

Abstract
Proteins possess unique molecular recognition capabilities and enzymatic activities, features that are usually tied to a particular tertiary structure. To make use of proteins for biotechnological and biomedical purposes, it is often required to enforce their tertiary structure in order to ensure sufficient stability under the conditions inherent to the application of interest. The introduction of intramolecular crosslinks has proven efficient in stabilizing native protein folds. Herein, we give an overview of methods that allow the macrocyclization of expressed proteins, discussing involved reaction mechanisms and structural implications.
Keywords: disulfide mimetics, expressed protein ligation, INCYPRO, split inteins, unnatural amino acids
Protein function is often tightly linked to a particular tertiary structure. For many protein applications it is required to stabilize the tertiary structure and prevent unfolding. Here, we summarize strategies that have been used to cyclize expressed proteins aiming for the stabilization of their native fold.

Introduction
Proteins possess unique folding characteristics and are central components of biological systems. The three‐dimensional structure of folded proteins is determined by constraints of backbone flexibility and by intramolecular interactions that involve both backbone and side chain atoms.[1] Usually, a particular tertiary structure is required for protein functionality, such as the engagement in molecular recognition processes or enzymatic activity. Proteins have been exploited for various biotechnological and biomedical applications, but their applicability is often hampered due to limited stability under the required conditions.[2] For example, thermal and chemical stress can result in protein unfolding and consequently loss of function. To increase the usability of proteins, it is therefore of central importance to stabilize their tertiary structure.[3] This has been achieved by optimizing intradomain interactions e. g. via directed evolution, consensus based mutagenesis, computational design or the introduction of unnatural amino acids.[4, 5, 6, 7, 8]
In nature, Protein tertiary structures are additionally stabilized by disulfide bridges between two closely aligned cysteines,[9] and in rare cases via head‐to‐tail cyclization.[10] The resulting proteins possess cyclic topologies and often show a reduced tendency to unfold under thermal and chemical stress. In particular, the incorporation of additional disulfide bonds has been pursued to engineer proteins with sometimes considerably increased stability.[9] However, disulfide engineering remains challenging since the short length of the cystine crosslink limits the number of suitable modification sites.[9, 11] This has triggered the search for alternative protein macrocyclization approaches which, to some extent, utilize methodologies originally developed for the synthesis of cyclic peptides.[12, 13] Compared to peptides, the cyclization of proteins, however, possesses additional challenges which are mainly linked to the larger molecular weight of proteins and the difficulties to introduce unnatural amino acids. Both aspects complicate the design of sufficiently selective and reactive crosslinking reagents. Further, compared to peptides, the crosslinking of folded proteins requires more structural insight. Herein, we give an overview of strategies that have been used to cyclize expressed proteins in order to stabilize their tertiary structure. We present the mechanisms of involved cyclization reactions and discuss structural implications.
Disulfide Rebridging
Strategies that take advantage of existing disulfide bridges and convert them into chemically modified crosslinks usually aim at the introduction of additional functionalities and/or the generation of chemically inert structures.[14, 15] Efforts have particularly focused on the rebridging of inter‐chain disulfides within antibodies[16] utilizing bifunctional crosslinkers.[17, 18, 19, 20, 21] The rebridging of intra‐chain disulfides has been less pursued. In general, the first step involves the reduction of the existing disulfide (1, Figure 1a) usually by treatment with tris(2‐carboxyethyl)phosphine (TCEP). The free cysteines (2) can then react with a bis‐electrophilic reagent (3) which results in intramolecular crosslink formation to provide the rebridged protein (4, Figure 1a).[22, 23, 24] One approach involves the use of an α,β‐unsaturated‐β’‐monosulfone (3 a, Figure 1b).[22] Crosslinking proceeds initially via thiol addition to the monosulfone, after which sulfinic acid is eliminated to form a double bond. The second thiol addition then provides the final crosslink (4 a). In the reported example, the monosulfone bis‐electrophile bears a poly(ethylene glycol) (PEG) chain, which can promote protein biostability. This approach was applied for the cyclization of e. g. the protein ligand interferon‐α2b and the enzyme L‐asparaginase, providing modified versions with retained tertiary structure and biological activity. In an alternative approach, 3,6‐dichloro‐1,2,4,5‐tetrazine (3 b, Figure 1b) was used as bis‐electrophile allowing reactions at relatively low pH value.[23] Tetrazine crosslinks (4 b) have been introduced into a thioredoxin (Trx) protein and were shown to be photolytically removable. In another strategy, a 3,3‐bis(bromomethyl)oxetane reagent (3 c) was used to rebridge disulfide bonds (4 c, Figure 1b) in detoxified diphtheria toxin CRM and the Trx protein.[24] Rebridging of the thiols with 3 c resulted in increased stability and bioactivity. Notably for the diphtheria toxin, it was shown that an inaccessible disulfide within the protein core was not modified under applied conditions.
Figure 1.
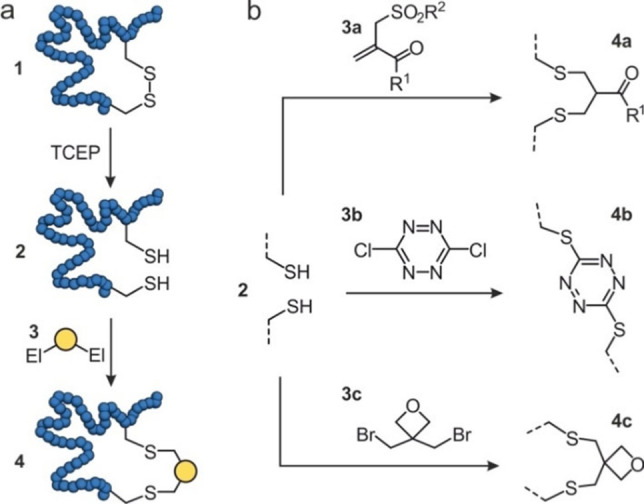
Schematic overview of disulfide rebridging: (a) A protein (blue) containing a disulfide (1) is reduced to thiols (2) by tris(2‐carboxyethyl)phosphine (TCEP), followed by a reaction with a bis‐electrophile (3) resulting in a rebridged protein (4). (b) Different bis‐electrophiles have been reported: α,β‐unsaturated‐β’‐monosulfone (3 a), 3,6‐dichloro‐1,2,4,5‐tetrazine (3 b), 3,3‐bis(bromomethyl)oxetane (3 c) to form new disulfide bridges 4 a‐4 c, respectively.
Head‐to‐Tail Cyclization
Head‐to‐tail (or backbone) cyclizations involve a lactam formation between the N‐ and C‐terminus of a protein. In an early proof of principle, head‐to‐tail cyclization was achieved for a bovine pancreatic trypsin inhibitor by the activation of its C‐terminus with a carbodiimide and subsequent intramolecular amide formation with the N‐terminal amine.[25] An alternative chemical cyclization approach involves the cyanylation of a C‐terminal cysteine, thereby converting it into a leaving group and facilitating head‐to‐tail cyclization with the N‐terminus.[26] This reaction was applied to cyclize dihydrofolate reductase, which resulted in increased stability towards thermal and chemical denaturation. The generation of activated protein C‐termini can also be accomplished using intein‐based strategies. Inteins are protein domains that possess auto‐cleavage activity and allow the fusion of their flanking N‐ and C‐terminal extein sequences. This process is called protein cis‐splicing and results in excision of the intein.[27] The so‐called expressed protein ligation (EPL) takes advantage of the ability of inteins to promote an N‐to‐S acyl transfer, thereby converting an amide bond (5, Figure 2a) into a thioester (6). Thereafter, reaction with a thiol promotes trans‐thioesterification and cleavage of the intein sequence (8). The N‐terminal cysteine then undergoes an intramolecular native chemical ligation with the C‐terminal thioester to provide the head‐to‐tail cyclized protein (7, Figure 2a). An early example of EPL‐mediated protein cyclization employed the Sce VMA intein for the cyclization of SH3 domains and of β‐lactamase which, in most cases, resulted in proteins with increased stability.[28, 29, 30] Using a different intein (Mxe GyrA), the effects of SH3 domain cyclization were analysed later in more detail revealing that cyclization‐induced strain can also diminish the stabilizing effect.[31]
Figure 2.

Schematic overview of intein‐based head‐to‐tail cyclizations (blue: protein of interest): (a) Expressed protein ligation (EPL) involving N‐to‐S acyl transfer to convert an amide bond (5) to a thioester (6). This is followed by trans‐thioesterification with a thiol to release the intein sequence (8, yellow) and subsequent intramolecular native chemical ligation to form the cyclic protein (7). (b) Split intein‐based cyclization first involves assembly of the N‐ and C‐terminal intein fragments (9, orange and yellow), after which an N‐to‐S acyl transfer takes place to form thioester 10. Next, trans‐esterification with an intramolecular nucleophile (Y=S or O) occurs to form a lactone intermediate (11) to release the C‐terminal intein fragment (12). Finally, Y‐to‐N acyl transfer cleaves the N‐terminal intein (13) and forms cyclic protein 14.
The head‐to‐tail ligation in EPL‐mediated cyclizations requires an N‐terminal cysteine which then spontaneously attacks the formed thioester (6 to 7, Figure 2a). This can limit cyclization efficiency. An intein‐based cyclization approach that supports the correct alignment of the N‐terminus applies protein trans‐splicing and is also called SICLOPPS (split intein circular ligation of peptides and proteins).[32] This protein cyclization takes advantage of inteins that assemble from two fragments (split inteins), which are fused to the protein N‐ and C‐terminus, respectively. If protein termini are located in proximity and if necessary appropriate linker sequences have been included, the split intein fragments assemble into the active splicing domain (9, Figure 2b). Similar to EPL, formation of a thioester (10) represents the initial step, which is then, however, followed by intramolecular trans‐esterification with a particular cysteine or serine side chain (Y=S or O). This results in the formation of a lactone intermediate (11) and release of the C‐terminal intein sequence (12, Figure 2b). Subsequently, the N‐terminal intein is cleaved under aspartimide (13) formation providing the cyclized protein (14).[32] To date, several split inteins from different organisms have been used for protein cyclization. For example, DnaB and DnaE split inteins were retrieved from the cyanobacterium Synechocystis sp. Strain PCC6803.[33, 34] Early examples involve the use of the DnaE split intein for the cyclization of a dihydrofolate reductase and a maltose binding protein.[32, 35] Based on the DnaB intein, a mini‐intein was engineered which has been utilized to cyclize various protein targets including the DnaB protein, a binary protein complex (FLINC4) and the enzyme Sortase A.[36, 37, 38] In all three examples, the thermostability was improved. For cyclized Sortase A, increased enzymatic activity was observed under chemical stress (3 M urea). Another example, using the PI‐Pful split intein from Pyrococcus furiosus, involves the cyclization of the green fluorescent protein (GFP) and of an endoglucanase both experiencing increased thermostability.[39, 40] Furthermore, a DnaE split intein was retrieved from Nostoc Punctiforme showing increased efficiency when compared to the DnaE intein from Synechocystis sp. Strain PCC6803.[41] This split intein has been utilized to cyclize granulocyte colony‐stimulating‐factor thereby enhancing its thermostability.[42]
Protein head‐to‐tail cyclization has also been performed using peptidases.[43, 44, 45] For example, cysteine transpeptidases recognize their target sequence to initially cleave a peptide bond within the protein (15) which results in the formation of an activated thioester (17, Figure 3a). If aligned appropriately, the N‐terminus of the target protein then attacks the thioester intramolecularly to provide the cyclic protein (18, Figure 3a). Transpeptidases used to generate cyclic proteins are Sortase A and Butelase 1. Sortase A from Staphylococcus aureus recognizes a five amino acid peptide sequence (L−P‐aa‐T−G, aa=any amino acid) and cleaves the amide between threonine (T) and glycine (G, 15 a, Figure 3b). The N‐terminus of the protein must contain an oligoglycine (15 a) for cyclization to occur (18 a, Figure 3b). Cyclization efforts with Sortase A were successful for different proteins, including Cre recombinase, GFP, human interferon α and granulocyte colony‐stimulating‐factor 3.[46, 47, 48] More recently, the robustness of Sortase A has been improved, which can be expected to support the future application of Sortase A for head‐to‐tail cyclizations.[49, 50] Butelase 1 from Clitoria ternetea recognizes a short peptide sequence (a3‐H−V; a3=asparagine (N) or aspartic acid (D)) and cleaves the amide bond between position a3 and histidine (H, 15 b, Figure 3c).[51] To facilitate cyclizations, the first N‐terminal amino acid (a1) can be any amino acid, except for proline (P), while the second amino acid (a2) must be isoleucine (I), leucine (L), valine (V) or cysteine (C, 15 b, Figure 3c).[52] Increased thermostability through Butelase 1 cyclization has been reported for an interleukin 1 receptor antagonist and a murine dihydrofolate reductase.[51, 53] It has been shown that cyclizations with Butelase 1 can occur with higher efficiency and faster kinetics, compared with Sortase A.[51]
Figure 3.
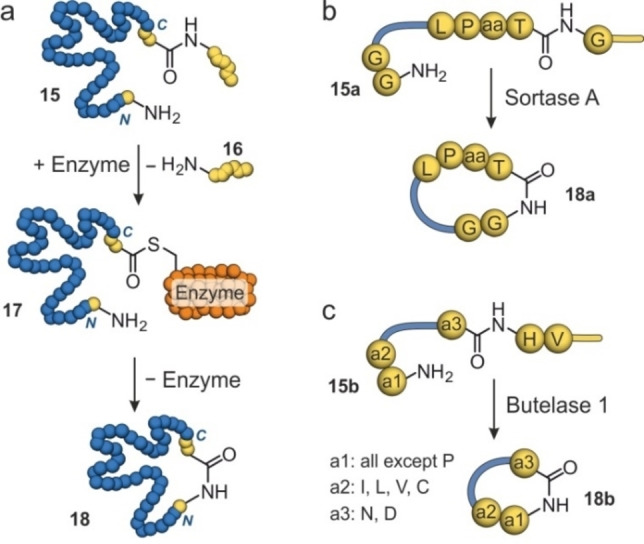
Schematic overview of enzymatic cyclization with Sortase A and Butelase 1: (a) The cysteine transpeptidase cleaves an amide bond in the recognition sequence of the protein (15, blue) forming a thioester intermediate (17) and releasing the C‐terminal sequence (16, yellow). Subsequently, the reaction of the N‐terminus with the thioester results in the cyclized protein (18). (b) Sortase A requires L−P‐aa‐T−G as the recognition motif and an oligoglycine at the N‐terminus (15 a) to generate a cyclic protein (18 a). (c) Butelase 1 requires a3‐H−V as the recognition motif and a1‐a2 at the N‐terminus (15 b) to generate a cyclic protein (18 b).
Isopeptide‐Mediated Cyclization
The pilin structures from Streptococcus pyogenes (Spy0128) undergo spontaneous isopeptide bond formation between a lysine and an asparagine side chain.[54] Both amino acids are embedded in a particular sequence context that facilitates the spatial alignment of a glutamic acid side chain which mediates amide formation. Taking advantage of this ligation reaction, a protein labelling strategy has been developed, which was later also applied to perform protein cyclizations. In this setup, the Pilin‐C (282 amino acids) and the Isopeptag (16 amino acids) sequences have been fused to the protein N‐ and C‐terminus, respectively (Figure 4).[55] In addition to the Pilin‐C/Isopeptag pair, other lysine and asparagine‐containing tags have been designed and used for protein cyclization, such as SnoopTag/SnoopCatcher (Figure 4).[56, 57] An alternative and frequently used pair of tags for protein cyclization involves the SpyTag (13 amino acids) and SpyCatcher (138 amino acids), originating from the collagen adhesin domain in Streptococcus pyogenes.[58] SpyTag utilizes an aspartic acid which undergoes isopeptide bond formation with a lysine side chain in the SpyCatcher (Figure 4). Several proteins have been cyclized with the SpyTag fused to their N‐terminus and SpyCatcher to the C‐terminus of the protein resulting in proteins with increased thermostability.[59, 60, 61, 62, 63, 64, 65] In some examples, SpyTag has been fused to the C‐terminus and SpyCatcher to the N‐terminus, also resulting in cyclic proteins with increased thermostability.[66, 67]
Figure 4.
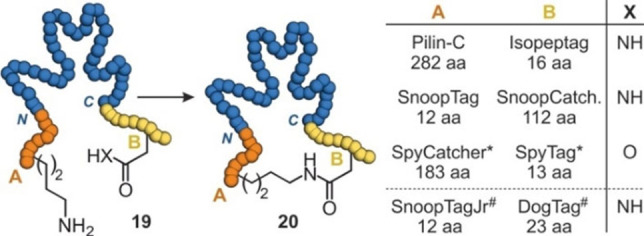
Isopeptide bond formation with different pairs of tags: (a) Protein (19, blue) with N‐ and C‐terminal tags containing a lysine (A, orange) and an asparagine or aspartic acid (B, yellow), respectively, is cyclized (20). (b) Table with pairs of tags including the corresponding numbers of involved amino acids. * SpyCatcher and SpyTag have been fused at both N‐ and C‐terminus. # Isopeptide formation requires the presence of SnoopLigase.
One study compares the performance of the three pairs Pilin‐C/Isopeptag, SpyTag/Catcher and SnoopTag/Catcher for the cyclization of β‐lactamase.[59, 60] In this study, cyclization with SpyTag/Catcher proceeded most efficiently and resulted in a β‐lactamase version with reduced tendency for aggregation at high temperatures. Other studies comparing protein cyclization via SpyTag/Catcher and SnoopTag/Catcher also observed increased tolerance towards thermal stress for the SpyTag/Catcher pair.[64, 68] More recently, the RrgA domain of Streptococcus pneumoniae has been split into three domains providing SnoopTagJr (12 amino acids), DogTag (23 amino acids) and SnoopLigase (104 amino acids). SnoopTagJr and DogTag were fused to the N‐ and C‐terminus of a protein, respectively (Figure 4). Only upon addition of SnoopLigase rapid protein cyclization was observed.[57] Notably, for the two tested enzymes, β‐lactamase and phytase C, the degree of stabilization with SnoopTagJr/DogTag exceeded what was earlier observed for the SpyTag/Catcher pair.[59, 60]
Unnatural Amino Acids
The introduction of unnatural amino acids (uaa) into proteins allows the installation of non‐proteinogenic functionalities.[69] One of the strategies to incorporate unnatural amino acids involves the suppression of the amber stop codon and its recognition by an orthogonal tRNA. This requires the availability of corresponding tRNA synthases and can come at the cost of lower expression yields.[70] While early examples focused on the introduction of bioorthogonal reactive groups, it has also been employed to incorporate functionalities that allow intramolecular reactions with natural amino acids (Y, Figure 5a). For example, artificial disulfide bridges have been installed by the introduction of thiol‐containing derivatives of tyrosine differing in the lengths of the thiol‐alkyl chain (21 a, Figure 5b). Disulfide formation with a cysteine in spatial proximity then resulted in crosslink formation (22 a).[71] This approach overcomes the geometrical constraints of natural cysteine disulfides, which only span relatively short distances. The introduction of this artificial disulfide in β‐lactamase increased its thermal stability. Notably, the degree of stabilization was dependent on the length of the alkyl linker providing increases in the protein melting temperature (T m) of up to 9 °C.
Figure 5.
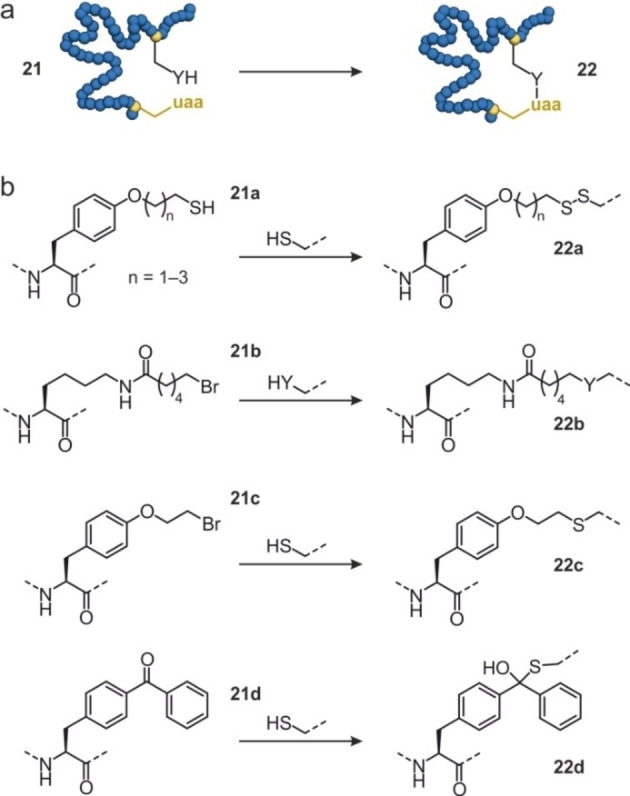
Protein cyclization utilizing unnatural amino acids: (a) Schematic overview of protein 21 with an unnatural amino acid (uaa) which undergoes an intramolecular reaction with a natural amino acid to form cyclic protein 22. (b) Overview of unnatural amino acids used for protein cyclization: O‐(2‐mercaptoalkyl)‐L‐tyrosine (21 a), (S)‐2‐amino‐6‐(6‐bromohexanamido)‐hexanoic acid (21 b), O‐2‐bromoethyl tyrosine (21 c) and (p‐benzoylphenyl)alanine (21 d). They react with ‐SH (cysteine) or ‐YH (cysteine, histidine or lysine) to form the new bridges 22 a–d.
Alternatively, electrophilic unnatural amino acids have been introduced to undergo crosslinking reactions with nearby nucleophilic natural residues (21 b‐d, Figure 5b).[72, 73, 74] For instance, (S)‐2‐amino‐6‐(6‐bromohexanamido)hexanoic acid (21 b, Figure 5b) was shown to react with cysteine, histidine and lysine (22 b).[72] This approach was used for the cyclization of an affibody exhibiting high crosslinking efficiency, however at the cost of possible inhomogeneities in the cyclized product. Later, a computational approach was employed to identify potential crosslinking positions in a myoglobin‐based cyclopropanation biocatalyst. After the incorporation of O‐2‐bromoethyl tyrosine (21 c, Figure 5b) and an appropriately aligned cysteine (22 c), crosslink formation was observed.[73] Depending on the crosslink position within the protein, different stabilizing effects were observed (T m increase of 4–10 °C). Notably, when combining two of these crosslinks, protein stability was further increased (T m increase of 17 °C). In addition, enhanced stability towards chemical denaturation was observed while retaining catalytic activity and stereoselectivity.[73] Intramolecular reaction with a cysteine side chain has also been achieved by incorporation of (p‐benzoylphenyl)alanine (21 d, Figure 5b) resulting in the formation of a hemithioketal crosslink (22 d). For a homoserine O‐succinyltransferase, cyclization resulted in considerably increased thermal stability (T m increase of 21 °C).[74]
In Situ Cyclization
The site‐specific crosslinking of proteins entirely composed of natural amino acids represents an appealing strategy. In an early example, 1,5‐difluoro‐2,4‐dinitrobenzene was used to bridge a pair of solvent exposed lysine residues in ribonuclease A thereby increasing the protein's thermal stability.[75, 76] Due to the high frequency of lysine on protein surfaces and the resulting selectivity issues, later, the targeting of cysteines moved into the focus. In analogy to the bicyclization of peptides,[77, 78, 79] C 3‐symmetric tris(bromomethyl)benzene was used to cyclize a de novo designed, artificial mini‐protein exposing three cysteines.[80] To facilitate the stabilization of natural tertiary structures, larger and water‐soluble tris‐electrophilic agents have been developed and used for the in situ cyclization of proteins (INCYPRO).[50, 81] Here, three surface‐exposed cysteines are introduced (23, Figure 6a), preferably within different secondary structure elements.[50, 81] Initially, a hydrophilic tris‐amine core (Z1) was decorated with chloroacetamide (24 a)[82, 83] and used to generate a bicyclic version of Sortase A. Notably, the INCYPRO reaction was selective for the three introduced cysteines tolerating the presence of an active‐site cysteine.[50] Bicyclic Sortase A exhibited increased thermal stability and performed labelling reactions under denaturing conditions. Later, a library of crosslinkers was composed using three different electrophiles (24 a‐24 c, Figure 6b) which were combined with three different C 3‐symmetric core structures (Z1‐Z3, Figure 6c).[81] The resulting nine tris‐electrophilic crosslinkers have then been used to cyclize the human KIX domain. All crosslinkers furnished bicyclic KIX domains with increased stability (T m increase of 19–29 °C), with hydrophilic crosslink Z1/25 a providing the most pronounced stabilizing effect. Interestingly, it was also found that the degree of protein stabilization correlated with the hydrophilicity of the crosslink.
Figure 6.
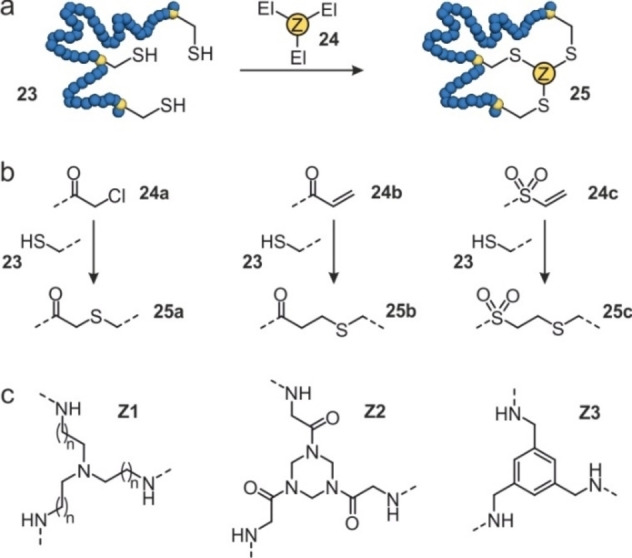
In situ cyclization of proteins (INCYPRO): (a) Schematic overview of protein (blue) with three cysteines (23) that can react with a tris‐electrophile (24) to form a bicyclic protein (25). (b) Chemical structures of the electrophiles attached to the core of the tris‐electrophile: chloroacetamide (24 a), acrylamide (24 b), and vinyl sulfonamide (24 c). They can react with cysteines to form 25 a–c. (c) Chemical structures of the core of the tris‐electrophile based on triethylamine (Z1), triazinane (Z2) and benzene (Z3).
Conclusions
To execute their biological function, proteins often require to adopt a folded state. For biotechnological and biomedical applications, it is usually necessary to stabilize the protein tertiary structure to prevent unfolding. Macrocyclization represents an efficient strategy towards the stabilization of protein tertiary structures. In fact, natural proteins already feature cyclic topologies, for example through the installation of disulfide bridges. Utilizing this feature, disulfide rebridging approaches have been pursued which usually do not aim for further tertiary structure stabilization, but for the selective introduction of additional functionalities or the generation of redox insensitive crosslinks. However, agents used for disulfide rebridging can also be expected to allow the crosslinking of newly installed cysteines. The remaining macrocyclization approaches described above aim for the installation of new intramolecular crosslinks and differ in particular in respect to i) the structural requirements for their introduction into the protein, ii) the level of modification caused within the protein, iii) the complexity of the methodology, and iv) the variability of the crosslink.
i) Structural requirements: All presented cyclization approaches require some degree of insight into the 3D structure of the protein. For example, head‐to‐tail cyclizations utilize protein termini that are located in proximity, and therefore in some cases linker sequences have to be introduced. Isopeptide bond‐mediated cyclizations are less sensitive to the orientation of the termini, as relatively large peptide tags are introduced. Crosslinking via unnatural amino acids or INCYPRO provides a large number of possible bridging sites, however, the protein structure or a precise homology model is usually required for the design process.
ii) Level of modification within the protein: With the exception of disulfide rebridging, all cyclization approaches require the variation of the protein sequence. The largest tags are introduced to facilitate isopeptide bond formation, while all other approaches usually only cause the variation or addition of a few amino acids. Intein‐based as well as enzymatic cyclization approaches involve specific amino acid sequences at the ligation site and sometimes require the introduction of additional linker sequences to allow head‐to‐tail ligation.
iii) Complexity of the methodology: Cyclizations via split inteins, isopeptide bond‐forming tags and unnatural amino acids mainly rely on biotechnological techniques required to prepare protein variants, while the actual crosslink formation proceeds spontaneously. The incorporation of unnatural amino acids for crosslinking involves additionally engineered cellular expression systems which tend to provide lower protein yields. The remaining approaches involve a separate crosslinking step.
iv) Variability of crosslink: The nature of the introduced crosslink can affect protein properties. For that reason, it may be advantageous to test various crosslink architectures also considering structures that go beyond the natural repertoire. Here, the use of unnatural amino acids and the INCYPRO approach are appealing. In particular, INCYPRO provides straight‐forward access to diverse linker structures.[81] In general, it can be expected that more cyclization strategies will be developed which allow the simultaneous introduction of an additional functionality (e. g. PEG chains[84] or photo‐switchable moieties[85, 86]).
To compare structural prerequisites of different protein cyclization approaches, we selected β‐lactamase and Sortase A as examples. Both are enzymes and therefore also illustrate effects of tertiary structure stabilization on enzymatic activity. β‐Lactamases are single domain enzymes (Figure 7a) that support bacterial resistance towards β‐lactam antibiotics.[87] In an early example, a Sce VMA derived intein was used for EPL‐mediated macrocyclization and resulted in the introduction of a 15 amino acid sequence between the original domain termini (Figure 7a, top).[30] The cyclic enzyme exhibited a 5 °C higher T m‐value, however, no activity tests at elevated temperatures were reported. Later a series of isopeptide‐mediated cyclizations were described using various tag systems. Here, in particular, the refolding after thermal denaturation was studied revealing a lower tendency for aggregation for the cyclic versions.[57, 59, 60] The smallest reported tag system (SnoopTagJr/DogTag, Figure 7a, middle) resulted in a β‐lactamase version retaining solubility and activity following heat treatment up to 100 °C.[57] Screening a library of β‐lactamases with deliberately positioned cysteines and randomly introduced thiol‐containing unnatural amino acids provided an enzyme containing a disulfide between tyrosine derivative SbuY and a cysteine (Figure 7a, bottom). Its T m‐value was increased by 9 °C when compared to the wildtype.[71] Even though, the crosslink was located opposite to the enzyme's active site, the cyclic version showed lower activity than the wildtype under native conditions. At 40 °C, however, the wildtype enzyme was inactive while the cyclic version retained its activity.
Figure 7.

Structures of cyclized enzymes (blue spheres: active site residues, yellow: protein termini, orange: crosslinking sites). Arrows indicate the ligation site for macrocyclization: (a) Crystal structure of β‐lactamase (TEM‐1, PDB ID 1fqg)[88] showing the introduced modifications resulting from macrocyclizations via EPL (top),[30] isopeptide formation (middle, introduction of 23 and 12 amino acids respectively)[57] and an unnatural amino acid (bottom, disulfide‐bridge between cysteine and tyrosine derivative SbuY at positions 65 and 184, respectively).[71] (b) NMR structure of Sortase A (PDB ID 1ija)[89] showing the introduced modifications resulting from macrocyclizations via a split intein (top),[38] enzymatic ligation (middle, 19‐mer linker sequence: GSSHHHHHHSSGLVPRGSH),[49] and INCYPRO (bottom, with Cys111, Cys149 and Cys177).[50]
Sortase A is a single domain enzyme (Figure 7b) that catalyses transpeptidation reactions and has been applied for protein labelling. To increase its performance under chemical and thermal stress, different cyclization strategies have been pursued. Fusion with a DnaB split intein has provided a cyclic version with a 9‐mer linker sequence (Figure 7b, top) exhibiting activity comparable to the wildtype enzyme under native conditions.[38] Under denaturing conditions, cyclic Sortase A showed higher activity that the wildtype enzyme (e. g. in the presence of 2.5 M urea, 36 % vs. 8 % residual activity). Later, a more active Sortase version was head‐to‐tail cyclized using an enzymatic ligation.[49] Here, a 26 amino acid sequence was introduced providing a cyclic enzyme (Figure 7b, middle) with slightly increased thermal stability (ΔT m=2 °C) when compared with its linear precursor. However, under chemical stress, the cyclic version showed significantly increased resistance (e. g. in the presence of 1 M guanidine hydrochloride, 9 % vs. <1 % residual activity). The bicyclization of Sortase A using the INCYPRO approach (Ap3*, Figure 7b, bottom) resulted in increased thermal stability (ΔT m=11 °C) and increased enzymatic activity above 55 °C when compared to the linear version.[50] Under denaturing conditions, bicyclic Sortase A showed considerably increased resistance (e. g. in the presence of 1 M guanidine hydrochloride, 36 % vs. <1 % residual activity).
Taken together, a variety of macrocyclization approaches is available, which may be selected based on the requirements of a particular application (e. g. straight‐forward implementation, requirement for minimal modification or large crosslink diversity). Importantly, the meaningful stabilization of a protein often requires the introduction of multiple crosslinks.[50, 73] This renders the INCYPRO approach appealing which directly provides bicyclic proteins (when starting from linear precursors). Also, it should be highlighted that many of the presented cyclization approaches are orthogonal to each other and could in principle be combined to achieve high stabilization effects. The presented cyclic proteins have mainly found application as biocatalysts or ligands for cellular receptors. There is growing evidence that stabilized tertiary structures can also allow the inhibition of protein‐protein interactions[90, 91, 92, 93] that are not addressable with classic peptidomimetic approaches.[94, 95] Therefore, it can be expected that cyclic proteins will find more fields of application in the future.
Conflict of interest
S.N. and T.N.G. are listed as inventors on a patent application related to the in situ cyclization of proteins, and they are co‐founders of Incircular BV.
Acknowledgements
T.N.G. is grateful for support by the European Research Council (ERC starting grant number 678623).
A. Haim, S. Neubacher, T. N. Grossmann, ChemBioChem 2021, 22, 2672.
References
- 1.Sali A., Shakhnovich E., Karplus M., Nature 1994, 369, 248–251. [DOI] [PubMed] [Google Scholar]
- 2.Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 3.Horne W. S., Grossmann T. N., Nat. Chem. 2020, 12, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reetz M. T., Angew. Chem. Int. Ed. 2013, 52, 2658–2666; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2720–2666. [Google Scholar]
- 5.Jost C., Plückthun A., Curr. Opin. Struct. Biol. 2014, 27, 102–112. [DOI] [PubMed] [Google Scholar]
- 6.Magliery T. J., Curr. Opin. Struct. Biol. 2015, 33, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman A. M., McNaughton B. R., Cell Chem. Biol. 2016, 23, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agostini F., Völler J. S., Koksch B., Acevedo-Rocha C. G., Kubyshkin V., Budisa N., Angew. Chem. Int. Ed. 2017, 56, 9680–9703; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9810–9703. [Google Scholar]
- 9.Dombkowski A. A., Sultana K. Z., Craig D. B., FEBS Lett. 2014, 588, 206–212. [DOI] [PubMed] [Google Scholar]
- 10.Cascales L., Craik D. J., Org. Biomol. Chem. 2010, 8, 5035–5047. [DOI] [PubMed] [Google Scholar]
- 11.Dani V. S., Ramakrishnan C., Varadarajan R., Protein Eng. Des. Sel. 2003, 16, 187–193. [DOI] [PubMed] [Google Scholar]
- 12.White C. J., Yudin A. K., Nat. Chem. 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]
- 13.Shinbara K., Liu W., van Neer R. H. P., Katoh T., Suga H., Front. Chem. 2020, 8, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuan S. L., Wang T., Weil T., Chem. Eur. J. 2016, 22, 17112–17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lühmann T., Mong S. K., Simon M. D., Meinel L., Pentelute B. L., Org. Biomol. Chem. 2016, 14, 3345–3349. [DOI] [PubMed] [Google Scholar]
- 16.Yamada K., Ito Y., ChemBioChem 2019, 20, 2729–2737. [DOI] [PubMed] [Google Scholar]
- 17.Schumacher F. F., Nobles M., Ryan C. P., Smith M. E. B., Tinker A., Caddick S., Baker J. R., Bioconjugate Chem. 2011, 22, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maruani A., Smith M. E. B., Miranda E., Chester K. A., Chudasama V., Caddick S., Nat. Commun. 2015, 6, 6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nunes J. P. M., Morais M., Vassileva V., Robinson E., Rajkumar V. S., Smith M. E. B., Pedley R. B., Caddick S., Baker J. R., Chudasama V., Chem. Commun. 2015, 51, 10624–10627. [DOI] [PubMed] [Google Scholar]
- 20.Sun S., Akkapeddi P., Marques M. C., Martínez-Sáez N., Torres V. M., Cordeiro C., Boutureira O., Bernardes G. J. L., Org. Biomol. Chem. 2019, 17, 2005–2012. [DOI] [PubMed] [Google Scholar]
- 21.Counsell A. J., Walsh S. J., Robertson N. S., Sore H. F., Spring D. R., Org. Biomol. Chem. 2020, 18, 4739–4743. [DOI] [PubMed] [Google Scholar]
- 22.Balan S., Choi J. W., Godwin A., Teo I., Laborde C. M., Heidelberger S., Zloh M., Shaunak S., Brocchini S., Bioconjugate Chem. 2007, 18, 61–76. [DOI] [PubMed] [Google Scholar]
- 23.Brown S. P., Smith A. B., J. Am. Chem. Soc. 2015, 137, 4034–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martínez-Sáez N., Sun S., Oldrini D., Sormanni P., Boutureira O., Carboni F., Compañón I., Deery M. J., Vendruscolo M., Corzana F., Adamo R., Bernardes G. J. L., Angew. Chem. Int. Ed. 2017, 56, 14963–14967; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15159–14967. [Google Scholar]
- 25.Goldenberg D. P., Creighton T. E., J. Mol. Biol. 1983, 165, 407–413. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi H., Arai M., Takenawa T., Sota H., Xie Q. H., Iwakura M., J. Biol. Chem. 2007, 282, 9420–9429. [DOI] [PubMed] [Google Scholar]
- 27.Paulus H., Chem. Soc. Rev. 1998, 27, 375–386. [Google Scholar]
- 28.Camarero J. A., Muir T. W., J. Am. Chem. Soc. 1999, 121, 5597–5598. [Google Scholar]
- 29.Camarero J. A., Fushman D., Sato S., Giriat I., Cowburn D., Raleigh D. P., Muir T. W., J. Mol. Biol. 2001, 308, 1045–1062. [DOI] [PubMed] [Google Scholar]
- 30.Iwai H., Plückthun A., FEBS Lett. 1999, 459, 166–172. [DOI] [PubMed] [Google Scholar]
- 31.Schumann F. H., Varadan R., Tayakuniyil P. P., Grossman J. H., Camarero J. A., Fushman D., Front. Chem. 2015, 3, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott C. P., Abel-Santos E., Wall M., Wahnon D. C., Benkovic S. J., Proc. Natl. Acad. Sci. USA 1999, 96, 13638–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H., Xu M. Q., Liu X. Q., Biochim. Biophys. Acta 1998, 1387, 422–432. [DOI] [PubMed] [Google Scholar]
- 34.Wu H., Hu Z., Liu X. Q., Proc. Natl. Acad. Sci. USA 1998, 95, 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans T. C., Martin D., Kolly R., Panne D., Sun L., Ghosh I., Chen L., Benner J., Liu X. Q., Xu M. Q., J. Biol. Chem. 2000, 275, 9091–9094. [DOI] [PubMed] [Google Scholar]
- 36.Williams N. K., Prosselkov P., Liepinsh E., Line I., Sharipo A., Littler D. R., Curmi P. M. G., Otting G., Dixon N. E., J. Biol. Chem. 2002, 277, 7790–7798. [DOI] [PubMed] [Google Scholar]
- 37.Jeffries C. M., Graham S. C., Stokes P. H., Collyer C. A., Guss J. M., Matthews J. M., Protein Sci. 2006, 15, 2612–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhulenkovs D., Jaudzems K., Zajakina A., Leonchiks A., Biochem. Eng. J. 2014, 82, 200–209. [Google Scholar]
- 39.Iwai H., Lingel A., Plückthun A., J. Biol. Chem. 2001, 276, 16548–16554. [DOI] [PubMed] [Google Scholar]
- 40.van Lieshout J. F. T., Gutiérrez O. N. P., Vroom W., Planas A., de Vos W. M., van der Oost J., Koutsopoulos S., Appl. Biochem. Biotechnol. 2012, 167, 2039–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwai H., Züger S., Jin J., Tam P. H., FEBS Lett. 2006, 580, 1853–1858. [DOI] [PubMed] [Google Scholar]
- 42.Miyafusa T., Shibuya R., Nishima W., Ohara R., Yoshida C., Honda S., ACS Chem. Biol. 2017, 12, 2690–2696. [DOI] [PubMed] [Google Scholar]
- 43.Purkayastha A., Kang T. J., Biotechnol. Bioprocess Eng. 2019, 24, 702–712. [Google Scholar]
- 44.Nuijens T., Toplak A., Schmidt M., Ricci A., Cabri W., Front. Chem. 2019, 7, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris K. S., Guarino R. F., Dissanayake R. S., Quimbar P., McCorkelle O. C., Poon S., Kaas Q., Durek T., Gilding E. K., Jackson M. A., Craik D. J., van der Weerden N. L., Anders R. F., Anderson M. A., Sci. Rep. 2019, 9, 10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antos J. M., Popp M. W. L., Ernst R., Chew G. L., Spooner E., Ploegh H. L., J. Biol. Chem. 2009, 284, 16028–16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Popp M. W., Dougan S. K., Chuang T. Y., Spooner E., Ploegh H. L., Proc. Natl. Acad. Sci. USA 2011, 108, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu J., Zhao W., Gao Y., Sun M., Wei Y., Deng H., Gao W., Biomaterials 2015, 47, 13–19. [DOI] [PubMed] [Google Scholar]
- 49.Zou Z., Mate D. M., Nöth M., Jakob F., Schwaneberg U., Chem. Eur. J. 2020, 26, 13568–13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pelay-Gimeno M., Bange T., Hennig S., Grossmann T. N., Angew. Chem. Int. Ed. 2018, 57, 11164–11170; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11334–11170. [Google Scholar]
- 51.Nguyen G. K. T., Kam A., Loo S., Jansson A. E., Pan L. X., Tam J. P., J. Am. Chem. Soc. 2015, 137, 15398–15401. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen G. K. T., Qiu Y., Cao Y., Hemu X., Liu C. F., Tam J. P., Nat. Protoc. 2016, 11, 1977–1988. [DOI] [PubMed] [Google Scholar]
- 53.Bi X., Yin J., Hemu X., Rao C., Tam J. P., Liu C. F., Bioconjugate Chem. 2018, 29, 2170–2175. [DOI] [PubMed] [Google Scholar]
- 54.Kang H. J., Coulibaly F., Clow F., Proft T., Baker E. N., Science 2007, 318, 1625–1628. [DOI] [PubMed] [Google Scholar]
- 55.Zakeri B., Howarth M., J. Am. Chem. Soc. 2010, 132, 4526–4527. [DOI] [PubMed] [Google Scholar]
- 56.Veggiani G., Nakamura T., Brenner M. D., Gayet R. V., Yan J., Robinson C. V., Howarth M., Proc. Natl. Acad. Sci. USA 2016, 113, 1202–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buldun C. M., Jean J. X., Bedford M. R., Howarth M., J. Am. Chem. Soc. 2018, 140, 3008–3018. [DOI] [PubMed] [Google Scholar]
- 58.Zakeri B., Fierer J. O., Celik E., Chittock E. C., Schwarz-Linek U., Moy V. T., Howarth M., Proc. Natl. Acad. Sci. USA 2012, 109, 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schoene C., Fierer J. O., Bennett S. P., Howarth M., Angew. Chem. Int. Ed. 2014, 53, 6101–6104; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6215–6104. [Google Scholar]
- 60.Schoene C., Bennett S. P., Howarth M., Sci. Rep. 2016, 6, 21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J., Wang Y., Wang X., Zhang D., Wu S., Zhang G., Biotechnol. Biofuels 2016, 9, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y., Tian J., Xiao Y., Wang Y., Sun H., Chang Y., Luo H., Biotechnol. Lett. 2019, 41, 987–994. [DOI] [PubMed] [Google Scholar]
- 63.Sun X. B., Cao J. W., Wang J. K., Lin H. Z., Gao D. Y., Qian G. Y., Park Y. D., Chen Z. F., Wang Q., Nat. Biotechnol. 2019, 49, 28–36. [DOI] [PubMed] [Google Scholar]
- 64.Gao D. Y., Sun X. B., Liu M. Q., Liu Y. N., Zhang H. E., Shi X. L., Li Y. N., Wang J. K., Yin S. J., Wang Q., J. Agric. Food Chem. 2019, 67, 6837–6846. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y., Chang Y., Jia R., Sun H., Tian J. W., Luo H., Yu H., Shen Z., Process Biochem. 2020, 95, 260–268. [Google Scholar]
- 66.Si M., Xu Q., Jiang L., Huang H., PLoS One 2016, 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu C., Xu Q., Huang H., Jiang L., Biosci. Biotechnol. Biochem. 2018, 82, 1473–1479. [DOI] [PubMed] [Google Scholar]
- 68.Xu Q., Ming D., Shi C., Lu D., Tang S., Jiang L., Huang H., Process Biochem. 2019, 80, 64–71. [Google Scholar]
- 69.Ren H., Biochem. Soc. Trans. 2020, 48, 1807–1817. [DOI] [PubMed] [Google Scholar]
- 70.Young T. S., Schultz P. G., J. Biol. Chem. 2010, 285, 11039–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu T., Wang Y., Luo X., Li J., Reed S. A., Xiao H., Young T. S., Schultz P. G., Proc. Natl. Acad. Sci. USA 2016, 113, 5910–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen X. H., Xiang Z., Hu Y. S., Lacey V. K., Cang H., Wang L., ACS Chem. Biol. 2014, 9, 1956–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moore E. J., Zorine D., Hansen W. A., Khare S. D., Fasan R., Proc. Natl. Acad. Sci. USA 2017, 114, 12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J. C., Liu T., Wang Y., Mehta A. P., Schultz P. G., J. Am. Chem. Soc. 2018, 140, 15997–16000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin S. H., Konishi Y., Denton M. E., Scheraga H. A., Biochemistry 1984, 23, 5504–5512. [DOI] [PubMed] [Google Scholar]
- 76.Weber P. C., Sheriff S., Ohlendorf D. H., Finzel B. C., Salemme F. R., Proc. Natl. Acad. Sci. USA 1985, 82, 8473–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Timmerman P., Beld J., Puijk W. C., Meloen R. H., ChemBioChem 2005, 6, 821–824. [DOI] [PubMed] [Google Scholar]
- 78.Heinis C., Rutherford T., Freund S., Winter G., Nat. Chem. Biol. 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- 79.Chen S., Bertoldo D., Angelini A., Pojer F., Heinis C., Angew. Chem. Int. Ed. 2014, 53, 1602–1606; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1628–1606. [Google Scholar]
- 80.Dang B., Wu H., Mulligan V. K., Mravic M., Wu Y., Lemmin T., Ford A., Silva D. A., Baker D., DeGrado W. F., Proc. Natl. Acad. Sci. USA 2017, 114, 10852–10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neubacher S., Saya J. M., Amore A., Grossmann T. N., J. Org. Chem. 2020, 85, 1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brauckhoff N., Hahne G., Yeh J. T.-H., Grossmann T. N., Angew. Chem. Int. Ed. 2014, 53, 4337–4340; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4425–4340. [Google Scholar]
- 83.Stiller C., Krüger D. M., Brauckhoff N., Schmidt M., Janning P., Salamon H., Grossmann T. N., ACS Chem. Biol. 2017, 12, 504–509. [DOI] [PubMed] [Google Scholar]
- 84.Xiao Q., Ashton D. S., Jones Z. B., Thompson K. P., Price J. L., RSC Chem. Biol. 2020, 1, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoppmann C., Lacey V. K., Louie G. V., Wei J., Noel J. P., Wang L., Angew. Chem. Int. Ed. 2014, 53, 3932–3936; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4013–3936. [Google Scholar]
- 86.Hoppmann C., Maslennikov I., Choe S., Wang L., J. Am. Chem. Soc. 2015, 137, 11218–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tooke C. L., Hinchliffe P., Bragginton E. C., Colenso C. K., Hirvonen V. H. A., Takebayashi Y., Spencer J., J. Mol. Biol. 2019, 431, 3472–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strynadka N. C. J., Adachi H., Jensen S. E., Johns K., Sielecki A., Betzel C., Sutoh K., James M. N. G., Nature 1992, 359, 700–705. [DOI] [PubMed] [Google Scholar]
- 89.Ilangovan U., Ton-That H., Iwahara J., Schneewind O., Clubb R. T., Proc. Natl. Acad. Sci. USA 2001, 98, 6056–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sadek J., Wuo M. G., Rooklin D., Hauenstein A., Hong S. H., Gautam A., Wu H., Zhang Y., Cesarman E., Arora P. S., Nat. Commun. 2020, 11, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adihou H., Gopalakrishnan R., Förster T., Guéret S. M., Gasper R., Geschwindner S., Carrillo García C., Karatas H., Pobbati A. V., Vazquez-Chantada M., Davey P., Wassvik C. M., Pang J. K. S., Soh B. S., Hong W., Chiarparin E., Schade D., Plowright A. T., Valeur E., Lemurell M., Grossmann T. N., Waldmann H., Nat. Commun. 2020, 11, 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hong S. H., Yoo D. Y., Conway L., Richards-Corke K. C., Parker C. G., Arora P. S., Proc. Natl. Acad. Sci. USA 2021, 118, e2101027118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wendt M., Bellavita R., Gerber A., Efrém N.-L., van Ramshorst T., Pearce N. M., Davey P. R. J., Everard I., Vazquez-Chantada M., Chiarparin E., Grieco P., Hennig S., Grossmann T. N., Angew. Chem. Int. Ed. 2021, 60, 13937–13944; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 14056–14063. [Google Scholar]
- 94.Pelay-Gimeno M., Glas A., Koch O., Grossmann T. N., Angew. Chem. Int. Ed. 2015, 54, 8896–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9022–8927. [Google Scholar]
- 95.Cunningham A. D., Qvit N., Mochly-Rosen D., Curr. Opin. Struct. Biol. 2017, 44, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]