

Abstract
Mitochondria are essential organelles because of their function in energy conservation. Here, we show an involvement of mitochondria in phytochrome‐dependent light sensing in fungi. Phytochrome photoreceptors are found in plants, bacteria, and fungi and contain a linear, heme‐derived tetrapyrrole as chromophore. Linearization of heme requires heme oxygenases (HOs) which reside inside chloroplasts in planta. Despite the poor degree of conservation of HOs, we identified two candidates in the fungus Alternaria alternata. Deletion of either one phenocopied phytochrome deletion. The two enzymes had a cooperative effect and physically interacted with phytochrome, suggesting metabolon formation. The metabolon was attached to the surface of mitochondria with a C‐terminal anchor (CTA) sequence in HoxA. The CTA was necessary and sufficient for mitochondrial targeting. The affinity of phytochrome apoprotein to HoxA was 57,000‐fold higher than the affinity of the holoprotein, suggesting a “kiss‐and‐go” mechanism for chromophore loading and a function of mitochondria as assembly platforms for functional phytochrome. Hence, two alternative approaches for chromophore biosynthesis and insertion into phytochrome evolved in plants and fungi.
Keywords: chromophore, heme, heme oxygenase, metabolon, phytochrome
Subject Categories: Metabolism; Microbiology, Virology & Host Pathogen Interaction; Organelles
Characterization of fungal heme oxygenases suggests a role for the mitochondrial outer membrane in chromophore production for light‐sensing phytochromes.

Introduction
Phytochrome is an evolutionarily conserved red‐light photosensor in plants, bacteria, and fungi (Lamparter, 2004; Lamparter et al, 2017; Yu & Fischer, 2019; Rockwell & Lagarias, 2020). While its functions have been well studied in plants, the analyses of bacterial phytochromes allowed to provide the first structural information of these proteins (Hughes et al, 1997; Vierstra & Zhang, 2011; Schmidt et al, 2018). Light controls developmental decisions as well as metabolic properties in fungi and in plants through the phytochrome (Blumenstein et al, 2005; Ulijasz & Vierstra, 2011). Fungal phytochrome is best studied in Aspergillus nidulans, but it is also present in many other fungi (Froehlich et al, 2005; Purschwitz et al, 2008; Schumacher, 2017; Corrochano, 2019; Igbalajobi et al, 2019; Yu & Fischer, 2019; Schumacher & Gorbushina, 2020). Furthermore, phytochrome has been recently described also as thermosensor in A. nidulans and in Arabidopsis thaliana (Jung et al, 2016; Legris et al, 2016; Yu et al, 2019). However, how the phytochrome activates downstream signaling cascades is fundamentally different in plants and in fungi. Thus, whereas in plants activated phytochrome shuttles into the nucleus to interact with transcription factors (phytochrome‐interacting factors, PIFs) (Pfeiffer et al, 2012; Pham et al, 2018; Oh et al, 2020), in fungi, phytochrome interacts in the cytoplasm with the phosphotransfer protein YpdA and promotes signal transduction through the HOG (high osmolarity glycerol) signaling pathway (Yu et al, 2016; Yu & Fischer, 2019). In addition, in fungi, a fraction of phytochrome acts inside the nucleus by interacting with several transcription factors and enzymes required for chromatin modification (Purschwitz et al, 2008; Hedtke et al, 2015; Rauscher et al, 2016).
A central component for the photosensory function of all phytochromes is a linear tetrapyrrole, derived from heme. Ring opening of heme molecules is achieved by heme oxygenases (HOs), which convert heme in an oxygen‐dependent reaction into biliverdin IXα, iron, and carbon monoxide. In Arabidopsis thaliana, four heme oxygenases have been described, all of which are nuclear encoded proteins with a signal peptide sequence at their N‐termini for translocation into chloroplasts (Davis et al, 1999; Muramoto et al, 1999; Davis et al, 2001; Gisk et al, 2010).
Despite the well‐established roles of phytochrome as light and temperature sensor in A. nidulans, the biosynthesis of the chromophore remains enigmatic. In the genome of A. nidulans, no good heme oxygenase candidate can be identified using the cyanobacterial, bacterial, or mammalian heme oxygenases as bait. Here, we studied two putative heme oxygenases from A. alternata. Alternaria alternata contains a blue‐light sensing system but used also phytochrome for light sensing (Igbalajobi et al, 2019; Igbalajobi et al, 2020). Surprisingly, deletion of either of the two putative heme oxygenase genes caused a “blind” phenotype. They are able to form homo‐ and heterodimers and physically interact with phytochrome. Both enzymes together with phytochrome are attached to the mitochondrial outer membrane, suggesting metabolon formation. This could be a general strategy to separate heme linearization from heme degradation. Our study revealed a novel function of mitochondria as chromophore‐assembly platforms for phytochrome.
Results
Two heme oxygenases are required for phytochrome function in A. alternata
Here, we describe two HOs from the fungus A. alternata (Fig 1). Overall sequence similarities to other HOs are low, and comparison among fungal, plant, and animal HOs revealed two monophyletic fungal clades represented by HoxA and HoxB from A. alternata (Fig 1A). These two clades are highly different to the clade of plants, bacteria, or mammals. In other heme oxygenases, a histidine residue is conserved and involved in heme binding (Ito‐Maki et al, 1995). Whereas A. alternata HoxA contains this histidine, it is lacking in HoxB (Fig 1B). Nevertheless, in both proteins a HemeO‐like superfamily domain is predicted (Fig 2A). In addition, a hydrophobic stretch of 21 amino acids was identified at the C‐terminus of HoxA. Because this region is required for mitochondrial association, it was named C‐terminal anchor (CTA) (see below).
Figure 1. Phylogenetic analysis of HOs.
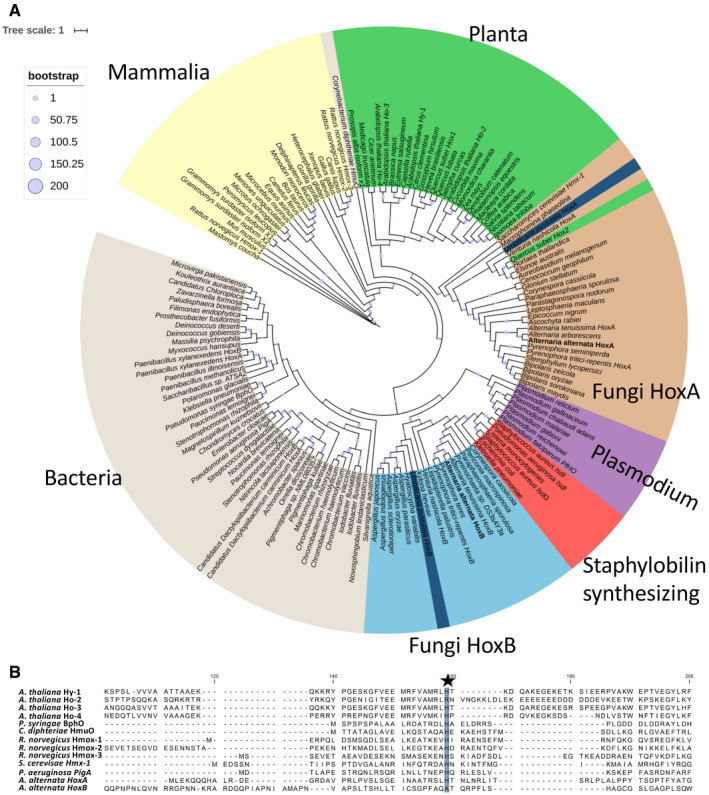
- A phylogenetic tree of 161 different HO sequences was calculated. HOs were aligned with Clustal Omega. Afterward, the sequences were trimmed by trimAL with gap threshold 0.8 and conservation percentage of 70%. The tree was calculated with PhyML with 200 bootstrap cycles and the AIK parameter switched on. Visualization was done in iTOL. Within the two fungal clades, two lichen‐derived sequences are shaded in dark blue.
- Alignment of the heme‐binding region of several HOs. The heme‐coordinating histidine is indicated with a star. HoxB lacks this histidine, like HO‐2 from A. thaliana.
Figure 2. Characterization of HoxA and HoxB from A. alternata .
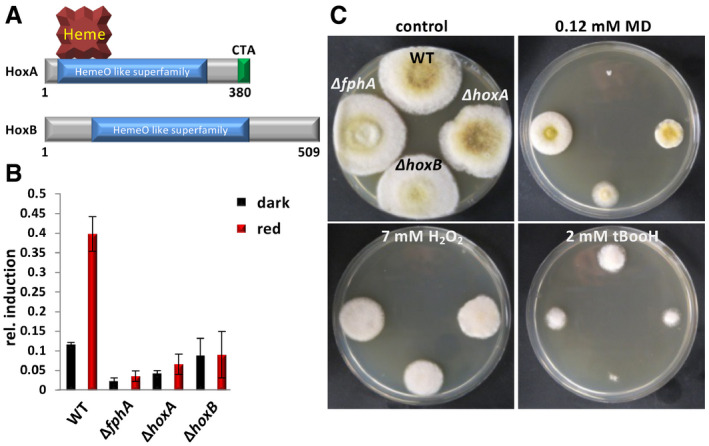
-
AScheme of HoxA and HoxB. The open reading frame of hoxA is interrupted by one 67 bp long intron (RNA‐seq data) and encodes a protein of 380 amino acids with a predicted mass of 42.5 kDa. The protein contains a heme‐binding pocket, a HemeO‐like superfamily domain and a C‐terminal putative membrane anchor (CTA). In comparison, hoxB is interrupted by a 50 bp intron. The protein (509 amino acids, 55.9 kDa) contains only the HemeO‐like superfamily domain.
-
BEffect of the deletion of hoxA or hoxB on red‐light induction of the ccgA gene. Alternaria alternata strains were grown for 36 h at 28°C. Samples were illuminated for 1 h in red light, while controls were kept in the dark. The amount of ccgA transcript was quantified by qRT–PCR analyses with the histone 2B gene as housekeeping gene. Error bars represent the standard deviation (n = 3).
-
CComparison of wild‐type colonies with phytochrome and hox‐deletion strains in the presence of 0.12 mM menadione (MD), 7 mM H2O2 or 2 mM tert‐butyl hydroperoxide (tBooH). The strains were incubated for 5 days at 28°C in the dark.
To functionally characterize hoxA and hoxB in A. alternata, both genes were deleted using CRISPR/Cas9 (Wenderoth et al, 2017; Wenderoth et al, 2019) (Appendix Fig S1). In either deletion strain, the induction of the red‐light regulated gene, ccgA, was drastically reduced, as much as in the phytochrome‐deletion strain (Fig 2B). To further link the deletion of hoxA or hoxB to the function of phytochrome (FphA), the stress behavior of the strains was tested. Stress was applied by adding hydrogen peroxide (H2O2), menadione (generates free radicals and superoxide), or tert‐butyl hydroperoxide (tBooH) (organic hydroperoxide) to the medium. Similar to phytochrome mutants (Yu et al, 2016; Igbalajobi et al, 2019), both hox‐deletion strains were more resistant toward menadione and H2O2 and more sensitive toward tBooH (Fig 2C). The higher stress resistance of the phytochrome mutant, and the hoxA and hoxB mutants, may be explained through the regulation of stress‐related genes, such as catalases, by phytochrome. It was shown that they are upregulated in the absence of phytochrome (Igbalajobi et al, 2019; Igbalajobi et al, 2020). In addition, there is evidence that under some stress conditions, human HO‐1 is proteolytically processed, shuttles into nuclei, and fulfills an unknown function there (Lin et al, 2007). Such an additional function in A. alternata may explain the slightly higher sensitivity of the hoxB mutant as compared to the hoxA or the fphA mutant. Taken together, the results suggest that HoxA and HoxB could provide the chromophore for phytochrome.
Most HO enzymes catalyze the conversion of heme to biliverdin IXα or other isomers, depending on the cleavage site. Biliverdin IXα is used by bacterial phytochromes as chromophore (Lamparter et al, 2003). To investigate whether HoxA and HoxB convert heme into a functional chromophore, several in vitro assays were performed. HoxA was expressed in E. coli without the hydrophobic C‐terminal membrane anchor (HoxA∆CTA) and HoxB in Pichia pastoris. HoxB overexpression in E. coli rendered an inactive protein (Appendix Fig S2). Spectral analyses of the enriched proteins did not show any evidence for heme binding already during the expression or the subsequent purification. We then added hemin and tested for heme binding. We started with a solution of 1 µM hemin and added HoxA or HoxB to a final concentration of 10 µM. The heme spectrum changed slightly after protein addition, suggesting heme binding. Afterward, the hemin concentration was stepwise increased up to 8 µM. At each step, absorption spectra were recorded. Binding of hemin by HoxA resulted in a peak at 408 nm. At 3 µM hemin, a shoulder appeared in the spectrum between 320 and 380 nm. This resembled free hemin, suggesting that not all HoxA molecules were active or able to bind the chromophore which could be caused by partial oligomerization (Appendix Fig S3A). In the corresponding experiment with HoxB, the spectra resembled the spectra of HoxA (with a peak at 410 nm), but the shoulder in the spectrum was more pronounced and increased faster (Appendix Fig S3B). Hence, both proteins are able to bind heme. Stoichiometries for heme binding were not determined as the protein preparations were not pure.
We then characterized the catalytic activity of HoxA and HoxB in the presence of ferredoxin as electron donor. 50 µM HoxA or HoxB, or 25 µM of each protein combined in one reaction, was incubated with 10 µM hemin and incubated for 10 min. When HoxA and HoxB were combined, HPLC analyses of the reaction products revealed a peak with a retention time 1–2.5 min larger than for the biliverdin standard (Appendix Fig S3C). The small difference of the retention time could point to minor modifications of the chromophore, although the spectrum matched well with the biliverdin standard (Appendix Fig S3D). In order to show that the produced biliverdin product is able to autocatalytically assemble with phytochrome, the photosensory domain (PGP, 68 kDa) of FphA was added to the assay above and spectra recorded every 30 s (Appendix Fig S3E). Whereas the Soret band decreased over time, the Q band increased, proving the functionality of the chromophore. The maxima of the resulting Pr form was 702 nm and resembled the maxima of biliverdin assembled FphA in the Pr form with 705 nm (Blumenstein et al, 2005). To test whether the in vivo activity of HOXs is the sum of HoxA and HoxB activities, we co‐expressed HoxA or HoxB alone or in combination along with the photosensory domain (PGP) of A. nidulans FphA in E. coli. In all cases, low expression protein levels were maintained to avoid oligomerization and inactivation of HoxB. As a control, we co‐expressed the Pseudomonas aeruginosa HOX, BphO (Blumenstein et al, 2005) (Fig 3A). In all four cases, functional photosensory domains were obtained (Fig 3B–E). The covalent insertion of the chromophore into PGP was further confirmed by Zn2+‐induced red fluorescence after protein separation in a polyacrylamide gel (Fig 3A, inset). Only about 5% of the amount of assembled PGP was obtained when A. alternata HoxA in combination with HoxB was used instead of the bacterial HO (Fig 3A). This amount was even lower, when only HoxA (1%) or HoxB (0.7%) was expressed, suggesting that the activity of HoxA and HoxB together is higher than the sum of both. The reason for the much higher activity of the bacterial enzyme in comparison with the fungal enzymes may be the lack of appropriate electron donors for the A. alternata enzymes.
Figure 3. Analysis of the HO enzymatic activities.
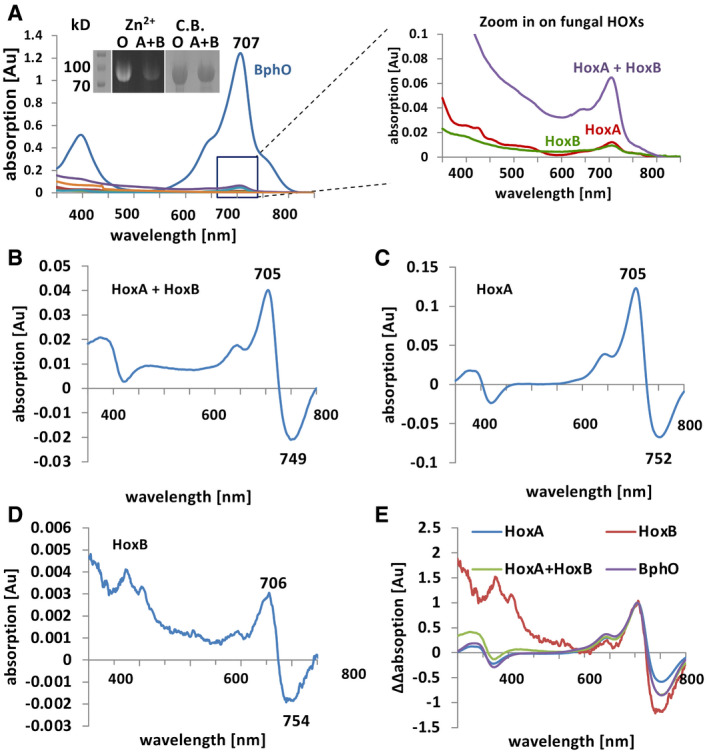
-
AUV–Vis spectra of the photosensory domain of A. nidulans FphA (PGP) after co‐expression together with bacterial HO (BphO) or A. alternata HoxA, HoxB, or HoxA plus HoxB overnight at 20°C in the dark. PGP was purified via the Strep‐tag system and concentrated before recording the spectra. Inset, left lane: Molecular mass markers stained with Coomassie Blue. Middle lanes: Zinc‐induced red fluorescence (labeled zinc, Zn2+) of equal amounts of PGP expressed in E. coli along with bacterial BphO (O) or A. alternata HoxA plus HoxB (A + B). Right lanes: Coomassie Blue staining (C.B.) of the polyacrylamide gel used for the Zn2+ fluorescence.
-
B–DRed/far‐red light induced difference spectrum of purified PGP expressed with HoxA and HoxB or with HoxA or HoxB alone after large‐scale batch fermentation and purification.
-
EOverlay of normalized (at 705 nm) difference spectra displayed in B‐D plus a difference spectrum of the photosensory domain in the presence of the bacterial HO BphO as in A.
Source data are available online for this figure.
Both heme oxygenases reside at the outer mitochondrial membrane
Next, the subcellular localization of A. alternata HoxA and HoxB was investigated by heterologous expression of GFP‐tagged versions in A. nidulans (Fig 4A). The observed GFP‐labeled structures resembled mitochondria, as confirmed with the MitoTracker co‐staining (Suelmann & Fischer, 2000). Therefore, we next tested whether the enzymes localize inside of mitochondria or are attached to the outer mitochondrial membrane (Fig 4B). Cells were fractionated by two sequential centrifugation steps. After the first low‐speed centrifugation, mitochondria should be still present in the supernatant (S1), whereas after the second higher‐speed centrifugation, mitochondria should be enriched in the pellet (P2). This pellet fraction was used to test the sensitivity of the proteins toward proteinase K treatment (P2 +PK). HoxA‐GFP and HoxB‐GFP were both detected in the mitochondrial fraction and were completely degraded after addition of proteinase K. As a control, we studied a mitochondrial matrix protein. We used the N‐terminal part of citrate synthase and fused it to GFP. The construct was used before for mitochondrial labeling (Suelmann & Fischer, 2000). The fusion protein has a molecular mass of 41 kDa, although the apparent molecular mass in the SDS–PAGE is less. After import, the protein has a predicted molecular mass of 35 kDa. In crude extracts, both bands, and in addition some degradation products, were detectable, suggesting that not all proteins are imported into mitochondria. After proteinase K treatment of the second pellet fraction, the 35 kDa protein band remained, plus a smaller degradation product. The small protein could be free GFP. From these experiments, we conclude that HoxA is attached to the outer mitochondrial membrane.
Figure 4. Localization of A. alternata HoxA and HoxB in A. nidulans .
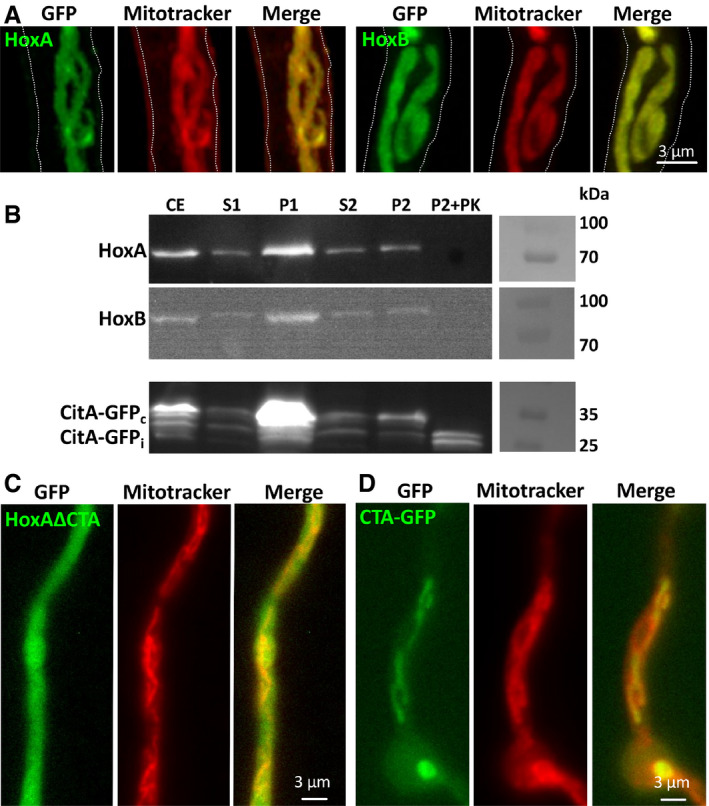
-
AStrains were grown overnight at 28°C. Mitochondria were stained for 30 min with MitoTracker Red CMOXRos. Microscopy was done with a LSM 900 Airyscan 2 (Zeiss). Pictures were taken in the GFP and the RFP channel and overlaid (merge).
-
BHoxA‐GFP and CitA‐GFP (ca. 170 amino acids from the N‐terminus of citrate synthase) were expressed overnight at 30°C in minimal medium with 2% threonine and 0.2% glucose. Mitochondria were incubated with 100 μg/ml proteinase K (PK) for 20 min on ice. Crude extract (CE), supernatant 1 (S1) containing mitochondria, pellet 1 (P1), supernatant 2 (S2), and pellet 2 (P2) with mitochondria were analyzed by Western Blot using anti‐GFP antibodies. CitA‐GFPc = cytoplasmic form and CitA‐GFPi = imported version.
-
C, DRole of the C‐terminal anchor in HoxA. Microscopy was done as in A using a fluorescent microscope without the Airyscan technology. C, Expression of GFP‐HoxA lacking the C‐terminal anchor (∆CTA) in A. alternata. D, Expression of GFP fused to the CTA motif.
Source data are available online for this figure.
A C‐terminal anchor (CTA) of HoxA is necessary and sufficient for mitochondrial targeting
None of the HOs comprises an N‐terminal mitochondrial targeting sequence. However, we identified a hydrophobic region at the C‐terminus of HoxA. To test whether this C‐terminal sequence is required for mitochondrial targeting, we removed the last 20 amino acids from the protein and expressed it as GFP fusion protein in A. nidulans. The truncated protein localized in the cytoplasm (Fig 4C). Next, we asked whether the C‐terminal sequence is sufficient for mitochondrial targeting and fused the sequence to GFP. Indeed, the GFP molecule labeled mitochondria. These results suggest that the C‐terminal sequence functions as an anchor for the protein.
Heme oxygenases form a complex with phytochrome
In order to test homo‐ or heterodimer formation of A. alternata HOs, bimolecular fluorescence complementation (BiFC, split‐YFP) was used. HoxA and HoxB were expressed as fusion proteins with the N‐terminal or the C‐terminal half of YFP in A. nidulans. Interaction of two proteins restores YFP fluorescence. We found that all three combinations, HoxA‐HoxA, HoxA‐HoxB, and HoxB‐HoxB, interacted at mitochondria. Moreover, the same results were obtained with the combination of HoxA or HoxB with phytochrome (Fig 5A). As a negative control, YFP‐N‐YpdA (Yu et al, 2016) was tested with either YFP‐C‐HoxA or YFP‐C‐HoxB. These combinations did not result in reconstitution of the fluorescent YFP protein.
Figure 5. Characterization of the HO‐phytochrome protein complex.
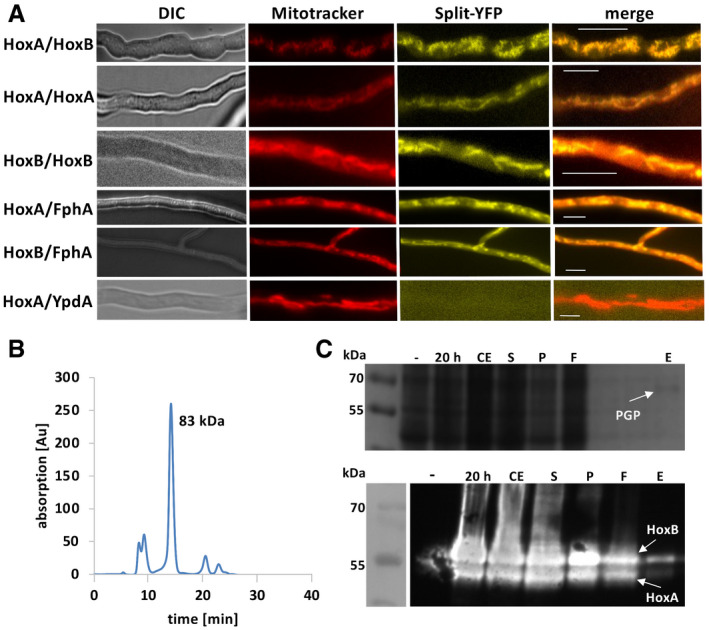
-
ABiFC (split‐YFP) analysis of HOs and phytochrome. Strains (SChS29‐33) were grown overnight at 28°C minimal medium with 2% threonine and 0.2% glucose. MitoTracker Red CMOXRos was used for mitochondrial staining. The MitoTracker signal was observed in the RFP and the split‐YFP signal in the YFP channel of the fluorescent microscope. Pictures in both channels were overlaid (merge). The left pictures show the same hyphae in differential interference contrast (DIC). The scale bar represents 10 µm.
-
BHoxA was expressed in E. coli 20 h at 15°C. Purification was performed with the Strep‐tag system. HoxA was analyzed by size‐exclusion chromatography. Absorption was measured at 280 nm.
-
CCo‐expression of the HOXs along with the photosensory domain of A. nidulans FphA (PGP) overnight at 20°C. PGP was purified via the Strep‐tag system and the eluate concentrated (upper panel). The lanes were loaded as follows: negative control before induction (−), 20 h after induction (20 h) crude extract (CE), supernatant (S), pellet (P), flow through (F), and eluate (E). The fractions were analyzed by Western blot using the anti‐His antibody (lower panel).
Source data are available online for this figure.
HoxA dimerization was confirmed by size‐exclusion chromatography (SEC) (Fig 5B). Calibration of the column was performed with cytochrome C, carbonic anhydrase, albumin, and alcohol dehydrogenase (Appendix Fig S3F). For HoxB SEC failed, because of impurities after enrichment from P. pastoris. As a further proof for protein–protein interaction, we co‐expressed HoxA, HoxB, and the photosensory domain of FphA (Strep‐tagged) in E. coli. After Strep‐tag purification, we identified HoxA and HoxB in the photosensory domain fraction (Fig 5C). To assess whether insertion of the chromophore into the photosensory domain influences its interaction with HoxA, which is proposed to be the key factor for assembly because of the membrane anchor, biolayer interferometry (BLI) was used for the analysis of interactions of the purified proteins (Fig 6, Appendix Fig S4). Association and dissociation curves were monitored, and the KD for the interaction of Apo‐PGP and HoxA was calculated to be 1.13 µM (Fig 6A) and for Holo‐PGP 64.6 mM (Fig 6B). The sudden increase of the signal after addition of the chromophore‐loaded PGP is due to the absorption of the chromophore (Fig 6B). The 57,000 times weaker interaction of the holoprotein suggests immediate dissociation from HoxA after chromophore insertion.
Figure 6. Analysis of the interaction of HO and the phytochrome photosensory domain in vitro .
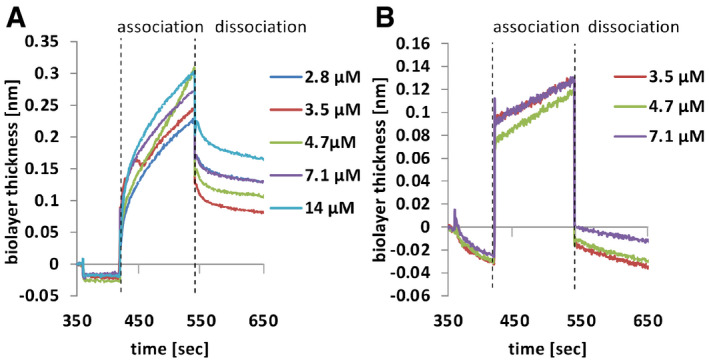
-
A, BBiolayer interferometry analysis with Apo‐PGP A and holo‐PGP B. Proteins were immobilized at a streptavidin sensor tip. Free binding sites were blocked with biotin. Association and dissociated kinetics were recorded at the indicated HoxA concentrations. K d values were calculated with the manufacturer’s software using a global fit for both experiments.
Discussion
Taken together our results, we propose the following model for the biosynthesis of functional phytochrome in fungi: The FphA apoprotein is translated in the cytoplasm and travels to the surface of mitochondria where it receives the chromophore (the electron donor for the heme oxidation is yet unknown). The holoprotein is then released back into the cytoplasm where it interacts with the phosphotransfer protein YpdA to induce the HOG pathway and ultimately the transcription factor AtfA. In addition, a fraction of Holo‐FphA is imported into the nucleus to control the activity of chromatin remodeling enzymes (Fig 7). Mitochondria can thus be considered as assembly platforms for phytochrome in fungi. Given that mitochondria arose in evolution before chloroplasts, our findings tempt us to speculate that chromophore biosynthesis at mitochondria is more ancient than in chloroplasts (Speijer et al, 2020).
Figure 7. Current model illustrating the role of mitochondria as chromophore‐assembly stations for phytochrome.
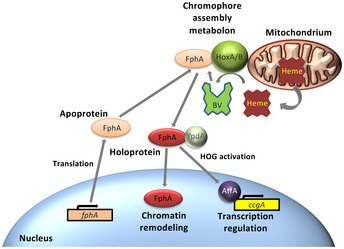
For details, see the Discussion.
In this work, we identified two HOs in A. alternata and showed catalytic activity for both. The activities when expressed in E. coli were quite low as compared to the activity of the bacterial HO used in our experiments. This may reflect their real activities in A. alternata, but it could also be that the HO activities in the fungus are significantly higher. One reason could be the lack of an appropriate electron donor in E. coli and in the in vitro experiments. The fact that HoxB undoubtedly produced active photosensory domains of FphA when co‐expressed in E. coli was surprising, because a conserved histidine is lacking. This may explain the lower activity as compared to HoxA. However, we found evidence that, in addition to its catalytic activity, HoxB could stimulate the HoxA activity. The mechanism of this stimulation remains to be determined, but it has been shown that human HO‐2 may have some chaperone functions (Vanella et al, 2013; Vanella et al, 2016). In addition, it was shown that human HO‐1 fulfills functions in nuclei under certain stress conditions (Lin et al, 2007). Further work is required to unravel whether such a specialization of the two enzymes exists in fungi.
The fact that phytochrome chromophore assembly in A. alternata occurs at the surface of mitochondria leads to the question why there and not in the cytoplasm? Heme biosynthesis is initiation and completed in mitochondria in animals and fungi. Glycine and succinyl‐CoA are converted in mitochondria to 5‐aminolevulinic acid, which is transported to the cytoplasm, further converted to coproporphyrinogen III, which in turn is re‐imported into mitochondria, where the final steps of heme biosynthesis occur. Heme is then either used in mitochondria for, e.g., cytochrome formation or exported into the cytoplasm (Kim et al, 2012). In plants, the final steps take place in chloroplasts. In the cytoplasm, heme is inserted into hemoproteins such as catalases. Likewise, HOs in A. alternata could use cytoplasmic heme to produce the phytochrome chromophore. HOs would thus compete for heme with the other enzymes which need the incorporation of a heme molecule for function. However, in the case of HOs, heme is catalytically converted and normally released for further degradation. Such degradation has to be also postulated for the A. alternata cytoplasm, because heme concentrations need to be controlled well, e.g., by high affinity heme‐binding proteins like peroxiredoxins, because free heme can produce free radicals through Fenton chemistry (Fenton, 1894; Gozzelino et al, 2010). Hence, A. alternata HoxA and HoxB would largely interfere with heme homeostasis. One way of separating heme degradation from chromophore biosynthesis is compartmentalization. In the case of heme oxidation for chromophore formation, further degradation of the linear tetrapyrrole has to be prevented. This could be the reason for the observed complex formation between HOs and phytochrome. This minimizes the chances that linearized heme is released and further degraded. This could be an example for the recently postulated metabolon concept (Piel et al, 2019). Metabolons are protein complexes of interacting enzymes in metabolic pathways. The created microenvironments facilitate substrate transport and specificity. In the case of phytochrome chromophore assembly, the protein complex could also be important for limitation of the enzymatic activity to the needs. If apo‐phytochrome binds to the HO complex, heme should be oxidized and transferred to form holo‐phytochrome. In the absence of apo‐phytochrome though, HO should be inactive in order to prevent uncontrolled degradation of heme. This could be achieved by modulation of the HO activity or through the accessibility of heme to the active center if phytochrome is missing from the metabolon. Supporting our metabolon model, protein complex formation between HO and phytochrome was also shown in the bacterium Pseudomonas syringae (Shah et al, 2012). Furthermore, in the pathogenic P. aeruginosa two heme oxygenases are expressed, one of which produces the chromophore for phytochrome and another one is used for heme degradation and iron acquisition (Wegele et al, 2004). Plants harbor also several HO with different catalytic activities, perhaps also serving distinct functions besides chromophore production (Davis et al, 2001).
Another reason for mitochondrial association of the phytochrome metabolon could be the need for an electron donor with a negative redox potential, such as ferredoxin. Although mitochondria are optimized organelles for electron flow, the localization of the phytochrome metabolon at the outside of mitochondria raises the question of how the electrons can be shuttled from the inner mitochondrial membrane toward the outside of the outer membrane. This remains to be resolved. Alternatively, cytoplasmic electron donors could be employed as it was shown for cytosolic iron/sulfur cluster biogenesis (Zhang et al, 2014). One challenge for future research will be to investigate whether the model proposed for fungal phytochrome biosynthesis may be generalized also to plant phytochrome.
Our results suggest that the two HOs of A. alternata are specialized enzymes for chromophore assembly at mitochondria. This special function may also explain the low enzymatic activity. One can speculate that for heme degradation, higher activities are to be expected. It will be the challenge of future research to decipher the interplay between the two HOs and to identify HOs for heme degradation and unravel their interplay in heme homeostasis and chromophore production.
Materials and Methods
Strains, plasmids, and culture conditions
Alternaria alternata ATC 66981 cultures were grown on modified Czapek Doth broth (mCDB) agar and incubated 1–12 days at 28°C. Supplemented minimal medium (MM) for A. nidulans was prepared as reported, and standard strain construction protocols were used (Käfer, 1977). Growth of Pichia pastoris was performed in BMGY or BMMY media according to the manufacturer’s protocol (Invitrogen). All strains are listed in Appendix Table S1, oligonucleotides in Appendix Table S2, and plasmids in Appendix Table S3.
Gene structure and deletion of hoxA and hoxB in A. alternata
The open reading frame of hoxA is interrupted by one 67 bp long intron (RNA‐seq data) and encodes a protein of 380 amino acids with a predicted mass of 42.5 kDa. In comparison, hoxB is also interrupted by one intron (50 bp) but contains only the HemeO‐like superfamily domain. The hoxB gene encodes a protein of 509 amino acids with a predicted mass of 55.9 kDa. We used two 20 bp protospacers adjacent to a 3′AGG protospacer‐adjacent motif (PAM) to target the beginning and the end of the gene. The protospacers were introduced into plasmids pFC332 and pFC330 by PCR and cloning. The resulting plasmids, which contain the Cas9‐coding sequence from Streptococcus pyogenes (codon optimized for Aspergillus niger) and the single‐guide RNA (sgRNA) targeting the genes of interest (hoxA and hoxB), were used for transformation of A. alternata SMW24 (ΔpyrG in ATCC66981 wild type). The hygromycin resistance and the pyrG auxotrophy cassettes residing in a self‐replicating plasmid (AMA plasmid) were used for selection. The plasmid was constructed using PCR on PFC334 with primers, which included the new protospacer. Subsequently, a builder reaction and the transformation was done as described, but in this case, two protospacers were used to target the beginning and the end of the gene of interest (Wenderoth et al, 2019).
RNA isolation
Alternaria alternata conidia were inoculated in 20–25 ml mCDB containing uracil and uridine in a Ø 3.5 cm Petri dish and incubated for 40 h in darkness at 28°C. Subsequently, mycelium was illuminated 1h with red light and controls were kept in the dark. The mycelia were harvested in dim‐red light and frozen immediately in liquid nitrogen. For the extraction, a "Fungal RNA Extraction Kit" from Omega was used. Disruption of the cells was performed by grinding in liquid nitrogen. To remove DNA contaminations, the RNA was treated with TURBO DNA‐free kit. After the treatment, RNA was diluted to 50 ng/μl with DEPC water. SensiFAST SYBR & Fluorescein One‐Step Kit from Bioline (Luckenwalde, Germany) was used for quantitative real‐time PCR. Each reaction was carried out using 25 μl containing 0.2 μM primers and 100 ng RNA. The program started with 10 min of the reverse transcription reaction at 45°C, followed by 2.5 min at 95°C for the inactivation of the reverse transcriptase and 40 cycles of polymerase chain reaction (10 s at 95°C and then 30 s at 58°C). To assess the quality of the resulting PCR product, melting curve analyses were carried out (80 cycles, 95–58°C with 10 s per step). The h2b gene was used for normalization. Each expression level is the average of three biological replicates. The error is shown as standard deviation for the replicates.
Expression of HOs in E. coli or P. pastoris
HoxA was cloned into the plasmid pASK. pASK contains an anhydrotetracycline‐inducible promoter (tet) and an ampicillin resistance cassette. For the expression, E. coli was grown at 37°C until OD600 0.8 was reached. Subsequently, the baffled flask was cooled down to 15°C and induction with 0.2 µg/ml AHT was performed for 20 h. The pellet of 1 l was resuspended in 10 ml extraction buffer and the cells ruptured using a French press at 1,500 psi. Cell debris was pelleted by centrifugation prior to the application to the column. Purification was done using the strep‐tag system with modified buffers: extraction buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.005% Triton X‐100, 1 mM PMSF, 1 pill for 10 ml pierce protease inhibitor cocktail EDTA free (Thermo Scientific), washing buffer (137 mM NaCl, 2.7 mM KCl, 50 mM Na2HPO4, 5 mM K2HPO4, pH 7.4 was adjusted with H3PO4), and elution buffer (50 mM Tris–HCl, 3 mM Desthiobiotin). For better solubility, the C‐terminal anchor was deleted. HoxB could not be purified using this system, because HoxB aggregated. In order to overcome this problem, HoxB‐strep was cloned to pPIC3.5K. pPIC3.5K is a vector containing a histidine auxotrophy marker and the methanol‐inducible AOX1 promoter for P. pastoris. P. pastoris GS115 was transformed with SacI linearized vector to insert into the AOX1 locus. The transformants were screened for mut+ phenotype. For protein expression, P. pastoris was grown in 50 ml BMGY overnight at 30°C. After incubation, cells were pelleted and 400 ml BMMY with OD600 1.0 were inoculated. Expression was performed for 2.5 days, while every 24 h methanol was added to maintain a concentration of 0.5% (v/v). 1 l culture was pelleted and resuspended in 20 mM MES pH 6. Cells were lysed using glass beads. Purification was done using a Mono S FPLC column. The protein eluted in a gradient ranging from 350 mM NaCl to 450 mM NaCl. HoxB was not as pure as HoxA after affinity chromatography. Further purification using the Strep‐tag failed.
Expression of phytochrome (FphA) or its photosensory domain (PGP) along with HOs in E. coli
We used the plasmids pASK for FphA PGP and pACYC Duet or pET for the Hox. pACYC Duet contains two MCS, while pET contains one MCS with an IPTG‐inducible T7 promoter. Cultures were grown in 500 ml LB at 37°C. After OD600 0.8 was reached, 1 mM IPTG was added to the culture to induce the HO. After 1 h, the culture was cooled down to 20°C and 0.2 µg/ml AHT was added to induce the induction of FphA PGP. FphA PGP was purified using French press and the strep‐tactin system (IBA). Subsequently, the protein was concentrated via vivaspin and the spectra were measured in the dark.
To improve the spectral results and in order to analyze the photoconvertability of PGP, we used a 5 l fermenter (Bioflo 115, Eppendorf) with the same protocol. Additionally, the fermenter was set to 400 rpm, pH 7.5, and an aeration rate of 5 l air per min. For co‐expression of HoxA and HoxB, we used the pACYC Duet plasmid. The protein samples were illuminated 2 min with red or far‐red light to photoconvert PGP between Pr and Pfr forms. Results are shown as difference spectra. This procedure doubled the yield of functional PGP in comparison with the expression in flask cultures. HoxA alone and the co‐expression of HoxA and HoxB with PGP yielded functional photoconvertible phytochrome, which suggests HoxA is sufficient.
Zinc‐induced red fluorescence
Covalent chromophore attachment has been verified by zinc‐induced red fluorescence as described previously (Berkelman & Lagarias, 1986). Free bilins as well as (denatured) biliproteins form fluorescent complexes with zinc ions that can be visualized under UV light. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was performed as described (Laemmli, 1970). Tris‐glycine running buffer as well as Tris‐buffers used for the 5% stacking gel and the 10% separating gel contained 1 mM zinc acetate. Zinc fluorescence was visualized by transillumination with UV light. Afterward, the same gel was Coomassie‐stained using ROTI®Blue quick (Carl Roth, Karlsruhe). Protein concentrations were determined using ROTI®Quant (Carl Roth, Karlsruhe) according to the manufacturer’s instructions.
Heme oxygenase assays
For the analysis of the catalytic activity of HoxA and HoxB, enriched enzymes were used. 50 µM HoxA or HoxB or 25 µM of each protein combined in one reaction was incubated with 10 µM hemin, 1.5 mg/ml BSA, 4.6 µM petF, 0.025 U/ml petH, 10 µM catalase, 5 mM tiron, 1.05 mM glucose‐6‐phosphate, 0.105 µM NADP+ and 0.15 U/ml glucose‐6‐phosphate‐dehydroxygenase, spinach ferredoxin (PetF, Sigma) (4.6 µM), and ferredoxin reductase (PetH, Sigma) (4.6 µM). Spectra from 300 nm to 1,100 nm were recorded every 30 s for 10 min (Photometer: 8453 UV visible System (Agilent)). For assays with phytochrome, 150 µg FphA was added per ml.
HPLC analysis
Hemeoxygenase assay products were pre‐purified. To this end, the sample was diluted 1:10 in 0.1% TFA immediately after the assay to stop the reaction. Next, Sep‐Pak C18 was equilibrated with subsequent addition of 3 ml acetonitrile, 3 ml H2O, 3 ml 0.1% TFA, 3 ml 10% methanol in 0.1% TFA, 3 ml acetonitrile, 3 ml H2O, and 3 ml 10% methanol in 0.1% TFA. The sample was added after the last step, and the cartridge was washed with 6 ml 0.1% TFA, 6 ml acetonitrile (20%):0.1%TFA (80%) and eluted with 1 ml acetonitrile. After drying using a SpeedVac, the sample was dissolved in 10 µl DMSO and diluted in 110 µl acetone (50%):20 mM formic acid (50%). The sample was applied to the Ultracarb 5 U column (Phenomenex). Bilins were detected at 350 and 650 nm.
Expression of HOs in A. nidulans
The expression of HoxA and/or HoxB in A. nidulans was achieved using the plasmid pMCB17apx as vector basis. The plasmid harbors a cloning site using AscI and PacI restriction enzymes. The cloned gene is under the control of the alcA promoter, which can be repressed by glucose and derepressed by glycerol and induced by threonine. It also encodes an N‐terminal tag of GFP, YFP‐C‐terminus, YFP‐N‐terminus, or HA.
Microscopy
For microscopic analysis, A. nidulans strains were grown 16 h at 28°C in MM containing 2% glycerol or 2% threonine with 0.2% glucose. Subsequently, the culture was stained for 30 min with MitoTracker Red CMXRos (M7512, Thermo Fisher) according to the manufacturer’s protocol. Fluorescence microscopy was performed using AxioImager Z1 (Zeiss), the software AxioVision V4.5, and Zen. Alternatively, we used the LSM 900 Airyscan 2 (Zeiss).
Mitochondrial fractionation
Aspergillus nidulans protoplasts were applied to the “Yeast Mitochondria Isolation Kit” (SigmA) and mitochondria isolated as described in the manufacturer’s protocol using detergent lysis (1:200 dilution). In the first centrifugational step, at 600× g mitochondria were obtained in the supernatant (S1). In the second centrifugational step, at 6,500× g mitochondria were sedimented in the pellet (P2). Proteins were analyzed in a Western blot using anti‐GFP antibodies (Roche). For precipitation of the GFP fusion protein (GFP trap), the protein extracts were incubated with the anti‐GFP antibody and protein G agarose (Roche).
BLI (biolayer interferometry)
For BLI assays, the BLItz system (FORTEBIO) was used. Streptavidin‐coated sensor tips were coated with purified 30 mg/ml Apo‐ or Holo‐PGP (tagged with streptavidin‐binding protein) in analysis buffer (50 mM Tris–HCl, pH 7.8, 300 mM NaCl, 10% glycerol, 0.05% Tween‐20). Free binding sites were blocked with 5 mM biotin. Measurements were performed with purified HoxA at the indicated concentrations. The baseline was measured for 60 s, binding of PGP to the sensor tip was done for 120 s and blocking for 180 s. The resulting new baseline was measured for 60 s. Association and dissociation were measured for 120 s. For calculating association and dissociation rates, global fit corrections were activated in the software. Resulting KD values are presented as mean of the values measured at the different concentrations.
Author contributions
CS performed most of the experiments. JH and KL contributed to the in vitro experiments. NK and TL supervised and discussed the spectroscopy. NF‐D supervised the biochemical experiments. FW and MB provided the infrastructure and support for the BLI experiments. ZY designed several experiments and supervised the work with A. alternata. RF supervised the entire project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by the German Science Foundation (DFG Fi‐459/19‐1). F.W. and M.B. were supported by the "Karlsruhe School of Optics and Photonics, DFG GSC 21”. We thank Luis Raupach, Linda Schlegel, and Birgit Schreckenberger for their technical assistance. We are grateful to Matthias Mack, Hochschule Mannheim, for providing us the P. pastoris expression system. We thank Gert Sonntag (Zeiss) for the opportunity and the help to use the LSM 900 Airyscan Microscope and Natalia Requena for critically reviewing the manuscript.
The EMBO Journal (2021) 40: e108083.
Contributor Information
Zhenzhong Yu, Email: yuzhenzhong@njau.edu.cn.
Reinhard Fischer, Email: reinhard.fischer@KIT.edu.
Data availability
This study includes no data deposited in external repositories.
References
- Berkelman TR, Lagarias JC (1986) Visualization of bilin‐linked peptides and proteins in polyacrylamide gels. Anal Biochem 156: 194–201 [DOI] [PubMed] [Google Scholar]
- Blumenstein A, Vienken K, Tasler R, Purschwitz J, Veith D, Frankenberg‐Dinkel N, Fischer R (2005) The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr Biol 15: 1833–1838 [DOI] [PubMed] [Google Scholar]
- Corrochano LM (2019) Light in the fungal world. Ann Rev Genet 53: 149–170 [DOI] [PubMed] [Google Scholar]
- Davis SJ, Bhoo SH, Durski AM, Walker JM, Vierstra RD (2001) The heme‐oxygenase family required for phytochrome chromophore biosynthesis is necessary for proper photomorphogenesis in higher plants. Plant Physiol 126: 656–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SJ, Kurepa J, Vierstra RD (1999) The Arabidopsis thaliana HY1 locus, required for phytochrome‐chromophore biosynthesis, encodes a protein related to heme oxygenases. Proc Natl Acad Sci USA 96: 6541–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton NJH (1894) Oxidation of tartaric acid in presence of iron. J Chem Soc 65: 899–910 [Google Scholar]
- Froehlich AC, Noh B, Vierstra RD, Loros J, Dunlap JC (2005) Genetic and molecular analysis of phytochromes from the filamentous fungus Neurospora crassa . Eukaryot Cell 4: 2140–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisk B, Yasui Y, Kohchi T, Frankenberg‐Dinkel N (2010) Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions. Biochem J 425: 425–434 [DOI] [PubMed] [Google Scholar]
- Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase‐1. Ann Rev Pharmacol Toxicol 50: 323–354 [DOI] [PubMed] [Google Scholar]
- Hedtke M, Rauscher S, Röhrig J, Rodriguez‐Romvero J, Yu Z, Fischer R (2015) Light‐dependent gene activation in Aspergillus nidulans is strictly dependent on phytochrome and involves the interplay of phytochrome and white collar‐regulated histone H3 acetylation. Mol Microbiol 97: 733–745 [DOI] [PubMed] [Google Scholar]
- Hughes J, Lamparter T, Mittmann F, Hartmann E, Gärtner W, Wilde A, Boerner T (1997) A prokaryotic phytochrome. Nature 386: 663 [DOI] [PubMed] [Google Scholar]
- Igbalajobi O, Gao J, Fischer R (2020) The HOG pathway plays different roles in conidia and hyphae during virulence of Alternaria alternata . Mol Plant Microbe Interact 33: 1405–1410 [DOI] [PubMed] [Google Scholar]
- Igbalajobi O, Yu Z, Fischer R (2019) Red‐ and blue‐light sensing in the plant pathogen Alternaria alternata depends on phytochrome and the white‐collar protein LreA. MBio 10: e00371–e1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito‐Maki M, Ishikawa K, Matera KM, Sato M, Ikeda‐Saito M, Yoshida T (1995) Demonstration that histidine 25, but not 132, is the axial heme ligand in rat heme oxygenase‐1. Arch Biochem Biophys 317: 253–258 [DOI] [PubMed] [Google Scholar]
- Jung JH, Domijan M, Klose C, Biswas S, Ezer D, Gao M, Khattak AK, Box MS, Charoensawan V, Cortijo Set al (2016) Phytochromes function as thermosensors in Arabidopsis . Science 354: 886–889 [DOI] [PubMed] [Google Scholar]
- Käfer E (1977) Meiotic and mitotic recombination in Aspergillus and its chromosomal aberrations. Adv Genet 19: 33–131 [DOI] [PubMed] [Google Scholar]
- Kim HJ, Khalimonchuk O, Smith PM, Winge DR (2012) Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim Biophys Acta 1823: 1604–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- Lamparter T (2004) Evolution of cyanobacterial and plant phytochromes. FEBS Lett 573: 1–5 [DOI] [PubMed] [Google Scholar]
- Lamparter T, Krauss N, Scheerer P (2017) Phytochromes from Agrobacterium fabrum . Photochem Photobiol 93: 642–655 [DOI] [PubMed] [Google Scholar]
- Lamparter T, Michael N, Caspani O, Miyata T, Shirai K, Inomata K (2003) Biliverdin binds covalently to agrobacterium phytochrome Agp1 via its ring A vinyl side chain. J Biol Chem 278: 33786–33792 [DOI] [PubMed] [Google Scholar]
- Legris M, Klose C, Burgie ES, Rojas CC, Neme M, Hiltbrunner A, Wigge PA, Schafer E, Vierstra RD, Casal JJ (2016) Phytochrome B integrates light and temperature signals in Arabidopsis . Science 354: 897–900 [DOI] [PubMed] [Google Scholar]
- Lin Q, Weis S, Yang G, Weng Y‐H, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz Fet al (2007) Heme oxygenase‐1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem 282: 20621–20633 [DOI] [PubMed] [Google Scholar]
- Muramoto T, Kohchi T, Yokota A, Hwang I, Goodman HM (1999) The Arabidopsis photomorphogenic mutant hy1 is deficient in phytochrome chromophore biosynthesis as a result of a mutation in a plastid heme oxygenase. Plant Cell 11: 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Park E, Song K, Bae G, Choi G (2020) PHYTOCHROME INTERACTING FACTOR8 inhibits phytochrome A‐mediated far‐red light responses in Arabidopsis . Plant Cell 32: 186–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Nagel MK, Popp C, Wust F, Bindics J, Viczian A, Hiltbrunner A, Nagy F, Kunkel T, Schäfer E (2012) Interaction with plant transcription factors can mediate nuclear import of phytochrome B. Proc Natl Acad Sci USA 109: 5892–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham VN, Kathare PK, Huq E (2018) Phytochromes and phytochrome interacting factors. Plant Physiol 176: 1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel RB 3rd, Dailey HA Jr, Medlock AE (2019) The mitochondrial heme metabolon: insights into the complex(ity) of heme synthesis and distribution. Mol Genet Metab 128: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschwitz J, Müller S, Kastner C, Schöser M, Haas H, Espeso EA, Atoui A, Calvo AM, Fischer R (2008) Functional and physical interaction of blue and red‐light sensors in Aspergillus nidulans . Curr Biol 18: 255–259 [DOI] [PubMed] [Google Scholar]
- Rauscher S, Pacher S, Hedtke M, Kniemeyer O, Fischer R (2016) A phosphorylation code of the Aspergillus nidulans global regulator VelvetA (VeA) determines specific functions. Mol Microbiol 99: 909–924 [DOI] [PubMed] [Google Scholar]
- Rockwell NC, Lagarias JC (2020) Phytochrome evolution in 3D: deletion, duplication, and diversification. New Phytol 225: 2283–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Sauthof L, Szczepek M, Lopez MF, Escobar FV, Qureshi BM, Michael N, Buhrke D, Stevens T, Kwiatkowski Det al (2018) Structural snapshot of a bacterial phytochrome in its functional intermediate state. Nat Commun 9: 4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J (2017) How light affects the life of Botrytis. Fungal Genet Biol 106: 26–41 [DOI] [PubMed] [Google Scholar]
- Schumacher J, Gorbushina A (2020) Light sensing in plant‐ and rock‐associated black fungi. Fungal Biol 124: 407–417 [DOI] [PubMed] [Google Scholar]
- Shah R, Schwach J, Frankenberg‐Dinkel N, Gärtner W (2012) Complex formation between heme oxygenase and phytochrome during biosynthesis in Pseudomonas syringae pv. tomato . Photochem Photobiol Sci 11: 1026–1031 [DOI] [PubMed] [Google Scholar]
- Speijer D, Hammond M, Lukes J (2020) Comparing early eukaryotic integration of mitochondria and chloroplasts in the light of internal ROS challenges: timing is of the essence. MBio 11: e00955‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suelmann R, Fischer R (2000) Mitochondrial movement and morphology depend on an intact actin cytoskeleton in Aspergillus nidulans . Cell Motil Cytoskel 45: 42–50 [DOI] [PubMed] [Google Scholar]
- Ulijasz AT, Vierstra RD (2011) Phytochrome structure and photochemistry: recent advances toward a complete molecular picture. Curr Opin Plant Biol 14: 498–506 [DOI] [PubMed] [Google Scholar]
- Vanella L, Barbagallo I, Tibullo D, Forte S, Zappala A, Li Volti G (2016) The non‐canonical functions of the heme oxygenases. Oncotarget 7: 69075–69086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanella L, Li Volti G, Guccione S, Rappazzo G, Salvo E, Pappalardo M, Forte S, Schwartzman ML, Abraham NG (2013) Heme oxygenase‐2/adiponectin protein‐protein interaction in metabolic syndrome. Biochem Biophys Res Comm 432: 606–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra RD, Zhang J (2011) Phytochrome signaling: solving the Gordian know with microbal relatives. T Plant Sci 16: 417–426 [DOI] [PubMed] [Google Scholar]
- Wegele R, Tasler R, Zeng Y, Rivera M, Frankenberg‐Dinkel N (2004) The heme oxygenase(s)‐phytochrome system of Pseudomonas aeruginosa . J Biol Chem 279: 45791–45802 [DOI] [PubMed] [Google Scholar]
- Wenderoth M, Garganese F, Schmidt‐Heydt M, Soukup ST, Ippolito A, Sanzani SM, Fischer R (2019) Alternariol as virulence and colonization factor of Alternaria alternata during plant infection. Mol Microbiol 112: 131–146 [DOI] [PubMed] [Google Scholar]
- Wenderoth M, Pinecker C, Voß B, Fischer R (2017) Establishment of CRISPR/Cas9 in Alternaria alternata . Fungal Genet Biol 101: 55–60 [DOI] [PubMed] [Google Scholar]
- Yu Z, Ali A, Igbalajobi OA, Streng C, Leister K, Krauss N, Lamparter T, Fischer R (2019) Two hybrid histidine kinases, TcsB and the phytochrome FphA, are involved in temperature sensing in Aspergillus nidulans . Mol Microbiol 112: 1814–1830 [DOI] [PubMed] [Google Scholar]
- Yu Z, Armant O, Fischer R (2016) Fungi use the SakA (HogA) pathway for phytochrome‐dependent light signaling. Nat Microbiol 1: 16019 [DOI] [PubMed] [Google Scholar]
- Yu Z, Fischer R (2019) Light sensing and responses in fungi. Nat Rev Microbiol 17: 25–36 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li H, Zhang C, An X, Liu L, Stubbe J, Huang M (2014) Conserved electron donor complex Dre2‐Tah18 is required for ribonucleotide reductase metallocofactor assembly and DNA synthesis. Proc Natl Acad Sci USA 111: E1695–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
This study includes no data deposited in external repositories.