

SUMMARY
Actinins are strain-sensing actin cross-linkers that are ubiquitously expressed and harbor mutations in human diseases. We utilize CRISPR, pluripotent stem cells, and BioID to study actinin interactomes in human cardiomyocytes. We identify 324 actinin proximity partners, including those that are dependent on sarcomere assembly. We confirm 19 known interactors and identify a network of RNA-binding proteins, including those with RNA localization functions. In vivo and biochemical interaction studies support that IGF2BP2 localizes electron transport chain transcripts to actinin neighborhoods through interactions between its K homology (KH) domain and actinin’s rod domain. We combine alanine scanning mutagenesis and metabolic assays to disrupt and functionally interrogate actinin-IGF2BP2 interactions, which reveal an essential role in metabolic responses to pathological sarcomere activation using a hypertrophic cardiomyopathy model. This study expands our functional knowledge of actinin, uncovers sarcomere interaction partners, and reveals sarcomere crosstalk with IGF2BP2 for metabolic adaptation relevant to human disease.
In brief
Ladha et al. combine BioID, human cardiomyocytes, and CRISPR-Cas9 to interrogate the actinin interactome. This reveals 324 actinin proximity partners, including RNA-binding proteins that bind transcripts encoding ETC functional components. Among these RNA-binding proteins, IGF2BP2 directly binds actinin, and actinin-IGF2BP2 interactions regulate ETC transcript localization and metabolic adaptation to sarcomere function.
Graphical Abstract
INTRODUCTION
Actinin proteins are ubiquitous spectrin family members that cross-link actin and are important for myriad cell functions, including adhesion, migration, contraction, and signaling (Liem, 2016). The four human actinin isoforms have distinct expression profiles (non-muscle ACTN1 and ACTN4, cardiac muscle ACTN2, and skeletal muscle ACTN3) but share homologous amino acid sequences that are organized into three structural domains: an actin-binding (AB) domain composed of two calponin-homology domains, a central rod domain containing four spectrin-like repeats (SRs), and a calmodulin-like domain (CaM) (Ribeiro et al., 2014). While it is thought that actinin evolved initially to regulate the early eukaryotic actin-based cytoskeleton (Virel and Backman, 2004), it has acquired more elaborate functions in vertebrates, including a mechanical role in the sarcomere, a specialized contractile system required for striated muscle function (Murphy and Young, 2015; Virel and Backman, 2004). Moreover, normal actinin function is important for multiple human organ systems as inheritance of ACTN1, ACTN2, and ACTN4 mutations have also been linked to congenital macrothrombocytopenia, hypertrophic cardiomyopathy (HCM), and focal segmental glomerulosclerosis of the kidney, respectively (Chiu et al., 2010; Kaplan et al., 2000; Kunishima et al., 2013).
In the cardiac sarcomere, alpha-actinin-2 (referred henceforth as actinin) is a major structural component of the Z-disk, where it is essential for sarcomere assembly and function (Chopra et al., 2018). Actinin regulates sarcomere assembly by providing a scaffold for protein-protein interactions (PPIs) such as with titin (TTN) and actin through CaM and AB domains, respectively (Chopra et al., 2018; Grison et al., 2017; Schultheiss et al., 1992). Secondary to these interactions, the thin and thick filament sarcomere structures are organized to promote twitch contraction. It has also been observed that while the Z-disk is one of the stiffest sarcomere structures, actinin demonstrates remarkably dynamic and complex molecular interactions (such as with the actin cytoskeleton) that are necessary for normal myocyte function (Sanger and Sanger, 2008). Moreover, actinin interaction partners have also been described to exit the sarcomere to execute critical roles in cell signaling, protein homeostasis, and transcriptional regulation (Lange et al., 2006). Secondary to the vast repertoire and dynamics of actinin molecular interactions, there remain extensive gaps in our knowledge of not only how actinin regulates sarcomere assembly and function but also within other cellular contexts enriched for actinin expression such as at focal adhesions in both striated and non-striated cells that may inform how actinin mutations cause human disease (Djinovic-Carugo et al., 2002; Foley and Young, 2014).
Here, we studied the actinin proximity interactome in CRISPR-engineered human cardiomyocytes using proximity-dependent biotinylation (BioID) (Roux et al., 2012). Our study employs induced pluripotent stem cell cardiomyocytes (iPSC-CMs) differentiated from human iPSCs that express actinin fused to the promiscuous biotinylating enzyme BirA* from the endogenous ACTN2 locus to provide physiological actinin expression levels and regulation. We identify 324 actinin proximity partners, including those that are dynamically regulated by sarcomere assembly, to reveal molecular insights into this process. Our study uncovers actinin interaction partners, including the RNA-binding protein IGF2BP2, which we observe to function in electron transport chain (ETC) transcript localization to actinin neighborhoods. Disruption of actinin-IGF2BP2 interactions revealed an essential role in metabolic adaptation to pathological sarcomere activation induced by an HCM mutation. Our study also provides a compendium of proteins that can be studied to reveal insights into sarcomere dynamics and other actinin functions.
RESULTS
BioID to identify actinin proximity partners through sarcomere assembly
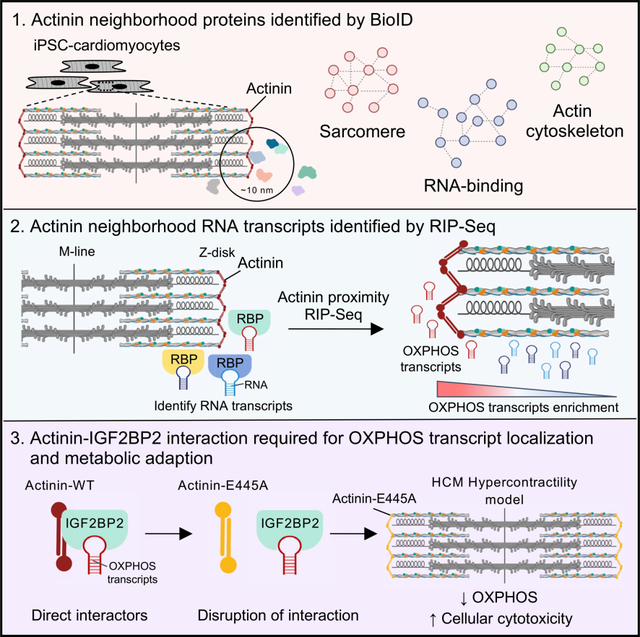
Actinin is an integral member of the actin cytoskeleton, where it interacts within multiprotein complexes such as Z-disks and focal adhesions (Figure 1A). To study actinin interactions, we utilized BioID (Gingras et al., 2019), which is a proximity-dependent tagging method using BirA*, a modified version of E. coli BirA that exhibits promiscuous lysine biotinylation within a 10 nm radius of expression (Figure 1B; Kim et al., 2014). Using CRISPR, we fused BirA* with a hemagglutinin (HA) tag to the C terminus of endogenous ACTN2 (Actinin-BirA*) in a human iPSC line (Figure S1A). Following differentiation to iPSC-CMs, expression levels of Actinin-BirA* fusion protein was similar to unmodified control iPSC-CMs that do no express Actinin-BirA* (non-BirA*) (Figure 1C). To verify that Actinin-BirA* functioned similarly to unmodified actinin, we utilized a cardiac microtissue (CMT) assay previously applied to study sarcomere mutations that cause heart failure (Cohn et al., 2019; Hinson et al., 2015). CMT twitch force was unaltered by Actinin-BirA* expression (Figure 1D; Video S1). We next verified localization of Actinin-BirA* to the Z-disk by both colocalization immunofluorescence (Figures 1E and S1B) and HA immunoprecipitation (Figure S1C) that demonstrated Actinin-BirA* pulled down with the Z-disk protein TCAP (Hayashi et al., 2004), but not the M-line protein myomesin (Auerbach et al., 1999). We observed that 50 μM biotin supplementation provided strong biotinylation signal in lysates obtained from Actinin-BirA* cells (Figures 1F and S1D), and biotinylated targets overlapped actinin (Figure 1E). Finally, we observed negligible biotinylation in biotin-pulsed wild-type (WT) control iPSC-CMs that do not express BirA* (Figure 1G).
Figure 1. BioID to identify actinin proximity partners through sarcomere assembly.
(A) Overview of actinin localization in cardiomyocytes including sarcomere Z-disk, focal adhesion, and cortical actin cytoskeleton.
(B) Overview of the BioID method using Actinin-BirA* fusion to study actinin-proximal protein networks.
(C) Actinin-BirA* and control non-BirA* iPSC-CM lysates were probed with anti-actinin and anti-GAPDH antibodies to identify (left) and quantify (right) Actinin-BirA* fusion (~130 kD) and endogenous actinin (~100 kD).
(D) Actinin-BirA* cardiac microtissues have similar twitch force compared to non-BirA* controls (n = 12–15) (scale bar, 150 μm).
(E) Confocal micrograph of Actinin-BirA* iPSC-CMs decorated with antibodies to actinin (red), streptavidin-AF488 (green), and DAPI DNA co-stain (blue) (scale bar, 10 μm).
(F) Overview of BioID experimental methods.
(G) iPSC-CM lysates were probed with streptavidin-horseradish peroxidase (HRP) to examine Actinin-BirA*-biotinylated proteins.
(H) Heatmap and hierarchical clustering of log2-transformed intensity values for the 324 Actinin-BirA* hits (L2FC ≥ 1 and FDR < 0.05 relative to control non-BirA*) from combined TMT experiments.
(I) Venn diagram of ACTN2 BioID protein dataset (red circle) and published ACTN interactors obtained from STRING where ACTN was bait (gray circle).
(J) 324 proteins from ACTN2 BioID were analyzed by GO and key enrichment terms are listed.
(K) ACTN2 BioID proteins in RNA-binding GO term were uploaded to STRING for subnetwork analysis. The resulting network was imported into Cytoscape (v. 3.7.2) and CLUSTER and BINGO features were utilized for clustering and GO term identification. Hits that were additionally studied are circled in black.
Data are n ≥ 3 (C and D); mean ± SEM; significance assessed by Student’s t test (C and D) or two-way ANOVA followed by a two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli to correct for multiple comparisons (H) and defined by p > 0.05 (ns) (C and D). See also Figure S1 and Video S1.
To identify actinin proximity partners, we subjected both biotin-pulsed Actinin-BirA* and control non-BirA* iPSC-CM lysates to streptavidin immunoprecipitation followed by tandem mass tag (TMT) quantitative proteomics (Table S1). Analysis of Actinin-BirA* versus control non-BirA* samples identified enrichment of 324 actinin proximity partners (log2 fold change [L2FC] ≥ 1 and false discovery rate [FDR] < 0.05; Figure 1H; Table S1). We confirmed 38% of previously known actinin interactors using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) (Szklarczyk et al., 2019; Figure 1I). Gene Ontology (GO) analysis of interactors revealed functions in cell adhesion, anchoring junction, actin cytoskeleton, contractile fiber, and RNA binding (Figure 1J; Table S1).
Z-disks develop from punctate Z-body precursors (Du et al., 2003), but the actinin interactions underlying this process are incompletely understood. To identify actinin neighborhoods underlying this transition, into Actinin-BirA* iPSCs, we knocked out cardiac troponin T (cTnT-KO; Figure S1E), because cTnT-KO iPSC-CMs assembly only Z-bodies unless cTnT is reintroduced by lentiviral transduction as described previously (Pettinato et al., 2020). After confirming that biotin-pulsed cTnT-KO and cTnT-WT iPSC-CMs exhibited the expected biotinylation of proteins overlapping Z-body and Z-disk structures (Figure S1F), respectively, we then performed TMT (Figure S1G; Table S1). We identified 24 actinin neighborhood proteins that were exclusively enriched in the Z-disk stage (L2FC ≥ 1 and FDR < 0.05; Table S1), which were significant for GO terms, including RNA binding, anchoring junction, and cadherin binding (Figure S1I; Table S1). Only SPHKAP, a membrane-anchoring protein associated with heart failure (Aye et al., 2012), was exclusive to Z-bodies. In addition, we identified 47 proteins that were more highly enriched in Z-disk relative to Z-body stages as defined by relative abundancies in cTnT-WT compared to cTnT-KO samples (L2FC ≥ 1 and FDR < 0.05; Table S1), which were enriched for GO terms, including contractile fiber and actin cytoskeleton (Figure S1J; Table S1). Among the 47 (Figure S1K), we observed that a muscle myosin (MYH7), as previously described (Rode et al., 2016), was one of the most highly enriched proteins in Z-disk relative to Z-body stages (Figure S1K), while non-muscle myosin (MYH10) was similar in both stages. In addition, we also identified enrichment of other late-assembling proteins, including TCAP and CSRP3 (White et al., 2014). The most enriched sarcomere assembly partner was SYNPO2L, a known binding partner of actinin that is critical to heart and skeletal muscle development (Beqqali et al., 2010). Taken together, we identified actinin interactomes through Z-body and Z-disk stages, including stage-specific RNA-binding proteins and stage-enriched myosins and late-assembling proteins.
We next focused our analysis on RNA-binding proteins in proximity to actinin, as these factors were identified in Z-disks (Figure S1I) and have been less studied relative to actin cytoskeletal and sarcomere interactors. Using GO analysis, our study identified 99 proteins with general RNA-binding functions (Figure 1J), including those with subfunctions in ribosomal assembly, transcription/translation, and RNA metabolism (Figure 1K). Proteins involved in RNA metabolism include regulators of RNA splicing, localization, stability, and translation (Van Nostrand et al., 2020). Except for SRSF1, which has been implicated in cardiac electrophysiology, and PCBP2, which has been implicated in hypertrophic signaling (Xu et al., 2005; Zhang et al., 2015), the cardiac functions of many of these proteins are unknown. Overall, our BioID study reveals a compendium of actinin interaction partners within cardiomyocytes through sarcomere assembly including a network of RNA-binding proteins with diverse functions including RNA localization.
Identification of transcripts bound to RNA-binding proteins in actinin neighborhoods
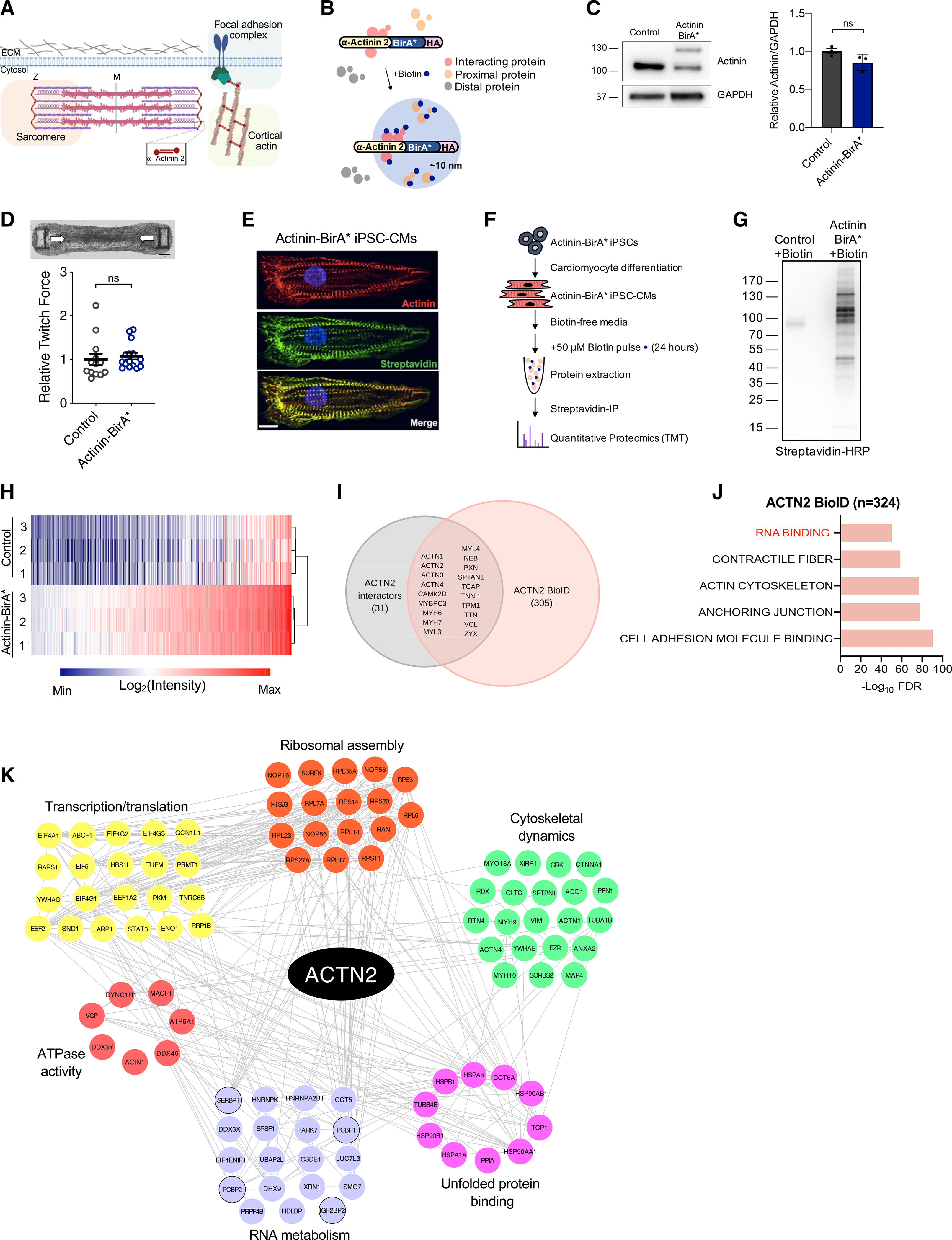
To reveal functional insights into actinin interactions with RNA-binding proteins, we next sought to identify gene transcripts bound to RNA-binding proteins in actinin neighborhoods. To do this, we performed RNA immunoprecipitation followed by sequencing (RIP-seq) (Tenenbaum et al., 2000) using streptavidin affinity purification to pull down RNA-binding proteins that were biotinylated because they were localized within ~10 nm of Actinin-BirA* (Figure 2A). Principal-component analysis (PCA) demonstrated clustering of Actinin-BirA* transcripts compared to non-BirA* controls (Figure 2B), and differential expression analysis identified 945 transcripts bound to biotinylated RNA-binding proteins (Table S2). GO analysis (Figure 2C) revealed enrichment of ETC and ribosome transcripts (Figures 2D and S2A). In contrast, we observed no enrichment for glycolysis transcripts (Figures 2E and S2B). 1,113 transcripts were depleted and clustered into GO terms for chromatin and nuclear functions (Figure 2C). Cytoskeletal transcripts, including those encoding thin and thick filament sarcomere proteins, were also depleted (Figures 2F and S2C). In summary, RIP-seq demonstrated that RNA-binding proteins that were in actinin neighborhoods bind specific ETC and ribosome transcripts, which provided preliminary evidence that these interactions could be important for metabolic and translational functions, respectively.
Figure 2. Identification of transcripts bound to RNA-binding proteins in actinin neighborhoods.
(A) Overview of modified RIP-seq protocol to study RNA transcripts bound to RNA-binding proteins in actinin neighborhoods by streptavidin affinity purification of RNA-binding proteins followed by RNA isolation and sequencing.
(B) PCA plot shows clustering of Actinin-BirA* RIP-seq samples relative to non-BirA* controls (n = 4).
(C) Volcano plot of RIP-seq data shaded red for 945 enriched transcripts (FDR < 0.05, L2FC ≥ 0.5) or blue for 1,113 depleted transcripts (FDR < 0.05, L2FC ≤ −0.5). Differentially enriched transcripts were analyzed by GO and enrichment terms are listed for enriched and depleted transcripts.
(D) ETC gene components spanning respiratory chain complex I-V proteins are enriched on average. Each circle represents a single ETC gene; black circle for statistically significant transcript.
(E) Glycolysis gene components are not enriched on average. Each circle represents a single glycolysis gene.
(F) Thin and thick filament sarcomere are depleted on average. Each circle represents a single sarcomere gene.
(G) qPCR validation of candidate RIP-seq hits (n = 4).
(H) Representative confocal micrograph of iPSC-CMs decorated with antibodies to actinin (red), DAPI DNA co-stain (blue), and RNA FISH probes against NDUFA1 (green) (scale bars: main image, 10 μm; inset, 5 μm).
(I) Representative confocal micrograph of iPSC-CMs decorated with antibodies to actinin (red), DAPI DNA co-stain (blue), and RNA FISH probes against NDUFA8 (cyan) (scale bars: main image, 10 μm; inset, 5 μm).
(J) Quantification of RNA proximity to actinin protein. Statistics are relative to TTN. Data are derived from n = 10 cells.
Data are mean ± SEM; significance assessed by Benjamini Hochberg method (C) or ANOVA using Holm-Sidak correction for multiple comparisons (G and J) and defined by p < 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****) unless otherwise stated. See also Figure S2.
Following qPCR validation of ETC transcript enrichment identified by RIP-seq (Figure 2G), we performed RNA fluorescence in situ hybridization (RNA FISH) with Imaris distance quantification (Figure S2D) to validate actinin neighborhood transcripts. We first conducted a control RNA FISH experiment excluding only the RNA probe to verify specificity of the RNA FISH signals (Figure S2E). Next, we found that RNA puncta encoding ETC component NDUFA1 were more likely to be localized near actinin protein relative to the sarcomere thick filament transcript TTN that served as a control because it was not RIP-seq enriched (Figures 2H, 2J, and S2J). Over 50% of NDUFA1 puncta overlapped actinin, but almost no puncta were greater than 1 μm from actinin. We confirmed specificity of the NDUFA1 probe by using small hairpin RNA (shRNA) to knock down NDUFA1 in iPSC-CMs, which demonstrated a reduction in NDUFA1 mRNA levels and RNA FISH puncta (Figures S2F–S2H). Similar to NDUFA1, we also confirmed actinin proximity for NDUFA8 transcripts (Figures 2I and 2J). We next assessed colocalization between NDUFA1 and NDUFA8 transcripts, which could reflect potential ETC transcript storage or processing mechanisms. While both NDUFA1 and NDUFA8 transcripts localized in actinin neighborhoods, they did not overlap each other (Figure S2I). Finally, we confirmed that two transcripts that were depleted by RIP-seq analysis, H1 family linker histone HIST1H1E and sarcoplasmic reticulum protein HRC, were not associated with actinin neighborhoods (Figures 2J, S2K, and S2L). Together, these data validate our RIP-seq study and confirm that specific ETC transcripts are localized in actinin neighborhoods.
Fine mapping actinin interactions with the RNA-binding protein IGF2BP2
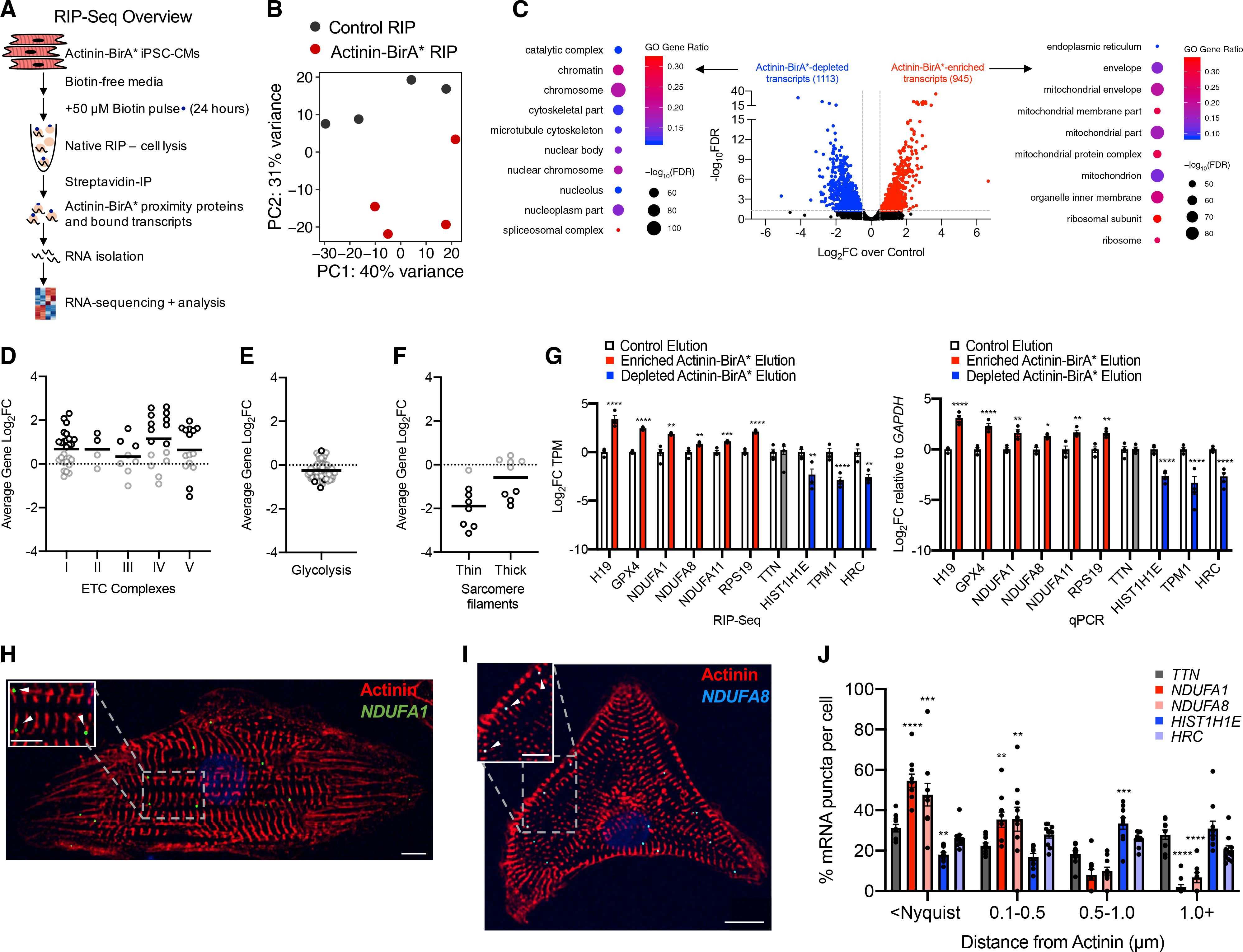
Informed by RIP-seq results that demonstrated ETC transcripts within actinin neighborhoods, we searched for RNA-binding proteins that could regulate this localization. We focused on candidate RNA-binding proteins with previously known metabolic functions, including IGF2BP2 (Saxena et al., 2007), PCBP1 (Ryu et al., 2017), PCBP2 (Frey et al., 2014), and SERBP1 (Muto et al., 2018; Figure 1K). After confirming actinin proximity for the four hits using streptavidin affinity purification followed by immunoblotting in iPSC-CM lysates (Figure 3A), we confirmed that actinin interacts directly or within a complex with three of the hits, including IGF2BP2, PCBP1, and PCBP2, using co-immunoprecipitation of Actinin-BirA* itself using an HA epitope antibody followed by hit-specific antibodies (Figure 3B). Only SERBP1 did not co-immunoprecipitate with actinin, which suggested a more complex interaction mechanism. We also studied the type of interaction between actinin and IGF2BP2, PCBP1, PCPB2, and SERBP1 using luciferase-based mammalian 2-hybrid (M2H) assays in HEK293T cells. We began by constructing expression vectors encoding full-length or previously identified structural domains of actinin and the four RNA-binding proteins. M2H using IGF2BP2 as “prey” and full-length actinin as “bait” resulted in luciferase activation, but not for PCBP1, PCBP2 or SERBP1 (Figure 3C). In addition, we observed colocalization of IGF2BP2 with actinin by immunofluorescence at a subset of iPSC-CM Z-disks (Figure 3D). Taken together, these studies demonstrated four candidate RNA-binding proteins in actinin neighborhoods, including IGF2BP2, that directly interact with actinin and localize to Z-disks.
Figure 3. Fine mapping actinin interactions with the RNA-binding protein IGF2BP2.
(A) Representative immunoblot probed with antibodies to IGF2BP2, PCPB1, PCPB2, and SERBP1 in streptavidin affinity-purified iPSC-CM lysates. Double band represents two IGF2BP2 splice products.
(B) Representative immunoblot probed with antibodies to IGF2BP2, PCPB1, PCPB2, and SERBP1 in anti-HA immunoprecipitated iPSC-CM lysates.
(C) M2H (conducted in HEK293T cells) results demonstrating that IGF2BP2 interacts with full-length actinin, but not PCBP1, PCPB2, or SERBP1.
(D) Representative confocal micrograph demonstrating partial colocalization of actinin (anti-actinin; red) with IGF2BP2 (anti-IGF2BP2; green) with DAPI DNA (blue) co-stain in iPSC-CMs (scale bars: main image, 10 μm; inset, 5 μm).
(E) M2H study conducted to fine map the direct interaction between actinin and IGF2BP2. IGF2BP2 was divided into RRM (residues 1–160) and KH (residues 161–600) domains to fine map the interaction.
(F) M2H results to map the direct interaction between the KH domain of IGF2BP2 and Actinin. Actinin was divided into AB (residues 1–275), rod (residues 276–750), and CaM domains (residues 751–895).
(G) M2H results to map IGF2BP2’s interaction within actinin rod domain through deletions of each spectrin repeat (SR).
(H) Ribbon structure PyMOL of actinin’s SR2 and SR3 domains (SR2 highlighted in red).
(I) Visualization of electrostatic potential of SR2 and SR3 domains. Actinin residue E445 circled in diagram and shaded yellow.
(J) M2H alanine scanning mutagenesis results within actinin SR2 domain with IGF2BP2 demonstrates partial disruption by E445A.
(K) M2H results demonstrating preservation of wild-type (WT) actinin interaction with E445A actinin.
All data are n = 3 and mean ± SEM; significance assessed by ANOVA using Holm-Sidak correction for multiple comparisons (C, E, F, G, J, and K) and defined by p > 0.05 (ns), p ≤ 0.0001 (****). See also Figure S3.
Using M2H, we then fine mapped the IGF2BP2-actinin interaction to IGF2BP2’s K homology (KH) domain, but not its RNA recognition motif (RRM) (Figure 3E), and IGF2BP2’s actinin-binding site to actinin’s rod domain, but not its AB or CaM domains (Figure 3F). As actinin’s rod domain consists of four SRs (Figure 3F; Ribeiro et al., 2014), we additionally studied single SR deletion constructs, which confirmed that both SR2 and SR3 were required for actinin-IGF2BP2 interactions, but not SR1 or SR4 (Figure 3G). To fine map the interaction between SR2 and SR3 with IGF2BP2, we utilized alanine scanning (Weiss et al., 2000), as we observed that smaller deletions within either SR2 or SR3 disturbed actinin dimerization as well as IGF2BP2 interactions, which is consistent with previous actinin dimerization studies (Djinović-Carugo et al., 1999). To establish a method to disrupt actinin-IGF2BP2 interactions, but not actinin dimerization, we took advantage of previous insights from structural studies of SR2 and SR3 domains (Ylänne et al., 2001). These domains contribute an acidic surface to actinin, which has been previously proposed as a protein interaction hub (Figures 3H and 3I; Ylänne et al., 2001). We designed a series of single alanine substitution mutations to disrupt the charge of acidic residues within SR2, after excluding residues that were predicted to contribute intrahelical interactions that could disrupt actinin dimerization or other actinin functions (Ylänne et al., 2001). M2H results of single alanine substitutions within this subset of acidic SR2 residues identified only glutamine 445 (E445) as necessary for IGF2BP2 interactions (Figures 3I and 3J) but did not disrupt actinin dimerization (Figure 3K). As E445A provided a model to specifically disrupt actinin-IGF2BP interactions, we did not study SR3 substitutions.
To validate M2H results for E445A, we next produced recombinant His-tagged WT and E445A actinin in E. coli that were independently immobilized on agarose beads as bait, followed by elution of IGF2BP2 prey from iPSC-CM lysates. Consistent with M2H results, E445A actinin partially disrupted IGF2BP2 interactions relative to WT actinin (Figure S3A). Taken together, these interaction studies (summarized in Figure S3B) implicate actinin as a proximity interaction hub for at least four RNA-binding proteins, including IGF2BP2, that directly bind actinin at SR2 and SR3 domains, which can be disrupted by the E445A mutation in actinin.
Actinin-IGF2BP2 interactions regulate ETC transcript localization to actinin neighborhoods and metabolic adaptation to hypercontractility in an HCM model
As IGF2BP2 binds ETC transcripts in other cell lines (Janiszewska et al., 2012), we tested whether IGF2BP2 could be necessary for ETC transcript localization to actinin neighborhoods in iPSC-CMs. We knocked down IGF2BP2 levels using an shRNA (Figure 4A), and performed RIP-qPCR focused on RIP-seq enriched ETC transcripts NDUFA1, NDUFA8, NDUFA11, and COX6B1. IGF2BP2 knockdown decreased streptavidin affinity-purified ETC transcripts (Figure 4B), but not total cellular ETC transcript levels (Figure 4C), demonstrating that IGF2BP2 is necessary for ETC transcript localization to actinin neighborhoods but not overall transcript stability. To confirm this result, we also sequentially precipitated transcripts by IGF2BP2 antibody to first collect all IGF2BP2-bound transcripts, followed by streptavidin affinity purification to study the subset of IGF2BP2-bound transcripts that have been biotinylated secondary to actinin proximity, which confirmed ETC transcript enrichment at IGF2BP2 within actinin neighborhoods (Figure S4A). As additional confirmation, we utilized RNA FISH to quantify NDUFA1 puncta relative to actinin after IGF2BP2 knockdown. IGF2BP2 knockdown resulted in greater distance between NDUFA1 puncta and actinin protein (Figures S4B and S4C). In summary, IGF2BP2 regulates the localization of ETC transcripts to actinin neighborhoods.
Figure 4. Actinin-IGF2BP2 interactions regulate ETC transcript localization to actinin neighborhoods and metabolic adaptation to hypercontractility in an HCM model.
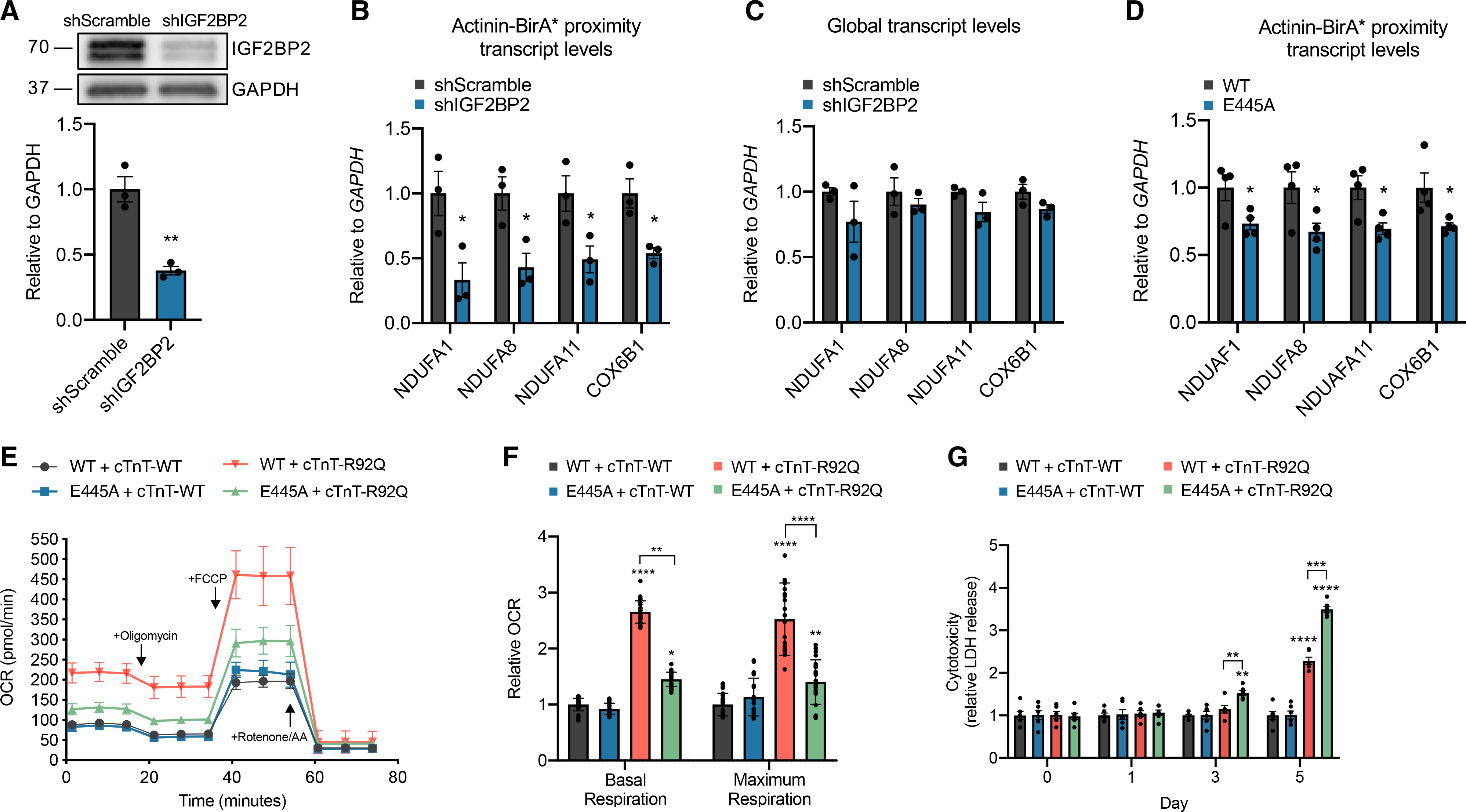
(A) Representative immunoblot of lysates from iPSC-CMs treated with shRNA targeting IGF2BP2 or scramble control probed with antibodies to IGF2BP2 and GAPDH.
(B) Streptavidin affinity purification of transcripts bound to RNA-binding proteins in actinin neighborhoods after IGF2BP2 knockdown, followed by qPCR of ETC transcripts NDUFA1, NDUFA8, NDUFA11, and COX6B1.
(C) qPCR analysis of global transcript levels of NDUFA1, NDUFA8, NDUFA11, and COX6B1 after IGF2BP2 knockdown.
(D) Streptavidin affinity purification of transcripts bound to actinin-proximal RNA-binding proteins with overexpressing lenti-Actinin-WT or lenti-Actinin-E445A, followed by qPCR of ETC transcripts NDUFA1, NDUFA8, NDUFA11, and COX6B1.
(E) Oxygen consumption rates after co-overexpression of either lenti-Actinin-WT or lenti-Actinin-E445A with lenti-cTnT-WT or lenti-cTnT-R92Q in iPSC-CMs.
(F) OCR parameter quantifications comparing lenti-Actinin-WT and lenti-Actinin-E445A with either lenti-cTnT-WT or lenti-cTnT-R92Q.
(G) LDH activity in conditioned media collected from iPSC-CMs expressing either lenti-Actinin-WT or lenti-Actinin-E445A with lenti-cTnT-WT or lenti-cTnT-R92Q in glucose-free media supplemented with galactose for 5 days.
Data are n ≥ 3; mean ± SEM; significance assessed by Student’s t test (A–D) or by ANOVA using Holm-Sidak correction for multiple comparisons (F and G) and defined by p < 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****). See also Figure S4.
We next interrogated the specific function of actinin-IGF2BP2 interactions in iPSC-CMs. To do this, we generated lentivirus encoding E445A or WT actinin, which has been previously utilized to study actinin functions in sarcomere assembly (Chopra et al., 2018; Toepfer et al., 2019). A multiplicity of infection (MOI) of 2 provided robust expression levels that replaced >80% of the endogenous protein (Figure S4D) and localized to Z-disks (Figure S4E). We first conducted RIP-qPCR to assess whether E445A disrupted ETC transcript localization to actinin neighborhoods. As expected by diminished actinin-IGF2BP2 interactions, E445A disrupted actinin neighborhood localization of NDUFA1, NDUFA8, NDUFA11, and COX6B1 (Figure 4D), though not to the extent of IGF2BP2 knockdown (Figure 4B), given that E445A does not completely abolish all IGF2BP2 interactions. Similarly, E445A resulted in greater distance between NDUFA1 puncta and actinin protein (Figure S4F) but to a lesser extent than IGF2BP2 knockdown (Figure S4C). Next, we studied the functional consequences of E445A on sarcomere structure and function. We found no difference in Z-disk length (Figure S4G), a previously utilized assay of sarcomere structure (Pettinato et al., 2020), and no difference in sarcomere contractile function using CMT assays (Figure S4H). To test whether E445A impacts oxidative phosphorylation as suggested by diminished ETC transcript localization to actinin neighborhoods, we quantified oxygen consumption rate (OCR) using a Seahorse analyzer (Figure 4E). OCR was also not different (Figure 4F). In summary, we could not identify baseline sarcomere or oxidative metabolic deficits induced by E445A actinin.
To test whether actinin-IGF2BP2 interactions could function in stress responses, we studied pathological sarcomere activation using a well-established HCM model resulting from a calcium-sensitizing mutation in cardiac troponin T (cTnT-R92Q) (Ferrantini et al., 2017; Javadpour et al., 2003; Pettinato et al., 2020). First, we co-transduced cTnT-R92Q or cTnT-WT along with E445A or WT actinin and measured OCR. We expected to observe increased OCR in response to sarcomere activation, because this process consumes substantial ATP (Lymn and Taylor, 1971), and cardiomyocytes predominantly utilize oxidative metabolism for ATP production (Stanley et al., 2005). cTnT-R92Q increased basal and maximum OCR responses relative to cTnT-WT (Figures 4E and 4F). However, in the presence of E445A actinin, the OCR response was disrupted relative to WT actinin (Figures 4E and 4F). To determine functional consequences of diminished OCR responsiveness to pathological sarcomere activation, we took advantage of previous knowledge that cells with disrupted OCR are less viable when glucose is replaced by galactose to force oxidative metabolism (Aguer et al., 2011; Robinson et al., 1992). To do this, we substituted glucose for galactose and measured iPSC-CM cytotoxicity by lactate dehydrogenase (LDH) release assays. When glucose was freely available, LDH release was negligible in conditioned media during the 5-day culture across all models (Figure S4I). However, in glucose-free media supplemented with galactose to force oxidative metabolism, we found that cTnT-R92Q induced earlier and greater LDH release when co-expressed with E445A actinin compared to WT actinin (Figure 4G). Taken together, these results demonstrate that actinin-IGF2BP2 interactions function in metabolic responses to sarcomere activation that are critical for cell survival in an HCM model.
DISCUSSION
The principal finding of this study is that actinin directly interacts with the RNA-binding protein IGF2BP2 in human cardiomyocytes. Disruption of this interaction impairs ETC transcript localization to actinin neighborhoods, oxidative metabolism, and cell survival in an HCM model associated with pathological sarcomere activation. Our study also provides a compendium of over 300 actinin proximity partners involved in numerous cellular functions, including cytoskeletal dynamics and cellular adhesion, as well as molecular insights into Z-disk assembly, including Z-body- and Z-disk-specific factors relevant to heart failure.
Our study prioritized establishing actinin functions in RNA biology that was inspired by our identification of a large network of RNA-binding proteins within actinin neighborhoods, including those with previous functions in RNA localization. In zebrafish, 71% of transcripts are localized in spatially distinct patterns (Lécuyer et al., 2007), which provides spatial and temporal regulation of gene expression such that local stimuli can regulate translation on-site (Martin and Ephrussi, 2009). While the sarcomere has been implicated in the regulation of RNA localization and functions (Lewis et al., 2018; Rudolph et al., 2019), we find that actinin serves as a PPI hub for several RNA-binding proteins and translational machinery, which we exploited to uncover a spatial relationship between ETC transcripts and actinin expression. Through follow-up studies, this PPI hub may explain mechanisms for the previous observation that intermyofibrillar mitochondria both reside adjacent to the Z-disk (Boncompagni et al., 2009) and have higher rates of oxidative phosphorylation (Ferreira et al., 2010). We observe that ETC transcript localization to actinin neighborhoods is dependent on IGF2BP2, an RNA-binding protein associated with type 2 diabetes by genome-wide association studies (Saxena et al., 2007). In our study, partial disruption of actinin-IGF2BP2 interactions by substituting glutamic acid to alanine at residue 445 resulted in both diminished oxygen consumption rates and cell survival following pathological sarcomere activation induced by an HCM mutation. While requiring further study, this interaction may also be relevant to the pathogenesis of diabetic cardiomyopathy (Jia et al., 2018), as IGF2BP2 knockout mice have been shown to exhibit impaired fatty acid oxidation in skeletal muscle, where actinin is also highly expressed (Regué et al., 2019), and may provide a lever to locally tune energy supply with energy consumption at the sarcomere.
Our study has important limitations, including the use of human iPSC-CMs. These cells empowered the opportunity to study sarcomere assembly in a human cellular context but do not achieve the maturity of adult cardiomyocytes. In the future, engineering and investigating mouse models with Actinin-BirA*, such as has been performed with TTN (Rudolph et al., 2020), could further refine and clarify Z-disk interactions in the healthy heart and diseases such as heart failure. Our study could also be extended to employ more dynamic approaches such as with the recently described TurboID enzyme (Branon et al., 2018), which exhibits improved biotinylation kinetics and labeling efficiencies compared to BirA*. Our RIP-seq approach is also limited by the inability to directly label actinin-proximal transcripts, which could be enabled by the recently developed method of APEX-RIP (Kaewsapsak et al., 2017). Finally, our determination of RNA transcript localization was performed using confocal microscopy, which is resolution limited relative to super-resolution microscopy methods.
In summary, we provide a catalog of actinin neighborhood partners, including those regulated by sarcomere assembly and molecules not well studied in sarcomere biology, including RNA-binding factors that regulate RNA localization. We identify how interactions between actinin and IGF2BP2 contribute to cardiomyocyte metabolic adaptations to stress, which we suggest could be a mode of localized metabolic regulation in proximity to the energy-consuming sarcomere. Further exploration and refinement of these interactions could reveal therapeutic targets and pathophysiology for diseases of the cardiomyocyte that cause heart failure, diabetes, and monogenic disorders associated with actinin mutations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for information and resources should be directed to the Lead Contact, Dr. J. Travis Hinson (travis.hinson@jax.org).
Materials availability
Materials generated in this study are available from the Lead Contact upon reasonable request.
Data and code availability
Genome sequencing datasets for RIP-sequencing are deposited at GEO under accession numbers: GSE144806. Quantitative proteomics data deposited to the ProteomeXchange Consortium identifier PXD018040.
No unique code was generated for the analysis.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PGP1 iPSCs (Coriell #GM23338) were used for all studies, which have been extensively characterized including whole genome sequencing and karyotyping as part of the ENCODE project (ENCBS368AAA). iPSCs were maintained on Matrigel-coated tissue culture plates (Corning 354230) in mTeSR1 (STEMCELL Technologies 85875). iPSCs were passaged at 80%–90% confluency utilizing Accutase (BD 561527) and 10 μM ROCK inhibitor Y-27632 (Tocris 1254). iPSC-CMs were directly differentiated by sequential modulation of Wnt/β-catenin signaling, as previously described (Cohn et al., 2019; Lian et al., 2013). On Day 13 of differentiation, iPSC-CMs were purified by metabolic enrichment using glucose-free DMEM (GIBCO 11966025) supplemented with 4 mM lactate (Sigma 71718) for 24–48 hours (Tohyama et al., 2013). Following selection, iPSC-CMs were trypsinized (GIBCO 25200056) and re-plated onto fibronectin-coated tissue culture plates (GIBCO 33016015). iPSC-CMs were maintained in RPMI-B27. For proximity-labeling experiments, Actinin-BirA* and control non-BirA* iPSC-CMs were maintained in DMEM (GIBCO 11965092) supplemented with homemade biotin-free B27 following metabolic enrichment (Gafni et al., 2013; Irie et al., 2015). For BioID studies, Actinin-BirA* and control non-BirA* iPSC-CMs were treated with 50 μM biotin for 24 hours followed by a 2-hour washout phase in biotin-free media before collection. All iPSC-CM analyses were performed on Day 25–30 unless otherwise noted.
METHOD DETAILS
Plasmid Cloning
The homologous recombination (HR) targeting vector (System Biosciences HR220PA-1) was modified prior to being used in genome-editing experiments. A gene fragment for BirA* (R118G)-HA tag was designed, obtained (IDT) and cloned into the parent vector via Gibson assembly (NEB E2621). ACTN2 ~800bp 5′ and 3′ HR arm gene fragments (IDT) were then cloned into the appropriate cloning sites (sequences in Table S3). All HR vector propagation steps were performed in DH5α E.coli (NEB C2987). For hairpin (shRNA) experiments, the pLKO.1 puro lentiviral plasmids containing either shScramble (Addgene 1864), shIGF2BP2 or a1 (sequence in Table S3) were assembled using T4 DNA ligase (NEB M0202S) and propagated in NEB Stable E.coli (NEB C3040). For overexpression experiments, PCR amplified products of actinin (primer sequences in Table S3) were cloned into a modified pLenti CMV backbone (Addgene 17448) using HIFI DNA Assembly Master Mix (NEB 26261S) and propagated in NEB Stable E.coli (NEB C3040) (Campeau et al., 2009). For cTnT expression, vectors were obtained as previously described (Pettinato et al., 2020).
CRISPR studies
Genome editing was performed utilizing a CRISPR/Cas9 protocol adapted from previous studies (Cohn et al., 2019; Hinson et al., 2015). 8×106 iPSCs were electroporated with 20 μg pCas9-GFP (Addgene 44719), 20 μg of the appropriate hU6-driven sgRNA (designed using https://zlab.bio/guide-design-resources), and 20 μg of the HR targeting vector to generate ACTN2-BirA* knock-in iPSCs (Ding et al., 2013). Electroporated cells were transferred to a Matrigel-coated 100 mm dish containing mTeSR1 and 10 μM Y-27632. The following day, selection was started with 50 μg/mL Hygromycin B (Invitrogen 10687010) to isolate single iPSC clones, which were then manually picked, expanded, and screened via Sanger sequencing. Isogenic knockout of TNNT2 was performed similarly, but in the absence of an HR vector and with GFP-based FACS enrichment in place of antibiotic selection.
Streptavidin immunoprecipitation
After biotin washout, Actinin-BirA* and control non-BirA* iPSC-CMs were rinsed three times with room temperature PBS. Cells were lysed with lysis buffer (50 mM Tris-Cl pH7.4, 500 mM NaCl, 0.2% SDS, 1x protease inhibitor, 1mM DTT, ddH2O). Lysates were sonicated (Branson 250) two times on ice with 2 minutes between each cycle (30 pulses, 30% duty cycle, output level3). Samples were centrifuged to pellet cell debris and cleared lysate were added to magnetic Dynabeads MyOne Streptavidin C1 beads (Invitrogen 65001), which were prewashed with PBS and lysis buffer. Immunoprecipitation was performed on a rotator overnight at 4°C. The following day, the beads were resuspended in wash buffer 1 (2% SDS, ddH2O), rotated for 8 minutes at room temperature, and then re-collected using a magnetic separation stand. The same steps were then repeated with wash buffer 2 (0.1% deoxycholic acid, 1% Triton X-100, 1mM EDTA, 500 mM NaCl, 50 mM HEPES pH7.5, ddH2O) and wash buffer 3 (0.5% deoxycholic acid, 0.5% NP-40, 1 mM EDTA, 250 mM LiCl, 10 mM Tris-Cl pH7.4, ddH2O). Finally, the washed beads were resuspended in PBS, snap frozen, and sent for quantitative proteomics.
HA immunoprecipitation
Actinin-BirA* and control non-BirA* iPSC-CMs were collected at Day 25 and HA-tag IP was conducted using reagents supplied in HA-tag IP/Co-IP Application set (Thermo Scientific 26180Y). The subsequent protein immunoblots were conducted as outlined in the immunoblot section.
RNA Immunoprecipitation (RIP)
After biotin washout, Actinin-BirA* and control non-BirA* iPSC-CMs were rinsed three times with room temperature PBS. Cells were lysed with ice-cold RIP buffer (150 mM KCl, 25 mM Tris-Cl pH7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, 1x protease inhibitor, 100 U/ml RNase inhibitor, RNase-free water). Cell debris was pelleted and cleared lysate was added to magnetic Dynabeads MyOne Streptavidin C1 beads (Invitrogen 65001), which were prewashed with PBS and RIP buffer. Samples and beads were incubated on a rotator overnight at 4°C. The following day, the beads were washed three times with ice-cold RIP buffer – all steps were performed in a 4°C room. Samples were then incubated with Proteinase K at 55°C for 30 minutes. Beads were resuspended in TRIzol (Invitrogen 15596018), RNA extraction was performed immediately followed by RNA-sequencing.
Tandem Immunoprecipitation
After biotin washout, Actinin-BirA* and control non-BirA* iPSC-CMs were rinsed three times with room temperature PBS and then lysed in RIP buffer. Cleared lysates were incubated with Dynabeads Protein G (Invitrogen 10003D) pre-bound to either IgG antibody (MBL PM035) or IGF2BP2 antibody (MBL RN008P). Samples and beads were incubated on a rotator overnight at 4°C. The following day, the Protein G beads were washed three times with RIP buffer and then incubated for 12 hours with rotation at 4°C in RIP buffer containing IMP2 peptide (Genescript). The supernatant was then mixed with magnetic Dynabeads MyOne Streptavidin C1 beads (Invitrogen 65001), which were prewashed with PBS and RIP buffer. The following day, the streptavidin beads were three times washed with ice-cold RIP buffer – all steps were performed in a 4°C room. Samples were then incubated with Proteinase K at 55°C for 30 minutes. Beads were resuspended in TRIzol, RNA extraction was performed immediately followed by cDNA synthesis (outlined in Quantitative PCR section).
QUANTITATIVE PROTEOMICS
Bead digestion
After conducting streptavidin immunoprecipitation, streptavidin beads were snap frozen and sent to the Thermo Fisher Center for Multiplexed Proteomics (TCMP) at Harvard Medical School. For each tandem mass tag (TMT) mass spectrometry experiment, three biological replicates per condition were submitted.
The beads were washed three times with 50 mM Tris buffer, pH 8.0 to remove any trace of detergents and unspecific binders followed by resuspension in 1M Urea, 50 mM Tris, pH 8.0 and initially digesting it with Trypsin (5 ng/μl) at 37°C for one hour under shaking. After initial trypsin incubation, samples were centrifuged briefly and the supernatant were collected in tubes. Beads were further washed three times with 1M Urea, 50 mM Tris, pH 8.0 and washed fractions were pooled in the same tubes and left to digest overnight at room temperature. The following morning, digested peptides were reduced first with 5 mM TCEP, followed by alkylation with 10 mM Iodoacetamide, quenching alkylation with 5 mM DTT and finally quenching the digestion process with TFA. Acidified digested peptide were desalted over C18 stagetip following protocol described before (Ong et al., 2002). Briefly, the tips were prepared placing a small disc of Empore material 3M in an ordinary pipette tip, preparing a single tip for each sample. Tips were cleaned and secured with Methanol, activated with 50% acetonitrile, 0.1% TFA, equilibrated with 0.1% TFA. Acidified digested sample was added to the column and finally washed twice with 0.1% TFA solution. Liquid was passed through the pipette tip with a centrifugation. Peptides were then eluted with 80% acetonitrile, 0.1% TFA buffer thrice and dried in a speedvac. Dried desalted nine peptides samples were reconstituted with 200 mM EPPS buffer, pH 8.0 and labeled with respective first 9 of a 10-plex tandem mass tag (TMT) reagent. The 9-plex labeling reactions were performed for 1 hour at room temperature. Modification of tyrosine residues with TMT was reversed by the addition of 5% hydroxyl amine for 15 minutes and the reaction was quenched with 0.5% TFA. Samples were combined, further desalted over stage-tip, finally eluted into an Autosampler Inserts (Thermo Scientific), dried in a speedvac and reconstituted with 5% Acetonitrile-5% TFA for MS analysis.
Liquid chromatorgraphy-MS3 spectrometry (LC-MS/MS)
Labeled peptide sample from the previous step was analyzed with an LC-MS3 data collection strategy (McAlister et al., 2014) on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) equipped with a Thermo Easy-nLC 1200 for online sample handling and peptide separations. Resuspended peptides from the previous step was loaded onto a 100 μm inner diameter fused-silica micro capillary with a needle tip pulled to an internal diameter less than 5 μm. The column was packed in-house to a length of 35 cm with a C18 reverse phase resin (GP118 resin 1.8 μm, 120 Å, Sepax Technologies). The peptides were separated using a 180 min linear gradient from 5% to 42% buffer B (90% ACN + 0.1% formic acid) equilibrated with buffer A (5% ACN + 0.1% formic acid) at a flow rate of 500 nL/min across the column.
The scan sequence for the Fusion Orbitrap began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, scan range of 350 – 1350 m/z, AGC target 1 × 106, maximum injection time 50 ms, dynamic exclusion of 90 s). The “Top10” precursors was selected for MS2 analysis, which consisted of CID (quadrupole isolation set at 0.7 Da and ion trap analysis, AGC 9 × 103, Collision Energy 35%, maximum injection time 80 ms). The top ten precursors from each MS2 scan were selected for MS3 analysis (synchronous precursor selection), in which precursors were fragmented by HCD prior to Orbitrap analysis (Collision Energy 55%, max. AGC 1 × 105, maximum injection time 120 ms, resolution 50,000 and isolation window set to 1.2 – 0.8).
LC-MS3 data analysis
A suite of in-house software tools were used for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. MS/MS spectra were searched using the SEQUEST (Eng et al., 1994) algorithm against a Uniprot composite human database (human release 2017–10) with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 50 ppm, fragment ion mass tolerance of 1.0 Da, static alkylation of cysteine (57.02146 Da), static TMT labeling of lysine residues and N-termini of peptides (229.162932 Da), and variable oxidation of methionine (15.99491 Da). Peptide spectral matches were filtered to a 1% false discovery rate (FDR) using the target-decoy strategy combined with linear discriminant analysis. The proteins were filtered to a < 1% FDR. TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Proteins were quantified only from peptides with a summed SN threshold of > 200 and MS2 isolation specificity of 0.5. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (< 200 summed signal-to-noise across 10 channels and < 0.5 precursor isolation specificity).
Before analysis, proteins previously reported to non-specifically bind BirA* were removed (Roux et al., 2018). For analysis, TMT raw intensity values (raw data in Table S1) underwent Log2-fold-change (L2FC) calculation and enrichment analysis. For actinin proteome analysis (Figure 1), data from experiments 1 and 2 were utilized to determine significantly enriched proteins by calculating the L2FC of proteins from Actinin-BirA* relative to control non-BirA*. Proteins considered to be significantly enriched were those with L2FC ≥ 1 and false discovery rate (FDR) < 0.05 (using two-way ANOVA followed by a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli to correct for multiple comparisons (Benjamini et al., 2006)). For sarcomere assembly actinin proteome analysis (Figure S1), data from experiment 2 was similarly analyzed to first determine enriched proteins using cTnT-WT-Actinin-BirA* relative to control non-BirA* (n = 294 hits with L2FC ≥ 1 and FDR < 0.05). These enriched proteins were then analyzed for sarcomere-dependency status by calculating the L2FC of proteins from cTnT-WT-Actinin-BirA* relative to cTnT-KO-Actinin BirA*, with significant hits being defined as those with L2FC ≥ 1 and FDR < 0.05 (n = 47). Additionally, Z-disk exclusive hits were those significantly enriched in cTnT-WT-Actinin-BirA* relative to control non-BirA*, but not in the cTnT-KO-Actinin-BirA* relative to control non-BirA* (n = 24 hits with L2FC ≥ 1 and FDR < 0.05). For visualization of data, Search Tool for the Retrieval of Interacting Genes/Proteins (STRING v. 11.0) was utilized for generation of interaction scores (proteins were not distinguished by isoform). Cytoscape (v.3.7.2) was utilized to generate interaction maps with an organic layout and the utilization of the CLUSTER and BINGO features for group clustering and appropriate GO term identification for each cluster. Quantitative proteomics data deposited to the ProteomeXchange Consortium identifier PXD018040.
Quantitative PCR, RNA sequencing, and analysis
cDNA was synthesized using Superscript III First-Strand synthesis (Invitrogen 18080–400). Gene-specific PCR primers were designed or identified from the literature and transcripts were quantified using Fast SYBR Green (Applied Biosystems 4385612) on a ViiA7 Real-Time PCR system (Applied Biosystems).
RNA Samples were sent for sequencing at the University of Connecticut Institute for Systems Genomics. RNA sequencing libraries were generated using Illumina TruSeq Stranded Total RNA library preparation (Ribo-Zero depletion for rRNA was utilized for RIP-seq). Illumina NextSeq 500/550 sequencing was conducted with the v2.5 300 cycle reagent kit 9 (High Output). Estimated total single end reads per sample = 30–35M 150bp PE reads. Samples sequences were aligned with STAR to the hg38 human genome. In order to look at differential expression, DESeq2 (Bioconductor) was utilized. Gene Set Enrichment Analysis (GSEA) was utilized to determine GO terms for datasets. Top 10 GO terms are presented. Furthermore, for ETC complexes, glycolysis, and sarcomere gene sets – GSEA gene sets were utilized and those with an average ≥ 5 TPM value were included in the dataset. TPM (transcripts per million) is a normalization technique for transcriptomic data to adjust for transcript count read depth and transcript length, such that all transcript counts are normalized by transcript length and then presented as a proportion out of 1 million counts. Genome sequencing datasets are deposited at GEO under accession numbers: GSE144806.
Protein Immunoblotting
Samples of interest were lysed in RIPA buffer (Cell Signaling 9806) containing protease inhibitor cocktail (Roche 11836170001), 1 mM PMSF, and phosphatase inhibitor (Pierce A32957), unless noted otherwise. Protein lysate concentrations were normalized using Pierce BCA (Thermo 23225), and then reduced and denatured in sample buffer (Thermo 39000). Lysates were separated on Bio-Rad 4%–20% Mini-PROTEAN TGX precast gels, transferred via Bio-Rad Trans-Blot Turbo onto PVDF membranes (Bio-Rad 1704272), blocked in TBS-T (TBS with 0.1% Tween-20) containing 5% BSA, and probed overnight at 4°C with primary antibody. The next day, blots were washed in TBS-T and probed with HRP-linked secondary antibody (Cell Signaling 7076; 7074; Streptavidin-HRP 3999S) for 1 hour. Signal detection was performed using ECL substrate (Thermo 34577; 34095) and a Bio-Rad ChemiDoc MP imaging system. Blot images were digitally processed and analyzed in ImageJ. The primary antibodies used were as follows: anti-actinin (isoforms 1–4) (Cell Signaling 6487), anti-HA (Cell Signaling 14793), anti-IGF2BP2 (MBL RN008P), anti-PBCBP1 (Abcam 74793), anti-PCBP2 (MBL RN250P), anti-SERBP1 (Abcam 55993), anti-TCAP (BD 612328), anti-FLAG (Cell Signaling 2368), anti-GAPDH (Cell Signaling 2118) and anti-MYOM (mMaC myomesin B4 was deposited to the DSHB by Perriard, J.-C.).
Immunofluorescence and RNA FISH
For standard immunofluorescence images, cells were fixed on coverslips (Fisherbrand 12–545-100) in PBS containing 4% paraformaldehyde (EMS 50–980-487) and then permeabilized and blocked in PBS-T (PBS containing 0.1% Triton-X) with 1% BSA (Fisher BP1605100). Cells were probed with primary antibody in PBS-T with BSA overnight at 4°C. The following primary antibodies were used:, anti-Actinin (Sigma A7811), anti-IGF2BP2 (Bethyl A303–316A), and anti-FLAG (Cell Signaling 2368). The next day coverslips were washed in PBS-T, incubated for 1 hour at room temperature with 1 μg/mL DAPI (Invitrogen D1306) and appropriate secondary antibody (Invitrogen A-11005 or A-11008) or streptavidin-488 (Invitrogen S11223), washed, and mounted onto slides (Corning 2948) in ProLong Diamond mountant (Invitrogen P36965). An Andor Dragonfly 500 confocal microscope system using a Zyla sCMOS camera and a Leica DMi8 63x oil immersion lens was used for imaging slides. Andor Fusion software was used to acquire images, which were converted to OME-TIFF format using Bitplane Imaris and analyzed in ImageJ.
ViewRNA direct fluorescence RNA in situ hybridization was conducted using the manufacturer’s protocol (Invitrogen 19887). The following human probes were utilized: NDUFA1 (Invitrogen VA6–3172657), NDUFA8 (Invitrogen VA1–3003564), TTN (Invitrogen VA6–3173894), HIST1H1E (Invitrogen VA6–3172001), and HRC (Invitrogen VA6–3172126). Final step of coverslip mounting and image acquisition are described above. For distance quantification, Imaris software was utilized. Images first underwent background subtraction to minimize background signal and allow for clear puncta visualization. The distance transformation tool on Imaris was used to quantify distance (in microns) of RNA puncta and actinin structures.
Mammalian-Two Hybrid
Actinin (full-length and domains), IGF2BP2 (full-length and domains), PCBP1, PBCBP2, and SERBP1 were amplified from human cDNA and cloned into the appropriate vectors (pACT or pBIND) provided by the CheckMate Mammalian Two-Hybrid kit (Promega E2440). To generate mutant variants of actinin, variant-specific mutagenic primers were designed (Agilent QuikChange) and paired with universal primers specific to pACT or pBIND vectors, such that PCR amplification would generate a mutant actinin split into two PCR fragments at the point of the mutation (sequences in Table S3). This strategy enables the use of a single, three-fragment Gibson Assembly reaction, avoiding potential polymerase errors that may go un-sequenced in classical PCR mutagenesis involving amplification of the entire backbone. The appropriate combination of edited pACT and pBIND vectors, in addition to the kit-provided pGluc5 vector, were transfected using PEI into HEK293T cells (ATCC CRL-3216), as outlined in the manufacturer’s protocol. In addition, the kit-provided positive and negative controls were transfected simultaneously for all experiments to verify proper signal production. Luciferase and Renilla activity were measured using manufacturer’s protocol (Promega E1910) utilizing BioTek’s Synergy 2 multi-mode microplate reader.
PyMOL Molecular Viewer
PyMOL version 2.4.0 was utilized to generate figures illustrating actinin’s ribbon structure and the plugin “APBS Electrostatics” was used to visualize electrostatic surfaces. The coordinates of the crystal structure of actinin’s rod domain were found on the Protein Data Bank (PDB ID: 1HCI). Protein structures were exported from PyMOL and presented in figures.
Lentivirus Production
HEK293T cells (ATCC CRL-3216) were seeded onto 150 mm plates in 20 mL DMEM (GIBCO 11965092) supplemented with 10% FBS (Gemini 100–106), GlutaMAX (GIBCO 35050061), and 1 mM sodium pyruvate (GIBCO 11360070). HEK293T cells were transfected with 18ug of lentiviral transfer vector, 12 μg psPAX2 (Addgene 12260), and 6 μg pCMV-VSV-G (Addgene 8454) (Stewart et al., 2003). Media was replenished the following day and virus-containing media was harvested at 48, 72, and 96 hours post-transfection, followed by concentration using PEG-6000 as previously described (Kutner et al., 2009). For titer determination - iPSCs were transduced with a serial dilution of lentivirus, followed by treating with appropriate antibiotic (1μg/ml puromycin), and counting resistant colony-forming units.
Individual Gene Knockdown and Overexpression Constructs
Lentiviral-based hairpins or overexpression constructs were transduced into iPSC-CMs at a MOI of 2 in RPMI-B27 (Sarbassov et al., 2005). Media was replenished the following day and cells were analyzed 7–10 days post transduction.
His Protein Interaction Pull-Down
For generation of bait bacterial expression vector, actinin (WT or E445A) was cloned into pNIC28-Bsa4 (Addgene 26103) (Savitsky et al., 2010). Vectors were transformed into BL21 (DE3) competent E. coli (NEB C2527I). A single colony was resuspended in 5 mL liquid culture with antibiotic and incubated at 37°C until OD600 reached 0.4–0.8. At this point IPTG (ThermoFisher R1171) induction at final concentration of 500 μM was begun for 4 hours at 37°C. For bait preparation, protocol from Pierce Pull-Down PolyHis Protein:Protein Interaction Kit (ThermoFisher 21277) was followed. Prey protein was prepared from day 30 iPSC-CMs. Pull-down was conducted using instructions and reagents provided in Pierce Pull-Down PolyHis Protein:Protein Interaction Kit (ThermoFisher 21277). Protein concentration of bait protein lysates were determined using BCA (ThermoFisher 23225) and equal amount of protein was loaded into columns for each condition.
OCR measurements using Seahorse
30,000 wild-type CMs were plated on Seahorse XFe96 Cell Culture Microplates (Agilent 101085–004) and lentiviral overexpression constructs (Actinin-WT or Actinin-E445A; cTnT-WT or cTnT-R92Q) were added the next day. The cells were cultured for 7 days before performing the Seahorse XF Cell Mito Stress Test (Agilent 103015–100) according to manufacturer’s protocol. The following final concentrations of compounds were injected: oligomycin (1 μM), FCCP (1 μM), and rotenone/antimycin A (0.5 μM).
Cardiac microtissue (CMT) assay
CMTs were generated as previously described (Cohn et al., 2019; Hinson et al., 2015). Briefly, polydimethylsiloxane (PDMS) (Corning Sylgard 184) cantilever devices were molded from SU-8 masters, which were embedded with fluorescent microbeads (Thermo F8820). Singularized iPSC-CMs were mixed with human cardiac fibroblasts and spun into PDMS devices containing a collagen-based ECM. For overexpression experiments, lentivirus was added to iPSC-CMs 7 days prior to generation of CMTs. CMT function was measured once tissues compacted and were visibly pulling the cantilevers. For acquisition of functional data, tissues were stimulated at 1 Hz with a C-Pace EP stimulator (IonOptix) with platinum wire electrodes and fluorescence images were taken at 25 Hz with a Andor Dragonfly microscope equipped with enclosed live-cell chamber (Okolabs). Displacement of fluorescent microbeads was tracked using the ImageJ ParticleTracker plug-in and twitch force was calculated. The cantilever spring constants were determined using the elastic modulus of PDMS and the dimensions of the tissue gauge devices previously described (Boudou et al., 2012).
LDH Release Cytotoxicity Assay
Wild-type CMs were plated and lentiviral overexpression constructs (Actinin-WT or Actinin-E445A + cTnT-WT or cTnT-R92Q) were added the next day. The cells were cultured for 7 days before being switched to glucose-free media (GIBCO 11966025) supplemented with 10 mM galactose (Sigma 59234). Media was collected and replenished on day 0, 1, 3, and 5. Cytotoxicity was determined utilizing LDH toxicity assay kit (ThermoFisher C20300).
QUANTIFICATION AND STATISTICAL ANALYSIS
Obtained data were analyzed and visualized using Microsoft Excel, GraphPad Prism or R. Data are presented as mean ± standard error of the mean (SEM) unless otherwise noted. Statistical comparisons were conducted as described in the text or figure legends. Statistical significance was defined by p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****) unless otherwise stated.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Alpha-actinin | Cell Signaling | Cat. #6487; RRID:AB_11179206 |
| GAPDH | Cell Signaling | Cat. #2118; RRID:AB_561053 |
| HA-Tag | Cell signaling | Cat. #14793; RRID:AB_2572291 |
| IGF2BP2 | MBL | Cat. #RN008P; RRID:AB_1570641 |
| PCBP1 | Abcam | Cat. #74793; RRID:AB_1281060 |
| PCBP2 | MBL | Cat. #RN250P; RRID:AB_1953052 |
| SERBP1 | Abcam | Cat. #55993; RRID:AB_882495 |
| TCAP | BD | Cat. #612328; RRID:AB_399643 |
| FLAG-Tag | Cell Signaling | Cat. #2368; RRID:AB_2217020 |
| MYOM | DSHB | Cat. #760349; RRID:AB_760349 |
| Streptavidin-HRP | Cell Signaling | Cat. #3999S; RRID:AB_10830897 |
| Goat anti-rabbit IgG HRP | Cell Signaling | Cat. #7074; RRID:AB_2099233 |
| Goat anti-mouse IgG HRP | Cell Signaling | Cat. #7076; RRID:AB_330924 |
| Alpha-actinin | Sigma | Cat. #A7811; RRID:AB_476766 |
| IGF2BP2 | Bethyl | Cat. #A303–316A; RRID:AB_10951966 |
| Streptavidin-488 | Invitrogen | Cat. #S11223; RRID:AB_2315383 |
| Goat anti-mouse 594 | Invitrogen | Cat. #A11005; RRID:AB_141372 |
| Goat anti-rabbit 488 | Invitrogen | Cat. #A11008; RRID:AB_143165 |
| NDUFA1 (RNA probe) | Invitrogen | Cat. #VA6–3172657 |
| NDUFA8 (RNA probe) | Invitrogen | Cat. #VA1–3003564 |
| TTN (RNA probe) | Invitrogen | Cat. #VA6–3173894 |
| HIST1H1E (RNA probe) | Invitrogen | Cat. #VA6–3172001 |
| HRC (RNA probe) | Invitrogen | Cat. #VA6–3172126 |
| IgG | MBL | Cat. #PM035; RRID:AB_10805234 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NextSeq 500/550 v2 | Illumina | Cat. #FC4042005 |
| HiFi DNA Assembly | New England BioLabs | Cat. #E2621 |
| T4 DNA ligase | New England BioLabs | Cat. #M0202S |
| Seahorse XF Cell Mito Stress | Agilent | Cat. #103015–100 |
| Qubit dsDNA kit | Invitrogen | Cat. #Q32854 |
| Fast SYBR Green Master Mix | Applied Biosystems | Cat. #4385612 |
| HA-tag IP/Co-IP Application set | Thermo Scientific | Cat. #26180Y |
| Superscript III First-Strand synthesis | Invitrogen | Cat. #18080–400 |
| ViewRNA in situ hybridization kit | Invitrogen | Cat. #19887 |
| CheckMate Mammalian Two-Hybrid kit | Promega | Cat. #E2440 |
| Dual-Luciferase Reporter assay system | Promega | Cat. #E1910 |
| Pull-down PolyHis Protein-protein interaction kit | ThermoFisher | Cat. #21277 |
| LDH toxicity assay kit | ThermoFisher | Cat. #C20300 |
|
| ||
| Experimental models: cell lines and animals | ||
|
| ||
| PGP1 human iPSCs | Coriell | Cat. #GM23338 |
| HEK293T | ATCC | Cat. #CRL3216 |
|
| ||
| Oligonucleotides | ||
|
| ||
| HR220PA | SBI | Cat. #HR220PA-1 |
| psPAX2 | Gift from Didier Trono | Addgene Cat. #12260 |
| pCMV-VSV-G | Stewart et al., 2003 | Addgene Cat. #8454 |
| pCas9-GFP | Ding et al., 2013 | Addgene Cat. #44719 |
| pLenti CMV GFP Puro | Campeau et al., 2009 | Addgene Cat. #17448 |
| pLKO.1 | Sarbassov et al., 2005 | Addgene Cat. #1864 |
| pNIC28-Bsa4 | Savitsky et al., 2010 | Addgene Cat. #26103 |
|
| ||
| Software and algorithms | ||
|
| ||
| STAR (v.2.5.4a) | GitHub | https://github.com/alexdobin/STAR |
| Molecular Signature Database | Broad | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| DESeq2 | GitHub | https://git.bioconductor.org/packages/DESeq2 |
| STRING (v. 11.0) | STRING | https://string-db.org/ |
| GraphPad Prism | GraphPad Software | http://graphad.com/scientfiic-software/prism |
| FlowJo | FLOWJO | https://flowjo.com |
| Fiji | NIH | https://fiji.sc |
| Cytoscape (v. 3.7.2) | Cytoscape | https://cytoscape.org/ |
| PyMOL (v. 2.4.0) | PyMOL | https://pymol.org/2/ |
| Imaris visualization and analysis software | BitPlane | Institutional license https://imaris.oxinst.com/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| IWP4 | Tocris | Cat. #5214 |
| CHIR99021 | Tocris | Cat. #4423 |
| Y-276932 | Tocris | Cat. #1254 |
| Fibronectin (human) | Corning | Cat. #33016015 |
| Matrigel GFR | Corning | Cat. #354230 |
| Biotin | ThermoFisher | Cat. #29129 |
| ROCK inhibitor Y-27632 | Tocris | Cat. #1254 |
| CHIR99021 | Cayman | Cat. #13122 |
| IMP2 peptide | Genescript | Custom order |
|
| ||
| Other | ||
|
| ||
| Accutase | BD Biosciences | Cat. #561527 |
| RPMI 1640 | ThermoFisher | Cat. #11875093 |
| DMEM (glucose-free) | GIBCO | Cat. #11966025 |
| Opti-MEM | GIBCO | Cat. #31985062 |
| Polyethylenimine (PEI) | Polysciences | Cat. #239662 |
| PEG-6000 | Millipore Sigma | Cat. #528877 |
| Trypsin-EDTA | GIBCO | Cat. #25200056 |
| Hygromycin | Invitrogen | Cat. #10687010 |
| TRIzol | Invitrogen | Cat. #15596018 |
| Dynabeads MyOne Streptavidin C1 beads | Invitrogen | Cat. #65001 |
| Dynabeads Proteins G | Invitrogen | Cat. #10003D |
| Puromycin | GIBCO | Cat. #A1113803 |
| B27 supplement | GIBCO | Cat. #17504044 |
| B27 supplement (-insulin) | GIBCO | Cat. #A1895601 |
| Penicillin/Streptomycin | GIBCO | Cat. #15140122 |
| GlutaMAX | GIBCO | Cat. #35050061 |
| Sodium lactate | Sigma | Cat. #71718 |
| mTeSR1 | STEMCELL Technologies | Cat. #85850 |
| DAPI | Invitrogen | Cat. #D1306 |
| Fetal bovine serum (heat-inactivated) | Gemini | Cat. #100–106 |
| PBS | GIBCO | Cat. #10010049 |
| PDMS | Corning | Cat. #4019862 |
| Florescent microbeads | ThermoFisher | Cat. #F8820 |
| Paraformaldehyde | EMS | Cat. #50–980-487 |
| 10 mM galactose | Sigma | Cat. #59234 |
| KOD Hot Start Master Mix | MilliporeSigma | Cat. #71842–4 |
| SuperSignal West Pico Chemiluminescent substrate | ThermoFisher | Cat. #35477 |
| SuperSignal West Femto Chemiluminescent substrate | ThermoFisher | Cat. #34095 |
| ProLong Gold Mountant | Invitrogen | Cat. #P36965 |
| 4–20% Mini-PROTEAN TGX gels | Bio-Rad | Cat. #4561095 |
| RTA PVDF Transfer kit | Bio-Rad | Cat. #1704272 |
| BSA, Fraction V | Fisher | Cat. #BP1605 |
| Pyruvate | GIBCO | Cat. #11360070 |
| Tris-Glycine SDS 10X | Fisher | Cat. #BP13414 |
| BL21 E.coli cells | New England BioLabs | Cat. #C2527I |
| IPTG | ThermoFisher | Cat. #R1171 |
| DH5a E.coli cells | New England BioLabs | Cat. #C2987 |
| Stbl3 E.coli cells | Invitrogen | Cat. #C737303 |
| NEB stable E.coli cells | New England BioLabs | Cat. #C3040 |
| 4mm cuvettes | Bio-Rad | Cat. #1652088 |
| RIPA buffer | Cell Signaling | Cat. #9806 |
| Protease inhibitor cocktail | Roche | Cat. #11836170001 |
| Phosphatase inhibitor | Pierce | Cat. #A32957 |
| BCA kit | ThermoFisher | Cat. #23225 |
| Protein sample buffer | ThermoFisher | Cat. #39000 |
|
| ||
| Deposited data | ||
|
| ||
| Proteomics data | ProteomeXchange | PXD018040 |
| RIP-seq data | GEO | GSE144806 |
Highlights.
BioID identifies 324 actinin proximity partners through sarcomere assembly
IGF2BP2 directly binds actinin’s rod domain through its KH domain
IGF2BP2-actinin interactions determine ETC transcript localization
Actinin-IGF2BP2 interactions regulate metabolic adaptation to sarcomere function
ACKNOWLEDGMENTS
This study could not have been completed without the assistance of Bo Reese (UConn Center for Genome Innovation) for RNA sequencing and RIP-seq and Anthony Carcio (The Jackson Laboratory for Genomic Medicine) for flow cytometry. TMT proteomics and quantitative analyses was supported by Sanjukta Thakurta from the Thermo Fisher Center for Multiplexed Proteomics at Harvard Medical School. Proteomics presentation was discussed and reviewed by Dr. Andy Greene of the Jackson Laboratory. Artwork was created using BioRender. Funding for this study was obtained from the National Institutes of Health (J.T.H., HL125807, HL142787, and EB028898), the American Heart Association (F.A.L., PRE35110005, and A.M.P., PRE34381021) and institutional start-up funds (UConn Health).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109512.
REFERENCES
- Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, and Harper ME (2011). Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 6, e28536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach D, Bantle S, Keller S, Hinderling V, Leu M, Ehler E, and Perriard JC (1999). Different domains of the M-band protein myomesin are involved in myosin binding and M-band targeting. Mol. Biol. Cell 10, 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye TT, Soni S, van Veen TA, van der Heyden MA, Cappadona S, Varro A, de Weger RA, de Jonge N, Vos MA, Heck AJ, and Scholten A (2012). Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol 52, 511–518. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Krieger AM, and Yekutieli D (2006). Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93, 491–507. [Google Scholar]
- Beqqali A, Monshouwer-Kloots J, Monteiro R, Welling M, Bakkers J, Ehler E, Verkleij A, Mummery C, and Passier R (2010). CHAP is a newly identified Z-disc protein essential for heart and skeletal muscle function. J. Cell Sci 123, 1141–1150. [DOI] [PubMed] [Google Scholar]
- Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT, and Protasi F (2009). Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 20, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, Zandstra PW, Epstein JA, Margulies KB, and Chen CS (2012). A microfabricated platform to measure and manipulate the mechanics of engineered cardiac microtissues. Tissue Eng. Part A 18, 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, and Semsarian C (2010). Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J. Am. Coll. Cardiol 55, 1127–1135. [DOI] [PubMed] [Google Scholar]
- Chopra A, Kutys ML, Zhang K, Polacheck WJ, Sheng CC, Luu RJ, Eyckmans J, Hinson JT, Seidman JG, Seidman CE, and Chen CS (2018). Force Generation via β-Cardiac Myosin, Titin, and α-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Dev. Cell 44, 87–96.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, Meredith E, Chen YS, Atamanuk K, Huey BD, and Hinson JT (2019). A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Reports 12, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, and Musunuru K (2013). Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djinović-Carugo K, Young P, Gautel M, and Saraste M (1999). Structure of the alpha-actinin rod: molecular basis for cross-linking of actin filaments. Cell 98, 537–546. [DOI] [PubMed] [Google Scholar]
- Djinovic-Carugo K, Gautel M, Ylänne J, and Young P (2002). The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett. 513, 119–123. [DOI] [PubMed] [Google Scholar]
- Du A, Sanger JM, Linask KK, and Sanger JW (2003). Myofibrillogenesis in the first cardiomyocytes formed from isolated quail precardiac mesoderm. Dev. Biol. 257, 382–394. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Ferrantini C, Coppini R, Pioner JM, Gentile F, Tosi B, Mazzoni L, Scellini B, Piroddi N, Laurino A, Santini L, et al. (2017). Pathogenesis of Hypertrophic Cardiomyopathy is Mutation Rather Than Disease Specific: A Comparison of the Cardiac Troponin T E163R and R92Q Mouse Models. J. Am. Heart Assoc 6, e005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA, and Amado F (2010). Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 10, 3142–3154. [DOI] [PubMed] [Google Scholar]
- Foley KS, and Young PW (2014). The non-muscle functions of actinins: an update. Biochem. J. 459, 1–13. [DOI] [PubMed] [Google Scholar]
- Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH, and Philpott CC (2014). Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 111, 8031–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, Kalma Y, Viukov S, Maza I, Zviran A, et al. (2013). Derivation of novel human ground state naive pluripotent stem cells. Nature 504, 282–286. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Abe KT, and Raught B (2019). Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol 48, 44–54. [DOI] [PubMed] [Google Scholar]
- Grison M, Merkel U, Kostan J, Djinović-Carugo K, and Rief M (2017). α-Actinin/titin interaction: A dynamic and mechanically stable cluster of bonds in the muscle Z-disk. Proc. Natl. Acad. Sci. USA 114, 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Arimura T, Itoh-Satoh M, Ueda K, Hohda S, Inagaki N, Takahashi M, Hori H, Yasunami M, Nishi H, et al. (2004). Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J. Am. Coll. Cardiol 44, 2192–2201. [DOI] [PubMed] [Google Scholar]
- Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et al. (2015). HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie N, Weinberger L, Tang WW, Kobayashi T, Viukov S, Manor YS, Dietmann S, Hanna JH, and Surani MA (2015). SOX17 is a critical specifier of human primordial germ cell fate. Cell 160, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, Radovanovic I, Rheinbay E, Provero P, and Stamenkovic I (2012). Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 26, 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadpour MM, Tardiff JC, Pinz I, and Ingwall JS (2003). Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. J. Clin. Invest. 112, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Hill MA, and Sowers JR (2018). Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res 122, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaewsapsak P, Shechner DM, Mallard W, Rinn JL, and Ting AY (2017). Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 6, e29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodríguez-Pérez JC, Allen PG, Beggs AH, and Pollak MR (2000). Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet 24, 251–256. [DOI] [PubMed] [Google Scholar]
- Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, and Roux KJ (2014). Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 111, E2453–E2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H, Chiba K, Tanaka H, Miyazaki K, Sakai M, et al. (2013). ACTN1 mutations cause congenital macrothrombocytopenia. Am. J. Hum. Genet 92, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, and Reiser J (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc 4, 495–505. [DOI] [PubMed] [Google Scholar]
- Lange S, Ehler E, and Gautel M (2006). From A to Z and back? Multicompartment proteins in the sarcomere. Trends Cell Biol. 16, 11–18. [DOI] [PubMed] [Google Scholar]
- Lécuyer E, Yoshida H, Parthasarathy N, Alm C, Babak T, Cerovina T, Hughes TR, Tomancak P, and Krause HM (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174–187. [DOI] [PubMed] [Google Scholar]
- Lewis YE, Moskovitz A, Mutlak M, Heineke J, Caspi LH, and Kehat I (2018). Localization of transcripts, translation, and degradation for spatiotemporal sarcomere maintenance. J. Mol. Cell. Cardiol 116, 16–28. [DOI] [PubMed] [Google Scholar]
- Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, and Palecek SP (2013). Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc 8, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem RK (2016). Cytoskeletal Integrators: The Spectrin Superfamily. Cold Spring Harb. Perspect. Biol 8, a018259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymn RW, and Taylor EW (1971). Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10, 4617–4624. [DOI] [PubMed] [Google Scholar]
- Martin KC, and Ephrussi A (2009). mRNA localization: gene expression in the spatial dimension. Cell 136, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AC, and Young PW (2015). The actinin family of actin cross-linking proteins - a genetic perspective. Cell Biosci. 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Sugihara Y, Shibakawa M, Oshima K, Matsuda T, and Nadano D (2018). The mRNA-binding protein Serbp1 as an auxiliary protein associated with mammalian cytoplasmic ribosomes. Cell Biochem. Funct 36, 312–322. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Pettinato AM, Ladha FA, Mellert DJ, Legere N, Cohn R, Romano R, Thakar K, Chen YS, and Hinson JT (2020). Development of a Cardiac Sarcomere Functional Genomics Platform to Enable Scalable Interrogation of Human TNNT2 Variants. Circulation 142, 2262–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regué L, Ji F, Flicker D, Kramer D, Pierce W, Davidoff T, Widrick JJ, Houstis N, Minichiello L, Dai N, and Avruch J (2019). IMP2 Increases Mouse Skeletal Muscle Mass and Voluntary Activity by Enhancing Autocrine Insulin-Like Growth Factor 2 Production and Optimizing Muscle Metabolism. Mol. Cell. Biol 39, e00528–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro E.d.A. Jr., Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjöblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, et al. (2014). The structure and regulation of human muscle α-actinin. Cell 159, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, and Wallace DC (1992). Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol 48, 122–126. [DOI] [PubMed] [Google Scholar]
- Rode C, Siebert T, Tomalka A, and Blickhan R (2016). Myosin filament sliding through the Z-disc relates striated muscle fibre structure to function. Proc. Biol. Sci 283, 20153030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Burke B, and May DG (2018). BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci 91, 19.23.11–19.23.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph F, Hüttemeister J, da Silva Lopes K, Jüttner R, Yu L, Bergmann N, Friedrich D, Preibisch S, Wagner E, Lehnart SE, et al. (2019). Resolving titin’s lifecycle and the spatial organization of protein turnover in mouse cardiomyocytes. Proc. Natl. Acad. Sci. USA 116, 25126–25136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph F, Fink C, Hüttemeister J, Kirchner M, Radke MH, Lopez Carballo J, Wagner E, Kohl T, Lehnart SE, Mertins P, and Gotthardt M (2020). Deconstructing sarcomeric structure-function relations in titin-BioID knock-in mice. Nat. Commun 11, 3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, and Philpott CC (2017). PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Invest 127, 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JM, and Sanger JW (2008). The dynamic Z bands of striated muscle cells. Sci. Signal 1, pe37. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, and Gileadi O (2010). High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol 172, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, et al. ; Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research (2007). Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316, 1331–1336. [DOI] [PubMed] [Google Scholar]
- Schultheiss T, Choi J, Lin ZX, DiLullo C, Cohen-Gould L, Fischman D, and Holtzer H (1992). A sarcomeric alpha-actinin truncated at the carboxyl end induces the breakdown of stress fibers in PtK2 cells and the formation of nemaline-like bodies and breakdown of myofibrils in myotubes. Proc. Natl. Acad. Sci. USA 89, 9282–9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, and Lopaschuk GD (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev 85, 1093–1129. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum SA, Carson CC, Lager PJ, and Keene JD (2000). Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. USA 97, 14085–14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toepfer CN, Sharma A, Cicconet M, Garfinkel AC, Mücke M, Neyazi M, Willcox JAL, Agarwal R, Schmid M, Rao J, et al. (2019). SarcTrack. Circ. Res 124, 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, et al. (2013). Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen JY, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virel A, and Backman L (2004). Molecular evolution and structure of alpha-actinin. Mol. Biol. Evol 21, 1024–1031. [DOI] [PubMed] [Google Scholar]
- Weiss GA, Watanabe CK, Zhong A, Goddard A, and Sidhu SS (2000). Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. USA 97, 8950–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Barro MV, Makarenkova HP, Sanger JW, and Sanger JM (2014). Localization of sarcomeric proteins during myofibril assembly in cultured mouse primary skeletal myotubes. Anat. Rec. (Hoboken) 297, 1571–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR Jr., Ye Z, Liu F, et al. (2005). ASF/SF2-regulated CaMKII-delta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120, 59–72. [DOI] [PubMed] [Google Scholar]
- Ylänne J, Scheffzek K, Young P, and Saraste M (2001). Crystal structure of the alpha-actinin rod reveals an extensive torsional twist. Structure 9, 597–604. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Si Y, Ma N, and Mei J (2015). The RNA-binding protein PCBP2 inhibits Ang II-induced hypertrophy of cardiomyocytes though promoting GPR56 mRNA degeneration. Biochem. Biophys. Res. Commun 464, 679–684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome sequencing datasets for RIP-sequencing are deposited at GEO under accession numbers: GSE144806. Quantitative proteomics data deposited to the ProteomeXchange Consortium identifier PXD018040.
No unique code was generated for the analysis.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.