

Abstract
The worldwide obesity epidemic has emerged as a major cause of insulin resistance and Type 2 diabetes. Chronic tissue inflammation is a well-recognized feature of obesity, and the field of immunometabolism has witnessed many advances in recent years. Here, we review the major features of our current understanding with respect to chronic obesity-related inflammation in metabolic tissues and focus on how these inflammatory changes affect insulin sensitivity, insulin secretion, food intake, and glucose homeostasis. There is a growing appreciation of the varied and sometimes integrated crosstalk between cells within a tissue (intraorgan) and tissues within an organism (interorgan) that supports inflammation in the context of metabolic dysregulation. Understanding these pathways and modes of communication has implications for translational studies. We also briefly summarize the state of this field with respect to potential current and developing therapeutics.
Introduction
The prevalence of type 2 diabetes mellitus (T2DM) is rapidly increasing on a global basis and has now reached epidemic proportions. The etiology of T2DM involves both insulin resistance and β-cell dysfunction, and one typically needs both defects in order to develop the full hyperglycemic diabetic state (referred to as the two-hit hypothesis) (Defronzo, 2009; Halban et al., 2014; Olefsky and Glass, 2010). Obesity is far and away the most common cause of insulin resistance in man; therefore, it is the ongoing obesity epidemic that is driving the parallel rise in the incidence of T2DM (Olefsky and Glass, 2010).
While insulin resistance and β-cell dysfunction co-exist in the great majority of T2DM patients, the relative contribution and temporal appearance of these two defects varies across different patients and populations. Chronic obesity-associated inflammation, particularly in adipose tissue and liver, is a widely reported contributor to insulin resistance (Hotamisligil, 2017), and recent evidence indicates that inflammation is also an important etiologic component of β-cell dysfunction in T2DM (Eguchi and Nagai, 2017). While there are many potential contributing mechanisms to both insulin resistance and β-cell dysfunction, in this review, we will focus our attention primarily on the role of chronic tissue inflammation in these metabolic defects. There are already many excellent recent reviews covering other potential non-inflammatory causes of insulin resistance and β-cell dysfunction (Czech, 2017; Newgard, 2017; Samuel and Shulman, 2016).
Several tissues or organs can make independent contributions to the etiology of T2DM. Chronic inflammation in the liver, muscle, adipose tissue, CNS, and gastrointestinal (GI) tract plays a role in achieving the final insulin-resistant, hyperglycemic diabetic state. This is depicted in Figure 1, which shows a variety of interconnections comprising an extensive interorgan crosstalk network. There are two aspects of consideration of immunometabolism. One is the effect of inflammation and immune responses on the control of systemic metabolism. The other focuses on metabolism within immune cells. Clearly, metabolic changes in immune cells have important effects on the function of these cell types (O’Neill et al., 2016; Puleston et al., 2017). Here, we focus on immunometabolism as it relates to the effects of chronic inflammation on metabolic events and will also discuss future directions in this rapidly growing area.
Figure 1. Obesity Is the Major Cause of Insulin Resistance in Humans and Is a Driver of the Global Type 2 Diabetes Mellitus Epidemic.
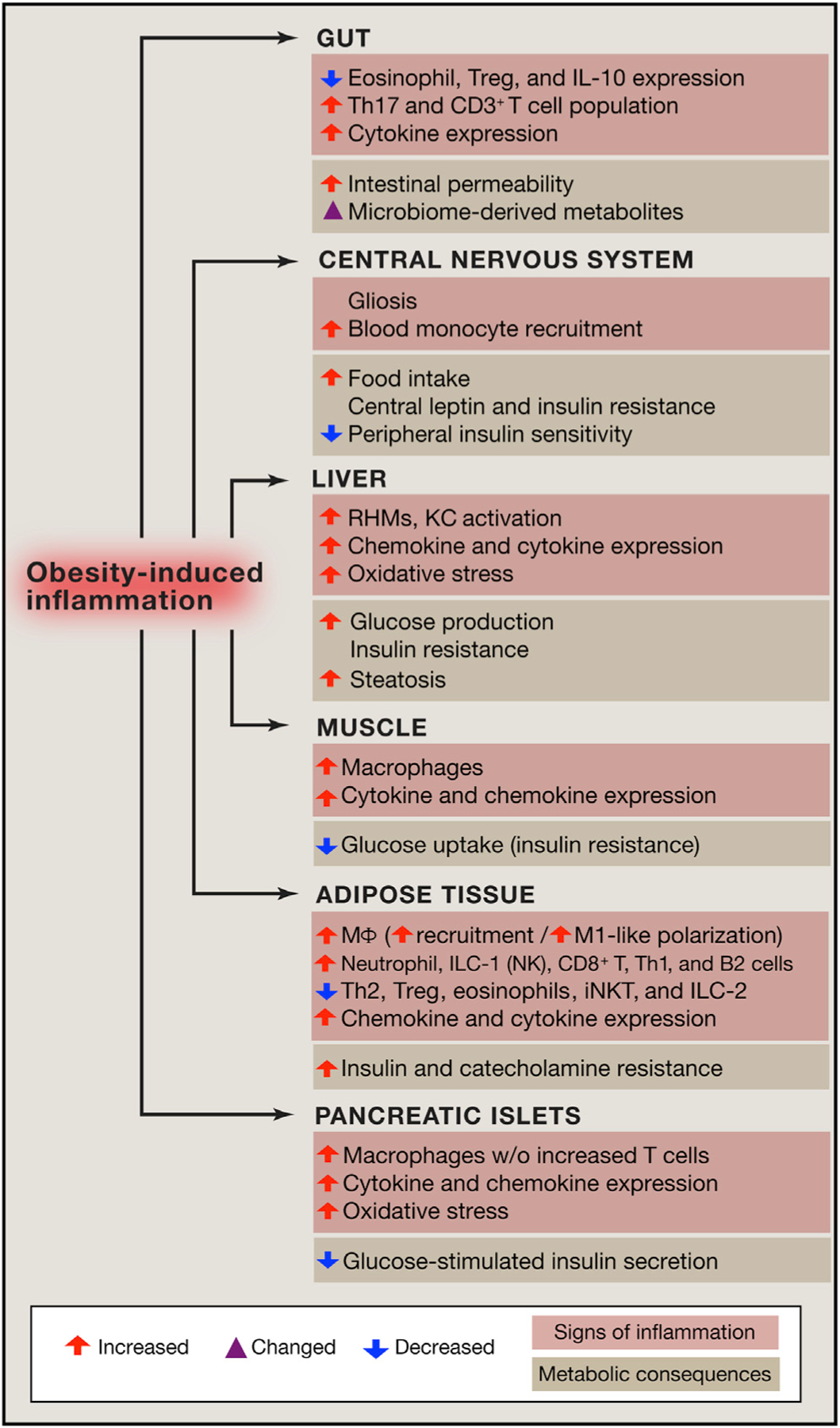
Obesity-induced chronic tissue inflammation is a key mechanism of the dys-metabolism that occurs in this condition. Chronic tissue inflammation induces a range of effects on adipose tissue, muscle, liver, pancreatic islets, the gut, and the CNS. These inflammatory changes contribute to insulin resistance (adipocytes, muscle, liver), decreased insulin secretion (islets), dysbiosis and intestinal permeability (gut), and increased food intake (CNS).
Establishing a Link between Chronic Inflammation and Metabolic Disease
The role of subacute chronic inflammation, particularly in adipose tissue and liver, has been widely studied with respect to its role in systemic insulin resistance (Hotamisligil, 2017). Recent evidence has also pointed toward the contribution of immune cell inflammation in islets in β-cell dysfunction (Eguchi and Nagai, 2017). With respect to both of these metabolic defects, obesity has emerged as an underlying etiology.
Anti-diabetic effects of anti-inflammatory treatments (e.g., salicylate and berberine) have been known for over 100 years (Ebstein, 1876; Lee et al., 2006; Yuan et al., 2001). However, over the last couple of decades, we have begun to understand the underlying cellular and physiologic mechanisms of how obesity-associated inflammation induces insulin resistance and glucose intolerance. The first studies to establish a concrete mechanistic link between inflammation and insulin resistance focused on tumor necrosis factor-α (TNF-α). In 1989, Grunfeld and Feingold observed that treatment of rodents with TNF-α led to adverse metabolic events, including increased serum glucose levels (Feingold et al., 1989). An additional seminal paper in the field discovered that TNF-α levels are elevated in adipose tissue of obese rodents and that TNF-α treatment causes insulin resistance in mice by activating IκB kinase β (IKKβ), while neutralization of circulating TNF-α improves insulin sensitivity (Hotamisligil et al., 1993; Yuan et al., 2001). Activation of other inflammatory kinases, such as Jun amino-terminal kinases (JNKs) is also a feature of the chronic obesity-induced tissue inflammatory state, and genetic ablation of JNK also leads to protection from obesity-induced inflammation and insulin resistance (Hirosumi et al., 2002). Increased proinflammatory signaling has subsequently been observed in the three classical insulin target tissues (fat, liver, and muscle), as well as in islets, the CNS, and the GI tract (Cai et al., 2005; De Souza et al., 2005; Fink et al., 2014; Hotamisligil et al., 1993; Lee et al., 2011b; Maedler et al., 2002; Weisberg et al., 2003; Winer et al., 2017; Xu et al., 2003).
While most of the direct evidence that inflammation is a key cause of insulin resistance and impaired glucose metabolism in obesity stems from rodent studies, there are several ex vivo studies with primary human adipocytes, hepatocytes, skeletal muscle (SM) cells, and islets that also support the idea that inflammation can cause decreased insulin sensitivity and impaired insulin secretion (Austin et al., 2008; Dietze et al., 2002; Löfgren et al., 2000; Maedler et al., 2002; Pardo Marques et al., 2016). However, the overall concept for a causal role of inflammation in insulin resistance still remains to be validated in man.
Tissue-Specific Events in Development of Insulin Resistance and Glucose Intolerance
Adipose Tissue
Adipose tissue can comprise white, brown, or beige adipocytes. White adipocytes are specialized to store excessive energy in the form of triglycerides and to release fatty acids (FAs) in times of energy deficiency. White adipose tissue (WAT) also releases a variety of secretory peptides and lipid metabolites that signal to other tissues (Deng and Scherer, 2010; Glass and Olefsky, 2012; Smith and Kahn, 2016), contributing to systemic energy and glucose homeostasis. In contrast to WAT, the major function of brown and beige adipocytes is to generate heat under cold stress, which is well reviewed elsewhere (Kajimura et al., 2015). Therefore, in this section, we will focus on inflammation and WAT.
Adipose Tissue Macrophages.
Adipose tissue inflammation occurs as early as 3–7 days after feeding high-fat diet (HFD) in mice and gradually increases, contributing to systemic insulin resistance and glucose intolerance (Lee et al., 2011b). During obesity, different types of both innate and adaptive immune cells accumulate in adipose tissue with increased adipocyte chemokine production (Lackey and Olefsky, 2016) and impaired macrophage emigration from adipose tissue (Ramkhelawon et al., 2014), preventing resolution of inflammation. Of the adipose tissue immune cells, macrophages are the most abundant and can comprise up to 40% of all stromal vascular cells in obese adipose tissue (Lumeng et al., 2007). They are a major source of proinflammatory cytokines, which have the potential to cause local and systemic insulin resistance (Xu et al., 2003). Indeed, numerous studies have shown that genetic manipulations that impair macrophage proinflammatory pathway activity protect against obesity-induced insulin resistance and glucose intolerance (Lackey and Olefsky, 2016).
The intracellular events leading to impaired insulin signaling after proinflammatory stimulation have been well studied and include activation of inhibitor of κB kinases (IKKs), nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), JNKs, and increased oxidative stress (Lackey and Olefsky, 2016). These pathways all trigger transcriptional activation of proinflammatory genes, decreased expression of adipocyte-specific genes, suppression of insulin-receptor action, and inhibitory phosphorylation or nitrosylation of insulin-signaling molecules (Hotamisligil, 2017; Xing et al., 1997).
Tissue macrophages are often classified based on their inflammatory state, with classically activated M1 proinflammatory macrophages versus alternatively activated M2 anti-inflammatory cells. While the designations M1 and M2 are often used to categorize macrophages, these definitions largely derive from in vitro studies (Sica and Mantovani, 2012). In the in vivo setting, it is likely that adipose tissue macrpophages (ATMs) exist across a polarization spectrum from the most inflammatory to anti-inflammatory phenotypes (Kratz et al., 2014; Li et al., 2010; Lumeng et al., 2008; Nguyen et al., 2007). Thus, the terms M1-like and M2-like are used to generally depict the proinflammatory state of recruited ATMs versus the anti-inflammatory state of resident ATMs. Most ATMs express the surface markers CD11b and F4/80. In obesity, the majority of recruited ATMs are also positive for CD11c, and ATMs expressing this marker typically exhibit increased expression of proinflammatory genes (Lumeng et al., 2007; Nguyen et al., 2007).
Other Innate Immune Cell Types.
While ATMs are the major effector cell type generating factors that cause insulin resistance, changes in several other innate immune cell types in obese adipose tissue contribute to initiation and/or progression of adipose tissue inflammation. For example, in lean adipose tissue, innate lymphoid type 2 (ILC2) cells secrete Th2 cytokines IL-5 and IL-13, stimulating eosinophil chemotaxis and maturation (Molofsky et al., 2013; Nussbaum et al., 2013). Activated eosinophils can repress adipose tissue inflammation by releasing IL-4 and IL-13, key drivers of the M2-like macrophage polarization state (Wu et al., 2011). During obesity, representation of ILC2 cells and eosinophils in adipose tissue declines with increasing adiposity, which can enhance obesity-induced inflammatory activation of ATMs (Molofsky et al., 2013). On the other hand, neutrophils are among the first responders recruited to adipose tissue in mice on HFD. They release neutrophil elastase (NE), which impairs insulin signaling by promoting IRS-1 degradation (Talukdar et al., 2012). Similar to ILC2 cells, ILC1 cells also reside in lean adipose tissue, but their number increases in obesity, contributing to adipose tissue inflammation (Boulenouar et al., 2017; Lee et al., 2016; Theurich et al., 2017).
Lymphocytes.
Several types of adaptive immune cells are also involved in obesity-induced adipose tissue inflammation. CD3+ T cells comprise the largest adipose-tissue immune-cell population next to macrophages, and their number is increased in HFD/obese mice (McNelis and Olefsky, 2014). T cells can be categorized into two groups, depending on their phenotypic expression of the surface coreceptors CD4 or CD8. Adipose tissue CD8+ T cell content is increased in obesity, and these cells release factors facilitating macrophage differentiation and chemotaxis (Nishimura et al., 2009).
CD4+ T cells can be further subcategorized based on their functional profile into proinflammatory T helper (Th)1 and Th17 cells, anti-inflammatory Th2 cells, and T regulatory (Treg) cells. The number of CD3+CD4+ Th1 cells is increased in obesity, and they contribute to adipose tissue inflammation by producing interferon γ (IFNγ) (Winer et al., 2009). In contrast, the number of CD3+CD4+ Th2 cells is decreased in obese adipose tissue (Winer et al., 2009).
Treg cells (CD3+CD4+FOXP3+) are a small subset of CD4+ T lymphocytes (5%–20% of CD3+CD4+ cells) that can control the activity of other T cells or the innate immune system, inhibiting inappropriate inflammation. Lean adipose tissue harbors a unique Treg population characterized by high peroxisome proliferator-activated receptor γ (PPARγ) expression, and these Tregs play an important role in maintaining adipose-tissue inflammatory tone and insulin sensitivity (Bapat et al., 2015; Feuerer et al., 2009). In obesity, the number or proportion of adipose tissue Treg cells declines, contributing to increased adipose tissue inflammation and insulin resistance (Feuerer et al., 2009). Deletion of PPARγ selectively in Treg cells attenuates the insulin-sensitizing effect of pioglitazone in HFD mice, suggesting that insulin-sensitizing effects of thiazolidinediones (TZDs) could be partially mediated through Treg PPARγ (Cipolletta et al., 2012). However, it has also been suggested that a gradual increase in the adipose tissue Treg cell population during the process of aging can exacerbate aging-associated insulin resistance (Bapat et al., 2015).
Invariant natural killer T (iNKT) cells are lipid-antigen-reactive T cells restricted by the major histocompatibility complex (MHC)-like molecule CD1d (Lynch, 2014). CD1d is highly enriched in adipocytes and hepatocytes, and iNKT cells comprise a subset of lymphocytes in normal adipose tissue (Huh et al., 2013). The number of these cells declines in obesity (Lynch, 2014).
B lymphocytes (mostly B2 cells) also accumulate and are activated in obese adipose tissue partly through the leukotriene B4 (LTB4/BLT1) system. B2 cells contribute to proinflammatory activation of ATMs and T cells (Winer et al., 2011; Ying et al., 2017b).
Collectively, the changes in content and activation state of immune cells within obese adipose tissue create a microenvironment to increase proinflammatory activation of ATMs and enhance adipose tissue inflammation. This leads to deleterious effects on insulin action in parenchymal cells, in this case adipocytes. Immune-cell-type-specific depletion and adoptive transfer experiment data fully support this idea. This represents a key mechanism of crosstalk within adipose tissue in which the immune cells involved in the inflammatory response induce adverse metabolic changes in the parenchymal cells, at least with respect to insulin resistance.
Triggers for Adipose Tissue Inflammation.
One of the initiating causes of adipocyte proinflammatory signaling is hypoxia within WAT, which occurs very early in the onset of obesity (Lee et al., 2011b, 2014). There are two components contributing to adipocyte hypoxia. The first and probably initiating one involves uncoupling of mitochondria oxidative metabolism as a result of elevated free fatty acids (FFAs) (Lee et al., 2014). FFAs can mediate uncoupled respiration by stimulating the mitochondrial adenine nucleotide translocase 2 (ANT2) (Lee et al., 2014; Shabalina et al., 2006). This leads to increased oxygen consumption within adipocytes, creating relative adipocyte hypoxia (Lee et al., 2014). This hypoxia is sensed by the hypoxia-inducible factor (HIF)/proline hydroxylase domain (PHD) system (Ivan and Kaelin, 2017). Specifically, hypoxia inhibits the activity of PHD1-3, causing decreased hydroxylation and increased stability of HIF-1α and HIF-2α protein, resulting in increased protein expression. In adipose tissue, the main metabolic culprit is HIF-1α, since increased HIF-1α levels stimulate the intra-adipocyte proinflammatory pathway, leading to chemokine release, monocyte influx, etc. (Jiang et al., 2011; Lee et al., 2011a; Lee et al., 2014; Sun et al., 2013). In contrast, the metabolic effects of HIF-2α are opposite to those of HIF-1α (Lee et al., 2014). As the process of obesity continues and enters a more chronic state, an imbalance between O2 perfusion and metabolic need emerges, creating a second cause of chronic adipose tissue hypoxia (Hosogai et al., 2007). Taken together, adipose tissue hypoxia in obesity promotes the chronic proinflammatory state.
The inflammatory state of adipose tissue can also be modified by various lipid species, including proinflammatory saturated fatty acids (SFAs) and anti-inflammatory ω3 FAs (Glass and Olefsky, 2012). SFAs are derived from dietary sources or through hydrolysis of adipocyte triglyceride stores, whereas ω3 FAs are essential FAs and are strictly dietary in origin. SFAs can work through TLR4 on adipocytes and macrophages to stimulate downstream inflammatory signaling (Holland et al., 2011; Saberi et al., 2009; Tao et al., 2017). In contrast, GPR120 serves as a receptor for ω3 FAs and activates a β arrestin-2-dependent pathway that directly inhibits inflammatory pathway activation (Oh et al., 2010). Other bioactive lipids, such as the eicosanoid pathway metabolite LTB4, which functions as an immune cell chemokine through its receptor BLT1, have been described (Li et al., 2015).
The Upside of Adipose Inflammation.
Inflammation is an important normal physiologic response, and not all the effects of adipose inflammation are metabolically adverse. Thus, inflammation is important for healthy adipose tissue remodeling and expansion. For example, factors released from macrophages can promote angiogenesis, which is necessary for tissue expansion (Corvera and Gealekman, 2014; Sun et al., 2012; Sung et al., 2013). Indeed, adipocyte-specific overexpression of an anti-inflammatory constitutive active form of inhibitor of κB (IκB) limits adipose tissue expansion, leading to ectopic lipid accumulation and glucose intolerance. Most likely, the acute phases of inflammation can have beneficial effects on tissue dynamics, whereas it is the chronic, subacute inflammatory state —as seen in established obesity—that has adverse metabolic effects (Kusminski et al., 2016).
Metabolically Normal and Abnormal Obesity.
Expansion of adipose tissue in times of caloric excess can serve a useful purpose by storing excess calories as triglycerides. From an evolutionary point of view, this plasticity of adipose tissue would allow early humans to tap into an endogenous source of energy during times of food deprivation. In Western cultures, this normal process has become maladaptive. Interestingly, a minority of overweight individuals do not manifest glucose intolerance and insulin resistance, and they are termed “metabolically normal obese” (MNO) (Appleton et al., 2013; Calori et al., 2011; Pajunen et al., 2011; Shea et al., 2011; van Vliet-Ostaptchouk et al., 2014). MNO subjects have less hepatic steatosis compared with metabolically abnormal obese (MAO) individuals (Fabbrini et al., 2013) and have less adipose tissue inflammation and less visceral adiposity than MAO subjects (Bigornia et al., 2012; Fabbrini et al., 2013, 2015). Long-term follow-up studies are scarce (Appleton et al., 2013; Bell et al., 2015), but it is possible that these MNO patients will eventually develop cardiovascular diseases (CVD) and metabolic abnormalities if the obesity persists over many years. Nevertheless, it would be of great interest to better understand the mechanistic determinants between metabolically normal and abnormal obese individuals.
This same phenomenon has been clearly shown in mouse models where obesity can be decoupled from adverse metabolic effects. For example, adiponectin overexpression in ob/ob mice exaggerates increased adipose tissue mass and body weight compared with ob/ob control mice. Nevertheless, these extremely obese mice are protected from glucose intolerance and adipose tissue inflammation (Kim et al., 2007). In a similar vein, NCoR is a PPARγ corepressor, and adipocyte-specific NCoR deletion leads to a constitutively active PPARγ state (Li et al., 2011). Although these mice develop increased adiposity, they do not develop glucose intolerance and are protected from HFD-induced insulin resistance (Li et al., 2011).
The immune cell response which accompanies caloric excess and the initial stages of obesity can also serve a beneficial adaptive role. For example, healthy adipose tissue expansion relies on angiogenesis to create adequate blood supply for the enlarging and proliferating adipocytes (Corvera and Gealekman, 2014; Sun et al., 2012; Sung et al., 2013). Macrophages, as well as other immune cells that populate obese adipose tissue, can be a source of angiogenic factors (Corvera and Gealekman, 2014). In this way, certain populations of immune cells or polarized macrophages can support beneficial adipose tissue expansion and remodeling (Kusminski et al., 2016; Wernstedt Asterholm et al., 2014). The extracellular matrix (ECM) is a key physical characteristic of normal and expanding adipose tissue, and ECM abnormalities, such as collagen deposition and fibrosis, are characteristic features of obese adipose tissue (Kusminski et al., 2016). Indeed, Khan et al. have already demonstrated that deletion of collagen VI, along with secretion of specific metalloproteases (MMPs), prevents formation of a restrictive fibrous collagen matrix, allowing physical space for healthy expansion of adipose tissue in times of caloric excess (Khan et al., 2009).
Liver
Kupffer Cells and Recruited Macrophages.
There are two major forms of macrophages in the liver: Kupffer cells (KCs) and recruited hepatic macrophages (RHMs) (Morinaga et al., 2015). KCs are resident macrophages that are important in hepatic inflammatory states and can account for up to 10% of all hepatic cells and 31% of the sinusoidal cells in normal liver (Bouwens et al., 1986). These cells are derived from the yolk sac and appear in the fetal liver during embryogenesis, where they ultimately proliferate and differentiate into recognizable KCs (Gomez Perdiguero et al., 2015; Naito et al., 1989). These cells retain the capacity to self-renew and might also be derived from local hepatic progenitor cells (Gomez Perdiguero et al., 2015; Schulz et al., 2012). In obesity, while absolute KC numbers remain relatively unchanged, these cells assume differential polarization states and can exist in a relatively proinflammatory M1-like state, in an anti-inflammatory M2-like state, or on the intervening spectrum (Wan et al., 2014). Therefore, the contribution of KCs to hepatic inflammation in obesity depends on the relative polarization state of the majority of these cells. In general, with obesity and steatosis, KCs secrete increased amounts of transforming growth factor (TGF) β, as well as other proinflammatory cytokines, and the overall tone of the entire KC population trends to the more proinflammatory end of the spectrum (Morinaga et al., 2015; Obstfeld et al., 2010).
RHMs are the major macrophage cell type that accumulates in the liver in obesity (Morinaga et al., 2015; Obstfeld et al., 2010). These cells are highly proinflammatory and characteristically express markers related to an M1-like phenotype and secrete TGFβ, as well as a variety of other cytokines, such as TNF-α, IL-1β, and IL-6 (Morinaga et al., 2015). During developing obesity, circulating monocytes are recruited to the liver through the actions of CCL2 (MCP1) and other standard chemokines, which are released from steatotic hepatocytes and KCs. In addition, RHMs appear to release their own set of chemokines, generating a feed-forward mechanism for ongoing replenishment of RHMs from circulating monocytes (Morinaga et al., 2015). The CCL2/CCR2 axis accounts for ~70% of the migration of monocytes into the liver (Morinaga et al., 2015). Co-culture studies indicate that conditioned medium derived from these proinflammatory RHMs can directly cause insulin resistance in primary hepatocytes. Thus, current evidence indicates that increased proinflammatory macrophage accumulation in the obese liver is mainly driven by a several-fold increase in RHM content.
Neutrophils.
Neutrophils are another proinflammatory cell type that can accumulate in the liver during the process of obesity (Talukdar et al., 2012). Neutrophils release a similar set of cytokines and chemokines as macrophages and contribute to the overall inflammatory process (Tsuda et al., 2004). In addition, neutrophils secrete NE, which exerts paracrine proinflammatory effects (Pham, 2006). NE can also be taken up by hepatocytes, where it leads to the degradation of IRS-2, promoting decreased insulin sensitivity (Talukdar et al., 2012). NE knockout (KO) mice show increased insulin sensitivity and decreased markers of inflammation in both the liver and adipose tissue (Talukdar et al., 2012).
Pathological Effects of Liver Inflammation.
With respect to the generation of hepatocyte insulin resistance, factors secreted from immune cells such as cytokines, Galectin-3, and eicosanoids (LTB4) can directly decrease hepatocyte insulin signaling (Hotamisligil et al., 1993; Li et al., 2015, 2016). In addition, the intrahepatocyte proinflammatory program also promotes insulin resistance as demonstrated by the fact that hepatocyte-specific deletion of IKKβ or JNK1 improves hepatic insulin sensitivity (Cai et al., 2005; Sabio et al., 2009).
In obesity, hepatic steatosis is nearly a uniform feature, and this is due to increased influx of adipocyte-derived FAs and dietary lipids (chylomicrons), as well as activation of lipogenesis and inhibition of fat oxidation pathways (Donnelly et al., 2005; Meex and Watt, 2017). Inflammatory cytokines, such as TNF-α, derived from immune cells in the liver can potentiate steatosis by enhancing hepatocyte lipogenic activity and increasing the enzymes involved in ceramide biosynthesis (Chavez and Summers, 2012). In turn, elevated intrahepatocyte ceramide levels inhibit Akt signaling, providing yet another mechanism for hepatocyte insulin resistance (Holland et al., 2011). Overnutrition can also induce hepatocyte endoplasmic reticulum (ER) stress, which further stimulates lipogenesis and activates hepatocyte proinflammatory signaling (Hotamisligil and Davis, 2016).
Hepatic steatosis can result in nonalcoholic fatty liver disease (NAFLD) and ultimately progress to nonalcoholic steatohepatitis (NASH) and fibrosis. The events underlying these transitions are of great interest, since NASH-related cirrhosis is a major metabolic disease state. Hepatic insulin resistance, steatosis, NASH, and fibrosis are highly interconnected and involve an extensive system of intraorgan crosstalk between hepatocytes, the various immune cell populations, and ultimately, stellate cells, which are responsible for fibrosis development (Koyama and Brenner, 2017). For example, once the intrahepatocyte inflammatory program is activated, these cells can secrete chemokines and cytokines, which activate resident KCs and cause migration of monocytes and neutrophils derived from the circulation (Koyama and Brenner, 2017; Son et al., 2007). These incoming immune cells are highly proinflammatory and secrete their own set of cytokines and chemokines, causing chemotaxis and accumulation of additional monocytes and neutrophils, setting up a feed-forward pathway. The cytokines secreted by KCs, RHMs, and neutrophils, particularly profibrogenic TGFβ1, can also activate quiescent stellate cells (Hellerbrand et al., 1999; Koyama and Brenner, 2017). Activated stellate cells are the main source of collagen secretion leading to hepatic fibrosis (Brenner et al., 2012). In terms of pathogenetic events, it is quite clear that there are multiple overlapping crosstalk mechanisms between liver cell types. For example, cytokines released from immune cells can feedback on hepatocytes to further stimulate lipogenesis with increased production of ceramides (Holland et al., 2011). Activated stellate cells can also elaborate cytokines and chemokines, which exacerbate steatosis and macrophage accumulation (Weiskirchen and Tacke, 2014). Thus, as in adipose tissue, the inflammatory response in the liver provides an intraorgan communication system where the immune cells promote hepatocyte insulin resistance and pathological sequelae.
Muscle
Since SM accounts for the great majority (70%–80%) of insulin-stimulated glucose disposal, SM insulin resistance plays a key role in the metabolic dysfunction of T2DM. There are several proposed mechanisms of SM insulin resistance that are unrelated to inflammation, such as ectopic fat distribution, mitochondrial dysfunction, and intrinsic defects in insulin action. This subject has been extensively reviewed elsewhere (Di Meo et al., 2017; Samuel and Shulman, 2016). In this section, we will focus on the possible role of SM inflammation as an etiologic factor in causing decreased muscle insulin sensitivity.
Numerous studies have demonstrated an increase in inflammatory markers in SM in obesity and diabetes (Fink et al., 2013; 2014; Hong et al., 2009; Patsouris et al., 2014). This includes activation of proinflammatory pathways within muscle cells themselves (Ciaraldi et al., 2016; Reyna et al., 2008; Varma et al., 2009). Since obesity is associated with large increases in proinflammatory immune cell accumulation in adipose tissue, circulating cytokines derived from adipose tissue could stimulate induction of SM proinflammatory pathways. In addition, there is an emerging literature that indicates that the same events that occur in visceral adipose tissue (VAT) might also occur in SM (Fink et al., 2013, 2014; Patsouris et al., 2014; Varma et al., 2009; Wu and Ballantyne, 2017). In the development of obesity, intermyocellular adipose tissue (IMAT) or perimuscular adipose tissue (PMAT) expands (Khan et al., 2015). This is the same process as “marbling” in beef products. Both IMAT and PMAT behave as adipose tissue depots, and during the process of obesity, these depots accumulate proinflammatory macrophages, as well as Th1 cells (Khan et al., 2015). In this way, the same events that occur in VAT can also occur in IMAT and PMAT. The macrophages and other immune cells in these SM fat depots secrete the usual set of cytokines, such as TNF-α, IL-1β, MCP1, etc. (Khan et al., 2015). This raises the possibility that local cytokines derived from IMAT and PMAT could directly cause decreased insulin sensitivity in SM. In addition, circulating cytokines and other factors that exist in the inflamed obese state could have the same or additive effects. As just one example, TNF-α or other cytokines can cause insulin resistance in myocytes (Austin et al., 2008), and the source of these cytokines could be IMAT, PMAT, or non-SM-associated adipose tissue depots (Khan et al., 2015). In obesity, myocytes can directly secrete chemokines, such as MCP-1 (Ciaraldi et al., 2016; Patsouris et al., 2014; Varma et al., 2009), but these chemokines can also be elaborated by IMAT and PMAT (Khan et al., 2015), mimicking the same situation that occurs in VAT in obesity. In this manner, an intraorgan crosstalk system can be established, comparable to what exists in adipose tissue and liver, in which factors secreted by local immune cells negatively affect insulin signaling in SM cells. Of course, the exact role of inflammation in the insulin resistance of SM remains to be quantitated, as there are other established non-inflammation-related mechanisms that can impair SM insulin sensitivity.
It is well known that a modest amount of weight reduction achieved by dietary means or bariatric procedures can dramatically improve SM insulin resistance. For example, Magkos et al. have shown that obese subjects who lose 5% of body weight exhibit marked increases in insulin sensitivity despite the fact that they remained obese—just less so (Magkos et al., 2016). The mechanisms underlying these clinical observations remain unsolved, but it is interesting to speculate that IMAT and PMAT may be uniquely sensitive to negative caloric balance. Thus, modest weight reduction might have a disproportionate beneficial effect on IMAT and PMAT.
Pancreatic Islets
Islet Macrophages and Inflammation.
Inflammation in pancreatic islets also contributes to β-cell dysfunction. Indeed, numerous studies have demonstrated that macrophage infiltration is increased in T2DM islets and that islet macrophage content generally correlates with the degree of β-cell dysfunction (Ehses et al., 2007; Kamata et al., 2014; Richardson et al., 2009; Zhao et al., 2003). Flow cytometry analyses of macrophages in isolated primary mouse islets revealed that obesity increases the number of Ly6c+ monocytes/macrophages and that proinflammatory cytokine expression is increased in islet macrophages from obese T2DM subjects and obese mice (Eguchi et al., 2012). Interestingly, incubation of primary islets or β cell lines with proinflammatory cytokines can cause increased β-cell apoptosis and decreased glucose-stimulated insulin secretion (GSIS) (Eguchi et al., 2012; Maedler et al., 2002). Moreover, co-culture of macrophages with β cells exacerbates the lipotoxic effects of chronic palmitate treatment, including decreased GSIS and decreased expression of the genes involved in β-cell differentiation and function (Eguchi et al., 2012). These effects are ameliorated by addition of neutralizing antibodies against the proinflammatory cytokines TNF-α and IL-1β in the culture media (Eguchi et al., 2012). Consistent with this, depletion of tissue macrophages by injection of clodronate liposomes improves glucose tolerance with increased GSIS in isolated islets in mouse models (Eguchi et al., 2012; Westwell-Roper et al., 2016). Together, these results suggest that macrophage-mediated inflammation participates in β-cell dysfunction by local release of cytokines in obese or T2DM states.
Lineage tracing studies indicate two different developmental origins for islet macrophages: yolk-sac-derived resident macrophages and bone-marrow-derived recruited macrophages (Schulz et al., 2012). In normal mouse islets, the yolk-sac-derived resident cells account for most of the islet macrophages (Schulz et al., 2012), while in obese mouse islets, macrophage content increases, and the majority of these cells express the blood monocyte marker Ly6C (CD11b+Ly6C+ population) (Eguchi et al., 2012), suggesting that the obesity-induced increased islet macrophage content involves recruitment of bone-marrow-derived blood monocytes into the islets. Consistent with this idea, type 2 diabetic islets express higher levels of chemokines compared with controls (Igoillo-Esteve et al., 2010). Moreover, primary human and mouse islets cultured in high-glucose and high-palmitate conditions produce increased amounts of chemokines, and conditioned media from these “metabolically stressed” islets stimulates monocyte chemotaxis (Eguchi et al., 2012; Igoillo-Esteve et al., 2010). These findings indicate that the metabolic stress induced by hyperglycemia and high FFA levels can trigger increased cytokine and chemokine production from β cells, causing increased blood monocyte recruitment with accumulation of M1-like islet macrophages.
Intracellular Mechanisms for Inflammation-Induced β-Cell Dysfunction.
The JNK and NF-κB signaling pathways operate in β cells to negatively regulate insulin secretion. For example, cytokine-induced NF-κB activation in β cells decreases expression of genes involved in β-cell differentiation and insulin secretion (Ardestani et al., 2011; Cardozo et al., 2001). Cytokine-induced JNK activation has also been implicated in decreased GSIS by suppressing the IRS/PI3K/Akt signaling pathway (Bouzakri et al., 2008; Kaneto et al., 2002; Kawamori et al., 2006; Martinez et al., 2008; Solinas et al., 2006).
β-cell mass may also be modulated by inflammatory pathways. For example, in vitro treatment with IL-1β or TNF-α induces β-cell apoptosis through activation of JNK and NF-κB pathways, while co-treatment with JNK or NF-κB specific inhibitors abolishes IL-1β-induced β-cell apoptosis (Bonny et al., 2001).
IL-1β.
One of the best-studied mechanisms by which islet macrophages can cause β-cell dysfunction is through IL-1β production. β cells express the IL-1 receptor and IL-1β expression is increased in islets in response to hyperglycemia and increased FFA levels (Maedler et al., 2002). While both β cells and macrophages can produce IL-1β, macrophages appear to be the major source of islet IL-1β production, along with many other pro-inflammatory cytokines, in obesity (Eguchi et al., 2012). Incubation of primary human or mouse islets, or β cell lines, with IL-1β decreases GSIS and increases apoptosis, while IL-1β neutralizing antibody protects against palmitate-induced β-cell dysfunction (Eguchi et al., 2012; Maedler et al., 2002).
Potential Beneficial Effects of Islet Macrophages.
Recent rodent studies have shown that islet resident macrophages are necessary for β-cell proliferation and maintenance of β-cell mass. Moreover, the acute post-prandial rise in IL-1β production can potentiate GSIS and may be necessary for normal glycemic control (Dror et al., 2017). Furthermore, other ligand/receptor systems previously implicated in immune cell chemotaxis such as fractalkine/CX3CR1 and SDF1/CXCR4, can increase GSIS and/or β-cell proliferation (Kayali et al., 2012; Lee et al., 2013; Yano et al., 2007). This raises the question of whether islet macrophages and inflammatory cytokines play a role in compensatory β-cell proliferation and/or insulin secretion in response to insulin resistance. In rodent models and obese T2DM subjects, β-cell proliferation may give rise to β cells with reduced expression of β-cell differentiation genes and decreased insulin secretory function (Accili et al., 2016; Butler et al., 2016; Cinti et al., 2016; Jonas et al., 1999; Rahier et al., 2008). Indeed, it is suggested that the cytokine-induced decrease in β-cell-specific gene expression can also lead to dedifferentiation into insulin-negative endocrine cells (Nordmann et al., 2017).
Central Nervous System
CNS Inflammation in Obesity.
Recent evidence indicates that the brain is neither isolated nor passive in its interactions with the immune system (Carson et al., 2006). It harbors a classical lymphatic system and actively interacts with the peripheral immune system through lymphatic vessels, the blood brain barrier (BBB), and choroid plexus (Guillemot-Legris and Muccioli, 2017). In normal healthy brain, the BBB limits entry of peripheral immune cells, and immune surveillance relies mainly on microglial cells. In obesity, increased expression of proinflammatory cytokines, along with increased accumulation of microglia (brain resident macrophages; microgliosis) and/or astrocytes (astrocytosis), occurs in different areas of the brain, including the cerebral cortex, cerebellum, hippocampus, amygdala, and hypothalamus (Guillemot-Legris and Muccioli, 2017). Obesity-associated brain inflammation has been implicated in decreased recognition, spatial memory, anxiety, and depression, as well as dysregulation of energy homeostasis (Guillemot-Legris and Muccioli, 2017).
Inflammation Affects Central Leptin and Insulin Resistance in Obesity.
The hypothalamus is key to regulation of food intake, energy expenditure, and the central regulation of glucose metabolism (Jais and Brüning, 2017). In obesity, it has been suggested that impaired hypothalamic sensing of leptin and/or insulin leads to dysregulation of food intake, energy expenditure, and glucose metabolism. It is possible that hypothalamic inflammation can contribute to insulin and leptin resistance (Jais and Brüning, 2017; Thaler and Schwartz, 2010). MRI and histologic analyses of the basomedial hypothalamus in humans shows that increased hypothalamic glial cell accumulation (gliosis) is associated with obesity and insulin resistance (Baufeld et al., 2016; Puig et al., 2015; Schur et al., 2015; Thaler et al., 2012; Cazettes et al., 2011). These results have been confirmed in diet-induced rodent models, along with activation of IKKβ/NF-κB and JNK and increased inflammatory cytokine expression. Inhibition of hypothalamic inflammation using intracerebro-ventricular (ICV) injection of TNF-α or TLR4-neutralizing antibodies can mitigate obesity-induced leptin resistance, whereas ICV injection of low-dose TNF-α induces hypothalamic leptin and insulin resistance (Mighiu et al., 2012; Romanatto et al., 2007). Taken together, these results suggest a role for CNS inflammation—at least in the hypothalamus—as a mediator of central leptin resistance, promoting weight gain.
Gastrointestinal Tract
Obesity Leads to Altered GI Microbiota Composition.
In addition to host-derived inflammatory signals that influence metabolism, the commensal microbiota of the GI tract also produce metabolites and byproducts that modulate inflammation and metabolism. In both rodents and humans, obesity is associated with dramatic changes in the composition of the GI microbiota, a condition termed dysbiosis (Ley et al., 2005; Turnbaugh et al., 2008; Turnbaugh et al., 2009; Turnbaugh et al., 2006). Interestingly, gnotobiotic (germ-free) mice display improved glucose tolerance and are protected against diet-induced adipose tissue inflammation and obesity unless colonized with bacteria from conventional animals (Bäckhed et al., 2007; Caesar et al., 2015). This provides evidence that microbiota play a role in the development of obesity, tissue inflammation, and metabolic dysfunction. In addition, treatment of obese rodents with broad-spectrum antibiotics leads to reduced inflammation, with improved glucose tolerance and insulin sensitivity (Cani et al., 2008; Hwang et al., 2015; Membrez et al., 2008). Fecal transplantation experiments in rodents and humans have demonstrated that microbiota can alter metabolic status and that this is transmissible. Fecal transplantation of gnotobiotic mice with fecal preparations from human twins that are discordant for obesity showed that mice receiving feces from the obese twin, but not the lean twin, developed increased weight gain and reduced insulin sensitivity (Ridaura et al., 2013). In addition, fecal transplants from obese humans into lean recipients leads to a decrease in systemic insulin sensitivity (Vrieze et al., 2012), although these effects were small, and further studies are needed. In either mice or humans, fecal transplants do not fully rescue the metabolic defects induced by obesity. This could indicate that the introduction of a healthy GI microbiota alone cannot restore host-derived signals from other tissues that govern metabolic status.
Microbiota-Produced Metabolites Influence Host Inflammation and Metabolism.
Numerous bioactive metabolites are produced by GI microbiota, of which short-chain fatty acids (SCFAs) are among the most abundant in the systemic circulation (Stevens and Hume, 1998). The SCFAs acetate, butyrate, and proprionate are produced through fermentation of dietary fiber and can function as signaling molecules, activating G-protein-coupled receptors (GPCRs), including GPR41 and GPR43, to modulate adiposity, inflammation, and insulin sensitivity (Canfora et al., 2015). Treatment of obese mice or humans with SCFAs or GPR41/43 agonists leads to reduced weight gain and adipose mass, as well as increased insulin secretion (Chambers et al., 2015; Gao et al., 2009; Lin et al., 2012; McNelis et al., 2015). However, the potential role of SCFAs in systemic inflammation is unclear, since reports have shown both anti-inflammatory and proinflammatory effects, depending on the cell types and conditions studied (Corrêa-Oliveira et al., 2016).
Gut microbes are also responsible for production of secondary and tertiary bile acids, which are efficiently reabsorbed by the host to enter the systemic circulation (Hylemon et al., 2009). In addition to their role in dietary fat absorption, bile acids activate several GPCRs and nuclear hormone receptors, including the bile acid receptor 1 (TGR5) and farnesoid X receptor (FXR), to modulate inflammation and metabolism (Pols et al., 2011; Wollam and Antebi, 2011). The composition of the bile acid pool is altered in obese and insulin-resistant rodents and humans (Brufau et al., 2010; Haeusler et al., 2013; Ridaura et al., 2013; Suhre et al., 2010; Yoshimoto et al., 2013), leading to changes in these metabolic signaling pathways. Indeed, treatment with bile acids or FXR agonists improves insulin sensitivity in mouse models of obesity (Cariou et al., 2006; Cipriani et al., 2010; Ma et al., 2006; Ma et al., 2013; Zhang et al., 2006). Bile acid activation of TGR5, a GPCR highly expressed in the intestine, also improves intestinal inflammation and glucose tolerance in HFD mice (Pols et al., 2011). However, bile acid metabolism is much different in mice compared to humans, making additional human studies necessary.
Obesity Leads to Increased GI Permeability and Endotoxemia.
Obesity-associated changes in circulating bacterially produced metabolites and byproducts can be due to increased intestinal permeability, as well as gut microbiota composition (Cani et al., 2007, 2008; de La Serre et al., 2010). This increased porosity is associated with alterations in tight junctions between cells, reduced thickness of the mucosal layer, and increased epithelial-cell uptake and translocation. This greater access allows increased levels of bacteria and inflammatory microbial byproducts to enter the circulation. Microbes and their products contain pathogen-associated molecular patterns (PAMPs), including the gram-negative bacterial cell wall component lipopolysaccharide (LPS), that are recognized by the host innate immune system through cognate pattern recognition receptors (Kumar et al., 2011). LPS activates toll-like receptor 4 (TLR4) to induce proinflammatory responses in immune cells, as well as insulin target cells (Saberi et al., 2009; Shi et al., 2006). In addition, LPS can alter the expression of tight junction proteins to increase intestinal permeability (Guo et al., 2013). HFD can induce TLR4-dependent translocation of gram-negative bacteria across the intestinal barrier, which might also contribute to decreased insulin sensitivity (Amar et al., 2011; Neal et al., 2006). Interestingly, even short-term changes in diet may lead to rapid alterations in gut permeability, as a single high-fat diet meal is associated with an acute rise in circulating LPS levels in humans (Erridge et al., 2007).
Modulation of the Host Immune System by GI Microbiota.
The host immune system itself is influenced by commensal microbiota and vice versa. Evidence of this can be found in germ-free mice, which exhibit numerous immune deficiencies both within and outside of the GI tract, including altered T cell populations (Kamada et al., 2013). Obese HFD mice display altered immune cell populations in the intestinal lamina propria, including a reduced proportion of Tregs, as well as elevated macrophage and reduced eosinophil levels—all of which may play a role in the increased intestinal inflammation observed in these animals (Johnson et al., 2015). Selective loss of Tregs leads to GI inflammation and altered gut microbiota composition (Josefowicz et al., 2012). Tregs produce interleukin 10 (IL-10), an anti-inflammatory cytokine that contributes to maintenance of immune homeostasis (Tanoue and Honda, 2012).
Intraorgan Crosstalk
In the initiation of obesity-induced inflammation, there are specific triggers that induce parenchymal cells, such as adipocytes, hepatocytes, and myocytes, to activate their intracellular proinflammatory programs with subsequent release of chemokines, such as MCP-1, LTB4, MIP-1α, and others. Since chemoattraction requires a concentration gradient, circulating chemokines are unlikely to be effective at driving tissue macrophage accumulation. Rather, chemotaxis requires a tissue source of chemokines, and this is typically derived from the parenchymal cells, such as adipocytes, myocytes, hepatocytes, or β cells. This occurs as a result of activation of parenchymal-cell proinflammatory pathways by various external triggering mechanisms, as depicted in Figure 2. These released chemokines then attract the initial wave of macrophages and other immune cells into the underlying tissue (Figure 2). These immune cells become activated and produce their own chemokines, which sets up a self-sustaining feed-forward process. Through the release of cytokines and other factors, these immune cells then influence the metabolic status of the tissue parenchymal cells to induce decreased insulin signaling, as well as other metabolic abnormalities.
Figure 2. Mechanisms of Intraorgan Cross-talk.
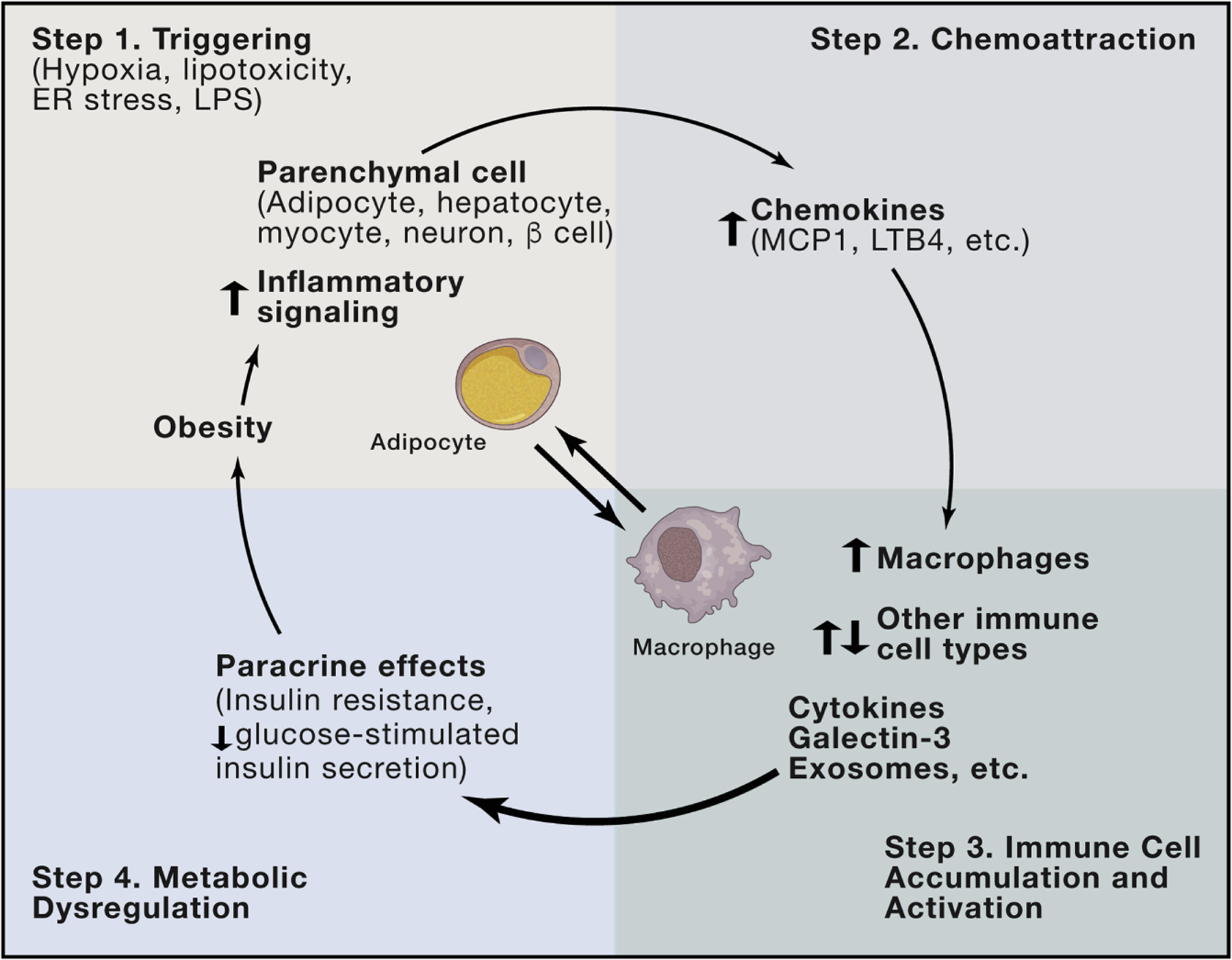
Obesity leads to various triggering events, such as ER stress, hypoxia, and lipotoxicity, which can initiate activation of proinflammatory pathways within tissue parenchymal cells (Step 1). As part of the activation mechanism in these parenchymal cells, they secrete a variety of chemokines (Step 2), which lead to chemotaxis and migration of macrophages, as well as other immune cell types, into the underlying tissue (Step 3). Overall, these immune cells take on a proinflammatory phenotype and secrete a number of factors (cytokines, galectin-3, miRNA-containing exosomes, etc.), which exert local paracrine effects to cause insulin resistance in adipocytes, hepatocytes, and myocytes, or decreased GSIS in β cells (Step 4).
In adipose tissue, there are a variety of immune cell types that accumulate in obesity, including macrophages, various T cell subsets, and B2 cells. All participate in an intricate immune cell communication network. With respect to insulin resistance, the major event is the accumulation and activation of proinflammatory macrophages, which release cytokines, galectin-3, leukotrienes, and other factors causing insulin resistance. The network of T and B cells has important effects to influence macrophage recruitment and activation, and macrophages are the most abundant immune cell type in these tissues. In this way, the proinflammatory, M1-like tissue macrophages are the final effector cells responsible for the adverse metabolic effects. In other words, while T and B cells have important effects to regulate macrophages, the macrophages are the major source of the factors that induce insulin resistance (Lee et al., 2011b; Subramanian et al., 2015). This central theme, depicted for adipose tissue in Figure 2, represents an important etiologic commonality across the other metabolic tissues.
The triggers that activate proinflammatory pathways and chemokine release within parenchymal cells are multifaceted and may differ across organs. For example, in adipose tissue, relative or absolute adipocyte hypoxia is an important trigger to increase HIF-1α and its downstream inflammatory program. Enlarging adipocytes due to triglyceride accumulation, ER stress, and perhaps signals from gastrointestinal factors that enter the circulation may all serve as co-contributors at different steps of this process. In the liver, hepatocytes can be initially activated by lipotoxicity, ER stress, or gut related factors, whereas hypoxia is probably not a major component. Some of these triggers directed towards the hepatocyte can induce apoptosis and/or necrosis. In this way, the hepatocyte locally releases chemokines that induce migration of monocytes to form RHMs and can also activate the resident KC macrophage population. In the muscle, a similar situation exists in which obesity-related triggers activate inflammatory programs within myocytes, while the majority of the immune cells in muscle accumulate in IMAT and PMAT. In these small localized intermyocyte adipose depots, the inflammatory process is probably quite similar to that which occurs in the more traditional adipose depots in obesity. Similar events have been reported in islets, where activation of β-cell proinflammatory pathways leads to chemokine release with immune cell recruitment and activation, contributing to β-cell dysfunction. Detailed studies of the liver, muscle, islets, and particularly adipose tissue have been conducted, and all point to this general theme. Oftentimes, these tissues are treated as isolated organs with respect to the inflammatory process, but stepping back to the more organismal level, these overall common themes are apparent.
Interorgan Crosstalk and Communication
There is an extensive interogan communication network bridging the classic insulin target tissues (adipose tissue, muscle, and liver) as well as the CNS, GI tract, and pancreatic islets (Figure 3). For example, there are several reports of adipocyte-specific KO models (Lee et al., 2014; Li et al., 2011; Mayoral et al., 2015; Qi et al., 2009), which lead to improved insulin resistance in liver and muscle. This high degree of interconnectedness makes it difficult to experimentally isolate initiating versus secondary events because of the interorgan feedback pathways that exist. The temporal sequence of the events that give rise to inflammation and insulin resistance are most likely on the order of minutes to hours rather than days to weeks, making it experimentally challenging to tease out a precise sequence from in vivo studies. Nevertheless, interorgan crosstalk is essential to the development of systemic metabolic abnormalities in obesity and diabetes.
Figure 3. Inflammation-Mediated Interorgan Crosstalk.
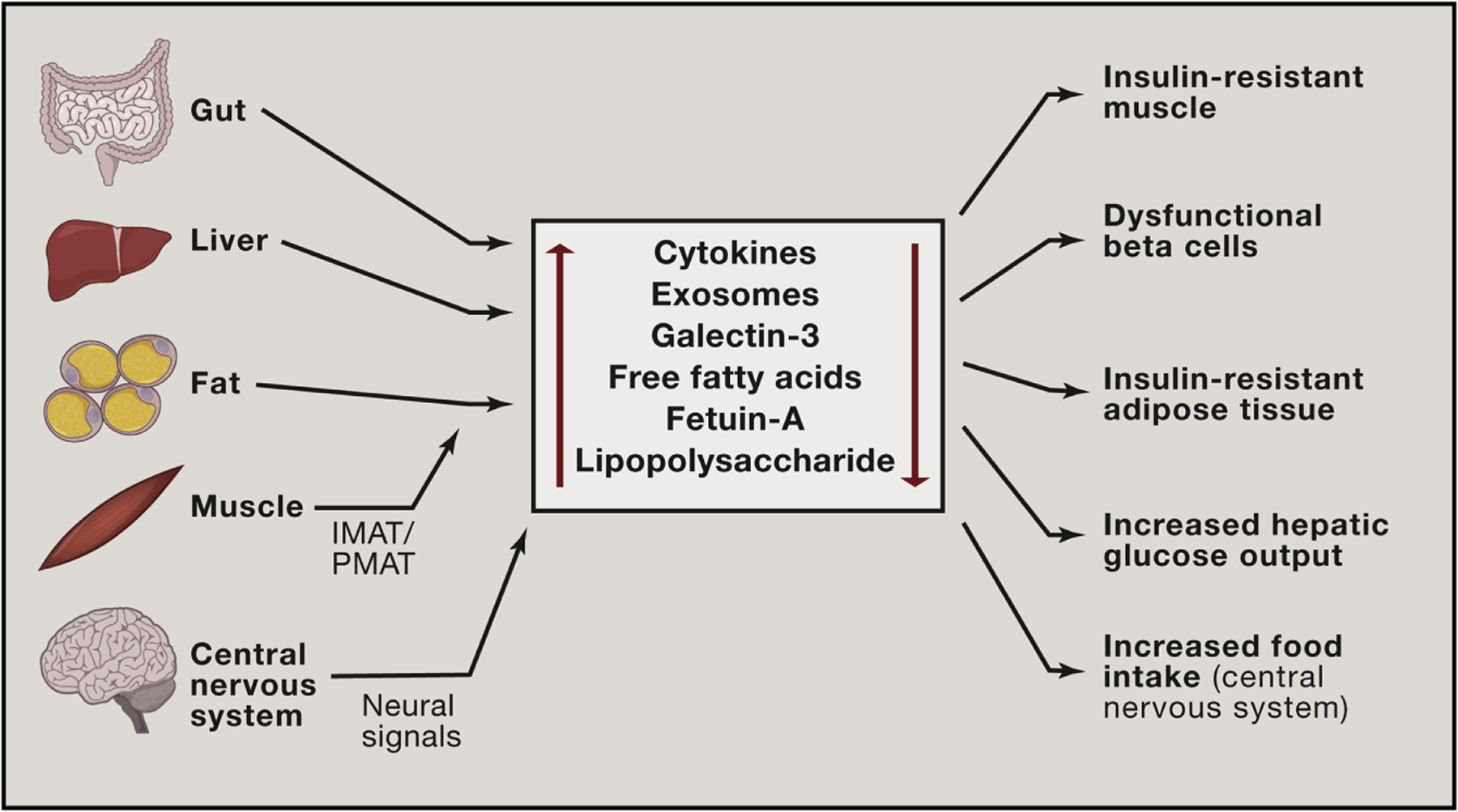
Obesity leads to chronic inflammation in metabolic tissues (liver, adipose tissue, and muscle). It also causes dysbiosis in the GI tract and gliosis in the CNS. Once these events are established, these tissues then elaborate a large number of secretory factors which not only act locally (paracrine effects), but can also enter the circulation to cause distal effects on insulin sensitivity, GSIS, and food intake. These factors, exemplified by cytokines, miRNA-containing exosomes, galectin-3, FFAs, fetuin A, and LPS do not act in isolation but can work in an additive or coordinated fashion on tissue sites of action. Each factor has the potential to influence production and secretion of some of the other factors, adding to the complexity of the integrated network comprising metabolic interorgan crosstalk.
As a general principle, interorgan communication is mediated by factors released from one tissue that travel through the circulation to influence distant organs. Additional crosstalk mechanisms can also include neural signals from the CNS. There are many substances that appear in the circulation from metabolic tissues, including cytokines, lipid species, adipokines, immune-cell secreted factors, microRNA (miRNA)-containing exosomes, GI-derived factors, myokines, and hepatokines (Cani et al., 2007; Chavez and Summers, 2012; Deng and Scherer, 2010; Hotamisligil et al., 1993; Meex and Watt, 2017; Thomou et al., 2017; Wu and Ballantyne, 2017). Collectively, they comprise a complex multifactorial system that likely works in a coordinated, integrated manner to affect systemic metabolism in the liver, muscle, adipose tissue, and pancreatic islets.
Chronic obesity-induced inflammation is certainly not the only cause of the metabolic abnormalities in obesity, but it is an important contributor to insulin resistance. Other factors that can influence systemic insulin resistance, such as lipotoxicity, ectopic lipid distribution, and non-inflammation-induced defects in insulin action, are the subject of a number of other excellent reviews (Czech, 2017; Newgard, 2017; Samuel and Shulman, 2016), and the reader is referred to these publications for further discussion.
TNF-α, IL-1β, and IL-6 are the most extensively studied cytokines in the context of metabolism. For example, TNF-α can cause insulin resistance (Hotamisligil et al., 1993), and the mechanisms of in vitro TNF-α-mediated decreased insulin signaling are discussed above (Adipose Tissue) and reviewed in (Hotamisligil, 2017). Most of these actions occur locally via paracrine effects, but circulating TNF-α levels can be elevated in obesity. Whether these levels are sufficient to cause systemic insulin resistance, particularly in humans, is still an active question. In rodents, it is clear that neutralization of TNF-α with anti-TNF-α antibodies leads to marked improvement of glucose tolerance and insulin sensitivity (Hotamisligil et al., 1993). However, whether these systemic effects of TNF-α translate to humans has not yet been demonstrated. IL-1β is another inflammatory cytokine that enters the circulation from inflamed tissues in obesity. However, IL-1β appears to have minimal effects on insulin sensitivity and exerts its primary actions on glucose homeostasis by affecting β-cell GSIS (Cavelti-Weder et al., 2012; Rissanen et al., 2012; Sloan-Lancaster et al., 2013; van Asseldonk et al., 2011). Another released cytokine is IL-6. While IL-6 can have acute insulin-like effects (Carey et al., 2006; Ellingsgaard et al., 2011; Pedersen and Febbraio, 2007), these do not persist in the chronic situation. Chronically elevated levels of IL-6 can promote inflammation and decreased insulin sensitivity (Franckhauser et al., 2008).
Galectin-3 is a member of a family of lectin molecules that can arise from immune cells. Recent evidence shows that circulating galectin-3 levels are elevated in human and rodent obesity and are correlated to the magnitude of insulin resistance in man (Li et al., 2016). Circulating galectin-3 is largely derived from macrophages and can lead to adverse metabolic effects. For example, galectin-3 has proinflammatory actions by promoting macrophage chemotaxis and directly stimulating proinflammatory pathways (Li et al., 2016). More importantly, galectin-3 can directly cause decreased insulin signaling in adipocytes, hepatocytes, and myocytes (Li et al., 2016). Galectin-3 interacts with the insulin receptor at an allosteric site, which does not change insulin binding but does interfere with insulin receptor signaling, leading to decreased downstream insulin actions (Li et al., 2016). Through these mechanisms, circulating galectin-3 can participate in the interorgan crosstalk network that downregulates insulin sensitivity in obesity.
Adiponectin and leptin are two major adipokines that have important effects on energy and metabolic homeostasis. Among its many effects, adiponectin can lead to insulin sensitization, while leptin is an important control point for food intake and energy expenditure. Although these adipokines might modulate the response to inflammatory factors, or in turn, inflammation could influence the actions of adiponectin/leptin, these adipokines are not viewed as a primary component of the inflammatory response.
More recent findings have added a new dimension to how interorgan crosstalk may occur in the context of immunometabolism. Both adipocytes and ATMs can release miRNA-containing exosomal vesicles that can enter the circulation to exert systemic effects (Thomou et al., 2017). There is clear evidence that exosomes secreted from one cell type can contain miRNAs that are transferred to other cell types (Tkach and Théry, 2016). miRNAs cause translational arrest of target mRNAs, influencing cellular metabolism (Bartel, 2009; Dumortier et al., 2013). Indeed, liver miR-143 can produce hepatic insulin resistance by inhibiting one of its target genes ORP8 (Jordan et al., 2011). In addition, expression of miR-222 is increased in obesity, and this miRNA can blunt insulin signaling by inhibiting GLUT4 expression (Esteves et al., 2017). Thomou et al. have specifically studied adipocyte miRNAs and found that adipocytes are a rich source of circulating exosomes (Thomou et al., 2017). They found that adipocyte-derived exosomes contain several hundred miRNAs and, in particular, noted that exosomes derived from brown adipocytes express high levels of miR-99b. These exosomes can be taken up by the liver where miR-99b restrains the expression of hepatic FGF21. This results in a glucose-intolerant phenotype, as would be predicted from FGF21 deficiency. In a similar vein, Ying et al. examined the effects of miRNA-containing exosomes from ATMs that were prepared from either obese or lean mice (Ying et al., 2017a). They found that in vivo treatment of lean recipient mice for 2 weeks with obese ATM exosomes led to transfer of miRNAs into insulin target tissues, with deterioration of glucose tolerance and decreased insulin sensitivity in muscle, liver, and adipose tissue. In reverse experiments, obese insulin-resistant mice were used as recipients and treated with ATM exosomes derived from lean mice. This treatment led to near normalization of glucose tolerance and insulin sensitivity. Taken together, these studies indicated a mechanism for interorgan cross-talk in which exosomes secreted from one cell type (e.g., adipocytes or ATMs) transport miRNAs to other metabolic tissues, where they regulate glucose metabolism and insulin sensitivity either positively or negatively. This represents an additional communication system to regulate metabolic homeostasis.
There are a number of other substances that arise from muscle (myokines) or liver (hepatokines) that may have possible effects on glucose homeostasis (Meex and Watt, 2017; Pedersen and Febbraio, 2012; Rai and Demontis, 2016), although reports on these factors are limited and remain an area of future investigation. For example, over 500 proteins are released from healthy liver, and 168 of these contain potential N-terminal secretory sequences. 20% of these proteins display altered secretion from steatotic hepatocytes compared with normal hepatocytes. Among these hepatokines are fetuin-A, fetuin-B, FGF21, sex hormone-binding globulin, angiopoietin-like protein 4, adropin, and retinol-binding protein-4 (Meex and Watt, 2017). One of the more well-studied hepatokines, fetuin-A is a 64-kDa glycoprotein mainly produced from liver, and circulating fetuin-A levels are increased in insulin-resistant humans (Pal et al., 2012; Stefan et al., 2006). Fetuin-A can also function as an adaptor protein, mediating SFA interactions with TLR4 and is necessary for SFA-induced TLR4 activation and inflammation (Pal et al., 2012). Another circulating liver-derived factor that has attracted great attention is FGF21. Rodent studies have shown that FGF21 administration can improve hyperglycemia and hyperlipidemia by increasing glucose uptake and β-oxidation in fat tissue and liver (BonDurant et al., 2017; Inagaki et al., 2007; Kharitonenkov et al., 2005). However, recent clinical trials in humans with FGF21 analogs showed marginal effects to decreased hyperglycemia, although potent triglyceride-lowering effects were observed (Gaich et al., 2013; Talukdar et al., 2016). Myokines, such as myostatin, myonectin, irisin, and brain-derived neutrophic factor have also been implicated in obesity or obesity-associated pathology (Wu and Ballantyne, 2017).
Gut-derived factors can participate in the metabolic communication network between tissues. In obesity, the GI epithelial layer becomes more permeable, and various bacterial products (like LPS) and bacteria themselves can “leak” into the circulation (Amar et al., 2011; Cani et al., 2007, 2008). Gut-derived circulating LPS might be particularly relevant to inflammation and immunometabolism, as blood levels of LPS are chronically elevated in obese humans and mice.
Thus, whether considering small molecules, small proteins, or exosomes, it is important to recognize that crosstalk between organs is an essential component of the body’s response to increasing obesity. Therapeutic attempts to mitigate glucose intolerance and insulin resistance are of high importance, and efforts in this direction will need to consider the interorgan communication networks and the whole-body response to chronic inflammation.
Therapeutic Implications
The role of chronic tissue inflammation in insulin resistance and diabetes has been shown in rodent models through genetic gain- and loss-of-function studies and by treatment with pharmacologic anti-inflammatory agents. However, this concept remains to be validated in humans. Clearly, genetic manipulations in humans are not feasible, and therefore, one must rely on pharmacologic treatment studies to assess the potential impact of chronic inflammation on metabolic responses. At this point, there are no clear-cut large clinical studies with anti-inflammatory therapeutics that have demonstrated robust improvements in either insulin sensitivity or hyperglycemia.
Salicylate or other salicylic acid derivatives are probably the best studied in humans (Goldfine and Shoelson, 2017). A small clinical research study showed that high-dose salicylate (salsalate) treatment of T2DM patients led to glucose lowering and improved insulin sensitivity (Goldfine et al., 2008). This led to larger-scale, more formalized clinical trials, which have shown reductions in HbA1c of 0.3%–0.5% in T2DM (Goldfine et al., 2010). Although these modest effects are encouraging, it should be noted that the mechanism of action of salicylate may be related to activation of AMPK (Hawley et al., 2012), which can cause metabolic improvements rather than direct anti-inflammatory actions.
Treatment with anti-IL-1β or anti-TNF-α antibodies has also been studied in humans, but with mixed results. Different anti-IL-1β approaches have shown small decreases in HbA1c levels, but the anti-diabetic mechanism relates to improved β-cell function (Cavelti-Weder et al., 2012; Rissanen et al., 2012; Sloan-Lancaster et al., 2013; van Asseldonk et al., 2011). Most likely, this is because IL-1β is not an important component of inflammation-induced insulin resistance, whereas IL-1β effects in islets can give rise to β-cell dysfunction. The studies showing glycemic lowering with IL-1β antibodies are of interest, since a very recent study has examined the effects of anti-IL-1β treatment on atherosclerosis (Ridker et al., 2017). This trial demonstrated a significant reduction in cardiovascular event recurrence as a result of anti-IL-1β antibody (canakinumab) treatment, indicating the potential for anti-inflammatory therapy in CVD. Measures of glycemia were not reported in the CANTOS trial.
Administration of anti-TNF-α antibodies is highly effective in rodent models of diabetes (Hotamisligil et al., 1993), but clinical results have been less compelling (Bernstein et al., 2006; Dominguez et al., 2005; Kiortsis et al., 2005; Ofei et al., 1996). Although large-scale placebo-controlled studies have not been reported, these agents have been used to treat many patients with various inflammatory diseases, some of whom had T2DM or metabolic syndrome. A few of these studies have noted modest reductions in glycemia and a decrease in incidence of T2DM development (Gonzalez-Gay et al., 2006; Kiortsis et al., 2005). However, most of these studies have proven negative (Bernstein et al., 2006; Dominguez et al., 2005; Ofei et al., 1996; Paquot et al., 2000; Wascher et al., 2011). Inflammation-induced insulin resistance involves multiple mechanisms, and these results may indicate that TNF-α is not a key component of this system in man. Alternatively, since adipose tissue levels of TNF-α are quite high, it’s possible that current anti-TNF-α biologics do not achieve sufficient interstitial concentrations to inhibit this pathway. A formal, randomized placebo-controlled trial will be necessary to address these issues.
TZDs are PPARγ agonists and are a well-known and efficacious class of anti-diabetic drugs (Olefsky, 2000). TZDs have clear-cut anti-inflammatory effects that might play a role in their insulin-sensitizing anti-diabetic actions (Chawla et al., 2001; Han et al., 2000; Ricote et al., 1998). However, TZDs have multiple additional effects that modulate glycemia and insulin sensitivity, such as stimulation of FGF21, adiponectin release, induction of multiple PPARγ target genes (such as GLUT4), and redistribution of adipose tissue fat storage (Lackey and Olefsky, 2016). It is currently not possible to tease out a potential component of TZD-mediated anti-inflammatory effects, which may make a potential contribution to the insulin sensitizing, anti-diabetic effects of these agents in humans.
IKKε and TBK1 are both induced by NF-κB activation in liver and fat on HFD (Chiang et al., 2009). Amlexanox is a small-molecule inhibitor of these kinases and treatment of obese mice with this compound leads to better glucose tolerance and insulin sensitivity, as well as decreased inflammation and weight loss (Reilly et al., 2013). Recent studies in humans show that Amlexanox treatment can improve glycemia and insulin sensitivity in obese individuals (Oral et al., 2017). While various inflammatory markers are lowered by treatment of mice or humans with Amlexanox, these may not be direct effects. It is possible that the beneficial effects of this compound on inflammation may be secondary to anti-obesity effects.
One anti-inflammatory approach that has had widespread use in humans involves ingestion of anti-inflammatory ω3 FAs. While there are numerous studies on this subject, clear conclusions are not evident (Lalia and Lanza, 2016). Many studies have shown cardiovascular benefits of ω3 FA supplementation, and some have shown modest improvements in insulin sensitivity or glycemia. On the other hand, there are also clinical studies showing negligible or no effects.
Despite there being a number of other promising anti-inflammatory targets, which might lead to metabolic benefit, none have advanced to the clinic, so it is too early to make any predictions. However, the LTB4/BLT1 system, macrophage-secreted galectin-3, and miRNA-containing exosomes are all worthy of consideration for therapeutic approaches, particularly since these mechanisms can be involved in the interorgan crosstalk.
In the clinical setting, any anti-inflammatory approach might carry the potential for unwanted suppression of certain aspects of the immune system with safety consequences. This should be carefully evaluated in any developmental program involving an anti-inflammatory approach toward the treatment of metabolic disease. These considerations could play into strategies to identify targets or drugs that might have anti-inflammatory, insulin-sensitizing effects. For example, since proximal signals that activate intracellular proinflammatory pathways stimulate a number of interconnected and overlapping mechanisms, an intervention directed at a distal inflammatory event might not be adequately efficacious because of the presence of redundant proinflammatory signaling networks. On the other hand, it is likely that the risk of unwanted infections and other side effects could be greater with inhibition of the wider network of inflammatory pathway signaling. It might be more compelling to target a specific aspect of the overall inflammatory pathway that is key to the development of insulin resistance. Furthermore, if there was a method to provide therapeutic-tissue selectivity, this would serve to ameliorate insulin resistance with less risk of unwanted side effects.
Since obesity is an underlying core mechanism for the development of insulin resistance, chronic tissue inflammation, and type 2 diabetes, any treatment that results in sustained weight reduction will be of obvious therapeutic benefit. Indeed, as little as 5% body-weight loss leads to substantial improvements in insulin sensitivity (Magkos et al., 2016). Progressive weight loss studies show that 11% body-weight reduction provides a near maximal effect to improve insulin sensitivity and inflammation with little further effect upon additional weight loss (Magkos et al., 2016). These clinical studies indicate that even modest reductions in obesity can have large beneficial metabolic consequences.
Currently, there are several pharmacologic anti-obesity treatments, lifestyle regimens, and surgical approaches that are clinically efficacious. However, recidivism (weight regain) is an ongoing problem with lifestyle and pharmacologic interventions, although weight loss after bariatric surgery has proven more durable. In the context of weight reduction, hyperglycemia, insulin resistance, and tissue inflammation are all improved, but it is not possible to tease out the potential contribution of tissue inflammation to the overall metabolic improvements, since there are so many other factors that change as result of weight reduction.
Given the strong etiologic component of obesity and its attendant consequences of insulin resistance as a cause of the rising prevalence of type 2 diabetes, it seems logical that therapeutic approaches aimed at improving insulin sensitivity are likely to have important clinical benefit. Such therapies would fall into the category of “disease-modifying” treatments, which should be particularly beneficial. Most current therapeutic approaches are aimed at improving the insulin secretion arm of the equation, with a number of effective treatments available. Therefore, it would seem logical that redoubling our collective efforts to identify new insulin-sensitizing drugs could lead to major therapeutic advances.
Finally, we are in the age of large-scale “omics” data, which holds the promise of precision or personalized medicine in sub-categories of diabetic patients. Although this is still a theory and tangible results have not yet been realized, this remains an exciting area of future research that might intersect with the design of clinical trials. Certainly, one could imagine a future in which specific anti-inflammatory approaches would work better in one class of T2DM patients versus another.
ACKNOWLEDGMENTS
This study was funded by the US National Institute of Diabetes and Digestive and Kidney Diseases (DK074868, DK063491, and DK101395).
REFERENCES
- Accili D, Talchai SC, Kim-Muller JY, Cinti F, Ishida E, Ordelheide AM, Kuo T, Fan J, and Son J (2016). When β-cells fail: lessons from dedifferentiation. Diabetes Obes. Metab 18 (Suppl 1), 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S, et al. (2011). Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol. Med 3, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton SL, Seaborn CJ, Visvanathan R, Hill CL, Gill TK, Taylor AW, and Adams RJ; North West Adelaide Health Study Team (2013). Diabetes and cardiovascular disease outcomes in the metabolically healthy obese phenotype: a cohort study. Diabetes Care 36, 2388–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardestani A, Sauter NS, Paroni F, Dharmadhikari G, Cho JH, Lupi R, Marchetti P, Oberholzer J, Conte JK, and Maedler K (2011). Retraction. J. Biol. Chem 290, 27532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin RL, Rune A, Bouzakri K, Zierath JR, and Krook A (2008). siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes 57, 2066–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Manchester JK, Semenkovich CF, and Gordon JI (2007). Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 104, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C, et al. (2015). Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baufeld C, Osterloh A, Prokop S, Miller KR, and Heppner FL (2016). High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 132, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JA, Hamer M, Sabia S, Singh-Manoux A, Batty GD, and Kivimaki M (2015). The natural course of healthy obesity over 20 years. J. Am. Coll. Cardiol 65, 101–102. [DOI] [PubMed] [Google Scholar]
- Bernstein LE, Berry J, Kim S, Canavan B, and Grinspoon SK (2006). Effects of etanercept in patients with the metabolic syndrome. Arch. Intern. Med 166, 902–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigornia SJ, Farb MG, Mott MM, Hess DT, Carmine B, Fiscale A, Joseph L, Apovian CM, and Gokce N (2012). Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr. Diabetes 2, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR, Walsh SA, Ornitz DM, and Potthoff MJ (2017). FGF21 Regulates Metabolism Through Adipose-Dependent and -Independent Mechanisms. Cell Metab. 25, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, and Schorderet DF (2001). Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50, 77–82. [DOI] [PubMed] [Google Scholar]
- Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O’Connor D, Stutte S, Tavakkoli A, Winters D, et al. (2017). Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity 46, 273–286. [DOI] [PubMed] [Google Scholar]
- Bouwens L, Baekeland M, De Zanger R, and Wisse E (1986). Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology 6, 718–722. [DOI] [PubMed] [Google Scholar]
- Bouzakri K, Ribaux P, Tomas A, Parnaud G, Rickenbach K, and Halban PA (2008). Rab GTPase-activating protein AS160 is a major downstream effector of protein kinase B/Akt signaling in pancreatic beta-cells. Diabetes 57, 1195–1204. [DOI] [PubMed] [Google Scholar]
- Brenner DA, Kisseleva T, Scholten D, Paik YH, Iwaisako K, Inokuchi S, Schnabl B, Seki E, De Minicis S, Oesterreicher C, and Taura K (2012). Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair 5 (Suppl 1), S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brufau G, Stellaard F, Prado K, Bloks VW, Jonkers E, Boverhof R, Kuipers F, and Murphy EJ (2010). Improved glycemic control with colese-velam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 52, 1455–1464. [DOI] [PubMed] [Google Scholar]
- Butler AE, Dhawan S, Hoang J, Cory M, Zeng K, Fritsch H, Meier JJ, Rizza RA, and Butler PC (2016). β-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of β-Cell Dedifferentiation and Degranulation. J. Clin. Endocrinol. Metab 101, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, and Bäckhed F (2015). Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 22, 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, and Shoelson SE (2005). Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med 11, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calori G, Lattuada G, Piemonti L, Garancini MP, Ragogna F, Villa M, Mannino S, Crosignani P, Bosi E, Luzi L, et al. (2011). Prevalence, metabolic features, and prognosis of metabolically healthy obese Italian individuals: the Cremona Study. Diabetes Care 34, 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfora EE, Jocken JW, and Blaak EE (2015). Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol 11, 577–591. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, and Burcelin R (2008). Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Heimberg H, Heremans Y, Leeman R, Kutlu B, Kruhøffer M, Ørntoft T, and Eizirik DL (2001). A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J. Biol. Chem 276, 48879–48886. [DOI] [PubMed] [Google Scholar]
- Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, et al. (2006). Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55, 2688–2697. [DOI] [PubMed] [Google Scholar]
- Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, et al. (2006). The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J. Biol. Chem 281, 11039–11049. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Doose JM, Melchior B, Schmid CD, and Ploix CC (2006). CNS immune privilege: hiding in plain sight. Immunol. Rev 213, 48–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen T, Dinarello CA, and Donath MY (2012). Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 35, 1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazettes F, Cohen JI, Yau PL, Talbot H, and Convit A (2011). Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res. 1373, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SE, MacDougall K, Preston T, Tedford C, Finlayson GS, et al. (2015). Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 64, 1744–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JA, and Summers SA (2012). A ceramide-centric view of insulin resistance. Cell Metab. 15, 585–594. [DOI] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, and Evans RM (2001). PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med 7, 48–52. [DOI] [PubMed] [Google Scholar]
- Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, et al. (2009). The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 138, 961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaraldi TP, Ryan AJ, Mudaliar SR, and Henry RR (2016). Altered Myokine Secretion Is an Intrinsic Property of Skeletal Muscle in Type 2 Diabetes. PLoS ONE 11, e0158209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, Marselli L, Suleiman M, Ratner LE, Marchetti P, and Accili D (2016). Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab 101, 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, and Mathis D (2012). PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani S, Mencarelli A, Palladino G, and Fiorucci S (2010). FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res 51, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, and Vinolo MA (2016). Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunology 5, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvera S, and Gealekman O (2014). Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim. Biophys. Acta 1842, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP (2017). Insulin action and resistance in obesity and type 2 diabetes. Nat. Med 23, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, and Raybould HE (2010). Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol 299, G440–G448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, and Velloso LA (2005). Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146, 4192–4199. [DOI] [PubMed] [Google Scholar]
- Defronzo RA (2009). Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58, 773–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, and Scherer PE (2010). Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. N Y Acad. Sci 1212, E1–E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo S, Iossa S, and Venditti P (2017). Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J. Endocrinol 233, R15–R42. [DOI] [PubMed] [Google Scholar]
- Dietze D, Koenen M, Röhrig K, Horikoshi H, Hauner H, and Eckel J (2002). Impairment of insulin signaling in human skeletal muscle cells by co-culture with human adipocytes. Diabetes 51, 2369–2376. [DOI] [PubMed] [Google Scholar]
- Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, Spohr C, Kober L, Vaag A, and Torp-Pedersen C (2005). Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J. Vasc. Res 42, 517–525. [DOI] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ (2005). Sources of fatty acids stored in liver and secreted via lipo-proteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest 115, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror E, Dalmas E, Meier DT, Wueest S, Thévenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, et al. (2017). Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol 18, 283–292. [DOI] [PubMed] [Google Scholar]
- Dumortier O, Hinault C, and Van Obberghen E (2013). MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab. 18, 312–324. [DOI] [PubMed] [Google Scholar]
- Ebstein W (1876). Zur therapie des Diabetes mellitus, insbesondere über die Anwendung des salicylsauren Natron bei demselben. Berliner Klinische Wochenschrift 13, 337–340. [Google Scholar]
- Eguchi K, and Nagai R (2017). Islet inflammation in type 2 diabetes and physiology. J. Clin. Invest 127, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, et al. (2012). Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 15, 518–533. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, et al. (2007). Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370. [DOI] [PubMed] [Google Scholar]
- Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, et al. (2011). Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med 17, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Attina T, Spickett CM, and Webb DJ (2007). A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr 86, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Esteves JV, Enguita FJ, and Machado UF (2017). MicroRNAs-Mediated Regulation of Skeletal Muscle GLUT4 Expression and Translocation in Insulin Resistance. J. Diabetes Res 2017, 7267910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, Chen Z, Finck BN, Han DH, Magkos F, et al. (2013). Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 145, 366–374, e361–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Yoshino J, Yoshino M, Magkos F, Tiemann Luecking C, Samovski D, Fraterrigo G, Okunade AL, Patterson BW, and Klein S (2015). Metabolically normal obese people are protected from adverse effects following weight gain. J. Clin. Invest 125, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Soued M, Staprans I, Gavin LA, Donahue ME, Huang BJ, Moser AH, Gulli R, and Grunfeld C (1989). Effect of tumor necrosis factor (TNF) on lipid metabolism in the diabetic rat. Evidence that inhibition of adipose tissue lipoprotein lipase activity is not required for TNF-induced hyperlipidemia. J. Clin. Invest 83, 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, and Mathis D (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink LN, Oberbach A, Costford SR, Chan KL, Sams A, Blüher M, and Klip A (2013). Expression of anti-inflammatory macrophage genes within skeletal muscle correlates with insulin sensitivity in human obesity and type 2 diabetes. Diabetologia 56, 1623–1628. [DOI] [PubMed] [Google Scholar]
- Fink LN, Costford SR, Lee YS, Jensen TE, Bilan PJ, Oberbach A, Blüher M, Olefsky JM, Sams A, and Klip A (2014). Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity (Silver Spring) 22, 747–757. [DOI] [PubMed] [Google Scholar]
- Franckhauser S, Elias I, Rotter Sopasakis V, Ferré T, Nagaev I, Andersson CX, Agudo J, Ruberte J, Bosch F, and Smith U (2008). Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia 51, 1306–1316. [DOI] [PubMed] [Google Scholar]
- Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, and Moller DE (2013). The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 18, 333–340. [DOI] [PubMed] [Google Scholar]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, and Ye J (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, and Olefsky JM (2012). Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 15, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, and Shoelson SE (2017). Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Invest 127, 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, and Shoelson SE (2008). Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin. Transl. Sci 1, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, and Shoelson SE; TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team (2010). The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med 152, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, and Rodewald HR (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J, and Llorca J (2006). Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin. Exp. Rheumatol 24, 83–86. [PubMed] [Google Scholar]
- Guillemot-Legris O, and Muccioli GG (2017). Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 40, 237–253. [DOI] [PubMed] [Google Scholar]
- Guo S, Al-Sadi R, Said HM, and Ma TY (2013). Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol 182, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Astiarraga B, Camastra S, Accili D, and Ferrannini E (2013). Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62, 4184–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L, and Weir GC (2014). β-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37, 1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KH, Chang MK, Boullier A, Green SR, Li A, Glass CK, and Quehenberger O (2000). Oxidized LDL reduces monocyte CCR2 expression through pathways involving peroxisome proliferator-activated receptor gamma. J. Clin. Invest 106, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, et al. (2012). The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, and Brenner DA (1999). The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol 30, 77–87. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, and Hotamisligil GS (2002). A central role for JNK in obesity and insulin resistance. Nature 420, 333–336. [DOI] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. (2011). Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest 121, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, et al. (2009). Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58, 2525–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, and Shimomura I (2007). Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2017). Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, and Davis RJ (2016). Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, and Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, Lee SK, Alfadda AA, Kim SS, Choi SH, et al. (2013). A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol. Cell. Biol 33, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Park YJ, Kim YR, Kim YN, Ka S, Lee HY, Seong JK, Seok YJ, and Kim JB (2015). Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J. 29, 2397–2411. [DOI] [PubMed] [Google Scholar]
- Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, and Dent P (2009). Bile acids as regulatory molecules. J. Lipid Res 50, 1509–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoillo-Esteve M, Marselli L, Cunha DA, Ladrière L, Ortis F, Grieco FA, Dotta F, Weir GC, Marchetti P, Eizirik DL, and Cnop M (2010). Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53, 1395–1405. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, et al. (2007). Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425. [DOI] [PubMed] [Google Scholar]
- Ivan M, and Kaelin WG Jr. (2017). The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Mol. Cell 66, 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jais A, and Brüning JC (2017). Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Invest 127, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, and Gonzalez FJ (2011). Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AM, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, and Olefsky JM (2015). High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS ONE 10, e0122195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patanè G, Laybutt R, Bonner-Weir S, and Weir GC (1999). Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem 274, 14112–14121. [DOI] [PubMed] [Google Scholar]
- Jordan SD, Krüger M, Willmes DM, Redemann N, Wunderlich FT, Brönneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Böttger T, et al. (2011). Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol 13, 434–446. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, and Rudensky AY (2012). Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, and Seale P (2015). Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 22, 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Seo SU, Chen GY, and Núñez G (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol 13, 321–335. [DOI] [PubMed] [Google Scholar]
- Kamata K, Mizukami H, Inaba W, Tsuboi K, Tateishi Y, Yoshida T, and Yagihashi S (2014). Islet amyloid with macrophage migration correlates with augmented β-cell deficits in type 2 diabetic patients. Amyloid 21, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, and Weir GC (2002). Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem 277, 30010–30018. [DOI] [PubMed] [Google Scholar]
- Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, and Yamasaki Y (2006). The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem 281, 1091–1098. [DOI] [PubMed] [Google Scholar]
- Kayali AG, Lopez AD, Hao E, Hinton A, Hayek A, and King CC (2012). The SDF-1α/CXCR4 axis is required for proliferation and maturation of human fetal pancreatic endocrine progenitor cells. PLoS ONE 7, e38721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, and Scherer PE (2009). Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol. Cell. Biol 29, 1575–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, Wang X, Shi ZZ, Lewis DE, Wu H, and Ballantyne CM (2015). Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int. J. Obes 39, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, et al. (2005). FGF-21 as a novel metabolic regulator. J. Clin. Invest 115, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. (2007). Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Invest 117, 2621–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, and Drosos AA (2005). Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann. Rheum. Dis 64, 765–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, and Brenner DA (2017). Liver inflammation and fibrosis. J. Clin. Invest 127, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 20, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kawai T, and Akira S (2011). Pathogen recognition by the innate immune system. Int. Rev. Immunol 30, 16–34. [DOI] [PubMed] [Google Scholar]
- Kusminski CM, Bickel PE, and Scherer PE (2016). Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat. Rev. Drug Discov 15, 639–660. [DOI] [PubMed] [Google Scholar]
- Lackey DE, and Olefsky JM (2016). Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol 12, 15–28. [DOI] [PubMed] [Google Scholar]
- Lalia AZ, and Lanza IR (2016). Insulin-Sensitizing Effects of Omega-3 Fatty Acids: Lost in Translation? Nutrients 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, et al. (2006). Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55, 2256–2264. [DOI] [PubMed] [Google Scholar]
- Lee KY, Gesta S, Boucher J, Wang XL, and Kahn CR (2011a). The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 14, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, et al. (2011b). Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60, 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Morinaga H, Kim JJ, Lagakos W, Taylor S, Keshwani M, Perkins G, Dong H, Kayali AG, Sweet IR, and Olefsky J (2013). The fractalkine/CX3CR1 system regulates β cell function and insulin secretion. Cell 153, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, et al. (2014). Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Kim MS, Pae M, Yamamoto Y, Eberlé D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, et al. (2016). Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab. 23, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, and Gordon JI (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102, 11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Lu M, Nguyen MT, Bae EJ, Chapman J, Feng D, Hawkins M, Pessin JE, Sears DD, Nguyen AK, et al. (2010). Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J. Biol. Chem 285, 15333–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, et al. (2011). Adipocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 147, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Maris M, Ofrecio JM, et al. (2015). LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med 21, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, Johnson AMF, Sears D, Shen Z, Cui B, et al. (2016). Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell 167, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HV, Frassetto A, Kowalik EJ Jr., Nawrocki AR, Lu MM, Kosinski JR, Hubert JA, Szeto D, Yao X, Forrest G, and Marsh DJ (2012). Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 7, e35240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löfgren P, van Harmelen V, Reynisdottir S, Näslund E, Rydén M, Rössner S, and Arner P (2000). Secretion of tumor necrosis factor-alpha shows a strong relationship to insulin-stimulated glucose transport in human adipose tissue. Diabetes 49, 688–692. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, and Saltiel AR (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest 117, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, DelProposto JB, Westcott DJ, and Saltiel AR (2008). Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L (2014). Adipose invariant natural killer T cells. Immunology 142, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Saha PK, Chan L, and Moore DD (2006). Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Invest 116, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Huang Y, Yan L, Gao M, and Liu D (2013). Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res 30, 1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, and Donath MY (2002). Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest 110, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, de Las Fuentes L, He S, Okunade AL, Patterson BW, and Klein S (2016). Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 23, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SC, Tanabe K, Cras-Méneur C, Abumrad NA, Bernal-Mizrachi E, and Permutt MA (2008). Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 57, 846–859. [DOI] [PubMed] [Google Scholar]
- Mayoral R, Osborn O, McNelis J, Johnson AM, Oh DY, Izquierdo CL, Chung H, Li P, Traves PG, Bandyopadhyay G, et al. (2015). Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab 4, 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis JC, and Olefsky JM (2014). Macrophages, immunity, and metabolic disease. Immunity 41, 36–48. [DOI] [PubMed] [Google Scholar]
- McNelis JC, Lee YS, Mayoral R, van der Kant R, Johnson AM, Wollam J, and Olefsky JM (2015). GPR43 Potentiates β-Cell Function in Obesity. Diabetes 64, 3203–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meex RCR, and Watt MJ (2017). Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol 13, 509–520. [DOI] [PubMed] [Google Scholar]
- Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, Corthesy I, Macé K, and Chou CJ (2008). Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 22, 2416–2426. [DOI] [PubMed] [Google Scholar]
- Mighiu PI, Filippi BM, and Lam TK (2012). Linking inflammation to the brain-liver axis. Diabetes 61, 1350–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, and Locksley RM (2013). Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med 210, 535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ, et al. (2015). Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64, 1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Yamamura F, Nishikawa S, and Takahashi K (1989). Development, differentiation, and maturation of fetal mouse yolk sac macrophages in cultures. J. Leukoc. Biol 46, 1–10. [DOI] [PubMed] [Google Scholar]
- Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, and Hackam DJ (2006). Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol 176, 3070–3079. [DOI] [PubMed] [Google Scholar]
- Newgard CB (2017). Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 25, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, and Olefsky JM (2007). A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem 282, 35279–35292. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med 15, 914–920. [DOI] [PubMed] [Google Scholar]
- Nordmann TM, Dror E, Schulze F, Traub S, Berishvili E, Barbieux C, Böni-Schnetzler M, and Donath MY (2017). The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep 7, 6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, and Locksley RM (2013). Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, Ables EV, and Ferrante AW Jr. (2010). C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 59, 916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofei F, Hurel S, Newkirk J, Sopwith M, and Taylor R (1996). Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45, 881–885. [DOI] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM (2000). Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J. Clin. Invest 106, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, and Glass CK (2010). Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol 72, 219–246. [DOI] [PubMed] [Google Scholar]
- Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N, Chenevert TL, Korytnaya E, Neidert AH, Hench R, et al. (2017). Inhibition of IKKε and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 26, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajunen P, Kotronen A, Korpi-Hyövälti E, Keinänen-Kiukaanniemi S, Oksa H, Niskanen L, Saaristo T, Saltevo JT, Sundvall J, Vanhala M, et al. (2011). Metabolically healthy and unhealthy obesity phenotypes in the general population: the FIN-D2D Survey. BMC Public Health 11, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, and Bhattacharya S (2012). Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med 18, 1279–1285. [DOI] [PubMed] [Google Scholar]
- Paquot N, Castillo MJ, Lefèbvre PJ, and Scheen AJ (2000). No increased insulin sensitivity after a single intravenous administration of a re-combinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J. Clin. Endocrinol. Metab 85, 1316–1319. [DOI] [PubMed] [Google Scholar]
- Pardo Marqués V, González-Rodríguez A, and Valverde AM (2016). New therapeutic targets for the treatment of insulin resistance based on the paracrine cross-talk between hepatocytes and Kupffer cells. Anales de la Real Academia Nacional de Farmacia 82, 200–209. [Google Scholar]
- Patsouris D, Cao JJ, Vial G, Bravard A, Lefai E, Durand A, Durand C, Chauvin MA, Laugerette F, Debard C, et al. (2014). Insulin resistance is associated with MCP1-mediated macrophage accumulation in skeletal muscle in mice and humans. PLoS ONE 9, e110653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, and Febbraio MA (2007). Point: Interleukin-6 does have a beneficial role in insulin sensitivity and glucose homeostasis. J. Appl. Physiol (1985) 102, 814–16. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, and Febbraio MA (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol 8, 457–465. [DOI] [PubMed] [Google Scholar]
- Pham CT (2006). Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol 6, 541–550. [DOI] [PubMed] [Google Scholar]
- Pols TW, Noriega LG, Nomura M, Auwerx J, and Schoonjans K (2011). The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol 54, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig J, Blasco G, Daunis-I-Estadella J, Molina X, Xifra G, Ricart W, Pedraza S, Fernández-Aranda F, and Fernández-Real JM (2015). Hypothalamic damage is associated with inflammatory markers and worse cognitive performance in obese subjects. J. Clin. Endocrinol. Metab 100, E276–E281. [DOI] [PubMed] [Google Scholar]
- Puleston DJ, Villa M, and Pearce EL (2017). Ancillary Activity: Beyond Core Metabolism in Immune Cells. Cell Metab. 26, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Saberi M, Zmuda E, Wang Y, Altarejos J, Zhang X, Dentin R, Hedrick S, Bandyopadhyay G, Hai T, et al. (2009). Adipocyte CREB promotes insulin resistance in obesity. Cell Metab. 9, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, and Henquin JC (2008). Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab 10 (Suppl 4), 32–42. [DOI] [PubMed] [Google Scholar]
- Rai M, and Demontis F (2016). Systemic Nutrient and Stress Signaling via Myokines and Myometabolites. Annu. Rev. Physiol 78, 85–107. [DOI] [PubMed] [Google Scholar]
- Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, et al. (2014). Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med 20, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, et al. (2013). An inhibitor of the protein kinases TBK1 and IKK-ε improves obesity-related metabolic dysfunctions in mice. Nat. Med 19, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A, and Musi N (2008). Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57, 2595–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, and Morgan NG (2009). Islet-associated macrophages in type 2 diabetes. Diabetologia 52, 1686–1688. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, and Glass CK (1998). The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79–82. [DOI] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFayden JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Rissanen A, Howard CP, Botha J, and Thuren T; Global Investigators (2012). Effect of anti-IL-1β antibody (canakinumab) on insulin secretion rates in impaired glucose tolerance or type 2 diabetes: results of a randomized, placebo-controlled trial. Diabetes Obes. Metab 14, 1088–1096. [DOI] [PubMed] [Google Scholar]
- Romanatto T, Cesquini M, Amaral ME, Roman EA, Moraes JC, Torsoni MA, Cruz-Neto AP, and Velloso LA (2007). TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient–effects on leptin and insulin signaling pathways. Peptides 28, 1050–1058. [DOI] [PubMed] [Google Scholar]
- Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, and Olefsky JM (2009). Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 10, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, and Davis RJ (2009). Prevention of steatosis by hepatic JNK1. Cell Metab. 10, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest 126, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. [DOI] [PubMed] [Google Scholar]
- Schur EA, Melhorn SJ, Oh SK, Lacy JM, Berkseth KE, Guyenet SJ, Sonnen JA, Tyagi V, Rosalynn M, De Leon B, et al. (2015). Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity (Silver Spring) 23, 2142–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina IG, Kramarova TV, Nedergaard J, and Cannon B (2006). Carboxyatractyloside effects on brown-fat mitochondria imply that the adenine nucleotide translocator isoforms ANT1 and ANT2 may be responsible for basal and fatty-acid-induced uncoupling respectively. Biochem. J 399, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea JL, Randell EW, and Sun G (2011). The prevalence of metabolically healthy obese subjects defined by BMI and dual-energy X-ray absorptiometry. Obesity (Silver Spring) 19, 624–630. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, and Flier JS (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest 116, 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, and Mantovani A (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, De Gaetano A, Berg JK, and Landschulz WH (2013). Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1β antibody, in patients with type 2 diabetes. Diabetes Care 36, 2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith U, and Kahn BB (2016). Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med 280, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Naugler W, Galimi F, Lee MS, and Karin M (2006). Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 103, 16454–16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son G, Iimuro Y, Seki E, Hirano T, Kaneda Y, and Fujimoto J (2007). Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol 293, G631–G639. [DOI] [PubMed] [Google Scholar]
- Stefan N, Hennige AM, Staiger H, Machann J, Schick F, Kröber SM, Machicao F, Fritsche A, and Häring HU (2006). Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care 29, 853–857. [DOI] [PubMed] [Google Scholar]
- Stevens CE, and Hume ID (1998). Contributions of microbes in vertebrate gastrointestinal tract to production and conservation of nutrients. Physiol. Rev 78, 393–427. [DOI] [PubMed] [Google Scholar]
- Subramanian M, Ozcan L, Ghorpade DS, Ferrante AW Jr., and Tabas I (2015). Suppression of Adaptive Immune Cell Activation Does Not Alter Innate Immune Adipose Inflammation or Insulin Resistance in Obesity. PLoS ONE 10, e0135842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K, Meisinger C, Döring A, Altmaier E, Belcredi P, Gieger C, Chang D, Milburn MV, Gall WE, Weinberger KM, et al. (2010). Metabolic footprint of diabetes: a multiplatform metabolomics study in an epidemiological setting. PLoS ONE 5, e13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA, and Scherer PE (2012). Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc. Natl. Acad. Sci. USA 109, 5874–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Halberg N, Khan M, Magalang UJ, and Scherer PE (2013). Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell. Biol 33, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung HK, Doh KO, Son JE, Park JG, Bae Y, Choi S, Nelson SM, Cowling R, Nagy K, Michael IP, et al. (2013). Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 17, 61–72. [DOI] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. (2012). Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med 18, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, et al. (2016). A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 23, 427–440. [DOI] [PubMed] [Google Scholar]
- Tanoue T, and Honda K (2012). Induction of Treg cells in the mouse colonic mucosa: a central mechanism to maintain host-microbiota homeostasis. Semin. Immunol 24, 50–57. [DOI] [PubMed] [Google Scholar]
- Tao C, Holland WL, Wang QA, Shao M, Jia L, Sun K, Lin X, Kuo Y-C, Johnson JA, Gordillo R, et al. (2017). Short-Term Versus Long-Term Effects of Adipocyte Toll-Like Receptor 4 Activation on Insulin Resistance in Male Mice. Endocrinology 158, 1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, and Schwartz MW (2010). Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology 151, 4109–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, et al. (2012). Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest 122, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurich S, Tsaousidou E, Hanssen R, Lempradl AM, Mauer J, Timper K, Schilbach K, Folz-Donahue K, Heilinger C, Sexl V, et al. (2017). IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab. 26, 171–184. [DOI] [PubMed] [Google Scholar]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, et al. (2017). Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, and Théry C (2016). Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 164, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, and Suzuki F (2004). Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 21, 215–226. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, and Gordon JI (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Bäckhed F, Fulton L, and Gordon JI (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Asseldonk EJ, Stienstra R, Koenen TB, Joosten LA, Netea MG, and Tack CJ (2011). Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J. Clin. Endocrinol. Metab 96, 2119–2126. [DOI] [PubMed] [Google Scholar]
- van Vliet-Ostaptchouk JV, Nuotio ML, Slagter SN, Doiron D, Fischer K, Foco L, Gaye A, Gögele M, Heier M, Hiekkalinna T, et al. (2014). The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: a collaborative analysis of ten large cohort studies. BMC Endocr. Disord 14, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma V, Yao-Borengasser A, Rasouli N, Nolen GT, Phanavanh B, Starks T, Gurley C, Simpson P, McGehee RE Jr., Kern PA, and Peterson CA (2009). Muscle inflammatory response and insulin resistance: synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am. J. Physiol. Endocrinol. Metab 296, E1300–E1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, et al. (2012). Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–16. [DOI] [PubMed] [Google Scholar]
- Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A, et al. (2014). M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 59, 130–142. [DOI] [PubMed] [Google Scholar]
- Wascher TC, Lindeman JH, Sourij H, Kooistra T, Pacini G, and Roden M (2011). Chronic TNF-α neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol. Med 17, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskirchen R, and Tacke F (2014). Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr 3, 344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, and Scherer PE (2014). Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 20, 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwell-Roper C, Denroche HC, Ehses JA, and Verchere CB (2016). Differential Activation of Innate Immune Pathways by Distinct Islet Amyloid Polypeptide (IAPP) Aggregates. J. Biol. Chem 291, 8908–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. (2009). Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med 15, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. (2011). B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med 17, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Dranse HJ, and Lam TK (2017). Immunologic impact of the intestine in metabolic disease. J. Clin. Invest 127, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollam J, and Antebi A (2011). Sterol regulation of metabolism, homeostasis, and development. Annu. Rev. Biochem 80, 885–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Ballantyne CM (2017). Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Invest 127, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, and Locksley RM (2011). Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H, Northrop JP, Grove JR, Kilpatrick KE, Su JL, and Ringold GM (1997). TNF alpha-mediated inhibition and reversal of adipocyte differentiation is accompanied by suppressed expression of PPARgamma without effects on Pref-1 expression. Endocrinology 138, 2776–2783. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, and Chen H (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest 112, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Liu Z, Donovan J, Thomas MK, and Habener JF (2007). Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes 56, 2946–2957. [DOI] [PubMed] [Google Scholar]
- Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A, Fu W, et al. (2017a). Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–384. [DOI] [PubMed] [Google Scholar]
- Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, Oh DY, Li P, Osborn O, and Olefsky JM (2017b). Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J. Clin. Invest 127, 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, and Shoelson SE (2001). Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293, 1673–1677. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, and Edwards PA (2006). Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 103, 1006–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HL, Lai FM, Tong PC, Zhong DR, Yang D, Tomlinson B, and Chan JC (2003). Prevalence and clinicopathological characteristics of islet amyloid in chinese patients with type 2 diabetes. Diabetes 52, 2759–2766. [DOI] [PubMed] [Google Scholar]