

Abstract
Comprehensive characterization of differentially spliced RNA transcripts with nanopore sequencing is limited by bioinformatics tools that are reliant on existing annotations. We have developed FLAME, a bioinformatics pipeline for alternative splicing analysis of gene-specific or transcriptome-wide long-read sequencing data. FLAME is a Python-based tool aimed at providing comprehensible quantification of full-length splice variants, reliable de novo recognition of splice sites and exons, and representation of consecutive exon connectivity in the form of a weighted adjacency matrix. Notably, this workflow circumvents issues related to inadequate reference annotations and allows for incorporation of short-read sequencing data to improve the confidence of nanopore sequencing reads. In this study, the Epstein-Barr virus long noncoding RNA RPMS1 was used to demonstrate the utility of the pipeline. RPMS1 is ubiquitously expressed in Epstein-Barr virus associated cancer and known to undergo ample differential splicing. To fully resolve the RPMS1 spliceome, we combined gene-specific nanopore sequencing reads from a primary gastric adenocarcinoma and a nasopharyngeal carcinoma cell line with matched publicly available short-read sequencing data sets. All previously reported splice variants, including putative ORFs, were detected using FLAME. In addition, 32 novel exons, including two intron retentions and a cassette exon, were discovered within the RPMS1 gene.
Keywords: bioinformatics, Epstein-Barr virus, RNA splicing, RPMS1
INTRODUCTION
Differential splicing by means of mutually exclusive exons, intron retention and alternative acceptor/donor splice sites brings considerable diversity to a single gene (Mollet et al. 2010). In the human transcriptome, estimations based on RNA sequencing data suggest that around 95% of multiexonic genes undergo alternative splicing (Pan et al. 2008). Human viruses utilizing the host transcription machinery for gene expression may likewise display substantial alternative RNA splicing. For instance, the temporal regulation of the viral gene expression in human papillomavirus is mediated by alternative RNA processing (Johansson and Schwartz 2013). Also, multiple Epstein-Barr virus (EBV) genes contain variably included exons and are subjected to extensive splicing (Farrell 2019).
The advent of short-read RNA sequencing technologies has provided the means to unbiasedly characterize transcriptomes with unprecedented speed and accuracy (Tang and Larsson 2017). Nonetheless, comprehension of exon connectivity at single-molecule level is irreversibly lost due to the fragmentation during library preparation. Although splice-junction reads or paired-end mapping of coupled reads to consecutive exons allow for efficient detection of splicing, these approaches fail to account for the relative abundance of alternatively spliced transcripts at full-length resolution. With the emergence of long-read sequencing technologies, it is now feasible to reveal the full spectrum of differential splicing at single-molecule level (Garalde et al. 2018). However, the Oxford Nanopore Technologies long-read sequencing methodology is to some extent afflicted by the relatively low accuracy and high incidence of indels, which entails uncertainty in distinguishing actual splice sites from artifacts (Byrne et al. 2017; Kovaka et al. 2019; Dohm et al. 2020). Available bioinformatics tools for long-read splicing analysis that combine the strengths of nanopore and short-read sequencing data, for example, FLAIR, have proven to facilitate correct alignment and annotation (Tang et al. 2020). However, existing tools encounter difficulties in properly handling a large accumulation of unannotated exon boundaries. In particular, stringent reliance on a given reference annotation requires laborious efforts to discover cryptic splice sites and discern extensively overlapping exons.
Here, we describe FLAME (full-length adjacency matrix and exon enumeration), a novel bioinformatics pipeline designed to generate a comprehensive catalog of RNA splicing, including unannotated splice sites and novel exons at single-molecule level. To that end, FLAME connects the dots between long-read and short-read sequencing as part of reliable de novo recognition of novel splice sites and exons. The program outputs sorted lists of both annotated and partly unannotated transcript variants. The latter are further processed to single out genuine exons on the basis of frequency, adjacent splice site dinucleotides and short-read support. Altogether, such a complete cataloging of splice variants, provided in a proper format, is conducive to adequately determine the relative frequencies of particular splicing patterns and uncover novel exons.
In this work, the EBV long noncoding RNA (lncRNA) RPMS1 was used to demonstrate the application of FLAME in detail. The tumorigenic nature of EBV is manifested in various lymphoid and epithelial malignancies. Studies have shown that RPMS1 is the most abundantly expressed polyadenylated viral RNA in EBV-associated nasopharyngeal carcinoma and gastric adenocarcinoma (GAC) (Raab-Traub et al. 1983; Tang et al. 2013). High expression of RPMS1 has also been shown in the EBV-positive nasopharyngeal carcinoma cell line C666-1 (Smith et al. 2000). The RPMS1 gene spans over 22 kb of the EBV genome and the 4 kb lncRNA RPMS1 contains seven main exons (I–VII) along with two minor cassette exons (Ia and Ib) (Marquitz et al. 2015). Alternative exons within RPMS1 have been reported in low-throughput studies (Yamamoto and Iwatsuki 2012). Within the introns of RPMS1, 44 microRNAs (miR-BARTs) are encoded and it has been proposed that the expression levels of these may be differentially regulated by alternating the splicing pattern (Edwards et al. 2008). These miR-BARTs have been observed in all proposed latency types of EBV-associated neoplasms (Qiu et al. 2011). In addition, minor circularized BARTs resulting from backsplicing of particular segments of RPMS1 have been described especially during reactivation (Toptan et al. 2018; Ungerleider et al. 2018). Moreover, putative ORFs have been described in RPMS1; however, the existence of these have remained controversial (Chen et al. 1999).
RESULTS AND DISCUSSION
Program framework
FLAME is a streamlined amalgamation of four main functions: (i) long-read categorization, (ii) splice variant enumeration, (iii) quantification of adjacent exon linkage and (iv) detection of novel splice sites and exons. These core functions are constructed of several interconnected subfunctions as described in Table 1. The general workflow is summarized in Figure 1 and organized as follows:
The initial long-read categorization utility determines whether the input reads align with the reference annotation. Reads are thus categorized as either annotated or incongruent with respect to any genomic annotation provided in gene transfer format (GTF). The annotation file is at the outset converted into a local variable based on filtering of general entries, thereby making the program applicable to both single- and multichromosomal organisms. All annotated exon ranges are subsequently translated from BED12 format into a numeric naming system established in the local reference variable. The exonic constituents of each and every annotated read are hence untangled and represented as a series of exon names. A read is categorized as incongruent if merely one exon departs from the local reference annotation file in terms of exon global start position, global end position or length. This three-factor approach (start, end, and length) entails awareness of overlapping exonic positions and allows for reads comprising intron retention events or alternative splice sites within previously annotated exons to be cataloged separately.
Sequences with identical exon arrangements are collapsed and quantified using the principle of item recognition. A quantitative description of all fully annotated patterns of splice variation is consequently obtained at single-molecule level.
Consecutive exon linkage is presented in the form of a weighted adjacency matrix. The relative inclusion rate and sequential arrangement of any given exon is thus specified. Moreover, any long-range exon coupling that may be derived from artifactual joining of primers appears clearly.
The program systematically catalogs all exon/intron boundaries in order to find any novel splice sites and exons. In brief, the network of subfunctions developed to facilitate detection of genuine splice sites and exons is oriented toward three aspects: (i) splice site usage frequency, (ii) presence of canonical splice site dinucleotides at the intron–exon junctions and (iii) incorporation of short-read evidence. These parameters are automatically compiled into one output file for subsequent manual assessment. Unannotated exons considered to be authentic can then be added to the local reference annotation file in order to recover incongruent reads by running an additional cycle of the workflow.
TABLE 1.
Description of FLAME functions

FIGURE 1.
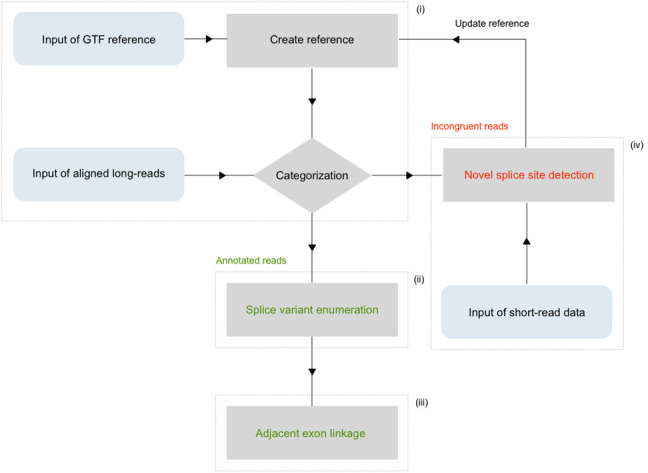
General organization of the pipeline. The four main functions are marked i–iv. (i) The create.ref function converts annotation written in GTF into a local variable for downstream use. Aligned long-reads are subsequently compared against the reference in the filter function. Transcript variants containing incongruent exons are singled out and unannotated splice sites are cataloged. (iv) In the following “novel splice site detection” function, unannotated splice site coordinates are quantified and set against a given fasta format reference to scan for adjacent canonical dinucleotides (GU/AG). This step also allows for optional incorporation of splice-junction reads from corresponding short-read data to raise the fidelity. Novel exons that are deemed true upon manual inspection are then added to the reference variable for repeated cycling through the filter function. (ii) Transcript variants that are congruent with the reference annotation are passed on to an enumeration operation, in which the relative and absolute abundance is determined. (iii) The concluding adjacency matrix displays large-scale consecutive exon connectivity.
FLAME is in its primary implementation intended for gene specific splicing analysis. However, the program allows for scalability to transcriptome-wide analysis of both native RNA and cDNA nanopore sequencing data. The global-wide module (FLAME-GLOW) outputs transcript variant quantification of well characterized genes. In the event that a vast proportion of reads aligning to a specific locus are incongruent with the reference annotation, the gene/s in question is/are flagged and itemized in a separate list for further assessment.
A case study on the splice variation of RPMS1
The EBV lncRNA RPMS1 was used as a case to present the different features of FLAME. Here, we combined nanopore long-read sequencing reads and publicly available short-read data sets of primary tumors and the EBV-positive nasopharyngeal carcinoma cell line C666-1 to resolve the splicing pattern of RPMS1 in transformed epithelial tissue. Total RNA was isolated from C666-1 and a GAC tumor tissue, in which EBV-RNA was detected using RT-qPCR (Supplemental Fig. 1). An adapted PCR-cDNA approach targeting RPMS1 was subsequently used to prepare full-length libraries for nanopore sequencing. In total, 186,738 and 164,000 raw reads were generated from the C666-1 and GAC library, respectively, on an Oxford Nanopore Technologies MinION device and aligned to the EBV genome. Filtering based on the global start position of RPMS1 eventually rendered 153,164 and 131,503 aligned reads in the C666-1 and GAC data set, respectively.
Inasmuch as PCR amplification and sequencing errors can overestimate obscure splicing events, a threshold limit value was set to diminish the number of truncated transcripts or otherwise artifactual sequences. The threshold limit value was here defined as the minimal number of supporting reads per transcript variant. We henceforth only considered transcript variants supported by ≥10 reads, hereinafter referred to as intermediate-confidence data. All transcripts supported by less than 10 reads were consequently omitted from further analysis. In addition, a high-confidence data set with a threshold limit value of ≥100 reads per transcript variant was implemented to more certainly rule out any technical artifacts. Given these thresholds, ∼12% and ∼18% of the total reads were discarded from the intermediate-confidence and high-confidence data sets, respectively. The parameter optimization of threshold limit values is presented in Supplemental Figure 2A–F.
To further remove any ambiguity concerning possible nanopore sequencing artifacts, matched short-read sequencing data was incorporated into the workflow. In total, publicly available short-read data from 106 EBV-positive nasopharyngeal carcinomas, 28 EBV-positive GAC tumors and one C666-1 data set (equivalent to 459,854; 36,290 and 3107 splice junction reads, respectively) was utilized (Supplemental Table 1). This library of splice-junction reads hence served as the frame of reference to determine the exact positions of unannotated exon boundaries discovered using nanopore sequencing. As illustrated in Figure 2, the coverage plots obtained from nanopore sequencing correlated with several noncanonical splice sites in the corresponding short-read data. Furthermore, based on the distribution of splice junctions across the RPMS1 gene, it was clear that the current RefSeq annotation (NC_007605.1) was inadequate. Firstly, the RefSeq annotation lacked exon Ia, Ib, and II. Moreover, the numerous splice-junction reads detected within exon V and VII indicated a complex pattern of splicing not captured by current annotation, which hampers the usefulness of any tool that is dependent on the provided reference.
FIGURE 2.

Alignment of long nanopore sequencing reads and splice junctions in corresponding short-read sequencing data sets. (A) Coverage plots of nanopore sequencing reads across RPMS1 in C666-1 and GAC. Bars display alignment to constitutive (I–VII) and cassette exons (Ia and Ib), and numbers below refer to position in the EBV genome. Changes in coverage peaks correspond to splicing. The current RefSeq annotation (NC_007605.1) does not include exon Ia, Ib, and II, nor the multitude alternative splice sites within exon III, V, and VII. (B) Detection of splice junction in short-read sequencing data sets. The number in the y-axis corresponds to the frequency of splice-junction reads. The coverage of nanopore reads across RPMS1 correlates with the extracted splice-junction positions in complementary short-read sequencing data sets. (NPC) Nasopharyngeal carcinoma.
It should be noted that a gene-specific PCR approach for nanopore sequencing does not account for diversity pertaining to alternative promoter usage and/or alternative polyadenylation. The extracted splice-junction reads from the short-read data sets revealed an unannotated splice site within the intron region upstream of exon V. Since this splice site does not have any coverage in the nanopore sequencing, we could infer that this splice site represents a 3′ splice site of a previously unknown transcription start site within the RPMS1 gene.
The NC_007605.1 reference annotation proved to be the limiting factor to describe the bulk of transcript variants generated by nanopore sequencing. Only 3.31% and 0.17% of the high-confidence reads from the C666-1 and GAC library, respectively, were fully annotated when the long-read categorization function was applied using the reference genome. As expected, however, exon Ia, Ib, and II were immediately flagged as authentic by the novel splice site detection function. These exons have previously been reported and are regarded as established amendments to the reference annotation. Exon Ia, Ib, and II were therefore added to the local reference variable. Utilizing this updated reference, FLAME categorized 12.03% of the C666-1 reads and 22 intermediate-confidence transcript variants as annotated. As regards the high-confidence data set, four transcript variants were captured, representing 12.92% of the reads. When the GAC library was used as input, 17 intermediate-confidence transcript variants were captured, yet only representing 5.23% of the reads. Similarly, 6.24% of the reads, distributed on six transcript variants, were retrieved in the high-confidence data set. Altogether, these numbers demonstrated that the available annotation was insufficient to account for the overwhelming majority of RPMS1 reads.
A central feature of FLAME is the capability to single out abundant unannotated splice sites from a large set of uncharacterized long-reads. The novel splice site detection function was therefore used to thoroughly sift through the large number of exon ranges that were incongruent with the RPMS1 reference annotation. The workflow was set up so that novel exons had to be supported by ≥10 reads and flanked by canonical splice site dinucleotides (GU/AG) in order to be deemed authentic. Moreover, the validity and exact position of all acceptor and donor sites had to be reinforced by complementary splice-junction reads from the short-read data. By using these criteria, FLAME distinguished 22 novel exons in the C666-1 library and 30 exons in the GAC library. In total, 32 novel elements were found, out of which 20 were common for both C666-1 and GAC (Fig. 3). All novel exons were ultimately merged into one reference file, which brought about an almost complete retrieval rate. Looking at the C666-1 intermediate-confidence data set, 95.70% of the reads and 245 transcript variants were now fully annotated, whereas 99.06% of the reads and 57 transcript variants were annotated in the high-confidence data set. With respect to the GAC library, corresponding numbers in the intermediate-confidence data set were 98.39% and 247 transcript variants, and 99.86% and 74 transcript variants in the high-confidence data set (Supplemental Table 2).
FIGURE 3.

Comprehensive annotation of RPMS1 in transformed epithelial tissue. The upper segment displays the starting reference annotation of RPMS1 as it is used in FLAME. Departures with respect to the RefSeq reference (addition of exon Ia, Ib, and II) are indicated. Moreover, exon III is by convention regarded as one constitutive exon, although it is divided into two constituents in the RefSeq reference. Alternative exons discovered by FLAME are displayed in the middle segment. The nomenclature and coordinates of all exons are listed in the attached table. Putative open reading frames are displayed in the bottom segment. Numbers on the X-axis refer to the global position in the EBV genome.
FLAME proved to be sensitive enough for detection of rare splicing events. A novel cassette exon spanning over 57 bp was discovered within the intron between exon VI and exon VII. This exon, designated exon VIa, was found to be mutually inclusive with exon VI and exon VIIa1 and occurred in three intermediate-confidence transcript variants in the GAC library. Notably, this exon was not detected in C666-1. Additionally, an intron retention event spanning over exon III and exon IV (r:III'IV) was observed in two intermediate-confidence transcript variants (corresponding to 0.09% of the reads) in the GAC library. r:III'IV was however significantly more frequent in the C666-1 library as it appeared in seven intermediate-confidence transcript variants (1.57% of reads) and three high-confidence transcript variants (1.50% of reads). In addition, another intron retention event spanning from exon IIIc to exon IV was observed in one intermediate-confidence transcript variant (0.02% of reads) in the C666-1 library. As illustrated in Figure 3, exon III is divided into two segments in the RefSeq annotation. Our analysis showed that exon IIIa and exon IIIc were virtually mutually inclusive and the 3′ end of exon IIIa and the 5′ end of exon IIIc were suboptimal splice sites, as exon III was found to be fully retained in 64.30% of high-confidence C666-1 reads and 64.52% of high-confidence GAC reads.
The spectrum of RPMS1 splice variants was largely attributable to alternative usage of multiple acceptor and donor splice sites within otherwise constitutive exons (exon III, exon V, and exon VII). The most common splicing patterns thus derived from several instances of variable 5′ and 3′ flanks of exons in multiplex combinations. The repertoire of transcript variants was thereby shifted to shorter transcripts due to splitting of long exons, as illustrated in Figure 4. Differential usage of fully contained exons; that is exon skipping, was of minor importance in this respect (Fig. 5). All alternative splice sites appeared to be relatively weak in terms of providing inter-exon connectivity as the canonical boundaries of the exons were with very few exceptions invariably maintained in the splice junctions between constitutive exons. Furthermore, there was an asymmetry in the numbers of novel acceptor and donor sites with an overweight toward acceptor sites. The relative strength of all acceptor and donor sites are depicted in Supplemental Figure 3. The four novel alternative donor sites within exon V were rarely consecutively connected to exon VI. Conversely, the seven alternative acceptor sites were rarely connected to exon IV. When viewed collectively, the alternative splice sites within exon V were used in 33.12% of C666-1 high-confidence reads and 35.50% of GAC high-confidence reads. Notably, exon Vg was observed to be a major splice site acceptor within both GAC and C666-1, thereby providing an intrinsically strong donor site for consecutive coupling to exon VI. Eight alternative acceptor sites and four alternative donor sites were detected within exon VII. These were used in 27.36% of the high-confidence reads in C666-1. In stark contrast, the alternative splice sites were used in 68.18% of the high-confidence reads in the GAC library. Among these, VIIa1 in conjunction with VIIf2 was most abundant. The spliceome of RPMS1 shows that for example, design of RT-PCR primers for RNA without the detailed knowledge of the gene transcript variants may generate amplicons which neglects a large proportion of the transcripts.
FIGURE 4.
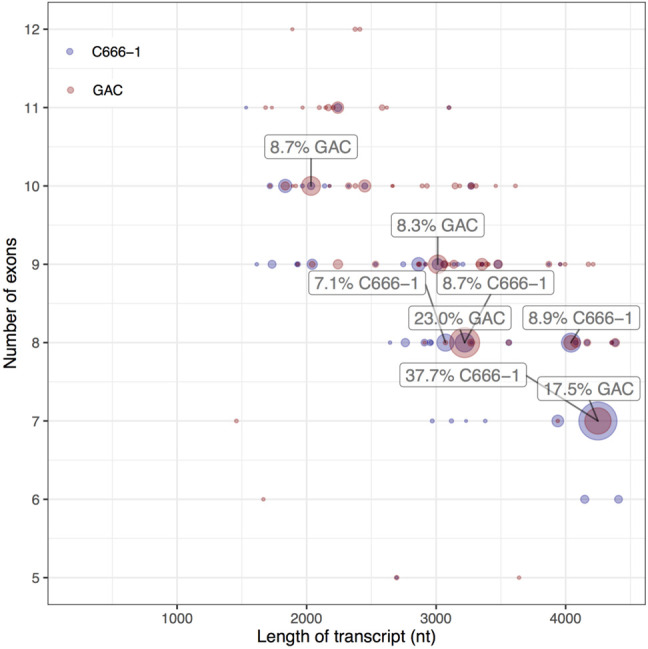
Diversity plot of high-confidence data. Each dot represents a unique transcript variant and the area correlates with the number of supporting reads expressed as percentage of fully annotated reads. The 4.2 kb splice form represents 37.7% and 17.5% of the reads in the C666-1 and GAC library, respectively. Splicing within long exons pulls the center of density toward shorter transcripts with more exons. Transcript variants comprising <5 exons (equivalent to 0.26% of the data) are not shown.
FIGURE 5.

Weighted adjacency matrices outlining the overall patterns of consecutive exon connectivity in all long-read sequences. Vertical shifting within a column represents alternative acceptor sites for a given donor site, whereas horizontal shifting within a row represents alternative donor sites for a given acceptor site. Long-range exon connectivity possibly resulting from artifactual joining of primers appears in the top left corner.
Given the high annotation rate using FLAME (>95%), a relative quantification of splice variants could be performed with improved precision as the full extent of alternative splicing events was considered. The 15 most abundant splice variants of RPMS1, supported by more than 1.2% of high-confidence reads in the C666-1 library and 1% in the GAC library, are delineated in Figure 6. The 4.2 kb variant of RPMS1 represented the paramount transcript in C666-1 and constituted 37.7% of high-confidence reads. It is worthwhile to notice, however, that this splice form only represented 17.5% of the high-confidence reads in GAC. The 15 most prevalent transcript variants, which together constituted 82.49% and 86.64% of high-confidence annotated reads in GAC and C666-1, respectively, contained 41 unique putative ORFs longer than five amino acids (Supplemental Table 3). In the transformed epithelial tissues all seven constitutive exons were represented in all of the 15 most common transcript variants. In contrast, in the Burkitt's lymphoma cell line Daudi, five noncanonical long-range splice junctions were observed among the 15 most common splice forms (Supplemental Fig. 4). The divergence of splice variants of RPMS1 in Daudi from the pattern observed in C666-1 and GAC could be cell type specific and/or related to the propensity for viral reactivation.
FIGURE 6.

The most abundant transcript variants of RPMS1. The 15 most prevalent splice forms of RPMS1, representing all variants supported by ≥1.2% of high-confidence reads in the C666-1 library and 1% in the GAC library, are illustrated. Quantification of the relative abundance is expressed as the percentage of annotated high-confidence reads. Numbers on the X-axis refer to the global position in the EBV genome.
Eight ORFs were only present in either of the epithelial libraries and the majority of these ORFs constituted less than five percent of the annotated reads. The suggested protein BARF0 (ORF_173) could be encoded from all transcripts in both GAC and C666-1. Previously described putative ORFs for RPMS1 (protein) and A73, but not RPMS1A (1.4%), were found to be represented by a large fraction (60.2% and 54.1%, respectively) of the annotated reads in GAC. In C666-1, from which most of these ORFs were discovered, RPMS1, RPMS1A, and A73 were present in 63.8%, 20.4%, and 20.5%, respectively, of the annotated reads.
Performance benchmarking using human and viral data sets
The computational efficiency and robustness of FLAME was evaluated against FLAIR, a pipeline for nanopore sequencing data analysis. Using different data sets, including both human and viral transcriptomes as well as various library preparation methods, the performance of FLAME was compared to FLAIR with respect to retrieval of annotated reads, assembling of transcript variants and runtime.
First, the performance of FLAIR was assessed on the RPMS1 data set presented above (Supplemental Table 4). When FLAIR was used with the RefSeq annotation to analyze the GAC data set, 549 reads (0.42%) were annotated and 11 full-length transcript variants were assembled on the basis of these. Despite expansion of the reference with all alternative exons obtained from the previous FLAME analysis, FLAIR was only able to annotate 4602 reads (3.50%) and assemble 26 full-length transcripts (compared with the 109,464 reads and 74 transcript variants annotated as high-confidence by FLAME). However, 16 out of the 26 alternative transcripts assembled by FLAIR were lacking both exon V and VI, and six transcripts were missing exon III, IV, V, and VI. This significant absence of otherwise established constitutive exons is notable and contradicts the coverage plot (Fig. 2). In addition, the intron between exon V and VI was retained in five cases. None of the transcript variants detected by FLAIR contained the canonical exon I–VII setup (Supplemental Fig. 5). In contrast, using FLAME, all of the high-confidence transcript variants present in more than 1% of the data set contained variants of the exon I–VII setup. The computational performance of FLAIR using the C666-1 data set was comparable in all material respects, although the retrieval rate of annotated reads actually deteriorated when the reference annotation was expanded with alternative exons. The computational processing time for the analysis of RPMS1 corresponded to 1255.49 CPU seconds and 52.24 CPU seconds for FLAIR and FLAME, respectively. Thus, FLAME performed alternative splicing analysis, including novel splice site detection, at 4.16% of the computational time it required for FLAIR to finish the analysis.
We next used FLAME-GLOW on a publicly available native RNA sequencing data set to identify adequately annotated genes for further comparison with FLAIR (Workman et al. 2019). The three housekeeping genes ACTB (beta-actin), B2M (beta-2 microglobulin), and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) were found to display high coverage and an acceptable proportion of annotated full-length transcripts using FLAME-GLOW. In total, 61%–73% of the reads mapping to these genes were fully annotated when FLAME-GLOW was applied. Corresponding numbers obtained from FLAIR were 16%–51% (Supplemental Table 5). Notably, for B2M, none of the 7 splice variants assembled by FLAIR contained the complete CDS (Supplemental Fig. 6). Lastly, FLAME was tested using data previously analyzed with FLAIR. Mutation of SF3B1 in the context of chronic lymphocytic leukemia has previously been shown to result in differential usage of one alternative 3′ splice site in the ERGIC3 gene (Tang et al. 2020). This finding was reproducible using FLAME; moreover, two additional alternative acceptor splice sites were detected within ERGIC3 (Supplemental Fig. 7).
Conclusions
Long-read sequencing technologies have opened up the possibility to analyze the splicing of full-length RNA transcripts in a high-throughput manner. Current tools for splicing analysis perform well for genes with an adequate reference genome, but strict dependence on a given annotation file renders the task of retrieving unannotated exons difficult. We have developed FLAME with the aim to provide a perspicuous description of the entire spliceome of any RNA transcript without reliance on existing annotation. To limit any deceiving impact of nanopore sequencing artifacts and compensate for inadequate reference annotations, FLAME allows for incorporation of complementary short-read data to rapidly and reliably define novel exons. FLAME-GLOW is suitable for global-wide identification of perturbed splicing in a large-scale context. FLAME is written in a single programming language and designed to rely on as few external packages, software and tools as possible, making the tool straightforward to use and almost self-sufficient in its implementation into different computational environments. Many facets of the program have been designed to allow for modularity, from the flexibility of implementing the user's own thresholds values, to the ability to use each individual subfunction independently with either direct command-line interaction or through scripting.
Here, we have analyzed the complex splice variation of the viral lncRNA RPMS1 and unveil a wealth of previously unknown splicing events. FLAME was compared with FLAIR and was shown to be 24 times faster and 24 times more efficient at processing the data. In this study, we only considered transcript variants supported by at least ten (intermediate-confidence) or a hundred (high-confidence) reads to minimize the number of false positive variants, while simultaneously reporting representative splicing patterns from the plethora of variants. Depending on sequencing quality/depth and purpose of the study, the threshold limit value can be adjusted to accommodate for the specific setting. Currently, no known molecular function has been attributed to RPMS1. Whether functional RNA domains exist within the RPMS1 RNA remains to be seen, but our study has now added additional possibilities for exploring alternative secondary RNA structures. Moreover, with a comprehensive list of putative ORFs it would be possible to confirm or rule out the protein coding ability of RPMS1 by unbiased approaches, for example, mass spectrometry. In summary, FLAME is a universal tool for accurate identification of novel exons and provides a comprehensive overview of the spliceome.
MATERIALS AND METHODS
Design and implementation of the pipeline
The FLAME software is available on https://github.com/marabouboy/FLAME.
Creating a reference
The create.ref function converts the input reference annotation from GTF into a list format. Certain entries are filtered out from the input reference file based on the following exclusion criteria: (a) the feature is classified as anything else but exon as feature type; (b) the feature does not contain the specific name of the targeted gene within transcript_id, gene_id or gene_name; (c) the feature is classified as a microRNA. These criteria allow for the input to be an entire chromosome or genome. The resulting list contains five features: (i) exon name based on start position, (ii) chromosome, (iii) exon global start position, (iv) exon global end position, and (v) exon length.
Long-read categorization
All irrelevant data is discarded in order to maximize the processing speed in the filter function. Temporarily saved relevant data includes: (a) BED12 start, which is the global start position; (b) BED12 blockCount, which is the number of exons in the read; (c) BED12 blockStarts, which is a comma-separated list of each exon start position relative to the long-read; and (d) BED12 blockSizes, which is a comma-separated list of each exon length. The following cross-referencing of input long-reads relies on three key principles. (i) Input long-read exon and corresponding annotated exon must be concordant with respect to global start position, exon length, and global end position. (ii) A variance function accounts for sequencing errors, base calling errors and insertion-deletion events. The default number of nucleotide variance is 20; however, this is customizable to fit different data sets including microexons. (iii) If any exon within a long-read does not find a matching annotated reference exon, the entire sequence is classified as incongruent. Conversely, the sequence is filtered into the annotated data set if all exons within the long-read find matching annotated reference exons. The translate function is crucial for efficient representation, storage and transfer of data. In this function, annotated reads are deciphered from read range format into predefined exon designations. This is to avoid the laborious process of scrutinizing the sizable set of information in BED format. Incongruent exons will remain untranslated as global read range.
Data representation of annotated reads
The quantify function collapses full-length exon permutations and quantifies the abundance of different transcript variants. This function relies on simple item detection and counting. If the inputted combination of exons exists in an array, the attached score of this particular isoform is raised by an increment of one. If the inputted combination of exons does not exist, the isoform is added to the array with an attached score of one. The annotated.adjmtx function displays the consecutive exon connectivity in a quantitative manner. The first exon specifies the i-dimension coordinate, while the preceding exon specifies the j-dimension coordinate.
Novel splice site and exon detection
The incongruent.separator function splits exon ranges into two separate splice sites. The splice sites will remain split when used in further analysis steps until the incongruent.adjmtx function is put into practice. The frequency.site function quantifies unannotated splice sites in an array data structure. A variance smoothing operator merges possible novel splice site positions that differ in ±m nucleotides (default = 2). The array data structure exists as raw data but could prove overwhelming. Therefore, a customizable threshold can be implemented via the frequency.thresh function. Consequently, only nucleotide positions that are represented by a certain percentage (default >1%) will be returned. The value of each potential splice site that surpasses the threshold is displayed in absolute numbers and percentage of incongruent reads.
The splice.signal function strengthens the validity of splice sites that exceed the threshold percentage. If a genomic reference file is presented, the potential novel splice sites are located and their neighboring nucleotides are processed with a range of ±3 nt. If any of the nucleotides within this seven nucleotide window, with the potential novel splice site signal being centered, contains the splice site dinucleotides (GU/AG), it is flagged.
The shortread function is dependent on the presence of a short-read sequencing file in either bam or sam format. The CIGAR string is processed so as to register the number of splice events. Once the cigar string registers the number of splice operators (N), it calculates the exact position of the splice site, and saves the position as a list variable. Once all the short-read sequences are processed and have been saved and quantified in the storage variable, said storage variable is cross-referenced with the list of splice site positions that passed the frequency analysis (frequency.thresh) percentage threshold.
The incongruent.adjmtx is the key function in the translation of novel splice sites into entire novel exons. The splice sites, generated from the frequency.site, frequency.thresh, splice.signal, and shortread functions can be used to generate a weighted adjacency matrix. The reads classified as incongruent are then processed to single out the exons that did not match the reference. These exons have their global start position interpreted as their column position and their global end position interpreted as their row position for which that position has its weight increased by an increment of one in the weighted adjacency matrix. This is done for each incongruent exon with the end result producing a table where the most frequent exon ranges have the highest weight. Manual determination of true novel exon or novel splice site is required as contextual information such as experiment design cannot be accessed through the input data. Throughout this work the frequency.thresh function was configured with a value of 1, meaning that only unannotated splice sites accounting for more than 1% of the incongruent reads were returned. Consequently, rare unannotated splicing events could be concealed from discovery. This problem was however circumvented by continuously updating the reference as part of iteratively cycling through the pipeline. Based on our experience it is sufficient with 3 to 4 cycles to reach saturation.
FLAME-GLOW
The transcriptome-wide module is a wrapper of already existing functions within the FLAME function library, with the addition of a single function segment. Briefly, FLAME-GLOW starts by extracting the name of each gene in the input reference file, and creating a database in Python list format. The pipeline then iterates through every gene name, initially applying three functions: create.ref, segment and filter. As explained previously, the create.ref creates a reference in list format for the current gene iteration. The main goal of the segment function is to extract the reads that are within the vicinity of the current gene iteration, with respect to the genomic position. The goal of this function is to optimize the program so as not needing to repeatedly process irrelevant reads, for example, reads that are located on a different genomic section from the current gene iteration. The filter function is then applied with the segmented reads as input and the create.ref output as reference. The function filter works in the exact same manner as previously described. However, post-filter, the ratio of reads classified as annotated compared to the total number of reads is calculated. Depending on a threshold, customizable with a default threshold of 25%, the gene iteration is processed differently. If the gene has an annotation ratio above the threshold, the reads are processed through the translate and quantify functions. However, if the gene falls below the threshold, it would be flagged as being worthy of a secondary analysis, recommended to be analyzed by the gene specific FLAME module. The reason for this threshold filtering is due to the assumption that if a gene is annotated well enough, the only interesting data would be the isoform quantification of the current gene iteration. Conversely, if a gene is not well annotated enough, the need for translating and quantifying the isoforms are superfluous as too many reads cannot be classified and quantified with the existing reference and would require a more in-depth analysis.
Cells
The nasopharyngeal carcinoma cell line C666-1 and the Burkitt's lymphoma cell line Daudi were grown in RPMI-1640 medium (Gibco) supplemented with 10% fetal calf serum and cultured at 37°C with 5% CO2. Total RNA was extracted using TRIzol reagent (Life Technologies) according to the supplier's instructions. The eluate was subjected to DNase treatment (TURBO DNA-free Kit [Thermo Fisher Scientific]) and then stored at −80°C.
Patient samples
The gastric resection material was collected within a translational collaboration named “Immunological biomarkers for gastric cancer” (ethical approval number 2010/176-10). Samples were collected between June 2011 and July 2012 at Hospital Escuela Dr. Roberto Calderón Gutierrez (GHERCG), in Managua, Nicaragua (Thorell et al. 2017). During the study period, 15 patients were enrolled and biopsies from eleven patients were obtained. Punch biopsies were taken immediately after the resection and thereafter placed in RNA later and instantly snap-frozen. RNA was extracted with TissueLyser disruption using the RNeasy Mini Kit (Qiagen). Five tumors were randomly selected and tested for EBV using RT-qPCR targeting RPMS1 (Fwd: 5′-GATGTTTTGCGCCTGGAAGTTG; Rev: 5′-TCTCCTCGGACATCCAGTGTC) and EBER-1 (Fwd: 5′-ACGCTGCCCTAGAGGTTTTG; Rev: 5′-AGACGGCAGAAAGCAGAGTC). GAPDH (Fwd: 5′-TCTCTGCTCCTCCTGTTCGA; Rev: 5′-GCCCAATACGACCAAATCC) served as an internal control.
Long-read sequencing
First strand cDNA synthesis was primed with an oligonucleotide targeting the sequence immediately upstream of the poly(A) signal (5′-TTGCATGTCTCACACCATGG). Approximately 2.5 µg of total RNA was incubated at 65°C for 5 min together with 20 pmol primer and 1 mM dNTP mix (Thermo Scientific), and thereafter instantaneously chilled on ice. The final reaction mixture was assembled in a total volume of 20 µL by adding 4 µL 5× RT Buffer (Thermo Scientific), 1 µL Maxima H Minus Reverse Transcriptase (Thermo Scientific) and 0.5 µL RNase OUT (Life Technologies), and incubated for 30 min at 55°C, 5 min at 85°C and then held at 4°C.
Full-length transcripts of RPMS1 were selected by PCR amplification using Q5 High-Fidelity 2× Master Mix (New England Biolabs) according to the manufacturer's protocol. A 2 µL-portion of the RT reaction mixture was carried into a total reaction volume of 25 µL and the following reaction was incubated at 98°C for 1 min prior to 18 cycles of [98°C for 10 sec, 66°C 15 sec, 72°C for 4 min], followed by a final extension at 72°C for 5 min and holding indefinitely at 4°C. For the RNA originating from the Daudi cells, the PCR was extended with 17 additional cycles. Resulting amplicons were purified by incubation with 0.8× Agencourt AMPure XP beads (Beckman Coulter), followed by two washes with 200 µL of 75% ethanol and resuspension in 25 µL nuclease-free water. The remaining DNA concentration was measured with Qubit Fluorometer (Qubit DNA HS Assay Kit).
Subsequent end-prep with NEBNext Ultra II End repair/dA-tailing Module (E7546), Agencourt AMPure XP bead binding and Oxford Nanopore Technologies adapter ligation with NEB Blunt/TA Ligase Master Mix (M0367) was performed following the Direct cDNA Sequencing (SQK-DCS109) protocol version DCS_9090_v109_revJ_14Aug2019. The adapted and tethered library was enriched using 0.4× Agencourt AMPure XP beads washed with 2 × 200 µL Adapter bead binding buffer, and finally eluted in 14 µL Elution buffer (Oxford Nanopore Technologies).
The two libraries were separately loaded on FLO-MIN106D R9 flow cells according to the manufacturer's specifications. The sequencing was performed on a MinION Mk1B device (MIN-101B) and operated through MinKNOW release 19.12.5. Raw data was base called using Guppy (3.6.1 + 249406c) configured with the high accuracy model (dna_r9.4.1_450bps_hac, default settings).
Data processing
Long-read aware RNA aligner minimap2 (https://doi.org/10.1093/bioinformatics/bty191, v2.17-r941) was used to map the sequences with parameters using the SPLICE preset of options and parameters while also specifying the exclusion of secondary alignment (Li 2018). The NCBI RefSeq for EBV was used as the reference for the alignment for both samples. The generated SAM-files were sorted, indexed and compressed into the binary form using the samtools toolkit (https://doi.org/10.1093/bioinformatics/btp352, v1.10) (Li et al. 2009). The generated bam-files were filtered so as to remove reads categorized as supplemental and/or secondary reads. Further filtering on the bam-files were performed so as to require the inclusion of RPMS1 exon 1. An additional filtering step was performed on the Daudi long-read data set as the raw sequencing reads contained a large proportion of primer related artifacts. This filtering step required the read to have a minimum read length of 1000 nt, which resulted in the filtering of 83.98% of the original 884,827 Daudi reads. These filtering steps were done through an in-house bash-script. The filtered bam-files were then converted into BED12 format using the bedtools toolkit (https://doi.org/10.1093/bioinformatics/btq033, v2.26.0) (Quinlan and Hall 2010). These two BED12-files were then used as input for FLAME.
The C666-1, nasopharyngeal carcinoma and GAC bulk RNA-seq samples were retrieved from EMBL-ENA (ENA study accession number PRJNA501807 [Edwards et al. 2008] and PRJNA397538 [Zhang et al. 2017]) and TCGA (samples that were classified as STAD and EBV positive [Cancer Genome Atlas Research Network 2014]), respectively. The data sets were then preprocessed by Prinseq (https://doi.org/10.1093/bioinformatics/btr026, Version 0.20.3) and TrimGalore (https://github.com/FelixKrueger/TrimGalore, Version 0.4.4) (Schmieder and Edwards 2011). The data sets were then aligned using STAR using the human HG38 (GRCh38) in fasta, the human annotation file in GTF format, the NCBI RefSeq EBV reference genome (NC_007605.1) in fasta format and the NCBI EBV annotation file in GTF format as reference. Specific parameters for all tools are available upon request.
FLAIR was used according to the developers’ instructions, using standard parameters for both the GAC and the C666-1 long-read RNA-seq, with each respective aforementioned short-read bulk RNA-seq pairing. FLAME was used with standard parameters for both the GAC and the C666-1 long-read RNA. The variance window was set at 20 nt both upstream and downstream, and the novel splice site detection had a frequency threshold of more than 1 percent of the incongruent long-read sequences. The comparison between FLAIR and FLAME was performed in the same system (Asus Vivobook S403F notebook with an Intel Core i7-8586U 1.80GHz quad core processor capable of up to 4.6 GHz of processing and 16 GB of memory).
Two data sets generated by Tang et al. (2020) (BioProject study accession number PRJNA369585), for which samples marked as Promethion WT 1 (SRR11142440) and Promethion MT 3 (SRR11142446) were used to represent the SF3B1 wild type and SF3B1 mutant, respectively. These two data sets were extracted and then aligned using Minimap2 using the SPLICE preset parameter while also specifying the exclusion of secondary alignment. The NCBI RefSeq fasta-file for the human genome (GRCh38) was used as the reference genome for the alignment of these data sets. The generated bam-files were filtered to remove reads categorized as supplementary and/or secondary reads as well as needing to be aligned within the genomic position of the ERGIC3 gene region (±1000 nt upstream/downstream). The filtered bam-files were then converted into BED12 format using the bedtools toolkit (v2.26.0). These two BED12-files were then used as input for the gene specific FLAME module.
We downloaded full native RNA long-read sequencing data generated by Workman et al. (2019) sequenced using ONT Nanopore MinION and base called using Guppy v4.2.2. This data was available from their Amazon Web Services storage servers using the bash wget function. The data set was then aligned using Minimap2 using the SPLICE preset parameter while also specifying the exclusion of secondary alignment. The NCBI RefSeq fasta-file for the human genome (GRCh38) was used as the reference genome for the alignment of this data set. Reads classified as secondary or supplementary reads were filtered out. The resulting bam-file was subsequently converted into BED12 format using the bedtools toolkit (v2.26.0). This file was used as input for the FLAME-GLOW module with standard parameters.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Ingemar Ernberg, Karolinska Institutet, Stockholm, Sweden, for the generous gift of the C666-1 cell line. We thank everyone involved in the Nicaraguan GAC sample collection initiative, especially Dr. Lawrence Paszat, Dr. Reyna Victoria Palacios Gonzáles, and surgeons Dr. Alvaro Lopez and Dr. Carlos Ruiz. The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The computations and data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC, project sens2018120) at Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) partially funded by the Swedish Research Council through grant agreement no. 2018-05973. We thank the Bioinformatics Core Facility at the Sahlgrenska Academy for bioinformatics analyses. This study was supported by grants from the Assar Gabrielssons Research Foundation, the BioCARE National Strategic Research Program at the University of Gothenburg, and the Wallenberg Centre for Molecular and Translational Medicine, University of Gothenburg, Sweden.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078800.121.
Freely available online through the RNA Open Access option.
REFERENCES
- Byrne A, Beaudin AE, Olsen HE, Jain M, Cole C, Palmer T, DuBois RM, Forsberg EC, Akeson M, Vollmers C. 2017. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun 8: 16027. 10.1038/ncomms16027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. 2014. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513: 202–209. 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Smith P, Ambinder RF, Hayward SD. 1999. Expression of Epstein-Barr virus BamHI-A rightward transcripts in latently infected B cells from peripheral blood. Blood 93: 3026–3032. 10.1182/blood.V93.9.3026.409k28_3026_3032 [DOI] [PubMed] [Google Scholar]
- Dohm JC, Peters P, Stralis-Pavese N, Himmelbauer H. 2020. Benchmarking of long-read correction methods. NAR Genom Bioinform 2: lqaaos7. 10.1093/nargab/lqaa037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RH, Marquitz AR, Raab-Traub N. 2008. Epstein-Barr virus BART microRNAs are produced from a large intron prior to splicing. J Virol 82: 9094–9106. 10.1128/JVI.00785-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell PJ. 2019. Epstein-Barr virus and cancer. Annu Rev Pathol 14: 29–53. 10.1146/annurev-pathmechdis-012418-013023 [DOI] [PubMed] [Google Scholar]
- Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, et al. 2018. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 15: 201–206. 10.1038/nmeth.4577 [DOI] [PubMed] [Google Scholar]
- Johansson C, Schwartz S. 2013. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat Rev Microbiol 11: 239–251. 10.1038/nrmicro2984 [DOI] [PubMed] [Google Scholar]
- Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. 2019. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20: 278. 10.1186/s13059-019-1910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquitz AR, Mathur A, Edwards RH, Raab-Traub N. 2015. Host gene expression is regulated by two types of noncoding RNAs transcribed from the Epstein-Barr virus BamHI A rightward transcript region. J Virol 89: 11256–11268. 10.1128/JVI.01492-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollet IG, Ben-Dov C, Felicio-Silva D, Grosso AR, Eleuterio P, Alves R, Staller R, Silva TS, Carmo-Fonseca M. 2010. Unconstrained mining of transcript data reveals increased alternative splicing complexity in the human transcriptome. Nucleic Acids Res 38: 4740–4754. 10.1093/nar/gkq197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Qiu J, Cosmopoulos K, Pegtel M, Hopmans E, Murray P, Middeldorp J, Shapiro M, Thorley-Lawson DA. 2011. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog 7: e1002193. 10.1371/journal.ppat.1002193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab-Traub N, Hood R, Yang CS, Henry B II, Pagano JS. 1983. Epstein-Barr virus transcription in nasopharyngeal carcinoma. J Virol 48: 580–590. 10.1128/jvi.48.3.580-590.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PR, de Jesus O, Turner D, Hollyoake M, Karstegl CE, Griffin BE, Karran L, Wang Y, Hayward SD, Farrell PJ. 2000. Structure and coding content of CST (BART) family RNAs of Epstein-Barr virus. J Virol 74: 3082–3092. 10.1128/JVI.74.7.3082-3092.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KW, Larsson E. 2017. Tumour virology in the era of high-throughput genomics. Philos Trans R Soc Lond B Biol Sci 372: 20160265. 10.1098/rstb.2016.0265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. 2013. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun 4: 2513. 10.1038/ncomms3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AD, Soulette CM, van Baren MJ, Hart K, Hrabeta-Robinson E, Wu CJ, Brooks AN. 2020. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11: 1438. 10.1038/s41467-020-15171-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorell K, Bengtsson-Palme J, Liu OH, Palacios Gonzales RV, Nookaew I, Rabeneck L, Paszat L, Graham DY, Nielsen J, Lundin SB, et al. 2017. In vivo analysis of the viable microbiota and Helicobacter pylori transcriptome in gastric infection and early stages of carcinogenesis. Infect Immun 85: e00031-17. 10.1128/IAI.00031-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toptan T, Abere B, Nalesnik MA, Swerdlow SH, Ranganathan S, Lee N, Shair KH, Moore PS, Chang Y. 2018. Circular DNA tumor viruses make circular RNAs. Proc Natl Acad Sci 115: E8737–E8745. 10.1073/pnas.1811728115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerleider N, Concha M, Lin Z, Roberts C, Wang X, Cao S, Baddoo M, Moss WN, Yu Y, Seddon M, et al. 2018. The Epstein Barr virus circRNAome. PLoS Pathog 14: e1007206. 10.1371/journal.ppat.1007206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman RE, Tang AD, Tang PS, Jain M, Tyson JR, Razaghi R, Zuzarte PC, Gilpatrick T, Payne A, Quick J, et al. 2019. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat Methods 16: 1297–1305. 10.1038/s41592-019-0617-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Iwatsuki K. 2012. Diversity of Epstein-Barr virus BamHI-A rightward transcripts and their expression patterns in lytic and latent infections. J Med Microbiol 61: 1445–1453. 10.1099/jmm.0.044727-0 [DOI] [PubMed] [Google Scholar]
- Zhang L, MacIsaac KD, Zhou T, Huang PY, Xin C, Dobson JR, Yu K, Chiang DY, Fan Y, Pelletier M, et al. 2017. Genomic analysis of nasopharyngeal carcinoma reveals TME-based subtypes. Mol Cancer Res 15: 1722–1732. 10.1158/1541-7786.MCR-17-0134 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.