

Summary
Using antibody arrays, we found that the RNA helicase DDX3 modulates the expression of secreted signaling factors in oral squamous cell carcinoma (OSCC) cells. Ribo-seq analysis confirmed amphiregulin (AREG) as a translational target of DDX3. AREG exerts important biological functions in cancer, including promoting cell migration and paracrine effects of OSCC cells and reprogramming the tumor microenvironment (TME) of OSCC in mice. DDX3-mediated translational control of AREG involves its 3′-untranslated region. Proteomics identified the signal recognition particle (SRP) as an unprecedented interacting partner of DDX3. DDX3 and SRP54 were located near the endoplasmic reticulum, regulated the expression of a common set of secreted factors, and were essential for targeting AREG mRNA to membrane-bound polyribosomes. Finally, OSCC-associated mutant DDX3 increased the expression of AREG, emphasizing the role of DDX3 in tumor progression via SRP-dependent, endoplasmic reticulum-associated translation. Therefore, pharmacological targeting of DDX3 may inhibit the tumor-promoting functions of the TME.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
DDX3-AREG axis promotes cancer progression through microenvironment remodeling
-
•
DDX3 activates AREG translation via binding to its 3′ UTR
-
•
DDX3 interacts with the signal recognition particle (SRP)
-
•
DDX3-SRP-mediated mRNA recruitment assists ER-associated translation
Molecular biology; Cell biology; Cancer
Introduction
Head and neck cancers encompass a heterogeneous group of tumors in the upper aerodigestive tract; the main type is oral squamous cell carcinomas (OSCCs), which arise primarily in the oral cavity (Peltanova et al., 2019). OSCC has a propensity to metastasize through lymphatics to regional lymph nodes. Cancer cells establish a myriad of interactions with components of their microenvironment, including vascular endothelial cells, infiltrating immune cells, and fibroblasts. The tumor microenvironment (TME) greatly influences cancer cell growth and invasion, immunogenicity, and even drug resistance. Via a complex network that includes secreted growth factors/cytokines and the extracellular matrix, cancer cells modify their stromal neighbors, which in turn impact tumor growth, metastasis, and response to therapy. Changes in the TME and immune surveillance represent a crucial hallmark of various types of cancer including OSCC (Eckert et al., 2016). Understanding how cancer cells produce factors that exert autocrine and paracrine effects on cancer progression is important for the development of targeted therapies.
Cancer cells have an increased demand for mRNA translational control to augment global or selective protein expression for rapid cell growth and quick adaption to environmental stresses (Robichaud et al., 2018). A number of the DEAD/H-box RNA helicases including DDX3 contribute to translation control in cancer cells (Sharma and Jankowsky, 2014). By means of its RNA helicase activity, DDX3 promotes the translation of mRNAs containing a long or structured 5′ untranslated region (UTR), as is the case for many oncogene and or chemokine transcripts (Lai et al., 2008; Soto-Rifo et al., 2012). Accordingly, depletion of DDX3 results in cell cycle arrest and reduced migration capacity of cancer cells and impairs the migration and phagocytosis of macrophages (Chen et al., 2015; Ku et al., 2019; Lai et al., 2010). DDX3 also participates in internal ribosome entry site-mediated translation of several viral and cellular mRNAs (Geissler et al., 2012; Han et al., 2020; Phung et al., 2019). Via this mechanism, DDX3 promotes the translation of microphthalmia-associated transcription factor (MITF) in melanoma cells (Phung et al., 2019). Moreover, our previous report revealed that DDX3 promotes the translation of a set of stress response oncogenic factors, including activating transcription factor 4 (ATF4), by counteracting the suppressive effect of upstream open reading frames and hence increases cancer cell invasion. Moreover, DDX3 can facilitate the assembly of functional 80S ribosomes independently of its ATPase activity (Geissler et al., 2012). Besides its role in translational control, DDX3 also participates in transcriptional activation and cellular signaling (Sharma and Jankowsky, 2014).
Genetic mutations or dysregulation of DDX3 has been found in various types of cancer (Bol et al., 2015). We previously reported that upregulation of DDX3 is associated with poor survival of patients with head and neck squamous cell carcinoma (HNSCC) (Chen et al., 2018). DDX3 is detected in the nucleus of normal cells but localizes predominantly in the cytoplasm of OSCC cells. Cytoplasmic localization of DDX3 implies a high demand of DDX3-mediated translational control in cancer cells. DDX3 is essential for the expression of ATF4 in OSCC cells, which promotes cell migration and invasion (Chen et al., 2018). In this study, we first observed that DDX3 was essential for the paracrine activity of OSCC cells. This finding promoted us to investigate whether DDX3 may regulate secretory pathways via translational control.
There is a role for DDX3 in promoting the translation of mRNAs that contain suppressive upstream open reading frames (Chen et al., 2018). Hence, a high level of DDX3 in cancer cells increases the expression of the stress response protein activating transcription factor 4 (ATF4) and thereby promotes metastasis. Here, we report that DDX3 regulates the translation of mRNAs encoding secreted signaling factors.
Secreted and membrane proteins are synthesized by endoplasmic reticulum (ER)-bound ribosomes and subsequently pass through the secretory pathway to their final destinations. During translation, the signal recognition particle (SRP) recognizes their N-terminal signal peptide emerging from the ribosome and leads ribosome-bound mRNAs to the translocation complex at the ER surface (Zhang and Shan, 2014). The nascent polypeptide is subsequently translocated into the ER lumen through the translocon. Signal peptide-independent pathways for targeting transcripts to the ER have also been identified (Chartron et al., 2016). ER membrane-associated RNA-binding proteins (RBPs), such as yeast She2p and mammalian p180 and AEG-1, can anchor those transcripts to the ER (Cui et al., 2012; Genz et al., 2013; Hsu et al., 2018), but how these RBPs select mRNAs is unclear. A type of secretion-enhancing cis-regulatory targeting element has been identified in yeast secreted/membrane protein-encoding mRNAs; such elements may stabilize target mRNAs and facilitate their translation and subsequent translocation and secretion (Cohen-Zontag et al., 2019). A recent study found that the ER-associated RBP, TIS11B, forms reticular granules that enrich the transcripts containing AU-rich elements in the 3′ UTR. HuR binds to such elements and recruits an effector protein for efficient transport of nascent membrane proteins through the secretory pathway to the plasma membrane (Berkovits and Mayr, 2015; Ma and Mayr, 2018). The details of how different RBPs modulate the translation of different sets of ER-targeted mRNAs also remain to be investigated.
In this study, we began to investigate the role of DDX3 in regulating secretory pathways in OSCC. We then found that DDX3 regulated the translation of secreted signaling factors and further explored the underlying mechanism and biological significance.
Results
DDX3 regulates the expression of secreted signaling factors including AREG
We have previously reported that DDX3 is essential for cell migration and invasion of SAS cells (an aggressive OSCC cell line), but whether it can influence the autocrine and paracrine signaling activity has not been characterized. In this study, we initially observed that SAS cells exhibited morphological alteration when cultured in SAS cell-derived conditioned medium (CM) (Figure 1A). However, such a change was not observed when using CM from SAS cells that had been transfected with DDX3 targeting siRNA (Figure 1A). This result indicated that DDX3 was essential for the paracrine signaling effect of SAS cells.
Figure 1.

DDX3 is required for the expression of AREG
(A) SAS cells were cultured in DMEM or conditioned medium from SAS cells or DDX3-depleted SAS cells. Immunoblotting showed the efficiency of siRNA (siD#1)-mediated DDX3 depletion.
(B) Antibody array analysis was performed by using the lysate of siRNA (siC or siD#1)-transfected SAS cells. AREG signals are framed. Right: The proteins of which the signals were downregulated or upregulated by ≥ 2-fold are listed. The complete list of antibodies on the array is shown in Figure S1A.
(C) Diagram shows that Ribo-seq and RNA-seq were performed in siC or siD-transfected SAS cells.
(D) Genomic organization and distribution of aligned Ribo-seq reads over the exons of AREG.
(E) Survival curves for patients with HNSCC with high or low AREG expression.
(F) Immunoblotting of AREG in the lysates of siC-transfected or siD#1-transfected OSCC cell lines as indicated.
(G) ELISA of secreted AREG in the medium of siRNA-transfected cells as in (F). Bar graph shows relative AREG level (mean ± SD; SAS/siC was set to 1); ∗p < 0.01.
To explore whether DDX3 modulates the expression of secreted signaling factors, we first used antibody arrays to examine the changes in protein levels of 60 cytokines/growth factors upon DDX3 depletion. In DDX3-knockdown SAS cells, the signals for 15 spots were decreased by ≥ 2-fold, whereas one spot was increased (Figure 1B). Meanwhile, we performed ribosome profiling (Ribo-seq) in DDX3-depleted SAS cells to determine whether DDX3 regulates the translation of any secreted factor identified above (Figure 1C). RNA sequencing (RNA-seq) revealed that knockdown of DDX3 altered the expression level of ∼2.5% of all identified transcripts. To reveal translational targets of DDX3, Ribo-seq read counts were normalized to those of RNA-seq for each gene. Hence, the translation of ∼4% of identified transcripts could potentially be affected by DDX3. Among the factors identified in the above antibody arrays, we confirmed that the translation of amphiregulin (AREG) was prominently downregulated upon DDX3 depletion (Figure 1D), whereas many other factors had very low read counts, so we were unable to discern whether their translation is modulated by DDX3. Nevertheless, except for AREG, Ribo-seq also revealed additional growth factors or cytokines as the translational targets of DDX3, including neuregulin-1 (NRG1), endothelin-1 (EDN1), etc. (Table S1).
We focused on AREG because it was identified in both antibody arrays and Ribo-seq analysis and has been considered a marker for poor prognosis of patients with HNSCC (Figure 1E) according to The Human Protein Atlas database. Using immunoblotting and ELISA, we confirmed that DDX3 knockdown reduced AREG expression in the cell lysate and culture medium of several OSCC cells including SAS cells (Figures 1F and 1G). Moreover, immunoblotting also confirmed that DDX3 regulated the expression of several other growth factors and immunoregulatory factors (Figure S1B), indicating an unprecedented role for DDX3 in regulating the expression of secreted signaling factors.
DDX3-regulated AREG acts as an autocrine factor for cell migration
Next, we investigated whether DDX3-regulated AREG expression has any functional effect on OSCC. Although AREG promotes cell proliferation and migration in various cancers (Berasain and Avila, 2014; Busser et al., 2011), overexpression of AREG in SAS cells, however, had no effect on cell proliferation (Figure S2A). Perhaps, the level of endogenous AREG was sufficient for optimal proliferation of SAS cells. Nevertheless, AREG overexpression induced mesenchymal cell-like membrane protrusions in SAS cells (Figure 2A), as observed in Figure 1A, whereas knockdown of AREG using short hairpin (sh) RNA abolished such a morphological change (Figure S2B). Recombinant AREG (rAREG) treatment also induced mesenchymal cell-like morphology (Figure S2C) and upregulated mesenchymal proteins, including vimentin (VIM), SLUG (SNAI2), actin alpha 2 (ATCA2), and fibroblast activation protein (FAP) in SAS cells and three other OSCC cell lines (GNM, HSC3, and OECM1) (Figure S2D). Moreover, we observed that CM derived from SAS cells could promote cell migration, whereas CM from DDX3-depleted SAS cells lost such an activity (Figure 2B, CM/shC and CM/shD). Supplement of rAREG into DDX3-depleted SAS CM restored cell migration (Figure 2B, CM/shD + rAREG). Therefore, DDX3-regulated AREG exerts an autocrine activity in promoting cell migration.
Figure 2.

The DDX3-AREG axis promotes cell migration, angiogenesis, and macrophage differentiation in vitro
(A) SAS cells were transfected with pP2A (vec) or pP2A-AREG for 72 hours. Immunoblotting of the cell lysates, cell morphology, and percentage of spindle cells are shown (mean ± SD; ∗p<0.01). Scale bars, 50 μm.
(B) Boyden chamber assay of SAS cells in DMEM or CM derived from control (shC) or DDX3-depleted (shD) SAS cells supplemented with or without 5 ng/ml rAREG (mean ± SD; ∗p<0.01). Immunoblotting was performed in shRNA-transfected SAS cells. Scale bar, 500 μm.
(C) Angiogenesis assay of EA.hy926 cells that were cultured in DMEM or CM as in (B). Bar graph quantifies tube formation by EA.hy926 cells (mean ± SD; ∗p<0.01). Scale bars, 500 μm.
(D) RT-qPCR analysis of the indicated transcripts in THP-1 cells cultured in DMEM or the various CM, as indicated (mean ± SD; ∗p<0.01).
(E) Schematic diagram shows experimental procedures for collection of CM used for angiogenesis assays. EA.hy926 cells were cultured in CM from THP-1 that had been cultured in CM from siC or siD-transfected SAS cells. Bar graph quantifies tube formation by EA.hy926 cells (mean ± SD; ∗p<0.01). Scale bar, 500 μm.
DDX3-regulated AREG exerts a paracrine activity in promoting angiogenesis and macrophage differentiation
Next, we evaluated whether DDX3-regulated AREG has a paracrine role. To analyze angiogenesis, we characterized capillary-like tube formation ability of human endothelial EA.hy926 cells. As above, we evaluated CM from control (CM/shC) or DDX3-depleted SAS cells (CM/shD) and AREG-supplemented CM from DDX3-depleted cells (CM/shD + AREG). SAS cell CM efficiently induced tube formation of human endothelial EA.hy926 cells, whereas knockdown of DDX3 abolished this activity (Figure 2C). Moreover, AREG antibody-treated CM also inhibited tube formation of EA.hy926 cells (Figure S2E), suggesting that AREG exerts an angiogenic effect through a paracrine pathway. rAREG restored the angiogenesis-promoting activity of CM derived from DDX3 knockdown cells (Figure 2C). Therefore, the DDX3-AREG axis likely contributes to tumor-induced angiogenesis.
Tumor-associated macrophages are abundant tumor-infiltrating immune cells in the TME and contribute to lymph node metastases and poor prognosis of OSCC (Weber et al., 2016). We first examined whether DDX3 modulates macrophage differentiation of human monocytic THP-1 cells. SAS cell CM induced the expression of macrophage marker genes (IL1B, IL23A, MAF, and VEGFA) in THP-1 cells (Figure 2D, CM/shC); knockdown of DDX3 or AREG reduced this activity by ∼50–80% (CM/shD and CM/shAREG). However, the observation that rAREG was insufficient to induce macrophage gene expression indicated that AREG-induced factors in SAS cells rather than AREG itself promoted macrophage differentiation (Figure S2F). Because M2-polarized tumor-associated macrophages in general promote tumorigenesis, we evaluated whether the DDX3-AREG pathway promotes M2 differentiation. We first observed that SAS cell CM could induce M2 marker (CD163, IL10, and MRC1) expression in phorbol ester-primed THP-1 cells in a DDX3-dependent manner (Figure S2G). On the other hand, phorbol ester-primed THP-1 cells expressed a higher level of M1 markers (HLADR, IL12B, and IL18) in DDX3 knockdown SAS CM than that of control CM (Figure S2H). Because M2 macrophages have angiogenic potential, we then functionally examined whether SAS CM-primed THP-1 cells could induce angiogenesis (Figure 2E, diagram). Indeed, CM of the aforementioned primed THP-1 cells efficiently induced tubularization of EA.hy926 cells, whereas the CM of THP-1 that had been cultured in DDX3-depleted SAS CM was incapable of doing so (Figure 2E). This result implied that the DDX3-AREG axis can potentiate THP-1 differentiation into M2 macrophages.
The DDX3-AREG axis is required for OSCC-mediated microenvironmental reprogramming in vivo
Next, we investigated whether the DDX3-AREG axis influences the TME in vivo using an allograft model. We evaluated several mouse OSCC cell lines, which were established from carcinogen-treated transgenic K14-EGFP-miR-211 C57BL/6 mice (Chen et al., 2019b). MOC-L1 was selected because this cell line was found to highly express AREG and potently activate macrophage differentiation (Figures S3A and S3B). Knockdown of DDX3 in MOC-L1 cells reduced both proliferation and migration (Figures S3C and S3D) and attenuated the ability of CM to induce tube formation and macrophage gene expression, as observed in DDX3-depleted SAS cells (Figures S3E and S3F). Next, DDX3-depleted MOC-L1 cells or control cells were transplanted into C57BL/6 mice (Figure 3A). Efficient depletion of DDX3 as well as AREG in siDDX3-transfected MOC-L1 tumors was reproducibly observed even at two weeks after transfection (Figure 3B). Control MOC-L1 tumors were larger in average (Figure 3C, siD#1) and had visible blood vessels compared with DDX3 knockdown tumors (Figure 3D). Administration of DDX3 knockown-MOC-L1 tumors with recombinant mouse AREG (rAREGm) restored the vasculature and tumor size (Figures 3C and 3D, siD#1 + rAREGm). Cell growth assays indicated that the DDX3-AREG axis did not directly affect cell proliferation in vitro (Figures S3G–S3I). Using periodic acid-Schiff to stain blood vessel, we confirmed the above observation on angiogenesis (Figure 3D, PAS). Immunofluorescence staining for the macrophage marker F4/80 revealed that control MOC-L1 tumors had a higher degree of macrophage infiltration than did DDX3-depleted tumors, and rAREGm treatment increased the intensity of macrophage staining in DDX3-depleted tumors (Figure 3D, F4/80). In contrast, immunostaining for the T-cell activation marker granzyme B indicated that infiltration of cytotoxic immune cells was exaggerated in DDX3-depleted tumors compared with the control, and rAREGm treatment attenuated such infiltration (Figure 3D, Granzyme B). This result indicated that the DDX3-AREG axis promoted macrophage infiltration and suppressed cytotoxic immunosurveillance in OSCC. Together, DDX3 exerts several tumor-promoting activities by regulating the expression of AREG (Figure 3E).
Figure 3.

The DDX3-AREG axis promotes angiogenesis and macrophage recruitment and suppresses cytotoxic immune cells in syngeneic tumors
(A) Experimental procedure for using a syngeneic graft model to evaluate the effect of DDX3 knockdown and rAREGm injection on tumor growth.
(B) Immunoblotting for DDX3 and GAPDH in the tumor extracts collected at 7 and 14 days after transplantation.
(C) Average size of the tumors from control (siC) or DDX3 knockdown (siD#1) MOC-L1 or the latter supplemented with rAREG (siD#1 + rAREGm). Bar graph shows average weight (mean ± SEM) of tumors as in (B). p < 0.01. Eight tumors from each frank of four mice in each group were analyzed.
(D) Representative images of each type of the tumors as in (C) with similar sizes are shown in the top image. Periodic acid-Schiff (PAS) and immunofluorescence staining of tumor sections. Arrowheads indicate the PAS-positive regions. Bar graphs show PAS intensity and the ratios of F4/80 or granzyme B-positive cells. Scale bars represent 1 mm and 100 μm in PAS and immunofluorescence images, respectively. Error bars represent mean ± SEM; ∗p < 0.01.
(E) A summary of the tumor-promoting effects of the DDX3-AREG axis.
DDX3 regulates the translation of AREG mRNA
The above result confirmed the important biological effect of the DDX3-AREG axis in tumor progression and microenvironmental remodeling. Next, we investigated the mechanism underlying how DDX3 may regulate the translation of AREG. We constructed humanized Renilla luciferase reporters containing either the 5′ or 3′ UTR or both UTRs of AREG (Figure 4A). After normalization with the control firefly luciferase, we observed that the 5′ UTR and 3’ UTR, respectively, suppressed and enhanced reporter translation (Figure 4B, siC). Knockdown of DDX3 decreased the translation of all the reporters without affecting their mRNA levels, indicating that both the 5′ and -3′ UTRs of AREG exert DDX3-dependent translation control (Figures 4B and S4A). RNAfold analysis revealed that the 5′ UTR forms extensive stem-loop structures (Figure S4B). Therefore, we tested the effect of wild-type and ATP hydrolysis (K230E) or RNA unwinding (S382L)-defective DDX3 (Yedavalli et al., 2004) in the translation of the 5′ UTR reporter in DDX3-depleted SAS cells. The result showed that only the wild type but not mutant DDX3 could moderately restore 5′ UTR-mediated reporter expression likely via the translational control (Figures 4C and S4C, 5′), indicating its activity in resolving secondary structures of the 5′ UTR. To our surprise, those two mutants were able to restore 3′ UTR reporter translation without affecting reporter mRNA level (Figures 4C and S4C, 3′) and AREG expression (Figure 4D). To further evaluate whether these two activities are dispensable for 3′ UTR-mediated translation, we tested the double mutant (S382L; K230E). This mutant rescued AREG expression in DDX3-depleted cells (Figure 4D, lane 5), emphasizing that DDX3 participates in 3′ UTR-mediated translation in an RNA helicase-independent manner. Therefore, DDX3 may regulate AREG mRNA translation via at least two distinct mechanisms (Figure 4E). This unprecedented and intriguing result prompted us to investigate the underlying mechanism.
Figure 4.

DDX3 activates AREG mRNA translation
(A) Schematic diagram showing humanized Renilla luciferase (hRL) reporters containing either or both of the 5′ and -3′ UTRs of AREG.
(B) In vivo translation assay using the indicated hRL reporters in siC- or siD#1-transfected SAS cells. The activity of human firefly luciferase (hFL) encoded by the same reporters was used as an internal control. Bar graph shows relative hRL/hFL activity (mean ± SD); ∗p < 0.01.
(C) Schematic diagram showing DDX3 in which two RecA-like domains are labeled in blue and violet and 11 conserved motifs (Q, I-VI), and the two mutations are indicated. An in vivo translation assay was performed using the indicated hRL reporters in endogenous DDX3-depleted (by siD#1) and exogenous DDX3 (wild-type or mutants)-overexpressing SAS cells. Bar graph shows relative hRL/hFL activity.
(D) SAS cells were transfected with siD#1 and the indicated DDX3 expression vectors. The lysates were analyzed by immunoblotting using antibodies as indicated.
(E) DDX3 modulates the translation of AREG mRNA via both its 5′ and -3′ UTR.
DDX3 in conjunction with the SRP controls the translation nearby the endoplasmic reticulum membrane
The above result that DDX3 may regulate the translation of AREG via 3′ UTR-mediated translation control prompted us to identify its interacting partners that participate in the expression of secreted proteins. We performed immunoprecipitation of endogenous DDX3 from SAS cells and analyzed the coprecipitates using mass spectrometry. After RNase digestion, coprecipitates were fractionated by electrophoresis. Thirteen visible bands were subjected to mass spectrometry analysis (Figure S5A). In total, 444 proteins were identified, and the 10 non-redundant proteins with the highest scores for each band are listed in Table S2. Gene Ontology analysis revealed enrichment of DDX3-interacting proteins in translation initiation and SRP-dependent cotranslational protein targeting to the membrane (Table S3). Using the technique “stable isotope labeling by amino acids in cell culture” followed by Gene Ontology analysis, we identified a similar set of DDX3-interacting partners (Table S4). We knocked down 22 candidate genes that are involved in mRNA processing and translation using shRNA and observed that depletion of SRP components (SRP9 and 68) reduced the translation of the AREG 5’+3′ UTR reporter to a level comparable to that of DDX3 knockdown (Figure 5A). Further examination revealed that depletion of either SRP component compromised the translation but not mRNA level of the 3′ UTR reporter (Figures 5B and S5B), whereas it had no effect on the 5′ UTR reporter (Figure S5C). All these SRPs were also required for the expression of endogenous AREG (Figure 5C). We deduced that DDX3 acts in conjunction with the SRP in 3′ UTR-mediated translation control.
Figure 5.

SRP proteins interact with DDX3 and activate AREG mRNA translation
(A) In vivo translation assays of the AREG 5’+3′ reporter in SAS cells that were transfected with the indicated shRNAs. Bar graphs show the hFL/hRL activity (mean ± SD) of shRNA transfectants relative to control cells. ∗p < 0.01.
(B) The experiment was performed as in (A), except that AREG 3′ reporter and shRNAs targeting DDX3 or SRP components were used (mean ± SD; ∗p<0.01).
(C) SAS cells were transfected with shRNA as in (B). Immunoblotting was performed using antibodies as indicated.
(D) Immunoprecipitation of SAS cell lysates using control IgG or anti-DDX3 in the absence or presence of RNase, followed by immunoblotting.
(E and F) Immunofluorescence co-staining for DDX3 with GRP94 (E) or SRP54 (F) in SAS cells. The framed region in the upper images is magnified in the lower images. The histograms of fluorescence intensity across the pink lines are shown to the right. Scale bars, 20 μm.
(G) The lysates of siRNA-transfected SAS cells were subjected to immunoblotting using antibodies as indicated.
In light of SRP54 as a key SRP factor (Wild et al., 2019), we evaluated its interaction with DDX3 and role in AREG expression. Immunoprecipitation of DDX3 or SRP54 from the SAS cell lysates followed by immunoblotting consistently revealed RNA-independent interaction between DDX3 and SRP54 (Figure 5D). Both of them also interacted with ribosomal protein RPL13, indicating their engagement in mRNA translation (Figure 5D). Indirect immunofluorescence revealed that endogenous DDX3 distributed to the regions surrounding the ER protein GRP94 and was located in proximity to SRP54 (Figures 5E and 5F), further supporting a role for DDX3 in ER-associated translation. Depletion of SRP54 also decreased the expression of DDX3-regulated secreted factors that we examined but had no effect on macrophage migration inhibitory factor (MIF) or E-cadherin (Figure 5G). Therefore, DDX3 likely controls the expression of secreted proteins in conjunction with the SRP via ER-associated translation.
DDX3 and SRP promote the association of target mRNAs to the ER membrane
We hypothesized that DDX3 recruits the SRP to the 3′ UTR of mRNAs encoding secreted proteins and facilitates their translation at the ER membrane. To test this hypothesis, we performed a pull-down assay using biotinylated AREG 3′ UTR. The result showed that DDX3 and SRP54/68 interacted with the AREG 3′ UTR (Figure 6A, lane 9). Depletion of DDX3 diminished the association of the SRP proteins with the AREG 3′ UTR (lane 10). Interestingly, depletion of either SRP protein also reduced the binding of DDX3 to the AREG 3′ UTR (lanes 11, 12), suggesting their interdependent association with target mRNAs. Using immunoprecipitation coupled with quantitative reverse transcription-PCR, we confirmed the association of DDX3 and SRP proteins with endogenous AREG mRNA in SAS cells (Figure 6B). Next, we evaluated whether DDX3 and/or SRP54/68 are essential for anchoring AREG mRNA on the membrane. Depletion of either factor reduced the membrane-to-cytosol partition ratio of AREG mRNA (Figure 6C). We also evaluated several other membrane-associated mRNAs, such as EDN1, NRG1, TGFA, and XBP1 (Figures 6C and S6A). Depletion of DDX3 resulted in the shift of these mRNAs but not CDH1 mRNA from the membrane to the cytosol to different extents (Figures 6C, S6B, and S6C). Finally, we examined whether the distribution of translation factors is also modulated by DDX3. Subcellular fractionation revealed that the translation initiation factor eIF2α and ribosomal proteins RPL13 and RPS3A, like DDX3 and SRP54, were partially present in the membrane fraction (Figure 6D, lane 2). Depletion of DDX3 shifted 20–40% of these factors from the membrane to the cytosol but had no significant effect on GRP94 (lanes 3, 4). A similar result was obtained with SRP54-depleted cells (lanes 5, 6). Together, these results indicated that DDX3 and the SRP tether a set of mRNAs to the ER membrane and recruit the translation machinery for ER-associated translation. To demonstrate the essential role of DDX3 and SRP on ER-associated AREG translation, sucrose gradient sedimentation of cytosol and membrane fractions was performed. The ribosome profiles along the gradient were similar between the control and knockdown cells (Figure 6E). We pooled the heavy and light polysomal fractions, respectively, and analyzed mRNA distribution. AREG mRNA was particularly enriched in the heavy fractions of membrane-associated polysomes, in sharp contrast to GAPDH mRNA (Figure 6F, compared siC bars). Notably, knockdown of DDX3 or SRP54 reduced the heavy-to-light ratio of AREG mRNA in membrane fraction (Figure 6F, AREG/Mem) but had no significant effect on GAPDH or cytosol AREG mRNA. Therefore, DDX3 plays a crucial role in recruiting target mRNAs as well as several translational factors onto the ER membrane (Figure 6G).
Figure 6.

DDX3 and SRP proteins are required for membrane association of target mRNAs
(A) Pull-down assay using the biotinylated AREG 3′ UTR RNA with lysates of siRNA-transfected SAS cells, followed by immunoblotting. Bar graph (mean ± SD; ∗p<0.01) shows relative levels of coprecipitation; siC was set to 1.
(B) RNA immunoprecipitation with SAS cell lysates using antibodies as indicated, followed by RT-qPCR analysis of the indicated mRNAs (mean ± SD; ∗p<0.01).
(C) RT-qPCR analysis of the indicated mRNAs in both the membrane and cytosol fractions of indicated siRNA-transfected SAS cells. Bar graph (mean ± SD; ∗p<0.01) shows the membrane-to-cytosol ratio of each mRNA in knockdown (siD#1, siSRP54, siSRP68) cells relative to control (siC).
(D) Immunoblotting of indicated proteins in the membrane and cytosol fractions from siRNA-transfected SAS cells. Bar graph shows the relative membrane/total (cytosol + membrane) ratio of the indicated proteins; siC was set to 1. Bars represent mean ± SD; ∗p < 0.01.
(E) Polysome profiles of the cytosol and membrane fractions of siRNA-transfected cells. Fractions from 40S to the first polysome (gray region) were collected as the light fractions; the following fractions (white region) were collected as the heavy fractions.
(F) RT-qPCR analysis of mRNAs from the heavy and light fractions of cytosol (Cyt) or membrane (Mem)-enriched polysomes. The heavy-to-light ratios of GAPDH and AREG in each sample were normalized to those of ACTB. ∗p < 0.01.
(G) DDX3 localizes proximally to the ER, where it associates with the SRP. DDX3/SRP cooperatively binds the 3′ UTR of target mRNAs and facilitates their translation by recruiting the translation machinery.
OSCC-derived DDX3 mutants confers a higher potential to induce AREG expression
Many of the cancer-associated mutations of DDX3, however, result in altered protein function rather than a loss of function (Bol et al., 2015). In TCGA PanCancer Atlas database, six missense mutations of DDX3 were identified among 523 HNSCC samples. Interestingly, three mutations exclusive to OSCC tumors occur at the negatively charged amino acids around the C-terminal region of the RNA helicase domain, including D521H, D558H, and E572Q (Figure 7A). In vitro translation assays revealed that these DDX3 mutants exhibited ∼20% higher activity than wild-type DDX3 to activate AREG 3′ UTR, whereas no difference was observed for 5′ UTR reporter translation (Figure 7B). These DDX3 mutants also exhibited higher activity than the wild type in promoting AREG expression in DDX3 knockdown cells but had no significant difference from the wild type with respect to the expression of Rac1, which involves helicase dependent and 5′ UTR-mediated translational control (Figure 7C). Therefore, the mutations may specifically enhance the pathway involved in AREG mRNA translation. Consistent with the above notion, immunoprecipitation of SRP54 revealed that it had greater affinity for the DDX3 mutants that were transiently expressed in SAS cells (Figure 7D). This result indicated that cancer-related DDX3 mutants have greater potential to create a pro-tumorigenic environment through activating AREG translation (Figure 7E).
Figure 7.

OSCC-derived DDX3 mutants have greater potential to induce AREG expression
(A) Schematic representation of human DDX3 protein as in Figure 4C, in which OSCC-derived mutations are indicated.
(B) In vivo translation assays were performed as Figure 4C, except that FLAG-tagged wild-type or DDX3 mutant was overexpressed. Bar graph shows relative hRL/hFL activity (mean ± SD); ∗p < 0.01.
(C) Immunoblotting of the lysates of SAS cells that were transfected with siD#1 and the indicated expression vectors.
(D) SAS cells were transfected with the expression vector encoding GFP-tagged wild-type or mutant DDX3 (in order to distinguish transiently expressed DDX3 from the endogenous). Immunoblotting of anti-SRP54-coimmunoprecipitates was performed using antibodies against GFP or SRP proteins as indicated. Bars represent IP/Input ratios of DDX3 blots (mean ± SD; ∗p<0.01).
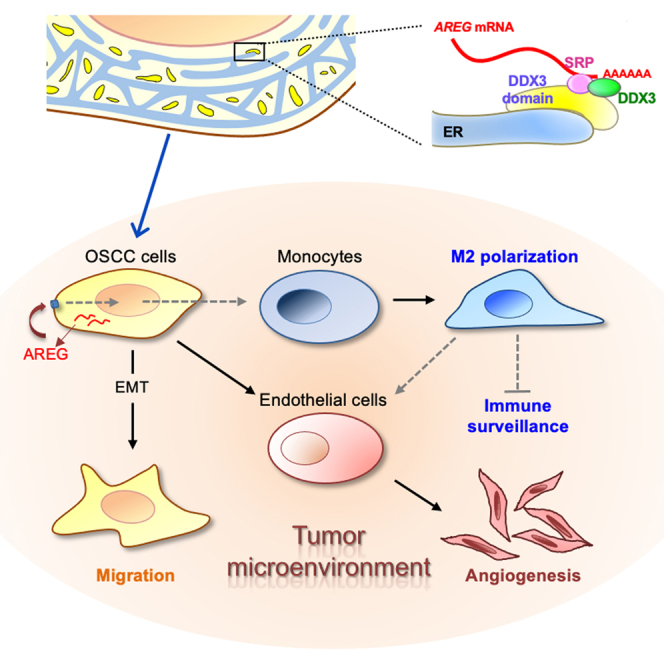
(E) OSCC-associated mutant DDX3 increases the expression of AREG, which exerts both autocrine and paracrine activities and hence promotes TME remodeling.
Discussion
Using both antibody arrays and Ribo-seq analysis, we identified a set of DDX3-regulated targets encoding secreted oncogenic and immunomodulatory factors in OSCC. Among them, we confirmed that the DDX3-AREG regulatory axis substantially contributes to tumorigenesis via both the autocrine and paracrine pathways. Moreover, DDX3 and its partner SRP are critical for the expression of AREG as well as other secreted signaling factors. Therefore, DDX3 promotes tumor progression by regulating the translation of TME factors.
DDX3 modulates the expression of secreted signaling factors via ER-associated translation
Previous reports have revealed that DDX3 participates in the translation of mRNAs that have structured elements, upstream open reading frames, or an internal ribosome entry site in the 5′ UTR (Chen et al., 2015, 2016, 2018; Han et al., 2020; Ku et al., 2019; Lai et al., 2008, 2010; Phung et al., 2019; Soto-Rifo et al., 2012). These mRNAs essentially encode intracellular proteins, many of which have oncogenic and/or metastatic function or act during the immune response. A recent report indicates that DDX3 localizes to mitochondria and is essential for mitochondrial translation and function (Heerma van Voss et al., 2018), but how DDX3 participates in the translational control of mitochondrial proteins is unclear. In this study, we found that DDX3 was present in the membrane-enriched subcellular fraction, localized close to the ER, and participated in the ER-associated translation of mRNAs encoding secretory proteins via its interaction with the SRP (Figures 1 and 5). DDX3 or SRP depletion reduced the expression of AREG. DDX3 and the SRP possibly bind to the 3′ UTR of target mRNAs in a cooperative manner and tether mRNAs on the ER membrane (Figure 6). DDX3 interacts with several translation initiation factors such as eIF4E and eIF3 (Lee et al., 2008; Shih et al., 2012). We hypothesize that the DDX3-SRP complex in ER-proximal granules recruits the translation machinery to translate mRNAs on the ER (Figure 6G). Our finding defines an unprecedented role for DDX3 in ER-associated translation.
In this study, we observed that the helicase activity of DDX3 was dispensable for its function in 3′ UTR-mediated translation (Figure 4). Indeed, it has been reported that the ATPase/helicase activity is nonessential for DDX3 function in promoting the assembly of functional 80S ribosomes and IRES-mediated translation and as an adapter molecule for activating Wnt-β-catenin signaling and promoting assembly of the NLRP3 inflammasomes (Cruciat et al., 2013; Geissler et al., 2012; Samir et al., 2019; Shih et al., 2008). Therefore, DDX3 may regulate different cellular functions including translation via both helicase-dependent and helicase-independent activities. Recently, several lines of evidence have indicated that the 3′ UTR of secreted/membrane protein-coding mRNAs can facilitate ER targeting and translation and even dictate the biological function of the encoded proteins (Berkovits and Mayr, 2015; Lee and Mayr, 2019; Ma and Mayr, 2018). The role of DDX3 in 3′ UTR-mediated and ER-associated translation is reminiscent of TIS11B, which recruits HuR to the 3′ UTR in ER-associated TIS11B-enriched granules. Through HuR, TIS11B assists SET association with the protein encoded by the AU-rich element-containing mRNAs, such as CD47 and CD274, which are critical for immune surveillance. It would be interesting to decipher whether DDX3 coordinates the translation of mRNAs encoding functionally related secreted proteins in cancer, immune cells, or any other secretory cells.
DDX3 modulates the TME via secreted signaling factors
Through translational control, DDX3 promotes the expression of cyclin E1, ATF4, and Rac1, which, respectively, participate in cell cycle progression, preventing apoptosis, stress adaptation, and cell migration (Chen et al., 2015, 2018; Lai et al., 2010; Phung et al., 2019). DDX3 also enhances Wnt-β-catenin signaling, a cancer driver, via multiple regulatory pathways (Chen et al., 2015; Cruciat et al., 2013). These results support an oncogenic role for DDX3 in various types of cancer. Nonetheless, DDX3 has also been implicated as a tumor suppressor, for which it suppresses the expression of the cyclin kinase inhibitor p21 and the epithelial-mesenchymal transition transcription factor Snail (Chao et al., 2006; Su et al., 2015). On the whole, previously determined targets of DDX3 essentially function intracellularly. In this study, we found that DDX3 regulates the expression of secreted proteins that can modulate cancer progression via both autocrine and paracrine signaling pathways (Figure 2). As a result, DDX3 promotes cancer cell migration and invasion through both the intracellular and extracellular signaling pathways. Moreover, a high level of DDX3 in cancer cells may promote angiogenesis and macrophage differentiation (Figures 2 and 3). M2-type macrophages further promote angiogenesis and tumor progression and may confer chemoresistance (Chen et al., 2019a). This result indicates that DDX3 amplifies its effects through secreted signaling factors. Notably, our Ribo-seq data revealed that DDX3 may modulate the acute-phase protein response, proinflammatory cytokines, and the HMGB1-mediated inflammatory response, indicating that a high level of DDX3 in cancer cells influences the immune response (Table S1). Therefore, the impact of DDX3 overexpression on cancer/tumor progression is greater than previously estimated.
The role of the DDX3-AREG axis in cancer
This study demonstrated a role of DDX3 in TME remodeling via secreted signaling factors including AREG. A previous report has indicated that AREG activates cell growth in several different cancer cells through EGFR activation (Busser et al., 2011). However, this function was negligible in cultured OSCC cells regardless of whether DDX3 was depleted or not (Figures S2A and S3G–S3I). Nevertheless, we observed that AREG partially rescued the size of DDX3 knockdown MOC-L1 tumors (Figure 3C), suggesting the DDX3-AREG pathway promoted tumor growth via remodeling the TME toward the pro-tumorigenic condition, in which increased nutrition or growth factor supplement and/or decreased immune surveillance facilitate tumor progression (Figure 3D).
It is notable that OSCC-derived mutants of DDX3 exhibited higher activity in AREG 3′ UTR reporter translation and AREG protein expression (Figure 7). Therefore, OSCC cells with a mutant DDX3 may be more aggressive as a consequence of increasing the expression of oncogenic and immunomodulatory factors that are secreted into the TME. Because the RNA helicase activity is not essential for the expression of such secreted factors (Figure 4), ATPase or helicase inhibitors may not completely abolish the oncogenic function of DDX3. Therefore, additional strategies such as downregulating DDX3 expression or decreasing granule formation may be applied to future anti-cancer therapies.
Limitations of the study
This study for the first time revealed the role of DDX3 in ER-associated mRNA translation, but questions such as whether its target mRNAs share any common feature in the 3′ UTR, how it exactly promotes mRNA translation in conjunction with the SRP, and whether it facilitates nascent protein targeting to the secretory pathway remain for future investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DDX3 | Santa Cruz | Cat#sc-365768; RRID:AB_10844621 |
| Actin | Merck | Cat#MABT1333 |
| αTubulin | GenScript | Cat#A01410; RRID:AB_1968943 |
| SRP54 | Santa Cruz | Cat#sc-393855; RRID:AB_1562699 |
| EIF2α | Santa Cruz | Cat#sc-133132 |
| GAPDH | ABclonal | Cat#AC002; RRID:AB_2736879 |
| Granzyme B | R&D Systems | Cat#AF1865; RRID:AB_2294988 |
| E-cadherin | BD Biosciences | Cat#610181; RRID:AB_397580 |
| Lamin A/C | Merck | Cat#SAB4200236; RRID:AB_10743057 |
| RAC1 | Merck | Cat#05-389; RRID:AB_309712 |
| AREG | ABclonal | Cat#A1860; RRID:AB_2763895 |
| NRG1 | ABclonal | Cat#A0687; RRID:AB_2757338 |
| IL1A | ABclonal | Cat#A2170; RRID:AB_2764188 |
| IL18 | ABclonal | Cat#A1115; RRID:AB_2861508 |
| VEGFA | ABclonal | Cat#A5708; RRID:AB_2766467 |
| SRP54 | ABclonal | Cat#A4126; RRID:AB_2765514 |
| MIF | ABclonal | Cat#A11231; RRID:AB_2861527 |
| RPL13 | ABclonal | Cat#A6723; RRID:AB_2767307 |
| RPS3A | ABclonal | Cat#A5885; RRID:AB_2766633 |
| IL1B | ABclonal | Cat#A19635; RRID:AB_2862708 |
| F4/80 | GeneTex | Cat#GTX26640; RRID:AB_385952 |
| GRP94 | GeneTex | Cat#GTX103232; RRID:AB_1950516 |
| VIM | GeneTex | Cat#GTX100619; RRID:AB_1952557 |
| SNAI2 | GeneTex | Cat#GTX128796; RRID:AB_2885815 |
| ACTA2 | GeneTex | Cat#GTX124505; RRID:AB_11166745 |
| FAP | GeneTex | Cat#GTX102732; RRID:AB_1950254 |
| SRP68 | Bethyl Laboratories | Cat#A303-955A; RRID:AB_2620304 |
| Goat anti-Rabbit IgG-heavy and light chain antibody, HRP-conjugated | Bethyl Laboratories | Cat#A120-101P; RRID:AB_67264 |
| Anti-Mouse IgG HRP linked whole antibody | Merck | Cat#GENA931-1ML |
| AccuBlotTM anti-Rabbit IgG-HRP | Royez | Cat#C04010 |
| AccuBlotTM anti-Mouse IgG-HRP | Royez | Cat#C04009 |
| Alexa Fluor 488 goat anti-Mouse IgG | Thermo Fisher | Cat#A11001; RRID:AB_2534069 |
| Alexa Fluor 568 goat anti-Rabbit IgG | Thermo Fisher | Cat#A11011; RRID:AB_143157 |
| AREG ELISA Kit (Human) | Aviva Systems | Cat#OKEH00009 |
| AREG ELISA Kit (Mouse) | Aviva Systems | Cat#OKCD05604 |
| HiYield Total RNA Extraction Kit | Arrowtec | Cat#YRT50 |
| ToolsQuant II Fast RT Kit | Biotools | Cat#KRT-BA06-2 |
| PerfeCta SYBR Green FastMix PCR Reagent | Quantabio | Cat#95072-012 |
| Human AREG/Amphiregulin Protein | LSBio | Cat#LS-G27177 |
| Mouse AREG /Amphiregulin Protein (Recombinant) | LSBio | Cat#LS-G137832 |
| Millicell hanging cell culture insert 24 well PET 8 μm | Merck | Cat#MCEP24H48 |
| μ-Slide Angiogenesis, ibiTreat, sterile | ibidi | Cat#81506 |
| Matrigel Growth Factor Reduced | Corning | Cat#354230 |
| Phorbol 12-myristate 13-acetate | Merck | Cat#P1585 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s modified Eagle’s medium (DMEM) | Thermo Fisher | Cat#11965092 |
| Fetal Bovine Serum (FBS) | Corning | Cat#35-010-CV |
| Penicillin and Streptomycin | Thermo Fisher | Cat#15140122 |
| L-glutamine | Thermo Fisher | Cat#25030081 |
| Trypsin | Thermo Fisher | Cat#15400054 |
| SILAC Metabolic Labeling Systems | Thermo Fisher | Cat#A33969 |
| Roswell Park Memorial Institute (RPMI) 1640 medium | Thermo Fisher | Cat#22400089 |
| Lipofectamine 3000 | Thermo Fisher | Cat#L3000015 |
| Lipofectamine 2000 | Thermo Fisher | Cat#11668500 |
| Trizol Reagent | Thermo Fisher | Cat#15596018 |
| Ribo-off rRNA Depletion Kit | Vazyme | Cat#N406 |
| NETNext Ultra Directional RNA Library Prep Kit for Illumina | New England BioLabs | Cat#E7760 |
| TRIS(Base) | Fisher Scientific | Cat#JT-4109-02 |
| Sodium Chloride | Merck | Cat# 31434 |
| Magnesium Chloride hexahydrate | Avantor | Cat#2444-01 |
| DTT | Merck | Cat#D0632 |
| Cycloheximide | Merck | Cat#C7698 |
| Triton X-100 | Merck | Cat#X100 |
| TURBO DNase | Thermo Fisher | Cat#AM2238 |
| Ambion RNase I | Thermo Fisher | Cat#AM2294 |
| T4 Polynucleotide Kinase | New England BioLabs | Cat# M0201S |
| T4 RNA ligase 2, truncated K227Q | New England BioLabs | Cat#M0351S |
| SuperScript III reverse transcriptase | Thermo Fisher | Cat#18080085 |
| CircLigase ssDNA Ligase | Lucigen | Cat#CL4111K |
| Phusion High-Fidelity DNA Polymerase | New England BioLabs | Cat#M0530S |
| AMPure XP for PCR Purification | Beckman Coulter | Cat#A63880 |
| Human Cytokine Antibody Array | Abcam | Cat#ab169818 |
| BCA Protein Assay Kit | ThermoFisher | Cat#23225 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling | Cat#5872 |
| RNaseA | Thermo Fisher | Cat#12091021 |
| Dynabeads Protein G | Thermo Fisher | Cat#10004D |
| RNasein | Promega | Cat#N2115 |
| Glycerol | Avantor | Cat#JT-2143-03 |
| SDS | Cyrusbioscience | Cat#101-151-21-3 |
| 2-mercaptoethanol | Merck | Cat#M3148 |
| Bromophenol Blue | Merck | Cat#B5525 |
| PRO-PREP Protein Extraction Solution | iNtRON Biotechnology | Cat#INT17081.1 |
| Periodic acid-Schiff Kit | Merck | Cat#395B |
| Cryo-Gel | Leica Biosystems | Cat#39475237 |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1980 |
| Potassium acetate | Merck | Cat#W292001 |
| HEPES potassium salt | Merck | Cat#H0527 |
| Magenesium acetate tetrahydrate | Merck | Cat#M5661 |
| EGTA | Merck | Cat#E3889 |
| Digitonin | Merck | Cat#D141 |
| NP-40 | Merck | Cat#56741 |
| Sodium deoxycholate | Merck | Cat#D6750 |
| Trizol LS reagent | Thermo Fisher | Cat#10296028 |
| T7 RNA polymerase | Promega | Cat#P2075 |
| Bio-16-UTP | Thermo Fisher | Cat#AM8452 |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher | Cat#65001 |
| Deposited data | ||
| RNA-seq and RIBO-seq data | This study | GEO: GSE160803 |
| Experimental models: cell lines | ||
| SAS | Gift from Cheng-Chi Chang | RRID: CVCL_1675 |
| Ca9-22 | Gift from Te-Chang Lee | RRID: CVCL_1102 |
| CAL27 | Gift from Te-Chang Lee | RRID: CVCL_1107 |
| FaDu | Gift from Te-Chang Lee | RRID: CVCL_1218 |
| GNM | Gift from Cheng-Chia Yu | RRID: CVCL_WL58 |
| HSC3 | Gift from Te-Chang Lee | RRID: CVCL_1288 |
| OECM1 | Gift from Te-Chang Lee | RRID: CVCL_6782 |
| SCC4 | Gift from Te-Chang Lee | RRID: CVCL_1684 |
| TW2.6 | Gift from Te-Chang Lee | RRID: CVCL_GZ05 |
| EA.hy926 | Gift from Te-Chang Lee | RRID: CVCL_3901 |
| MOC-L1 | Shu-Chun Lin | RRID: CVCL_A9X3 |
| MOC-L2 | Shu-Chun Lin | RRID: CVCL_A9X4 |
| MOC-L3 | Shu-Chun Lin | RRID: CVCL_A9X5 |
| MOC-L4 | Shu-Chun Lin | RRID: CVCL_A9X6 |
| THP-1 | Gift from Li-Min Huang | RRID: CVCL_0006 |
| Experimental models: organisms/strains | ||
| C57BL/6 | National Applied Research Laboratories, Taipei, Taiwan, ROC | Stock number: RMRC11005 |
| Oligonucleotides | ||
| siC sense: GCGCUUCUACCAAAUACACUUGAUA | Thermo Fisher | N/A |
| siD#1 sense: CCUAGACCUGAACUCUUCAGAUAAU | Thermo Fisher | Cat# MSS236324 |
| siD#2 sense: GGGAGAAAUUAUCAUGGGAAACAUU | Thermo Fisher | Cat# MSS236325 |
| siD#3 sense: CACCAACGAGAGAGUUGGCAGUACA | Thermo Fisher | Cat# MSS236326 |
| siSRP54 sense: CAACAACAUGUUCAAAGCUAGCAUA | GenePharma | N/A |
| siSRP68 sense: CCCUUGAGGACAAGUUGGAACAGAA | GenePharma | N/A |
| Sense sequence for shRNA, see Table S5 | Integrated DNA Technologies | N/A |
| Primers, see Table S6 | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pcDNA3.1(+) | Thermo Fisher | V79020 |
| Software and algorithms | ||
| Human genome version hg38/GRCh38.p13 | Ensembl | http://asia.ensembl.org/Homo sapiens/Info/Index |
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Cutadapt | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| TopHat2 | (Kim et al., 2013) | http://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflink and Cuffdiff 2 | (Trapnell et al., 2013) | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/ |
| The Human Protein Atlas database (AREG and cancer prognosis) | (Uhlen et al., 2017) | https://www.proteinatlas.org/ENSG00000109321-AREG/pathology |
| TCGA PanCancer Atlas database (HNSCC) | (Gao et al., 2013) | https://www.cbioportal.org/study/summary?id=hnsc_tcga_pan_can_atlas_2018 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Woan-Yuh Tarn (wtarn@ibms.sinica.edu.tw).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Information about the cell lines, mouse strain and reagents used in the study is given in key resources table.
Cell culture and transfection
THP-1 and other cells (SAS, Ca9-22, CAL27, FaDu, GNM, HSC3, OECM1, SCC4, TW2.6, EA.hy926, MOC-L1, MOC-L2, MOC-L3, and MOC-L4) were respectively cultured in RPMI 1640 and DMEM supplemented with 10% (v/v) FBS at 37°C with a humidified atmosphere of 5% CO2, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. For SILAC (see below), SAS cells were grown and treated in the SILAC Metabolic Labeling Systems for more than 10 passages according to the manufacturer’s instruction. Complete replacement of 13C and 15N-labeled amino acids was confirmed by mass spectrometry. Transfection of siRNA and plasmid DNA was performed by using Lipofectamine 3000 and Lipofectamine 2000, respectively according to manufacturer’s instructions.
Cell growth assay
To measure cell growth, equal amounts of cells were seeded into 12-well plates between 24-72 hours after transfection. The cell number was counted between 24-72 hours after seeding by using the Luna Automated Cell Counter (Logos Biosystems).
Conditioned medium
Cells were transfected with siRNA or an overexpression plasmid for 48 hrs or a knockdown plasmid for 72 hrs. The medium was subsequently replaced with fresh serum-free (SF) or 10% FBS-containing (complete) DMEM. After 24 hrs, conditioned medium (CM) was collected 24. SF CM was used for the tube formation assay, and complete CM was used for the Boyden chamber assay and THP-1 differentiation. To collect THP-1 CM for the angiogenesis assay, THP-1 cells were cultured in the SAS complete CM for 72 hrs and subsequently in SF DMEM for 48 hrs.
Boyden chamber assay
Cells were seeded into 24-well Millicell inserts (8 μm pore) with 8×104 cells in 200 μl SF DMEM per insert. The inserts were placed in wells containing complete DMEM as control or various SAS CM and cultured for 24 hrs. Cells on the upper sites were removed and cells on the bottom sites were fixed and stained with crystal violet followed by microscopic examination. Staining intensity representing cell migration efficiency was quantified by using ImageJ software.
Angiogenesis assay
Tube formation assay was performed using μ-Slide Angiogenesis according to manufacturer’s instructions. Briefly, wells of the slides were filled with 10 μl of Matrigel Growth Factor Reduced, and 15,000 EA.hy926 cells were seeded onto the Matrigel in 50 μl of SF DMEM or CM. After 24 hrs, images of cells were obtained using IX71 inverted research microscope (Olympus). Tube numbers were automatically counted by using Angiogenesis Analyzer plugin for ImageJ and normalized to the tube numbers of cells cultured in SF DMEM.
THP-1 differentiation and polarization
THP-1 cells grown to approximately 1×106 cells/ml were collected by centrifugation and then resuspended in DMEM or CM with 5×105 cells/ml and cultured for 72 hrs. For phorbol 12-myristate 13-acetate (PMA) treatment, THP-1 cells at a density of 5×105 cells/ml were cultured in the presence of 100 nM of PMA for 48 hrs and then the medium was changed to DMEM or CM for 24 hrs. Cells were harvested, and RNA was extracted for RT-qPCR analysis.
Syngeneic graft model
Academia Sinica Institutional Animal Care and Use Committee approved the experiments. Each flank of 3-week-old male C57BL/6 mice was injected with 1×107 siRNA-transfected MOC-L1 cells (derived from a male C57BL/6 mouse) that were suspended in 200 μl of PBS. To treat tumors with recombinant mouse AREG (rAREGm), 80 ng of rAREGm were co-injected into each flank while transplantating. One week after transplantation, additional 20-ng rAREGm in 100-μl PBS was intratumorally injected into mock or rAREGm treatment groups. For immunoblotting, tumors were isolated 1 week and 2 weeks after injection, and tumor proteins were homogenized and extracted in PRO-PREP Protein Extraction Solution. For Periodic acid-Schiff or immunofluorescence staining, tumors were isolated 2 weeks after transplantation. Tumors were then frozen in liquid nitrogen, embedded in Cryo-Gel and cut to 12 μm sections by using Cryostat (Leica Biosystems).
Method details
Information about the plasmids, antibodies and reagents used in the study is given in key resources table.
Plasmid constructs
The pP2A plasmid was derived from pcDNA3.1 containing an in frame FLAG epitope, the porcine teschovirus-1 2A peptide and EGFP (Chen et al., 2018) and used for construction of overexpression vectors. The pP2A-DDX3 (wild-type, S382LMe, K230E) vectors were previously described (Chen et al., 2015). OSCC-associated DDX3 mutations (D521H, D558H, E572Q) were generated from the pP2A-DDX3 (wild type) vector by using PCR-based mutageneis. The pP2A-AREG vector was constructed by inserting human AREG cDNA from SAS cells. In all of the DDX3 overexpression vectors, synonymous substitutions in the siD#1-targeting site were made to avoid knockdown in the compensation experiments. For transient knockdown of DDX3 partners, each of the gene-specific shRNA sequences was constructed into the pAAVEMBL-CB-EGFP vector as described (Chen et al., 2016). The AREG expression vector was constructed by cloning the human AREG cDNA 5’ in-frame with the FLAG tag into pP2A. To construct the in vivo translation reporters, the 5’ UTR (210 nt) and 3’ UTR (265 nt) sequences of AREG were respectively inserted upstream and downstream of the humanized renilla luciferase (hRL) gene of psiCHEK-2 (pCH, Promega.
RNA-seq and Ribo-seq
Total RNA was extracted from control or DDX3-depleted SAS cells using Trizol Reagent according to the manufacturer’s instructions. Ribosomal RNA (rRNA) was depleted using the Ribo-off rRNA Depletion Kit. Library preparation was performed using the NETNext Ultra Directional RNA Library Prep Kit for Illumina according to the manufacturer’s instructions. The quality and quantity of the libraries were respectively analyzed by Fragment Analyzer Automated CE System (Advanced Analytical Technologies) and Qubit Fluorometer (ThermoFisher). Both RNA-seq and Ribo-seq (see below) were performed on the Illumina sequencing platform (NextSeq 500) following the manufacturer’s instructions, and assisted by Insight Genomics (Tainan, Taiwan).
The procedure of Ribo-seq was moderately modified from the protocol as described (Ingolia et al., 2012). Briefly, control or DDX3-depleted cells were lysed with the polysome buffer containing 20 mM Tris-HCl pH7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 200 μg/ml cycloheximide (CHX), 1% (v/v) Triton X-100 and 25 U/ml Turbo DNase. The lysates were subsequnetly digested with RNase I. After purification, rRNA was depleted using the Ribo-off rRNA Depetion Kit. Ribosome-protected fragments (RPF) were purified with PAGE and dephosphorylated with T4 Polynucleotide Kinase. Linkers were ligated to RPFs using T4 RNA ligase 2, truncated K227Q and the cDNA was synthesized using SuperScript III reverse transcriptase. The cDNA samples were circularized by CircLigase ssDNA Ligase, amplified by Phusion High-Fidelity DNA Polymerase and purified using AMPure XP for PCR Purification.
For analysis of the sequencing data, Cutadapt was used to remove the short adapter ‘CTGTAGGCACCATCAAT‘ from the RNAseq and Riboseq samples. Bowtie and TopHat2 were used to map the reads to the human genome version hg38/GRCh38.p13 with specific settings ‘--no-coverage-search --segment-length 14 --segment-mismatches 1’, and calculate the mapped reads statistically. The reads per kilobase pase million (RPKM) of each protein-coding gene for each sample, and the differential expressions of genes between various samples were calculated with Cufflinks and Cuffdiff 2 based on the protein coding gene annotation file provided from Ensembl database. After Cuffdiff normalization and elimination of the genes with no read coverage, the transcripts with siC RPKM higher than 1, p value lower than 0.05 and log2(siD/siC) larger than 1 and lower than 1 were selected for analysis.
Antibody array
Profiling of growth factors/cytokines in control or DDX3-depleted SAS cells was performed using Human Cytokine Antibody Array according to the manufacturer’s instructions. Briefly, cells were lysed in Cell Lysis Buffer. Protein concentration of the lysates was evaluated using the BCA Protein Assay Kit. For hybridization, 1 mg of the lysate proteins in 1 ml of the blocking buffer were incubated with array slides overnight at 4°C. The slides were then sequentially incubated with Biotinylated Antibody Cocktail at 4°C overnight, and subsequently HRP-Conjugated Streptavidin Solution at 4°C overnight with thorough washing after each step. Images were captured using LAS-3000 Imager (Fujifilm), and the intensity of blots was analyzed using ImageJ.
Immunoprecipitation, RNA immunoprecipitation and mass spectrometry
Cell lysates were collected in the immunoprecipitation (IP) buffer (50 mM Tris-HCl pH7.5, 150 mM NaCl, 0.5% Triton X-100, 5 mM MgCl2) containing Protease/Phosphatase Inhibitor Cocktail. 400 ng/μl RNase A and 0.5 unit/μl RNasin were added for removing RNA-mediated interaction and RNA immunoprecipitation (RIP), respectively. For IP or RIP, 1 mg of cell lysate proteins were incubated with 3 μg of antibody that was bound onto Dynabeads Protein G in 1 ml of protease/phosphatase inhibitor-containing IP buffer at 4°C for 2 hrs. After washing with 1 ml of IP buffer for 5 times, beads were suspended in the sample buffer (125 mM Tris-HCl pH6.8, 11% glycerol, 2.4% SDS, 3% 2-mercaptoethanol, 0.01% Bromophenol Blue) or Trizol for immunoblotting or for RNA extraction, respectively. For mass spectrometry analysis of DDX3-interacting proteins, 5 mg of cell lysate proteins and 15 μg of DDX3 antibody were used. Protein bands separated by using SDS-PAGE were cut, digested by trypsin, purified and subjected for LC-ESI/MS/MS analysis. In the SILAC experiment, light and heavy isotope-labeled SAS cell lysates were immunoprecipitated without and with RNase A, respectively. After IP, samples were mixed, digested by trypsin, purified and subjected for 2D-LC-ESI/MS/MS. Ratios of heavy to light amplitude of proteins higher than 2 were picked to represent RNA-independent interacting proteins of DDX3.
ELISA analysis
The AREG level of culture medium was measured by using the AREG ELISA Kit according to manufacturer’s instructions. Briefly, 100-μl DMEM or CM was added to each well of the AREG microplate and incubated at 37°C for 1 hr. After discarding the medium, wells were incubated with 100 μl of 1× Biotinylated AREG Detector Antibody at 37°C for 1 hr. After thorough wash with 1× Wash Buffer, wells were incubated with 100 μl of 1× Avidin-HRP Conjugated at 37°C for 30 min. After thorough wash with 1× Wash Buffer, wells were incubated with 90 μl of TMB Substrate at 37°C in the dark for 20 min. Finally, 50 μl of Stop solution was added, and optical absorbance at 450 nm was measured immediately by using SpectraMax 190 Microplate Reader (Molecular Devices).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNAs were extracted using the HiYield Total RNA Extraction Kit and then subjected to DNase digestion on beads according to manufacturer's instructions. RNAs were reversely transcribed using the ToolsQuant II Fast RT Kit according to manufacturer’s instructions. cDNAs was analyzed by qPCR using PerfeCta SYBR Green FastMix PCR Reagent in LightCycler480 instrument (Roche).
Indirect immunofluorescence, confocal microscopy and image analysis
Immunofluorescence staining was performed as previously described (Chen et al., 2012). Confocal imaging with a Plan-Apochromat 100x/1.4 Oil objective was carried out by using ZEISS LSM with Airyscan super-resolution mode. Histograms of fluorescence intensities were produced by using ImageJ.
In vivo translation assay
For knockdown experiments, SAS cells were transfected with siRNA or shRNA 48 hours prior to reporter transfection. For reporter transfection, 0.05 μg of pCH or pCH-AREG UTR reporters and 0.45 μg of pP2A or DDX3 overexpression vectors were used. Luciferase assay was performed by using the Dual-Luciferase Reporter Assay System 24 hours after reporter transfection. The activity of humanized firefly luciferase encoding by the same pCH reporters was used for references. The same cell lysates were used for RT-qPCR and immnunoblotting analysis.
Cellular fractionation and polysome fractionation
Fractionation of the cytosol and membrane fractions was performed as described previously (Jagannathan et al., 2011) with minor modification. Briefly, SAS cells with 80–90% confluent in a 10-cm plate were washed with PBS, treated with PBS containing 50 μg/ml CHX and then incubated with 0.5 ml of permeabilization buffer (110 mM KOAc, 25 mM K-HEPES pH7.2, 2.5 mM Mg(OAc)2, 1 mM EGTA, 0.3% digitonin, 1 mM DTT, Protease/Phosphatase Inhibitor Cocktail, 40 U/ml RNasin, 50 μg/ml CHX) for 5 min to collect cytosol lysate. Cells were then washed with wash buffer (110 mM KOAc, 25 mM K-HEPES pH7.2, 2.5 mM Mg(OAc)2, 1 mM EGTA, 0.004% digitonin, 1 mM DTT), and then incubated with 0.5 ml of lysis buffer (400 mM KOAc, 25 mM K-HEPES pH7.2, 15 mM Mg(OAc)2, 1% NP-40, 0.5% sodium deoxycholate, 1 mM DTT, Protease/Phosphatase Inhibitor Cocktail, 40 U/ml RNasin, 50 μg/ml CHX) for 5 min to collect membrane lysate. Both lysates were centrifuged at 7500×g for 10 min and supernatants were collected as the cytosol and membrane fractions. To perform polysome fractionation, 0.4 ml of the samples were overlayed on the 15–40% sucrose gradient and centrifuged at 38,000 rpm for 2 hrs. Polysome fractions were collected and monitored via an automatic gradient fractionator (ISCO). Nine fractions from 40S to the first polysome and the following nine fractions were collected as the light and heavy fractions, respectively. Trizol LS reagent was used for RNA extraction from the fractions.
Biotin-labeled RNA affinity selection
Biotin-labeled AREG 3′ UTR RNA was synthesized by using T7 RNA polymerase according to the manufacturer's instructions except that 3.2 mM MgCl2 and 1 mM Bio-16-UTP were additionally added. RNA was recovered by using Trizol reagent and the quality of RNA was checked by electrophoresis. Cell lysates collected in the pulldown buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl2 and 0.1% NP-40) supplemented with Protease/Phosphatase Inhibitor Cocktail and 40 U/ml RNasin. Pulldown was performed in a final volume of 200 μl containing 10 μl of Dynabeads MyOne Streptavidin C1 and 1 mg of lysate with or without 0.5 μg of biotin-labeled AREG 3′ UTR RNA at room temperature for 4 hrs. After thorough wash with IP buffer, immunoblotting was performed to reveal RNA-associated proteins.
Quantification and statistical analysis
In all experiments shown in Figures, average values and standard deviations were obtained from at least three independent experiments. Statistical analysis was performed using Student's t-test.
Acknowledgments
We thank the Core Facilities of the Institute of Biomedical Sciences, Academia Sinica, for their technical assistance; these facilities included the Proteomics Core, Light Microscopy, DNA Sequencing, Experimental Animal Facility, and Pathology Core. We also thank Joseph Ta-Chien Tseng for his valuable advice on sample preparation and data analysis of polysome profiling. Funding was provided by grant IBMS-CRC107-P03 from the Institute of Biomedical Sciences, Academia Sinica.
Author contributions
H.-H.C. designed and performed the majority of the experiments (including cell culture, transfection, cell proliferation assays, Boyden chamber assays, angiogenesis assays, RT-qPCR analysis, in vitro translation assays, data mining of Ribo-seq and proteomics, immunofluorescence staining, affinity selection experiments), interpreted the data, and wrote the manuscript. H.-I.Y. performed experiments for Figures 1B, 1F, 1G, 2B–2D, 3, all panels, 5A, 5C, 5G, 6B, and 7D, and carried out morphology analysis of SAS cells and vector construction. R.R. assisted with vector construction and experimental analyses. S.-L.L. and S.-H.W. performed Ribo-seq data analysis. Y.-F.C. and S.-C.L. provided mouse OSCC cell lines. M.-H.Y. provided critical suggestions on experimental design. W.-Y.T. devised the study, supervised the experiments, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103086.
Supplemental information
Data and code availability
-
•
RNA-seq and RIBO-seq data have been deposited at GEO and are publicly available after publication. Accession numbers are listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required for analysis of the data reported in this paper is available from the lead contact upon request.
References
- Berasain C., Avila M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014;28:31–41. doi: 10.1016/j.semcdb.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Berkovits B.D., Mayr C. Alternative 3' UTRs act as scaffolds to regulate membrane protein localization. Nature. 2015;522:363–367. doi: 10.1038/nature14321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol G.M., Xie M., Raman V. DDX3, a potential target for cancer treatment. Mol. Cancer. 2015;14:188. doi: 10.1186/s12943-015-0461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser B., Sancey L., Brambilla E., Coll J.L., Hurbin A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta. 2011;1816:119–131. doi: 10.1016/j.bbcan.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Chao C.H., Chen C.M., Cheng P.L., Shih J.W., Tsou A.P., Lee Y.H. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 2006;66:6579–6588. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- Chartron J.W., Hunt K.C., Frydman J. Cotranslational signal-independent SRP preloading during membrane targeting. Nature. 2016;536:224–228. doi: 10.1038/nature19309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.H., Yu H.I., Chiang W.C., Lin Y.D., Shia B.C., Tarn W.Y. hnRNP Q regulates Cdc42-mediated neuronal morphogenesis. Mol. Cell Biol. 2012;32:2224–2238. doi: 10.1128/MCB.06550-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.H., Yu H.I., Cho W.C., Tarn W.Y. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene. 2015;34:2790–2800. doi: 10.1038/onc.2014.190. [DOI] [PubMed] [Google Scholar]
- Chen H.H., Yu H.I., Tarn W.Y. DDX3 modulates neurite development via translationally activating an RNA Regulon involved in Rac1 activation. J. Neurosci. 2016;36:9792–9804. doi: 10.1523/JNEUROSCI.4603-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.H., Yu H.I., Yang M.H., Tarn W.Y. DDX3 activates CBC-eIF3-Mediated translation of uORF-containing oncogenic mRNAs to promote metastasis in HNSCC. Cancer Res. 2018;78:4512–4523. doi: 10.1158/0008-5472.CAN-18-0282. [DOI] [PubMed] [Google Scholar]
- Chen Y., Song Y., Du W., Gong L., Chang H., Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J. Biomed. Sci. 2019;26:78. doi: 10.1186/s12929-019-0568-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.F., Liu C.J., Lin L.H., Chou C.H., Yeh L.Y., Lin S.C., Chang K.W. Establishing of mouse oral carcinoma cell lines derived from transgenic mice and their use as syngeneic tumorigenesis models. BMC Cancer. 2019;19:281. doi: 10.1186/s12885-019-5486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Zontag O., Baez C., Lim L.Q.J., Olender T., Schirman D., Dahary D., Pilpel Y., Gerst J.E. A secretion-enhancing cis regulatory targeting element (SECReTE) involved in mRNA localization and protein synthesis. PLoS Genet. 2019;15:e1008248. doi: 10.1371/journal.pgen.1008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat C.M., Dolde C., de Groot R.E., Ohkawara B., Reinhard C., Korswagen H.C., Niehrs C. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-beta-catenin signaling. Science. 2013;339:1436–1441. doi: 10.1126/science.1231499. [DOI] [PubMed] [Google Scholar]
- Cui X.A., Zhang H., Palazzo A.F. p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol. 2012;10:e1001336. doi: 10.1371/journal.pbio.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert A.W., Wickenhauser C., Salins P.C., Kappler M., Bukur J., Seliger B. Clinical relevance of the tumor microenvironment and immune escape of oral squamous cell carcinoma. J. Transl. Med. 2016;14:85. doi: 10.1186/s12967-016-0828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler R., Golbik R.P., Behrens S.E. The DEAD-box helicase DDX3 supports the assembly of functional 80S ribosomes. Nucl. Acids Res. 2012;40:4998–5011. doi: 10.1093/nar/gks070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genz C., Fundakowski J., Hermesh O., Schmid M., Jansen R.P. Association of the yeast RNA-binding protein She2p with the tubular endoplasmic reticulum depends on membrane curvature. J. Biol. Chem. 2013;288:32384–32393. doi: 10.1074/jbc.M113.486431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Sun S., Li P., Liu Q., Zhang Z., Dong H., Sun M., Wu W., Wang X., Guo H. Ribosomal protein L13 promotes IRES-driven translation of foot-and-mouth disease virus in a helicase DDX3-dependent manner. J. Virol. 2020;94 doi: 10.1128/JVI.01679-19. e01679-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerma van Voss M.R., Vesuna F., Bol G.M., Afzal J., Tantravedi S., Bergman Y., Kammers K., Lehar M., Malek R., Ballew M. Targeting mitochondrial translation by inhibiting DDX3: a novel radiosensitization strategy for cancer treatment. Oncogene. 2018;37:63–74. doi: 10.1038/onc.2017.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.C., Reid D.W., Hoffman A.M., Sarkar D., Nicchitta C.V. Oncoprotein AEG-1 is an endoplasmic reticulum RNA-binding protein whose interactome is enriched in organelle resident protein-encoding mRNAs. RNA. 2018;24:688–703. doi: 10.1261/rna.063313.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan S., Nwosu C., Nicchitta C.V. Analyzing mRNA localization to the endoplasmic reticulum via cell fractionation. Methods Mol. Biol. 2011;714:301–321. doi: 10.1007/978-1-61779-005-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku Y.C., Lai M.H., Lo C.C., Cheng Y.C., Qiu J.T., Tarn W.Y., Lai M.C. DDX3 participates in translational control of inflammation induced by infections and injuries. Mol. Cell Biol. 2019;39:e00285-18. doi: 10.1128/MCB.00285-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.C., Chang W.C., Shieh S.Y., Tarn W.Y. DDX3 regulates cell growth through translational control of cyclin E1. Mol. Cell Biol. 2010;30:5444–5453. doi: 10.1128/MCB.00560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.C., Lee Y.H., Tarn W.Y. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol. Biol. Cell. 2008;19:3847–3858. doi: 10.1091/mbc.E07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.S., Dias A.P., Jedrychowski M., Patel A.H., Hsu J.L., Reed R. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucl. Acids Res. 2008;36:4708–4718. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Mayr C. Gain of additional BIRC3 protein functions through 3'-UTR-mediated protein complex formation. Mol. Cell. 2019;74:701–712.e9. doi: 10.1016/j.molcel.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W., Mayr C. A membraneless organelle associated with the endoplasmic reticulum enables 3'UTR-mediated protein-protein interactions. Cell. 2018;175:1492–1506.e19. doi: 10.1016/j.cell.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17 10.14806/ej.14817.14801.14200 [Google Scholar]
- Peltanova B., Raudenska M., Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol. Cancer. 2019;18:63. doi: 10.1186/s12943-019-0983-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phung B., Ciesla M., Sanna A., Guzzi N., Beneventi G., Cao Thi Ngoc P., Lauss M., Cabrita R., Cordero E., Bosch A. The X-linked DDX3X RNA helicase dictates translation reprogramming and metastasis in melanoma. Cell Rep. 2019;27:3573–3586.e7. doi: 10.1016/j.celrep.2019.05.069. [DOI] [PubMed] [Google Scholar]
- Robichaud N., Sonenberg N., Ruggero D., Schneider R.J. Translational control in cancer. Cold Spring Harb Perspect. Biol. 2018;11:a032896. doi: 10.1101/cshperspect.a032896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samir P., Kesavardhana S., Patmore D.M., Gingras S., Malireddi R.K.S., Karki R., Guy C.S., Briard B., Place D.E., Bhattacharya A. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature. 2019;573:590–594. doi: 10.1038/s41586-019-1551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D., Jankowsky E. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit. Rev. Biochem. Mol. Biol. 2014;49:343–360. doi: 10.3109/10409238.2014.931339. [DOI] [PubMed] [Google Scholar]
- Shih J.W., Tsai T.Y., Chao C.H., Wu Lee Y.H. Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene. 2008;27:700–714. doi: 10.1038/sj.onc.1210687. [DOI] [PubMed] [Google Scholar]
- Shih J.W., Wang W.T., Tsai T.Y., Kuo C.Y., Li H.K., Wu Lee Y.H. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 2012;441:119–129. doi: 10.1042/BJ20110739. [DOI] [PubMed] [Google Scholar]
- Soto-Rifo R., Rubilar P.S., Limousin T., de Breyne S., Decimo D., Ohlmann T. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J. 2012;31:3745–3756. doi: 10.1038/emboj.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.Y., Lin T.C., Lin Y.F., Chen M.H., Lee C.H., Wang H.Y., Lee Y.C., Liu Y.P., Chen C.L., Hsiao M. DDX3 as a strongest prognosis marker and its downregulation promotes metastasis in colorectal cancer. Oncotarget. 2015;6:18602–18612. doi: 10.18632/oncotarget.4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M., Zhang C., Lee S., Sjostedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Weber M., Iliopoulos C., Moebius P., Buttner-Herold M., Amann K., Ries J., Preidl R., Neukam F.W., Wehrhan F. Prognostic significance of macrophage polarization in early stage oral squamous cell carcinomas. Oral Oncol. 2016;52:75–84. doi: 10.1016/j.oraloncology.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Wild K., Juaire K.D., Soni K., Shanmuganathan V., Hendricks A., Segnitz B., Beckmann R., Sinning I. Reconstitution of the human SRP system and quantitative and systematic analysis of its ribosome interactions. Nucl. Acids Res. 2019;47:3184–3196. doi: 10.1093/nar/gky1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedavalli V.S., Neuveut C., Chi Y.H., Kleiman L., Jeang K.T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Zhang X., Shan S.O. Fidelity of cotranslational protein targeting by the signal recognition particle. Annu. Rev. Biophys. 2014;43:381–408. doi: 10.1146/annurev-biophys-051013-022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq and RIBO-seq data have been deposited at GEO and are publicly available after publication. Accession numbers are listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required for analysis of the data reported in this paper is available from the lead contact upon request.