

Abstract
Many epigenetic modifications occur in glioma, in particular the histone‐deacetylase class proteins play a pivotal role in glioma development, driving the proliferation rate and the invasiveness of tumor cells, and modulating the tumor microenvironment. In this study, we evaluated the role of the histone deacetylase HDAC8 in the regulation of the immune response in glioma and tumor growth. We found that inhibition of HDAC8 by the specific inhibitor PCI‐34051 reduces tumor volume in glioma mouse models. We reported that HDAC8 modulates the viability and the migration of human and murine glioma cells. Interestingly, HDAC8 inhibition increases the acetylation of alpha‐tubulin, suggesting this epigenetic modification controls glioma migration. Furthermore, we identify HDAC8 as a key molecule that supports a poorly immunogenic tumor microenvironment, modulating microglial phenotype and regulating the gene transcription of NKG2D ligands that trigger the Natural Killer cell‐mediated cytotoxicity of tumor cells. Altogether, these results identify HDAC8 as a key actor in glioma growth and tumor microenvironment, and pave the way to a better knowledge of the molecular mechanisms of immune escape in glioma.
Keywords: epigenetic, glioma, histone‐deacetylase 8 protein, microglia, natural killer cell, natural killer group 2D (NKG2D) ligands, PCI‐34051
Main Points
HDAC8 modulates the viability of human and murine glioma cells and tumor migration through a–tubulin acetylation.
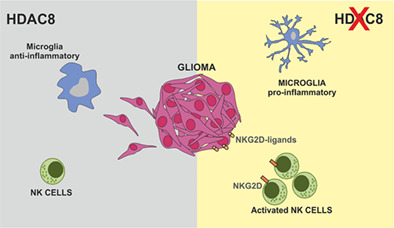
HDAC8 supports a poorly immunogenic tumor microenvironment modulating microglial phenotype and inhibiting NK cell cytotoxic activity.
1. INTRODUCTION
Among the brain neoplasms, glioma is the most diffuse tumor, and its prognosis is significantly correlated with pathological grades. Glioblastoma (GBM) (classified as >IV grade of glioma by World Health Organization [WHO]) has high proliferation rate and cell invasiveness in surrounding parenchyma, promoting tumor relapse (Stupp et al., 2009). Despite the advance in surgery, radiation and immunotherapy patient prognosis is poor, with an overall survival rate of about 14 months (Preusser et al., 2011). One of the main hallmarks of glioma is its molecular diversity, due to the genomic heterogeneity and instability and to the epigenetic modifications that occur during glioma growth and development (Phillips et al., 2020; Xu et al., 2021). Epigenomic reprogramming is one of the fundamental drivers of glioma.
For example, isocitrate dehydrogenases (IDH) 1 is a key metabolic enzyme that generates nicotinamide adenine dinucleotide phosphate (NADPH), and the mutational status of IDH1 genes induces differences in methylation patterns in malignant glioma, which correlate with distinct clinical characteristics (Shabason et al., 2011).
Another important epigenetic modulator identified in the last years and correlated with different kinds of cancers is the methyltransferase Enhancer of Zeste 2 (EZH2). EZH2 is a component of the polycomb repressive complex 2 (PRC2) and catalyzes the methylation of lysine 27 of histone H3, a modification associated with gene silencing. EZH2 is expressed predominantly in proliferating cells, and is required for hematopoietic stem cell function and lymphocyte development (Nutt et al., 2020). Recent studies demonstrated that EZH2 is amplified in a variety of solid tumor, and elevated EZH2 activity is associated with poor prognosis (Helin & Dhanak, 2013; Varambally et al., 2002). These data have suggested that EZH2 inhibition could be a potential anti‐cancer strategy. Indeed, the drug Tazemetostat, a potent EZH2‐inhibitor, was recently approved for the treatment of multiple types of hematological and solid tumors (Kim & Roberts, 2016; Marsch & Jimeno, 2020; Mohammad et al., 2017; Rugo et al., 2020), supporting the hypothesis. Furthermore, EZH2 plays a central role also in glioma, promoting tumor cell invasion, migration and proliferation (Chen et al., 2021; Suvà et al., 2009), and driving microglial cells toward an anti‐inflammatory, pro‐tumor phenotype (Qiao et al., 2016; Yin et al., 2017).
In particular, in the last years, many studies focussed their attention on the histone‐deacetylase (HDAC) proteins that cause global chromatin condensation and transcriptional repression, as well as the deacetylation of cytoplasmatic target proteins. In this way, HDAC proteins modulate several pathways involved in glioma development and tumor microenvironment (Eyüpoglu et al., 2016; Lee et al., 2015; Was et al., 2019). The use of different HDAC inhibitors (HDACi) in clinical trials induces the cell‐cycle arrest and apoptosis in tumor cells, inhibits angiogenesis and the generation of reactive oxygen species (ROS) (Lee et al., 2015). Among the HDAC proteins, HDAC8, a zinc‐dependent class I protein, has been implicated in different diseases and cancers (Chakrabarti et al., 2015). In physiological condition, HDAC8 selectively deacetylates the histone H3 lysine 27 (H3K27Ac), and acts on various non‐histone proteins, such as the estrogen receptor alpha (ERα) and cortical actin‐binding protein (cortactin), controlling energy homeostasis, microtubule organization, chromatid separation, and many other processes involved in cellular homeostasis (Li et al., 2014; Wilson et al., 2010). Furthermore, HDAC8 overexpression is observed in different types of cancer such as breast, lung, neuroblastoma, T cell lymphoma, and GBM (Balasubramanian et al., 2008; Nakagawa et al., 2007; Oehme, Deubzer, Lodrini, et al., 2009; Oehme, Deubzer, Wegener, et al., 2009; Zhang et al., 2014), suggesting a role in tumor development. Nevertheless, few information are available on the role of HDAC8 in glioma. Recent observations indicate that HDAC8 regulates human glioma cell viability in vitro, and modulates the levels of O6‐methyl‐guanine DNA methyltransferase (MGMT), which correlates with Temozolomide resistance (Santos‐Barriopedro et al., 2019).
Among the cells that infiltrate glioma, the glioma‐associated microglia/macrophages (GAMs) play a crucial role, constituting almost 30% of the cells in the tumor mass (Graeber et al., 2002; Markovic et al., 2009). Indeed, glioma releases many cytokines and chemokines that attract and revert GAM phenotypes in a way that supports tumor growth and development, playing an active part in the enhancement of the invasive and proliferative properties of glioma, further contributing to the immunosuppressive phenotype of the tumor microenvironment (Coniglio & Segall, 2013; Markovic et al., 2005). Many studies identified a plethora of GAM phenotypes, with additional differences between mice and humans (Szulzewsky et al., 2016). In particular, murine GAMs express several genes with anti‐inflammatory properties, and many research efforts aim at promoting the expression of pro‐inflammatory genes which could be translated in therapy for a better patient prognosis (Pyonteck et al., 2013). In addition, glioma is frequently infiltrated by Natural Killer (NK) cells, even if actively suppressed by the tumor microenvironment (Burger et al., 2019; Golán et al., 2018). Interestingly, recent studies demonstrated that HDAC8 inhibition drives microglial phenotype (Lin et al., 2019) and increases NK cell activity and the number of interferon‐ γ (IFN‐γ) producing NK cells (Watters et al., 2021).
In this article, we investigated the involvement of HDAC8 in the control of glioma growth. In particular, we show that specific inhibition of HDAC8 with the compound PCI‐34051 reduces the human and murine tumor growth, both in vitro and in vivo. Furthermore, HDAC8 inhibition or silencing by siRNA abolishes the migration of glioma cells and increases the acetylation of the alpha‐tubulin. HDAC8 inhibition also switches GAMs cells, identified as CD11b+ cells (Gabrusiewicz et al., 2016), isolated from glioma bearing mice and from GBM patients, toward a pro‐inflammatory/anti‐tumoral phenotype. In addition, ChIP analyses on PCI‐34051‐treated glioma cells demonstrate that HDAC8 regulates the gene expression of natural killer group 2D (NKG2D)‐ligands h60, ulbp1/mult1, and rae1, involved in triggering the cytotoxicity of NKG2D receptor‐expressing NK cells (Garofalo et al., 2020). Accordingly, we report that HDAC8 inhibition enhances the cytotoxic activity of NK cells in vitro, and increases the infiltration of NKG2D+ and CD69+ NK cells in the tumor mass, in vivo (Garofalo et al., 2015; Nath et al., 2019).
Altogether these results highlight a central role of HDAC8 in glioma development and in the control of tumor microenvironment, and provide additional information on the epigenetic mechanisms activated by glioma to escape the host immune response.
2. MATERIALS AND METHODS
2.1. Synthetic route and characterization of PCI‐34051
Scheme of the synthesisa
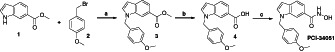
aReagents and conditions: a) NaH, anhydrous DMF, 0°C to rt; b) 2 N KOH, THF/MeOH 1:1, rt; c) (1) O‐(tetrahydro‐2H‐pyran‐2‐yl)hydroxylamine (OTX), EDCI, anhydrous DCM, rt; (2) 4 N HCl in dioxane, anhydrous MeOH, −10°C.
General: Melting points were determined on a Büchi 530 melting point apparatus and are uncorrected. 1H‐NMR spectra were recorded at 400 MHz on a Bruker AC 400 spectrometer; chemical shifts are reported in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). All compounds were routinely checked by TLC, 1H‐ and 13C‐NMR. TLC was performed on aluminum‐backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulphate. Elemental analysis has been used to determine the purity of the described compounds, which is >95%. Analytical results are within ±0.40% of the theoretical values. All chemicals were purchased from Merck KGaA, Darmstadt (Germany) or from TCI Europe N.V., Zwijndrecht (Belgium), and were of the highest purity.
Procedure for the synthesis of methyl 1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxylate (3)
Commercially available 1H‐indole‐6‐carboxylic acid methyl ester 1 (0.6 g, 3.42 mmol) in 2 mL of anhydrous DMF was added dropwise to a suspension of sodium hydride (0.09 g, 3.77 mmol) in 2 mL of anhydrous DMF at 0°C and after 20 minutes 4‐methoxybenzyl bromide 2 (0.6 mL, 4.11 mmol) was added dropwise at the same temperature. The reaction was then kept stirring at room temperature for 2.5 h and afterwards diluted with water (50 mL) and followed by extraction with ethyl acetate (3 × 75 mL). The organic layer was washed brine (50 mL), dried over Na2SO4, filtered and concentrated. The remaining residue was chromatographed (ethyl acetate/hexane, 1:15) to provide methyl 1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxylate 3 as a colorless solid (0.8 g, 79% yield); mp: 89–91°C; 1H NMR (400 MHz, CDCl3) δ 3.70 (s, 3H, —OCH 3), 3.85 (s, 3H, —COOCH 3), 5.24 (s, 2H, —CH 2—), 6.49 (d, 1H, J = 3.2 Hz, aromatic proton), 6.76 (d, 2H, J = 8.4 Hz, aromatic protons), 7.01 (d, 2H, J = 8.4 Hz, aromatic protons), 7.17 (d, 2H, J = 2.8 Hz, aromatic protons), 7.57 (d, 1H, J = 8.4 Hz, aromatic proton), 7.72 (d, 1H, J = 8.4 Hz, aromatic proton), 8.04 (s, 1H, aromatic proton), 13C NMR (100 MHz, DMSO) δ. 50.60, 55.30, 103.94, 111.73, 114.16, 114.20, 121.97, 122.94, 127.82, 128.02, 128.46, 129.94, 130.23, 130.25, 132.53, 137.62, 159.15, 167.64; Elemental analysis: calculated: C: 73.20%, H: 5.80%, N: 4.74%, O: 16.25%, found: C: 73.28%, H: 5.83%, N: 4.75%, O: 16.14%.
Procedure for the synthesis of 1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxylic acid (4)
Methyl 1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxylate 3 (0.765 g, 2.59 mmol) was dissolved in a mixture of 12 mL THF/MeOH (1:1) and then 4 mL of 2 N KOH solution was added and heated at 60°C for 5 h. Upon completion of the reaction, the mixture was quenched with 2 N HCl solution at 0°C. The precipitated solid was filtered off, washed with distilled water and dried to obtain pure 1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxylic acid 4 (0.7 g, 96% yield); mp: 163–165°C; 1H NMR (400 MHz, DMSO) δ 3.70 (s, 3H, —OCH 3), 5.44 (s, 2H, —CH 2—), 6.56 (d, 1H, J = 2.8 Hz, aromatic proton), 6.88 (d, 2H, J = 8.4 Hz, aromatic protons), 7.15 (d, 2H, J = 8.4 Hz, aromatic protons), 7.62 (s, 2H, aromatic protons), 7.72 (d, 1H, J = 2.8 Hz, aromatic proton), 8.08 (s, 1H, aromatic proton), 12.60 (bs, 1H, —COOH); 13C NMR (100 MHz, DMSO) δ 50.60, 55.30, 103.94, 111.73, 114.16, 121.97, 122.94, 127.82, 128.02, 128.46, 129.94, 130.23, 130.25, 133,52, 137.62, 159.15, 167.64; Elemental analysis: calculated: C: 72.58%, H: 5.37%, O: 4.98%, N: 17.06%, found: C: 72.47%, H: 5.39%, N: 4.96%, O: 17.18%.
Procedure for the synthesis of N‐hydroxy‐1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxamide (PCI‐34051).
The 1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxylic acid 4 (0.4 g, 1.42 mmol) and O‐(tetrahydro‐2H‐pyran‐2‐yl)hydroxylamine (OTX) (0.335 g, 2.84 mmol) were dissolved in anhydrous DCM, then EDCI (0.409 g, 2.13 mmol) was added portion‐wise. Upon completion of the reaction, the organic solvent was removed and the crude loaded on a flash chromatography column (ethyl acetate/hexane, 1:1.5). The so obtained protected hydroxamate (0.450 g, 1.18 mmol) was immediately deprotected by reacting with 4 N HCl dioxane (2.95 mL, 1.18 mmol) in anhydrous MeOH (20 mL) at −10°C. After 40 min stirring at −10°C, the reaction was complete and subsequently quenched with Na2CO3 saturated solution (50 mL) followed by extraction with ethyl acetate (3 × 75 mL). The organic layer was washed brine (25 mL), dried over Na2SO4, filtered and concentrated. The obtained solid was triturated with hexane and filtered to obtain a pink solid that was recrystallized by toluene to obtain the pure PCI‐34051 (N‐hydroxy‐1‐(4‐methoxybenzyl)‐1H‐indole‐6‐carboxamide); mp: 143–145°C; 1H NMR (400 MHz, DMSO) δ 3.70 (s, 3H, —OCH 3), 5.37 (s, 2H, —CH 2—), 6.50 (d, 1H, J = 2.8 Hz, aromatic proton), 6.87 (d, 2H, J = 8.8 Hz, aromatic protons), 7.18 (d, 2H, J = 8.8 Hz, aromatic protons), 7.45 (d, 1H, J = 8.4 Hz, aromatic proton), 7.55 (d, 1H, J = 8.4 Hz, aromatic proton), 7.60 (d, 1H, J = 2.8 Hz, aromatic proton), 7.94 (s, 1H, aromatic proton), 9.10 (bs, 1H, ‐CONHOH), 11.13 (bs, 1H, —CONHOH) ppm; 13C NMR (100 MHz, DMSO) δ 49.07, 55.51, 101.52, 103.67, 109.92, 114.43, 120.41, 128.90, 130.44, 132.05, 135.52, 142.67, 159.06, 161.26, 173.61, 177.37, 203.80 ppm; Elemental analysis: calculated: C: 68.91%, H: 5.44%, N: 9.45%, O: 16.20%; found: C: 68.85%, H: 5.42%, N: 9.48% O: 16.25%.
2.2. Materials
All Culture media, fetal bovine serum (FBS), goat serum, penicillin G, streptomycin, glutamine, Na pyruvate, recombinant human EGF, Thermo Script RT‐PCR System, anti‐Acetyl‐alpha Tubulin (6‐11B‐1), anti‐Acetyl‐alpha Tubulin lys40 Abs and Hoechst (#33342, RRID:AB_10626776) were from GIBCO Invitrogen (Carlsbad, CA). Glucose, hematoxylin, eosin, Percoll, Papain (#P3125), phosphate buffered‐saline (PBS) tablet (#P4417), Bovine Serum Albumine (BSA) and deoxyribonuclease I were from Sigma‐Aldrich (Milan, Italy). Transwell inserts were from BD Labware (Franklin Lakes, NJ). Ki67 (#12202, RRID:AB 2620142) Ab were from Cell Signaling (Danvers, MA). NKp46 (M20) (#sc‐18,161, RRID: AB_2149152) antibody (Ab) was from Santa Cruz biotechnology (Santa Cruz, CA). Anti‐alpha tubulin (ab52866) and anti‐TMEM119 Abs were from Abcam (Cambridge, UK). Secondary Abs were from DAKO (Milan, Italy). CXCL12 was from Peprotech. LPS is from Immunotools (Friesoythe, Germany). IFN‐γ were from Immunological Sciences (Rome, Italy). Microbeads CD11b+ were from Miltenyi Biotec (Bologna, Italy). CD45, CD69, CD107a, NK1.1 Abs were from eBioscience Inc. (San Diego, CA). Rabbit anti‐Iba1 from Wako (VA).
2.3. Mice and cell lines
Experiments described in the present work were approved by the Italian Ministry of Health in accordance with the guidelines on the ethical use of animals from the European Community Council Directive of September 22, 2010 (2010/63/EU). We used C57BL/6 (wt) mice from Charles River Laboratories (Calco, Italy) and C57BL/6‐Prf1 < tm1Sdz > /J (Cat# JAX:002407) (prf ko) transgenic mice from Jackson Laboratory (BarHarbor, ME). We always used 2‐month‐old male mice.
The GL261 glioma cell line (RRID:CVCL Y003; kindly provided by Dr. Serena Pellegatta, Istituto Di Ricovero e Cura a Carattere Scientifico, Besta, Milan, Italy) was cultured in DMEM supplemented with 20% heat inactivated FBS, 100 IU/mL penicillin G, 100 μg/mL streptomycin, 2.5 μg/mL amphotericin B, 2 mM glutamine, and 1 mM sodium pyruvate. The GL261‐CD133+ cells (obtained as described previously in Garofalo et al., 2017) were maintained in DMEM with 20 ng/mL, fibroblast growth factor‐2, 20 ng/mL EGF and Heparin 10Uml. U87MG (RRID:CVCL_0022), primary human glioma cells (obtained from patients at the IRCCS Neuromed), CT2A cell lines (established by Thomas N. Seyfried), primary murine microglia, and astrocytes, and primary human astrocytes (Thermo Fisher N7805100) were cultured in DMEM supplemented with 10% FBS. Human GBM samples were from patients who gave their informed consent to the use of tissues for research purposes. Primary human GBM cells were obtained as described previously (Sciaccaluga et al., 2010). Human iPSC‐derived neurons, obtained as described previously (Hill et al., 2016), were provided by Dr. Alessandro Rosa (Sapienza University of Rome).
2.4. Intracranial injection of glioma and mice treatment
Male C57BL/6 mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and placed in a stereotaxic head frame. Animals were stereotactically injected with 7.5 × 104 CT2A: a median incision of ~1 cm was made, a burr hole was drilled in the skull, and cells were injected 2 mm lateral (right) and 1 mm anterior to the bregma in the right striatum. Cell suspensions, in PBS (4 μL) were injected with a Hamilton syringe at a rate of 1 mL/min at 3 mm depth. Starting at 7 days after glioma injection, mice were randomly grouped for the treatments and were intraperitoneally (i.p.) injected with PCI‐34051 or vehicle daily. After 17 days, animals were sacrificed for different analyses.
2.5. Histopathological evaluation of tumor volume and invasiveness
After 17 days from glioma cells injection, brains were isolated for morphological evaluation of tissues and fixed in 4% buffered formaldehyde. Coronal brain sections (20 μm) were prepared by standard procedures and stained with hematoxylin and eosin. A section every 100 μm was collected, and the tumor area was evaluated using Image Tool 3.00. For analysis of tumor invasiveness, glioma cells protruding more than 150 μm from the main tumor mass were counted in at least 20 fields, obtained from six slices per mice.
2.6. Survival analysis
Following injection of the glioma cells, mice were treated with PCI‐34051 or vehicle as previously described and were monitored daily. The endpoint was defined by the lack of physical activity or death. The probability of survival was calculated using the Kaplan–Meier method, and statistical analysis was performed using a log‐rank test.
2.7. Immunostaining
Seventeen days after injection of CT2A cells, mice were sacrificed and the brains fixed in 4% formaldehyde. Subsequently the brains were incubated in 30% sucrose solution for 48 h and then snap frozen. Cryostat sections (10 μm) were boiled for 10 min in citrate buffer, pH 6.0, at 95–100°C, then rinse in PBS, and incubated with 3% goat serum in 0.3% Triton X‐100 for 1 h at room temperature, and then overnight at 4°C with specific antibodies in PBS containing 1% goat serum and 0.1% Triton X‐100. The sections were stained with the following primary Abs: Ki67 (1:50), anti Iba1 (1:500). After several washes, sections were stained with the fluorophore‐conjugated antibody and Hoechst (1:3000) for nuclei visualization and analyzed by fluorescence microscope.
2.8. Image acquisition and data analysis
Images were digitized using a CoolSNAP camera (Photometrics) coupled to an ECLIPSE Ti‐S microscope (Nikon) and processed using MetaMorph 7.6.5.0 image analysis software (Molecular Devices). Brain slices were scanned by consecutive fields of vision (10X objective lens) to build a single image per section. The percentage of positive cells was measured as the ratio of the area occupied by fluorescent cells versus the total tumor area (by converting pixels to square millimeters). For comparison between different treatments, at least 12 coronal sections per brain around the point of injection were analyzed.
2.9. Cell viability in vitro
To assess the viability of cells exposed to different concentrations of PCI‐34051, murine and human tumors and normal cells (13 × 104/cm2) were treated with PCI‐34051 (1, 5, or 10 μM) for 24, 48, and 72 h. Cell viability was determined by MTT assay. Results are expressed as percentage of cell survival, taking as 100% the cells treated with vehicle.
2.10. Boyden chamber chemotaxis assays
The CT2A cells or primary microglia were incubated with PCI‐34051 (1, 5, 10 μM) for 48 h. After that, cells were trypsinized, preincubated in chemotaxis medium (DMEM without glutamine, 100 IU/mL penicillin G, 100 μg/mL streptomycin, 0.1% BSA, and 25 mM HEPES, pH 7.4) supplemented with anti‐mitotic AraC 5 μm for 15 min and plated (4 × 104 cells) on poly‐L‐lysine‐coated Transwells (8 μm pore size filters) in this same medium. The lower chamber contained EGF (100 ng/mL), CXCL12 (50 ng/mL), GCM, or vehicle. After 4 h (microglia) or 18 h (CT2A), cells were fixed with trichloroacetic acid. Cells adhering to the upper side of the filter were scraped off and cells on the lower side were stained with a solution containing 50% isopropanol, 1% formic acid, and 0.5% (w/v) brilliant blue R 250. For each membrane, the stained cells were counted in at least 20 fields with a 40X objective. Experiments were done in six repeats and performed at least four times.
2.11. Western blot analyses
Cells were stimulated with PCI‐34051 (10 μM) for 48 h. The same amount of proteins (20 μg/sample) was loaded onto 7.5% SDS polyacrylamide gel and transferred to nitrocellulose paper at 4°C for 2 h. Blots were incubated for 1 h with 5% non‐fat dry milk or 3% BSA in Tris‐buffered saline containing 0.2% Tween 20 to block nonspecific binding sites and then incubated overnight at 4°C with specific primary Abs. After washing, membranes were incubated with HRP‐conjugated secondary Abs and immunoreactivity was detected by ECL. Densitometric analysis of immunoreactive bands was performed using Chemi‐Doc XRS and Quantity One software (Bio‐Rad).
2.12. Primary microglial and astrocyte cultures and microglial polarization
Microglia cultures were obtained from mixed glia cultures derived from the cerebral cortices of post‐natal day 0–1 (P0–P1) C57BL/6 mice, as described (Garofalo et al., 2017). In brief, cortices were chopped and digested in 15 U/mL papain for 20 min at 37°C. Cells (5 × 105 cells/cm2) were plated on flasks coated with poly‐L‐lysine (100 mg/mL) in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. After 7–9 days, cells were shaken for 2 h at 37°C to detach and collect microglial cells. These procedures gave almost pure microglial cell populations. The remaining attached cells were astrocytes. Primary microglia cultures were treated for 48 h with LPS 100 ng/mL + IFN‐γ 20 ng/mL, or IL‐4 20 ng/mL to induce cell polarization.
2.13. Form factor calculation
IL‐4 and LPS + IFN‐γ treated microglia were seeded on glass coverslips, with or without PCI‐34051 (10 μM) for 24 h, fixed and stained with anti‐Iba1 (1:500) for 1 h together with Hoechst. Fluorescent images were processed using MetaMorph 7.6.5.0 software (Molecular Device, USA) and Form Factor was calculated according the formula 4π area/perimeter.
2.14. Isolation of CD11b + cells and extraction of total RNA
Glioma bearing C57BL/6 were anesthetized and decapitated. Brains were removed, brain tissues were cut into small pieces and single‐cell suspension was achieved in Hank's balanced salt solution (HBSS). The tissue was further mechanically dissociated using a glass wide‐tipped pipette and the suspension was applied to a 30 μm cell strainer (Miltenyi Biotec). Cells were processed immediately for MACS MicroBead separation. CD11b+ cells were magnetically labeled with CD11b MicroBeads. Among CD11b+ cells, population including myeloid and NK cells, GAMs constituted the higher percentage, in brain tumor (Gabrusiwicz et al., 2016). The cell suspension was loaded onto a MACS Column placed in the magnetic field of a MACS Separator and the negative fraction was collected. After removing the magnetic field, CD11b+ cells were eluted as a positive fraction. The purity of CD11b+ cell fraction was verified by immunofluorescence and flow cytometry (FACS) and was 99%” (Garofalo et al., 2017). After sorting the positive and negative fractions, total RNA was isolated with Trizol reagent, and processed for real‐time PCR. The quality and yield of RNAs were verified using the NANODROP One system (Thermo Scientific).
2.15. Primary human GBM
Tumor specimens were obtained from the Neurological Science Department of Sapienza Medical School and from IRCCS Neuromed, from primary GBM in adult patients who gave informed consent to the research proposals. All the information about the primary GBM cells used were reported in D'Alessandro et al., 2019. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Policlinico Umberto I and Neuromed. Ethic code: 23 April 2015, Rif.3623 Prot.2061/15, Ethics Committee of Policlinico Umberto I; 31 July 2012, Prot. 7/12, Ethics Committee of Neuromed. Tissues were processed within half an hour from surgical resection. Histopathological typing and tumor grading were done according to the WHO criteria, resulting as grade IV.
2.16. Isolation of CD11b + cells from human GBM
Tumor specimens obtained from adult patients with GBM who gave informed consent to the research proposals (IRCCS Neuromed) were dissociated and treated for 48 h with PCI‐34051 (10 μM) or vehicle. Tissues were then processed as described above to isolate CD11b+ cells, live cells were assessed by flow cytometry (FACS) (91 ± 3%), and mRNAs were analyzed by rtPCR for gene expression.
2.17. Real‐time PCR (rtPCR)
CD11b+ cells sorted from contra‐ and ipsilateral hemisphere of injected mice, GBM patients, cultured primary microglia or glioma cells, were lysed in Trizol reagent for isolation of RNA. Reverse transcription reaction was performed in a thermocycler (MJ Mini Personal Thermal Cycler; Biorad) using IScript TM Reverse Transcription Supermix (Biorad) according to the manufacturer's protocol, under the following conditions: incubation at 25°C for 5 min, reverse transcription at 42°C for 30 min, inactivation at 85°C for 5 min. Real‐time PCR (rtPCR) was carried out in a I‐Cycler IQ Multicolor rtPCR Detection System (Biorad) using SsoFast EvaGreen Supermix (Biorad) according to the manufacturer's instructions. The PCR protocol consisted of 40 cycles of denaturation at 95°C for 30 s and annealing/extension at 60°C for 30 s. For quantification analysis, the comparative Threshold Cycle (Ct) method was used. The Ct values from each gene were normalized to the Ct value of GAPDH in the same RNA samples. Relative quantification was performed using the 2‐ΔΔCt method (Schmittgen & Livak, 2008) and expressed as fold change in arbitrary values. The used primers were listed in Table S1.
2.18. HDAC8 silencing by siRNA
CT2A cells were infected by HDAC8 siRNA particles (provided by Santa Cruz Biotechnology). Cells (5 × 104 or 2 × 105) were plated in 96‐well or 24‐well plates, respectively, and infected for 48 h according to the manufacturer's instructions. Transduced cells were selected with 2 μg/mL puromycin. Knockdown efficiency was evaluated by RT‐PCR.
2.19. RT‐PCR
Total RNA was isolated from CT2A cells incubated with siRNA‐HDAC8 or control siRNA for 48 h, as reported. DNA contamination was removed according to the manufacturer's protocol. 500 ng of RNA was reverse transcribed using the Thermo Script RT‐PCR System protocol and the cDNAs were amplified by PCR with specific primers: mouse hdac8 fw 5‐ATCTCAATGATGCTGTCCTGG‐3 and rev 5‐ CATGATCTGGGATCTCAGAGG‐3; mouse actin fw 5‐TCACCCACACTGTGCCCAT‐3 and rev 5‐ACAGAGTACTTGCGCTCAGGA‐3. The PCR was as follows: DNA was denatured for 5 min at 94°C and sequences were amplified for 35 cycles for hdac8 94°C for 45 s; 60°C for 55 s; 72°C for 10 min; for actin 94°C 30 s; 57°C 2 min; 72°C 40 s, followed by the last extension step at 72°C for 10 min. A MJ Mini Thermal Cycler (Bio‐Rad) was used for all reactions. Products were analyzed on 1.2% agarose gels stained by ethidium bromide.
2.20. Chromatin immunoprecipitation assay (ChIP)
ChIP assays were performed as described (Bellissimo et al., 2017) using an anti‐trimethyl‐Histone H3 (Lys4) antibody (H3K4me3) (Cell Signaling; cat#9751S). Genomic regions on Ulbp1, H60a and Rae1, were amplified using primers designed by the Primer 3 software. PCR was performed using the SYBR green dye detection method and the following primers: Ulbp1 Fw‐TCCCTAGTAGGCAGCAGGAA; Rev‐CCCTGGACAGAAACCAAAGA. Rae1a Fw‐GGCCGCTGTAGTCAGTTACC; Rev‐GCGAAGCTGGAGTGTCTGTC. H60a Fw‐GCACCCACTTCATCCTCTGT; Rev‐CACCGTCCCTTCATCTCACT. GAPDH Fw‐CACTCCAAGGACTCCTCGTC; Rev‐ CCTCTCTCATTTCCCCTCCT. Values obtained for each immunoprecipitated sample were quantified versus the respective input (1:50 dilution) and calculated following the 2‐ΔΔCt method. Relative enrichment was obtained by normalizing over GAPDH promoter values.
2.21. Immunofluorescence and FACS analysis
Cells isolated from the indicated organs, were washed and resuspended in staining buffer (PBS without Ca2+ Mg2+, BSA 0,5%, EDTA 2 mM and NaN3 0,025%). After 10 min incubation with anti‐CD16/32 (clone 24G2), cells were stained with the indicated monoclonal Abs for 25 min at 4°C. IFN‐γ production expression was analyzed by intracellular staining using BD Cytofix/CytopermTM Fixation/Permeabilization Kit after 4 h incubation with Brefeldin A. Samples were analyzed by flow cytometry using a FACSCanto II (BD Biosciences), and data were elaborated using Diva Version 6.1.3 (BD Biosciences) or FlowJo Version 7.6 software (TreeStar).
2.22. Cytotoxicity and degranulation assay
Cell viability of CT2A was determined by MTT assay. Results are expressed as percentage of cell survival, taking as 100% the cells not incubated with NK cells (C). For the degranulation assay, enriched NK cells from spleen of healthy C57BL/6 or prf ko mice were stimulated overnight with IL‐15 (25 ng/mL), washed in PBS and co‐incubated for 4 h at 37°C with CT2A cells at the indicated Effector:Target (E:T) ratio in the presence of Monensin 100 μM and FITC‐conjugated anti‐mouse CD107a or anti‐ IgG. Degranulating NK cell populations were identified by FACS analysis after staining with fluorochrome conjugated anti‐NK1.1 (PE), ‐CD3 (PerCP‐cy5.5) mAbs.
2.23. Statistical analysis
Data are shown as the mean ± SEM. Statistical significance was assessed by Student's t‐test or one‐way ANOVA for parametrical data, as indicated; Holm–Sidak test was used as a post hoc test; Mann–Whitney Rank test and Kruskal–Wallis for non‐parametrical data, followed by Dunn's or Tukey's post hoc tests. For multiple comparisons, multiplicity‐adjusted p‐values are indicated in the corresponding figures (*P 0.05, **P 0.01). For the Kaplan–Meier analysis of survival, the log‐rank test was used. Statistical analyses comprising calculation of degrees of freedom were done using Sigma Plot 11.0, Imaris; Origin 7, and Prism 7 software.
3. RESULTS
3.1. HDAC8 inhibition reduces glioma growth and prolongs mice survival
Before investigating the effect of HDAC8 inhibition on glioma growth, we analyzed the expression of HDAC8 in different human GBM cell lines, in primary cells obtained from GBM patients, and in murine glioma cells. We also investigated HDAC8 expression in human normal astrocytes, human microglia collected from surgery of epileptic patients, and human neurons derived from induced pluripotent stem cells (iPSCs), and in murine astrocytes, microglia, and neurons. Data shown in Figure 1(a) demonstrated that all the tested cells express HDAC8. Interestingly, primary cells obtained from patients, GBM19 and GBM41R, express higher levels of HDAC8 (Figure 1(a)). To investigate the effect of HDAC8 inhibition in glioma development, we treated glioma‐bearing mice with increasing concentrations of the selective HDAC8 inhibitor PCI‐34051 (daily i.p. injection), starting 7 days after brain transplantation of glioma cells (see scheme in Figure 1(b)). Note that the highest dose of PCI‐34051 used for in vivo treatment, that is 40 mg/kg, is not toxic for mice (Rettig et al., 2015). We demonstrated that the blockade of HDAC8 reduces glioma growth in a concentration‐dependent manner (Figure 1(b)), and increases the mean survival time of glioma‐bearing mice, in comparison with vehicle‐treated mice (Figure 1(c)). Interestingly, the blockade of HDAC8 reduced the proliferation rate of glioma cells in vivo (indicated as by a reduction of Ki67+ cells in the tumor area) (Figure 1(d)). We demonstrated that this effect is at least in part due to a direct action of HDAC8 inhibition in human and murine glioma cell, since a single application of PCI‐34051 (1, 5, or 10 μM) reduces cell viability in cultured cells (Figure 1(e)). Same results on glioma growth were obtained when cells were treated every 24 h with PCI‐34051 (Figure S1(a)). No effects were observed in murine primary astrocytes and microglia (Figure S1(b)), or if we treated CT2A cells with the inactive intermediate 3 in the synthetic process of the PCI‐34051 (iPCI‐34051, as indicate in methods) (Figure 1(f)). To further validate our data, we used a different approach silencing HDAC8 in murine glioma cells with a specific siRNA. Data reported in Figure 1(g) show that siRNA treatment reduced HDAC8 expression of 50 ± 0.12%, in comparison with control siRNA (c‐siRNA) treatment, also affecting CT2A cell viability (Figure 1(h)).
FIGURE 1.
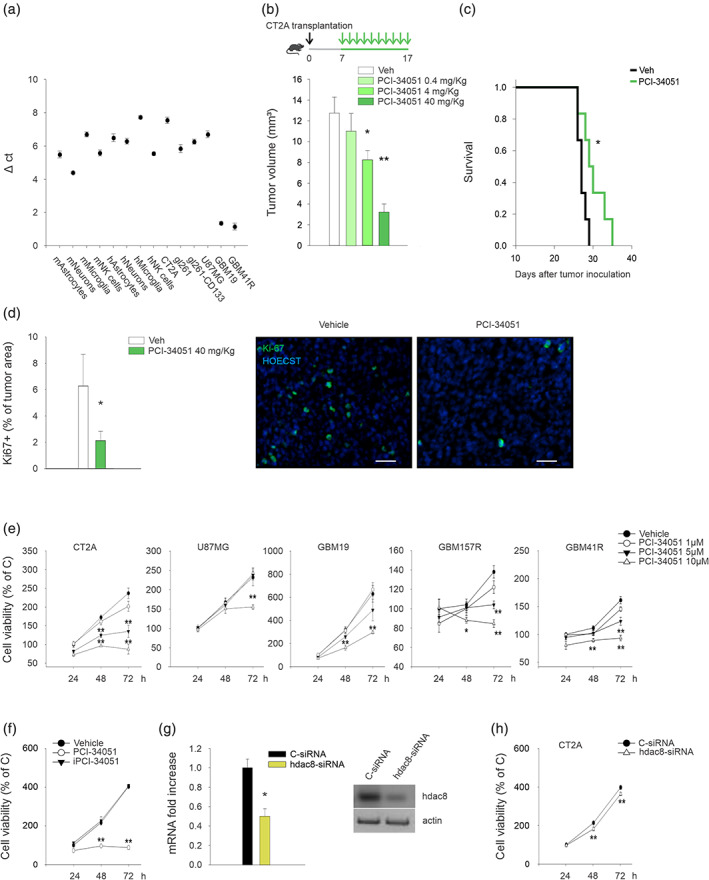
HDAC8 inhibition affects glioma growth and increases mice survival. (a) RT–PCR analyses of hdac8 expression in tumor cells, normal cells, and human tissues from patients (n = 3). (b and c) mean tumor volume (b) and Kaplan–Meier curve (c) in CT2A‐bearing mice. Mice were treated with PCI‐34051, as indicated in the scheme on top (n = 7–8 mice per treatment; *P < 0.05 **P < 0.01, one‐way ANOVA). (d) Data show the Ki67+ cells in brain tumors (expressed as the mean area ± S.E.M. percentage of the tumor) at 17 days after glioma implantation in mice treated with PCI‐34051 40 mg/kg, as indicated (n = 4 mice per treatment; *P < 0.05, Student's t test). Representative immunofluorescence images of proliferating Ki67+ cells (green) under the two experimental conditions are shown on the right. Scale bars, 50 μm. (e) Growth curves of human and murine glioma cells treated with PCI‐34051 for the indicated time points. The results are expressed as percentage of vehicle‐treated cells ± S.E.M. (n = 4–7; *P < 0.05 **P < 0.01, one‐way ANOVA). (f) Growth curves of CT2A glioma cells treated with PCI‐34051 or iPCI‐34051 10 μM for the indicated time points. The results are expressed as percentage of vehicle‐treated cells ± S.E.M. (n = 3; **P < 0.01, one‐way ANOVA). (g) RT‐PCR analysis of hdac8 expression in CT2A cells transfected with c‐ or hdac8‐siRNA for 48 h. one representative western blot analysis of HDAC8 protein in silenced cells is shown on the right. (n = 4; *P < 0.05, one‐way ANOVA). (h) Growth curves of CT2A glioma cells transfected with c‐ or hdac8‐siRNA. The results are expressed as percentage of c‐siRNA treated cells ± S.E.M. (n = 4; **P < 0.01, one‐way ANOVA)
3.2. HDAC8 regulates alpha‐tubulin acetylation driving glioma cell migration
To investigate if HDAC8 inhibition reduces tumor growth affecting by glioma cell migration, we treated human and murine glioma cells with the selective HDAC8 inhibitor PCI‐34051 at different concentrations (1, 5, or 10 μM). We demonstrated that HDAC8 inhibition reduces the basal movement of human U87MG and GBM157 cells, and abolishes the migration induced by EGF and CXCL12 (Figure 2(a)). No effects were observed treating tumor cells with iPCI‐34051 (Figure 2(b)). To further investigate the involvement of HDAC8 in glioma cell migration, we silence HDAC8 by siRNA in CT2A cells. Figure 2(b) reports that siRNA treatment abolished EGF‐induced migration in comparison to c‐siRNA treatment.
FIGURE 2.
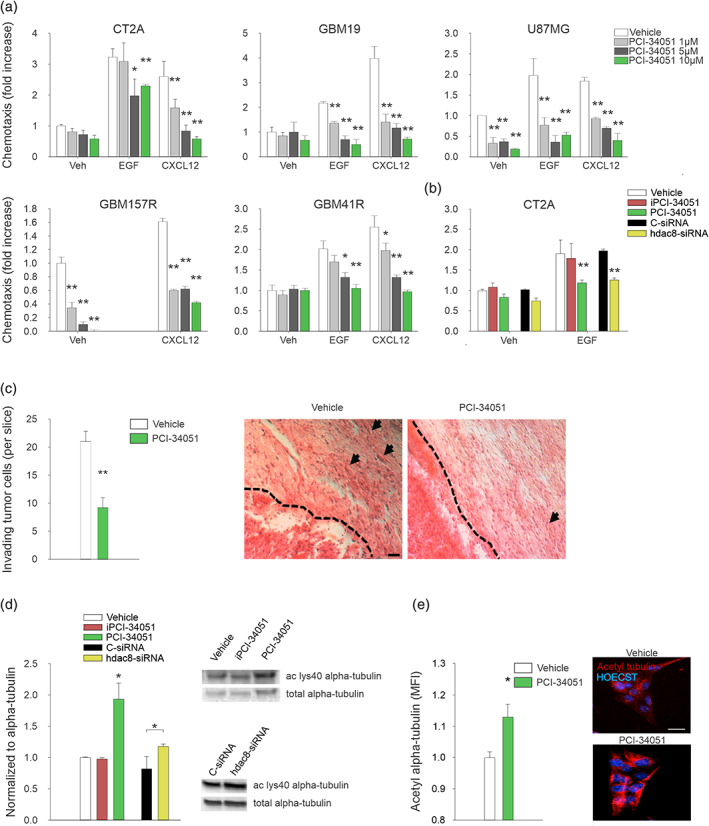
HDAC8 inhibition reduces glioma cell migration and invasion, and increases alpha‐tubulin acetylation. (a) Human and murine glioma cell chemotaxis toward vehicle, EGF (100 ng/mL) or CXCL12 (50 ng/mL), preincubated with PCI‐34051 at the indicated doses. Results are expressed as the fold increase in comparison with vehicle ± S.E.M. (n = 3–6; *P < 0.05 **P < 0.01, one‐way ANOVA). (b) CT2A glioma cell chemotaxis toward vehicle, EGF (100 ng/mL) preincubated with PCI‐34051 10 μM, iPCI‐34051 10 μM or transfected with c‐ or hdac8‐siRNA. Results are expressed as the fold increase in comparison with vehicle ± S.E.M. (n = 4; **P < 0.01, one‐way ANOVA). (c) Mean number (± S.E.M.) of glioma cells invading the brain parenchyma for >150 μm (n = 5 mice per condition; **P < 0.01, Student's t test). Right, representative coronal brain sections stained with hematoxylin and eosin. Black arrows indicate invading glioma cells beyond the main tumor border (dashed line). Scale bars, 20 μm. (d and e) analysis of acetylated alpha‐tubulin protein by western blot (ac‐lys40) (d) and immunofluorescence (e) in glioma cells incubated with vehicle or PCI‐34051 10 μM, iPCI‐34051 10 μM or transfected with c‐ or hdac8‐siRNA for48 h. Results are expressed as the fold increase in comparison with vehicle‐treated glioma cells (n = 4–7; *P < 0.05, Student's t test). Right, representative blot and images of alpha‐tubulin acetylation in glioma cells under the two experimental conditions. Scale bars, 5 μm
Further, PCI‐34051 treatment reduces tumor cell invasiveness in brain parenchyma, calculated by the number of glioma cells that invade the surrounding parenchyma more than 150 μm from tumor mass in vivo, at day 17 after glioma transplantation (Figure 2(c)).
A recent study indicates that HDAC8 regulates the acetylation levels of alpha‐tubulin acting on lysine 40 (ac‐lys40), and controls cervical cancer cells migration (Vanaja et al., 2018). To understand whether this mechanism is also involved on the effect of HDAC8 on glioma cell invasiveness and migration, we treated CT2A cells with PCI‐34051 and measured ac‐lys40 and total alpha‐tubulin acetylation. Data reported in Figure 2(d)–(e) show that after 48 h of incubation with 10 μM PCI‐34051, the acetylation level of alpha‐tubulin, analyzed by western blot (Figures 2(D), S2(a) and full blot in Figure S2(b)) and immunofluorescence (Figure 2(e)), increased. To further investigate the involvement of HDAC8 in the acetylation of the alpha‐tubulin, we performed western blot analysis of ac‐lys40 alpha‐tubulin after genetic knowdown of HDAC8 by specific siRNA in CT2A cells. We reported that siRNA treatment increases the ac‐lys40 alpha‐tubulin of 35 ± 6% (Figure 2(d)). These data suggest that HDAC8 controls glioma cell migration by regulating alpha‐tubulin acetylation.
3.3. HDAC8 regulates microglial invasion and plasticity in glioma
Since GAMs recruitment to glioma mass represents a key mechanism to support tumor growth, we investigated whether HDAC8 inhibition also affected GAM migration. At this aim, primary microglia obtained from mice pups (P0‐2), were treated with PCI‐34051 to investigate its effects on EGF‐ and glioma cell‐conditioned medium (GCM) induced chemotaxis. Data obtained demonstrate that HDAC8 inhibition totally abolished microglial cell chemotaxis (Figure 3(a)). Furthermore, in mice treated with PCI‐34051 (40 mg/kg), the number of Iba1+ cells (a marker of microglia/macrophages) infiltrating tumor mass was reduced (Figure 3(b)). To assess if the reduced myeloid cell numbers was specifically due to microglia reduction, we performed immunostaining for TMEM119, a microglial specific marker (Bennett et al., 2016). Data reported in Figure 3(c) show that TMEM119+ cells are reduced in tumor area after HDAC8 inhibition, similarly to Iba1+ cell reduction, highlighting a principal effect on microglial cells.
FIGURE 3.

HDAC8 regulates microglia migration and morphology dynamic change in different activation states. (a) Microglia chemotaxis toward the control medium (vehicle), EGF (100 ng/mL) or CT2A‐glioma conditioned medium (CT2A), pretreated with vehicle or PCI‐34051 10 μM for 48 h. results are expressed as fold increases in comparison with vehicle (n = 5; * < 0.05, one‐way ANOVA). (b and c) the mean (± S.E.M.) area of Iba1+ cells (b) and TMEM119+ cells (c) (as % of the tumor area) 17 days after CT2A transplantation in mice treated with PCI‐34051 40 mg/kg, as indicated. Right, representative immunofluorescences of Iba1+ and TMEM119+ cells glioma infiltrated (green) (n = 4 mice per condition, **P < 0.01, Student's t test). Scale bars, 20 μm. (d) RT‐PCR analysis of hdac8 expression in primary microglia incubated with vehicle, IL‐4 or LPS + IFN‐γ for 48 h (n = 7–4, **P < 0.01, Student's t test). For boxplots, the center line, boxes and whiskers represent the median, inner quartiles, and rest of the data distribution, respectively. (e) Primary microglia incubated with vehicle, IL‐4 or LPS + IFN‐γ for 48 h, in presence or absence of PCI‐34051 10 μM. Form factor values (calculated as indicated in methods) are shown as mean ± S.E.M (n = 5, *P < 0.05, one‐way ANOVA). Right, representative immunofluorescences of microglia stained with Iba1 (green). Scale bars, 10 μm
We also investigated the role of HDAC8 on the modulation of GAMs phenotype. We first analyzed the expression of HDAC8 in primary microglial cells treated with different cytokines to mimic different states of activation. We observed that HDAC8 expression was specifically reduced only upon LPS and IFN‐γ stimulation, that mimics a pro‐inflammatory phenotype (Tam & Ma, 2014) (Figure 3(d)).
In addition, microglia are dynamic cells that change morphology with functional activity, even if there is no direct shape‐function relation (Wright‐Jin & Gutmann, 2019). We investigated the role of HDAC8 in affecting microglia shape upon LPS + IFN‐γ or IL‐4 (that mimic an anti‐inflammatory phenotype) treatment. Data shown in Figure 3(e) reported that blockade of HDAC8 affected morphological changes only in IL‐4 ‐treated microglia.
To further investigate the role of HDAC8 in shaping GAMs phenotype in vivo, glioma‐bearing mice were treated for 17 days with PCI‐34051 (40 mg/kg) starting 7 days after glioma cell transplantation. Mice were sacrificed and CD11b+ cells were isolated from the ipsi‐ or contra‐lateral cerebral hemispheres for the analysis of gene expression by rtPCR. We confirmed that tumor cell injection increased the expression of anti‐inflammatory genes (arg‐1, retnla, chil3, and tgfβ, see Gabrusiewicz et al., 2011) in CD11b+ cells from th‐e glioma‐bearing hemisphere and demonstrated that this effect is completely abolished by PCI‐34051 treatment, with a simultaneous increase of pro‐inflammatory genes (Figure 4(a)).
FIGURE 4.

HDAC8 modulates gene expression in GAMs. (a) rtPCR of anti‐ (arg‐1, chil3, tgfβ and retnla) and pro‐inflammatory (tnfα, il1β, nos2, and cd86) genes in CD11b+ cells sorted from ipsi‐ and contralateral hemisphere of CT2A‐bearing mice treated with vehicle or PCI‐34051 40 mg/kg, as indicated. Data are the mean ± S.E.M. versus vehicle contra (n = 6 mice per condition, *P < 0.05 **P < 0.05, one‐way ANOVA). (b) rtPCR analysis of anti‐ (arg‐1, chil3, tgfβ and retnla) and pro‐inflammatory (tnfα, il1β, nos2 and cd86) genes in primary microglia incubated with vehicle, IL‐4 or LPS + IFN‐γ for 48 h, in presence or not of PCI‐34051 10 μM. Data are the mean ± S.E.M. expressed as fold increased in comparison with vehicle‐treated microglia (n = 5, **P < 0.01, one‐way ANOVA). (c) rtPCR of human pro‐ (cxcl10, nos2, and il12a) and anti‐inflammatory (cd163, mmp12 and tgfβ) genes in CD11b+ cells sorted from patient‐derived GBM tissue, after tissue treatment with PCI‐34051(10 μM, 48 h) or vehicle. Above: Scheme of human GBM treatment. Data are the mean ± S.E.M., n = 5, *P < 0.05 versus vehicle, Student's t‐test
To evaluate if HDAC8 activity directly regulates GAMs gene expression, we analyzed the effect of PCI‐34051 in primary microglia culture in vitro. As expected, LPS + IFN‐γ treatment increased the expression of pro‐inflammatory genes, while IL‐4 increased the expression of anti‐inflammatory genes and reduced the pro‐inflammatory ones (Figure 4(b)). Data reported in Figure 4(b) show that PCI‐34051 treatment differently modulated gene expression as a function of microglial phenotype, with the reduction of some anti‐inflammatory genes (arg‐1 and retnla), and the increase of some pro‐inflammatory genes (il1β, nos2, and cd86), only in IL‐4 stimulated microglia (Figure 4(b)). No effects on anti‐ and pro‐inflammatory gene expression were observed in microglia stimulated with LPS + IFN‐γ or vehicle (Figure 4(b)).
To investigate the effect of HDAC8 inhibition on human GAMs, we incubated human GBM tissues with PCI‐34051 (10 μM) or vehicle for 48 h and then analyzed the expression of pro‐ and anti‐inflammatory genes in the CD11b+ cell population. Figure 4(c) shows that PCI‐34051 treatment decreased the expression of the anti‐inflammatory genes cd163 and mmp12, and increased the expression of the pro‐inflammatory genes nos2 and cxcl10.
Altogether these results indicate that HDAC8 drives murine and human GAMs toward an anti‐inflammatory phenotype, because its inhibition with PCI‐34051 stimulates the expression of pro‐inflammatory genes.
3.4. HDAC8 inhibition increases the expression of NKG2D ligands, making glioma cells putative targets of NK cell‐mediated cytotoxicity
Making the tumor cells desirable targets for cytotoxic T and NK cells is one main effort of the research in the last years. We hypothesized that the transcription of ligands of the NKG2D receptor, that triggers the cytotoxicity of NK cells, was affected by HDAC8 activity. To verify if the expression of the ulbp1, h60a, and rae1 genes is transcriptionally activated upon HDAC8 inhibition, we performed ChIP assays. Specifically, we evaluated a marker of transcriptional promoter activation, for example, trimethylated histone H3 lysine 4 (H3K4me3), in CT2A glioma cells treated with PCI‐34051 (10 μM) for 24 h. As shown in Figure 5(a), this treatment leads to a significant increase of H3K4me3 level in h60, ulbp1 and rae1 promoters. Accordingly, increased transcript levels for these genes were also observed following treatment with PCI‐34051 (Figure S3(a)).
FIGURE 5.
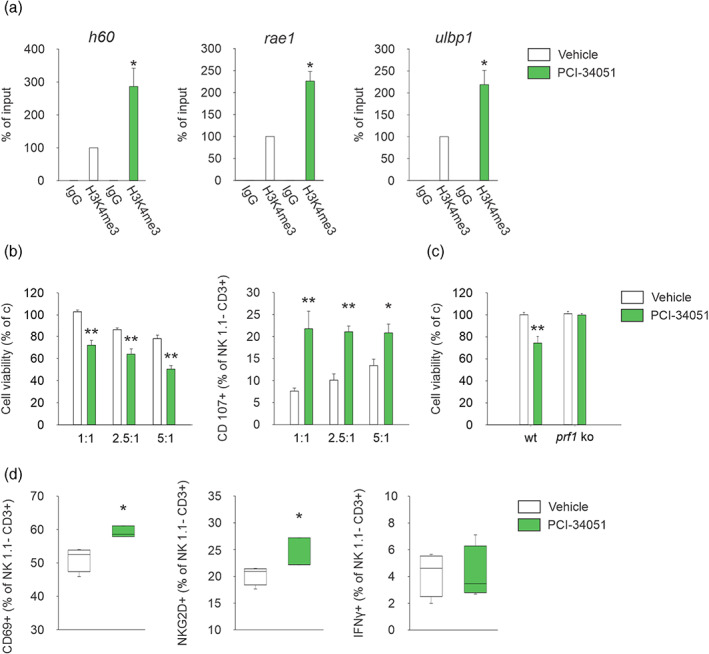
HDAC8 inhibition increases NKG2D ligands in glioma cells boosting NK cell cytotoxicity. (a) ChIP experiments performed on CT2A cells treated or not with the HDAC8 inhibitor for 24 h using the indicated antibody (n = 3; *P < 0.05, Student's t‐test). (b) NK cells were incubated with CT2A cells pretreated with PCI‐34051 or vehicle for 48 h. degranulation of NK cells (having subtracted basal degranulation) in a 1:1, 2.5:1, and 5:1 E:T ratio with CT2A cells was assessed by FACS analysis of CD107a+ cells (right panel); cell viability in glioma cells is shown in the left panel (n = 6, *P < 0.05 **P < 0.01, one‐way ANOVA). Error bars show mean ± SEM. (c) NK cells, isolated from the spleen of wt or prf1 ko mice, were incubated with CT2A cells pretreated with PCI‐34051 or vehicle for 48 h. Glioma cell viability in a 2.5:1 E:T ratio is shown as mean ± S.E.M. (n = 6, **P < 0.01, one‐way ANOVA). (d) Percentage of CD69+, NKG2D+ and IFN‐γ+, cells in the CD3−/NK1.1+ cell population obtained from the brain of vehicle or PCI‐34051 treated mice (n = 4; *P < 0.05, Student's t‐test). For boxplots, the center line, boxes and whiskers represent the median, inner quartiles, and rest of the data distribution, respectively
To support a possible role for HDAC8 in modulating the cytotoxic activity against glioma, we performed NK cell‐mediated cytotoxic assays. We observe that co‐incubation with NK cells affected the viability of PCI‐34051‐treated glioma cells and resulted in an increased percentage of CD107a+ (a lysosomal marker of active degranulation) NK cells (Figure 5(b)). To investigate the molecular mechanisms involved in the cytotoxic activity of NK cells dependent on HDAC8, we evaluated the role of perforin (prf1), a protein involved in the formation of lytic holes. At this aim, NK cells isolated from prf1 ko mice were tested against PCI‐34051‐treated‐glioma cells, in a cytotoxicity assay. Results shown in Figure 5(c) demonstrated that the absence of perforin abolished NK cell‐mediated tumor death induced by HDAC8 inhibition.
Similar results were obtained treating glioma‐bearing mice with PCI‐34051, where we observed an increase of activated CD69+ and NKG2D+ NK cells infiltrating the tumor mass (Figure 5(d)). No variations in IFN‐γ levels were observed (Figure 5(d)).
These results demonstrate that HDAC8 activity plays relevant role in reducing glioma cell recognition and killing by NK cells.
4. DISCUSSION
Many epigenetic modifications occur in glioma resulting in substantial molecular heterogeneity that confers advantages for tumor development (Phillips et al., 2020). Among the enzymes involved, the histone deacetylases regulate different hallmarks of glioma pathogenesis, such as proliferation, invasiveness, and formation of new vessels (Pastorino et al., 2019; Zhao et al., 2020). HDACi fascinated researchers in almost all fields of cancer biology: five of them have been approved by FDA for clinical use and many others tentered clinical trials as single agents or in combination therapy for a variety of cancer diseases (Ecker et al., 2013; Lee et al., 2015). Among the HDAC proteins, the role of HDAC8 in brain tumors is poorly understood, although it is overexpressed in different types of solid cancers (Balasubramanian et al., 2008; Nakagawa et al., 2007; Oehme, Deubzer, Lodrini, et al., 2009; Oehme, Deubzer, Wegener, et al., 2009; Zhang et al., 2014). Herein, we demonstrated that HDAC8 plays a pivotal role in glioma growth, since its inhibition with PCI‐34051 reduced the tumor volume in a glioma mouse model. A recent article report that the PCI‐34051 may have limited permeability in the brain on wild type mice (Wang et al., 2014), and we cannot exclude that the effect of PCI‐34051 in vivo was, at least in part, due to the peripheral inhibition with an indirect effect on the tumor. However, it should be considered that in the glioma context, the blood–brain barrier (BBB) is compromised, and glioma cells create a blood‐tumor barrier (BTB) with altered permeability (Arvanitis et al., 2020). Here we demonstrated that the reduction of tumor volume observed in vivo could be at least in part due to a direct action of HDAC8 on glioma cells, confirming previous results with a different HDAC8 inhibitor, N1‐(2,5‐dimethoxyphenyl)‐N8‐hydroxyoctanediamide (N25) (Zhang et al., 2014) and by HDAC8 siRNA silencing (Santos‐Barriopedro et al., 2019).
One of the most important hallmarks of GBM is its invasiveness in the surrounding parenchyma, which increases the tendency to relapse after surgery, chemo‐ and radio‐therapy in GBM‐patients (Preusser et al., 2011). We hypothesized that HDAC8 drives the glioma cell migration and invasiveness regulating the acetylation level of the alpha‐tubulin. In fact, the HDAC8 inhibition increased the acetylated alpha‐tubulin abolishing the human and murine glioma cell migration. It was reported that alpha‐tubulin acetylation is an important epigenetic modification for regulating microtubule architecture and cell migration (Li et al., 2014) and its level is mainly governed by opposing actions of alpha‐tubulin acetyltransferase 1 (ATAT1) and HDAC6‐8 (Li et al., 2014; Picci et al., 2020; Vanaja et al., 2018). Here, we reported for the first time a relation between HDAC8, alpha‐tubulin acetylation, and migration in glioma. This is different from what reported for commercial PCI‐34051, where no effects on alpha‐tubulin are reported. However, our results are corroborated by siRNA HDAC8 silencing, thus confirming the role of HDAC8 on alpha‐tubulin acetylation. Moreover, we demonstrated that HDAC8 inhibition abolished the migration of microglia toward GCM, suggesting that HDAC8 is related to microglial migration to the tumor site, and regulates the acetylation level of alpha‐tubulin also in microglial cells.
Glioma creates a microenvironment that supports its own growth, impairing NK cell recruitment and activity (Burger et al., 2019) and switching GAMs toward a predominant anti‐inflammatory phenotype (Szulzewsky et al., 2016). Attempt to reprogram the tumor microenvironment to counteract glioma is one of the main efforts of bio‐medical research in the last years. In particular, the ‘re‐education’ of GAMs cells toward a pro‐inflammatory, anti‐tumor phenotype successfully reduces tumor size and increases the mean survival times of tumor‐bearing animals (Mieczkowski et al., 2015). Here, we showed that HDAC8 plays a pivotal role in the activation state of GAMs in glioma microenvironment, since its inhibition totally abolished the expression of anti‐inflammatory/pro‐tumoral genes and increased pro‐inflammatory genes in both murine CD11b+ cells in the glioma‐bearing hemisphere and human CD11b+ cells from GBM‐patients. Interestingly, the direct inhibition of HDAC8 in cultured murine microglial cells in vitro has a minor effect on the regulation of pro‐ and anti‐inflammatory gene expression in the different activation states, suggesting an indirect action of HDAC8 inhibition in vivo. Furthermore, our data report that HDAC8 inhibition modulates morphology and gene expression only in IL‐4‐treated microglia in vitro, suggesting that HDAC8 is fundamental mainly in the regulation of the anti‐inflammatory microglial activity. In addition, the pro‐inflammatory microglia stimulated with LPS + IFN‐γ strongly reduces the HDAC8 expression per se.
Making the tumor cells desirable targets for cytotoxic T and NK cells is one main effort of the research in the last years. Epigenetic dysregulation of NKG2D ligand genes is a significant contributor to immune escape in glioma (Zhang et al., 2016). In particular, HDAC proteins regulate cell surface expression of NKG2D ligands with a calcium‐dependent mechanism (Jensen et al., 2013). We identify HDAC8 as a key regulator that affects the transcription of ligands for the NKG2D receptor that triggers the cytotoxicity of NK cells. Specifically, HDAC8 leads to a significant increase of H3K4me3 level in h60, ulbp1, and rae1 promoters, enhancing the NK cell cytotoxicity against glioma, and increasing the CD69+ and activated NKG2D+ NK cells infiltrating the glioma. rtPCR analyses revealed that the expression of Ulbp1 is not changed at 24 h of PCI‐34051 treatment. However, it is possible that additional epigenetic modifications or binding of specific transcription factors are necessary for the full activation of this promoter upon treatment with PCI‐34051 leading to mRNA increase. Moreover, it is also reasonable to hypothesize that the various promoters may present different treatment time requirement to cause mRNA induction after promoter activation.
Altogether our results describe that HDAC8 plays a central role in glioma growth, regulating cell viability and the migration through alpha‐tubulin acetylation. Furthermore, HDAC8 supports an immunosuppressive environment, regulating the gene transcription of NKG2D‐ligands and the microglial phenotype.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
A.M.: performed most of the experimental work and wrote the article; G.C.: contributed to many experimental activities for mice manipulation; G.F.: performed ChIP experiments; F.F.: supervised and performed ChIP experiments; S.V.: produced and provided PCI‐34051; A.M.: produced and provided PCI‐34051; V.E.: provided human GBM tissues; A.S.: provided human GBM tissues; G.B.: performed FACS analyses and immune cell activity assays; A.S.: supervised experiments on NK cells; C.L.: supervised all the experimental work and wrote the manuscript; S.G.: ideated and supervised all the experimental work and wrote the article.
Supporting information
Supplementary Information S1
ACKNOWLEDGMENTS
We are grateful to Alessandro Rosa to provide the human iPSC‐derived neurons.
This work was supported by AIRC 22329 2018 and NKINALS ARISLA 2019 to S.G.; AIRC 2016 (n. 19162) to A.M.; AIRC IG 2018‐ID. 21406 project and Istituto Pasteur Italia‐Fondazione Cenci Bolognetti to F.F.; PRIN 2017, AIRC 2019, Ministero della Salute RF2018 to C.L. Open Access Funding provided by Universita degli Studi di Roma La Sapienza within the CRUI‐CARE Agreement.
Mormino, A., Cocozza, G., Fontemaggi, G., Valente, S., Esposito, V., Santoro, A., Bernardini, G., Santoni, A., Fazi, F., Mai, A., Limatola, C., & Garofalo, S. (2021). Histone‐deacetylase 8 drives the immune response and the growth of glioma. Glia, 69(11), 2682–2698. 10.1002/glia.24065
Cristina Limatola and Stefano Garofalo have equally contributed as last author.
Funding information Associazione Italiana per la Ricerca sul Cancro, Grant/Award Numbers: Fellowship 22329 2018, IG 19162 2016, IG 21406 2018, IG CL 2019; Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica, Grant/Award Number: Pilot NKINALS 2019; Ministero della Salute, Grant/Award Number: CL 2018; PRIN ‐ Progetti di ricerca di interesse nazionale, Grant/Award Number: CL 2017
Contributor Information
Cristina Limatola, Email: cristina.limatola@uniroma1.it.
Stefano Garofalo, Email: stefano.garofalo@uniroma1.it.
DATA AVAILABILITY STATEMENT
All data are available in the main text or the supplementary materials. The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Arvanitis, C. D., Ferraro, G. B., & Jain, R. K. (2020). The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nature Reviews Cancer [Internet], 20, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian, S., Ramos, J., Luo, W., Sirisawad, M., Verner, E., & Buggy, J. J. (2008). A novel histone deacetylase 8 (HDAC8)‐specific inhibitor PCI‐34051 induces apoptosis in T‐cell lymphomas. Leukemia, 22, 1026–1034. [DOI] [PubMed] [Google Scholar]
- Bellissimo, T., Ganci, F., Gallo, E., Sacconi, A., Tito, C., De Angelis, L., Pulito, C., Masciarelli, S., Diso, D., Anile, M., Petrozza, V., Giangaspero, F., Pescarmona, E., Facciolo, F., Venuta, F., Marino, M., Blandino, G., & Fazi, F. (2017). Thymic epithelial tumors phenotype relies on miR‐145‐5p epigenetic regulation. Molecular Cancer, 16, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. L., Bennett, F. C., Liddelow, S. A., Ajami, B., Zamanian, J. L., Fernhoff, N. B., Mulinyawe, S. B., Bohlen, C. J., Adil, A., Tucker, A., Weissman, I. L., Chang, E. F., Li, G., Grant, G. A., Hayden Gephart, M. G., & Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113, E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger, M. C., Zhang, C., Harter, P. N., Romanski, A., Strassheimer, F., Senft, C., Tonn, T., Steinbach, J. P., & Wels, W. S. (2019). CAR‐engineered NK cells for the treatment of Glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Frontiers in Immunology, 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti, A., Oehme, I., Witt, O., Oliveira, G., Sippl, W., Romier, C., Pierce, R. J., & Jung, M. (2015). HDAC8: A multifaceted target for therapeutic interventions. Trends in Pharmacological Sciences [Internet], 36, 481–492. [DOI] [PubMed] [Google Scholar]
- Chen, Y. N., Hou, S. Q., Jiang, R., Sun, J. L., Cheng, C. D., & Qian, Z. R. (2021). EZH2 is a potential prognostic predictor of glioma. Journal of Cellular and Molecular Medicine, 25, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coniglio, S. J., & Segall, J. E. (2013). Review: Molecular mechanism of microglia stimulated glioblastoma invasion. Matrix Biology [Internet], 32, 372–380. [DOI] [PubMed] [Google Scholar]
- D'Alessandro, G., Monaco, L., Catacuzzeno, L., Antonangeli, F., Santoro, A., Esposito, V., Franciolini, F., Wulff, H., & Limatola, C. (2019). Radiation increases functional KCa3.1 expression and invasiveness in glioblastoma. Cancers (Basel), 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker, J., Witt, O., & Milde, T. (2013). Targeting of histone deacetylases in brain tumors. CNS Oncology, 2, 359–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyüpoglu, Y. I., & Savaskan, E. N. (2016). Epigenetics in brain tumors: HDACs take center stage. Current Neuropharmacology, 14, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrusiewicz, K., Ellert‐Miklaszewska, A., Lipko, M., Sielska, M., Frankowska, M., & Kaminska, B. (2011). Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS One, 6, e23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, Liebelt BD, Yaghi NK, Ezhilarasan R, Huang N, Weinberg JS, Prabhu SS, Rao G, Sawaya R, Langford LA, … Heimberger AB. (2016). Glioblastoma‐infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 1(2), e85841. 10.1172/jci.insight.85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo, S., Cocozza, G., Porzia, A., Inghilleri, M., Raspa, M., Scavizzi, F., Aronica, E., Bernardini, G., Peng, L., Ransohoff, R. M., Santoni, A., & Limatola, C. (2020). Natural killer cells modulate motor neuron‐immune cell cross talk in models of amyotrophic lateral sclerosis. Nature Communications [Internet], 11, 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo, S., D'Alessandro, G., Chece, G., Brau, F., Maggi, L., Rosa, A., Porzia, A., Mainiero, F., Esposito, V., Lauro, C., Benigni, G., Bernardini, G., Santoni, A., & Limatola, C. (2015). Enriched environment reduces glioma growth through immune and non‐immune mechanisms in mice. Nature Communications, 6, 6623. 10.1038/ncomms7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo, S., Porzia, A., Mainiero, F., Di Angelantonio, S., Cortese, B., Basilico, B., Pagani, F., Cignitti, G., Chece, G., Maggio, R., Tremblay, M. E., Savage, J., Bisht, K., Esposito, V., Bernardini, G., Seyfried, T., Mieczkowski, J., Stepniak, K., Kaminska, B., … Limatola, C. (2017). Environmental stimuli shape microglial plasticity in glioma. eLife, 6, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golán, I., De La Fuente, L. R., & Costoya, J. A. (2018). NK cell‐based glioblastoma immunotherapy. Cancers (Basel), 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber, M. B., Scheithauer, B. W., & Kreutzberg, G. W. (2002). Microglia in brain tumors. Glia, 40, 252–259. [DOI] [PubMed] [Google Scholar]
- Helin, K., & Dhanak, D. (2013). Chromatin proteins and modifications as drug targets. Nature, 502, 480–488. [DOI] [PubMed] [Google Scholar]
- Hill, E., Nagel, D., Parri, R., & Coleman, M. (2016). Stem cell‐derived astrocytes: Are they physiologically credible? The Journal of Physiology, 594, 6595–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, H., Hagemann‐Jensen, M., Lauridsen, F., & Skov, S. (2013). Regulation of NKG2D‐ligand cell surface expression by intracellular calcium after HDAC‐inhibitor treatment. Molecular Immunology, 53, 255–264. [DOI] [PubMed] [Google Scholar]
- Kim, K. H., & Roberts, C. W. M. (2016). Targeting EZH2 in cancer. Nature Medicine, 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, P., Murphy, B., Miller, R., Menon, V., Banik, N. L., Giglio, P., Lindhorst, S. M., Varma, A. K., Vandergrift, W. A., 3rd, Patel, S. J., & Das, A. (2015). Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Research, 35, 615–625. [PMC free article] [PubMed] [Google Scholar]
- Li, J., Chen, S., Cleary, R. A., Wang, R., Gannon, O. J., Seto, E., & Tang, D. D. (2014). Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. American Journal of Physiology‐Cell Physiology, 307, C288–C295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F. L., Yen, J. L., Kuo, Y. C., Kang, J. J., Cheng, Y. W., Huang, W. J., & Hsiao, G. (2019). HDAC8 inhibitor WK2‐16 therapeutically targets lipopolysaccharide‐induced mouse model of neuroinflammation and microglial activation. International Journal of Molecular Sciences, 20(2), 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic, D. S., Glass, R., Synowitz, M., Van Rooijen, N., & Kettenmann, H. (2005). Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease‐2. Journal of Neuropathology and Experimental Neurology, 64, 754–762. [DOI] [PubMed] [Google Scholar]
- Markovic, D. S., Vinnakota, K., Chirasani, S., Synowitz, M., Raguet, H., Stock, K., Sliwa, M., Lehmann, S., Kälin, R., Van Rooijen, N., Holmbeck, K., Heppner, F. L., Kiwit, J., Matyash, V., Lehnardt, S., Kaminska, B., Glass, R., & Kettenmann, H. (2009). Gliomas induce and exploit microglial MT1‐MMP expression for tumor expansion. Proceedings of the National Academy of Sciences of the United States of America, 106, 12530–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, S., & Jimeno, A. (2020). Tazemetostat for the treatment of multiple types of hematological malignancies and solid tumors. Drugs Today (Barc), 56, 377–387. [DOI] [PubMed] [Google Scholar]
- Mieczkowski, J., Kocyk, M., Nauman, P., Gabrusiewicz, K., Sielska, M., Przanowski, P., Maleszewska, M., Rajan, W. D., Pszczolkowska, D., Tykocki, T., Grajkowska, W., Kotulska, K., Roszkowski, M., Kostkiewicz, B., & Kaminska, B. (2015). Down‐regulation of IKKβ expression in glioma‐infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget, 6, 33077–33090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad, F., Weissmann, S., Leblanc, B., Pandey, D. P., Højfeldt, J. W., Comet, I., Zheng, C., Johansen, J. V., Rapin, N., Porse, B. T., Tvardovskiy, A., Jensen, O. N., Olaciregui, N. G., Lavarino, C., Suñol, M., De Torres, C., Mora, J., Carcaboso, A. M., & Helin, K. (2017). EZH2 is a potential therapeutic target for H3K27M‐mutant pediatric gliomas. Nature Medicine [Internet], 23, 483–492. [DOI] [PubMed] [Google Scholar]
- Nakagawa, M., Oda, Y., Eguchi, T., Aishima, S. I., Yao, T., Hosoi, F., Basaki, Y., Ono, M., Kuwano, M., Tanaka, M., & Tsuneyoshi, M. (2007). Expression profile of class I histone deacetylases in human cancer tissues. Oncology Reports, 18, 769–774. [PubMed] [Google Scholar]
- Nath, P. R., Pal‐Nath, D., Mandal, A., Cam, M. C., Schwartz, A. L., & Roberts, D. D. (2019). Natural killer cell recruitment and activation are regulated by CD47 expression in the tumor microenvironment. Cancer Immunology Research, 7, 1547–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt, S. L., Keenan, C., Chopin, M., & Allan, R. S. (2020). EZH2 function in immune cell development. Biological Chemistry, 401, 933–943. [DOI] [PubMed] [Google Scholar]
- Oehme, I., Deubzer, H. E., Lodrini, M., Milde, T., & Witt, O. (2009). Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opinion on Investigational Drugs, 18, 1605–1617. [DOI] [PubMed] [Google Scholar]
- Oehme, I., Deubzer, H. E., Wegener, D., Pickert, D., Linke, J. P., Hero, B., Kopp‐Schneider, A., Westermann, F., Ulrich, S. M., Von Deimling, A., Fischer, M., & Witt, O. (2009). Histone deacetylase 8 in neuroblastoma tumorigenesis. Clinical Cancer Research, 15, 91–99. [DOI] [PubMed] [Google Scholar]
- Pastorino, O., Teresa Gentile, M., Mancini, A., Del Gaudio, N., Di Costanzo, A., Bajetto, A., Franco, P., Altucci, L., Florio, T., Patrizia Stoppelli, M., & Colucci‐D'amato, L. (2019). Histone deacetylase inhibitors impair vasculogenic mimicry from glioblastoma cells. Cancers (Basel), 11, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, R. E., Soshnev, A. A., & Allis, C. D. (2020). Epigenomic reprogramming as a driver of malignant Glioma. Cancer Cell [Internet], 38, 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picci, C., Wong, V. S. C., Costa, C. J., McKinnon, M. C., Goldberg, D. C., Swift, M., Alam, N. M., Prusky, G. T., Shen, S., Kozikowski, A. P., Willis, D. E., & Langley, B. (2020). HDAC6 inhibition promotes α‐tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Experimental Neurology [Internet], 328, 113281. [DOI] [PubMed] [Google Scholar]
- Preusser, M., De Ribaupierre, S., Wöhrer, A., Erridge, S. C., Hegi, M., Weller, M., & Stupp, R. (2011). Current concepts and management of glioblastoma. Annals of Neurology, 70, 9–21. [DOI] [PubMed] [Google Scholar]
- Pyonteck, S. M., Akkari, L., Schuhmacher, A. J., Bowman, R. L., Sevenich, L., Quail, D. F., Olson, O. C., Quick, M. L., Huse, J. T., Teijeiro, V., Setty, M., Leslie, C. S., Oei, Y., Pedraza, A., Zhang, J., Brennan, C. W., Sutton, J. C., Holland, E. C., Daniel, D., & Joyce, J. A. (2013). CSF‐1R inhibition alters macrophage polarization and blocks glioma progression HHS public access author manuscript. Nature Medicine [Internet], 19, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, Y., Kang, K., Giannopoulou, E., Fang, C., & Ivashkiv, L. B. (2016). IFN‐γ induces histone 3 lysine 27 Trimethylation in a small subset of promoters to stably silence gene expression in human macrophages. Cell Reports [Internet], 16, 3121–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig, I., Koeneke, E., Trippel, F., Mueller, W. C., Burhenne, J., Kopp‐Schneider, A., Fabian, J., Schober, A., Fernekorn, U., Von Deimling, A., Deubzer, H. E., Milde, T., Witt, O., & Oehme, I. (2015). Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid‐mediated differentiation. Cell Death and Disease [Internet], 6, e1657–e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugo, H. S., Jacobs, I., Sharma, S., Scappaticci, F., Paul, T. A., Jensen‐Pergakes, K., & Malouf, G. G. (2020). The promise for histone Methyltransferase inhibitors for epigenetic therapy in clinical oncology: A narrative review. Advances in Therapy [Internet], 37, 3059–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Barriopedro, I., Li, Y., Bahl, S., & Seto, E. (2019). Hdac8 affects mgmt levels in glioblastoma cell lines via interaction with the proteasome receptor adrm1. Genes and Cancer, 10, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T. D., & Livak, K. J. (2008). Analyzing real‐time PCR data by the comparative CT method. Nature Protocols, 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Sciaccaluga, M., Fioretti, B., Catacuzzeno, L., Pagani, F., Bertollini, C., Rosito, M., Catalano, M., D'Alessandro, G., Santoro, A., Cantore, G., Ragozzino, D., Castigli, E., Franciolini, F., & Limatola, C. (2010). CXCL12‐induced glioblastoma cell migration requires intermediate conductance Ca2+−activated K+ channel activity. American Journal of Physiology‐Cell Physiology, 299, 175–184. [DOI] [PubMed] [Google Scholar]
- Shabason, J. E., Tofilon, P. J., & Camphausen, K. (2011). Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. Journal of Cellular and Molecular Medicine, 15, 2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp, R., Hegi, M. E., Mason, W. P., van den Bent, M. J., Taphoorn, M. J., Janzer, R. C., Ludwin, S. K., Allgeier, A., Fisher, B., Belanger, K., Hau, P., Brandes, A. A., Gijtenbeek, J., Marosi, C., Vecht, C. J., Mokhtari, K., Wesseling, P., Villa, S., Eisenhauer, E., … Mirimanoff, R. O. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncology [Internet], 10, 459–466. [DOI] [PubMed] [Google Scholar]
- Suvà, M. L., Riggi, N., Janiszewska, M., Radovanovic, I., Provero, P., Stehle, J. C., Baumer, K., Le Bitoux, M. A., Marino, D., Cironi, L., Marquez, V. E., Clément, V., & Stamenkovic, I. (2009). EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Research, 69, 9211–9218. [DOI] [PubMed] [Google Scholar]
- Szulzewsky, F., Arora, S., de Witte, L., Ulas, T., Markovic, D., Schultze, J. L., Holland, E. C., Synowitz, M., Wolf, S. A., & Kettenmann, H. (2016). Human glioblastoma‐associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia, 64, 1416–1436. [DOI] [PubMed] [Google Scholar]
- Tam, W. Y., & Ma, C. H. E. (2014). Bipolar/rod‐shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Scientific Reports, 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja, G. R., Ramulu, H. G., & Kalle, A. M. (2018). Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Communication and Signaling: CCS, 16, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally, S., Dhanasekaran, S. M., Zhou, M., Barrette, T. R., Kumar‐Sinha, C., Sanda, M. G., Ghosh, D., Pienta, K. J., Sewalt, R. G. A. B., Rubin, M. A., & Chinnaiyan, A. M. (2002). The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature, 419, 624–629. [DOI] [PubMed] [Google Scholar]
- Wang, C., Eessalu, T. E., Barth, V. N., Mitch, C. H., Wagner, F. F., Neelamegam, R., Schroeder, F. A., Holson, E. B., Haggarty, S. J., & Jacob, M. (2014). Design, synthesis, and evaluation of hydroxamic acid‐based molecular probes for in vivo imaging of histone deacetylase (HDAC) in brain. American Journal of Nuclear Medicine and Molecular Imaging, 4, 29–38. [PMC free article] [PubMed] [Google Scholar]
- Was, H., Krol, S. K., Rotili, D., Mai, A., Wojtas, B., Kaminska, B., & Maleszewska, M. (2019). Histone deacetylase inhibitors exert anti‐tumor effects on human adherent and stem‐like glioma cells. Clinical Epigenetics, 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters, J. M., Wright, G., Smith, M. A., Shah, B., & Wright, K. L. (2021). Histone deacetylase 8 inhibition suppresses mantle cell lymphoma viability while preserving natural killer cell function. Biochemical and Biophysical Research Communications [Internet], 534, 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, B. J., Tremblay, A. M., Deblois, G., Sylvain‐Drolet, G., & Giguère, V. (2010). An acetylation switch modulates the transcriptional activity of estrogen‐related receptor α. Molecular Endocrinology, 24, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright‐Jin, E. C., & Gutmann, D. H. (2019). Microglia as dynamic cellular mediators of brain function. Trends in Molecular Medicine [Internet], 25, 967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L., Chen, Y., Huang, Y., Sandanaraj, E., Yu, J. S., Lin, R. Y. T., Dakle, P., Ke, X. Y., Chong, Y. K., Koh, L., Mayakonda, A., Nacro, K., Hill, J., Huang, M. L., Gery, S., Lim, S. W., Huang, Z., Xu, Y., Chen, J., … Phillip, K. H. (2021). Topography of transcriptionally active chromatin in glioblastoma. Science Advances, 7, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y., Qiu, S., Li, X., Huang, B., Xu, Y., & Peng, Y. (2017). EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. Journal of Neuroinflammation, 14, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S., Bin, H. W., Wu, L., Wang, L. Y., Ye, L. B., & Feng, B. H. (2014). A novel suberoylanilide hydroxamic acid histone deacetylase inhibitor derivative, N25, exhibiting improved antitumor activity in both human U251 and H460 cells. Asian Pacific Journal of Cancer Prevention, 15, 4331–4338. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Rao, A., Sette, P., Deibert, C., Pomerantz, A., Kim, W. J., Kohanbash, G., Chang, Y., Park, Y., Engh, J., Choi, J., Chan, T., Okada, H., Lotze, M., Grandi, P., & Amankulor, N. (2016). IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro‐Oncology, 18, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, G., Jia, J., Wang, L., Zhang, Y., Yang, H., Lu, Y., Yu, R., Liu, H., & Zhu, Y. (2020). Local delivery of minocycline and vorinostat targets the tumor microenvironment to inhibit the recurrence of glioma. Oncotargets and Therapy, 13, 11397–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information S1
Data Availability Statement
All data are available in the main text or the supplementary materials. The data that support the findings of this study are available from the corresponding author upon reasonable request.