

Abstract
Toll‐like receptors (TLRs) are a class of membrane‐spanning proteins of host cells. TLR2 and TLR4 are displayed on the surface of macrophages, neutrophils and dendritic cells and recognise structurally conserved microbial signatures defined as Pathogen associated molecular patterns (PAMPs). C3H mice are susceptible to tick‐borne pathogens; Lyme disease causing Borrelia burgdorferi that manifests arthritis and carditis and Apicomplexan protozoan, Babesia microti (Bm) that causes significant parasitemia associated with erythrocytopenia and haemoglobinuria. B. burgdorferi lacks typical TLR4 ligand lipopolysaccharides (LPS) and Bm TLR ligand(s) remain unknown. Only Borrelia lipoproteins that signal through TLR2 are established as PAMPs of these pathogens for TLR2/TLR4. Infection of C3H mice with each pathogen individually resulted in increase in the percentage of splenic B, T and FcR+ cells while their co‐infection significantly diminished levels of these cells and caused increased B. burgdorferi burden in the specific organs. The most pronounced inflammatory arthritis was observed in co‐infected C3H/HeJ mice. Parasitemia levels and kinetics of resolution of Bm in both mice strains were not significantly different. Transfected HEK293 cells showed pronounced signalling by B. burgdorferi through TLR2 and to some extent by TLR4 while Bm and infected erythrocytes did not show any response confirming our results in mice.
Keywords: Babesia microti, Babesiosis, Borrelia burgdorferi, co‐infection, inflammation, Lyme disease, toll like receptor 4
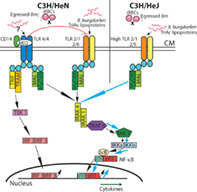
TLR4 stimulation in C3H/HeN mice by B. burgdorferi by unknown (indicated by a question mark) ligand(s) early in infection causes suppression of TLR2 expression. An increase in TLR2 expression in C3H/HeJ mice in the absence of a functional TLR4 possibly enhances signaling by Tri‐Acylated lipoproteins of colonized B. burgdorferi resulting in an increase in inflammatory Lyme arthritis, particularly during co‐infection that is associated with higher lymphocytes infiltration and hyperplasia of synovium in B. burgdorferi infected, and co‐infected C3H/HeJ mice. In vitro TLR2 signaling by Lyme spirochetes, and the lack of signaling through TLR2/TLR4 by egressed Bm or iRBCs (marked by a cross) support this model.
Abbreviations
- ACK
Ammonium‐Chloride–Potassium
- BSKII
Barbour‐Stoenner‐Kelly‐II
- GPI
glycosylphosphatidylinositol
- i.p.
intraperitoneal
- IACUC
Institutional Animal Care and Use Committee
- IVIS
in vivo imaging system
- NK
natural killer
- PAMP
pathogen associated molecular patterns
- PRRs
pattern recognition receptors
- qPCR
real‐time quantitative polymerase chain reaction
- RBCs
red blood cells
- RS
rabbit serum
- s.c.
subcutaneous
- SCID
severe combined immunodeficient
- TLR
toll like receptor; CDC, Centers for Disease Control and Prevention; FAM, Fluorescein amidite; TET, Tetrachlorofluorescein; EDTA, Ethylenediaminetetraacetic acid; FACS, Fluorescence‐activated cell sorting; ELISA, Enzyme‐linked immunosorbent assay
1.
Take Away.
Live imaging allows detection of Lyme spirochetes colonization in different organs at different stages of infection of mice.
Exacerbation of TLR2 signaling by B. burgdorferi and inflammation occurs during co‐infection with B. microti in TLR4‐defective host.
B. microti PAMPs including its GPI anchor on merozoites surface fail to signal host cells through TLR2/TLR4.
2. INTRODUCTION
Lyme disease is the most prevalent tick‐borne infection in Europe and North America. The CDC estimates that 300,000 to >400,000 cases of Lyme disease and ~2,000 cases of human babesiosisk occur in the USA annually and Lyme spirochetes infect ~65,000 people in Europe every year (Fallahi, Elia, & Bonatti, 2017; Kugeler, Schwartz, Delorey, Mead, & Hinckley, 2021). Borrelia burgdorferi infection could result in systemic Lyme disease that affects skin, musculoskeletal system, heart, joints and neuronal system (Steere, 2001). Babesia microti (Bm) and B. divergens are the major cause of babesiosis in the USA and Europe, respectively. Babesia species are intra‐erythrocytic tick‐borne Apicomplexan protozoan parasites that cause increased lysis of red blood cells that is associated with anaemia, jaundice and splenomegaly (Homer, Aguilar‐Delfin, Telford 3rd, Krause, & Persing, 2000). Healthy individuals infected with Bm remain asymptomatic such that transfusion of donated blood of these people to immunocompromised recipients often leads to transfusion‐associated babesiosis and results in high morbidity and mortality (Herwaldt et al., 2011; Krause et al., 1998; Krause et al., 2008).
Co‐infections of Ixodes species ticks with B. burgdorferi and Bm have been increasing steadily over the years (Hersh et al., 2014; Lommano, Bertaiola, Dupasquier, & Gern, 2012; Moutailler et al., 2016; Piesman et al., 1986; Schulze, Jordan, Healy, & Roegner, 2013). The same reservoir host(s) and tick vector feeding habits determine the spread of these pathogens to humans (Swanson, Neitzel, Reed, & Belongia, 2006). Although co‐infections with various tick‐borne pathogens have also been increasing in humans in regions of North America and Europe (Aliota et al., 2014; Caulfield & Pritt, 2015; Hersh et al., 2014; Horowitz et al., 2013; Jahfari et al., 2016; Moutailler et al., 2016; Panczuk, Tokarska‐Rodak, Koziol‐Montewka, & Plewik, 2016; Primus et al., 2018; Schlachter, Chan, Marras, & Parveen, 2017; Tomasiewicz, Chmielewska‐Badora, Zwolinski, & Murias‐Brylowska, 2016), the most common co‐infection in the Northeastern United States occur with B. burgdorferi and Bm, accounting for ~80% of tick‐borne concurrent infections by these pathogens (Swanson et al., 2006). Co‐infections with Babesia species and Lyme disease causing B. burgdorferi sensu lato group of spirochetes are also emerging as a serious problem in Europe in addition to the United States (Diuk‐Wasser, Vannier, & Krause, 2016; Dunn et al., 2014; Knapp & Rice, 2015; Rizzoli et al., 2014). Epidemiological studies demonstrate that Bm‐B. burgdorferi co‐infected patients exhibit persistence of more diverse and intense symptoms (Krause et al., 1996; Krause et al., 2002; Krause et al., 2003). Therefore, a comprehensive evaluation of the effect of co‐infections on overall disease severity affected by host factors remains a great urgency.
Early murine model studies showed contradictory outcomes of Bm‐B. burgdorferi co‐infections as relevant to severity of Lyme disease (Coleman, LeVine, Thill, Kuhlow, & Benach, 2005; Moro et al., 2002). Susceptibility of the host and pathogenicity of infection depends on virulence factors of microbes and the genetic background of the host with respect to the modulating effect of the immune system (Benoit et al., 2010; Gronberg‐Hernandez, Sunden, Connolly, Svanborg, & Wullt, 2011; Wells et al., 2003). To address the effect of selected mouse strain on infection with tick‐borne pathogens (Barthold, Beck, Hansen, Terwilliger, & Moody, 1990; Borggraefe et al., 2006; Coleman et al., 2005; Vannier et al., 2004), we were prompted to use two different derivatives of C3H mice to investigate the pathogenesis of B. burgdorferi and Bm. Similar to other pathogens, infection of the host by Bm and B. burgdorferi triggers an early innate immune response that is usually initiated by the recognition of pathogen‐associated molecular patterns (PAMPs) through responsive pattern recognition receptors (PRRs) in host cells that provides the first line of defence against invading pathogens (Baravalle et al., 2010; Bose, 1994; W. C. Brown, Norimine, Knowles, & Goff, 2006; Bulut, Faure, Thomas, Equils, & Arditi, 2001; Cervantes et al., 2011; Love, Schwartz, & Petzke, 2014; Oosting et al., 2010; Rahman, Shering, Ogden, Lindsay, & Badawi, 2016; Suarez et al., 2011; Werling & Jungi, 2003; Wooten et al., 2002a, 2002b). PAMPs‐mediated signalling through PRRs is crucial for many aspects of microbial pathogenesis such as recruitment of inflammatory cells to the infected tissue, expression of pro‐inflammatory mediators, modulation of innate and adaptive immunity and eventual clearance of infecting organisms. Excessive activation of PRRs is harmful with a significant pathology caused by systemic or localised inflammation in hosts (Heine & Lien, 2003; Takeuchi & Akira, 2010) and can lead to chronic disease or death.
Mating of female BALB and male DBA mice resulted in the development of the current inbred C3H strain. The C3H strain was introduced to the Charles River and Jackson Laboratory in the 1970s. Expression of PRRs may differ across host genetic background causing varying degrees of pathogenicity by the infecting microbe to affect disease severity (Barthold et al., 1990; Borggraefe et al., 2006). A classic case of differing expression of Toll Like Receptor 4 (TLR4) in the same genetic background is the C3H/HeN mice that retain a functional TLR4 receptor, and the C3H/HeJ mice in which a spontaneous point mutation in the TLR4 gene Tlr4 Lps‐d rendered it dysfunctional (Totemeyer, Foster, Kaiser, Maskell, & Bryant, 2003). In the United States, the suppliers Charles River and Jackson Laboratory offer either C3H/HeN or C3H/HeJ strains but not both. Both C3H/HeN and C3H/HeJ mice are susceptible to infection with B. burgdorferi, and manifest severe lesions like polyarthritis and carditis (Armstrong, Barthold, Persing, & Beck, 1992; Barthold, 1991; G. Wang et al., 2001). These strains of mice are also susceptible to Bm and exhibit pronounced parasitemia, anaemia and splenomegaly similar to that observed in humans (Coleman et al., 2005; Djokic et al., 2019; 2018). The most established PAMP recognised by TLR4 receptor is lipopolysaccharide (LPS), a component of the outer membrane of Gram‐negative bacteria. TLR4/myeloid differentiation factor 2 (MD‐2) complex recognises LPS to induce an innate immune response (Chow, Young, Golenbock, Christ, & Gusovsky, 1999; Nagai et al., 2002; Schromm et al., 2001; Shimazu et al., 1999; Y. Wang et al., 2016). Therefore, it is not surprising that the C3H/HeJ mice are more susceptible to infections with several Gram‐negative bacteria such as E. coli and S. typhimurium (Hagberg et al., 1984; Hagberg, Briles, & Eden, 1985). B. burgdorferi outer membrane lacks classical LPS, and hence does not typically stimulate a typical TLR4 mediated immune response (Takayama, Rothenberg, & Barbour, 1987); however, B. burgdorferi expresses a large number of lipoproteins that contribute to the major inflammatory pathway by activating monocytes/macrophages, neutrophils, dendritic cells, lymphocytes, endothelial cells and fibroblasts through TLR2 signalling (Cervantes et al., 2011; Gautam et al., 2012; Lasky, Pratt, Hilliard, Jones, & Brown, 2016; Lien et al., 1999; Liu, Montgomery, Barthold, & Bockenstedt, 2004; Lorenz, Mira, Cornish, Arbour, & Schwartz, 2000; Morr, Takeuchi, Akira, Simon, & Muhlradt, 2002; Rupprecht et al., 2007; Salazar et al., 2005; Salazar et al., 2009; Wooten et al., 2002a; Wooten & Weis, 2001).
Glycosylphosphatidylinositol (GPI) anchor on merozoites surface of a Bm related Apicomplexan parasite belonging to Plasmodium species have been reported to signal through both TLR2 and TLR4 that depends on GPI composition and could contribute to severe, cerebral and placental malaria (Gowda, 2007; Mockenhaupt et al., 2006a, 2006b; Seixas, Moura Nunes, Matos, & Coutinho, 2009). Only limited information is available for GPI anchored merozoite proteins of Babesia species and was reported not to induce stimulation of pro‐inflammatory cytokines (Debierre‐Grockiego et al., 2019; Delbecq et al., 2002; Nathaly Wieser, Schnittger, Florin‐Christensen, Delbecq, & Schetters, 2019). The role of TLR4 in other protozoan parasites' pathogenesis (Campos et al., 2001; Debierre‐Grockiego et al., 2007) led us to hypothesise that Bm's GPI anchors can potentially cause TLR4 and/or TLR2 stimulation. Unlike Plasmodium species and other protozoa, GPI composition in Bm proteins is not yet described, such that contribution of TLR2 and TLR4 for Bm remains to be investigated.
Our extensive analysis of the splenic immune response in young and older C3H/HeJ mice infected with Bm, B. burgdorferi or co‐infected with both pathogens at early (pre‐peak parasitemia) stage of infection suggested that stimulation of innate immune response by B. burgdorferi likely causes reduction in peak parasitemia in the co‐infected mice (Djokic, Primus, Akoolo, Chakraborti, & Parveen, 2018). An obvious question arose whether Bm and B. burgdorferi infections separately, or together in non‐functional TLR4 containing C3H/HeJ versus conventional TLR4 possessing C3H/HeN mice show different host responses and respective disease manifestations. Following infection with Bm, the first line of defence is mediated by macrophages, producing TNF‐α, and by NK cells reactive oxygen species and nitric oxide, and IFN‐γ (Homer et al., 2000). Control of Bm infections at early and later stage is facilitated by activation of IFN‐γ producing CD4 T cells that enhances killing of Babesia species by macrophages (Djokic, Primus, et al., 2018; Igarashi et al., 1999; Skariah et al., 2017). Co‐infection with B. burgdorferi and Bm results in exacerbation of Lyme disease manifestations in patients (Krause et al., 1996) but host factors involved in this enhancement in humans have not been assessed until now. In this study, we investigated the impact of signalling in the presence, or absence of functional TLR4 on the pathogenesis of B. burgdorferi and Bm infections individually, and during co‐infections in C3H mice. We further confirmed these results and determined contributions of TLR2 and TLR4 signalling by each pathogen individually using the transfected human embryonic kidney (HEK) 293 cells in vitro.
3. RESULTS
3.1. Bm parasitemia levels are not significantly affected by TLR4 functional attenuation in C3H mice
To determine the effect of the dysfunctional TLR4 on infectivity, we monitored Bm parasitemia in C3H/HeJ and C3H/HeN mice infected singly or co‐infected with B. burgdorferi. The onset of parasitemia appeared earlier in the C3H/HeN (seventh day) than in C3H/HeJ mice (10th day) with net gain in Bm parasitemia. Due to similar kinetics of replication, peak parasitemia for C3H/HeN mice was on day 12 and for C3H/HeJ mice 1 day later, that is, day 13, post‐infection and the clearance of iRBCs by immune system was also slightly faster in C3H/HeN mice. In addition, parasitemia in C3H/HeJ mice, unlike C3H/HeN mice, remained detectable by microscopic examination until 20th day in Bm‐infected mice as well as those co‐infected with B. burgdorferi (Figure 1). The peak parasitemia levels between these two sets of mice were comparable when they were either infected by Bm alone (43.2 ± 4.9% in HeJ vs. 40.2 ± 2.4% in HeN mice) or together with B. burgdorferi (36.0 ± 3.7% in HeJ vs. 33.5 ± 5.3% in HeN mice); however, it is clear that peak parasitemia in both C3H/HeJ and C3H/HeN co‐infected mice was significantly lower than that in respective Bm infected mouse strain. These results are reproducible. Even in the co‐infected C3H/HeJ mice, parasitemia remains detectable by microscopic examination longer than in co‐infected C3H/HeN mice. Similar pattern was also observed in Bm infected respective strains of mice (Figure 1).
FIGURE 1.
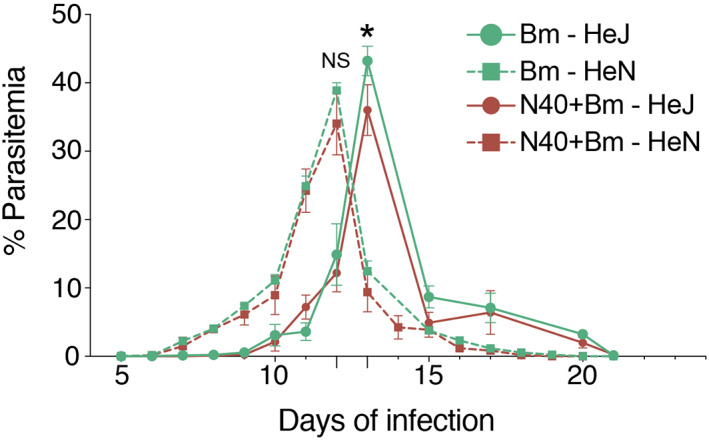
TLR4 deficiency prolongs survival of B microti in mouse blood. Parasitemia determined in 10 C3H/HeJ and C3H/HeN mice infected with Bm (104 infected erythrocytes injected i.p.) alone, or together with B. burgdorferi (103 spirochetes injected s.c. per mouse) by microscopic examination of Giemsa‐stained thin blood smears at regular intervals. Each point represents average parasitemia in each group of mice (mean ± SD) with peak parasitemia significantly higher (p < .05) in Bm infected than in respective co‐infected mouse strain but not significantly different between C3H/HeJ and C3H/HeN mice due to each infection (NS). Onset of parasitemia in Bm infected and co‐infected C3H/HeN appeared earlier than in C3H/HeJ mice (7th vs. 10th day post‐infection) but it cleared slightly faster (on 19th and 18th day of infection, respectively) than that in C3H/HeJ mice in which parasitemia was detectable in both Bm infected and co‐infected mice until 20th day post‐infection
3.2. Colonisation of the head region, hearts, joints and skin by B. burgdorferi at 2 weeks of infection increases in the Bm co‐infected respective mouse strains
Our previous experiments have shown that peak Bm parasitemia and highest B. burgdorferi tissue colonisation occur at 2 weeks of infection in the C3H/HeJ strain (Djokic et al., 2019). To determine the impact of the inherent differences between C3H/HeN and C3H/HeJ strains on colonisation of different organs by bioluminescent B. burgdorferi N40 strain in the presence or absence of co‐infecting Bm, light emission was used as a measure of tissue colonisation detected by live imaging of mice at 2 weeks of infection. We detected an overall higher colonisation levels by N40 in mice co‐infected with Bm as compared to those infected with B. burgdorferi alone in the respective mouse strains (Figure 2a,b). To further plot comparative levels of light emission in all B. burgdorferi infected mice, we determined total body bioluminescence radiance of infected (Figure 2a,b) and uninfected mice (Figure 2c). Net radiance in infected mice after deduction of average radiance from five uninfected controls was calculated (Figure 2d). As apparent in the images shown (Figure 2a,b), total radiance was significantly higher in C3H/HeJ compared to C3H/HeN mice when infected only with N40 (Figure 2d, t = 9.1, p < .0001, 95% CI: 3,982–6,372). Difference in total radiance between co‐infected C3H/HeJ and C3H/HeN mice was also significant with slightly higher radiance in C3H/HeJ mice; however, difference was not as pronounced as between respective N40‐infected mice (t = 3.1, p = .006, 95% CI: 1,308–6,608). Difference between net bioluminescence in co‐infected C3H/HeN mice compared to respective N40 infected strain was more pronounced (t = 6.8, p < .0001, 95% CI: 2,894–5,460) than difference observed in co‐infected and N40‐infected C3H/HeJ mice. This was because overall radiance was higher even in N40 infected C3H/HeJ group making its difference from co‐infected mice not as high as observed in C3H/HeN mice (Figure 2d, t = 2.4, p = .03, 95% CI: 349–5,567). In addition, co‐infected C3H/HeN mice showed comparable bioluminescence radiance to N40‐infected C3H/HeJ mice (t = 0.2, p = .81, 95% CI: −727 to 579).
FIGURE 2.
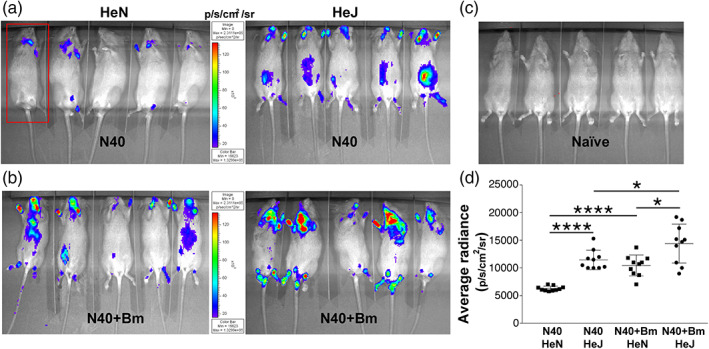
Colonisation of organs by B. burgdorferi in C3H/HeJ and C3H/HeN mice infected with N40 alone and co‐infected with Bm at 2 weeks of infection. (a) Representative real‐time images of five C3H/HeJ (HeJ) and C3H/HeN (HeN) mice infected with bioluminescent N40 strain using IVIS‐200 displaying bioluminescence as a semi‐quantitative indicator of B. burgdorferi colonisation in different tissues of mice. Bioluminescence radiance in the whole body of each mouse (as marked in one mouse by a red rectangle) was also measured. (b) Overall, light emission in five mice each, representing colonisation of organs and spirochetal burden in co‐infected C3H/HeJ and C3H/HeN mice at 2 weeks post‐infection. (c) A set of five uninfected mice was also imaged at the same time with average bioluminescence radiance measured to be (483.23 p/s/cm2/sr). (d) Average bioluminescence radiance from five uninfected mice was deducted from the radiance obtained for each infected mouse (as marked in 2a and 2b) and net radiance values data for 10 mice for each infection is presented. Horizontal lines indicate the net mean radiance in each group of infected mice. Statistical analysis was conducted using a two‐tailed unpaired student t tests for unequal variance to determine significant difference between the paired groups (*p < .05, ****p < .0001)
3.3. Colonisation of the specific organs by B. burgdorferi persists in singly infected, and Bm co‐infected animals at 3 weeks of infection
By 3 weeks of infection, the specific adaptive response develops against each pathogen and Bm parasitemia becomes undetectable microscopically (Figure 1). Difference in the light emission in joints and head of two strains of mice were higher in C3H/HeJ mice when infected with N40 alone (Figure 3a). Similarly, co‐infected C3H/HeJ strain showed slightly higher colonisation of joints as detected by bioluminescence (Figure 3b). In fact, net bioluminescence radiance, after deduction of average radiance obtained from five uninfected mice (Figure 3c), remained significantly higher in co‐infected versus N40‐infected C3H/HeJ (t = 4.9, p = .0001, 95% CI: 812–2,044) and C3HHeN mice (t = 4.8, p = .0001, 95% CI: 705–1,807); however, difference in average radiance between N40 infected C3H/HeJ and co‐infected C3H/HeN animals was not statistically significant (Figure 3d). We also observed an overall decline in colonisation by B. burgdorferi at 3 weeks compared to that at 2 weeks of respective infections in each strain of mice (Figures 2d and 3d). N40 survival and tissue colonisation were further confirmed by recovering live spirochetes consistently when injection site, left ear and bladder from both strains of mice were recovered and grown in B. burgdorferi medium at 3 weeks post‐infection (data not shown).
FIGURE 3.
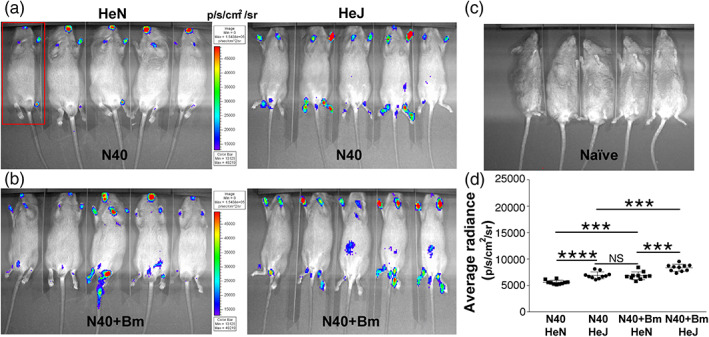
Colonisation of mice organs by B. burgdorferi as detected by light emission due to the presence of live bioluminescent N40 strain at 3 weeks of infection. (a) Representative real‐time images of five C3H/HeJ (HeJ) and C3H/HeN (HeN) mice infected with bioluminescent N40 strain using IVIS‐200 displaying bioluminescence in the head and joints regions. Bioluminescence radiance in the whole body of each mouse (as marked in one mouse by a red rectangle) was also measured. (b) Bioluminescence detection in co‐infected C3H/HeJ and C3H/HeN mice. (c) Live imaging of a set of five uninfected mice at the same time point. (d) Average bioluminescence radiance from five uninfected mice was deducted from the radiance obtained for each infected mouse and net radiance data for each mouse is shown. Horizontal lines indicate the comparison of net mean radiance in each group of infected mice. Difference between average bioluminescence radiance in N40‐infected C3H/HeJ and co‐infected C3H/HeN mice was not statistically significant (NS). Statistical analysis was conducted using a two‐tailed unpaired student t tests for unequal variance to determine significant difference between the paired groups (***p < .001, ****p < .0001)
3.4. Higher B. burgdorferi burden in TLR4 depleted C3H/HeJ than in C3H/HeN mice at 3 weeks post‐infection result in increased pathology
After euthanasia at 3 weeks post‐infection, we determined the effect of C3H/HeN and C3H/HeJ genotypes, in the context of functional TLR4 presence, on colonisation of N40 when present alone, or with Bm in co‐infected mice. The mice left hind legs (from hip to ankle), whole brain and heart together with major vasculature (aorta and left vena cava) of mice were recovered and a duplex qPCR assay performed to quantify the spirochete levels normalised to mouse DNA content. The right hind legs including knee and tibiotarsus joints were used for histopathological examination. In mice infected with N40 alone, significantly higher spirochete loads were observed in the tissues of C3H/HeJ mice (dysfunctional TLR4) as compared to TLR4 competent C3H/HeN mice by qPCR (Figure 4a) confirming the results obtained by live imaging (Figure 3). In these mice, the burden of spirochetes in each of three organs was ~2‐fold higher in C3H/HeJ mice. In Bm and N40 co‐infected mice, spirochetes burden was again higher in C3H/HeJ as compared to C3H/HeN mice in all three organs examined (Figure 4a), supporting our live imaging results (Figure 3). Co‐infected mice had significantly higher numbers of spirochetes in brains and joints than singly infected mice irrespective of the mouse strain. The level of colonisation of heart between N40 infected and co‐infected mice were almost comparable in respective strains (Figure 4a, Not significant or p < .05). Burden of spirochetes was also higher in the hearts of both N40‐infected and co‐infected C3H/HeJ than in C3H/HeN mice.
FIGURE 4.
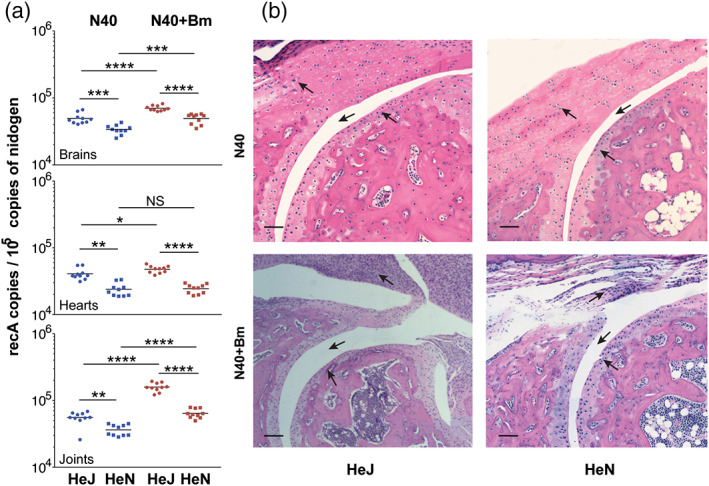
Borrelia burgdorferi strain N40 colonisation levels determined by qPCR and resulting pathology in C3H/HeJ and C3H/HeN mice. (a) Burden of B. burgdorferi in tissues as detected by duplex qPCR assay from 10 mice for each group using recA primers and molecular beacon probes with mouse nidogen amplicon copy number used for normalisation of spirochete quantities. Horizontal lines represent the mean B. burgdorferi recA copy number per 105 copies of nidogen genes. Statistical analysis was conducted using a two‐tailed unpaired student t tests for unequal variance to determine significant difference between the paired groups (NS‐Not significant, *p < .05, **p < .01, ***p < .001, ****p < .0001). (b) Severe arthritis in tibiotarsal joint manifested by synovial hyperplasia and erosion of cartilage, higher lymphocytic infiltration and change in synovial space (arrows) were observed in co‐infected mice as compared to B. burgdorferi‐infected mice of both strains. Bar represents a size of 100 μm
We sought to evaluate if increased colonisation by N40 during co‐infection also enhanced pathology in mice possessing functional or non‐functional TLR4. The joint sections were scored in a blinded manner to determine arthritis severity. Scoring of inflammation was based on infiltration of lymphocytes in the synovium and perisynovium, hyperplasia of the synovium, erosion or cellular infiltration of articular cartilage, increased synovial space (marked by arrows in the Figure 4b) and so forth and severity of arthritis were ranked on a scale from (−), that is, similar to uninfected mice, to (+++) for highly inflamed joints (Table 1). We observed inflammation of tibiotarsus in both B. burgdorferi infected C3H/HeJ and C3H/HeN mice (Figure 4b and Table 1) with a slight increase in pathology in C3H/HeJ mice reflecting the impact of higher burden of N40 in this mouse strain. More importantly, highest lymphocyte infiltration and hyperplasia of synovium were observed in co‐infected C3H/HeJ mice compared to all other infected mice representing most pronounced inflammatory arthritis in these mice.
TABLE 1.
Histopathological scoring of joints of infected mice
| Experimental groups | Numbera of mice with each inflammation score for joints | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Knee | Tibiotarsus | ||||||||
| − | +/− | + | − | +/− | + | ++ | +++ | ||
| C3H/HeJ | B. Burgdorferi | 1 | 2 | 2 | 0 | 0 | 1 | 4 | 0 |
| C3H/HeN | B. Burgdorferi | 2 | 1 | 2 | 0 | 0 | 3 | 1 | 1 |
| C3H/HeJ | B. burgdorferi + Bm | 1 | 0 | 4 | 0 | 0 | 0 | 3 | 2 |
| C3H/HeN | B. burgdorferi + Bm | 0 | 2 | 3 | 0 | 0 | 2 | 1 | 2 |
| C3H/HeJ | Bm | 5 | 0 | 0 | 5 | 0 | 0 | 0 | 0 |
| C3H/HeN | Bm | 2 | 2 | 0 | 4 | 0 | 0 | 0 | 0 |
Number of infected mice in each group of five infected mice, except four Bm infected C3H/HeN mice that showed a specific inflammation score for knee and tibiotarsus. Inflammation scoring was based upon infiltration of lymphocytes in the synovium and perisynovium, hyperplasia of the synovium, erosion or cellular infiltration of articular cartilage, increased synovial space and the severity of the lesions was ranked on a scale from no inflammation (−) that is equivalent to uninfected mice, to maximum inflammation (+++).
3.5. Modulation of splenic immune responses by infection with Bm and B. burgdorferi separately, or together at 3 weeks post‐infection of C3H/HeJ and C3H/HeN mice
To define the effect of functional TLR4 absence or presence in C3H/HeJ and C3H/HeN mice, respectively, on the host immune response to Bm and B. burgdorferi infections, we studied the changes in different splenic myeloid and lymphoid cells of infected mice by quantification of total T cells (CD3+), natural killer (NK1.1+)/FcR+ cells, macrophages (F4/80+) and B (CD19+) cells. Changes in both myeloid and lymphoid splenic cells percentages in infected versus the naïve mice are shown (Figure 5). Since C3H mice lack the NK1.1 marker, it is likely that after B cells sorting by flow cytometry, NK1.1 label gated all phagocytic cells possessing FcR. A significant increase in macrophages was observed in all Bm infected and co‐infected mice compared to N40 infected and naïve mice (Figure 5a). Interestingly, macrophage percentage increase in co‐infected C3H/HeN strain was higher compared to both strains of mice infected with B. burgdorferi and Bm individually (Figure 5a). Total phagocytic (FcR+) cells increased significantly in all infected C3H/HeJ mice but only in N40 infected C3H/HeN mice (Figure 5b). Co‐infected mice showed decreased in FcR+ cells in spleen as compared to N40 infected mice. We did not observe significant differences in percentages of macrophages between C3H/HeN and C3H/HeJ mice infected either with B. burgdorferi or Bm individually, while co‐infection resulted in significant increases in percentages of macrophages in both strains of mice (Figure 5a). Thus, effect of infection with each pathogen is different on macrophage versus FcR+ phagocytic cells, both of which participate in innate immune response.
FIGURE 5.
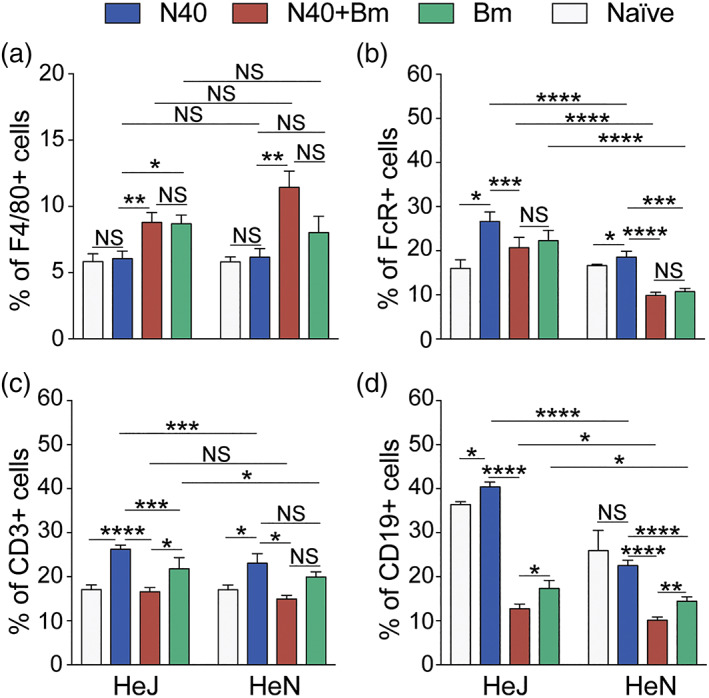
Modulation of splenic immune responses in mice by infection with B microti and N40 separately, or simultaneously. (a) Single cell suspension of mice spleen infected with N40, Bm and N40 + Bm were prepared and stained with fluorophore‐conjugated antibodies against F4/80+ (macrophages), (b) FcR+ phagocytic cells, (c) CD3+ (T cells), and (d) CD19+ (B cells), followed by FACS analysis. Higher percentages of B, FcR+ phagocytic and T cells were observed in C3H/HeJ mice. Proportions of various immune cells from spleens of mice from each experimental group analysed by FACS are expressed as mean ± SD. Statistical analysis was conducted using a two‐tailed unpaired student t tests for unequal variance to determine significant difference between the paired groups (NS‐Not Significant, *p < .05, **p < .01, ***p < .001, ****p < .0001)
In both strains of mice infected with B. burgdorferi alone, we observed a significant increase in the percentages of total T cells; however, increase was higher in C3H/HeJ as compared to C3H/HeN mice (Figure 5c). The percentage of B cells increased significant only in N40 infected C3H/HeJ mice (Figure 5d). Changes in B and T cells in Bm infected, or co‐infected mice were not significant relative to naïve mice (Figure 5c,d). All co‐infected mice showed a significant decrease in the percentages of T and B cells as compared to B. burgdorferi infected and even Bm infected mice in most cases, particularly in TLR4 defective C3H/HeJ strain (Figure 5b–d). T cells percentages in co‐infected mice were comparable in both mouse strains although their percentage diminished relative to B. burgdorferi infected mice (Figure 5c). Increases in FcR+ cells after B. burgdorferi infection were not as high in C3H/HeN mice as C3H/HeJ mice and decline in co‐infected mice was also relatively lower in the former. No change in FcR+ and T cells percentage was detected in co‐infected C3H/HeN mice as compared to naïve mice.
3.6. Signalling of TLR2/TLR4 transfected HEK293 cells with B. burgdorferi and Bm infected RBCs
Enhanced response reported through TLR2 in the absence of TLR4 (Mu, Pennock, Humphreys, Kirschning, & Cole, 2005) suggests either increased expression and/or enhanced responsiveness of TLR2 to B. burgdorferi lipoproteins. Since such a response to Lyme spirochetes could be localised to the specific regions of organs, such as in the heart base and synovium of joints, quantification of each TLR by real‐time PCR and their response to PAMPs in the particular regions is very difficult. Therefore, we decided to use a clean system of transfected cell line to evaluate signalling by PAMPs of B. burgdorferi and potentially Bm. Parental HEK293 cells do not express either TLR2 or TLR4. We used transfected HEK293 cells that stably express TLR2‐YFP, or TLR4‐CFP + Flag‐tagged MD‐2 and included control cells (pCDNA3 vector transfected) in our experiments that were generously provided by Dr. Golenbock (Latz et al., 2002). We confirmed the expression of respective TLRs by microscopy (Figure 6a,b). As compared to the medium treated HEK293 transfected cells (control), TLR2‐transfected cells showed significant increase in production of IL‐8 after stimulation by B. burgdorferi and caused induction of pro‐inflammatory cytokines production even when 20 μg/ml Polymyxin B was included in treatment medium (Figure 6d,h, and data not shown). Signalling of TLR4 by B. burgdorferi moderately increased IL‐8 production; however, it was significantly higher than the control cell line treated with the same dose of spirochetes (Figure 6d,h). Neither TLR2 nor TLR4 signalling was observed in cells treated with Bm infected and diluted whole blood of mice that also possessed a few egressed parasites suggesting minimal levels or no Bm PAMPs were available for stimulation of these two TLRs (Figure 6e). Treatment of TLR transfected cell lines with 1.25 × 107 Bm parasites released by saponin treatment in the presence of Polymyxin B also did not show any stimulation (Figure 6f) indicating that either mature merozoites egressed from iRBCs are required for stimulation of TLRs on monocytes, macrophages, neutrophils, natural killer (NK) and dendritic cells during infection or Bm merozoites do not display PAMP on their surface for TLR signalling. A similar pattern was observed with respect to TNF‐α production by these cell line (Figure 6f–h) although levels of this cytokine were significantly lower than that observed for IL‐8.
FIGURE 6.
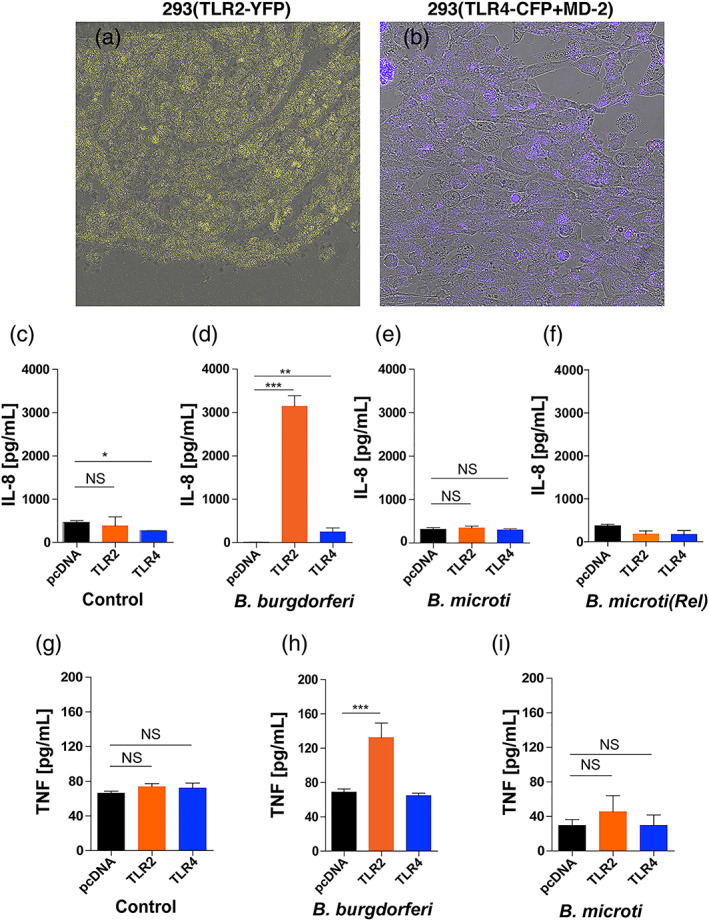
Response of TLR2/TLR4 transfected HEK293 cells to B. burgdorferi N40 strain and Bm‐infected mouse blood. (a and b) Yellow fluorescence and Cyan fluorescence associated with TLR2‐YFP and TLR4‐CFP + MD‐2 transfected HEK293 cells confirmed expression of respective TLRs. (c and d) As compared to control (6c, medium only) treatment, exposure of transfected HEK293 cells to 2 × 107 B. burgdorferi for ~16 hr resulted in high stimulation of TLR2 and moderate but significant level of stimulation of TLR4 to produce pro‐inflammatory IL‐8 cytokine (6d). (e and f) Neither treatment with 1 × 106 RBCs of Bm infected mouse blood (~3.1 × 105 iRBCs) nor released (Rel) Bm parasites suspended in 20 μg/ml Polymyxin B containing treatment medium stimulated TLR2 or TLR4 to produce pro‐inflammatory IL‐8 cytokine. (g–i) Significant TNF‐α (TNF) stimulation was only observed by B. burgdorferi N40 strain treatment of HEK293 cells transfected with TLR2. Again, Bm did not shown stimulation neither of TLR2 nor TLR4 expressed in HEK293 cells. Statistical analysis was conducted using a two‐tailed unpaired student t tests for unequal variance to determine significant difference between the paired groups (NS‐Not significant, *p < .05, **p < .01, ****p < .0001)
4. DISCUSSION
Transmission of Bm and B. burgdorferi occurs primarily through the bite of infected Ixodes species ticks. Babesia species were identified as infectious organisms in 1893 and babesiosis was first detected in humans in the USA in 1969 (Western et al., 1970; Ouhelli and Schein, 1988); however, delayed establishment of babesiosis in the endmeic regions for tick‐borne diseases including Northeastern United States has been attributed to less efficient transmission of Bm by ticks compared to B. burgdorferi (Krause et al., 2006). Interestingly, acquisition of Bm from the white‐footed mice by the tick vector improves when mice are co‐infected with a highly infectious B. burgdorferi (Dunn et al., 2014). Diuk‐Wasser et al. reported that incidence of babesiosis increases particularly in the regions where B. burgdorferi is already prevalent, thus suggesting that ongoing presence of B. burgdorferi in reservoir hosts and ticks promotes spread and establishment of Bm infection in the endemic regions (Diuk‐Wasser et al., 2014). As a result, co‐infections with these pathogens have started emerging in different regions of the World (Mayne, 2011, 2015, Anderson et al., 1991; Krause et al., 1991; Mitchell et al., 1996; Belongia, 2002, Swanson et al., 2006, Primus et al., 2018, Welc‐Faleciak et al., 2010). This study was undertaken to better understand signalling of TLR2, in the presence or absence of functional TLR4, by PAMPs of both pathogens and resulting host immune responses in the absence of other mitigating factors. Therefore, a more controlled, needle‐injection method with precise infection doses was used in our experiments. Although it does not reflect tick bite‐mediated inoculation route, advantgae of this method is that it eliminates the effect of variability introduced during natural infection by ticks, which may not inoculate consistent dose of each pathogen in each set of inbred mice, thus making comparison rather difficult without using a large number of animals.
Despite evidence of high susceptibility of both TLR4 competent C3H/HeN mice and dysfunctional TLR4 possessing C3H/HeJ mice to Bm and B. burgdorferi infections, a comparative study of the pathogenesis of infections in these strains has not been conducted until now. We sought to examine the effect of TLR4 on Bm and B. burgdorferi when inoculated separately, or together. Although TLR4 ligand has not been identified in either Bm or B. burgdorferi; GPI glycolipid abundantly present in the membranes of many pathogenic protozoa has been identified as a possible TLR4 ligand (Debierre‐Grockiego et al., 2007). We decided to address whether TLR4 is important for these tick‐borne infections by assessing the differences in kinetics of growth patterns of Bm in blood, and burdens of B. burgdorferi in organs of the infected or co‐infected C3H/HeJ and C3H/HeN mice. Overall, lower Bm peak parasitemia was observed in co‐infected mice as compared to Bm‐infected mice, but difference in peak parasitemia between two strains of C3H mice was not significant (Figure 1). Unlike the study by Coleman et al. (Coleman et al., 2005), we observed differences in B. burgdorferi colonisation of different organs of co‐infected C3H mice (Figures 1, 2, 3, 4) that resulted in enhancement of Lyme disease pathology.
Both TLR4 and TLR2 activation has been shown to play critical roles in pathogenesis of arthritis (Abdollahi‐Roodsaz et al., 2013; Campo et al., 2011). Abundant B. burgdorferi surface lipoproteins signal host cells primarily through TLR2 heterodimer and resulting in stimulation of proinflammatory cytokines production (Cervantes et al., 2011; Lien et al., 1999; Salazar et al., 2009); however, other TLRs have also been implicated in disease and tolerance to infection (Cervantes et al., 2011; Cervantes et al., 2013; Gebbia, Coleman, & Benach, 2004; Hirschfeld et al., 1999; Salazar et al., 2003; Salazar et al., 2005; Shin et al., 2008). The role of TLR4 in control of the B. burgdorferi infections was based upon the observation that TLR4 defective bone marrow derived macrophages (BMDMs) from C3H/HeJ mice produced less TNF‐α in response to B. burgdorferi infection, compared to BMDMs from normal TLR4 containing C3H/FeJ mice (Glickstein & Coburn, 2006). Consistent with the known requirement of TLR4 for full function of innate immune system, our results show a significant increase in overall N40 strain burden in C3H/HeJ mice at 2 weeks post‐infection (Figure 2). Previously, B. burgdorferi infection in rhesus monkeys after TLR4 depletion, or after inhibition of its functional activity showed opposite effect on inflammatory responses in the neuronal tissues (Parthasarathy & Philipp, 2015). Control of B. burgdorferi infection was found to be dependent upon phagocytosis mediated killing through spirochete lipoproteins signalling after recognition of TLR2 (Hirschfeld et al., 1999), while higher proinflammatory cytokine production could attenuate B. burgdorferi infection in TLR4 sufficient mice by a still unknown PAMP (Glickstein & Coburn, 2006). Signalling through mainly TLR2, and to a lesser extent TLR4 by B. burgdorferi to produce proinflammatory cytokines was confirmed by our in vitro experiments using the specific TLR‐transfected HEK 293 cells (Figure 6). The absence of functional TLR4 in C3H/HeJ mice could be responsible for preventing early bacterial clearance and lead to increased tissue invasion by B. burgdorferi compared to that in C3H/HeN mice while spirochetes burden in various organs becomes almost comparable at 3 weeks post‐infection after adaptive immune response was established (Figures 2 and 3).
Higher burden of spirochetes was previously reported to also reflect more severe arthritis in infected patients and a similar pattern was also observed in mice (Diuk‐Wasser et al., 2016; Krause et al., 1996; Moro et al., 2002). Indeed, we observed higher pathology in C3H/HeJ than C3H/HeN mice when co‐infected with Bm (Figure 4). Although genotype‐based resistance of some mouse strains to Lyme arthritis irrespective of B. burgdorferi quantities in joints had been observed (Bramwell et al., 2014; C. R. Brown & Reiner, 1999; Pahl, Kuhlbrandt, Brune, Rollinghoff, & Gessner, 1999; Wooten & Weis, 2001), N40 burden correlated with the severity of Lyme arthritis within the same strain of mice in our study. Bm infection of mice activates CD4+ T cells and results in production of IFN‐γ that could further activate macrophages leading to Th1 response stimulation (Igarashi et al., 1999). In our recent studies in young C3H/HeJ mice, Th1 cells proliferation and response demonstrated by intracellular IFN‐γ production were most pronounced during early acute stage of infection (before peak parasitemia, on day 11) with Bm. These results indicated that Th1 cells likely play an important role in resolution of parasitemia (Djokic, Primus, et al., 2018). A significant increase in Th1 cells in N40 infected and co‐infected mice at this time point suggested that these CD4+ cells likely contributed to the clearance of both pathogens during acute phase of infection. There was no significant change in CD19+ B cells in infected C3H/HeJ mice at the early phase of infection in that study (Djokic, Primus, et al., 2018).
In our previous study, we observed a significant increase in myeloid cell numbers in C3H/HeJ infected mice at acute phase with total macrophage numbers increased in both Bm infected, and N40 co‐infected mice relatively more than in N40 infected mice (Djokic, Primus, et al., 2018). The activation of macrophages also results in TNF‐α, reactive oxygen species and nitric oxide production (Homer et al., 2000), which together enhance their microbicidal activity. A significant proliferation of FcR+, representing primarily phagocytic cells in infected C3H/HeJ mice suggests that these cells may also be involved in clearance of both B. burgdorferi and Bm at the acute phase of infection. Although C3H/HeJ macrophages fail to induce inflammatory cytokines in response to microbial TLR4 stimulants like LPS (Hoshino et al., 1999), the role of PAMPS of Bm in TLR‐NF‐kβ activation pathway to cause macrophages activation soon after infection is not known. The absence of detectable differences in Bm parasitemia development and resolution in Bm infected C3H/HeJ and C3H/HeN mice alone, or with B. burgdorferi, suggests that TLR2 and TLR4 signalling do not contribute significantly to Bm clearance. Our results agree with previous study by Skariah and coworkers, who showed that MyD88 deficiency did not affect Bm parasitemia in mice (Skariah et al., 2017). Furthermore, the lack of stimulation of TLR2 and TLR4 in transfected HEK293 cells to the whole blood from Bm‐infected C3H mice, which contained iRBCs as well as egressed parasites that should display GPI‐anchored proteins on their surface such as BmGPI12 (Thekkiniath et al., 2018; Thekkiniath et al., 2019), in our in vitro experiment confirmed the lack of response of these PRRs to Bm (Figure 6). These results also confirmed our in vivo data.
Previous studies have shown enhanced activity of TLR2 in the hosts deficient in TLR4 (Mu et al., 2005). It is, therefore, expected that the activity of TLR2 in C3H/HeJ mice could also be higher during B. burgdorferi infection due to the absence of TLR4, thus increasing inflammation in this mouse strain. TLRs form a crucial link between innate and adaptive immunity by up‐regulating the ability of accessory cells to generate immune responses mediated by T and B lymphocytes (Iwasaki & Medzhitov, 2010). In fact, we observed significantly higher levels of B and T cells in C3H/HeJ strain at 3 weeks of infection with B. burgdorferi that greatly diminished in the presence of Bm (Figure 5). Slight delay in resolution of Bm infection in TLR4‐defective mice can be attributed to their somewhat attenuated adaptive immune response.
To summarise, our results demonstrate that TLR4 may also play an indirect role in modulating immune response and pathology during B. burgdorferi infections. We propose that activation of TLR4 by unknown ligands from co‐infecting pathogens in C3H/HeN mice, more importantly from B. burgdorferi, could down regulate TLR2 heterodimeric receptor, as described previously (Mu et al., 2005), particularly in peripheral blood nucleated cells early in infection, thus reducing signalling through MyD88 dependent (or independent) pathway, which then suppresses inflammatory immune responses (Figures 4, 5, 6). In the absence of functional TLR4 in C3H/HeJ mice, unrestrained stimulation could occur through TLR2 signalling due to the abundance of lipoproteins on spirochetes surface, causing more pronounced inflammatory response in B. burgdorferi infected and co‐infected C3H/HeJ mice as compared to C3H/HeN mice (graphical abstract). The effect of Bm PAMPS on TLR2/TLR4 signalling‐mediated innate immune response in both strains of mice during infection, whether present alone or with B. burgdorferi, appears minimal (Figure 1). These results were further confirmed using transfected HEK293 cell lines in vitro (Figure 6) suggesting the absence of display of Bm PAMPs, such as GPI anchored proteins, on iRBCs surface.
5. EXPERIMENTAL PROCEDURES
5.1. Animal studies
The Newark Institutional Animal Care and Use Committee (IACUC) reviewed and approved the protocol number D‐14011‐A1 under which this study was conducted at Rutgers University New Jersey Medical School. Guidelines of the Animal Welfare Act, The Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals and Public Health Service Policy were followed in this study.
5.2. Culture and maintenance of B. burgdorferi and Bm
Bm (ATCC30221) was first inoculated in C3H severe combined immunodeficient (SCID) mice to obtain inoculum for subsequent experiments. To determine parasitemia, a drop of blood sample was collected from the infected mouse by tail bleed to prepare a smear. Thin blood smears were air‐dried, fixed with absolute methanol for approximately 5 min and then stained using Giemsa solution diluted 1:2 in water for 15 min. To estimate parasitemia, Giemsa stained blood smears were examined under a light microscope and the number of infected red blood cells counted in a total of 20–50 fields at high magnification (×10 and ×100 ocular magnification oil immersion objective lenses). Parasitemia was expressed as a percentage of infected erythrocytes among total red blood cells counted. Bioluminescent B. burgdorferi N40 strain (N40 D10/E9 clone derivative) carrying a firefly luciferase gene (Bbluc) was used in this study (Chan, Alter, Barthold, & Parveen, 2015). Spirochetes were cultured at 33°C in Barbour‐Stoenner‐Kelly‐II (BSK‐II) medium supplemented with 6% rabbit serum (BSKII‐RS).
5.3. Infection of mice with B. burgdorferi and Bm
Three‐week old C3H/HeN and C3H/HeJ mice were purchased from reputed vendors (Charles River and Jackson, respectively) and after acclimatisation for one‐week, 35 mice of each strain were randomly divided into four experimental groups. A group of five mice of each strain was kept uninfected, and 10 mice were used for each infection group. One experimental group of each mouse strain was injected with B. burgdorferi N40 alone, second group received both B. burgdorferi and Bm and third group was inoculated with Bm alone. To infect mice for experimental purpose, blood was collected from infected SCID mice and the number of parasitised RBCs calculated, as described above. Briefly, total number of RBCs per ml was determined using a haemocytometer. The number of infected RBCs was then determined by multiplying percent parasitemia with calculated total RBCs per ml of blood. The number of parasitised RBCs was then adjusted to 105 per ml in 1xPBS and 100 μl injected in each mouse intraperitoneally (i.p.), that is, 104 infected Red Blood Cells (iRBCs) inoculated per mouse. The spirochete numbers were adjusted to 104 per ml of medium and 100 μl (103 B. burgdorferi) of inoculum was used for injection of each mouse subcutaneously (s.c.) on the dorso‐lateral aspect of the right thigh. Our previous studies showed that subcutaneous injection of B. burgdorferi suspended in BSKII‐RS allows sufficient time for live‐imaging of mice (Chan et al., 2015). Since BSKII‐RS may affect Bm adversely, we used different sites of infection for these pathogens such that consistent infection dose with healthy pathogens is ensured. Thus, we were able to conduct a well‐controlled study with fewer variables introduced, which is not possible when natural, tick‐borne infection procedure is employed.
5.4. Monitoring of infected mice
Infected mice were monitored closely for both B. burgdorferi and Bm infection progression for up to 21 days post‐infection, after which they were euthanised and evaluated for tissue colonisation and disease pathology. Mice infected with Bm were monitored for parasitemia regularly by examination of Giemsa stained blood smears. Colonisation by bioluminescent B. burgdorferi was monitored by live imaging at 2 and 3 weeks of infection using In Vivo Imaging System 200 (IVIS‐200, Perkin Elmer, MA) and images captured. Briefly, after isoflurane anaesthesia, each mouse was injected with 200 μl PBS containing 30 mg/ml of D‐luciferin intraperitoneally. Images were captured with 3 min exposure. At each stage, five uninfected mice were also imaged as controls for bioluminescence quantification after injection of the substrate and net radiance reported for infected mice was determined after deduction of average value from these controls (483.23 p/s/cm2/sr). After 21 days of infection, mice were imaged, heparinated blood collected by cardiac puncture and animals were then euthanised. Plasma was recovered for antibody response determination.
5.5. Analysis of tissue colonisation and disease pathology
To eliminate microbiome on skin surface, mice were soaked in Betadine for 30–40 min followed by submersion in 70% ethyl alcohol for 30 min and then dissected in biosafety hood and organs removed aseptically. The skin at the injection site, ear and urinary bladder were transferred to tubes containing BSK‐II + RS medium and antibiotic mixture allowing Borreliae (Sigma‐Aldrich, MO) growth and restricting contaminants growth. Borrelia burgdorferi were allowed to grow at 33°C to recover live spirochetes to ensure that tissue colonisation has occurred by N40 strain. Cultures were monitored for up to 14 days and live spirochetes observed by dark field microscopy. DNA was isolated from the left hind leg, whole brain and heart together with major vasculature (aorta and left vena cava) and used for PCR quantification for B. burgdorferi recA amplicon using the specific molecular beacon probes tagged with FAM fluorophore in the duplex assay developed in our laboratory (Chan, Marras, & Parveen, 2013). Mouse DNA was quantified by determining nidogen amplicon copy number using the specific molecular beacon tagged with TET fluorophore for normalisation of B. burgdorferi copy number to calculate relative spirochetes burden in each organ. Aseptically removed spleens were used to isolate splenocytes for flow cytometry.
5.6. Histopathology
Right hind legs of infected mice were fixed in 10% neutral buffered formalin for at least 48 hours after which bone tissue in joints was decalcified by 10% EDTA‐decalcification protocol. Then, 5 μm thick sections were cut by microtome, mounted on slides and stained with haematoxylin and eosin. Histopathological examination of sections of the tibiotarsus joint and knee were independently conducted in a blinded manner and scored according to the established criteria.
5.7. Isolation of T cells and flow cytometry
Spleens were aseptically harvested from infected mice, weighed and single cell suspensions of the splenocytes obtained by slicing the organ into small pieces and straining into 50 ml conical tubes through a 70 μm nylon sterile cell strainer. The cells were then washed with PBS by centrifugation at 1,500 rpm and RBCs lysed by Ammonium‐Chloride–Potassium (ACK) lysis buffer (Thermofisher #A10492201). The splenocytes were resuspended in FACS buffer (PBS +5%FBS) at 106 cells/ml and labelled. Total splenocytes population was incubated for 30 min in the dark at 4°C in order to label B cells with Brilliant violet 421 conjugated anti‐mouse CD19 antibodies (Bio legend, #115537), macrophages with BV605 conjugated anti‐mouse F4/80 antibodies (Bio legend, #123133), all FcR+ cells with APC‐Cy7 conjugated anti‐mouse NK1.1 antibodies (Bio legend #108724), T cells with PE‐Cy7 conjugated anti‐mouse CD3 antibodies (Bio legend #100220). We used anti‐mouse NK1.1 monoclonal mouse IgG2a antibodies (PK136) that can bind to all cells displaying FcR I, FcR II and/or FcR III. Since NK1.1 marker is lacking in C3H mice, these antibodies helped us quantify all splenic phagocytes. The cells were washed with PBS containing 5% FBS by centrifugation and resuspended in FACS buffer. BD LSRFortessa™ X‐20 (BD Biosciences) driven by software FACS DiVa (BD Biosciences) was used for FACS data collection for the stained cells. Acquired data were analysed using FlowJo, Version 10.3 software.
5.8. In vitro stimulation of transfected HEK293 cells
The HEK293 cells were cultivated in 1:1 mixture of Dulbecco's Modified Eagle's Medium (DMEM) and Ham's F12 medium supplemented with 10% FBS and P/S mixture with G418 included for selection of transfection plasmids, that is, pCDNA3 vector, plasmid containing TLR2‐YFP fusion, TLR4‐CFP fusion + flag tagged MD‐2 in incubator set at 37°C and 5% CO2. Confirmation of expression of respective TLRs by confocal microscopy was achieved by detecting fluorescence associated with respective fusion proteins. Heparinated Bm infected blood from C3H/HeJ mouse (30.7% parasitemia) was collected. After growing cells in 24 well plate (4 replicates), they were washed once with PBS and then added 500 μl DMEM high glucose culture media with 10% FBS without any antibiotics, that is, treatment medium (control), or the same medium containing 2 × 107 N40 per well or 1 × 106 RBCs (~3.1 × 105 iRBCs) per well per 500 μl. In addition, Bm parasites were isolated by treatment of RBCs from infected mice with 0.15% saponin prepared in PBS by incubating on ice for 30 min followed by centrifugation at 2,000 ×g to remove RBC debris. Released parasites pellet was obtained by centrifugation of supernatant at 10,000 ×g for 10 min. The pellet was then suspended in treatment medium containing 20 μg/ml Polymyxin B to eliminate the effect of LPS contamination, if any. Transfected 293 cell lines were also treated with N40 in Polymyxin B containing medium (not shown) or 1.25 × 107 released Bm parasites suspension per well per 500 μl. Plates were centrifuged in Beckman‐Coulter Allegra‐x14 bench top centrifuge at 900 ×g and incubated overnight in 5% CO2 incubator at 37°C for TLR stimulation. Supernatants were collected the next day (~16 hr incubation) and analysed for cytokines secretion using ELISA kits from R & D Systems (Minneapolis, USA) following manufacturer's protocol. Experiment was conducted three times with four replicates for each treatment. Data from a representative experiment are presented here.
5.9. Statistical analysis
All data collected were analysed by GraphPad prism, version 7 for Windows (GraphPad Software, San Diego, CA) and are presented as mean ± standard deviation (SD). Comparisons between two groups were made using unpaired, two‐tailed Student t test with unequal variance, whereas ordinary one‐way ANOVA assuming Gaussian distribution was used to determine significant difference between paired C3H/HeJ and C3H/HeN strain results. Values below 0.05 were considered significant for a paired group samples comparison at 95% confidence interval.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
All authors contributed significantly to this work. Nikhat Parveen conceived the study and Vitomir Djokic and Lavoisier Akoolo designed and conducted all animal experiments. Lavoisier Akoolo started this study and Vitomir Djokic analysed data and both Vitomir Djokic and Sandra C. Rocha prepared figures for the manuscript. Vitomir Djokic and Lavoisier Akoolo scored sections of heart and joint for inflammatory response. Nikhat Parveen and Sandra C. Rocha conducted in vitro stimulation experiments and cytokine release assays. Nikhat Parveen wrote the manuscript. All authors have read and approved the manuscript for submission.
ACKNOWLEDGEMENTS
We acknowledge valuable technical assistance provided by the technical Director, Sukhwinder Singh of Flow cytometry core facility of Rutgers New Jersey Medical School; Luke Fritzky and Joel Pierre for assistance in organ samples preparation, sectioning and staining for the histopathological examination of tissues, and Sharanjeet Atwal of Public Health Research Institute for capturing fluorescent microscopic images of HEK293 cells. We thank Dr James Theis for critical reading of this manuscript. This work was supported by the National Institutes of Health (R01AI089921 and R01AI137425) and New Jersey Health Foundation grants to Nikhat Parveen.
Akoolo, L., Djokic, V., Rocha, S. C., & Parveen, N. (2021). Pathogenesis of Borrelia burgdorferi and Babesia microti in TLR4‐Competent and TLR4‐dysfunctional C3H mice. Cellular Microbiology, 23(9), e13350. 10.1111/cmi.13350
Lavoisier Akoolo and Vitomir Djokic contributed equally to this work.
Funding information National Institute of Allergy and Infectious Diseases, Grant/Award Number: R01AI089921; National Institutes of Health, National Institute of Allergy and Infectious Diseases, Grant/Award Number: R01AI137425
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study. All information that is part of this study are included in the manuscript.
REFERENCES
- Abdollahi‐Roodsaz, S., Koenders, M. I., Walgreen, B., Bolscher, J., Helsen, M. M., van den Bersselaar, L. A., … van den Berg, W. B. (2013). Toll‐like receptor 2 controls acute immune complex‐driven arthritis in mice by regulating the inhibitory Fcgamma receptor IIB. Arthritis and Rheumatism, 65(10), 2583–2593. 10.1002/art.38087 [DOI] [PubMed] [Google Scholar]
- Aliota, M. T., Dupuis, A. P., 2nd, Wilczek, M. P., Peters, R. J., Ostfeld, R. S., & Kramer, L. D. (2014). The prevalence of zoonotic tick‐borne pathogens in Ixodes scapularis collected in the Hudson Valley, New York state. Vector Borne and Zoonotic Diseases, 14(4), 245–250. 10.1089/vbz.2013.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, A. L., Barthold, S. W., Persing, D. H., & Beck, D. S. (1992). Carditis in Lyme disease susceptible and resistant strains of laboratory mice infected with Borrelia burgdorferi . The American Journal of Tropical Medicine and Hygiene, 47(2), 249–258. [DOI] [PubMed] [Google Scholar]
- Anderson, J. F., Mintz, E. D., Gadbaw, & J. J. , Magnarelli, L. A. (1991). Babesia microti, human babesiosis, and Borrelia burgdorferi in Connecticut. Journal of Clinical Microbiology, 29(12), 2779–2783. 10.1128/jcm.29.12.2779-2783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belongia, E. A. (2002). Epidemiology and Impact of Coinfections Acquired from Ixodes Ticks. Vector‐Borne and Zoonotic Diseases, 2(4), 265–273. 10.1089/153036602321653851. [DOI] [PubMed] [Google Scholar]
- Baravalle, M. E., Thompson, C., de Echaide, S. T., Palacios, C., Valentini, B., Suarez, C. E., … Echaide, I. (2010). The novel protein BboRhop68 is expressed by intraerythrocytic stages of Babesia bovis . Parasitology International, 59(4), 571–578. 10.1016/j.parint.2010.07.008 [DOI] [PubMed] [Google Scholar]
- Barthold, S. W. (1991). Infectivity of Borrelia burgdorferi relative to route of inoculation and genotype in laboratory mice. The Journal of Infectious Diseases, 163, 419–420. [DOI] [PubMed] [Google Scholar]
- Barthold, S. W., Beck, D. S., Hansen, G. M., Terwilliger, G. A., & Moody, K. D. (1990). Lyme borreliosis in selected strains and ages of laboratory mice. The Journal of Infectious Diseases, 162(1), 133–138. [DOI] [PubMed] [Google Scholar]
- Benoit, V. M., Petrich, A., Alugupalli, K. R., Marty‐Roix, R., Moter, A., Leong, J. M., & Boyartchuk, V. L. (2010). Genetic control of the innate immune response to Borrelia hermsii influences the course of relapsing fever in inbred strains of mice. Infection and Immunity, 78(2), 586–594. 10.1128/IAI.01216-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggraefe, I., Yuan, J., Telford, S. R., 3rd, Menon, S., Hunter, R., Shah, S., … Vannier, E. (2006). Babesia microti primarily invades mature erythrocytes in mice. Infection and Immunity, 74(6), 3204–3212. 10.1128/IAI.01560-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, R. (1994). Polyclonal antibody characterization of Babesia caballi antigens. International Journal for Parasitology, 24(4), 511–517. [DOI] [PubMed] [Google Scholar]
- Bramwell, K. K., Ma, Y., Weis, J. H., Chen, X., Zachary, J. F., Teuscher, C., & Weis, J. J. (2014). Lysosomal beta‐glucuronidase regulates Lyme and rheumatoid arthritis severity. The Journal of Clinical Investigation, 124(1), 311–320. 10.1172/JCI72339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C. R., & Reiner, S. L. (1999). Genetic control of experimental Lyme arthritis in the absence of specific immunity. Infection and Immunity, 67(4), 1967–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, W. C., Norimine, J., Knowles, D. P., & Goff, W. L. (2006). Immune control of Babesia bovis infection. Veterinary Parasitology, 138(1–2), 75–87. 10.1016/j.vetpar.2006.01.041 [DOI] [PubMed] [Google Scholar]
- Bulut, Y., Faure, E., Thomas, L., Equils, O., & Arditi, M. (2001). Cooperation of toll‐like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein a lipoprotein: Role of toll‐interacting protein and IL‐1 receptor signaling molecules in toll‐like receptor 2 signaling. Journal of Immunology, 167(2), 987–994. [DOI] [PubMed] [Google Scholar]
- Campo, G. M., Avenoso, A., Nastasi, G., Micali, A., Prestipino, V., Vaccaro, M., … Campo, S. (2011). Hyaluronan reduces inflammation in experimental arthritis by modulating TLR‐2 and TLR‐4 cartilage expression. Biochimica et Biophysica Acta, 1812(9), 1170–1181. 10.1016/j.bbadis.2011.06.006 [DOI] [PubMed] [Google Scholar]
- Campos, M. A., Almeida, I. C., Takeuchi, O., Akira, S., Valente, E. P., Procopio, D. O., … Gazzinelli, R. T. (2001). Activation of toll‐like receptor‐2 by glycosylphosphatidylinositol anchors from a protozoan parasite. Journal of Immunology, 167(1), 416–423. 10.4049/jimmunol.167.1.416 [DOI] [PubMed] [Google Scholar]
- Caulfield, A. J., & Pritt, B. S. (2015). Lyme disease coinfections in the United States. Clinics in Laboratory Medicine, 35(4), 827–846. 10.1016/j.cll.2015.07.006 [DOI] [PubMed] [Google Scholar]
- Cervantes, J. L., Dunham‐Ems, S. M., La Vake, C. J., Petzke, M. M., Sahay, B., Sellati, T. J., … Salazar, J. C. (2011). Phagosomal signaling by Borrelia burgdorferi in human monocytes involves toll‐like receptor (TLR) 2 and TLR8 cooperativity and TLR8‐mediated induction of IFN‐beta. Proceedings of the National Academy of Sciences of the United States of America, 108(9), 3683–3688. 10.1073/pnas.1013776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes, J. L., La Vake, C. J., Weinerman, B., Luu, S., O'Connell, C., Verardi, P. H., & Salazar, J. C. (2013). Human TLR8 is activated upon recognition of Borrelia burgdorferi RNA in the phagosome of human monocytes. Journal of Leukocyte Biology, 94(6), 1231–1241. 10.1189/jlb.0413206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, K., Alter, L., Barthold, S. W., & Parveen, N. (2015). Disruption of bbe02 by insertion of a luciferase gene increases transformation efficiency of Borrelia burgdorferi and allows live imaging in Lyme disease susceptible C3H mice. PLoS One, 10(6), e0129532. 10.1371/journal.pone.0129532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, K., Marras, S. A., & Parveen, N. (2013). Sensitive multiplex PCR assay to differentiate Lyme spirochetes and emerging pathogens Anaplasma phagocytophilum and Babesia microti . BMC Microbiology, 13, 295. 10.1186/1471-2180-13-295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, J. C., Young, D. W., Golenbock, D. T., Christ, W. J., & Gusovsky, F. (1999). Toll‐like receptor‐4 mediates lipopolysaccharide‐induced signal transduction. Journal of Biological Chemistry, 274(16), 10689‐10692. 10.1074/jbc.274.16.10689 [DOI] [PubMed] [Google Scholar]
- Coleman, J. L., LeVine, D., Thill, C., Kuhlow, C., & Benach, J. L. (2005). Babesia microti and Borrelia burgdorferi follow independent courses of infection in mice. The Journal of Infectious Diseases, 192(9), 1634–1641. 10.1086/496891 [DOI] [PubMed] [Google Scholar]
- Diuk‐Wasser, M. A., Liu, Y., Steeves, T. K., Folsom‐O'Keefe, C., Dardick, K. R., Lepore, T., … Krause, P. J. (2014). Monitoring human babesiosis emergence through vector surveillance. New England, USA. Emerging Infectious Diseases, 20(2), 225–231. 10.3201/eid2002.130644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debierre‐Grockiego, F., Campos, M. A., Azzouz, N., Schmidt, J., Bieker, U., Resende, M. G., … Schwarz, R. T. (2007). Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii . Journal of Immunology, 179(2), 1129–1137. 10.4049/jimmunol.179.2.1129 [DOI] [PubMed] [Google Scholar]
- Debierre‐Grockiego, F., Smith, T. K., Delbecq, S., Ducournau, C., Lantier, L., Schmidt, J., … Cornillot, E. (2019). Babesia divergens glycosylphosphatidylinositols modulate blood coagulation and induce Th2‐biased cytokine profiles in antigen presenting cells. Biochimie, 167, 135–144. 10.1016/j.biochi.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbecq, S., Precigout, E., Vallet, A., Carcy, B., Schetters, T. P., & Gorenflot, A. (2002). Babesia divergens: Cloning and biochemical characterization of Bd37. Parasitology, 125(Pt 4), 305–312. 10.1017/s0031182002002160 [DOI] [PubMed] [Google Scholar]
- Diuk‐Wasser, M. A., Vannier, E., & Krause, P. J. (2016). Coinfection by Ixodes tick‐borne pathogens: Ecological, epidemiological, and clinical consequences. Trends in Parasitology, 32(1), 30–42. 10.1016/j.pt.2015.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djokic, V., Akoolo, L., Primus, S., Schlachter, S., Kelly, K., Bhanot, P., & Parveen, N. (2019). Protozoan parasite Babesia microti subverts adaptive immunity and enhances Lyme disease severity. Froniters in Microbiology, 10, 1596. 10.3389/fmicb.2019.01596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djokic, V., Akoolo, L., & Parveen, N. (2018). Babesia microti infection changes host spleen architecture and is cleared by a Th1 immune response. Frontiers in Microbiology, 9, 1–11. 10.3389/fmicb.2018.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djokic, V., Primus, S., Akoolo, L., Chakraborti, M., & Parveen, N. (2018). Age‐related differential stimulation of immune response by Babesia microti and Borrelia burgdorferi during acute phase of infection affects Disease severity. Frontiers in Immunology, 9, 2891. 10.3389/fimmu.2018.02891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, J. M., Krause, P. J., Davis, S., Vannier, E. G., Fitzpatrick, M. C., Rollend, L., … Diuk‐Wasser, M. A. (2014). Borrelia burgdorferi promotes the establishment of Babesia microti in the northeastern United States. PLoS One, 9(12), e115494. 10.1371/journal.pone.0115494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallahi, P., Elia, G., & Bonatti, A. (2017). Interferon‐gamma‐induced protein 10 in Lyme disease. La Clinica Terapeutica, 168(2), e146‐e150. 10.7417/CT.2017.1997 [DOI] [PubMed] [Google Scholar]
- Gautam, A., Dixit, S., Embers, M., Gautam, R., Philipp, M. T., Singh, S. R., … Dennis, V. A. (2012). Different patterns of expression and of IL‐10 modulation of inflammatory mediators from macrophages of Lyme disease‐resistant and ‐susceptible mice. PLoS One, 7(9), e43860. 10.1371/journal.pone.0043860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbia, J. A., Coleman, J. L., & Benach, J. L. (2004). Selective induction of matrix metalloproteinases by Borrelia burgdorferi via toll‐like receptor 2 in monocytes. The Journal of Infectious Diseases, 189(1), 113–119. [DOI] [PubMed] [Google Scholar]
- Glickstein, L. J., & Coburn, J. L. (2006). Short report: Association of macrophage inflammatory response and cell death after in vitro Borrelia burgdorferi infection with arthritis resistance. The American Journal of Tropical Medicine and Hygiene, 75(5), 964–967. [PubMed] [Google Scholar]
- Gowda, D. C. (2007). TLR‐mediated cell signaling by malaria GPIs. Trends in Parasitology, 23(12), 596–604. 10.1016/j.pt.2007.09.003 [DOI] [PubMed] [Google Scholar]
- Gronberg‐Hernandez, J., Sunden, F., Connolly, J., Svanborg, C., & Wullt, B. (2011). Genetic control of the variable innate immune response to asymptomatic bacteriuria. PLoS One, 6(11), e28289. 10.1371/journal.pone.0028289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg, L., Briles, D. E., & Eden, C. S. (1985). Evidence for separate genetic defects in C3H/HeJ and C3HeB/FeJ mice, that affect susceptibility to gram‐negative infections. Journal of Immunology, 134(6), 4118–4122. [PubMed] [Google Scholar]
- Hagberg, L., Hull, R., Hull, S., McGhee, J. R., Michalek, S. M., & Svanborg Eden, C. (1984). Difference in susceptibility to gram‐negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infection and Immunity, 46(3), 839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine, H., & Lien, E. (2003). Toll‐like receptors and their function in innate and adaptive immunity. International Archives of Allergy and Immunology, 130(3), 180–192. 10.1159/000069517 [DOI] [PubMed] [Google Scholar]
- Hersh, M. H., Ostfeld, R. S., McHenry, D. J., Tibbetts, M., Brunner, J. L., Killilea, M. E., … Keesing, F. (2014). Co‐infection of blacklegged ticks with Babesia microti and Borrelia burgdorferi is higher than expected and acquired from small mammal hosts. PLoS One, 9(6), e99348. 10.1371/journal.pone.0099348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwaldt, B. L., Linden, J. V., Bosserman, E., Young, C., Olkowska, D., & Wilson, M. (2011). Transfusion‐associated babesiosis in the United States: A description of cases. Annals of Internal Medicine, 155(8), 509–519. 10.1059/0003-4819-155-8-201110180-00362 [DOI] [PubMed] [Google Scholar]
- Hirschfeld, M., Kirschning, C. J., Schwandner, R., Wesche, H., Weis, J. H., Wooten, R. M., & Weis, J. J. (1999). Cutting edge: Inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll‐like receptor 2. Journal of Immunology, 163(5), 2382–2386. [PubMed] [Google Scholar]
- Homer, M. J., Aguilar‐Delfin, I., Telford, S. R., 3rd, Krause, P. J., & Persing, D. H. (2000). Babesiosis. Clinical Microbiology Reviews, 13(3), 451–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz, H. W., Aguero‐Rosenfeld, M. E., Holmgren, D., McKenna, D., Schwartz, I., Cox, M. E., & Wormser, G. P. (2013). Lyme disease and human granulocytic anaplasmosis coinfection: Impact of case definition on coinfection rates and illness severity. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America, 56(1), 93–99. 10.1093/cid/cis852 [DOI] [PubMed] [Google Scholar]
- Hoshino, K., Takeuchi, O., Kawai, T., Sanjo, H., Ogawa, T., Takeda, Y., … Akira, S. (1999). Cutting edge: Toll‐like receptor 4 (TLR4)‐deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. Journal of Immunology, 162(7), 3749–3752. [PubMed] [Google Scholar]
- Igarashi, I., Suzuki, R., Waki, S., Tagawa, Y., Seng, S., Tum, S., … Toyoda, Y. (1999). Roles of CD4(+) T cells and gamma interferon in protective immunity against Babesia microti infection in mice. Infection and Immunity, 67(8), 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki, A., & Medzhitov, R. (2010). Regulation of adaptive immunity by the innate immune system. Science, 327(5963), 291–295. 10.1126/science.1183021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahfari, S., Hofhuis, A., Fonville, M., van der Giessen, J., van Pelt, W., & Sprong, H. (2016). Molecular detection of tick‐borne pathogens in humans with tick bites and erythema Migrans, in The Netherlands. PLoS Neglected Tropical Diseases, 10(10), e0005042. 10.1371/journal.pntd.0005042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause, P. J., Foley, D. T., Burke, G. S., Christianson, D., Closter, L., Spielman, A., & Tick‐Borne Disease Study Group (2006). Reinfection and relapse in early Lyme disease. American Journal of Tropical Medicine and Hygiene, 75(6), 1090–1094. 10.4269/ajtmh.2006.75.1090. [DOI] [PubMed] [Google Scholar]
- Krause, P. J., Telford, S. R., Ryan, R., Hurta, A. B., Kwasnik, I., Luger, S., Niederman, J., Gerber, M., & Spielman, A. (1991). Geographical and temporal distribution of babesial infection in Connecticut.. Journal of Clinical Microbiology, 29(1), 1–4. 10.1128/jcm.29.1.1-4.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp, K. L., & Rice, N. A. (2015). Human coinfection with Borrelia burgdorferi and Babesia microti in the United States. Journal of Parasitology Research, 2015, 587131. 10.1155/2015/587131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause, P. J., Gewurz, B. E., Hill, D., Marty, F. M., Vannier, E., Foppa, I. M., … Spielman, A. (2008). Persistent and relapsing babesiosis in immunocompromised patients. Clinical Infectious Diseases, 46(3), 370–376. 10.1086/525852 [DOI] [PubMed] [Google Scholar]
- Krause, P. J., McKay, K., Gadbaw, J., Christianson, D., Closter, L., Lepore, T., … Spielman, A. (2003). Increasing health burden of human babesiosis in endemic sites. The American Journal of Tropical Medicine and Hygiene, 68(4), 431–436. [PubMed] [Google Scholar]
- Krause, P. J., McKay, K., Thompson, C. A., Sikand, V. K., Lentz, R., Lepore, T., … Deer‐Associated Infection Study, G . (2002). Disease‐specific diagnosis of coinfecting tickborne zoonoses: Babesiosis, human granulocytic ehrlichiosis, and Lyme disease. Clinical Infectious Diseases, 34(9), 1184–1191. 10.1086/339813 [DOI] [PubMed] [Google Scholar]
- Krause, P. J., Spielman, A., Telford, S. R., 3rd, Sikand, V. K., McKay, K., Christianson, D., … Persing, D. H. (1998). Persistent parasitemia after acute babesiosis. The New England Journal of Medicine, 339(3), 160–165. 10.1056/NEJM199807163390304 [DOI] [PubMed] [Google Scholar]
- Krause, P. J., Telford, S. R., 3rd, Spielman, A., Sikand, V., Ryan, R., Christianson, D., … Persing, D. H. (1996). Concurrent Lyme disease and babesiosis. Evidence for increased severity and duration of illness. JAMA, 275(21), 1657–1660. [PubMed] [Google Scholar]
- Kugeler, K. J., Schwartz, A. M., Delorey, M. J., Mead, P. S., & Hinckley, A. F. (2021). Estimating the frequency of Lyme Disease diagnoses, United States, 2010‐2018. Emerging Infectious Diseases, 27(2), 616–619. 10.3201/eid2702.202731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky, C. E., Pratt, C. L., Hilliard, K. A., Jones, J. L., & Brown, C. R. (2016). T cells exacerbate Lyme Borreliosis in TLR2‐deficient mice. Frontiers in Immunology, 7, 468. 10.3389/fimmu.2016.00468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz, E., Visintin, A., Lien, E., Fitzgerald, K. A., Monks, B. G., Kurt‐Jones, E. A., … Espevik, T. (2002). Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll‐like receptor 4‐MD‐2‐CD14 complex in a process that is distinct from the initiation of signal transduction. The Journal of Biological Chemistry, 277(49), 47834–47843. [DOI] [PubMed] [Google Scholar]
- Lien, E., Sellati, T. J., Yoshimura, A., Flo, T. H., Rawadi, G., Finberg, R. W., … Golenbock, D. T. (1999). Toll‐like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. The Journal of Biological Chemistry, 274(47), 33419–33425. [DOI] [PubMed] [Google Scholar]
- Liu, N., Montgomery, R. R., Barthold, S. W., & Bockenstedt, L. K. (2004). Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi‐infected mice. Infection and Immunity, 72(6), 3195–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommano, E., Bertaiola, L., Dupasquier, C., & Gern, L. (2012). Infections and coinfections of questing Ixodes ricinus ticks by emerging zoonotic pathogens in Western Switzerland. Applied and Environmental Microbiology, 78(13), 4606–4612. 10.1128/AEM.07961-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, E., Mira, J. P., Cornish, K. L., Arbour, N. C., & Schwartz, D. A. (2000). A novel polymorphism in the toll‐like receptor 2 gene and its potential association with staphylococcal infection. Infection and Immunity, 68(11), 6398–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, A. C., Schwartz, I., & Petzke, M. M. (2014). Borrelia burgdorferi RNA induces type I and III interferons via toll‐like receptor 7 and contributes to production of NF‐kappaB‐dependent cytokines. Infection and Immunity, 82(6), 2405–2416. 10.1128/IAI.01617-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockenhaupt, F. P., Cramer, J. P., Hamann, L., Stegemann, M. S., Eckert, J., Oh, N. R., … Schumann, R. R. (2006b). Toll‐like receptor (TLR) polymorphisms in African children: Common TLR‐4 variants predispose to severe malaria. The Journal of Communicable Diseases, 38(3), 230–245. [PubMed] [Google Scholar]
- Mockenhaupt, F. P., Cramer, J. P., Hamann, L., Stegemann, M. S., Eckert, J., Oh, N. R., … Schumann, R. R. (2006a). Toll‐like receptor (TLR) polymorphisms in African children: Common TLR‐4 variants predispose to severe malaria. Proceedings of the National Academy of Sciences of the United States of America, 103(1), 177–182. 10.1073/pnas.0506803102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro, M. H., Zegarra‐Moro, O. L., Bjornsson, J., Hofmeister, E. K., Bruinsma, E., Germer, J. J., & Persing, D. H. (2002). Increased arthritis severity in mice coinfected with Borrelia burgdorferi and Babesia microti . The Journal of Infectious Diseases, 186(3), 428–431. 10.1086/341452 [DOI] [PubMed] [Google Scholar]
- Morr, M., Takeuchi, O., Akira, S., Simon, M. M., & Muhlradt, P. F. (2002). Differential recognition of structural details of bacterial lipopeptides by toll‐like receptors. European Journal of Immunology, 32(12), 3337–3347. [DOI] [PubMed] [Google Scholar]
- Moutailler, S., Valiente Moro, C., Vaumourin, E., Michelet, L., Tran, F. H., Devillers, E., … Vayssier‐Taussat, M. (2016). Co‐infection of ticks: The rule rather than the exception. PLoS Neglected Tropical Diseases, 10(3), e0004539. 10.1371/journal.pntd.0004539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, P. D., Reed, K. D., & Hofkes, J. M. (1996). Immunoserologic evidence of coinfection with Borrelia burgdorferi, Babesia microti, and human granulocytic Ehrlichia species in residents of Wisconsin and Minnesota. Journal of clinical microbiology, 34(3), 724–727. 10.1128/jcm.34.3.724-727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, H. H., Pennock, N. D., Humphreys, J., Kirschning, C. J., & Cole, B. C. (2005). Engagement of toll‐like receptors by mycoplasmal superantigen: Downregulation of TLR2 by MAM/TLR4 interaction. Cellular Microbiology, 7(6), 789–797. 10.1111/j.1462-5822.2005.00511.x [DOI] [PubMed] [Google Scholar]
- Mayne, P. (2011). Emerging incidence of Lyme borreliosis, babesiosis, bartonellosis, and granulocytic ehrlichiosis in Australia. International Journal of General Medicine, 2011(4), 845–852. 10.2147/ijgm.s27336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne, P. (2014). Clinical determinants of Lyme borreliosis, babesiosis, bartonellosis, anaplasmosis, and ehrlichiosis in an Australian cohort. International Journal of General Medicine, 2015(8), 15–26. 10.2147/ijgm.s75825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, Y., Akashi, S., Nagafuku, M., Ogata, M., Iwakura, Y., Akira, S., … Miyake, K. (2002). Essential role of MD‐2 in LPS responsiveness and TLR4 distribution. Nature Immunology, 3(7), 667–672. 10.1038/ni809 [DOI] [PubMed] [Google Scholar]
- Nathaly Wieser, S., Schnittger, L., Florin‐Christensen, M., Delbecq, S., & Schetters, T. (2019). Vaccination against babesiosis using recombinant GPI‐anchored proteins. International Journal for Parasitology, 49(2), 175–181. 10.1016/j.ijpara.2018.12.002 [DOI] [PubMed] [Google Scholar]
- Oosting, M., Berende, A., Sturm, P., Ter Hofstede, H. J., de Jong, D. J., Kanneganti, T. D., … Joosten, L. A. (2010). Recognition of Borrelia burgdorferi by NOD2 is central for the induction of an inflammatory reaction. The Journal of Infectious Diseases, 201(12), 1849–1858. 10.1086/652871 [DOI] [PubMed] [Google Scholar]
- Ouhelli, H., & Schein, E. (1988). Effect of temperature on transovarial transmission of Babesia bigemina (Smith and Kilborne, 1893) in Boophilus annulatus (Say, 1821). Veterinary Parasitology, 26(3‐4), 229–235. 10.1016/0304-4017(88)90091-x. [DOI] [PubMed] [Google Scholar]
- Pahl, A., Kuhlbrandt, U., Brune, K., Rollinghoff, M., & Gessner, A. (1999). Quantitative detection of Borrelia burgdorferi by real‐time PCR. Journal of Clinical Microbiology, 37(6), 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panczuk, A., Tokarska‐Rodak, M., Koziol‐Montewka, M., & Plewik, D. (2016). The incidence of Borrelia burgdorferi, Anaplasma phagocytophilum and Babesia microti coinfections among foresters and farmers in eastern Poland. Journal of Vector Borne Diseases, 53(4), 348–354. [PubMed] [Google Scholar]
- Parthasarathy, G., & Philipp, M. T. (2015). Inflammatory mediator release from primary rhesus microglia in response to Borrelia burgdorferi results from the activation of several receptors and pathways. Journal of Neuroinflammation, 12, 60. 10.1186/s12974-015-0274-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piesman, J., Mather, T. N., Donahue, J. G., Levine, J., Campbell, J. D., Karakashian, S. J., & Spielman, A. (1986). Comparative prevalence of Babesia microti and Borrelia burgdorferi in four populations of Ixodes dammini in eastern Massachusetts. Acta Tropica, 43(3), 263–270. [PubMed] [Google Scholar]
- Primus, S., Akoolo, L., Schlachter, S., Gedroic, K., Rojtman, A. D., & Parveen, N. (2018). Efficient detection of symptomatic and asymptomatic patient samples for Babesia microti and Borrelia burgdorferi infection by multiplex qPCR. PLoS One, 13(5), e0196748. 10.1371/journal.pone.0196748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, S., Shering, M., Ogden, N. H., Lindsay, R., & Badawi, A. (2016). Toll‐like receptor cascade and gene polymorphism in host‐pathogen interaction in Lyme disease. Journal of Inflammation Research, 9, 91–102. 10.2147/JIR.S104790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli, A., Silaghi, C., Obiegala, A., Rudolf, I., Hubalek, Z., Foldvari, G., … Kazimirova, M. (2014). Ixodes ricinus and its transmitted pathogens in urban and Peri‐urban areas in Europe: New hazards and relevance for public health. Frontiers in Public Health, 2, 251. 10.3389/fpubh.2014.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht, T. A., Kirschning, C. J., Popp, B., Kastenbauer, S., Fingerle, V., Pfister, H. W., & Koedel, U. (2007). Borrelia garinii induces CXCL13 production in human monocytes through toll‐like receptor 2. Infection and Immunity, 75(9), 4351–4356. 10.1128/IAI.01642-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar, J. C., Duhnam‐Ems, S., La Vake, C., Cruz, A. R., Moore, M. W., Caimano, M. J., … Radolf, J. D. (2009). Activation of human monocytes by live Borrelia burgdorferi generates TLR2‐dependent and ‐independent responses which include induction of IFN‐beta. PLoS Pathogens, 5(5), e1000444. 10.1371/journal.ppat.1000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar, J. C., Pope, C. D., Moore, M. W., Pope, J., Kiely, T. G., & Radolf, J. D. (2005). Lipoprotein‐dependent and ‐independent immune responses to spirochetal infection. Clinical and Diagnostic Laboratory Immunology, 12(8), 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar, J. C., Pope, C. D., Sellati, T. J., Feder, H. M., Jr., Kiely, T. G., Dardick, K. R., … Lyme Disease, N. (2003). Coevolution of markers of innate and adaptive immunity in skin and peripheral blood of patients with erythema migrans. Journal of Immunology, 171(5), 2660–2670. [DOI] [PubMed] [Google Scholar]
- Schlachter, S., Chan, K., Marras, S. A. E., & Parveen, N. (2017). Detection and differentiation of Lyme spirochetes and other tick‐borne pathogens from blood using real‐time PCR with molecular beacons. Methods in Molecular Biology, 1616, 155–170. 10.1007/978-1-4939-7037-7_10 [DOI] [PubMed] [Google Scholar]
- Schromm, A. B., Lien, E., Henneke, P., Chow, J. C., Yoshimura, A., Heine, H., … Golenbock, D. T. (2001). Molecular genetic analysis of an endotoxin nonresponder mutant cell line: A point mutation in a conserved region of MD‐2 abolishes endotoxin‐induced signaling. The Journal of Experimental Medicine, 194(1), 79–88. 10.1084/jem.194.1.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze, T. L., Jordan, R. A., Healy, S. P., & Roegner, V. E. (2013). Detection of Babesia microti and Borrelia burgdorferi in host‐seeking Ixodes scapularis (Acari: Ixodidae) in Monmouth County, New Jersey. Journal of Medical Entomology, 50(2), 379–383. [DOI] [PubMed] [Google Scholar]
- Seixas, E., Moura Nunes, J. F., Matos, I., & Coutinho, A. (2009). The interaction between DC and Plasmodium berghei/chabaudi‐infected erythrocytes in mice involves direct cell‐to‐cell contact, internalization and TLR. European Journal of Immunology, 39(7), 1850–1863. 10.1002/eji.200838403 [DOI] [PubMed] [Google Scholar]
- Shimazu, R., Akashi, S., Ogata, H., Nagai, Y., Fukudome, K., Miyake, K., & Kimoto, M. (1999). MD‐2, a molecule that confers lipopolysaccharide responsiveness on toll‐like receptor 4. The Journal of Experimental Medicine, 189(11), 1777–1782. 10.1084/jem.189.11.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, O. S., Isberg, R. R., Akira, S., Uematsu, S., Behera, A. K., & Hu, L. T. (2008). Distinct roles for MyD88 and toll‐like receptors 2, 5, and 9 in phagocytosis of Borrelia burgdorferi and cytokine induction. Infection and Immunity, 76(6), 2341–2351. 10.1128/IAI.01600-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skariah, S., Arnaboldi, P., Dattwyler, R. J., Sultan, A. A., Gaylets, C., Walwyn, O., … Mordue, D. G. (2017). Elimination of Babesia microti is dependent on Intraerythrocytic killing and CD4+ T cells. Journal of Immunology, 199(2), 633–642. 10.4049/jimmunol.1601193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere, A. C. (2001). Lyme disease. The New England Journal of Medicine, 345(2), 115–125. [DOI] [PubMed] [Google Scholar]
- Suarez, C. E., Laughery, J. M., Bastos, R. G., Johnson, W. C., Norimine, J., Asenzo, G., … Goff, W. L. (2011). A novel neutralization sensitive and subdominant RAP‐1‐related antigen (RRA) is expressed by Babesia bovis merozoites. Parasitology, 138(7), 809–818. 10.1017/S0031182011000321 [DOI] [PubMed] [Google Scholar]
- Swanson, S. J., Neitzel, D., Reed, K. D., & Belongia, E. A. (2006). Coinfections acquired from Ixodes ticks. Clinical Microbiology Reviews, 19(4), 708–727. 10.1128/CMR.00011-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama, K., Rothenberg, R. J., & Barbour, A. G. (1987). Absence of lipopolysaccharide in the Lyme disease spirochete, Borrelia burgdorferi . Infection and Immunity, 55(9), 2311–2313. 10.1128/IAI.55.9.2311-2313.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi, O., & Akira, S. (2010). Pattern recognition receptors and inflammation. Cell, 140(6), 805–820. 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- Thekkiniath, J., Kilian, N., Lawres, L., Gewirtz, M. A., Graham, M. M., Liu, X., … Ben Mamoun, C. (2019). Evidence for vesicle‐mediated antigen export by the human pathogen Babesia microti . Life Science Alliance, 2(3), e201900382. 10.26508/lsa.201900382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thekkiniath, J., Mootien, S., Lawres, L., Perrin, B. A., Gewirtz, M., Krause, P. J., … Ben Mamoun, C. (2018). BmGPAC, an antigen capture assay for detection of active Babesia microti infection. Journal of Clinical Microbiology, 56(10). e00067–18. 10.1128/JCM.00067-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasiewicz, K., Chmielewska‐Badora, J., Zwolinski, J., & Murias‐Brylowska, E. (2016). Analysis of main T‐cell subsets and activated T supressor/cytotoxic cells in patients with Borrelia burgdorferi s. lato only infection and co‐infections with Anaplasma phagocytophilum, Bartonella spp. and Babesia microti . Annals of Agricultural and Environmental Medicine, 23(1), 111–115. 10.5604/12321966.1196864 [DOI] [PubMed] [Google Scholar]
- Totemeyer, S., Foster, N., Kaiser, P., Maskell, D. J., & Bryant, C. E. (2003). Toll‐like receptor expression in C3H/HeN and C3H/HeJ mice during Salmonella enterica serovar typhimurium infection. Infection and Immunity, 71(11), 6653–6657. 10.1128/iai.71.11.6653-6657.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier, E., Borggraefe, I., Telford, S. R., Menon, S., Brauns, T., Spielman, A., … Wortis, H. H. (2004). Age‐associated decline in resistance to Babesia microti is genetically determined. Journal of Infectious Diseases, 189(9), 1721–1728. 10.1086/382965 [DOI] [PubMed] [Google Scholar]
- Wang, G., Ojaimi, C., Iyer, R., Saksenberg, V., McClain, S. A., Wormser, G. P., & Schwartz, I. (2001). Impact of genotypic variation of Borrelia burgdorferi sensu stricto on kinetics of dissemination and severity of disease in C3H/HeJ mice. Infection and Immunity, 69(7), 4303–4312. 10.1128/Iai.69.7.4303-4312.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Su, L., Morin, M. D., Jones, B. T., Whitby, L. R., Surakattula, M. M., … Beutler, B. (2016). TLR4/MD‐2 activation by a synthetic agonist with no similarity to LPS. Proceedings of the National Academy of Sciences of the United States of America, 113(7), E884‐893. 10.1073/pnas.1525639113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, C. A., Ravasi, T., Faulkner, G. J., Carninci, P., Okazaki, Y., Hayashizaki, Y., … Hume, D. A. (2003). Genetic control of the innate immune response. BMC Immunology, 4, 5. 10.1186/1471-2172-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling, D., & Jungi, T. W. (2003). TOLL‐like receptors linking innate and adaptive immune response. Veterinary Immunology and Immunopathology, 91(1), 1–12. [DOI] [PubMed] [Google Scholar]
- Western, K. A., Benson, G. D., Gleason, N. N., Healy, G. R., & Schultz, M. G. (1970). Babesiosis in a massachusetts resident. New England Journal of Medicine, 283(16), 854–856. 10.1056/nejm197010152831607. [DOI] [PubMed] [Google Scholar]
- Welc‐Falęciak, R., Hildebrandt, A., & Siński, E., & (2010). Co‐infection with Borrelia species and other tick‐borne pathogens in humans: two cases from Poland. Annals of Agricultural and Environmental Medicine, 17(2), 309–313. [PubMed] [Google Scholar]
- Wooten, R. M., Ma, Y., Yoder, R. A., Brown, J. P., Weis, J. H., Zachary, J. F., … Weis, J. J. (2002a). Toll‐like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi . Journal of Immunology, 168(1), 348–355. [DOI] [PubMed] [Google Scholar]
- Wooten, R. M., Ma, Y., Yoder, R. A., Brown, J. P., Weis, J. H., Zachary, J. F., … Weis, J. J. (2002b). Toll‐like receptor 2 plays a pivotal role in host defense and inflammatory response to Borrelia burgdorferi . Vector Borne and Zoonotic Diseases, 2(4), 275–278. 10.1089/153036602321653860 [DOI] [PubMed] [Google Scholar]
- Wooten, R. M., & Weis, J. J. (2001). Host‐pathogen interactions promoting inflammatory Lyme arthritis: Use of mouse models for dissection of disease processes. Current Opinion in Microbiology, 4(3), 274–279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study. All information that is part of this study are included in the manuscript.