

Abstract
Confucius said, “Good tools are prerequisite to the successful execution of a job”. Among his many groundbreaking achievements, Dr. Leland W. K. Chung established several widely used prostate cancer (PCa) cell lines, including C4-2, C4-2B, and ARCaP. These cellular models have been pivotal tools to enhance our understanding of the biology of PCa progression and assist in the discovery of new strategies to treat metastatic, castration-resistant PCa. Recent studies in the ARCaP PCa progression model uncovered epithelial protein lost in neoplasm (EPLIN), an actin-binding protein with an indispensable role in the maintenance of epithelial structures, as a negative regulator of epithelial-mesenchymal transition. Clinical evidence further supports the potential role of EPLIN in controlling metastasis in PCa and other solid tumors. In this article, we review the current understanding of the biology of EPLIN and the ARCaP model in the discovery of new agents for the prevention and treatment of PCa metastasis.
Keywords: EPLIN, EMT, PCa, metastasis, chemoresistance
Introduction
This article is a part of a special issue of the AJCEU dedicated to Dr. Leland W. K. Chung. The selection of the ARCaP epithelial-mesenchymal transition (EMT) model for this article is particularly poignant because the story of its discovery was one of Dr. Chung’s favorites, and in our opinion, a prime example of his brilliant and unconventional thinking. The original ARCaP cells were actually an unexpected by-product from an effort to collect soluble factors in ascites fluid. “When we passed by the sink and saw several centrifuge tubes containing the precipitates, a thought hit us”, recalled Dr. Chung, with a satisfactory smile. “Why not give it a try”? Thus was the first step to isolating the unique ARCaP cells. Dr. Chung’s team further established ARCaP variants with distinct metastatic capabilities and tissue preference. Among them, ARCaPE and ARCaPM have been the most widely used for their representative EMT and neuroendocrine characteristics. To this day, the treasure box of ARCaP cells as a new tool in prostate cancer research remains an area for exploration.
Prostate cancer metastasis and EMT
At the cellular and molecular levels, prostate cancer (PCa) metastasis can be driven by a combination of hereditary and somatic gene alterations in a variety of signaling pathways [1-3]. EMT is a major mechanism by which epithelial cells acquire plasticity and stemness [4-11]. Complete conversion to a mesenchymal phenotype can lead to enhanced migratory and invasive capacities and allow cancer cells to migrate away from their original epithelial layer. Therefore, EMT may play a critical role in the initial stages of metastasis [12,13]. Understanding the biological alterations during EMT could provide insight into the mechanisms underlying PCa metastasis and therapeutic resistance.
ARCaP cells provide a relevant preclinical model for the study of EMT and PCa metastasis
A major limitation in the field of PCa research is that, despite numerous attempts to establish immortalized cell lines from human tumor tissues, relevant models that can closely recapitulate the diverse molecular subtypes of PCa, particularly metastatic and therapeutic-resistant diseases, are still lacking [14-16]. Among the currently available human PCa cell lines, DU145 and PC-3 have been widely used in the study of EMT and metastasis. However, both cell lines express only low levels of epithelial markers, such as E-cadherin, and do not exhibit a morphology typical of epithelial-like cells [17,18]. In addition, although the two cell lines can form bone metastases following intracardiac injection, they mainly cause osteolytic lesions in mouse skeletons [19].
The ARCaP (androgen-repressed prostate cancer) cell line was established by Drs. Haiyen E. Zhau and Leland W. K. Chung’s group from the ascites fluid of a patient with metastatic PCa [20,21]. ARCaP cells are tumorigenic with 100% incidence in intact or castrated athymic male mice. When the cells were implanted orthotopically, metastases were observed in multiple organs, including lymph nodes, lung, kidney, pancreas, liver, and bones. Unlike DU145 or PC-3, ARCaP cells elicit both osteoblastic and osteolytic responses in mouse bones, mimicking a distinct pathological feature of PCa metastasis [22]. Drs. Zhau and Chung’s team further established multiple ARCaP sublines from metastatic tissues following the intracardiac injection of parental ARCaP cells in athymic nude mice. These subclones exhibit similar cytogenetic patterns but distinct morphology; they also differ in their growth rates, migratory, invasive, and metastatic potentials, and drug sensitivity. Similar to the establishment of C4-2 and C4-2B lines, the isolation and identification of multiple ARCaP sublines from metastatic tissues highlighted an important role of tumor-microenvironment interaction in the acquisition of tissue tropism in metastatic PCa cells. Two sublines, i.e., ARCaPE and ARCaPM, display the most striking differences. ARCaPE cells display typical epithelial-like morphology and have a relatively low metastatic propensity (12.5%) after intracardiac injection. The lineage-derived ARCaPM cells recovered from mouse bone have spindle-shaped fibroblastic morphology associated with increased expression of vimentin and reduced expression of epithelial markers. Importantly, the switch in morphology and gene expression in ARCaPM cells is associated with increased metastatic propensity to skeletal (100%) and soft tissues [23]. ARCaPE/ARCaPM cells provide an excellent model that can undergo spontaneous and predictable bone metastasis in vivo and exhibit the molecular and behavioral characteristics of EMT in vitro. Since the first publication in 2006 describing the two cell lines, at least 40 published studies used ARCaPE/ARCaPM cells and identified several novel factors involved in the control of EMT, cellular plasticity, and metastasis [8,24-31]. In this review, we discuss epithelial protein lost in neoplasm (EPLIN) and its potential function in EMT, tumor progression and therapeutic resistance.
Biological function of EPLIN
EPLIN was initially identified as a cytoskeletal protein in epithelial cells [32]. The human EPLIN gene is located on chromosome 12q13.12. Two EPLIN isoforms, the 600-amino-acid EPLIN-α and 759-amino-acid EPLIN-β, are generated from alternative RNA processing: the β isoform consists of all 11 exons, and the EPLIN-α mRNA consists of exons 4-11 [33]. Both EPLIN isoforms contain a single, centrally located LIM domain, allowing the protein to dimerize with itself or associate with other proteins. At least two actin-binding domains are present at both the amino- and carboxyl-termini, which allow EPLIN to effectively cross-link and bundle actin filaments and lower the monomer dissociation rate constant at the pointed end of actin filaments and stabilize them from depolymerization. Additionally, EPLIN-α can inhibit Arp2/3 complex-mediated actin nucleation by increasing the lag at the outset of polymerization. Therefore, the inhibitory effect of EPLIN on reorganization of the actin cytoskeleton can be two-fold, i.e., affecting both assembly (nucleation) and disassembly (stability) of filamentous actin [34,35].
EPLIN plays an indispensable role in the maintenance of epithelial phenotypes, directly binding α-catenin and connecting the E-Cadherin/β-Catenin-α-Catenin complex and the actin filaments in the epithelium. These physical connections are critical to stabilization of the circumferential actin belt at adherens junctions (AJs) and regulation of apical-basal polarity in epithelial cells, including mechanosensitivity [36,37]. In vascular endothelium, EPLIN exerts a similar function by directly interacting with α-catenin anchored to the endothelial-specific VE-cadherin-β-catenin complex and promoting vinculin junctional recruitment, thereby reinforcing the cohesion of cell-cell junctions and stabilizing the capillary structures [38-40]. Interestingly, it appears that EPLIN-β is highly expressed in aortic and shear stress-loaded endothelial cells to induce and stabilize stress fibers, whereas EPLIN-α is increased in growing and migrating cells to control protrusion dynamics of actin-driven membrane protrusions, specifically classical lamellipodia (cLP) and junction-associated intermittent lamellipodia (JAIL). The distinct functions of EPLIN isoforms in the fine-tuning of actin dynamics are consistent with a lower EPLIN-β turnover rate compared to EPLIN-α [41].
EPLIN depletion induces cytokinesis failure and formation of multinucleation and aneuploidy [42], suggesting that EPLIN may be involved in the regulation of cytokinesis and genomic stability. During membrane ingression and cleavage furrow formation, EPLIN interacts with at least two cytoskeletal systems, i.e., the actin-myosin II contractile ring and septin filaments, for a complete cytokinesis. EPLIN is required for the recruitment of RhoA, Cdc42, and ACAT-related protein required for viability 1 (Arv1) to the cleavage furrow. Although both EPLIN-α and -β localize to the cleavage furrow during cytokinesis, EPLIN-β may have a stronger interaction with Arv1 via the amino terminus [43].
Zhang et al. recently reported an unexpected role of EPLIN in lipid regulation. A causal single-nucleotide variant of the EPLIN gene was identified in a remote Chinese family with inherited low low-density lipoprotein cholesterol (LDL-C) and reduced cholesterol absorption. In a mouse model, EPLIN protein is highly expressed in the brush border of the small intestine and binds two proteins essential for cholesterol absorption, i.e., NPC1-like intracellular cholesterol transporter 1 (NPC1L1) and myosin Vb. EPLIN knockout mice displayed reduced cholesterol absorption and low LDL-C levels [44,45].
The evolving role of EPLIN in cancer progression
EPLIN was initially identified as a protein that is highly expressed in normal epithelial cells but its expression is frequently reduced in human cancer cells and primary tumors. Ectopic overexpression of EPLIN-α or -β suppresses the in vitro and in vivo growth of certain cancer cell lines, including MDA-MB-231 and PC-3 [32,46-48]. These observations suggested that EPLIN may serve as a tumor suppressor and play a role in the early stages of tumor progression, such as tumorigenesis and primary tumor growth. The underlying mechanism of EPLIN as a tumor suppressor remains largely unknow. Recent work from the Fujita group showed that EPLIN may be directly involved in a cancer preventive process, i.e., the apical elimination of transformed cells from the epithelial layer. In RasV12-transformed cells, EPLIN is upregulated and interacts with caveolin-1, plectin and paxillin, thereby promoting microtubule acetylation and facilitating the apical extrusion of RasV12 cells [49,50].
To gain unbiased insight into the molecular mechanisms underlying PCa EMT, we performed quantitative proteomic analyses to profile protein expression patterns in the ARCaP EMT model. Among 76 unique proteins differentially expressed in ARCaPE and ARCaPM cells, EPLIN-β was found to be markedly reduced in ARCaPM cells. Consistently, immunohistochemical expression of EPLIN was high in ARCaPE tumors but significantly reduced in ARCaPM tumors. These data suggested a potential connection between EPLIN, EMT and invasive phenotypes in PCa cells. A similar correlation between EPLIN expression and invasiveness was observed in other experimental models of PCa and squamous cell carcinoma of the head and neck (SCCHN) [11].
To determine the function of EPLIN in PCa EMT and invasiveness, we established ARCaPE cells with temporary or permanent knockdown of EPLIN, which demonstrated spindle-shaped and mesenchymal-like morphology. EPLIN depletion caused the disassembly of cellular stress fibers with a concomitant gain of actin foci and formation of prominent membrane ruffles. These data indicated that the loss of EPLIN expression may allow PCa cells to undergo dynamic remodeling of the actin cytoskeleton and to acquire migratory and invasive capabilities. EPLIN knockdown led to similar phenotypic changes in other cancer lines, such as LNCaP, PC-3, and MCF-7. Taken together, these results provided the first observation that EPLIN may be a negative regulator of EMT and invasiveness in human cancer cells [11]. This hypothesis was further supported by several studies from other groups [48,51-56].
At the molecular level, EPLIN regulates a subset of genes involved in EMT, stemness, actin cytoskeleton remodeling, invasion and metastasis, adhesion and extracellular matrix remodeling, and growth factor signaling. EPLIN depletion in ARCaPE cells led to a remarkable increase in the subpopulation carrying the CD44high/CD24negative marker [11]. In contrast to previous observations that EPLIN depletion usually resulted in increased proliferation in certain human cancer cell lines [32,46-48], EPLIN shRNA-expressing ARCaPE cells (ARCaPE-shEPLIN) have slower in vitro proliferation and in vivo tumor growth than the control cells (ARCaPE-shCtrl) ([11] and unpublished results). Another unexpected consequence of EPLIN depletion is that ARCaP-shEPLIN cells display significantly higher resistance to chemotherapeutics, including docetaxel and doxorubicin (by ~8-fold and ~4.4-fold, respectively) [11]. These results indicated that in addition to acting as a tumor suppressor, EPLIN may also play an important role in the acquisition of stemness and therapeutic resistance.
Expression of EPLIN in human cancers
In the first study of EPLIN expression in human cancer specimens, the Jiang group found that compared with normal tissues, EPLIN-α expression is reduced in low-grade breast cancer, and is further decreased in high-grade tumors. EPLIN-α downregulation is associated with poor prognosis and increased incidence of recurrence and mortality [46]. Subsequent studies observed a similar expression pattern of EPLIN-α in other cancer types [48,54]. Reduced EPLIN-α expression is associated with local advanced esophageal cancer as well as lymphatic metastasis [48]. EPLIN transcripts are decreased in colon, lung and breast cancers compared with corresponding normal tissues [53]. In gastric cancer, EPLIN expression was significantly lower in the T3 + T4 stage group than in the combined T1 + T2 stage group and could be an independent prognostic factor of disease-free survival and overall survival. EPLIN expression is also significantly associated with responsiveness to neoadjuvant chemotherapy [57]. In a cohort of oral tongue squamous cell carcinoma (OTSCC) patients, high expression of EPLIN protein was significantly associated with longer survival [58]. Low EPLIN expression in melanoma was correlated with increased invasion and metastases [51].
We explored the clinical significance of EPLIN in human cancer progression. In a published study, analyses of four independent sets of microarray data on clinical PCa revealed that EPLIN transcripts were expressed at a similar level in primary tumors and normal prostatic tissues, but were remarkably reduced in metastatic tumors [11]. We further analyzed the expression of EPLIN transcripts using PCa Transcriptome Atlas (PCTA), a database comprised of 1,321 clinical specimens from 38 PCa cohorts [59]. EPLIN expression in primary tumors was significantly reduced when compared with that of benign prostatic glands (fold change = -0.142; P < 0.001), and further reduced in metastatic, castration-resistant PCa (fold change = -0.357; P < 0.001) (Figure 1).
Figure 1.

EPLIN transcript profile in benign prostatic glands, primary PCa and metastatic, castration-resistant PCa.
Immunohistochemistry (IHC) expression of EPLIN was evaluated in matched pairs of tumor tissue specimens from primary tumors and lymph node metastases from PCa, breast cancer, colorectal cancer and SCCHN. EPLIN expression was markedly reduced in lymph node metastases compared to their matched primary tumors [11]. In another study, EPLIN expression was evaluated in a prostate tissue microarray (TMA) composed of 69 primary PCa cases with variable Gleason scores (grade groups) and 10 cases composed of benign prostatic glands and stroma (Figure 2A). EPLIN protein was highly expressed in all the examined benign prostate tissues (60.0% 3+, 40.0% 2+). The overall positive rate of EPLIN was 94.2% in cancer tissues (43.5% 3+, 23.2% 2+, 27.5% 1+). The IHC score for EPLIN expression in high-grade tumors (Gleason scores 8-10/grade groups 4-5; 1.82 ± 0.15) was significantly lower that than in benign prostatic glands (2.6 ± 0.16; P = 0.02) or low-grade tumors (Gleason score ≤ 7/grade groups 1-2; 2.43 ± 0.19; P = 0.01). There was no statistically significant difference between EPLIN expression in benign prostatic glands and low-grade PCa (P = 0.59) (Figure 2B). Collectively, EPLIN expression at both the RNA and protein levels is downregulated in PCa, and its reduction is further associated with high-grade and metastatic, castration-resistant PCa. These results suggested that EPLIN downregulation may serve as a predictor of tumor aggressiveness and poor prognosis in PCa patients.
Figure 2.
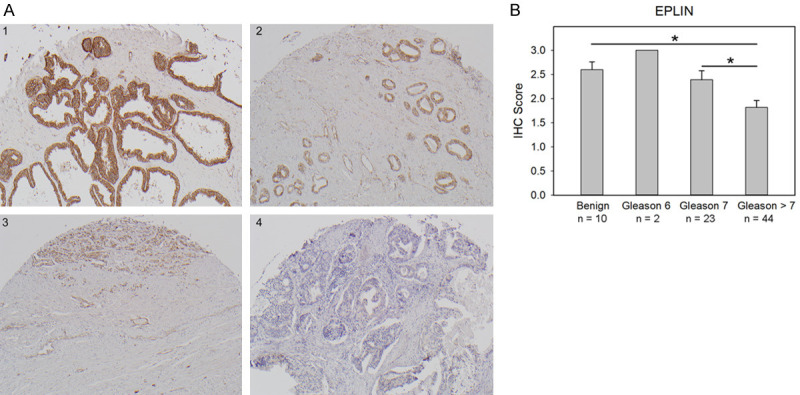
IHC expression of EPLIN in benign prostatic glands and PCa. A. Representative IHC staining of EPLIN in human prostate TMA: (1) benign tissue; (2) Gleason score 6 (3 + 3) tumor; (3) Gleason score 7 (4 + 3) tumor; (4) Gleason score 9 (4 + 5) tumor. B. IHC intensity of EPLIN in prostate TMA with different Gleason scores (*P < 0.05). Refer to Supplementary Information.
EPLIN signaling in human cancer cells
The underlying mechanisms by which EPLIN regulates the proliferation, migration, and invasion in cancer cells remain largely unknown. In melanoma cells, EPLIN downregulation leads to activation of insulin-like growth factor 1 receptor (IGF-1R)-Akt/signal transducer and activator of transcription 3 (Stat3) signaling, followed by an increase in Slug and consecutive downregulation of E-cadherin [51]. EPLIN is capable of regulating the expression and phosphorylation of paxillin, FAK and Src in PCa cells, thereby affecting their aggressive characteristics [56].
Our recent studies have uncovered a novel function of EPLIN in the regulation of chemoresistance. EPLIN depletion in ARCaPE cells activates noncanonical EZH2 signaling, consisting of Stat3, S-phase protein kinase 2 (SKP2), ATP-binding cassette B1 (ABCB1, p-glycoprotein), and survivin, that allows PCa cells to evade chemotherapeutics. C4-2B-TaxR cells, a highly docetaxel-resistant PCa mimicking acquired chemoresistance [60], appear to activate this common pathway for their survival. However, how EPLIN regulates EZH2 signaling is not clear.
Regulation of EPLIN expression
Several regulatory mechanisms of EPLIN transcription have been identified. A fragment within the first intron ranging from position -43,729 to -43,233 upstream of the EPLIN-α transcriptional start site may be responsible for its induction by wild-type p73 isoforms. p73-dependent EPLIN transcription can be completely inhibited by co-expression of transactivation-deficient N-terminally truncated isoforms of p73 (DNp73) [51]. Two consensus p53-binding motif sequences have been identified in the 4th intron and downstream region of the EPLIN gene, respectively. Consistently, the EPLIN mRNA level in breast and colorectal cancers containing mutant p53 is significantly reduced than that in cancers containing wild-type p53 [53]. The 3’-untranslated region (UTR) of EPLIN mRNA has the targeting sequence of the seed region of an oncogenic miRNA, miR-93-5p; EPLIN has been experimentally validated as a target of miR-93-5p and mediates the oncogenic effect of miR-93-5p in endothelial cells [61]. The human EPLIN-α promoter contains a serum response factor (SRF) binding site, and the EPLIN-β promoter has putative binding sites for Oct-1, Sp1, and AP1. Consistently, serum stimulation activates SRF and induced the transcription of EPLIN-α but not EPLIN-β [33].
We reported that epidermal growth factor (EGF) promotes robust EMT in ARCaPE cells by activating extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent phosphorylation, ubiquitination, and degradation of EPLIN protein. Two serine residues (serines 362 and 604) were further identified as the ERK1/2 phosphorylation sites [62]. A later study found that hCDC14A, a key regulator of cell migration and adhesion, counteracts EGF-induced rearrangements of actin cytoskeleton by dephosphorylating EPLIN at serines 362 and 604, thereby maintaining the expression of E-cadherin and α/β-catenin at cell-cell adhesions. hCDC14A-mediated EPLIN dephosphorylation could reduce actin dynamics and restrict tumor malignancy [63].
We predicted a putative PEST sequence (RASSLSESSPPK) within the EPLIN protein that may be involved in its post-translational regulation [62]. Supporting this model, a chimeric fusion apoptosis inhibitor 2 (API2)-mucosa-associated lymphoid tissue translocation gene 1 (MALT1) protein cleaves EPLIN-α at the major cleavage site arginine 206 and at the minor cleavage site lysine 289, thereby disrupting the tumor suppressor function of EPLIN-α in B cells. Intriguingly, API2-MALT1 paracaspase-mediated EPLIN-α proteolysis produces a LIM domain-only (LMO)-containing fragment with oncogenic properties in lymphomagenesis [64].
EPLIN was identified as a Rab40b-Cullin5-specific substrate in breast cancer cells. EPLIN binding with GTP-bound Rab40b-CRL5 via the Rab40b SOCS box leads to EPLIN ubiquitylation, disassociation of EPLIN of Rab40b and EPLIN complex, and eventual degradation by the proteasome. Rab40b-Cullin5-mediated EPLIN degradation decreases its subcellular localization at the leading edge of lamellipodia, allowing EPLIN accumulation at stress fibers and invadopodia [65].
EPLIN and the ARCaP model in the discovery of new agents for the prevention and treatment of PCa metastasis
Epidemiological studies support that soy-rich diets could play an important role in the modulation of PCa incidence and mortality [66-68]. A meta-analysis of five cohort studies and eight case-control studies demonstrated an inverse relationship between PCa mortality and dietary consumption of genistein (4’,5,7-trihydroxyisoflavone), a major isoflavone in soy products [69]. In ARCaPE and ARCaPM cells, genistein effectively inhibited their in vitro invasion through Boyden chambers and enhanced the adhesion on fibronectin-coated plates (Figure 3A). Intriguingly, genistein markedly activated EPLIN-β transcription in a dose-dependent manner in both PC-3 and ARCaPM cells (Figure 3B, 3C), as confirmed at the protein level (Figure 3D). Consistently, administration of genistein via the intraperitoneal route significantly increased both EPLIN and E-cadherin at the tissue level in C4-2 tumors (Figure 3E). These data revealed EPLIN as a novel molecular target of genistein that may contribute to the inhibitory effect of genistein on PCa metastasis in preclinical models [67,70-75].
Figure 3.
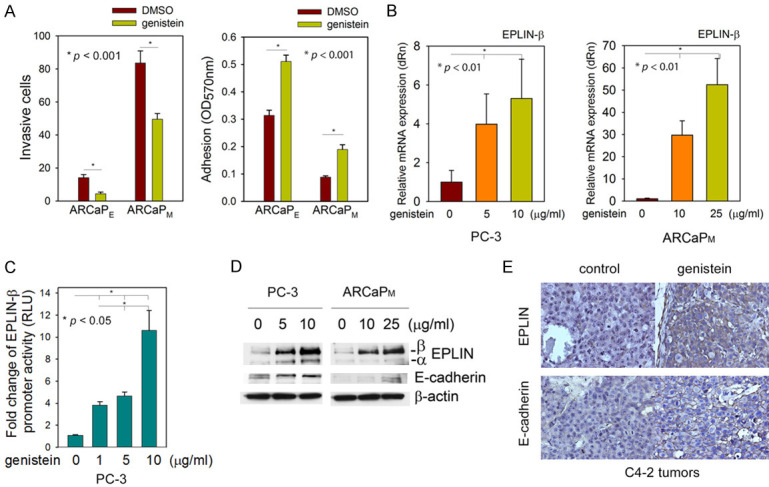
EPLIN is a molecular target of genistein in metastatic PCa cells. A. Genistein inhibits the in vitro invasion in transwell experiments (18 h) and increases the in vitro adhesion on fibronectin-coated plates (6 h) in ARCaPE and ARCaPM cells. B. Genistein treatment (48 h) induces EPLIN-β mRNA in PC-3 and ARCaPM cells, as determined by qRT-PCR. C. Genistein activates the human EPLIN-β promoter in a dose-dependent manner (48 h). D. Genistein dose-dependently induces EPLIN protein expression (72 h). E. Genistein (50 mg/kg, intraperitoneal injection, 4 times per week, 4-week treatment) increases EPLIN and E-cadherin in C4-2 subcutaneous tumors (n = 6 per group). Refer to Supplementary Information.
Docetaxel resistance is a major obstacle in the treatment of metastatic, castration-resistant PCa [76-81]. To discover novel drug candidates for targeting chemoresistant PCa, which is highly heterogeneous, we developed a sequential phenotypic screening platform that consists of two independent cellular models of chemoresistant PCa, i.e., ARCaPE-shEPLIN and C4-2B-TaxR [82]. We proposed that ARCaPE-shEPLIN represents a subpopulation of cancer cells that gain stemness and exhibit intrinsic chemoresistance via EMT [11], and that C4-2B-TaxR cells (developed by Dr. Allen C. Gao’s group) mimic the progression of acquired docetaxel resistance [60]. Small-molecule inhibitors of chemoresistant cancer were analyzed through this screening platform based on their capability of selectively inducing cell death only in ARCaPE-shEPLIN and C4-2B-TaxR cells but not in their parental counterparts.
LG1980, an aminobisphosphonate-conjugated compound, was identified as a specific and effective inhibitor of chemoresistant PCa cells. In a recent report [82], we described the in vitro and in vivo activities of LG1980 against chemoresistant PCa. Mechanistic studies uncovered LG1980 as a novel inhibitor of embryonic ectoderm development (EED) that interrupts EED-EZH2 interactions and induces degradation of key PRC2 components, including EZH2, EED and SUZ12, subsequently inhibiting noncanonical EZH2 signaling. As a lead compound, LG1980 is well-tolerated in mice and has an excellent safety pharmacology profile. In several xenograft models, LG1980 effectively suppresses the in vivo growth of chemoresistant PCa and synergistically enhances the efficacy of docetaxel chemotherapy.
Using LG1980 as a positive control, we recently conducted a larger-sized screening of potential lead compounds against chemoresistant PCa cells. At least nine FDA-approved, non-oncology drugs were found to have high selectivity and potency in ARCaPE-shEPLIN and C4-2B-TaxR cells. Most of these candidates effectively inhibited the in vivo growth of chemoresistant PCa xenografts and demonstrated excellent safety profiles in animals (data not shown). These drugs are well positioned to be repurposed as novel treatments for metastatic and chemoresistant PCa.
Concluding remarks
Since the discovery of EPLIN as a cytoskeletal protein about two decades ago, there has been greatly increased interest in the biology and clinical implications of EPLIN. EPLIN is not only well recognized as an indispensable component of cytoskeleton dynamics and cell-cell adhesions in various cell types, but also as a potential signaling molecule that plays important roles in human physiology and pathology, including cancer. The role of EPLIN in cancer progression has expanded from that of a tumor suppressor, which may mainly function at early disease stages, to a potential suppressor of metastasis, acting at later stages. Currently available clinical data, although still limited, points to a common pattern that low EPLIN expression is associated with advanced disease, worse response to chemotherapy and poor prognosis in multiple cancer types. While most mechanistic research has focused on the well-acknowledged role of EPLIN in the remodeling of the cytoskeleton and cell-cell adhesion, a few recent studies, including ours, demonstrated that EPLIN may affect multiple signaling pathways implicated in EMT, stemness, and metastasis. A key remaining question is how and when EPLIN exerts these diverse functions in different phases of tumor progression. There is an expanding list of EPLIN-interacting proteins that have been discovered during the past decade [52], which may provide important clues to the underlying mechanisms by which EPLIN controls tumorigenesis and progression towards metastasis and therapeutic resistance.
For translational science, understanding the regulatory mechanism of EPLIN expression could identify novel agents (such as genistein) to upregulate EPLIN in cancer cells and prevent or delay the metastatic process. Our recent work demonstrated the use of the ARCaP EMT model in the discovery of a novel function of EPLIN in cancer progression and new therapeutic agents that selectively and potently target chemoresistant PCa. Elucidation of the mechanisms of action of these drug candidates obtained from the ARCaP phenotypic screening platform could reveal a cancer-relevant EPLIN signaling network and identify new “druggable” therapeutic targets for the treatment of metastatic PCa.
Tribute to Leland W. K. Chung, PhD
Daqing Wu, PhD: Dr. Leland W. K. Chung was my dear mentor and friend. Leland was an outstanding educator who thoughtfully nurtured the minds of his students, fully supporting the growth and development of their careers as future scientists. He taught us how to see the hidden meanings and connections in seemingly random and messy results. Whether arriving in his office with “good” data or “bad” data, we came out with big smiles, as his trust in our potential and talents was unwavering, and he always encouraged us to strive for the highest standards possible. Leland’s open-mindedness and ability to synthesize information and exude positivity was greatly inspiring.
Throughout his career, Leland chose to be affiliated with clinical departments, and the reason was simple: “As a cancer researcher, you need to follow real-world problems and find ways to help patients who are dying every day”. His quest to find a cure for cancer was fueled by his dedication, his innovative methods, and his ability to build research teams and collaborate with others. Leland was a great leader and a true visionary. The breadth of his personal and professional networks demonstrated how scientists, engineers, and clinicians must together with industry work closely to address not only cancer but broader societal issues as well. As a cancer survivor himself, Leland positioned himself as an entrepreneur at the end of his career and put enormous energy and effort into his start-up company, DaZen (“Great Kindness”) Theranostics. When we last spoke, he clearly articulated his vision for DaZen’s future and his dream of finding a cure for cancer, with his characteristic enthusiasm and optimism. Yes, this was Leland.
On the wall in Leland’s office was a framed picture of the space shuttle Columbia. Leland and his wife Haiyen witnessed Columbia blasted off from the Kennedy Space Center on January 16, 2003 carrying an experiment designed by Leland and his team. The experiment was to test an innovative idea about how prostate cancer cells, as an “organoid”, behave in a three-dimensional setting under zero gravity. However, on February 1, 2003, when making re-entry, the shuttle was lost, with all seven dedicated astronauts whom Leland got to know from their close collaboration. There was a deep sadness and admiration when he talked about these people and their brave adventures into the unknown. Today, we share the same feelings when we reflect on Leland and his legacy. Like the Columbia crew, Dr. Leland W. K. Chung was here to show what a person with curiosity, talent, a passion for the common good, an infectious smile, and a warm heart could bring to this world. We miss this gentleman, and we are grateful to him for enriching our lives.
Adeboye O. Osunkoya, MD: Leland Chung, PhD will always have a very special place in my memory for the inspiration he gave me during the early phase of my academic career, prior to his departure from Emory. He was one of those that I met during the interview process for my first and current Faculty position here at Emory back in 2006. Apart from the obvious great potential collaborations we would have, his very warm personality and constant smile definitely put me at ease during the interview, and I was convinced that we would have a great collaborative experience together at Emory. Within the first couple of weeks of my arrival at Emory as a newly minted Faculty in July 2007, Leland had already reached out to me to set up a research meeting with his wife and research partner Haiyen E. Zhau, PhD to discuss further about some of their research projects and potential areas of collaboration. The rest as they say is history. I know so many others had a similar experience with him, because he was constantly surrounded by mentees. I will definitely miss him. Rest in peace my great friend/collaborator and “gentle giant of urology research”.
Acknowledgements
The authors are grateful to the editors for the opportunity to contribute to this special issue of the AJCEU dedicated to Dr. Leland W. K. Chung. We sincerely appreciate Dr. Haiyen E. Zhau for her review and advice. We thank Drs. Shumin Zhang, Haian Fu and Yuhong Du at Emory University School of Medicine for the genistein and high-throughput screening studies, Dr. Allen C. Gao at University of California Davis for providing C4-2B-TaxR cells, and Winship Cancer Institute Cancer Tissue and Pathology core for immunohistochemistry. We also thank Drs. Jer-Tsong Hsieh (University of Texas Southwestern Medical Center), Shian-Ying Sung (Taipei Medical University), and Shareen Iqbal (The Centers for Disease Control and Prevention) for sharing their collections of Dr. Chung’s pictures (Supplementary Figure 1). Dr. Rhea-Beth Markowitz at Georgia Cancer Center and Alyssa Y. Wu at Emory College of Arts and Sciences provided editorial assistance. This work was supported by National Cancer Institute grants R21CA164612-01A1 and R41CA217491-01A1, Georgia Research Alliance VentureLab grant, Emory University Research Committee Award, and Winship Cancer Institute Prostate Cancer Research Pilot Grant (D. Wu).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.De La Taille A, Vacherot F, Salomon L, Druel C, Gil Diez De Medina S, Abbou C, Buttyan R, Chopin D. Hormone-refractory prostate cancer: a multi-step and multi-event process. Prostate Cancer Prostatic Dis. 2001;4:204–212. doi: 10.1038/sj.pcan.4500534. [DOI] [PubMed] [Google Scholar]
- 2.Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, Smith BA, Cheng C, Tsai BL, Cheng D, Huang J, Kurdistani SK, Graeber TG, Witte ON. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science. 2018;362:91–95. doi: 10.1126/science.aat5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, Long HW, Xu B, Brown M, Loda M, Sawyers CL, Ellis L, Goodrich DW. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 6.Thiery JP, Huang R. Linking epithelial-mesenchymal transition to the well-known polarity protein Par6. Dev Cell. 2005;8:456–458. doi: 10.1016/j.devcel.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Zhau HE, Odero-Marah V, Lue HW, Nomura T, Wang R, Chu G, Liu ZR, Zhou BP, Huang WC, Chung LW. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis. 2008;25:601–610. doi: 10.1007/s10585-008-9183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 10.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Wang X, Osunkoya AO, Iqbal S, Wang Y, Chen Z, Muller S, Chen Z, Josson S, Coleman IM, Nelson PS, Wang YA, Wang R, Shin DM, Marshall FF, Kucuk O, Chung LW, Zhau HE, Wu D. EPLIN downregulation promotes epithelial-mesenchymal transition in prostate cancer cells and correlates with clinical lymph node metastasis. Oncogene. 2011;30:4941–4952. doi: 10.1038/onc.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728:23–34. doi: 10.1016/j.mrrev.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27:2192–2206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pienta KJ, Abate-Shen C, Agus DB, Attar RM, Chung LW, Greenberg NM, Hahn WC, Isaacs JT, Navone NM, Peehl DM, Simons JW, Solit DB, Soule HR, VanDyke TA, Weber MJ, Wu L, Vessella RL. The current state of preclinical prostate cancer animal models. Prostate. 2008;68:629–639. doi: 10.1002/pros.20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hensley PJ, Kyprianou N. Modeling prostate cancer in mice: limitations and opportunities. J Androl. 2012;33:133–144. doi: 10.2164/jandrol.111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saranyutanon S, Deshmukh SK, Dasgupta S, Pai S, Singh S, Singh AP. Cellular and molecular progression of prostate cancer: models for basic and preclinical research. Cancers (Basel) 2020;12:2651. doi: 10.3390/cancers12092651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontana F, Raimondi M, Marzagalli M, Sommariva M, Limonta P, Gagliano N. Epithelial-to-mesenchymal transition markers and CD44 isoforms are differently expressed in 2D and 3D cell cultures of prostate cancer cells. Cells. 2019;8:143. doi: 10.3390/cells8020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burton LJ, Hawsawi O, Loyd Q, Henderson V, Howard S, Harlemon M, Ragin C, Roberts R, Bowen N, Gacii A, Odero-Marah V. Association of epithelial mesenchymal transition with prostate and breast health disparities. PLoS One. 2018;13:e0203855. doi: 10.1371/journal.pone.0203855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Ameri AH, Wang S, Jansson KH, Casey OM, Yang Q, Beshiri ML, Fang L, Lake RG, Agarwal S, Alilin AN, Xu W, Yin J, Kelly K. EGR1 regulates angiogenic and osteoclastogenic factors in prostate cancer and promotes metastasis. Oncogene. 2019;38:6241–6255. doi: 10.1038/s41388-019-0873-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhau HY, Chang SM, Chen BQ, Wang Y, Zhang H, Kao C, Sang QA, Pathak SJ, Chung LW. Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci U S A. 1996;93:15152–15157. doi: 10.1073/pnas.93.26.15152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhau HE, Li CL, Chung LW. Establishment of human prostate carcinoma skeletal metastasis models. Cancer. 2000;88:2995–3001. doi: 10.1002/1097-0142(20000615)88:12+<2995::aid-cncr15>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, Gasser TC, Mihatsch MJ. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Wang R, Xie ZH, Odero-Marah V, Pathak S, Multani A, Chung LW, Zhau HE. Prostate cancer metastasis: role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate. 2006;66:1664–1673. doi: 10.1002/pros.20488. [DOI] [PubMed] [Google Scholar]
- 24.Wu D, Zhau HE, Huang WC, Iqbal S, Habib FK, Sartor O, Cvitanovic L, Marshall FF, Xu Z, Chung LW. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene. 2007;26:5070–5077. doi: 10.1038/sj.onc.1210316. [DOI] [PubMed] [Google Scholar]
- 25.Wang R, Xu J, Mabjeesh N, Zhu G, Zhou J, Amin M, He D, Marshall FF, Zhau HE, Chung LW. PrLZ is expressed in normal prostate development and in human prostate cancer progression. Clin Cancer Res. 2007;13:6040–6048. doi: 10.1158/1078-0432.CCR-07-0640. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Zhau HE, Osunkoya AO, Iqbal S, Yang X, Fan S, Chen Z, Wang R, Marshall FF, Chung LW, Wu D. Vascular endothelial growth factor regulates myeloid cell leukemia-1 expression through neuropilin-1-dependent activation of c-MET signaling in human prostate cancer cells. Mol Cancer. 2010;9:9. doi: 10.1186/1476-4598-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo SI, Gera L, Zhau HE, Qian WP, Iqbal S, Johnson NA, Zhang S, Zayzafoon M, Stewart J, Wang R, Chung LW, Wu D. BKM1740, an acyl-tyrosine bisphosphonate amide derivative, inhibits the bone metastatic growth of human prostate cancer cells by inducing apoptosis. Clin Cancer Res. 2008;14:6198–6206. doi: 10.1158/1078-0432.CCR-08-1023. [DOI] [PubMed] [Google Scholar]
- 28.Josson S, Sharp S, Sung SY, Johnstone PA, Aneja R, Wang R, Gururajan M, Turner T, Chung LW, Yates C. Tumor-stromal interactions influence radiation sensitivity in epithelial- versus mesenchymal-like prostate cancer cells. J Oncol. 2010;2010:232831. doi: 10.1155/2010/232831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Josson S, Matsuoka Y, Chung LW, Zhau HE, Wang R. Tumor-stroma co-evolution in prostate cancer progression and metastasis. Semin Cell Dev Biol. 2010;21:26–32. doi: 10.1016/j.semcdb.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He H, Yang X, Davidson AJ, Wu D, Marshall FF, Chung LW, Zhau HE, Wang R. Progressive epithelial to mesenchymal transitions in ARCaP E prostate cancer cells during xenograft tumor formation and metastasis. Prostate. 2010;70:518–528. doi: 10.1002/pros.21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He H, Davidson AJ, Wu D, Marshall FF, Chung LW, Zhau HE, He D, Wang R. Phorbol ester phorbol-12-myristate-13-acetate induces epithelial to mesenchymal transition in human prostate cancer ARCaPE cells. Prostate. 2010;70:1119–1126. doi: 10.1002/pros.21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maul RS, Chang DD. EPLIN, epithelial protein lost in neoplasm. Oncogene. 1999;18:7838–7841. doi: 10.1038/sj.onc.1203206. [DOI] [PubMed] [Google Scholar]
- 33.Chen S, Maul RS, Kim HR, Chang DD. Characterization of the human EPLIN (epithelial protein lost in neoplasm) gene reveals distinct promoters for the two EPLIN isoforms. Gene. 2000;248:69–76. doi: 10.1016/s0378-1119(00)00144-x. [DOI] [PubMed] [Google Scholar]
- 34.Song Y, Maul RS, Gerbin CS, Chang DD. Inhibition of anchorage-independent growth of transformed NIH3T3 cells by epithelial protein lost in neoplasm (EPLIN) requires localization of EPLIN to actin cytoskeleton. Mol Biol Cell. 2002;13:1408–1416. doi: 10.1091/mbc.01-08-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maul RS, Song Y, Amann KJ, Gerbin SC, Pollard TD, Chang DD. EPLIN regulates actin dynamics by cross-linking and stabilizing filaments. J Cell Biol. 2003;160:399–407. doi: 10.1083/jcb.200212057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abe K, Takeichi M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A. 2008;105:13–19. doi: 10.1073/pnas.0710504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi K, Ishiuchi T, Takeichi M. Mechanosensitive EPLIN-dependent remodeling of adherens junctions regulates epithelial reshaping. J Cell Biol. 2011;194:643–656. doi: 10.1083/jcb.201104124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chervin-Petinot A, Courcon M, Almagro S, Nicolas A, Grichine A, Grunwald D, Prandini MH, Huber P, Gulino-Debrac D. Eplin interacts with alpha-catenin and actin filaments in endothelial cells and in vitro stabilizes the vascular capillary network. J Biol Chem. 2011;287:7556–72. doi: 10.1074/jbc.M111.328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abu Taha A, Schnittler HJ. Dynamics between actin and the VE-cadherin/catenin complex: novel aspects of the ARP2/3 complex in regulation of endothelial junctions. Cell Adh Migr. 2014;8:125–135. doi: 10.4161/cam.28243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases. 2014;5:1–15. doi: 10.4161/21541248.2014.973760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taha M, Aldirawi M, Marz S, Seebach J, Odenthal-Schnittler M, Bondareva O, Bojovic V, Schmandra T, Wirth B, Mietkowska M, Rottner K, Schnittler H. EPLIN-alpha and -beta isoforms modulate endothelial cell dynamics through a spatiotemporally differentiated interaction with actin. Cell Rep. 2019;29:1010–1026. e1016. doi: 10.1016/j.celrep.2019.09.043. [DOI] [PubMed] [Google Scholar]
- 42.Chircop M, Oakes V, Graham ME, Ma MP, Smith CM, Robinson PJ, Khanna KK. The actin-binding and bundling protein, EPLIN, is required for cytokinesis. Cell Cycle. 2009;8:757–764. doi: 10.4161/cc.8.5.7878. [DOI] [PubMed] [Google Scholar]
- 43.Sundvold H, Sundvold-Gjerstad V, Malerod-Fjeld H, Haglund K, Stenmark H, Malerod L. Arv1 promotes cell division by recruiting IQGAP1 and myosin to the cleavage furrow. Cell Cycle. 2016;15:628–643. doi: 10.1080/15384101.2016.1146834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang YY, Fu ZY, Wei J, Qi W, Baituola G, Luo J, Meng YJ, Guo SY, Yin H, Jiang SY, Li YF, Miao HH, Liu Y, Wang Y, Li BL, Ma YT, Song BL. A LIMA1 variant promotes low plasma LDL cholesterol and decreases intestinal cholesterol absorption. Science. 2018;360:1087–1092. doi: 10.1126/science.aao6575. [DOI] [PubMed] [Google Scholar]
- 45.Donato LJ. LIMA1: a newly identified player in the field of cholesterol control. Clin Chem. 2018;64:1792–1793. doi: 10.1373/clinchem.2018.294645. [DOI] [PubMed] [Google Scholar]
- 46.Jiang WG, Martin TA, Lewis-Russell JM, Douglas-Jones A, Ye L, Mansel RE. Eplin-alpha expression in human breast cancer, the impact on cellular migration and clinical outcome. Mol Cancer. 2008;7:71. doi: 10.1186/1476-4598-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanders AJ, Martin TA, Ye L, Mason MD, Jiang WG. EPLIN is a negative regulator of prostate cancer growth and invasion. J Urol. 2011;186:295–301. doi: 10.1016/j.juro.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Sanders AJ, Zhang L, Jiang WG. EPLIN-alpha expression in human oesophageal cancer and its impact on cellular aggressiveness and clinical outcome. Anticancer Res. 2012;32:1283–1289. [PubMed] [Google Scholar]
- 49.Ohoka A, Kajita M, Ikenouchi J, Yako Y, Kitamoto S, Kon S, Ikegawa M, Shimada T, Ishikawa S, Fujita Y. EPLIN is a crucial regulator for extrusion of RasV12-transformed cells. J Cell Sci. 2015;128:781–789. doi: 10.1242/jcs.163113. [DOI] [PubMed] [Google Scholar]
- 50.Kasai N, Kadeer A, Kajita M, Saitoh S, Ishikawa S, Maruyama T, Fujita Y. The paxillin-plectin-EPLIN complex promotes apical elimination of RasV12-transformed cells by modulating HDAC6-regulated tubulin acetylation. Sci Rep. 2018;8:2097. doi: 10.1038/s41598-018-20146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steder M, Alla V, Meier C, Spitschak A, Pahnke J, Furst K, Kowtharapu BS, Engelmann D, Petigk J, Egberts F, Schad-Trcka SG, Gross G, Nettelbeck DM, Niemetz A, Putzer BM. DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell. 2013;24:512–527. doi: 10.1016/j.ccr.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 52.Wu D. Epithelial protein lost in neoplasm (EPLIN): beyond a tumor suppressor. Genes Dis. 2017;4:100–107. doi: 10.1016/j.gendis.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohashi T, Idogawa M, Sasaki Y, Tokino T. p53 mediates the suppression of cancer cell invasion by inducing LIMA1/EPLIN. Cancer Lett. 2017;390:58–66. doi: 10.1016/j.canlet.2016.12.034. [DOI] [PubMed] [Google Scholar]
- 54.Liu R, Martin TA, Jordan NJ, Ruge F, Ye L, Jiang WG. Epithelial protein lost in neoplasm-alpha (EPLIN-alpha) is a potential prognostic marker for the progression of epithelial ovarian cancer. Int J Oncol. 2016;48:2488–2496. doi: 10.3892/ijo.2016.3462. [DOI] [PubMed] [Google Scholar]
- 55.Zeng J, Jiang WG, Sanders AJ. Epithelial protein lost in neoplasm, EPLIN, the cellular and molecular prospects in cancers. Biomolecules. 2021;11:1038. doi: 10.3390/biom11071038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins RJ, Morgan LD, Owen S, Ruge F, Jiang WG, Sanders AJ. Mechanistic insights of epithelial protein lost in neoplasm in prostate cancer metastasis. Int J Cancer. 2018;143:2537–2550. doi: 10.1002/ijc.31786. [DOI] [PubMed] [Google Scholar]
- 57.Gong W, Zeng J, Ji J, Jia Y, Jia S, Sanders AJ, Jiang WG. EPLIN expression in gastric cancer and impact on prognosis and chemoresistance. Biomolecules. 2021;11:547. doi: 10.3390/biom11040547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wirsing AM, Bjerkli IH, Steigen SE, Rikardsen O, Magnussen SN, Hegge B, Seppola M, Uhlin-Hansen L, Hadler-Olsen E. Validation of selected head and neck cancer prognostic markers from the pathology atlas in an oral tongue cancer cohort. Cancers (Basel) 2021;13:2387. doi: 10.3390/cancers13102387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.You S, Knudsen BS, Erho N, Alshalalfa M, Takhar M, Al-Deen Ashab H, Davicioni E, Karnes RJ, Klein EA, Den RB, Ross AE, Schaeffer EM, Garraway IP, Kim J, Freeman MR. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016;76:4948–4958. doi: 10.1158/0008-5472.CAN-16-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Y, Liu C, Nadiminty N, Lou W, Tummala R, Evans CP, Gao AC. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol Cancer Ther. 2013;12:1829–1836. doi: 10.1158/1535-7163.MCT-13-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang L, Zhao L, Zan Y, Zhu Q, Ren J, Zhao X. MiR-93-5p enhances growth and angiogenesis capacity of HUVECs by down-regulating EPLIN. Oncotarget. 2017;8:107033–107043. doi: 10.18632/oncotarget.22300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang S, Wang X, Iqbal S, Wang Y, Osunkoya AO, Chen Z, Chen Z, Shin DM, Yuan H, Wang YA, Zhau HE, Chung LW, Ritenour C, Kucuk O, Wu D. Epidermal growth factor promotes protein degradation of epithelial protein lost in neoplasm (EPLIN), a putative metastasis suppressor, during epithelial-mesenchymal transition. J Biol Chem. 2013;288:1469–1479. doi: 10.1074/jbc.M112.438341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen NP, Uddin B, Hardt R, Ding W, Panic M, Lucibello I, Kammerer P, Ruppert T, Schiebel E. Human phosphatase CDC14A regulates actin organization through dephosphorylation of epithelial protein lost in neoplasm. Proc Natl Acad Sci U S A. 2017;114:5201–5206. doi: 10.1073/pnas.1619356114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nie Z, Du MQ, McAllister-Lucas LM, Lucas PC, Bailey NG, Hogaboam CM, Lim MS, Elenitoba-Johnson KS. Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat Commun. 2015;6:5908. doi: 10.1038/ncomms6908. [DOI] [PubMed] [Google Scholar]
- 65.Linklater ES, Duncan ED, Han KJ, Kaupinis A, Valius M, Lyons TR, Prekeris R. Rab40-Cullin5 complex regulates EPLIN and actin cytoskeleton dynamics during cell migration. J Cell Biol. 2021;220:e202008060. doi: 10.1083/jcb.202008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Von Low EC, Perabo FG, Siener R, Muller SC. Review. Facts and fiction of phytotherapy for prostate cancer: a critical assessment of preclinical and clinical data. In Vivo. 2007;21:189–204. [PubMed] [Google Scholar]
- 67.de Souza PL, Russell PJ, Kearsley JH, Howes LG. Clinical pharmacology of isoflavones and its relevance for potential prevention of prostate cancer. Nutr Rev. 2010;68:542–555. doi: 10.1111/j.1753-4887.2010.00314.x. [DOI] [PubMed] [Google Scholar]
- 68.Jacobsen BK, Knutsen SF, Fraser GE. Does high soy milk intake reduce prostate cancer incidence? The Adventist Health Study (United States) Cancer Causes Control. 1998;9:553–557. doi: 10.1023/a:1008819500080. [DOI] [PubMed] [Google Scholar]
- 69.Hwang YW, Kim SY, Jee SH, Kim YN, Nam CM. Soy food consumption and risk of prostate cancer: a meta-analysis of observational studies. Nutr Cancer. 2009;61:598–606. doi: 10.1080/01635580902825639. [DOI] [PubMed] [Google Scholar]
- 70.Pavese JM, Farmer RL, Bergan RC. Inhibition of cancer cell invasion and metastasis by genistein. Cancer Metastasis Rev. 2010;29:465–482. doi: 10.1007/s10555-010-9238-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y, Che M, Bhagat S, Ellis KL, Kucuk O, Doerge DR, Abrams J, Cher ML, Sarkar FH. Regulation of gene expression and inhibition of experimental prostate cancer bone metastasis by dietary genistein. Neoplasia. 2004;6:354–363. doi: 10.1593/neo.03478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lakshman M, Xu L, Ananthanarayanan V, Cooper J, Takimoto CH, Helenowski I, Pelling JC, Bergan RC. Dietary genistein inhibits metastasis of human prostate cancer in mice. Cancer Res. 2008;68:2024–2032. doi: 10.1158/0008-5472.CAN-07-1246. [DOI] [PubMed] [Google Scholar]
- 73.Pavese JM, Krishna SN, Bergan RC. Genistein inhibits human prostate cancer cell detachment, invasion, and metastasis. Am J Clin Nutr. 2014;100(Suppl 1):431S–436S. doi: 10.3945/ajcn.113.071290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shafiee G, Saidijam M, Tayebinia H, Khodadadi I. Beneficial effects of genistein in suppression of proliferation, inhibition of metastasis, and induction of apoptosis in PC3 prostate cancer cells. Arch Physiol Biochem. 2020:1–9. doi: 10.1080/13813455.2020.1717541. [DOI] [PubMed] [Google Scholar]
- 75.Wang G, Zhang D, Yang S, Wang Y, Tang Z, Fu X. Co-administration of genistein with doxorubicin-loaded polypeptide nanoparticles weakens the metastasis of malignant prostate cancer by amplifying oxidative damage. Biomater Sci. 2018;6:827–835. doi: 10.1039/c7bm01201b. [DOI] [PubMed] [Google Scholar]
- 76.van Dodewaard-de Jong JM, Verheul HM, Bloemendal HJ, de Klerk JM, Carducci MA, van den Eertwegh AJ. New treatment options for patients with metastatic prostate cancer: what is the optimal sequence? Clin Genitourin Cancer. 2015;13:271–279. doi: 10.1016/j.clgc.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Agarwal N, Di Lorenzo G, Sonpavde G, Bellmunt J. New agents for prostate cancer. Ann Oncol. 2014;25:1700–1709. doi: 10.1093/annonc/mdu038. [DOI] [PubMed] [Google Scholar]
- 78.Omlin A, Pezaro C, Gillessen Sommer S. Sequential use of novel therapeutics in advanced prostate cancer following docetaxel chemotherapy. Ther Adv Urol. 2014;6:3–14. doi: 10.1177/1756287213509677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCain J. Drugs that offer a survival advantage for men with bone metastases resulting from castration-resistant prostate cancer: new and emerging treatment options. P T. 2014;39:130–143. [PMC free article] [PubMed] [Google Scholar]
- 80.Kroon J, Kooijman S, Cho NJ, Storm G, van der Pluijm G. Improving taxane-based chemotherapy in castration-resistant prostate cancer. Trends Pharmacol Sci. 2016;37:451–462. doi: 10.1016/j.tips.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 81.Tu SM, Lin SH. Current trials using bone-targeting agents in prostate cancer. Cancer J. 2008;14:35–39. doi: 10.1097/PPO.0b013e318161d32d. [DOI] [PubMed] [Google Scholar]
- 82.Li X, Gera L, Zhang S, Chen Y, Lou L, Wilson LM, Xie ZR, Sautto G, Liu D, Danaher A, Mamouni K, Yang Y, Du Y, Fu H, Kucuk O, Osunkoya AO, Zhou J, Wu D. Pharmacological inhibition of noncanonical EED-EZH2 signaling overcomes chemoresistance in prostate cancer. Theranostics. 2021;11:6873–6890. doi: 10.7150/thno.49235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.