

Abstract
Background
Barth syndrome (BTHS) is a rare X-linked recessive disorder characterized by clinical features including cardiomyopathy, skeletal myopathy, neutropenia, growth delay, and exercise intolerance. It is often considered to be a paediatric disease, owing to most cases being diagnosed during childhood and mortality being the highest during the first few years of life.
Case summary
We report a case of dilated cardiomyopathy due to BTHS in a 27-year-old adult male patient, who initially presented with lightheadedness, dyspnoea, orthopnoea, and bilateral lower extremity oedema. Key findings from investigations included leukopenia, prolonged QTc interval, reduced left ventricular ejection fraction (LVEF), global enlargement of all heart chambers, patent coronary arteries, and mild pulmonary hypertension. The patient was diuresed to euvolemia and discharged with a LifeVest. Guideline-directed medical therapy was initiated and uptitrated as an outpatient. A repeat echocardiogram 2 years after initial presentation showed marked improvement in LVEF.
Discussion
It is possible that there are adult patients with idiopathic cardiomyopathy, which may be attributable to BTHS. In the absence of an obvious underlying cause, with the appropriate historical information, clinical exam, laboratory investigations, and imaging findings, BTHS should be considered as a likely cause of non-ischaemic cardiomyopathy.
Keywords: Barth syndrome, TAZ gene, Tafazzin, Cardiomyopathy, Neutropenia, Case report
For the podcast associated with this article, please visit https://academic.oup.com/ehjcr/pages/podcast.
Learning points
Barth syndrome (BTHS) is a genetic condition with a wide spectrum of clinical features including cardiomyopathy, neutropenia, exercise intolerance, growth delay, 3-methylglutaconic aciduria, and hypocholesterolemia.
In the absence of an obvious underlying cause, with the appropriate historical information, clinical exam, laboratory investigations, and imaging findings, BTHS should be considered as a likely cause of non-ischaemic cardiomyopathy.
Introduction
Barth syndrome (BTHS) is a rare X-linked recessive condition, first described by Peter Barth in 1983, with the classic triad of cardiomyopathy, skeletal myopathy, and neutropenia.1 The pathogenesis is related to mutations in the tafazzin (TAZ) gene, responsible for cardiolipin remodelling, with consequent alteration of normal mitochondrial activity in tissues with high energy demand, such as heart and skeletal muscle.2 BTHS is a multi-system disorder most commonly affecting children, with the highest mortality occurring during the first few years of life.3 This makes it an important entity for geneticists, obstetricians, paediatricians, and cardiologists to be aware of. Families of adult patients living with BTHS have been previously reported, possibly owing to improvements in treatment of cardiomyopathy, but the prevalence is low due to high childhood mortality.2 Initial diagnosis of the disease in adults, however, remains remarkably rare. We present a rare case of a 27-year-old adult man with BTHS and highlight the importance of considering it as a cause of non-ischaemic cardiomyopathy in young adults, if there is no obvious underlying cause.
Timeline
| Presentation | A 27-year-old man presented with lightheadedness, dyspnoea, orthopnoea, and bilateral lower extremity oedema |
| History |
No similar problems in the past No significant past medical history Family history of Barth syndrome No alcohol or recreational drug use Active cigarette smoker |
| Physical exam | Hypotension, tachycardia, jugular venous distension, bilateral lower extremity oedema |
| Diagnostic workup | Leukopenia, neutropenia, monocytosis, hypocholesterolemia |
| Electrocardiogram: sinus tachycardia, left atrial enlargement, prolonged QTc interval | |
| Echocardiogram: left ventricular ejection fraction (LVEF) 15–20%, global enlargement of all chambers, mild pulmonary hypertension | |
|
Left heart catheterization: patent coronary arteries Right heart catheterization: mean pulmonary artery pressure 16 mmHg, cardiac index 2.02 L/min/m2 |
|
| Non-ischaemic cardiomyopathy workup, diuresis to euvolemia | |
| Discharge |
LifeVest wearable defibrillator Medications: furosemide, carvedilol, sacubitril-valsartan |
| Outpatient follow-up | Cardiology follow-up; addition of spironolactone to medication regimen |
| Cardiac magnetic resonance imaging non-suggestive of infarction, infiltrate, or other abnormal enhancement; diagnosis confirmed by genetic testing | |
| Regular follow-up with advanced heart failure cardiologist | |
| 2 years later | Echocardiogram: LVEF improved to 45–50% |
Case presentation
A 27-year-old man presented to the emergency room with complaints of lightheadedness, dyspnoea, orthopnoea, and bilateral lower extremity oedema over the course of a few weeks. His symptoms were associated with chest discomfort and he denied similar past episodes. He had no significant past medical history. Family history was remarkable for a brother diagnosed with BTHS by genetic testing. The patient denied alcohol use and recreational drug use. He endorsed active cigarette smoking (0.33 packs per day). On presentation, the patient was afebrile with a temperature of 36.7°C, hypotensive to 86/54 mmHg, tachycardic to 104 beats per minute (b.p.m.), breathing at a rate of 18 breaths per minute, with an oxygen saturation of 96% on room air. Physical examination was remarkable for diminished breath sounds at the lung bases bilaterally, tachycardia with regular rhythm, jugular venous distension to the earlobe, and 2+ pitting oedema in the lower extremities bilaterally. Complete blood count revealed leukopenia (2.9 k/μL; reference range: 4.0–10.8 k/μL), neutropenia (27.7%; reference range: 43.0–75.0%), and monocytosis (37.1%; reference range: 3.0–12.0%). Lipid panel showed total cholesterol of 91 mg/dL (reference range: 120–200 mg/dL), with a calculated LDL of 44 mg/dL (reference range: 0–99 mg/dL), and an HDL of 22 mg/dL (reference range: 40–59 mg/dL). Initial electrocardiogram showed sinus tachycardia with a ventricular rate of 128 b.p.m., criteria for left atrial enlargement, and a prolonged QTc interval of 589 ms (Figure 1). The patient was admitted to the hospital for further evaluation, diagnostic interventions, and treatment. Echocardiographic exam revealed enlargement of all four chambers (Figure 2 and Video 1), with significant dilatation of the left ventricle (end-diastolic diameter 6.0 cm, end-systolic diameter 5.8 cm) and severely reduced left ventricular ejection fraction (LVEF) of 15–20%. Additionally, mild pulmonary hypertension was present. Ischaemic workup with coronary angiography revealed patent vessels (Figure 3 and Video 2), and ventriculogram showed an LVEF of 20%. Right heart catheterization was remarkable for pulmonary arterial pressure of 26/11 mmHg (mean of 16 mmHg), a calculated Fick cardiac output of 3.18 L/min (normal range: 4–8 L/min) and a cardiac index of 2.02 L/min/m2 (normal range 2.5–4 L/min/m2). HIV testing was negative, ethanol level was within normal limits, thyroid stimulating hormone (TSH) was mildly elevated at 5.12 μIU/mL (reference range: 0.4–4.0 μIU/mL), with free T4 within normal limits at 1.16 ng/dL. Based on the history of present illness, family history, physical examination findings, laboratory investigations, and diagnostic imaging results, the patient could likely have a diagnosis of BTHS. He was carefully diuresed to euvolemia, discharged in stable condition with a LifeVest wearable defibrillator for 3 months, and furosemide 20 mg per orum (PO) daily. He was also started on guideline-directed medical therapy including carvedilol 3.125 mg PO twice a day and sacubitril-valsartan 24–26 mg PO twice a day. At a 6-month follow-up visit, the patient had no further complaints, remained compliant with his discharge medications, and spironolactone 12.5 mg PO daily was added. An outpatient cardiac magnetic resonance imaging study with and without contrast for evaluation of infiltrative disease and other causes of non-ischaemic cardiomyopathy was non-suggestive of myocardial infarction, infiltrates, or other abnormal enhancement by late gadolinium enhancement imaging. The patient’s diagnosis was confirmed with outpatient genetic testing for TAZ gene mutation, and he continued to regularly follow-up with an advanced heart failure cardiologist. A repeat echocardiogram (Video 3) 2 years after the initial hospitalization showed evidence of cardiac remodelling with improvement in left ventricular size (end-diastolic diameter 5.02 cm, end-systolic diameter 4.06 cm) and LVEF (45–50%).
Figure 1.

Electrocardiogram showing sinus tachycardia with a ventricular rate of 128 b.p.m., criteria for left atrial enlargement, and a prolonged QTc interval of 589 ms.
Figure 2.
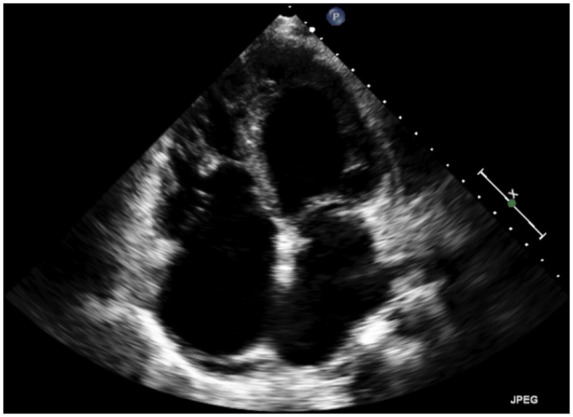
Initial echocardiogram (apical window) revealing global enlargement of all four cardiac chambers.
Figure 3.

Coronary angiogram revealing patent coronary arteries.
Discussion
BTHS is a rare X-linked recessive condition, with <200 living males worldwide, caused by disabling mutations or deletions in the TAZ gene.2 These genetic alterations contribute to the pathophysiology of BTHS by reducing remodelling of cardiolipin, a principal phospholipid component of the inner mitochondrial membrane, in turn compromising the function of the electron transport chain and aerobic respiration.4 This leads to prolonged deficiency in ATP production capacity, the underlying process of tissue pathology that eventually manifests as cardiomyopathy including dilated cardiomyopathy or left ventricular non-compaction.4,5 Clinically, BTHS is characterized by a broad spectrum of features including dilated cardiomyopathy, left ventricular non-compaction, prolonged QTc interval, exercise intolerance, growth delay, 3-methylglutaconic aciduria, and hypocholesterolemia. Several other rare cardiomyopathic phenotypes have also been previously described including endocardial fibroelastosis, hypertrophic cardiomyopathy, and an apical form of hypertrophic cardiomyopathy.2 Other non-cardiac sequalae of BTHS may include attention deficits, recurrent aphthous ulcers, sore gums, delayed puberty, and recurrent infections.2 Our patient demonstrated several common clinical features of BTHS including dilated cardiomyopathy, QTc prolongation, neutropenia with compensatory monocytosis, and hypocholesterolemia. Neutropenia is usually one of the earliest manifestations of the disease, and present in 90% of patients.2
Prompt diagnosis is crucial, leading to early intervention for patients, and consequently earlier steps towards improving their quality of life. It has been previously reported that routine use of cardiac medications for patients with cardiac complications of BTHS resulted in progressive improvement of the LVEF.6 Additionally, the prognostic implications of an earlier diagnosis are very well supported by lower mortality outcomes in prospectively identified patients with proactive management of disease.2,7
The diagnosis of BTHS can be considered based on clinical features, and molecular testing of TAZ mutations would be confirmatory. In the USA, each year ∼10 patients are diagnosed with BTHS.2 There is, however, growing evidence that the disease quite possibly may be underdiagnosed, including a mutation analysis study which identified five unrelated families with BTHS in one hospital over the course of a 7-year period.8 Considering these findings, it is possible that there are adult patients with idiopathic cardiomyopathy, which may be attributable to BTHS. In the absence of an obvious underlying cause, with the appropriate historical information, clinical exam, laboratory investigations, and imaging findings, BTHS should be considered as a likely cause of non-ischaemic cardiomyopathy. Additionally, such patients would require a multidisciplinary team approach in management, due to the wide variety of organ systems which can be potentially involved.
Lead author biography

Shiavax Rao is a resident physician at the MedStar Health Internal Medicine Residency Program in Baltimore, Maryland (USA). He graduated with his M.D. degree from Saba University School of Medicine. Following residency training, he is interested in pursuing a fellowship in Cardiovascular Disease.
Supplementary material
Supplementary material is available at European Heart Journal - Case Reports online.
Slide sets: A fully edited slide set detailing this case and suitable for local presentation is available online as Supplementary data.
Consent: The authors confirm that written consent for submission and publication of this case report including images and associated text has been obtained from the patient in line with COPE guidance.
Conflict of interest: None declared.
Funding: None declared.
Supplementary Material
References
- 1.Barth PG, Scholte HR, Berden JA, Van Der Klei-Van Moorsel JM, Luyt-Houwen IEM, Van’T Veer-Korthof ETh. et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 1983;62:327–355. [DOI] [PubMed] [Google Scholar]
- 2.Clark SLN, Bowron AB, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N. et al. Barth syndrome. Orphanet J Rare Dis 2013;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz FM. et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A 2004;126A:349–354. [DOI] [PubMed] [Google Scholar]
- 4.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJA. et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 2000;279:378–382. [DOI] [PubMed] [Google Scholar]
- 5.Dudek J, Maack C.. Barth syndrome cardiomyopathy. Cardiovasc Res 2017;113:399–410. [DOI] [PubMed] [Google Scholar]
- 6.Reynolds S. Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach. J Multidiscip Healthc 2015;8:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huhta JC, Pomerance HH, Barness EG.. Clinicopathologic conference: Barth syndrome. Fetal Pediatr Pathol 2005;24:239–254. [DOI] [PubMed] [Google Scholar]
- 8.Cantlay AM, Shokrollahi K, Allen JT, Lunt PW, Newbury-Ecob RA, Steward CG.. Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 1999;135:5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.