

Abstract
The cytochrome P450 (CYP) 2D6 enzyme exhibits large interindividual differences in metabolic activity. Patients are commonly assigned a CYP2D6 phenotype based on their CYP2D6 genotype, but there is a lack of consensus on how to translate genotypes into phenotypes, causing inconsistency in genotype‐based dose recommendations. The aim of this study was to quantify and compare the impact of different CYP2D6 genotypes and alleles on CYP2D6 metabolism using a large clinical data set. A population pharmacokinetic (popPK) model of tedatioxetine and its CYP2D6‐dependent metabolite was developed based on pharmacokinetic data from 578 subjects. The CYP2D6‐mediated metabolism was quantified for each subject based on estimates from the final popPK model, and CYP2D6 activity scores were calculated for each allele using multiple linear regression. The activity scores estimated for the decreased function alleles were 0.46 (CYP2D6*9), 0.34 (CYP2D6*10), 0.01 (CYP2D6*17), 0.65 (CYP2D6*29), and 0.21 (CYP2D6*41). The CYP2D6*17 and CYP2D6*41 alleles were thus associated with the lowest CYP2D6 activity, although only the difference to the CYP2D6*9 allele was shown to be statistically significant (p = 0.02 and p = 0.05, respectively). The study provides new in vivo evidence of the enzyme function of different CYP2D6 genotypes and alleles. Our findings suggest that the activity score assigned to CYP2D6*41 should be revisited, whereas CYP2D6*17 appears to exhibit substrate‐specific behavior. Further studies are needed to confirm the findings and to improve the understanding of CYP2D6 genotype–phenotype relationships across substrates.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Genotype‐based prediction of patients’ cytochrome P450 (CYP) 2D6 phenotype is commonly used to guide dosing of CYP2D6 substrates, but consensus on how to translate genotypes into phenotypes is lacking.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study aimed to quantify the CYP2D6 activity exhibited by different CYP2D6 genotypes and alleles through the population pharmacokinetic modeling of tedatioxetine.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The study provides new evidence of the in vivo function of different CYP2D6 genotypes and alleles from a high‐quality clinical data set. We found low enzyme activity associated with CYP2D6*17 and CYP2D6*41, implying that lower activity scores might better reflect the activity of these alleles in the metabolism of tedatioxetine.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Our findings contribute to the existing evidence of low metabolic activity of CYP2D6*41 and would support a discussion of downgrading the activity score for this allele. The low activity observed for CYP2D6*17 underlines the challenge of assigning a universal phenotype to this allele and further studies are needed to understand its substrate‐specific behavior.
INTRODUCTION
The cytochrome P450 (CYP) 2D6 enzyme is involved in the metabolism of numerous therapeutic drugs. The enzyme has attracted considerable attention due to its polymorphic nature that causes substantial interindividual variability in enzyme activity.
More than 100 allele variants of the CYP2D6 gene have been identified causing either normal, decreased, or no function of the CYP2D6 enzyme. Carriers of two nonfunctional alleles represent 0.4%–6.0% of the population, depending on ethnicity, and are classified as CYP2D6 poor metabolizers (PMs).1 At the other end of the spectrum are carriers of duplicated functional alleles on one chromosome in combination with a normal function allele who are assigned the ultra‐rapid metabolizer (UM) phenotype.2 The UMs constitute approximately 1.4%–11.5% of the population, depending on ethnicity.1
Between the PMs and UMs are the intermediate metabolizers (IMs) and normal metabolizers (NMs). Although there is generally consensus on which CYP2D6 genotypes to translate into PM and UM, the assignment of genotypes to the IM and NM phenotypes remain subject for debate.
Particularly decreased function alleles present a challenge for IM and NM phenotype assignment. Decreased function alleles are commonly assigned the same functional value, although studies have shown that the alleles display different levels of metabolic activity.3, 4, 5 A further complication is that some decreased function alleles (e.g., CYP2D6*10 and CYP2D6*17) have been shown to exhibit varying degrees of metabolic activity depending on the substrate studied.6, 7
To standardize the way CYP2D6 genotypes are translated into phenotypes, the Clinical Pharmacogenomics Implementation Consortium (CPIC) and the Dutch Pharmacogenomics Working Group (DPWG) recently published a joint consensus guideline.2 The guideline presented a new harmonized CYP2D6 genotype–phenotype translation scheme and encouraged researchers to report their findings using this standardized translation method in the future. One of the recommendations included in the guideline was to downgrade the activity score of the CYP2D6*10 allele to reflect a lower metabolic activity compared with other decreased function alleles.
This recommendation stands in contrast to findings from our recent study of vortioxetine.8 Using rich pharmacokinetic (PK) data of vortioxetine and its CYP2D6‐dependent metabolite, we quantified the in vivo CYP2D6 activity of 1140 CYP2D6‐genotyped subjects through population PK (popPK) modeling.8 The results showed that the CYP2D6*10 allele was associated with significantly higher activity compared with the decreased function alleles CYP2D6*17 (p = 0.01) and CYP2D6*41 (p = 0.02) in the metabolism of vortioxetine. We also found that carriers of one fully functional allele in combination with one null function allele had 77% higher CYP2D6 activity compared with carriers of two decreased function alleles (p < 0.001), although both diplotypes are translated into the same functional level according to the consensus guideline.
These findings highlight some of the challenges associated with phenotype assignment of CYP2D6 genotypes involving decreased function alleles. To improve our understanding of the behavior of these alleles, more research on different CYP2D6 substrates is needed, preferably using high‐quality clinical data.
Tedatioxetine (Lu AA24530) is a multitarget compound that was in development by H. Lundbeck A/S for the treatment of major depressive disorder (MDD). However, development of the compound was terminated as the development of other drug candidates was advanced.
Although tedatioxetine is no longer in development, data from completed clinical studies offer a unique opportunity to study CYP2D6 genotype–phenotype relationships as tedatioxetine is a sensitive CYP2D6 substrate.
Figure 1 shows the proposed metabolic pathways of tedatioxetine and the CYP450 enzymes involved. The major metabolic route of tedatioxetine is through oxidation to the metabolite Lu AA37208 via an intermediate, Lu AA37209. The primary enzyme involved in this metabolic pathway in vitro is CYP2D6, although a minor involvement of CYP2C19 cannot be excluded (internal data, H. Lundbeck A/S).
FIGURE 1.
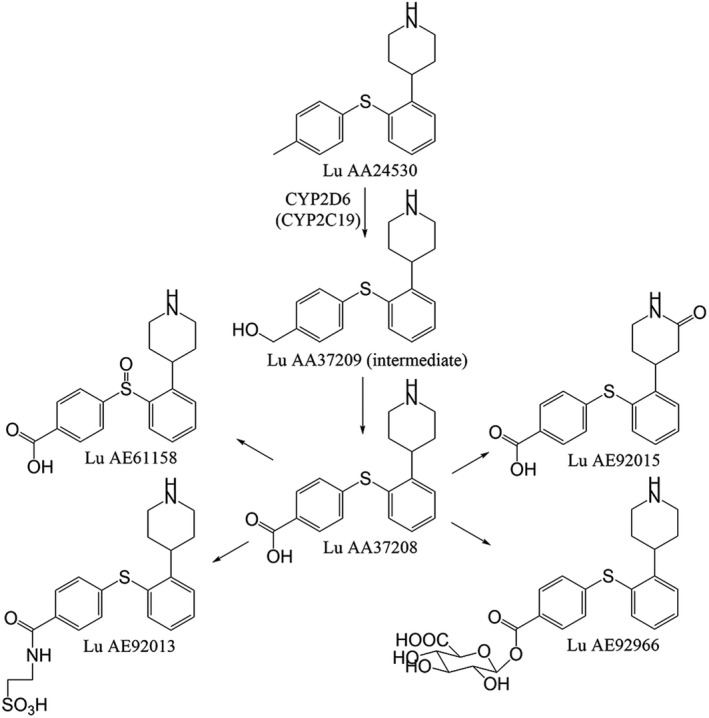
Biotransformation of tedatioxetine
Data from clinical studies of tedatioxetine showed mean oral clearances of 18 L/h for CYP2D6 PMs, 40 L/h for IMs, 60 L/h for NMs, and 77 L/h for UMs, and approximately 80% of the total clearance has been estimated to be mediated via CYP2D6. Following oral administration, tedatioxetine has shown a slow absorption rate with a median time to maximum plasma concentration (tmax) of approximately 5–6 h. The tmax observed for the metabolite Lu AA37208 was similar or shorter, indicating the presence of presystemic metabolism (internal data, H. Lundbeck A/S).
Seven clinical studies including 578 CYP2D6 genotyped subjects have been completed where PK samples of both tedatioxetine and Lu AA37208 were collected.
The objective of the current study was to develop a joint popPK model of tedatioxetine and Lu AA37208 with the aim of estimating the CYP2D6‐mediated metabolism of individuals carrying different CYP2D6 genotypes.
METHODS
Studies and subjects
Data from six phase I studies and one phase II study with oral administration of tedatioxetine were pooled for a popPK analysis. All studies were approved by ethical committees, and all subjects provided written informed consent prior to any study‐related procedures.
In total, 220 healthy subjects and 358 patients with MDD were included in the data set. An overview of the clinical studies and subject characteristics is provided in Table 1.
TABLE 1.
Characteristics of the clinical studies and subjects included in the population pharmacokinetic analysis
| Study description | N | Doses (mg) | PK sampling time points | PGx panela |
|---|---|---|---|---|
| Single dose study (healthy subjects) | 50♂/14♀ | 2–60 (SD) | 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, 120, 168, and 216 h | A |
| Multiple dose study (healthy subjects) | 78♂/22♀ | 5–50 (MD) | Day 1: 2, 3, 4, 5, 6, 8, 12, and 24 h, predose prior to steady state and 0, 2, 3, 4, 5, 6, 8, 12, 24, 48, and 72 h at steady state | A |
| PET study (healthy subjects) | 18♂ | 25, 35, and 50 (SD) | 2, 4, 8, 10, 12, 24, 48, 72, and 96 h | B |
| DDI study omeprazole (healthy subjects) | 12♂/12♀ | 15 and 5 (MD) | 0, 2, 3, 4, 5, 6, 8, 12, and 24 h at steady state and predose prior to steady state | C |
| ADME (healthy subjects) | 6♂ | 50 (SD) | 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 96, 120, 144, and 168 h | D |
| Bioavailability study (healthy subjects) | 8♂ | 50 (SD) | 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48, 72, 96, 144, 192, 240, and 312 h | E |
| Dose‐finding study (patients with MDD) | 139♂/219♀ | 5, 10, and 20 | Weeks 1, 3, and 6 | F |
| Continuous characteristic | N | Median | IQR | Range |
|---|---|---|---|---|
| Age (years) | 578 | 37 | 28–50 | 18–80 |
| Weight (kg) | 578 | 72 | 61–82 | 42–140 |
| Height (cm) | 578 | 170 | 163–177 | 146–202 |
| BMI (kg/m2) | 578 | 24 | 22–27 | 17–48 |
| LBM (kg) | 578 | 51 | 45–58 | 33–83 |
| Creatinine clearance (ml/min)b | 575 | 98 | 83–117 | 28–313 |
| Categorical characteristic | Frequency | Description |
|---|---|---|
| CYP2D6 phenotypec | 11/291/200/32/44 | UM/NM/IM/PM/missing |
| Sex | 311/267 | Males/females |
| Race | 442/31/88/17 | Caucasian/Black or African American/Asian/Other |
| aPGx panel | N | CYP2D6 variant alleles tested |
|---|---|---|
| A | 164 | CYP2D6*2, *3, *4, *5, *6, *9, *10, *16, *17, *29, *41 and gene duplication |
| B | 18 | CYP2D6*2, *3, *4, *5, *6, *10, *17, *41 and gene duplication |
| C | 24 | CYP2D6*3, *4, *5, *6, *10, *41 |
| D | 6 | CYP2D6*3, *4, *5, *6, *9, *10, *17, *41 and gene duplication |
| E | 8 | CYP2D6*3, *4, *5, *6 |
| F | 358 | CYP2D6*2, *4, *5, *6, *9, *10, *41 and gene duplication |
Abbreviations: ADME, absorption, distribution, metabolism, excretion; BMI, body mass index; DDI, drug‐drug interaction; IM, intermediate metabolizer; IQR, interquartile range; LBM, lean body mass; MD, multiple dose; MDD, major depressive disorder; NM, normal metabolizer; PET, positron emission tomography; PGx, pharmacogenomics; PM, poor metabolizer; SD, single dose; UM, ultra‐rapid metabolizer.
PGx: Pharmacogenetics.
Creatinine clearance estimated by the Cockcroft–Gault formula.
CYP2D6 genotype characteristics are provided in Table S1.
Three subjects did not have data available on creatinine clearance. For these subjects, the median value (98 ml/min) was imputed. A total of 44 subjects were missing data on CYP2D6 genotype and phenotype. These subjects were included in the popPK analysis but not in the subsequent CYP2D6 genotype–phenotype analysis.
CYP2D6 genotyping
CYP2D6 genotyping was performed at four external laboratories, and different panels were used across the studies (see Table 1). Six of the studies (N = 570) used genotyping assays allowing detection of at least the following allele variants: CYP2D6*3, CYP2D6*4, CYP2D6*5, CYP2D6*6, CYP2D6*10, and CYP2D6*41. Some studies also tested for CYP2D6*9, CYP2D6*16, CYP2D6*17, and/or CYP2D6*29. The wild‐type allele (CYP2D6*1) was assigned when no variant alleles were identified. Five of the studies (N = 546) tested for gene duplication (denoted “XN”), but none of the assays provided information on which allele was duplicated or the actual number of gene copies.
CYP2D6 genotypes were translated into an activity score and predicted CYP2D6 phenotype according to the consensus guideline from the CPIC and DPWG.2 For heterozygous duplicated genotypes where the two alleles had different functionalities (e.g., *1/*4XN or *1/*10XN), no activity score was assigned.
An overview of the CYP2D6 genotypes identified along with their associated activity scores and predicted phenotypes is provided in Table S1.
PK data
In the phase I studies, dense PK samples were collected from each subject, whereas a maximum of three PK samples were collected over six weeks from each patient with MDD in the phase II (dose‐finding) study.
The concentrations of tedatioxetine and its metabolite, Lu AF37208, in the plasma samples were quantified using a liquid chromatography with tandem mass spectrometry method validated according to good laboratory practice. Plasma concentration values below the lower limit of quantification were excluded from the analysis data set. For the drug‐drug interaction studies, only samples following monotherapy of tedatioxetine were included in the popPK analysis. The analysis data set comprised a total of 5373 quantifiable plasma concentrations of tedatioxetine and 5449 quantifiable plasma concentrations of Lu AF37208.
PopPK analysis
A popPK model describing the PK of tedatioxetine and Lu AF37208 simultaneously was developed using nonlinear mixed effect modeling in NONMEM® (ICON Development Solutions, Version 7.4). The Markov Chain Monte Carlo stochastic approximation expectation maximization method followed by importance sampling was used for minimization.
Different structural models were tested to describe the joint PK of tedatioxetine and Lu AF37208. Initial popPK analyses of the two compounds alone showed that two‐compartment models best described the disposition of each compound individually. Therefore, the initial structural model tested was a four‐compartment model including central and peripheral compartments for both tedatioxetine and Lu AA37208.
However, this model did not converge successfully, which was thought to be due to an early peak in the plasma concentration of the metabolite, Lu AA37208. To account for this, an extra compartment was added to reflect presystemic formation of Lu AA37208. This enabled the model to converge successfully.
Interindividual variability (IIV) of the model parameters was modeled using exponential error terms and covariance between selected parameters was tested. Different residual error models were tested including proportional, additive, and a combination of the two. The same residual error estimate was used for tedatioxetine and Lu AA37208.
Each model was evaluated by different diagnostic tools including the objective function value (OFV), Akaike information criterion (AIC), condition number, and goodness‐of‐fit plots. In addition, the precision of the parameter estimates, measured by relative standard error, was used to assess and compare models.
The influence of covariates on PK parameters was investigated using stepwise forward inclusion followed by backward elimination. During forward inclusion, covariates were added to the model if they resulted in a significant OFV reduction (p < 0.01, corresponding to an OFV reduction of 6.64 for one degree of freedom). Significant covariates were added to the model in a stepwise manner until a significant reduction of OFV could no longer be obtained. When no more covariates could be added to the model, stepwise backward deletion was performed. To justify the inclusion of each covariate in the final model, a stricter significance level of p = 0.001 (corresponding to a ΔOFV of 10.83 for one degree of freedom) was used in the backward‐elimination process.
To evaluate the precision of the final model parameters, nonparametric bootstrap analyses was performed where 245 bootstrap data sets were created and analyzed with the final popPK model.
The final model was also evaluated by visual predictive check (VPC) plots based on 500 simulated data sets. The plots were assessed visually for agreement between the observed plasma concentrations from the original data and model simulated plasma concentrations.
Statistical analysis
The activity associated with the individual CYP2D6 alleles was assessed using a multiple linear regression analysis. Indicator variables reflecting the number of each variant allele in each CYP2D6 genotype were used as predictors and the estimated CYP2D6 activity was the outcome variable.
Different regression models were tested using both untransformed and log‐transformed CYP2D6 activity estimates and pooling null function alleles and fully functional alleles, respectively. Each regression model was evaluated by the visual inspection of residual plots, and nested models were compared using the AIC and analysis of variance. The coefficients of the individual alleles in the final regression model were compared using Wald tests.
All statistical analyses were performed using the open source software environment, R (Version 3.5.1) run under RStudio.
Calculation of activity scores
An activity score was calculated for each CYP2D6 allele based on the results from the final multiple linear regression model. The activity associated with the null function alleles (CYP2D6*3, CYP2D6*4, CYP2D6*5, CYP2D6*6, and CYP2D6*16) was assumed to reflect a non–CYP2D6‐mediated formation of the metabolite and was therefore fixed to zero, whereas the activity for the fully functional alleles (CYP2D6*1 and CYP2D6*2) was fixed to one. The activity score for each allele was calculated as the relative activity to the fully functional alleles adjusted by the contribution of the null alleles:
where the β’s denote the back‐transformed coefficients from the final regression model. Confidence intervals (CIs) for each allele were estimated using a nonparametric bootstrap analysis with 10,000 samples.
RESULTS
PopPK analysis
The final popPK model is illustrated in Figure 2 (see Supplementary Materials for the final NONMEM control stream). The model was parameterized in terms of a rate constant for presystemic metabolism of tedatioxetine to Lu AA37208 (k g,met), absorption rate constants for tedatioxetine (k a) and Lu AA37208 (k a,met), oral clearances for tedatioxetine and Lu AA37208 (CL and CLmet), volumes of distribution for central compartments for tedatioxetine and Lu AA37208 (V3 and V5), volumes of distribution for peripheral compartments for tedatioxetine and Lu AA37208 (V4 and V6), intercompartmental clearances (Q and Q met), and a lag‐time parameter.
FIGURE 2.
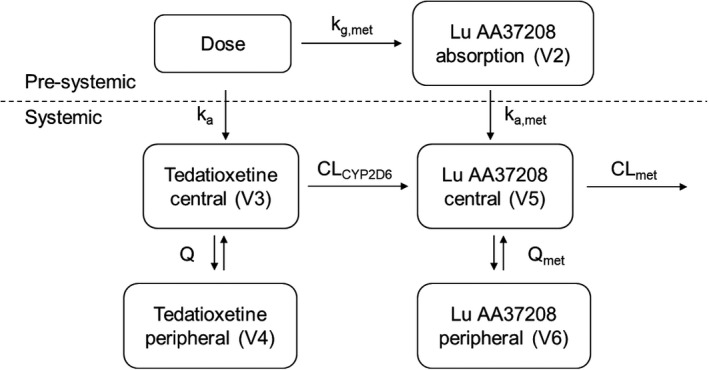
Structure of the population pharmacokinetic model of tedatioxetine and its metabolite, Lu AA37208. The model is parameterized by a rate constant for presystemic formation of Lu AA37208 (kg,met), absorption rate constants for tedatioxetine (ka) and Lu AA37208 (ka,met), central (V3, V5) and peripheral (V4, V6) compartments, intercompartmental clearances (Q, Q met), and two clearance parameters (CLCYP2D6, CLmet). CYP2D6, cytochrome P450 2D6
IIV was modeled on the model parameters k g,met, k a, k a,met, CL, V3, CLmet, and V5, and covariances were included between CL‐V3 and CLmet‐V5. Residual error was modeled as proportional and the same sigma estimate was used for tedatioxetine and Lu AA37208.
In the first forward‐inclusion step, food on k a,met resulted in the most significant OFV reduction (−596 points) and was therefore included in the model. In the second step, age on CLmet was the only relationship that resulted in a significant OFV drop (−14 points). No other covariate relationships resulted in model convergence, and the full model thus included food on k a,met and age on CLmet.
During backward elimination, removal of each of the covariates resulted in a significant increase in OFV (>10.83 points). Both covariates were therefore retained in the final model.
The parameter estimates for the final popPK model are summarized in Table 2. Goodness‐of‐fit and prediction‐corrected visual predictive check plots are presented in Figures S1 and S2.
TABLE 2.
Parameter estimates from the final population pharmacokinetic model of tedatioxetine and Lu AA37208
| Model parameter | Estimate (%RSE) | IIV (%RSE) (shrinkage) | 95% CI |
|---|---|---|---|
| Absorption rate constant, tedatioxetine (k a) (h−1) | 0.195 (6.4) | 69.79 (10.5) (33.7%) | 0.16–0.23 |
| Rate constant formation of Lu AA37208 (k g,met) (h−1) | 0.0972 (8.2) | 92.41 (13.3) (30.1%) | 0.05–0.11 |
| Absorption rate constant, Lu AA37208 (k a,met) (h−1) fasted | 11.1 (35.6) | 226.50 (17.5) (45.2%) | 0.56–25.8 |
| Absorption rate constant, Lu AA37208 (k a,met) (h−1) fed | 0.0286 (29.4) | – | 0.02–0.25 |
| Lag‐time (ALAG) (h) | 0.652 (0.7) | – | 0.52–0.71 |
| Volume of distribution, central compartment, tedatioxetine (V3) (L) | 1,380 (4.3) | 42.78 (8.7) (36.2%) | 1170–1880 |
| Clearance, tedatioxetine (CLCYP2D6) (L/h) | 30.5 (6.6) | 83.49 (8.3) (8.20%) | 30.0–39.2 |
| Volume of distribution, peripheral compartment, tedatioxetine (V4) (L) | 507 (0.8) | – | 470–704 |
| Intercompartmental clearance, tedatioxetine (Q) (L/h) | 39.1 (0.8) | – | 32.3–63.9 |
| Volume of distribution, central compartment, Lu AA37208 (V5) (L) | 33.1 (5.9) | 68.56 (19.5) (7.80%) | 10.0–38.5 |
| Clearance, Lu AA37208 (CLmet) (L/h) | 11.9 (3.4) | 55.05 (4.0) (6.62%) | 11.5–12.6 |
| Volume of distribution, peripheral compartment, Lu AA37208 (V6) (L) | 12.2 (8.1) | – | 13.4–22.6 |
| Intercompartmental clearance, Lu AA37208 (Q met) (L/h) | 0.940 (0.7) | – | 1.14–1.95 |
| Age on CLmet | −0.0830 (20.0) | – | −0.13 to −0.04 |
| Covariance ω(CL, V3) | 0.079 | – | 0.02–0.38 |
| Covariance ω(CLmet, V5) | 0.372 | – | −0.08 to 0.55 |
| Residual error (proportional)a | 23.6 (0.3) | – | 22.3–24.5 |
%RSE indicates the relative standard error expressed as percentage of the parameter estimate. IIV indicates the interindividual variability expressed as the coefficient of variation calculated as . 95% CI indicates the confidence interval from bootstrap analysis.
Expressed as the coefficient of variation calculated as.
Most model parameters were estimated with good precision; only the point estimates for V6 and Q met were outside the 95% CI from the bootstrap analysis.
All parameters associated with the absorption phase (k a, k g,met, k a,met) had ETA shrinkage >30%, which was largely driven by the sparse phase II data (see Figure S3). This may be explained by a limited number of plasma samples collected in the absorption phase from the patients in the phase II study, leading to shrinkage.
CYP2D6 activity estimates
The CYP2D6 activity was estimated for each subject using the individual parameter estimates (empirical Bayes estimates) from the final popPK model.
The extent of Lu AA37208 formed systemically was estimated by the parameter CLCYP2D6. A fraction reflecting the amount of Lu AA37208 formed presystemically (F met) was calculated based on the estimates of the absorption rate constant for tedatioxetine (k a) and the rate constant for presystemic formation of Lu AA37208 (k g,met) from the final popPK model:
To account for both presystemic and systemic metabolite formation, the total CYP2D6 activity was calculated as the product of the two estimates, that is, F met × CLCYP2D6.
Figure 3 shows the distribution of the individual estimates of the presystemic CYP2D6 activity (F met), the systemic CYP2D6 activity (CLCYP2D6), and the total CYP2D6 activity (F met × CLCYP2D6) colored by the subjects’ predicted CYP2D6 phenotype. A bimodal distribution, characteristic for CYP2D6 activity, was clearly seen for the total (F met × CLCYP2D6) and systemic (CLCYP2D6) CYP2D6 activity. This pattern was not observed to the same extent for the presystemic CYP2D6 activity (F met), which might be explained by a poor estimation of the absorption parameters for some individuals.
FIGURE 3.
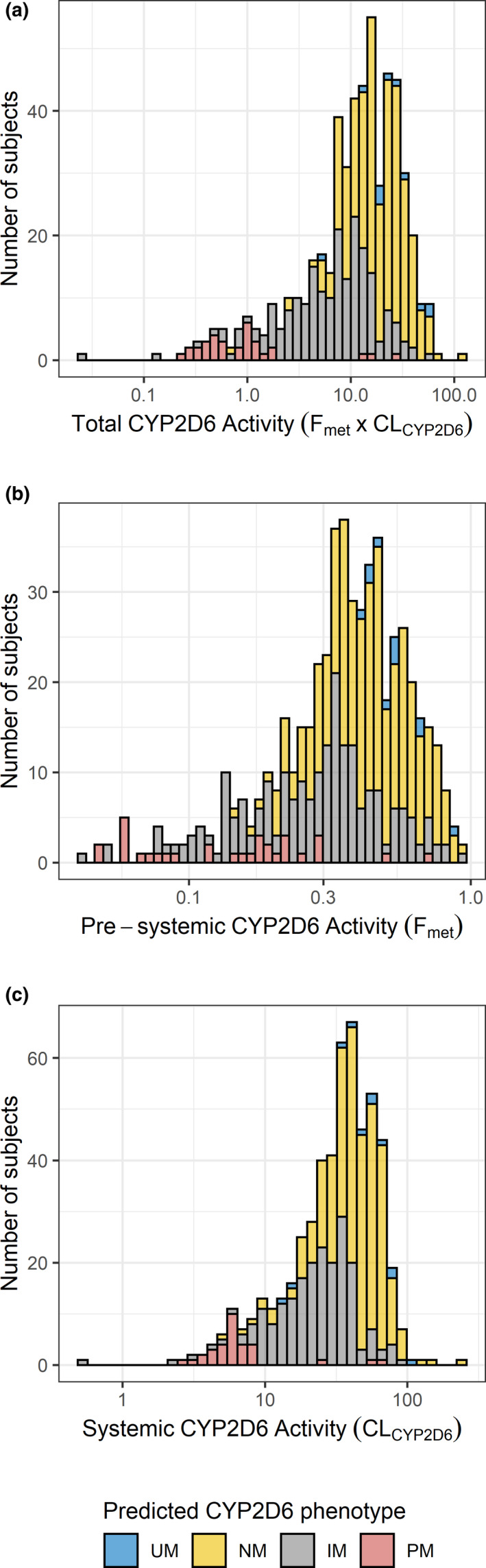
Distribution of individual estimates of (a) total CYP2D6 activity (Fmet × CLCYP2D6), (b) presystemic CYP2D6 activity (Fmet) and (c) systemic CYP2D6 activity (CLCYP2D6) colored by predicted CYP2D6 phenotype. CYP2D6, cytochrome P450 2D6; IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; UM, ultrarapid metabolizer
The median (interquartile range) total CYP2D6 activity was 0.86 (0.46–1.14) for CYP2D6 PMs, 7.52 (3.66–12.07) for IMs, 19.39 (12.44–28.75) for NMs, and 24.92 (19.21–41.99) for UMs. The median CYP2D6 activity for UMs was thus more than 29‐fold higher than the median activity of the PMs.
CYP2D6 activity scores
Figure 4 shows the estimated total CYP2D6 activity plotted against activity scores assigned based on the CPIC/DPWG consensus guideline2 (see Table S1 for details).
FIGURE 4.
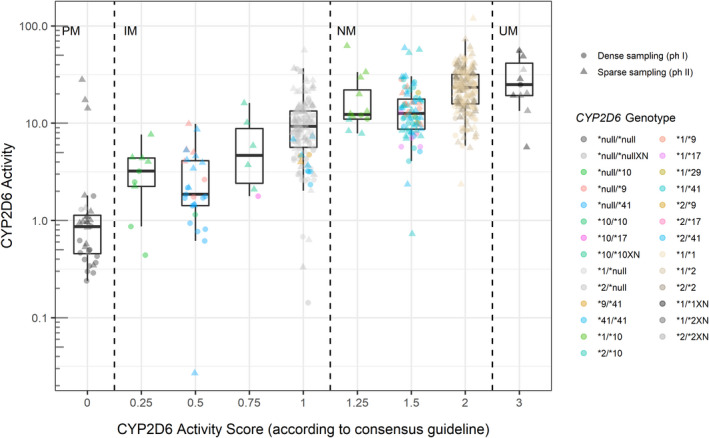
Boxplots and scatterplots of individual CYP2D6 activity estimates (Fmet × CLCYP2D6) according to subjects’ CYP2D6 activity score (consensus definition). Colors indicate individual CYP2D6 genotypes, and shapes indicate whether the estimate is based on dense (circles) or sparse (triangles) pharmacokinetic sampling. CYP2D6, cytochrome P450 2D6
There appeared to be a reasonably good correlation between CYP2D6 activity estimates and activity scores. However, the activity score 0.25 (assigned to CYP2D6*null/*10) was associated with CYP2D6 activity estimates comparable with or higher than those of activity score 0.5 (assigned to CYP2D6*null/*9 and CYP2D6*null/*41). Similarly, the CYP2D6 activity of the genotypes assigned the activity score 1.25 (CYP2D6*1/*10 and CYP2D6*2/*10) was at level with the activity estimated for the genotypes assigned an activity score of 1.5 (CYP2D6*1/*9, CYP2D6*1/*17, CYP2D6*1/*29, CYP2D6*1/*41, CYP2D6*2/*9, CYP2D6*2/*17, CYP2D6*1/*41).
The CYP2D6 activity estimates originating from sparse PK sampling (indicated by triangular shapes in Figure 4) accounted for most of the outliers within each CYP2D6 activity score. This was particularly evident for activity scores of zero (PM) where three outliers with high CYP2D6 activity were identified. None of the four individuals had extreme values of covariates, which could explain the discrepancy. A plausible explanation for the outliers was a poor estimation of the parameter F met for these individuals as they did not have data available from the absorption phase.
The contribution of individual CYP2D6 alleles to the CYP2D6 activity was estimated by a multiple linear regression model. The results from the final regression model were used to calculate CYP2D6 activity scores relevant for tedatioxetine (see the Methods section).
In the final multiple linear regression model, the CYP2D6 activity estimates were log‐transformed, and the null function (CYP2D6*null: *3, *4, *5, *6, *16) and fully functional alleles (CYP2D6*full: *1, *2) were pooled into two groups as this was shown not to deteriorate model fit. The results from the final linear regression model are presented in Table 3.
TABLE 3.
Estimated CYP2D6 activity for individual CYP2D6 alleles based on multiple linear regression model
| Allele | n b | CYP2D6 activity estimatec | CYP2D6 activity score | 95% CIa |
|---|---|---|---|---|
| CYP2D6*full | 655 | 1.25 | 1 | – |
| CYP2D6*null | 247 | 0.32 | 0 | – |
| CYP2D6*9 | 21 | 0.75 | 0.46 | 0.29–0.60 |
| CYP2D6*10 | 37 | 0.63 | 0.34 | 0.17–0.49 |
| CYP2D6*17 | 6 | 0.33 | 0.01 | −0.14 to 0.13 |
| CYP2D6*29 | 3 | 0.92 | 0.65 | 0.40–1.03 |
| CYP2D6*41 | 99 | 0.52 | 0.21 | 0.12–0.30 |
CI, confidence interval; CYP2D6, cytochrome P450 2D6.
Number of alleles (sum of indicator variables from multiple linear regression).
The CYP2D6 activity estimates were log‐transformed in the multiple linear regression analysis. The table presents the exponentially back‐transformed regression coefficients.
95% confidence intervals calculated using nonparametric bootstrap with 10,000 samples.
A sensitivity analysis excluding the three outliers in the PM group (activity score zero) provided similar results (see Table S2).
The estimated activity score for the CYP2D6*29 allele was slightly higher than what would be expected for a decreased function allele, whereas the activity score of the CYP2D6*17 allele was at level with the null function alleles. It should be noted that these estimates were only based on a limited number of individuals (N = 3 and N = 6, respectively), but that these individuals all originated from phase I studies using dense PK sampling enabling good estimation of PK parameters.
Comparisons of the activity scores estimated for the decreased function alleles showed a significant difference between CYP2D6*29 and CYP2D6*17 (p = 0.04), CYP2D6*9 and CYP2D6*17 (p = 0.02), and borderline significance for the comparison of CYP2D6*9 and CYP2D6*41 (p = 0.05). None of the other comparisons reached statistical significance (p < 0.05).
DISCUSSION
The objective of this analysis was to quantify the CYP2D6 activity of individuals carrying different CYP2D6 genotypes through popPK modeling of tedatioxetine and its metabolite, Lu AA37208, and to estimate the activity associated with individual CYP2D6 alleles.
The analysis included carriers of five different decreased function alleles: CYP2D6*9, CYP2D6*10, CYP2D6*17, CYP2D6*29, and CYP2D6*41. All of the decreased function alleles showed a reduction of CYP2D6 activity compared with fully functional alleles ranging from 35%–99%.
The results showed a good correlation of the CYP2D6 activity estimates from the popPK analysis with CYP2D6 activity scores. However, individuals carrying the downregulated CYP2D6*10 allele (i.e., activity scores 0.25 and 1.25) had higher CYP2D6 activity estimates than expected based on their activity score. This suggests that in the metabolism of tedatioxetine, assigning a lower activity score to CYP2D6*10 compared with other decreased function alleles may not be appropriate.
This is in line with findings from our recent study of vortioxetine where the CYP2D6*10 allele was associated with significantly higher activity compared with the decreased function alleles CYP2D6*17 and CYP2D6*41.8 In the vortioxetine study, an activity score of 0.37 was estimated for CYP2D6*10, which is very similar to the activity score estimated in the current analysis (0.34). These findings suggest that the activity level of the CYP2D6*10 allele in the metabolism of vortioxetine and tedatioxetine is similar.
A surprising finding was that the activity score estimated for the CYP2D6*17 allele was only 0.01, that is, comparable to the activity level of null function alleles. Although the estimate was only based on six individuals, it supports the hypothesis of substrate‐specific behavior of CYP2D6*17.
The CYP2D6*17 allele is characterized by four single nucleotide polymorphisms causing three amino acid substitutions: T107I, R296C, and S486T.9 These changes are believed to affect residues involved in substrate recognition,10 which may explain the substrate‐specific behavior of the CYP2D6*17 allele.
Although CYP2D6*17 is normally interpreted as a reduced function allele, in vitro studies have demonstrated a pronounced substrate‐dependent activity of the allele6, 7 while clinical data have shown a normal or increased metabolic capacity of CYP2D6*17 carriers in the metabolism of risperidone.11 These findings underline the challenge of assigning a universal phenotype to CYP2D6*17, and further research across different substrates is needed to improve our understanding of this allele.
Contrary to CYP2D6*17 and CYP2D6*10, the mutations characterizing the CYP2D6*41 allele cause a reduced enzyme expression rather than changes to substrate binding sites.12 Consequently, the activity of CYP2D6*41 is not expected to be substrate dependent.
In the current study, we estimated an activity score of 0.21 for the CYP2D6*41 allele. This is in line with findings from our recent report on vortioxetine, where an activity score of 0.21 was estimated for CYP2D6*41 using a similar methodology.8
Low metabolic activity of the CYP2D6*41 allele has previously been reported by several authors.4, 13, 14 In a study of 1003 Norwegian patients, CYP2D6*41 carriers were found to have significantly lower CYP2D6 activity compared with carriers of CYP2D6*9 and CYP2D6*10 measured by the metabolic ratio of O/N‐desmethylvenlafaxine.4 Furthermore, a study of 114 patients with tamoxifen‐treated premenopausal breast cancer showed that CYP2D6*41 carriers had endoxifen levels comparable with PMs.13 Recently, authors found evidence that the functional impact of CYP2D6*41 on dose‐adjusted serum levels of patients treated with perphenazine was similar to that of null function alleles.14
Collectively, these results indicate that the CYP2D6*41 allele causes more than a 50% reduction in enzyme activity across a range of substrates and a reduction of the activity score to, for example, 0.25, may better reflect the in vivo activity of the allele.
Evidence suggests that CYP2D6 activity may be affected by MDD through involvement of the hypothalamic pituitary adrenal axis. Both endogenous and exogenous glucocorticoids have been shown to induce CYP2D6 expression in human hepatocytes and in rodent livers in vivo.15, 16 In the covariate analysis of the current study, disease state (healthy vs. MDD) on CYP2D6‐mediated clearance was not identified as a significant covariate. Availability of MDD disease biomarkers would have allowed for a more thorough investigation of the influence of MDD pathophysiology on CYP2D6 activity and the collection of biomarkers (e.g., serum cortisol concentrations) should therefore be considered for future studies.
When associating CYP2D6 phenotypes with genotypes, findings may be affected by the CYP2D6 variant alleles included in the genotyping test panel. In the absence of extensive genotyping or sequencing, rare allele variants may elude detection, and incorrect genotypes may be assigned to individuals. For example, several hybrids and tandems interfere with the CYP2D6*10 allele, and consequently null function alleles such as CYP2D6*36, CYP2D6*57, and CYP2D6*68 will default to CYP2D6*10 unless interrogated.17 Similarly, the null function allele CYP2D6*40 will often be identified as CYP2D6*17, and several alleles will automatically be assigned as CYP2D6*1 (wild type) if not detected.1
In our study, only the most frequent CYP2D6 alleles were tested for, and different panels were used across the studies (see Table 1). It is therefore possible that some alleles in our data set were misclassified, which could bias the results. Furthermore, not all genotyping panels included tests for gene duplication, and 32 subjects were therefore not tested for the presence of multiple gene copies, which could also affect results.
The final popPK model described the data well and was considered stable and reliable based on different diagnostic criteria. The popPK model was based on both dense and sparse PK data originating from seven clinical studies.
The CYP2D6 activity estimates originating from sparse data accounted for most outliers. This might be explained by a poor estimation of the absorption related parameters for the sparse data, as samples from the absorption phase were lacking for these individuals. This hypothesis is supported by the ETA plots in Figure S3, where it appears that shrinkage for the absorption‐related parameters was largely driven by the individuals with sparse data.
Despite generating outliers, the estimates based on sparse data were generally in good agreement with the estimates originating from dense PK data for comparable CYP2D6 genotypes. This indicates that sparse data, despite its limitations, can be used to generate reliable estimates of CYP2D6 activity, particularly in the presence of dense PK data, which facilitates model stabilization.
When quantifying the CYP2D6 activity, it was assumed that the formation of Lu AA37208 from tedatioxetine was exclusively mediated via CYP2D6. However, based on in vitro studies, a minor contribution of CYP2C19 cannot be excluded. As a potential refinement, CYP2C19 genotypes could have been tested as covariates in the popPK model. Unfortunately, CYP2C19 genotypes were not collected in the clinical studies.
In conclusion, the CYP2D6 activity of subjects with a diverse selection of CYP2D6 genotypes was successfully quantified through popPK modeling of tedatioxetine and Lu AA37208. The CYP2D6*10 and CYP2D6*41 alleles were associated with similar activity levels as estimated for vortioxetine, whereas the CYP2D6*17 allele showed an activity level close to that of null function alleles. It should be noted that our results are based on the metabolism of tedatioxetine, and extrapolation of the findings to other CYP2D6 substrates may not be straightforward. Further investigations of other CYP2D6 substrates using high‐quality clinical data are therefore warranted to improve personalized pharmacotherapy with these drugs.
CONFLICT OF INTEREST
Frederiksen T, Areberg J and Schmidt E are employed by H. Lundbeck A/S. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
T.F., J.A., E.S., T.B.S., and K.B. wrote the manuscript. J.A. and E.S. designed the research. T.F. and J.A. performed the research. T.F., J.A., T.B.S., and K.B. analyzed the data.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Supplementary Materials
ACKNOWLEDGMENTS
This research was funded by Innovation Fund Denmark and H. Lundbeck A/S. The data underlying the popPK analysis were provided by H. Lundbeck A/S and Takeda Pharmaceutical Company Ltd. The authors acknowledge Lars Lau Raket for statistical support.
Frederiksen T, Areberg J, Schmidt E, Stage TB, Brøsen K. Cytochrome P450 2D6 genotype–phenotype characterization through population pharmacokinetic modeling of tedatioxetine. CPT Pharmacometrics Syst Pharmacol. 2021;10:983–993. 10.1002/psp4.12635
Funding information
The research was funded by Innovation Fund Denmark and H. Lundbeck A/S.
REFERENCES
- 1.Gaedigk A, Sangkuhl K, Whirl‐Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19:69‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caudle KE, Sangkuhl K, Whirl‐Carrillo M, et al. Standardizing CYP2D6 genotype to phenotype translation: consensus recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin Transl Sci. 2019;13:116‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hicks JK, Swen JJ, Gaedigk A. Challenges in CYP2D6 phenotype assignment from genotype data: a critical assessment and call for standardization. Curr Drug Metab. 2014;15:218‐232. [DOI] [PubMed] [Google Scholar]
- 4.Haslemo T, Eliasson E, Jukic MM, Ingelman‐Sundberg M, Molden E. Significantly lower CYP2D6 metabolism measured as the O/N‐desmethylvenlafaxine metabolic ratio in carriers of CYP2D6*41 versus CYP2D6*9 or CYP2D6*10: a study on therapeutic drug monitoring data from 1003 genotyped Scandinavian patients. Br J Clin Pharmacol. 2019;85:194‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goetz MP, Sangkuhl K, Guchelaar HJ, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and tamoxifen therapy. Clin Pharmacol Ther. 2018;103:770‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen H, He MM, Liu H, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos. 2007;35:1292‐1300. [DOI] [PubMed] [Google Scholar]
- 7.Bogni A, Monshouwer M, Moscone A, et al. Substrate specific metabolism by polymorphic cytochrome P450 2D6 alleles. Toxicol Vitr. 2005;19:621‐629. [DOI] [PubMed] [Google Scholar]
- 8.Frederiksen T, Areberg J, Schmidt E, Bjerregaard Stage T, Brøsen K. Quantification of in vivo metabolic activity of CYP2D6 genotypes and alleles through population pharmacokinetic analysis of vortioxetine. Clin. Pharmacol. Ther. 2020;109:150‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masimirembwa C, Persson I, Bertilsson L, Hasler J, Ingelman‐Sundberg M. A novel mutant variant of the CYP2D6 gene (CYP2D6*17) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br J Clin Pharmacol. 1996;42:713‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin Pharmacokinet. 2009;48:761‐804. [DOI] [PubMed] [Google Scholar]
- 11.Cai WM, Nikoloff DM, Pan RM, et al. CYP2D6 genetic variation in healthy adults and psychiatric African‐American subjects: implications for clinical practice and genetic testing. Pharmacogenomics J. 2006;6:343‐350. [DOI] [PubMed] [Google Scholar]
- 12.Toscano C, Toscano C, Klein K, et al. Impaired expression of CYP2D6 in intermediate metabolizers carrying the *41 allele caused by the intronic SNP 2988G>A: evidence for modulation of splicing events. Pharmacogenet. Genomics. 2006;16:755‐766. [DOI] [PubMed] [Google Scholar]
- 13.Thorén L, Lindh JD, Ackehed G, et al. Impairment of endoxifen formation in tamoxifen‐treated premenopausal breast cancer patients carrying reduced‐function CYP2D6 alleles. Br J Clin Pharmacol. 2020;87:1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waade RB, Solhaug V, Høiseth G. Impact of CYP2D6 on serum concentrations of flupentixol, haloperidol, perphenazine and zuclopenthixol. Br J Clin. Pharmacol. 2021;87:2228‐2235. [DOI] [PubMed] [Google Scholar]
- 15.Ayyar VS, Almon RR, DuBois DC, Sukumaran S, Qu J, Jusko WJ. Functional proteomic analysis of corticosteroid pharmacodynamics in rat liver: Relationship to hepatic stress, signaling, energy regulation, and drug metabolism. J Proteomics. 2017;160:84‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng L, Chen Y, Wang Y, et al. MicroRNA hsa‐miR‐370‐3p suppresses the expression and induction of CYP2D6 by facilitating mRNA degradation. Biochem. Pharmacol. 2017;140:139‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nofziger C, Turner AJ, Sangkuhl K, et al. PharmVar GeneReview: CYP2D6. Clin Pharmacol Ther. 2019; 107:154‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Supplementary Materials