

Abstract
Ultra‐electron‐deficient azaacenes were synthesized via Buchwald‐Hartwig coupling of ortho‐diaminoarenes with chlorinated mellophanic diimide followed by oxidation of the intermediate N,N’‐dihydro compounds with MnO2 or PbO2. The resulting cata‐annulated bisimide azaacenes have ultrahigh electron affinities with first reduction potentials as low as −0.35 V recorded for a tetraazapentacene. Attempts to prepare a tetrakis(dicarboximide)tetraazaheptacene resulted in the formation of a symmetric butterfly dimer.
Keywords: azaacenes, butterfly dimer, bisimides, electron acceptors, heteroacenes
How high can you go?Cata‐annulation of dicarboximides onto azaacenes lowers the lowest unoccupied molecular orbitals and increases reduction potentials up to −0.35 V vs. Fc/Fc+, but does not stabilize a tetrakis(dicarboximide)tetraazaheptacene which dimerized regioselectively to its butterfly adduct.
There is a constant push in materials science to prepare organic materials with extreme and attractive properties for uses that range from solar cells and n‐channel materials to spintronics. Attachment of bisimides to organic cores is known to a) increase their solubility,[1] b) boost their electron affinity,[2] c) stabilize radical anion states[3] and d) push their λ max into the red.[4] While there have been reports of the juxtaposition of arenes with bisimides,[5] the multiple cata‐annulation of azaacenes with imides has not been described despite the potential to furnish materials with unprecedented properties. Here we present the synthesis and characterization of novel azaacene derivatives with ultrahigh electron affinities (−3.88 eV to −4.35 eV) and near‐IR absorptions. These features are enabled by the presence of two or four bisimide rings at ends of the azaacenes.
Previously, Cao et al. prepared electron‐accepting and chromophoric heteroacenes by nucleophilic aromatic substitution of dichloro mellophanic diimide (MDI‐Cl2 ) with ortho dinucleophiles.[6] Here, we couple MDI‐Cl2 [7] with aromatic diamines[8] to explore novel electron‐deficient azaacenes. Buchwald‐Hartwig coupling of diamines 6–9 with MDI‐Cl2 (Scheme 1) using Pd‐RuPhos(G1)[9] as catalyst and Cs2CO3 as base furnished 1‐H2 , 2‐H2 , 3‐H2 and 4‐H2 in 43 %–87 % yield, respectively. Although the diamine 10 did not couple under these conditions,[10] switching to Pd2(dba)3 with RuPhos[11] in Hünig's base as solvent gave 5‐H2 in 60 % yield (Scheme 1). The Pd‐catalyst is necessary for this coupling since its omission leads to decomposition of the starting diamines. Oxidation of the dihydro compounds with MnO2 or PbO2 furnishes the heteroacenes in 92 %‐97 %. Oxidation was more difficult for the larger and/or more electron‐poor starting N,N’‐dihydroazaacenes. Compounds 1‐H2 and 2‐H2 could be oxidized with MnO2 at room temperature (rt) while 3‐H2 needed to be heated to 50 °C to achieve oxidation. The even more electron‐poor 4‐H2 could only be oxidized by PbO2 at 70 °C in chloroform. For 5 a, PbO2 at rt was sufficiently oxidizing, but the reaction had to be carried out under N2 because reduction back to 5‐H2 occurred spontaneously in air, similar to what has been previously observed for similarly electron‐poor dicyanodiazahexacene[12] and hexaazahexacene.[8a] This spontaneous reduction is a testament to the high electron affinities of 4 a and 5 a.
Scheme 1.

Synthesis of 1 a–5 a via Buchwald‐Hartwig coupling of 6–10 with MDI‐Cl2 and the subsequent oxidation of the dihydro species. [Pd]: Pd‐RuPhos(G1), i. Cs2CO3, toluene, 16 h 120 °C, ii. Hünig's base, 120 °C, 16 h.
Compounds 1 a–5 a display red‐shifted absorption p‐bands compared to those of the unsubstituted azaacenes (Figure 1; 4545 cm−1 (1 a) to 2521 cm−1 (3 a)), with 3 a and 5 a being NIR absorbers. The absorption bands between 300 and 400 nm arise from the bisimide derivatization. Absorption maxima increase with azaacene size (series 1 a–3 a) and 3 a displays a λ max of 908 nm.
Figure 1.
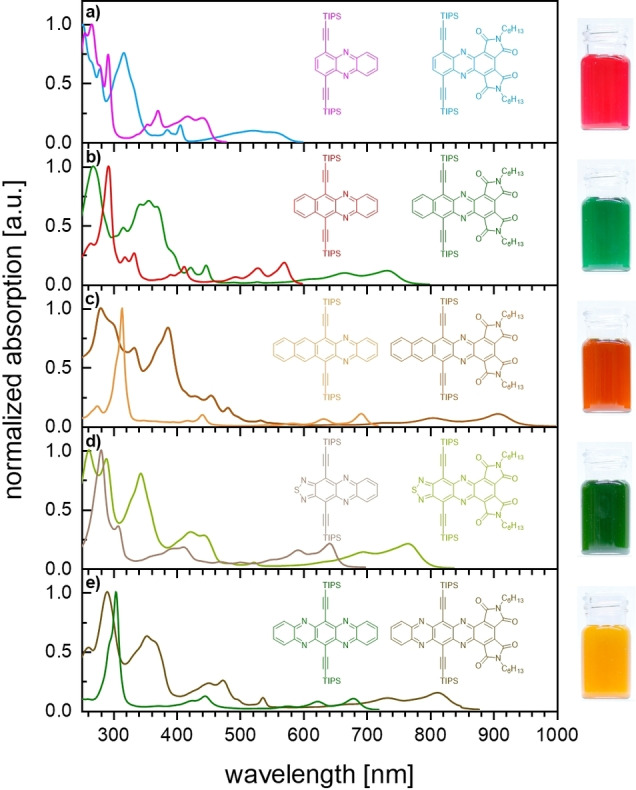
Left: Absorption spectra of azaacene bisimides and their parent compounds in n‐hexane. Right: Photographs of the azaacene bisimides in dichloromethane. From top to bottom: 1 a to 5 a.
First reduction potentials and electron affinities (cyclic voltammetry, CV, Figure S2, Supporting Information) also increase with azaacene size and number of nitrogen atoms. Interestingly, cata‐condensed bisimides are more powerful than CN‐substituents, as evidenced by a comparison of the first reduction potentials of 3 a (−0.62 V) to that of a dicyanodiazaacene (−0.7 V).[12] The even more electron accepting 4 a and 5 a are reduced at −0.36 V and −0.35 V (vs. Fc/Fc+), respectively. Together, the electrochemical data suggest that 2 a, 3 a, 4 a and 5 a should be isolable as air‐stable radical anions due to their high electron affinities.[13] DFT‐calculated energy gaps are in agreement with optical gaps and the LUMO energies correlate with reduction potentials linearly (Table 1). Bisimide substitution lowers LUMO energies of azaacenes between 0.52 eV (4 a/4 b) and 0.87 eV (5 a/5 b) (Table 1), while the effect on the HOMO positions is less pronounced (0.26 eV for 4 a/4 b; 0.51 eV for 5 a/5 b), an observation that is consistent with the red‐shifted absorption bands. The FMOs (Figure 2) explain this qualitatively: Orbital coefficients of the LUMO extend to the bisimides, whereas those of the HOMO reside on the acene backbone and are thus less affected by annulation. In contrast to benzannulated acenes, NICS(1)zz calculations show that the outer rings of the azaacene core are aromatic (δ=−17.6 to −21.5 ppm) and an integral part of the acene backbone, whereas the maleimides are antiaromatic (δ=8.70 to 9.39 ppm) (see Figure S7, Supporting Information).
Table 1.
Optical, electrochemical and quantum‐chemical data of the azaacene bisimides and their parent compounds.
|
Compound |
E(0/−) [V][a] |
EA [eV][b] |
ELUMO, DFT [eV][c] |
IP [eV][d] |
EHOMO, DFT [eV][c] |
gapDFT [eV] |
λmax, abs [nm] |
opt. gap [eV][e] |
|---|---|---|---|---|---|---|---|---|
|
1a |
−0.82 |
−3.88 |
−3.92 |
−5.93 |
−6.27 |
2.35 |
550 |
2.05 |
|
1b [f] |
−1.68 |
−3.02 |
−3.08 |
−5.72 |
−5.97 |
2.88 |
440 |
2.67 |
|
2a |
−0.67 |
−4.03 |
−4.04 |
−5.59 |
−5.83 |
1.79 |
733 |
1.56 |
|
2b [f] |
−1.23 |
−3.47 |
−3.35 |
−5.59 |
−5.54 |
2.20 |
570 |
2.09 |
|
3a |
−0.62 |
−4.08 |
−4.10 |
−5.47 |
−5.50 |
1.40 |
908 |
1.31 |
|
3b [f] |
−1.05 |
−3.65 |
−3.50 |
−5.40 |
−5.25 |
1.75 |
692 |
1.74 |
|
4a |
−0.36 |
−4.34 |
−4.37 |
−5.89 |
−6.03 |
1.66 |
766 |
1.55 |
|
4b [g] |
−0.83 |
−3.87 |
−3.85 |
−5.73 |
−5.77 |
1.82 |
642 |
1.86 |
|
5a |
−0.35 |
−4.35 |
−4.30 |
−5.79 |
−5.80 |
1.50 |
813 |
1.44 |
|
5b [h] |
−0.79 |
−3.67 |
−3.43 |
−5.49 |
−5.29 |
1.86 |
680 |
1.82 |
[a] First reduction potentials measured by cyclic voltammetry (CV) in dichloromethane with Bu4NPF6 as the electrolyte against Fc/Fc+ as an internal standard at 0.2 V s−1. The offset of the exact values is estimated by adjusting the ferrocene redox couple to −5.10 eV[14] on the Fermi scale, for azaacenes EA=−(E 0/− [vs. Fc+/Fc]+4.70 eV) gives a much better empirical fit of almost all CV data to DFT calculations.[12, 15] A table referencing ferrocene as −5.10 eV can be found in the Supporting Information (Table S1). [b] Calculated from CV measurements EA=−(E 0/− [vs. Fc+/Fc]+4.7 eV).[14] [c] Obtained from quantum‐chemical calculations with DFT/B3LYP/def2‐TZVP. [d] IP=E A ‐ opt. gap. [e] Calculated from λ onset, abs. [f] CV data and quantum‐chemical calculations taken from Ref. [16] and adjusted for Fc/Fc+=−4.70 eV. [g] CV data and quantum‐chemical calculations taken from Ref. [17] and adjusted for Fc/Fc+=−4.70 eV. [h] CV data and quantum‐chemical calculations taken from Ref. [18] and adjusted for Fc/Fc+=−4.70 eV.
Figure 2.
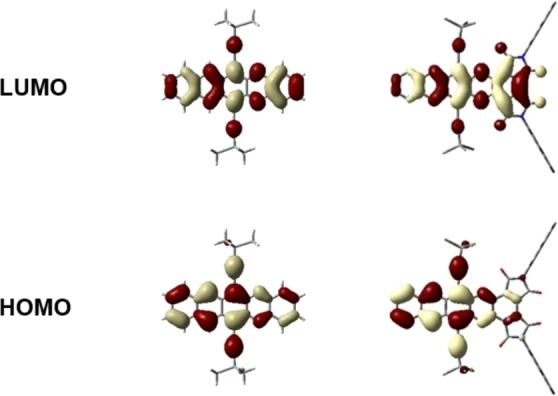
Comparison of the DFT‐calculated HOMOs and LUMOs [B3LYP, def2TZVP] of 3 a* to 3 b* (TIPS‐substituents were replaced by TMS‐substituents).
Single crystals X‐ray analysis of 2 a reveals molecules packed into a chiral helix of trigonal symmetry (Figure 3). Intermolecular interactions are dominated by the imide groups; carbonyl oxygens are in close contact with neighbouring partially positively charged carbon atoms. The one‐helix turn distance between the azaacene planes is 7.46 Å. The non‐overlapping acene backbones exhibit end‐to‐end tilts of 15°. The absolute structure could not be determined as there are two possible enantiomers for the chiral space group (Table S7, Supporting Information).
Figure 3.
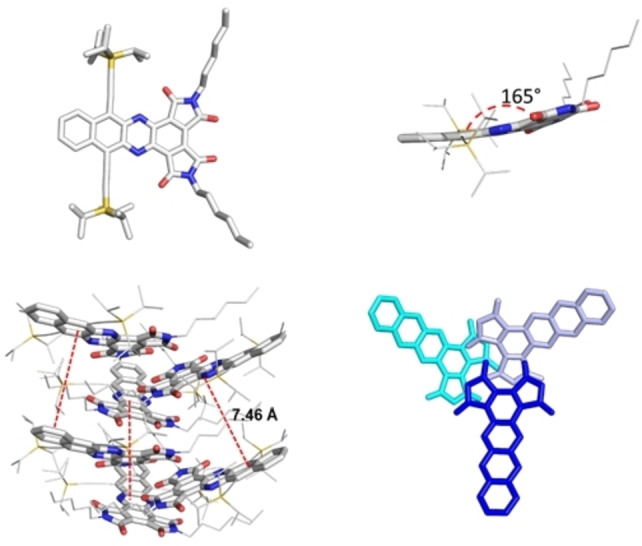
Crystal structure and packing of 2 a. TIPS‐ethynyl and hexyl substituents were either reduced in size or omitted for clarity.
To evaluate whether azaheptaacenes would exhibit similarly improved properties, Müllen's tetraaminoanthracene 11 [8f] was coupled to MDI‐Cl2 using a Buchwald‐Hartwig reaction to yield the tetrahydro compound 12‐H4 . PbO2 oxidation of 12‐H4 in dry DCM furnished 13, a dimer of the intended product 12, as a green solid. 1H NMR spectroscopy suggests a non‐centrosymmetric constitution of C2h symmetry, represented either by the head‐to‐tail [4+4] dimer as shown in Scheme 2 or its head‐to‐head isomer (Figure S1, Supporting Information). This phenomenon is similar to the products of photochemical dimerization of TIPS‐pentacene.[19] Attempts to isolate 12 were unsuccessful, indicating that the dimerization is fast on the experimental timescale.
Scheme 2.

Attempted synthesis of a tetraazaheptacene 12 and isolation of its [4+4] degradation product 13 (one geometric isomer shown).
High‐resolution MALDI mass spectrometry confirms the dimeric constitution, in addition to providing evidence that retro cycloaddition to the heptacene 12 occurs under these conditions. The absorption spectrum of 13 is a close match to that of 2 a (Figure S5, Supporting Information), which can be rationalized by noting that the largest conjugated chromophore in 13 is structurally analogous to 2 a.
Conclusion
The addition of bisimides to the terminal ring of azaacenes using a simple coupling‐oxidation sequence results in near‐IR absorbers with high electron affinities up to −4.35 eV. Doubly or quadruply bisimide‐substituted azapentacenes are stable, processable, yet amorphous materials that should be almost ideal materials for solar cells, and, upon shortening the hexyl chains, may also give attractive n‐channel semiconductors for thin film transistors, which we will report upon in the future.
Experimental Section
General Procedure (GP)
The corresponding ortho‐diamine (1.00 eq.), MDI‐Cl2 (1.00 eq.), caesium carbonate (3.00 eq) and Pd‐RuPhos G1 (5 mol %) were placed in a dry Schlenk tube under Argon atmosphere. Then dry and degassed toluene (1 mL per 50 mg diamine) was added and the reaction was stirred at 120 °C for 16 h. The reaction mixture was diluted with DCM and water was added. The phases were separated and the aqueous layer was extracted with methylene chloride (3×10 mL). The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure.
2,5‐Dihexyl‐8,11‐bis((triisopropylsilyl)ethynyl)‐7,12‐dihydrodipyrrolo[3,4‐a:3′,4′‐c]phenazine‐1,3,4,6(2H,5H)‐tetraone (1‐H2)
The GP was applied to 3,6‐bis((triisopropylsilyl)ethynyl)benzene‐1,2‐diamine (51.7 mg, 110 μmol, 1.00 eq.) and MDI‐Cl2 (50.0 mg, 110 μmol, 1.00 eq.), using caesium carbonate (108 mg, 331 μmol, 3.00 eq.) and Pd‐RuPhos (G1) (4.50 mg, 5.51 μmol, 0.05 eq.). The resulting crude product was purified by column chromatography (SiO2 PE/DCM 1 : 1) yielding 1‐H2 as a red solid (40.2 mg, 47.3 μmol, 43 %). 1H NMR (400 MHz, CDCl3) δ [ppm]=8.49 (s, 2H), 6.69 (s, 2H), 3.59 (t, J=7.0 Hz, 4H), 1.62 (quin, J=6.6 Hz, 4H), 1.32–1.11 (m, 58H), 0.92–0.83 (m, 7H). 13C NMR (101 MHz, CDCl3) δ [ppm]=167.8, 163.9, 136.6, 131.0, 126.4, 120.8, 110.0, 109.0, 101.9, 99.4, 38.2, 31.5, 28.3, 26.5, 22.6, 18.8, 14.2, 11.4. HRMS (MALDI+, DCTB): m/z calcd. for C50H72N4O4Si2: [M]+ 848.5092, found: 848.5085, correct isotope distribution. IR (ATR) [cm−1]=3320, 2927, 2859, 2363, 1759, 1691, 1540, 1380, 802, 776, 675, 660, 619, 609, 595, 442, 410. m.p.=180 °C.
2,5‐Dihexyl‐8,11‐bis((triisopropylsilyl)ethynyl)dipyrrolo[3,4‐a:3′,4′‐c]phenazine‐1,3,4,6(2H,5H)‐tetraone (1 a)
1‐H2 (30.0 mg, 35.3 μg, 1.00 eq.) was dissolved in methylene chloride and an excess of manganese dioxide was added. The reaction mixture was stirred at room temperature until TLC showed full consumption of the dihydro species. The mixture was filtered and the solvent was removed under reduced pressure to yield 1 a as a red solid (28.1 mg, 33.2 μmol, 94 %).1H NMR (600 MHz, CDCl3) δ [ppm]=8.14 (s, 2H), 3.83 (t, J=7.2 Hz, 4H), 1.79–1.72 (m, 4H), 1.35–1.24 (m, 54H), 0.90–0.87 (m, 6H). 13C NMR (151 MHz, CDCl3) δ [ppm]=164.6, 164.5, 145.2, 139.7, 137.9, 134.4, 130.2, 125.6, 103.6, 102.0, 38.7, 31.3, 28.3, 26.4, 22.4, 18.8, 14.1, 11.5. HRMS (MALDI+, DCTB): m/z calcd. for C50H71N4O4Si2: [M+H]+ 847.5008, found: 847.5038, correct isotope distribution. IR (ATR) [cm−1]=2923, 2864, 1771, 1722, 1713, 1463, 1398, 1364, 1064, 996, 881, 791, 676, 659, 589, 462, 456, 418. m.p.=174 °C.
2,5,13,16‐Tetrahexyl‐9,20‐bis([tri(propan‐2‐yl)silyl]ethynyl)‐7,11,18,22‐tetrahydrotetrapyrrolo[3,4‐h:3,4‐h′:3,4‐j:3,4‐j′]benzo[1,2‐b:4,5‐b′]diphenazine‐1,3,4,6,12,14,15,17(2H,5H,13H,16H)‐octone (12‐H4)
The GP was applied to Müllen's 9,10‐bis((triisopropylsilyl)ethynyl)‐anthracene‐2,3,6,7‐tetraaminiumchloride (100 mg, 134 μmol, 1.00 eq.) and MDI‐Cl2 (137 mg, 302 μmol, 2.25 eq), using caesium carbonate (875 mg, 2.69 mmol, 20.0 eq.) and PdRuPhos (G1) (11.0 mg, 13.4 μmol, 0.10 eq.). The resulting crude product was washed with methanol and PE and then purified via GPC yielding 12‐H4 as a dark purple solid (59.3 mg, 42.9 μmol, 32 %). 1H NMR (600 MHz, CD2Cl2) δ [ppm]=8.46 (s, 4H), 7.36–7.34 (m, 4H), 3.63 (br t, J=7.2 Hz, 8H), 1.68–1.63 (m, 8H), 1.34–1.29 (m, 66H), 0.90 (br t, J=6.9 Hz, 12H). 13C NMR (151 MHz, CD2Cl2) δ [ppm]=168.8, 164.0, 134.4, 131.3, 129.6, 120.00, 114.7, 111.4, 108.9, 102.9, 31.7, 28.7, 26.8, 22.8, 19.0, 14.1, 11.9. HRMS (MALDI+, DCTB): m/z calcd. for C80H103 N8O8Si2: [M+H]+ 1359.7432, found: 1359.7429, correct isotope distribution. IR (ATR) [cm−1]=3345, 2924, 2854, 2362, 2341, 2331, 1769, 1698, 1685, 1570, 1498, 1465, 1364, 1278, 778, 758, 733, 691, 678, 550. m.p.=>330 °C.
Butterfly‐Dimer (13)
12‐H4 (2.00 mg, 1.47 μmol, 1.00 eq) was dissolved in DCM (0.5 mL) in a nitrogen glove box. Then an excess of PbO2 was added. The reaction was stirred at rt for 1 d. The reaction mixture was filtered to yield 13 as a green solid (2.60 mg, 0.96 μmol, 65 %). 1H NMR (600 MHz, CD2Cl2) δ [ppm]=9.51 (s, 4H), 6.64–6.47 (m, 4H), 3.81 (br t, J=7.3 Hz, 8H), 3.70–3.61 (m, 8H), 1.80–1.73 (m, 8H), 1.70–1.46 (m, 126H), 1.45–1.39 (m, 14H), 0.91–0.87 (m, 24H). 13C NMR (151 MHz, CD2Cl2) δ [ppm]=165.7, 165.0, 164.3, 164.1, 159.6, 142.3, 141.3, 139.6, 139.0, 135.2, 135.1, 133.6, 131.1, 131.0, 129.8, 122.3, 110.5, 101.1, 57.8, 39.2, 39.0, 32.0, 31.8, 29.0, 28.8, 27.1, 27.0, 23.1, 23.0, 19.6, 19.5, 14.4, 12.3. HRMS (MALDI+, DCTB): m/z calcd. for C160H196N16O16Si4: [M/2]⋅+ 1354.7046, found: 1354.7027, correct isotope distribution. IR (ATR) [cm−1]=2955, 2922, 2853, 2359, .2341, 1773, 1725, 1458, 1391, 1376, 1365, 1259, 1083, 1014, 880, 702, 679, 670, 661, 403.
Crystallographic data
Deposition Number(s) 2080832 (2 a) and 2080833 (3‐H 2) contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank the SFB 1249 for generous support. L.A. thanks the “Studienstiftung des Deutschen Volkes” for a scholarship. S.M.L. and D.C. were supported by the National Science Foundation under Grant No. (NSF 1954975) and the Donors of the American Chemical Society Petroleum Research Fund (57163‐UNI1). Open access funding enabled and organized by Projekt DEAL.
M. Elter, L. Ahrens, S. M. Luo, F. Rominger, J. Freudenberg, D. D. Cao, U. H. F. Bunz, Chem. Eur. J. 2021, 27, 12284.
Contributor Information
Dr. Jan Freudenberg, Email: freudenberg@oci.uni-heidelberg.de.
Prof. Dennis D. Cao, Email: dcao@macalester.edu.
Prof. Uwe H. F. Bunz, Email: uwe.bunz@oci.uni-heidelberg.de.
References
- 1.Chen L., Li C., Müllen K., J. Mater. Chem. C 2014, 2, 1938–1956. [Google Scholar]
- 2.
- 2a.Nowak-Król A., Würthner F., Org. Chem. Front. 2019, 6, 1272–1318; [Google Scholar]
- 2b.Suraru S.-L., Würthner F., Angew. Chem. Int. Ed. 2014, 53, 7428–7448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7558–7578; [Google Scholar]
- 2c.Shukla J., Mukhopadhyay P., Eur. J. Org. Chem. 2019, 2019, 7770–7786. [Google Scholar]
- 3.
- 3a.Ji L., Shi J., Wei J., Yu T., Huang W., Adv. Mater. 2020, 32, 1908015; [DOI] [PubMed] [Google Scholar]
- 3b.Tajima K., Matsuo K., Yamada H., Seki S., Fukui N., Shinokubo H., Angew. Chem. Int. Ed. 2021, 60, 14060–14067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 14179–14186. [Google Scholar]
- 4.
- 4a.Katsuta S., Tanaka K., Maruya Y., Mori S., Masuo S., Okujima T., Uno H., Nakayama K.-i., Yamada H., Chem. Commun. 2011, 47, 10112–10114; [DOI] [PubMed] [Google Scholar]
- 4b.Yin J., Zhang K., Jiao C., Li J., Chi C., Wu J., Tetrahedron Lett. 2010, 51, 6313–6315. [Google Scholar]
- 5.Zhao K., Yu F., Liu W., Huang Y., Said A. A., Li Y., Zhang Q., J. Org. Chem. 2020, 85, 101–107. [DOI] [PubMed] [Google Scholar]
- 6.Luo S. M., Stellmach K. A., Ikuzwe S. M., Cao D. D., J. Org. Chem. 2019, 84, 10362–10370. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.Surry D. S., Buchwald S. L., Chem. Sci. 2011, 2, 27–50; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b.Hartwig J. F., Angew. Chem. Int. Ed. 1998, 37, 2046–2067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2154–2177. [Google Scholar]
- 8.
- 8a.Lindner B. D., Engelhart J. U., Tverskoy O., Appleton A. L., Rominger F., Peters A., Himmel H.-J., Bunz U. H. F., Angew. Chem. Int. Ed. 2011, 50, 8588–8591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8747–8750; [Google Scholar]
- 8b.Lindner B. D., Engelhart J. U., Märken M., Tverskoy O., Appleton A. L., Rominger F., Hardcastle K. I., Enders M., Bunz U. H. F., Chem. Eur. J. 2012, 18, 4627–4633; [DOI] [PubMed] [Google Scholar]
- 8c.Miao S., Brombosz S. M., Schleyer P. v. R., Wu J. I., Barlow S., Marder S. R., Hardcastle K. I., Bunz U. H. F., J. Am. Chem. Soc. 2008, 130, 7339–7344; [DOI] [PubMed] [Google Scholar]
- 8d.Appleton A. L., Miao S., Brombosz S. M., Berger N. J., Barlow S., Marder S. R., Lawrence B. M., Hardcastle K. I., Bunz U. H. F., Org. Lett. 2009, 11, 5222–5225; [DOI] [PubMed] [Google Scholar]
- 8e.An C., Guo X., Baumgarten M., Cryst. Growth Des. 2015, 15, 5240–5245; [Google Scholar]
- 8f.Xia D., Guo X., Chen L., Baumgarten M., Keerthi A., Müllen K., Angew. Chem. Int. Ed. 2016, 55, 941–944; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 953–956. [Google Scholar]
- 9.Biscoe M. R., Fors B. P., Buchwald S. L., J. Am. Chem. Soc. 2008, 130, 6686–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bunz U. H. F., Engelhart J. U., Chem. Eur. J. 2016, 22, 4680–4689. [DOI] [PubMed] [Google Scholar]
- 11.Milne J. E., Buchwald S. L., J. Am. Chem. Soc. 2004, 126, 13028–13032. [DOI] [PubMed] [Google Scholar]
- 12.Müller M., Beglaryan S. S., Koser S., Hahn S., Tverskoy O., Rominger F., Bunz U. H. F., Chem. Eur. J. 2017, 23, 7066–7073. [DOI] [PubMed] [Google Scholar]
- 13.Reiss H., Ji L., Han J., Koser S., Tverskoy O., Freudenberg J., Hinkel F., Moos M., Friedrich A., Krummenacher I., Lambert C., Braunschweig H., Dreuw A., Marder T. B., Bunz U. H. F., Angew. Chem. Int. Ed. 2018, 57, 9543–9547; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9688–9692. [Google Scholar]
- 14.Cardona C. M., Li W., Kaifer A. E., Stockdale D., Bazan G. C., Adv. Mater. 2011, 23, 2367–2371. [DOI] [PubMed] [Google Scholar]
- 15.Engelhart J. U., Lindner B. D., Schaffroth M., Schrempp D., Tverskoy O., Bunz U. H. F., Chem. Eur. J. 2015, 21, 8121–8129. [DOI] [PubMed] [Google Scholar]
- 16.Biegger P., Stolz S., Intorp S. N., Zhang Y., Engelhart J. U., Rominger F., Hardcastle K. I., Lemmer U., Qian X., Hamburger M., Bunz U. H. F., J. Org. Chem. 2015, 80, 582–589. [DOI] [PubMed] [Google Scholar]
- 17.Lindner B. D., Paulus F., Appleton A. L., Schaffroth M., Engelhart J. U., Schelkle K. M., Tverskoy O., Rominger F., Hamburger M., Bunz U. H. F., J. Mater. Chem. C 2014, 2, 9609–9612. [Google Scholar]
- 18.Miao S., Appleton A. L., Berger N., Barlow S., Marder S. R., Hardcastle K. I., Bunz U. H. F., Chem. Eur. J. 2009, 15, 4990–4993. [DOI] [PubMed] [Google Scholar]
- 19.Coppo P., Yeates S. G., Adv. Mater. 2005, 17, 3001–3005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information