

Abstract
Background
Coronary artery disease is a major public health problem affecting both developed and developing countries. Acute coronary syndromes include unstable angina and myocardial infarction with or without ST‐segment elevation (electrocardiogram sector is higher than baseline). Ventricular arrhythmia after myocardial infarction is associated with high risk of mortality. The evidence is out of date, and considerable uncertainty remains about the effects of prophylactic use of lidocaine on all‐cause mortality, in particular, in patients with suspected myocardial infarction.
Objectives
To determine the clinical effectiveness and safety of prophylactic lidocaine in preventing death among people with myocardial infarction.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) (2015, Issue 3), MEDLINE Ovid (1946 to 13 April 2015), EMBASE (1947 to 13 April 2015) and Latin American Caribbean Health Sciences Literature (LILACS) (1986 to 13 April 2015). We also searched Web of Science (1970 to 13 April 2013) and handsearched the reference lists of included papers. We applied no language restriction in the search.
Selection criteria
We included randomised controlled trials assessing the effects of prophylactic lidocaine for myocardial infarction. We considered all‐cause mortality, cardiac mortality and overall survival at 30 days after myocardial infarction as primary outcomes.
Data collection and analysis
We performed study selection, risk of bias assessment and data extraction in duplicate. We estimated risk ratios (RRs) for dichotomous outcomes and measured statistical heterogeneity using I2. We used a random‐effects model and conducted trial sequential analysis.
Main results
We identified 37 randomised controlled trials involving 11,948 participants. These trials compared lidocaine versus placebo or no intervention, disopyramide, mexiletine, tocainide, propafenone, amiodarone, dimethylammonium chloride, aprindine and pirmenol. Overall, trials were underpowered and had high risk of bias. Ninety‐seven per cent of trials (36/37) were conducted without an a priori sample size estimation. Ten trials were sponsored by the pharmaceutical industry. Trials were conducted in 17 countries, and intravenous intervention was the most frequent route of administration.
In trials involving participants with proven or non‐proven acute myocardial infarction, lidocaine versus placebo or no intervention showed no significant differences regarding all‐cause mortality (213/5879 (3.62%) vs 199/5848 (3.40%); RR 1.02, 95% CI 0.82 to 1.27; participants = 11727; studies = 18; I2 = 15%); low‐quality evidence), cardiac mortality (69/4184 (1.65%) vs 62/4093 (1.51%); RR 1.03, 95% CI 0.70 to 1.50; participants = 8277; studies = 12; I2 = 12%; low‐quality evidence) and prophylaxis of ventricular fibrillation (76/5128 (1.48%) vs 103/4987 (2.01%); RR 0.78, 95% CI 0.55 to 1.12; participants = 10115; studies = 16; I2 = 18%; low‐quality evidence). In terms of sinus bradycardia, lidocaine effect is imprecise compared with effects of placebo or no intervention (55/1346 (4.08%) vs 49/1203 (4.07%); RR 1.09, 95% CI 0.66 to 1.80; participants = 2549; studies = 8; I2 = 21%; very low‐quality evidence). In trials involving only participants with proven acute myocardial infarction, lidocaine versus placebo or no intervention showed no significant differences in all‐cause mortality (148/2747 (5.39%) vs 135/2506 (5.39%); RR 1.01, 95% CI 0.79 to 1.30; participants = 5253; studies = 16; I2 = 9%; low‐quality evidence). No significant differences were noted between lidocaine and any other antiarrhythmic drug in terms of all‐cause mortality and ventricular fibrillation. Data on overall survival 30 days after myocardial infarction were not reported. Lidocaine compared with placebo or no intervention increased risk of asystole (35/3393 (1.03%) vs 14/3443 (0.41%); RR 2.32, 95% CI 1.26 to 4.26; participants = 6826; studies = 4; I2 = 0%; very low‐quality evidence) and dizziness/drowsiness (74/1259 (5.88%) vs 16/1274 (1.26%); RR 3.85, 95% CI 2.29 to 6.47; participants = 2533; studies = 6; I2 = 0%; low‐quality evidence). Overall, safety data were poorly reported and adverse events may have been underestimated. Trial sequential analyses suggest that additional trials may not be needed for reliable conclusions to be drawn regarding these outcomes.
Authors' conclusions
This Cochrane review found evidence of low quality to suggest that prophylactic lidocaine has very little or no effect on mortality or ventricular fibrillation in people with acute myocardial infarction. The safety profile is unclear. This conclusion is based on randomised controlled trials with high risk of bias. However (disregarding the risk of bias), trial sequential analysis suggests that additional trials may not be needed to disprove an intervention effect of 20% relative risk reduction. Smaller risk reductions might require additional higher trials.
Plain language summary
Prophylactic lidocaine for myocardial infarction
Review question We reviewed the clinical effectiveness and safety of prophylactic lidocaine for myocardial infarction.
Background Coronary artery disease is a major public health problem that affects both developed and developing countries. Acute coronary syndromes include unstable angina and myocardial infarction with or without ST‐segment elevation (electrocardiogram sector is higher than baseline). Ventricular arrhythmia after myocardial infarction is associated with high risk of mortality. The evidence is out of date, and considerable uncertainty remains about the effects of prophylactic lidocaine use on all‐cause mortality, in particular, in patients with suspected myocardial infarction.
Study characteristics We identified 37 trials conducted between 1969 and 1999. The evidence is current up to April 2015. Trials were conducted in Australia, Belgium, Brazil, Canada, Denmark, France, Germany, Italy, New Zealand, Northern Ireland, Norway, Poland, Sweden, Switzerland, The Netherlands, United Kingdom and United States of America and included 11,948 participants. Trials were conducted in pre‐hospital and in‐hospital settings and included individuals with or without proved acute myocardial infarction. Some trials did not limit results to acute myocardial infarction only. Lidocaine was given by intravenous (bolus and/or infusion) and intramuscular (alone or in combination with intravenous dosage) routes. Overall, trials included small sample sizes and reported low numbers of events. All trials had high risk of bias. Ten trials were sponsored by the pharmaceutical industry.
Key results In people who had known or suspected heart attack, we found that lidocaine compared with placebo, no intervention or any other antiarrhythmic drug had very small or no effects on death, cardiac death and ventricular fibrillation.
Quality of evidence Our confidence in the results of this review is low because the included trials that we synthesised were of low quality (overestimation of benefits and underestimation of harms) and were conducted with a small number of participants, leading to imprecision of results.
Summary of findings
Summary of findings 1. Lidocaine compared with placebo or no intervention for acute myocardial infarction.
| Lidocaine compared with placebo or no intervention for acute myocardial infarction | ||||||
| Patient or population: patients with acute myocardial infarction Settings: pre‐hospital and in‐hospital Intervention: lidocaine Comparison: placebo or no intervention | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo or no intervention | Lidocaine | |||||
|
All‐cause mortality Follow‐up: ranging between unknown and 1 month |
34 per 1000 | 35 per 1000 (29 to 42) | RR 1.02 (0.85 to 1.27) | 11727 (18 studiesa) | ⊕⊕⊝⊝ Lowb,c | |
|
Cardiac mortality Follow‐up: ranging between unknown and 1 month |
15 per 1000 | 16 per 1000 (11 to 23) | RR 1.03 (0.70 to 1.50) | 8277 (12 studiesa) | ⊕⊕⊝⊝ Lowb,d | |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | No trial assessed this outcome |
|
Ventricular fibrillation
Electrocardiogram Follow‐up: ranging between unknown and 1 month |
21 per 1000 | 16 per 1000 (11 to 23) | RR 0.78 (0.55 to 1.12) | 10115 (16 studiesa) | ⊕⊕⊝⊝ Lowb,e | |
|
Adverse events (AEs; adverse drug reaction): asystole Follow‐up: ranging between unknown and 1 month |
4 per 1000 |
9 per 1000 (5 to 17) |
RR 2.32 (1.26 to 4.26) | 6826 (4 studiesa) |
⊕⊝⊝⊝ Verylowb,f,g | |
|
Adverse events (AEs; adverse drug reaction): sinus bradycardia Follow‐up: ranging between unknown and 1 month |
41 per 1000 | 44 per 1000 (27 to 73) | RR 1.09 (0.66 to 1.80) | 2549 (8 studiesa) |
⊕⊝⊝⊝ Verylowb,h,i | |
|
Adverse events (AEs; adverse drug reaction): drowsiness/dizziness Follow‐up: ranging between unknown and 1 month |
13 per 1000 |
48 per 1000 (29 to 81) |
RR 3.85 (2.29 to 6.47) | 2533 (6 studiesa) | ⊕⊕⊝⊝ Lowj.k.l | |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aTrials include participants with proven or unproven acute myocardial infarction. bDowngraded two levels because of limitations in trial design or execution (high attrition bias). cAssumed risk was gotten from control group risk (3.4%). dAssumed risk was gotten from control group risk (1.5%). eAssumed risk was gotten from control group risk (2.1%). fAssumed risk was gotten from control group risk (0.41%). gDowngraded one level because of imprecision (low number of events with an impact on the precision of effect estimates). hAssumed risk was gotten from control group risk (4.1%). iDowngraded one level because of imprecision (low number of events with an impact on the precision of effect estimates). jDowngraded one level because of limitations in trial design or execution (high attrition bias). kDowngraded one level because of imprecision (low number of events with an impact on the precision of effect estimates). lAssumed risk was gotten from control group risk (1.3%).
Summary of findings 2. Lidocaine compared with disopyramide for myocardial infarction.
| Lidocaine compared with disopyramide for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: disopyramide | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Disopyramide | Lidocaine | |||||
| All‐cause mortality Follow‐up: ranging between 12 hours and 24 hours | 70 per 1000 | 98 per 1000 (33 to 291) | RR 1.39 (0.47 to 4.13) | 144 (2 studies) | ⊕⊝⊝⊝ Verylowa,b,c | |
| Cardiac mortality Follow‐up: ranging between 12 hours and 24 hours | 41 per 1000 | 42 per 1000 (9 to 200) | RR 1.02 (0.21 to 4.87) | 144 (2 studies) | ⊕⊝⊝⊝ Verylowa,b,d | |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | No trial assessed this outcome |
|
Ventricular fibrillation Follow‐up: 12 hours |
79 per 1000 |
26 per 1000 (3 to 242) |
RR 0.32 (0.04 to 2.97) | 76 (1 study) |
⊕⊝⊝⊝ Verylowe,f,g | |
|
Adverse events (AEs; adverse drug reaction): asystole Follow‐up: 12 hours |
26 per 1000 |
9 per 1000 (0 to 209) |
RR 0.33 (0.01 to 7.93) | 76 (1 study) | ⊕⊝⊝⊝ Verylowe,f,h | |
|
Adverse events (AEs; adverse drug reaction):
sinoatrial block Follow‐up: 24 hours |
30 per 1000 | 28 per 1000 2 to 438 |
RR 0.94 (0.06 to 14.47) |
68 (1 study) |
⊕⊝⊝⊝ Verylowi,j,k | |
|
Adverse events (AEs; adverse drug reaction):
cardiac blocks (high‐degree atrioventricular block and bundle) Follow‐up: 24 hours |
152 per 1000 | 86 per 1000 (23 to 330) | RR 0.57 (0.15 to 2.18) | 68 (1 study) |
⊕⊝⊝⊝ Verylowj,k,l | |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in trial design or execution (high attrition bias). bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (7%). dAssumed risk was gotten from control group risk (4.1%). eDowngraded one level because of limitations in trial design. fDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). gAssumed risk was gotten from control group risk (7.9%). hAssumed risk was gotten from control group risk (2.6%). iDowngraded one level because of limitations in trial design or execution (high attrition bias). jDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). kAssumed risk was gotten from control group risk (3%). lAssumed risk was gotten from control group risk (15.2%).
Summary of findings 3. Lidocaine compared with tocainide for myocardial infarction.
| Lidocaine compared with tocainide for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: tocainide | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Tocainide | Lidocaine | |||||
| All‐cause mortality Follow‐up: 48 hours | 62 per 1000 | 77 per 1000 (5 to 1000) | RR 1.23 (0.08 to 17.83) | 29 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Cardiac mortality Follow‐up: 48 hours | 62 per 1000 | 77 per 1000 (5 to 1000) | RR 1.23 (0.08 to 17.83) | 29 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | Neither Keefe 1986 nor Rehnqvist 1983 assessed this outcome |
| Ventricular fibrillation | See comment | See comment | See comment | See comment | See comment | Keefe 1986 reported no participants with VF. Rehnqvist 1983 did not mention this outcome |
| Adverse events (AEs; adverse drug reaction): any adverse event | 444 per 1000 |
751 per 1000 (476 to 1000) |
RR 1.69 (1.07 to 2.68) | 69 (2 studies) | ⊕⊝⊝⊝ Very lowa,b,d | As the result of severe inconsistencies regarding reporting data on adverse events, we preferred to show the evidence using this approach |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in trial design and execution of trials. bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (6.3%). dAssumed risk was gotten from control group risk (44.4%).
Summary of findings 4. Lidocaine compared with mexiletine for myocardial infarction.
| Lidocaine compared with mexiletine for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: mexiletine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Mexiletine | Lidocaine | |||||
|
All‐cause mortality Follow‐up: 48 hours |
83 per 1000 | 28 per 1000 (1 to 621) | RR 0.33 (0.01 to 7.45) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
|
Cardiac mortality Follow‐up: 48 hours |
83 per 1000 | 28 per 1000 (1 to 621) | RR 0.33 (0.01 to 7.45) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | Neither Horowitz 1981 nor Rolli 1981 assessed this outcome |
|
Ventricular fibrillation Follow‐up: 48 hours |
See comment | See comment | RR 3.00 (0.13 to 67.06) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b | No events in the control group |
| Adverse events (AEs; adverse drug reaction): atrioventricular block Follow‐up: 48 hours | 83 per 1000 | 28 per 1000 (1 to 621) | RR 0.33 (0.01 to 7.45) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Adverse events (AEs; adverse drug reaction): composite neurological adverse event (confusion, vertigo, nystagmus, vomiting and diplopia) Follow‐up: between 3 hours and 48 hoursd | 459 per 1000 |
289 per 1000 (74 to 1000) |
RR 0.63 (0.16 to 2.47) | 74 (2 studies) | ⊕⊝⊝⊝ Very lowa,b,e | |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in the trial design. bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (8.3%). dHorowitz 1981 and Rolli 1981 used 'composite neurological adverse' terms for reporting this adverse event. eAssumed risk was gotten from control group risk (45.9%).
Summary of findings 5. Lidocaine compared with propafenone for myocardial infarction.
| Lidocaine compared with propafenone for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: propafenone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Propafenone | Lidocaine | |||||
| All‐cause mortality | See comment | See comment | See comment | See comment | See comment | No trial assessed this outcome |
| Cardiac mortality | See comment | See comment | See comment | See comment | See comment | No trial assessed this outcome |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | No trial assessed this outcome |
|
Ventricular fibrillation Follow‐up: 24 hours |
See comment | See comment | RR 3.00 (0.14 to 65.90) | 20 (1 study) | ⊕⊝⊝⊝ Very lowa,b | Control group had no event |
| Adverse events (AEs; adverse drug reaction): heart failure Follow‐up: 24 hours | See comment | See comment | RR 6.38 (0.32 to 127.77) | 64 (1 study) | ⊕⊝⊝⊝ Very lowa,b | Control group had no event |
| Adverse events (AEs; adverse drug reaction): bilateral bundle branch block Follow‐up: 24 hours | 28 per 1000 | 12 per 1000 (1 to 279) | RR 0.43 (0.02 to 10.06) | 64 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Adverse events (AEs; adverse drug reaction): neuropsychiatric disturbances Follow‐up: 24 hours | See comment | See comment | RR 6.95 (0.86 to 55.94) | 84 (2 studies) | ⊕⊝⊝⊝ Very lowa,b | Control group had no event |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in trial design and execution of the trial. bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (2.8%).
Summary of findings 6. Lidocaine compared with amiodarone for myocardial infarction.
| Lidocaine compared with amiodarone for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: amiodarone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Amiodarone | Lidocaine | |||||
|
All‐cause mortality Follow‐up: not stated |
See comment | See comment | See comment | See comment | See comment | Capucci 1985 did not assess this outcome |
| Cardiac mortality Follow‐up: not stated | See comment | See comment | See comment | See comment | See comment | Capucci 1985 did not assess this outcome |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | Capucci 1985 did not assess this outcome |
|
Ventricular fibrillation Follow‐up: not stated |
See comment | See comment | RR 3.44 (0.18 to 46.11) | 25 (1 study) | ⊕⊝⊝⊝ Very lowa,b | No ventricular fibrillation in control group |
| Bradycardia Follow‐up: not stated | 100 per 1000 | 23 per 1000 (1 to 512) | RR 0.23 (0.01 to 5.12) | 25 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Hypotension Follow‐up: not stated | 200 per 1000 | 28 per 1000 (2 to 520) | RR 0.14 (0.01 to 2.60) | 25 (1 study) | ⊕⊝⊝⊝ Very lowa,b,d | |
| Diplopia plus sleepiness Follow‐up: not stated | See comment | See comment | RR 2.06 (0.09 to 46.11) | 25 (1 study) | ⊕⊝⊝⊝ Very lowa,b | No diplopia in control group |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in trial design. bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (10%). dAssumed risk was gotten from control group risk (20%).
Summary of findings 7. Lidocaine compared with dimethylammonium for myocardial infarction.
| Lidocaine compared with dimethylammonium for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: dimethylammonium | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Dimethylammonium | Lidocaine | |||||
| All‐cause mortality Follow‐up: unclear | See comment | See comment | See comment | See comment | See comment | Bergdahl 1978 did not assess this outcome |
| Cardiac mortality Follow‐up: unclear | See comment | See comment | See comment | See comment | See comment | Bergdahl 1978 did not assess this outcome |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | Bergdahl 1978 did not assess this outcome |
| Ventricular fibrillation Follow‐up: unclear | See comment | See comment | See comment | See comment | See comment | Bergdahl 1978 did not assess this outcome |
| Hypotension Follow‐up: unclear | 312 per 1000 | 266 per 1000 (88 to 809) | RR 0.85 (0.28 to 2.59) | 31 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| Tachycardia Follow‐up: unclear | 500 per 1000 | 30 per 1000 (0 to 500) | RR 0.06 (0.00 to 1.0) | 31 (1 study) | ⊕⊝⊝⊝ Very lowa,b,d | |
| Bradycardia Follow‐up: unclear | 62 per 1000 | 22 per 1000 (1 to 505) | RR 0.35 (0.02 to 8.08) | 31 (1 study) | ⊕⊝⊝⊝ Very lowa,b,e | |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in trial design or execution (high attrition bias). bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (31.3%). dAssumed risk was gotten from control group risk (50%). eAssumed risk was gotten from control group risk (6.3%).
Summary of findings 8. Lidocaine compared with aprindine for myocardial infarction.
| Lidocaine compared with aprindine for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: aprindine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Aprindine | Lidocaine | |||||
|
All‐cause mortality Follow‐up: 3 days |
See comment | See comment | See comment | See comment | See comment | Depaepe 1974 did not assess this outcome |
| Cardiac mortality Follow‐up: 3 days | See comment | See comment | See comment | See comment | See comment | Depaepe 1974 did not assess this outcome |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | Depaepe 1974 did not assess this outcome |
| Ventricular fibrillation Follow‐up: 3 days | See comment | See comment | See comment | See comment | See comment | Depaepe 1974 did not mention this outcome |
| Coma Follow‐up: 3 days | See comment | See comment | RR 3.00 (0.13 to 67.06) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b | No coma in control group |
| Seizures Follow‐up: 3 days | See comment | See comment | RR 5.00 (0.27 to 94.34) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b | No seizures in control group |
| Agitation Follow‐up: 3 days | 167 per 1000 | 33 per 1000 (2 to 628) | RR 0.20 (0.01 to 3.77) | 24 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded one level because of limitations in trial design. bDowngraded two levels because of imprecision (small sample and very low number of events with an impact on the precision of effect estimates). cAssumed risk was gotten from control group risk (16.7%).
Summary of findings 9. Lidocaine compared with pirmenol for myocardial infarction.
| Lidocaine compared with pirmenol for myocardial infarction | ||||||
| Patient or population: patients with myocardial infarction Settings: in‐hospital Intervention: lidocaine Comparison: pirmenol | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo or no intervention | Lidocaine | |||||
|
All‐cause mortality Follow‐up: 24 hours |
See comment | See comment | See comment | See comment | See comment | Cuendet 1988 did not assess this outcome |
| Cardiac mortality Follow‐up: 24 hours | See comment | See comment | See comment | See comment | See comment | Cuendet 1988 did not assess this outcome |
| Overall survival at 30 days after myocardial infarction | See comment | See comment | See comment | See comment | See comment | Cuendet 1988 did not assess this outcome |
| Ventricular fibrillation Follow‐up: 24 hours. | See comment | See comment | See comment | See comment | See comment | Cuendet 1988 did not assess this outcome |
|
Safety (AEs; adverse drug reaction): any adverse event Follow‐up: 24 hours |
500 per 1000 |
555 per 1000 (235 to 1000) |
RR 1.11 (0.47 to 2.60) | 19 (1 studya) |
⊕⊝⊝⊝ Verylowb,c,d | |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aTrial includes participants with proven or unproven acute myocardial infarction. bDowngraded one level because of limitations in the trial design. cDowngraded two levels because of imprecision (very small sample size and very low number of events with an impact on the precision of effect estimates). dAssumed risk was gotten from control group risk (50%).
Background
See Appendix 1 for a medical and epidemiological glossary.
Description of the condition
Coronary artery disease is a major public health problem (Gaziano 2006; Leys 2001; Manson 1996; Watkins 2004) that affects both developed and developing countries (Braunwald 2001; Gaziano 2006). The burden of coronary artery disease depends on several modifiable and non‐modifiable risk factors and varies by geographical region (Alter 2008; Bainey 2009; Giannakoulas 2009; Goldenberg 2008; Gorter 2007; Goyal 2006; Kerr 2008; Lloyd‐Williams 2008; Steptoe 2007). The epidemiology of coronary artery disease has been reviewed widely, as have methods of prevention (Labarthe 1998; Manson 1996).
Acute coronary syndromes include unstable angina and myocardial infarction with or without ST‐segment elevation (electrocardiogram sector is higher than baseline) (Kolansky 2009). Acute myocardial infarction is the most important clinical entity of acute coronary syndromes; its definition is based on troponin elevation together with ischaemic symptoms, typical electrocardiogram changes or imaging evidence of loss of viable myocardium (Thygesen 2008). The epidemiology and burden of acute myocardial infarction have been described widely (al‐Adsani 2000; Cabadés 2007; Ljung 2006; Manson 1996; Pop 2004; Rich 2006; Roger 2007).
The most frequent complications of acute myocardial infarction are cardiac arrhythmias, conduction abnormalities and left ventricular systolic dysfunction (heart failure). Of these, ventricular arrhythmias are associated with the worst prognosis for people with acute myocardial infarction (Henkel 2006; Hreybe 2009; Khairy 2003; Piccini 2008; Rahimi 2006; Singla 2008; Velazquez 2004; Weir 2006; Wolfe 1991). Ventricular arrhythmias after myocardial infarction are associated with high risk of mortality (Henkel 2006). Between 3% and 10% of uncomplicated acute myocardial infarctions will be affected by ventricular fibrillation (Noneman 1978). Mortality is due mainly to sudden death, which is caused by acute ventricular tachyarrhythmia, often triggered by acute coronary events that may occur in persons without known cardiac disease or in association with structural heart disease (Bayés de Luna 1989Huikuri 2001). Ventricular fibrillation is the most frequent ventricular tachyarrhythmia, and it usually occurs secondary to ventricular tachyarrhythmia (Bayés de Luna 1989). The presence of arrhythmias in patients with acute myocardial infarction is associated with a poor prognosis (Volpi 1990).
In the era of thrombolysis, early or late primary ventricular fibrillation in patients with first acute myocardial infarction is an independent predictor of in‐hospital mortality (Volpi 1998). After adjustment for other variables, patients with ventricular arrhythmias continue to be at significantly higher risk of 30‐day or one‐year mortality, both of which are significantly increased in patients with sustained ventricular fibrillation or ventricular tachyarrhythmia after myocardial infarction as compared with patients without these arrhythmias (Al‐Khatib 2003).
Although different schemes have been used to classify death according to presumed mechanisms, considerable evidence shows that between one‐quarter and one‐half of cardiac deaths are sudden and are due to arrhythmia (Crystal 2003; Gardner 2000; Goldstein 1986; Koplan 2009; Kuch 2009; Myerburg 1986; Solomon 2005). Thus prevention of sudden death is an important clinical goal (Crystal 2003).
Description of the intervention
Since the 1950s, lidocaine, a local anaesthetic, has been used to control ventricular arrhythmias associated with myocardial infarction and cardiac surgery (Hitchcock 1959). For decades, lidocaine was used as a standard intravenous antiarrhythmic agent to prevent complications such as ventricular tachyarrhythmia and ventricular fibrillation after myocardial infarction (Harrison 1989). The dosage of lidocaine therapy was established by Aps et al., who recommended "a bolus (75‐100 mg) followed by 4 mg/min for 30 minutes, 2 mg/min for two hours, and 1 mg/min thereafter" for patients affected with uncomplicated myocardial infarction (Aps 1976). Asystole is associated with lidocaine use (Applebaum 1986; Hill 1973; Manyari‐Ortega 1978; Sadikot 1997), although evidence shows that use of lidocaine may not be associated with increased mortality rates (Alexander 1999). Lidocaine does not interact with the autonomic nervous system (Anderson 1984) but causes toxicity of the central nervous system (seizures, tremor, dysarthria, altered levels of consciousness and nystagmus), some of which is associated with high blood levels of lidocaine (Brunton 2008). Lidocaine is also known as lignocaine, but in this review, we use the name lidocaine.
Lidocaine is not used much anymore in high‐income countries, but it continues to be used in many low‐income countries (Reyes Caorsi 2006), and it is recommended in guidelines on management of patients with myocardial infarction (Anonimous 2006) and patients with ventricular arrhythmias and the prevention of sudden cardiac death (Zipes 2006). Furthermore, many published studies were conducted to explore this issue (Piccini 2011; Tagawa 2008; Takaya 2009).
How the intervention might work
Lidocaine is an antiarrhythmic drug of type IB Vaugham‐Williams classification that works by inhibiting rapid sodium channels (a characteristic effect of this class of drugs) (Brunton 2008; Collinsworth 1974). Details of the electrophysiological effects of lidocaine on the heart are presented by Collinsworth 1974. These effects, which were observed in animal studies, briefly include the following: decreased automaticity of pacemaker tissue and sinoatrial node, increased ventricular fibrillation threshold and increased atrioventricular node conduction time according to dosage (Collinsworth 1974). The antiarrhythmic mechanism and efficacy of lidocaine are related to extracellular potassium concentration (Collinsworth 1974). Lidocaine may affect sinus node conduction or function (Klein 1975; Lippestad 1971).
Why it is important to do this review
Numerous randomised controlled trials were conducted to assess the clinical effectiveness and safety of lidocaine in preventing ventricular tachyarrhythmia and ventricular fibrillation among patients with myocardial infarction. In general, lidocaine used to reduce rates of ventricular tachyarrhythmia and ventricular fibrillation is beneficial but is associated with adverse effects (hypotension, neurological complications and other problems) that might be related to dosage. However, five systematic reviews used meta‐analysis to verify no evidence of benefit in reducing the mortality rate (De Silva 1981; Hine 1989; MacMahon 1988; Sadowski 1999; Teo 1993). These reviews, published between 1981 and 1999, are now more than 10 years out of date. An update of the evidence is required for the following reasons.
Reviews consistently reported high clinical heterogeneity between trials but did not report tools used to assess risk of bias (De Silva 1981; Hine 1989; MacMahon 1988; Sadowski 1999; Teo 1993).
Currently, the I2 statistic is the favoured method for assessing statistical heterogeneity (Higgins 2003), although previously the Chi2 test was applied (De Silva 1981; Hine 1989; MacMahon 1988; Sadowski 1999; Teo 1993).
Different summary measures such as Peto odds ratios (MacMahon 1988), odds ratios (Sadowski 1999) and risk differences (Hine 1989) showed no significant differences between non‐surrogate clinical outcomes such as death. Use of risk ratio as a summary statistic for meta‐analysis with binary data may have revealed significant differences in mortality (Deeks 2002). Only De Silva 1981 used risk ratios, but the endpoint was incidence of ventricular fibrillation.
Systematic reviews did not conduct sensitivity analyses (trials with low risk of bias vs trials with high risk of bias).
Overall, the main outcome was a surrogate marker: ventricular extrasystole/ventricular fibrillation. Although choosing a surrogate marker is not strictly inappropriate, this is not currently recommended (Schünemann 2009).
We did not include trials comparing lidocaine versus any other antiarrhythmic drug. These trials should be included because indirectness is a reason for reducing confidence in the evidence (Guyatt 2008).
Hine 1989 excluded trials that were not published in the English language. This decision may have led to oversampling of statistically significant studies (i.e. language bias) (Borenstein 2009).
MacMahon 1988 and Teo 1993 did not include trials of cross‐over design. De Silva 1981, Hine 1989 and Sadowski 1999 included cross‐over trials; however, they did not report the methods used for analysis.
In addition, lidocaine is used in low‐income countries (Anonimous 2006; Reyes Caorsi 2006).
In conclusion, the evidence is out of date and considerable uncertainty remains about the effects of prophylactic lidocaine use in all‐cause mortality, in particular, in patients with suspected acute myocardial infarction. This Cochrane review seeks to update current knowledge and resolve uncertainties. The research question is this: "What is the clinical effectiveness and safety of prophylactic lidocaine for preventing death in people with acute myocardial infarction?"
Objectives
To determine the clinical effectiveness and safety of prophylactic lidocaine in preventing death among people with acute myocardial infarction.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials irrespective of design (parallel and cross‐over) or publication status (unpublished or published as an article, an abstract or a letter). We applied no language, country or sample size limitations and included trials conducted in a hospital or community setting, or both. We also applied no limits with respect to period of follow‐up, pre‐hospital or in‐hospital setting, lidocaine use or bolus with or without infusion.
Types of participants
Adults (≥ 18 years) with acute myocardial infarction. We applied no restrictions by definition of acute myocardial infarction.
Types of interventions
As acute myocardial infarction requires different medical and non‐medical treatments (i.e. primary intervention), lidocaine is considered a complementary intervention. Thus, for the purpose of this review, eligible trials compared the same primary interventions with and without lidocaine.
Intervention
Lidocaine. We applied no restrictions by route of administration (intravenous, intra‐muscular or both) or dose.
Comparison
Placebo. Standard care or antiarrhythmic drug alone or in any combination.
Types of outcome measures
Primary outcomes
All‐cause mortality.
Cardiac mortality.
Overall survival at 30 days after myocardial infarction (MI), which was defined as the proportion of survivors in a group. The proportion of persons in a specified group alive at the beginning of the time interval who survive to the end of the interval (Porta 2008).
Secondary outcomes
Ventricular fibrillation: assessed by counting how many participants developed this arrhythmia.
Adverse events: numbers and types of adverse events defined as any untoward medical occurrences not necessarily having a causal relationship with treatment. We reported separately on adverse events that led to treatment discontinuation and those that did not lead to treatment discontinuation. We defined a serious adverse event according to the International Conference on Harmonisation (ICH) Guidelines (ICH‐GCP 1997) as any event that at any dose resulted in death, was life‐threatening, required in‐patient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability or was a congenital anomaly/birth defect, and any important medical event that may have jeopardised the patient or required intervention to prevent it. We considered all other adverse events as non‐serious.
Search methods for identification of studies
Electronic searches
We searched the following electronic databases to find reports of relevant randomised controlled trials.
Cochrane Central Register of Controlled Trials (CENTRAL) (2015, Issue 3 of 12).
MEDLINE (Ovid, 1946 to Week 1 April 2015).
EMBASE Classic and EMBASE (Ovid, 1947 to 10 April 2015).
Latin American Caribbean Health Sciences Literature (LILACS) (13 April 2015).
Web of Science (Thomson Reuters, 1970 to 13 April 2015).
We used Cochrane sensitive‐maximising RCT filters to search MEDLINE and EMBASE (Lefebvre 2011). Appendix 2 shows the search strategies.
Searching other resources
We searched the Clinical Trials Search Portal of the World Health Organization (http://apps.who.int/trialsearch/) for ongoing and unpublished trials.
We also searched the following websites.
We checked the reference lists of all trials identified by the above methods, and we imposed no language restrictions.
Data collection and analysis
We summarised data using standard methodologies of The Cochrane Collaboration (Higgins 2011).
Selection of studies
Two review authors (AM‐C, DS‐R) independently assessed each reference identified by the search against the inclusion criteria. Through discussion, we resolved disagreements that arose. We retrieved in full references that appeared to meet the inclusion criteria for further independent assessment by two review authors.
Data extraction and management
One review author (AM‐C) independently extracted data from included trials using a spreadsheet data extraction form; the other review author (DS‐R) checked entered data for accuracy. We extracted the following data: eligibility criteria, demographics (age, sex, country), characteristics of included patients (treatment setting, lidocaine use (dosage, administration route)), types of control comparison treatments and outcomes. We discussed discrepancies between review authors to reach final consensus and used a pre‐formed sheet (Zavala 2006).
Assessment of risk of bias in included studies
Three review authors (AM‐C, DS‐R, VA) independently assessed the risk of bias of each included trial using the domain‐based evaluation as described in the Cochrane Handbook for Systematic Reviews of Interventions, Section 5.1.0 (Higgins 2011; Lundh 2012; Savović 2012; Wood 2008). Two review authors (of AM‐C, DS‐R, VA) checked the assessment. Review authors discussed discrepancies and achieved consensus.
The definition of each classification is given below.
Generation of allocation sequence
Low risk of bias: if the allocation sequence was generated by a computer or a random number table, drawing of lots, tossing of a coin, shuffling of cards or throwing dice.
Unclear risk of bias: if the trial was described as randomised but the method used for allocation sequence generation was not described.
High risk of bias: if a system involving dates, names or admittance numbers was used for allocation of participants. These studies are known as quasi‐randomised and were excluded from the review when beneficial effects were assessed.
Allocation concealment
Low risk of bias: if allocation of participants involved a central independent unit, an on‐site locked computer, identical‐appearing numbered drug bottles or containers prepared by an independent pharmacist or investigator, or sealed envelopes.
Unclear risk of bias: if the trial was described as randomised but the method used to conceal the allocation was not described.
High risk of bias: if the allocation sequence was known to investigators who assigned participants, or if the study was quasi‐randomised. The latter studies were excluded from the review when beneficial effects were assessed.
Blinding (or masking)
We assessed each trial (as low, unclear or high risk) with regard to the following levels of blinding.
Blinding of clinician (person delivering treatment) to treatment allocation.
Blinding of participant to treatment allocation.
Blinding of outcome assessor to treatment allocation.
Incomplete outcome data
Low risk of bias: if numbers of and reasons for dropouts and withdrawals in all intervention groups were described, or if it was specified that no dropouts or withdrawals occurred.
Unclear risk of bias: if the report gave the impression that no dropouts or withdrawals occurred, but this was not specifically stated.
High risk of bias: if numbers of or reasons for dropouts and withdrawals were not described.
We further examined the percentage of dropouts overall in each trial and per randomisation arm, and we evaluated from published information whether intention‐to‐treat analysis was performed or could be performed.
Selective outcome reporting
Low risk of bias: if pre‐defined or clinically relevant and reasonably expected outcomes were reported on.
Unclear risk of bias: if not all pre‐defined or clinically relevant and reasonably expected outcomes were reported on or were not reported on fully, or if it was unclear whether data on these outcomes were recorded.
High risk of bias: if one or more clinically relevant and reasonably expected outcomes were not reported on; data on these outcomes were likely to have been recorded.
Other bias
Low risk of bias: if the trial appeared to be free of other components that could put it at risk of bias.
Unclear risk of bias: if the trial may or may not be free of other components that could put it at risk of bias.
High risk of bias: if other factors in the trial could put it at risk of bias.
Overall risk of bias
We made explicit judgements about whether randomised controlled trials were at high risk of bias, according to the criteria given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We assessed risk of bias as high if any of the above domains were unclear or had high risk of bias.
Trials that showed adequate generation of allocation sequence, allocation concealment, blinding and handling of incomplete outcome data, and no selective outcome reporting, and that were without other risks of bias were considered trials with low risk of bias. We explored the impact of the risk of bias by undertaking subgroup analyses.
One review author (AM‐C) entered the information using RevMan 2011 software. Two review authors (DS‐R, VA) checked the entered data.
Measures of treatment effect
For each binary outcome such as all‐cause mortality, cardiac mortality, ventricular fibrillation, adverse events and adverse drug reactions, we calculated the risk ratio (RR) with 95% confidence interval (CI).
We would have attempted to assess time‐to‐event outcomes and overall survival at 30 days by using the hazard ratio (HR) with 95% CI if included trials had reported this outcome. This will be done in future updates if trials report this outcome.
We planned to include cross‐over trials, but none were available. If cross‐over trials become available in the future, we will use the inverse variance method to pool data from these trials and will apply the Becker‐Balagtas marginal estimated odds ratio to summarise ventricular fibrillation (Elbourne 2002).
Dealing with missing data
We assessed the percentage of dropouts for each included trial and for each intervention group, and we evaluated whether an intention‐to‐treat (ITT) analysis had been performed or could have been performed from available published information.
To undertake an ITT analysis, we sought data from trial authors on numbers of participants in treatment groups, irrespective of compliance and whether or not participants were later thought to be ineligible or otherwise excluded from treatment or lost to follow‐up. If this information was not forthcoming, we undertook a complete participant analysis, knowing that it may be biased.
Assessment of heterogeneity
We quantified statistical heterogeneity using the I2 statistic, which describes the percentage of total variation across trials due to heterogeneity rather than to sampling error (Higgins 2003). We considered statistical heterogeneity to be present if I2 was greater than 50% (Higgins 2011).
Assessment of reporting biases
We assessed publication bias and other bias by using a funnel plot (Sterne 2011). We assessed publication bias for all‐cause mortality and ventricular fibrillation using Comprehensive Meta‐Analysis software (CMA 2005).
Data synthesis
We used random‐effects methods to determine pool estimates and 95% confidence intervals.
Trial sequential analysis
Meta‐analysis of cumulative data may run the risk of random errors ('play of chance') due to sparse data and repetitive analyses of cumulative data (Brok 2008; Brok 2009; Thorlund 2009; Thorlund 2010; Thorlund 2011a; Wetterslev 2008; Wetterslev 2009). To assess risks of random error in our cumulative meta‐analyses, we conducted diversity‐adjusted trial sequential analyses based on the proportion with the outcome in the control group; an a priori set relative risk reduction of 20%; alpha of 5% and beta of 20%; and squared diversity in the meta‐analysis (CTU 2011; Thorlund 2009; Thorlund 2011b).
Subgroup analysis and investigation of heterogeneity
Despite statistical heterogeneity less than 50% for primary outcomes, we conducted the following pre‐planned subgroup analyses.
Route of administration of lidocaine (intravenous vs intramuscular).
Pre‐hospital setting lidocaine use versus In‐hospital setting lidocaine use.
Doses of lidocaine.
We were not able to conduct subgroup analysis by age and gender; congestive heart failure, cardiogenic shock or bradycardia/atrioventricular block before randomisation.
We performed subgroup analyses for primary outcomes.
We had planned to conduct meta‐regression analyses. However, we did not use this approach because we found low statistical heterogeneity in meta‐analyses for primary outcomes.
Furthermore, we conducted the following post hoc subgroup analyses (undertaken after results of the studies had been compiled).
Acute myocardial infarction patients only.
Trials without suspicion of industry bias versus trials with suspicion of industry bias.
Sensitivity analysis
We would have used the following procedures (and will apply these in future updates, if possible) in conducting sensitivity analysis to compare trials having 'low risk of bias' versus trials having 'high risk of bias' (Higgins 2011). As all included trials were rated as having high risk of bias, we were not able to conduct sensitivity analysis as planned.
We conducted a sensitivity analysis in the following way.
Repeating the analysis while taking attrition bias into consideration (best‐worst case scenario and worst‐best case scenario).
'Summary of findings' tables
We used the principles of the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) system (Guyatt 2008; Guyatt 2008b) in our review to assess the quality of the body of evidence associated with specific outcomes (all‐cause mortality, cardiac mortality, overall survival at 30 days after myocardial infarction, ventricular fibrillation, adverse events) and constructed a 'Summary of findings (SoF)' table using GRADE software. The GRADE approach is used to appraise the quality of a body of evidence according to the extent to which one can be confident that an estimate of effect or association reflects the item being assessed. Assessment of the quality of a body of evidence considers within‐study risk of bias (methodological quality), directness of the evidence, heterogeneity of the data, precision of effect estimates and risk of publication bias.
Results
Description of studies
Results of the search
We identified 1230 references by using our search strategies. Thirty‐seven trials (45 references) involving 11,948 participants met our inclusion criteria (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988). See Figure 1 for details.
1.
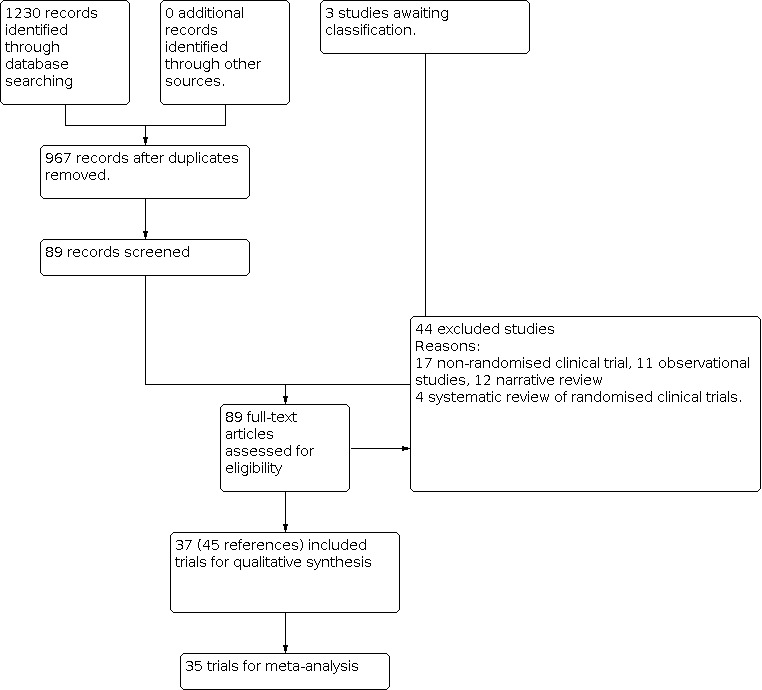
Study flow diagram.
Included studies
Tables of Characteristics of included studies show detailed descriptions of the studies.
Lidocaine and populations assessed in included trials
The 37 randomised controlled trials reported comparisons between lidocaine and several different control interventions.
Twenty‐four trials compared lidocaine versus placebo (without or with co‐interventions). Comparisons included saline solution (Chopra 1971; Dunn 1985; Lie 1978; NNLIT 1992; Rossi 1976; Sandler 1976; Valentine 1974; Wennerblom 1982) and 5% dextrose solution (Baker 1971; Kostuk 1969; Lie 1974; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987). Characteristics of controls were not sufficiently described in ALIT 1985; Bennett 1970; Darby 1972; Hargarten 1990; Kuck 1985; Rademaker 1986; Sadowski 1999; Solimene 1983; and Wyse 1988.
Thirteen trials compared lidocaine versus another antiarrhythmic drug (with or without co‐interventions). Comparisons included disopyramide (Horowitz 1981; Pedersen 1986; Ronnevik 1987; Sbarbaro 1979), amiodarone (Capucci 1985), dimethylammonium chloride (Bergdahl 1978), pirmenol (Cuendet 1988), mexiletine (Rolli 1981), aprindine (Depaepe 1974), propafenone (Rehnqvist 1984; Touboul 1988) and tocainide (Keefe 1986; Rehnqvist 1983).
Co‐interventions used most often in experimental and control groups were lidocaine (Baker 1971; Horowitz 1981; Pharand 1995; Pitt 1971), oxygen (Bergdahl 1978; Keefe 1986), hydromorphone or pentazocine (Bergdahl 1978), defibrillation (Capucci 1985; Lie 1974; NNLIT 1992; Valentine 1974), electroversion (Horowitz 1981; Solimene 1983), mexiletine (Horowitz 1981), pacemaker (Pitt 1971), subcutaneous heparin (Keefe 1986; Poprawski 1987), nitroglycerin (Keefe 1986; Poprawski 1987; Sadowski 1999), morphine sulphate (Keefe 1986), furosemide (Keefe 1986; Ronnevik 1987), intracoronary thrombolysis (Kuck 1985; NNLIT 1992; Sadowski 1999), digitalis (Ronnevik 1987) and atropine (Sandler 1976; Wennerblom 1982). Twenty trials did not report use of a co‐intervention (ALIT 1985; Bennett 1970; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Kostuk 1969; Lie 1978; O'Brien 1973; Pedersen 1986; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Rossi 1976; Sbarbaro 1979; Touboul 1988; Wyse 1988).
Twenty‐six trials used intravenous lidocaine (Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Depaepe 1974; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Sadowski 1999; Sbarbaro 1979; Solimene 1983; Wyse 1988, six used intramuscular lidocaine (ALIT 1985; Lie 1978; Rossi 1976; Sandler 1976; Valentine 1974; Wennerblom 1982) and three used intravenous and intramuscular lidocaine (Darby 1972; Dunn 1985; NNLIT 1992). Two studies used intravenous and oral routes (Rehnqvist 1983; Touboul 1988).
The follow‐up period varied between trials: one hour (ALIT 1985; Lie 1978), two hours (Sbarbaro 1979), three hours (Chopra 1971; Dunn 1985; NNLIT 1992; Wennerblom 1982), 12 hours (Pedersen 1986; Solimene 1983), 24 hours (Cuendet 1988; Rehnqvist 1983; Rehnqvist 1984; Ronnevik 1987; Touboul 1988; Wyse 1988), 48 hours (Baker 1971; Bennett 1970; Darby 1972; Horowitz 1981; Keefe 1986; Kostuk 1969; Lie 1974; O'Brien 1973; Pharand 1995; Pitt 1971; Rademaker 1986; Sadowski 1999), 72 hours (Depaepe 1974), 504 hours (Rossi 1976) and 720 hours (Valentine 1974). Four trials did not report follow‐up (Capucci 1985; Hargarten 1990; Poprawski 1987; Sandler 1976), and for three trials, the follow‐up period was unclear (Bergdahl 1978; Kuck 1985; Rolli 1981).
Diagnostic criteria for myocardial infarction varied among included trials. Five trials used World Health Organization criteria (Baker 1971; Poprawski 1987; Rolli 1981; Sandler 1976; Valentine 1974); nine trials used Lawrie's criteria (ALIT 1985; Bennett 1970; Capucci 1985; Dunn 1985; Kuck 1985; Pedersen 1986; Rehnqvist 1983; Sadowski 1999; Wennerblom 1982); and 15 trials used clinical signs, electrocardiograms and laboratory enzymes alone or in combination (Chopra 1971; Depaepe 1974; Hargarten 1990; Keefe 1986; Lie 1974; Lie 1978; Pharand 1995; Pitt 1971; Rademaker 1986; Rehnqvist 1984; Ronnevik 1987; Rossi 1976; Solimene 1983; Touboul 1988; Wyse 1988). Eight trials had unclear diagnostic criteria or did not report them (Bergdahl 1978; Cuendet 1988; Darby 1972; Horowitz 1981; Kostuk 1969; NNLIT 1992; O'Brien 1973; Sbarbaro 1979).
Included trials were conducted in participants with proved or suspected myocardial infarction. Seventeen trials included participants with confirmed acute myocardial infarction (Baker 1971; Bennett 1970; Capucci 1985; Chopra 1971; Darby 1972; Depaepe 1974; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; O'Brien 1973; Pitt 1971; Rehnqvist 1984; Rossi 1976; Solimene 1983; Touboul 1988), and 20 trials included participants with acute myocardial infarction or suspected acute myocardial infarction (ALIT 1985; Bergdahl 1978; Cuendet 1988; Dunn 1985; Hargarten 1990; Horowitz 1981; NNLIT 1992; Pedersen 1986; Pharand 1995; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rolli 1981; Ronnevik 1987; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Valentine 1974; Wennerblom 1982; Wyse 1988).
Three trials were conducted in a pre‐hospital setting (ALIT 1985; NNLIT 1992; Wennerblom 1982), 31 in a hospital setting (Baker 1971; Bergdahl 1978; Depaepe 1974; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Valentine 1974; Wyse 1988) and three in both settings (Bennett 1970; Dunn 1985; Hargarten 1990).
Thirty‐one trials reported participants' age. Overall, the mean age of participants was older than 50 years (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Sadowski 1999; Sandler 1976; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988). Six trials did not report participants' age (Darby 1972; Kostuk 1969; O'Brien 1973; Rossi 1976; Sbarbaro 1979; Solimene 1983). Thirty‐one trials reported the percentage of included male participants, which was 75.06 ± 11.58 (minimum 50, maximum 95, median 76) (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Sadowski 1999; Sandler 1976; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988), and six trials did not report the gender of participants (Kostuk 1969; O'Brien 1973; Pedersen 1986; Rossi 1976; Sbarbaro 1979; Solimene 1983).
Trial locations
Included trials were conducted between 1969 and 1999 in 17 countries: three in Australia (Horowitz 1981; Pitt 1971; Valentine 1974), one in Belgium (Depaepe 1974), one in Brazil (Solimene 1983), four in Canada (Chopra 1971; Kostuk 1969; Rademaker 1986; Wyse 1988), one in Denmark (Pedersen 1986), one in France (Touboul 1988), one in Germany (Kuck 1985), three in Italy (Capucci 1985; Rolli 1981; Rossi 1976), one in New Zealand (O'Brien 1973), one in Northern Ireland (Dunn 1985), two in Norway (NNLIT 1992; Ronnevik 1987), two in Poland (Poprawski 1987; Sadowski 1999), four in Sweden (Bergdahl 1978; Rehnqvist 1983; Rehnqvist 1984; Wennerblom 1982), one in Switzerland (Cuendet 1988), three in The Netherlands (ALIT 1985; Lie 1974; Lie 1978), four in the United Kingdom (Baker 1971; Bennett 1970; Darby 1972; Sandler 1976) and four in the United States of America (Hargarten 1990; Keefe 1986; Pharand 1995; Sbarbaro 1979).
Trial methods
The mean sample size was 357.22 ± 994.08 (minimum 19, maximum 6024, median 150). One trial reported sample size estimation a priori (NNLIT 1992). Thirty‐six trials were conducted without sample size estimated a priori (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988).
Thirty‐six trials used a parallel study design (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Solimene 1983; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988). Thirty‐two trials used two comparison groups (ALIT 1985; Baker 1971; Bergdahl 1978; Capucci 1985; Chopra 1971; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Solimene 1983; Valentine 1974; Wennerblom 1982; Wyse 1988), two trials three comparison groups (Bennett 1970; Touboul 1988) and two trials four comparison groups (Sandler 1976; Sbarbaro 1979). One trial had a cross‐over design (Sbarbaro 1979).
Excluded studies
We excluded 43 studies for the following reasons: non‐randomised controlled trials (Bernard 1972; Bertini 1993; Bleifeld 1973; Church 1972; Diederich 1979; Fehmers 1972; Garratt 1998; Gonzalez 1977; Leone 1991; Miller 1973; Mogensen 1971; Riabokon' 1980; Ryden 1973; Singh 1976; Szeplaki 1973; Szeplaki 1976; Wojtala 1982), systematic reviews of randomised controlled trials (De Silva 1981; Hine 1989; MacMahon 1988; Teo 1993), observational studies (Beloev 1983; Campbell 1978; Destuelles 1969; Egre 1981; Gianelly 1967; Mazur 1982; Pentecost 1981; Ruano 1989; Shih 1995; Wyman 2004) and narrative reviews (Antman 1992; Bernard 1970; Campbell 1980; Campbell 1983; Formichev 1995; Goodman 1979; Iosava 1982; Jaffe 1992; Lechleitner 1987; Noneman 1978; Oltmanns 1979; Ribner 1979). See the Characteristics of excluded studies table.
Studies awaiting classification
Three references were considered as 'Studies awaiting classification' (Bolinska 1971; Hopperstead 1980; Knight 1973). See Characteristics of studies awaiting classification for details. These three studies lacked an abstract indicating whether they were randomised trials. We were not able to find the addresses of study authors and were not able to find their full‐text articles.
Risk of bias in included studies
See Figure 2 and Figure 3 for details.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
Risk of bias arising from the method of generation of the allocation sequence was considered low in three trials (Depaepe 1974; Hargarten 1990; Sbarbaro 1979). Thirty‐four studies had an unclear risk of bias for this domain (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Dunn 1985; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Solimene 1983; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988).
Allocation concealment
Risk of bias arising from the method of allocation concealment was considered low in one trial (Valentine 1974). Thirty‐six trials had unclear risk of bias for this domain (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Wennerblom 1982; Wyse 1988).
Blinding
Risk of bias due to lack of blinding of participants and personnel was rated as low in seven trials (Kostuk 1969; Lie 1974; Lie 1978; NNLIT 1992; Rossi 1976; Valentine 1974; Wennerblom 1982). Risk of bias of blinding was high in 30 trials (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kuck 1985; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Wyse 1988).
In two trials, outcome assessment was clearly reported as blinded and detection bias was considered low (ALIT 1985; Wennerblom 1982). Blinding was unclear or was not performed in 35 trials and risk of detection bias was considered high (Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Valentine 1974; Wyse 1988).
Incomplete outcome data
Risk of attrition bias was rated as low in nine trials (Bennett 1970; Darby 1972; Depaepe 1974; Dunn 1985; Horowitz 1981; Lie 1974; NNLIT 1992; Pedersen 1986; Wyse 1988). Risk of attrition bias was rated as high in 28 trials (ALIT 1985; Baker 1971; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Hargarten 1990; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1978; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Valentine 1974; Wennerblom 1982).
Selective reporting
Risk of selective outcome reporting bias was rated as low in 22 trials (ALIT 1985; Baker 1971; Bennett 1970; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Touboul 1988; Valentine 1974; Wennerblom 1982) and high in 15 trials (Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Kostuk 1969; Kuck 1985; Lie 1974; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Sandler 1976; Sbarbaro 1979; Solimene 1983; Wyse 1988).
Other potential sources of bias
Risk of other bias was rated as high in all trials because of bias in presentation of data, design bias or industry bias (ALIT 1985; Baker 1971; Bennett 1970; Bergdahl 1978; Capucci 1985; Chopra 1971; Cuendet 1988; Darby 1972; Depaepe 1974; Dunn 1985; Hargarten 1990; Horowitz 1981; Keefe 1986; Kostuk 1969; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pedersen 1986; Pharand 1995; Pitt 1971; Poprawski 1987; Rademaker 1986; Rehnqvist 1983; Rehnqvist 1984; Rolli 1981; Ronnevik 1987; Rossi 1976; Sadowski 1999; Sandler 1976; Sbarbaro 1979; Solimene 1983; Touboul 1988; Valentine 1974; Wennerblom 1982; Wyse 1988).
Ten trials had potential industry bias (Bennett 1970; Bergdahl 1978; Chopra 1971; Depaepe 1974; Keefe 1986; Lie 1974; Pitt 1971; Rademaker 1986; Rossi 1976; Sbarbaro 1979).
Accordingly, all trials were considered as having high risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9
Primary outcomes
All‐cause mortality
Lidocaine versus placebo or no intervention
Meta‐analysis of 18 trials involving participants with proven or non‐proven acute myocardial infarction, comparing lidocaine versus placebo or no intervention, showed no significant differences in all‐cause mortality (213/5879 (3.62%) vs 199/5848 (3.40%); RR 1.02, 95% CI 0.82 to 1.27; participants = 11727; I2 = 15%; P value = 0.86; low‐quality evidence) (ALIT 1985; Baker 1971; Bennett 1970; Chopra 1971; Darby 1972; Dunn 1985; Hargarten 1990; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987; Rossi 1976; Sadowski 1999; Valentine 1974; Wennerblom 1982). See Analysis 1.1. A funnel plot revealed no evidence of publication bias for this outcome (Figure 4). Trial sequential analysis shows that 14 trials provided evidence that lidocaine is not able to induce a 20% RR reduction in all‐cause mortality compared with placebo or no intervention, if we disregard risks of bias (Figure 5).
1.1. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 1: All‐cause mortality (participants with proven or non‐proven acute myocardial infarction)
4.
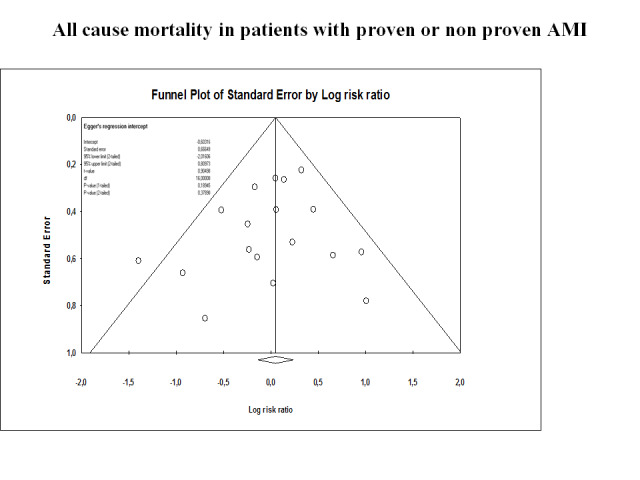
Funnel plot on all‐cause mortality in 18 lidocaine vs placebo or no intervention trials
Funnel plot of data from the meta‐analysis evaluating the effects of lidocaine compared with placebo for preventing all‐cause mortality in patients with proven or not proven acute myocardial infarction (18 trials). This figure shows low risk of publication bias. Individual circles represent point estimates of the included randomised controlled trials. The pattern of distribution simulates an inverted funnel. Larger trials are closer and upper to the pooled estimate. Effect sizes of smaller trials are lower and are more or less symmetrically distributed around the pooled estimate.
5.

Trial sequential analysis on all‐cause mortality in 18 lidocaine vs placebo or no intervention trials
Trial sequential analysis of lidocaine vs placebo or no intervention on all‐cause mortality in participants with or without proven myocardial infarction based on the diversity‐adjusted required information size (DARIS) of 25,777 participants. This DARIS was calculated on the basis of a proportion of participants with suspected myocardial infarction of 3.40% in the control group; RRR of 20% in the experimental intervention group; alpha (α) of 5%; beta (β) of 20%; and diversity of 22%.
The cumulative Z‐curve (blue line) did not cross the conventional alpha 5% boundaries (green lines) at any time. After the 14th trial, the cumulative Z‐curve crosses the trial sequential monitoring boundary for futility. Accordingly, although only 45.5% (11,727/25,777) of the DARIS has been obtained, we can reject an intervention effect of 20% or larger. This implies that no additional trials may be needed to disprove an intervention effect of 20% relative risk reduction if bias can be ignored. Smaller risk reductions may require additional trials with larger sample sizes.
Heterogeneity for this critical endpoint was low, as conveyed by I2 values. However, because of the large number of trials and the importance of determining the effect of prophylactic lidocaine on all‐cause mortality in individuals with proven acute myocardial infarction, we conducted many subgroup analyses for this population.
Subgroup analyses involving acute myocardial infarction patients only
Meta‐analysis of 16 trials comparing lidocaine versus placebo or no intervention revealed no differences regarding all‐cause mortality (148/2747 (5.38%) vs 135/2506 (5.38%); RR 1.01, 95% CI 0.79 to 1.30; participants = 5253; I2 = 9%; P value = 0.92) (ALIT 1985; Baker 1971; Bennett 1970; Chopra 1971; Darby 1972; Dunn 1985; Hargarten 1990; Lie 1974; Lie 1978; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987; Rossi 1976; Valentine 1974; Wennerblom 1982). See Analysis 1.2. Figure 6 shows no evidence of publication bias outcome.
1.2. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 2: All‐cause mortality (subgroup analysis by acute myocardial infarction‐only participants)
6.
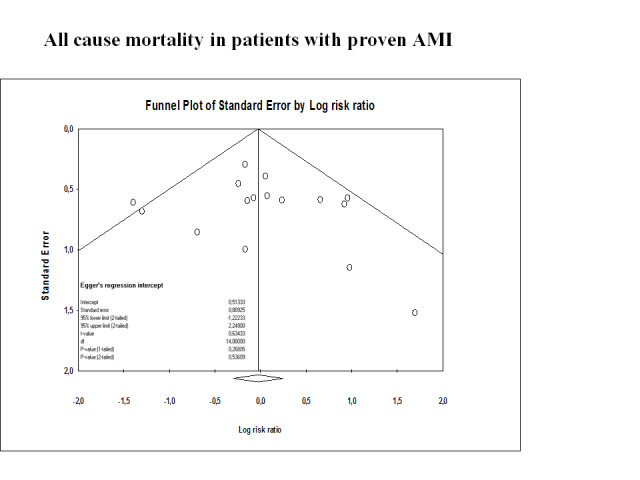
Funnel plot on all‐cause mortality in participants with proven acute myocardial infarction in 16 lidocaine vs placebo or no intervention trials
Funnel plot of data from the meta‐analysis of effects of lidocaine compared with placebo for preventing all‐cause mortality in individuals with proven acute myocardial infarction (16 trials). This figure shows low risk of publication bias. The circles show point estimates of the included randomised controlled trials. The pattern of distribution simulates an inverted funnel. Each half of the funnel plot includes eight trials. Larger trials are closer and upper to the pooled estimate. Effect sizes of the smaller trials are lower and are more or less symmetrically distributed around the pooled estimate. The right half of the funnel plot (near the bottom corner) shows two smaller trials with higher standard error and far of the point estimate.
Trial sequential analysis shows that 13 trials provided evidence that lidocaine is not able to induce a 25% RR reduction in all‐cause mortality among participants with myocardial infarction compared with placebo or no intervention, if we disregard risks of bias (Figure 7).
7.
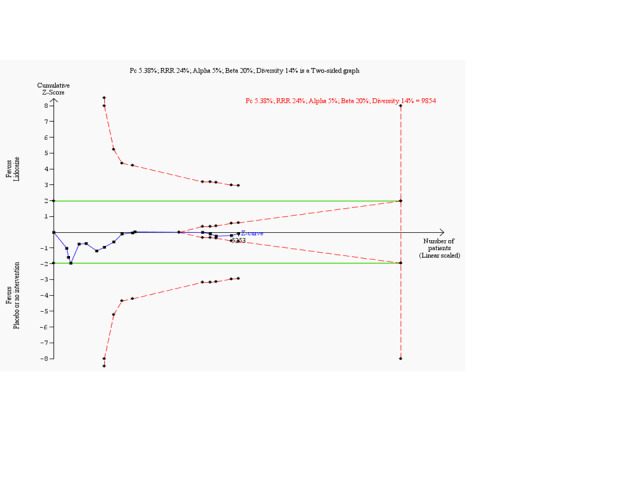
Trial sequential analysis on all‐cause mortality among participants with myocardial infarction in 16 lidocaine vs placebo or no intervention trials
Trial sequential analysis of lidocaine vs placebo or no intervention on all‐cause mortality in participants with myocardial infarction based on the diversity‐adjusted required information size (DARIS) of 9854 participants. This DARIS was calculated on the basis of a proportion of participants with mortality by myocardial infarction of 5.38% in the control group; post hoc selected RRR of 25% in the experimental intervention group; alpha (α) of 5%; beta (β) of 20%; and diversity of 14%. The cumulative Z‐curve (blue line) did not cross the conventional alpha 5% boundaries (green lines). After the 12th trial, the cumulative Z‐curve crossed the trial sequential monitoring boundary for futility. Accordingly, although only 53.30% (5253/9854) of the DARIS has been obtained, we can reject an intervention effect of 25% or larger. Had we calculated the DARIS on the basis of a more realistic RRR like 20% (as we had planned) or less, the obtained information would represent a smaller part of the DARIS. Accordingly, the boundaries for futility would not have been crossed in such scenarios. Therefore, risk reduction of 20% or less may require additional trials with larger sample sizes.
Subgroup analysis of trials according to administration route of lidocaine
Meta‐analysis of nine trials administering lidocaine by the intravenous route showed no significant differences regarding all‐cause mortality when lidocaine was compared with placebo or with no intervention (92/1100 (8.36%) vs 70/942 (7.43%); RR 1.16, 95% CI 0.82 to 1.63; participants = 2042; I2 = 14%; P value = 0.40) (Baker 1971; Bennett 1970; Chopra 1971; Hargarten 1990; Lie 1974; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987). Meta‐analysis of five trials administering lidocaine by the intramuscular route showed no significant differences regarding all‐cause mortality when lidocaine was compared with placebo or with no intervention (41/1436 (2.85%) vs 53/1368 (3.87%); RR 0.77, 95% CI 0.50 to 1.17; participants = 2804; I2 = 4%; P value = 0.22) (ALIT 1985; Lie 1978; Rossi 1976; Valentine 1974; Wennerblom 1982). Meta‐analysis of two trials administering lidocaine by the intramuscular route showed no significant differences regarding all‐cause mortality when lidocaine was compared with placebo or with no intervention (15/211 (7.11%) vs 12/196 (6.12%); RR 1.17, 95% CI 0.56 to 2.42; participants = 407; I2 = 0%; P value = 0.68) (Darby 1972; Dunn 1985). Tests for subgroup differences showed no significant differences (I2 = 16.3%; P value = 0.92). See Analysis 1.3.
1.3. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 3: All‐cause mortality in acute myocardial infarction‐only participants (subgroup analysis by administration route for lidocaine)
Subgroup analysis of trials with infusion administration only compared with trials with bolus and infusion administrations
Meta‐analysis of three trials administering lidocaine by infusion showed no significant differences regarding all‐cause mortality only when lidocaine was compared with placebo or with no intervention (16/229 (6.98%) vs 22/237 (9.28%); RR 0.85, 95% CI 0.33 to 2.17; participants = 466; I2 = 40%; P value = 0.73) (Baker 1971; Pharand 1995; Pitt 1971). Meta‐analysis of six trials administering lidocaine by both bolus and infusion approaches revealed no significant differences regarding all‐cause mortality when lidocaine was compared with placebo or with no intervention (76/871 (8.72%) vs 48/705 (6.81%); RR 1.30, 95% CI 0.92 to 1.83; participants = 1576; I2 = 0%; P value = 0.14) (Bennett 1970; Chopra 1971; Hargarten 1990; Lie 1974; O'Brien 1973; Poprawski 1987). Tests for subgroup differences showed no significant differences (I2 = 0%; P value = 0.40). See Analysis 1.4.
1.4. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 4: All‐cause mortality (subgroup analysis according to intravenous administration)
Subgroup analysis according to bolus lidocaine dose
One trial found no significant differences in all‐cause mortality when lidocaine administered by bolus up to 50 mg was compared with placebo or with no intervention (7/39 (17.95%) vs 4/43 (9.30%); RR 1.93, 95% CI 0.61 to 6.09; participants = 82; P value = 0.26) (Chopra 1971). One bolus of 60 mg of lidocaine does not produce statistically significant differences when compared with to placebo or with no intervention in terms of all‐cause mortality (25/249 (10.04%) vs 8/125 (6.4%); RR 1.57, 95% CI 0.73 to 3.38; participants = 374; P value = 0.25) (Bennett 1970). Meta‐analysis of two trials comparing a bolus of 75 mg of lidocaine versus placebo or no intervention showed no significant differences regarding all‐cause mortality (34/240 (14.17%) vs 24/232 (10.34%); RR 1.49, 95% CI 0.70 to 3.16; participants = 472; I2 = 42%; P value = 0.30) (O'Brien 1973; Poprawski 1987). One trial found no statistically significant differences in all‐cause mortality when lidocaine administered by bolus at a dose of 100 mg was compared with placebo or with no intervention (8/107 (7.48%) vs 10/105 (9.52%); RR 0.79, 95% CI 0.32 to 1.91; participants = 212; P value = 0.59) (Lie 1974). At a dose of 1 mg/kg, lidocaine did not significantly affect all‐cause mortality when compared with placebo or no intervention (2/236 (0.85%) vs 2/200 (1%); RR 0.85, 95% CI 0.12 to 5.96; participants = 436; P value = 0.87) (Hargarten 1990). Tests for subgroup differences showed no significant differences (I2 = 0%; P value = 0.70). See Analysis 1.5.
1.5. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 5: All‐cause mortality (subgroup analysis according to bolus‐lidocaine dose)
Subgroup analysis according to number of lidocaine boluses at any dose
Meta‐analysis of four trials assessing one bolus of lidocaine at any dose versus placebo or no intervention showed no statistically significant differences regarding all‐cause mortality (51/549 (9.29%) vs 26/419 (6.20%); RR 1.47, 95% CI 0.90 to 2.38; participants = 968; I2 = 5%; P value = 0.12) (Bennett 1970; Chopra 1971; Lie 1974; O'Brien 1973). Administration of two boluses at any dose of lidocaine versus placebo or no intervention did not significantly affect all‐cause mortality in participants with acute myocardial infarction (27/322 (8.38%) vs 22/286 (7.69%); RR 1.19, 95% CI 0.72 to 1.95; participants = 608; I2 = 0%; P value = 0.49) (Hargarten 1990; Poprawski 1987). See Analysis 1.6.
1.6. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 6: All‐cause mortality (subgroup analysis according to number of lidocaine boluses)
Subgroup analysis according to intravenous infusion dose of lidocaine
Meta‐analysis of three trials comparing infusion of lidocaine between 1 mg/min and 1.5 mg/min versus placebo or no intervention showed no significant differences regarding all‐cause mortality (32/370 (8.64%) vs 14/248 (5.64%); RR 1.45, 95% CI 0.71 to 2.95; participants = 618; I2 = 11%; P value = 0.31) (Baker 1971; Bennett 1970; Pharand 1995). Meta‐analysis of six trials comparing infusion of lidocaine between 2 mg/min and 3 mg/min versus placebo or no intervention also showed no statistically significant differences regarding all‐cause mortality (60/730 (8.21%) vs 56/694 (8.1%); RR 1.08, 95% CI 0.72 to 1.62; participants = 1424; I2 = 20%; P value = 0.72) (Chopra 1971; Hargarten 1990; Lie 1974; O'Brien 1973; Pitt 1971; Poprawski 1987). See Analysis 1.7.
1.7. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 7: All‐cause mortality (subgroup analysis according to intravenous infusion dose)
Subgroup analysis according to clinical setting
Meta‐analysis of two trials performed in a pre‐hospital setting showed no statistically significant differences regarding all‐cause mortality when lidocaine was compared with placebo or with no intervention (12/1034 (1.16%) vs 11/955 (1.15%); RR 1.00, 95% CI 0.46 to 2.19; participants = 1989; I2 = 0%; P value = 1.00) (ALIT 1985; Wennerblom 1982). Meta‐analysis of 11 trials performed in a hospital setting also showed no statistically significant differences regarding all‐cause mortality when lidocaine was compared with placebo or with no intervention (106/1120 (9.46%) vs 113/1130 (10%); RR 0.95, 95% CI 0.69 to 1.32; participants = 2250; I2 = 30%; P value = 0.77) (Baker 1971; Chopra 1971; Darby 1972; Lie 1974; Lie 1978; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987; Rossi 1976; Valentine 1974). Meta‐analysis of three trials performed in both pre‐hospital and hospital settings similarly showed no significant differences in all‐cause mortality when lidocaine was compared with placebo or with no intervention (30/593 (5.1%) vs 11/421 (2.61%); RR 1.53, 95% CI 0.77 to 3.02; participants = 1014; I2 = 0%; P value = 0.22) (Bennett 1970; Dunn 1985; Hargarten 1990). Tests for subgroup differences showed no significant differences (I2 = 0%; P value = 0.47). See Analysis 1.8.
1.8. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 8: All‐cause mortality (subgroup analysis by clinical setting)
Subgroup analysis of trials without risk of industry bias compared with trials with risk of industry bias
Meta‐analysis of 11 trials without risk of industry bias showed no significant differences in all‐cause mortality when lidocaine was compared with placebo or with no intervention (96/2145 (4.48%) vs 79/1972 (4.01%); RR 1.09, 95% CI 0.82 to 1.44; participants = 4117; I2 = 0%; P value = 0.54) (ALIT 1985; Baker 1971; Darby 1972; Dunn 1985; Hargarten 1990; Lie 1978; O'Brien 1973; Pharand 1995; Poprawski 1987; Valentine 1974; Wennerblom 1982). Meta‐analysis of five trials at risk of industry bias showed no significant differences in all‐cause mortality when lidocaine was compared with placebo or with no intervention (52/602 (8.64%) vs 56/534 (10.49%); RR 0.84, 95% CI 0.44 to 1.58; participants = 1136; I2 = 58%; P value = 0.58) (Bennett 1970; Chopra 1971; Lie 1974; Pitt 1971; Rossi 1976). Tests for subgroup differences showed no significant differences (I2 = 0%; P value = 0.45). See Analysis 1.9.
1.9. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 9: All‐cause mortality (subgroup analysis according to non‐suspected trials with industry bias compared with suspected trials with industry bias)
Sensitivity analyses taking attrition into consideration
Of the 18 trials (11,727 participants) combined for this outcome, three trials (17% (3/18)) reported exact numbers of participants with missing events in the intervention and control groups (Bennett 1970; Darby 1972; NNLIT 1992). Two trials involving 614 participants reported information for this outcome overall (Dunn 1985; Lie 1974). Thirteen trials did not report information for this outcome in any comparison group (ALIT 1985; Baker 1971; Chopra 1971; Hargarten 1990; Lie 1978; O'Brien 1973; Pharand 1995; Pitt 1971; Poprawski 1987; Rossi 1976; Sadowski 1999; Valentine 1974; Wennerblom 1982). Thus, three trials reported missing data for intervention groups and control groups involving 6.60% of participants (774/11,727). Furthemore, these three trials involved 18.8% of events in the experimental group (40/213) and 13.6% of events in the control group (27/199).
'Best‐worse case' scenario
In a best‐worst case scenario analysis, we found no statistically significant differences in proportions of all‐cause mortality (40/448 (8.93%) vs 48/326 (14.72%); RR 0.57, 95% CI 0.30 to 1.08; participants = 774; I2 = 49%; P value < 0.08).
'Worst‐best case' scenario
In a worst‐best case scenario analysis, we found no statistically significant differences in proportions of all‐cause mortality (100/448 (22.32%) vs 27/326 (8.28%); RR 2.20, 95% CI 1.02 to 4.73; participants = 774; I2 = 67%; P value = 0.04).
See Analysis 1.10.
1.10. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 10: All‐cause mortality (sensitivity analysis by attrition bias)
Lidocaine versus disopyramide
Meta‐analysis of two trials comparing lidocaine versus disopyramide showed no significant differences in terms of all‐cause mortality (7/73 (9.59%) vs 5/71 (7.04%); RR 1.39, 95% CI 0.47 to 4.13; participants = 144; I2 = 0%; P value = 0.55; very‐low‐quality evidence) (Pedersen 1986; Ronnevik 1987). See Analysis 2.1.
2.1. Analysis.

Comparison 2: Lidocaine vs disopyramide, Outcome 1: All‐cause mortality
Sensitivity analyses taking attrition into consideration
Pedersen 1986 and Ronnevik 1987 reported the exact numbers of participants with missing events in lidocaine and disopyramide groups.
'Best‐worse case' scenario
In a best‐worst case scenario analysis, we found no significant differences in proportions of participants for all‐cause mortality (7/73 (9.59%) vs 12/71 (16.90%); RR 0.49, 95% CI 0.08 to 3.02; participants = 144; I2 = 62%; P value = 0.44).
'Worst‐best case' scenario
In a worst‐best case scenario analysis, we found no significant differences in proportions of participants for all‐cause mortality (15/73 (20.54%) vs 5/71 (7.04%); RR 2.75, 95% CI 1.05 to 7.20; participants = 144; I2 = 0%; P value = 0.04).
See Analysis 2.2.
2.2. Analysis.

Comparison 2: Lidocaine vs disopyramide, Outcome 2: All‐cause mortality (sensitivity analysis by risk of attrition bias)
Lidocaine versus tocainide
One trial comparing lidocaine versus tocainide showed no significant differences in risk of all‐cause mortality (1/13 (7.6%) vs 1/16 (6.25%); RR 1.23, 95% CI 0.08 to 17.83; participants = 29; P value = 0.88; very‐low‐quality evidence) (Keefe 1986). See Analysis 3.1.
3.1. Analysis.

Comparison 3: Lidocaine vs tocainide, Outcome 1: All‐cause mortality
Sensitivity analyses taking attrition into consideration
Keefe 1986 reported the exact numbers of participants with missing events in lidocaine and tocainide groups.
'Best‐worse case' scenario
In a best‐worst case scenario analysis, we found no statistically significant differences in proportions of participants for all‐cause mortality (1/13 (7.7%) vs 2/16 (12.5%); RR 0.62, 95% CI 0.06 to 6.05; participants = 29; P value = 0.68).
'Worst‐best case' scenario
In a worst‐best case scenario analysis, we found no statistically significant differences in proportions of participants for all‐cause mortality (2/13 (15.4%) vs 1/16 (6.25%); RR 2.46, 95% CI 0.25 to 24.21; participants = 29; P value = 0.44).
See Analysis 3.2.
3.2. Analysis.

Comparison 3: Lidocaine vs tocainide, Outcome 2: All‐cause mortality (sensitivity analysis by risk of attrition bias)
Lidocaine versus mexiletine
No significant differences in risk of all‐cause mortality were noted between lidocaine and mexiletine (0/12 (0%) vs 1/12 (8.33%); RR 0.33, 95% CI 0.01 to 7.45; participants = 24; P value = 0.49; very‐low‐quality evidence) (Horowitz 1981). See Analysis 4.1.
4.1. Analysis.

Comparison 4: Lidocaine vs mexiletine, Outcome 1: All‐cause mortality
Cardiac mortality
Lidocaine versus placebo or no intervention
Meta‐analysis of 12 trials showed no significant differences regarding cardiac mortality in participants with or without proved acute myocardial infarction when lidocaine was compared with placebo or with no intervention (69/4184 (1.65%) vs 62/4093 (1.51%); RR 1.03, 95% CI 0.70 to 1.50; participants = 8277; I2 = 12%; P value = 0.90; low‐quality evidence) (ALIT 1985; Baker 1971; Bennett 1970; Chopra 1971; Darby 1972; Lie 1974; Lie 1978; NNLIT 1992; Pharand 1995; Pitt 1971; Valentine 1974; Wennerblom 1982). See Analysis 1.11. Figure 7 shows no evidence of publication bias outcome. Trial sequential analysis shows 11 trials provided evidence showing that lidocaine is not able to induce a 30% RR reduction in cardiac mortality compared with placebo or with no intervention, if we disregard risks of bias (Figure 8).
1.11. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 11: Cardiac mortality
8.
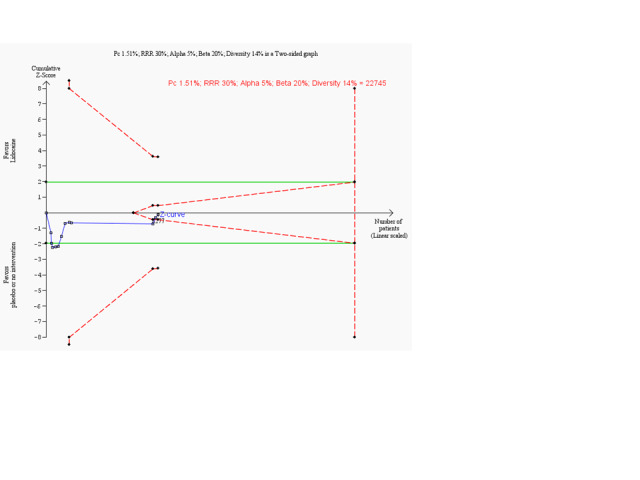
Trial sequential analysis on cardiac mortality in 12 lidocaine vs placebo or no intervention trials
Trial sequential analysis of lidocaine vs placebo or no intervention on cardiac mortality in participants with or without proven myocardial infarction based on the diversity‐adjusted required information size (DARIS) of 22,745 participants. This DARIS was calculated on the basis of a proportion of participants with cardiac mortality among those with suspected myocardial infarction of 1.51% in the control group; post hoc selected RRR of 30% in the experimental intervention group; alpha (α) of 5%; beta (β) of 20%; and diversity of 14%. The cumulative Z‐curve (blue line) crossed the conventional alpha of 5% (green line) after 3 trials suggested harm. After 10 trials, however, the cumulative Z‐curve (blue line) crossed the trial sequential monitoring boundary for futility. Accordingly, after only 36.4% (8277/22,745) of the DARIS had been obtained, we were able to reject an intervention effect of 30% or larger. Had we calculated the DARIS on the basis of a more realistic RRR like 20% (as originally planned) or less, the obtained evidence would represent a smaller part of the DARIS. Accordingly, boundaries for futility would not have been crossed in such scenarios. Therefore, risk reductions of 20% or less may require additional trials with larger sample sizes.
Subgroup analysis of trials not suspected to be at risk of industry bias versus trials suspected to be at risk of industry bias
Meta‐analysis of eight trials without risk of industry bias showed no significant differences regarding cardiac mortality among participants with and those without proved acute myocardial infarction when lidocaine was compared with placebo or with no intervention (35/3681 (0.95%) vs 42/3706 (1.13%); RR 0.85, 95% CI 0.52 to 1.39; participants = 7387; I2 = 14%; P value = 0.51) (ALIT 1985; Baker 1971; Darby 1972; Lie 1978; NNLIT 1992; Pharand 1995; Valentine 1974; Wennerblom 1982). Meta‐analysis of four trials at risk of industry bias showed no significant differences regarding cardiac mortality when lidocaine was compared with placebo or with no intervention (34/503 (6.76%) vs 20/387 (5.16%); RR 1.36, 95% CI 0.77 to 2.39; participants = 890; I2 = 0%; P value = 0.29) (Bennett 1970; Chopra 1971; Lie 1974; Pitt 1971). Tests for subgroup differences showed no significant differences (I2 = 34.4%; P value = 0.22). See Analysis 1.12.
1.12. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 12: Cardiac mortality (sensitivity analysis according to non‐suspected trials of industry bias versus suspected trials of industry bias)
Lidocaine versus disopyramide
Regarding cardiac mortality, one meta‐analysis of two trials found no significant differences between lidocaine and placebo (3/71 (4.23%) vs 3/73 (4.10%); RR 1.02, 95% CI 0.21 to 4.87; participants = 144; I2 = 0%; P value = 0.98; very‐low‐quality evidence) (Pedersen 1986; Ronnevik 1987). See Analysis 2.3.
2.3. Analysis.

Comparison 2: Lidocaine vs disopyramide, Outcome 3: Cardiac mortality
Lidocaine versus tocainide
One trial comparing lidocaine with tocainide showed no significant differences in risk of cardiac mortality (1/13 (7.7%) vs 1/16 (6.25%); RR 1.23, 95% CI 0.08 to 17.83; participants = 29; P value = 0.88; very‐low‐quality evidence) (Keefe 1986). See Analysis 3.3.
3.3. Analysis.

Comparison 3: Lidocaine vs tocainide, Outcome 3: Cardiac mortality
Lidocaine versus mexiletine
No significant differences in risk of cardiac mortality were noted between lidocaine and mexiletine (0/12 (0%) versus 1/12 (8.33%); (RR 0.33, 95% CI 0.01 to 7.45; participants = 24; P = 0.49, very low quality evidence) (Horowitz 1981). See Analysis 4.2.
4.2. Analysis.

Comparison 4: Lidocaine vs mexiletine, Outcome 2: Cardiac mortality
Overall survival at 30 days after myocardial infarction
Trials did not assess this outcome.
Secondary outcomes
Ventricular fibrillation
Lidocaine versus placebo or no intervention
Meta‐analysis of 16 trials showed no significant differences between lidocaine and placebo or no intervention regarding prophylaxis of ventricular fibrillation in participants with or without proven acute myocardial infarction (76/5128 (1.48%) vs 103/4987 (2.07%); RR 0.78, 95% CI 0.55 to 1.12; participants = 10115; I2 = 18%; P value = 0.18; low‐quality evidence) (ALIT 1985; Baker 1971; Bennett 1970; Chopra 1971; Darby 1972; Dunn 1985; Hargarten 1990; Kuck 1985; Lie 1974; Lie 1978; NNLIT 1992; O'Brien 1973; Poprawski 1987; Sadowski 1999; Solimene 1983; Valentine 1974). See Analysis 1.13. Figure 9 shows no evidence of publication bias. Trial sequential analysis reveals that 16 trials provided evidence to show that lidocaine is not able to induce a 30% RR reduction in cardiac mortality compared with placebo or with no intervention, if we disregard risks of bias (Figure 10).
1.13. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 13: Ventricular fibrillation
9.
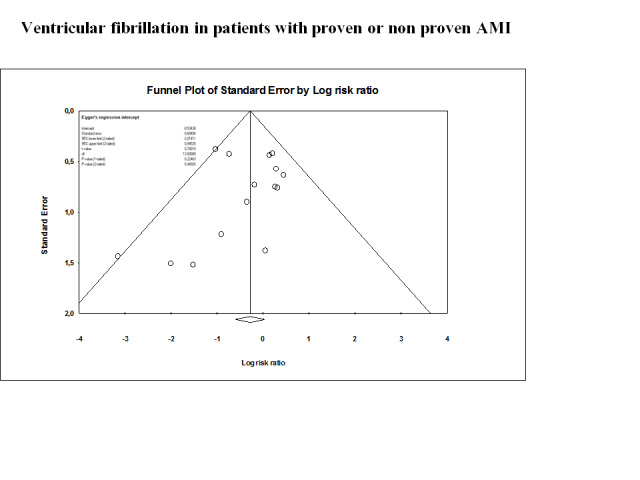
Funnel plot of data from the meta‐analysis of effects of lidocaine compared with placebo for preventing ventricular fibrillation in individuals with proven or non‐proven acute myocardial infarction (15 trials). This figure shows low risk of publication bias. Circles show point estimates of the included randomised controlled trials. The pattern of distribution simulates an inverted funnel. Trials are symmetrically distributed in each of the halves. Larger trials are closer and upper to the pooled estimate. Effect sizes of the smaller trials are lower and are more or less symmetrically distributed around the pooled estimate.
10.
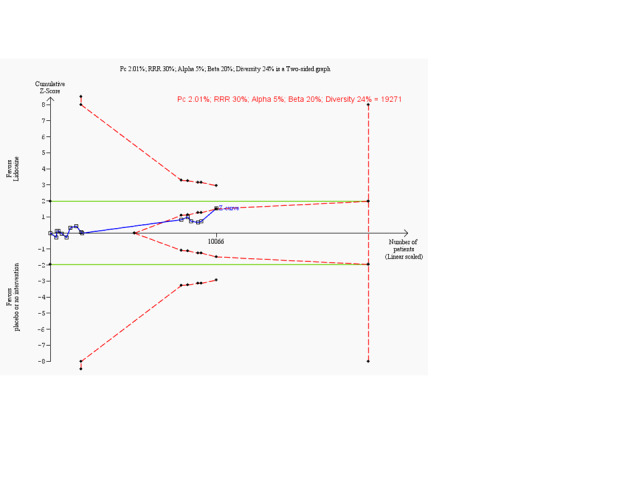
Trial sequential analysis on prevention of ventricular fibrillation in 15 lidocaine vs placebo or no intervention trials
Trial sequential analysis of lidocaine vs placebo or no intervention on prevention of ventricular fibrillation in participants with or without proven myocardial infarction based on the diversity‐adjusted required information size (DARIS) of 19,271 individuals. This DARIS was calculated on the basis of a proportion of participants with ventricular fibrillation of 2.01% in the control group; post hoc selected RRR of 30% in the experimental intervention group; alpha (α) of 5%; beta (β) of 20%; and diversity of 24%. The cumulative Z‐curve (blue line) did not cross conventional alpha 5% boundaries (green lines). After the 10th trial, the cumulative Z‐curve crosses the trial sequential monitoring boundary for futility. Accordingly, after only 52.2% (10,066/19,271) of the DARIS had been obtained, we were able to reject an intervention effect of 30% or larger. Had we calculated the DARIS on the basis of a more realistic RRR like 20% (as originally planned) or less, the obtained evidence would represent a smaller portion of the DARIS. Accordingly, the boundaries for futility would not have been crossed in such scenarios. Therefore, risk reductions of 20% or less may require additional trials with larger sample sizes.
Lidocaine versus disopyramide
One trial comparing lidocaine versus disopyramide provided very‐low‐quality evidence regarding ventricular fibrillation (1/38 (2.6%) vs 3/38 (7.9%); RR 0.33, 95% CI 0.04 to 3.06; participants = 76; P value = 0.33) (Pedersen 1986). See Analysis 2.4.
2.4. Analysis.

Comparison 2: Lidocaine vs disopyramide, Outcome 4: Ventricular fibrillation
Lidocaine versus tocainide
Two trials assessed this comparison (Keefe 1986; Rehnqvist 1983). However, Keefe 1986 reported that no participants experienced ventricular fibrillation. On the other hand, Rehnqvist 1983 did not mention this outcome.
Lidocaine versus mexiletine
No significant differences in risk of ventricular fibrillation were noted between comparison groups (1/12 (8.33%) vs 0/12 (0%); RR 3.00, 95% CI 0.13 to 67.06; participants = 24; P value = 0.49; very‐low‐quality evidence) (Horowitz 1981). See Analysis 4.3.
4.3. Analysis.

Comparison 4: Lidocaine vs mexiletine, Outcome 3: Ventricular fibrillation
Lidocaine versus amiodarone
No significant differences in risk of ventricular fibrillation were noted between comparison groups (2/15 (13.33%) vs 0/10 (0%); RR 3.44, 95% CI 0.18 to 64.88; participants = 25; P value = 0.41; very‐low‐quality evidence) (Capucci 1985). See Analysis 6.1.
6.1. Analysis.

Comparison 6: Lidocaine vs amiodarone, Outcome 1: Ventricular fibrillation
Lidocaine versus propafenone
No significant differences in risk of ventricular fibrillation were noted between comparison groups (1/10 (1%) vs 0/10 (0%); RR 3.00, 95% CI 0.14 to 65.90; participants = 20; P value = 0.49; low‐quality evidence) (Rehnqvist 1984). See Analysis 5.1.
5.1. Analysis.

Comparison 5: Lidocaine vs propafenone, Outcome 1: Ventricular fibrillation
Adverse events
Cardiovascular adverse events
Lidocaine versus placebo or no intervention
Lidocaine significantly increased the risk of asystole over placebo or no intervention (35/3393 (1.03%) vs 14/3443 (0.41%); RR 2.32, 95% CI 1.26 to 4.26; participants = 6826; I2 = 0%; very‐low‐quality evidence) (ALIT 1985; Darby 1972; Dunn 1985; NNLIT 1992). No significant differences were noted between lidocaine and placebo or no intervention regarding sinus bradycardia (55/1346 (4.09%) vs 49/1203 (4.07%); RR 1.09, 95% CI 0.66 to 1.80; participants = 2549; I2 = 21%; P value = 0.74; very‐low‐quality evidence) (Bennett 1970; Darby 1972; Dunn 1985; Hargarten 1990; Rademaker 1986; Sandler 1976; Touboul 1988; Wennerblom 1982); bundle branch block (83/853 (9.73%) vs 75/733 (10.23%); RR 1.07, 95% CI 0.80 to 1.44; participants = 1586; I2 = 0%; P value = 0.64) (Bennett 1970; Darby 1972; Sadowski 1999; Touboul 1988); non‐complete atrioventricular block (78/888 (8.79%) vs 75/773 (9.70%); RR 1.01, 95% CI 0.75 to 1.37; participants = 1661; I2 = 0%%; P value = 0.93) (Bennett 1970; Darby 1972; Sadowski 1999; Sandler 1976); complete atrioventricular block (13/443 (2.93%) vs 5/315 (1.59%); RR 1.77, 95% CI 0.66 to 4.78; participants = 758; I2 = 0%; P value = 0.26) (Bennett 1970; Darby 1972; Sandler 1976); unknown grade atrioventricular block (66/919 (7.18%) vs 56/808 (6.93%); RR 1.12, 95% CI 0.75 to 1.67; participants = 1727; I2 = 7%; P value = 0.49) (Bennett 1970; O'Brien 1973; Sadowski 1999; Wennerblom 1982); pulmonary edema (83/868 (9.56%) vs 71/762 (9.32%); RR 1.08, 95% CI 0.80 to 1.46; participants = 1630; I2 = 0%; P value = 0.51) (Bennett 1970; Darby 1972; Sadowski 1999; Wennerblom 1982); cardiogenic shock (77/868 (8.87%) vs 73/762 (9.58%); RR 1.04, 95% CI 0.77 to 1.41; participants = 1630; I2 = 0%; P value = 0.79) (Bennett 1970; Darby 1972; Sadowski 1999; Wennerblom 1982); hypotension (87/814 (10.69%) vs 88/885 (9.94%); RR 1.07, 95% CI 0.81 to 1.41; participants = 1699; I2 = 0%; P value = 0.59) (Darby 1972; NNLIT 1992; Rossi 1976; Sadowski 1999; Wennerblom 1982); cardiac arrest (76/1149 (6.61%) vs 76/1181 (6.43%); RR 1.03, 95% CI 0.77 to 1.39; participants = 2330; I2 = 0%; P value = 0.85) (Hargarten 1990; Sadowski 1999); and heart failure (134/851 (15.74%) vs 170/800 (21.25%); RR 0.91, 95% CI 0.63 to 1.33; participants = 1751; I2 = 62%; P value = 0.64) (Dunn 1985; Pharand 1995; Rossi 1976; Sadowski 1999). See Analysis 1.14.
1.14. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 14: Cardiovascular adverse events
Lidocaine versus disopyramide
No significant differences were noted between lidocaine and disopyramide regarding pulmonary oedema (2/73 (2.73%) vs 4/71 (5.63%); RR 0.62, 95% CI 0.12 to 3.10; participants = 144; I2 = 0%; P value = 0.56) (Pedersen 1986; Ronnevik 1987); cardiogenic shock (2/38 (5.26%) vs 1/38 (2.63%); RR 2.00, 95% CI 0.19 to 21.14; participants = 76; P value = 0.56) (Pedersen 1986); asystole (0/38 (0%) vs 1/38 (2.63%); RR 0.33, 95% CI 0.01 to 7.93; participants = 76; P value = 0.50; very‐low‐quality evidence) (Pedersen 1986); sinoatrial block (1/35 (2.85%) vs 1/33 (3.03%); RR 0.94, 95% CI 0.06 to 14.47; participants = 68; P value = 0.97; very‐low‐quality evidence) (Ronnevik 1987) and cardiac block (high‐degree atrioventricular block and bundle branch block) (3/35 (8.57%) vs 5/33 (15.15%); RR 0.57, 95% CI 0.15 to 2.18; participants = 68; P value = 0.51; very‐low‐quality evidence) (Ronnevik 1987). See Analysis 2.5.
2.5. Analysis.

Comparison 2: Lidocaine vs disopyramide, Outcome 5: Cardiovascular adverse events
Lidocaine versus tocainide
Meta‐analysis of two trials shows increased risk of any adverse event in the lidocaine group compared with the tocainide group (25/33 (75.75%) vs 16/36 (44.44%); RR 1.69, 95% CI 1.07 to 2.68; participants = 69; I2 = 17%); very‐low‐quality evidence) (Keefe 1986; Rehnqvist 1983). See Analysis 3.4.
3.4. Analysis.

Comparison 3: Lidocaine vs tocainide, Outcome 4: Adverse events
Lidocaine versus mexiletine
No significant differences were noted between lidocaine and mexiletine groups in terms of cardiogenic shock (0/12 (0%) vs 1/12 (8.33%); RR 0.33, 95% CI 0.01 to 7.45; participants = 24; P value = 0.49); incomplete atrioventricular block (0/12 (0%) vs 1/12 (8.33%); RR 0.33, 95% CI 0.01 to 7.45; participants = 24; P value = 0.49) and pulmonary oedema (3/12 (25%) vs 2/12 (16.66%); RR 1.50, 95% CI 0.30 to 7.43; participants = 24; P value = 0.62; very‐low‐quality evidence) (Horowitz 1981). See Analysis 4.4.
4.4. Analysis.

Comparison 4: Lidocaine vs mexiletine, Outcome 4: Adverse events
Lidocaine versus propafenone
One trial showed no significant differences between lidocaine and propafenone regarding heart failure (2/28 (7.14%) vs 0/36 (0%); RR 6.38, 95% CI 0.32 to 127.77; participants = 64; P value = 0.23; very‐low‐quality evidence) and bilateral bundle branch block (0/28 (0%) vs 1/36 (2.78%); RR 0.43, 95% CI 0.02 to 10.06; participants = 64; P value = 0.60; very‐low‐quality evidence) (Touboul 1988). See Analysis 5.2.
5.2. Analysis.

Comparison 5: Lidocaine vs propafenone, Outcome 2: Adverse events
Lidocaine versus amiodarone
Capucci 1985 found no significant differences between lidocaine and amiodarone regarding bradycardia (0/15 (0%) vs 1/10 (10%); RR 0.23, 95% CI 0.01 to 5.12; participants = 25; P value = 0.35; very‐low‐quality evidence) and hypotension (0/15 (0%) vs 12/10 (20%); RR 0.14, 95% CI 0.01 to 2.60; participants = 25; P value = 0.19; very‐low‐quality evidence). See Analysis 6.2.
6.2. Analysis.

Comparison 6: Lidocaine vs amiodarone, Outcome 2: Adverse events
Lidocaine versus dimethylammonium
One trial found no significant differences between lidocaine and dimethylammonium in terms of hypotension (4/15 (26.7%) vs 5/16 (31.25%); RR 0.85, 95% CI 0.28 to 2.59; participants = 31; P value = 0.78; very‐low‐quality evidence); rise in blood pressure (0/15 (0%) vs 5/16 (31.25%); RR 0.10, 95% CI 0.01 to 1.61; participants = 31; P value = 0.10; very‐low‐quality evidence); tachycardia (0/15 (0%) vs 8/16 (50%); RR 0.06, 95% CI 0.00 to 1.00; participants = 31; P value = 0.05; very‐low‐quality evidence); and bradycardia (0/15 (0%) vs 1/16 (6.25%); RR 0.35, 95% CI 0.02 to 8.08; participants = 31; P value = 0.52; very‐low‐quality evidence) (Bergdahl 1978). See Analysis 7.1.
7.1. Analysis.

Comparison 7: Lidocaine vs dimethylammonium, Outcome 1: Adverse events
Neurological adverse events
Lidocaine versus placebo or no intervention
Meta‐analysis of three trials showed no significant differences between lidocaine and placebo or no intervention in terms of seizures (4/3248 (0.12%) vs 0/3263 (0%); RR 3.58, 95% CI 0.59 to 21.85; participants = 6481; I2 = 0%; P value = 0.17) (ALIT 1985; Poprawski 1987; Rademaker 1986). Lidocaine, compared with placebo or with no intervention, increased significantly the risk of dizziness/drowsiness (74/1259 (5.88%) vs 16/1274(1.25%); RR 3.85, 95% CI 2.29 to 6.47; participants = 2533; I2 = 0%; low‐quality evidence) (Hargarten 1990; Lie 1974; Lie 1978; NNLIT 1992; Pharand 1995; Rademaker 1986). Lidocaine and placebo or no intervention do not differ significantly regarding nausea/vomiting (30/245 (12.24%) vs 24/240 (10%); RR 1.62, 95% CI 0.45 to 5.89; participants = 485; I2 = 64%; I2 = 64%; P value = 0.46) (Pharand 1995; Rademaker 1986). Risk of speech disturbances is not statistically significant in the lidocaine group compared with the placebo or no intervention group (16/438 (3.65%) vs 1/431 (0.23%); RR 4.34, 95% CI 1.00 to 18.81; participants = 869; I2 = 0%; P value = 0.05) (Lie 1974; Pharand 1995; Poprawski 1987; Rademaker 1986). Comparison groups did not differ in terms of confusion (17/3386 (0.50%) vs 6/3423 (0.17%); RR 2.44, 95% CI 0.76 to 7.81; participants = 6809; I2 = 21%; P value = 0.13) (O'Brien 1973; Pharand 1995; Rademaker 1986) or agitation (3/186 (0.50%) vs 2/186 (0.17%); RR 1.35, 95% CI 0.26 to 7.06; participants = 372; I2 = 0%; P value = 0.73) (Pharand 1995; Poprawski 1987). Two trials reporting overall neurological adverse events showed no significant differences between comparison groups (22/307 (7.17%) vs 11/295 (3.73%); RR 2.24, 95% CI 0.44 to 11.31; participants = 602; I2 = 73%; P value = 0.33) (Dunn 1985; Pharand 1995). See Analysis 1.15.
1.15. Analysis.

Comparison 1: Lidocaine vs placebo or no intervention, Outcome 15: Neurological adverse events
Lidocaine versus disopyramide
No significant differences between comparison groups were noted in risk of confusion (3/35 (8.57%) vs 0/33 (0%); RR 6.61, 95% CI 0.35 to 123.30; participants = 68; P value = 0.21; very‐low‐quality evidence) (Ronnevik 1987). See Analysis 2.6.
2.6. Analysis.

Comparison 2: Lidocaine vs disopyramide, Outcome 6: Neurological adverse events
Lidocaine versus mexiletine
Meta‐analysis of two trials revealed very‐low‐quality evidence when lidocaine was compared with mexiletine regarding risk of composite neurological adverse events (nausea/vomiting, confusion, vertigo, nystagmus) (9/37 (24.32%) vs 17/37 (45.94%); RR 0.63, 95% CI 0.16 to 2.47; participants = 74; I2 = 26%; P value = 0.51) (Horowitz 1981; Rolli 1981). See Analysis 4.4.
Lidocaine versus propafenone
Very‐low‐quality evidence was found when lidocaine was compared with propafenone regarding mental or neurological symptoms (6/38 (15.78%) vs 0/46 (0%); RR 6.95, 95% CI 0.86 to 55.94; participants = 84; I2 = 0%; P value = 0.07) (Rehnqvist 1984; Touboul 1988). See Analysis 5.2.
Lidocaine versus amiodarone
No significant differences in risk of diplopia/sleepiness were noted between groups (1/15 (6.67%) vs 0/10 (0%); RR 2.06, 95% CI 0.09 to 46.11; participants = 25; P value = 0.65; very‐low‐quality evidence) (Capucci 1985). See Analysis 6.2.
Lidocaine versus dimethylammonium
One trial found very‐low‐quality evidence when lidocaine was compared with dimethylammonium regarding risks of nausea and vomiting (1/15 (6.66%) vs 7/16 (43.75%); RR 0.15, 95% CI 0.02 to 1.10; participants = 31; P value = 0.06); vertigo (1/156.67%) vs 0/16 (0%); RR 3.19, 95% CI 0.14 to 72.69; participants = 31; P value = 0.47) and paraesthesia (0/15 (0%) vs 7/16 (43.75%); RR 0.07, 95% CI 0.00 to 1.14; participants = 31; P value = 0.06) (Bergdahl 1978). See Analysis 7.1.
Lidocaine versus aprindine
Very‐low‐quality evidence was found when lidocaine was compared with aprindine in terms of coma (1/12 (8.33%) vs 0/12 (0%); RR 3.00, 95% CI 0.13 to 67.06; participants = 24; P value = 0.49); seizures (2/12 (16.67%) vs 0/12 (0%); RR 5.00, 95% CI 0.27 to 94.34; participants = 24; P value = 0.28); agitation (0/12 (0%) vs 2/12 (16.67%); RR 0.20, 95% CI 0.01 to 3.77; participants = 24; P value = 0.28) and disturbances of speech (2/12 (16.67%) vs 0/12 (0%); RR 5.00, 95% CI 0.27 to 94.34; participants = 24, P = 0.28) (Depaepe 1974). See Analysis 8.1.
8.1. Analysis.

Comparison 8: Lidocaine vs aprindine, Outcome 1: Adverse events
Lidocaine versus pirmenol
One trial found very‐low‐quality evidence when lidocaine was compared with pirmenol regarding adverse events (5/9 (55.55%) vs 5/10 (50%); RR 1.11, 95% CI 0.47 to 2.60; participants = 19; P value = 0.81) (Cuendet 1988). See Analysis 9.1.
9.1. Analysis.

Comparison 9: Lidocaine vs pirmenol, Outcome 1: Adverse event
Discussion
Summary of main results
This Cochrane systematic review on prophylactic lidocaine for myocardial infarction found 37 randomised controlled trials incorporating 11,948 participants. Trials reported comparisons between lidocaine versus placebo or no intervention, as well as versus eight antiarrhythmic drugs (i.e., disopyramide, tocainide, mexiletine, propafenone, amiodarone, dimethylammonium chloride, aprindine and pirmenol). Overall, trials had high risks of bias and were underpowered. Ninety‐seven per cent of trials (36/37) did not report an a priori sample size estimation. Drug companies sponsored at least 10 trials, suggesting potential risk of industry bias. Trials were conducted in 17 countries (Australia, Belgium, Brazil, Canada, Denmark, France, Germany, Italy, New Zealand, Northern Ireland, Norway, Poland, Sweden, Switzerland, The Netherlands, United Kingdom and United States of America), in general in pre‐hospital and/or hospital settings. Included participants had proven or non‐proven acute myocardial infarction.
We were able to meta‐analyse data for all‐cause mortality. One meta‐analysis of 18 trials involved participants with proven or non‐proven acute myocardial infarction; investigators compared lidocaine versus placebo or no intervention and found no statistically significant differences between comparison groups (Table 1). A second meta‐analysis combined two trials and compared lidocaine versus disopyramide. Researchers found no significant differences between antiarrhythmic drugs (Table 2). Non‐pooled trials examining lidocaine versus tocainide or mexiletine did not differ significantly in terms of all‐cause mortality (Table 3; Table 4, respectively).
We were able to meta‐analyse data from 12 trials on cardiac mortality, which showed that lidocaine did not result in significant differences in cardiac mortality compared with placebo or no intervention (Table 1). Meta‐analysis of two trials revealed no significant differences between lidocaine and disopyramide in reducing cardiac mortality (Table 2). Similarly, lidocaine did not differ significantly from tocainide and mexiletine in terms of cardiac mortality (Table 3; Table 4, respectively).
Lidocaine compared with placebo or no intervention, disopyramide, mexiletine and propafenone did not significantly reduce the proportions of participants developing ventricular fibrillation (Table 1; Table 2; Table 4; Table 5).
Lidocaine compared with placebo or no intervention significantly increased risks of asystole, drowsiness and dizziness (Table 1). No significant differences were noted between lidocaine and disopyramide, tocainide, mexiletine, propafenone, amiodarone, dimethylammonium and aprindine in terms of adverse events ‐ cardiovascular or neurological (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8). However, safety data were poorly reported overall, and adverse events may be underestimated. No trials reported data on overall survival at 30 days after myocardial infarction.
Overall completeness and applicability of evidence
This Cochrane review found evidence suggesting that prophylactic lidocaine for myocardial infarction is not useful in preventing all‐cause mortality nor ventricular fibrillation. However, this conclusion is based on randomised controlled trials with high risk of bias. Furthermore, the safety profile of lidocaine is unclear from data reported in the included trials.
We conducted subgroup analyses of participants with proven acute myocardial infarction including administration route, infusion administration only compared with bolus and infusion administrations, bolus lidocaine dose, number of lidocaine boluses at any dose, intravenous infusion doses of lidocaine and clinical setting. Furthermore, we performed sensitivity analyses that included trials without risk of industry bias versus trials with risk of industry bias, while taking attrition into consideration. Both types of analyses were conducted for all‐cause mortality. Results show consistency and are based on data from trials that included a broad range of participants with different co‐morbidities, for whom different treatment approaches were provided. Although these aspects could be considered as a threat to applicability, consistency in results derived from our analyses shows that the included trials may represent a broad spectrum of patients with low and high risk of mortality.
We tried to identify all published and unpublished data, as well as ongoing studies, to warrant confidence in the completeness of data gathered in the review. However, we cannot rule out that calculated effects are overestimated as the result of poor methodological quality (design, analysis) and small sample size of randomised controlled trials. Furthermore, we cannot rule out an underestimation of safety findings.
Figure 5, Figure 7, Figure 8 and Figure 10 seem to present overly optimistic considerations regarding which intervention effects can be proved or disproved; these illustrations show that lidocaine could have effects that would be not only statistically significant but clinically significant as well. However, much larger trials are needed to prove or disprove these effects.
Quality of the evidence
Grades of Recommendation, Assessment, Development and Evaluation (GRADE) assessments were conducted on outcomes of meta‐analyses and non‐pooled trials. No trials were graded as providing strong evidence, primarily because small sample sizes were used (even after meta‐analysis), and because studies were found to have high risk of bias due to lack of adequate randomisation methods, lack of blinding, high attrition, unclear reporting of outcomes and other biases such as industry bias and bias in the presentation of data. Furthermore, we graded evidence as low or very low in quality because of imprecision in clinically relevant outcomes.
See Table 1, Table 2, Table 3, Table 4, Table 5, Table 6, Table 7 and Table 8 for complete assessments and the rationale for ratings.
We suspected 10 trials to be at potential risk of industry bias (Bennett 1970; Bergdahl 1978; Chopra 1971; Depaepe 1974; Keefe 1986; Lie 1974; Pitt 1971; Rademaker 1986; Rossi 1976; Sbarbaro 1979). This review conducted a subgroup analysis of trials at risk of industry bias versus trials without risk of industry bias that examined lidocaine versus placebo or no intervention. Review authors were not able to detect significant differences between subgroups in risk of all‐cause mortality (Analysis 1.9).
Trials with high risk of bias generate SPIN randomised controlled trials, which distort results presented by using specific reporting strategies, whatever their motive, to highlight that the experimental treatment is beneficial, despite a statistically non‐significant difference for the primary outcome, or to distract the reader from statistically non‐significant results when published reports of randomised controlled trials present such results for primary outcomes (Boutron 2010).
This Cochrane review has identified the following issues, which may be particularly relevant to consider as future trials are planned. Overall, information on all‐cause mortality and cardiac mortality has been found to be inconsistent because included trials did not appropriately discriminate between participants with proven and non‐proven myocardial infarction, and because investigators used different outcome definitions for 'ventricular arrhythmias' and reported outcomes inconsistently. Researchers should adopt an agreed upon set of core outcomes for each medical condition (Clarke 2007) with the goal of reducing the impact of outcome reporting bias (Kirkham 2010).
The impact of outcome reporting bias may be reduced if investigators adopt the recommendations of the Patient‐Centered Outcomes Research Institute (PCORI) (PCORI 2012), an independent, non‐profit organisation established by the US Congress to conduct research with the goal of providing information about the best available evidence required to make informed decisions. Research conducted by PCORI is intended to help patients better understand available prevention, treatment and care options, and the science that supports those options (Basch 2012; Gabriel 2012; Selby 2012).
Potential biases in the review process
A systematic review process involves a group of biases called 'significance‐chasing biases', such as publication bias and selective outcome reporting bias (Ioannidis 2010). Publication bias represents a major threat to the validity of a systematic review, particularly a review that includes small trials. However, this Cochrane review is at low risk of publication bias because a meticulous trial search was conducted by research authors, which ensured identification of randomised controlled trials reported in English and non‐English languages. Also, review authors found no evidence of asymmetry in the funnel plot prepared for all‐cause mortality among participants with proven or non‐proven myocardial infarction (Figure 4; Figure 6) and ventricular fibrillation (Figure 9).
Selective outcome reporting bias is seen in suppression of information on specific outcomes and is similar to publication bias in whole studies or trials, in that ‘negative’ results remain unpublished (Ioannidis 2010). We were surprised to find that many trials did not provide data on all‐cause mortality, ventricular fibrillation or safety. The authors of this Cochrane review observed that 38% of included randomised controlled trials are at high risk of selective outcome reporting. For example, adverse events were reported in nine trials comparing lidocaine versus placebo or no intervention, and all‐cause mortality was reported in 18 trials assessing this comparison. This indicates that safety data for 50% (11,727/2) of randomly assigned participants in trials comparing lidocaine versus placebo or no intervention remain unknown.
Agreements and disagreements with other studies or reviews
Overall, our results are similar to those of other, non‐Cochrane reviews (De Silva 1981; Hine 1989; MacMahon 1988; Sadowski 1999; Teo 1993). These five reviews differ from one another in their eligibility criteria, and from this Cochrane review in the following ways: (1) inclusion of non‐randomised clinical trials by MacMahon 1988 (one; Singh 1976); Hine 1989 (two; Bleifeld 1973; Mogensen 1971 and Sadowski 1999 (three; Bleifeld 1973; Mogensen 1971; Singh 1976); (2) inclusion by MacMahon 1988 and Sadowski 1999 of one trial with bias in the presentation of data (Wyse 1988; this trial randomly assigned participants to lidocaine and placebo, but published results compared two approaches: prophylactic vs selective); (3) inability of Cochrane review authors to extract data from Kostuk 1969 and Sandler 1976 (because these trials were at high risk of selective outcome reporting, i.e. investigators did not report all‐cause mortality or ventricular fibrillation data; however, both trials were included by Sadowski 1999); (4) inclusion by MacMahon 1988 and Sadowski 1999 of data from a trial with bias in the presentation of data (Wyse 1988; we considered this trial to have high risk of bias in selective outcome reporting for the above mentioned reason); (5) assessment by MacMahon 1988 of data from Dunn 1985 on lidocaine use through intramuscular and intravenous bolus; (6) publication bias of one meta‐analysis due to exclusion of non‐English language trials (Hine 1989); (7) inclusion by Cochrane review authors of head‐to‐head comparisons of lidocaine versus other antiarrhythmic drugs such as disopyramide, tocainide, mexiletine, propafenone, amiodarone, dimethylammonium chloride, aprindine and pirmenol; and, finally, (8) inclusion in this Cochrane systematic review of additional trials (not included in the non‐Cochrane reviews) comparing lidocaine versus placebo or no intervention (Poprawski 1987; Rossi 1976; Solimene 1983; inclusion of these additional studies allowed us to obtain more accurate estimates for our outcomes of interest). Despite these differences, all systematic reviews have reached similar results for the most relevant outcomes, showing no significant effects derived from prophylactic lidocaine use on all‐cause mortality, cardiac mortality and ventricular fibrillation. However, a limitation of this Cochrane review was introduced by trials that included participants both with and without myocardial infarction. Trial authors must report data on the entire study population and on those with proven myocardial infarction to reduce uncertainty.
Authors' conclusions
Implications for practice.
This Cochrane review provides low‐quality evidence to suggest that prophylactic lidocaine leads to very little or no effect on all‐cause mortality and ventricular fibrillation following myocardial infarction. These results are based on 37 trials (11,948 participants) comparing lidocaine at any dosage and route of administration versus placebo or no intervention or antiarrhythmic drugs. Included trials showed no benefit for preventing death among individuals with acute myocardial infarction at low or high risk of death. Results are based on the findings of randomised controlled trials at high risk of bias, and safety data remain unclear. Therefore, we conclude that based on this systematic review and meta‐analyses comparing lidocaine versus placebo, no intervention or other antiarrhythmic drugs, prescription of lidocaine prophylactically is not justified in acute myocardial infarction.
Implications for research.
Trial sequential analysis suggests that no additional trials may be needed to disprove an intervention effect of 20% relative risk reduction for assessing benefits of prophylactic lidocaine in myocardial infarction. Smaller risk reductions might require higher trials. Potential trials should include clinical outcomes such as all‐cause mortality, ventricular fibrillation and adverse events. Trials should be designed according to the SPIRIT statement (Chan 2013) and reported according to the CONSORT statement to improve the quality of reporting of efficacy and harms in clinical research (Ioannidis 2004Moher 2010). Future trials should be planned in accordance with the recommendations of the Patient‐Centered Outcomes Research Initiative (Basch 2012; Gabriel 2012; McKinney 2012).
What's new
| Date | Event | Description |
|---|---|---|
| 21 September 2021 | Review declared as stable | No new studies since the review was published in 2015 (search up to 1 June 2020) and no known ongoing studies. |
History
Protocol first published: Issue 6, 2010 Review first published: Issue 8, 2015
Acknowledgements
We want to express our gratitude to peer reviewers for improving the quality of the protocol of this Cochrane review.
Special thanks to Mrs. Maroussia Tzanova, Dr. Vasiliy Vlassov, Dr. Sonia Jaramillo, Dr. Andreas Lundin and Dr. Jolanta Sabbat for helping with translations to the English language.
Our gratitude to Karen Hovhannisyan, who helped us obtain one Danish randomised controlled trial.
We express our special thanks to Dr. Ricardo Hidalgo for contributions to the protocol of this review.
Appendices
Appendix 1. Glossary of clinical and epidemiological terms
| Terms | DEFINITION | REFERENCE | ||||
| Acute coronary syndrome | An episode of myocardial ischemia that generally lasts longer than a transient anginal episode and ultimately may lead to myocardial infarction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Amiodarone | An antianginal and antiarrhythmic drug. It increases the duration of ventricular and atrial muscle action by inhibiting Na,K‐activated myocardial adenosine triphosphatase. A decrease in heart rate and in vascular resistance results. . | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Anterior wall myocardial infarction | Myocardial infarction in which the anterior wall of the heart is involved. Anterior wall myocardial infarction is often caused by occlusion of the left anterior descending coronary artery. It can be categorised as anteroseptal or anterolateral wall myocardial infarction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Antiarrhythmic treatment | Agents used for the treatment or prevention of cardiac arrhythmias. They may affect the polarisation‐repolarisation phase of the action potential, its excitability or refractoriness or impulse conduction or membrane responsiveness within cardiac fibers. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Arrhythmias | Any disturbances of the normal rhythmical beating of the heart or myocardial contraction. Cardiac arrhythmias can be classified by abnormalities in heart rate, disorders of electrical impulse generation or impulse conduction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Atrioventricular block | Impaired impulse conduction from heart atria to heart ventricles. AV block can mean delayed or completely blocked impulse conduction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Atropine | An alkaloid, originally from Atropa belladonna. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| B | ||||||
| Bradycardia | Cardiac arrhythmias characterised by excessively slow heart rate, usually below 50 beats per minute in human adults. These arrhythmias can be classified broadly into sinoatrial node dysfunction and atrioventricular blocks. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Bundle branch block | Form of heart block in which electrical stimulation of heart ventricles is interrupted at one of the branches of the bundle of His, thus preventing simultaneous depolarisation of the 2 ventricles. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| C | ||||||
| Calcium antagonist drugs | A class of drugs that act by selective inhibition of calcium influx through cell membranes or on release and binding of calcium in intracellular pools. As they are inducers of vascular and other smooth muscle relaxation, these agents are used in drug therapy for hypertension and cerebrovascular spasms, as myocardial protective agents and in relaxation of uterine spasms. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Cardiac complexes, premature | A group of cardiac arrhythmias in which cardiac contractions are not initiated at the sinoatrial node. They include both atrial and ventricular premature beats, and are also known as extra or ectopic heartbeats. Their frequency is increased in individuals with heart disease. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Cardiogenic shock | Shock resulting from diminution of cardiac output in heart disease. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Central nervous system | The main information‐processing organs of the nervous system, consisting of brain, spinal cord and meninges. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Congestive heart failure | A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic needs of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction) or sudden overload beyond capacity. Chronic heart failure is more common than acute heart failure, which results from sudden insult to cardiac function, such as myocardial infarction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Consciousness disorders | Organic mental disorders characterised by impairment of the ability to maintain awareness of self and environment, and to respond to environmental stimuli. Dysfunction of the cerebral hemispheres or of the brain stem reticular formation may result in this condition. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Coronary care unit | The hospital unit in which patients with acute cardiac disorders receive intensive care. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Creatine phosphokinase | A transferase that catalyses formation of phosphocreatine from ATP + creatine. The reaction stores ATP energy as phosphocreatine. Three cytoplasmic isoenzymes have been identified in human tissues: the MM type from skeletal muscle, the MB type from myocardial tissue and the BB type from nervous tissue and from a mitochondrial isoenzyme. Macro‐creatine kinase refers to creatine kinase complexed with other serum proteins. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Cyanosis | A bluish or purplish discoloration of the skin and mucous membranes due to an increase in the amount of deoxygenated haemoglobin in the blood or a structural defect in the haemoglobin molecule. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| D | ||||||
| Disopyramide | A class I antiarrhythmic agent (one that interferes directly with depolarisation of cardiac membrane and thus serves as a membrane‐stabilising agent) with a depressant action on the heart, similar to that of guanidine. It possesses anticholinergic and local anaesthetic properties. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Dysrhythmias | Any disturbances of normal rhythmic beating of the heart or myocardial contraction. Cardiac arrhythmias can be classified by abnormalities in heart rate, disorders of electrical impulse generation or impulse conduction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| E | ||||||
| Electrocardiography | Recording of the moment‐to‐moment electromotive forces of the heart as projected onto various sites on the body's surface, delineated as a scalar function of time. The recording is monitored by tracing on slow‐moving chart paper or by observing on a cardioscope, which is a cathode ray tube display. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| F | ||||||
| Furosemide | A benzoic‐sulfonamide‐furan. This diuretic with fast onset and short duration is used for oedema and chronic renal insufficiency. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| H | ||||||
| Heart failure | A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic needs of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction) or sudden overload beyond capacity. Chronic heart failure is more common than acute heart failure, which results from sudden insult to cardiac function, such as myocardial infarction. | MeSH Database from PubMed U.S. National Library of Medicine |
||||
| Hypokalemia | Abnormally low potassium concentration in the blood. This may result from potassium loss by renal secretion or by the gastrointestinal route, as by vomiting or diarrhoea. It may manifest clinically by neuromuscular disorders ranging from weakness to paralysis, by electrocardiographic abnormalities (depression of the T wave and elevation of the U wave), by renal disease and by gastrointestinal disorders. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Hypotension | Abnormally low blood pressure that can result in inadequate blood flow to the brain and other vital organs. Common symptom is dizziness, but greater negative impacts on the body occur when deprivation of oxygen and nutrients is prolonged. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| I | ||||||
| Inferior wall myocardial infarction | Myocardial infarction involving the inferior wall of the heart. This is often caused by occlusion of the right coronary artery. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Infusion | The administration of liquid medication, nutrient or other fluid through a route other than the alimentary canal, usually over minutes or hours, by gravity flow or often by infusion pumping. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| L | ||||||
| Lidocaine | A local anaesthetic and cardiac depressant used as an antiarrhythmia agent. Its actions are more intense and its effects more prolonged than those of procaine, but its duration of action is shorter than that of bupivacaine or prilocaine. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Loss of consciousness | Loss of the ability to maintain awareness of self and environment combined with markedly reduced responsiveness to environmental stimuli. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| M | ||||||
| Mexiletine | Antiarrhythmic agent pharmacologically similar to lidocaine. It may have some anticonvulsant properties. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Morphine | The principal alkaloid in opium and the prototype opiate analgesic and narcotic. Morphine has widespread effects in the central nervous system and on smooth muscle. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| N | ||||||
| Nausea | An unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning and various enteroviruses. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| P | ||||||
| Pacemaker | A device designed to stimulate, by electrical impulses, contraction of the heart muscles. It may be temporary (external) or permanent (internal or internal‐external). | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Paraesthesia | Subjective cutaneous sensations (e.g. cold, warmth, tingling, pressure) experienced spontaneously in the absence of stimulation. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Prajmalium | A derivative of the rauwolfia alkaloid ajmaline. It is an anti‐arrhythmia agent but may cause liver damage. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Premature ventricular complexes | A type of cardiac arrhythmia with premature contractions of the heart ventricles. It is characterised by the premature QRS complex on ECG that is of abnormal shape and great duration (generally >129 msec). It is the most common form of all cardiac arrhythmias. Premature ventricular complexes have no clinical significance, except in concurrence with heart diseases. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Procainamide | A derivative of procaine with less central nervous system action. It acts as a non‐nucleoside inhibitor of DNA methylation and has led to systemic lupus erythematosus. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Propafenone | An antiarrhythmia agent that is particularly effective in ventricular arrhythmias. It also has weak beta‐blocking activity. The drug is generally well tolerated. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Pulmonary oedema | Excessive accumulation of extravascular fluid in the lung, an indication of a serious underlying disease or disorder. Pulmonary oedema prevents efficient pulmonary gas exchange in the pulmonary alveoli, and can be life‐threatening. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Pump failure (heart failure) | A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic needs of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction) or sudden overload beyond the capacity of the heart. Chronic heart failure is more common than acute heart failure, which results from sudden insult to cardiac function, such as myocardial infarction. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| S | ||||||
| Saline solution | Hypertonic sodium chloride solution. A solution having an osmotic pressure greater than that of physiological salt solution (0.9 g NaCl in 100 mL purified water). | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Seizures | Clinical or subclinical disturbances of cortical function due to a sudden, abnormal, excessive, and disorganised discharge of brain cells. Clinical manifestations include abnormal motor, sensory and psychic phenomena. Recurrent seizures are usually referred to as epilepsy or "seizure disorder". |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Shock | A pathological condition manifested by failure to perfuse or oxygenate vital organs. | MeSH Database from PubMed
U.S. National Library of Medicine |
||||
| Streptokinase | Streptococcal fibrinolysin. An enzyme produced by haemolytic streptococci. It hydrolyaes amide linkages and serves as an activator of plasminogen. It is used in thrombolytic therapy and is in mixtures with streptodornase. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Sudden death | The abrupt cessation of all vital bodily functions, manifested by permanent loss of total cerebral, respiratory and cardiovascular functions. Death results from an unexpected circulatory arrest, usually due to a cardiac arrhythmia within an hour of onset of symptoms. |
MeSH Database from PubMed U.S. National Library of Medicine Zipes 2006 |
||||
| Sustained ventricular tachycardia | This is a ventricular tachyarrhythmia > 30 seconds in duration and/or requiring termination due to haemodynamic compromise in less than 30 seconds. It can be both, monomorphic, with a stable single QRS morphology, or, polymorphic, with a changing or multiform QRS morphology at cycle length between 600 and 180 milliseconds. | Zipes 2006 | ||||
| T | ||||||
| Tachycardia | Abnormally rapid heartbeat, usually with a heart rate above 100 beats per minute for adults. Tachycardia accompanied by disturbance in cardiac depolarisation (cardiac arrhythmia) is called tachyarrhythmia. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Tocainide | An antiarrhythmic agent that exerts potential‐ and frequency‐dependent block of sodium channels. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Torsade de pointes | A malignant form of polymorphic ventricular tachycardia that is characterised by heart rate between 200 and 250 beats per minute, and QRS complexes with changing amplitude and twisting of the points. This term also describes the syndrome of tachycardia with prolonged ventricular repolarisation, long QT intervals exceeding 500 milliseconds or bradycardia. Torsades de pointes may be self limited or may progress to ventricular fibrillation. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| V | ||||||
| Vasodilatation | The physiological widening of blood vessels by relaxing of the underlying vascular smooth muscle. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Venous pressure | The blood pressure in the veins. It is usually measured to assess filling pressure to the heart ventricle. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Ventricular extrasystole | A type of cardiac arrhythmia with premature contractions of the heart ventricles. It is characterised by the premature QRS complex on ECG that is of abnormal shape and great duration (generally > 129 msec). It is the most common form of all cardiac arrhythmias. Premature ventricular complexes have no clinical significance except in concurrence with heart disease. |
MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Ventricular fibrillation | Potentially lethal cardiac arrhythmia that is characterised by unco‐ordinated extremely rapid firing of electrical impulses (400‐600/min) in heart ventricles. Such asynchronous ventricular quivering or fibrillation prevents any effective cardiac output and results in unconsciousness (syncope). It is one of the major electrocardiographic patterns seen with cardiac arrest. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Ventricular flutter | A potentially lethal cardiac arrhythmia characterised by an extremely rapid, haemodynamically unstable ventricular tachycardia (150‐300 beats/min) with a large oscillating sine‐wave appearance. If untreated, ventricular flutter typically progresses to ventricular fibrillation. | MeSH Database from PubMed U.S. National Library of Medicine | ||||
| Ventricular tachycardia | An abnormally rapid ventricular rhythm usually in excess of 150 beats per minute. It is generated within the ventricle below the bundle of HIS, as autonomic impulse formation or reentrant impulse conduction. Depending on the origin, onset of ventricular tachycardia can be paroxysmal (sudden) or non‐paroxysmal, its wide QRS complexes can be uniform or polymorphic and ventricular beating may be independent of atrial beating (AV dissociation). | MeSH Database from PubMed U.S. National Library of Medicine | ||||
Appendix 2. Search strategies (13 April 2015)
CENTRAL
#1 MeSH descriptor lidocaine this term only #2 lidocain* in All Text #3 lignocain* in All Text #4 xylocain* in All Text #5 (#1 or #2 or #3 or #4) #6 MeSH descriptor myocardial infarction explode all trees #7 myocardial next infarct* in All Text #8 heart next infarct* in All Text #9 (coronary in All Text near/3 syndrome* in All Text) #10 heart next attack in All Text #11 (#6 or #7 or #8 or #9 or #10) #12 (#5 and #11)
MEDLINE Ovid
1. Lidocaine/
2. lidocain*.tw.
3. lignocain*.tw.
4. xylocain*.tw.
5. 1 or 2 or 3 or 4
6. exp Myocardial Infarction/
7. (coronary adj3 syndrome*).tw.
8. heart attack.tw.
9. heart infarct*.tw.
10. myocardial infarct*.tw.
11. 6 or 7 or 8 or 9 or 10
12. 5 and 11
13. randomized controlled trial.pt.
14. controlled clinical trial.pt.
15. randomized.ab.
16. placebo.ab.
17. drug therapy.fs.
18. randomly.ab.
19. trial.ab.
20. groups.ab.
21. 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20
22. exp animals/ not humans.sh.
23. 21 not 22
24. 12 and 23
EMBASE Ovid
1. lidocaine/
2. lidocain*.tw.
3. lignocain*.tw.
4. xylocain*.tw.
5. 1 or 2 or 3 or 4
6. exp heart infarction/
7. (coronary adj3 syndrome*).tw.
8. heart attack.tw.
9. heart infarct*.tw.
10. myocardial infarct*.tw.
11. 6 or 7 or 8 or 9 or 10
12. 5 and 11
13. random$.tw.
14. factorial$.tw.
15. crossover$.tw.
16. cross over$.tw.
17. cross‐over$.tw.
18. placebo$.tw.
19. (doubl$ adj blind$).tw.
20. (singl$ adj blind$).tw.
21. assign$.tw.
22. allocat$.tw.
23. volunteer$.tw.
24. crossover procedure/
25. double blind procedure/
26. randomized controlled trial/
27. single blind procedure/
28. 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27
29. (animal/ or nonhuman/) not human/
30. 28 not 29
31. 12 and 30
LILACS
lidocain$ or lignocain$ or xylocain$ [Words] and infarct$ or attack$ [Words]
Web of Science – with Conference Proceedings
#13 #12 AND #11
#12 TS=(random* or blind* or allocat* or assign* or trial* or placebo* or crossover* or cross‐over*)
#11 #10 AND #4
#10 #9 OR #8 OR #7 OR #6 OR #5
#9 TS=cardia* infarct*
#8 TS=myocardial infarct*
#7 TS=heart infarct*
#6 TS=heart attack*
#5 TS=(coronary SAME syndrome*)
#4 #3 OR #2 OR #1
#3 TS=xylocain*
#2 TS=lignocain*
#1 TS=lidocain*
Data and analyses
Comparison 1. Lidocaine vs placebo or no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality (participants with proven or non‐proven acute myocardial infarction) | 18 | 11727 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.82, 1.27] |
| 1.2 All‐cause mortality (subgroup analysis by acute myocardial infarction‐only participants) | 16 | 5253 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.79, 1.30] |
| 1.3 All‐cause mortality in acute myocardial infarction‐only participants (subgroup analysis by administration route for lidocaine) | 16 | 5253 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.79, 1.30] |
| 1.3.1 Intravenous route | 9 | 2042 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.82, 1.63] |
| 1.3.2 Intramuscular route | 5 | 2804 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.17] |
| 1.3.3 Both routes | 2 | 407 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.56, 2.42] |
| 1.4 All‐cause mortality (subgroup analysis according to intravenous administration) | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4.1 Trials with infusion only | 3 | 466 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.33, 2.17] |
| 1.4.2 Trials with bolus followed by infusion | 6 | 1576 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.92, 1.83] |
| 1.5 All‐cause mortality (subgroup analysis according to bolus‐lidocaine dose) | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.5.1 Up to 50 mg | 1 | 82 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [0.61, 6.09] |
| 1.5.2 60 mg | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.73, 3.38] |
| 1.5.3 75 mg | 2 | 472 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [0.70, 3.16] |
| 1.5.4 100 mg | 1 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.32, 1.91] |
| 1.5.5 1 mg/kg‐p | 1 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.12, 5.96] |
| 1.6 All‐cause mortality (subgroup analysis according to number of lidocaine boluses) | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.6.1 One bolus | 4 | 968 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.90, 2.38] |
| 1.6.2 Two bolus | 2 | 608 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.72, 1.95] |
| 1.7 All‐cause mortality (subgroup analysis according to intravenous infusion dose) | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.7.1 1 mg/min to 1.5 mg/min | 3 | 618 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.71, 2.95] |
| 1.7.2 2 mg/min to 3 mg/min | 6 | 1424 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.72, 1.62] |
| 1.8 All‐cause mortality (subgroup analysis by clinical setting) | 16 | 5253 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.79, 1.30] |
| 1.8.1 Pre‐hospital setting lidocaine use | 2 | 1989 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.46, 2.19] |
| 1.8.2 Hospital setting lidocaine use | 11 | 2250 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.69, 1.32] |
| 1.8.3 Lidocaine use in both pre‐hospital and hospital settings | 3 | 1014 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.77, 3.02] |
| 1.9 All‐cause mortality (subgroup analysis according to non‐suspected trials with industry bias compared with suspected trials with industry bias) | 16 | 5253 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.79, 1.30] |
| 1.9.1 Trials non‐sponsored by industry | 11 | 4117 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.82, 1.44] |
| 1.9.2 Trials sponsored by industry | 5 | 1136 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.44, 1.58] |
| 1.10 All‐cause mortality (sensitivity analysis by attrition bias) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.10.1 Best‐worst case scenario | 3 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.30, 1.08] |
| 1.10.2 Worst‐best case scenario | 3 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 2.20 [1.02, 4.73] |
| 1.11 Cardiac mortality | 12 | 8277 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.70, 1.50] |
| 1.12 Cardiac mortality (sensitivity analysis according to non‐suspected trials of industry bias versus suspected trials of industry bias) | 12 | 8277 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.70, 1.50] |
| 1.12.1 Trials non‐sponsored by drug company | 8 | 7387 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.52, 1.39] |
| 1.12.2 Trials sponsored by drug company | 4 | 890 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.77, 2.39] |
| 1.13 Ventricular fibrillation | 16 | 10115 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.55, 1.12] |
| 1.14 Cardiovascular adverse events | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.14.1 Asystole | 4 | 6826 | Risk Ratio (M‐H, Random, 95% CI) | 2.32 [1.26, 4.26] |
| 1.14.2 Sinus bradycardia | 8 | 2549 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.66, 1.80] |
| 1.14.3 Bundle branch block | 5 | 1586 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.80, 1.44] |
| 1.14.4 Non‐complete atrioventricular block | 4 | 1661 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.75, 1.37] |
| 1.14.5 Complete atrioventricular block | 3 | 758 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [0.66, 4.78] |
| 1.14.6 Unknown grade atrioventricular block | 4 | 1727 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.75, 1.67] |
| 1.14.7 Pulmonary oedema | 4 | 1630 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.80, 1.46] |
| 1.14.8 Cardiogenic shock | 4 | 1630 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.77, 1.41] |
| 1.14.9 Hypotension | 5 | 1699 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.81, 1.41] |
| 1.14.10 Cardiac arrest | 2 | 2330 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.77, 1.39] |
| 1.14.11 Heart failure | 4 | 1751 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.63, 1.33] |
| 1.15 Neurological adverse events | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.15.1 Seizures | 3 | 6481 | Risk Ratio (M‐H, Random, 95% CI) | 3.58 [0.59, 21.85] |
| 1.15.2 Drowsiness/Dizziness | 5 | 2533 | Risk Ratio (M‐H, Random, 95% CI) | 3.85 [2.29, 6.47] |
| 1.15.3 Nausea/Vomiting | 2 | 485 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [0.45, 5.89] |
| 1.15.4 Speech disturbances | 4 | 869 | Risk Ratio (M‐H, Random, 95% CI) | 4.34 [1.00, 18.81] |
| 1.15.5 Confusion | 4 | 6809 | Risk Ratio (M‐H, Random, 95% CI) | 2.44 [0.76, 7.81] |
| 1.15.6 Agitation | 2 | 372 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.26, 7.06] |
| 1.15.7 Global adverse events in central nervous system | 2 | 602 | Risk Ratio (M‐H, Random, 95% CI) | 2.24 [0.44, 11.31] |
Comparison 2. Lidocaine vs disopyramide.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 All‐cause mortality | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.47, 4.13] |
| 2.2 All‐cause mortality (sensitivity analysis by risk of attrition bias) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.2.1 Best‐worst case scenario | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.08, 3.02] |
| 2.2.2 Worst‐best case scenario | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 2.75 [1.05, 7.20] |
| 2.3 Cardiac mortality | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.21, 4.87] |
| 2.4 Ventricular fibrillation | 1 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.06] |
| 2.5 Cardiovascular adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.5.1 Pulmonary oedema | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.12, 3.10] |
| 2.5.2 Cardiogenic shock | 1 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.19, 21.14] |
| 2.5.3 Asystole | 1 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.93] |
| 2.5.4 Sinoatrial block | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.06, 14.47] |
| 2.5.5 Cardiac blocks (high‐degree atrioventricular block and bundle branch block) | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.15, 2.18] |
| 2.6 Neurological adverse events | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 6.61 [0.35, 123.30] |
Comparison 3. Lidocaine vs tocainide.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.08, 17.83] |
| 3.2 All‐cause mortality (sensitivity analysis by risk of attrition bias) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.2.1 Best‐worst case scenario | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.06, 6.05] |
| 3.2.2 Worst‐best case scenario | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.46 [0.25, 24.21] |
| 3.3 Cardiac mortality | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.08, 17.83] |
| 3.4 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.4.1 Any adverse event | 2 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 1.69 [1.07, 2.68] |
Comparison 4. Lidocaine vs mexiletine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 All‐cause mortality | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.45] |
| 4.2 Cardiac mortality | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.45] |
| 4.3 Ventricular fibrillation | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.13, 67.06] |
| 4.4 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.4.1 Cardiogenic shock | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.45] |
| 4.4.2 Atrioventricular block | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.45] |
| 4.4.3 Pulmonary oedema | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.30, 7.43] |
| 4.4.4 Composite neurological adverse event (confusion, vertigo, nystagmus and diplopia) | 2 | 74 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.16, 2.47] |
Comparison 5. Lidocaine vs propafenone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Ventricular fibrillation | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 5.2 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.2.1 Heart failure | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 6.38 [0.32, 127.77] |
| 5.2.2 Bilateral bundle branch block | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.02, 10.06] |
| 5.2.3 Neuropsychiatric disturbances | 2 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 6.95 [0.86, 55.94] |
Comparison 6. Lidocaine vs amiodarone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Ventricular fibrillation | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 3.44 [0.18, 64.88] |
| 6.2 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.2.1 Bradycardia | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.01, 5.12] |
| 6.2.2 Hypotension | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.60] |
| 6.2.3 Diplopia plus sleepiness | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 2.06 [0.09, 46.11] |
Comparison 7. Lidocaine vs dimethylammonium.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1.1 Hypotension | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.28, 2.59] |
| 7.1.2 Rise in blood pressure | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 0.10 [0.01, 1.61] |
| 7.1.3 Tachycardia | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 1.00] |
| 7.1.4 Bradycardia | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.02, 8.08] |
| 7.1.5 Nausea/Vomiting | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 1.10] |
| 7.1.6 Paraesthesia | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.14] |
| 7.1.7 Vertigo | 1 | 31 | Risk Ratio (M‐H, Random, 95% CI) | 3.19 [0.14, 72.69] |
Comparison 8. Lidocaine vs aprindine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1.1 Coma | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.13, 67.06] |
| 8.1.2 Seizures | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.27, 94.34] |
| 8.1.3 Agitation | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 3.77] |
| 8.1.4 Disturbance of speech | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.27, 94.34] |
Comparison 9. Lidocaine vs pirmenol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Adverse event | 1 | 19 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.47, 2.60] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
ALIT 1985.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Duration of the study: 33 months Country: The Netherlands Follow‐up: unclear | |
| Participants | Enrolled: 7026 Randomly assigned, N = 6024
Age, mean, years (standard deviation)
Gender, male, % (n/N)
Inclusion criteria: suspected to have acute myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine: 400 mg, intramuscular route Control group: not stated | |
| Outcomes | Mortality Incidence of ventricular fibrillation Frequent termination of ventricular tachycardia | |
| Notes | Sample size calculation a priori: not reported
Sponsor: The Netherlands Heart Fundation
Role of sponsor: not reported Trial conduction dates: 16 September 1986 and 17 June 1983 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...was thus randomized... " (page 1106) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ´‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "...observers who were blinded to randomization" (page 1106) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Baker 1971.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: England Follow‐up period: 48 hours | |
| Participants | Enrolled: 91 Randomly assigned: N = 44 (acute myocardial infarction within 48 hours before admission)
Age, % (n/N)
Gender, male, % (n/N)
Inclusion criterion: patients with acute myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine: continuous infusion of 1.5 mg of lidocaine per minute in 5% dextrose solution Placebo: 5% dextrose solution: continuous infusion alone at same speed as intervention Co‐intervention: "additional lidocaine, either as bolus injection or as increased infusion doses, was given to four patients" (page 53) |
|
| Outcomes | Mortality Incidence and types of dysrhythmias | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...numbered according to a randomized sequence" (page 2) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | "... cards in sealed envelopes number ..." (page 2) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | " a double‐blind trial" (page 1) Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified |
| Other bias | High risk | Design bias (Porta 2008) |
Bennett 1970.
| Study characteristics | ||
| Methods | Parallel design (3 arms) Country: USA Follow‐up period: 48 hours | |
| Participants | Randomly assigned: N = 374
Age ≥ 50 years, %
Age < 50 years, %
Gender, male, %: 70 Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine (2 arms)
Placebo: quote "no lidocaine" (page 910). Nature of control not reported Co‐intervention: not described |
|
| Outcomes | Mortality Incidence of ventricular arrhythmias | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Astra Chemicals Ltd
Role of sponsor: not reported Trial conduction data: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Prearranged code held by a member of nursing staff" (page 910) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal from study, % (n/N)
Reasons
|
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) Industry bias |
Bergdahl 1978.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Sweden Follow‐up period: unclear | |
| Participants | Randomly assigned: N = 31
Age ≥ 50 years, % (n/N)
Age < 50 years, % (n/N)
Gender, male, % (n/N)
Inclusion criterion: ventricular arrhythmias had not been controlled or recurred 0.5 to 24 hours after initiation of lidocaine treatment Exclusion criteria (1 of the following)
|
|
| Interventions | Lidocaine group: 50 mg (bolus) and infusion at 3 mg/min Dimethylammonium chloride: infusion (600 mg in 200 mL saline solution). Infusion time: 30 minutes; first 3 participants; and 60 minutes; next 13 participants Co‐interventions: oxygen 4 Lts/min (nasal catheter), hydromorphone or pentazocine (endovenous) for relief pain | |
| Outcomes | Type and frequency of side effects | |
| Notes | A priori sample size estimation: not reported Sponsor: Hässle AB, Gothenburg Sweden Role of sponsor: supplied the drugs studied and performed the analysis of plasma drug concentrations Trial conduction dates: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were allocated randomly" (page 311) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal from study
|
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study Comments: This study did not include important outcomes (mortality and ventricular fibrillation) |
| Other bias | High risk | Design bias (Porta 2008) Industry bias |
Capucci 1985.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Italy Follow‐up period: not reported | |
| Participants | Randomly assigned: N = 25
Age, years, mean (standard deviation not reported)
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine group: initial bolus of 1 mg/kg, in a pump infusion at a dose of 10 mg/min for 20 minutes. Maintenance dose of 1.5 mg/min
Amiodarone group: initial bolus of 5 mg/kg in 2 minutes followed by a second bolus of 150 mg after 3 minutes (if previous dose was insufficient). Maintenance dose: 1.8 g/24 hours in continuous infusion pump Co‐intervention: defibrillation |
|
| Outcomes | Number and type of ventricular premature beats Number and duration of episodes of ventricular tachycardia Appearance of ventricular fibrillation Blood pressure at baseline, every 2 minutes to 10 minutes from beginning of infusion, every 5 minutes from 11 to 60 minutes, every 2 hours in the remaining 23 hours Symptoms and/or clinical signs of congestive heart failure Electrocardiographic parameters at baseline, at 60 minutes and 24 hours after infusion of the drug |
|
| Notes | A priori sample size estimation: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported in such a study Comments: This study did not report mortality |
| Other bias | High risk | Design bias (Porta 2008) |
Chopra 1971.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Canada Follow‐up period: unclear | |
| Participants | Enrolled: 805 Randomly assigned: N = 82
Age, years
Gender, male, % (n/N)
Inclusion criteria (≥ 1 of the following types of ventricular ectopic activity within 72 hours of infarction)
Exclusion criteria
|
|
| Interventions | Lidocaine group Single rapid intravenous injection of 50 mg of lidocaine (first bolus)
Infusion was continued for 24 hours and then tapered off by a reduction of the infusion rate to half the previous rate for 2 hours. In participants whose ectopic activity was not suppressed by first or second bolus, no further treatment was given Placebo: normal saline solution under the same parameters as for the intervention group Co‐interventions: not reported |
|
| Outcomes | Mortality Incidence of major ventricular arrhythmias Effectiveness of intravenous lidocaine in suppressing ventricular ectopic activity after acute myocardial infarction | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Pharmaceutical Manufacturing Co. Ltd.
Role of sponsor: supplied lidocaine, saline ampoules and randomisation code Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | " ... according to a randomised numerical code, ... " (page 668) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the present double‐blind trial" (page 668) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study Comment: This study did not discuss safety |
| Other bias | High risk | Design bias (Porta 2008) Industry bias |
Cuendet 1988.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Switzerland Follow‐up period: 24 hours | |
| Participants | Randomly assigned: N = 19
Age, years, mean (SE or SD)
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria: not reported |
|
| Interventions | Lidocaine, infusion at mean dose of 42 (8.8) µg/min/kg Pirmenol, infusion at mean dose of 6.1 (1.6) µg/min/kg Co‐interventions: not reported | |
| Outcomes | Prevalence of ventricular arrhythmias (non‐ventricular fibrillation). Safety |
|
| Notes | A priori sample size estimation: no Sponsor: not reported Data were taken from "Resumés du XVIII. Congres de I Union Therapeutique Internationale" (date: unclear) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | " ...have been randomised..." (page 158) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double blind randomized study" (page 158) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported in such a study
Comment: This study did not report mortality and ventricular fibrillation ≥ 1 outcomes of interest in the review are reported incompletely, so they cannot be entered into a meta‐analysis Quote: "...side effects have been observed in 10 pts, 5 in each group, but interruption of treatment was not necessary" (page 158) |
| Other bias | High risk | Design bias (Porta 2008) Bias in presentation of data (Porta 2008) |
Darby 1972.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: United Kigndom Follow‐up period: 48 hours | |
| Participants | Enrolled: 322 Randomly assigned: N = 203
Age, years: not reported Gender, male, %: 79.3 (both groups) Inclusion criterion: myocardial infarction in preceding 48 hours Exclusion criteria
|
|
| Interventions | Lidocaine: 200 mg, intramuscular Injection in emergency department, and infusion of 2 mg lidocaine/min for 48 hours on arrival at the coronary care unit Control: no routine antiarrhythmic treatment; no details supplied Co‐intervention: not stated | |
| Outcomes | Mortality Incidence of ventricular arrhythmias (ventricular extrasystole, ventricular fibrillation, ventricular tachycardia) | |
| Notes | Sample size estimation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...randomly consigned..." (page 818) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal from study
Lidocaine group: 11% (11/103) Reasons
Control group: no reported withdrawal |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Depaepe 1974.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Belgium Follow‐up period: 3 days | |
| Participants | Randomly assigned patients: N = 24
Age, years, mean (standard error)
Gender, male, % (n/N)
Inclusion criterion: Participants with acute myocardial infarction confirmed by cardiac enzyme dosage (creatinine phosphokinase, serum glutamic oxaloacetic transaminase, lactate dehydrogenase but not troponin) and for which symptoms occurred within 36 hours Exclusion criteria (≥ 1 of the following were present)
|
|
| Interventions | Lidocaine, hours 0 to 0.5: 2 mg/min = 60 mg 0.5 to 24: 2 mg/min = 2800 mg 24 to 48: 2 mg/min = 2800 mg 48 to 72: 2 mg/min = 2800 mg Aprinidine, intravenously, hours 0 to 0.5: 2 mg/min = 200 mg 0.5 to 24: 2 mg/min = 200 mg 24 to 48: 2 mg/min = 200 mg 48 to 72: 2 mg/min = 200 mg Co‐intervention: not reported |
|
| Outcomes | Mortality Safety |
|
| Notes | Sample size estimation a priori: not reported
Sponsor: A. Christiaens, S.A.
Role of sponsor: aprindine provided freely by Christiaens Pharmaceutical Company Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "...were randomly divided in two groups" (page 412) Comment: They used a random number table |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals from study Lidocaine group: 10% (1/10) Reasons Neurological coma |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) Comment: This trial did not report ventricular fibrillation |
| Other bias | High risk | Design bias (Porta 2008) Industry bias |
Dunn 1985.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Northern Ireland Follow‐up period: not reported | |
| Participants | Randomly assigned: N = 425 Withdrawal from study: 7.3% (31/425) Eight participants who had a proved myocardial infarction did not enter the study (page 354). Twenty‐three patients did not fulfil entry criteria Analysed, % (n/N)
Age: 56 years (both groups); not reported by comparison groups Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria (≥ 1 of the following was present)
|
|
| Interventions | Lidocaine, 300 mg, intramuscular route, followed by lidocaine, 100 mg, by intravenous bolus over 3 minutes Placebo: normal saline (equivalent volume of normal saline) | |
| Outcomes | Incidence of ventricular fibrillation, sustained ventricular tachycardia, warning arrhythmias Incidence of central nervous system side effects, hypotension, tachycardia, bradycardia, asystole | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: November 1981 to February 1983 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | " ...we undertook a double‐blind randomised trial..." (page 354) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals from study: 7.3% (31/425) Eight patients who had a proved myocardial infarction did not enter the study (page 354) Twenty‐three patients did not fulfil entry criteria
Comment: Trial authors did not report lost participants by comparison group |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Bias of presentation data, design bias (Porta 2008) |
Hargarten 1990.
| Study characteristics | ||
| Methods | Parallel‐design (2 arms) Country: USA Follow‐up: unclear | |
| Participants | Randomly assigned: N = 1427
Age, years, mean (standard error or standard deviation unclear) (total group)
Gender, male, % (n/N) Total group: 50.17 (716/1427) Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine, intravenous (IV), initial bolus of 1 mg/kg; simultaneous 2 mg/min IV drip to maintain therapeutic blood levels. Ten minutes after first dose of lidocaine, second bolus of 0.5 mg/kg to prevent decrease to below therapeutic range Control group: not detailed Co‐intervention: not reported |
|
| Outcomes | Incidence of sudden death Incidence of warning arrhythmia | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: January 1984 to January 1988 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "... was generated from a table of numbers from the Minitab software package of Perkin‐Elmer 3230 Supermini Computer" (page 82) |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Bias in presentation data (Porta 2008) |
Horowitz 1981.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Australia Follow‐up period: 48 hours | |
| Participants | Randomly assigned: N = 24
Age, years, mean (standard error)
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine: 100 mg/bolus, infusion of 3 mg/min for 1 hour; thereafter, reduced to 2 mg/min after 1 hour Mexiletine: 200 mg/bolus, infusion of 1 mg/min for 1 hour; thereafter, reduced to 0.5 mg/min after 1 hour Co‐interventions: electroversion, bolus of mexiletine (200 mg) or lidocaine (100 mg) (page 410) | |
| Outcomes | Ocurrence of complex ventricular tachyarrhythmia Comment: Trial did not assess the outcome explicitly | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Austin Hospital Research Foundation Role of sponsor: support of the study Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... were randomised and allocated to receive..." (page 410) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ "Patients who had persisted multifocal ventricular extrasystoles or ventricular tachycardia or ventricular fibrillation... were given additional bolus of 200 mg of mexiletine or 100 mg of lignocaine" (page 410) This trial did not report whether the co‐intervention (additional bolus) was allocated concealment or not |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal from study, % (n/N)
Reasons
|
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) Comment: This trial did report safety Quote: "the greater efficacy of mexiletine was not associated with increased drug toxicity" (page 409) |
| Other bias | High risk | Design bias and bias in presentation of data (Porta 2008) |
Keefe 1986.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Follow‐up: 48 hours Country: USA | |
| Participants | Randomly assigned: N = 29
Age, years, mean (SD)
Gender, male, % (n/N)
Inclusion criterion: acute myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine, 100 mg over 2 minutes, was given, followed by 60 mg over 15 minutes, then 1000 mg every 6 hours for 48 hours; additional bolus of 100 mg could be given for breakthrough arrhythmias and the maintenance dose of lidocaine reduced to 1000 mg over 8 hours for signs of toxicity, for 3 to 7 days Tocainide, 250 mg over 2 minutes, 500 mg over 15 minutes, then 500 mg every 6 hours for 48 hours; bolus of 250 mg of tocainide could be given for breakthrough arrhythmias and the infusion lengthened to 500 mg over 8 hours for signs of toxicity, for 3 to 7 days Co‐interventions: oxygen by nasal cannula, subcutaneous heparin, nitroglycerin, morphine sulphate and furosemide |
|
| Outcomes | Prophylaxis of ventricular arrhythmias | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Merck Sharp and Dohme, West Point, Pennsylvania
Role of sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... with a randomized block sign design with two treatment groups" (page 528) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal from study Overall: 24.13% (7/29): "Of the 29 patients who entered the study, 22 completed the infusion portion of the study and 18 entered the oral phase..." (page 528) On page 530, table III, this was reported: "withdrawn because of adverse effects"
Inconsistency is evident between information published on page 528 and page 530 regarding withdrawal |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) Bias in the presentation of data (Porta 2008) Industry bias |
Kostuk 1969.
| Study characteristics | ||
| Methods | Parallel design (2 arms)
Country: Canada Follow‐up: 48 hours |
|
| Participants | Enrolled: 95 Randomly assigned: N = 65
Age: not stated Gender: not stated Inclusion criteria: acute myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine 1 mg/min (infusion rate) Placebo: 5% glucose in water (at similar infusion rate) Co‐intervention: not reported | |
| Outcomes | Prophylactic benefit in prevention of ventricular arrhythmias | |
| Notes | Source of data: abstract of scientific sessions Sample size calculation a priori: not reported Sponsor: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "in random fashion..." (page III‐125) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Pts received, unknown to the nurses, either..." (page III‐125) 'pts' means patients |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study Comments: This trial does not report mortality, ventricular fibrillation |
| Other bias | High risk | Design bias (Porta 2008) |
Kuck 1985.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Germany Follow‐up period: unclear | |
| Participants | Randomly assigned: N = 49
Age, years, mean (standard error or deviation not reported explicitly)
Gender, male, % (n/N)
Inclusion criterion: patients with acute myocardial infarction Exclusion criterion: not stated |
|
| Interventions | Lidocaine, initial bolus of 200 mg following intravenous infusion of 2 mg/min Control: no lidocaine; no details were stated Co‐intervention: intracoronary thrombolysis | |
| Outcomes | Prophylactic of ventricular tachyarrhythmias following recanalisation of an occluded coronary artery | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...were randomized into two groups..." (page 807) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study Comments: This trial reported neither mortality nor ventricular fibrillation |
| Other bias | High risk | Design bias (Porta 2008) |
Lie 1974.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: The Netherlands Follow‐up period: 48 hours | |
| Participants | Enrolled: 225 Randomly assigned: N = 212
Age, years, mean
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine, initial bolus of 100 mg, followed by infusion of 3 mg/min
Placebo: 5% glucose and water Co‐interventions: defibrillation |
|
| Outcomes | Preventing primary ventricular fibrillation | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Ottenvanger (Astra) Pharmaceutical
Role of sponsor: not reported Trial conduction dates: June 1973 to September 1974 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... on the basis of randomization" (page 1324) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The patients were not informed whether they might or might not receive lidocaine..." (page 1324) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding assessors to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals from the study Post randomisation Overall: 6% (13/212) Reasons
|
| Selective reporting (reporting bias) | High risk | One clinically relevant and reasonably expected outcome (mortality) was not reported, and data on that outcome were likely to have been recorded Comment: Side effects in control group were not reported |
| Other bias | High risk | Design bias (Porta 2008) Industry bias |
Lie 1978.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: The Netherlands Follow ‐up: 1 hour | |
| Participants | Enrolled: 321 Randomly assigned: N = 300
Age, years
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine group: lidocaine 300 mg intramuscular in a 10% solution Placebo: "sodium chloride 0.65 percent and water in the deltoid muscle..." (page 487) | |
| Outcomes | Incidence of major ventricular arrhythmias Mortality | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...randomized patients received..." (page 487) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The nature of the injected solution was unknown to the medical and nursing staff" (page 487) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawls from the study
|
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
NNLIT 1992.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Norwegian countries Follow‐up period: 3 hours | |
| Participants | Enrolled: 204 Randomly assigned: N = 197
Proved diagnosis of acute myocardial infarction: 63% (125/197)
Age ≥ 50 years, % (n/N)
Age < 50 years, % (n/N(
Gender, male, % (n/N)
Inclusion criteria: suspected of myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine, 100 mg, intravenous bolus, followed by 300 mg intramuscular injection Placebo group: physiological saline solution Co‐interventions: defibrillation, thrombolytic and streptokinase therapy | |
| Outcomes | Prevention of ventricular fibrillation | |
| Notes | Sample size calculation a priori: yes (Berntsen 1991)
This trial was stopped early ("the study had to be terminated at this time") (page 1479)
Sponsor: Norwegian Council on Cardiovascular Disease and the Laerdal Fundation.
Role of sponsor: not reported Trial conduction dates: 19 March 1988 to 12 July 1991 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | " randomly allocated... " (page 1479) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The nature of the trial material contained in each packet was unknown to all involved parties" (page 1479) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals from the study, % (n/N)
|
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
O'Brien 1973.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: New Zealand Follow‐up period: 48 hours | |
| Participants | Randomly assigned: N = 300
Age: not reported Gender (male): not reported Inclusion criterion: proven myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine, 75 mg, first bolus. Infusion 2.5 mg/min in 5% dextrose at 1 mL/min Placebo: 5% dextrose solution Co‐intervention: not reported | |
| Outcomes | Reducing the frequent of ventricular fibrillation and ventricular tachycardia | |
| Notes | Sample size calculation a priori: not reported Sponsor: not reported Trial conduction dates: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "allocation.... by means of random selection" (page 36) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ Comments: This was described as double blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Pedersen 1986.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Denmark Study phase: III Follow‐up: 12 hours | |
| Participants | Randomly assigned: N = 76
Age, years
Gender: not stated Inclusion criteria
Exclusion criteria
|
|
| Interventions | Disopyramide (Norpace): bolus injection 150 mg + infusion 24 mg/h. Infusion regulated depending on effect and side effects to between 30 to 90 mL/h Lidocaine: bolus injection 100 mg + infusion 100 mg/h. Infusion regulated depending on effects and side effects to between 30 to 90 mL/h Co‐interventions: not stated |
|
| Outcomes | Not defined in Methods Ventricular extrasystoles Death |
|
| Notes | A priori sample size estimation: no Sponsor: not stated Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "...double blind study plan" (page 1) Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss after lidocaine: 7.89% (3/38) Loss after disopyramide: 5.26% (2/38) |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified |
| Other bias | High risk | Design bias (Porta 2008) |
Pharand 1995.
| Study characteristics | ||
| Methods | Parallel design (2 arms)
Country: USA Follow‐up: 40 hours |
|
| Participants | Randomly assigned: N = 200
Age, years, mean (standard deviation)
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine 2 gr in 500 mL of 5% dextrose in water: infusion for a period of 40 hours Placebo: 5% dextrose in water Co‐intervention group: Quote: "As a part of the standard practice in the Emergency Department of our hospital, patients received an intravenous lidocaine bolus" (page 472) |
|
| Outcomes | Efficacy and safety for prophylaxis of ventricular arrhythmias in patients with uncomplicated acute myocardial infarction | |
| Notes | Sample size calculation a priori: no
Sponsor: not reported Trial conduction dates: March 1990 to November 1992 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomized..." (page 472) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "This was a double‐blind, randomised placebo‐controlled trial" (page 471) Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient reporting of attrition/exclusions to permit judgement of ‘low risk’ or ‘high risk’ (e.g. number randomly assigned not stated, no reasons for missing data provided) |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Bias of presentation data, design bias (Porta 2008) |
Pitt 1971.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Australia. Follow‐up: 48 hours | |
| Participants | Randomly assigned: N = 222 Class 1 (participants without haemodynamic disturbances): 113
Class 2 (hypotension blood pressure < 90 mmHg after relief of pain, or left ventricular failure): 109
Age, years (range) Class 1
Class 2
Gender, male, % (n/N) Class 1
Class 2
Inclusion criteria: not reported Exclusion criteria
|
|
| Interventions | Lidocaine: 2.5 mg/min for 48 hours in 5% dextrose
For the first half of the trial, all participants receiving lidocaine were given an intravenous bolus injection of 75 to 100 mg, but this was not routinely administered in the second half of the trial Placebo: dextrose 5% by intravenous infusion Co‐interventions: pacemaker and lidocaine in control group because of the development of ventricular tachyarrhythmia |
|
| Outcomes | Mortality Fequency of ventricular tachyarrhythmia | |
| Notes | Sample size calculation a priori: no
Sponsor: Astra Chemicals Pty. Ltd.
Rol of the sponsor: not reported Trial conduction dates: 5 January 1968 to 30 June 1970 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly allotted..." (page 613) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "165 patients were excluded..." (page 613) It is unclear whether these exclusions occurred before or after randomisation |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Bias of presentation data, design bias (Porta 2008) Industry bias |
Poprawski 1987.
| Study characteristics | ||
| Methods | Parallel design (2 arms)
Country: Poland Follow‐up (hours): not stated |
|
| Participants | Randomly assigned: N = 172
Age, years, mean (range)
Gender, male, % (n/N)
Inclusion criterion: patients with acute myocardial infarction by World Health Organization and International Cardiology Society Criteria, 1979 Exclusion criteria
|
|
| Interventions | Lidocaine intravenous. 75 mg over 1 minute immediately on admission – no later than within 8 hours of onset of pain, then 3 doses intravenous 50 mg each over 1 minute, followed by 2 mg/min intravenous infusion pump Placebo: 5% glucose solution Co‐interventions: routine administration of nitroglycerin and heparin intravenously |
|
| Outcomes | Endpoints reported for the 2 arms
|
|
| Notes | Comment: This trial was written in Polish. Therefore, we describe some details here
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomly divided in two groups" (page 667) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient reporting of attrition/exclusions to permit judgement of ‘low risk’ or ‘high risk’ |
| Selective reporting (reporting bias) | Unclear risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Rademaker 1986.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Canada Follow‐up period: 48 hours | |
| Participants | Randomly assigned: 285
Proven acute myocardial infarction: 75 participants Comparison group not given Age, years, mean (standard error or standard deviation), not stated
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine, initial 100 mg intravenous bolus given over 3 to 5 minutes, followed by 3 mg/min intravenous by infusion pump and a second 100 mg intravenous bolus 30 minutes after the first bolus Placebo: no stated details Co‐intervention: not reported |
|
| Outcomes | Safety | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Astra Pharmaceuticals supplied lidocaine Trial conduction dates: July 1980 to December 1984 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...were randomized in a double‐blind manner..." (page 72) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study. Investigators did not assess mortality and ventricular fibrillation. Furthemore, safety data were reported incompletely, so they cannot be entered into a meta‐analysis |
| Other bias | High risk | Bias of presentation data, design bias (Porta 2008) Industry bias |
Rehnqvist 1983.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Sweden Follow‐up period: 24 hours | |
| Participants | Randomly assigned: N = 40
Age, years, mean (range)
Gender, male, % (n/N)
Inclusion criteria (≥ 1 of the following high‐grade ventricular arrhythmias had to be present for inclusion)
Exclusion criteria
|
|
| Interventions | Tocainide: bolus injection of 750 mg over 15 minutes, immediately followed by 800 mg orally and thereafter 400 mg TID Lidocaine: bolus injection of 75 mg followed by continuous infusion at a rate of 2 mg/min Co‐interventions: not reported |
|
| Outcomes | Suppressing premature ventricular contractions | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "were randomized to treatment" (page 22) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "... in an open fashion" (page 22) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "... in an open fashion" (page 22) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study Comment: Trial does not assess mortality and ventricular fibrillation |
| Other bias | High risk | Bias of presentation data and design bias (Porta 2008) |
Rehnqvist 1984.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Sweden Follow‐up period: 24 hours | |
| Participants | Randomly assigned: N = 20
Age, years: 61 (both groups) Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine
Propafenone
Co‐interventions: not reported |
|
| Outcomes | Reduction in premature ventricular complexes | |
| Notes | A priori sample size estimation: not reported
Sponsor: not reported. Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were randomly allocated..." (page 527) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "... in an open fashion" (page 22) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "... in an open fashion" (page 22) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal from study Propafenone group: 30% (3/10) Lidocaine group: not reported Reason Increasing numbers of premature ventricular complexes |
| Selective reporting (reporting bias) | High risk | "The study report fails to include results for a key outcome that would be expected to have been reported such a study" This study did not report mortality |
| Other bias | High risk | Bias of presentation data, design bias (Porta 2008) |
Rolli 1981.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Italia Follow‐up period: 3 hours | |
| Participants | Randomly assigned: N = 50
Age, years (standard error or standard deviation), not stated
Gender, male, % (n/N)
Inclusion criteria: not stated Exclusion criteria: not stated |
|
| Interventions | Mexiletine: bolus endovenous 2 mg/kg in 5 minutes, followed by a continuous infusion of 500 mg in the next 3 hours (250 mg the first hour, and the other 250 mg the next 2 hours) Maintenance intravenous infusion (0.5 to 1 mg/min) depending on therapeutic response Lidocaine group: bolus intravenous 2 mg/kg, continued to an infusion of 2 mg/min. Continuous infusion of 5% dextrose solution alone at same speed as intervention Co‐intervention: not reported | |
| Outcomes | Ventricular arrhythmia | |
| Notes | A priori sample size estimation: not reported Sponsor: not reported Trial conduction dates: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...randomly allocated into two groups" (page 468) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study reported information about mortality and safety Comment: Trial did not assess ventricular fibrillation |
| Other bias | High risk | Bias in presentation of data, design bias (Porta 2008) |
Ronnevik 1987.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Norway Follow‐up period: 24 hours | |
| Participants | Randomly assigned: N = 68
Age, years
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Disopyramide: intravenous bolus of 150 mg (100 mg to persons < 60 kg), followed by a constant infusion of 30 mg/h for 24 hours
Lidocaine: intravenous bolus injection of 100 mg (75 mg to persons < 60 kg), followed by a constant infusion of 3 mg/h for 24 hours
Co‐interventions
|
|
| Outcomes | Death Ventricular arrhythmias |
|
| Notes | A priori sample size estimation: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...were randomised to disopyramide and..." (page 30) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals from study
Overall: 15% (10/68) Disopyramide: 15.1% (5/33) Reasons
Lidocaine: 14.2% (5/35) Reasons
|
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Rossi 1976.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Italy Follow‐up period: 3 weeks | |
| Participants | Randomly assigned: N = 246
Age, years: not reported Gender, male: not reported Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine by an intramuscular injection of 250 mg Placebo: physiological solution and intervention in equal volumes Co‐intervention: not reported |
|
| Outcomes | Mortality Incidence of severe arrhythmias | |
| Notes | Sample size calculation a priori: not reported
Sponsor: Astra Company
Role of sponsor: Lidocaine and saline solutions have been packaged for the double‐blind by Astra Company Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "and was randomised... " (page 221) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The contents of the syringe [are] kept secret until after the search. The distribution of the drug or placebo was randomised at the time of the package" (page 221) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | All clinically relevant and reasonably expected outcomes were reported |
| Other bias | High risk | Design bias (Porta 2008) Industry bias |
Sadowski 1999.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Follow‐up period: 48 hours Country: Poland | |
| Participants | Randomly assigned: N = 903
703 randomly assigned to streptokinase plus heparin or heparin alone Age, years, mean
Gender total group, male, %: 79.84 Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine, intravenously as four 50‐mg boluses at 2‐minute intervals, followed by continuous infusion of 3 mg/min for 12 hours, then 2 mg/min for 36 hours Control group: no lidocaine Co‐interventions: streptokinase, 1.5 million unit intravenous infusion titrated to maintain an activated partial thromboplastin time 2 to 2.5 times normal for 48 hours. All patients received intravenous nitroglycerin, 20 to 150 µg/min, titrated to control blood pressure and heart rate | |
| Outcomes | Mortality Ventricular fibrillation Asystole Atrioventricular block | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: "...between 1986 and 1987" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... were randomly assigned in a 2×2 factory design..." (page 793) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) However, safety data were reported incompletely, so they cannot be entered into a meta‐analysis |
| Other bias | High risk | Design bias and bias in data presentation (Porta 2008) |
Sandler 1976.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: United Kingdom Follow‐up period: unclear | |
| Participants | Randomly assigned: N = 181
Age, years: not reported Gender: not reported Inclusion criteria: patients with suspected myocardial infarction Exclusion criteria
|
|
| Interventions | Lidocaine was given by an intramuscular injection of 200 mg or 300 mg Placebo: physiological solution and intervention in equal volumes Co‐intervention: atropine 0.6 mg by intravenous injection | |
| Outcomes | Incidence of arrhythmias | |
| Notes | A priori sample size estimation: not reported Sponsor: not reported Trial conduction dates: not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... according to a randomised allocation..." (page 564) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information about the blinding level process to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | Quote: "The study report fails to include results for a key outcome that would be expected to have been reported for such a study" Comments: Mortality and ventricular fibrillation were not reported in this study |
| Other bias | High risk | Design bias and bias in data presentation (Porta 2008). |
Sbarbaro 1979.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: USA Follow‐up period: 2 hours | |
| Participants | Randomly assigned: N = 26
Patients with acute myocardial infarction
Age, years: not reported for patients with acute myocardial infarction Gender, male: not reported for patients with acute myocardial infarction Inclusion criteria
Exclusion criteria
|
|
| Interventions | Dysopyramide 2 mg/kg over 15 minutes, then 2 mg/kg over 45 minutes, then 0.4 mg/kg per hour maintenance Lidocaine 75 mg or 100 mg bolus injection, then 3 or 4/min maintenance infusion, with additional bolus injection as indicated clinically Co‐intervention: not given |
|
| Outcomes | Incidence of arrhythmias | |
| Notes | A priori sample size estimation: not reported
Sponsor: Searle Labboratories
Role of the sponsor: supplied the drug and provided determination of drug blood levels Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "... to a modification of a computer‐based randomization scheme" (page 514). |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "The investigators and the patient´s physician were aware of which agent [was] being administered" (page 514) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated Comment: This trial included only 4 participants with acute myocardial infarction |
| Selective reporting (reporting bias) | High risk | One or more outcomes of interest in the review are reported incompletely, so they cannot be entered into a meta‐analysis Comment: This trial included only 4 participants with acute myocardial infarction. However, researchers did not provide information on these individuals |
| Other bias | High risk | Design bias and bias in data presentation (Porta 2008) Industry bias |
Solimene 1983.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Brazil Follow‐up period: 12 hours | |
| Participants | Randomly assigned: N = 43
Age, years, mean (standard error or standard deviation: not stated)
Gender, male, % (n/N)
Inclusion criteria: based on clinic criteria and electrocardiogram (no additional details) Exclusion criteria: not stated |
|
| Interventions | Lidocaine
Control group: no lidocaine Co‐intervention: cardioversion |
|
| Outcomes | Ventricular extrasystoles Ventricular tachycardia Ventricular fibrillation | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported
Trial conduction dates: not stated E‐mail was sent to the main trial author |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... os pacientes foram seleccionados, ao acaso,..." (page 377) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | High risk | The study report fails to include results for a key outcome that would be expected to have been reported for such a study This study did not report mortality data |
| Other bias | High risk | Design bias and bias in data presentation (Porta 2008). |
Touboul 1988.
| Study characteristics | ||
| Methods | Parallel design (3 arms) Country: France Follow‐up period: 24 hours | |
| Participants | 112 enrolled Randomly assigned: N = 89
Age, years
Gender, male, % (n/N)
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine: intravenous as a bolus injection of 100 mg, followed by an infusion of 2 mg/min Propafenone: bolus of 105 mg, followed by 300 mg orally every 8 hours Placebo: no details of its nature given Co‐intervention group: not reported |
|
| Outcomes | Suppression of complex arrhythmias, couples and ventricular tachycardia | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: April 1985 to March 1986 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "... were randomly assigned to treatments" (page 1189) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "A double blind, placebo‐controlled trial" (page 1188) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawals from study
Excluded from analysis: 20% (23/112)
Comment: It is unknown whether these exclusions were treated Reasons No myocardial infarction; defective Holter recording By comparison groups: not reported Withdrawals among remaining participants: 8% (7/89)
|
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias and bias in data presentation (Porta 2008) |
Valentine 1974.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Trial duration: 15 months Follow‐up: 30 days Country: Australia | |
| Participants | Randomly assigned (class 1 or 2): N = 269
Age, years
Gender, male, %
Inclusion criteria: chest pain with a provisional diagnosis of acute cardiac infarction Exclusion criteria
|
|
| Interventions | Lidocaine 300 mg intramuscular (10% solution) Placebo: physiological saline Co‐intervention: defibrillation |
|
| Outcomes | Mortality | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "...trial randomly..." (page 1327) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Low risk | "... a reply‐paid envelope and another sealed envelope containing the code..." (page 1327) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "participating doctors remained ignorant of the nature of the trial material injected..." (page 1327) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Wennerblom 1982.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Sweden Follow‐up period: 3 hours | |
| Participants | Enrolled: 407 Randomly assigned: N = 150
Age, years
Gender, male, %: 60 Inclusion criteria
Exclusion criteria
|
|
| Interventions | Lidocaine: intramuscular injection of 300 mg Placebo: physiological solution and intervention in equal volumes Co‐intervention: atropine |
|
| Outcomes | Mortality Incidence of ventricular arrhythmias | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were allocated at random..." (page 516) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "...double blind" (page 516) "Each dose was prepared in advance by person not involved in the study..." (page 517) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The arrhythmia analysis... was done by one of the authors 'blindly'..." (page 517) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The report gave the impression that no dropouts or withdrawals had occurred, but this was not specifically stated |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available, but it is clear that published reports describe all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon) |
| Other bias | High risk | Design bias (Porta 2008) |
Wyse 1988.
| Study characteristics | ||
| Methods | Parallel design (2 arms) Country: Canada Follow‐up period: 24 hours | |
| Participants | Randomly assigned: N = 333
Age, years, mean (standard error)
Gender, male, %
Inclusion criteria
Excluded criteria
|
|
| Interventions | Lidocaine: 100 mg intravenous loading infusion given over 3 to 5 minutes, followed by 3 mg/min continuous intravenous maintenance. An identical 100 mg intravenous infusion was administered 30 minutes after the first loading infusion. Dosage was adjusted on a milligram‐per‐kilogram basis for participants < 50 kg or > 90 kg Control: no information given about this issue | |
| Outcomes | Safety of lidocaine therapy in participants with acute myocardial infarction | |
| Notes | Sample size calculation a priori: not reported
Sponsor: not reported Trial conduction dates: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "On randomization, drug (placebo or lidocaine) was administered..." (page 508) Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the allocation concealment process to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "...(placebo or lidocaine) was administered in a double blind manner" (page 508) Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout from the study
Reasons
|
| Selective reporting (reporting bias) | High risk | One or more outcomes of interest in the review are reported incompletely, so they cannot be entered into a meta‐analysis "in mortality rate (selective=3%, prophylactic=5%, p=NS)" (page 507) This trial reported results by approach (prophylaxis vs selective) rather than by lidocaine vs placebo |
| Other bias | High risk | Design bias and bias in data presentation (Porta 2008) |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Antman 1992 | Narrative review |
| Beloev 1983 | Observational study |
| Bernard 1970 | Narrative review |
| Bernard 1972 | Controlled clinical trial |
| Bertini 1993 | It was reported as a "randomised controlled trial" (page 668). However, the sequence generation was conducted by an inappropriate method (born in odd years) |
| Bleifeld 1973 | It was reported as a "randomised controlled trial" (page 119). However, the sequence generation was conducted by an inappropriate method (dates of birth) |
| Campbell 1978 | Observational study |
| Campbell 1980 | Narrative review |
| Campbell 1983 | Narrative review |
| Church 1972 | It was reported as a "randomised controlled trial" (page 139). However, the sequence generation was conducted by an inappropriate method (birthday) |
| De Silva 1981 | Systematic review of randomised clinical trials |
| Destuelles 1969 | Observational study |
| Diederich 1979 | It was reported as a "randomised controlled trial" (page 1007). However, the sequence generation was conducted by an inappropriate method (sequence generated by participant admission to the hospital) |
| Egre 1981 | Observational study |
| Fehmers 1972 | Controlled clinical trial |
| Formichev 1995 | Narrative review |
| Garratt 1998 | Non‐randomised clinical trial |
| Gianelly 1967 | Observational study |
| Gonzalez 1977 | Controlled clinical trial |
| Goodman 1979 | Narrative review |
| Hine 1989 | Systematic review of randomised clinical trials |
| Iosava 1982 | Narrative review |
| Jaffe 1992 | Narrative review |
| Kudenchuk 2013 | Observational study |
| Lechleitner 1987 | Narrative review |
| Leone 1991 | Non‐randomised clinical trials |
| MacMahon 1988 | Systematic review on randomised clinical trials |
| Mazur 1982 | Observational study |
| Miller 1973 | Controlled clinical trial |
| Mogensen 1971 | It was reported as a "randomised controlled trial" (page 41). However, the sequence generation was conducted by an inappropriate method (date of birth) |
| Noneman 1978 | Narrative review |
| Oltmanns 1979 | Narrative review |
| Pentecost 1981 | Observational study |
| Riabokon' 1980 | Controlled clinical trial |
| Ribner 1979 | Narrative review |
| Ruano 1989 | Observational study |
| Ryden 1973 | It was reported as a "randomised controlled trial" (page 1125). However, the sequence generation was conducted by an inappropriate method (date of birth) |
| Shih 1995 | Observational study |
| Singh 1976 | Controlled clinical trial |
| Szeplaki 1973 | Controlled clinical trial |
| Szeplaki 1976 | Controlled clinical trial |
| Teo 1993 | Systematic review of randomised clinical trials |
| Wojtala 1982 | Non‐randomised clinical trial |
| Wyman 2004 | Observational study |
Characteristics of studies awaiting classification [ordered by study ID]
Bolinska 1971.
| Methods | Unknown |
| Participants | Acute myocardial infarct |
| Interventions | Lidocaine |
| Outcomes | Unknown |
| Notes |
Hopperstead 1980.
| Methods | Unknown |
| Participants | Patients with acute myocardial infarction |
| Interventions | Prophylactic lidocaine |
| Outcomes | Unknown |
| Notes |
Knight 1973.
| Methods | Unknown |
| Participants | Myocardial infarction |
| Interventions | Prophylactic lidocaine |
| Outcomes | Unknown |
| Notes |
Differences between protocol and review
Based on the Consolidated Standards of Reporting Trials (CONSORT) statement, we changed the term "Safety" to "Adverse Events". Safety may be considered as substantive evidence of absence of harm. This term is often misused when evidence of harm is simply absent (Ioannidis 2004).
We did not conduct a cumulative meta‐analysis to assess the influence of individual studies (Egger 2001). We prefer to conduct trial sequential analyses to assess risks of random error in our cumulative meta‐analyses (CTU 2011).
We conducted two additional subgroup analyses involving only patients with acute myocardial infarction and trials without suspicion of industry bias versus trials with suspicion of industry bias. We considered both analyses to be of clinical importance.
As all included trials were rated as having high risk of bias, we were not able to conduct sensitivity analyses to compare trials assigned 'low risk of bias' versus trials assigned 'high risk of bias', as planned.
A priori, we used a fixed‐effect model in combining data. However, we used a random‐effects model to minimise sources of variance.
Contributions of authors
Arturo Martí‐Carvajal conceived of and drafted the review with comments from Daniel Simancas, Vidhu Anand and Shirikant Bangdiwala. Arturo Martí‐Carvajal serves as contact author for this review.
Sources of support
Internal sources
-
Universidad Tecnológica Equinoccial, Ecuador
Partially funded.
External sources
-
Iberoamerican Cochrane Center, Spain
Academic.
-
Cochrane Heart Group, UK
Academic.
Declarations of interest
In 2004, Arturo Martí‐Carvajal was employed by Eli Lilly to run a four‐hour workshop on 'How to critically appraise clinical trials on osteoporosis and how to teach this'. This activity was not related to his work with The Cochrane Collaboration or to any Cochrane review.
In 2007, Arturo Martí‐Carvajal was employed by Merck to run a four‐hour workshop on 'How to critically appraise clinical trials and how to teach this'. This activity was not related to his work with The Cochrane Collaboration or to any Cochrane review.
Vidhu Anand: none known.
Shrikant Bangdiwala: The presentation of the results of the sensitivity analysis of the review may be but are in my view unlikely to be informed by results of a grant from the European Commission on "Evaluation and development of measures to uncover and overcome bias due to non‐publication of clnical trials".
Daniel Simancas‐Racines: none known.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
ALIT 1985 {published data only}
- Koster RW, Dunning AJ. Prehospital prevention of ventricular-fibrillation in acute myocardial infarction - Lidocaine Intervention Trial Amsterdam. Circulation 1983;68(4):275. [Google Scholar]
- Lidocaine Intervention Trial Amsterdam. Intramuscular lidocaine for prevention of lethal arrhythmias in the prehospitalization phase of acute myocardial infarction. New England Journal of Medicine 1985;313:1105-10. [PMID: 3900727 ] [DOI] [PubMed] [Google Scholar]
Baker 1971 {published data only}
- Baker JA, Collins JV, Evans TR. Prophylaxis of ventricular dysrhythmias following acute myocardial infarction: a double-blind trial of continuous intravenous infusion of lignocaine. Guy's Hospital Reports 1971;120:1-7. [PMID: ] [PubMed] [Google Scholar]
Bennett 1970 {published data only}
- Bennett MA, WilnerJM, Pentecost BL. Controlled trial of lignocaine in prophylaxis of ventricular arrhythmias complicating myocardial infarction. Lancet 1970;2:909-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bergdahl 1978 {published data only}
- Bergdahl B, Karlsson E, Magnusson JO, Sonnhag C. Lidocaine and the quaternary ammonium compound QX-572 in acute myocardial infarction. A comparative study. Acta Medica Scandinavica 1978;204(4):311-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Capucci 1985 {published data only}
- Capucci A, Melandri G, Mantovani B, Maresta A, Magnani B. Intravenous administration of lidocaine and amiodarone in patients with acute myocardial infarction. Giornale Italiano de Cardiologia 1985;15:285-9. [PMID: ] [PubMed] [Google Scholar]
Chopra 1971 {published data only}
- Chopra MP, Portal RW, Aber CP, Chopra MP, Portal RW, Aber CP. Lignocaine therapy after acute myocardial infarction. British Medical Journal 1969;1(5638):213-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra MP, Thadani D, Portal RW, Aber CP. Lignocaine therapy for ventricular ectopic activity after acute myocardial infarction: a double blind trial. British Medical Journal 1971;3:668-70. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cuendet 1988 {published data only}
- Cuendet BP. Pirmenol versus lidocaine in the treatment of ventricular arrhythmias at the acute phase of myocardial infarction. Double blind randomized study. Therapie 1988;43(2):158. [Google Scholar]
Darby 1972 {published data only}
- Darby S, Cruickshank JC, Bennett MA, Pentecost BL. Trial of combined intramuscular and intravenous lignocaine in prophylaxis of ventricular tachyarrhythmias. Lancet 1972;1(7755):817-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Depaepe 1974 {published data only}
- Depaepe A, Bohyn P, De Maertelaere M, Dernier J, Cloetens W, De Mey D. Comparison of aprindine (AC 1802) and lidocaine by intravenous route during the acute phase of myocardial infarct [Comparaison aprindine (AC 1802)-lidocaine par voie intraveineuse a la phase aiguë de l'infarctus du myocarde]. Acta Cardiologica 2974;Suppl 18:423-32. [PMID: 4138738 ] [PubMed] [Google Scholar]
Dunn 1985 {published data only}
- Dunn HM, McComb JM, Kinney CD, Campbell NP, Shanks RG, MacKenzie G, et al. Prophylactic lidocaine in the early phase of suspected myocardial infarction. American Heart Journal 1985;110:353-62. [PMID: 3895875 ] [DOI] [PubMed] [Google Scholar]
- Dunn HM, McComb JM. Prophylactic lignocaine in early phase of suspected myocardial infarction [abstract]. Irish Journal Medical Sci 1984;153(2):81-2. [Google Scholar]
Hargarten 1990 {published data only}
- Hargarten K, Chapman PD, Stueven HA, Waite EM, Mateer JR, Haecker P, et al. Prehospital prophylactic lidocaine does not favorably affect outcome in patients with chest pain. Annals of Emergency Medicine 1990;19:1274-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Horowitz 1981 {published data only}
- Goble AJ, Horowitz JD, Morris P. A comparative trial of mexiletine and lignocaine in the treatment of ventricular tachyarrhythmias after acute myocardial infarction. Australian & New Zealand Journal of Medicine 1980;10(3):392-3. [DOI] [PubMed] [Google Scholar]
- Horowitz JD, Anavekar SN, Morris PM, Goble AJ, Doyle AE, Louis WJ. Comparative trial of mexiletine and lignocaine in the treatment of early ventricular tachyarrhythmias after acute myocardial infarction. Journal of Cardiovascular Pharmacology 1981;3(3):409-19. [PMID: ] [DOI] [PubMed] [Google Scholar]
Keefe 1986 {published data only}
- Keefe DL, Williams S, Torres V, Flowers D, Somberg JC. Prophylactic tocainide or lidocaine in acute myocardial infarction. American Journal of Cardiology 1986;57:527-31. [PMID: 3082175 ] [DOI] [PubMed] [Google Scholar]
Kostuk 1969 {published data only}
- Kostuk WJ, Beanlands DS. Prophylactic lidocaine in acute myocardial infarction. Circulation 1969;39 Suppl III:125. [Google Scholar]
Kuck 1985 {published data only}
- Kuck KH, Bleifeld W, Mathey DG. Uneffective Lidocain prophylaxis of reperfusion arrhythmia in patients with acute myocardial infarction. Zeitschrift fur Kardiologie 1984;73(Suppl 1):64. [Google Scholar]
- Kuck KH, Jannasch B, Schluter M, Schofer J, Mathey DG. Ineffective use of lidocaine in preventing reperfusion arrhythmias in patients with acute myocardial infarct [Inneffektive lidocaineprophylaxe von reperfusions arrhythmien bei patienten mit akutem myokardinfarkt]. Zeitschrift fur Kardiologie 1985;74(3):185-90. [PMID: ] [PubMed] [Google Scholar]
Lie 1974 {published data only}
- Lie KI, Wellens HJ, Van Capelle FJ, Durrer D, Lie KI, Wellens HJ, et al. Lidocaine in the prevention of primary ventricular fibrillation. A double-blind, randomized study of 212 consecutive patients. New England Journal of Medicine 1974;291(25):1324-6. [DOI] [PubMed] [Google Scholar]
- Lie KI, Wellens HJ, Van CFJ, Durrer D. A double blind randomized study of lidocaine in preventing primary ventricular fibrillation. Circulation 1974;50(4 Suppl 3). [Google Scholar]
Lie 1978 {published data only}
- Lie KI, Liem KL, Durrer D. A double blind randomized study of intramuscular lidocaine in preventing primary ventricular fibrillation. American Journal of Cardiology 1977;39(2):275. [DOI] [PubMed] [Google Scholar]
- Lie KI, Liem KL, Louridtz WJ, Janse MJ, Willebrands AF, Durrer D. Efficacy of lidocaine in preventing primary ventricular fibrillation within 1 hour after a 300 mg intramuscular injection. A double-blind, randomized study of 300 hospitalized patients with acute myocardial infarction. American Journal of Cardiology 1978;42:486-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
NNLIT 1992 {published data only}
- Berntsen RF, Rasmussen K. Lidocaine to prevent ventricular fibrillation in the prehospital phase of suspected acute myocardial infarction: the North-Norwegian Lidocaine Intervention Trial. American Heart Journal 1992;124:1478-83. [PMID: ] [DOI] [PubMed] [Google Scholar]
O'Brien 1973 {published data only}
- O'Brien KP, Taylor PM, Croxson RS. Prophylactic lignocaine in hospitalized patients with acute myocardial infarction. The Medical Journal of Australia 1973;2 Suppl:36-7. [PMID: ] [PubMed] [Google Scholar]
Pedersen 1986 {published data only}
- Pedersen-Bjergaard O, Lindeneg O, Svendsen TL, Enk B, Fridberg M. Disopyramide and lignocaine in the treatment of ventricular arrhythmia in patients with acute ischemic heart disease. Ugeskrift for Laeger 1986;148:1349-51. [PMID: ] [PubMed] [Google Scholar]
Pharand 1995 {published data only}
- Pharand C, Kluger J, O'Rangers E, Ujhelyi M, Fisher J, Chow M. Lidocaine prophylaxis for fatal ventricular arrhythmias after acute myocardial infarction. Clinical Pharmacology and Therapeutics 1995;57:471-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pitt 1971 {published data only}
- Pitt A, Lipp H, Anderson ST. Lignocaine given prophylactically to patients with acute myocardial infarction. Lancet 1971;1(7700):612-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Poprawski 1987 {published data only}
- Poprawski K, Krug H, Ochotny R, Piszczek I, Jankowski J, Straburzyńska-Migaj E, et al. Effectiveness of lidocaine in the prevention of primary ventricular fibrillation in acute myocardial infarction. Multiple initial intravenous doses of the drug. Kardiologia Polska 1987;30:661-8. [PMID: ] [PubMed] [Google Scholar]
Rademaker 1986 {published data only}
- Rademaker AW, Kellen J, Tam YK, Wyse DG. Character of adverse effects of prophylactic lidocaine in the coronary care unit. Clinical Pharmacology and Therapeutics 1986;40(1):71-80. [PMID: ] [DOI] [PubMed] [Google Scholar]
Rehnqvist 1983 {published data only}
- Rehnqvist N, Erhardt L, Ericsson CG, Olsson G, Svensson G, Sjögren A. Comparative study of tocainide and lidocaine in patients admitted for suspected acute myocardial infarction. Acta Medica Scandinavica 1983;214:21-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Rehnqvist 1984 {published data only}
- Rehnqvist N, Ericsson CG, Eriksson S, Olsson G, Svensson G. Comparative investigation of the antiarrhythmic effect of propafenone (Rytmonorm) and lidocaine in patients with ventricular arrhythmias during acute myocardial infarction. Acta Medica Scandinavica 1984;16:525-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Rolli 1981 {published data only}
- Rolli A, Bonatti V, Finardi A, Favaro L, Botti G. Comparative study of the anti-arrhythmic activity of mexiletine and lidocaine in ventricular hyperkinetic arrhythmias [Studio comparativo tra l'efficacia antiaritmica della mexiletina e della lidocaina nelle aritmie ipercinetiche ventricolari]. Giornale Italiano di Cardiologia 1981;11(4):468-76. [PMID: ] [PubMed] [Google Scholar]
Ronnevik 1987 {published data only}
- Ronnevik PK, Gundersen T, Abrahamsen AM. Tolerability and antiarrhythmic efficacy of disopyramide compared to lignocaine in selected patients with suspected acute myocardial infarction. European Heart Journal 1987;8(1):19-24. [PMID: ] [PubMed] [Google Scholar]
Rossi 1976 {published data only}
- Rossi P, Lombardi M, Lotto A, Puddu V. Intramuscular injection of lidocaine in prevention of complications and mortality in acute myocardial infarction: double blind study on 246 cases [La lidocaina per via intramuscolare nella prevenzione delle compliczioni e della mortalitá nell´infarto miocardico acuto: Studio a doppio cieco di 246 casi]. Giornale Italiano di Cardiologia 1976;6:220-4. [PMID: ] [PubMed] [Google Scholar]
Sadowski 1999 {published data only}
- Sadowski ZP, Alexander JH, Skrabucha B, Dyduszynski A, Kuch J, Nartowicz E, et al. Multicenter randomized trial and a systematic overview of lidocaine in acute myocardial infarction. American Heart Journal 1999;137:792-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sandler 1976 {published data only}
- Sandler G, Rey N, Amonkar J. Prophylactic intramuscular lidocaine in myocardial infarction. Current Therapeutic Research 1976;20:563-71. [Google Scholar]
Sbarbaro 1979 {published data only}
- Sbarbaro JA, Rawling DA, Fozzard HA. Suppression of ventricular arrhythmias with intravenous disopyramide and lidocaine: efficacy comparison in a randomized trial. American Journal of Cardiology 1979;44(3):513-20. [PMID: ] [DOI] [PubMed] [Google Scholar]
Solimene 1983 {published data only}
- Solimene MC, Bellotti G, Ramires JA, Lage S, Silva LA, Barchi CA, et al. Value of lidocaine in the prevention of ventricular arrhythmias in the acute phase of myocardial infarct [Valor da lidocaína na profilaxia das disritmias ventriculares na fase aguda do infarto do miocárdio]. Arquivos Brasileiros de Cardiologia 1983;40(6):377-81. [PMID: ] [PubMed] [Google Scholar]
Touboul 1988 {published data only}
- Touboul P, Moleur P, Mathieu MP, Vrancea F, Ferry S, Kirkorian G, et al. A comparative evaluation of the effects of propafenone and lidocaine on early ventricular arrhythmias after acute myocardial infarction. European Heart Journal 1988;9:1188-93. [PMID: ] [DOI] [PubMed] [Google Scholar]
Valentine 1974 {published data only}
- Valentine PA, Frew JL, Mashford ML, Sloman JG. A double-blind trial of lidocaine in acute myocardial infarction. The Medical Journal of Australia 1973;11(Suppl):43-5. [PubMed] [Google Scholar]
- Valentine PA, Frew JL, Mashford ML, Sloman JG. Lidocaine in the prevention of sudden death in the pre-hospital phase of acute infarction. A double-blind study. New England Journal of Medicine 1974;19:1327-31. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wennerblom 1982 {published data only}
- Wennerblom B, Holmberg S, Rydén L, Wedel H. Antiarrhythmic efficacy and side-effects of lidocaine given in the prehospital phase of acute myocardial infarction. European Heart Journal 1982;3:516-24. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wyse 1988 {published data only}
- Wyse DG, Kellen J, Rademaker A. Prophylactic versus selective lidocaine for early ventricular arrhythmias of myocardial infarction. Journal of the American College of Cardiology 1988;12:507-13. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Antman 1992 {published data only}
- Antman EM, Berlin JA. Declining incidence of ventricular fibrillation in myocardial infarction. Implications for the prophylactic use of lidocaine. Circulation 1992;86(3):764-73. [PMID: ] [DOI] [PubMed] [Google Scholar]
Beloev 1983 {published data only}
- Beloev I, Tomov I, Dzhonov A, Orbetsov M, Stankusheva G. Effect of the use of lidocaine on patients with acute myocardial infarct. Vutreshni Bolesti 1983;22(3):88-95. [PMID: ] [PubMed] [Google Scholar]
Bernard 1970 {published data only}
- Bernard R, Stacquez A, Denolin H, Bernard R, Stacquez A, Denolin H. [Preventive treatment of arrhythmia in acute phase of myocardial infarct]. [French]. Annales de Cardiologie et d Angeiologie 1970;19(1):62-4. [PubMed] [Google Scholar]
Bernard 1972 {published data only}
- Bernard R, Lewinson H, Renta C, Thys JP. Hemodynamic effects of xylocaine and ajmaline in myocardial infarction. Archives des Maladies du Coeur et des Vaisseaux 1972;65(10):1215-8. [PMID: ] [PubMed] [Google Scholar]
Bertini 1993 {published data only}
- Bertini G, Giglioli C, Rostagno C, Conti A, Russo L, Taddei T, et al. Early out-of-hospital lidocaine administration decreases the incidence of primary ventricular fibrillation in acute myocardial infarction. The Journal of Emergency Medicine 1993;11:667-72. [PMID: 8157902 ] [DOI] [PubMed] [Google Scholar]
Bleifeld 1973 {published data only}
- Bleifeld W, Merx KW, Heinrich KW, Effert S. Controlled trial of prophylactic treatment with lidocaine in acute myocardial infarction. European Journal of Clinical Pharmacology 1973;6:119-26. [PMID: ] [DOI] [PubMed] [Google Scholar]
Campbell 1978 {published data only}
- Campbell NP, Kelly JG, Adgey AA, McDevitt DG, Pantridge JF, Campbell NP, et al. Observations on intravenous administration of lignocaine in patients with myocardial infarction. British Heart Journal 1978;40(12):1371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Campbell 1980 {published data only}
- Campbell RWF. Placebo controlled study of prophylaxis of ventricular arrhythmias in acute myocardial infarction. American Heart Journal 1980;100(6 II):995-9. [DOI] [PubMed] [Google Scholar]
Campbell 1983 {published data only}
- Campbell RWF. Treatment and prophylaxis of ventricular arrhythmias in acute myocardial infarction. American Journal of Cardiology 1983;52:55c-9. [DOI] [PubMed] [Google Scholar]
Church 1972 {published data only}
- Church G, Biern R. Prophylactic lidocaine in acute myocardial infarction. Circulation 1972;46 Suppl 2:139. [Google Scholar]
De Silva 1981 {published data only}
- DeSilva RA, Hennekens CH, Lown B, Casscells W. Lignocaine prophylaxis in acute myocardial infarction: an evaluation of randomised trials. Lancet 1981;2(8251):855-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Destuelles 1969 {published data only}
- Desruelles J, Gerard A, Waucampt JJ, Goethals S, Decalf A. Value of xylocaine in coronary thrombosis. Archives des Maladies du Coeur et des Vaisseaux 1969;62(2):243-6. [PMID: ] [PubMed] [Google Scholar]
- Desruelles J, Waucampt JJ, Goethals S, Decalf A. Value of lignocaine in coronary thrombosis (and especially in rhythm disorders in the initial phase of myocardial infarct). Lille Medical 1969;14(10):1219-21. [PubMed] [Google Scholar]
Diederich 1979 {published data only}
- Diederich KW, Fassl H, Djonlagic H, Oltmanns D, Floor-Wieringa A. Lidocaine prophylaxis in the pre-hospital phase of acute myocardial infarction. Deutsche Medizinische Wochenschrift 1979;104(28):1006-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Egre 1981 {published data only}
- Djiane P, Egre A, Perchicot B, Bory M, Serradimigni A, Bruguerolle B, et al. Use of intramuscular lidocaine in the acute stage of myocardial infarction. Archives des Maladies du Coeur et des Vaisseaux 1981;74(8):931-8. [PMID: ] [PubMed] [Google Scholar]
- Egre A, Djiane P, Serradimigni A, Bruguerolle B, Valli M, Bouyard P. Utilization of lidocaine in the acute phase of myocardial infarction [Utilisation de la lidocaine a la phase aigue de l'infarctus du myocarde]. Therapie 1981;36(1):71-9. [PMID: ] [PubMed] [Google Scholar]
Fehmers 1972 {published data only}
- Fehmers MC, Dunning AJ, Fehmers MC, Dunning AJ. Intramuscularly and orally administered lidocaine in the treatment of ventricular arrhythmias in acute myocardial infarction. American Journal of Cardiology 1972;29(4):514-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Fehmers MC, Daatselaar JJ, Dunning AJ. Intramuscular and oral administration of lidocaine for the treatment of ventricular arrhythmias in myocardial infarct. Nederlands Tijdschrift voor Geneeskunde 1972;116(29):1214-20. [PubMed] [Google Scholar]
Formichev 1995 {published data only}
- Fomichev VI, Preobrazhenskiï DV. Prophylactic use of lidocaine in the acute period of myocardial infarction. Klinicheskaia Meditsina 1995;73:17-20. [PMID: 7474809] [PubMed] [Google Scholar]
Garratt 1998 {published data only}
- Garratt KN, Holmes DR Jr, Molina-Viamonte V, Reeder GS, Hodge DO, Bailey KR, et al. Intravenous adenosine and lidocaine in patients with acute myocardial infarction. American Heart Journal 1998;136(2):196-204. [DOI] [PubMed] [Google Scholar]
Gianelly 1967 {published data only}
- Gianelly R, Groeben JO, Spivack AP, Harrison DC. Effect of lidocaine on ventricular arrhythmias in patients with coronary heart disease. New England Journal of Medicine 1967;277(23):1215-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gonzalez 1977 {published data only}
- Gonzalez Carmona V, Cheja Chaya N, Villegas Bermudes J, Gonzalez Carmona V, Cheja Chaya N, Villegas Bermudes J. Use of lidocaine in acute myocardial infarction [Uso de lidocaína en el infarto agudo del miocardio]. Archivos del Instituto de Cardiologia de Mexico 1977;47(5):604-11. [PubMed] [Google Scholar]
Goodman 1979 {published data only}
- Goodman SL, Geiderman JM, Bernstein IJ. Prophylactic lidocaine in suspected acute myocardial infarction. JACEP 1979;8(6):221-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hine 1989 {published data only}
- Hine LK, Laird N, Hewitt P, Chalmers TC. Meta-analytic evidence against prophylactic use of lidocaine in acute myocardial infarction. Archives of Internal Medicine 1989;149:2694-8. [PMID: ] [PubMed] [Google Scholar]
Iosava 1982 {published data only}
- Iosava KV, Areshidze TK, Lezhava MG, Gaprindashvili TG. Effectiveness of lidocaine use in the initial period of acute myocardial infarct. Kardiologiia 1982;22(12):82-5. [PMID: ] [PubMed] [Google Scholar]
Jaffe 1992 {published data only}
- Jaffe AS. Prophylactic lidocaine for suspected acute myocardial infarction? Heart Disease and Stroke 1992;1(4):179-83. [PMID: ] [PubMed] [Google Scholar]
Kudenchuk 2013 {published data only}
- Kudenchuk PJ, Newell C, White L, Fahrenbruch C, Rea T, Eisenberg M. Prophylactic lidocaine for post resuscitation care of patients with out-of-hospital ventricular fibrillation cardiac arrest. Resuscitation 2013;84(11):1512-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lechleitner 1987 {published data only}
- Lechleitner P, Dienstl F. Preventive use of lidocaine in the prehospital phase of acute myocardial infarct. Wiener Medizinische Wochenschrift 1987;137(10-11):216-21. [PMID: ] [PubMed] [Google Scholar]
Leone 1991 {published data only}
- Leone A, Mori L, Bertanelli F, Fabiano P. Life-threatening arrhythmias after intravenous lidocaine alone or with magnesium in myocardial infarction complicated by ventricular fibrillation. Singapore Medical Journal 1991;32:169-70. [PMID: ] [PubMed] [Google Scholar]
MacMahon 1988 {published data only}
- MacMahon S, Collins R, Peto R, Koster RW, Yusuf S. Effects of prophylactic lidocaine in suspected acute myocardial infarction. An overview of results from the randomized, controlled trials. JAMA 1988;260:1910-6. [PMID: ] [PubMed] [Google Scholar]
Mazur 1982 {published data only}
- Mazur NA, Riabokon' OS, Banshchikov GT. Prevention of ventricular fibrillation using lidocaine in the acute period of a myocardial infarct. Biulleten Vsesoiuznogo Kardiologicheskogo Nauchnogo Tsentra Amn SSSR 1982;5(2):59-64. [PMID: ] [PubMed] [Google Scholar]
Miller 1973 {published data only}
- Miller RR, Hilliard G, Lies JE, Massumi RA, Zelis R, Mason DT, et al. Hemodynamic effects of procainamide in patients with acute myocardial infarction and comparison with lidocaine. American Journal of Medicine 1973;55(2):161-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mogensen 1971 {published data only}
- Mogensen L. Ventricular tachyarrhythmias and lignocaine prophylaxis in acute myocardial infarction. Acta Medica Scandinavica 1971;513:1-80. [PMID: ] [PubMed] [Google Scholar]
Noneman 1978 {published data only}
- Noneman JW, Rogers JF. Lidocaine prophylaxis in acute myocardial infarction. Medicine 1978;57(6):501-15. [PMID: ] [DOI] [PubMed] [Google Scholar]
Oltmanns 1979 {published data only}
- Oltmanns D, Lubben C, Pentz R, Siegers CP. Plasma levels of lidocaine after intramuscular injection and subsequent infusion in patients with acute myocardial infarction. International Journal of Clinical Pharmacology, Therapy, and Toxicology 1982;20(12):582-4. [PMID: ] [PubMed] [Google Scholar]
- Oltmanns D, Siegers CP. Pharmacokinetics of lidocaine after intramuscular injection in patients with acute myocardial infarction. Zeitschrift fur Kardiologie 1979;68(3):131-6. [PMID: ] [PubMed] [Google Scholar]
Pentecost 1981 {published data only}
- Pentecost BL, De Giovanni JV, Lamb P, Cadigan PJ, Evemy KL, Flint EJ, et al. Reappraisal of lignocaine therapy in management of myocardial infarction. British Heart Journal 1981;45(1):42-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Riabokon' 1980 {published data only}
- Riabokon' OS, Piotrovskii VK, Smirnova EB, Metelitsa VI, Mazur NA. Comparative clinical study of trimecaine and lidocaine as anti-arrhythmia agents in myocardial infarct. Kardiologiia 1980;20(10):40-2. [PMID: ] [PubMed] [Google Scholar]
Ribner 1979 {published data only}
- Ribner HS, Isaacs ES, Frishman WH, Ribner HS, Isaacs ES, Frishman WH. Lidocaine prophylaxis against ventricular fibrillation in acute myocardial infarction. Progress in Cardiovascular Diseases 1979;21(4):287-313. [PMID: 368880 ] [DOI] [PubMed] [Google Scholar]
Ruano 1989 {published data only}
- Ruano Marco M, Lacueva Moya V, Garcia Pardo J, Martin Montes N, Miquel Servet J, Rodriguez P. Lignocaine prophylaxis for reperfusion arrhythmias during treatment with streptokinase in acute myocardial infarction. Lancet 1989;2(8667):872-3. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ryden 1973 {published data only}
- Ryden L, Waldenstrom A, Ehn L, Holmberg S, Husaini M. Comparison between effectiveness of intramuscular and intravenous lignocaine on ventricular arrhythmia complicating acute myocardial infarction. British Heart Journal 1973;35(11):1124-31. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shih 1995 {published data only}
- Shih RD, Hollander JE, Burstein JL, Nelson LS, Hoffman RS, Quick AM. Clinical safety of lidocaine in patients with cocaine-associated myocardial infarction. Annals of Emergency Medicine 1995;26:702-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Singh 1976 {published data only}
- Singh JB, Kocot SL. A controlled trial of intramuscular lidocaine in the prevention of premature ventricular contractions associated with acute myocardial infarction. American Heart Journal 1976;91:430-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Singh JB, Kocot SL. A controlled trial of intramuscular lidocaine in the prevention of premature ventricular contractions associated with acute myocardial infarction. Circulation 1973;48(4 Suppl):28. [DOI] [PubMed] [Google Scholar]
- Singh JB, Kocot SL. Controlled trial of Intramuscular lidocaine in prevention of premature ventricular contractions associated with acute myocardial-infarction. Circulation 1973;48(4):8-8. [DOI] [PubMed] [Google Scholar]
Szeplaki 1973 {published data only}
- Szeplaki S, Szilagyi A, Boszormenyi E, Barsi B, Mate K, Kulcsar M, et al. Anti-ischemic effect of lidocaine in the acute phase of myocardial infarct without shock. Orvosi Hetilap 1974;115(12):671-5. [PMID: ] [PubMed] [Google Scholar]
- Szeplaki S, Szilagyi A, Szeplaki Z, Boszormenyi E, Barsi B, Mate K, et al. Anti-ischemic effect of lidocaine in the acute state of myocardial infarction. Agressologie 1973;14(5):339-45. [PMID: ] [PubMed] [Google Scholar]
Szeplaki 1976 {published data only}
- Szeplaki S, Szilaigyi A, Harsanyi A, Szeplaki Z, Mate K, Kulcsar M, et al. Lidocaine prophylaxis in preinfarction angina and in the reactive stage of myocardial infarction. Agressologie 1976;17(4):245-50. [PMID: ] [PubMed] [Google Scholar]
Teo 1993 {published data only}
- Teo KK, Yusuf S, Furberg CD. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction. An overview of results from randomized controlled trials. JAMA 1993;270:1589-95. [PMID: ] [PubMed] [Google Scholar]
Wojtala 1982 {published data only}
- Wojtala M, Stopczyk M, Wojtala M, Stopczyk M. Relationship between lidocaine-induced suppression of premature excitation and reduced possibility of the appearance of ventricular fibrillation in myocardial infarction. Kardiologia Polska 1982;25(7-8):647-53. [PMID: ] [PubMed] [Google Scholar]
Wyman 2004 {published data only}
- Wyman MG, Wyman RM, Cannom DS, Criley JM. Prevention of primary ventricular fibrillation in acute myocardial infarction with prophylactic lidocaine. American Journal of Cardiology 2004;94:545-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Bolinska 1971 {published data only}
- Bolinska H, Kuczborski M, Bolinska H, Kuczborski M. Lignocaine in the treatment of arrhythmia in acute myocardial infarct. Wiadomosci Lekarskie 1971;24(19):1851-4. [PMID: ] [PubMed] [Google Scholar]
Hopperstead 1980 {published data only}
- Hopperstead LO, Myers MH. Prophylactic lidocaine in the early management of acute myocardial infarction. The Journal of the Maine Medical Association 1980;71:77-81. [PMID: 7365324 ] [PubMed] [Google Scholar]
Knight 1973 {published data only}
- Knight AL. Prophylactic lidocaine in myocardial infarction. Journal of Occupational Medicine 1973;15(7):604. [PMID: ] [PubMed] [Google Scholar]
Additional references
al‐Adsani 2000
- al-Adsani A, Memon A, Peneva A, Baidas G. Clinical epidemiology of acute myocardial infarction in Kuwait. Acta Cardiologica 2000;55:17-23. [PMID: ] [DOI] [PubMed] [Google Scholar]
Alexander 1999
- Alexander JH, Granger CB, Sadowski Z, Aylward PE, White HD, Thompson TD, et al. Prophylactic lidocaine use in acute myocardial infarction: incidence and outcomes from two international trials. The GUSTO-I and GUSTO-IIb Investigators. American Heart Journal 1999;137:799-805. [PMID: ] [DOI] [PubMed] [Google Scholar]
Al‐Khatib 2003
- Al-Khatib SM, Stebbins AL, Califf RM, Lee KL, Granger CB, White HD, et al. Sustained ventricular arrhythmias and mortality among patients with acute myocardial infarction: results from the GUSTO-III trial. American Heart Journal 2003;145:515-21. [PMID: ] [DOI] [PubMed] [Google Scholar]
Alter 2008
- Alter DA, Stukel TA, Newman A. The relationship between physician supply, cardiovascular health service use and cardiac disease burden in Ontario: supply-need mismatch. The Canadian Journal of Cardiology 2008;24:187-93. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderson 1984
- Anderson JL. Current understanding of lidocaine as an antiarrhythmic agent: a review. Clinical Therapeutics 1984;6:125-41. [PMID: ] [PubMed] [Google Scholar]
Anonimous 2006
- Sociedad Uruguaya de Cardiología. Consenso uruguayo de manejo del infarto agudo de miocardio con elevación del segmento ST. Revista Uruguaya de Cardiología 2006;21(1):48-95. [Google Scholar]
Applebaum 1986
- Applebaum D, Halperin E. Asystole following a conventional therapeutic dose of lidocaine. The American Journal of Emergency Medicine 1986;4:143-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Aps 1976
- Aps C, Bell JA, Jenkins BS, Poole-Wilson PA, Reynolds F. Logical approach to lignocaine therapy. British Medical Journal 1976;1(6000):13-5. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bainey 2009
- Bainey KR, Jugdutt BI. Increased burden of coronary artery disease in South-Asians living in North America. Need for an aggressive management algorithm. Atherosclerosis 2009;204:1-10. [PMID: ] [DOI] [PubMed] [Google Scholar]
Basch 2012
- Basch E, Aronson N, Berg A, Flum D, Gabriel S, Goodman SN, Helfand M, et al. Methodological standards and patient-centeredness in comparative effectiveness research: the PCORI perspective. JAMA 2012;307(15):1636-40. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bayés de Luna 1989
- Bayés de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. American Heart Journal 1989;117:151-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Berntsen 1991
- Berntsen RF, Rasmussen K, Bjornstad JF. Monitoring a randomized clinical trial for futility: the north-Norwegian lidocaine intervention trial. Statistics in Medicine 1991;10(3):405-12. [PMID: ] [DOI] [PubMed] [Google Scholar]
Borenstein 2009
- Borenstein M, Hedges LV, Higgins JPT, Rothstein HR. Publication Bias. In: Borenstein M, Hedges LV, Higgins JPT, Rothstein HR, editors(s). Introduction to Meta-Analysis. First edition. West Sussex, United Kingdom: Wiley, 2009:277-92. [Google Scholar]
Boutron 2010
- Boutron I, Dutton S, Ravaud P, Altman DG. Reporting and interpretation of randomized controlled trials with statistically nonsignificant results for primary outcomes. JAMA 2010;303(20):2058-64.. JAMA 2010;303(10):2058-64. [PMID: ] [DOI] [PubMed] [Google Scholar]
Braunwald 2001
- Braunwald E. Heart Disease: A Textbook of Cardiovascular Medicine. 6th edition. Philadelphia: W. B. Saunders Company, 2001. [Google Scholar]
Brok 2008
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta-analyses. Journal of Clinical Epidemiology 2008;61:763-9. 2008;61(8):763-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta-analyses may be inconclusive - Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta-analyses. International Journal of Epidemiology 2009;38(1):287-98. [PMID: ] [DOI] [PubMed] [Google Scholar]
Brunton 2008
- Brunton LL, Parker KL, Blumenthal DK, Buxton ILO. Goodman & Gilman’s Manual of Pharmacology and Therapeutics. 11th edition. New York: The McGraw-Hill Companies, Inc, 2008. [DOI: 10.1036/0071443436] [DOI] [Google Scholar]
Cabadés 2007
- Cabadés O'Callaghan A. The REGICOR registry and the epidemiology of myocardial infarction in Spain: forging a path. Revista Española de Cardiología 2007;60:342-5. [PMID: ] [PubMed] [Google Scholar]
Chan 2013
- Chan AW, Tetzlaff JM, Gøtzsche PC, Altman DG, Mann H, Berlin JA, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ 2013;346:e7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Clarke 2007
- Clarke M. Standardising outcomes for clinical trials and systematic reviews. Trials 2007;8:39. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
CMA 2005 [Computer program]
- Comprehensive Meta-analysis. Borenstein M, Hedges L, Higgins J, Rothstein H. Englewood NJ, USA: Biostat, 2005.
Collinsworth 1974
- Collinsworth KA, Kalman SM, Harrison DC. The clinical pharmacology of lidocaine as an antiarrhythymic drug. Circulation 1974;50:1217-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Crystal 2003
- Crystal E, Connolly SJ, Dorian P. Prevention and treatment of life-threatening ventricular arrhythmia and sudden death. In: Yusuf S, Cairns JA, Camm AJ, Fallen EL, Gersh BJ, editors(s). Evidence-Based Cardiology. 2nd edition. London: BMJ Books, 2003:577-86. [Google Scholar]
CTU 2011 [Computer program]
- TSA - Trial Sequential Analysis. Copenhagen Trial Unit. http://ctu.dk/tsa/ (accessed 29 September 2011), 2011.
Deeks 2002
- Deeks JJ. Issues in the selection of a summary statistic for meta-analysis of clinical trials with binary outcomes. Statistics in Medicine 2002;21:1575–1600. [DOI: 10.1002/sim.1188] [PMID: ] [DOI] [PubMed] [Google Scholar]
Egger 2001
- Egger M, Smith GD, Altman D, editors. Systematic Reviews in Health Care. Second edition. London: BMJ Publishing Group, 2001. [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31:140-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gabriel 2012
- Gabriel SE, Normand SL. Getting the methods right - the Foundation of Patient-Centered Outcomes Research. New England Journal of Medicine 2012;367(9):787-90. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gardner 2000
- Gardner MJ, Leather R, Teo K. Prevention of sudden death from ventricular arrhythmia. Epidemiology. Canadian Journal of Cardiology 2000;16:10-2. [PMID: ] [PubMed] [Google Scholar]
Gaziano 2006
- Gaziano T, Reddy KS, Paccaud F, Horton S, Chaturvedi V. Cardiovascular disease. In: Jamison DT, Breman JG, Measham AR, Alleyne G, Claeson M, Evans DB et al, editors(s). Disease Control Priorities in Developing Countries. 2nd edition. Washington (DC): IBRD/The World Bank and Oxford University Press, 2006. [Google Scholar]
Giannakoulas 2009
- Giannakoulas G, Dimopoulos K, Engel R, Goktekin O, Kucukdurmaz Z, Vatankulu MA, et al. Burden of coronary artery disease in adults with congenital heart disease and its relation to congenital and traditional heart risk factors. American Journal of Cardiology 2009;103:1445-50. [PMID: ] [DOI] [PubMed] [Google Scholar]
Goldenberg 2008
- Goldenberg I, Moss AJ, Ryan D, Pietrasik G, Zareba W, McNitt S, et al. Cumulative burden of atherosclerotic risk genotypes and the age at onset of a first myocardial infarction: a case-only carriership approach. Annals of Noninvasive Electrocardiology 2008;13:287-94. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Goldstein 1986
- Goldstein S. The epidemiology of tachyfibrillation in sudden death. Pacing and Clinical Electrophysiology 1986;9 (Pt 2):1339-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gorter 2007
- Gorter PM, Visseren FL, Algra A, Graaf Y, SMART Study Group. The impact of site and extent of clinically evident cardiovascular disease and atherosclerotic burden on new cardiovascular events in patients with Type 2 diabetes. The SMART study. Diabetic Medicine 2007;24:1352-60. [PMID: ] [DOI] [PubMed] [Google Scholar]
Goyal 2006
- Goyal A, Yusuf S. The burden of cardiovascular disease in the Indian subcontinent. The Indian Journal of Medical Research 2006;124:235-44. [PMID: ] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, and GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008b
- Guyatt G, Oxman AD, Kunz R, Vist GE, Falck‐Ytter Y, Schunemann HJ. What is "quality of evidence" and why is it important to clinicians. BMJ 2008;336:995-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Harrison 1989
- Harrison DC. Arrhythmia prophylaxis after acute myocardial infarction: a decade of controversy. Cardiovascular Drugs and Therapy 1989;2:783-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Henkel 2006
- Henkel DM, Witt BJ, Gersh BJ, Jacobsen SJ, Weston SA, Meverden RA, et al. Ventricular arrhythmias after acute myocardial infarction: a 20-year community study. American Heart Journal 2006;151:806-12. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration. www.cochrane-handbook.org 2011.
Hill 1973
- Hill GJ. Lidocaine asystole. JAMA 1973;224:401. [PMID: ] [PubMed] [Google Scholar]
Hitchcock 1959
- Hitchcock P, Keown KK. The management of cardiac arrhythmias during cardiac surgery. Southern Medical Journal 1959;52:702-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hreybe 2009
- Hreybe H, Saba S. Location of acute myocardial infarction and associated arrhythmias and outcome. Clinical Cardiology 2009;32:274-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Huikuri 2001
- Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. New England Journal of Medicine 2001;345:1473-82. [PMID: ] [DOI] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice 1997 CFR & ICH Guidelines. PA 19063-2043, USA: Barnett International/PAREXEL, 1997. [Google Scholar]
Ioannidis 2004
- Ioannidis JP, Evans SJ, Gotzsche PC, O'Neill RT, Altman DG, Schulz K, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Annals of Internal Medicine 2004;141(10):781-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ioannidis 2010
- Ioannidis JP. Meta-research: the art of getting it wrong. Research Synthesis Methods 2010;10(3-4):169-84. [DOI] [PubMed] [Google Scholar]
Kerr 2008
- Kerr AJ, McLachlan A, Furness S, Broad J, Riddell T, Jackson R, et al. The burden of modifiable cardiovascular risk factors in the coronary care unit by age, ethnicity, and socioeconomic status - PREDICT CVD-9. The New Zealand Medical Journal 2008;121:20-33. [PMID: ] [PubMed] [Google Scholar]
Khairy 2003
- Khairy P, Thibault B, Talajic M, Dubuc M, Roy D, Guerra PG, et al. Prognostic significance of ventricular arrhythmias post-myocardial infarction. The Canadian Journal of Cardiology 2003;19:1393-404. [PMID: ] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [PMID: ] [DOI] [PubMed] [Google Scholar]
Klein 1975
- Klein HO, Jutrin I, Kaplinsky E. Cerebral and cardiac toxicity of a small dose of lignocaine. British Medical Journal 1975;37:775-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kolansky 2009
- Kolansky DM. Acute coronary syndromes: morbidity, mortality, and pharmacoeconomic burden. The American Journal of Managed Care 2009;15(Suppl 2):36-41. [PMID: ] [PubMed] [Google Scholar]
Koplan 2009
- Koplan BA, Stevenson WG. Ventricular tachycardia and sudden cardiac death. Mayo Clinic Proceedings 2009;84:289-97. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kuch 2009
- Kuch M, Janiszewski M, Mamcarz A, Cudnoch-Jedrzejewska A, Duzniewski M. Major adverse cardiac event predictors in survivors of myocardial infarction with asymptomatic left ventricular dysfunction or chronic heart failure. Medical Science Monitor 2009;15:40-8. [PMID: ] [PubMed] [Google Scholar]
Labarthe 1998
- Labarthe DR. Epidemiology and Prevention of Cardiovascular Disease. Gaithersburg, Maryland: Aspen Publishers, 1998. [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane-handbook.org.
Leys 2001
- Leys D. Atherothrombosis: a major health burden. Cerebrovascular Diseases 2001;11(Suppl 2):1-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lippestad 1971
- Lippestad CT, Forfang K. Production of sinus arrest by lignocaine. British Medical Journal 1971;1(5748):537. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ljung 2006
- Ljung R, Hallqvist J. Accumulation of adverse socioeconomic position over the entire life course and the risk of myocardial infarction among men and women: results from the Stockholm Heart Epidemiology Program (SHEEP). Journal of Epidemiology and Community Health 2006;60:1080-4. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lloyd‐Williams 2008
- Lloyd-Williams F, O'Flaherty M, Mwatsama M, Birt C, Ireland R, Capewell S. Estimating the cardiovascular mortality burden attributable to the European Common Agricultural Policy on dietary saturated fats. Bulletin of the World Health Organization 2008;86:535-41. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. Art. No: MR000033. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Manson 1996
- Manson JE, Ridker PM, Gaziano JM, Hennekens. Myocardial Infarction: Epidemiologic Overview. In: Manson JE, Ridker PM, Gaziano JM, Hennekens, editors(s). Prevention of Myocardial Infarction. New York: Oxford University Press, 1996:3-31. [Google Scholar]
Manyari‐Ortega 1978
- Manyari-Ortega DE, Brennan FJ. Lidocaine-induced cardiac asystole. Chest 1978;74:227-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
McKinney 2012
- McKinney M. 'Time is of the essence'. PCORI moves to implement comparative effectiveness research, funding. Modern Health Care 2012;42(5):12-3. [PMID: ] [PubMed] [Google Scholar]
Moher 2010
- Moher D, Hopewell S, Schulz KF, Montori V, Gotzsche PC, Devereaux PJ. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Myerburg 1986
- Myerburg RJ. Epidemiology of ventricular tachycardia/ventricular fibrillation and sudden cardiac death. Pacing and Clinical Electrophysiology 1986;9 (Pt 2):1334-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
PCORI 2012
- Patient-Centered Outcomes Research Institute (PCORI). Preliminary draft methodology report: “Our questions, our decisions: Standards for patient‐centered outcomes research”. http://www.pcori.org/assets/Preliminary-Draft-Methodology-Report.pdf (accessed 10 September 2013) 2012;(1-61).
Piccini 2008
- Piccini JP, Berger JS, Brown DL. Early sustained ventricular arrhythmias complicating acute myocardial infarction. The American Journal of Medicine 2008;121:797-804. [PMID: ] [DOI] [PubMed] [Google Scholar]
Piccini 2011
- Piccini JP, Schulte PJ, Pieper KS, Mehta RH, White HD, Van de Werf F, et al. Antiarrhythmic drug therapy for sustained ventricular arrhythmias complicating acute myocardial infarction. Critical Care Medicine 2011;39(1):78-83. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pop 2004
- Pop C, Pop L, Dicu D. Epidemiology of acute myocardial infarction in Romanian county hospitals: a population-based study in the Baia Mare district. Romanian Journal of Internal Medicine 2004;42:607-23. [PMID: ] [PubMed] [Google Scholar]
Porta 2008
- Porta M (Editor). A Dictionary of Epidemiology. Fifth edition. New York: Oxford University Press, 2008. [Google Scholar]
Rahimi 2006
- Rahimi K, Watzlawek S, Thiele H, Secknus MA, Hayerizadeh BF, Niebauer J, et al. Incidence, time course, and predictors of early malignant ventricular arrhythmias after non-ST-segment elevation myocardial infarction in patients with early invasive treatment. European Heart Journal 2006;27:1706-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
RevMan 2011 [Computer program]
- Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre: The Cochrane Collaboration, 2011.
Reyes Caorsi 2006
- Reyes Caorsi W. Reflexiones sobre las guías clínicas. A propósito del caso de la lidocaína. Revista Uruguaya de Cardiología 2006;21(3):267-9. [Google Scholar]
Rich 2006
- Rich MW. Epidemiology, clinical features, and prognosis of acute myocardial infarction in the elderly. The American Journal of Geriatric Cardiology 2006;15:7-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Roger 2007
- Roger VL. Epidemiology of myocardial infarction. The Medical Clinics of North America 2007;91:537-52. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sadikot 1997
- Sadikot R, Patel N, Smith E, Bissett J, Talley JD. Lidocaine-induced cardiac asystole. The Journal of the Arkansas Medical Society 1997;93:410-1. [PMID: ] [PubMed] [Google Scholar]
Savović 2012
- Savović J, Jones HE, Altman DG, Harris RJ, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429-38. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schünemann 2009
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Interpreting results and drawing conclusions. In: Higgins JPT, Green S, editors(s). Cochrane Handbook for Systematic Reviews of Interventions. West Sussex, England: Wiley-Blackwell, 2009:359-87. [Google Scholar]
Selby 2012
- Selby JV, Beal AC, Frank L. The Patient-Centered Outcomes Research Institute (PCORI) national priorities for research and initial research agenda. JAMA 2012;307(15):1583-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Singla 2008
- Singla I, Hreybe H, Saba S. Risk of death and recurrent ventricular arrhythmias in survivors of cardiac arrest concurrent with acute myocardial infarction. Indian Pacing and Electrophysiology Journal 2008;8:5-13. [PMID: ] [PMC free article] [PubMed] [Google Scholar]
Solomon 2005
- Solomon SD, Zelenkofske S, McMurray JJ, Finn PV, Velazquez E, Ertl G, Valsartan in Acute Myocardial Infarction Trial (VALIANT) Investigators. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. New England Journal of Medicine 2005;352:2581-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Steptoe 2007
- Steptoe A, Shamaei-Tousi A, Gylfe A, Henderson B, Bergström S, Marmot M. Socioeconomic status, pathogen burden and cardiovascular disease risk. Heart 2007;93:1567-70. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ 2011;343:d4002. [PMID: ] [DOI] [PubMed] [Google Scholar]
Tagawa 2008
- Tagawa T, Nakao K, Nakamura A, Yamasaki N, Tsuchiya T, Nagayasu T. [How to treat arrhythmias in thoracic surgery]. Kyobu Geka. The Japanese Journal of Thoracic Surgery 2008;61(8 Suppl):715-20. [PMID: ] [PubMed] [Google Scholar]
Takaya 2009
- Takaya T, Okamoto M, Yodoi K, Hata K, Kijima Y, Nakajima H, et al. Torsades de Pointes with QT prolongation related to donepezil use. Journal of Cardiology 2009;54(3):507-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta-analyses. International Journal of Epidemiology 2009;38(1):276-86. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thorlund 2010
- Thorlund K, Anema A, Mills E. Interpreting meta-analysis according to the adequacy of sample size. An example using isoniazid chemoprophylaxis for tuberculosis in purified protein derivative negative HIV-infected individuals. Clinical Epidemiology 2010;2:57-66. [PMID: 20865104 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011a
- Thorlund K, Imberger G, Walsh M, Chu R, Gluud C, Wetterslev J, et al. The number of patients and events required to limit the risk of overestimation of intervention effects in meta-analysis - a simulation study. PLoS One 2011;6(10):e25491. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011b [Computer program]
- User Manual for Trial Sequential Analysis (TSA). Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. http://ctu.dk/tsa/files/tsa_manual.pdf 2011 (accessed 30 April 2012), 2011.
Thygesen 2008
- Thygesen K, Alpert JS, Jaffe AS, White HD. Diagnostic application of the universal definition of myocardial infarction in the intensive care unit. Current Opinion in Critical Care 2008;14:543-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Velazquez 2004
- Velazquez EJ, Francis GS, Armstrong PW, Aylward PE, Diaz R, O'Connor CM, VALIANT registry. An international perspective on heart failure and left ventricular systolic dysfunction complicating myocardial infarction: the VALIANT registry. European Heart Journal 2004;25:1911-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Volpi 1990
- Volpi A, Cavalli A, Santoro E, Tognoni G. Incidence and prognosis of secondary ventricular fibrillation in acute myocardial infarction. Evidence for a protective effect of thrombolytic therapy. GISSI Investigators. Circulation 1990;82:1279-88. [PMID: PubMed: 2205418] [DOI] [PubMed] [Google Scholar]
Volpi 1998
- Volpi A, Cavalli A, Santoro L, Negri E. Incidence and prognosis of early primary ventricular fibrillation in acute myocardial infarction - results of the Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico (GISSI-2) database. American Journal of Cardiology 1998;82:265-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Watkins 2004
- Watkins LO. Epidemiology and burden of cardiovascular disease. Clinical Cardiology 2004;27(Suppl 3):2-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weir 2006
- Weir RA, McMurray JJ. Epidemiology of heart failure and left ventricular dysfunction after acute myocardial infarction. Current Heart Failure Reports 2006;3:175-80. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta-analysis. Journal of Clinical Epidemiology 2008;61(1):64-75. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in a random-effects meta-analysis. BMC Medical Research Methodology 2009;9:86. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolfe 1991
- Wolfe CL, Nibley C, Bhandari A, Chatterjee K, Scheinman M. Polymorphous ventricular tachycardia associated with acute myocardial infarction. Circulation 1991;84:1543-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Juni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008;336(7644):601-5. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zavala 2006 [Computer program]
- Sheet to enter data for performing a Cochrane review. Zavala D, Martí A, Peña-Martí G, Comunian G. Valencia: Universidad de Carabobo, 2006. http://www.cochrane.fcs.uc.edu.ve/hrs/.
Zipes 2006
- Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, American College of Cardiology, American Heart Association Task Force, European Society of Cardiology Committee for Practice Guidelines. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death?executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). European Heart Journal 2006;27:2099-140. [PMID: ] [DOI] [PubMed] [Google Scholar]