures 1D and S2).
Hyperinflammatory cell subtypes in patients with COVID-19, sepsis, and HIV infection
To characterize the inflammatory responses of patients with these 3 infectious diseases, we defined an inflammatory score for each cell according to its expression of literature-supported cytokines and a set of inflammatory response-related genes (Table S2). A total of 10 cell subtypes were defined as hyperinflammatory cells (see Method details for details), including 5 monocyte subtypes, 3 T cell subtypes, 1 neutrophil subtype, and 1 megakaryocyte subtype (Figures 2 A and S3A). UMAP analysis showed that the inflammatory score was obviously elevated in monocytes of COVID-19 and HIV patients compared to healthy donors, excepting a small proportion of neutrophils (Figure 2B). In contrast, little change in inflammatory score was observed in the sepsis samples (Figure 2B).
Figure 2.

Characterization of inflammation signatures in COVID-19, sepsis, and HIV infection
(A) UMAP embeddings of cells colored by ten hyperinflammatory cell subtypes.
(B) The bar plots representing the inflammatory score of each hyperinflammatory cell subtype (top) and UMAP embeddings of cells colored by inflammatory score (bottom) in the COVID-19, sepsis, HIV infection, and HD groups. Error bars in the bar plots represent the 95% confidence intervals.
(C) UMAP embeddings showing the gene expression of TNF, IL6, and IL1B in the PBMCs from COVID-19, sepsis, HIV infection, and HD groups.
(D) Unsupervised hierarchical clustering of the Pearson correlation coefficients (PCCs) of the relative normalized gene expression of ten hyperinflammatory cell subtypes in the COVID-19, sepsis, and HIV infection. Color bars indicate the subtype and disease state (see legend for key).
(E) Heatmaps of the upregulated (left) and downregulated (right) biological pathways in COVID-19, sepsis, and HIV infection patients versus HDs.
(F) Heatmap of relative normalized gene expression of the enriched gene modules from weighted gene correlation network analysis (WGCNA). Color bars indicate the disease groups and subtypes (left, see legend for key). Top, a WGCNA dendrogram. Bottom, WGCNA gene modules. Mono, monocytes; Neu, neutrophils; Mega, megakaryocytes.
This analysis supports that monocytes may be a major source of cytokine storms, as reported in previous studies (Guo et al., 2020; Zhou et al., 2020). Consistently, canonical proinflammatory cytokines were highly expressed in monocytes of patients compared to other cell subtypes (Figure 2C). IL1B and IL6 showed the highest expression levels in HIV patients, followed by COVID-19, with the lowest levels in sepsis patients. To further explore inflammatory responses, we conducted unsupervised hierarchical clustering by applying relative gene expression changes against the healthy donors in these hyperinflammatory cell subtypes (Figure 2D; see Method details for details). The hyperinflammatory cell subtypes were clustered together according to disease, rather than cell type (Figures 2D and S3B), findings similar to the results of an integrated analysis of COVID-19 and influenza infection patients (Lee et al., 2020).
We also used Gene Ontology (GO) analysis to investigate disease-specific and shared biological pathways among the 3 diseases (see Method details for details). The top 10 biological pathways most enriched in the DEGs of each group are shown (Figure 2E). Cytokine and chemokine receptor genes (i.e., CXCR4, TRAF1, IFNGR1) and nuclear transcription factors (TFs; i.e., NFKB1, RUNX1, XBP1) were specifically upregulated in COVID-19. Leukemia inhibitory factor (LIF) is known to play an important role in cytokine storms in the lungs during pneumonia (Foronjy et al., 2014; Quinton et al., 2012), and we found that the response to the LIF pathway was specifically enriched in PBMCs in COVID-19 patients. STAT1, IFI30, and class I HLA genes were specifically upregulated in HIV infection. Genes encoding ubiquinone oxidoreductase subunits were specifically upregulated in sepsis. In addition, ISG15, ISG20, IFI6, IL1B, and TNF, genes that are involved in the defense response to viruses and the type I IFN signaling pathway, were upregulated in both COVID-19 and HIV infection. SLC25A5, CDKN2D, and ZNF385A, which are involved in the regulation of apoptotic signaling, were downregulated in both COVID-19 and HIV infection, suggesting viral infection-induced apoptosis of PBMCs (Cao and Li, 2020; Selliah and Finkel, 2001; Zhang et al., 2020b). Both the downregulated and upregulated gene sets in both COVID-19 and sepsis were enriched in antigen processing and presentation genes (e.g., HLA-DRA and CD74), indicating varied alterations in different types of antigen-presenting cells (APCs).
We conducted weighted gene correlation network analysis (WGCNA) to further identify the contribution of each cell type to each disease at the molecular level and identified four GMs (Figure 2F; Table S3). We found that T cell subtypes shared a common GM (GM2) (Figure 2F). CCL5, CSF1R, S100A8, and IFITM3 were modularized in T cell subtypes of all 3 diseases (GM2, Figure 2F) and were involved in T cell proliferation and various cytokine-mediated cell pathways (Figure S3C). Interestingly, we found 3 disease-specific GMs (GM1, GM3, and GM4), all of which showed that the greatest portion of variation compared to healthy donors were concentrated in the inflammatory monocyte subtypes (Figures 2F and S3D). Specifically, in the COVID-19 group, TNFSF13B, IRAK2 and APP, which function in cytokine-mediated signaling and platelet degranulation, were modularized in monocytes (GM1; Figure 2F). In the sepsis group, GRN, SRP14, and CSTB, which are involved in mitochondrial ATP synthesis and oxidative phosphorylation, were modularized in monocytes (GM4, Figure 2F). In the HIV infection group, ISG15, IFI30, IFI6, IFIT1, JAK2, and STAT1, which have functions related to the IFN response and viral genome replication, were modularized in monocytes (GM3; Figure 2F).
These results imply that monocytes are the cell type responsible for the greatest portion of the differences in immune responses among these diseases, while T cells may play a common role in host responses to all of these infections. We also observed consistent patterns of cytokine gene expression in these diseases. In monocytes, TNFSF13, PDGFA, and TNFSF13B were highly expressed in COVID-19. FTH1, IL18, and S100A12 were highly expressed in sepsis. OSM, CCL4, TNF, CXCL3, and IL1RN were highly expressed in HIV infection (Figure S3E; see Method details for details). Thus, distinct cytokines are secreted by monocytes in response to these infections, which may serve as hallmarks of these infectious diseases. In total, we identified 10 hyperinflammatory cell subtypes, and the differences in inflammatory responses among these 3 infections was dominated by monocytes.
Distinct inflammatory signatures of monocytes in patients with COVID-19, sepsis, and HIV infection
We identified 7 clusters of monocytes, namely, Mono-CD14-CCL3, Mono-CD14-HLA, Mono-CD14-S100A8-RETN, Mono-CD14-S100A8-CD163, Mono-CD14-IFITM3, Mono-CD14-CD16, and Mono-CD16 (Figure 1B). To further dissect the immune signatures in monocytes among COVID-19, sepsis, and HIV patients, we conducted differential expression analysis and GO enrichment analysis to identify the DEGs and biological functions of these 7 monocyte subtypes. Notably, Mono-CD14-IFITM3 and Mono-CD14-CD16 are two monocyte subtypes specific to COVID-19 patients (Figure 1D) that initiate IFN signaling in response to infection (Figure S4A). Both Mono-CD14-HLA and Mono-CD16 expressed high levels of MHC class II molecules (HLA-DRA, HLA-DRB1, and HLA-DQB1, Figure 3 A). However, Mono-CD14-HLA was simultaneously enriched in three diseases, whereas Mono-CD16 was enriched in sepsis and HIV but depleted in COVID-19 patients (Figure 1D). We found that the antigen processing and presentation pathways and cellular response to IFN-γ were enriched in Mono-CD14-HLA (Figure S4A). This suggested that the Mono-CD14-HLA subtype apparently represents a “classical monocyte subtype” that is normally activated in response to infection.
Figure 3.

Monocytes in COVID-19 share distinct signatures with sepsis and HIV infection
(A) Heatmap of the gene expression of the differentially expressed genes (DEGs) (top 20) for each monocyte subtype, arranged by unsupervised hierarchical clustering. Hyperinflammatory cell subtypes are marked in red.
(B and D) Heatmaps showing K-means clustering of relative normalized gene expression of DEGs from the comparison between different states and HDs in MS1-like monocytes (B) and Mono-CD14-CCL3 monocytes (D) from the COVID-19, sepsis, and HIV infection groups.
(C and E) Violin plots of selected inflammation-associated genes for Mono-CD14-CCL3 subtypes (C) and MHC class II genes for MS1-like subtypes (E), which are colored by median expression.
(F) PHATE dimensionality reduction of Mono-CD14-HLA, Mono-CD14-CCL3, Mono-CD14-S100A8-RETN, and Mono-CD14-S100A8-CD163 subtypes from COVID-19 patients (left) and pseudotime trajectories, indicating the potential transition of monocyte subtypes (right; Path1, Mono-CD14-HLA to Mono-CD14-CCL3; Path2, Mono-CD14-HLA to MS1-like monocytes).
(G) PHATE dimensionality reduction showing Palantir-imputed gene expression of Path 1 marker genes (IL1B, IL6, and TNF) and Path 2 marker genes (HLA-DRA, ALOX5AP, and RETN).
(H and I) Line plots for gene expression trends of enriched transcription factors (TFs) and corresponding target genes with pseudotime trajectories along Path1 (H) and Path2 (I).
In (E), (F), and (G), MS1-like monocytes including Mono-CD14-S100A8-RETN and Mono-CD14-S100A8-CD163 subtypes; in (A), (B), and (C), representative genes are shown on the right side.
Mono-CD14-CCL3 is marked by high expression of the proinflammatory cytokines IL1B and CCL3, which may be reminiscent of cytokine storm-correlated monocytes in COVID-19 (Guo et al., 2020). Consistent with this conjecture, Mono-CD14-CCL3 is significantly enriched in cytokine-mediated signaling pathways (Figure S4A). We also found that the proportion of Mono-CD14-CCL3 cells was significantly increased in COVID-19 and HIV patients (Figure S4B). To uncover the potential immune features of Mono-CD14-CCL3 across the three diseases, we performed pairwise differential expression analysis in samples from different states of COVID-19, sepsis, HIV, and healthy donors. We characterized five disease-specific and cross-disease gene signatures using k-means clustering in Mono-CD14-CCL3 cells (Figure 3B). DEGs in C2 (shared between COVID-19 and HIV) included the proinflammatory cytokines TNF and IL1B and chemokines CCL3 and CXCL3, which have been reported to be correlated with cytokine storms in COVID-19 patients (Guo et al., 2020; Huang et al., 2020). Further GO analysis indicated that the DEGs in C2 exhibited significant enrichment for cytokine-mediated signaling pathways and type I IFN responses (Figure S4C). Although these results showed that Mono-CD14-CCL3 was associated with cytokine storms, inflammatory cytokines were expressed differently in the 3 diseases (Figure 3C): the sepsis samples did not show high levels of proinflammatory cytokine, chemokine, or interferon-stimulated genes (ISGs) expression compared to healthy donors. Thus, we identified Mono-CD14-CCL3 as a specific monocyte state that may contribute to cytokine storms in COVID-19 and HIV patients.
Mono-CD14-S100A8-CD163 and Mono-CD14-S100A8-RETN were marked by ALOX5AP and RETN, genes which have been linked to immunosuppressive MS1 monocytes in sepsis (Reyes et al., 2020a). We found that Mono-CD14-S100A8-CD163 and Mono-CD14-S100A8-RETN significantly expressed gene signatures derived from MS1-like monocytes (Reyes et al., 2020a) and presented low expression of MHC class II genes (Figure S4D), which are considered to be anergic in sepsis (Venet et al., 2020). Therefore, we defined Mono-CD14-S100A8-CD163 and Mono-CD14-S100A8-RETN as MS1-like monocytes. Furthermore, we found that the proportion of MS1-like monocytes was elevated in COVID-19 and sepsis patients compared with healthy donors (Figure S4E).
Similar to the pairwise differential expression analysis in Mono-CD14-CCL3 cells, we obtained 6 gene signatures in MS1-like monocytes (Figure 3D). DEGs in C3 were shared between COVID-19 and sepsis and showed high expression of calprotectin-encoding genes (S100A8, S100A9), RETN, and ALOX5AP. The DEGs in pattern C3 were enriched for functional annotations related to neutrophil activation and enhanced mitochondrial respiratory complex biogenesis (Figure S4F). Studies have shown that mitochondrial biogenesis is stimulated after mitochondrial damage in order to promote the maintenance of healthy mitochondria (Kraft et al., 2019; Zhang et al., 2018). Mitochondrial malfunction occurs in patients with sepsis and is one of the potential pathogeneses of sepsis-induced multiple organ dysfunction (Kohoutová et al., 2018; Mantzarlis et al., 2017). Unexpectedly, major histocompatibility complex (MHC) class II genes were highly expressed in the MS1-like monocytes of patients with HIV infection (Figure 3E). These results suggest that the immunosuppression of MS1-like monocytes is a characteristic that may be shared between COVID-19 and sepsis patients.
SARS-CoV-2 infection can result in abnormal differentiation of the myeloid compartment, such as sustained production of circulating cytokines (e.g., IL-6, TNF-α) and functional abnormalities caused by decreased expression of HLA-DR in monocytes (Giamarellos-Bourboulis et al., 2020; Silvin et al., 2020). Schulte-Schrepping et al. (2020) further described the dynamic changes of monocyte phenotypes in longitudinal COVID-19 patients’ samples, including the dynamic shifts of inflammatory (HLA-DR high) and immunosuppressive (HLA-DR low) monocyte subtypes. In the present study, we hypothesized that hyperinflammatory monocytes and immunosuppressive monocytes may be derived from the abnormal activation of normally activated monocytes in response to SARS-CoV-2 infection. Pursuing this, we conducted pseudotime analysis with Palantir (Setty et al., 2019) (Figure 3F). The pseudotime ordering included two lineages: Path 1 (from Mono-CD14-HLA to Mono-CD14-CCL3) and Path 2 (from Mono-CD14-HLA to MS1-like monocytes), along which different patterns of gene clusters were dynamically downregulated and upregulated (Figure S5A). For example, HLA-DRA, ALOX5AP, and RETN were obviously downregulated with pseudotime along Path 2, indicating an immunosuppressive state, whereas the proinflammatory cytokines IL6, IL1B, and TNF were upregulated with pseudotime along Path 1, denoting a hyperinflammatory response (Figure 3G).
We next used SCENIC (Aibar et al., 2017) to predict the TFs that may dynamically regulate the genes following Path 1 and 2 (Figure S5B). We observed that the gene expression of IL6 and ISG15 was upregulated in Path 1; simultaneously, their upstream TFs (FOS, JUN, ATF3, and IRF1) were significantly enriched by SCENIC and showed co-upregulated gene expression (Figure 3H and S5B), in accordance with the enhancement of cytokine-related pathways in Path 1 (Figure S5A). CIITA, a transcriptional coactivator, has been reported as a key regulator of all MHC class II genes (Greer et al., 2003). CIITA and its enriched upstream regulators (ELF1, ETV6, BCLAF1) showed co-downregulated expression, illustrating the dynamic loss of MHC class II molecules with pseudotime along Path 2 (Figure 3I). Collectively, these results support that monocytes in COVID-19 presented a pathogenetic mechanism in response to SARS-CoV-2 infection wherein the Mono-CD14-CCL3 subtype presented a hyperinflammatory state and MS1-like monocytes manifested immunosuppressive phenotypes.
Three distinct PBMC profiles in COVID-19 patients correlate with inflammatory signatures in monocytes
COVID-19 patients at different disease states showed distinct immune features. We took advantage of our broad set of COVID-19 samples to investigate whether the 10 hyperinflammatory cell subtypes identified may informatively related to the disease heterogeneity of the sampled COVID-19 patients. To capture the co-variation in gene expression programs among patients, we transformed the single-cell expression matrix into a sample-level matrix and then utilized unsupervised analysis to perform dimensionality reduction, clustering, and pseudo-stage analysis (Figure 4 A; see Method details for details). Our basic working hypothesis was that the expression of inflammatory signatures would somehow reflect the development of COVID-19 disease, and we visualized the COVID-19 patient samples into a trajectory. Each patient sample was reduced to a single dot, and the dots were aligned (by the pseudo-stage) within identified clusters.
Figure 4.

Heterogeneity analysis of COVID-19 patients illustrates a “three-stage” model associated with inflammatory signatures in monocytes
(A) A workflow schematic for the heterogeneity analysis of COVID-19 patients.
(B) UMAP embeddings of samples from COVID-19 patients, colored by cluster (top) and pseudostage (bottom). Pie plots within the top panel show the proportion of sample states for each cluster.
(C) Illustration of the proportion analysis of the ten hyperinflammatory cell subtypes in COVID-19 patients. Top, heatmap of cell-type proportions for each hyperinflammatory cell subtype in COVID-19 patients. The color bar above the heatmap indicates different clusters. Middle, heatmaps of the ordered pseudostage and corresponding inflammatory score in samples from COVID-19 patients. Bottom, boxplots of the cell proportion of each sample for these four subtypes (from left to right: T-CD4-LTB-S100A4, Mono-CD14-S100A8-RETN, Mono-CD14-S100A8-CD163, Mono-CD14-CCL3) from the indicated clusters (cluster 1, n = 93; cluster 2, n = 37; cluster 3, n = 27).
(D) Top, boxplots of the cell proportions of samples for each subtype (from left to right: Mono-CD14-CCL3, Mono-CD14-S100A8-RETN, Mono-CD14-S100A8-CD163) in different clusters (cluster 1, n = 8; cluster 2, n = 7; cluster 3, n = 21) from severe active patients. Bottom, boxplots of the signature scores of each subtype in different clusters from severe active patients.
(E) Diagram of biological pathways and genes enriched in samples of cluster 3 from severe active patients. Network edges represent gene-pathway associations, and edges of different pathways are indicated by different colors. The significance of the pathways is indicated by circle size. The color bar (from blue to red) represents fold change values in gene expression levels, p values were assessed by clusterProfiler’s built-in function “enrichGO” with default parameters.
In (C) and (D), statistical significance was evaluated with Student’s t test. The boxplots show the mean and interquartile range (IQR), with whiskers extending to 1.5 × IQR.
We identified three clusters (cluster 1, cluster 2, and cluster 3) of samples from individuals with different disease states along the pseudo-stage and detected clear differences in the inflammatory responses of these clusters (Figure 4B). We then developed a “three-stage model” for understanding the state of monocyte-mediated inflammatory responses in these different groups of patients that is directly based on the subtype proportions and DEGs of our data from cluster 1, cluster 2, and cluster 3. We found that the composition of different clusters presented a diverse distribution (Figure 4B). Specifically, cluster 1 was mainly composed of samples from healthy donors and convalescent patients, and cluster 2 contained most of the samples from convalescent patients and a minority of samples from patients with active disease, while cluster 3 was mainly the samples of patients with severe active disease (Figure 4B). Notably, samples in cluster 3 were significantly enriched in inflammation-related genes, such as IL1B, TNF, and CCL3 (Figure S6A). These results indicated that peripheral inflammation is quite heterogeneous among COVID-19 patients.
Next, we analyzed the composition of immune cells in the three clusters and found distinct enrichment of different cell subtypes. The T-CD4-LTB-S100A4 subtype was enriched in cluster 1 samples; two MS1-like monocyte subtypes (Mono-CD14-S100A8-CD163 and Mono-CD14-S100A8-RETN) were enriched in cluster 2 samples, and megakaryocytes and Mono-CD14-CCL3 were highly enriched in cluster 3 samples (Figure 4C). These results suggested that the difference in inflammation levels in patients may be caused by differential abundance of certain immune cell subtypes. To further explore the relationship between these immune cell subtypes and clusters of samples from patients, we compared the expression of the identified marker genes of these cell subtypes among samples from the clusters 1–3. We found that samples from cluster 2 had the highest expression of MS1-like monocyte markers, while samples from cluster 3 overexpressed markers of Mono-CD14-CCL3 (Figure S6B). These findings indicated that the clusters of COVID-19 samples represent 3 informative stages (three-stage) reflecting distinct monocyte inflammatory signatures.
To further probe the heterogeneity of the inflammatory response in patients with severe active disease, we first compared the cellular composition and expression of inflammatory markers in Mono-CD14-CCL3 and MS1-like monocytes of samples from three clusters. We found that cluster 3 had the highest inflammatory level and the highest proportion of the Mono-CD14-CCL3 subtype. In contrast, the proportion and expression of the signatures of two MS1-like subtypes (Mono-CD14-S100A8-CD163 and Mono-CD14-S100A8-RETN) were elevated in samples from patients with severe active disease in cluster 2. Cluster 1 showed no enrichment in the signatures or proportions of these monocytes (Figure 4D).
We then performed DEG analysis and GO enrichment analysis to reveal the molecular factors and pathways, which may help differentiate clusters 1–3. We noticed enrichment for T cell functional pathways associated with adaptive immune response to infection were enriched in patients of cluster 1 patients, sepsis-associated terms including complement and coagulation cascades were activated in cluster 2 patients, and inflammatory genes and processes were overexpressed in severe patients from cluster 3 (Figure S6C). In addition, IL6, IL1B, CCL3, and other factors, which are involved in several inflammatory responses, were upregulated in patients from cluster 3 (Figure 4E), suggesting inflammation. Genes associated with immunosuppression in sepsis, such as S100A8, S100A9, SLC30A1, IL18, RETN, etc. (Gao et al., 2017; Reyes et al., 2020a), were highly expressed in patients from cluster 2 (Figure S6D). We further assessed the expression level of cytokines in different pseudostage clusters and found that CCL3, TNF, IL1B, and IL6 were significantly increased in cluster 3 (Figure S6E). We also confirmed the high expression levels of these cytokines in plasma from matched patients in cluster 3 (Figure S6F). Indeed, some of these profiled cytokines are currently being developed for anti-COVID-19 clinical treatment worldwide (IL-6 receptor blockade, chiCTR2000029765; IL-1 receptor antagonist, ClinicalTrials.gov: NCT04594356).
Collectively, these results illustrate a “three-stage” heterogeneity of COVID-19 associated with inflammatory signatures in monocytes, especially for those with severe active disease. This information may help lead to a better understanding of the mechanisms of cytokine storms in COVID-19 and may help guide therapeutic choices COVID-19 patients in the clinic.
Discussion
Most cases of SARS-CoV-2 infection manifest only mild disease, but ∼10%–20% of cases progress into a severe state with acute respiratory distress syndrome (ARDS) or severe pneumonia. A previous study reported that 28% of fatal COVID-19 cases presented aggressive inflammation in which excessive pro-inflammatory cytokines were produced (Zhang et al., 2020a). In this study, we collected single-cell transcriptome datasets from several cohorts of patients with different inflammatory infectious diseases (COVID-19, sepsis, and HIV infection). Through integrative analysis, we uncovered the immune landscapes of these diseases and identified both common and distinct inflammatory signatures. We conclude that inflammatory signatures were generally upregulated in PBMCs from COVID-19 and HIV patients but displayed only minor changes in sepsis. Immune responses to different infections varied differently among different PBMCs, especially in monocytes. Furthermore, we found a subtype of monocytes (Mono-CD14-CCL3) that may drive the hyperinflammatory response shared by patients with COVID-19 and HIV infection. Additionally, we identified another two subtypes of monocytes (MS1-like monocytes: Mono-CD14-S100A8-CD163 and Mono-CD14-S100A8-RETN) associated with COVID-19 and sepsis, which might lead to immune suppression. Thus, our integrative analysis of monocyte subpopulations provided insights into the mechanism of pathogenesis in COVID-19.
A recent study, which examined a cohort of seven COVID-19 patients (Wilk et al., 2020), reported that peripheral monocytes do not express substantial amounts of pro-inflammatory cytokines. In the present study, we detected that patients with COVID-19 and HIV infection both show enrichment for a subset of monocytes (“Mono-CD14-CCL3”) that express high levels of inflammatory signatures, which may drive hyperinflammatory responses. Moreover, several studies (Chevrier et al., 2020; Zhou et al., 2020) have confirmed at the protein level that circulating monocytes highly express pro-inflammatory cytokines. Importantly, when we re-analyzed another large-scale COVID-19 dataset (Stephenson et al., 2021), we again detected substantial expression of pro-inflammatory cytokine genes (TNF, IL1B, CCL3 etc.) by a subset of peripheral monocytes, lending support to the idea that peripheral monocytes can apparently contribute cytokine storms in COVID-19 (Figures S7A and S7B; see Method details for details).
Recently, a mass-spectrometry-based study showed two distinct stages of pathogenesis during COVID-19 disease progression (Tian et al., 2020), and extensive evidence points to a key role for cytokine storms in severe COVID-19 (Del Valle et al., 2020; Hu et al., 2021, Jose and Manuel, 2020; Kox et al., 2020). Other studies have indicated that immune suppression may also contribute to the development of this disease (Reyes et al., 2020b; Tian et al., 2020). In the present study, we observed a “three-stage” heterogeneity in our analyses of COVID-19 patients. In particular, patients in cluster 2 with severe COVID-19 showed high expression of MS1-like signatures associated with immune suppression, patients in cluster 3 showed high expression of inflammatory cytokines, and patients in cluster 1 showed enrichment for adaptive T cell immunity function response to infection (Figure S7C). Intriguingly, COVID-19 patients in cluster 2 shared immunosuppressive phenotypes with sepsis patients (Figure S7C). As treatments to elevate host immunity, IL-7, anti-PD-1, and thymosin alpha-1 have shown promising results in trials for the sepsis indication (Hotchkiss et al., 2013a; Payen et al., 2013), so these agents may aid the recovery of this group of COVID-19 patients. In contrast, COVID-19 patients in cluster 3 had excessive inflammatory response phenotypes that appeared similar to those of HIV patients (including cytokine storm signatures, TNF, IL1RN, IL6, CCL3, and IL1B) (Figure S7C). It is notable that immunosuppressive agents such as tocilizumab (targeting the IL-6 receptor) have been reported to eliminate sustained inflammation in COVID-19 patients (Guaraldi et al., 2020; Stone et al., 2020). Our analysis revealed that, in addition to IL-6, the IL1B, TNF, CCL3, and IL-10 genes and their corresponding receptors could be potential targets for COVID-19 patients with accompanying cytokine storm. The most effective treatment for COVID-19 may depend on whether the patient is in a hyperinflammatory or immunosuppressive state.
The large number of clinical samples included in our integrated analysis allowed us to investigate the pathogenesis and heterogeneity of COVID-19 patients. However, the COVID-19 patients analyzed in our study were all from China, and the HIV and sepsis data included a few samples from patients with other ethnic backgrounds. Therefore, our findings may be limited by the genetic diversity of the included patients and by the number of patients with other infectious diseases. Heterogeneity analysis of COVID-19 patients was performed based on single-cell transcriptomes in PBMCs, but data from complementary technologies, such as single-cell proteomics, could help to verify and extend our conclusions. Our work revealed the occurrence of both hyperinflammation and immunosuppression in COVID-19 patients and revealed immunity-related trends in common with two other infectious diseases, data which can help promote drug development and individualized treatments for COVID-19 patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Data files for single-cell RNA sequencing from COVID-19 patients (processed data) | Ren et al., 2021 | Gene Expression Omnibus: GSE158055 |
| Data files for single-cell RNA sequencing from COVID-19 patients (raw data) | Ren et al., 2021 | The Genome Sequence Archive (GSA), and the access number is in the process |
| Data files for single-cell RNA sequencing from sepsis patients (processed data) | Reyes et al., 2020a | The Broad Institute Single Cell Portal: SCP548 |
| Data files for single-cell RNA sequencing from HIV-1 infection patients (processed data) | Kazer et al., 2020 | The Broad Institute Single Cell Portal: SCP256 |
| Software and Algorithms | ||
| Harmony | Korsunsky et al., 2019 | https://github.com/immunogenomics/harmony |
| Scrublet | Wolock et al., 2019 | https://github.com/swolock/scrublet |
| Scanpy | Wolf et al., 2018 | https://github.com/pachterlab/kb_python |
| Enricher | Kuleshov et al., 2016 | https://maayanlab.cloud/Enrichr/ |
| PHATE | Moon et al., 2019 | https://github.com/KrishnaswamyLab/PHATE |
| Palantir | Setty et al., 2019 | https://github.com/dpeerlab/Palantir |
| SCENIC | Aibar et al., 2017 | https://github.com/aertslab/SCENIC |
| scran | Haghverdi et al., 2018 | https://bioconductor.org/packages/release/bioc/html/scran.html |
| Monocle3 | Cao and Li, 2020 | https://github.com/cole-trapnell-lab/monocle3 |
| custom analysis code | This paper | Zenodo: https://zenodo.org/record/5498909 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kun Qu (qukun@ustc.edu.cn).
Materials availability
This study did not generate new unique reagents.
Method details
Integrating the datasets and identifying the cell clusters
Before integrating the datasets collected from different pieces of literature and cohorts, a series of preprocessing steps were performed. We first filtered out cells with fewer than 500 detected genes. We also removed potential doublets using Scrublet (Wolock et al., 2019). We ran the typical Scrublet workflow to calculate the doublet scores and score threshold using the default parameters except that expected_doublet_rate was set to 0.08. Then, we obtained the top 2000 most variable genes of each dataset after removing all the mitochondrial, ribosomal, and immunoglobulin genes. The top 2000 most frequently occurring genes were retained for the integrated analysis. We ran Harmony (Korsunsky et al., 2019) with PCA embeddings (30 PCs) as input, using the default parameters (except setting theta = 1.5), to eliminate the batch effects among sixteen datasets (14 COVID-19 datasets, one HIV-1 infection dataset, and one bacterial sepsis dataset, the technical covariates). Subsequently, the Harmony embeddings were applied to identify the clusters using scanpy (Wolf et al., 2018). In the first round of clustering, major cell types were identified by Louvain clustering with resolution = 0.5, including T cells (CD4+ T, CD8+ T, gamma-delta T (gdT)), NK cells, MKI67+ lymphocytes, B cells, plasma cells, monocytes (Mono), dendritic cells (DC), neutrophils (Neu), and megakaryocytes (Mega). In the second round of clustering, T cells, B cells, plasma cells, mono cells, and DCs were further subclustered, with resolutions ranging from 0.1 to 0.8.
Characterization of hyperinflammatory cell subtypes
Similar to our last work (Ren et al., 2021), we downloaded a gene set termed “HALLMARK_INFLAMMATORY_RESPONSE” from MSigDB (Liberzon et al., 2015) and a manually curated gene set of literature-supported cytokines in COVID-19, sepsis, and HIV patients. (Table S2). The inflammatory score was calculated with the built-in function “scanpy.tl.score_genes” in Scanpy. We performed the Mann-Whitney rank test (single-tailed, greater) for the scores of each pair of cell subtypes. Ten cell subtypes (T-CD4-FOS, T-CD4-LTB-S100A4, T-CD8-SLC4A10, Mega, Mono-CD14-CCL3, Mono-CD14-HLA, Mono-CD14-S100A8-RETN, Mono-CD14-S100A8-CD163, Mono-CD16, Neu) were defined as hyperinflammation-associated clusters with statistical significance (P-value < 0.0001) in all three infectious diseases.
Differential expression analysis
To identify the DEGs in different clusters or disease states, we performed differential expression analysis using the built-in function “scanpy.tl.rank_genes_groups” in scnapy and then screened the DEGs with the following thresholds (exceptional cases are explained separately): fold change ≥ 2, P-value ≤ 0.01 and fraction of cells expressing the genes in both clusters > 10%. In Figures 3B and 3C, pairwise comparisons of HIV (hyperacute, acute), sepsis (cohort 1, cohort 2), COVID-19 (moderate, severe), and healthy donors were performed for Mono-CD14-CCL3 and MS1-like monocytes, respectively. Then, we utilized the k-means clustering algorithm to determine the relative average gene expression of DEGs to identify the gene signatures.
Enrichment analysis
Briefly, we performed GO analysis using Enrichr (Kuleshov et al., 2016). As proposed in a previous study (Lee et al., 2020), in Figure 2E, the hyperinflammatory cell subtypes in COVID-19, sepsis, and HIV were separately compared to the same cell subtypes in healthy donors. The DEGs of the hyperinflammatory cell subtypes in each disease were combined. Then, the “common up” or “common down” gene sets at the disease level were defined by intersecting the upregulated or downregulated DEGs for COVID-19, sepsis, and HIV. Figure S4A shows the top 100 DEGs of each monocyte subtype that were used for GO enrichment analysis.
Weighted correlation network analysis
We performed WGCNA (Langfelder and Horvath, 2008) using the relative average expression matrix of the hyperinflammatory cell subtypes (The normalized gene expression values of the genes in COVID-19, sepsis, and HIV infection were divided by the values in healthy donors and log2-transformed (pseudo-count = 1)). The genes included in the matrix were derived from the top 10 biological pathway-related genes of each group in Figure 2E. We chose 10 as the soft thresholding power based on the criterion of approximate scale-free topology and then constructed a signed topological overlap matrix (TOM). Next, we applied dynamic tree cutting (minModuleSize = 25) to cluster the TOM and merged the close modules with default parameters, which resulted in eight gene modules. We finally visualized these gene modules by unsupervised hierarchical clustering of relative normalized gene expression of hyperinflammatory cell subtypes.
Based on the WGCNA result, we further profiled the cytokine expression in COVID-19, sepsis, and HIV infection. Only those cytokines which were expressed by at least 20% of cells in at least one cell subset were included in this analysis. Note that the total number of neutrophils is only 5020, comprising 0.46% of all cells (1,080,252 cells) (N = 3046, 1918, 0, 56 cells in COVID-19, healthy control, sepsis, and HIV samples, respectively). The total number of megakaryocytes is only 1,2767, consisting of 1.2% of all cells (1,2168, 534, 34, 31 cells in COVID-19, healthy control, sepsis, and HIV samples, respectively). It bears emphasis that the cell numbers for these subtypes is quite limited, and is extremely limited in the sepsis and HIV samples; these cell numbers are far from enough to support downstream comparisons among these three infections with statistical power.
Trajectory inference of transition in monocytes
Dimensionality reduction was performed based on Harmony embeddings by using PHATE, a dimensionality-reduction method that is capable of capturing both local and global structures (Moon et al., 2019). We conducted trajectory inference using Palantir (Setty et al., 2019) with default parameters. We also obtained the MAGIC-imputed gene expression matrix in Palantir, which was further used to determine the trends of gene expression. As implemented in Palantir, clustering of the gene expression trends was generated based on the Phenograph algorithm.
Gene regulatory network analysis
To explore the dynamics of regulon activity during the transition of monocytes, we applied the SCENIC (Aibar et al., 2017) algorithm to these cells of two trajectory paths. We built a coexpression network to target TFs with the R package GENIE3. DNA motif analysis was performed with RcisTarget, after which each cell was scored by AUCell.
Heterogeneity analysis of COVID-19 patients
We applied unbiased methods (Ren et al., 2021) to develop a workflow to uncover the heterogeneity of inflammation in COVID-19 patients. Since we identified ten hyperinflammatory cell subtypes that may contribute significantly to inflammatory heterogeneity, our analysis focused on these cell subtypes. We first merged the cells by calculating the average gene expression for each sample (samples with less than 1000 cells were filtered). Then, we calculated the variable genes in each dataset, counted the frequency of the variable genes across datasets, and selected the top 2000 genes for downstream analysis. We further performed principal component analysis (PCA) for dimensionality reduction and applied mutual nearest neighbor (MNN) analyses to eliminate the potential batch effects among datasets (Haghverdi et al., 2018). We utilized the Leiden clustering method to cluster samples of COVID-19 patients and applied reversed graph embedding (RGE) to uncover the potential pseudostages to link the clusters of patients, which were originally proposed to unveil complicated trajectories in single cells (Qiu et al., 2017).
Label mapping analysis
We re-analyzed another recently published single-cell transcriptome of PBMCs from COVID-19 patients. We downloaded the h5ad file with clustering annotation from https://www,covid19cellatlas.org/. we used the ingest function in scanpy (Wolf et al., 2018) to project the cell annotations in this study to the unannotated data based on the PCA embeddings.
Quantification and statistical analysis
DEGs between conditions in this study were calculated with the Wilcoxon rank-sum test (two-tailed). In Figure S3A, significance was evaluated by the Mann-Whitney rank test (single-tailed, greater). In Figure S4B, the Mann-Whitney rank test (single-tailed, greater (left) or less (right)) was conducted to assess the significance of gene signatures. In Figure S4C, the Wilcoxon rank-sum test was utilized to identify the significance of cell subtype proportion shifts among states in each cell type. For all boxplots in Figure 4 and Figure S6, Student’s t test was applied to evaluate the significance of differences between conditions. In Figure S2, the significance of cell type composition shifts between disease states and healthy donors was calculated by the Wilcoxon rank-sum test (two-tailed).
Acknowledgments
This work was supported by the National Key R&D Program of China (2020YFA0112200 to K.Q.); the National Natural Science Foundation of China grants (91940306, T2125012, 31970858, and 31771428 to K.Q., 31700796 to C.G., and 81871479 to J.L.); the CAS Project for Young Scientists in Basic Research YSBR-005 (to K.Q.); and the Fundamental Research Funds for the Central Universities (YD2070002019, WK9110000141, and WK2070000158 to K.Q. and WK9100000001 to J.L.). We thank the USTC supercomputing center and the School of Life Science Bioinformatics Center for providing supercomputing resources for this project. We thank the CAS interdisciplinary innovation team for helpful discussion.
Author contributions
K.Q. and C.G. conceived and supervised the project; N.L., C.J., and P.C. performed the data analysis with help from Z.S., W.S., H.X., M.F., and X.Y.; K.Q., C.G., N.L., C.J., and P.C. interpreted data with help from L.Z., X.G., and J.F.; K.Q., N.L., C.J., and P.C. wrote the manuscript with the help from C.G., J.L., and all the other authors.
Declaration of interests
Jingwen Fang is the chief executive officer of HanGen Biotech. The other authors declare no competing interests.
Published: September 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109793.
Supplemental information
Data and code availability
The published article includes all datasets generated or analyzed during this study. And the analysis codes supporting the current study have been publicly deposited at Zenodo: https://zenodo.org/record/5498909. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza F.A., Salluh J.I., Japiassu A.M., Soares M., Assis E.F., Gomes R.N., Bozza M.T., Castro-Faria-Neto H.C., Bozza P.T. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit. Care. 2007;11:R49. doi: 10.1186/cc5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W., Li T. COVID-19: towards understanding of pathogenesis. Cell Res. 2020;30:367–369. doi: 10.1038/s41422-020-0327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S., Zurbuchen Y., Cervia C., Adamo S., Raeber M.E., de Souza N., Sivapatham S., Jacobs A., Bachli E., Rudiger A., et al. A distinct innate immune signature marks progression from mild to severe COVID-19. Cell Rep. Med. 2020;2:100166. doi: 10.1016/j.xcrm.2020.100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collora J.A., Liu R., Albrecht K., Ho Y.C. The single-cell landscape of immunological responses of CD4+ T cells in HIV versus severe acute respiratory syndrome coronavirus 2. Curr. Opin. HIV AIDS. 2021;16:36–47. doi: 10.1097/COH.0000000000000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle D.M., Kim-Schulze S., Huang H.H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foronjy R.F., Dabo A.J., Cummins N., Geraghty P. Leukemia inhibitory factor protects the lung during respiratory syncytial viral infection. BMC Immunol. 2014;15:41. doi: 10.1186/s12865-014-0041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M.L., Shive C.L., Nguyen T.P., Younes S.A., Panigrahi S., Lederman M.M. Cytokines and T-Cell Homeostasis in HIV Infection. J. Infect. Dis. 2016;214(Suppl 2):S51–S57. doi: 10.1093/infdis/jiw287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Zhao L., Wang H., Xie E., Wang X., Wu Q., Yu Y., He X., Ji H., Rink L., et al. Metal transporter Slc39a10 regulates susceptibility to inflammatory stimuli by controlling macrophage survival. Proc. Natl. Acad. Sci. USA. 2017;114:12940–12945. doi: 10.1073/pnas.1708018114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P., et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer S.F., Zika E., Conti B., Zhu X.S., Ting J.P.Y. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 2003;4:1074–1082. doi: 10.1038/ni985. [DOI] [PubMed] [Google Scholar]
- Guaraldi G., Meschiari M., Cozzi-Lepri A., Milic J., Tonelli R., Menozzi M., Franceschini E., Cuomo G., Orlando G., Borghi V., et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol. 2020;2:e474–e484. doi: 10.1016/S2665-9913(20)30173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Li B., Ma H., Wang X., Cai P., Yu Q., Zhu L., Jin L., Jiang C., Fang J., et al. Single-cell analysis of two severe COVID-19 patients reveals a monocyte-associated and tocilizumab-responding cytokine storm. Nat. Commun. 2020;11:3924. doi: 10.1038/s41467-020-17834-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss R.S., Monneret G., Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013;13:260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss R.S., Monneret G., Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Huang S., Yin L. The cytokine storm and COVID-19. J. Med. Virol. 2021;93:250–256. doi: 10.1002/jmv.26232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose R.J., Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir. Med. 2020;8:e46–e47. doi: 10.1016/S2213-2600(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazer S.W., Aicher T.P., Muema D.M., Carroll S.L., Ordovas-Montanes J., Miao V.N., Tu A.A., Ziegler C.G.K., Nyquist S.K., Wong E.B., et al. Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat. Med. 2020;26:511–518. doi: 10.1038/s41591-020-0799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohoutová M., Dejmek J., Tůma Z., Kuncová J. Variability of mitochondrial respiration in relation to sepsis-induced multiple organ dysfunction. Physiol. Res. 2018;67(Suppl 4):S577–S592. doi: 10.33549/physiolres.934050. [DOI] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kox M., Waalders N.J.B., Kooistra E.J., Gerretsen J., Pickkers P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA. 2020;324:1565–1567. doi: 10.1001/jama.2020.17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft B.D., Chen L., Suliman H.B., Piantadosi C.A., Welty-Wolf K.E. Peripheral Blood Mononuclear Cells Demonstrate Mitochondrial Damage Clearance During Sepsis. Crit. Care Med. 2019;47:651–658. doi: 10.1097/CCM.0000000000003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1) doi: 10.1093/nar/gkw377. W90-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.S., et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5:eabd1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantzarlis K., Tsolaki V., Zakynthinos E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxid. Med. Cell. Longev. 2017;2017:5985209. doi: 10.1155/2017/5985209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon K.R., van Dijk D., Wang Z., Gigante S., Burkhardt D.B., Chen W.S., Yim K., Elzen A.V.D., Hirn M.J., Coifman R.R., et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 2019;37:1482–1492. doi: 10.1038/s41587-019-0336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muema D.M., Akilimali N.A., Ndumnego O.C., Rasehlo S.S., Durgiah R., Ojwach D.B.A., Ismail N., Dong M., Moodley A., Dong K.L., et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. 2020;18:81. doi: 10.1186/s12916-020-01529-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pairo-Castineira E., Clohisey S., Klaric L., Bretherick A.D., Rawlik K., Pasko D., Walker S., Parkinson N., Fourman M.H., Russell C.D., et al. GenOMICC Investigators. ISARIC4C Investigators. COVID-19 Human Genetics Initiative. 23andMe Investigators. BRACOVID Investigators. Gen-COVID Investigators Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- Payen D., Monneret G., Hotchkiss R. Immunotherapy - a potential new way forward in the treatment of sepsis. Crit. Care. 2013;17:118. doi: 10.1186/cc12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S.F., Ho Y.C. SARS-CoV-2: a storm is raging. J. Clin. Invest. 2020;130:2202–2205. doi: 10.1172/JCI137647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017;14:979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton L.J., Mizgerd J.P., Hilliard K.L., Jones M.R., Kwon C.Y., Allen E. Leukemia inhibitory factor signaling is required for lung protection during pneumonia. J. Immunol. 2012;188:6300–6308. doi: 10.4049/jimmunol.1200256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy K.E., Brakenridge S.C., Francois B., Daix T., Deutschman C.S., Monneret G., Jeannet R., Laterre P.F., Hotchkiss R.S., Moldawer L.L. Immunotherapies for COVID-19: lessons learned from sepsis. Lancet Respir. Med. 2020;8:946–949. doi: 10.1016/S2213-2600(20)30217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184:1895–1913 e1819. doi: 10.1016/j.cell.2021.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M., Filbin M.R., Bhattacharyya R.P., Sonny A., Mehta A., Billman K., Kays K.R., Pinilla-Vera M., Benson M.E., Cosimi L.A., et al. Induction of a regulatory myeloid program in bacterial sepsis and severe COVID-19. bioRxiv. 2020 doi: 10.1101/2020.09.02.280180. [DOI] [Google Scholar]
- Riedemann N.C., Guo R.F., Ward P.A. The enigma of sepsis. J. Clin. Invest. 2003;112:460–467. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd K.E., Johnson S.C., Agesa K.M., Shackelford K.A., Tsoi D., Kievlan D.R., Colombara D.V., Ikuta K.S., Kissoon N., Finfer S., et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395:200–211. doi: 10.1016/S0140-6736(19)32989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte W., Bernhagen J., Bucala R. Cytokines in sepsis: potent immunoregulators and potential therapeutic targets--an updated view. Mediators Inflamm. 2013;2013:165974. doi: 10.1155/2013/165974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selliah N., Finkel T.H. Biochemical mechanisms of HIV induced T cell apoptosis. Cell Death Differ. 2001;8:127–136. doi: 10.1038/sj.cdd.4400822. [DOI] [PubMed] [Google Scholar]
- Setty M., Kiseliovas V., Levine J., Gayoso A., Mazutis L., Pe’er D. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 2019;37:451–460. doi: 10.1038/s41587-019-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A., Chapuis N., Dunsmore G., Goubet A.G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey A.R., Norris P.J., Qin L., Haygreen E.A., Taylor E., Heitman J., Lebedeva M., DeCamp A., Li D., Grove D., et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009;83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Cambridge Institute of Therapeutic Immunology and Infectious Disease-National Institute of Health Research (CITIID-NIHR) COVID-19 BioResource Collaboration Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/s41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J.H., Frigault M.J., Serling-Boyd N.J., Fernandes A.D., Harvey L., Foulkes A.S., Horick N.K., Healy B.C., Shah R., Bensaci A.M., et al. BACC Bay Tocilizumab Trial Investigators Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020;383:2333–2344. doi: 10.1056/NEJMoa2028836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Satija R. Integrative single-cell analysis. Nat. Rev. Genet. 2019;20:257–272. doi: 10.1038/s41576-019-0093-7. [DOI] [PubMed] [Google Scholar]
- Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., et al. ISB-Swedish COVID19 Biobanking Unit Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/j.cell.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teigler J.E., Leyre L., Chomont N., Slike B., Jian N., Eller M.A., Phanuphak N., Kroon E., Pinyakorn S., Eller L.A., et al. RV254/RV217 study groups Distinct biomarker signatures in HIV acute infection associate with viral dynamics and reservoir size. JCI Insight. 2018;3:e98420. doi: 10.1172/jci.insight.98420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W., Zhang N., Jin R., Feng Y., Wang S., Gao S., Gao R., Wu G., Tian D., Tan W., et al. Immune suppression in the early stage of COVID-19 disease. Nat. Commun. 2020;11:5859. doi: 10.1038/s41467-020-19706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien P.C., Choi A.I., Zolopa A.R., Benson C., Tracy R., Scherzer R., Bacchetti P., Shlipak M., Grunfeld C. Inflammation and mortality in HIV-infected adults: analysis of the FRAM study cohort. J. Acquir. Immune Defic. Syndr. 2010;55:316–322. doi: 10.1097/QAI.0b013e3181e66216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venet F., Demaret J., Gossez M., Monneret G. Myeloid cells in sepsis-acquired immunodeficiency. Ann. N Y Acad. Sci. 2020 doi: 10.1111/nyas.14333. Published online March 23, 2020. [DOI] [PubMed] [Google Scholar]
- Wen W., Su W.R., Tang H., Le W.Q., Zhang X.P., Zheng Y.F., Liu X.X., Xie L.H., Li J.M., Ye J.G., et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6:18. doi: 10.1038/s41421-020-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Feng Y.W., Yao Y.M. Potential therapy strategy: targeting mitochondrial dysfunction in sepsis. Mil. Med. Res. 2018;5:41. doi: 10.1186/s40779-018-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Zhou X., Qiu Y., Song Y., Feng F., Feng J., Song Q., Jia Q., Wang J. Clinical characteristics of 82 cases of death from COVID-19. PLoS ONE. 2020;15:e0235458. doi: 10.1371/journal.pone.0235458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Y., Wang X.M., Xing X., Xu Z., Zhang C., Song J.W., Fan X., Xia P., Fu J.L., Wang S.Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- Zhou Y.G., Fu B.Q., Zheng X.H., Wang D.S., Zhao C.C., Qi Y.J., Sun R., Tian Z.G., Xu X.L., Wei H.M. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020;7:998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Yang P., Zhao Y., Zhuang Z., Wang Z., Song R., Zhang J., Liu C., Gao Q., Xu Q., et al. Single-Cell Sequencing of Peripheral Mononuclear Cells Reveals Distinct Immune Response Landscapes of COVID-19 and Influenza Patients. Immunity. 2020;53:685–696.e3. doi: 10.1016/j.immuni.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. And the analysis codes supporting the current study have been publicly deposited at Zenodo: https://zenodo.org/record/5498909. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.