

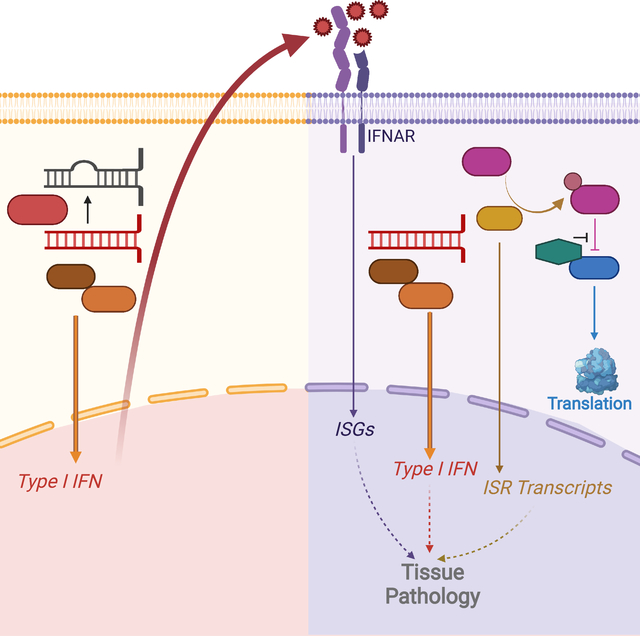
Summary
The RNA deaminase ADAR1 is an essential negative regulator of the RNA sensor MDA5, and loss of ADAR1 function triggers inappropriate activation of MDA5 by self-RNAs. Mutations in ADAR, the gene that encodes ADAR1, cause human immune diseases, including Aicardi-Goutières Syndrome (AGS). However, the mechanisms of MDA5-dependent disease pathogenesis in vivo remain unknown. Here, we generated mice with a single amino acid change in ADAR1 that models the most common human ADAR AGS mutation. These Adar-mutant mice developed lethal disease that required MDA5, the RIG-I-like receptor LGP2, type I interferons, and the eIF2α kinase PKR. A small molecule inhibitor of the integrated stress response (ISR) that acts downstream of eIF2α phosphorylation prevented immunopathology and rescued the mice from mortality. These findings place PKR and the ISR as central components of immunopathology in vivo and identify therapeutic targets for treatment of human diseases associated with the ADAR1-MDA5 axis.
Keywords: ADAR1, Aicardi-Goutières Syndrome, MDA5, LGP2, Interferons, PKR, Integrated Stress Response
eTOC:
Mutations in ADAR1 cause human immune diseases, including Aicardi-Goutières Syndrome (AGS), but mechanisms of disease pathogenesis remain unknown. Using a mouse model of ADAR1 mutation, Maurano et. al. define a pathway linking MDA5 and LGP2 to PKR and the Integrated Stress Response (ISR). Pharmacological inhibition of the ISR prevents disease, establishing its role in immunopathology caused by ADAR1 mutations.
Graphical Abstract
Introduction
The RIG-I-like receptors (RLRs) MDA5 and RIG-I are important for the rapid detection of viral RNAs and the initiation of the type I interferon (IFN) response, which restricts viral replication and spread (Goubau et al., 2013). However, this mode of defense comes with the risk of potential recognition of self-RNAs and chronic activation of the IFN response. Negative regulation of these nucleic acid sensors is therefore essential to prevent inappropriate recognition of endogenous RNAs by the RLRs (Crowl et al., 2017).
The RNA-editing enzyme Adenosine Deaminase Acting on RNA 1 (ADAR1) edits endogenous double-stranded RNAs (dsRNAs) to prevent chronic activation of MDA5 and IFN production in response to self-RNA (George et al., 2016; Liddicoat et al., 2015; Mannion et al., 2014; Pestal et al., 2015). ADAR1 is expressed as two distinct isoforms: the constitutively expressed 110 kDa ADAR1 p110, and the IFN-inducible 150 kDa ADAR1 p150 (Patterson and Samuel, 1995). ADAR1 edits mRNA coding sequences (Hartner et al., 2004), micro RNAs (Yang et al., 2006), viral RNAs (Tenoever et al., 2007), and, most frequently, inverted repeats of SINE retroelements in noncoding RNA regions (Osenberg et al., 2010). The inosine products of ADAR1 deamination are read as guanosine by the translational machinery, so RNA editing by ADAR1 can result in non-synonymous coding changes. Inosines also disrupt the dsRNA structures necessary for MDA5 activation (Ahmad et al., 2018; Liddicoat et al., 2015). In vitro, MDA5 binds to these same regions - predominantly inverted repeats of SINEs - in cells lacking ADAR1 (Ahmad et al., 2018; Chung et al., 2018; Ishizuka et al., 2019). The p150 isoform of ADAR1 is specifically responsible for MDA5 regulation, whereas the p110 isoform contributes to multiorgan development independent of MDA5 regulation (Pestal et al., 2015).
Over 200 distinct mutations in the ADAR gene have been identified that cause human diseases associated with a type I IFN response, including Aicardi-Goutières syndrome (AGS), Bilateral Striatal Necrosis (BSN), and Dyschromatosis Symmetrica Herediteria (DSH; Hayashi and Suzuki, 2013; Livingston et al., 2014; Rice et al., 2012). Despite considerable progress in understanding the molecular mechanisms of AGS and related diseases, these conditions remain untreatable and incurable, underscoring the need to identify new therapeutic targets for intervention.
Three mouse models of ADAR1 mutation have been instrumental in defining the relationship between ADAR1 RNA editing and the MDA5-MAVS pathway: Adar-null mice that lack both ADAR1 isoforms (Hartner et al., 2004; Hartner et al., 2009; Wang et al., 2004), an Adar p150 deficient mouse that lacks ADAR1 p150 but retains ADAR1 p110 (Ward et al., 2011), and an Adar point mutant mouse that disrupts deaminase activity but retains ADAR1 protein expression (Liddicoat et al., 2015). Moreover, CRISPR targeting of the ADAR gene in human cells has been used to further explore consequences of ADAR1 loss (Ahmad et al., 2018; Chung et al., 2018; Gannon et al., 2018; Pestal et al., 2015), and ADAR1 targeting was recently identified as a strategy to enhance innate immune responses in tumor cells (Ishizuka et al., 2019). However, characterization of the ADAR1-MDA5 regulatory axis in vivo has been hampered by the embryonic lethality caused by the null alleles of Adar in mice, which can only be rescued by simultaneous disruption of the genes that encode MDA5 or MAVS (Liddicoat et al., 2015; Mannion et al., 2014; Pestal et al., 2015). Thus, study of the relationship between ADAR1 and MDA5 in live mice has been impossible using current models.
To address these limitations and to enable the identification of AGS disease mechanisms in vivo, we generated a mouse that models the most common ADAR allele found in AGS: a nonsynonymous point mutation that converts a proline to an alanine at position 193 in the human ADAR1 protein (P193A; P195A in mice). This mutation is located within the Zα domain that is unique to the p150 isoform, and it is present at a remarkably high allele frequency of ~1/360 in humans of northern European ancestry (Crow et al., 2015; Rice et al., 2012). Using these mice, we showed that Adar P195A paired with a null allele of Adar causes complete, postnatal, MDA5-dependent mortality. We further defined essential requirements for the RLR LGP2, type I IFNs, and the eIF2α kinase PKR in disease progression. Finally, we showed that therapeutic inhibition of the Integrated Stress Response (ISR) downstream of PKR was sufficient to completely prevent disease. Together, these data reveal effector mechanisms downstream of MDA5 activation that contribute to immunopathology in vivo, with implications for treatment of human diseases caused by ADAR mutation.
Results
The Adar P195A mouse model
Over 60% of AGS patients with ADAR mutations carry the P193A allele as a compound heterozygote with either a frameshift mutation or a mutation in the deaminase domain of ADAR1 (Crow et al., 2015; Rice et al., 2012). No AGS patients have been identified who are homozygous for ADARP193A. We hypothesized that this was because homozygosity of P193A mutation would be incompatible with life, similar to the phenotype of total ADAR1 loss in mice (Hartner et al., 2004; Wang et al., 2004). To determine how this mutation impacts ADAR1 function and self-RNA detection, we used CRISPR targeting of fertilized mouse oocytes to generate mice carrying the orthologous P195A mutation in the endogenous Adar locus (Figure 1A). We intercrossed AdarP195A/+ mice and identified live births of Adar+/+, AdarP195A/+, and AdarP195A/P195A mice at the expected Mendelian ratios (Figure 1B). We tracked the survival and weights of AdarP195A/+ and AdarP195A/P195A mice and found them to be indistinguishable from wild type controls (Figure 1C, 1D). This suggests that the absence of known ADARP193A/P193A AGS patients might be due to a lack of disease. We then measured expression of Adar mRNA in cerebellum and in bone marrow-derived macrophages (BMMs) and found that the AdarP195A mutation did not impact Adar mRNA levels in resting cells or after treatment with recombinant IFN beta (IFNβ; Figure S1). Moreover, the protein expression levels, inducibility, and nuclear and cytosolic distribution of the ADAR1 p110 and p150 isoforms were unaffected by the AdarP195A mutation (Figure S1). Thus, the AdarP195A mutation is not pathogenic on its own.
Figure 1. Adar P195A mice model the most common Aicardi-Goutieres Syndrome mutation.

(A) Schematic of the structure of ADAR1 protein, and the location of the P195A mutation. Zα, Zβ, = Z-DNA binding domains, NES = Nuclear export signal, NLS = nuclear localization signal.
(B) Percentage of mice of the indicated genotype from intercrosses of AdarP195A/+ mice. (n=140 pups)
(C) Survival of Adar+/+ (n=23), AdarP195A/+ (n=47), AdarP195A/P195A (n=48) mice.
(D) Weights of mice at 23 days of age. Adar+/+ (n= 6F,12M), AdarP195A/+ (n= 13F,14M), AdarP195A/P195A (n= 8F, 9M). Bar represents the mean. See also Figure S1.
Recapitulation of AGS patient genotypes causes severe disease in Adar P195A mice
Because the ADARP193A mutation in AGS is invariably found as a compound heterozygote with a more severe ADAR allele (Crow et al., 2015; Rice et al., 2012), we intercrossed AdarP195A/P195A mice with either Adar+/− or Adar p150+/− mice to model the combinations of ADAR alleles found in AGS patients. We found that both AdarP195A/− mice and AdarP195A/p150− mice were born at frequencies that matched the recovery of heterozygous mice from crosses of the parental Adar-null and Adar p150-null alleles (Figure S2A, S2B). However, and in contrast to the AdarP195A/P195A mice (Figure 1C), we noted complete postnatal mortality when the P195A mutation was paired with either the full Adar null allele or the Adar p150-null allele (Figure 2A, 2B). Mortality in the AdarP195A/− mice (median survival 21 days) progressed more rapidly than in the AdarP195A/p150− mice (median survival 40 days; Figure 2A, 2B). Next, we performed these same intercrosses of the AdarP195A mutation with the Adar-null and Adar p150-null alleles on an Ifih1−/− (MDA5 KO) background and observed complete rescue from mortality of both AdarP195A/− Ifih1−/− and AdarP195A/p150−Ifih1−/− mice (Figure 2C, 2D). Consistent with the essential contribution of MDA5 to disease, we found that the AdarP195A/− and AdarP195A/p150− mice were severely runted at weaning compared to littermate controls, and that this runting was entirely MDA5-dependent (Figure 2E, 2F). We also noted that heterozygosity for Ifih1 delayed mortality, revealing a gene dosage-specific effect of MDA5 expression on disease (Figure S2C, S2D). Taken together, these data demonstrate that the AdarP195A mice recapitulate the human ADAR genotypes found in AGS and develop severe disease that is driven by MDA5. More broadly, they represent Adar-mutant mice that are born with intact MDA5 signaling, allowing the dissection of disease mechanisms in vivo.
Figure 2. Recapitulation of AGS patient genotypes causes severe disease in AdarP195A mice.

(A-B) Survival of Ifihi1+/+ mice of the indicated genotypes: Adarr+/+ (n=14), Adar+/− (n=21), AdarP195A/Adar+ (n=41), AdarP195A/Adar− (n=9); Adar p150+/+ (n=15), Adar p150+/− (n=6), AdarP195A/p150+ (n=34), AdarP195A/p150− (n=25).
(C-D) Survival of Ifihi1−/− mice of the indicated genotypes: Adar+/+ (n=18), Adar+/− (n=22), AdarP195A/Adar+ (n=14), AdarP195A/Adar- (n=19); Adar p150+/+ (n=17), Adar p150+/− (n=24), AdarP195A/p150+ (n=25), AdarP195A/p150- (n=28).
(E-F). Weights of mice of the indicated genotypes, measured at 23 days. Bars represent mean. Male and female mice are pooled in (E) because there was no significant difference by sex. Adar+/+ (n=2); Adar+/− (n=3); AdarP195A/Adar+ (n=4); AdarP195A/Adar− (n=5); Adar p150+/+ (n=5M, 6F); p150+/− (n=5M, 5F); AdarP195A/p150+ (n=11M, 10F); AdarP195A/p150− (n=6M, 4F); AdarP195A/p150+ (n=9M, 8F); AdarP195A/p150− (n=11M, 8F). See also Figure S2
To better understand the causes of runting and mortality in this model, we performed necropsies on sex-matched AdarP195A/p150− and AdarP195A/p150+ littermates to evaluate the pathologies associated with disease. We focused these and all additional analyses on the AdarP195A/p150− mice because they survived longer than AdarP195A/− mice (Figure 2A, 2B). In our initial assessment, the clearest histological defects were found in the kidney and liver. The kidney exhibited glomerular mesangial matrix expansion that increased with age, and the liver exhibited extensive microvesicular cytoplasmic vacuolation (Figure 3A–3C). In contrast to the Trex1−/− mouse model of AGS that is driven by the cGAS-STING DNA sensing pathway rather than by MDA5-MAVS RNA sensing (Gall et al., 2012; Gao et al., 2015; Gray et al., 2015; Stetson et al., 2008), we found no substantial inflammatory immune cell infiltrates in these tissues (Figure 3A–3C). We additionally identified abnormal architecture of the spleens in AdarP195A/p150− mice, characterized by lymphoid depletion (Figure 3C). We developed a histological scoring approach to quantitate these pathologies across several mice per genotype. AdarP195A/p150− mice had significantly higher pathological scores in all three organs, and these scores were normalized to control scores in AdarP195A/p150− Ifih1−/− mice (Figure 3D–3F). Additionally, AdarP195A/p150− mice exhibited significant deposition of IgG in kidney glomeruli (Figure 3G). As an independent measure of liver function, we found that serum alkaline phosphatase (ALP) was elevated, and serum albumin was reduced in AdarP195A/p150− mice compared to controls (Figure S2E). We next grew primary BMMs from AdarP195A/p150− mice on Ifih1+/+ and Ifih1−/− backgrounds. The AdarP195A/p150− BMMs did not spontaneously express elevated Ifnb mRNA, but treatment of these cells with recombinant IFNβ protein instigated robust, MDA5-dependent Ifnb transcription (Figure 3H), as has been previously observed in ADAR-null human cell lines (Ahmad et al., 2018). Together, these data demonstrate severe, MDA5-dependent pathologies and aberrant type I IFN expression in AdarP195A/p150− mice.
Figure 3. AdarP195A/p150− mice develop organ-specific pathology.

(A) Representative histology of kidney and liver from AdarP195A/p150− mice measured at 23 days of age.
(B) Representative histology of kidney and liver from AdarP195A/p150−Ifih1−/− mice measured at 23 days of age.
(C) Representative histology of kidney, liver, and spleen from AdarP195A/p150− mice measured at 147 days of age. Arrows indicate glomeruli in kidneys, asterisk indicates region of cytoplasmic vacuolation in liver with small arrow showing microvesicular vacuolation and large arrow showing macrovesicular vacuolation.
(D-F) Histological scores in the kidney, liver, and spleen, measured at 23 days of age. Adar p150+/+ (n=5); AdarP195A/p150+ (n=5); AdarP195A/p150− (n=6); AdarP195A/p150+Ifih1−/− (n=4); AdarP195A/p150Ifih1−/− (n=5).
(G) Representative (of 3 of each genotype) immunofluorescence of the kidney for AdarP195A/p150− and AdarP195/p150+ mice. DAPI is blue. IgG is red.
(H) Ifnb transcript measured by qRT-PCR in BMMs of the indicated genotypes, with and without 24 hours of treatment with recombinant mouse IFNβ. AdarP195A/p150+ (n=4); AdarP195A/p150− (n=4); AdarP195A/p150+Ifih1−/− (n=3); AdarP195A/p150−Ifihi1−/− (n=5).
We performed mRNA-Seq analyses comparing age-matched AdarP195A/p150− mice and AdarP195A/p150+ controls to quantitate global changes in gene expression. We analyzed liver and kidney because of the specific pathologies we uncovered in these tissues (Figure 3). We also analyzed cerebellum to compare changes in gene expression in the brains of these mice, given the localization of AGS pathology in the brain. Focusing on well-curated interferon-stimulated genes (ISGs), specifically genes in the GO term ‘response to type I interferon’, we carried out a gene set enrichment analysis and found significant up-regulation of this gene set in all three tissues (Figure 4A–4C). The extent of the ISG signatures varied among tissues, with cerebellum showing the most significant increases in ISG expression (adjusted p=0.009), followed by kidney (adjusted p=0.002) and then liver (adjusted p=0.007; Figure 4A–4C). We then performed quantitative RT-PCR of selected ISGs in all three of these tissues, comparing additional AdarP195A/p150− mice and AdarP195A/p150+ controls, on both Ifih1+/+ and Ifih1−/− backgrounds. Consistent with the complete rescue from mortality and pathology in AdarP195A/p150−Ifih1−/− mice (Figures 2 and 3), we found that the elevated ISG expression was also entirely MDA5-dependent (Figure 4D–4F). Thus, AdarP195A/p150− mice recapitulate the MDA5-dependent ISG signatures found in the blood and cells of AGS patients (Rice et al., 2013).
Figure 4. AdarP195A/p150− mice have an MDA5-dependent interferon signature.

(A-C) Expression data for ISGs, defined by the GO term ‘response to type I interferon,’ were evaluated in the liver, kidney, and cerebellum of 23 day old AdarP195A/p150− mice, plotting the log2 fold change over matched AdarP195A/p150+ control mice. ISGs that were not significantly changed are shown in bright red; significant expression changes are shown in dark red.
(D-F) Expression of ISGs identified in A-C, measured by TaqMan qPCR, in 23 day old mice of the indicated genotypes. AdarP195A/p150+ (n=6); AdarP195A/p150− (n=7); AdarP195A/p150+Ifihi1−/− (n=4); Adar P195A/p150− Ifih1−/− (n=6).
Genetic dissection of the AdarP195A/p150− phenotype in vivo reveals essential effectors of pathology
The development of postnatal, lethal, MDA5-dependent disease in AdarP195A/p150− mice allowed us to further characterize how aberrant MDA5 signaling results in pathology in vivo. To do this, we performed a series of crosses to test the contributions of additional signaling pathways. For each cross, we monitored survival, weights, and ISG signatures. We started these analyses by examining the contribution of the third RIG-I-like receptor, LGP2, which is encoded by the Dhx58 gene. The role of LGP2 in antiviral immunity is less well understood than that of MDA5 and RIG-I, in part because LGP2 lacks the Caspase Activation and Recruitment domains (CARDs) that are essential for RIG-I and MDA5 signaling through MAVS. Moreover, a role for LGP2 in any immune pathology has not been previously reported. Numerous studies have demonstrated that LGP2 interacts with MDA5 (Deddouche et al., 2014), modulates the formation of MDA5 filaments on dsRNAs (Bruns et al., 2014; Duic et al., 2020), and is important for antiviral responses to RNA viruses that specifically activate MDA5 (Satoh et al., 2010). We intercrossed AdarP195A/P195A mice with Adar p150+/− mice on a Dhx58−/− background, analyzing AdarP195A/p150− mice and comparing them to AdarP195A/p150+ littermate controls. We found that LGP2 deficiency completely rescued the AdarP195A/p150− mice from mortality, restored their weights to normal, and prevented the ISG signature in cerebellum, liver, and kidney (Figure 5A–5C). These findings place LGP2, together with MDA5, at the apex of the signaling pathway that links ADAR1 dysfunction to disease.
Figure 5. Identification of a genetic pathway linking the AdarP195A mutation to disease.

(A) Survival of Dhx58−/− mice of the indicated genotype: Adar p150+/+ (n=3), Adar p150+/− (n=8), AdarP195A/p150+ (n=5), AdarP195A/p150− (n=12).
(B) Weights, measured at 23 days, of AdarP195A/p150−Dhx58−/− mice (n=7), as a percentage of the average weight of age- and sex-matched AdarP195A/p150+Dhx58−/− (n=7) control mice.
(C) Expression of the indicated ISGs measured by TaqMan qRT-PCR, normalized to HPRT, in the cerebellum, liver, or kidney, comparing AdarP195A/p150−Dhx58−/− mice (n=8) to AdarP195A/p150+Dhx58−/− controls (n=6).
(D) Survival of Ifnar1−/− mice of the indicated genotype: Adar p150+/+ (n=3), Adar p150+/− (n=7), AdarP195A/p150+ (n=4), AdarP195A/p150− (n=16).
(E) Weights, measured at 23 days, of AdarP195A/p150−Ifnar1−/− (n=19) mice, as a percentage of the average weight of age- and sex-matched AdarP195A/p150+Ifnar1−/− (n=6) control mice.
(F) Expression of the indicated ISGs measured by TaqMan qRT-PCR, normalized to HPRT, in the cerebellum, liver, or kidney, comparing AdarP195A/p150−Ifnar1−/−(n=8) mice to AdarP195A/p150+Ifnar1−/− (n=5) controls.
(G) Survival of Eif2ak2−/− mice of the indicated genotype: AdarP195A/p150+ (n=4), or AdarP195A/p150− (n=7).
(H) Weights, measured at 23 days, of AdarP195A/p150−Eif2ak2−/− mice (n=6), as a percentage of the average weight of age- and sex-matched AdarP195A/p150+Eif2ak2−/− control (n=4)mice.
(I) Expression of the indicated ISGs measured by TaqMan qRT-PCR, normalized to HPRT, in the cerebellum, liver, or kidney, comparing AdarP195A/p150−Eif2ak2−/− mice(n=4) to AdarP195A/p150+Eif2ak2−/− controls (n=4). (See also figures S3 and S4)
Next, we tested the importance of the ISG signatures in AdarP195A/p150− mice by crossing them to Ifnar1−/− mice that lack the type I interferon receptor. We found that IFNAR1 deficiency also completely rescued all aspects of disease (Figure 5D–5F), similar to the rescue of Trex1−/− mice on an Ifnar1−/− background (Stetson et al., 2008).
We evaluated the in vivo contribution of the dsRNA-activated eIF2α kinase PKR (encoded by the Eif2ak2 gene) to disease in AdarP195A/p150− mice. PKR is activated by similar dsRNA structures as those that activate MDA5/LGP2, and the activation of PKR in ADAR1-deficient cells has been demonstrated in both mouse and human cell lines (Chung et al., 2018; George et al., 2016; Ishizuka et al., 2019; Li et al., 2010). Additionally, prior studies have identified a role for PKR as an essential cofactor for MDA5-dependent type I IFN responses (Pham et al., 2016; Schulz et al., 2010). However, a specific contribution of PKR to in vivo immunopathology has not been described. We found that AdarP195A/p150−Eif2ak2−/− mice were completely protected from mortality and weight loss (Figure 5G–5H). However, the ISG signature remained significantly elevated in cerebellum and liver from these mice (Figure 5I). These findings implicate PKR in disease caused by ADAR1 dysfunction in vivo, and they place PKR as an essential downstream effector, rather than an activator, of the MDA5/LGP2-dependent IFN response in this model.
We examined the contribution of the endonuclease RNase L to disease in the AdarP195A/p150− mouse model. RNase L is activated by the 2’−5’ oligoadenylate (2–5A) products of the dsRNA-activated OAS enzymes (Kristiansen et al., 2011; Zhou et al., 1993), which are IFN-inducible nucleotidyltransferases that resemble cGAS in overall structure and catalytic mechanism (Civril et al., 2013). Once activated by 2–5A, RNase L cleaves cellular and viral RNAs, which limits viral replication and restricts mRNA translation (Hornung et al., 2014). RNase L has been previously implicated as a key effector that mediates cell death downstream of ADAR disruption in human cell lines (Daou et al., 2020; Li et al., 2017). We found that RNase L deficiency had no impact on the mortality or weight loss in AdarP195A/p150− mice (Figure S3), suggesting that the contribution of RNase L to disease in this model might be more subtle or cell type specific.
Together, this genetic dissection of the Adar P195A/p150− mouse model reveals essential contributors to in vivo immunopathology and places them in a hierarchy that links MDA5- and LGP2-dependent IFN responses to PKR-dependent effector mechanisms that drive disease.
The ISR is responsible for pathology and mortality caused by Adar mutation
Based on the complete rescue of PKR-deficient AdarP195A/p150− mice, we explored the contribution of PKR-dependent effector mechanisms to disease in more detail. PKR is one of four metazoan eIF2α kinases that couple diverse perturbations in cellular homeostasis to a program called the Integrated Stress Response (ISR), which restricts new protein synthesis and results in the transcriptional induction of genes that can either restore homeostasis or cause cell death, depending on the strength and duration of the insult (Costa-Mattioli and Walter, 2020; Harding et al., 2003). The eIF2 GTPase is responsible for the delivery of the initiator methionyl tRNA to the ribosome to commence mRNA translation at the AUG start codon. GTP hydrolysis releases eIF2 from the ribosome-mRNA complex, after which the eIF2 must be recycled from its GDP-bound inactive form into its GTP-bound active form in order to initiate a new round of mRNA translation (Hinnebusch and Lorsch, 2012). Phosphorylation of the α subunit of eIF2 on serine 51 prevents this recycling of the eIF2 complex by the guanine nucleotide exchange factor (GEF) eIF2B, and it results in a reduction of most canonical mRNA translation initiation (Krishnamoorthy et al., 2001). However, certain mRNAs that contain unusual arrangements of AUG start codons in their 5’ untranslated regions become selectively translated after eIF2α phosphorylation (Sachs et al., 1997). These include the transcription factor ATF4, which induces the expression of ISR-activated genes (Harding et al., 2000).
The ISR gene expression program has been extensively defined in cell lines, and in vivo in mice that harbor hypomorphic mutations in Eif2b5, one of the genes that encodes the eIF2B complex (Wong et al., 2019). EIF2B gene mutations in humans cause a lethal leukoencephalopathy called Vanishing White Matter disease (VWM) that is driven by a chronic ISR (Leegwater et al., 2001; van der Knaap et al., 2002). Using a curated ISR gene expression signature defined in the Eif2b5-mutant mouse model of VWM (Wong et al., 2019), we examined our mRNA-Seq data and found significant up-regulation of the ISR gene set in AdarP195A/p150− mice (Figure 6A-C). The ISR gene set was significantly elevated in liver (adjusted p=0.05) and kidney (adjusted p=0.02) but was not significantly increased in cerebellum (adjusted p=0.99; Figure 6A–6C). Taken together with the ISG analyses (Figure 3), the liver and kidney exhibited elevation of both ISG and ISR gene sets, but the cerebellum exhibited only the ISG signature (Figure 6A–6C).
Figure 6. An ISR gene expression signature is evident in AdarP195A/p150− mice.

(A-C) Expression data for ISR gene set genes in the liver, kidney, and cerebellum of 23 day old AdarP195A/p150− mice (n=7), plotting the log2 fold change over control AdarP195A/p150+ (n=5) mice. ISR genes that are not significantly changed are shown in bright red; significant expression changes are shown in dark red.
(D-F) Expression of ISR transcripts identified in the livers of rescued mice, measured by TaqMan qRT-PCR, and compared among the indicated genotypes. Each data point represents an individual mouse. AdarP195A/p150+ (n=6); AdarP195A/p150− (n=7); AdarP195A/p150+Ifihi1−/− (n=4); AdarP195A/p150−Ifih1−/− (n=6); AdarP195A/p150+Dhx58−/− (n=7); AdarP195A/p150−Dhx58−/− (n=8); AdarP195A/p150+Ifnar1−/− (n=4); AdarP195A/p150− Ifnar1−/− (n=7); AdarP195A/p150+Eif2ak2−/− (n=4); AdarP195A/p150−Eif2ak2−/− mice (n=4).
We selected three of the most robustly induced ISR genes in the livers of AdarP195A/p150− mice (Asns, Cdkn1a, and Hmox1) and evaluated their expression by quantitative RT-PCR, comparing the affected mutant mice to all of the rescued genotypes that we identified in Figures 2 and 5. We found that induction of ISR gene expression required MDA5, LGP2, IFNAR1, and PKR (Figure 6D–6F), which precisely mirrored the rescue from mortality and pathology in all of these genotypes, even more specifically than the ISG signature that remained elevated in PKR-deficient AdarP195A/p150− mice (Figure 5I). Similar ISR transcripts were previously reported to be elevated in human cells targeted for ADAR1 (Gannon et al., 2018), but their direct connection of the ISR to PKR remained untested. To confirm that human cells exhibited the same PKR-dependent upregulation of the ISR that we identified in AdarP195A/p150− mice, we used lentiCRISPR to target the human ADAR and EIF2AK2 (PKR) genes in A549 lung carcinoma cells. We found that disruption of either the p150 isoform or both isoforms of human ADAR1 resulted in robust induction of ISR transcripts after treatment with recombinant human IFNβ, and that this upregulation was entirely dependent on PKR (Figure 7G).
Figure 7. Pharmacological Inhibition of the ISR rescues AdarP195A/p150− mice.

(A) Survival of mice on control chow: AdarP195A/p150+ (n=23), AdarP195A/p150− (n=10); versus survival of mice on 2BAct chow: AdarP195A/p150+ (n=17), AdarP195A/p150− (n=17).
(B) Weights of AdarP195A/p150− mice on control chow AdarP195A/p150+ (n=12), AdarP195A/p150− (n=9) or 2BAct chow AdarP195A/p150+ (n=16), AdarP195A/p150− (n=7), as a percentage of average weight of age- and sex-matched AdarP195A/p150+ mice on control chow.
(C) Representative histology of kidney, liver, and spleen of untreated, Eif2ak2−/−, and 2BAct-treated mice of the indicated genotypes.
(D) Histological scores of the kidneys, livers, and spleens of 2BAct-treated mice at day 23. AdarP195A/p150+ (n=7), AdarP195A/p150− (n=8). Arrows indicate glomeruli, asterisk indicates region of cytoplasmic vacuolation, and oval indicates periarteriolar lymphoid sheath (white pulp of spleen).
(E) ISG expression in the kidneys and livers of AdarP195A/p150+ (n=5) and AdarP195A/p150−(n=7) mice treated with 2BAct.
(F) ISR transcript expression in the liver of AdarP195A/p150+ (n=5) and AdarP195A/p150− (n=7) mice treated with 2BAct. Bars represents the mean in all graphs for B-F.
(G) Human A549 cells were transduced simultaneously with two lentiCRISPR constructs, each containing a distinct selectable marker. The first construct (puromycin-resistant) encoded gRNAs targeting just the p150 isoform of ADAR1, both isoforms of ADAR1, or an H1 nontargeting control. The second construct (blasticidin resistant) encoded either an H1 nontargeting control gRNA or a gRNA targeting the human EIFAK2 (PKR) gene. After selection in puromycin and blasticidin, the cells were treated with 1000U recombinant human IFNb for 72 hours, followed by measurement of the indicated genes by qRT-PCR. n=3 independent transduced populations for each group. Data are representative of 2 independent repeats.
A small molecule called Integrated Stress Response Inhibitor (ISRIB) stabilizes the eIF2B complex and activates its GEF function, rendering eIF2B resistant to the inhibitory effects of eIF2α phosphorylation (Sekine et al., 2015; Sidrauski et al., 2013; Sidrauski et al., 2015; Tsai et al., 2018; Zyryanova et al., 2018). More recently, a similarly potent and brain-penetrant analog of ISRIB with improved in vivo pharmacokinetics and pharmacodynamics called 2BAct was shown to prevent all aspects of VWM in a mouse model of Eif2b5 mutation, including brain pathology and induction of ISR genes (Wong et al., 2019). We tested whether the ISR inhibitor 2BAct could impact disease in the AdarP195A/p150− mouse model. To do this, we formulated 2BAct into mouse chow (Wong et al., 2019), and we placed breeders on 2BAct-containing or control chow two days after timed matings. We maintained the mice on this regimen through birth and nursing of pups, and then continued treatment of the mice following weaning. We tracked survival and disease phenotypes for 125 days after birth, which is three times the median survival of unmanipulated AdarP195A/p150− mice (Figure 2). We found that dietary introduction of 2BAct nearly completely rescued the AdarP195A/p150− mice from mortality, and it restored the mice to normal weights (Figure 7A-7B). Additionally, we found that the control chow for this experiment, which differed in nutrient and fat content from the typical diet used for all of our other mouse crosses, resulted in accelerated mortality of AdarP195A/p150− mice (Figure S6), further emphasizing the rescue of 2BAct-treated AdarP195A/p150− mice. 2BAct-treated AdarP195A/p150− mice were indistinguishable from controls when examined by tissue pathology in liver, kidney, and spleen, both at weaning and at the end of the 125-day treatment (Figure 7C-7D). However, the ISGs remained significantly elevated in 2BAct-treated AdarP195A/p150− mice (Figure 7E), but mRNA expression of the three ISR genes was restored to control levels (Figure 7F). We further verified in vitro that ISRIB treatment did not affect the IFN response to diverse innate immune stimuli in primary murine cells (Figure S4). However, ISRIB did blunt the ISR in ADAR-targeted human cells (Figure S5). Thus, therapeutic amelioration of the ISR is sufficient to prevent mortality and pathology in this Adar-mutant mouse model, revealing an essential IFN-dependent effector mechanism that contributes to disease in vivo.
Discussion
We introduce a mouse model of an AGS Adar mutation that develops postnatal, MDA5-dependent mortality. We use this model to delineate the genetic pathways responsible for pathology and we reveal a therapeutic approach that completely prevents disease, with implications for our understanding of disease mechanisms and targets for intervention in the human immune disorders caused by ADAR mutation.
Our studies of the AdarP195A/p150− mice offer insights into the links between ADAR1 dysfunction, MDA5, and disease manifestations. First, we show that the RLR LGP2 is essential for the MDA5-mediated antiviral response in this model, revealing a target for therapeutic intervention. Prior in vitro studies of the biochemical mechanisms of MDA5 filament formation on dsRNAs in the context of ADAR1 editing have focused exclusively on interactions between synthesized dsRNAs and purified recombinant MDA5 (Ahmad et al., 2018). Because LGP2 can modulate the size of MDA5 filaments and stabilize smaller MDA5-dsRNA complexes (Bruns et al., 2014; Duic et al., 2020), the size and composition of dsRNAs that are competent to trigger LGP2/MDA5 in vivo in the context of ADAR1 dysfunction might be distinct from those defined in vitro. Second, our findings reveal essential contributors to disease that were not appreciated in prior studies of the Adar-null alleles in mouse models. Specifically, neither IFNAR1 deficiency nor PKR deficiency rescued Adar-null mice to birth (Pestal et al, unpublished data; and Wang et al., 2004). However, we have now found that both IFNAR1 and PKR are essential for disease in the AdarP195A/p150− mouse model (Figure 5). This likely reflects the severity of the null alleles of Adar compared to the AdarP195A point mutation, together with the essential function of ADAR1 that is independent of its role in regulating MDA5 (Pestal et al., 2015). Unlike the Adar-null alleles, which eliminate both functions of ADAR1, the AdarP195A mouse model isolates the specific MDA5 regulatory roles of the p150 isoform of ADAR1.
Comparisons of AdarP195A/p150− mice to other mouse models of ADAR1 Z-alpha domain mutations will also increase our understanding of ADAR1 regulation. Two recently submitted manuscripts describe a mouse model of ADAR1 mutation in which two predicted RNA-binding residues (N175 and Y179) were simultaneously mutated to alanines (Tang et al 2020; de Reuver et al, 2020). A human N173S mutation that is homologous to mouse N175 has been found in AGS, but a corresponding Y179 mutation has not been observed in humans. Similar to our findings with homozygous AdarP195A/P195A mice (Figure 1), no overt pathology or survival defect was observed in these AdarmZα/mZα mice. They did, however, observe an upregulation of ISGs. The lack of pathology and mortality in AdarmZα/mZα mice emphasizes that chronic upregulation of IFN is insufficient to drive pathology on its own, and it demonstrates that the Z-alpha domain mutations of ADAR1 are only pathogenic when paired with a null allele of Adar, as we observed in our comparisons of AdarP195A/P195A and AdarP195A/p150− mice (Figure 1, 2). A third recently submitted manuscript describes a W197A model of ADAR1 z-alpha domain mutation, which has not been observed in humans (Nakahama et al, 2020). Unlike AdarmZα/mZα mice, Nakahama et. al. observed MDA5-dependent runting and mortality of homozygous AdarW197A/W197A mice. It will be interesting to determine whether these mice also have an upregulation of ISR that correlates with reduced survival.
By modeling the disposition of human ADAR AGS mutations in mice, we have established a genetic pathway that links ADAR1 dysfunction to disease. In this pathway, MDA5 and now LGP2 are the initiating sensors that are required to detect unedited self-RNAs in AdarP195A/p150− mice. Next, the MDA5- and LGP2-dependent type I interferon response drives all aspects of disease pathology. Finally, PKR is an essential IFN-dependent effector that mediates the disease manifestations and mortality. Whereas PKR is clearly important for antiviral defense and is targeted by virus-encoded antagonists (Elde et al., 2009), PKR has not previously been implicated in self RNA-mediated immune pathology in vivo.
We have found that the ISR inhibitor 2BAct prevents mortality and all aspects of disease pathology in AdarP195A/p150− mice. Moreover, we identify an ISR gene expression signature in affected mice that is more specifically correlated with genetic and therapeutic rescue than the ISG signature. Specifically, the ISGs remain significantly elevated in PKR-deficient AdarP195A/p150− mice and in 2BAct-treated AdarP195A/p150− mice, but the loss of the ISR signature correlates perfectly with all of the genetic and therapeutic rescues that we have defined. These findings have a number of important implications. First, we directly implicate the ISR as the cause of immune pathology. Second, we reveal a potential therapeutic path to treat human diseases caused by ADAR1 dysfunction that targets a specific effector mechanism while leaving other antiviral responses intact. Third, we identify a gene expression signature that could be harnessed to explore the contributions of the ADAR1-MDA5/LGP2-PKR axis to additional immune diseases in which IFNs and MDA5 have been implicated, including type I diabetes and systemic lupus erythematosus (Nejentsev et al., 2009; Robinson et al., 2011). More broadly, we speculate that the link between the ISR and immune pathology might also apply to conditions in which the ISR is activated by distinct mechanisms, including the endoplasmic reticulum stress sensor PERK, the heme sensor HRI, and the nutrient sensor GCN2 (Costa-Mattioli and Walter, 2020). All of these kinases share eIF2α as their principal target, and all induce the ISR. In other words, because the ISR contributes to immune pathology downstream of PKR in this model of ADAR1 mutation, it might also drive a similar pathology downstream of diverse stresses that are not typically thought of as immune in origin. Indeed, ISR gene expression signatures have been identified in tissue samples from human patients with multiple sclerosis (MS; Way and Popko, 2016). Current models of MS envision the ISR as a cytoprotective mechanism that slows disease progression by preventing the loss of myelin-producing oligodendrocytes (Way and Popko, 2016). However, our findings raise the alternative possibility that the ISR might contribute to disease pathology. We propose that further study of protective versus pathogenic contributions of the ISR will illuminate new ways to classify and treat human immune diseases.
In summary, we have developed and characterized a mouse model of ADAR1 dysfunction that recapitulates human AGS genotypes, we have revealed contributors to disease, and we have provided proof-of-concept for a therapeutic approach targeting the ISR as an essential contributor to immune pathology in vivo.
Limitations of the Study
One important difference between human AGS patients with ADARP193A mutations and the AdarP195A/p150− mouse model is that AGS is characterized by severe neurological changes and psychomotor dysfunction, but we did not observe any overt neurological defects in the mice. We speculate that the distinct tissues and organs that display pathology in the mouse model versus the human disease reflect differences in the identity, abundance, and cell type specificity of the endogenous RNA substrates of ADAR1 that become potent MDA5 ligands when ADAR1-mediated RNA editing is compromised.
METHODS
RESOURCE AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Daniel Stetson (stetson@uw.edu).
Materials availability
The Adar P195A mouse line and plasmids generated in this study will be made available to research labs, accompanied by a standard material transfer agreement for non-commercial use.
Data and code availability
The mRNA-Seq datasets generated during this study have been deposited at GEO and are publicly available as of the date publication. Accession number is listed in the key resources table
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-β Actin | SIGMA | cat # A5441 |
| ADAR1 Antibody (15.8.6) | Santa Cruz Biotechnology | cat # sc-73408 |
| Bacterial and Virus Strains | ||
| Encephalomyocarditis virus (EMCV)-K | Gorman et al, Nat Immunol. 2017 Jul; 18(7): 744–752 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2BAct mouse chow | Wong et al., eLife 2019; 8:e42940 | N/A |
| Envigo Teklad Global 14% Protein Rodent Maintenance Diet | Envigo | Cat # 2914 |
| BioServ Rodent Reproductive Diet | Bio Serv | Product# S3823P |
| Recombinant mouse Interferon beta protein | R&D Systems | Cat # 12405-1 |
| Recombinant human interferon beta protein | R&D Systems | Cat # 11415-1 |
| ISRIB | SIGMA | Cat # SML0843 |
| Critical Commercial Assays | ||
| Taq Man Gene Expression Assays | Thermo Fisher | Cat # 4351372 |
| Deposited Data | ||
| RNA Seq raw data of P195A/p150+ and P195A/p150− liver, kidney, and cerebellum | GEO | GSE162583 |
| Experimental Models: Cell Lines | ||
| C57BL/6 primary MEFs, (mouse embryonic fibroblasts) | Gray EE et al, J Immunol 2015 195:1939 | N/A |
| HEK 293T cells | ATCC | CRL-3216 |
| A549 cells | ATCC | CCL-185 |
| mouse bone marrow macrophages, C57BL/6J | Gray EE et al, J Immunol 2015 195:1939 | N/A |
| Experimental Models: Organisms/Strains | ||
| mouse: Adar P195A: C57BL/6J | this paper | N/A |
| mouse: Eif2ak2−/−: C57BL/6J-Eif2ak2tm1Jcbe | Nakamura et al. Cell Rep. 2015 Apr 14; 11(2): 295–307. | N/A |
| mouse: Adar p150+/−: C57BL/6J | Pestal et al. Immunity 2015 Nov 17;43(5):933–44. | N/A |
| mouse: Adar+/−: C57BL/6J | Pestal et al. Immunity 2015 Nov 17;43(5):933–44. | N/A |
| mouse: Dhx58−/−: C57BL/6J-Dhx58tm1Itl | Suthar et al. Immunity 2012 Aug 24; 37(2): 235–248. | N/A |
| mouse: Ifih1−/−: C57BL/6J-Ifih1tm1.1Cln | Gitlin et al. Proc Natl Acad Sci 2006 May 30;103(22):8459–64. | N/A |
| mouse: RnaseI−/−: C57BL/6J-RnaseItm1Slvm | Zhou et al. EMBO. 1997 Nov 3;16(21):6355–63. | N/A |
| mouse: Ifnar1−/−: C57BL/6J-Ifnar1tm1Agt | Kolumam et al. J Exp Med. 2005 Sep 5;202(5):637–50. | N/A |
| Oligonucleotides | ||
| See Table S1 for qPCR primer sequences | See table S1 | N/A |
| See Table S2 for guide RNA sequences | See table S2 | N/A |
| Recombinant DNA | ||
| pRRL lentiCRISPR vectors. Guide RNA sequences in Table S2 | Gray et al. Immunity. 2016 Aug 16;45(2):255–66 | N/A |
| Software and Algorithms | ||
| Prism v 9.0 | GraphPad | N/A |
| Adobe Photoshop Elements 2020 | Adobe | N/A |
| Galaxy FASTQ Trimmer tool v1.0.0 | N/A | N/A |
| FastqMcf (v1.1.2) | N/A | N/A |
| STAR aligner (v2.4.2a) | N/A | N/A |
| HTSeq-count (v0.4.1) | N/A | N/A |
| PICARD (v1.134) | N/A | N/A |
| FASTQC (v0.11.3) | N/A | N/A |
| Samtools (v1.2) | N/A | N/A |
| HTSeq-count (v0.4.1) | N/A | N/A |
| DESeq2 | Love et al., Genome Biol 2014 15,550 | N/A |
| fgsea | Koretkevich et al., 2019 | https://github.com/ctlab/fgsea/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
AdarP195A mutant mice were generated using previously described techniques (Henao-Mejia et al., 2016), using the sgRNA target sequence GAAGGGGGAAACCTCCTTTGTGG, and the oligo DNA to replace the sequence, ATCAATCGTATTTTGTACTCCCTGGAAAAGAAGGGAAAGCTGCACAGAGGAAGGGGGAAACCTGCCTTGTGGAGCCTTGTGCCCTTGAGTCAGGCTTGGACTCAGCCCCCTGGAGTTGTGAATCCAGAT. In brief, C57BL6/J oocytes were microinjected with Cas9 complexed with gRNA for WT Adar sequence and ssDNA donor template containing the desired sequence. Oocytes were implanted into a pseudopregnant CD-1 females. Founder pups born were screened by the Surveyor assay. Mice positive by the Surveyor assay were genotyped using TaqMan SNP genotyping, primers: WT: AAACCTCCT/ZEN/TTGTGGAGCCTT, Mut: AAACCTGCCTTGTGGAGCCTT. Mice positive by SNP genotyping were then sequenced to confirm fidelity of the introduced mutation. Mice were bred to C57BL6/J mice to confirm germline transmission, and AdarP195A/+ mice were then crossed to one another or to other genetically modified mice.
Adarfl/fl mice (Hartner et al., 2009) were generously provided by Stuart Orkin, and were bred to Cre-expressing mice to generate the Adar-null allele as described previously (Pestal et al., 2015). Adar p150+/− gametes (Ward et al., 2011) were generously provided by Michael Oldstone (Pestal et al., 2015). Ifih1−/− mice (Gitlin et al., 2006) were generously provided by Michael Gale, Jr. Dhx58−/− mice (Suthar et al., 2012) were generously provided by Michael Gale, Jr. Ifnar1−/− mice (Muller et al., 1994) were backcrossed 14 generations to C57BL/6J (Kolumam et al., 2005). Eif2ak2−/− mice (Abraham et al., 1999), backcrossed at least 10 generations to C57BL/6J, were generously provided by Gӧkhan Hotamışlıgil. Rnasel−/− mice (Zhou et al., 1997) were generously provided by Robert Silverman. Both male and female mice were used for all experiments; we observed no difference in survival between male and femal mutant mice. All mice were maintained in a specific pathogen-free (SPF) barrier facility at the University of Washington, and all experiments were done in accordance with the Institutional Animal Care and Use Committee guidelines of the University of Washington. Mice were weighed at 23 days of age. Numbers of each sex are noted and represented separately except where indicated because no difference by sex was observed. Mice were fed the institutional breeder cage diet (BioServ, Figures 1–6), or Envigo 14% protein rodent chow with or without 2BAct (Figure 7). Details of the chow formulations are listed in Figure S6.
Primary bone marrow macrophages (BMM) and mouse embryonic fibroblasts (MEFs)
Bone marrow of male and female 6–12 week old mice (unless otherwise noted), was harvested, and plated in complete RPMI containing MCSF. Mouse embryos between d E12–14 were harvested, and genotyped by PCR. All cells were maintained in 5% CO2 conditions.
HEK 293T clonal lines
ADAR knock out and non-targeted 293T cells were previously published (Pestal et al, 2015). In brief: 293T cells (female) were transduced with Cas9-sgRNA lentivirus targeting ADAR1. After three days of selection, cells were plated at limiting dilution to produce single cell clones. Clones that grew out were screened by Western blot for expression of p150 and p110 isoforms of ADAR1, and targeting verified by sequencing.
A549 cells
Human A549 cells (male) were confirmed by STR profiling and screened for Mycoplasma by PCR. lentiCRISPR targeting for ADAR1 or nontargeting control as above was verified by sequencing and TIDE analysis (Brinkman et al., 2014).
METHOD DETAILS
Histology and Pathology
For all histological analyses, sex-matched littermate mice were euthanized in accordance with IACUC protocols by CO2 asphyxiation followed by cardiac puncture. Mice were washed with PBS and then fixed in 10% neutral-buffered formalin. Tissues were routinely processed, embedded in paraffin, cut into approximately 4 μm sections and hematoxylin and eosin stained. Slides of kidney, liver, and spleen were also stained with periodic acid-Schiff. Additionally, select kidney sections were stained with Congo Red and Masson’s trichrome.
Tissues were evaluated and scored by a board-certified veterinary pathologist (J.M.S), who was blinded to genotype and experimental manipulation for all groups except for the initial group of mice subjected to phenotyping of multiple organs including decalcified cross section of skull, lungs, heart, kidney, liver, spleen, pancreas, lymph nodes, reproductive tract, stomach, and intestines. These juvenile mice evaluated initially in an unblinded fashion were re-scored blindly prior to manuscript preparation. For subsequent mice, histological analysis was focused on the kidney, liver, and spleen.
For the kidney, expansion of the glomerular mesangial matrix was scored from 0–4, with 0 = normal; 1 = minimal; 2 = mild; 3 = moderate (with accompanying tubular protein); and 4 = severe histological changes. An extent score was also given for the kidney, with 1 representing <10%; 2 = 10–32%; 3 = 33–65%; and 4 representing > 66% of glomeruli affected. For the liver, microvesicular and lesser macrovesicular cytoplasmic vacuolation were scored from 0–5, with 0 = normal; 1 = minimal changes affecting only a small region (< 5%) of the liver; 2 = mild changes throughout the liver but without enlargement of hepatocytes, coalescing lesions, or necrosis; 3 = mild to moderate cytoplasmic vacuolation throughout liver with enlargement of hepatocytes but no necrosis or loss of parenchyma; 4 = moderate, coalescing throughout liver with multifocal mild regions of loss of parenchyma or necrosis; and 5 = severe with moderate multifocal regions of cavitation and necrosis. Liver inflammation was also scored on a 0–5 scale with 0 = fewer than 2 small microgranulomas per section; 1 = minimal scattered inflammation or microgranulomas; 2 = mild (less than 5% of parenchyma involved); 3 = mild to moderate (11–30%); 4 = moderate (31–60%); and 5 = severe (affecting >60% of parenchyma). Lymphoid depletion of the spleen was scored on a scale of 0–3 with 0 = none; 1 = mild; 2 = moderate; 3 = severe.
Representative images were taken using NIS-Elements BR 3.2 64-bit and plated in Adobe Photoshop Elements. Image white balance, lighting, brightness and contrast were adjusted using auto corrections applied to the entire image. Final magnification is stated.
For measurement of alkaline phosphatase (ALP) and albumin in serum, blood was collected by cardiac puncture from mice at 23 days of age and stored in SST Tubes at 4C until analysis. Samples were run at 2x dilution on a Siemens Atellica 1600 (Siemens Healthineers, Germany).
Immunofluorescence microscopy
Spleens were frozen in Optimal Cutting Temperature (OCT) media (Sakura). 5 μm sections of fresh frozen tissue were fixed in 100% acetone at −20°C for 10 min and then stained with anti-mouse IgG Alexa Fluor 488 (Invitrogen) and DAPI. Stained slides were mounted with Prolong Gold antifade reagent (Life Technologies), imaged with a Nikon Eclipse 90i microscope, and analyzed with Adobe Photoshop software. Three mice of each genotype were evaluated.
mRNASeq and Analysis
Total RNA was added directly to lysis buffer from the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara), and reverse transcription was performed followed by PCR amplification to generate full-length amplified cDNA. Sequencing libraries were constructed using the NexteraXT DNA sample preparation kit (Illumina) to generate Illumina-compatible barcoded libraries. Libraries were pooled and quantified using a Qubit® Fluorometer (Life Technologies). Dual-index, single-read sequencing of pooled libraries was carried out on a HiSeq2500 sequencer (Illumina) with 58-base reads, using HiSeq v4 Cluster and SBS kits (Illumina) with a target depth of 5 million reads per sample.
Reads were processed using workflows managed on the Galaxy platform. Reads were trimmed by 1 base at the 3’ end, and then trimmed from both ends until base calls had a minimum quality score of at least 30 (Galaxy FASTQ Trimmer tool v1.0.0). FastqMcf (v1.1.2) was used to remove any remaining adapter sequence. To align the trimmed reads, we used the STAR aligner (v2.4.2a) with the GRCm38 reference genome and gene annotations from ensembl release 91. Gene counts were generated using HTSeq-count (v0.4.1). Quality metrics were compiled from PICARD (v1.134), FASTQC (v0.11.3), Samtools (v1.2), and HTSeq-count (v0.4.1). Libraries with less than 2 million mapped reads per sample were not included in the analysis, and analysis was restricted to genes with non-zero counts in all libraries (n=8,843 genes). We carried out normalization and tested for differential expression using DESeq2 (Love et al., 2014). Gene set enrichment analysis was carried out using fgsea (Koretkevich et al., 2019), ranking genes by the sorted p values from the differential expression test for each tissue. Gene sets tested included all GO categories, Kegg pathway gene sets, Hallmark gene sets, and Reactome gene sets. We also included the manually curated ISR target gene set as defined previously (Wong et al., 2019). In total, 7084 gene sets were evaluated, and multiple testing correction was done using the Benjamini-Hochberg method.
Quantitative RT-PCR
Mice were euthanized by CO2 asphyxiation and cardiac puncture. Organs were immediately immersed in Trizol and frozen at −80C until later processing. To extract RNA, organs were mashed on ice, then resuspended in Trizol by passage through 25G needles. Samples were spun down briefly (5 min at 5000 g), and the supernatant processed by the Direct-zol RNA MiniPrep kit (Genesee Scientific) per the manufacturer’s instructions with an additional dry spin after disposing of the final wash to prevent EtOH carryover. Complementary DNA (cDNA) was generated using EcoDry double primed premix (Clontech). Transcript expression was measured using the ThermoFisher TaqMan Gene expression assays listed in Table S1.
In vitro treatments
IFN:
Bone marrow was harvested from age- and sex-matched mice and grown/differentiated in RPMI supplemented with 10% fetal calf serum, L-glutamine, penicillin/streptomycin, sodium pyruvate, HEPES, and M-CSF for 7 days. 105 BMM were plated in 12 well plates and rested overnight, then stimulated with 100U/mL recombinant murine IFNβ (Sigma, I9032) or equivalent volume of water for 24 hours. Cells were then harvested in Trizol before RNA purification via Directzol kits, and cDNA generation using Ecodry kits as described above. qPCR was performed using iTaq supermix on the Bio-Rad CFX96 Real-Time System, and primers with sequences listed in Table S1.
Ligands and infections:
MEFs were plated in 1 mL of complete media at 105 cells/12 well and BMM plated at 106/12 well and rested overnight. Cells received 4 μg of CT DNA complexed with 4 μl of Lipofectamine 2000, or 1 μg of RIG-I ligand or Poly I:C complexed with 1 μl of Lipofectamine. Cells were infected with EMCV at an MOI of 1. Cells received 0.5 uM Thapsigargin (Invitrogen) in DMSO. Cells treated with ISRIB (SIGMA) received 5uM ISRIB in DMSO at the same time as the indicated stimulation.
Western Blots:
105 MEF were plated in a 6 well plate and rested overnight, then stimulated with 100U recombinant murine IFNβ (Sigma, I9032) or equivalent volume of water for 24 hours. The NE-PERS kit instructions (Thermo Fisher) were followed completely, separated on acrylamide gels, transferred to membranes for western blot (00.45 μM pore size), and blotted with the indicated primary and secondary antibodies using standard approaches. Western blot images were acquired using a BioRad Chemidoc and associated software.
lentiCRISPR targeting
For CRISPR-Cas9 gene targeting, we generated pRRL lentiviral vectors in which a U6 promoter drives expression of a gRNA and a synthetic MND promoter drives expression of Cas9, a T2A peptide, and either a puromycin or blasticidin resistance cassette. gRNA sequences are listed in Table S2. VSV-G pseudotyped, self-inactivating lentivirus was prepared by transfecting a 60 to 80% confluent 10-cm plate of HEK 293T cells with 1.5 μg of pVSV-G expression vector, 3 μg of pMDLg/pRRE, 3 μg of pRSV-Rev, and 6 μg of pRRL lentiCRISPR vectors using poly(ethyleneimine) (Sigma). Medium was replaced 24 hours after transfection and harvested 24 hours later for filtration with a 0.45-μm filter (Steriflip, Millipore). Approximately 2×105 A549 cells in 6-well dishes were transduced simultaneously with 1.5 mL of lentiviral supernatant for each target. Media on the transfected 293T producer cells was replaced and A549 transduction repeated 24 hours later. Selection with 5 μg/ml puromycin and 10μg/ml blasticidin was begun the day after the last transduction of A549s. Cells were selected for 5 full days, at which point kill controls exhibited complete cell death as assessed by eye. Recombinant human IFNβ was added on the 6th day of selection. Cells were harvested 72 hours later.
2BAct treatment
AdarP195A/P195A mice were intercrossed with Adar p150+/− mice. 48 hours after mating, the breeders were switched to 2BAct chow, formulated to achieve a 2Bact concentration of 300 ppm (300 μg 2BAct/g of meal) as previously described (Wong et al., 2019). 2BAct chow was contract manufactured with Envigo. The placebo diet was Teklad 2014 without added 2BAct compound. Breeders and pups were maintained on 2BAct until termination of survival experiments at 125 days. Male and female sex matched littermates were weighed at 23 days of age. Littermate mice of both sexes were harvested for histological and mRNA expression analysis as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Quantitative data were visualized and analyzed using GraphPad Prism software. Differences in survival were assessed by the Log-rank (Mantel-Cox) Test. Observed and expected birth rates by genotypes were compared using the Chi-square test. Weights and mRNA expression measured by qPCR were compared between genotypes within each cross using unpaired t-tests. Significance is indicated as follows in all figures: ns=not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Number of mice in each group is indicated in figure legends for main figures, and in the figure for Supplemental figures. Replicate number for each in vitro experiment is in the figure legend.
Supplementary Material
Table S2: lentiCRISPR guide RNA target sequences, related to Figure 7.
Highlights:
AdarP195A/p150− mice model ADAR1 mutations found in human Aicardi-Goutières Syndrome
Disease in AdarP195A/p150− mice requires MDA5, LGP2, the type I IFN receptor, and PKR
AdarP195A/p150− mice develop PKR-dependent activation of Integrated Stress Response (ISR)
Pharmacological inhibition of the ISR in vivo prevents all aspects of disease
Acknowledgements
We thank Stuart Orkin, Michael Oldstone, Michael Gale, Jr, Gӧkhan Hotamışlıgil, and Robert Silverman for providing mice used in this study. We are grateful to the University of Washington Histology and Imaging Core and B. Johnson for histology preparation, and to Vivian Gersuk and the Benaroya Research Institute Genomics Core for mRNA-Seq. We are grateful to Leonel Joannas for expertise in the generation of the Adar P195A mice, to Sarah Miller for expert mouse colony management, and to all members of the Stetson lab for insightful discussions. This work was supported by NIH R01 AI084914 (D.B.S.); and T32 AI106677 and NIH F31 AI40432 (M.M.) D.B.S. is a Faculty Scholar of the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
C.S. is an employee of Calico Life Sciences and is listed as an inventor on a patent application WO2017193063 describing 2BAct. D.B.S. is a co-founder and shareholder of Danger Bio, LLC, and a scientific advisor for Related Sciences, LLC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, and Hur S (2018). Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 172, 797–810 e713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns AM, Leser GP, Lamb RA, and Horvath CM (2014). The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol Cell 55, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, and Rice CM (2018). Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, and Hopfner KP (2013). Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167A, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowl JT, Gray EE, Pestal K, Volkman HE, and Stetson DB (2017). Intracellular Nucleic Acid Detection in Autoimmunity. Annu Rev Immunol 35, 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daou S, Talukdar M, Tang J, Dong B, Banerjee S, Li Y, Duffy NM, Ogunjimi AA, Gaughan C, Jha BK, et al. (2020). A phenolic small molecule inhibitor of RNase L prevents cell death from ADAR1 deficiency. Proc Natl Acad Sci U S A 117, 24802–24812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deddouche S, Goubau D, Rehwinkel J, Chakravarty P, Begum S, Maillard PV, Borg A, Matthews N, Feng Q, van Kuppeveld FJ, and Reis e Sousa C (2014). Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. Elife 3, e01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duic I, Tadakuma H, Harada Y, Yamaue R, Deguchi K, Suzuki Y, Yoshimura SH, Kato H, Takeyasu K, and Fujita T (2020). Viral RNA recognition by LGP2 and MDA5, and activation of signaling through step-by-step conformational changes. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Child SJ, Geballe AP, and Malik HS (2009). Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457, 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr., Barber GN, and Stetson DB (2012). Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity 36, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, et al. (2018). Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat Commun 9, 5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, and Chen ZJ (2015). Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112, E5699–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Ramaswami G, Li JB, and Samuel CE (2016). Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J Biol Chem 291, 6158–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, and Reis ESC (2013). Cytosolic sensing of viruses. Immunity 38, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, and Stetson DB (2015). Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol 195, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, and Seeburg PH (2004). Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279, 4894–4902. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, and Orkin SH (2009). ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, and Suzuki T (2013). Dyschromatosis symmetrica hereditaria. J Dermatol 40, 336–343. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, and Lorsch JR (2012). The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Hartmann R, Ablasser A, and Hopfner KP (2014). OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol 14, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, and Hinnebusch AG (2001). Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol 21, 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, and Hartmann R (2011). The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J Interferon Cytokine Res 31, 41–47. [DOI] [PubMed] [Google Scholar]
- Leegwater PA, Vermeulen G, Konst AA, Naidu S, Mulders J, Visser A, Kersbergen P, Mobach D, Fonds D, van Berkel CG, et al. (2001). Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet 29, 383–388. [DOI] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, and Weiss SR (2017). Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wolff KC, and Samuel CE (2010). RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 396, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, et al. (2014). A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet 51, 76–82. [DOI] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, and Todd JA (2009). Rare Variants of IFIH1, a Gene Implicated in Antiviral Responses, Protect Against Type 1 Diabetes. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg S, Paz Yaacov N, Safran M, Moshkovitz S, Shtrichman R, Sherf O, Jacob-Hirsch J, Keshet G, Amariglio N, Itskovitz-Eldor J, and Rechavi G (2010). Alu sequences in undifferentiated human embryonic stem cells display high levels of A-to-I RNA editing. PLoS One 5, e11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JB, and Samuel CE (1995). Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15, 5376–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, and Stetson DB (2015). Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marie IJ, and Levy DE (2016). PKR Transduces MDA5-Dependent Signals for Type I IFN Induction. PLoS Pathog 12, e1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. (2013). Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol 12, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. (2012). Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, Mikolaitis RA, Guerrero G, Utset TO, Drevlow BE, et al. (2011). Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. J Immunol 187, 1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB, Sarnow P, and Hentze MW (1997). Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell 89, 831–838. [DOI] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, and Takeuchi O (2010). LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A 107, 1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, Takeuchi O, Akira S, Kaufman RJ, and Reis e Sousa C (2010). Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, and Ron D (2015). Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, McGeachy AM, Ingolia NT, and Walter P (2015). The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, and Maniatis T (2007). Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 315, 1274–1278. [DOI] [PubMed] [Google Scholar]
- Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, Frost A, and Walter P (2018). Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Leegwater PA, Konst AA, Visser A, Naidu S, Oudejans CB, Schutgens RB, and Pronk JC (2002). Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol 51, 264–270. [DOI] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, and Nishikura K (2004). Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279, 4952–4961. [DOI] [PubMed] [Google Scholar]
- Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, and Oldstone MB (2011). RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci U S A 108, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, and Popko B (2016). Harnessing the integrated stress response for the treatment of multiple sclerosis. Lancet Neurol 15, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, LeBon L, Basso AM, Kohlhaas KL, Nikkel AL, Robb HM, Donnelly-Roberts DL, Prakash J, Swensen AM, Rubinstein ND, et al. (2019). eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Hassel BA, and Silverman RH (1993). Expression cloning of 2–5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72, 753–765. [DOI] [PubMed] [Google Scholar]
- Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, Harding HP, Allen F, Parts L, Fromont C, et al. (2018). Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 359, 1533–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: lentiCRISPR guide RNA target sequences, related to Figure 7.
Data Availability Statement
The mRNA-Seq datasets generated during this study have been deposited at GEO and are publicly available as of the date publication. Accession number is listed in the key resources table
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-β Actin | SIGMA | cat # A5441 |
| ADAR1 Antibody (15.8.6) | Santa Cruz Biotechnology | cat # sc-73408 |
| Bacterial and Virus Strains | ||
| Encephalomyocarditis virus (EMCV)-K | Gorman et al, Nat Immunol. 2017 Jul; 18(7): 744–752 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2BAct mouse chow | Wong et al., eLife 2019; 8:e42940 | N/A |
| Envigo Teklad Global 14% Protein Rodent Maintenance Diet | Envigo | Cat # 2914 |
| BioServ Rodent Reproductive Diet | Bio Serv | Product# S3823P |
| Recombinant mouse Interferon beta protein | R&D Systems | Cat # 12405-1 |
| Recombinant human interferon beta protein | R&D Systems | Cat # 11415-1 |
| ISRIB | SIGMA | Cat # SML0843 |
| Critical Commercial Assays | ||
| Taq Man Gene Expression Assays | Thermo Fisher | Cat # 4351372 |
| Deposited Data | ||
| RNA Seq raw data of P195A/p150+ and P195A/p150− liver, kidney, and cerebellum | GEO | GSE162583 |
| Experimental Models: Cell Lines | ||
| C57BL/6 primary MEFs, (mouse embryonic fibroblasts) | Gray EE et al, J Immunol 2015 195:1939 | N/A |
| HEK 293T cells | ATCC | CRL-3216 |
| A549 cells | ATCC | CCL-185 |
| mouse bone marrow macrophages, C57BL/6J | Gray EE et al, J Immunol 2015 195:1939 | N/A |
| Experimental Models: Organisms/Strains | ||
| mouse: Adar P195A: C57BL/6J | this paper | N/A |
| mouse: Eif2ak2−/−: C57BL/6J-Eif2ak2tm1Jcbe | Nakamura et al. Cell Rep. 2015 Apr 14; 11(2): 295–307. | N/A |
| mouse: Adar p150+/−: C57BL/6J | Pestal et al. Immunity 2015 Nov 17;43(5):933–44. | N/A |
| mouse: Adar+/−: C57BL/6J | Pestal et al. Immunity 2015 Nov 17;43(5):933–44. | N/A |
| mouse: Dhx58−/−: C57BL/6J-Dhx58tm1Itl | Suthar et al. Immunity 2012 Aug 24; 37(2): 235–248. | N/A |
| mouse: Ifih1−/−: C57BL/6J-Ifih1tm1.1Cln | Gitlin et al. Proc Natl Acad Sci 2006 May 30;103(22):8459–64. | N/A |
| mouse: RnaseI−/−: C57BL/6J-RnaseItm1Slvm | Zhou et al. EMBO. 1997 Nov 3;16(21):6355–63. | N/A |
| mouse: Ifnar1−/−: C57BL/6J-Ifnar1tm1Agt | Kolumam et al. J Exp Med. 2005 Sep 5;202(5):637–50. | N/A |
| Oligonucleotides | ||
| See Table S1 for qPCR primer sequences | See table S1 | N/A |
| See Table S2 for guide RNA sequences | See table S2 | N/A |
| Recombinant DNA | ||
| pRRL lentiCRISPR vectors. Guide RNA sequences in Table S2 | Gray et al. Immunity. 2016 Aug 16;45(2):255–66 | N/A |
| Software and Algorithms | ||
| Prism v 9.0 | GraphPad | N/A |
| Adobe Photoshop Elements 2020 | Adobe | N/A |
| Galaxy FASTQ Trimmer tool v1.0.0 | N/A | N/A |
| FastqMcf (v1.1.2) | N/A | N/A |
| STAR aligner (v2.4.2a) | N/A | N/A |
| HTSeq-count (v0.4.1) | N/A | N/A |
| PICARD (v1.134) | N/A | N/A |
| FASTQC (v0.11.3) | N/A | N/A |
| Samtools (v1.2) | N/A | N/A |
| HTSeq-count (v0.4.1) | N/A | N/A |
| DESeq2 | Love et al., Genome Biol 2014 15,550 | N/A |
| fgsea | Koretkevich et al., 2019 | https://github.com/ctlab/fgsea/ |