

Summary
Eggplant (Solanum melongena L.) is an important horticultural crop and one of the most widely grown vegetables from the Solanaceae family. It was domesticated from a wild, prickly progenitor carrying small, round, non‐anthocyanic fruits. We obtained a novel, highly contiguous genome assembly of the eggplant ‘67/3’ reference line, by Hi‐C retrofitting of a previously released short read‐ and optical mapping‐based assembly. The sizes of the 12 chromosomes and the fraction of anchored genes in the improved assembly were comparable to those of a chromosome‐level assembly. We resequenced 23 accessions of S. melongena representative of the worldwide phenotypic, geographic, and genetic diversity of the species, and one each from the closely related species Solanum insanum and Solanum incanum. The eggplant pan‐genome contained approximately 51.5 additional megabases and 816 additional genes compared with the reference genome, while the pan‐plastome showed little genetic variation. We identified 53 selective sweeps related to fruit color, prickliness, and fruit shape in the nuclear genome, highlighting selection leading to the emergence of present‐day S. melongena cultivars from its wild ancestors. Candidate genes underlying the selective sweeps included a MYBL1 repressor and CHALCONE ISOMERASE (for fruit color), homologs of Arabidopsis GLABRA1 and GLABROUS INFLORESCENCE STEMS2 (for prickliness), and orthologs of tomato FW2.2, OVATE, LOCULE NUMBER/WUSCHEL, SUPPRESSOR OF OVATE, and CELL SIZE REGULATOR (for fruit size/shape), further suggesting that selection for the latter trait relied on a common set of orthologous genes in tomato and eggplant.
Keywords: Solanum melongena; genome assembly; whole‐genome resequencing; pan‐genome; pan‐plastome; domestication; single nucleotide polymorphism; indel, eggplant wild relatives
Significance Statement
Chromosome conformation capture (Hi‐C) was applied for improving a short read‐based genome assembly of eggplant (Solanum melongena). The enhanced assembly, combined with resequencing data of accessions representing the genetic and phenotypic diversity of the species and of its closest wild relatives, allowed the construction of the first eggplant pan‐genome and pan‐plastome and the identification of selective sweeps and candidate genes likely involved in eggplant domestication and breeding.
INTRODUCTION
With a worldwide production of more than 54 megatons, common eggplant (Solanum melongena L.) is an important horticultural crop and the third most cultivated Solanaceae species, after potato (Solanum tuberosum L.) and tomato (Solanum lycopersicum L.) (FAO). Within the genus Solanum, eggplant and its relatives are part of subgenus Leptostemonum, collectively known as the ‘spiny solanum’ group (Vorontsova et al., 2013). Species from the eggplant clade, such as the direct wild ancestor Solanum insanum L. and the sister species Solanum incanum L., are closely related to S. melongena. More distant species from the Anguivi grade include the two other cultivated eggplant species: the scarlet eggplant (Solanum aethiopicum L.) and the gboma eggplant (Solanum macrocarpon L.) (Acquadro et al., 2017; Barchi et al., 2019a; Vorontsova et al., 2013).
Within the ‘spiny solanum’ group, non‐anchored genome sequences are available for S. melongena (v1.0, Hirakawa et al., 2014) and S. aethiopicum (Song et al., 2019). A chromosome‐anchored genome sequence (v3.0; Barchi et al., 2019b) and a chromosome‐level (CL) assembly (Wei et al., 2020a) of two different lines have been recently released for S. melongena, but improvement of the former sequence is still needed, due to the low proportion of anchored genes.
Common eggplant exhibits a wide range of phenotypic variation (Cericola et al., 2014; Portis et al., 2015) and metabolic diversity (Kaushik et al., 2017). Similar to other species, individual cultivars/accessions of eggplant are expected to contain genes absent in the reference genome and vice versa (referred to as presence/absence variants [PAVs]). These variants are expected to affect phenotypic traits, and can be revealed by resequencing of different accessions. Within the Solanaceae, pan‐genome resequencing projects have been undertaken in pepper (Capsicum annuum L.) (Ou et al., 2018), tomato (Gao et al., 2019) and S. aethiopicum (Song et al., 2019).
Quantitative trait locus (QTL) mapping in eggplant has been undertaken using germplasm sets as well as progeny populations from both intra‐ and interspecific crosses (Barchi et al., 2012; Barchi et al., 2018; Doganlar et al., 2002b; Frary et al., 2003; Frary et al., 2014; Mangino et al., 2020; Mangino et al., 2021; Miyatake et al., 2012; Nunome et al., 2001; Portis et al., 2014; Salgon et al., 2018; Toppino et al., 2016; Toppino et al., 2020; Wei et al., 2020b). Several QTLs for eggplant fruit weight, shape, and color have been found at orthologous positions with respect to those from tomato, potato, or pepper, suggesting a common genetic basis for some fruit traits in Solanaceae (Chapman, 2019; Doğanlar et al., 2014; Doganlar et al., 2002a; Mangino et al., 2021).
Sequencing of whole plastomes and selected plastid regions allows the construction of phylogenies at the species and, occasionally, at the subspecies level (Lakušić et al., 2013; Magdy et al., 2019; Parks et al., 2009). Plastid DNA consists of a circular chromosome comprised of a pair of inverted repeats (IRs) separated by two single‐copy regions. Currently, 145 Solanaceae plastid genomes are available in the National Center for Biotechnology Information (NCBI) database, including those of S. melongena, S. incanum, S. aethiopicum, and S. macrocarpon. The availability of a large number of chloroplast sequences may contribute to the development of reproducible molecular markers for DNA barcoding, population‐based studies, phylogeography, and domestication genetics.
Here, we report an improved, highly contiguous S. melongena reference genome (v4.0), obtained by scaffolding of v3.0 by the help of 3D chromosome conformation capture (Hi‐C) information. In total, 23 S. melongena accessions, representative of the genetic and phenotypic diversity of the species, plus one accession each from the closest wild relatives S. insanum and S. incanum (non‐melongena species [NMSs]) were resequenced to deduce a first eggplant pan‐genome and pan‐plastome. We used this information to estimate the genetic variability of nuclear and plastid sequences, identify additional genes absent in the reference genome (i.e., PAVs), compare the congruence of nuclear‐ and plastid‐based phylogenies, and identify selective sweeps (SSs) associated with the selection of key agronomic traits in S. melongena.
RESULTS
Assembly of the Smel v4.0 genome
A total of 118191133 paired‐end reads were generated, and after adapter trimming and quality control, 26 759 980 valid Hi‐C links were identified (Figure S1(a); Table S1). The Hi‐C data were first combined with published genetic map information to assign scaffolds to chromosomes using a novel iterative approach which resulted in a large reduction of the size of the ‘unknown’ chromosome chr. 0 (from 22.5% to 4.4% of the total assembly). The scaffolds of the final v3.0 assembly were super‐scaffolded with optical maps, introducing long stretches of N bases and considerably altering scaffold size and order. We applied Hi‐C to the pre‐optical map scaffold set, ultimately improving the order and significantly reducing the proportion of N bases (from 28.2% to 9.1%). Following Hi‐C‐based automated scaffold ordering and manual editing with the aid of Hi‐C contact plots, Hi‐C asymmetry plots (Himmelbach et al., 2018), and the positions of genetic map markers on the pseudomolecules, a near‐optimal final order was found, which clearly improved the quality of the genome (Table 1; Figure S1(b)). Compared with v3.0, the percentage of anchored genes in v4.0 increased to 95.6% and the benchmarking against universal single‐copy orthologs (BUSCO; Simão et al., 2015) revealed 96.9% representation (Table 1). These values are comparable to those of the recent CL assembly (Wei et al., 2020a). The size of individual chromosomes, which for some in our v3.0 was dissimilar from the one of the CL assembly, in v4.0 was highly comparable (Table S2). The CL and v4.0 assemblies exhibited high collinearity (Figure S2(a)), with just three minor intrachromosomal inversions in chrs. 2, 10, and 11. A comparison of the v4.0 and CL annotations showed a slight predominance of v4.0 proteins with similar lengths to those of their Arabidopsis homologs, as well as a slightly lower fraction of v4.0 proteins with no hit in the Arabidopsis annotation (Figure S2(b)).
Table 1.
Metrics of publicly available eggplant genome assemblies: Smel v1.0 from Hirakawa et al. (2014), S. aethiopicum from Song et al. (2019), Smel v3.0 from Barchi et al. (2019b), Smel CL from Wei et al. (2020a), and Smel v4.0 (this paper)
| Smel v1.0 | S. aethiopicum | Smel v3.0 | Smel CL | Smel v4.0 | |
|---|---|---|---|---|---|
| Size of assembly (Gb) | 0.83 | 1.02 | 1.21 | 1.07 | 1.16 |
| Contig N50 (kb) | 14.3 | 25.2 | 678.7 | 5260 | 702.9 |
| Scaffold N50 (Mb) | 0.065 | 516.1 | 2.9 | 89.64 | 92.1 |
| % Ns | 4.75 | – | 28.23 | 0.38 | 9.10 |
| % anchored | – | – | 77.5% | 92.7% | 95.6% |
| Protein‐coding genes | 85 446 | 34 906 | 34 916 | 36 582 | 34 916 |
| % anchored genes | 0% | 0% | 81.4% | 97.4% | 96.6% |
| Annotated BUSCO genes | 74.8% | 77.8% | 96.9% | 94.2% | 96.9% |
Pan‐genome construction and presence/absence variants
In addition to the reference line ‘67/3’, 25 additional accessions were chosen on the basis of their genotype, phenotype, and geographic distribution, being representative of approximately 3600 genotyped accessions from a worldwide collection (http://www.g2p‐sol.eu/G2P‐SOL‐gateway.html), exhibiting very diverse morphology with respect to fruit shape, pigmentation, and presence of prickles (Figure 1; Table S3). Resequencing data for nine of them were already available (Gramazio et al., 2019), while the remaining 16 were subjected to paired‐end (2×150 bp) Illumina sequencing, reaching an average coverage of 25‐fold after cleaning, trimming, and removal of non‐plant contaminants (Table S3). The genome for each accession was de novo assembled using Megahit into contigs larger than 500 bp. The final genome sizes ranged from 826 to 999 Mb and the N50 value from 3 to 26.8 kb (Table S3). All assembled contigs were compared to the reference genome to identify novel sequences. After removing redundancies, these novel sequences were rechecked by removing contaminant sequences from non‐green plants, resulting in the identification of approximately 51.5 Mb of additional sequences (available at https://solgenomics.net/).
Figure 1.
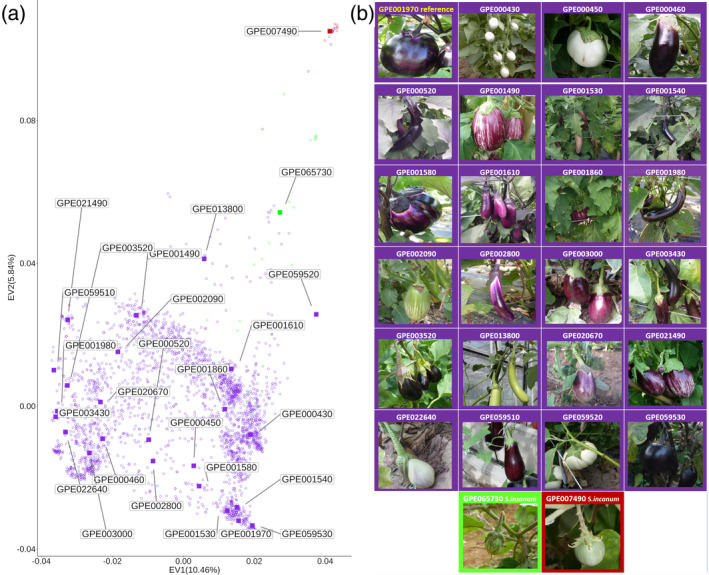
Accessions included in the eggplant pan‐genome.
(a) PCA of 24 eggplant accessions from the G2P‐SOL project, genotyped with a previously developed 5k SNP panel (Barchi et al., 2019a). Resequenced accessions are depicted as squares. Solanum melongena entries are shown in purple, S. incanum entries in red, and S. insanum entries in green.
(b) Fruit phenotypes of the resequenced accessions.
A total of 816 protein‐coding genes with Annotation Edit Distance (AED) score < 0.5 were predicted in the novel sequences using Maker‐P. After Interproscan analysis, 439 novel genes contained at least a Pfam domain, and 631 contained Pfam or Panther domains. Overall, 365 new genes could be annotated with gene ontology (GO). The eggplant pan‐genome, including reference and new genome sequences, had a total size of 1.21 Gbp and contained 35 732 protein‐coding genes. A total of 41 non‐reference genes were covered by reads of the reference line ‘67/3’ with a coverage fraction greater than 95%, suggesting that they were not previously assembled in the reference genome. In total, 460 genes from the reference genome were not covered by reads in at least one of the accessions in this study (Table S4). Figure 2(a) shows how the pan‐genome size was modeled by iterative random sampling of accessions. The model suggested an open pan‐genome with a projected pan‐genome size of 39 437 genes based on the Chao estimator (Chao, 1987). Indeed, the fitting of the curve in Figure 2(b) to a power law (265.833n −0.59) indicates that adding another genome sequence would add 40 ± 4 additional genes (95% confidence interval) to the pan‐genome.
Figure 2.
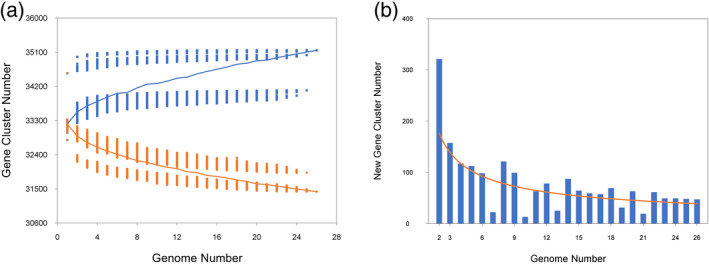
The eggplant pan‐genome.
(a) Gene accumulation curves of the pan‐genome (blue) and the core genome (orange). These curves describe the number of new genes (pan‐genome) and genes in common (core genome) obtained by adding a new genome to a previous set. The blue vertical lines denote the pan‐genome size for each genome for comparison. The orange vertical lines show the core genome size for each genome for comparison. The curve is the least squares fit of the power law for the average values.
(b) Curve (orange) for the number of new genes at each increase in the number of genomes.
The pan‐genome genes were categorized based on their presence frequencies: 31 424 core genes were shared by all 26 accessions, 922 softcore genes were shared by 25 accessions, 1556 shell genes were shared by 2–24 accessions, and 1246 cloud genes were present just in one accession (Gao et al., 2019; Gordon et al., 2017) (Table S5). The core and softcore groups contained highly conserved genes, whereas the shell and cloud groups contained the so‐called flexible genes. The reference genome v4.0 contained the majority of highly conserved genes (only 86 from the additional contigs) and around 78% of the flexible genes. Compared with the entire pan‐genome, significant GO term enrichments were observed for the different pan‐genome gene categories by using AGRIGO SEACOMPARE (Table S6). Several GO terms highly enriched in the flexible category were related to photosynthesis, protein synthesis (ribosome, structural constituent of ribosome, ribosomal subunit), and ATP biosynthetic processes. The flexible photosynthesis genes included several genes (accD, petA, petB, ndhD, psaA‐C, psbA‐E, rbcL, ycf‐2) probably resulting from differential organellar insertions in the nuclear genome of different accessions.
Pan‐plastome construction and analysis
The complete chloroplast genomes of the 26 accessions were successfully assembled (Table S7) and submitted to GenBank (accession numbers MW384824–MW384851). They presented a typical quadripartite circular structure, composed of one long single copy (LSC) region, one short single copy (SSC) region, and two IRs (Figure 3(a)). In total, 79 protein‐coding genes, 29–30 tRNA genes (with trnG‐UCC present only in S. incanum), and 4 rRNA genes were identified (Table S8). Three pseudogenes (infA, ycf1, rps19) and 14 intron‐containing genes (rps16, atpF, rpoC1, ycf3, rps12, rpl2, clpP, ndhB, ndhA, trnA‐UGC, trnI‐GAU, trnK‐UUU, trnL‐UAA, trnV‐UAC) were also identified. The rps12 gene was predicted to be trans‐spliced, with the 5′‐end located in the LSC region and the duplicated 3′‐end in the IR region. The trnK‐UUU had the largest intron, encompassing the matK gene, with 2513 bp, while the trnI‐GAU had the shortest intron, with just 15 bp. Some genes were predicted to have an alternative start codon, like GTG (for psbC and rps19) or ATA (petD).
Figure 3.
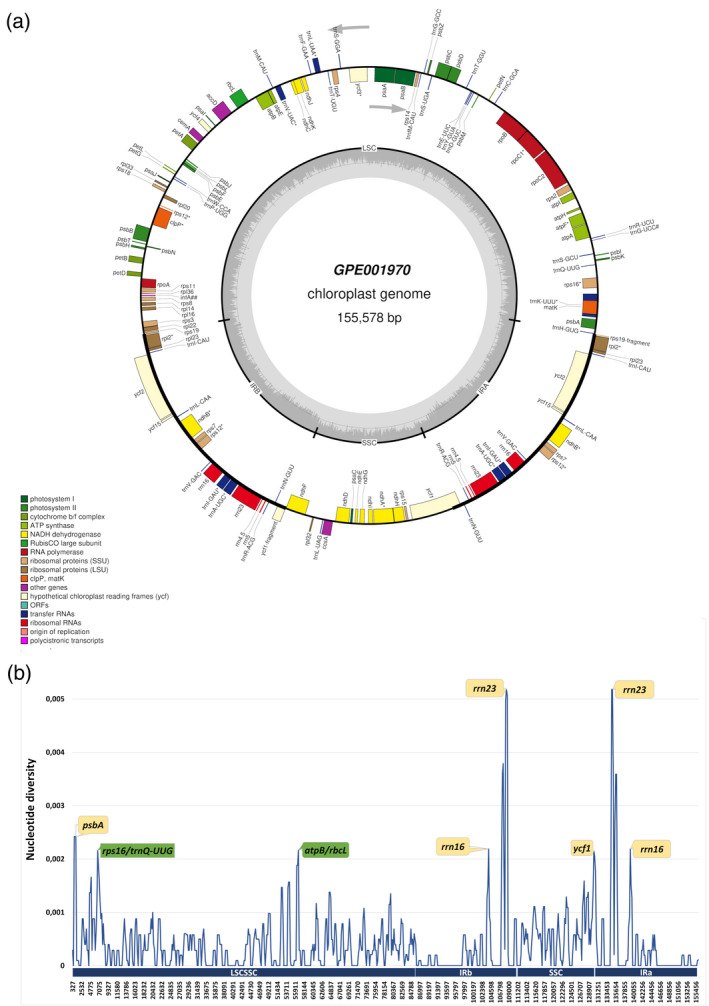
The eggplant pan‐plastome.
(a) Circular gene map of the chloroplast genome from the reference line GPE001970. Genes drawn inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise. The darker gray in the inner circle represents GC content.
(b) Nucleotide diversity (Pi) values in the eggplant pan‐plastome. Mutational hotspots with Pi values >0.002 in the intergenic spacers (IGSs) are shown in green, and those within genes are shown in yellow.
The aligned matrix of the 32 available plastomes (26 from this study and six from Aubriot et al. (2018)) was analyzed using DNAsp 6, resulting in the identification of 154 743 invariable and 449 variable sites, of which 186 were singletons and 263 were parsimony‐informative. The total number of single‐nucleotide polymorphisms (SNPs) in coding regions was 343.
The whole chloroplast genome sequences of all the accessions in the study were compared using the mVISTA program (Figure S3). The comparison revealed few differences in the ycf1 gene and in the trnA‐trnR intergenic region among the chloroplast genomes of the S. melongena accessions, with the exclusion of GPE001530. The number of differences increased by extending the analyses to S. insanum, the species most similar to S. melongena, and to S. incanum (see intergenic regions psaA‐ycf3 and rpl32‐trnL as examples). Expectedly, non‐coding regions, in particular the intergenic spacers (IGSs), exhibited a higher divergence than coding regions.
The nucleotide variability (Pi) of the chloroplast genome was calculated (Figure 3(b)). The average value of Pi was relatively low (0.00039). The IR regions exhibited a lower variability (Pi = 0.00036) than LSC (Pi = 0.00037) and SSC (Pi = 0.00060) regions (Figure 3(b)). Five mutational hotspots with high Pi values (>0.002) were identified, involving IGSs as well as genes (psbA, rrn16, rrn23, ycf1).
A total of 2374 plastid simple sequence repeat (SSRs) were identified across the 32 Solanum accessions (Figure S4(a)), ranging from 60 for MH283713 to 66 for GPE059520 and GPE065730. The majority were mono‐nucleotide (A/T), followed by di‐ and tri‐nucleotide (AT/TA and AAT/ATT) repeats. By contrast, the tri‐nucleotide motif AAC/GTT and the tetra‐nucleotide AAAG/CTTT was only found in one S. incanum chloroplast. The analysis of the 32 plastomes, using REPUTER, identified a total of 1174 repeats (30–79 bp) (Figure S4(b)). The proportions of repeats located in non‐coding regions were higher than in coding regions, with ycf2 showing the highest rearrangements.
Nuclear genetic diversity
The sequence reads of the 26 accessions (including the reference line ‘67/3’) were aligned to the pan‐genome sequence, yielding 15004464 SNPs/indels (final SNP dataset), with 246 391 in exons and 577665 in introns. In the reference line ‘67/3’, 127 944 sites were heterozygous and 4322 indels were identified (Table S9). For the remaining accessions, the number of SNPs (including both homozygous and heterozygous) ranged from 97 913 for S. melongena GPE059530 to 6 047 125 for S. incanum, while the number of indels ranged from 4663 for S. melongena GPE059530 to 301172 for S. incanum.
Within the S. melongena accessions, the genomic residual heterozygosity was generally low, with the highest value being 0.04%. Of the two NMSs, S. insanum showed a higher level of homozygosity than S. incanum. The same trend was observed for the heterozygosity at the SNP/indel level, with S. melongena accessions showing no more than 5.46% of heterozygous sites compared to 22.30% of S. insanum and 34.33% of S. incanum (Table S9). The distribution of SNPs varied according to the species and the chromosomes (Figure S5; Table S10).
The maximum likelihood (ML) dendrograms based on the pan‐genome and pan‐plastome sequences (Figure 4(a)) grouped the accessions into two main branches. One branch includes all the S. melongena accessions together with their direct ancestor S. insanum, while a second branch includes S. incanum. Solanum insanum resulted closely related to two S. melongena accessions (GPE059520 and GPE000430) in both the nuclear and plastid phylogenies; both accessions are small‐fruited and are characterized by low vigor and a semi‐prostrate growth habit which is typical of some S. insanum accessions (Ranil et al., 2017) although they, unlike S. insanum, have no or few prickles (Table S3).
Figure 4.
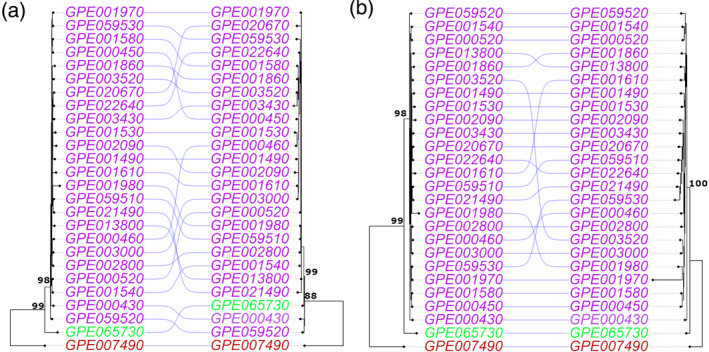
(a) Comparison of maximum likelihood phylogenetic trees, based on the nuclear (left) and plastidial (right) SNPs. (b) Comparison of maximum likelihood phylogenetic trees based on the nuclear SNPs (left) and PAVs (right). Solanum melongena entries are shown in purple, S. incanum entries in red, and S. insanum entries in green. Bootstrap values for main branches are also reported.
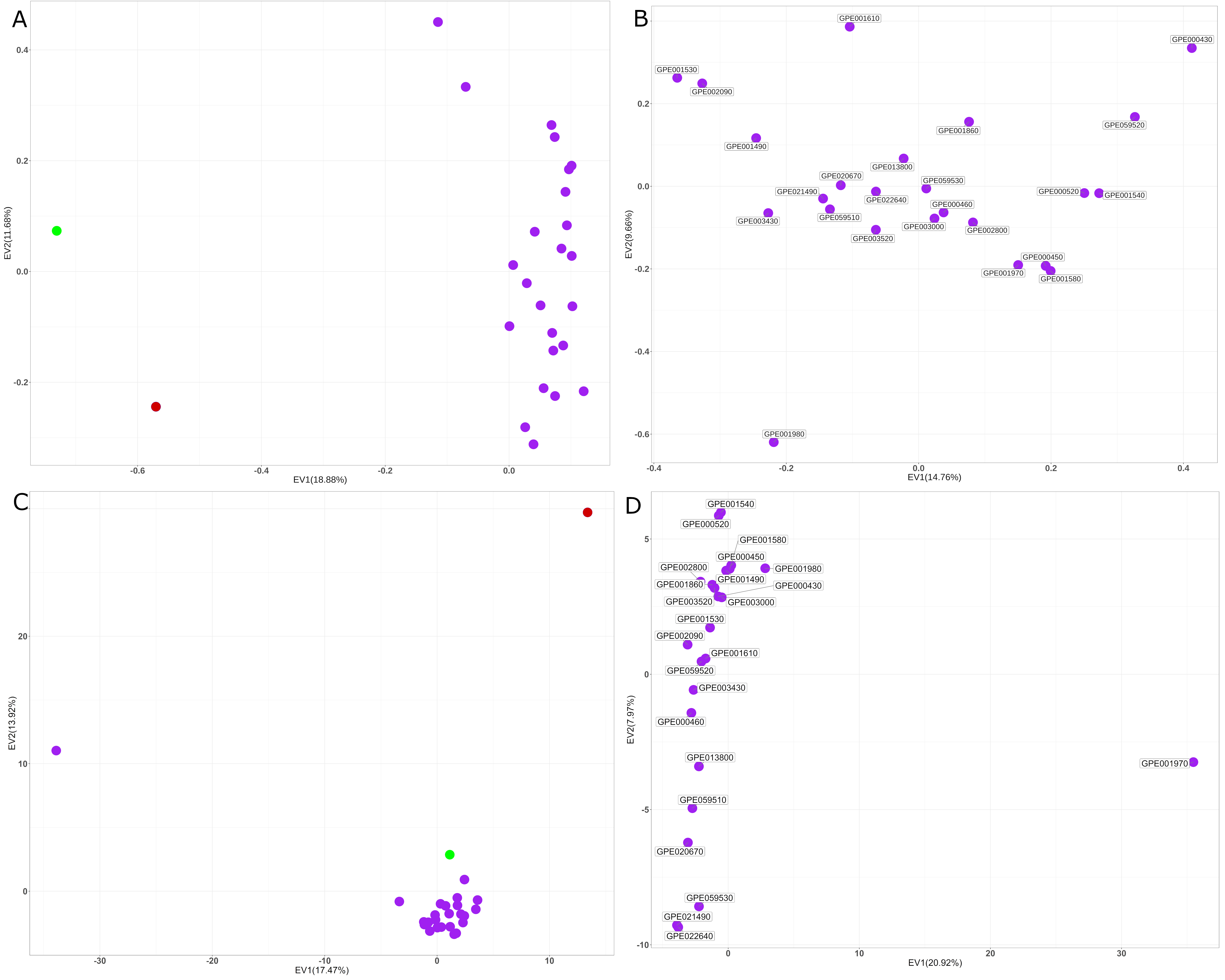
The principal component analysis (PCA) based on the whole pan‐genome SNP dataset (Figure S6(a)) largely confirmed the grouping of genotypes obtained in the ML‐based dendrogram. The first and second principal components account, respectively, for 18.88% and 11.68% of the genetic variation. Eggplant accessions are clearly separated from NMSs by the first component of the PCA. By restricting the PCA to the 24 S. melongena accessions, a higher separation was obtained (Figure S6(b)), and the first and second principal axes accounted, respectively, for 14.76% and 9.66% of the genetic variation. Almost all accessions group together, with the exclusion of GPE001980.
Nucleotide diversity (π) was 0.000502 for the S. melongena accessions and 0.00204 for NMSs. Linkage disequilibrium (LD) decay in S. melongena accessions stabilized at about 0.41 at a distance of approximately 1.5 Mb (Figure S7), while in the whole panel of accessions, LD decay stabilized at about 0.35 at 1.4 Mb.
Phylogenetic analysis and PCA using the PAV matrix largely agree with what was previously observed based on the SNP information. The ML dendrogram (Figure 4(b)) identified two main branches; the first one included all the S. melongena accessions and the S. insanum accession, while S. incanum was assigned to the second one. In the PCA graph (Figure S6(c)), the first and second principal axes account for 17.47% and 13.92% of the genetic variation, respectively. The closest species to S. melongena accessions is its wild ancestor S. insanum. Again, by restricting the analysis to the 24 S. melongena accessions a clear separation among them was obtained (Figure S6(d)). GPE001970, the reference ‘67/3’ line, shows a larger distance from the others in the PAV‐based tree (Figure 5(b) ) and PCA (Figure S6(d)). This is probably an artifact, due to the higher read coverage available following deeper sequencing of this accession (150‐fold) with consequently around 1000 PAVs identified only in this accession.
Figure 5.
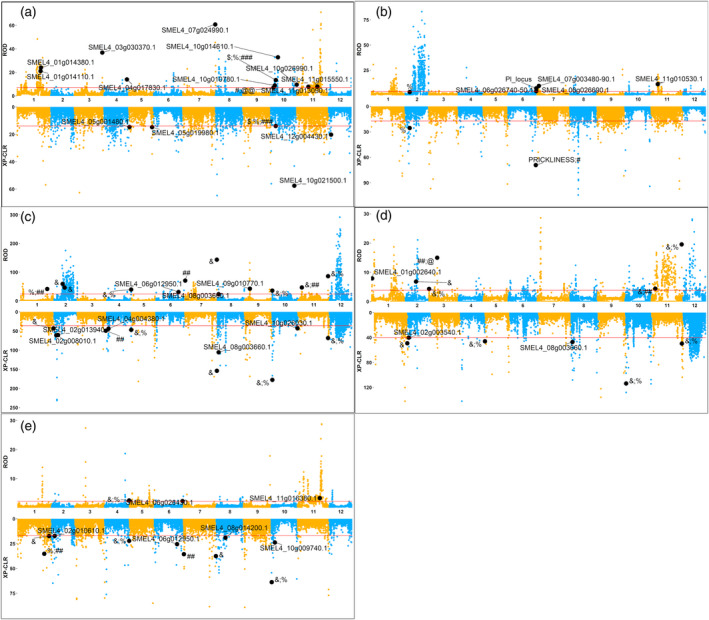
Plot of the π ratio (i.e., ROD; top) and XP‐CLR (bottom) across the eggplant genome.
(a) Non‐anthocyanic versus anthocyanic fruits (NAvsA).
(b) Prickly versus non‐prickly fruits (PvsNP).
(c) Round versus long fruits (RvsL).
(d) Round versus oval fruits (RvsO).
(e) Long versus oval fruits (LvsO). The positions of previously discovered quantitative trait loci (QTLs) and quantitative trait nucleotides (QTNs) ($Barchi et al., 2012; ###Cericola et al., 2014; &Portis et al., 2014; #Frary et al., 2014; %Portis et al., 2015; @@Toppino et al., 2016; ##Wei et al., 2020b; @Wei et al., 2020a; PI locus from Miyatake et al., 2020) as well as candidate genes (Table 2) located in selective sweep regions are shown. Red lines represent cutoff values for the top 5% and 1% of ROD and XP‐CLR scores, respectively.
Selective sweeps
The resequenced accessions show contrasting phenotypes for a series of phenotypic traits, such as fruit pigmentation (FP), presence/absence of prickles (PR), and fruit shape (FS) (Figure 1; Table S3). Given the lack of noticeable population structure in the accessions (Figure 4), we decided to conduct a search for potential selection signatures by comparing the genomes of eggplant accessions contrasting for these traits. Five different comparisons were made: accessions with non‐anthocyanic versus anthocyanic fruits (NAvsA), prickly versus non‐prickly fruits (PvsNP), round versus long fruits (RvsL), round versus oval fruits (RvsO), and long versus oval fruits (LvsO). Two commonly used indicators, the reduction in average nucleotide diversity (ROD or π ratio) and the cross‐population composite likelihood ratio test (XP‐CLR; Chen et al., 2010) were used to identify selective sweeps (SSs), at cutoff values of 5% and 1%, respectively (Figure 5; Table S11). For each comparison, the SSs with top 5% ROD or 1% XP‐CLR values covered, on average, 1458 (4.17%) and 1014 (2.90%) genes, respectively (Table S12).
We also searched for previously detected QTLs and/or quantitative trait nucleotides (QTNs) (Barchi et al., 2012; Cericola et al., 2014; Frary et al., 2014; Portis et al., 2014; Portis et al., 2015; Toppino et al., 2016; Wei et al., 2020b) (Figure 5). We also searched for candidate genes putatively involved in the control of the examined phenotypic traits, mapping in the SSs regions (Table 2); this resulted in the identification of 53 SSs spanning 35.3 Mb (Figure 5). No PAVs putatively involved in the control of the traits in study were found to map to any of the SS regions identified.
Table 2.
Selected candidate genes identified within selective sweep regions for the traits studied
| Trait | Gene ID | ROD 5% | XPCLR 1% | Annotation | Note |
|---|---|---|---|---|---|
| Fruit pigmentation | SMEL4_01g014380.1 | Yes | No | ABCC4: ABC transporter C family member 4 | Homolog of Vitis ABCC1 |
| SMEL4_01g014110.1 | Yes | No | PER64: Peroxidase 64 | Homolog of Vitis Peroxidase 31 | |
| SMEL4_01g014400.1 | Yes | No | ABCC4: ABC transporter C family member 4 | Homolog of Vitis ABCC1 | |
| SMEL4_03g030370.1 | Yes | No | Flavonol synthase/flavanone 3‐hydroxylase (Fragment) | Homolog of Arabidopsis FLS | |
| SMEL4_04g017830.1 | Yes | No | PER9: Peroxidase 9 | Homolog of Vitis Peroxidase 31 | |
| SMEL4_05g001480.1 | No | Yes | CHI3: Probable chalcone–flavonone isomerase 3 | Homolog to Arabidopsis CHI3 | |
| SMEL4_05g019980.1 | No | Yes | PER46: Peroxidase 46 | Homolog of Vitis Peroxidase 31 | |
| SMEL4_07g024990.1 | Yes | No | ABCC3: ABC transporter C family member 3 | Homolog of Vitis ABCC1 | |
| SMEL4_10g010780.1 | Yes | No | CHI: Chalcone–flavonone isomerase | Homolog of petunia CHI | |
| SMEL4_10g014610.1 | Yes | No | MYB14: Transcription factor MYB14 | Homolog of Medicago MYB14 | |
| SMEL4_10g021500.1 | No | Yes | DTX21: Protein DETOXIFICATION 21 | Homolog of Vitis MATE1 | |
| SMEL4_11g013090.1 | Yes | No | Putative anthocyanidin reductase | Homolog of Vitis ANR | |
| SMEL4_11g015550.1 | Yes | No | DFR1: Dihydroflavonol 4‐reductase (fragment) | Homolog of Medicago DFR | |
| SMEL4_10g010780.1 | Yes | No | CHI: Chalcone–flavonone isomerase | Ortholog of Arabidopsis TT5 | |
| SMEL4_10g026990.1 | Yes | No | MYB4: Transcription repressor MYB4 (AtMYBL2) | Eggplant MYBL1 | |
| SMEL4_12g004430.1 | No | Yes | DTX29: Protein DETOXIFICATION 29 | Homolog of Vitis MATE1 | |
| Prickliness | SMEL4_06g026690.1 | Yes | No | NUDT19: Nudix hydrolase 19 chloroplastic | PI locus |
| SMEL4_06g026740.1 | Yes | No | GATA11: GATA transcription factor 11 | PI locus | |
| SMEL4_06g026750.1 | Yes | No | ARF18: Auxin response factor 18 | PI locus | |
| SMEL4_07g003480.1 | Yes | No | MYB82: Transcription factor MYB82 | Homolog of Arabidopsis Gl1 | |
| SMEL4_07g003490.1 | Yes | No | MYB82: Transcription factor MYB82 | Homolog of Arabidopsis Gl1 | |
| SMEL4_11g010530.1 | Yes | No | GIS2: Zinc finger protein GIS2 | Homolog of Arabidopsis GLS2 | |
| Fruit shape | SMEL4_01g002640.1 | Yes | No | PCR2: Protein PLANT CADMIUM RESISTANCE 2 | Homolog of tomato FW2.2 |
| SMEL4_02g003540.1 | No | Yes | CNR1: Cell number regulator 1 | Ortholog of tomato FW2.2 | |
| SMEL4_02g008010.1 | No | Yes | OFP7: Transcription repressor OFP7 | Ortholog to tomato ovate | |
| SMEL4_02g010610.1 | No | Yes | WUS: Protein WUSCHEL | Ortholog of tomato LC/WUS | |
| SMEL4_02g013940.1 | No | Yes | PCR1: Protein PLANT CADMIUM RESISTANCE 1 | Homolog of tomato FW2.2 | |
| SMEL4_02g015770.1 | No | Yes | WOX9: WUSCHEL‐related homeobox 9 | Homolog of tomato LC/WUS | |
| SMEL4_02g015780.1 | No | Yes | WOX8: WUSCHEL‐related homeobox 8 | Homolog of tomato LC/WUS | |
| SMEL4_04g004380.1 | No | Yes | IQD1: Protein IQ‐DOMAIN 1 | Homolog of tomato SUN | |
| SMEL4_06g012950.1 | No | Yes | IQD14: Protein IQ‐DOMAIN 14 | Homolog of tomato SUN | |
| SMEL4_06g026430.1 | Yes | No | YAB2: Putative axial regulator YABBY 2 | Homolog of tomato FAS | |
| SMEL4_08g003660.1 | Yes | Yes | YAB4: Protein YABBY 4 | Homolog of tomato FAS | |
| SMEL4_08g014200.1 | No | Yes | IQD1: Protein IQ‐DOMAIN 1 | Homolog of tomato SUN | |
| SMEL4_09g010770.1 | Yes | No | FAF3: Protein FANTASTIC FOUR 3 | Homolog of tomato CSR | |
| SMEL4_10g009740.1 | No | Yes | OFP2: Transcription repressor OFP2 | Ortholog of tomato suppressor of ovate | |
| SMEL4_10g026030.1 | No | Yes | PCR12: Protein PLANT CADMIUM RESISTANCE 9 | Homolog of tomato FW2.2 | |
| SMEL4_11g016380.1 | Yes | No | IQD1: Protein IQ‐DOMAIN 1 | Homolog of tomato SUN |
Several candidate genes encoding proteins potentially involved in fruit anthocyanin pigmentation were identified in the SS regions (Figure 5; Table 2), including homologs of biosynthetic genes (FLAVONOL SYNTHASE [FLS], CHALCONE ISOMERASE [CHI], ANTHOCYANIDIN REDUCTASE [ANR], DIHYDROFLAVONOL 4‐REDUCTASE [FRR]) and genes encoding multidrug and toxin extrusion (MATE) and ATP‐binding cassette (ABC) transporters mediating anthocyanin sequestration in other species (Francisco et al., 2013; Jaakola et al., 2002; Pérez‐Díaz et al., 2014). Candidate genes also included homologs of Prx31, which encodes a peroxidase involved in anthocyanin degradation (Movahed et al., 2016), and of genes encoding transcription factors involved in anthocyanin/proanthocyanidin regulation, such as MYB14 (Hancock et al., 2012) and SmelMYBL1 (Moglia et al., 2020). Several QTLs and QTNs controlling fruit anthocyanin pigmentation co‐localized with the SS on chr. 10 (Figure 5).
Genes encoding transcription factors putatively involved in trichome/prickle formation were spotted within the SS regions identified for this trait, including homologs of the Arabidopsis GLABRA1 (GL1) and GLABROUS INFLORESCENCE STEMS2 (GIS2) genes (Figure 5; Table 2). QTNs for fruit calyx and stem prickles map on SSs located on chrs. 2, 6, and 8, as well as the PRICKLINESS marker (Gramazio et al., 2014) and the recently identified PI locus (Miyatake et al., 2020), both falling within a SS on chr. 6 (Figure 5).
Several homologs of genes controlling fruit size/shape in tomato were identified in SS regions involved in fruit shape, including FW2.2, YABBY/FASCIATED (FAS), LOCULE NUMBER (LC)/WUSCHEL (Muños et al., 2011), OVATE, IQ‐domain/SUN (IQD), SUPPRESSOR OF OVATE (SOV), and CELL SIZE REGULATOR (CSR; Mu et al., 2017) (Figure 5; Table 2). QTNs and QTLs previously identified on several chromosomes and controlling, among others, fruit shape, length, and diameter (on chrs. 1, 2, 3, 4, 7, 10, 11, and 12), weight (on chrs. 1, 2, 3,10, 11, and 12), and yield (on chrs. 2 and 3) were syntenic to the identified SS regions (Figure 5).
DISCUSSION
Hi‐C retrofitting of an eggplant short read assembly
The precipitous development of next‐generation sequencing technologies has increased the numbers of novel genome assemblies deposited each year in public databases enormously. Presently, 10 277 draft eukaryotic genome assemblies are listed in the NCBI GenBank database, the vast majority of which are based on short read technologies. Recently, CL assemblies, based on a combination of short and long reads, optical mapping, 10× Genomics, and Hi‐C scaffolding, have started to emerge. This is the case for eggplant, for which a short read‐ and optical mapping‐based chromosome‐anchored assembly (v3.0) was released in 2019 (Barchi et al., 2019b), and a CL assembly, based on a combination of Illumina, Nanopore, 10× Genomics, and Hi‐C scaffolding, was released in 2020 (Wei et al., 2020a).
Through the simple scaffolding of the v3.0 assembly using chromosome conformation capture (HI‐C), we have increased its quality to a level comparable to that of the CL assembly in terms of contiguity, the fraction of Ns, and numbers of anchored and positioned genes. Up to 1.11 Gb of sequences, containing 96.4% of the predicted genes, have been anchored to the 12 chromosomes, and the size of chr. 0 (i.e., the unanchored sequences) has been reduced to just approximately 51 Mb, smaller than the one of the CL assembly. BlastP comparison with the Arabidopsis proteome and BUSCO analysis revealed that the quality of the v3.0/v4.0 annotation is comparable, or better than, that of the CL one, apart from tandemly repeated genes (e.g., NBS genes) and repetitive elements, which are probably underrepresented in v4.0. Thus, the methods developed here can be used for the inexpensive retrofitting of thousands of short read‐based genome assemblies without having to rerun the corresponding annotations, which have been used for a long time and were often manually improved by the respective scientific communities.
An eggplant pan‐genome and pan‐plastome
A pan‐genome represents the entire gene set for a species and includes core genes, present in all individuals, as well as genes absent in one or more individuals (Golicz et al., 2016). We constructed an eggplant pan‐genome of 26 accessions, of which 24 were of the cultivated S. melongena and one each from two closely related wild species (S. insanum and S. incanum). About 52 Mb of additional sequences were captured, including 816 protein‐coding genes. These values are lower than those of other Solanaceae pan‐genomes containing a much larger number of resequenced accessions, for example, 351 Mb and 4873 genes in 725 tomato accessions (Gao et al., 2019); and 956 Mb and 6984 genes in 383 pepper accessions (Ou et al., 2018). The eggplant pan‐genome contains >88% of core genes, similar to Brassica oleracea (Golicz et al., 2016), but higher than tomato (74.2%; Gao et al., 2019), Arabidopsis (70%; Contreras‐Moreira et al., 2017), and Brassica napus (62%; Hurgobin et al., 2018). These differences are due to the genetic diversity of each taxon, as well as to the number of accessions and species included in each pan‐genome project. Based on the data shown here, we postulate a 400‐accession pan‐genome of S. melongena to contain up to 5249 additional genes with respect to the reference genome, with the 400th genome still providing eight new genes, while the core genome would be composed of 31 227 genes. This number of additional genes is slightly higher than the one predicted using the Chao estimator (4521), which gives a conservative estimate of pan‐genome size.
The size of the chloroplast genome was similar to the ones reported for pepper, tomato, and potato (Kahlau et al., 2006; Magdy et al., 2019; Wu, 2016) and its sequence was highly conserved. However, few faster‐evolving loci, namely ycf1 and the intergenic region trnA‐trnR, might be of use for DNA barcoding purposes (Hollingsworth et al., 2009). A high level of conservation was also observed for GC content and gene numbers, which were similar to those of the other cultivated Solanaceae (Zhang et al., 2018b). One tRNA (trnG‐UCC) was complete in the S. incanum accession, suggesting a species‐specific loss. The ycf1 gene starts in the SSC region, but its sequence crosses the SSC/IRa boundary, causing a duplication of the 3′‐end portion in IRb, thus producing a <1000‐bp ycf1 pseudogene, similar to what was observed in tomato, potato, and Nicotiana (Chung et al., 2006). In addition to IRa extending into the ycf1 gene, IRb also extends into the rps19 gene, creating a duplication of various lengths of the 5′‐end of the rps19 gene at the IRa/LSC border. Only one copy of rps19 is still functional, while the other turned into a pseudogene, as observed in other Solanaceae (Chung et al., 2006). A similar situation is observed for infA, coding for translation initiation factor 1, which represents a classic example of chloroplast‐to‐nucleus gene transfer; it is a pseudogene in eggplant and in at least 17 other Solanaceae species examined, suggesting the gene was lost in the common ancestor of this family (Millen et al., 2001).
Genetic diversity
The average heterozygosity in S. melongena accessions was less than 0.03%, slightly lower than previously reported (Barchi et al., 2019a; Gramazio et al., 2019), confirming the prevalent autogamy exhibited by the species. In the two NMSs (S. insanum and S. incanum), heterozygosity was on average higher (0.29%), consistent with their partial allogamy (Acquadro et al., 2017; Daunay et al., 2001b; Vorontsova and Knapp, 2016). Chromosome 2 was the richest in SNPs, probably reflecting the inclusion among the S. melongena accessions of the breeding line ‘305E40’ (GPE001980), carrying a large introgression from S. aethiopicum in chr. 2 (Portis et al., 2014; Toppino et al., 2008).
Based on the whole SNP dataset, S. melongena accessions clustered separately from the NMS accessions, in both the ML dendrogram and the PCA graph. The ML tree based on the PAV matrix provided similar results, with S. insanum, the direct wild ancestor of S. melongena (Knapp et al., 2013), showing a similar separation from eggplant accessions. On the other side, the tree we constructed on the basis of the chloroplast genome sequences was sufficient to correctly separate eggplant from S. incanum, while S. insanum is more closely related to two S. melongena accessions (GPE059520 and GPE000430) which are small‐fruited, have low vigor, and exhibit semi‐prostrate growth.
Selective sweeps and domestication
Several previous studies highlighted a decrease of genetic diversity in domesticated species compared to their wild relatives (Bellucci et al., 2014; Gao et al., 2019; Liu et al., 2019), especially in autogamous species. Although only one S. insanum (the direct progenitor of S. melongena) and one S. incanum (a close relative) accession were used in the analysis, the nucleotide diversity (π) for S. melongena suggests that the cultivated species underwent a reduction in diversity (πinsInc/πeggplant = 4.06), attributable to genetic bottlenecks during domestication. This is in line with a recent estimate of a 47% loss of genetic diversity following domestication in the species (Page et al., 2019).
Several SSs related to different phenotypic traits were identified (Tables S11 and S12). The stringent criteria used (top 5% ROD and/or top 1% XP‐CLR) allowed to narrow down the numbers of candidate genes falling within SSs for each trait. On average, for each scored trait, the SSs defined by either of the two criteria comprised 2400 (6.87%) genes, while those defined by both criteria comprised 276 (0.79%) genes. Although the exact locus targeted by selection cannot be determined from sweep data alone, the overlap between QTLs, QTNs, and candidate genes from previous studies and SSs is suggestive of which traits have been shaped by positive selection during eggplant evolution, and in some cases can be tied to known candidate genes. The absence of PAVs within SSs for the traits under study suggests that breeding for these traits acted by selecting for allelic variants, rather than by introducing or deleting individual genes.
Anthocyanin biosynthesis is one of the most studied pathways in plants. Its control is strongly dependent on the tissue, developmental stage, and environment, and has been partially elucidated in eggplant (Barchi et al., 2019b; Moglia et al., 2020; Xiao et al., 2018; Zhang et al., 2014). Modern eggplant varieties present anthocyanin pigmentation of the peel, whereas ancestral genotypes present non‐anthocyanic (white or green) fruits. By comparing genotypes with white/green versus purple fruits, a SS on chr. 10 was identified, which contains the transcriptional repressor gene SmelMYBL1 (Moglia et al., 2020) as well as QTLs and QTNs for flower color and anthocyanin accumulation (Barchi et al., 2012; Cericola et al., 2014; Frary et al., 2014; Portis et al., 2015; Toppino et al., 2016). SmelMYBL1 encodes an R3 MYB transcription factor and represents a novel component of the eggplant bHLH, MYB, WD40 (BMW) regulatory complex, where it probably competes with MYB activators of anthocyanin gene transcription (SmelANT1 and SmelAN2). Other candidate genes localized on different chromosomes include homologs of anthocyanin biosynthetic genes (ANR, FLS, CHI, FRR) and ABC transporters mediating vacuolar transport of anthocyanins in Vitis vinifera (Francisco et al., 2013). In addition, homologs of V. vinifera Prx31, which encodes a peroxidase involved in anthocyanin degradation (Movahed et al., 2016), and MATE1, encoding proanthocyanidin transporter (Pérez‐Díaz et al., 2014), as well as a gene encoding an R2R3‐MYB transcription factor (MYB14) involved in the regulation of proanthocyanidins in legumes (Hancock et al., 2012). Prickles are a distinctive trait of eggplant and its wild relatives, the Solanum subgenus Leptostemonum (spiny solanums) comprising approximately 450 species (Knapp et al., 2019). Although prickly eggplant types are preferred in certain regions on the basis of their perceived superior organoleptic quality, prickles are generally considered an undesirable trait and they have been counter‐selected in several modern varieties (Daunay et al., 2001a). It is thought that plant prickles are modified glandular trichomes and that their biogenesis in controlled by transcription factors from the MYB, bHLH, WD40, WRKY, and C2H2 zinc finger families (Wang et al., 2019). Indeed, by comparing prickled and non‐prickled genotypes, we identified several SSs containing genes encoding homologs of Arabidopsis transcription factors involved in trichome initiation, such as GL1 and GIS2 (Wang et al., 2019; Zhang et al., 2021). SSs on chr. 6 fall in proximity of previously identified QTLs and QTNs as well as the morphological marker PRICKLINESS (Frary et al., 2014; Gramazio et al., 2014; Portis et al., 2015) and the recently identified Pl locus involved in prickle formation in eggplant (Miyatake et al., 2020; Zhang et al., 2021), suggesting that these regions have been the targets of human selection for presence/absence of prickles.
Fruit size and shape are key traits for breeding and are controlled by several genes. In tomato, genes controlling fruit size and shape include CELL SIZE REGULATOR (CSR), FW2.2, KLUH/FW3.2, OVATE FAMILY PROTEIN (OFP), SOV, TONNEAU RECRUTING MOTIF (TRM5), SUN/IQD, LC/WUSCHEL, and FAS/YABBY (reviewed in van der Knaap and Østergaard, 2018). Fruit shape SSs in the resequenced accessions contain several homologs of CSR, FW2.2, OVATE, SOV, SUN/IQD, LC/WUSCHEL, and FAS/YABBY. In particular, the eggplant orthologs of FW2.2, OVATE, LC/WUSCHEL, SOV, and CSR are localized in SSs for fruit shape on chrs. 2, 10, and 12. LC, together with FAS, controls locule number and, consequently, flat shape in tomato. LC and FAS mutations arose early during tomato domestication, and contributed to the man‐made selection for increased fruit size (van der Knaap and Østergaard, 2018; Muños et al., 2011; Rodríguez et al., 2011). Our data suggest that selection involving eggplant LC occurred during the selection of modern large and more elongated fruit types from the original small and round‐fruited ones. CSR encodes an uncharacterized protein whose clade has expanded in the Solanaceae family and the large‐fruited allele is found in S. lycopersicum var. cerasiforme, suggesting that the selection at this locus was critical to the early domestication of tomato (Mu et al., 2017). Again, our data appear to confirm that selection at the eggplant ortholog had an influence on fruit shape. Additional QTLs and QTNs previously identified (Mangino et al., 2021; Portis et al., 2014, 2015; Wei et al., 2020b) and controlling fruit shape and size, as well as a recently identified region controlling fruit length (Wei et al., 2020a), fall within some of the SSs identified. However, we have not found, within the high‐confidence SS intervals for fruit shape, any SUN gene homologs like the one reported by Wei et al. (2020a) on chr. 3. This might be due to the absence of alleles of this gene in our resequenced accessions, which do not include extremely elongated fruit shaped accessions like in the study of Wei et al. (2020a).
In conclusion, the improved assembly of the eggplant reference genome and the first eggplant pan‐genome and pan‐plastome described in this paper add depth and completeness to our genomic understanding of eggplant and its breeding history. The identification of SSs and candidate genes for key eggplant agronomic traits lays the foundations for an initial understanding of the genomic events underlying domestication and selection of this important vegetable species.
EXPERIMENTAL PROCEDURES
Generation of the v4.0 assembly
Hi‐C library construction and sequencing was conducted in the ‘67/3’ S. melongena accession (G2P‐SOL code GPE001970) previously sequenced (Barchi et al., 2019b), as reported earlier (Padmarasu et al., 2019). Adapter trimming, quality control, and validation of Hi‐C fragments were conducted as previously described (Mascher et al., 2017) using the scripts as implemented in the TRITEX pipeline (Monat et al., 2019).
Hi‐C contact scores between 200 000‐bp bins were produced via binning and normalization of the interbin contact scores as described by Hu et al. (2012), also implemented using the TRITEX pipeline (Monat et al., 2019). Two sets of scaffolds produced in eggplant assembly v3.0 were used for producing the updated assembly (v4.0): (i) the Illumina scaffolds from v3.0 and (ii) the superscaffolds generated by the optical mapping, plus the Illumina scaffolds not incorporated into a superscaffold. Superscaffolds were given initial chromosome assignments using the mapped positions and linkage groups of the genetic map markers generated by Barchi et al. (2019a). For the set of markers mapped to a superscaffold, if the most commonly represented linkage group outnumbered the second most common by >2:1, then the v3.0 scaffolds comprising the superscaffold were initially assigned to the chromosome associated with that linkage group. We applied a three‐phase process to optimize the chromosome assignments of the v3.0 scaffolds:
‘Capture’ phase: Hi‐C links between chromosome‐assigned and unassigned scaffolds were tabulated, and unassigned scaffolds were assigned to a chromosome if (1) there were at least five Hi‐C links involving the scaffold and other chromosome‐assigned scaffolds and (2) the number of links joining to scaffolds assigned to the most commonly linked chromosome was at least 0.6 times the number of links to the next most commonly linked chromosome.
‘Correction’ phase: The assignments of all scaffolds were tested for consistency with the Hi‐C information. Every scaffold’s assignment was tested in the same way as during the capture phase, as though it was unassigned.
‘Agreement’ phase: Scaffolds were tested to check for disagreements between the Hi‐C‐based assignments and the original, map‐based assignments. Scaffolds with more than three genetic map markers and for which the Hi‐C‐based and the map‐based chromosome assignments disagreed were selected in order to remove their chromosome assignments.
The scaffolds were arranged into a tentative initial pseudomolecule scaffold arrangement for each chromosome (known as ‘A Golden Path’ [AGP]) using the Burton et al. (2013) method as implemented in the TRITEX pipeline and by exploiting the homologous AGP positions of genetic map markers available from several publicly available eggplant genetic mapping studies (Fukuoka et al., 2012; Hirakawa et al., 2014; Miyatake et al., 2012; Nunome et al., 2009). Manual editing of the order then proceeded in 10 rounds by informed trial‐and‐error, until a near‐optimal AGP was found, as revealed by visual inspection of the Hi‐C contact matrices (Figure 1), Hi‐C asymmetry plots (Himmelbach et al., 2018), and aligned genetic map marker positions. Genome annotation was lifted from v3.0 to v4.0 using flo (Pracana et al., 2017, Table S13).
For comparative purposes, v4.0 assembly was aligned against the CL assembly using minimap2 (Li, 2018) and plotted with dotPlotly (https://github.com/tpoorten/dotPlotly). Finally, our proteome (34 916 proteins) and the one recently released (36 568 proteins; Wei et al., 2020a), alongside those of tomato (ITAG4.1, 34 688 proteins), potato (ITAG v1, 35004 proteins; The Tomato Genome Consortium, 2012), pepper (PGA v2, 35 884 proteins Kim et al., 2017) and the eggplant annotation generated in Hirakawa et al. (2014) (Sme_r2.5.1, 42 035 proteins), were compared against the high‐quality Arabidopsis nuclear proteome (TAIR10, 27 206 proteins; Lamesch et al., 2012, p. 10) by BlastP (e‐value = 10−3), and the relative length ratios of the best protein pairs were computed.
Resequencing, assembly, and pan‐genome/pan‐plastome construction and annotation
Published genome sequencing data of nine S. melongena accessions and one S. incanum accession (Barchi et al., 2019b; Gramazio et al., 2019) were retrieved from the NCBI Sequence Read Archive (SRA) database. Genome sequences of a total of 16 additional genotypes available from a worldwide collection (http://www.g2p‐sol.eu/G2P‐SOL‐gateway.html) and including 15 S. melongena genotypes and one S. insanum genotype were generated here. Genomic DNA was extracted from a single seedling from each of these 16 accessions using a Qiagen plant Mini‐Prep kit. Paired‐end libraries with insert sizes of approximately 350 bp were constructed using the NEBNext DNA Library Prep kit (Illumina Inc.) according to the manufacturer’s instructions and sequenced on an Illumina Novaseq platform using the paired‐end 2×150 bp mode. The sequencing raw data of the 16 newly sequenced accessions are available at NCBI SRA (BioProject ID PRJNA649091).
Adapters and low‐quality sequences were trimmed from raw Illumina reads using Scythe (v0.994, https://github.com/vsbuffalo/scythe) to filter out contaminant substrings, and Sickle (v1.33, https://github.com/najoshi/sickle) was used to remove reads with poor‐quality ends. Reads were error‐corrected using the Spades tool (v3.13) (Bankevich et al., 2012). Finally, kraken2 (Wood et al., 2019) was used to remove contaminant reads from bacterial, archaeal, and viral genomes (MiniKraken DB_8GB) as well as from fungi (complete fungi sequences, https://www.ncbi.nlm.nih.gov/refseq/).
High‐quality cleaned Illumina reads from 25 samples (with the exclusion of the reference line) were de novo assembled using Megahit (v.1.2.9) (Li et al., 2015) with default parameters. The assembled contigs with lengths of >500 bp were kept and then aligned to the eggplant v4.0 genome of the line ‘67/3’ using minimap2 (v.2.17‐r954‐dirty) (Li, 2018) with setting ‘asm10’ for S. melongena and ‘asm20’ for other species. Contigs with no reliable alignments were kept as unaligned contigs. For contigs containing the reliable alignments, if they also contained continuous unaligned regions longer than 500 bp, the unaligned regions were extracted as unaligned sequences. The cleaned non‐reference sequences from all accessions were combined. The redundant sequences were consolidated into unique contigs using CD‐HIT (v.4.8.1) (Fu et al., 2012) and the non‐redundant contigs were finally searched against the GenBank nucleotide database using BlastN (Camacho et al., 2009). Sequences with best hits from outside the green plants or covered by known plant mitochondrial or chloroplast genomes were possible contaminations and hence were removed.
The final non‐redundant non‐reference sequences and the v4.0 assembly were merged into an eggplant pan‐genome. Curve fitting of the pan‐genome was performed using a power‐law regression based on Heaps’ law (y = A pan x Bpan + C pan), where y is the pan‐genome size, x the genome number, and A pan, B pan, and C pan the fitting parameters, as previously described (Rasko et al., 2008; Tettelin et al., 2005, 2008). Fitting was conducted with PanGP software (Zhao et al., 2014). B pan was equivalent to the γ‐parameter used by Tettelin et al. (2005, 2008) in estimating an open or closed pan‐genome. When 0 < B pan < 1, the size of the pan‐genome increases unboundedly with sequential additions of new genomes, thus indicating an open pan‐genome. Conversely, when B pan < 0 or B pan > 1, the trajectory approaches a plateau as further genomes are added, indicating a closed pan‐genome. Curve fitting of the core genome was performed using an exponential regression model (y = A core e ( B core x ) + C core), where y is the core genome size, x the genome number, and A core, B core, and C core the fitting parameters. Estimation of pan‐genome features was done using micropan (Snipen and Liland, 2015).
A custom repeat library was constructed and used to annotate the novel genome sequences using MAKER‐P (v.3.01.01) (Campbell et al., 2014) with Augustus (v.3.3.2) (Stanke et al., 2006) and SNAP (version 2006‐07‐28) (Bromberg and Rost, 2007). The gene models identified with an AED of <0.5 were checked using InterProScan 5 (v. 5.44–79.0) (Jones et al., 2014). Genes were functionally annotated by comparing their protein sequences against the SwissProt Viridiplantae database (The UniProt Consortium, 2014) and the InterPro domain database. GO enrichment was calculated using AGRIGO V2 (Tian et al., 2017) as well as with AGRIGO cross‐comparison of SEA (SEACOMPARE) to identify common and unique GO enrichment terms.
SNP calling, variant annotation, polymorphism estimates, LD decay, PAVs, and selective sweeps
The sequences were mapped to the pan‐genome using the Burrows‐Wheeler Aligner (BWA) program (v. 0.7.17–r1188) and the ‘mem’ command with default parameters (Li, 2013). BAM files were processed and used for the SNP calling using bcftools mpileup/call/norm utilities (v. 1.10.2) (Li, 2011) with default parameters except for the use of the multiallelic calling model (−m option), minimum mapping quality (Q = 20), and filtering out multimapping events (−q > 1). Bcftools was applied to generate the final SNP dataset using a read depth of 10 as a cutoff and a max missing rate of 30% for retaining a SNP at the genotype level.
SNPs/indels were counted and analyzed using bcftools. The estimation of the heterozygous level of each genome (i.e., ‘genome level’) was calculated by considering, for each accession, the ratio between the number of SNPs/indels (called heterozygous state) and the ungapped size of the pan‐genome, deprived of Ns (1.17 Gb) as previously reported (Acquadro et al., 2020). Vcftools (Danecek et al., 2011) was applied to count the number of SNPs along the reference genome using a sliding window approach (1‐Mb windows, sliding in 500‐kb steps). The results were plotted with R v4.0.3 (R Core Team, 2020).
RAxML‐NG (v.0.9) (Kozlov et al., 2019) was used to construct an ML tree and branch supports were obtained with an MRE‐based bootstrapping test (Pattengale et al., 2010). A PCA using the final SNP dataset was obtained with the SNPrelate (Zheng et al., 2012) program; SNPs with a minor allele frequency (MAF) of <0.05 were excluded. LD plots were calculated with PopLDdecay (Zhang et al., 2018a), while Vcftools was used to calculate genetic diversity (π) and population differentiation (F ST), which was estimated using Weir and Cockerham (Weir and Cockerham, 1984) values, using a sliding window approach (200‐kb windows, sliding in 100‐kb steps); SNPs with an MAF of <0.05 were excluded.
SSs in S. melongena were identified for five contrasting fruit traits: (i) NAvsA; (ii) PvsNP; (iii) LvsO (length/width ratio > 2 versus length/width ratio = 1–2); (iv) RvsL (length/width ratio = approximately 1 versus length/width ratio > 2); and (v) RvsO (length/width ratio = approximately 1 versus length/width ratio = 1–2). Firstly, we calculated the ROD using a sliding window approach (200‐kb windows, sliding in 100‐kb steps; SNPs with an MAF of <0.05 were excluded) in Vcftools between S. melongena accessions showing one of the five contrasting phenotypes in study. In parallel, an implementation (https://github.com/hardingnj/xpclr) of the XP‐CLR (Chen et al., 2010) was also used. For each chromosome, the XP‐CLR score was calculated in 50‐kb windows with parameters ‘‐‐maxsnps 400, ‐‐rrate 1e‐8 ‐‐ld0 0.7’. Then, SS regions were identified as regions identified in the top 5% values for ROD statistics and/or in the top 1% of XP‐CLR scores. Adjacent regions within the same phenotypic trait were merged with the bedtools suite (Quinlan and Hall, 2010).
Genome reads from each accession were aligned to the pan‐genome using BWA‐MEM (Li, 2013) with default parameters. The presence or absence of each gene in each accession was determined using SGSGeneLoss (v.0.1) (Golicz et al., 2015). In brief, for a given gene in a given accession, if less than 20% of its exon regions were covered by at least two reads (minCov = 2, lostCutoff = 0.2), this gene was considered as absent in that accession; otherwise it was considered present. An ML phylogenetic tree was constructed based on the binary PAV data with 1000 bootstraps using IQ‐TREE (Minh et al., 2020). A co‐phylogenetic plot of the ML dendrograms based on the pan‐genome and PAV sequences was obtained using the R package phytools (Revell, 2012).
Chloroplast genome assembly and annotation
Chloroplast genomes of the resequenced accessions, together with the one from the S. melongena reference line ‘67/3’, were assembled using GetOrganelle (Jin et al., 2018) and annotated using GeSeq (Tillich et al., 2017). The same annotation pipeline was applied to already available eggplant chloroplast sequences, including those of three S. melongena (Aubriot et al., 2018; Ding et al., 2016), two S. incanum, and one S. insanum sequence from Aubriot et al. (2018). A circular plastome map representing S. melongena ‘67/3’ lines was drawn using OrganellarGenome DRAW (Greiner et al., 2019).
All plastomes were aligned using MAFFT v7 (Katoh and Standley, 2013). A phylogenetic tree by the ML method using the IQ‐TREE software (Nguyen et al., 2015) was constructed; branch supports were obtained with the ultrafast bootstrap (Hoang et al., 2018). A co‐phylogenetic plot of the ML dendrograms based on the pan‐genome and pan‐plastome sequences was obtained using the R package phytools (Revell, 2012).
Hypervariable regions were identified with sliding window analysis in DNASP v6 (Rozas et al., 2017). The mVISTA program (Frazer et al., 2004) in Shuffle‐LAGAN (Brudno et al., 2003) mode was used to compare the cp genomes using ‘67/3’ as a reference. The top 10 most variable non‐coding regions of high Pi value were counted by potentially informative characters following Shaw et al. (2005).
The SciRoKo tool (Kofler et al., 2007) was used to search for SSR loci (i.e., mono‐, di‐, tri‐, tetra‐, penta‐ and hexanucleotide repeats). The minimum numbers (thresholds) of repeats were 10, 5, 4, 3, 3, and 3 for mono‐, di‐, tri‐, tetra‐, penta‐, and hexanucleotides, respectively. REPUTER (Kurtz et al., 2001) was used to assess the number and location of repeat sequences, including direct (forward), inverted (palindromic), complement, and reverse repeats. For all the repeat types, the constraints set in REPUTER were (i) a minimum repeat size of 30 bp and (ii) 90% greater sequence identity with Hamming distance equal to 3 (i.e., the gap size between repeats had a maximum length of 3 bp). All overlapping repeats were removed from the final results.
AUTHOR CONTRIBUTIONS
JP, SL, NS, GLR, and GG conceived the study. SP produced the Hi‐C library. MTRW performed the genome assembly. LT provided materials. EP contributed producing sequencing data. LB produced and analyzed resequencing data. LB and GG wrote the manuscript. All authors critically revised and approved the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Figure S1. Hi‐C scaffolding of the v3.0 eggplant genome assembly. (a) In silico inferred distribution of valid (valid restriction site‐associated, religated) and invalid (‘paired‐end’ [PE]) Hi‐C fragment lengths in the Hi‐C dataset (Monat et al., 2019). (b) Hi‐C contact frequency plots showing the improvement in chromosome‐scale contiguity between assembly versions v3.0 (Barchi et al., 2019) and v4.0 (current study). Log Hi‐C link counts between 200‐Mbp bins are given after normalization (see the Experimental Procedures section). Stark discontinuities in the contact frequencies are evidence of missing sequences or incorrectly arranged scaffolds.
{kind=link}
Figure S2. (a) Synteny analysis of S. melongena ‘67/3’ assembly v4.0 (on the x‐axis) and the S. melongena CL assembly (y‐axis). (b) Histogram of pairwise comparisons of predicted protein length ratios with their best hit in the Arabidopsis TAIR10 annotation (BlastP, e‐value = 10−3).
{kind=link}
Figure S3. Comparison of 32 chloroplast genomes using mVISTA. Complete cp genomes were compared, with GPE001970 as a reference. Blue block: conserved genes, sky‐blue block: tRNA and rRNA, red block: conserved non‐coding sequences (CNSs). White represents regions with sequence variation.
Figure S4. Numbers of (a) SSRs and (b) repeats identified in the 32 chloroplast sequences. Colors depend on the motifs (a) or size (b) of the repeats identified.
{kind=link}
Figure S5. Distribution of single‐nucleotide polymorphisms (SNPs) along the reference genome sequence. The SNP distribution for all the accessions in the study is shown in orange, the SNP distribution only for eggplant is shown in purple, and the SNP distribution only for NMSs is shown in blue (S. insanum and S. incanum).
{kind=link}
Figure S6. Principal component analysis (PCA) visualization of the genetic relationships among the accessions of eggplant as well as cultivated and wild related species, based on (a) the whole set of SNPs identified; (b) the whole set of SNPs identified limited to S. melongena; (c) PAVs; and (d) PAVs limited to S. melongena. Solanum melongena entries are shown in purple, S. incanum entries in red, and S. insanum entries in green.
{kind=link}
Figure S7. Linkage disequilibrium (LD) decay in all accessions (orange) and S. melongena (purple). r 2 estimates were plotted against the physical distance.
{kind=link}
Table S1. Hi‐C metrics.
Table S2. Chromosome metrics of Smel v3.0 and v4.0 and their comparison with Smel CL.
Table S3. Phenotypic characteristics, geographical origin, and genome assembly metrics of the eggplant accessions.
Table S4. Pan‐genome genes present or absent following read mapping in the reference line GPE001970 (‘67/3’).
Table S5. Different categories of genes in the eggplant pan‐genome.
Table S6. Gene ontology (GO) term enrichments for different pan‐genome gene categories using AGRIGO SEACOMPARE.
Table S7. Assembly metrics of the eggplant pan‐plastome. The asterisk (*) indicates already assembled chloroplasts.
Table S8. List of genes in the eggplant pan‐plastome.
Table S9. SNPs identified in the pan‐genome sequences for all 26 accessions.
Table S10. SNP distribution per chromosome in the 26 accessions.
Table S11. Highest ROD and XP‐CLR values for the 5 breeding traits in study in the selective sweeps identified.
Table S12. Selective sweeps number and size and genes number for the five breeding traits in study identified with ROD top 5% and/or XP‐CLR top 1%.
Table S13. Conversion of gene models between annotations v3.0 and v4.0.
ACKNOWLEDGMENTS
This work was supported by the European Commission, Horizon 2020 G2P‐SOL project (grant agreement no. 677379). We gratefully acknowledge Ines Walde and Axel Himmelbach for technical assistance.
DATA AVAILABILITY STATEMENT
All Raw Illumina data were deposited at NCBI SRA (BioProject ID PRJNA649091). The v4.0 and pan‐genome assemblies and annotations are available at https://solgenomics.net/. Chloroplast genomes were submitted to GenBank (accession numbers MW384824–MW384851). Seeds of the accessions used in the present study can be obtained from COMAV (jprohens@btc.upv.es) or CREA (laura.toppino@crea.gov.it).
REFERENCES
- Acquadro, A., Barchi, L., Gramazio, P., Portis, E., Vilanova, S., Comino, C.et al. (2017) Coding SNPs analysis highlights genetic relationships and evolution pattern in eggplant complexes M. Singh, ed., 12, e0180774. 10.1371/journal.pone.0180774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acquadro, A., Barchi, L., Portis, E., Nourdine, M., Carli, C., Monge, S.et al. (2020) Whole genome resequencing of four Italian sweet pepper landraces provides insights on sequence variation in genes of agronomic value. Scientific Reports, 10, 9189. 10.1038/s41598-020-66053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubriot, X., Knapp, S., Syfert, M.M., Poczai, P. & Buerki, S. (2018) Shedding new light on the origin and spread of the brinjal eggplant (Solanum melongena L.) and its wild relatives. American Journal of Botany, 105, 1175–1187. 10.1002/ajb2.1133. [DOI] [PubMed] [Google Scholar]
- Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S.et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19, 455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi, L., Acquadro, A., Alonso, D.et al. (2019a) Single Primer Enrichment Technology (SPET) for high‐throughput genotyping in tomato and eggplant germplasm. Front. Plant Sci., 10, 10.3389/fpls.2019.0100510.3389/fpls.2019.01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi, L., Lanteri, S., Portis, E., Valè, G., Volante, A., Pulcini, L.et al. (2012) A RAD tag derived marker based eggplant linkage map and the location of QTLs determining anthocyanin pigmentation. PLoS One, 7, e43740. 10.1371/journal.pone.0043740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi, L., Pietrella, M., Venturini, L., Minio, A., Toppino, L., Acquadro, A.et al. (2019b) A chromosome‐anchored eggplant genome sequence reveals key events in Solanaceae evolution. Scientific Reports, 9, 11769. 10.1038/s41598-019-47985-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi, L., Toppino, L., Valentino, D., Bassolino, L., Portis, E., Lanteri, S.et al. (2018) QTL analysis reveals new eggplant loci involved in resistance to fungal wilts. Euphytica, 214, 20. 10.1007/s10681-017-2102-2. [DOI] [Google Scholar]
- Bellucci, E., Bitocchi, E., Ferrarini, A., Benazzo, A., Biagetti, E., Klie, S.et al. (2014) Decreased nucleotide and expression diversity and modified coexpression patterns characterize domestication in the common bean. The Plant Cell, 26, 1901–1912. 10.1105/tpc.114.124040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg, Y. & Rost, B. (2007) SNAP: predict effect of non‐synonymous polymorphisms on function. Nucleic acids research, 35, 3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno, M., Malde, S., Poliakov, A., Do, C.B., Couronne, O., Dubchak, I.et al. (2003) Glocal alignment: finding rearrangements during alignment. Bioinformatics (Oxford, England), 19(Suppl 1), i54–62. 10.1093/bioinformatics/btg1005. [DOI] [PubMed] [Google Scholar]
- Burton, J.N., Adey, A., Patwardhan, R.P., Qiu, R., Kitzman, J.O. & Shendure, J. (2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology, 31, 1119–1125. 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K.et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M.S., Law, MeiYee, Holt, C., Stein, J.C., Moghe, G.D., Hufnagel, D.E.et al. (2014) MAKER‐P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant physiology, 164, 513–524. 10.1104/pp.113.230144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cericola, F., Portis, E., Lanteri, S., Toppino, L., Barchi, L., Acciarri, N.et al. (2014) Linkage disequilibrium and genome‐wide association analysis for anthocyanin pigmentation and fruit color in eggplant. BMC genomics, 15, 896. 10.1186/1471-2164-15-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, A. (1987) Estimating the population size for capture‐recapture data with unequal catchability. Biometrics, 43, 783–791. 10.2307/2531532. [DOI] [PubMed] [Google Scholar]
- Chapman, M.A. (Ed.) (2019) The Eggplant Genome. Cham, Switzerland: Springer International Publishing. [Google Scholar]
- Chen, H., Patterson, N. & Reich, D. (2010) Population differentiation as a test for selective sweeps. Genome Research, 20, 393–402. 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, H.J., Jung, J.D., Park, H.W., Kim, J.H., Cha, H.W., Min, S.R.et al. (2006) The complete chloroplast genome sequences of Solanum tuberosum and comparative analysis with Solanaceae species identified the presence of a 241‐bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Reports, 25, 1369–1379. 10.1007/s00299-006-0196-4. [DOI] [PubMed] [Google Scholar]
- Contreras‐Moreira, B., Cantalapiedra, C.P., García‐Pereira, M.J., Gordon, S.P., Vogel, J.P., Igartua, E.et al. (2017) Analysis of plant pan‐genomes and transcriptomes with GET_HOMOLOGUES‐EST, a clustering solution for sequences of the same species. Frontiers in Plant Science, 8, 184. 10.3389/fpls.2017.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P., Auton, A., Abecasis, G., Albers, C.a., Banks, E., DePristo, M.a.et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daunay, M.C., Lester, R.N. & Ano, G. (2001a) Cultivated eggplants. In: Charier, A., Jacquot, M., Hamon, S. & Nicolas, D. (Eds.) Tropical Plant Breeding. Oxford: Oxford University Press, pp. 200–225. [Google Scholar]
- Daunay, M.C., Lester, R.N., Gebhardt, C., Hennart, J.W. & Jahn, M. (2001b) Genetic resources of eggplant (Solanum melongena) and allied species: a new challenge for molecular geneticists and eggplant breeders. In R.G. van den Berg and G.W.M. Barendse, van der Weerden G.M., eds. Solanaceae V: Advances in Taxonomy and Utilization. pp. 251‐274. Nijmegen: Nijmegen University Press. [Google Scholar]
- Ding, Q.‐X., Liu, J. & Gao, L. (2016) The complete chloroplast genome of eggplant (Solanum melongena L.). Mitochondrial DNA Part B, 1, 843–844. 10.1080/23802359.2016.1186510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doğanlar, S., Frary, A., Daunay, M.‐C., Huvenaars, K., Mank, R. & Frary, A. (2014) High resolution map of eggplant (Solanum melongena) reveals extensive chromosome rearrangement in domesticated members of the Solanaceae. Euphytica, 198, 231–241. 10.1007/s10681-014-1096-2. [DOI] [Google Scholar]
- Doganlar, S., Frary, A., Daunay, M.‐C.‐C., Lester, R.N. & Tanksley, S.D. (2002a) A comparative genetic linkage map of eggplant (Solanum melongena) and its implications for genome evolution in the Solanaceae. Genetics, 161, 1697–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doganlar, S., Frary, A., Daunay, M.‐C.‐C., Lester, R.N. & Tanksley, S.D. (2002b) Conservation of gene function in the Solanaceae as revealed by comparative mapping of domestication traits in eggplant. Genetics, 161, 1713–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO, authors. http://faostat3.fao.org/home/E.org/
- Francisco, R.M., Regalado, A., Ageorges, A.et al. (2013) ABCC1, an ATP Binding cassette protein from grape berry, transports anthocyanidin 3‐o‐glucosides. The Plant Cell, 25, 1840–1854. 10.1105/tpc.112.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frary, A., Doganlar, S., Daunay, M.C. & Tanksley, S.D. (2003) QTL analysis of morphological traits in eggplant and implications for conservation of gene function during evolution of solanaceous species. TAG Theoretical and Applied Genetics, 107, 359–370. 10.1007/s00122-003-1257-5. [DOI] [PubMed] [Google Scholar]
- Frary, A., Frary, A., Daunay, M.‐C., Huvenaars, K., Mank, R. & Doğanlar, S. (2014) QTL hotspots in eggplant (Solanum melongena) detected with a high resolution map and CIM analysis. Euphytica, 197, 211–228. 10.1007/s10681-013-1060-6. [DOI] [Google Scholar]
- Frazer, K.A., Pachter, L., Poliakov, A., Rubin, E.M. & Dubchak, I. (2004) VISTA: computational tools for comparative genomics. Nucleic acids research, 32, W273–W279. 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. (2012) CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28, 3150–3152. 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka, H., Miyatake, K., Nunome, T., Negoro, S., Shirasawa, K., Isobe, S.et al. (2012) Development of gene‐based markers and construction of an integrated linkage map in eggplant by using Solanum orthologous (SOL) gene sets. Theoretical and Applied Genetics, 125, 47–56. 10.1007/s00122-012-1815-9. [DOI] [PubMed] [Google Scholar]
- Gao, L., Gonda, I., Sun, H., Ma, Q., Bao, K., Tieman, D.M.et al. (2019) The tomato pan‐genome uncovers new genes and a rare allele regulating fruit flavor. Nature Genetics, 51, 1044–1051. 10.1038/s41588-019-0410-2. [DOI] [PubMed] [Google Scholar]
- Golicz, A.A., Bayer, P.E., Barker, G.C., Edger, P.P., Kim, HyeRan, Martinez, P.A.et al. (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nature Communications, 7, 1–8. 10.1038/ncomms13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz, A.A., Martinez, P.A., Zander, M., Patel, D.A., Van De Wouw, A.P., Visendi, P.et al. (2015) Gene loss in the fungal canola pathogen Leptosphaeria maculans . Functional and Integrative Genomics, 15, 189–196. 10.1007/s10142-014-0412-1. [DOI] [PubMed] [Google Scholar]
- Gordon, S.P., Contreras‐Moreira, B., Woods, D.P., Des Marais, D.L., Burgess, D., Shu, S.et al. (2017) Extensive gene content variation in the Brachypodium distachyon pan‐genome correlates with population structure. Nature Communications, 8, 2184. 10.1038/s41467-017-02292-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramazio, P., Prohens, J., Plazas, M., Andújar, I., Herraiz, F.J., Castillo, E.et al. (2014) Location of chlorogenic acid biosynthesis pathway and polyphenol oxidase genes in a new interspecific anchored linkage map of eggplant. BMC Plant Biology, 14, 350. 10.1186/s12870-014-0350-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramazio, P., Yan, H., Hasing, T., Vilanova, S., Prohens, J. & Bombarely, A. (2019) Whole‐genome resequencing of seven eggplant (Solanum melongena) and one wild relative (S. incanum) accessions provides new insights and breeding tools for eggplant enhancement. Frontiers . Plant Science, 10, 10.3389/fpls.2019.01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner, S., Lehwark, P. & Bock, R. (2019) OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Research, 47, W59–W64. 10.1093/nar/gkz238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, K.R., Collette, V., Fraser, K., Greig, M., Xue, H., Richardson, K.et al. (2012) Expression of the R2R3‐MYB transcription factor TaMYB14 from Trifolium arvense activates proanthocyanidin biosynthesis in the legumes Trifolium repens and Medicago sativa . Plant Physiology, 159, 1204–1220. 10.1104/pp.112.195420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelbach, A., Ruban, A., Walde, I., Šimková, H., Doležel, J., Hastie, A.et al. (2018) Discovery of multi‐megabase polymorphic inversions by chromosome conformation capture sequencing in large‐genome plant species. The Plant Journal, 96, 1309–1316. 10.1111/tpj.14109. [DOI] [PubMed] [Google Scholar]
- Hirakawa, H., Shirasawa, K., Miyatake, K., Nunome, T., Negoro, S., Ohyama, A.et al. (2014) Draft genome sequence of eggplant (Solanum melongena L.): the representative solanum species indigenous to the old world. DNA Research : An International Journal for Rapid Publication of Reports on Genes and Genomes, 21, 649–660. 10.1093/dnares/dsu027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, D.T., Chernomor, O., von Haeseler, A., Minh, B.Q. & Vinh, L.S. (2018) UFBoot2: improving the ultrafast bootstrap approximation. Molecular Biology and Evolution, 35, 518–522. 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth, P.m., Forrest, L.l., Spouge, J.l., Hajibabaei, M., Ratnasingham, S., van der Bank, M.et al. (2009) A DNA barcode for land plants. Proceedings of the National Academy of Sciences of the United States of America, 106, 12794–12797. 10.1073/pnas.0905845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, M., Deng, K., Selvaraj, S., Qin, Z., Ren, B. & Liu, J.S. (2012) HiCNorm: removing biases in Hi‐C data via Poisson regression. Bioinformatics, 28, 3131–3133. 10.1093/bioinformatics/bts570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurgobin, B., Golicz, A.A., Bayer, P.E.et al. (2018) Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus . Plant Biotechnology Journal, 16, 1265–1274. 10.1111/pbi.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola, L., Määttä, K., Pirttilä, A.M., Törrönen, R., Kärenlampi, S. & Hohtola, A. (2002) Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiology, 130, 729–739. 10.1104/pp.006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, J.‐J., Yu, W.‐B., Yang, J.‐B., Song, Y., Yi, T.‐S. & Li, D.‐Z. (2018) GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data, bioRxiv, 256479. 10.1101/256479. [DOI] [Google Scholar]
- Jones, P., Binns, D., Chang, H.‐y., Fraser, M., Li, W., McAnulla, C.et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics (Oxford, England), 30, 1236–1240. 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlau, S., Aspinall, S., Gray, J.C. & Bock, R. (2006) Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. Journal of Molecular Evolution, 63, 194–207. 10.1007/s00239-005-0254-5. [DOI] [PubMed] [Google Scholar]
- Katoh, K. & Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution, 30, 772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik, P., Gramazio, P., Vilanova, S., Raigón, M.D., Prohens, J. & Plazas, M. (2017) Phenolics content, fruit flesh colour and browning in cultivated eggplant, wild relatives and interspecific hybrids and implications for fruit quality breeding. Food Research International, 102, 392–401. 10.1016/j.foodres.2017.09.028. [DOI] [PubMed] [Google Scholar]
- Kim, S., Park, J., Yeom, S.‐I., Kim, Y.‐M., Seo, E., Kim, K.‐T.et al. (2017) New reference genome sequences of hot pepper reveal the massive evolution of plant disease‐resistance genes by retroduplication. Genome Biology, 18, 210. 10.1186/s13059-017-1341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp, S., Aubriot, X. & Prohens, J. (2019) Eggplant (Solanum melongena L.): taxonomy and relationships. In: Chapman, M.A. (Ed.) The Eggplant Genome. Compendium of Plant Genomes. Cham: Springer International Publishing, pp. 11–22. 10.1007/978-3-319-99208-2_2. [DOI] [Google Scholar]
- Knapp, S., Vorontsova, M.S. & Prohens, J. (2013) Wild Relatives of the eggplant (Solanum melongena L.: Solanaceae): new understanding of species names in a complex group. PLoS One, 8, e57039. 10.1371/journal.pone.0057039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, R., Schlötterer, C. & Lelley, T. (2007) SciRoKo: a new tool for whole genome microsatellite search and investigation. Bioinformatics, 23, 1683–1685. 10.1093/bioinformatics/btm157. [DOI] [PubMed] [Google Scholar]
- Kozlov, A.M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. (2019) RAxML‐NG: a fast, scalable and user‐friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, 35, 4453–4455. 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, S., Choudhuri, J.V., Ohlebusch, E., Schleiermacher, C., Stoye, J. & Giegerich, R. (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research, 29, 4633–4642. 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakušić, D., Liber, Z., Nikolić, T., Surina, B., Kovačić, S., Bogdanović, S.et al. (2013) Molecular phylogeny of the Campanula pyramidalis species complex (Campanulaceae) inferred from chloroplast and nuclear non‐coding sequences and its taxonomic implications. Taxon, 62, 505–524 10.12705/623.1. [DOI] [Google Scholar]
- Lamesch, P., Berardini, T.Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R.et al. (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Research, 40, D1202–D1210. 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D., Liu, C.‐M., Luo, R., Sadakane, K. & Lam, T.‐W. (2015) MEGAHIT: an ultra‐fast single‐node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, 31, 1674–1676. 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- Li, H. (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics (Oxford, England), 27, 2987–2993. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. Genomics, 1303. [Google Scholar]
- Li, H. (2018) Minimap2: pairwise alignment for nucleotide sequences I. Birol, ed. Bioinformatics, 34, 3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W., Chen, L., Zhang, S., Hu, F., Wang, Z., Lyu, J.et al. (2019) Decrease of gene expression diversity during domestication of animals and plants. BMC Evolutionary Biology, 19, 19. 10.1186/s12862-018-1340-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdy, M., Ou, L., Yu, H., Chen, R., Zhou, Y., Hassan, H.et al. (2019) Pan‐plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Horticulture Research, 6, 108. 10.1038/s41438-019-0191-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangino, G., Plazas, M., Vilanova, S., Prohens, J. & Gramazio, P. (2020) Performance of a set of eggplant (Solanum melongena) lines with introgressions from its wild relative S. incanum under open field and screenhouse conditions and detection of QTLs. Agronomy, 10, 467. 10.3390/agronomy10040467. [DOI] [Google Scholar]
- Mangino, G., Vilanova, S., Plazas, M., Prohens, J. & Gramazio, P. (2021) Fruit shape morphometric analysis and QTL detection in a set of eggplant introgression lines. Scientia Horticulturae, 282, 110006. 10.1016/j.scienta.2021.110006. [DOI] [Google Scholar]
- Mascher, M., Gundlach, H., Himmelbach, A., Beier, S., Twardziok, S.O., Wicker, T.et al. (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature, 544, 427–433. 10.1038/nature22043. [DOI] [PubMed] [Google Scholar]
- Millen, R.S., Olmstead, R.G., Adams, K.L.et al. (2001) Many parallel losses of infa from chloroplast dna during angiosperm evolution with multiple independent transfers to the nucleus. The Plant Cell, 13, 645–658. 10.1105/tpc.13.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh, B.Q., Schmidt, H.A., Chernomor, O., Schrempf, D., Woodhams, M.D., von Haeseler, A.et al. (2020) IQ‐TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37, 1530–1534. 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake, K., Saito, T., Negoro, S., Yamaguchi, H., Nunome, T., Ohyama, A.et al. (2012) Development of selective markers linked to a major QTL for parthenocarpy in eggplant (Solanum melongena L.). TAG Theoretical and Applied Genetics, 124, 1–11. 10.1007/s00122-012-1796-8. [DOI] [PubMed] [Google Scholar]
- Miyatake, K., Saito, T., Nunome, T., Yamaguchi, H., Negoro, S., Ohyama, A.et al. (2020) Fine mapping of a major locus representing the lack of prickles in eggplant revealed the availability of a 0.5‐kb insertion/deletion for marker‐assisted selection. Breeding Science, 70, 438–448. 10.1270/jsbbs.20004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moglia, A., Florio, F.E., Iacopino, S.et al. (2020) Identification of a new R3 MYB type repressor and functional characterization of the members of the MBW transcriptional complex involved in anthocyanin biosynthesis in. eggplant (S. melongena L.) S. Aceto, ed. PLoS One, 15, e0232986. 10.1371/journal.pone.0232986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monat, C., Padmarasu, S., Lux, T., Wicker, T., Gundlach, H., Himmelbach, A.et al. (2019) TRITEX: chromosome‐scale sequence assembly of Triticeae genomes with open‐source tools. Genome Biology, 20, 284. 10.1186/s13059-019-1899-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahed, N., Pastore, C., Cellini, A., Allegro, G., Valentini, G., Zenoni, S.et al. (2016) The grapevine VviPrx31 peroxidase as a candidate gene involved in anthocyanin degradation in ripening berries under high temperature. Journal of Plant Research, 129, 513–526. 10.1007/s10265-016-0786-3. [DOI] [PubMed] [Google Scholar]
- Mu, Q., Huang, Z., Chakrabarti, M., Illa‐Berenguer, E., Liu, X., Wang, Y.et al. (2017) Fruit weight is controlled by Cell Size Regulator encoding a novel protein that is expressed in maturing tomato fruits. PLOS Genetics, 13, e1006930. 10.1371/journal.pgen.1006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muños, S., Ranc, N., Botton, E., Bérard, A., Rolland, S.Duffé, P.et al. (2011) Increase in tomato locule number is controlled by two single‐nucleotide polymorphisms located near WUSCHEL . Plant Physiology, 156, 2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T., Schmidt, H.A., von Haeseler, A. & Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunome, T., Ishiguro, K., Yoshida, T. & Hirai, M. (2001) Mapping of fruit shape and color development traits in eggplant (Solanum melongena L.) based on RAPD and AFLP markers. Breeding science, 51, 19–26. [Google Scholar]
- Nunome, T., Negoro, S., Kono, I., Kanamori, H., Miyatake, K., Yamaguchi, H.et al. (2009) Development of SSR markers derived from SSR‐enriched genomic library of eggplant (Solanum melongena L.). Theoretical and applied genetics, 119, 1143–1153. 10.1007/s00122-009-1116-0. [DOI] [PubMed] [Google Scholar]
- Ou, L., Li, D., Lv, J., Chen, W., Zhang, Z., Li, X.et al. (2018) Pan‐genome of cultivated pepper (Capsicum ) and its use in gene presence‐absence variation analyses. New Phytologist, 220, 360–363. 10.1111/nph.15413. [DOI] [PubMed] [Google Scholar]
- Padmarasu, S., Himmelbach, A., Mascher, M. & Stein, N. (2019) In Situ Hi‐C for plants: an improved method to detect long‐range chromatin interactions. In: Chekanova, J.A. & Wang, H.‐L.‐V. (Eds.) Plant Long Non‐Coding RNAs: Methods and Protocols. Methods in Molecular Biology. New York: Springer, pp. 441–472. 10.1007/978-1-4939-9045-0_28. [DOI] [PubMed] [Google Scholar]
- Page, A., Gibson, J., Meyer, R.S. & Chapman, M.A. (2019) Eggplant domestication: pervasive gene flow, feralization, and transcriptomic divergence. Molecular Biology and Evolution, 36, 1359–1372. 10.1093/molbev/msz062. [DOI] [PubMed] [Google Scholar]
- Parks, M., Cronn, R. & Liston, A. (2009) Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biology, 7, 84. 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattengale, N.D., Alipour, M., Bininda‐Emonds, O.R.P., Moret, B.M.E. & Stamatakis, A. (2010) How many bootstrap replicates are necessary? In Journal of Computational Biology. Mary Ann Liebert, Inc. Rochelle, NY, pp. 337–354. 10.1089/cmb.2009.0179. [DOI] [PubMed] [Google Scholar]
- Pérez‐Díaz, R., Ryngajllo, M., Pérez‐Díaz, J., Peña‐Cortés, H., Casaretto, J.A., González‐Villanueva, E.et al. (2014) VvMATE1 and VvMATE2 encode putative proanthocyanidin transporters expressed during berry development in Vitis vinifera L. Plant Cell Reports, 33, 1147–1159. 10.1007/s00299-014-1604-9. [DOI] [PubMed] [Google Scholar]
- Portis, E., Barchi, L., Toppino, L., Lanteri, S., Acciarri, N., Felicioni, N.et al. (2014) QTL mapping in eggplant reveals clusters of yield‐related loci and orthology with the tomato genome. PLoS One, 9, e89499. 10.1371/journal.pone.0089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portis, E., Cericola, F., Barchi, L., Toppino, L., Acciarri, N., Pulcini, L.et al. (2015) Association mapping for fruit, plant and leaf morphology traits in eggplant. PLoS One, 10, e0135200. 10.1371/journal.pone.0135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracana, R., Priyam, A., Levantis, I., Nichols, R.A. & Wurm, Y. (2017) The fire ant social chromosome supergene variant Sb shows low diversity but high divergence from SB. Molecular Ecology, 26, 2864–2879. 10.1111/mec.14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A.R. & Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England), 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2020) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ranil, R.h.g., Prohens, J., Aubriot, X., Niran, H.m.l., Plazas, M., Fonseka, R.m.et al. (2017) Solanum insanum L. (subgenus Leptostemonum Bitter, Solanaceae), the neglected wild progenitor of eggplant (S. melongena L.): a review of taxonomy, characteristics and uses aimed at its enhancement for improved eggplant breeding. Genetic Resources and Crop Evolution, 64, 1707–1722. 10.1007/s10722-016-0467-z. [DOI] [Google Scholar]
- Rasko, D.A., Rosovitz, M.j., Myers, G.S.A., Mongodin, E.F., Fricke, W.F., Gajer, P.et al. (2008) The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. Journal of Bacteriology, 190, 6881–6893. 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell, L.J. (2012) phytools: an R package for phylogenetic comparative biology (and other things): phytools: R package . Methods in Ecology and Evolution, 3, 217–223. 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- Rodríguez, G.R., Muños, S., Anderson, C., Sim, S.‐C., Michel, A., Causse, M.et al. (2011) Distribution of SUN, OVATE, LC, and FAS in the tomato germplasm and the relationship to fruit shape diversity. Plant Physiology, 156, 275–285. 10.1104/pp.110.167577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., Ferrer‐Mata, A., Sánchez‐DelBarrio, J.C., Guirao‐Rico, S., Librado, P., Ramos‐Onsins, S.E.et al. (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34, 3299–3302. 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- Salgon, S., Raynal, M., Lebon, S., Baptiste, J.M., Daunay, M.C., Dintinger, J.et al. (2018) Genotyping by sequencing highlights a polygenic resistance to Ralstonia pseudosolanacearum in eggplant (Solanum melongena L.). International Journal of Molecular Sciences, 19, 357. 10.3390/ijms19020357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, J., Lickey, E.B., Beck, J.T., Farmer, S.B., Liu, W., Miller, J.et al. (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany, 92, 142–166. 10.3732/ajb.92.1.142. [DOI] [PubMed] [Google Scholar]
- Simão, F.A., Waterhouse, R.M., Ioannidis, P., Kriventseva, E.V. & Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Snipen, L. & Liland, K.H. (2015) Micropan: an R‐package for microbial pan‐genomics. BMC Bioinformatics, 16, 79. 10.1186/s12859-015-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, B.o., Song, Y., Fu, Y., Kizito, E.B., Kamenya, S.N., Kabod, P.N.et al. (2019) Draft genome sequence of Solanum aethiopicum provides insights into disease resistance, drought tolerance, and the evolution of the genome. GigaScience, 8, giz115. 10.1093/gigascience/giz115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S. & Morgenstern, B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research, 34, W435–W439. 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin, H., Masignani, V., Cieslewicz, M.j., Donati, C., Medini, D., Ward, N.l.et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan‐genome". Proceedings of the National Academy of Sciences, 102(39), 13950–13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin, H., Riley, D., Cattuto, C. & Medini, D. (2008) Comparative genomics: the bacterial pan‐genome. Current Opinion in Microbiology, 11, 472–477. 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- The Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature, 485, 635–641. 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2014) UniProt: a hub for protein information. Nucleic Acids Research, 43, D204–212. 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, T., Liu, Y., Yan, H., You, Q., Yi, X., Du, Z.et al. (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Research, 45, W122–W129. 10.1093/nar/gkx382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht‐Jones, E.S., Fischer, A., Bock, R.et al. (2017) GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Research, 45, W6–W11. 10.1093/nar/gkx391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toppino, L., Barchi, L., Lo Scalzo, R.et al. (2016) Mapping Quantitative Trait Loci affecting biochemical and morphological fruit properties in eggplant (Solanum melongena L.). Frontiers in Plant Science. 10.3389/fpls.2016.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toppino, L., Barchi, L., Mercati, F., Acciarri, N., Perrone, D., Martina, M.et al. (2020) A new intra‐specific and high‐resolution genetic map of eggplant based on a RIL Population, and location of QTLs related to plant anthocyanin pigmentation and seed vigour. Genes, 11, 745. 10.3390/genes11070745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toppino, L., Vale, G. & Rotino, G.L. (2008) Inheritance of Fusarium wilt resistance introgressed from Solanum aethiopicum Gilo and Aculeatum groups into cultivated eggplant (S. melongena) and development of associated PCR‐based markers. Molecular Breeding, 22, 237–250. [Google Scholar]
- van der Knaap, E. & Østergaard, L. (2018) Shaping a fruit: developmental pathways that impact growth patterns. Seminars in Cell & Developmental Biology, 79, 27–36. 10.1016/j.semcdb.2017.10.028. [DOI] [PubMed] [Google Scholar]
- Vorontsova, M.S. & Knapp, S. (2016) A revision of the “spiny Solanums,” Solanum subgenus Leptostemonum (Solanaceae), in Africa and Madagascar. Systematic Botany Monographs, Vol. 99, pp. 1–432. [Google Scholar]
- Vorontsova, M.S., Stern, S., Bohs, L. & Knapp, S.(2013) African spiny Solanum (subgenus Leptostemonum, Solanaceae): a thorny phylogenetic tangle. Botanical Journal of the Linnean Society, 173, 176–193. 10.1111/boj.12053. [DOI] [Google Scholar]
- Wang, Z., Yang, Z. & Li, F. (2019) Updates on molecular mechanisms in the development of branched trichome in Arabidopsis and nonbranched in cotton. Plant Biotechnology Journal, 17, 1706–1722. 10.1111/pbi.13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Q., Wang, J., Wang, W., Hu, T., Hu, H. & Bao, C. (2020a) A high‐quality chromosome‐level genome assembly reveals genetics for important traits in eggplant. Horticulture Research, 7, 1–15. 10.1038/s41438-020-00391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Q., Wang, W., Hu, T., Hu, H., Wang, J. & Bao, C. (2020b) Construction of a SNP‐based genetic map using SLAF‐Seq and QTL analysis of morphological traits in eggplant. Frontiers in Genetics, 11, 178. 10.3389/fgene.2020.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B.S. & Cockerham, C.C. (1984) Estimating F‐statistics for the analysis of population structure. Evolution, 38, 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wood, D.E., Lu, J. & Langmead, B. (2019) Improved metagenomic analysis with Kraken 2. Genome Biology, 20, 257. 10.1101/762302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z. (2016) The completed eight chloroplast genomes of tomato from Solanum genus. Mitochondrial DNA Part A, 27, 4155–4157. 10.3109/19401736.2014.1003890. [DOI] [PubMed] [Google Scholar]
- Xiao, X.O., Lin, Wq., Li, K., Feng, X.F., Jin, H. & Zou, Hf. (2018). Transcriptome analyses reveal anthocyanin biosynthesis in eggplants. PeerJ Preprints, 6:e27289v1 [Google Scholar]
- Zhang, C., Dong, S.‐S., Xu, J.‐Y., He, W.‐M. & Yang, T.‐L. (2018a) PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics, 35, 1786–1788. 10.1093/bioinformatics/bty875. [DOI] [PubMed] [Google Scholar]
- Zhang, L., Sun, H., Xu, T., Shi, T., Li, Z. & Hou, W. (2021) Comparative transcriptome analysis reveals key genes and pathways involved in prickle development in eggplant. Genes, 12, 341. 10.3390/genes12030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R., Zhang, L., Wang, W., Zhang, Z., Du, H., Qu, Z.et al. (2018b) Differences in codon usage bias between photosynthesis‐related genes and genetic system‐related genes of chloroplast genomes in cultivated and wild solanum species. International Journal of Molecular Sciences, 19, 3142. 10.3390/ijms19103142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Hu, Z., Chu, G., Huang, C., Tian, S., Zhao, Z.et al. (2014) Anthocyanin accumulation and molecular analysis of anthocyanin biosynthesis‐associated genes in eggplant (Solanum melongena L.). Journal of Agricultural and Food Chemistry, 62, 2906–2912. 10.1021/jf404574c. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., Jia, X., Yang, J., Ling, Y., Zhang, Z., Yu, J.et al. (2014) PanGP: a tool for quickly analyzing bacterial pan‐genome profile. Bioinformatics (Oxford, England), 30, 1297–1299. 10.1093/bioinformatics/btu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X., Levine, D., Shen, J., Gogarten, S.M., Laurie, C. & Weir, B.S. (2012) A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–3328. 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Hi‐C scaffolding of the v3.0 eggplant genome assembly. (a) In silico inferred distribution of valid (valid restriction site‐associated, religated) and invalid (‘paired‐end’ [PE]) Hi‐C fragment lengths in the Hi‐C dataset (Monat et al., 2019). (b) Hi‐C contact frequency plots showing the improvement in chromosome‐scale contiguity between assembly versions v3.0 (Barchi et al., 2019) and v4.0 (current study). Log Hi‐C link counts between 200‐Mbp bins are given after normalization (see the Experimental Procedures section). Stark discontinuities in the contact frequencies are evidence of missing sequences or incorrectly arranged scaffolds.
Figure S2. (a) Synteny analysis of S. melongena ‘67/3’ assembly v4.0 (on the x‐axis) and the S. melongena CL assembly (y‐axis). (b) Histogram of pairwise comparisons of predicted protein length ratios with their best hit in the Arabidopsis TAIR10 annotation (BlastP, e‐value = 10−3).
Figure S3. Comparison of 32 chloroplast genomes using mVISTA. Complete cp genomes were compared, with GPE001970 as a reference. Blue block: conserved genes, sky‐blue block: tRNA and rRNA, red block: conserved non‐coding sequences (CNSs). White represents regions with sequence variation.
Figure S4. Numbers of (a) SSRs and (b) repeats identified in the 32 chloroplast sequences. Colors depend on the motifs (a) or size (b) of the repeats identified.
Figure S5. Distribution of single‐nucleotide polymorphisms (SNPs) along the reference genome sequence. The SNP distribution for all the accessions in the study is shown in orange, the SNP distribution only for eggplant is shown in purple, and the SNP distribution only for NMSs is shown in blue (S. insanum and S. incanum).
Figure S6. Principal component analysis (PCA) visualization of the genetic relationships among the accessions of eggplant as well as cultivated and wild related species, based on (a) the whole set of SNPs identified; (b) the whole set of SNPs identified limited to S. melongena; (c) PAVs; and (d) PAVs limited to S. melongena. Solanum melongena entries are shown in purple, S. incanum entries in red, and S. insanum entries in green.
Figure S7. Linkage disequilibrium (LD) decay in all accessions (orange) and S. melongena (purple). r 2 estimates were plotted against the physical distance.
Table S1. Hi‐C metrics.
Table S2. Chromosome metrics of Smel v3.0 and v4.0 and their comparison with Smel CL.
Table S3. Phenotypic characteristics, geographical origin, and genome assembly metrics of the eggplant accessions.
Table S4. Pan‐genome genes present or absent following read mapping in the reference line GPE001970 (‘67/3’).
Table S5. Different categories of genes in the eggplant pan‐genome.
Table S6. Gene ontology (GO) term enrichments for different pan‐genome gene categories using AGRIGO SEACOMPARE.
Table S7. Assembly metrics of the eggplant pan‐plastome. The asterisk (*) indicates already assembled chloroplasts.
Table S8. List of genes in the eggplant pan‐plastome.
Table S9. SNPs identified in the pan‐genome sequences for all 26 accessions.
Table S10. SNP distribution per chromosome in the 26 accessions.
Table S11. Highest ROD and XP‐CLR values for the 5 breeding traits in study in the selective sweeps identified.
Table S12. Selective sweeps number and size and genes number for the five breeding traits in study identified with ROD top 5% and/or XP‐CLR top 1%.
Table S13. Conversion of gene models between annotations v3.0 and v4.0.
Data Availability Statement
All Raw Illumina data were deposited at NCBI SRA (BioProject ID PRJNA649091). The v4.0 and pan‐genome assemblies and annotations are available at https://solgenomics.net/. Chloroplast genomes were submitted to GenBank (accession numbers MW384824–MW384851). Seeds of the accessions used in the present study can be obtained from COMAV (jprohens@btc.upv.es) or CREA (laura.toppino@crea.gov.it).