

Abstract
Capturing the heterogeneous phenotypes of microbial populations at relevant spatiotemporal scales is highly challenging. Here, we present par-seqFISH, a transcriptome-imaging approach that records gene-expression and spatial context within microscale assemblies at a single-cell and molecule resolution. We apply this approach to the opportunistic pathogen, Pseudomonas aeruginosa, analyzing ~600,000 individuals across dozens of conditions in planktonic and biofilm cultures. We identify numerous metabolic and virulence-related transcriptional states that emerge dynamically during planktonic growth, as well as highly spatially resolved metabolic heterogeneity in sessile populations. Our data reveal that distinct physiological states can coexist within the same biofilm, just several microns away, underscoring the importance of microenvironments. Our results illustrate the complex dynamics of microbial populations and present a new way of studying them at high resolution.
Introduction
Life exists in context. Cells within microbial populations and communities are typically closely associated with one another in multicellular biofilms, whether found within infected tissues, attached to diverse surfaces, or forming assemblages in the deep sea (1, 2). Natural microbiota and infectious bacteria generally exist in biofilm aggregates that are on the order of several dozen microns and which can contain many interacting species (3–5). Despite the ubiquity of the biofilm lifestyle in both natural and manmade habitats, understanding what life is like within it for individual microbes has proven highly challenging. While single-cell level activities have been tracked at high spatial resolution using a variety of approaches in diverse contexts (6–8), we have been unable to resolve the hundreds if not thousands of concurrent activities that characterize microbial life at relevant spatiotemporal scales. What we understand about microbial life literally has been limited by our ability to see.
Despite this limitation, it has become clear in recent years that extreme phenotypic heterogeneity defines the microbial experience (9, 10). This is as true for isogenic populations as it is for complex biofilm communities. Clonemates sampled from the same environment often display significant differences that are thought to result from stochastic gene-expression and variable environmental factors (9, 11, 12). The detection of phenotypic diversity even in seemingly well-mixed environments such as chemostats (11, 13) also serves as a powerful reminder that life at the microscale may inhabit far more diverse niches than are readily apparent. Phenotypic diversity has been rationalized as providing microbes with a fitness advantage in an unpredictable world (9, 14). In addition, specialized functions have been proposed to underpin collective interactions such as division of labor (9, 15–17). However, little is still known about the range of possible cellular phenotypic states and their roles in most biological processes.
What triggers such phenotypic plasticity, and are there underlying “rules” that govern any patterns that may exist at the microscale? In sessile communities, both clonal or multispecies, biological activities give rise to changing chemical gradients that create a range of local microenvironments (18, 19). Furthermore, spatial organization enables different conflicting metabolic states or species to co-exist via physical separation, increasing the potential for diversity and allowing for new interactions to emerge (10, 20–23). Indeed, natural communities often contain many interacting species that assemble into intricate spatial structures. These microscale assemblies can promote interactions between species and represent a key ecosystem feature (23, 24). Yet a wide gulf—limited by technology—still separates such observations from a coherent conceptual framework to explain the rules governing microbial ecology.
Recent advances in imaging methods provide a means to chart the physical associations between different species in natural environments (4, 25–27). However, interpreting these maps remains challenging without additional functional information on the physiological states and activities of relevant community members. In contrast, recent adaptations of single-cell RNA-sequencing (scRNA-seq) approaches to free-living bacteria provide a powerful means of exploring their phenotypic landscape (28–30). Yet these approaches do not preserve the spatial context of analyzed cells and are therefore limited in their capacity to address single and multispecies biofilms. Thus, a major gap exists in our ability to account for both spatial and functional complexity, limiting progression toward a high-resolution understanding of microbial life.
Single-molecule fluorescence in situ hybridization (FISH) based technologies have been used to measure gene-expression directly within native tissues, recording both spatial and functional information. While these methods have shed important light on single-cell heterogeneity, they have been traditionally limited to measuring the expression of only a few genes at a time (31–34). In addition to this limited throughput, single-gene measurements do not provide a means to capture coordinated cellular responses—the molecular “fingerprint” of multiple biological activities that underpin distinct physiological states. Recent advances in combinatorial mRNA labeling and sequential FISH (seqFISH) now allow for hundreds and even thousands of genes to be analyzed within the same sample at a sub-micron resolution (35–37). Until now, seqFISH has been used in mammalian systems to expose the physical organization of cell states within tissues (35–39). We reasoned that the high spatial resolution of these modern transcriptome-imaging techniques also had the potential to illuminate the microscale organization of microbial populations and communities.
In this study, we adapted and further developed seqFISH for studying bacteria, measuring the expression of hundreds of genes within individual cells while also capturing their spatial context. We utilized Pseudomonas aeruginosa planktonic and biofilm populations to demonstrate how different cellular functions are coordinated in time and space. Our proof-of-concept work illustrates how the ability to observe transcriptional activities at the microscale permits insights into the spatiotemporal regulation and coordination of critical life processes, enabling hitherto unrecognized, transient physiological states to be identified and new hypotheses to be generated. These findings represent the tip of the iceberg and the opportunities for discovery our approach enables promise to reveal new insights about the rules governing microbial ecology.
Results
A sequential mRNA-FISH framework for studying bacterial gene-expression.
Combinatorial mRNA labeling requires that each measured mRNA molecule be individually resolved. However, this is much more challenging in bacteria due to the small size of their cells, as many different mRNA molecules occur in close proximity and cannot be resolved using standard fluorescent microscopy. We therefore utilized a nonbarcoded seqFISH approach (40).
In seqFISH, target mRNAs are first hybridized with a set of primary, non-fluorescent probes, which are flanked by short sequences uniquely assigned per gene (Fig. 1A). Specific genes can be turned “ON” via a secondary hybridization with short fluorescently labeled “readout” probes, complementary to the gene-specific flanking sequences (Fig. 1A). Several genes can be measured at once using a set of readout probes labeled with different fluorophores. Importantly, these short fluorescent readout probes can be efficiently stripped and washed away from the sample without affecting the primary probes (41) (Fig. 1A). Thus, once expression is measured, fluorescence can be turned OFF and a new set of genes can be measured by introducing a new set of readout probes (Fig. 1B). This 2-step design allows for potentially hundreds of genes to be measured sequentially, one after the other in the same sample, using automated microscopy (Fig. 1B). The individual gene mRNA-FISH data can be combined into spatially resolved multigene profiles at the single-bacterium level (Fig. 1B).
Fig. 1. Parallel and sequential mRNA-FISH in bacteria.

(A) seqFISH probe design scheme. Primary probes contain unique sequences (Si) that are read by secondary probes (colored wands). Each gene is read by a unique probe and its fluorescence can be turned “ON” or “OFF”. (B) mRNA-FISH applied sequentially to the same sample. In each cycle, a new set of secondary readout probes are introduced. Raw fluorescence data is shown on the right and the detected local spot maxima are shown in the spot detection image. Merged spots for many genes shown in shuffled colors. (C) Combinatorial labeling can be used to encode species taxonomy using 16S rRNA, or to enable the parallel study of (D) bacteria grown in different conditions.
Due to the diffraction limit and the small size of bacteria, mRNA-FISH fluorescent signals (appearing as spots within cells) can contain overlapping mRNA molecules that cannot be spatially resolved in standard microscopes. Thus, counting the number of spots within a bacterial cell severely underestimates expression levels. This problem can be overcome by integrating the fluorescent intensity per spot, which scales linearly with the number of mRNAs. Fluorescent intensity can be converted to discrete mRNA counts by measuring the characteristic intensity of a single transcript. This analog to digital conversion approach has been shown to provide a wide dynamic range in bacteria (33, 42).
We developed seqFISH in the study of Pseudomonas aeruginosa, an opportunistic human pathogen and a severe cause of morbidity and mortality in cystic fibrosis (CF) patients (43, 44). We generated a probe library targeting a set of 105 marker genes that capture many core physiological aspects of this pathogen (table S1 and S2). These included genes involved in biosynthetic capacity (ribosome and RNA-polymerase subunits), anerobic physiology (fermentation and denitrification pathways), stress responses (oxidative and nutrient limitation), cellular signaling (c-di-GMP), biofilm matrix components, motility (flagella and T4P), all major quorum-sensing (QS) systems, as well as multiple antibiotic resistance and core virulence factors. In addition, to control for false positives, we designed probes targeting three different negative control genes that do not exist in Pseudomonas (fig. S1).
Parallel and sequential mRNA-FISH in single bacterial cells
To test our bacterial seqFISH approach, we first studied P. aeruginosa grown in well-understood batch culture conditions. We performed a growth curve experiment in LB medium, where key parameters such as cell density, growth rate, and oxygen levels change in a predictable manner. We collected 11 time points representing the lag phase, exponential growth, and stationary phase and imaged the expression of 105 genes within them simultaneously in 2 days (Fig. 2A). Independent imaging of these 11 samples in a serial manner would have taken ~3 weeks of automated microscopy time.
Fig. 2. Parallel seqFISH (par-seqFISH) of an LB growth curve experiment.
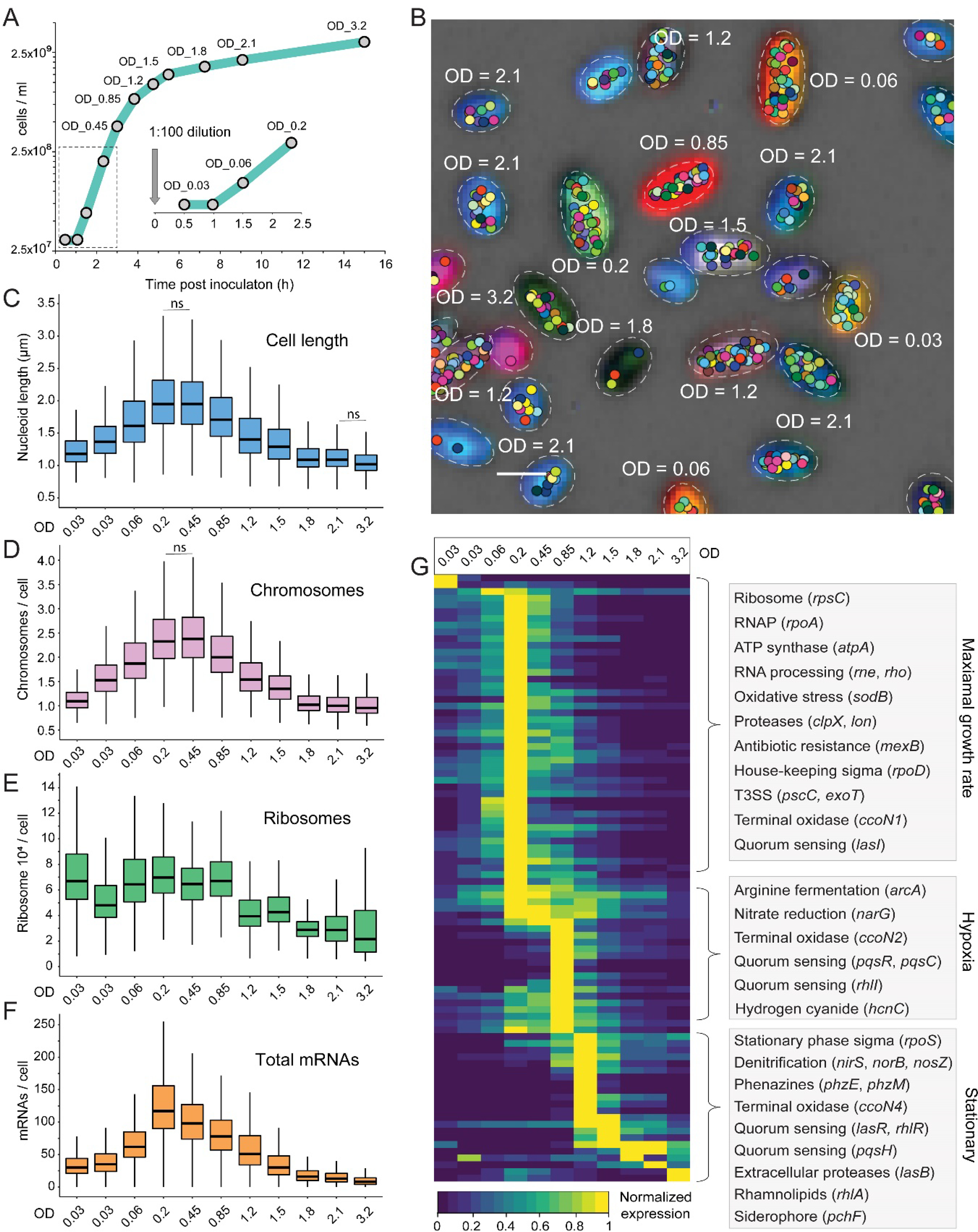
(A) The sampled LB growth curve. Collected time points are indicated with gray circles. A zoom-in shows the sampled lag phase. The presented cfu/ml were estimated using OD600 values (OD600 = 1.0 reporting on ~109 cfu/ml (104)). The OD600 values are indicated over each time-point. (B) Demultiplexed bacteria and their mRNAs. The merged, raw Ribo-Tag 16S rRNA fluorescence is shown for a representative region. Different barcodes (16S combinations) result in unique colors that visually report the condition of which they originated from (indicated with the corresponding OD600 value). Ellipses fitted to the segmented cell boundaries are shown. The mRNA spots (fitted position of maximal intensity) for all genes per cell are shown in unique colors per gene. Each spot may represent more than one mRNA copy. (C-F) Condition specific distributions of nucleoid length, chromosome copy, ribosome levels and total mRNAs detected across our gene set. Distributions contain all demultiplexed cells per condition and are significantly different from their previous time-point unless otherwise noted (Wilcoxon, p < 0.001). (G) Heatmap showing average gene expression normalized to the maximal value for each gene across all conditions. Highlighted gene groups and their functions are indicated on the right.
To perform simultaneous imaging, we developed an efficient multiplexing method that enables parallel seqFISH experiments (par-seqFISH). We designed a set of primary probes targeting the 16S rRNA (Ribo-Tags), which contain unique combinations of flanking sequences (barcodes), that serve as the “readout” in a seqFISH run (Fig. 1, C and D, and table S3). In principle, this multiplexing approach can be applied to studying combinations of different species or for pooling bacteria from different growth conditions (Fig. 1C). We validated the latter application by individually labeling the 16S rRNAs of each of the 11 growth curve samples with unique Ribo-Tags. The samples were pooled, collectively hybridized with the 105 gene probe library, and subjected to sequential hybridizations to measure gene-expression and to decode cell identity (Fig. 2B). We acquired expression profiles for >50,000 individual P. aeruginosa cells, over 91.8% of which were unambiguously decoded and assigned to the condition from which they originated (Fig. 2B). We estimate the false positive decoding rate at 0.04% (1 in 2500 cells) by counting the number of hits for barcodes left out of the experiment, demonstrating both high efficiency and accuracy for par-seqFISH.
In addition to acquiring mRNA expression profiles, our imaging-based platform permits concurrent tracking of key information such as cell size and shape, and can be combined with functional stains, markers and/or immunofluorescence measurements (45). This opens up the possibility of correlating particular expression profiles at the single cell level with integrative physiological or cell biological parameters. We applied a 4′,6-diamidino-2-phenylindole (DAPI) stain as a part of the par-seqFISH experiment and used DAPI fluorescence to estimate the nucleoid size and chromosome copy per cell. Comparing cells at different stages of growth shows that both nucleoid size (estimating cell size) and chromosome number distributions follow identical trends, in agreement with the P. aeruginosa literature (46) (Fig. 2, C and D). We also estimated ribosome abundance using 16S rRNA fluorescence. Notably, the distribution of this metric differed significantly from that of the chromosome parameters, displaying contrasting intensities at different stages of lag phase, increased variability at deep stationary and a delay in signal decline during the shift from exponential growth to stationary phase (Fig. 2E). In contrast, the total number of mRNAs per cell (estimated by our 105 genes) differentiates each time point along the growth curve, reaching a maxima and minima at the fastest and slowest growth rates, respectively (Fig. 2F). These data further support the accuracy of our par-seqFISH multiplexing approach and demonstrate the unique ability of this method to integrate single cell gene-expression with global parameters.
To examine whether our expression profiles faithfully capture known physiological processes that occur during culture development, we grouped the cells according to their decoded conditions and calculated their average gene expression profiles. We find a temporally resolved expression pattern associated with different stages of growth (Fig. 2G). For example, genes representing high replicative/biosynthetic capacity such as those involved in RNA and protein biosynthesis reach their peak expression during maximal division rate but decreased between 90 to 250-fold in stationary phase (Fig. 2G). In contrast, stress factors involved in stationary phase adaptation and nutrient limitation peak at low division rates and higher cell densities (Fig. 2G). QS signal production, receptor expression and target activation reflect the known hierarchical QS regulatory network (47). Notably, the expression of anaerobic metabolism genes occurred in two stages: early induction of the fermentation and nitrate/nitrite reduction genes in the entry to stationary phase, in which hypoxic conditions emerge, followed by expression of the remaining denitrification pathway at lower predicted oxygen levels (48) (Fig. 2G). Furthermore, the shift from aerobic to anaerobic metabolism was accompanied by sequential exchanges in terminal oxidase identities, from ccoN1 to ccoN2 and finally, ccoN4, concomitantly with the induction of phenazine biosynthesis (49, 50) (Fig. 2G).
Notably, repeated mRNA measurements of the same genes in independent and spaced hybridization rounds were well correlated, both in average expression and at single-bacterium levels (Pearson R = 0.86, 0.89 and 0.9, for sigX, rpsC and rpoS, respectively). In addition, the three negative control genes had an average false positive rate of 0.002 transcripts per cell (fig. S1). We also found a good correlation between par-seqFISH and previous RNA-Seq experiments conducted under similar conditions (51) (Pearson, R=0.79–0.84), as well as a strong correlation between close time-points along the growth curve (fig. S2). Together, these results further validate the accuracy of our multiplexing method and demonstrate that our marker genes capture diverse transcriptional states across a wide range of physiological conditions.
Transient emergence of physiologically distinct sub-populations during LB growth
Phenotypic diversity in clonal populations can generate distinct sub-populations that adjust to dynamic environmental changes and specialize in different tasks at different times, setting a fertile ground for bet-hedging behaviors and complex interactions (9, 15, 52). The single-cell resolution and high sensitivity of seqFISH has the potential to shed light on this important yet largely unexplored aspect of microbial life.
We applied Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction and unsupervised clustering to identify distinct transcriptional cell-states in our single-cell expression data (29, 53). The cell clusters detected by this analysis chart the phenotypic landscape in LB growth, from the perspective of our chosen marker genes. Analyzing the 11 time points together, we detect 20 clusters (representing different sub-populations) with diverse predicted functional capabilities. These included among others, differential replicative capacity, exoproduct biosynthesis, and virulence factor production (Fig. 3, A and B). We find that the sampled populations of most of the growth conditions are partitioned into multiple co-existing sub-groups with distinct expression profiles (Figs. 3A and S2, and table S4). Notably, our data suggest that the degree of dispersion within this expression space (estimating phenotypic diversity) varies significantly between conditions and is elevated during stationary phase (fig. S3 and table S4).
Fig. 3. Single-bacterium analysis reveals physiologically distinct dynamic sub-populations.
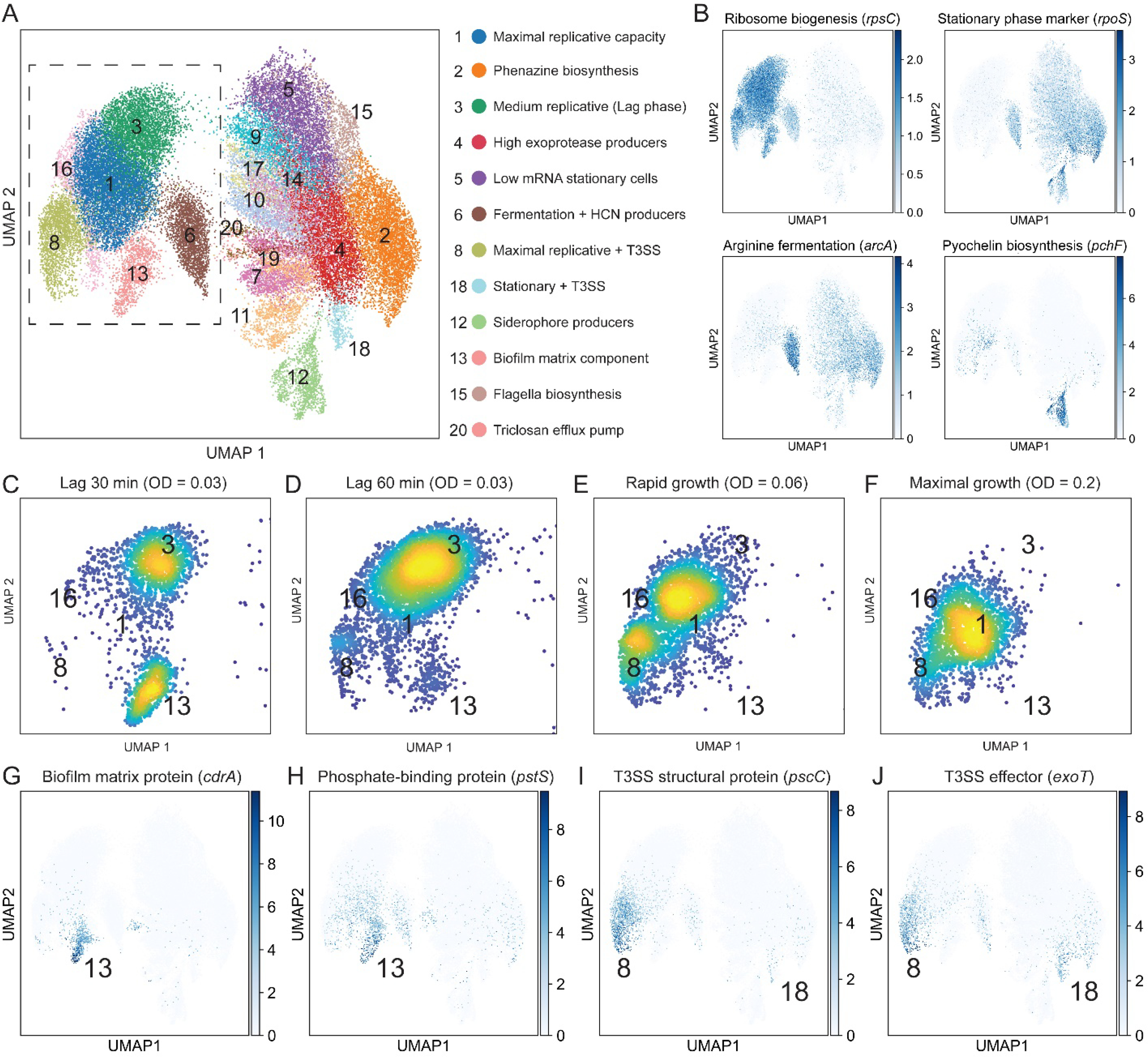
(A) UMAP analysis using cells from all 11 time points. Identified clusters are shown in different colors and are indexed by group size. Specific group and their enriched functions are shown to the right. (B) Gene expression overlays for four genes that report on metabolic state, stationary phase progression and exoproduct biosynthesis. The color map shows the normalized expression scaled to unit variance. Cells from all 11 time points are displayed in the plot (C-F) Density scatter plots of cells from individual conditions in a zoom-in of the UMAP (dashed box in panel A). The clusters are indicated by their index. (G-J) Gene expression overlays shown as in B.
Our growth condition-specific analysis revealed intriguing dynamics during lag phase progression. It could be expected that lag phase cultures will follow a steady ribosome accumulation as the cells progress toward exponential growth and maximal ribosome content (54). In contrast, we found a relative decline in the average ribosomal rRNA levels: early lag phase populations (30 min post dilution) had a higher signal than the more advanced lag culture (60 min post dilution; Fig. 2E). These differences appear to be rooted in the transient emergence and disappearance of an early lag sub-population with exceptionally high levels of 16S rRNA (cluster 13; comprising 34.6% of the population in early lag; Figs. 3, C to F, and S3, and table S4). In agreement with the deviation in the rRNA signal, this sub-population also shows a proportional increase in total mRNA counts. However, its size and chromosome copy distributions were not elevated (fig. S4; cluster 13 vs. 3).
Beyond illuminating the extent of heterogeneity in seemingly well mixed cultures and classifying subpopulations into particular types, seqFISH can directly connect global cell-specific parameters such as ribosome levels or cell shape to particular gene-expression signatures. For example, a closer examination of the metabolically hyperactive sub-population revealed a 186-fold enrichment in cdrA expression relative to the rest of the population (Fig. 3G). The cdrA gene encodes a major adhesive protein component of the P. aeruginosa biofilm matrix (55, 56). Expression of cdrA is commonly used as a reporter for cyclic diguanylate monophosphate (c-di-GMP) levels, a key signaling molecule involved in surface attachment (16). In addition, this sub-population also displays a 30-fold enrichment in pstS expression, which encodes for the phosphate-binding component of the pstSCAB phosphate uptake system (Fig. 3H). PstS has been previously detected in extracellular appendages of P. aeruginosa and has been suggested to provide an adhesion phenotype to intestinal epithelial cells (57). In support of this non-canonical role, pstS was recently suggested to confer a similar adherence phenotype in Acinetobacter baumannii, another human pathogenic bacterium (58).
A second example from our dataset of the type of fine-grained information seqFISH can provide comes from the temporal expression of genes involved in virulence factor production. Single cell variation in virulence factor production has been suggested as a mechanism for division of labor during infection (17). P. aeruginosa employs a variety of virulence factors to overcome the host immune response (44), including the type 3 secretion system (T3SS) that translocate toxins (effectors) directly into host cells (59). Our gene set monitors two T3SS structural genes (pscC and pcrD) and two main effectors (exoT and exoY), all of which are encoded in different operons (60). We detected two different types of sub-populations with enriched T3SS related genes, suggesting a unique division of cells into virulent and avirulent states (Fig. 3, I and J). The first group transiently appears during exponential growth and constitutes 8–30% of the population (Fig. 3, C to F, I and J, and table S4). This group expresses both the secretion system genes (86-fold enrichment) and the effectors (28-fold). In contrast, the second group appears 3–4 divisions later, close to the replicative minima at stationary phase, and occupies only ~2.7% of cells (table S4). This sub-population is strongly enriched for the two effectors (average 26-fold; Fig. 3, I and J) but only mildly so for the secretion system (6-fold), as compared with the earlier group.
We can potentially reconcile these observations as follows: P. aeruginosa has been shown to contain approximately 1–3 T3SS units per cell under inducing conditions (61). Thus, successive divisions following T3SS expression will result in rapid dilution of the T3SS+ group. Assuming the inheritance of the T3SS and effectors is uncoupled, then T3SS+ stationary phase cells are likely to lose their effectors during division and are predicted to be “inactive”. Thus, an intriguing hypothesis is that P. aeruginosa invests in the costly T3SS+ sub-population during “times of plenty” (rapid growth) and specifically expresses the effectors at stationary to “reload” and maintain this sub-population following division-based dilution, just prior to growth arrest. Together, these examples underscore the power of seqFISH to suggest hypotheses that can be tested going forward.
Spatial transcriptomics at a single-cell resolution in P. aeruginosa biofilms
Though much can be learned by applying seqFISH to planktonic cultures, in many contexts, bacteria exist in biofilms (1, 2). Variation in local environmental conditions and the effect of spatially confined metabolic activities in biofilm populations can promote the emergence of chemically distinct microenvironments and phenotypes (10, 19). We reasoned that seqFISH’s capacity to record transcriptional activities with micron resolution would be particularly useful in shedding light on these processes.
The P. aeruginosa biofilm mode of life is particularly important in chronic infections such as those residing in the airways of individuals with CF (62, 63). Accordingly, having used LB to validate bacterial seqFISH, we switched to synthetic cystic fibrosis sputum medium (SCFM) for our biofilm studies (64). Briefly, bacteria were incubated in coverslip attached microwells and the medium was replaced every several hours. Using biofilms that were allowed to develop for 10 or 35 hours, we imaged hundreds of aggregates ranging in size from several bacteria to tens-of-thousands of tightly bound members (Fig. 4, A and B). As a reference for cellular physiological states, we also performed a planktonic growth curve experiment in SCFM. We applied par-seqFISH multiplexing to image 10 time points matching those sampled in the planktonic LB experiment. We find a similar degree of heterogeneity in SCFM and LB grown populations (Fig. 4D). We extracted the physical coordinates of individual bacterial cells within microaggregates, acquiring a microscale spatial expression profile for ~365,000 surface attached bacteria (Fig. 4, A and B). In addition, we collected single-cell expression data for ~218,000 planktonic cells.
Fig. 4. Spatial transcriptomics in P. aeruginosa biofilms at a single cell resolution.
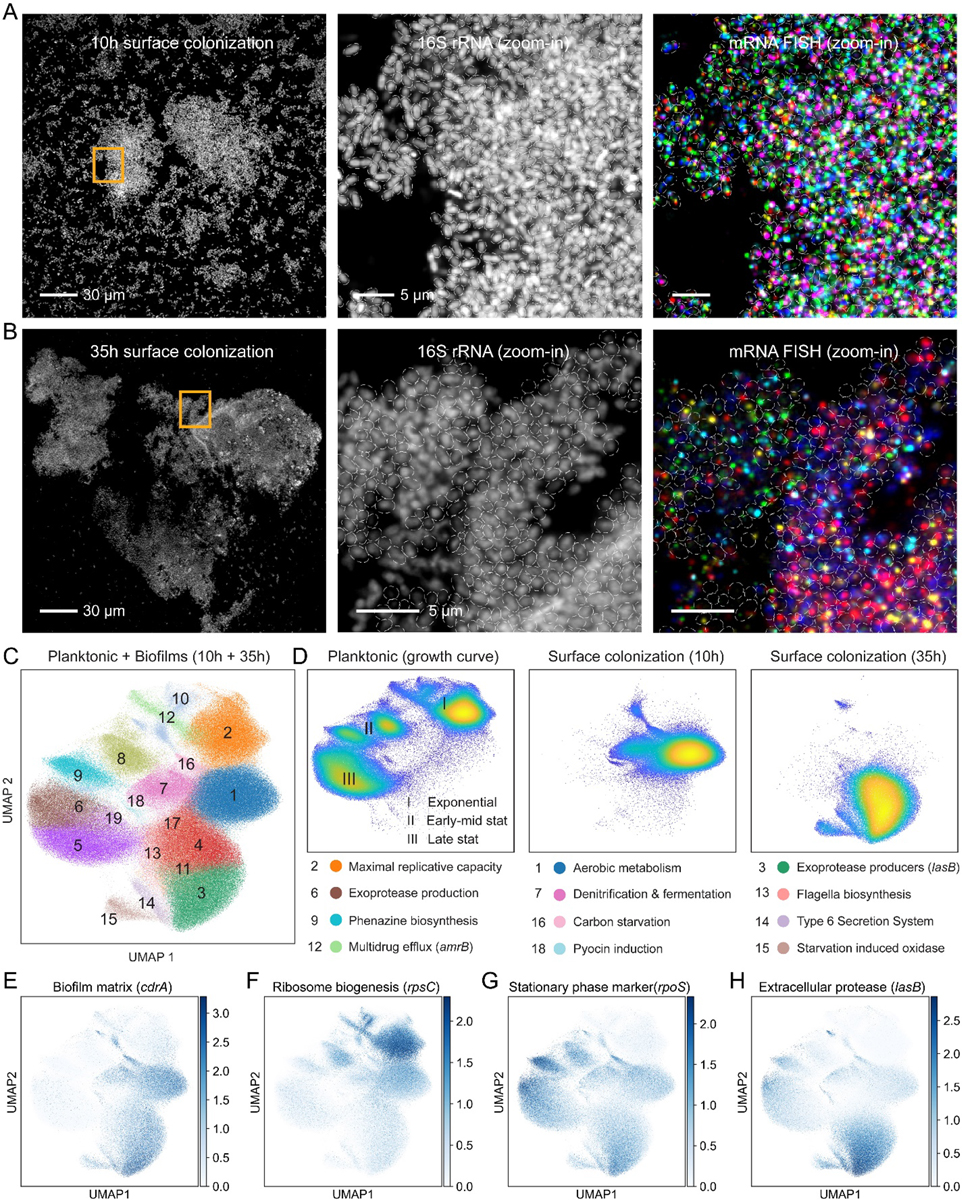
(A) A representative field of view collected during a 10h surface colonization experiment showing cells via 16S rRNA fluorescence (gray). A zoom-in (orange box) shows the cell segmentation masks depicted as white ellipses. The 16S rRNA signal and mRNA-FISH data for several genes are shown in different colors. (B) A 35h experiment field is shown in an identical manner to panel A. Scale bar length is annotated within the figure. (C) Joint UMAP cluster analysis of biofilm and planktonic experiments. Planktonic cells are shown for all time points collected (D) UMAP scatter plots showing cells from either planktonic or biofilm experiments as indicated. Below, a highlighted set of UMAP clusters associated with each experiment is annotated with enriched functions. (E-HGHH) UMAP overlay with specific gene data. The color map shows the normalized expression scaled to unit variance. Cells from the liquid experiment and both 10h and 35h biofilms are displayed together.
A basic question we sought to answer was the extent to which transcriptional responses are unique to the biofilm lifestyle. We performed a joint UMAP analysis using both biofilm and planktonic samples (Fig. 4C). These different modes of growth cluster into independent groups in expression space, reflecting their significant physiological differences (Fig. 4D). Ribosome and RNAP subunit expression in the planktonic experiment correlated strongly with growth rate, as observed in LB (Fig. 4D). Examining these marker genes in the biofilm-derived cells places the average replicative capacity of the 10h and 35h biofilm populations at roughly equal to those of early-mid and late stationary planktonic populations, respectively (Fig. 4F). Expression of the stationary phase master regulator, rpoS further supports this classification (Fig. 4G). However, biofilm cells also have unique expression profiles that distinguish them from liquid cultures. For example, the matrix component gene, cdrA, is uniformly expressed in both 10 and 35h biofilms but repressed in most planktonic cells (Fig. 4E). In addition, compared with stationary liquid cells, our data indicates that early biofilms (10h) have higher expression of sigX (5.1-fold), a transcription factor recently implicated in biofilm formation (65), mexB (>4.5-fold), of the mexA-mexB-oprM antibiotic efflux system, and an increase in the 3’−5’ exonuclease, polynucleotide phosphorylase (pnp) (7.5-fold). Comparing the 35h biofilm to the stationary cells, we find a 3.3-fold increase in the extracellular protease, lasB, but reduced expression of other proteases such lasA (3-fold lower), as well as aprA and the rhamnolipid biosynthesis gene, rhlA (~10-fold lower). Notably, these genes are quorum-sensing (QS) regulated and our liquid cultures expressed both lasA and rhlA at later time points than lasB, suggesting these differences may reflect the age of the biofilm rather than features that define the biofilm state per se.
In situ analysis of biofilm specific functions
The above data demonstrate that seqFISH can capture both cell states and their physical position directly within intact biofilms, providing an opportunity to examine known and new processes that contribute to biofilm development from a quantitative and highly spatially resolved perspective. To illustrate this, we focused on the expression patterns of representative genes known to define critical stages in biofilm development such as attachment, maturation and exclusion of competitors.
Motility systems such as the flagella and the type 4 pilus (T4P) are major determinant of surface colonization subsequent biofilm formation (66–68). Recent work identified an asymmetric division process coined “Touch-Seed-and-Go”, in which flagellated mother cells first attach to a surface and then produce un-flagellated daughter cells that contain the T4P. This c-di-GMP dependent phenotypic diversification enables the mother “spreader” cell to spawn multiple adherent “seed” populations (69). This is thought to be mainly regulated by surface sensing (69). However, how such motility-based division of labor affects the organization of biofilms at stages beyond surface attachment remains unknown.
We examined the spatial expression patterns of the major flagellum and T4P components, fliC and pilA, respectively in the early surface colonization experiment (10h biofilm). An abundant “checkerboard” like pattern is evident, in which cells express high levels of either fliC or pilA but generally not both (Fig. 5A). We find that the highly expressing fliC+ and pilA+ sub-populations (>3 standard deviations above the population mean) represent a total of ~4% of all cells in our experiment (2% for each sub-group), yet the double positive sub-population (fliC+/pilA+) only constitutes 0.07%. This pattern occurs uniformly across most aggregates, both small groups (~tens of cells) and in large sets that contain thousands of cells. In contrast, the older 35h biofilms show lower expression of pilA but contain a sparse but uniform distribution of fliC+ cells, suggesting that biofilm associated bacteria invest in a costly motility apparatus despite being spatially confined (Fig. 5B), effectively, the bacterial equivalent of purchasing a sports car during a midlife crisis. Examining the expression of fliC and pilA in our paired planktonic experiment we find a similar mutually exclusive pattern (a roughly equal ~2% of both single positive groups and 0.15% of the double positive cells) (Fig. 5C). Thus, in contrast to the current model, our planktonic control experiment suggests that the asymmetric distribution of motility systems is unlikely to be directly regulated by surface sensing (Fig. 5C); such a conclusion would not be possible without the means to compare transcriptional activities at the single cell level.
Fig. 5. Spatial expression patterns for motility and pyocin related genes.

(A-B) Representative regions from the 10h and 35h biofilm experiments, cells are shown in via 16S rRNA fluorescence (gray) and overlayed with raw mRNA-FISH fluorescence for different genes as indicated. (C) planktonic cells from the pair liquid experiments. Cells are shown via DAPI and expression as indicated (D-E) 10h aggregate showing R2-pyocin expression. (F) Enrichment of R2-Pyocin mRNA near strong induction sites (cell with 99.5th percentile pyocin expression). X-axis shows the number of cells closest to an induction site that were analyzed (neighborhood size; center cell was excluded). Y-axis shows the enrichment in each neighborhood relative to the total population. A non-pyocin control gene is shown (rpoA). (G) Examples of mRNA R-pyocin transcript and ribosome polar localization as indicated in the legends.
Beyond initial surface attachment, bacteria must establish a strong foothold for colony development as well as outcompete resident microbes. One strategy that potentially address both needs is the utilization of phage tail-like bacteriocins, broadly called tailocins (70). These elements are thought to be adapted from prophages and are applied as narrow-spectrum toxins for kin exclusion (70, 71). However, in contrast with antibiotics, these phage tail-like structures are released into the environment via explosive lysis events that kill the producer and spray the toxin locally to inhibit nearby competitors (72, 73). This event also releases extracellular DNA that integrates into the biofilm matrix, structurally supporting biofilm maturation (72, 74). Yet how this “sacrificial” process is regulated within developing biofilms is not well understood.
Our UMAP analysis identified a sub-population (cluster 18; Fig. 4C) exhibiting >1000-fold enrichment in expression of the R2-pyocin operon (Pseudomonas tailocin), represented by the PA14_08150 gene. This UMAP cluster was enriched ~4-fold in 10h biofilm derived cells (0.45% of the entire population), suggesting pyocin induction is upregulated during surface attachment. Furthermore, we find an 11-fold higher expression of the DNA-repair gene, recA, in agreement with its role in inducing pyocin expression (75). Visualizing the expression of the pyocin producers, we find that induction events are spread across various microaggregates regions but often appear in local clusters (Fig. 5, D and E). Indeed, we find a ~37-fold average spatial enrichment in pyocin expression in the immediate vicinity of strong induction sites as compared with the general population (Fig. 5F). This enrichment decayed rapidly as a function of neighborhood size, suggesting a highly localized effect (Fig. 5F).
In addition to reporting multigene expression profiles, seqFISH also reports the physical position of measured mRNA molecules at a sub-micron resolution. During this analysis we noticed that R2-pyocin transcript fluorescence generally appeared as two spots. Upon closer examination, we discovered that this mRNA is strongly localized to the two cell poles (Fig. 5G). The 16S rRNA fluorescent signal in these pyocin producers show identical polarization, a rare pattern not observed in neighboring non-inducing cells (Fig. 5G). These data suggest that ribosomes and the R2-pyocin transcript are mobilized following induction and spatially co-localize. In contrast, the expression of recA did not follow this pattern, suggesting a pyocin-specific effect (Fig. 5G). Notably, a recent study discovered an identical polar localization for two different Pseudomonas protegens R-tailocins at the protein level (73). Together, these data hint at a potentially evolutionary conserved RNA-dependent mechanism for R-tailocin protein polar localization. We hypothesize that the spatially correlated ribosomal enrichment may provide efficient local translation and particle accumulation prior to cell lysis.
Temporal evolution of metabolic heterogeneity during biofilm development.
Beyond resolving transcriptional activities that contribute to biofilm developmental processes, seqFISH can reveal how biofilm cells metabolically respond to subtle changes in their local microenvironment. Chemical heterogeneity is a key feature of spatially structured environments, and metabolic heterogeneity characterizes mature biofilms (10, 18, 19, 76). Yet until now, it has been impossible to capture the development of fine-grained metabolic structure across multiple suites of genes at different times.
To map biofilm metabolic development, we focused on genes whose regulation and functions are well understood. In particular, we focused on catabolic genes whose gene products enable energy conservation under different oxygen concentrations. Oxygen is a central and dynamic factor that influences metabolic activity in bacterial biofilms (10, 19, 77, 78). Local oxygen availability can vary significantly within structured environments and is biotically shaped within biofilms (18, 77, 79). P. aeruginosa can survive under anaerobic conditions by fermenting different substrates and/or denitrifying (50, 80, 81). Accordingly, monitoring the expression of these catabolic genes as well as others that are co-regulated with them provides a means to track local oxygen availability and its dynamic effects on biofilm metabolic coordination.
How quickly and over what spatial scales do biofilm cells metabolically differentiate? Following the uspL gene, which was strongly induced during hypoxic conditions and correlated with anaerobic fermentation and denitrification genes in our planktonic growth experiments, we observed surprisingly heterogeneous responses to oxygen depletion over just a few microns in young (10h) biofilms (Fig. 6A). Notably, uspL expression is strongly spatially correlated with multiple anaerobic markers (fig. S5), indicating that this gene reports on local anaerobic activities. A closer examination of these putative hypoxic sites showed a frequent anti-correlation of uspL with multiple genes that are otherwise uniformly expressed in 10h biofilms, appearing as co-localized but reversed expression patches (Fig. 6B). Among the anti-correlated functions are the TCA cycle gene, sucC, and replicative capacity genes such as RNAP and ribosome subunits (Fig. 6B and figs. S4 and S5). However, exceptions to this anti-correlation were also visible (fig. S6).
Fig. 6. Oxygen availability shapes microscale metabolic heterogeneity in biofilms.

(A-E) Representative 10h biofilms. Cells are shown via 16S rRNA FISH fluorescence (gray) and overlayed with raw mRNA-FISH fluorescence for different genes as indicated in each panel. White circles highlight regions of interest. (F) Cells painted according to their UMAP derived metabolic state as indicated in the panel legends (also see fig. S7 clusters, 0, 8, 12 and 15), showing co-localization of multiple metabolic states within a given region.
Can the metabolic heterogeneity revealed by oxygen-responsive marker genes provide an entry point for the discovery of more nuanced cellular responses at the microscale? Our spatial correlation analysis revealed an intriguing association between anaerobic metabolism genes, such as the denitrification pathway (narG-nirS-norB-nosZ), and the oxidative stress response genes katA, katB and sodM, encoding for the inducible catalases and an Mn-dependent superoxide dismutase, respectively (82–84) (Fig. 6, C and D, and figs, S4 and S5). Nitrite respiring P. aeruginosa produce the highly toxic intermediate nitric oxide (NO) (85). Indeed, KatA was recently demonstrated to play a role in protection from NO-associated stress (84), suggesting that these sub-aggregate regions correspond to microenvironments with high NO levels. In agreement with this hypothesis, we find that the stress response pattern is also spatially correlated with heat-shock protease expression, including the membrane protease, ftsH, which was found to play an important role in survival under anoxic conditions (86) (Figs. 6E and S4). These data highlight how contrasting physiological states can be established just a few microns away early in biofilm development.
These coordinated expressions patterns for particular genes led us to hypothesize that these patterns reflected the spatiometabolic distribution of distinct physiological “states” across the biofilm. To test this hypothesis we conducted a targeted UMAP analysis using only the 10h biofilm cells (fig. S7). We identified two main anaerobic sub-populations corresponding to denitrification and fermentation dominated metabolic states, representing 11.8% and 7.2% of all cells in the experiment, respectively (fig. S7). In addition, we detected a smaller sub-population of denitrifying cells (2.4% of cells) with 5.3-fold average increase in the oxidative stress factors katB, sodM, and ahpF, which encodes for an alkyl hydroperoxide reductase (87). Relative to the main denitrifying sub-group, stressed cells have lower expression of the denitrification pathway (~4-fold) and a >2-fold reduction in replicative capacity marker levels (rpoA, rpsC and atpA), in support of a potentially damaged state. Projecting these single-cell metabolic states over their respective biofilm positions showed a strong overlap with the above predicted hypoxic pockets, supporting our hypothesis and revealing that multiple metabolic states can co-exist in the same patch (Figs. 6F and S5).
Given the extent of transcriptional heterogeneity manifest in young biofilms, we wondered whether such heterogeneity would persist as biofilms aged. We speculated that the higher cell densities and more committed spatial structuring of mature biofilms might favor larger scale metabolic zonation. We therefore examined the spatial expression patterns in a 35h biofilm experiment.
In contrast to the spatial variation in aerobic and anaerobic metabolic processes seen in 10h biofilms, 35h biofilms have ~50-fold lower average expression of the denitrification pathway genes nar-nirs-norB-nosZ. Indeed, these genes are known to be repressed by the las and rhl QS-systems, indicating P. aeruginosa is programmed to shut down denitrification at high cell densities (80, 88). However, in addition to this complete and co-regulated pathway, P. aeruginosa also encodes an independent periplasmic nitrate reductase (nap) (89). Intriguingly, the napA gene is expressed in a spatially uniform manner but at low level in the 35h aggregates, a pattern that was closely shared with the uspL gene, which together are expressed in 20.3% (+/− 5.5%) of the measured cells within each individual aggregate (Figs. 7A and S7). NapA has been implicated in maintaining redox homeostasis under oxygen limitation (78) and the uspL paralogue, uspK, was shown to play a role in survival under such conditions (86, 90). At first blush, these results suggest that as aggregate cell mass grows, survival physiology dominates over growth-promoting processes, on average. Yet we also find substantial and large-scale spatial heterogeneity in certain genes, such as the replicative capacity markers, which are highly expressed in 17.7% (+/− 10.9%) of aggregate cells (Figs. 7B and S7), and lasB, encoding a QS-regulated extracellular protease, which is expressed at similarly high levels in 43.5% (+/− 6.1%) of the cells (Figs. 7C and S7). These data suggest that a single 35h microaggregate can contain regions with distinct physiological states and virulence-related activities. Finally, that metabolism dynamically shapes the microenvironment leads to the prediction that differences in local nutrient availability will be reflected in heterogenous transcriptional activities over small spatial scales (10). We see evidence of this phenomenon in our data when focusing on carbon metabolism, for example. Where replicative capacity appears to be high and carbon is presumably replete, we see co-expression of the TCA cycle gene (sucC) (Fig. 7, B to D). However, when carbon is limiting, bacteria can utilize the glyoxylate shunt (GS), which bypasses the oxidative decarboxylation steps of the TCA. The GS provides an alternative metabolic pathway for utilizing acetate and fatty acids as carbon sources (91, 92). In the GS, carbon flux is redirected by isocitrate lyase (ICL) which competes with the TCA enzyme isocitrate dehydrogenase (ICD) for isocitrate. Because ICD has a much lower Km it must be enzymatically inactivated via phosphorylation for the carbon flux to be redirected to the GS (92). However, little is still known about the transcriptional regulation of these pathways (93). Our gene set contains both the GS gene, aceA, as well as a downstream TCA cycle gene, sucC. While these genes are often co-expressed, we find that only the GS marker, aceA, is expressed in predicted lower energetic capacity biofilm zones (Figs. 7D and S7), suggesting these subregions experience carbon limitation. In support of this hypothesis, these regions also express the tightly regulated terminal oxidase gene, coxA, which is transcriptionally induced by carbon starvation, a condition in which it promotes survival (86, 94) (Figs. 7D and S7). Together aceA and coxA expressing cells covered up to 43% of an aggregate cell mass in our experiment. This is just one example of the type of coherent spatiometabolic stratification pattern seqFISH can reveal at a given moment in time.
Fig. 7. Functional zonation in a single microaggregate.

(A-D) A P. aeruginosa 35h aggregate. Bacteria are shown via 16S rRNA FISH fluorescence (gray) and are overlaid with raw mRNA-FISH fluorescence for different genes as described in the panel legends.
Discussion
Until now, our ability to capture the dynamic metabolic activities of microbial populations and communities at small spatial scales has been limited to tracking just a few parameters. This technical limitation has restricted our ability to observe and understand the features that define these ubiquitous associations. In this work, our analysis of P. aeruginosa populations has shown that par-seqFISH can reveal a high degree of transcriptional heterogeneity spanning multiple dimensions, from the subcellular to the microscale. Moreover, by tracking the temporal and spatial dynamics of cellular states in sub-populations, our results demonstrate that spatial transcriptomics can provide new insights into how bacteria sustain functional diversity.
The high temporal and spatial resolution enabled by par-seqFISH permitted us to make unexpected discoveries. For example, in planktonic cultures, we observed the short-lived temporal emergence of two T3SS+ populations: a large group, that appeared in exponential phase and expressed all the needed T3SS components, and a second, ~10-fold smaller group that emerged during mid-stationary and expressed the effectors but not the secretion system. The estimated 3–4 divisions that separate these sub-population correlate with their size differences, suggesting these two sub-populations could represent the same T3SS+ population, just at different stages of growth. We hypothesize that the specific expression of effector genes in the stationary T3SS+ sub-population serves to replenish the effectors lost by the diluting effect of cell divisions. If true, this would mean that P. aeruginosa not only generates heterogenous sub-populations but can also actively maintains their functional capabilities. Such an observation would not have been possible without the ability to measure the expression many genes within the same cell.
In P. aeruginosa biofilms, despite striking levels of metabolic heterogeneity, coherent co-expression patterns also emerged. We found a strong spatial correlation between denitrification genes and oxidative-stress factors that suggest that local denitrification results in NO toxicity. This hypothesis is based on the expression of the inducible peroxidase, katA, which is known to be upregulated by NO under anaerobic conditions and to alleviate NO toxicity (84). We also observed overlapping induction of other factors such as katB (83) and the superoxide dismutase, sodM (82), suggesting they may also play protective roles. Notably, these patterns were highly spatially confined, suggesting that NO toxicity did not propagate to neighboring cells, even those just a few microns beyond. However, it remains unclear how such hydrogen peroxide and superoxide detoxifying enzymes protect cells from NO. It is known that NO interacts with relevant oxidants to produce reactive nitrogen species (RNS) such as peroxynitrite (ONOO−) (95). Thus, perhaps these oxidative-stress response factors act by limiting the pool of oxidants available for RNS production. Various RNS cause diverse types of cellular damage including the chemical modification of proteins, specifically cysteine and tyrosine residues (95). Our data points toward elevated expression of cellular proteases in NO stressed regions. We therefore suggest that these proteases act to detoxify cells by eliminating damaged proteins, a hypothesis that remains to be tested in future studies.
The T3SS+ and NO-related results are but two examples of the type of unexpected findings revealed by the seqFISH approach that were uniquely enabled by the method. Much remains to be done to follow up these and other observations, yet they illustrate the power of single-cell spatial transcriptomics to literally and figuratively change our perspective. Importantly, we note that the par-seqFISH multiplexing approach, which we developed to increase the throughput of seqFISH for single-cell analysis, can potentially be applied in various other ways, both in synthetic and natural communities. For example, because par-seqFISH is based on 16S rRNA labels (Ribo-Tags), it could in principle be used to encode bacterial taxonomy. Recently, a conceptually similar and exciting method for combinatorial labeling of taxonomy was introduced in a biogeographical study of the human microbiome (25). In principle, the par-seqFISH strategy could be readily extended to capture similar or higher level of taxonomic complexity, as well as adding the currently missing feature of mRNA expression. We predict that this future application will be useful in providing a functional measure for interpreting spatial associations between microbial species.
Together, this study presents a highly multiplexed and spatially resolved single-cell analysis of the transcriptional landscape of bacterial populations. Future application of this approach at greater temporal resolution or in other biofilm models could help shed light on biofilm functional organization and development. Furthermore, extension of this approach to natural and clinical samples could provide important insights into the conditions experienced by microbes in more complex environments and the coordinated physiological responses that emerge in turn. A critical next step will be to develop methods to chart the environmental conditions that contextualize expression patterns observed in any given case. Understanding the roles that spatial and temporal heterogeneity play in microbial populations and communities represents an exciting and important research aim for modern microbiology. The results presented in this manuscript provide a new way of addressing these defining features on our way to understanding microbial life on its own terms.
Materials and methods
Bacterial strains and growth conditions
P. aeruginosa strain UCBPP-PA14 was grown aerobically with shaking at 250 rpm in lysogeny broth (LB) (Difco) or on LB agar plates at 37°C. SCFM was made as previously described (64). For the growth curve experiments, an overnight LB culture was washed twice using fresh growth media (either LB or SCFM) and then diluted 1:100 into 100 ml prewarmed fresh media. The cultures were grown at 37°C with shaking at 250 rpm and collected at various time points as indicated in Fig. 2A. The SCFM samples were collected cell densities identical to the LB experiment, except the OD600 = 3.2 sample was omitted. Collected samples were immediately fixed in ice-cold 2% paraformaldehyde (PFA) and were incubated on ice for 1.5h in the dark, and then washed twice with 1x PBS. Samples were resuspended in 70% EtOH and incubated at −20°C for at 24h to permeabilize the cells. Surface colonization was performed by washing and diluting an LB overnight culture 1:100 into fresh SCFM and dispensing 100 μl into coverslip attached open incubation chambers (Electron Microscopy Sciences, 70333–42). The coverslips were incubated in parafilm sealed sterile petri dishes at 37°C and the media was gently exchanged every 4 hours. A damp Kimwipe was placed in the petri dish to control media evaporation. During the overnight stage of the 35h experiment, the media was exchanged only once after 8h. Biofilm experiments were collected by gently exchanging the SCFM with 100 μl ice cold 2% PFA solution and incubating the sample at 4°C for 1.5h. The samples were washed twice with 1x PBS, resuspended in 70% EtOH and incubated overnight at 4°C and prepared for seqFISH as described below the following day.
SeqFISH probe design and library generation
Primary probes were designed as 30 nt stretches in a GC range of 45–65%. Probe sequences containing more than four consecutive base repeats were removed. The remaining probes were compared to the reference genome using blast and any probe with non-specific binding of at 18nt or more was discarded. Negative control genes were selected from the P1 phage genome (NC_005856.1) with the same criteria. Each selected gene was covered by 12–20 nonoverlapping probes randomly selected from the gene probe set. The probes were designed as a 30nt mRNA binding region flanked by overhangs composed of four repeats of the secondary hybridization sequence (complementary to a designated fluorescent readout probe; table S2). Thus, it is estimated that during secondary hybridization, each mRNA is covered by 48–80 fluorescent readout probes (12–20 × 4), on par with previous mRNA-FISH experiments in bacteria (33, 42).
A library of 1,763 probes targeting 105 P. aeruginosa genes and three negative controls were designed (tables S1 and S2). Additional flanking sequences were added to the primary probe sequences to enable library amplification via PCR (Forward 5’- TTTCGTCCGCGAGTGACCAG-3’ and reverse 5’-CAACGTCCATGTCGGGATGC-3’). The primary probe set was purchased as oligoarray complex pool from Twist Bioscience and constructed as previously described (36) (table S2). Briefly, a set of 9 PCR cycles were used to amplify the designated probe sequences from the oligo pool. The amplified PCR products were purified using the QIAquick PCR Purification Kit (28104; Qiagen) according to the manufacturer’s instructions. The PCR products were used as the template for in vitro transcription (E2040S; NEB) followed by reverse transcription (EP7051; Thermo Fisher). Then, the single-stranded DNA (ssDNA) probes were alkaline hydrolyzed with 1 M NaOH at 65 °C for 15 min to degrade the RNA templates, followed by 1 M acetic acid neutralization. Next, to clean up the probes, we performed ethanol precipitation to remove stray nucleotides, phenol–chloroform extraction to remove protein, and used Zeba Spin Desalting Columns (7K MWCO) (89882; Thermo Fisher) to remove residual nucleotides and phenol contaminants. Readout probes were designed as previously described and ordered from Integrated DNA Technologies (IDT) (36).
Ribo-Tag probes were designed to target the same region in the 16S rRNA gene according to the criteria described above, but with a 28nt binding regions. Each probe sequence was flanked with two secondary sequences selected out a set of six that were dedicated to multiplexing (table S3). An additional 16S rRNA probe was generated as a standard between all multiplexed samples and was hybridized to an independent region of the 16S rRNA (table S3). This probe provided an additional reference and was used to register images from different channels (see below).
Coverslip functionalization
Coverslips were cleaned with a plasma cleaner on a high setting (PDC-001, Harrick Plasma) for 5 min, followed by immersion in 1% bind-silane solution (GE; 17–1330-01) made in pH 3.5 10% (v/v) acidic ethanol solution for 30 min at room temperature. The coverslips were washed with 100% ethanol three times and dried in an oven at >90 °C for 30 min. The coverslips were then treated with 100 μg μl−1 of poly-D-lysine (P6407; Sigma) in water for at least one hour at room temperature, followed by three rinses with water. Coverslips were air-dried and kept at −20°C for no longer than 2 weeks before use.
Parallel seqFISH
Independent fixed samples were individually hybridized with 16S rRNA labels, washed and then pooled into a single mixture that was hybridized with the gene probe library and prepared for imaging. Approximately 108 cells were collected from each sample into a microcentrifuge, pelleted via centrifugation (6,000 rpm) and then resuspended in 20 μl H20 with 6 nM of the designated 16S rRNA label (sample specific) and another 6 nM of a shared reference 16S rRNA probe (table S3). Each sample was then mixed with prewarmed 30 μl of primary hybridization buffer (50% formamide, 10% dextran sulfate and 2× SSC) via gentle pipetting, incubated at 37°C for >16 h, washed twice with 100 μl wash buffer (55% formamide and 0.1% Triton-X 100 in 2× SSC; 5 min 8,000 rpm for the viscous hybridization buffer) and then incubated at 37°C in 100 μl wash buffer for 30 min to remove non-specific probe binding. Samples were washed twice with 100 μl 2x SSC and pooled together into a new microcentrifuge in equal volumes. The mixture was pelleted and resuspended in 40 μl H20 and 10 μl of the mixture was added to 10 μl gene probe library mixture and mixed well with prewarmed 30 μl primary hybridization buffer. The hybridizations were incubated for >16 h at 37°C and were washed and prepared as described above. The final mixture was resuspended in 20–25 μl 1x PBS and 5–10 μl were gently spotted at the center of the coverslip and incubated at RT for 10 min to allow the cells to sediment and bind the surface. The coverslips were centrifuged for 5 min at 1,000 rpm to create a smooth and dense cell monolayer. The cells were immobilized using a hydrogel as previously described (36) and stained with 10 μl ml−1 DAPI (D8417; Sigma) for 5 min before imaging so that cells could be visualized.
In biofilm experiments, the fixed and permeabilized surface attached microaggregates were air dried, covered with a hydrogel and hybridized with the gene library and a rRNA probes in one single reaction, as described above.
seqFISH imaging
All seqFISH experiments were performed using a combined imaging and automated fluidics delivery system as previously described (36). DAPI stained samples mounted on coverslips were connected to the fluidic system. The ROIs were registered using the DAPI fluorescence and a set of sequential secondary hybridizations, washes and imaging was performed.
Each hybridization round contained three unique 15-nt readouts probes each conjugated to either Alexa Fluor 647 (A647), Cy3B and Alexa Fluor 488 (A488). All readout probes were ordered from Integrated DNA Technologies and prepared into 500 nM stock solutions. Each serial probe mixture was prepared in EC buffer (10% ethylene carbonate (E26258; Sigma), 10% dextran sulfate (D4911; Sigma), 4× SSC). Hybridizations were incubated with the sample for 20 min to allow for secondary probe binding. The samples were then washed to remove excess readout probes and to limited non-specific binding using ~300 μl of 10% formamide wash buffer (10% formamide and 0.1% Triton X-100 in 2× SSC). Samples were then rinsed with ~200 μl of 4× SSC and then stained with DAPI solution (10 μg ml−1 of DAPI, 4× SSC). Lastly, an anti-bleaching buffer solution made was flowed through the samples (10% (w/v) glucose, 1:100 diluted catalase (Sigma C3155), 0.5 mg ml−1 glucose oxidase (Sigma G2133) and 50 mM pH 8 Tris-HCl in 4× SSC). Imaging was performed with a Leica DMi8 microscope equipped with a confocal scanner unit (Yokogawa CSU-W1), a sCMOS camera (Andor Zyla 4.2 Plus), a 63× oil objective lens (Leica 1.40 NA) and a motorized stage (ASI MS2000). Lasers from CNI and filter sets from Semrock were used. Snapshots were acquired using 647-nm, 561-nm, 488-nm and 405-nm fluorescent channels with 0.5-μm z-steps for all experiments with the exception of the 35h biofilm experiment in which 1.0-μm z-steps were collected. After imaging, readout probes were stripped using 55% wash buffer (55% formamide and 0.1% Triton-X 100 in 2× SSC) that was flowed through for 1 min, followed by an incubation time of 15 min before rinsing with 4× SSC solution. This protocol: serial hybridizations, imaging and signal quenching steps, was repeated for ~40 rounds to capture 16S rRNA for multiplexing, mRNA expression and background signal. The integration of automated fluidics delivery system and imaging was controlled via μManager (96).
Image analysis demultiplexing and gene-expression measurement
Maximal projection images were generated using ImageJ (97) for DAPI and 16S rRNA and hybridization rounds were registered using the DAPI fluorescence. Aberrations between fluorophores were corrected by alignment of 16S rRNA signals across all channels. Cells were segmented using the DAPI signal with SuperSegger using the 60XPa configuration (98) and filtered using custom scripts to eliminate odd shapes, autofluorescent or low signal components.
For par-seqFISH demultiplexing, the background (no readouts) and 16S rRNA fluorescent intensity for each relevant secondary readout probe was measured within segmented cell boundaries to provide a signal-to-background score for each readout. The cells were classified according to the positive readout combinations (table S3). The level of false positives was estimated by counting the number of cells classified into combinations left out of the experiment.
The mRNA-FISH data was analyzed using Spätzcells (42). Briefly, spots were detected as regional maxima with intensity greater than a threshold value that was set using the negative control genes and were fit with a 2D gaussian model. The integrated intensity of the spot and the position of its estimated maxima were determined (42). Spots were assigned to cells using the cell segmentation masks (42). In biofilm experiments spots were assigned to cells in a z-section sensitive manner. Deviating spots maxima positions that did not overlap a cell boundary were tested against the flanking z-sections to identify their cell of origin. If no cell was detected the spots were discarded. All predicted low expression genes (defined as gene with spots in less than 30% of all cells) were identified and the distribution of their spot intensities was fit with a gaussian mixture model to identify the characteristic intensity of a single mRNA, normalized to the number of probes used for the specific gene. The median characteristic single-mRNA signal was then calculated using all low expression genes for each fluorophore (A647, A488 and cy3B). The variation between different genes labeled with the same fluorophore was low, with a coefficient of variation of 18–21%. This median characteristic value was used to transform fluorescent intensity into to discrete mRNA counts per gene within each cell. The A488 characteristic signal was corrected by a factor of 1.5 to account for its lower intensity in our system. In each cell, the total intensity of each gene was calculated by summing the intensities of all spots. The total value was normalized by the characteristic value for a single mRNA in the corresponding fluorophore.
Single-cell expression analysis and cell biological parameter calculations
Single-cell UMAP analysis was performed using Scanpy v1.7.0 (99). Genes detected at consistently low levels were excluded from the analysis. These included pilY1, flgK, nasA, algU, purF, phzH, phzS and pslG (table S1). We followed the standard Scanpy normalization and scaling, dimensionality reduction, and clustering as described in the Scanpy tutorial, minus the high variance gene selection and without a library size normalization. We used 15 neighbors and 15 and 17 PCA components, for the LB and merged SCFM analyses, respectively. Clustering was performed using the Leiden method. Jupyter notebooks with chosen parameters, run lines, output files and source data are available at (105).
Cell nucleoid size was calculated using the segmentation mask. A chromosome score was calculated as the median DAPI intensity multiplied by the nucleoid size. The median chromosome score was calculated for the last time point in our LB experiment (deep stationary; OD600 = 3.2). Because most cells in this stage are in a non-dividing state, we set this value as a reference for a single chromosome copy. We then normalized the scores of all cells in the experiment using this value, as seen in Fig. 2. In addition to using Ribo-Tags to label cells from different conditions, we also hybridized another region in the 16S rRNA with a probe that was shared across all samples (table S3; described above). We used this reference signal to compare the 16S rRNA intensity between cells from different conditions. We measured the median 16S rRNA signal per cells and multiplied it by the nucleoid size (which completely overlaps the 16S signal and estimates cell size). In E. coli, maximal ribosome numbers appear at the maximal growth rate and have been estimated at 72,000 (100). The median rRNA score was calculated for the maximal growth (OD600 = 0.2) and normalized to 72,000 as in E. coli for a rough estimate (Fig. 2).
Image analysis in surface colonization experiments
Images were registered as described above and segmentation was performed using the pixel classification workflow in Ilastik (101). We trained the Ilastik classification model with background, cell boundaries and cell bodies, using the 16S rRNA signal. We find that Ilastik performs extremely well. However, in high density regions, segmentation often resulted in over-connectivity due to incorrect 3D overlaps. We disconnected such cells clusters. The binary masks (segmentation output) were thinned, and all 3D connected components (CCs) were re-calculated. This reduced spurious connections. Then, all CCs traversing more than 2.5 μm were set aside for re-evaluation for potential over-connections. For each such 3D component we examined each z-slice at a time and identified all 2D CCs. We removed overly large or curved blobs, which represent segmentation artefacts that often incorrectly connect distinct cells across z-sections. In addition, for each 2D detected component we calculated its orientation and overlap with components in the previous flanking z-section. If this component exhibited a significant change in its orientation (the direction it is pointing) we disconnected it from the component below. We then continued the analysis using the newly oriented component as a seed. Cell clusters that could not be properly disentangled were removed from the analysis. At the end of the analysis the cell 3D masks were re-thickened.
We conducted bulk neighborhood analysis where we studied the immediate neighborhoods associated with high expression of a specific gene. For the gene of interest, we identified all top 99th percentile cells (99.5th for the pyocin specific analysis), denoted as “center cells”. Using the 3D centroid coordinates of center cells we identified their closest neighbors within a specified distance (up to 10 μm for pyocins and 3 μm for the rest). We then collected up to k closest cells (up to 5–300 neighbors in the pyocin analysis to view the enrichment decay and up to 5 for the rest of the genes). All of the neighborhood cells selected (not including the center cells) were then analyzed in bulk together and their mean gene-expression was calculated and compared to the population (minus all center cells not used). We conducted this analysis across all genes and performed a Pearson correlation analyses to identify spatially correlating genes (fig. S5).
RNA-Seq analysis
We analyzed previously published RNA-Seq datasets (51), in which WT P. aeruginosa PA14 was grown in LB media to OD600 values similar to those collected in our experiment (OD600=1.1 and OD600 = 2.0 in the RNA-Seq experiment). The raw sequencing data was aligned to the P. aeruginosa reference genome using bowtie2 (102) and the reads were assigned to genes using featureCounts (103). The Reads per Kilobase per Million (RPKM) values were calculated for all genes and then compared with the average seqFISH expression profile of each LB grown sample (fig. S2).
Supplementary Material
Acknowledgments
We thank George A. O’Toole and Marvin Whiteley for help with designing the gene set. We also thank Megan Bergkessel and Rotem Sorek and members of the Newman lab for critically reading the manuscript. We thank the Newman lab for fruitful discussions and comments, particularly Megan Bergkessel for assistance with RNA-Seq analysis.
Funding:
grants to DKN from the NIH (1R01AI127850-01A1 and 1R01HL152190-01) and ARO (W911NF-17-1-0024) supported this work. LC was funded by the Allen Frontier group. DD was supported by the Rothschild foundation, EMBO Long-Term, and the Helen Hay Whitney postdoctoral fellowships, as well as a Geobiology Postdoctoral Fellowship from the Division of Geological and Planetary Sciences, Caltech.
Footnotes
References
- 1.Flemming H-C, Wuertz S, Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol 17, 247–260 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M, Marrie TJ, Bacterial biofilms in nature and disease. Annu. Rev. Microbiol 41, 435–464 (1987). [DOI] [PubMed] [Google Scholar]
- 3.DePas WH, Starwalt-Lee R, Van Sambeek L, Ravindra Kumar S, Gradinaru V, Newman DK, Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. MBio. 7, e00796–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mark Welch JL, Rossetti BJ, Rieken CW, Dewhirst FE, Borisy GG, Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 113, E791–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaber JA, Triffo WJ, Suh SJ, Oliver JW, Hastert MC, Griswold JA, Auer M, Hamood AN, Rumbaugh KP, Pseudomonas aeruginosa forms biofilms in acute infection independent of cell-to-cell signaling. Infect. Immun 75, 3715–3721 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorth P, Spero MA, Livingston J, Newman DK, Quantitative Visualization of Gene Expression in Mucoid and Nonmucoid Pseudomonas aeruginosa Aggregates Reveals Localized Peak Expression of Alginate in the Hypoxic Zone. MBio. 10, e02622–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatzenpichler R, Krukenberg V, Spietz RL, Jay ZJ, Next-generation physiology approaches to study microbiome function at single cell level. Nat. Rev. Microbiol 18, 241–256 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chadwick GL, Jiménez Otero F, Gralnick JA, Bond DR, Orphan VJ, NanoSIMS imaging reveals metabolic stratification within current-producing biofilms. Proc. Natl. Acad. Sci. USA 116, 20716–20724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackermann M, A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol 13, 497–508 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Evans CR, Kempes CP, Price-Whelan A, Dietrich LEP, Metabolic Heterogeneity and Cross-Feeding in Bacterial Multicellular Systems. Trends Microbiol. 28, 732–743 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiber F, Littmann S, Lavik G, Escrig S, Meibom A, Kuypers MMM, Ackermann M, Phenotypic heterogeneity driven by nutrient limitation promotes growth in fluctuating environments. Nat. Microbiol. 1, 16055 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Schreiber F, Ackermann M, Environmental drivers of metabolic heterogeneity in clonal microbial populations. Curr. Opin. Biotechnol 62, 202–211 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Kopf SH, McGlynn SE, Green-Saxena A, Guan Y, Newman DK, Orphan VJ, Heavy water and (15) N labelling with NanoSIMS analysis reveals growth rate-dependent metabolic heterogeneity in chemostats. Environ. Microbiol 17, 2542–2556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veening J-W, Smits WK, Kuipers OP, Bistability, epigenetics, and bet-hedging in bacteria. Annu. Rev. Microbiol 62, 193–210 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Rosenthal AZ, Qi Y, Hormoz S, Park J, Li SH-J, Elowitz MB, Metabolic interactions between dynamic bacterial subpopulations. Elife. 7, e33099. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armbruster CR, Lee CK, Parker-Gilham J, de Anda J, Xia A, Zhao K, Murakami K, Tseng BS, Hoffman LR, Jin F, Harwood CS, Wong GC, Parsek MR, Heterogeneity in surface sensing suggests a division of labor in Pseudomonas aeruginosa populations. Elife. 8, e45084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diard M, Garcia V, Maier L, Remus-Emsermann MNP, Regoes RR, Ackermann M, Hardt W-D, Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature. 494, 353–356 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Stewart PS, Franklin MJ, Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6, 199–210 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Stewart PS, Diffusion in biofilms. J. Bacteriol 185, 1485–1491 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfsberg E, Long CP, Antoniewicz MR, Metabolism in dense microbial colonies: 13C metabolic flux analysis of E. coli grown on agar identifies two distinct cell populations with acetate cross-feeding. Metab. Eng 49, 242–247 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Kotte O, Volkmer B, Radzikowski JL, Heinemann M, Phenotypic bistability in Escherichia coli’s central carbon metabolism. Mol. Syst. Biol 10, 736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bocci F, Suzuki Y, Lu M, Onuchic JN, Role of metabolic spatiotemporal dynamics in regulating biofilm colony expansion. Proc. Natl. Acad. Sci. USA. 115, 4288–4293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadell CD, Drescher K, Foster KR, Spatial structure, cooperation and competition in biofilms. Nat. Rev. Microbiol 14, 589–600 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Cordero OX, Datta MS, Microbial interactions and community assembly at microscales. Curr. Opin. Microbiol 31, 227–234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi H, Shi Q, Grodner B, Lenz JS, Zipfel WR, Brito IL, De Vlaminck I, Highly multiplexed spatial mapping of microbial communities. Nature. 588, 676–681 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tropini C, Earle KA, Huang KC, Sonnenburg JL, The gut microbiome: connecting spatial organization to function. Cell Host Microbe. 21, 433–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilbert SA, Mark Welch JL, Borisy GG, Spatial ecology of the human tongue dorsum microbiome. Cell Rep 30, 4003–4015.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imdahl F, Vafadarnejad E, Homberger C, Saliba A-E, Vogel J, Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat. Microbiol 5, 1202–1206 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Kuchina A, Brettner LM, Paleologu L, Roco CM, Rosenberg AB, Carignano A, Kibler R, Hirano M, DePaolo RW, Seelig G, Microbial single-cell RNA sequencing by split-pool barcoding. Science. 371 (2021), doi: 10.1126/science.aba5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blattman SB, Jiang W, Oikonomou P, Tavazoie S, Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat. Microbiol 5, 1192–1201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S, Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Femino AM, Fay FS, Fogarty K, Singer RH, Visualization of single RNA transcripts in situ. Science. 280, 585–590 (1998). [DOI] [PubMed] [Google Scholar]
- 33.So L-H, Ghosh A, Zong C, Sepúlveda LA, Segev R, Golding I, General properties of transcriptional time series in Escherichia coli. Nat. Genet 43, 554–560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi HMT, Beck VA, Pierce NA, Next-generation in situ hybridization chain reaction: higher gain, lower cost, greater durability. ACS Nano. 8, 4284–4294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X, RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eng C-HL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan G-C, Cai L, Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L, Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, Rubinstein ND, Hao J, Regev A, Dulac C, Zhuang X, Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 362 (2018), doi: 10.1126/science.aau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah S, Lubeck E, Zhou W, Cai L, In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lignell A, Kerosuo L, Streichan SJ, Cai L, Bronner ME, Identification of a neural crest stem cell niche by Spatial Genomic Analysis. Nat. Commun 8, 1830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah S, Takei Y, Zhou W, Lubeck E, Yun J, Eng C-HL, Koulena N, Cronin C, Karp C, Liaw EJ, Amin M, Cai L, Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell. 174, 363–376.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skinner SO, Sepúlveda LA, Xu H, Golding I, Measuring mRNA copy number in individual Escherichia coli cells using single-molecule fluorescent in situ hybridization. Nat. Protoc 8, 1100–1113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, Duan K, Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm Med. 16, 174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malhotra S, Hayes D, Wozniak DJ, Cystic Fibrosis and Pseudomonas aeruginosa: the Host-Microbe Interface. Clin. Microbiol. Rev 32 (2019), doi: 10.1128/CMR.00138-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takei Y, Yun J, Zheng S, Ollikainen N, Pierson N, White J, Shah S, Thomassie J, Suo S, Eng C-HL, Guttman M, Yuan G-C, Cai L, Integrated spatial genomics reveals global architecture of single nuclei. Nature. 590, 344–350 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vallet-Gely I, Boccard F, Chromosomal organization and segregation in Pseudomonas aeruginosa. PLoS Genet. 9, e1003492 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee J, Zhang L, The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell. 6, 26–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Price-Whelan A, Dietrich LEP, Newman DK, Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J. Bacteriol 189, 6372–6381 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jo J, Cortez KL, Cornell WC, Price-Whelan A, Dietrich LE, An orphan cbb3-type cytochrome oxidase subunit supports Pseudomonas aeruginosa biofilm growth and virulence. Elife. 6, e30205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arai H, Regulation and Function of Versatile Aerobic and Anaerobic Respiratory Metabolism in Pseudomonas aeruginosa. Front. Microbiol 2, 103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz S, Eckweiler D, Bielecka A, Nicolai T, Franke R, Dötsch A, Hornischer K, Bruchmann S, Düvel J, Häussler S, Elucidation of sigma factor-associated networks in Pseudomonas aeruginosa reveals a modular architecture with limited and function-specific crosstalk. PLoS Pathog. 11, e1004744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finkel SE, Long-term survival during stationary phase: evolution and the GASP phenotype. Nat. Rev. Microbiol 4, 113–120 (2006). [DOI] [PubMed] [Google Scholar]
- 53.McInnes L, Healy J, Melville J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv, 1802.03426 (2018). [Google Scholar]
- 54.Bosdriesz E, Molenaar D, Teusink B, Bruggeman FJ, How fast-growing bacteria robustly tune their ribosome concentration to approximate growth-rate maximization. FEBS J. 282, 2029–2044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR, Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol 75, 827–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reichhardt C, Wong C, Passos da Silva D, Wozniak DJ, Parsek MR, CdrA Interactions within the Pseudomonas aeruginosa Biofilm Matrix Safeguard It from Proteolysis and Promote Cellular Packing. MBio. 9, e01376–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaborina O, Holbrook C, Chen Y, Long J, Zaborin A, Morozova I, Fernandez H, Wang Y, Turner JR, Alverdy JC, Structure-function aspects of PstS in multi-drug-resistant Pseudomonas aeruginosa. PLoS Pathog. 4, e43 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gil-Marqués ML, Labrador Herrera G, Miró Canturri A, Pachón J, Smani Y, Pachón-Ibáñez ME, Role of PstS in the Pathogenesis of Acinetobacter baumannii Under Microaerobiosis and Normoxia. J. Infect. Dis 222, 1204–1212 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Hauser AR, The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol 7, 654–665 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, Sorek R, Lory S, The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 8, e1002945 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lombardi C, Tolchard J, Bouillot S, Signor L, Gebus C, Liebl D, Fenel D, Teulon J-M, Brock J, Habenstein B, Pellequer J-L, Faudry E, Loquet A, Attrée I, Dessen A, Job V, Structural and Functional Characterization of the Type Three Secretion System (T3SS) Needle of Pseudomonas aeruginosa. Front. Microbiol 10, 573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Høiby N, Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol 44, 547–558 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Høiby N, Ciofu O, Johansen HK, Song Z, Moser C, Jensen PØ, Molin S, Givskov M, Tolker-Nielsen T, Bjarnsholt T, The clinical impact of bacterial biofilms. Int. J. Oral Sci 3, 55–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palmer KL, Aye LM, Whiteley M, Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol 189, 8079–8087 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gicquel G, Bouffartigues E, Bains M, Oxaran V, Rosay T, Lesouhaitier O, Connil N, Bazire A, Maillot O, Bénard M, Cornelis P, Hancock REW, Dufour A, Feuilloley MGJ, Orange N, Déziel E, Chevalier S, The extra-cytoplasmic function sigma factor sigX modulates biofilm and virulence-related properties in Pseudomonas aeruginosa. PLoS One. 8, e80407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burrows LL, Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu. Rev. Microbiol 66, 493–520 (2012). [DOI] [PubMed] [Google Scholar]
- 67.O’Toole GA, Kolter R, Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol 30, 295–304 (1998). [DOI] [PubMed] [Google Scholar]
- 68.Belas R, Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol. 22, 517–527 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Laventie B-J, Sangermani M, Estermann F, Manfredi P, Planes R, Hug I, Jaeger T, Meunier E, Broz P, Jenal U, A Surface-Induced Asymmetric Program Promotes Tissue Colonization by Pseudomonas aeruginosa. Cell Host Microbe. 25, 140–152.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Ghequire MGK, De Mot R, The tailocin tale: peeling off phage tails. Trends Microbiol. 23, 587–590 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Bobay L-M, Touchon M, Rocha EPC, Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 111, 12127–12132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turnbull L, Toyofuku M, Hynen AL, Kurosawa M, Pessi G, Petty NK, Osvath SR, Cárcamo-Oyarce G, Gloag ES, Shimoni R, Omasits U, Ito S, Yap X, Monahan LG, Cavaliere R, Ahrens CH, Charles IG, Nomura N, Eberl L, Whitchurch CB, Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat. Commun 7, 11220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vacheron J, Heiman CM, Keel C, Live cell dynamics of production, explosive release and killing activity of phage tail-like weapons for Pseudomonas kin exclusion. Commun. Biol 4, 87 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS, Extracellular DNA required for bacterial biofilm formation. Science. 295, 1487 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Brazas MD, Hancock REW, Ciprofloxacin induction of a susceptibility determinant in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49, 3222–3227 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Povolotsky TL, Keren-Paz A, Kolodkin-Gal I, Metabolic microenvironments drive microbial differentiation and antibiotic resistance. Trends Genet. 37, 4–8 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Wessel AK, Arshad TA, Fitzpatrick M, Connell JL, Bonnecaze RT, Shear JB, Whiteley M, Oxygen limitation within a bacterial aggregate. MBio. 5, e00992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dietrich LEP, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, Newman DK, Bacterial community morphogenesis is intimately linked to the intracellular redox state. J. Bacteriol 195, 1371–1380 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cowley ES, Kopf SH, LaRiviere A, Ziebis W, Newman DK, Pediatric cystic fibrosis sputum can be chemically dynamic, anoxic, and extremely reduced due to hydrogen sulfide formation. MBio. 6, e00767 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoon SS, Hennigan RF, Hilliard GM, Ochsner UA, Parvatiyar K, Kamani MC, Allen HL, DeKievit TR, Gardner PR, Schwab U, Rowe JJ, Iglewski BH, McDermott TR, Mason RP, Wozniak DJ, Hancock REW, Parsek MR, Noah TL, Boucher RC, Hassett DJ, Pseudomonas aeruginosa Anaerobic Respiration in Biofilms. Dev. Cell 3, 593–603 (2002). [DOI] [PubMed] [Google Scholar]
- 81.Eschbach M, Schreiber K, Trunk K, Buer J, Jahn D, Schobert M, Long-term anaerobic survival of the opportunistic pathogen Pseudomonas aeruginosa via pyruvate fermentation. J. Bacteriol 186, 4596–4604 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hassett DJ, Charniga L, Bean K, Ohman DE, Cohen MS, Response of Pseudomonas aeruginosa to pyocyanin: mechanisms of resistance, antioxidant defenses, and demonstration of a manganese-cofactored superoxide dismutase. Infect. Immun 60, 328–336 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown SM, Howell ML, Vasil ML, Anderson AJ, Hassett DJ, Cloning and characterization of the katB gene of Pseudomonas aeruginosa encoding a hydrogen peroxide-inducible catalase: purification of KatB, cellular localization, and demonstration that it is essential for optimal resistance to hydrogen peroxide. J. Bacteriol 177, 6536–6544 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Su S, Panmanee W, Wilson JJ, Mahtani HK, Li Q, Vanderwielen BD, Makris TM, Rogers M, McDaniel C, Lipscomb JD, Irvin RT, Schurr MJ, Lancaster JR, Kovall RA, Hassett DJ, Catalase (KatA) plays a role in protection against anaerobic nitric oxide in Pseudomonas aeruginosa. PLoS One. 9, e91813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cutruzzolà F, Frankenberg-Dinkel N, Origin and Impact of Nitric Oxide in Pseudomonas aeruginosa Biofilms. J. Bacteriol 198, 55–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Basta DW, Bergkessel M, Newman DK, Identification of fitness determinants during energy-limited growth arrest in Pseudomonas aeruginosa. MBio. 8, e01170–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ, Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J. Bacteriol 182, 4533–4544 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Toyofuku M, Nomura N, Fujii T, Takaya N, Maseda H, Sawada I, Nakajima T, Uchiyama H, Quorum sensing regulates denitrification in Pseudomonas aeruginosa PAO1. J. Bacteriol 189, 4969–4972 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Alst NE, Sherrill LA, Iglewski BH, Haidaris CG, Compensatory periplasmic nitrate reductase activity supports anaerobic growth of Pseudomonas aeruginosa PAO1 in the absence of membrane nitrate reductase. Can. J. Microbiol 55, 1133–1144 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schreiber K, Boes N, Eschbach M, Jaensch L, Wehland J, Bjarnsholt T, Givskov M, Hentzer M, Schobert M, Anaerobic survival of Pseudomonas aeruginosa by pyruvate fermentation requires an Usp-type stress protein. J. Bacteriol 188, 659–668 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dolan SK, Welch M, The glyoxylate shunt, 60 years on. Annu. Rev. Microbiol 72, 309–330 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Crousilles A, Dolan SK, Brear P, Chirgadze DY, Welch M, Gluconeogenic precursor availability regulates flux through the glyoxylate shunt in Pseudomonas aeruginosa. J. Biol. Chem 293, 14260–14269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dolan SK, Pereira G, Silva-Rocha R, Welch M, Transcriptional regulation of central carbon metabolism in Pseudomonas aeruginosa. Microb Biotechnol 13, 285–289 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kawakami T, Kuroki M, Ishii M, Igarashi Y, Arai H, Differential expression of multiple terminal oxidases for aerobic respiration in Pseudomonas aeruginosa. Environ. Microbiol. 12, 1399–1412 (2010). [DOI] [PubMed] [Google Scholar]
- 95.Stern AM, Zhu J, An introduction to nitric oxide sensing and response in bacteria. Adv Appl Microbiol. 87, 187–220 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N, Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol Chapter 14, Unit14.20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stylianidou S, Brennan C, Nissen SB, Kuwada NJ, Wiggins PA, SuperSegger: robust image segmentation, analysis and lineage tracking of bacterial cells. Mol. Microbiol 102, 690–700 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Wolf FA, Angerer P, Theis FJ, SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Milo R, Jorgensen P, Moran U, Weber G, Springer M, BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, Eren K, Cervantes JI, Xu B, Beuttenmueller F, Wolny A, Zhang C, Koethe U, Hamprecht FA, Kreshuk A, ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Zhang Y, Hu Z, Combined treatment of Pseudomonas aeruginosa biofilms with bacteriophages and chlorine. Biotechnol. Bioeng 110, 286–295 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Dar D, Dar N, Cai L, Newman DK. Data for the paper: “Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution”. Zenodo (2021); 10.5281/zenodo.4767568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dar D, Dar N, Cai L, Newman DK. Imaging data for the paper: “Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution”. Zenodo (2021); 10.5281/zenodo.4771778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.