

Abstract
Deregulation of the epigenome underlies oncogenesis in numerous primary brain tumours in children and young adults. In this review, we describe how recurrent mutations in isocitrate dehydrogenases or histone 3 variants (oncohistones) in gliomas, expression of the oncohistone-mimic EZHIP in a subgroup of ependymoma, and epigenetic alterations in other embryonal tumours promote oncogenicity. We review the proposed mechanisms of cellular transformation, current tumorigenesis models and their link to development. We further stress the narrow developmental windows permissive to their oncogenic potential and how this may stem from converging effects deregulating Polycomb Repressive Complex 2 function and targets. As altered chromatin states may be reversible, improved understanding of aberrant cancer epigenomes could orient the design of effective therapies.
Keywords: epigenome, Glioma, isocitrate dehydrogenases, polycomb repressive complex 2, EZH inhibiting protein EZHIP
Altered Development at the core of Pediatric and Young Adult Brain Tumours
Primary brain tumours (BT) are a significant cause of morbidity and mortality among children and young adults. These neoplasms are traditionally classified according to their presumed cell of origin using histopathological features, into high-grade (grades III and IV) or low-grade (grades I and II) tumours, and into specific prognostic subgroups by select markers [1]. Tumours of presumed glial origin include gliomas (see Glossary), the most frequent malignant BT (~80%), and ependymomas (EPN). Tumours of neuronal origin include embryonal tumours such as medulloblastomas (MB), atypical teratoid/rhabdoid tumours (ATRT) and embryonal tumours with multilayered rosettes (ETMR). Genomic analysis and molecular profiling helped refine their categorization into prognostic subgroups based on molecular profiles and DNA methylation patterns[2–11]. A central trend emerging from these studies is the deregulation of the epigenome during development at the core of BT pathogenesis. Indeed, many driver events display tight spatiotemporal patterns in the brain, promote altered epigenetic states, and associate with specific lineages of origin as shown in recent single-cell transcriptomic studies[12–14]. We will review herein the main epigenetic alterations identified in subgroups of high-grade gliomas (HGG) and posterior fossa group A EPN (PFA-EPN). We will detail evidence showing these alterations promote tumorigenesis in experimental models, and are required for tumour initiation, progression, and maintenance of oncogenic states. We highlight how deregulation of Polycomb Repressive Complex 2 (PRC2) targets involved in cell fate specification is shared among these entities, and by other aggressive embryonal brain tumours. Epigenomic changes are reversible, and in-depth understanding of altered chromatin states may provide needed alternate therapeutic strategies in these deadly brain cancers.
DNA methylation, Histone 3 (H3) post-translational modifications (PTM), PRC, and their interplay with chromatin across development.
Cytosine DNA methylation (5mC) and histone PTM form a reversible code which helps regulate virtually all processes that act on, or depend on DNA, including replication and repair, regulation of gene expression, and maintenance of centromeres and telomeres. H3 variants are a core component of the nucleosome, the fundamental unit of chromatin, and can be modulated by a large variety of covalent PTMs mostly occurring in the N-terminal tails, which associate with repressive (heterochromatin) or active (euchromatin) states (reviewed in [15]). In this review, we focus on methylation and acetylation of specific lysine (K) residues on the H3 tail. The trimethylation of either H3K9 or H3K27 (H3K9me3/H3K27me3) establish heterochromatin and silence transcription, viewed as either constitutive (H3K9me3) or facultative (H3K27me3). The dimethylated states of these residues, H3K9me2/H3K27me2, potentially play different roles than their trimethylated counterparts[16, 17]. They are deposited in distinct areas of chromatin from H3K27me3 or H3K9me3 and appear to favour repression, but not as strongly as what is observed in trimethylated domains[16, 17]. H3K27 is uniquely methylated by PRC2 through the catalytic subunit Enhancer of Zester Homolog 1 or 2 (EZH1/2), in complex with other core subunits [18]. H3K27 lysine demethylases 6A and 6B (KDM6A/B) oppose the effects of PRC2 on H3K27. Polycomb Repressive Complex 1 (PRC1) recognizes H3K27me3, ubiquitylates histone 2A K119, and contributes to chromatin compaction and repression of gene expression. In mammalian cells, PRC2 is recruited to high affinity cell-type specific nucleation sites including unmethylated CpG island (CGIs) promoters. From there the complex deposits and spreads the H3K27me2/me3 marks throughout adjacent domains[18–20]. Several other mechanisms, including interaction with RNA and reciprocal interplay with PRC1, can influence PRC2 recruitment to chromatin and its localization[21].
As heterochromatin is maintained and inherited across cell divisions, activation of repressed promoters requires the demethylation of H3K27me3, eviction of PRC2 and occupancy by transcription-favouring complexes. These transitions involve Complex Proteins Associated with Set1 (COMPASS) and BRG1/BRM Associated Factors (BAF) complexes that compete with PRC2/1 for occupancy and activation of target genes [15, 22]. Within COMPASS complexes, lysine methyltransferases of the Mixed Lineage Leukemia (MLL) family deposit H3K4me3 at expressed promoters and H3K4me1 at active enhancer regions[15]. These elements also acquire H3K27ac, a PTM read by bromodomain-containing protein complexes favouring transcriptional activation. Active gene bodies and intergenic euchromatin are marked by H3K36me2. Genic regions acquire H3K36me3 uniquely via SET Domain containing 2 (SETD2) recruitment after splicing of genes’ first intron sequences [23]. The recognition of H3K36me2 and H3K36me3 by DNA Methyltransferases 3A and 3B (DNMT3A and DNMT3B), respectively, result in 5mC deposition throughout euchromatin [24], while promoter DNA methylation is generally associated with gene silencing. H3K36me2/3 and 5mC can limit the spread of H3K27me2/3 through hindrance of PRC2 activity [18]. Mutual exclusivity of active and repressive marks (H3K27ac and H3K27me2/3) and indirect antagonism by regulation of methyltransferases (PRC2 and H3K36me2/3) help establish and preserve distinct domains of permissive and repressive chromatin. The antagonism between PRC2/PRC1 and COMPASS are part of an evolutionarily conserved epigenetic memory system that maintains active and repressed promoter states, important during stem cell, embryonic, and brain development [25]. The transition from expansion of pluripotent stem cells to neurogenesis and further differentiation into neural and glial lineages is thus characterized by major epigenetic changes, where redistribution of the H3K27me3 mark plays a major role [25]. Deregulation of PRC2 is an oncogenic mechanism shared by numerous drivers in the aggressive brain tumors that will be described below.
Pediatric and young adult HGGs: a story of altered lysines on histone 3
IDH-mutant gliomas.
Recurrent somatic heterozygous mutations in isocitrate-dehydrogenase (IDH) 1 and 2 in young adult gliomas affect specific residues of the enzymes, mainly R132 of IDH1 and R172 of IDH2, and result in loss of native function of the wild-type enzyme. IDH1/2 mutations are early events in gliomas [26], and are maintained in recurrences after treatment and in progression to HGG [27, 28]. IDH-mutant tumours occur mainly in the frontal brain and are uniformly deadly when they associate with TP53 alterations and loss-of-function alterations in the chromatin remodeler and H3.3 chaperone, α-thalassemia/mental retardation-X-linked (ATRX) [6, 29] (Figure 1). IDH-mutants show neomorphic activity and generate an oncometabolite 2-hydroxyglutarate (2-HG) that competitively inhibits Ten-Eleven Translocation (TET1/2) DNA demethylases as well as Jumonji-domain histone demethylases (KDMs). TET1/2 inhibition by IDH mutants produces a CpG island methylator phenotype (CIMP) characterized by DNA hypermethylation in tumours [30]. However, exogenous introduction of IDH mutants into cells does not consistently induce CIMP, suggesting a time-dependent clonal evolutionary process in tumours [31]. IDH mutants also variably induce gain of H3K4me3, H3K9me3, H3K27me3 and H3K36me3 through KDM inhibition[32, 33]. Recent data suggests that H3K27me3 dysregulation through KDM6A/B demethylase silencing may potentially play a greater role in transcriptional silencing of key PRC2 target genes than the 5mC CIMP in IDH-mutant tumours [34] Nonetheless, the specific epigenetic mechanisms by which IDH mutants favour the acquisition of CIMP [32, 35], or alter expression of genes associated with proliferation and stemness, including PRC2 target genes[36, 37], remain the subject of active investigations (Figure 2).
Figure 1. Brain tumour subgroups associated with epigenetic remodeling.
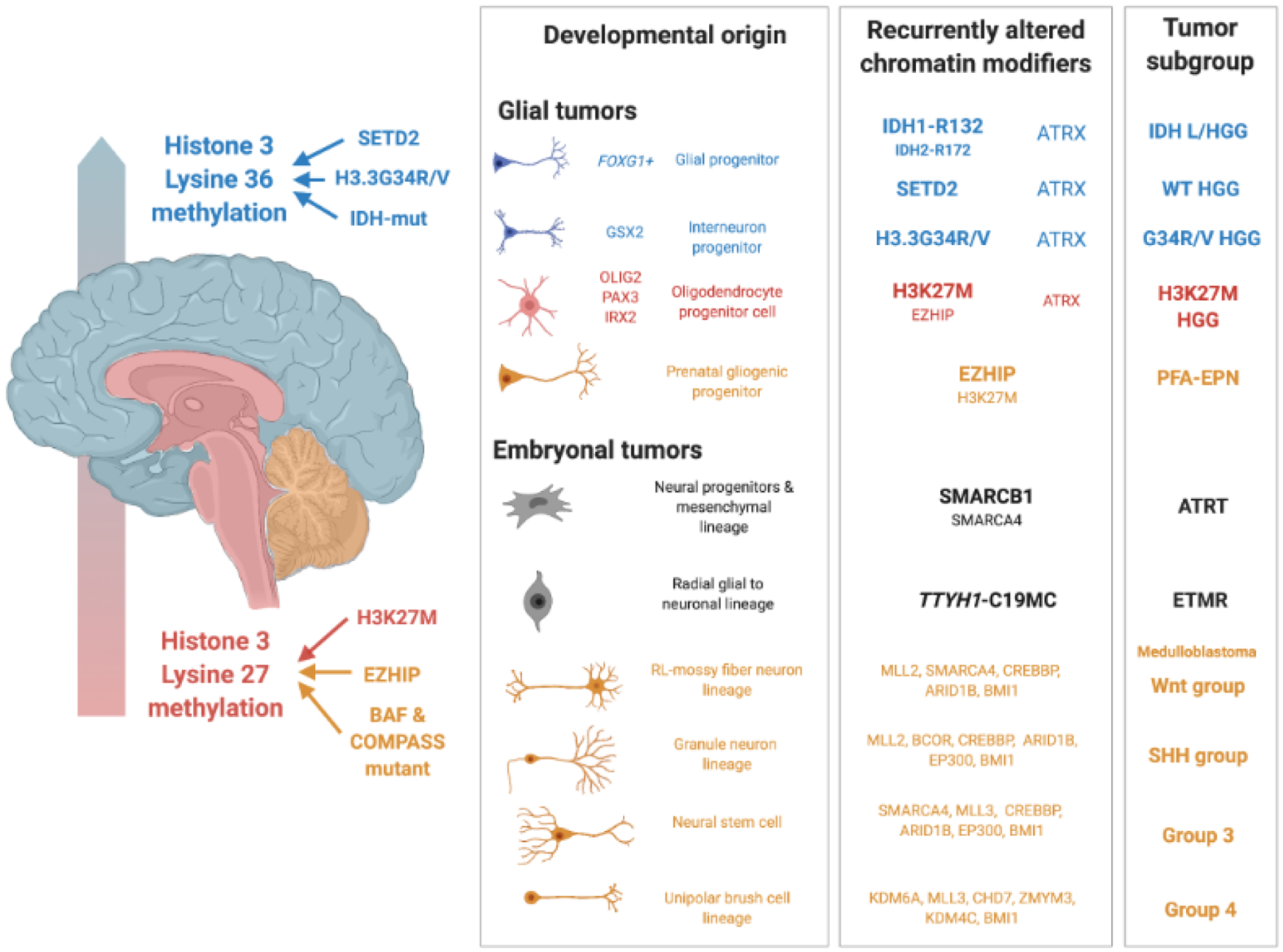
Brain tumours are broadly stratified as glial or embryonal, and by regions of occurrence in the forebrain (blue), midbrain and pons (red) or cerebellum (yellow). Tumours arising in early development are located in the midbrain, pons and cerebellum events and are characterized by events altering the methylation of histone 3 lysine 27 (H3K27). H3 lysine 27-methione (H3K27M) mutations and EZHIP are likely functionally interchangeable yet associate with either oligodendroglial fates in midline high-grade glioma (HGGs) or radial glial progenitors in posterior fossa group A ependymomas (PFA-EPN). Low and High-Grade Glioma (L/HGG) subgroups of later childhood and young adults target the cortical hemispheres, and carry H3.3G34R/V, isocitrate dehydrogenase 1 or 2 mutations (IDH1-R132/IDH2-R172) and/or loss of SET domain containing 2 (SETD2) function. These events converge on altering higher methylation states at H3K36, with implications for altered polycomb complex distribution. Subgroup-specific lineage markers demarcate driver mutation associations with glial cell types. Atypical teratoid/rhabdoid tumours (ATRT) driven by SMARBC1 or occasionally SMARCA4 loss arise from early neural or mesenchymal progenitors in the brain. Embryonal tumours with multilayered rosettes (ETMR)present unique DNA methylation landscape likely tied to the TTYH1-C19MC fusion event, with their transcriptomes recapitulating a neuronal lineage. Four MB subgroups match different neuronal lineages from the cerebellum and carry a variety of mutations in epigenetic targets.
Figure 2. Targets of chromatin modifiers altered in brain tumours.
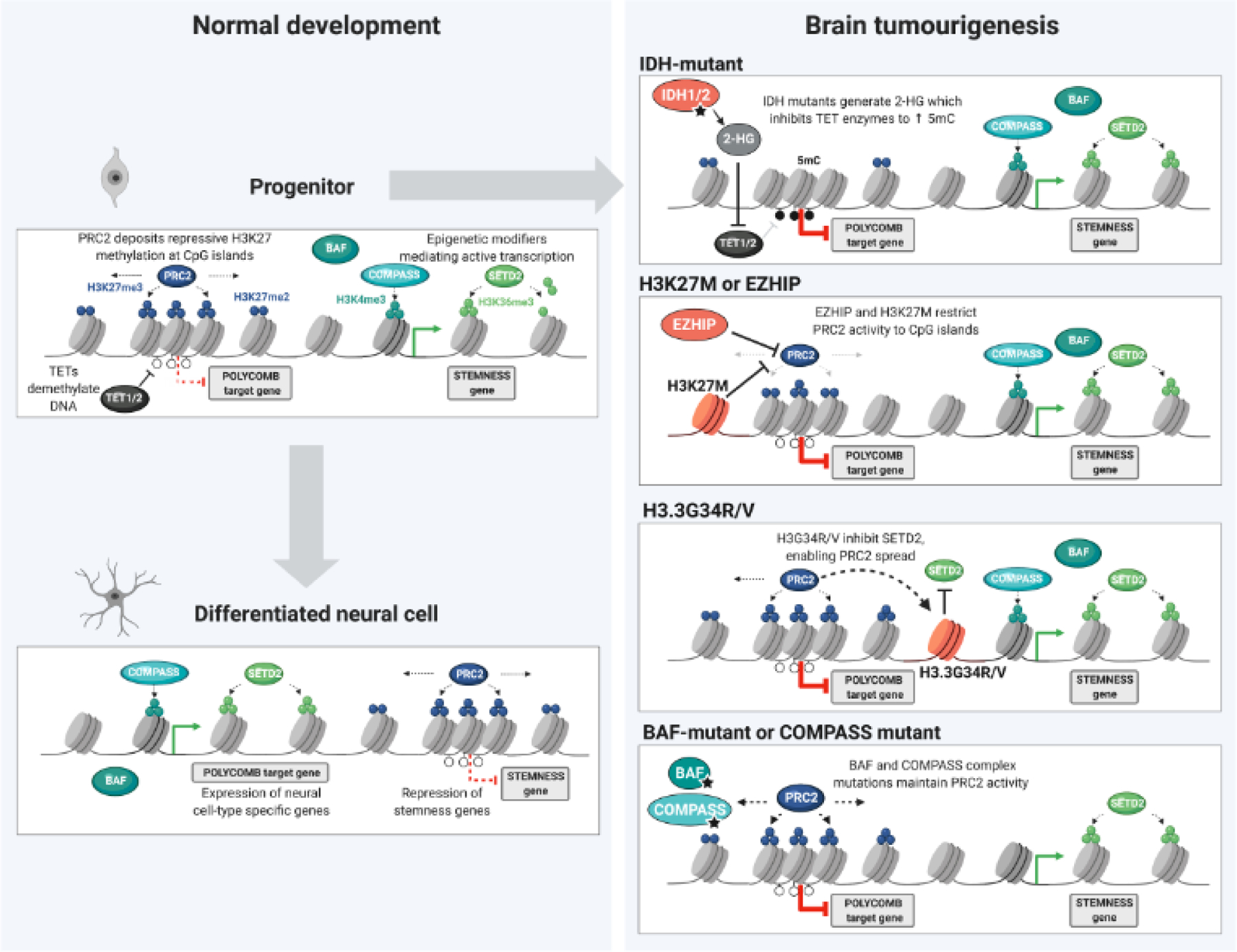
Appropriate differentiation of neural lineages requires remodeling of chromatin landscapes through the interplay of polycomb repressive complex 2 (PRC2), acting in opposition to BRG1/BRM associated factors (BAF) complexes and Complex Proteins associated with Set1 (COMPASS). Abnormal PRC2 occupancy at genomic loci involved in fate specification, and expression of stemness associated genes, can stall differentiation and promote tumorigenesis linked to enhanced self-renewal. In stem cells, polycomb targets are prone to acquisition of repressive 5 methyl-cytosine through the inhibition of histone and DNA demethylases. Restraining the spread of PRC2 through H3K27M or Enhancer of Zeste Homologs Inhibitory Protein (EZHIP) aberrant expression maintains repression of specific polycomb targets and an active chromatin at stemness-associated genes. Deposition of H3.3G34R/V mutant histone prevents SET Domain containing 2 (SETD2) deposition of H3K36me3, thereby facilitating PRC2 redistribution to genic regions. The loss of BAF and COMPASS functions further prevents the conversion from repressed PRC2 targets to an active state, and is associated with stalled neural differentiation.
H3-mutant gliomas.
Disruptions to tumour chromatin states are caused by recurrent somatic mutations at specific residues in H3 variants in HGG affecting children and young adults [2, 7, 9, 10, 38]. Similar to IDH mutations, H3 mutations, termed oncohistones, show neuroanatomical specificity and associate with distinct DNA methylation patterns, age distributions and specific molecular alterations[7, 8]. Lysine 27 to methionine amino acid substitutions occur in any H3 variant (H3K27M) in gliomas located in the thalamus, midbrain and occasionally the spinal cord. These mutations predominate in children and young adults[39] and very rarely target older patients[40]. Glycine 34 to arginine or valine substitutions specifically target noncanonical H3.3 (H3.3G34R/V) and are identified in adolescent/young adult HGG located in the temporo-parietal cortex[41]. These mutations are mutually exclusive with each other and with IDH-mutant tumours and define unique biological subgroups of gliomas (Figure 1). They highlight how changing selective pressures can drive gliomagenesis in different developmental contexts.
H3K27M mutations.
H3K27M mutations, the presumed initiating genetic lesion, are clonal and observed in a high proportion of midline gliomas[39, 42]. Rare case reports identify H3K27M as the sole initial genetic alteration in seemingly low-grade tumours which subsequently transform to HGG upon acquisition of other genetic alterations[43]. Early cooperating partner mutations with this subgroup include somatic loss of p53 function or related pathway members (PPM1D), and mutually exclusive gain-of-function mutations or genetic amplification of growth factor receptors involved in brain development, namely ACVR1/ALK2 in the pons, FGFR1 and EGFR in the thalamus and cerebellum, and PDGFRA across all brain regions[2, 39, 44–46].
H3K27M are dominant negative inhibitors of PRC2 activity. Even though mutant histones account for only 3–10% of all H3, they diminish PRC2 activity in the majority of the H3 pool, across models spanning from C. elegans to human tumours[41, 47–54]. Indeed, lysine-to-methionine and lysine-to-isoleucine mutants of H3 impair the catalytic activities of methyltransferases acting on the respective mutant residue, including H3K4, K9, K27 and K36 [41]. H3K27M affects PRC2 deposition of K27me2/3[41, 47, 49, 50, 55, 56] and diminishes EZH2 automethylation [57], which may impact PRC2 function beyond transient association with the mutant H3 [53]. Structural resemblance of K-M and K-I mutants to suitable substrates of lysine methyltransferase catalytic domains plausibly accounts for their inhibitory effect [58]. However, the nature of the physical interaction between these oncohistones and target methyltransferases in the context of chromatin remains unclear. There is residual H3K27me3/me2 deposition on chromatin of H3K27M-expressing cells, indicating that the mutant does not result in complete loss of PRC2 function[50–52, 59]. Some models propose that H3K27M physically sequesters PRC2 on mutant nucleosomes on chromatin based on increased binding of the complex to H3K27M[58], while others suggest the reverse and no increased affinity of this complex to H3K27M-mutant nucleosomes compared to wild-type [56, 60]. Regardless, the strength of associations between PRC2 and mutant peptides, mutant histones, or native chromatin substrates largely depends on the experimental context across these biochemical studies[41, 52, 56, 58, 60, 61]. Other hypotheses of H3K27M mode of action favour residual recruitment of PRC2 mainly to its strong affinity sites[51], or suggest its exclusion from its nucleation sites by heterotypic H3K27M-K27ac nucleosomes, which may create new oncogenic promoter/enhancer landscapes [52], consistent with the increased H3K27ac levels in H3.3K27M mutant cells[41, 49, 52, 59]. The discovery of the H3K27M-mimic EZH inhibiting protein (EZHIP) challenges the view that PRC2 is inhibited by its association with a H3K27M-containing chromatin substrate, since EZHIP does not incorporate into chromatin and yet can still affect PRC2’s catalytic activity [50, 59, 62]. Furthermore, incorporation of H3K27M in the nucleosome in various chromatin domains does not preclude the presence of a fraction of labile H3 mutants capable of transient association with PRC2 in the vicinity of H3K27me3-marked domains.
The loss of EZH2 automethylation and impaired allosteric EED activation may explain the prolonged impairment of EZH2/1 catalysis, in the absence of continual association with H3K27M. H3K27me3 deposition is normally slow to catalyze[19, 20] and allosteric activation of the complex through autophosphorylation of EZH2 or EED stimulation facilitates the production and spread of this PTM. In the presence of H3K27M, PRC2 can be recruited at its nucleation sites where it deposits H3K27me3 but is unable to mediate distal spreading of the mark[50, 53] (Figure 2). The more rapidly deposited and abundant H3K27me2 can spread but is restrained to domains amenable to PRC2, normally marked by H3K27me3 in H3 wild-type cells and delineated by boundaries with H3K36me2 [49, 50]. Importantly, the spread of H3K27me2 indicates that PRC2 is able to move on chromatin beyond its nucleation sites in the presence of H3K27M, albeit to a lesser extent than in cells not carrying the mutation. In all, H3K27M limits the capacity of PRC2 to deposit H3K27me3 beyond select nucleation sites. The loss of distal deposition of this PTM results in promoter de-repression, particularly of lowly expressed genes and of repeat elements, that can diversely affect stemness, proliferation and cellular stress responses[50, 51, 59, 63, 64]. Residual H3K27me3 deposition at specific CGIs plays a role in the oncogenic phenotype. This contributes to explain why complete loss of PRC2 is never observed in gliomas and why pharmacological EZH2 inhibition selectively harms H3K27M mutants[50, 51, 65]. Indeed, dynamic spread of PRC2 and distribution of H3K27me3 on chromatin play a major role in the regulation of gene expression programs during development[15, 25]. Preserved but restricted occupancy of PRC2, and likely the reader complex PRC1, to local nucleation sites contribute to maintaining repression of numerous genes, including developmental regulators [50]. These loci include Homeobox gene clusters that are repressed in H3K27M mouse models [51, 63, 66], and are depleted of strong enhancer features in H3K27M compared to H3 wild-type HGGs [59]. The low frequency mutations among pediatric HGGs in subunits of BAF complexes and in KDM6A (H3K27 demethylase) [39] further propose tumorigenic effects due to strengthened PRC2 target repression. Ultimately, this continuous repression at select CGIs may, in cooperation with H3K27M partner mutations, favour stemness and impair differentiation of tumour cells in an oligodendrocyte progenitor cell state, the presumed lineage of origin inferred from single-cell studies [12, 14] and in enhancer landscape profiles of tumours [67].
H3K27M has effects that further extend downstream of H3K27 methylation to other chromatin modifications, which may also play a role in transformation[49, 53, 59]. Lacking heterochromatin establishment, intergenic regions of H3K27M mutants acquire H3K27me1 [49], H3K27ac [59], and elevated H3K36me2 in some contexts[49, 53]. Redistribution of H3K27me1 from active genic to intergenic regions suggests that H3K27M-stalled PRC2 is limited in its spatial distribution. Gain of H3K9me3 in regions losing H3K27me2/3 propose alternative forms of heterochromatin compensate for chromatin integrity. In all, these findings mirror what has been observed for the other oncohistone, H3K36M[68]: the mutation affects the PTM and global deposition of the residue it occurs on; it also promotes genome wide redistribution of other antagonistic PTM, which may in turn participate in oncogenicity.
Findings in K-to-M H3-mutants suggest a model whereby the extent of H3K27me3 spread determines the recruitment and the strength of repression and chromatin compaction by the reader PRC1. In H3K27M, restriction of H3K27me3 to PRC2 nucleation sites may lead to persistent strong PRC1 repression, while the reverse is observed in H3K36M: undue intergenic spread of H3K27me3 following H3K36me2 loss leads to PRC1 dilution of and de-repression of its target genes [68]. More work is needed to further unravel the effects of H3K27M, what modifications are to be targeted to reverse its oncogenic potential of this mutation, and what underlies its need for specific partnership genetic alterations.
H3.3G34R/V mutations.
H3.3G34R/V account for 30% of HGGs in adolescents and young adults. At the molecular level, these HGG almost invariably carry mutations in ATRX and TP53, and cluster distinctly from other glioma entities based on DNA methylation [9] (Figure 1). Transcriptomic analysis of G34R/V HGGs reveals these mutations occur in interneuron progenitors during early development[69, 70] and that G34R/V or W mutations may affect splicing[70, 71]. Activation of the MAPK pathway, largely through PDGFRA overexpression and gain-of-function mutations, provides the aberrant astrocyte characteristics that contributes to their labeling as gliomas, and promotes tumorigenicity. The lineage of origin may facilitate PDGFRA co-option through a chromatin loop connecting PDGFRA to regulatory elements of the interneuron progenitor core transcription factor GSX2, thereby facilitating PDGFRA overexpression and possible subsequent mutation of this gene [69].
In contrast to H3K27M that affects both canonical H3.1/2 and H3.3 variants, G34R/V occur exclusively on non-canonical H3.3 and have been suggested to act in cis, with the mutant histone affecting PTMs solely on the mutant peptide, without a global effect on WT histone [41]. Substituting glycine 34 for a variety of larger sized residues hinders the trimethylation of the adjacent K36 residue[41, 72, 73] and reader protein recognition of H3.3K36me3[74]. H3.3G34R/V can also inhibit the KDM4 family of demethylases, thus indirectly altering H3K9 and H3K36 methylation [75]. On H3.3G34 mutant histones, decreased H3K36me3 correlates with the gain of the antagonistic mark, H3K27me3[24, 69, 76]. In HGGs, this effect appears to cooperatively impair interneuron differentiation[69] (Figure 2). Other recent studies further emphasize that forebrain progenitors are the one to be exquisitely amenable to H3.3G34R/V oncogenic effects, compared to other lineages/brain locations[70, 77]. The creation of new focal H3K27me3 domains by H3.3G34 mutations parallels patterns of chromatin in giant cell tumours of the bone[69], which universally carry H3.3G34W/R/L mutations[78]. The contribution of PRC2 in the oncogenic process is further substantiated by experiments in which H3.3G34W-K27R double mutants, which are unable to acquire H3K27me3, are less effective in promoting tumour development in mouse mesenchymal stem cells[24].
The HIRA chaperone deposits H3.3 in the bodies of actively transcribed genes, while the ATRX/DAXX chaperone complex facilitates its deposition in pericentromeric and telomeric heterochromatin. These selective deposition patterns likely restrict potential cis-regulatory elements amenable to be repressed by H3.3G34 mutations. ATRX loss-of-function in H3.3G34R/V HGGs may thus drive enhanced remodeling of histone methylation, through increased amounts of H3.3, including mutant H3.3, deposited in euchromatin by HIRA. Collectively, a key role for PRC2 emerges in mediating tumorigenesis. H3.3G34R/V can lead to PRC2 redistribution and the creation of ectopic H3K27me3 domains, which are likely an initial requirement for transformation in H3.3G34 mutants.
H3 and IDH wild-type HGGs.
In the brain midline, rare H3 wild-type HGG have IDH hotspot mutations [79], or MYCN amplification [45], or ectopic expression of the oncohistone mimic EZHIP, which is normally seen in PFA-EPN (see below) [80]. In hemispheric pediatric and young adult HGGs, loss-of-function mutations in the H3K36 methyltransferase SETD2 and in rare cases in the H3.3K36me3 reader ZMYND11 have been identified and occur in association to TP53 and ATRX loss [38, 81], suggesting that the loss of H3K36 methylation may parallel the effects of mutually exclusive H3.3G34R/V mutations (Figure 1).
In summary, midline HGG are primarily defined by decreased H3K27me2/3, due to H3K27M (~85%) or aberrant expression of EZHIP (10%), while hemispheric HGGs show genetic alterations affecting the methylation of H3K36 and frequent loss-of-function alterations in ATRX. ATRX mutations strongly associate with thalamic H3K27M tumours and hemispheric HGGs, but not pontine HGGs. More work is needed to better understand the role of this cooperating partner mutation shared between oncohistones and IDH mutations and how its loss promotes tumour formation in gliomas.
PFA-EPN
Ependymomas are classified into subgroups by DNA methylation profiles [82]. Of these, PFA-EPN occur in the hindbrain, affect mainly younger children, and show similar dismal prognosis as H3K27M-mutant HGG. Tumours display CIMP characteristics but lack recurrent genetic alterations [83]. Recently, abnormal expression of EZHIP was shown to occur in ~95% of PFA-EPN and to induce decreased H3K27me3 levels[84], while mutually exclusive H3K27M mutations were found in the remaining ~5% [84, 85] (Figure 1). EZHIP was subsequently characterized as a factor associating with PRC2, promoting its allosteric inhibition [3, 55, 62, 84, 86]. The sole region of sequence conservation across species in this gene encodes a 12 amino acid peptide sequence, which mimics the H3 tail with a methionine in the site analogous to lysine 27, mirroring H3K27M. This domain potently impairs catalysis of H3K27me2/3 when EZHIP associates in complex with PRC2 [55, 62], and correlates with significant loss of H3K27me3 previously noted in PFA-EPN tumours, as well as oocytes where EZHIP is normally expressed [87, 88]. EZHIP is not incorporated into chromatin but acts similarly to K27M and inhibits PRC2, globally reducing H3K27me3 levels, thus strengthening the hypothesis that transient association of the oncohistone with PRC2 is sufficient for inhibition of the complex. Functionally interchangeable effects are presumed from rare cases of PFA-EPNs carrying H3K27M mutations, and histone-WT midline HGGs displaying EZHIP expression. However, PFA-EPNs arise in a unique context of early brain development linked with hypoxia and resembling prenatal gliogenic progenitors [13], whose growth and epigenome depend on low oxygen conditions [89]. The dependence of PFA-EPN tumour cells on residual PRC2 activity [89] parallels that of H3K27M-HGGs, as cells likely require the repression of specific H3K27me3-marked promoters. EZHIP is a requirement of PFA-EPN cells to maintain loss of H3K27me2/3, and in some contexts, proliferative fitness [86]. This gene is normally expressed solely in the placenta and germline [88]. Mechanisms by which tumours co-opt its expression remain unclear, and likely involve loss of promoter 5mC and responses to hypoxia [84, 86, 89]. In all, PFA-EPNs present a unique case of tumour progression involving metabolic-epigenetic crosstalk hinging on abnormal EZHIP expression and leading to the restraint of PRC2 at its nucleation sites.
Embryonal BT with epigenetic drivers dysregulate PRC2 targets
Subunits of BAF complexes are frequently mutated in diverse cancers including ATRTs. In these embryonal BT, homozygous loss of SMARCB1 (also known as SNF5, BAF47 or INI1) or rarely of SMARCA4, are the sole recurrent genetic alterations identified in tumours (Figure 1). In close to 30% of ATRTs, and mainly in younger children, de novo germline heterozygous SMARCB1 mutations predispose patients to these rhabdoid tumours. Integrated expression and DNA methylation partition ATRTs into 3 molecular entities [90, 91]. SMARCB1 loss disrupts the antagonism between BAF and PRC2 complexes, leading to widespread deposition of H3K27me3 at PRC2 target genes [92]. H3K27me3-mediated repression at neuronal differentiation genes reflects the inability of cells to evict PRC2 in the absence of SMARCB1, likely favouring self-renewal [93] (Figure 2). Preliminary clinical data in rhabdoid tumours indicate sensitivity to pharmacological inhibition of EZH2, supporting the importance of H3K27me3 in the maintenance of tumorigenesis.
Recurrent mutations in chromatin modifiers have been identified in the 4 major MB subgroups (Figure 1). These include MLL2/3, SMARCA4, ARID1A, and KDM6A, highlighting alterations of COMPASS, PRC2 and BAF complexes in these tumours[94]. Mutation of MLL2/3 methyltransferases results in loss of H3K4me3, while KDM6A alterations increase H3K27me3 levels. Given that KDM6A can be a component of COMPASS and can associate with BAF complexes[95], which compete with PRC2 on target promoters, tumours with these mutations may fail to transition repressed polycomb targets to an active state, thus promoting a stem-cell state. Moreover, a PRC2 signature is identified in group 3 and 4 MB, which further emphasizes the importance of enforcing repressive chromatin in these BT for disease progression.
In ETMR a fusion between TTHY1 and the C19MC microRNA cluster leads to expression of this microRNA cluster, which is normally restricted to the trophoblast [5] (Figure 1). These microRNAs promote overexpression of fetal brain-specific isoforms of the de novo DNA methyltransferases DNMT3B and DNMT3A, leading to aberrant DNA methylation patterns, including at promoters of PRC2 targets, and an oncogenic state which recapitulates early neurogenesis[49]. Single-cell transcriptomics describe ETMR cells in self-renewing states that are unable to progress along their programmed differentiation path, helping explain their histology resembling undifferentiated neural tubes[12]. Here also, disruption of PRC2 distribution to its nucleation sites by aberrant DNA methylation may play a role in cellular transformation.
Establishment and reversibility of tumorigenic epigenomes
Mouse models have facilitated appreciation of the contexts in which IDH, H3K27M and SMARCB1 mutations can promote BT development (Figure 3). Introduction of IDH1-R132H establishes proliferative and infiltrative progenitors of the subventricular zone [31], and metabolic changes favoring glioma initiation[33]. In cooperation with PDGFRA overexpression and CDKN2A, PTEN and ATRX loss, IDH mutants accelerate glioma progression and establish tumour expression and 5mC profiles resembling human IDH-mutant tumours (Figure 3). The unique catalytic activity of IHD1-R132H renders it targetable by small molecule inhibition as blocking 2-HG production promotes glial differentiation[96] (Figure 4). Numerous small molecule inhibitors of mutant IDH are currently under clinical investigation for IDH-mutant tumours, and their clinical value for glioma patients is becoming better established.
Figure 3. Epigenetic drivers potentiate tumour development in murine models.
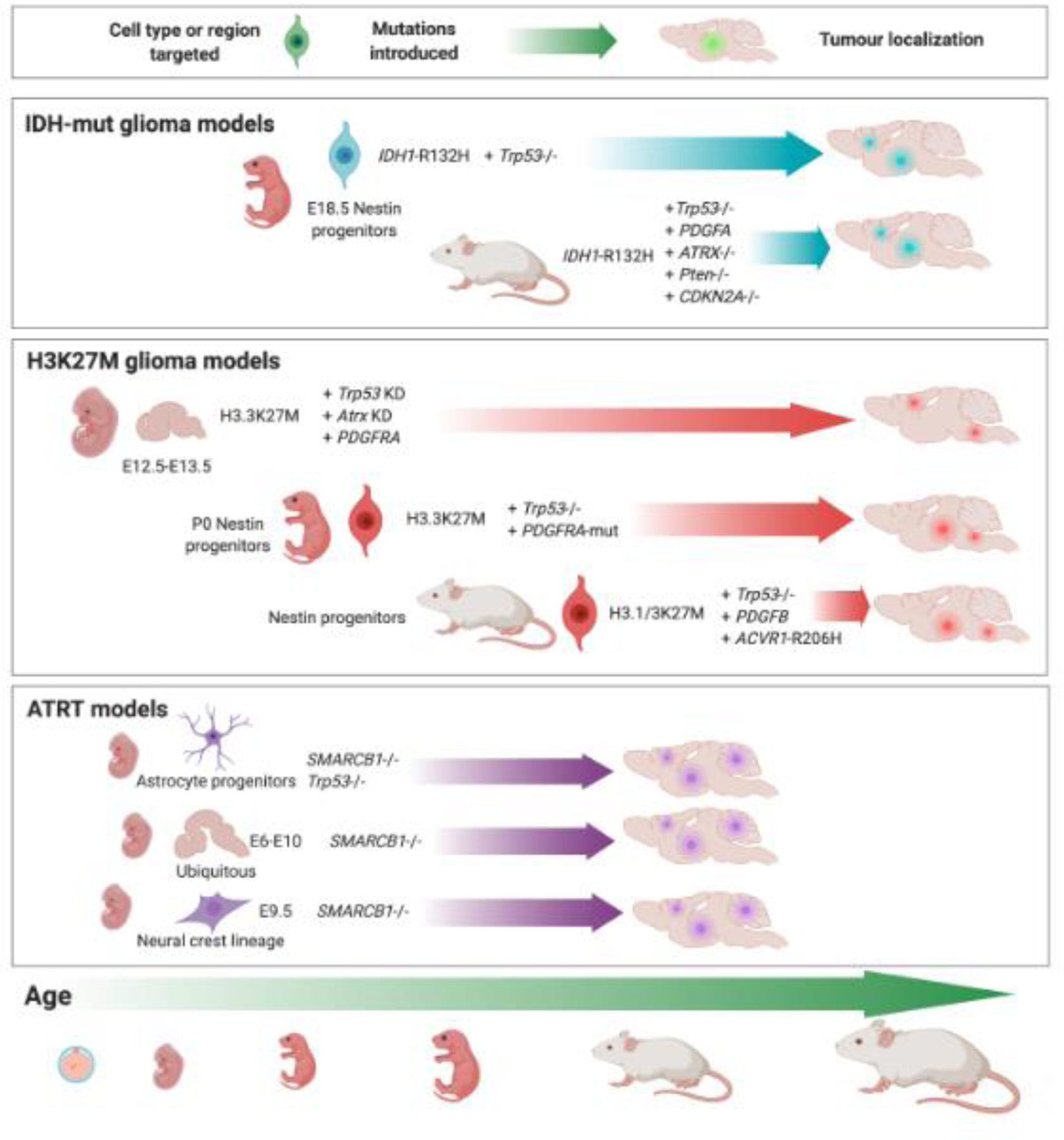
The introduction of epigenetic driver events in mice has revealed the dependencies of tumour formation on time windows, combinations of cooperating oncogenic stimuli, and cell type, lineage or brain region in which they are introduced. In H3K27M, added Trp53 loss is sufficient to induce HGGs. In other H3K27M mouse models, in addition to loss of p53 function, additional PDGFRA/PDGFB overexpression or activating mutations, or ACVR1-R206H appear needed to accelerate disease onset and induce features of human HGG. IDH1-R132H mutant cells also requires Trp53 loss for tumorigenicity and can promote tumours resembling human disease from late embryonic development to adulthood. Loss of Smarcb1 transforms neural progenitors either in combination with Trp53 loss, or on its own between embryonic development days 6–10.
Figure 4. Manipulation of driver events determines reversibility of tumour epigenomes.
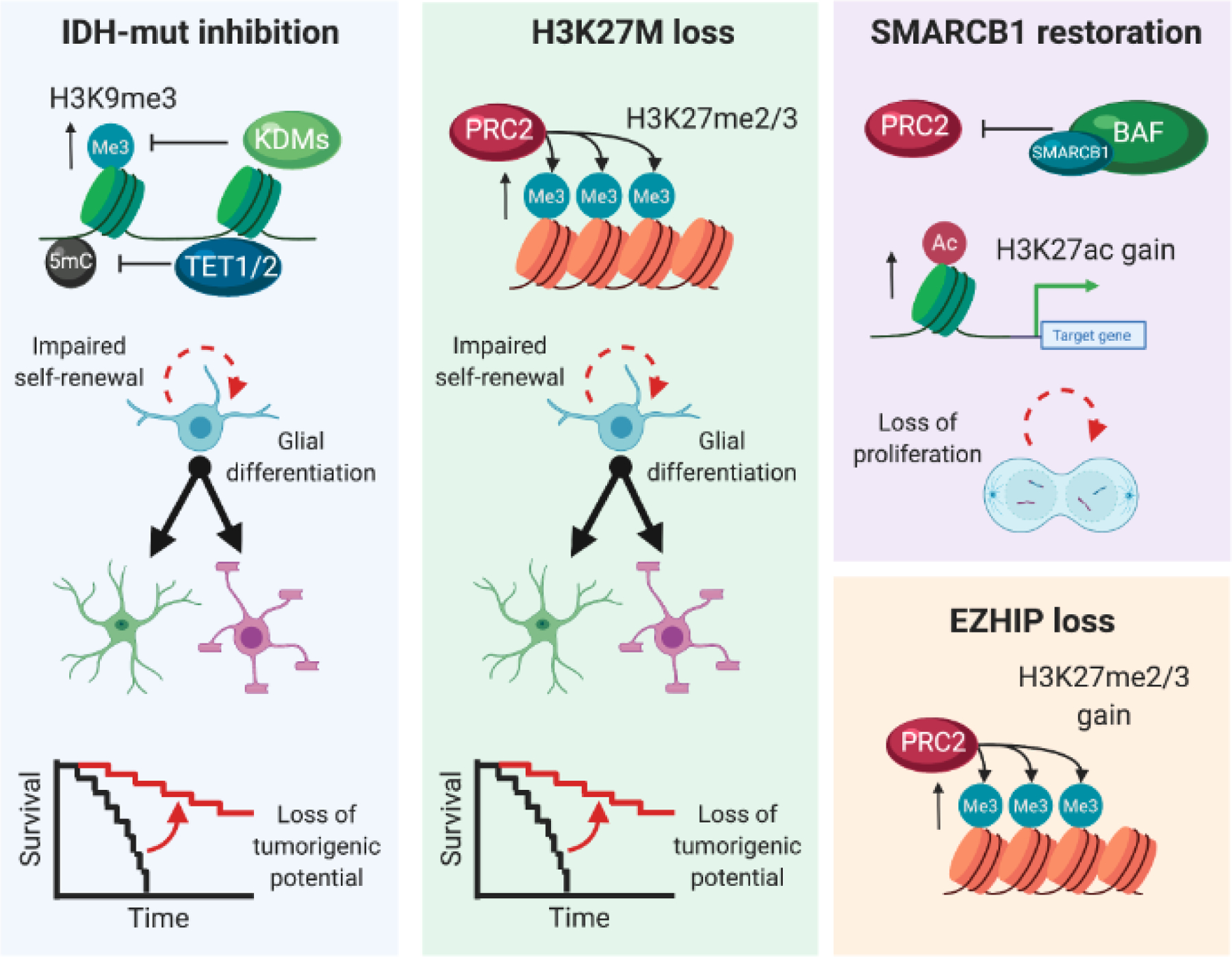
The manipulation of driver events remodeling the epigenome serves to characterize the reversibility of their effects, and the dependence of transformed cells on their function to proliferate, self-renew, and form tumours. The inhibition of 2-HG production by IDH1-R132H using a small molecule drug elevates 5-hydroxymethylcytosine, diminishes histone methylation and promotes glial differentiation in vitro, while delaying tumour burden in xenografts. The knockout or knockdown of the H3F3A-K27M mutant allele in tumour-derived cell lines restores PRC2 activity and H3K27me2/3deposition, renders the cells capable of enhanced glial differentiation and delays or abolishes the potential to form xenograft tumours. Similar trends are observed upon knockout of EZHIP, variably diminishing cell proliferation in some contexts. The restoration of SMARCB1 expression in ATRT lines reversed the loss of H3K27ac to reactivate silenced genes and decrease proliferation.
Targeted expression of H3K27M cannot drive HGG development alone and requires cooperation with p53 loss in specific developmental windows [66]. PDGFRA overexpression and activating mutations accelerate tumour onset, high-grade features and glial histology [63, 66] (Figure 3). These 3 events impair astrocytic differentiation of neural progenitor cells [48]. H3K27M can also cooperate with Acvr1 and Pik3ca signaling that help establish a glial progenitor state [97]. The restricted developmental windows for H3K27M tumorigenesis may relate to the extent of PRC2 spread in the progenitor of origin and its responsiveness to sustained mitogenic signaling. H3K27M mutation is required for initiation, progression and maintenance of impaired H3K27me2/3 spread and the oncogenic potential of cells [50, 65], as it actively impairs their progression to mature glial states [12] (Figure 4). In preclinical models, efforts to target H3K27M HGGs have included inhibition of PRC1[98], PRC2[51], reversal of H3K27me3 loss by inhibition of KDM6B[99], histone deacetylase (HDAC)[100], DNA methylation[59] and bromodomain inhibitors[52], metabolic targeting [101, 102], or combination therapies of the above pathways[103]. Current clinical trials targeting the epigenome are still in their infancy. They include the use of histone deacetylase (HDAC) inhibitors or DNA demethylating agents, and local delivery of EZH2 inhibitors[104] as there are no brain-penetrant compounds currently available. A clinical trial using a CART-cell directed against GD2 is ongoing based on promising pre-clinical data[105]. The DNA demethylating agent 5-azacytidine is also in clinical testing for PFA-EPNs [106]. H3.3G34R/V mutations are weakly tumour-promoting in mice, on their own and require association to Tp53 loss and further enhanced by Pdgfra overexpression[69]. Removal of the mutation allows cells to further mature across the interneuron lineage even if, in contrast to H3K27M, this does not impact tumorigenic potential. Careful targeting of these mutations to the more difficult ventral forebrain (where the proposed interneuron progenitor cells of origin reside) instead of the current models derived from the dorsal forebrain will better elucidate the oncogenic potential of H3.3G34R/V and the role of its associated drivers.
In embryonal tumours, epigenetic models are mainly available for ATRTs. Targeted loss of Smarcb1 in combination to Tp53 loss in astrocytic lineages [107], or ubiquitous loss of the gene exquisitely between E6 and E10 led to BT resembling human ATRT [108]. The narrow temporal window for oncogenic manifestations of Smarcb1 loss was further emphasized, when loss of this gene in the neural crest lineage prior to E12.5 also led to rhabdoid tumours [109] (Figure 3). SMARCB1 absence is required for the continued fitness of ATRT cells. Its restoration favors gain of H3K27ac and diminished proliferation, impaired growth and increased cell cycle arrest [93] (Figure 4). Loss of BAF complex function thus likely overcomes barriers to transformation beyond lineage differentiation.
Concluding remarks
The scope of brain tumour epigenomes illustrates diverse paths to remodeling of chromatin landscapes. While different routes can be associated with specific lineages of origin across space and time, the silencing of developmental genes by PRC2 emerges as a recurrent pattern. Restraining PRC2 spread can both derepress targets covered by its spread, and repress promoters at nucleation sites via H3K27M mutations, likely resembling the effect of endogenous EZHIP in specific tissues of early development. In H3.3G34R/V HGGs, focal H3K27me3 gain can arise from selective loss of H3.3K36me3. Loss of DNA demethylase activity in IDH mutants, or abnormal DNA methylation in the context of ETMR associates with promoter silencing, while the loss of the BAF complexes and H3K27 demethylases in several BT entities plausibly results in failure to activate PRC2 targets. Improved understanding of the interplay between PRC2/1, H3K36 and 5mC, histone and DNA demethylases, BAF and COMPASS complexes during development and cellular differentiation will be essential to illuminate the role played by these mutations and what are the essential targets they ultimately disrupt. These investigations will be crucial in establishing hierarchies and kinetics of deposition during lineage commitment and cellular differentiation. Also, links between PRC1/2, COMPASS and the 3D nucleosome structure for activating or repressing gene expression is emerging. These likely establish windows of opportunity for potential oncogenic co-option of the cis-regulatory landscape, as recently shown for H3.3G34R HGGs. Rare cancer drivers may relate to broader trends, as the CGI hypermethylation of developmental PRC2 targets are frequent in numerous cancers, and a CIMP acquisition in aging IDH-WT cells can be a requirement for transformation[110]. Preserving epigenome integrity must therefore consider selective forces acting to maintain or remodel chromatin over development, from conception to old age. Strong associations between driver events and tumour subgroups imply that select chromatin landscapes are vulnerable to transformation, yet also reversible. Further progress in carefully charting the interplay of these epigenetic marks, their writers and readers will be fruitful paths in taking basic science to clinical application. The generation of brain-penetrant epigenetic modulators, especially for PRC2 subunits, will enable testing of their achievable therapeutic benefits.
Outstanding Questions Box.
What constraints define windows of opportunity for driver mutations to initiate tumourigenesis? The cell states amenable to transformation may present specific proliferative cycling, histone variant expression, polycomb complex distribution and tolerance to chromatin reorganization in cooperation with partnering oncogenic stimuli. What contribution does 3D chromatin architecture play in facilitating oncogenic effects of these mutations?
What is the role of ATRX and TP53 loss in contributing to the oncogenic effects of H3 and IDH mutations? Why is their prevalence high in tumours of the thalamus and cerebral cortex? What is the impact of older age in their acquisition? What is the role of other partnering mutations to H3K27M and H3.3G34R/V?
What drives EZHIP’s abnormal expression in gliomas, including what fosters loss of promoter DNA methylation? This gene has a very specific spatiotemporal pattern of expression and its regulation is poorly understood. Additional data is needed to further understand its role of stalling PRC2 spread in germ cells and tumours.
What regulatory function is served by subcomplex diversity over development (PRC2.1-2 and PRC1.1-6), factors determining recruitment and domain propagation spreading H3K27me2/3, and landscapes of redistribution of the PRC2-opposing marks H3K9, H3K36 and 5mC?
Will brain penetrant epigenetic therapeutic modulators, including EZH2 and/or EED inhibitors, benefit patients with these brain tumours? What are synergistic drug combinations in these diseases? The generation of reliable preclinical animal and human models of these diseases is warranted to fully address these therapeutic needs.
Highlights.
Multiple brain tumours of children and young adults are defined by driver events altering chromatin
Functional characterization of these drivers uncovers convergent effects that remodel landscapes of polycomb repressive complexes
Driver events can potentiate tumour development and stall the appropriate differentiation of neural lineages
Dynamic reversibility of polycomb complex target regulation implicates potential for therapies to overcome tumour self-renewal
Acknowledgements
This work was performed within the context of the International CHildhood Astrocytoma INtegrated Genomic and Epigenomic (ICHANGE) consortium. It was supported by funding from: A Large-Scale Applied Research Project grant from Genome Quebec, Genome Canada, the Government of Canada, and the Ministère de l’Économie, de la Science et de l’Innovation du Québec, US National Institutes of Health (NIH grant P01-CA196539); the Canadian Institutes for Health Research (CIHR grant MOP-286756 and FDN-154307) and support from the Fondation Charles Bruneau and We Love You Connie Foundation. NJ is a member of the Penny Cole Laboratory. Figures are created with BioRender.com. The authors declare no competing interests.
Glossary
- BAF
BRG1/BRM Associated Factors (BAF) is a group of multi-protein ATP-dependent chromatin remodeling complexes of humans, with variants shared among eukaryotes. They are also referred to as mammalian SWItch/Sucrose Non-Fermentable (mSWI/SNF). These complexes establish chromatin accessibility through destabilization and rearrangement of nucleosome-DNA interactions.
- Ependymoma
A type of glioma located in the brain or spinal cord thought to arise from ependymal cells. Ependymomas occur predominantly in children and are classified in subgroups by clinical and molecular features.
- EZHIP
Enhancer of Zeste Homologs (EZH) Inhibiting Protein (EZHIP) is a protein with a structural domain resembling the H3K27M oncohistone, which inhibit the catalytic activity of EZH2 and decrease the production and deposition on chromatin of H3K27me2/3. This gene is found in eutherian mammals, where it is normally expressed in germ cells. Its abnormal expression has been identified in a few cancers (ependymomas, gliomas and endometrial cancers) and is presumed to be oncogenic similar to H3K27M.
- Glioma
A group of brain tumours classified as glial in origin by histological features and the most common primary central nervous malignancy. Gliomas encompass a variety of heterogeneous biological subgroups and are traditionally further sub-divided in low-grade (grades I and II) and high-grade (grade III and IV) tumours based on morphology, proliferation index, and the level of angiogenesis and necrosis in the tumour.
- Oncohistones
Recurrent somatic heterozygous mutations in histone 3 encoding genes that associate with specific cancers. Experimental evidence propose that they drive cancer initiation through altering histone methylation and chromatin regulation. The oncohistones established thus far as cancer drivers include histone 3 lysine 27 and lysine 36 to methionine or isoleucine (H3K27M/I; H3K36M/I), and histone variant 3.3 glycine 34 to arginine, valine or tryptophan (H3.3G34R/V/W) mutations.
- Polycomb repressive complexes (PRC) 1 and 2
-
Important developmental multi-protein complexes that regulate a plethora of cellular processes, including X-chromosome inactivation, genomic imprinting, cell cycle regulation, stem cell biology, and cancer. This functional diversity is achieved by a variety of PcG complexes that are assembled in a developmental-stage- and cell-specific manner to modify chromatin at their target genes via histone-modifying or chromatin remodeling activities. Historically, based on biochemical purification experiments from Drosophila melanogaster, the polycomb machinery has been subdivided into PRC1 and PRC2.
PRC1 is subdivided into canonical and non-canonical complexes that share core proteins composed of proteins (RING1A or RING1B), which have E3 ubiquitin ligase activity mediating ubiquitination of histone H2A on lysine 119 (H2AK119ub), and one of the six Polycomb group ring-finger domain proteins (PCGF1–PCGF6). PRC1 binds to H3K27me3 and mediates chromatin compaction.
PRC2 is a multiprotein complex responsible for the methylation of histone 3 lysine 27. Together with polycomb repressive complex 1, the methylation of H3K27 establishes context-dependent gene silencing. The core subunits of mammalian PRC2 include Enhancer of Zester Homolog 1 or 2 (EZH1/2) Embryonic Ectoderm Development (EED), Suppressor of Zeste Homolog 12 (SUZ12) and Retinoblastoma Binding Protein 4 or 7 (RBBP4/7).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Louis DN et al. (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6), 803–20. [DOI] [PubMed] [Google Scholar]
- 2.Fontebasso AM et al. (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46 (5), 462–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hubner JM et al. (2019) EZHIP / CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khuong-Quang DA et al. (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124 (3), 439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleinman CL et al. (2014) Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46 (1), 39–44. [DOI] [PubMed] [Google Scholar]
- 6.Liu XY et al. (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124 (5), 615–25. [DOI] [PubMed] [Google Scholar]
- 7.Schwartzentruber J et al. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482 (7384), 226–31. [DOI] [PubMed] [Google Scholar]
- 8.Sturm D et al. (2016) New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5), 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sturm D et al. (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 (4), 425–37. [DOI] [PubMed] [Google Scholar]
- 10.Wu G et al. (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44 (3), 251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capper D et al. (2018) DNA methylation-based classification of central nervous system tumours. Nature 555 (7697), 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jessa S et al. (2019) Stalled developmental programs at the root of pediatric brain tumors. Nat Genet 51 (12), 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vladoiu MC et al. (2019) Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572 (7767), 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filbin MG et al. (2018) Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360 (6386), 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuettengruber B et al. (2017) Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171 (1), 34–57. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari KJ et al. (2014) Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 53 (1), 49–62. [DOI] [PubMed] [Google Scholar]
- 17.Jih G et al. (2017) Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature 547 (7664), 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu JR et al. (2019) PRC2 is high maintenance. Genes Dev 33 (15–16), 903–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oksuz O et al. (2018) Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol Cell 70 (6), 1149–1162 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hojfeldt JW et al. (2018) Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol 25 (3), 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laugesen A et al. (2019) Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol Cell 74 (1), 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alfert A et al. (2019) The BAF complex in development and disease. Epigenetics Chromatin 12 (1), 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberg DN et al. (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain SU et al. (2020) Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc Natl Acad Sci U S A 117 (44), 27354–27364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albert M and Huttner WB (2018) Epigenetic and Transcriptional Pre-patterning-An Emerging Theme in Cortical Neurogenesis. Front Neurosci 12, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe T et al. (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174 (4), 1149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juratli TA et al. (2012) IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol 108 (3), 403–10. [DOI] [PubMed] [Google Scholar]
- 28.Jones DTW et al. (2019) The Power of Human Cancer Genetics as Revealed by Low-Grade Gliomas. Annu Rev Genet 53, 483–503. [DOI] [PubMed] [Google Scholar]
- 29.Jiao Y et al. (2012) Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3 (7), 709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noushmehr H et al. (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17 (5), 510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bardella C et al. (2016) Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 30 (4), 578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu C et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483 (7390), 474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki M et al. (2012) IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488 (7413), 656–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Court F et al. (2019) Transcriptional alterations in glioma result primarily from DNA methylation-independent mechanisms. Genome Res 29 (10), 1605–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turcan S et al. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483 (7390), 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Figueroa ME et al. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18 (6), 553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turcan S et al. (2018) Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet 50 (1), 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fontebasso AM et al. (2013) Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol 125 (5), 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackay A et al. (2017) Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4), 520–537 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleinschmidt-DeMasters BK and Mulcahy Levy JM (2018) H3 K27M-mutant gliomas in adults vs. children share similar histological features and adverse prognosis. Clin Neuropathol 37 (2018) (2), 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis PW et al. (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340 (6134), 857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nikbakht H et al. (2016) Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7, 11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joyon N et al. (2017) K27M mutation in H3F3A in ganglioglioma grade I with spontaneous malignant transformation extends the histopathological spectrum of the histone H3 oncogenic pathway. Neuropathol Appl Neurobiol 43 (3), 271–276. [DOI] [PubMed] [Google Scholar]
- 44.Sievers P et al. (2021) A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol 23 (1), 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buczkowicz P et al. (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46 (5), 451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu G et al. (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46 (5), 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bender S et al. (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24 (5), 660–72. [DOI] [PubMed] [Google Scholar]
- 48.Funato K et al. (2014) Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346 (6216), 1529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harutyunyan AS et al. (2020) H3K27M in Gliomas Causes a One-Step Decrease in H3K27 Methylation and Reduced Spreading within the Constraints of H3K36 Methylation. Cell Rep 33 (7), 108390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harutyunyan AS et al. (2019) H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun 10 (1), 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mohammad F et al. (2017) EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23 (4), 483–492. [DOI] [PubMed] [Google Scholar]
- 52.Piunti A et al. (2017) Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23 (4), 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stafford JM et al. (2018) Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4 (10), eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delaney K et al. (2019) H3.3K27M-induced chromatin changes drive ectopic replication through misregulation of the JNK pathway in C. elegans. Nat Commun 10 (1), 2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jain SU et al. (2020) H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol Cell 80 (4), 726–735 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herz HM et al. (2014) Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345 (6200), 1065–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee CH et al. (2019) Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev 33 (19–20), 1428–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Justin N et al. (2016) Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krug B et al. (2019) Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27 M Gliomas. Cancer Cell 35 (5), 782–797 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X et al. (2017) Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol 24 (12), 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarthy JF et al. (2020) Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jain SU et al. (2019) PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1), 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larson JD et al. (2019) Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 35 (1), 140–155 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pajovic S et al. (2020) Epigenetic activation of a RAS/MYC axis in H3.3K27M-driven cancer. Nat Commun 11 (1), 6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Silveira AB et al. (2019) H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol 137 (4), 637–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pathania M et al. (2017) H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 32 (5), 684–700 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagaraja S et al. (2019) Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol Cell 76 (6), 965–980 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu C et al. (2016) Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352 (6287), 844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khazaei S et al. (2020) H3.3 G34W Promotes Growth and Impedes Differentiation of Osteoblast-Like Mesenchymal Progenitors in Giant Cell Tumor of Bone. Cancer Discov 10 (12), 1968–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Funato K et al. (2021) Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lutsik P et al. (2020) Globally altered epigenetic landscape and delayed osteogenic differentiation in H3.3-G34W-mutant giant cell tumor of bone. Nat Commun 11 (1), 5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fang J et al. (2018) Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proc Natl Acad Sci U S A 115 (38), 9598–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yadav RK et al. (2017) Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wen H et al. (2014) ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508 (7495), 263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voon HPJ et al. (2018) Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat Commun 9 (1), 3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang TY et al. (2020) Effects of H3.3G34V mutation on genomic H3K36 and H3K27 methylation patterns in isogenic pediatric glioma cells. Acta Neuropathol Commun 8 (1), 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bressan RB et al. (2021) Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Behjati S et al. (2013) Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45 (12), 1479–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang L et al. (2014) Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet 46 (7), 726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sievers P et al. (2020) A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salloum R et al. (2017) Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas. Acta Neuropathol Commun 5 (1), 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pajtler KW et al. (2015) Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5), 728–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mack SC et al. (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506 (7489), 445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pajtler KW et al. (2018) Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136 (2), 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ryall S et al. (2017) H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33 (7), 1047–1051. [DOI] [PubMed] [Google Scholar]
- 86.Piunti A et al. (2019) CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci Adv 5 (7), eaax2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bayliss J et al. (2016) Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 8 (366), 366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ragazzini R et al. (2019) EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat Commun 10 (1), 3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Michealraj KA et al. (2020) Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 181 (6), 1329–1345 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johann PD et al. (2016) Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3), 379–393. [DOI] [PubMed] [Google Scholar]
- 91.Torchia J et al. (2016) Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 30 (6), 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wilson BG et al. (2010) Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 (4), 316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Erkek S et al. (2019) Comprehensive Analysis of Chromatin States in Atypical Teratoid/Rhabdoid Tumor Identifies Diverging Roles for SWI/SNF and Polycomb in Gene Regulation. Cancer Cell 35 (1), 95–110 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Northcott PA et al. (2017) The whole-genome landscape of medulloblastoma subtypes. Nature 547 (7663), 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Narayanan R et al. (2015) Loss of BAF (mSWI/SNF) Complexes Causes Global Transcriptional and Chromatin State Changes in Forebrain Development. Cell Rep 13 (9), 1842–54. [DOI] [PubMed] [Google Scholar]
- 96.Rohle D et al. (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340 (6132), 626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fortin J et al. (2020) Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 37 (3), 308–323 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Balakrishnan I et al. (2020) Senescence Induced by BMI1 Inhibition Is a Therapeutic Vulnerability in H3K27 M-Mutant DIPG. Cell Rep 33 (3), 108286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hashizume R et al. (2014) Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 20 (12), 1394–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grasso CS et al. (2015) Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 21 (6), 555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khan A et al. (2021) Dual targeting of polyamine synthesis and uptake in diffuse intrinsic pontine gliomas. Nat Commun 12 (1), 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bonner ER et al. (2020) Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin GL et al. (2019) Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med 11 (519). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sasaki T et al. (2020) Convection-Enhanced Delivery of Enhancer of Zeste Homolog-2 (EZH2) Inhibitor for the Treatment of Diffuse Intrinsic Pontine Glioma. Neurosurgery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mount CW et al. (2018) Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med 24 (5), 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sandberg DI et al. (2019) Infusion of 5-Azacytidine (5-AZA) into the fourth ventricle or resection cavity in children with recurrent posterior Fossa Ependymoma: a pilot clinical trial. J Neurooncol 141 (2), 449–457. [DOI] [PubMed] [Google Scholar]
- 107.Ng JM et al. (2015) Generation of a mouse model of atypical teratoid/rhabdoid tumor of the central nervous system through combined deletion of Snf5 and p53. Cancer Res 75 (21), 4629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Han ZY et al. (2016) The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun 7, 10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vitte J et al. (2017) Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat Commun 8 (1), 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tao Y et al. (2019) Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and Braf(V600E)-Induced Tumorigenesis. Cancer Cell 35 (2), 315–328 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]