

Abstract
Human vaginal microorganisms play an important role in maintaining good health throughout the human life cycle. An imbalance in the vaginal microbiota is associated with an increased risk of pelvic inflammatory disease (PID). This study aimed to characterize and compare vaginal microbial profiles of premenopausal Korean women with and without PID. 74 Korean premenopausal female vaginal samples were obtained; 33 were from healthy women (a control group) and 41 from PID patients. Vaginal fluid samples were collected from the vaginal wall and posterior cervix and then analyzed by 16S ribosomal ribonucleic acid (rRNA) gene-based amplicon sequencing. Results showed a significant difference between the vaginal microbial communities of the two groups (Jensen-Shannon, p = 0.014; Bray-Curtis, p = 0.009; Generalized UniFrac, p = 0.007; UniFrac, p = 0.008). Lactobacillus accounted for the highest percentage (61.0%) of the control group but was significantly decreased (34.9%) in PID patients; this was the most significant difference among all bacterial communities (p = 0.028, LDA effect size = 5.129). In addition, in the PID patient group, species diversity significantly increased (Simpson, p = 0.07) as the proportion of various pathogens increased evenly, resulting in a polymicrobial infection. Similarly, lactate, which constituted the highest percentage of the organic acids in the control group, was significantly decreased in the PID patient group (p = 0.04). The present study’s findings will help understand PID from the microbiome perspective and are expected to contribute to the development of more efficient PID diagnosis and treatment modalities.
Keywords: vaginal microflora, pelvic inflammatory disease, 16S rRNA amplicon sequencing, premenopausal, Korean
Introduction
Pelvic inflammatory disease (PID) is an infection of the upper genital tract (the uterus, fallopian tubes, or ovaries), occurring predominantly in sexually active young women (Curry et al. 2019). PID includes inflammation of the inner lining of the uterus (endometritis) and infection/inflammation of the fallopian tubes (salpingitis). If untreated, PID can cause severe and long-term complications, including tubal factor infertility (TFI), chronic pelvic pain, recurrent PID (Haggerty et al. 2016), ectopic pregnancy, and infertility. In addition, these complications could lead to severe and lasting damage to the female reproductive organs (Wang et al. 2018). Therefore, early diagnosis and treatment of PID are essential in preventing complications (Jennings and Krywko 2020).
In particular, in the United States, acute PID remains a leading gynecologic cause for hospitalization, with 1 million people diagnosed every year (Walker and Wiesenfeld 2007). Approximately 4.1% of sexually active young women in the United States receive lifelong PID treatment, and approximately 40% of PID diagnoses have symptoms, and 10–20% of cases complicate to infertility or ectopic pregnancy (Reekie et al. 2018)
The etiology of PID is mainly associated with sexually transmitted microorganisms such as Chlamydia trachomatis, Neisseria gonorrhoeae, Mycoplasma genitalium, and Gram-negative bacteria (Revzin et al. 2016); however, other cervical, enteric, bacterial vaginosis-associated, and respiratory pathogens, including Mycobacterium tuberculosis, may be involved (Curry et al. 2019). In virgin women, PID is rare, and it is hypothesized that lower genital, urinary tract, gastrointestinal tract, and skin wounds are the sites of origin for PID, from which the infection spreads directly or ascends through the lower genital tract to the upper genital tract (Cho et al. 2017). In addition, vaginal microorganisms consistent with bacterial vaginosis (BV) are also known risk factors for upper genital tract infectious diseases like PID in gynecological and obstetric patients (Sharma et al. 2014; Graspeuntner et al. 2018).
Diagnosis and management of PID are challenging because of the varying signs and symptoms and a polymicrobial etiology. Therefore, broad-spectrum antimicrobial agents have been mainly used for treatment. Unfortunately, this empirical treatment results in antibiotic resistance and side effects such as allergic reactions and bowel disease (Gradison 2012). Thus, the treatment of women with acute PID hinges on recognizing the polymicrobial etiology of the infectious process (Walker and Wiesenfeld 2007). However, classical methods of culturing pathogens or detecting the nucleic acids of pathogens in PID patients cannot distribute all the microbes in the environment (Graspeuntner et al. 2018).
Molecular methods such as next-generation sequencing (NGS) are actively employed to characterize human microbiota in patients and healthy individuals (Virtanen et al. 2017). In particular, the meta-genomics method based on 16S RNA has recently gained popularity owing to its ability to detect unculturable microorganisms and analyze whole microorganisms in the environment (Wang et al. 2018). This change has enabled analysis of a wide range of intravaginal microbiomes, infections, and diseases (Fettweis et al. 2012), including vaginal microbiome (Ravel et al. 2011), bacterial vaginitis (BV) (Srinivasan et al. 2012), PID (Wang et al. 2018), and changes in vaginal microbial distribution during pregnancy and the postpartum period (MacIntyre et al. 2015).
Female genital tract microbiota plays a crucial role in maintaining health, and an imbalance of the microbiota has been associated with an increased risk of pelvic infections. In addition, the microbial environment of the vagina differs between races, suggesting that vaginal microbiome analysis should be done by race (Fettweis et al. 2014).
Here, we performed an NGS-based microbiome analysis of the vaginal microbiome for Korean premenopausal PID patients and healthy women, and reported the results of the profiling analysis.
Experimental
Materials and Methods
Patients and sample collection. A sampling of female vaginal fluid was done from January 2020 to February 2020 in the Gynecology Department of Soonchunhyang University Cheonan Hospital affiliated with the Probiotics Microbiome Convergence Center in Soonchunhyang University, Korea. The sampling was approved by the Ethics Committee of Soonchunhyang University Cheonan Hospital (eIRB) (IRB No. 2019-10-017-005). A total of 74 premenopausal women aged 18 to 50 years were enrolled in this study: 41 premenopausal PID patients and 33 premenopausal normal control women. The diagnosis was made clinically based on the CDC PID diagnostic criteria (Crossman 2006). All participants were informed of sampling procedures and risks. They agreed with all laboratory tests and were provided with written informed consent. Samples of PID patients were collected when abnormal vaginal discharge was reported regardless of fertility, body mass index (BMI), underlying disease, or gynecologic disease. Abnormal vaginal discharge was considered when a patient described a discharge with abnormal color or appearance, foul odor, itching, and burning that necessitated examination and treatment. Vaginal swabs were collected by gently rubbing the entire vaginal wall and posterior cervix using a 10 cm long cotton swab with sterile normal saline and a cotton swab provided by the commercial STD (sexually transmitted disease) polymerase chain reaction (PCR) and culture kit. The sampling was done in an independent space by one female gynecologist after confirming patients eligible for PID screening (Table I). Samples of normal control women were collected in the same way after confirming that they did not fit the criteria of PID. All samples were collected at room temperature. They were immediately transferred in a sterile normal saline container provided by the STD PCR kit and culture kit and then sent to the laboratory immediately for microbiome analysis.
Table I.
Clinical profiles of PID patients (n = 41) and women from control group (n = 33).
| Group | Age (years) (mean ± SD) |
Parity (mean ± SD) |
Body mass index (mean ± SD) |
Symptom | Number of women | % |
|---|---|---|---|---|---|---|
| PID patients | 35.5 ± 2.4 | 3.3 ± 0.2 | 29.5 ± 6.9 | Foul odor (Fish and rotten) | 25 | 61.0 |
| Itching and burning sensation | 11 | 26.8 | ||||
| Abnormal color of discharge | 5 | 12.2 | ||||
| Total | 41 | 100 | ||||
| Control group | 39.4 ± 3.2 | 3.1 ±0.2 | 30.1 ± 4.3 | No observable abnormality | 33 | 100 |
| Total | 33 | 100 |
SD – standard deviation
Microbe detection using STD multiplex real-time PCR and culture methods. Real-time PCR was performed to detect eight STD-related microorganisms: Mycoplasma hominis, Ureaplasma parvum, Ureaplasma urealyticum, Gardnerella vaginalis, C. trachomatis, N. gonorrhoeae, Trichomonas vaginalis, and Candida albicans. Nucleic acid was extracted from the vaginal fluid using a MagNA Pure 96 (Roche Diagnostics, Germany). Multiplex real-time PCR was performed on a CFX96 real-time PCR detection system (CFX96; Bio-Rad, USA) using the following PCR conditions: primary denaturation at 95°C for 15 minutes; 50 cycles of denaturation at 95°C for 30 seconds, annealing at 60°C for 1 minute, and extension at 72°C for 30 seconds; final cooling down at 55°C for 30 seconds. Then, STD diagnosis was performed as previously reported and interpreted as positive or negative (Kriesel et al. 2016). Microorganisms were also cultured for conventional identification using a sterile transport swab with agar gel (COPAN, Brescia, Italy). Only cultures with a growth of ≥ 1.5 × 103 colony-forming unit (CFU)/ml were considered positive.
PCR amplification of 16S rRNA genes. 1 ml of vaginal secretion diluted with saline solution was transferred in a Lysing Matrix B tube with 0.1-mm-diameter glass beads (MP Biomedicals, USA). Bead beating was carried out using a FastPrep-24 5G instrument (MP Biomedicals, USA) at a speed of 6.0 m/s for 30 seconds. The V4 region of the 16S rRNA gene was amplified using primers containing overhang sequences compatible with the Illumina Nextera XT index. The forward primer sequence was 515F (5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGC-CAGCMGCCGCGGTAA-3’), and the reverse was 806R (5’-GTCTCGTGGGCTCGGAGATGTGTATAAGA-GACAGGGACTACHVGGGTWTCTAAT-3’). Among the 515F and 806R primer sequences, the first 33 and 34 sequences are overhang sequences, and the sequences connected by dashes are V4 locus-specific sequences. Moreover, this primer set was designed to specifically amplify from 515 bp to 806 bp of the 16S rRNA gene. Accordingly, the expected size of the PCR product was confirmed by gel electrophoresis as 359 bp, and in the case of the template-free control, it was confirmed that the target sequence was not amplified after PCR. All PCR reactions were carried out using a 2X KAPA HiFi HotStart ReadyMix (Kapa Biosystems, USA). The final volume of each sample was 25 µl; 12.5 µl of 2X KAPA HiFi HotStart ReadyMix reagent, 5 µl of each primer at a concentration of 1 µM, and 2.5 µl (5 ng/µl) of template DNA. Reactions were run with the following cycling parameters: initial denaturation at 95°C for 3 min; 25 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s; and a final extension at 72°C for 5 min.
PCR product clean up. A PCR plate was centrifuged at 1,000 g for 1 minute at 20°C to collect condensation. AMPure XP beads (Beckman Coulter, High Wycombe, UK) were vortexed for 30 seconds, and 20 µl of them was added to each PCR plate well. The entire volume was gently pipetted up and down ten times, followed by incubation at room temperature for 5 minutes. Next, the PCR plate was placed on a magnetic stand for 2 minutes. With the amplicon PCR plate on the magnetic stand, supernatants were removed using a multichannel pipette, and beads were washed twice with freshly prepared 80% ethanol. Then the beads were air-dried for 10 minutes, the amplicon PCR plate was removed from the magnetic stand, and 52.5 µl of 10 mM Tris (pH 8.5) was added to each well of the amplicon PCR plate. After the mixture was gently pipetted up and down ten times, beads were fully resuspended. The plate was incubated at room temperature for 2 minutes and then placed on the magnetic stand for 2 minutes. Using a multichannel pipette, 50 µl of the supernatant was transferred from the amplicon PCR plate to a new 96-well PCR plate.
Index PCR using 16S amplicons for library construction. According to the manufacturer’s protocol, the amplicon library was prepared using Nextera XT DNA Library Prep Kit (Illumina, USA). First, 5 µl of DNA, Nextera XT Index Primer 1 (N7xx), Nextera XT Index Primer 2 (S5xx), 25 µl of 2X KAPA HiFi HotStart ReadyMix (KAPA Biosystems, USA), and 10 µl of PCR Grade water were mixed by pipetting. Then, a PCR was conducted using the following program: initial denaturation at 95°C for 30 s; 8 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s; and a final extension step at 72°C for 5 min. Lastly, each pool was cleaned.
Sequencing and data analysis. The sample was diluted from 1 nM to 50 pM with 10 mM Tris (pH 8.5). After adding a 10% PhiX Control library (Illumina, USA), the library was then loaded onto an iSeq-100 reagent cartridge (Illumina, USA) and sequenced on an iSeq-100 platform (Illumina, USA) to generate 300 bp paired-end reads. Sequencing data were analyzed using the EzBioCloud server (http://www.ezbiocloud.net). Raw read processing began with quality checking, and filtering of low-quality (< Q25) reads in the Trimmomatic (version. 0.32) (Bolger et al. 2014). After passing quality check (QC), paired-end sequence data were merged using the fastq_mergepairs command in VSEARCH (version 2.13.4) with default parameters (Rognes et al. 2016). The primers were then trimmed to a similarity cutoff of 0.8 using Myers and Miller’s alignment algorithm (Myers and Miller 1988). Amplicons not encoding 16S rRNA were detected by nhmmer in the HMMER software (package ver. 3.2.1) (Wheeler and Eddy 2013). After the unique reads were extracted, the redundant reads were clustered with the unique reads by VSEARCH’s derep_fulllength command (Rognes et al. 2016). EzBioCloud’s 16S rRNA database (Yoon et al. 2017) was used for a taxonomic assignment using VSEARCH’s userarch_global command (Rognes et al. 2016), followed by more precise pairwise alignment (Myers and Miller 1988). Next, chimeric reads were filtered from reads with < 97% similarity by reference-based chimera detection using the UCHIME algorithm (Edgar et al. 2011), and the EzBioCloud’s non-chimeric 16S rRNA database. After chimera filtering, reads not identified at the species level in the EzBioCloud database were compiled, then de-novo clustering was performed using the cluster_fast command (Rognes et al. 2016) to generate additional OTUs. Moreover, OTUs with single reads were excluded from further analysis. The 16S rRNA amplicon sequences were deposited in Sequence Read Archive (SRA) database with accession number (BioProject ID: PRJNA745060). The SRA is accessible at https://www.ncbi.nlm.nih.gov/bioproject/745060.
Metabolomics analysis. Vaginal samples were eluted from swabs using methanol and subjected to targeted liquid chromatography (LC) mass spectrometry (MS)-based metabolomics, as previously described (Yuan et al. 2012). Briefly, an LC-MS analysis was performed using Exion LC AD coupled with the X500B QTOF system (AB Sciex Pte. Ltd, USA). Samples were injected into an ACQUITY UPLC BEH HILIC column (2.1 × 50 mm, 1.7 µm). The mobile phase comprised phase A (water with 10 mM ammonium formate) and phase B (methanol). At the same time, to ensure the accuracy of mass detection, auto-calibration was performed every five injections.
Statistical analysis. Alpha diversity indices were analyzed based on ACE (Chao and Lee 1992), Chao1 (Chao 1987), Jackknife (Burnham and Overton 1979), Shannon/Simpson (Magurran 2013), NPShannon (Chao and Shen 2003), and Phylogenetic diversity (Faith 1992). Beta diversity distances based on Jenson-Shannon (Lin 1991), Bray-Curtis (Beals 1984), Generalized UniFrac (Chen et al. 2012), and Fast Uni-Frac (Hamady et al. 2010) were analyzed. Using functional profiles predicted by the PICRUSt (Ye and Doak 2009) and MinPath (Langille et al. 2013) algorithms, taxonomic and functional biomarkers were found by statistical comparison algorithms of the LEfSe (Segata et al. 2011) and Kruskal-Wallis H tests (Wallis 1952). The Student’s t-test was performed to evaluate the statistical significance in comparing metabolite concentrations between groups.
Results
STD multiplex real-time PCR and culture method for PID patients. By STD multiplex real-time PCR, 38 (92.7%) of the 41 subjects tested positive for at least one microorganism (%Table II). The most prevalent pathogens were U. urealyticum (24.4%, n = 10/41) and U. parvum (24.4%, n = 10/41), followed by M. hominis (22.0%, n = 9/41), C. trachomatis (14.6%, n = 6/41), N. gonorrhoeae (2.4%, n = 1/41), and T. vaginalis (2.4%, n = 1/41). In the culture method, Staphylococcus aureus, Streptococcus pyogens, Pseudomonas koreensis, C. albicans, Escherichia coli, and Pseudomonas putida were detected in 9 patients (21.9%, n = 9/41).
Table II.
Identification of microbes using STD multiplex PCR and culture method for PID patients.
| No. | STD PCR method | Culture method | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MH | UP | UU | GV | CT | NG | TV | CA | ||
| 1 | + | – | – | + | – | – | – | – | – |
| 2 | + | + | – | + | + | – | – | – | – |
| 3 | – | – | + | + | – | – | – | – | – |
| 4 | – | + | – | + | – | – | – | – | – |
| 5 | – | – | + | + | – | – | – | – | – |
| 6 | – | + | – | + | – | – | – | – | – |
| 7 | – | – | + | + | – | – | – | – | – |
| 8 | + | + | – | + | + | + | – | – | – |
| 9 | – | – | – | + | + | – | + | – | – |
| 10 | – | – | + | + | – | – | – | – | – |
| 11 | + | + | – | + | + | – | – | – | – |
| 12 | + | – | + | + | – | – | – | – | Staphylococcus aureus |
| 13 | – | – | – | – | – | – | – | – | Streptococcus pyogens |
| 14 | – | – | – | – | – | – | – | + | – |
| 15 | + | + | – | + | – | – | – | – | Pseudomonas koreensis |
| 16 | + | – | + | + | + | – | – | – | – |
| 17 | – | – | – | + | – | – | – | – | – |
| 18 | – | – | – | – | – | – | – | – | – |
| 19 | – | – | – | – | – | – | – | + | – |
| 20 | – | + | – | + | – | – | – | – | – |
| 21 | + | – | – | + | – | – | – | – | – |
| 22 | – | – | – | – | – | – | – | + | Candida albicans |
| 23 | – | – | – | + | – | – | – | – | Escherichia coli |
| 24 | – | – | + | + | – | – | – | – | – |
| 25 | – | + | – | + | + | – | – | – | – |
| 26 | – | – | – | + | – | – | – | – | – |
| 27 | – | – | – | + | – | – | – | – | – |
| 28 | – | – | – | – | – | – | – | + | Candida albicans |
| 29 | – | – | – | – | – | – | – | – | – |
| 30 | – | – | – | + | – | – | – | – | Pseudomonas putida |
| 31 | – | – | – | – | – | – | – | + | Candida albicans |
| 32 | – | – | + | + | – | – | – | – | – |
| 33 | + | + | – | + | – | – | – | – | – |
| 34 | – | – | – | + | – | – | – | – | Escherichia coli |
| 35 | – | – | + | + | – | – | – | – | – |
| 36 | – | – | – | + | – | – | – | – | – |
| 37 | – | – | – | + | – | – | – | – | – |
| 38 | – | – | – | + | – | – | – | – | – |
| 39 | – | + | – | + | – | – | – | – | – |
| 40 | – | – | – | + | – | – | – | – | – |
| 41 | – | – | + | + | – | – | – | – | – |
MH – Mycoplasma hominis, UP – Ureaplasma parvum, UU – Ureaplasma urealyticum,
GV – Gardnerella vaginalis, CT – Chlmamydia trachomatis, NG – Neisseria gonorrhea,
TV – Trichomonas vaginalis, CA – Candida albicans
Microbial diversity in vaginal metagenomes of normal women and PID patients. %Table III shows the average taxonomic compositions at the phylum, class, order, and family levels for the control group and the PID patient group. At the phylum level, Firmicutes accounted for the highest percentage, followed by Actinobacteria, Proteobacteria, Bacteroides, and Fusobacteria; the composition for each phylum was 79.4%, 13.0%, 4.6%, and 1.4% for the control group and 56.9%, 23.2%, 12.8%, 4.7%, and 2.4% for the PID patient group, respectively. Actinobacteria was significantly decreased in the PID patient group compared to the control group, while the other four Firmicutes phylum were significantly increased in the PID patient group compared to the control group. Bacilli, which correspond to Firmicutes phylum, accounted for the highest percentage at the class level, and it was significantly decreased in the PID patient group (53.7%) compared to the control group (77.7%). In addition, the percentage of Tissierellia of the same phylum was slightly higher in the PID patient group than in the control group; 1.0% and 1.5%, respectively. Other classes were slightly increased in the patient group compared to the control group. Lactobacillaceae occupied the highest percentage at the family level and was 27.9% less in the control group than in the PID patient group; 63.7% and 35.8%. Apart from the Bacilli and Lactobacillales, the percentage of all the other orders was slightly higher in the PID patient group than in the control group. Lactobacillaceae occupied the highest percentage at the family level and was 27.9% less in the control group than in the PID patient group; 63.7% and 35.8%, respectively. Streptococcaceae was decreased in the PID group compared to the control group; 2.6% and 0%, respectively. Contrary to the Lactobacillaceae, the percentage of Enterococcaceae in the same order was higher in the PID patient group than in the control group; 8.1% and 13.4%, respectively. For all the families apart from those in the Lactobacillales order, the percentages of the PID patient group were similar or higher than those of the control group except for the Moraxellaceae family.
Table III.
Distributions of bacterial community at different taxonomic levels (phylum, class, order, and family).
| Phylum | Con. | PID | Class | Con. | PID | Order | Con. | PID | Family | Con. | PID |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lactobacillaceae | 63.7 | 35.8 | |||||||||
| Lactobacillales | 74.5 | 50.2 | Enterococcaceae | 8.1 | 13.4 | ||||||
| Firmicutes | 79.4 | 56.9 | Bacilli | 77.7 | 53.7 | Streptococcaceae | 2.6 | 0 | |||
| Bacillales | 3.1 | 3.4 | Staphylococcaceae | 3.1 | 3.1 | ||||||
| Tissierellia | 1.0 | 1.5 | Tissierellales | 1.0 | 1.5 | Peptoniphilaceae | 1.0 | 1.5 | |||
| Actinobacteria | 13.0 | 23.2 | Actinobacteria | 10.7 | 16.7 | Bifidobacteriales | 9.8 | 15.8 | Bifidobacteriaceae | 9.8 | 15.8 |
| Coriobacteriia | 2.3 | 6.6 | Coriobacteriales | 2.3 | 6.6 | Coriobacteriaceae | 2.3 | 6.6 | |||
| Gammaproteobacteria | 4.1 | 11.0 | Pseudomonadales | 2.7 | 1.5 | Moraxellaceae | 2.4 | 0 | |||
| Proteobacteria | 4.6 | 12.8 | Enterobacterales | 1.4 | 9.4 | Enterobacteriaceae | 0 | 9.2 | |||
| Betaproteobacteria | 0 | 1.6 | Burkholderiales | 0 | 1.6 | Oxalobacteraceae | 0 | 1.3 | |||
| Bacteroidetes | 1.4 | 4.7 | Bacteroidia | 1.3 | 4.6 | Bacteroidales | 1.3 | 4.6 | Prevotellaceae | 1.2 | 3.7 |
| Fusobacteria | 1.2 | 2.4 | Fusobacteria | 1.2 | 2.4 | Fusobacteriales | 1.2 | 2.4 | Leptotrichiaceae | 1.2 | 2.2 |
Unit – %, those with less than 1% share are not included
The average taxonomic compositions at the genus level are shown in Fig. 1. In the control women group, Lactobacillus accounted for the highest percentage (61.0%), followed by Gardnerella (8.4%), Enterococcus (7.7%), Staphylococcus (3.0%), Lactobacillaceae_uc (2.7%), Streptococcus (2.6%), Acinetobacter (2.3%), Atopobium (2.2%), Prevotella (1.2%), Sneathia (1.2%), and Bifidobacterium (1.1%). In the PID patient group, Lactobacillus (34.9%), Lactobacillaceae_uc (0%), Acinetobacter (2.3%), and Staphylococcus (2.9%) were significantly decreased compared to the control group. On the other hand, Gardnerella (13.9%), Enterococcus (13.1%), Atopobium (6.0%), Prevotella (3.4%), Sneathia (2.2%), and Bifidobacterium (1.5%) were increased in the PID patient group compared to the control group. Specifically, Escherichia and Herbaspirillum were not found in the control group but were identified in PID patients; 9.7% and 1.3%, respectively. In addition, the sum of the proportions of genera with a distribution of less than 1% was 6.6% in the control group but significantly higher in the PID patient group and equal to 12.0%.
Fig. 1.

Genus-level vaginal microbiome compositions in women of control group and PID patients. Data for minor orders with a relative abundance < 1% are not shown.
Correlation between normal control women and PID patients. In the control group, microbial richness tended to be higher than in the PID patient group, but the difference was not statistically significant (Ace, p = 0.274; Chao1, p = 0.289; Jackknife, p = 0.267; No. of identified species, p = 0.299) (Fig. 2). As for microbial diversity, the Simpson index was significantly decreased in the PID patient group compared to the control group, but there was no significant difference in the NPShannon, Shannon, and phylogenetic diversity indices (NPShanon, p = 0.251; Shannon, p = 0.091; Simpson, p = 0.007; Phylogenetic diversity, p = 0.373) (Fig. 3). The difference in bacterial communities between the healthy women and PID patients was also analyzed using the principal coordinate analysis (PCoA) plots based on Jensen-Shannon divergence, Bray-Curtis, Generalized UniFrac, UniFrac and presented in two and three dimensions, respectively (Fig. 4A and 4B). The microbiome PCoA in the vagina shows that most vaginal samples from PID patients (light green/red) are placed in the lower right region of the ordination, whereas most vaginal samples from normal women (blue/purple) are placed in the upper left region. Beta set-significance analysis showed significant differences in genera or species between the PID patients and the control women group (Fig. 4C). This result indicates that PID characterizes women’s microbiome composition. Cluster analysis based on Unweighted Pair Group Method with Arithmetic means (UPGMA) hierarchical clustering analysis also showed that vaginal samples from PID patients and normal women were grouped separately (Fig. 5). Samples from the same group showed that they were usually clustered together.
Fig. 2.

Boxplot of Species richness indices.
A) Ace (p = 0.274), B) Chao1 (p = 0.289), C) Jackknife (p = 0.267) indices and D) The number of OTUs (p = 0.299) reflects the diversity of OTU in samples. Bars indicate the median, and the hinges represent the lower and upper quartiles. In panels (A-D), no statistically significant differences were observed between the normal control women and PID patients. The Wilcoxon rank-sum test was used to determine the species richness.
Fig. 3.

Boxplot of Species diversity indices.
NP Shannon (p = 0.251), Shannon (p = 0.091), Simpson (p = 0.007), Phylogenetic diversity (p = 0.373) reflect the diversity of OTU in samples. Bars indicate the median, and the hinges represent the lower and upper quartiles. The Wilcoxon rank-sum test was used to determine the diversity index. *p < 0.05; **p < 0.01
Fig. 4.
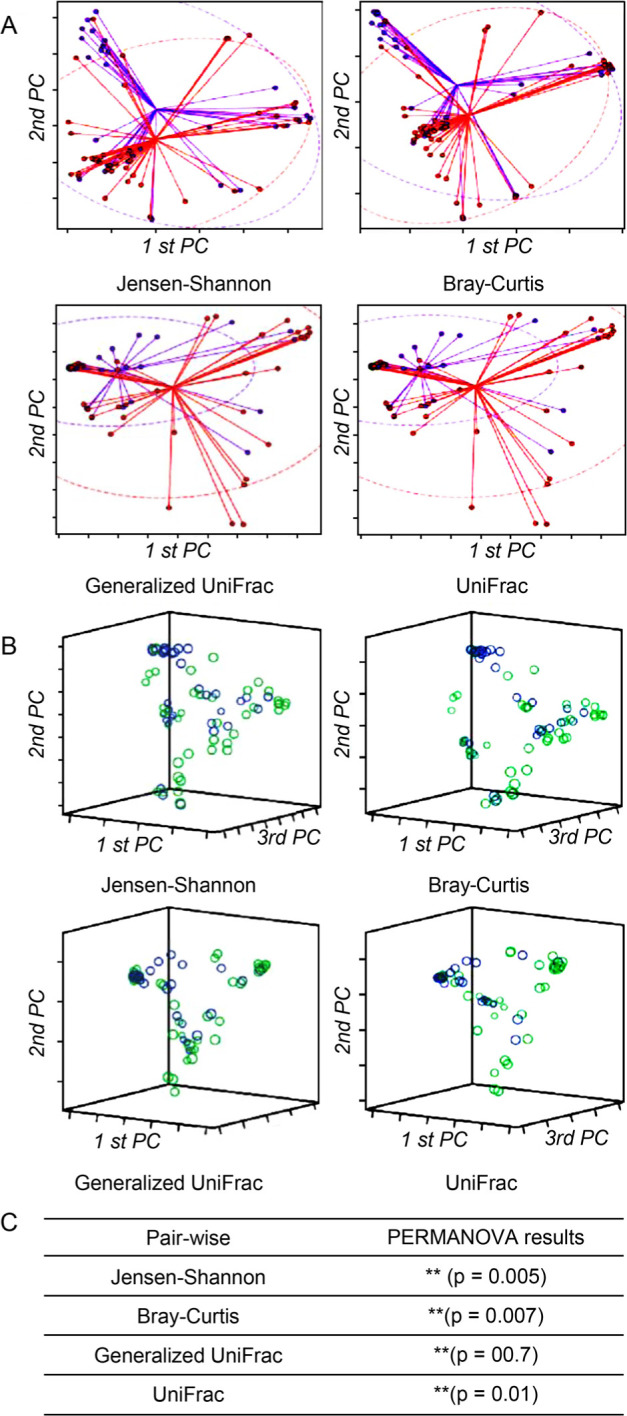
Beta diversity of microbial communities based on Jensen-Shannon divergence, Bray-Curtis, Generalized UniFrac, and UniFrac.
A) PCoA plots were produced as an ellipse in two dimensions based on a 95% confidence interval. B) PCoA plots were also presented in 3D. The blue/purple color indicates normal control women, and the light green/red color indicates PID patients, respectively. C) Permutational multivariate analysis of variance (PERMANOVA) results demonstrated the beta set-significance between the PID patient and normal control women groups. **p < 0.01.
Fig. 5.

Clustering using the Unweighted Pair Group Method with Arithmetic mean (UPGMA).
Taxonomic biomarker discovery. Based on Kruskal-Wallis H test results, there were significant differences in two phyla, two classes, three orders, six families, and 11 genera (%Table IV). Firmicutes phylum, Bacilli class, Lactobacillales order, Lactobacillaceae family, Moraxellaceae family, Lactobacillaceae_uc genus, Gardnerella genus, and Lactobacillus genus had a distribution of more than 1% in control and PID patient groups. The distributions of Firmicutes phylum (control, 79.38%; PID, 56.87%), Bacilli class (control, 77.74%; PID, 53.65%), Lactobacillales order (control, 74.52%; PID, 50.17%), Lactobacillaceae family (control, 63.70%; PID, 35.76%), Lactobacillaceae_uc genus (control, 2.67%; PID, 0.84%), Lactobacillus genus (control, 61.03%; PID, 34.92%), and Moraxellaceae family (control, 2.40%; PID, 0.68%) were decreased in the PID patient group compared to the control group. On the contrary, the distributions of Gardnerella genus (control, 8.36%; PID, 13.89%) were higher in the PID patient group than in the control group. The taxons with linear discriminant analysis (LDA) effect size exceeding five included Firmicutes (5.09916), Bacilli (5.12693), Lactobacillales (5.13602), Lactobacillaceae (5.15948), and Lactobacillus (5.12933) (Fig. 6). For the Moraxellaceae family (4.05126) and Gardnerella genus (4.54975), the LDA effect size was greater than 4 and less than 5.
Table IV.
The Kruskal-Wallis H tests and LEfSe analysis of the associations between normal control women and PID patients.
| Taxon rank | Taxon name | p-value | LDA effect size |
Control (%) |
PID patients (%) |
| Phylum | Firmicutes | 0.04364 | 5.09916 | 79.38 | 56.87 |
| Saccharibacteria_TM7 | 0.02254 | 3.25735 | 0.42 | 0.02 | |
| Class | Bacilli | 0.04089 | 5.12693 | 77.74 | 53.65 |
| Saccharimonas_c | 0.02254 | 3.21094 | 0.42 | 0.02 | |
| Lactobacillales | 0.04089 | 5.13602 | 74.52 | 50.17 | |
| Order | Saccharimonas_o | 0.02254 | 3.24345 | 0.42 | 0.02 |
| Propionibacteriales | 0.00628 | 2.90945 | 0.24 | 0.13 | |
| Lactobacillaceae | 0.01377 | 5.15948 | 63.70 | 35.76 | |
| Moraxellaceae | 0.04593 | 4.05126 | 2.40 | 0.68 | |
| Family | Pseudomonadaceae | 0.00272 | 3.56834 | 0.28 | 0.83 |
| Saccharimonas_f | 0.02254 | 3.23883 | 0.42 | 0.02 | |
| Propionibacteriaceae | 0.00628 | 2.91443 | 0.24 | 0.12 | |
| Yersiniaceae | 0.04997 | 2.84709 | 0.06 | 0.16 | |
| Lactobacillus | 0.02881 | 5.12933 | 61.03 | 34.92 | |
| Gardnerella | 0.02706 | 4.54975 | 8.36 | 13.89 | |
| Lactobacillaceae_uc | 0.00128 | 3.98329 | 2.67 | 0.84 | |
| Pseudomonas | 0.00272 | 3.54428 | 0.28 | 0.83 | |
| Parvimonas | 0.00445 | 3.31257 | 0.00 | 0.50 | |
| Genus | Enterobacteriaceae_uc | 0.01355 | 3.29397 | 0.15 | 0.50 |
| AF125206_g | 0.02254 | 3.25111 | 0.42 | 0.02 | |
| Megasphaera | 0.03362 | 3.04489 | 0.14 | 0.37 | |
| Yersinia | 0.04997 | 2.85166 | 0.06 | 0.16 | |
| Cutibacterium | 0.01311 | 2.85157 | 0.19 | 0.11 | |
| Actinotignum | 0.02287 | 2.50200 | 0.06 | 0.00 |
Only those with a p-value less than 0.05 were summarized
% refers to the percentage of distribution for each group
LDA – linear discriminant analysis
Fig. 6.

Taxonomic abundance with an LDA effect size of more than 5.
Vaginal organic acids by metabolomics analysis.Fig. 6 shows the quantitative analysis results for organic acids. In the control group, lactate concentration was the highest; 98.3% of the total organic acid content was analyzed, followed by pyruvate, 4-hydroxyphenylacetate, 2-hydroxylsovalerat, succinate, benzoate, isovalerate, butyrate, and malonate. In particular, the lactate concentration in the PID patient group was 93.5%, which was significantly reduced compared to the control group (p = 0.04), while pyruvate, isovalerate, and malonate were slightly decreased in the PID patient group compared to the control group. 4-hydroxyphenylacetate (p = 0.0063) and 2-hydroxylsovalerate were significantly increased (p = 0.0015) in the PID patient group compared to the control group, while succinate, benzoate, and butyrate were slightly increased in the PID patient group compared to the control group.
Discussion
Sequencing-based metagenomics research on the human microbiome has seen a marked increase since the launch of the Human Microbiome Project by the US National Institute of Health in 2008 (Turnbaugh et al. 2007). Metagenomics is now a widely used technique that has revolutionized the study of the microbiota owing to its ability to generate a comprehensive catalog of microbial sequences in a wide range of ecological niches within large hosts such as humans (Martin et al. 2014). Its application has been reported by numerous published scientific studies (Callahan et al. 2019) and meta-analyses (White et al. 2011).
Fig. 7.
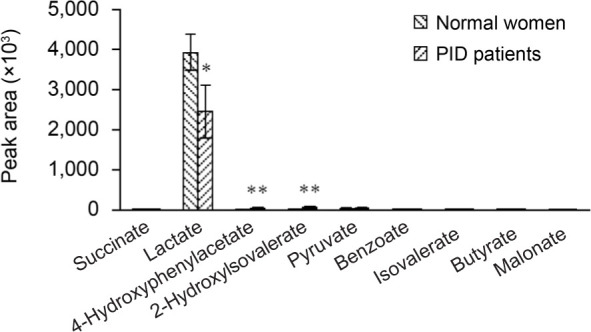
The relative level of organic acids in normal control women and PID patients. In the PID patient group, lactate was significantly decreased compared to the control group, whereas the concentrations of 4-hydroxyphenylacetate and 2-hydroxylsovalerate were significantly increased. *p < 0.05; **p < 0.01.
Since genital tract microbiota plays a crucial role in maintaining health, profiling the entire vaginal microbiota in disease states is essential for diagnosis (Ravel et al. 2011). However, conventional diagnostic methods can detect only a few specific pathogens and analyze a small fraction of the entire microbial community (Li et al. 2019). In our present study, there were cases in which patients with clinically diagnosed PID did not match the positive results of certain sexually transmitted microorganisms. In addition, PID management is a challenge since some treatments exhibit their therapeutic effects by inhibiting these specific pathogens, therefore not being suitable for the treatment of the underlying cause of PID (Taylor et al. 2011). To overcome PID diagnosis and treatment challenges, we performed a vaginal 16S rRNA amplicon sequencing analysis for the whole vaginal microbial community in PID patients and normal control women.
Our beta diversity analysis results showed a statistically significant difference between the control and the PID patient groups. This difference could be attributed to many Lactobacillus in the control group and significantly fewer Lactobacillus in the PID patient group. Lactobacillus dominates and maintains vaginal health through several mechanisms, including inhibiting pathogens, and it is believed that such a significant decrease in Lactobacillus leads to opportunistic infections and overgrowth of various pathogens (Witkin and Linhares 2017). This theory explains the difference in species diversity in the present study, consequently resulting in a polymicrobial infection. Our findings are consistent with a previous report of varying species diversity between healthy women and PID patients due to infection by different pathogens in PID patients (Sharma et al. 2014).
In the control group, Lactobacillus dominated, but the species richness showed a similar trend to that of the PID group. This result probably occurred because Lactobacillus, which has antibacterial activity, is dominant, and various pathogenic microorganisms are inhibited and colonized at a very low density. Therefore, if the balance of the vaginal microbiome is disrupted by prolonged antibiotic treatment, Lactobacillus decreases, leading to pathogen overgrowth, consequently causing disease (Eade et al. 2012). This phenomenon highlights the challenges of PID antibiotic therapy. Conventional antibiotics that inhibit only specific microorganisms destroy normal microbial communities, resulting in other diseases (Larsen and Monif 2001). The Lactobacillus treatment has been recently proposed as an alternative to conventional antibiotics; they may restore the disrupted vaginal flora. This principle is consistent with our present study analysis results (Reid et al. 2001).
In the vagina, Lactobacillus produces lactic acid, an essential antibacterial substance, and maintains a microbiota while suppressing microorganisms that cause dysbiosis (Tachedjian et al. 2017). In the present study, we confirmed that lactic acid constituted a very high proportion of the organic acids in the control group, explaining the low number of pathogens in the control group compared to that of the PID patient group. Based on these findings, it is suggested that Lactobacillus and lactic acid concentration could be essential factors in diagnosing and treating PID.
There is a strong positive correlation between PID and STD-related pathogens, and these pathogens play a vital role in the balance of the microbiome in the vagina (Loeper et al. 2018). The vaginal microbiome is highly associated with vaginal infections other than PID, including bacterial vaginosis, vulvar candidiasis, sexually transmitted diseases, and human immunodeficiency virus (HIV) infection (van de Wijgert 2017; Eastment and McClelland 2018). Furthermore, changes in the vaginal microbiota can lead to severe gynecological problems such as pregnancy loss, preterm labor, and low conception rates (Bracewell-Milnes et al. 2018). The present study’s findings will help understand PID from the microbiome perspective and are expected to contribute to the development of more efficient PID diagnosis and treatment modalities. In addition, these findings are expected to expand the understanding of a wide range of gynecological diseases. However, considering the limitation that only the distribution analysis of vaginal bacteria was performed in this study, additional studies on fungi and viruses should be conducted to understand more relationships between the vaginal ecosystem and PID. Understanding the distribution of microorganisms, metabolites, gene expression analysis, and their overall correlation is also required.
Acknowledgments
This research was supported financially by the Ministry of Trade, Industry, and Energy (MOTIE), Korea, under the “Regional industry-based organization support program” (reference number P0001942) supervised by the Korea Institute for Advancement of Technology (KIAT). In addition, this study was also supported by Soonchunhyang University Research Fund.
Footnotes
Conflict of interest
The authors do not report any financial or personal connections with other persons or organizations, which might negatively affect the contents of this publication and/or claim authorship rights to this publication.
Literature
- Beals EW. Bray-curtis ordination – an effective strategy for analysis of multivariate ecological data. Adv Ecol Res. 1984;14:1–55. 10.1016/S0065-2504(08)60168-3 [DOI] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014August01; 30(15): 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracewell-Milnes T, Saso S, Nikolaou D, Norman-Taylor J, Johnson M, Thum MY. Investigating the effect of an abnormal cervico-vaginal and endometrial microbiome on assisted reproductive technologies: A systematic review. Am J Reprod Immunol. 2018November;80(5):e13037. 10.1111/aji.13037 [DOI] [PubMed] [Google Scholar]
- Burnham KP, Overton WS. Robust estimation of population-size when capture probabilities vary among animals. Ecology. 1979October;60(5):927–936. 10.2307/1936861 [DOI] [Google Scholar]
- Callahan BJ, Wong J, Heiner C, Oh S, Theriot CM, Gulati AS, McGill SK, Dougherty MK. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019October10;47(18):e103. 10.1093/nar/gkz569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A, Lee SM. Estimating the number of classes via sample coverage. J Am Stat Assoc. 1992March;87(417):210–217. 10.1080/01621459.1992.10475194 [DOI] [Google Scholar]
- Chao A, Shen TJ. Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environ Ecol Stat. 2003;10(4):429–443. 10.1023/A:1026096204727 [DOI] [Google Scholar]
- Chao A. Estimating the population size for capture-recapture data with unequal catchability. Biometrics. 1987December;43(4):783–791. 10.2307/2531532 [DOI] [PubMed] [Google Scholar]
- Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD, Collman RG, Bushman FD, Li H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012August15;28(16):2106–2113. 10.1093/bioinformatics/bts342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HW, Koo YJ, Min KJ, Hong JH, Lee JK. Pelvic inflammatory disease in virgin women with tubo-ovarian abscess: A single-center experience and literature review. J Pediatr Adolesc Gynecol. 2017April;30(2):203–208. 10.1016/j.jpag.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Crossman SH. The challenge of pelvic inflammatory disease. Am Fam Physician. 2006March1;73(5):859–864. [PubMed] [Google Scholar]
- Curry A, Williams T, Penny ML. Pelvic inflammatory disease: Diagnosis, management, and prevention. Am Fam Physician. 2019September15;100(6):357–364. [PubMed] [Google Scholar]
- Eade CR, Diaz C, Wood MP, Anastos K, Patterson BK, Gupta P, Cole AL, Cole AM. Identification and characterization of bacterial vaginosis-associated pathogens using a comprehensive cervical-vaginal epithelial coculture assay. PLoS One. 2012November15;7(11):e50106. 10.1371/journal.pone.0050106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastment MC, McClelland RS. Vaginal microbiota and susceptibility to HIV. AIDS. 2018March27;32(6):687–698. 10.1097/QAD.0000000000001768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011August15;27(16):2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61(1):1–10. 10.1016/0006-3207(92)91201-3 [DOI] [Google Scholar]
- Fettweis JM, Brooks JP, Serrano MG, Sheth NU, Girerd PH, Edwards DJ, Strauss JF, Jefferson KK, Buck GA. The Vaginal Microbiome Consortium. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology. 2014October01;160(10):2272–2282. 10.1099/mic.0.081034-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettweis JM, Serrano MG, Sheth NU, Mayer CM, Glascock AL, Brooks JP, Jefferson KK, Buck GA. Vaginal Microbiome Consortium (additional members). Species-level classification of the vaginal microbiome. BMC Genomics. 2012December;13(S8) Suppl 8:S17. 10.1186/1471-2164-13-S8-S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradison M. Pelvic inflammatory disease. Am Fam Physician. 2012April15;85(8):791–796. [PubMed] [Google Scholar]
- Graspeuntner S, Bohlmann MK, Gillmann K, Speer R, Kuenzel S, Mark H, Hoellen F, Lettau R, Griesinger G, König IR, et al. Microbiota-based analysis reveals specific bacterial traits and a novel strategy for the diagnosis of infectious infertility. PLoS One. 2018January9; 13(1):e0191047. 10.1371/journal.pone.0191047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty CL, Totten PA, Tang G, Astete SG, Ferris MJ, Norori J, Bass DC, Martin DH, Taylor BD, Ness RB. Identification of novel microbes associated with pelvic inflammatory disease and infertility. Sex Transm Infect. 2016September;92(6):441–446. 10.1136/sextrans-2015-052285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010January;4(1):17–27. 10.1038/ismej.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings LK, Krywko DM. Pelvic inflammatory disease [Internet]. Treasure Island (USA): Statpearls; 2020[cited 2021 Jun 10]. Available from https://www.ncbi.nlm.nih.gov/books/NBK499959 [PubMed] [Google Scholar]
- Kriesel JD, Bhatia AS, Barrus C, Vaughn M, Gardner J, Crisp RJ. Multiplex PCR testing for nine different sexually transmitted infections. Int J STD AIDS. 2016December;27(14):1275–1282. 10.1177/0956462415615775 [DOI] [PubMed] [Google Scholar]
- Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952December;47(260):583–621. 10.1080/01621459.1952.10483441 [DOI] [Google Scholar]
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013September;31(9):814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen B, Monif GRG. Understanding the bacterial flora of the female genital tract. Clin Infect Dis. 2001February15;32(4):e69–e77. 10.1086/318710 [DOI] [PubMed] [Google Scholar]
- Li T, Liu ZH, Li K, Bai HH. Evaluation of the vaginal microbiome in clinical diagnosis and management of vaginal infectious diseases. Chin Med J (Engl). 2019May5;132(9):1100–1103. 10.1097/CM9.0000000000000211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. Divergence measures based on the Shannon entropy. IEEE Trans Inf Theory. 1991January;37(1):145–151. 10.1109/18.61115 [DOI] [Google Scholar]
- Loeper N, Graspeuntner S, Rupp J. Microbiota changes impact on sexually transmitted infections and the development of pelvic inflammatory disease. Microbes Infect. 2018October;20(9-10):505–511. 10.1016/j.micinf.2018.02.003 [DOI] [PubMed] [Google Scholar]
- MacIntyre DA, Chandiramani M, Lee YS, Kindinger L, Smith A, Angelopoulos N, Lehne B, Arulkumaran S, Brown R, Teoh TG, et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci Rep. 2015August;5(1):8988. 10.1038/srep08988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magurran AE. Measuring biological diversity. Hoboken (USA): Wiley-Blackwell; 2013. [Google Scholar]
- Martín R, Miquel S, Langella P, Bermúdez-Humarán LG. The role of metagenomics in understanding the human microbiome in health and disease. Virulence. 2014April;5(3):413–423. 10.4161/viru.27864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Miller W. Optimal alignments in linear space. Comput Appl Biosci. 1988March;4(1):11–17. 10.1093/bioinformatics/4.1.11 [DOI] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. 2011March15;108(Supplement 1):4680–4687. 10.1073/pnas.1002611107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reekie J, Donovan B, Guy R, Hocking JS, Kaldor JM, Mak DB, Pearson S, Preen D, Stewart L, Ward J, et al. Chlamydia and Reproductive Health Outcome Investigators; Chlamydia and Reproductive Health Outcome Investigators. Risk of pelvic inflammatory disease in relation to chlamydia and gonorrhea testing, repeat testing, and positivity: A population-based cohort study. Clin Infect Dis. 2018January18;66(3):437–443. 10.1093/cid/cix769 [DOI] [PubMed] [Google Scholar]
- Reid G, Beuerman D, Heinemann C, Bruce AW. Probiotic Lactobacillus dose required to restore and maintain a normal vaginal flora. FEMS Immunol Med Microbiol. 2001December;32(1):37–41. 10.1111/j.1574-695X.2001.tb00531.x [DOI] [PubMed] [Google Scholar]
- Revzin MV, Mathur M, Dave HB, Macer ML, Spektor M. Pelvic inflammatory disease: multimodality imaging approach with clinical-pathologic correlation. Radiographics. 2016September;36(5):1579–1596. 10.1148/rg.2016150202 [DOI] [PubMed] [Google Scholar]
- Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016October18;4: e2584. 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma H, Tal R, Clark N, Segars J. Microbiota and pelvic inflammatory disease. Semin Reprod Med. 2014January3;32(01):043–049. 10.1055/s-0033-1361822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Hoffman NG, Morgan MT, Matsen FA, Fiedler TL, Hall RW, Ross FJ, McCoy CO, Bumgarner R, Marrazzo JM, et al. Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS One. 2012June18;7(6):e37818. 10.1371/journal.pone.0037818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachedjian G, Aldunate M, Bradshaw CS, Cone RA. The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res Microbiol. 2017November;168(9-10):782–792. 10.1016/j.resmic.2017.04.001 [DOI] [PubMed] [Google Scholar]
- Taylor BD, Ness RB, Darville T, Haggerty CL. Microbial correlates of delayed care for pelvic inflammatory disease. Sex Transm Dis. 2011May;38(5):434–438. 10.1097/OLQ.0b013e3181ffa7c7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007October;449 (7164): 804–810. 10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wijgert JHHM. The vaginal microbiome and sexually transmitted infections are interlinked: consequences for treatment and prevention. PLoS Med. 2017December27;14(12):e1002478. 10.1371/journal.pmed.1002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen S, Kalliala I, Nieminen P, Salonen A. Comparative analysis of vaginal microbiota sampling using 16S rRNA gene analysis. PLoS One. 2017July19;12(7):e0181477. 10.1371/journal.pone.0181477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CK, Wiesenfeld HC. Antibiotic therapy for acute pelvic inflammatory disease: the 2006 Centers for Disease Control and Prevention sexually transmitted diseases treatment guidelines. Clin Infect Dis. 2007April01;44(Supplement_3):S111–S122. 10.1086/511424 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Zhang Q, Chen H, Feng Y. Characterization of pelvic and cervical microbiotas from patients with pelvic inflammatory disease. J Med Microbiol. 2018October01;67(10):1519–1526. 10.1099/jmm.0.000821 [DOI] [PubMed] [Google Scholar]
- Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013October01;29(19):2487–2489. 10.1093/bioinformatics/btt403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BA, Creedon DJ, Nelson KE, Wilson BA. The vaginal microbiome in health and disease. Trends Endocrinol Metab. 2011October; 22(10):389–393. 10.1016/j.tem.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin SS, Linhares IM. Why do lactobacilli dominate the human vaginal microbiota? BJOG. 2017March;124(4):606–611. 10.1111/1471-0528.14390 [DOI] [PubMed] [Google Scholar]
- Ye Y, Doak TG. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLOS Comput Biol. 2009August14;5(8):e1000465. 10.1371/journal.pcbi.1000465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol. 2017May01;67(5):1613–1617. 10.1099/ijsem.0.001755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012May;7(5):872–881. 10.1038/nprot.2012.024 [DOI] [PMC free article] [PubMed] [Google Scholar]