

Abstract
Ewing sarcomas are driven by EWS-ETS fusions, most commonly EWS-FLI1, which promote widespread metabolic reprogramming, including activation of serine biosynthesis. We previously reported that serine biosynthesis is also activated in Ewing sarcoma by the scaffolding protein menin through as yet undefined mechanisms. Here, we investigated whether EWS-FLI1 and/or menin orchestrate serine biosynthesis via modulation of ATF4, a stress-response gene that acts as a master transcriptional regulator of serine biosynthesis in other tumors. Our results show that in Ewing sarcoma, ATF4 levels are high and that ATF4 modulates transcription of core serine synthesis pathway (SSP) genes. Inhibition of either EWS-FLI1 or menin leads to loss of ATF4, and this is associated with diminished expression of SSP transcripts and proteins. We identified and validated an EWS-FLI1 binding site at the ATF4 promoter, indicating that the fusion can directly activate ATF4 transcription. In contrast, our results suggest that menin-dependent regulation of ATF4 is mediated by transcriptional and post-transcriptional mechanisms. Importantly, our data also reveal that the downregulation of SSP genes that occurs in the context of EWS-FLI1 or menin loss is indicative of broader inhibition of ATF4-dependent transcription. Moreover, we find that menin inhibition similarly leads to loss of ATF4 and the ATF4-dependent transcriptional signature in MLL-rearranged B-cell acute lymphoblastic leukemia, extending our findings to another cancer in which menin serves an oncogenic role.
Implications:
These studies provide new insights into metabolic reprogramming in Ewing sarcoma and also uncover a previously undescribed role for menin in the regulation of ATF4.
Keywords: Ewing sarcoma, menin, ATF4 pathway, leukemia, serine biosynthesis
Introduction
Ewing sarcoma is an aggressive bone and soft tissue tumor and the second most common pediatric bone tumor. Despite maximally intensive therapy, Ewing sarcoma remains lethal for a third of patients, and survival rates for patients with metastatic or relapsed disease are dismal [1–3]. Furthermore, the current standard of care leaves patients with profound and devastating long-term toxicities [4–6]. Ewing sarcoma is defined by the presence of EWS-ETS fusion proteins, most commonly EWS-FLI1, which results from a t(11;22) chromosomal translocation, fusing the N-terminal domain of EWSR1 to the DNA-binding domain of FLI1 [2]. This tumor-initiating event leads to malignant transformation of the cell of origin, which is presumed to be a stem or progenitor cell of mesoderm or neural crest lineage [7]. EWS-FLI1 initiates and maintains tumorigenicity through diverse functions that converge on disruption of normal transcriptional regulation and splicing [8–11]. In particular, EWS-ETS proteins promote widespread epigenetic and transcriptional reprogramming by binding and creating de novo enhancers at GGAA microsatellites in intergenic regions and by direct promoter binding [8–10]. Significantly, aside from the presence of the pathognomonic gene fusions, Ewing sarcoma tumors are genomically quiet, with few other recurrent mutations [12–14]. Thus, the molecular mechanisms underlying tumor initiation and progression are inherently linked to the function of EWS-ETS proteins.
Metabolic reprogramming is essential for tumor cell survival under conditions of stress, including both cell intrinsic (e.g. genotoxic, proliferative, protein folding) and extrinsic (e.g. hypoxia, nutrient deprivation, growth constraint) stressors. In recent years it has become apparent that tumor cells orchestrate complex and interconnected enzymatic programs to retain metabolic homeostasis under conditions of stress, including the regulation of amino acid biosynthesis pathways [15]. Serine is a non-essential amino acid that, in addition to its proteinogenic role, is used for the maintenance of redox, epigenetic, and proliferative homeostasis. Significantly, many cancers display enhanced activation of and dependence on the serine synthesis pathway (SSP) for tumor survival and propagation [16]. Recent studies from our group and others have reported that Ewing sarcomas are highly dependent on SSP activity, and that 3-phosphoglycerate dehydrogenase (PHGDH), the rate-limiting enzyme in the pathway, is highly overexpressed by Ewing sarcoma cell lines and patient tumors [17–20]. Knockdown of EWS-FLI1 leads to loss of SSP gene expression and pathway activation, revealing that the fusion contributes directly or indirectly to SSP activation [17, 19, 20]. In addition, we reported that the scaffolding protein and trithorax complex protein menin also maintains SSP activity in Ewing sarcoma cells, and that this is mediated in part through epigenetic activation of PHGDH [18]. Nevertheless, the precise molecular mechanisms that control hyperactivation of the SSP in Ewing sarcoma remain to be elucidated.
In melanoma and breast cancers, the SSP is often activated as a result of amplification of the PHGDH locus [21, 22]. In contrast, in non-small cell lung cancer and other solid tumors that lack PHGDH amplification, activation of the SSP has been attributed to Activating Transcription Factor 4 (ATF4), a master transcriptional regulator of amino acid metabolism and stress responses [15, 16, 23–27]. ATF4 orchestrates a distinct gene expression program that allows cancer cells to adapt to an increased demand for metabolites and macromolecules, supporting growth and survival in harsh environments [15]. In the current work, we investigated whether ATF4 contributes to SSP activation in Ewing sarcoma and whether EWS-FLI1- and/or menin-mediated effects on this metabolic pathway involve ATF4. Our findings provide evidence that ATF4 does indeed act as a central node in SSP modulation in Ewing sarcoma, acting downstream of both EWS-FLI1 and menin. Importantly, we also find that the effects of menin on the ATF4-SSP response are not limited to Ewing sarcoma but are also evident in mixed lineage leukemia (MLL)-rearranged (MLLr) leukemia cells. These results provide novel insights into the molecular mechanisms by which cellular metabolism is rewired in Ewing sarcoma. In addition, they reveal a previously undescribed connection between menin and ATF4.
Materials and Methods
Cell Lines
Ewing sarcoma cell lines were obtained from the Children’s Oncology Group (COG) cell bank (cogcell.org). U2OS, HT-1080, SW 1353, and HEK 293FT cells were obtained directly from ATCC. Human marrow stromal cells (hMSCs) were kindly provided by Dr. Kurt Hankenson at the University of Michigan. Cells were not cultured past 20 passages. A673, A4573, and TC32 cells were cultured in RPMI 1640 (Corning). CHLA10 cells were cultured in IMDM media (Gibco), supplemented with 1X Insulin-Transferrin-Selenium (Gibco) and 20% fetal bovine serum. 293FT, HT-1080, and SW 1353 cell lines were cultured in DMEM medium, high glucose (Gibco). DMEM media for SW 1353 cells was supplemented with 1% antibiotic-antimycotic (Gibco) without additional glutamine. U2OS cells were cultured in McCoy’s 5A medium (Gibco). hMSCs were cultured in MEM Alpha medium (Gibco) supplemented with 1% antibiotic-antimycotic (Gibco) and 20% fetal bovine serum. All media were supplemented with 10% fetal bovine serum (FBS—Atlas Biologicals) and 1% L-glutamine (Life Technologies), unless otherwise noted, and grown at 37°C, 5% CO2. Identities were confirmed by short tandem repeat (STR) profiling. All cell lines were routinely tested for mycoplasma contamination as described previously [18].
Chemical Synthesis of Menin-MLL Inhibitors and Treatments
MI-503 and MI-3454 were prepared using the synthetic procedures previously reported [28, 29]. RNA from MI-3454-treated RS4;11 and MOLM13 leukemia cell lines was provided by Dr. Jolanta Grembecka’s laboratory at the University of Michigan. Cells were treated as previously published, with 25 or 50 nM MI-3454 for 7 days [29]. For MI-503 treatment studies, cells were plated in 100 mm dishes, media was changed the following day and supplemented with 3 µM MI-503 (IC50) or DMSO control, and cells were collected after 96 hours. For Ewing sarcoma cell lines, cells were plated at the following densities: 0.75 X 106 (A673), 0.25 X106 (A4573), and 3.25 X 106 (TC32) cells/100 mm dish.
Generation of Cell Lines Expressing shRNA, or Over-expressing ATF4
For the following plasmids: pLKO.1 shNS (non-silencing control), shATF4, shFLI1, and shKMT2B (Sigma-Aldrich MISSION shRNA library); pLenti CMV/Puro empty and mAtf4 (Addgene); and TRIPZ Inducible Lentiviral shNS and shMEN1 (Dharmacon); concentrated (10X) lentivirus production was carried out by the University of Michigan Vector Core facility or prepared in the laboratory following standard procedures [30]. In brief, plasmids were co-transfected with pCD/NL-BH*DDD (Addgene) and pMD2.G (Addgene) plasmids into 293FT packaging cells using PEI. 24 hours post-transfection, sodium butyrate was added to plates for 6 hours. Viral supernatant was collected 48 hours after transfection and filtered through 40 µM filters. Ewing sarcoma cell lines were plated in complete media in 100 mm dishes. Viral supernatant was added to the media at 1X concentration and removed after 24-hour incubation. Cells were selected by supplementing the media with 1.5 µg/mL puromycin (Gibco) for 48 hours before collection. TRIPZ inducible shNS and shMEN1 cells were grown in tetracycline (tet)-free FBS-containing media (Takara Bio). Viral supernatant was added to cells in serum-free media, and media was supplemented with 5 mL complete tet-free media 6–8 hours post-transduction. Media was replaced with puromycin-selective media 48 hours post-transduction. Cells underwent two rounds of transduction with inducible shNS and shMEN lentivirus to maximize efficiency. Doxycycline was added to cells at 1 µg/mL and replenished every 24 hours, and shRNA expression was monitored by expression of RFP. See Supplementary Materials for shRNA sequences and plasmid information. The mAtf4 plasmid was kindly provided by Dr. Lewis Cantley’s laboratory at Weill Cornell Medicine. mAtf4 has sufficient sequence homology to hATF4 and has been used previously in other human cancer studies [23].
Proliferation Assays
For Trypan blue exclusion proliferation assays, shATF4 knockdown and shNon-silencing (shNS) control cells were seeded at 100,000 cells per dish in 60 mm dishes after puromycin selection. Floating and adherent cells were collected at day 3 and day 6 after plating, and viable cells were quantified using a Countess Automated Cell Counter (Invitrogen). Real-time cell analysis (RTCA) of cell proliferation was monitored using the xCELLigence DP system (Agilent). Before cell seeding, E-plates were coated with 0.2% gelatin and equilibrated for 1 hour at 37°C, 5% CO2 with 100 µL of media per well. 5 × 103 (A673), or 3 × 103 (A4573) cells were plated per well to a final volume of 200 µL, and the plate was equilibrated for 30 minutes.
Western Blotting
Cells were detached with trypsin-EDTA (0.05%) (Gibco), washed with PBS (Gibco), and lysed in RIPA buffer (Thermo Fisher) supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail tablets (Roche). Cleared supernatants were subjected to protein quantification by DC (detergent compatible) protein assay (Bio-Rad). Western blot was performed using the Bio-Rad Mini-PROTEAN Tetra System. Proteins were resolved by SDS-PAGE (4–15%), transferred to 0.45 µM nitrocellulose membranes, and blocked in 1:1 TBS Odyssey Blocking Buffer (LI-COR) and 1X TBST for 1 hour at room temperature. Membranes were incubated rotating overnight at 4°C with primary antibody. See Supplementary Materials for list of antibodies and dilutions. Membranes were then washed 4 times for 5 minutes each in TBST and probed with secondary antibody IRDye 680RD Goat anti-Mouse IgG 1:15,000, IRDye 800CW Goat anti-Rabbit IgG 1:10,000 (LI-COR), or donkey anti-Rabbit IgG peroxidase-linked secondary (Cytiva) 1:10,000. Quantification was performed using LI-COR Image Studio software or Bio-rad Image Lab Software.
Quantitative Real-Time PCR
Total RNA was extracted from cells at the same time as protein collection using Quick-RNA MiniPrep Kit (Zymo Research), and RNA was subjected to reverse transcription using iSCRIPT cDNA Synthesis Kit (Bio-Rad) following the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was performed using fluorescent universal SYBR-Green Supermix (Bio-Rad) for designed primers or TaqMan Fast Universal PCR Master Mix (Applied Biosystems) with TaqMan probes. Analysis was performed in triplicate using the Light-Cycler 480 System (Roche) and average Cp values were normalized relative to the geometric mean of two housekeeping genes (18s and HPRT, or 18s and EEF1A1 for SYBR; 18s and B2M for TaqMan Assay). Primer sequences and TaqMan probes are provided in Supplementary Table S1.
Chromatin Immunoprecipitation (ChIP) Quantitative PCR
Chromatin immunoprecipitation (ChIP) to assess enrichment for ATF4, H3K4me3, and EWS-FLI1 was performed using the Zymo-Spin ChIP kit (Zymo Research) with Protein G-Dynabeads (Life Technologies) following the manufacturer’s protocol. Briefly, 5 million cells were harvested for ChIP (1 million cells per 1 mL ChIP reaction). Cells were formaldehyde fixed and chromatin was fragmented using the Biorupter Pico Sonicator (Diagenode) (30s on/30s off for 30 cycles). Chromatin was incubated with the following antibodies: 0.34 µg/IP anti-ATF4 (ATF-4 (D4B8) Rabbit mAb, Cell Signaling 11815), 0.5 µg/IP anti-H3K4me3 ChIP-seq grade (Diagenode C15410003–50), 5 µg/IP anti-FLI1 ChIP Grade (abcam ab15289), 5 µg/IP Rabbit IgG polyclonal isotype control (Abcam ab37415), overnight on a rotator at 4°C. Antibody-chromatin complexes were incubated with Protein G-Dynabeads for 2 hours. Washes, elution, reverse crosslinking, and proteinase K digestion were carried out per manufacturer’s protocol. DNA was eluted in 20 µL, and ChIP-qPCR was performed as described above. Primer sequences for the ATF4 and SSP gene promoters, and negative control regions are detailed in Supplementary Table S1.
Motif Analysis
Bromouridine sequencing (Bru-Seq) of MI-503-treated Ewing sarcoma cell lines was performed previously [18]. Transcripts with absolute fold change greater than 2.0 and mean RPKM > 0.5 were categorized as differentially expressed. Promoter regions of downregulated genes (fold change < −2.0), were analyzed for enriched motifs using the findMotifs.pl function from the HOMER analysis suite. Genes were searched within 2 Kb of their transcription start site for motifs of 8, 10, or 12 nucleotides in length to identify enriched motifs in gene promoters [31].
Microarrays, RNA-Seq, and ChIP-Seq Datasets
The mRNA expression of ATF4 in Ewing sarcoma patient tumors was determined using three previously published microarray datasets: GSE63157 [32], GSE34620 [33], and GSE17679 [34]. RNA-sequencing data for EWS-FLI1 knockdown are from three independent studies, publicly available at GSE122535 [35], GSE103843 [17], and GSE61950 [8]. To determine the differentially expressed genes between the experimental groups, read counts were analyzed with DESeq2 package (v 1.22.2) in R. Heatmap for GSE122535 [35] was generated using normalized expression values from DESeq2 and the Morpheus matrix visualization and analysis software (Morpheus, https://software.broadinstitute.org/morpheus). Bromouridine sequencing (nascent RNA-seq) of MI-503-treated Ewing sarcoma cells was re-analyzed from previously reported data [18]. RNA-sequencing for VTP-50469 treatment in RS4;11 and MOLM13 cell lines and CRISPR-mediated menin knockout in MOLM13 cells are publicly available at GSE127506 and GSE137813, respectively [36]. The ChIP-seq datasets we acquired for FLI1, H3K27Ac, and H3K4me3 are publicly available at NCBI Gene Expression Omnibus (GEO) under the GEO accession: GSE61953 [8], and NCBI Sequencing Read Archive (SRA) under the accession number SRA096176 and accessible at: http://www.medical-epigenomics.org/papers/tomazou2015/ [37]. Data was visualized using the Integrative Genomics Viewer (IGV) or UCSC Genome Browser platforms, respectively, at each region of interest.
Statistical Analysis
All data is plotted using Prism (GraphPad) and bar plots represent the mean ± SEM. Prism was used to perform two-tailed, unpaired t-test analysis. P< 0.05 was considered statistically significant. For volcano plots of RNA-seq data, differential expression values greater than 2-fold or less than −2-fold and adjusted P< 0.05 were considered significant, and cutoffs are indicated on plot.
Results:
ATF4 regulates serine biosynthesis pathway expression in Ewing sarcoma
To determine the molecular mechanism by which the SSP is activated in Ewing sarcoma, we first investigated ATF4, given that it regulates the pathway in other cancers [16, 23–27]. Gene expression studies of tumor biopsies show that ATF4 transcript expression is highly variable in Ewing sarcoma tumors in vivo (Fig. 1A). Our studies of Ewing sarcoma cell lines in vitro revealed similarly variable expression of ATF4 mRNA and protein, but levels were generally higher in Ewing than non-Ewing cells under ambient conditions (Fig. 1B). Given its status as a highly dynamic and stress-induced transcription factor, this variability in expression is not unexpected [38]. Knockdown of ATF4 in Ewing sarcoma cells led to reduced cell proliferation, as determined by direct cell counting (Fig. 1C) and real-time proliferation assays (Supplementary Fig. S1A), indicating that, in standard culture conditions, ATF4 supports the proliferative state. The SSP generates serine de novo by diverting the 3-phosphogycerate intermediate from glycolysis via three enzymatic steps requiring the enzymes PHGDH, phosphoserine aminotransferase (PSAT1), and phosphoserine phosphatase (PSPH) [39]. Loss of ATF4 was accompanied by marked and reproducible downregulated expression of all three core SSP transcripts (Fig. 1D) and proteins (Fig. 1E). Chromatin immunoprecipitation qPCR (ChIP-qPCR) studies confirmed that ATF4 binding is enriched at the PHGDH and PSAT1 promoters, supporting direct transcriptional regulation of these genes by ATF4 (Fig. 1F). Of note, we were unable to successfully design primers capturing the PSPH promoter region due to high sequence homology with other regions in the genome, which we believe has precluded other groups from similarly investigating ATF4 binding at this region [23, 24]. Having shown that loss of ATF4 resulted in loss of SSP gene expression, we next transduced cells with a mouse Atf4 cDNA overexpression construct (ATF4 OE) and discovered that forced over-expression of the protein resulted in a further increase in SSP genes and proteins (Fig. 1G). These results demonstrate that in Ewing sarcoma, like other cancers, ATF4 directly regulates expression of core SSP genes.
Figure 1. ATF4 Modulates Serine Biosynthesis Pathway Genes in Ewing sarcoma.
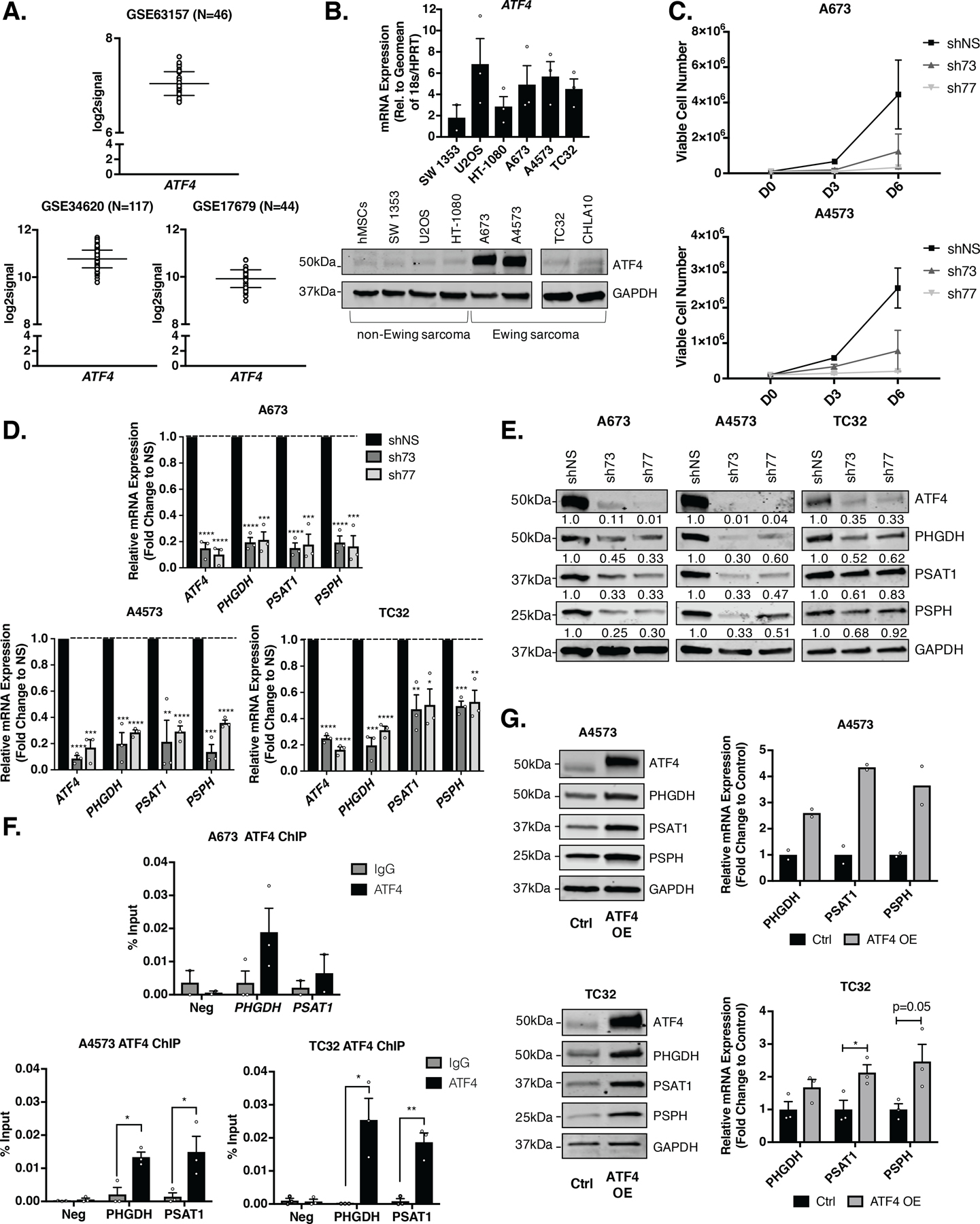
A,ATF4 gene expression in patient tumors from three independently published Ewing sarcoma datasets [32–34]. B, ATF4 mRNA and protein levels in Ewing sarcoma cell lines and non-Ewing mesenchymal cell lines (N=3). C, Trypan blue exclusion proliferation assay after shRNA knockdown of ATF4 (N=2). D, qRT-PCR and E, representative western blot of SSP (PHGDH, PSAT1, and PSPH) mRNA and protein (A673 & A4573– 30 µg, TC32– 50 µg) in Ewing sarcoma cell lines after 96 hours of shATF4 knockdown (N=3). F, Chromatin immunoprecipitation qRT-PCR (ChIP-qPCR) for ATF4 at PHGDH and PSAT1 gene promoters. Negative control is a promoter region in chr2 without ATF4 binding (N=3). G, Representative western blots and qRT-PCR for SSP expression in ATF4 overexpressing (OE) Ewing sarcoma cells (A4573– 30 µg, TC32– 50 µg) (N=2, N=3). Error bars represent SEM from independent biological replicates. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; Two-tailed t-test.
EWS-FLI1 directly binds to the ATF4 gene promoter
Although a role for EWS-FLI1 in regulation of the SSP has been described previously [17, 19, 20], and direct EWS-FLI1 binding to regulatory regions of all SSP genes has been reported [17], the mechanistic basis of fusion-mediated SSP activation is controversial and has yet to be fully elucidated. To begin to decipher whether EWS-FLI1 may function to regulate the SSP, at least in part via ATF4, we stably knocked down EWS-FLI1 using FLI1-targeted lentiviral shRNA in three Ewing sarcoma cell lines (A673, A4573, TC32). Ewing sarcomas do not express wildtype FLI1, permitting targeted knockdown of the fusion. Our results confirmed the published data, demonstrating that loss of EWS-FLI1 leads to downregulation of SSP gene and protein expression (Fig. 2A-B) [17, 19, 20]. In addition, following EWS-FLI1 knockdown, cells show a concomitant and time-dependent loss of ATF4 and ATF4 target gene expression (Fig. 2C-D). These findings were corroborated in a fourth Ewing sarcoma cell line, CHLA10 (Supplementary Fig. S2A). In all cell lines, loss of ATF4 protein was coincident with loss of gene expression, suggesting that EWS-FLI1 may regulate ATF4 transcription. To address whether EWS-FLI1 might directly bind and activate the ATF4 locus, we interrogated public ChIP-seq data for potential EWS-FLI1 binding sites in the ATF4 promoter and more distal GGAA microsatellites that may act as de novo enhancers [40]. This analysis revealed a strong EWS-FLI1 binding peak in the ATF4 promoter region, a region that was also defined by this group to be among a core set of EWS-FLI1 binding sites (Fig. 2E) [8]. Consistent with gene activation, the ATF4 promoter was also enriched with H3K4me3 and H3K27Ac marks (Fig. 2E). In contrast, enrichment of EWS-FLI1 was not consistently evident at the PHGDH promoter or the other SSP gene promoters, despite the presence of the active chromatin-associated histone modifications (Fig. 2E and Supplementary Fig. S2B) [8, 37]. Preferential binding of EWS-FLI1 to the ATF4 promoter and the absence of binding at the PHGDH promoter was validated by ChIP-qPCR in A673 cells in comparison to the EWS-FLI1-negative U2OS osteosarcoma cell line (Fig. 2F). Together these data demonstrate that EWS-FLI1 positively modulates expression of ATF4 mRNA transcription, and that this modulation is likely to, at least in part, mediate fusion-dependent regulation of the SSP in Ewing sarcoma.
Figure 2. EWS-FLI1 Directly Binds to and Activates ATF4.
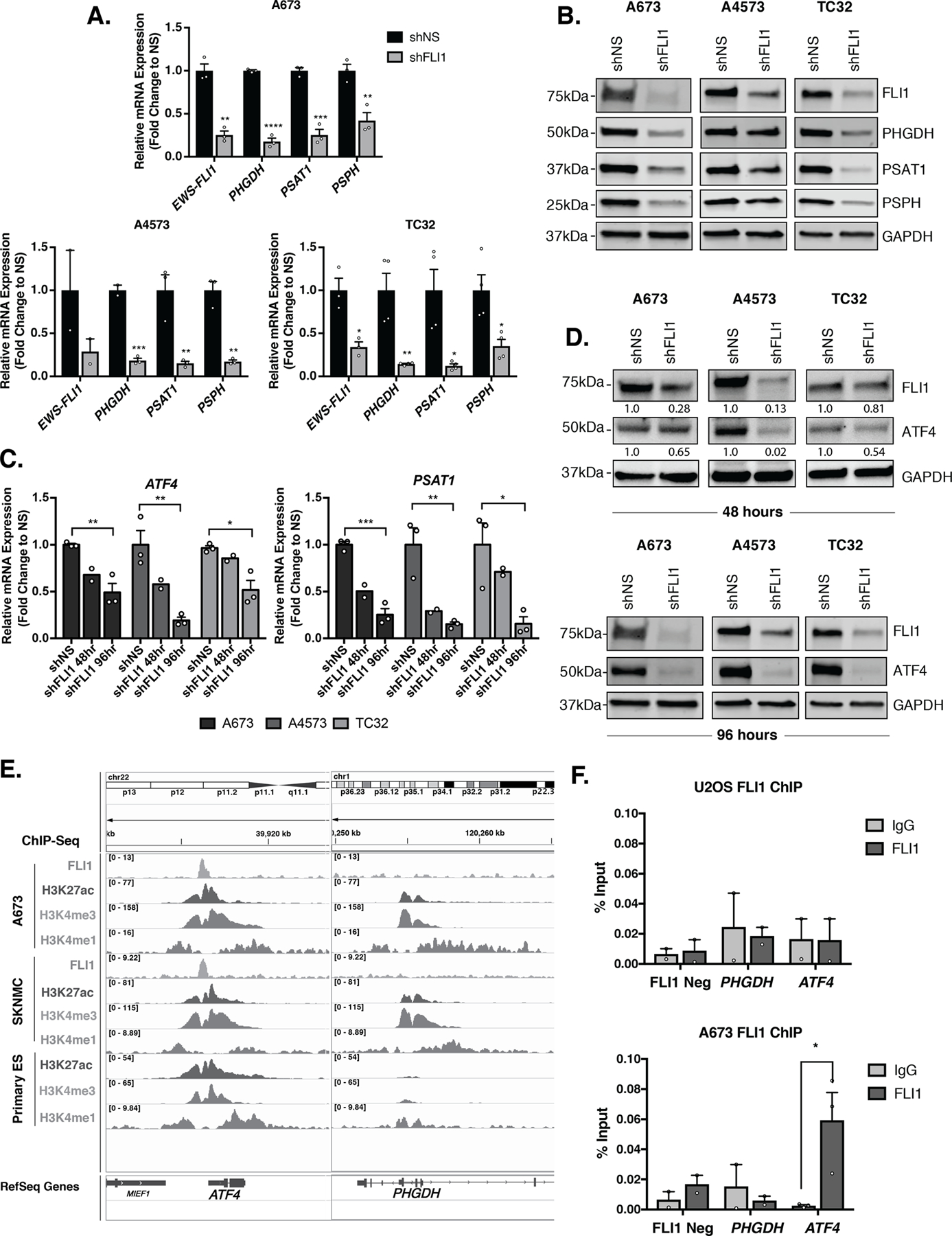
A, qRT-PCR and B, representative western blot (30 µg) for FLI1 (EWS-FLI1), and SSP (PHGDH, PSAT1, and PSPH) mRNA and protein levels after 96 hours of EWS-FLI1 knockdown (N=3, N=4). C, qRT-PCR for ATF4 and PSAT1 mRNA at 48 (N=2) and 96 (N=3) hours post-shFLI1 knockdown in A673, A4573, and TC32 cells. D, Representative western blot (30 µg) for FLI1 (EWS-FLI1) and ATF4 protein levels after 48 and 96 hours of FLI1 (EWS-FLI1) knockdown (N=3). Western blots depicted for FLI1 and GAPDH are the same as those presented in panel B and are reproduced here for ease of comparison. E, Integrative Genomics Viewer (IGV) screenshot of publicly available ChIP-seq tracks for FLI1 (EWS-FLI1), H3K27Ac, and H3K4me3 at the ATF4 and PHGDH gene promoters in Ewing sarcoma cell lines and primary tumors [8]. F, ChIP-qPCR for FLI1 (EWS-FLI1) at the PHGDH and ATF4 gene promoters (N=3). Negative control is a region in chr2 without EWS-FLI1 binding. Error bars represent SEM from independent biological replicates. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; Two-tailed t-test.
Menin inhibition leads to loss of ATF4 and associated downregulation of the SSP
Menin is a ubiquitously expressed scaffolding protein whose best characterized function is its role as an MLL binding partner in the context of epigenetic trithorax complexes [41]. Disruption of menin: MLL interactions in MLLr leukemias leads to tumor regression, and menin inhibition has also been shown to slow the growth of some solid tumors, including prostate cancer and Ewing sarcoma [28, 42, 43]. Our previously published work demonstrated that pharmacologic and genetic menin loss-of-function in Ewing sarcoma leads to downregulation of SSP gene expression and activity [18]. We first compared menin protein expression in a panel of EWS-FLI1 fusion-positive Ewing sarcoma cell lines and other malignant and non-malignant mesenchymal cells: human mesenchymal stem cells (hMSCs), immortalized fibroblasts (293FT), chondrosarcoma (SW 1353), osteosarcoma (U2OS), and fibrosarcoma (HT-1080). As we previously published, menin expression is higher in Ewing sarcoma cell lines than in non-malignant human mesenchymal stem cells (hMSCs) [43]. Levels of menin are also high in immortalized fibroblasts (293FT) and U2OS cells but are low in soft tissue sarcoma cell lines (HT-1080, SW 1353) (Fig. 3A). To determine whether menin inhibition influences ATF4 expression, we exposed cells to the menin inhibitor MI-503, as previously described [18, 43]. Consistent with our published data [43], treatment with MI-503 suppressed cell growth, albeit to a lesser degree than ATF4 knockdown (Supplementary Fig. S1A-B). In addition, MI-503 induced a time-dependent reduction in menin protein (Fig. 3B, Supplementary Fig. S3A-B), with no change in menin mRNA (Supplementary Fig. S3D) [43]. Given the consistent and reproducible loss of menin protein at 96 hours, we next assessed levels of ATF4 and SSP genes at this timepoint. As shown, menin loss was accompanied by a dramatic reduction in ATF4 mRNA and protein, and by reduction in SSP gene and protein expression, as previously reported (Fig. 3B-D) [18]. Moreover, ChIP-qPCR studies confirm that ATF4 enrichment at the PHGDH and PSAT1 promoters is diminished following menin inhibition (Fig. 3E). Thus, inhibition of menin leads to loss of ATF4, loss of ATF4-binding at SSP gene promoters, and diminished SSP gene transcription. Importantly, levels of EWS-FLI1 were unaffected (Supplementary Fig. S3A-B), revealing that menin inhibition does not inhibit ATF4 via downregulation of the fusion protein.
Figure 3. Menin Inhibition Leads to Loss of ATF4 in Ewing sarcoma.
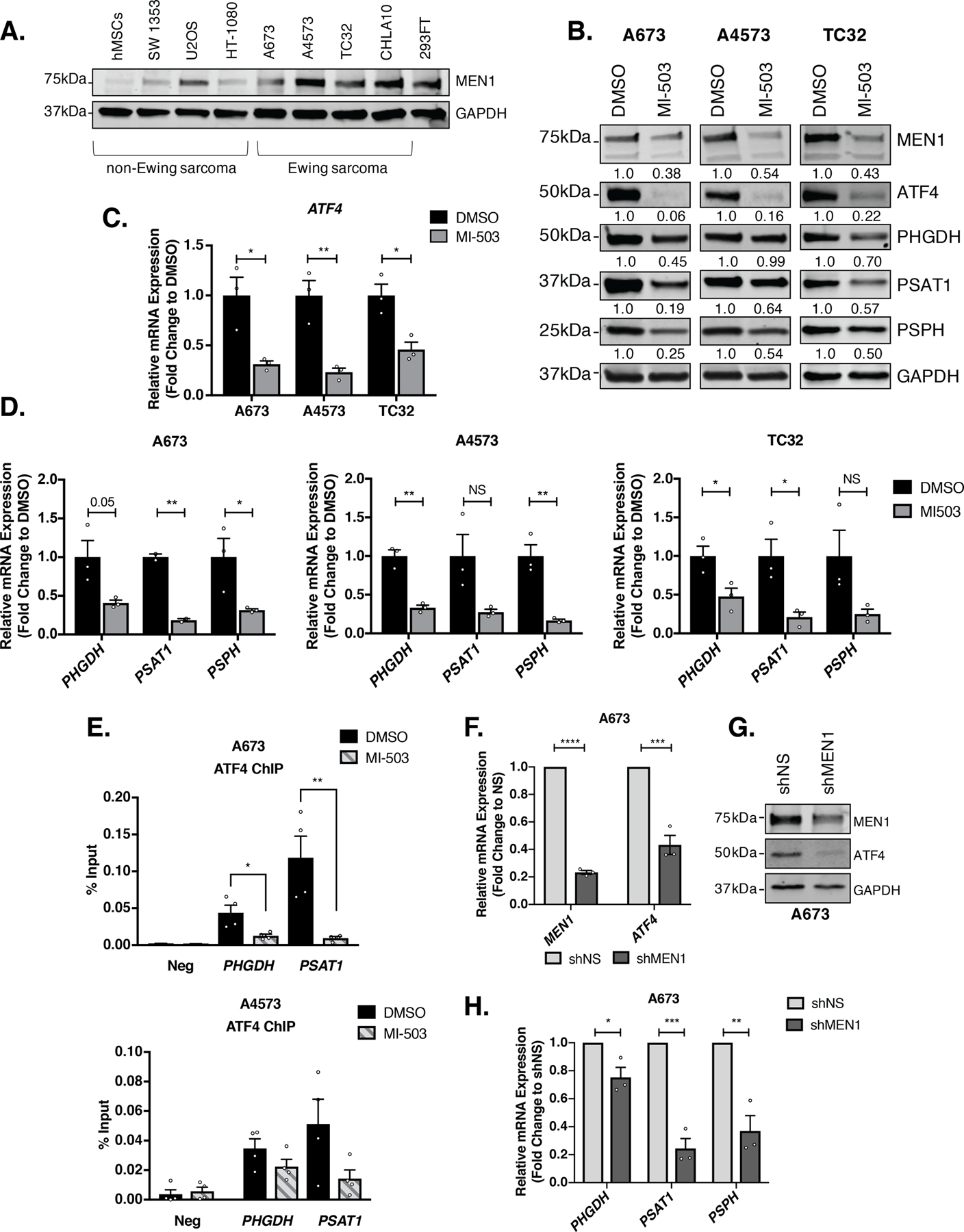
A, Representative western blot (50 µg) of MEN1 in a panel of Ewing sarcoma and non-Ewing sarcoma cell lines. B, Representative western blot (A673 & A4573– 30 µg, TC32– 50 µg) for ATF4 and SSP (PHGDH, PSAT1, and PSPH) protein after 96 hours of treatment with 3 µM MI-503 or DMSO control (N=3). C, qRT-PCR for ATF4 and D, SSP (PHGDH, PSAT1, and PSPH) mRNA in three Ewing sarcoma cell lines after 96 hours of treatment with 3 µM MI-503 or DMSO (N=3). E, ChIP-qPCR for ATF4 at PHGDH and PSAT1 gene promoters after 96 hours of 3 µM MI-503 treatment compared to DMSO control (N=4). F, qRT-PCR for MEN1 and ATF4 mRNA after 72 hours of doxycycline (dox)-inducible MEN1 knockdown (N=3). G, Representative western blot (30 µg) of MEN1 and ATF4 in A673 dox-inducible shNS or shMEN1 cells after 72 hours treatment with dox. H, qRT-PCR for SSP mRNA after MEN1 knockdown (N=3). Error bars represent SEM from independent biological replicates. * p<0.05; **p<0.01; *** p<0.001; **** p<0.0001; Two-tailed t-test.
In our experience, stable knockdown of menin in Ewing sarcoma cell lines is difficult to achieve [43]. Therefore, to validate MI-503 studies, we generated cells harboring a doxycycline-inducible shRNA to permit acute silencing of the gene. In comparison to MI-503 treatment, shRNA knockdown of menin led to a much less pronounced effect on cell proliferation (Supplementary Fig. S1C); however, even with a modest knockdown of menin, Ewing sarcoma cells displayed loss of ATF4 mRNA and protein (Fig. 3F-G) and downregulation of SSP gene expression (Fig. 3H). Together, these data support the hypothesis that menin, either directly or indirectly, contributes to maintenance of ATF4 expression in Ewing sarcoma cells.
To address whether menin may function to regulate ATF4 through its role in the epigenetic trithorax complexes, we measured H3K4me3 levels in Ewing sarcoma cell lines treated with MI-503. No reduction in global H3K4me3 was observed in inhibitor-treated cells after 96 hours, despite marked reduction in ATF4 at this time point (Fig. 4A). Importantly, targeted ChIP-qPCR studies of the ATF4 gene promoter confirmed that local H3K4me3 enrichment was also unaffected by MI-503 (Fig. 4B). Menin’s interaction with either MLL1 or MLL2 (KMT2A or KMT2B) methyltransferases is required for the deposition of H3K4me3 by trithorax complexes at gene promoters [41]. We previously reported that knockdown of MLL1 does not have a significant impact on expression of PHGDH, PSAT1, and PSPH [18]. To examine whether the impact of menin inhibition on ATF4 and SSP gene expression could be mediated by interaction with MLL2 rather than MLL1, we knocked down KMT2B by shRNA. As shown, knockdown of MLL2 also had no significant impact on ATF4 or SSP gene expression (Fig. 4C). Thus, these data suggest that acute inhibition of menin may lead to downregulation of ATF4 by trithorax complex-independent mechanisms.
Figure 4. Menin Inhibition Downregulates ATF4 Expression in an H3K4me3-Independent Manner.
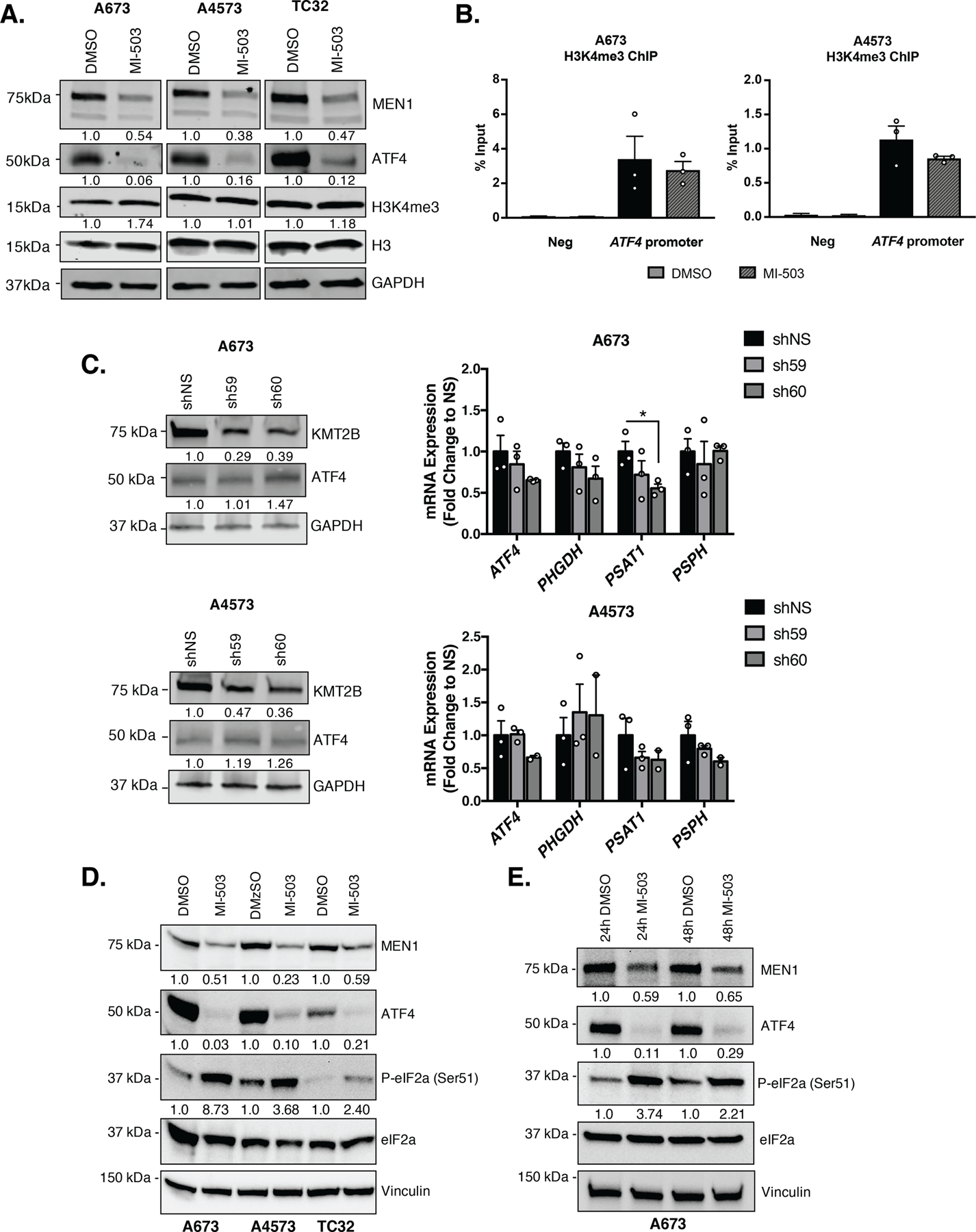
A, Representative western blot (A673 & A4573– 30 µg, TC32– 50 µg) of MEN1, ATF4, H3K4me3 and total H3 levels after 96 hours of treatment with 3 µM MI-503 or DMSO control. B, ChIP-qPCR for H3K4me3 enrichment at the ATF4 gene promoter after 96 hours of MI-503 treatment compared to DMSO control. Negative control is a gene desert region in chr2 without H3K4me3 enrichment (N=3). C, Representative western blot of MLL2 (KMT2B), MEN1, and ATF4 protein after knockdown of MLL2, and qRT-PCR for ATF4 and SSP (PHGDH, PSAT1, PSPH) mRNA after KMT2B knockdown (N=3). D, Representative western blot for MEN1, ATF4, phosphorylated-eIF2a (Ser51) and total eIF2a after 96 hours of 3 µM MI-503 treatment in A673, A4573, and TC32 cells (N=3). E, Representative western blot for P-eIF2a (Ser51) and total eIF2a after 24 and 48 hours of 3 µM MI-503 treatment in A673 cells (N=2). Error bars represent SEM from independent biological replicates. * p<0.05; Two-tailed t-test.
To begin to address potential alternate mechanisms of ATF4 regulation by menin, we performed a timecourse study. As shown, our findings revealed that, although transcript and protein levels were both reduced by MI-503 in A673 cells, ATF4 protein was very rapidly downregulated despite more modest effects on transcript expression (Supplementary Fig. S3A-C). Reductions in ATF4 protein were accompanied by reduction in ATF4 target gene, PSAT1 (Supplementary Fig. S3C). The striking and early loss of ATF4 protein in A673 cells led us hypothesize that, in addition to its effects on gene transcription, inhibition of menin might also influence post-transcriptional regulation of ATF4. ATF4 protein levels are tightly regulated by multiple post-transcriptional mechanisms, the most well characterized of which is the integrated stress response (ISR). In response to diverse cell stressors, including endoplasmic reticulum stress, amino acid deprivation, and others, stress-sensing kinases activate the ISR by inducing phosphorylation of eukaryotic initiation factor 2-alpha (eIF2a) at serine 51 [15]. In response to increased P-eIF2a, cells shut down global protein translation and selectively upregulate translation of ATF4. Therefore, we exposed cells to MI-503 and measured P-eIF2a (Ser51). As shown, and in direct contrast to our expectations, MI-503 treated cells showed a reproducible increase in P-eIF2a (Ser51), coincident with loss of ATF4 protein at timepoints as early as 24 hours (Fig. 4D-E). Thus, acute inhibition of menin in Ewing sarcoma cells leads to activation of the ISR, but the expected increase in ATF4 protein is blocked. Ongoing studies are investigating the mechanism underlying this block and whether the paradoxical loss of ATF4 protein downstream of ISR activation is a direct consequence of menin inhibition or an indirect effect mediated by feedback pathways.
EWS-FLI1 and menin loss induce broad downregulation of ATF4-dependent gene expression
The marked and reproducible reduction of ATF4 expression in the context of both EWS-FLI1 and menin inhibition led us to hypothesize that broader ATF4-dependent transcriptional programs beyond just the SSP might be impacted by modulation of either or both of these proteins. In particular, we wondered about expression of adaptive genes that are activated downstream of ATF4 in the context of stress responses such as the unfolded protein response, amino acid response, and the oxidative stress response [15]. To address this, we interrogated previously published RNA-seq data that were generated from Ewing sarcoma cells following either EWS-FLI1 knockdown or menin inhibition [8, 17, 18, 35]. As shown, EWS-FLI1 knockdown in A673 cells [35] led to downregulation of ATF4, PHGDH, PSAT1, and PSPH, as well as other known ATF4-modulated stress response genes such as ASNS, TRIB3, NARS1, and MTHFD2 (Fig. 5A). This expression profile was at least partially restored upon rescue with an RNAi-resistant EWS-FLI1 cDNA, confirming the role of the fusion in transcriptional regulation of both ATF4 and multiple of its downstream targets. Consistent with this finding, two additional and independent RNA-seq studies [8, 17] corroborate the finding that loss of EWS-FLI1 leads to diminished expression of an ATF4 gene signature (Supplementary Fig. S4A-B). We validated these sequencing-based studies using qRT-PCR, confirming that acute knockdown of EWS-FLI1 leads to profound and reproducible downregulation of ATF4 as well as multiple ATF4-regulated stress response genes (Fig. 5B).
Figure 5. EWS-FLI1 Inhibition in Ewing sarcoma Broadly Inhibits an ATF4-dependent Gene Expression Program.
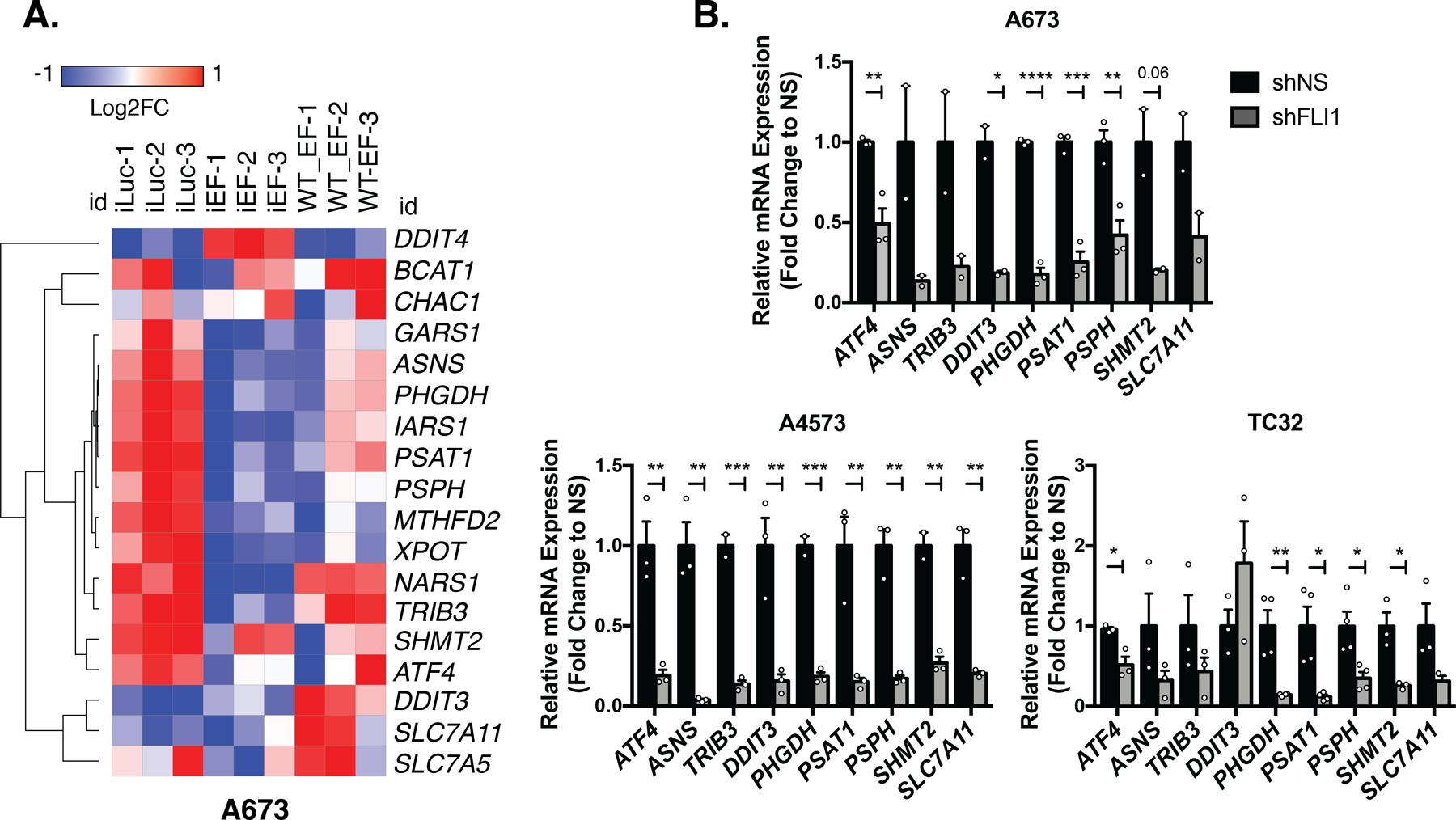
A, Heatmap of Log2Fold Change in expression from publicly available RNA-seq data of shRNA knockdown of EWS-FLI1 (iEF), luciferase-targeting control (iLuc), and wildtype EWS-FLI1 cDNA (WT-EF) rescue in A673 cells [35]. B, qRT-PCR validation of ATF4 and select ATF4 target genes after 96 hours of FLI1 (EWS-FLI1) knockdown (N=3, N=4). Error bars represent SEM from independent biological replicates. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; Two-tailed t-test.
Next, we analyzed previously published nascent RNA-seq data from MI-503-treated Ewing sarcoma cell lines [18]. These data show that numerous ATF4 target genes, including TRIB3, SESN2, ASNS, CHAC1, and SLC7A11 were reproducibly downregulated in the context of acute menin inhibition (Fig. 6A-B). Furthermore, bioinformatic analysis of transcription factor binding sites in promoters of significantly downregulated genes revealed ATF4 binding sites to be most highly and significantly over-represented (Fig. 6C). These unbiased genomic studies were validated by qRT-PCR analysis of independently treated Ewing sarcoma cells (Fig. 6D).
Figure 6. Menin Inhibition Identifies an ATF4-dependent Gene Expression Signature.
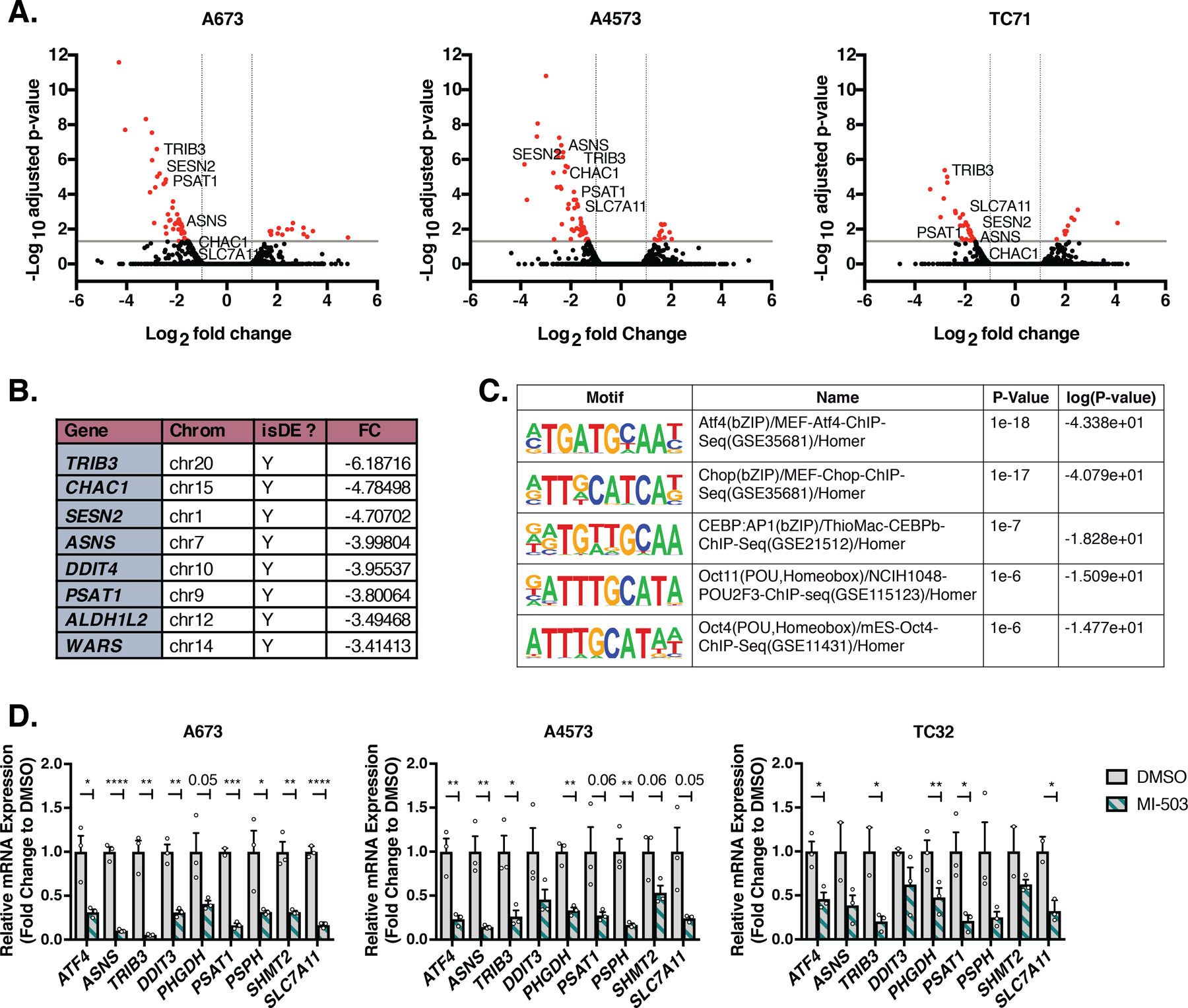
A, Volcano plot of global changes in mRNA levels from previously published RNA-seq of MI-503-treated A673, A4573, and TC71 Ewing sarcoma cells [18]. Cells were treated with 3 µM vehicle (MI-NC) or MI-503 for 72 hours and analyzed by bromouridine sequencing (Bru-seq). Genes marked in red show <−2-fold change in gene expression and adjusted p<0.05. B, Top downregulated genes from combined Bru-seq data of all three cell lines after 3 µM MI-503 treatment for 72 hours [18]. C, Top transcription factor motifs identified using the HOMER software by motif analysis of promoters of genes downregulated in (A) from three independent cell lines (A673, A4573. TC71). D, qRT-PCR of ATF4 and select ATF4 target genes after 96 hours of treatment with 3 µM MI-503 or DMSO control (N=3). Error bars represent SEM from independent biological replicates. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; Two-tailed t-test.
All together, these data lead us to conclude that both EWS-FLI1 and menin regulate ATF4 expression and activity in Ewing sarcoma cells, and that acute inhibition of either protein leads to a loss of the ATF4-dependent stress response gene signature.
Menin inhibition impacts ATF4 and its target genes in MLLr leukemia
To our knowledge, menin has not previously been implicated in regulation of ATF4 or the ATF4-mediated, stress-associated transcriptional signature. Menin is broadly recognized to function as a tumor suppressor gene, largely as a consequence of its role in familial cancers in preventing endocrine tumor development. However, it also plays an essential pro-tumorigenic role in the context of MLLr leukemia [41]. Indeed, the tumorigenicity of MLLr leukemias is profoundly dependent on menin, given its function as an obligate binding partner for oncogenic MLL-fusion proteins, and menin inhibition is currently being evaluated as a tumor-specific, biologically targeted therapy in these diseases [29, 36]. To address whether menin influences ATF4 and its target genes in MLLr leukemia cells, we exposed two MLLr leukemia cell lines – MV4;11, a B-cell acute lymphoblastic leukemia (ALL) cell line, and MOLM13, an acute myeloid leukemia (AML) cell line – to MI-3454, a next-generation and more potent pharmacologic inhibitor of menin through its interference with menin: MLL-fusion protein interactions [29]. Remarkably, and consistent with menin inhibition in Ewing sarcoma cells, ATF4 and ATF4 target gene expression were significantly reduced in MV4;11 cells at nanomolar drug concentrations that inhibit leukemogenesis (Fig. 7A). By comparison, although some effect was also apparent in MOLM13 cells, the impact was much less pronounced and was mainly evident only at the higher dose of inhibitor (Fig. 7B). To complement these results, we interrogated publicly available RNA-Seq data from MLLr cells that were exposed to a different, independently developed menin: MLL inhibitor, VTP50469 [36]. VTP50469-treated B-cell ALL cells (RS4;11) showed a modest reduction in expression of ATF4 and its target genes (Fig. 7C). In contrast, VTP50469 induced no demonstrable effect on the ATF4 transcriptional signature in MOLM13 cells (Fig. 7D). Intriguingly, however, CRISPR/Cas9-mediated knockout of menin in MOLM13 cells [36] was associated with a significant downregulation of several ATF4 target genes, including PSAT1, PHGDH, ASNS, and DDIT3 (Fig. 7E). Thus, these data suggest that menin may also contribute to regulation of ATF4 and its stress-response transcriptional signature in MLLr leukemia.
Figure 7. Menin Inhibition Downregulates ATF4 Activity in MLLr Leukemia Cell Lines.
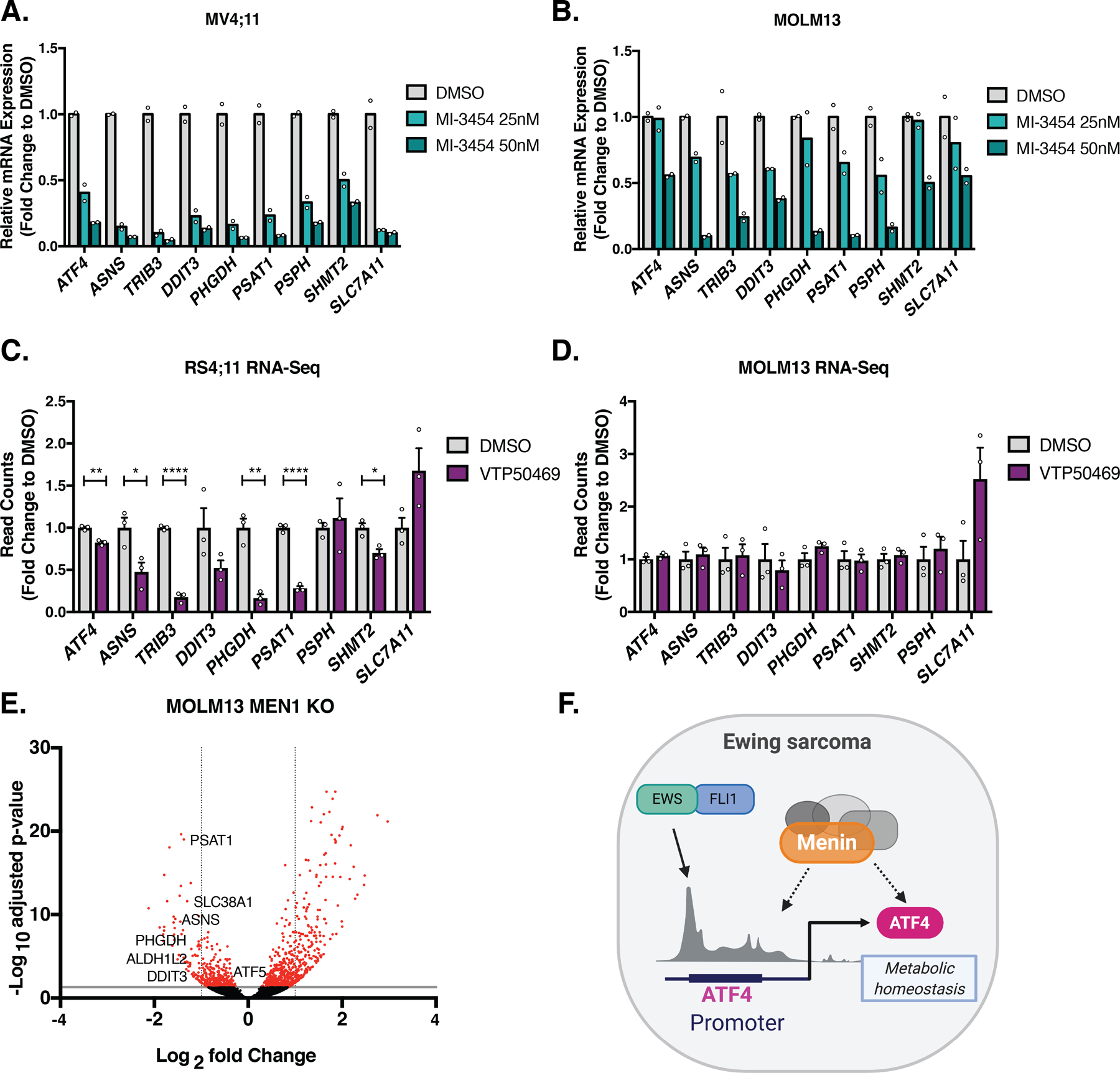
A, qRT-PCR of ATF4 and select ATF4 target genes in MV4;11 B-ALL and B, MOLM13 AML cell lines after 7 days of treatment with 25 and 50 nM MI-3454, a next-generation menin inhibitor, or DMSO control (N=2). C, Histogram of publicly available RNA-seq in RS4;11, and D, MOLM13 cell lines after 7 days of treatment with 330 nM VTP50469 (N=3) [36]. E, Volcano plot of publicly available RNA-seq data from MOLM13 cells following 6 days of MEN1 knockout [36]. Upregulated genes with adjusted p > 30 are excluded. Genes marked in red show <−2-fold change in gene expression and p <0.05. F, Working model in which the EWS-FLI1 oncogene in Ewing sarcoma and high menin expression converge on regulation of ATF4. Error bars represent SEM from independent biological replicates. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; Two-tailed t-test.
Discussion
Hyperactivation of the SSP has been identified as a central metabolic dependency in multiple different solid tumors [16], and we and others have demonstrated this pathway to be of key biologic and translational relevance to Ewing sarcoma [17–20]. The mechanisms of SSP activation differ in different tumor types and both EWS-FLI1 and menin have been implicated in the context of Ewing sarcoma [17–20]. In tumors that lack genomic amplification of the PHGDH locus, hyperactivation of the SSP is most often under the control of master transcriptional regulators, in particular ATF4 [15]. ATF4 is a ubiquitously expressed stress response gene whose function is critically important for maintenance of metabolic homeostasis in both normal and cancer cells, especially under conditions where cells are subjected to the stress of limited resources, such as glucose, amino acids, and oxygen [15]. Although metabolic reprogramming is evident in Ewing sarcoma, ATF4 has not previously been implicated in mediating this process in these tumors [17–19, 44, 45]. Our current findings provide evidence that ATF4 directly binds to and maintains SSP gene expression in Ewing sarcoma and lend support for the hypothesis that ATF4 acts downstream of both EWS-FLI1 and menin in their respective regulation of the SSP (Fig. 7F). Moreover, our data show that, in addition to their impact on the SSP, maintenance of ATF4 by EWS-FLI1 and menin contributes to expression of a broader ATF4-dependent transcriptional program. To our knowledge, a link between ATF4 and either EWS-FLI1 or menin has not been previously described. Although prior work suggested that EWS-FLI1 does not regulate ATF4 [17, 19], our studies demonstrate direct binding of the ATF4 promoter by the fusion, confirming previously published ChIP-seq data [8, 37]. In addition, we find that knockdown of the fusion leads to diminished expression of ATF4 mRNA, which is also consistent with some prior studies [8, 17, 35]. Notably, we and others have observed that, although EWS-FLI1 knockdown universally leads to reduced ATF4 expression in Ewing sarcoma cells, the extent of loss is highly influenced by the cell line, time, cell culture media, and other technical and biologic variables. We speculate that this variability explains the contradictory findings of prior published studies and is indicative of the dynamic and multi-faceted nature of ATF4 regulation and of its central role in maintaining cell homeostasis. In support of this, we observed that knockdown of ATF4 is deleterious to Ewing sarcoma cell growth, and that cells with forced over-expression of murine ATF4 (mAtf4) rapidly down-regulate transcription of endogenous ATF4 in order to maintain homeostatic levels of the protein (data not shown). In addition, we observed that ATF4 mRNA expression is exquisitely sensitive to changes in the microenvironment that would result in cell stress, including changes in pH, confluence, and nutrient availability. Thus, we conclude that, although EWS-FLI1 directly binds and activates the ATF4 promoter, other transcription factors that contribute to ATF4 regulation under conditions of cell stress, such as NRF2 [15, 23] or MYC [46], may rapidly compensate for reductions in mRNA that are associated with loss of the fusion.
Although EWS-FLI1 and other related EWS-ETS fusions are unique to Ewing sarcoma, menin is a ubiquitously expressed scaffolding protein that has context-dependent roles in both tumor suppression and tumor promotion. In MLLr leukemia, inhibition of menin leads to downregulation of tumor-promoting oncogenes, such as HOXA9 and MEIS1, that are under the control of MLL fusions, and this is accompanied by differentiation of leukemic blasts and tumor involution [28]. In Ewing sarcoma, our prior work showed that menin contributes to tumorigenicity through mechanisms that are not clearly linked to cellular differentiation and are instead associated with cell metabolism, in particular maintenance of the SSP [18, 43]. In the current work, we have discovered that the mechanism of menin-mediated regulation of SSP gene expression is, at least in part, mediated by ATF4. In addition, the loss of ATF4 and SSP gene expression that results from menin inhibition is accompanied by reduced expression of multiple stress-response genes that are under the transcriptional control of ATF4. Significantly, analysis of menin inhibitor-treated MLLr leukemia cells revealed a similar loss of ATF4 and ATF4 target gene expression. These findings suggest that the contribution of menin to regulation of ATF4 is not unique to Ewing sarcoma.
Menin’s functions in normal and cancer cell biology are diverse and dependent on its highly cell type- and context-dependent protein interaction partners [41]. Although the exact molecular mechanisms of menin action in ATF4 regulation remain to be fully elucidated, our studies suggest that they are independent of its interactions with MLL proteins in the trithorax complexes. Enrichment of H3K4me3 at the ATF4 gene promoter remains robust at a time point when MI-503 has induced profound loss of ATF4 protein expression. In addition, acute knockdown of MLL1 or MLL2 (KMT2A or KMT2B) does not reproduce the effect of menin inhibition [18]. Indeed, although the ATF4 transcript is reproducibly reduced by menin inhibition, we observed that loss of ATF4 protein is often more immediate and more pronounced. This suggests that menin may also contribute to post-transcriptional regulation ATF4. This novel hypothesis is supported by our observation that MI-503 leads to activation of the ISR, a key regulator of ATF4 protein translation. However, in direct contrast to expectations, activation of the ISR by MI-503 is associated with loss rather than gain of ATF4 protein. The reasons for this disconnect remain unclear and we are now investigating how menin inhibition blocks ATF4 upregulation in the context of ISR activation. Of note, it has been shown that mTORC1 can regulate ATF4 mRNA stability and ATF4 translation [47], an ISR-independent mechanism for ATF4 regulation that we are also exploring.
Menin inhibitors are now being tested in early phase clinical trials for MLLr leukemia (registered at www.clinicaltrials.gov as #NCT04067336 and #NCT04065399). Their efficacy as anti-leukemic agents are linked to their induction of blast cell differentiation and death, downstream of menin: MLL inhibition. Significantly, however, there is now emerging evidence from several groups, including with our own, that menin inhibitors may have additional effects on cell metabolism. Specifically, menin inhibitors have recently been shown to alter the cellular energetics and glycolysis of breast cancer cells, as well as to induce ferroptosis, an iron-dependent form of oxidative stress-induced cell death in ovarian and other carcinoma cells [48, 49]. Intriguingly, the demonstration of effects of menin inhibition on ferroptosis were uniquely observed with MI-503 and its related compounds and not reproduced with the VTP-50469 compound [49]. This suggests that the biologic impact of menin inhibitors on cell metabolism and cell death may be distinct. This is noteworthy for Ewing sarcoma, where VTP-50469 was recently shown to have no impact on tumor cell viability in vitro or tumorigenicity in vivo [50]. We speculate that differential effects of MI-503 on cell metabolism, possibly via selective inhibition of ATF4 and induction of oxidative stress, may explain the differences in the response of Ewing sarcoma cells to different menin inhibitors. A direct comparison between different modes of menin inhibition, pharmacologic and genetic, and a deep mechanistic evaluation of the effect of each of these approaches on menin and its interaction partners is now required to fully resolve why, how, and in which cellular contexts, menin inhibition influences cellular metabolism.
In summary, through an in-depth analysis of published sequencing data and new mechanistic studies, we provide the first evidence that EWS-FLI1 and menin converge to maintain SSP gene expression through activation of ATF4. In addition, our data show that this regulation of ATF4 has downstream effects on expression of multiple ATF4 target genes that are important for maintenance of cellular homeostasis under conditions of stress. Finally, we show that menin-mediated regulation of this ATF4-dependent transcriptional program may also be functionally important in MLLr leukemias.
Supplementary Material
Acknowledgements:
We would like to thank members of the Lawlor, Lyssiotis, Grembecka and Cierpicki labs for intellectual input, as well as the University of Michigan Rogel Cancer Center Vector and Sequencing Cores. We gratefully acknowledge Dr. Lewis Cantley for the Atf4 expression plasmid. Illustration was created with BioRender.com. Support for this work was provided by the following sources: National Institute of Health: F31 CA254079 (JJ), R01 CA218116 (ERL), T32 CA009676 (AA), K00 CA234810 (AGH); R01 CA200660 (JG); R37CA237421, R01CA248160, R01CA244931 (CAL); and P50 CAP30CA046592 to the Rogel Cancer Center. The authors also gratefully acknowledge additional funding support from the Rogel Cancer Center (collaborative research grant to ERL and CAL; CaRSIP summer internship to AG & EH), and the U-M Department of Pediatrics Charles Woodson Collaborative Research Award and Russell G. Adderley endowment.
Financial Support:
This work was supported by the National Institute of Health: F31 CA254079 (JJ), R01 CA218116 (ERL), T32 CA009676 (AA), K00 CA234810 (AGH); R01 CA200660 (JG); R37 CA237421, R01 CA248160, R01 CA244931 (CAL); and P50 CAP30CA046592 to the Rogel Cancer Center. The authors also gratefully acknowledge additional funding support from the Rogel Cancer Center (collaborative research grant to ERL and CAL; CaRSIP summer internship to AG & EH), and the U-M Department of Pediatrics Charles Woodson Collaborative Research Award and Russell G. Adderley endowment.
Footnotes
Conflict of interest: Dr. Grembecka and Dr. Cierpicki receive research support from Kura Oncology, Inc. and have equity ownership in the company.
References
- 1.Balamuth NJ and Womer RB, Ewing’s sarcoma. Lancet Oncol, 2010. 11(2): p. 184–92. [DOI] [PubMed] [Google Scholar]
- 2.Lawlor ER and Sorensen PH, Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit Rev Oncog, 2015. 20(3–4): p. 155–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Womer RB, et al. , Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol, 2012. 30(33): p. 4148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong GT, et al. , Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol, 2014. 32(12): p. 1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginsberg JP, et al. , Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst, 2010. 102(16): p. 1272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn P., et al. , Long-term cause-specific mortality in survivors of adolescent and young adult bone and soft tissue sarcoma: a population-based study of 28,844 patients. Cancer, 2014. 120(15): p. 2334–42. [DOI] [PubMed] [Google Scholar]
- 7.Lin PP, Wang Y, and Lozano G., Mesenchymal Stem Cells and the Origin of Ewing’s Sarcoma. Sarcoma, 2011. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riggi N., et al. , EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell, 2014. 26(5): p. 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomazou EM, et al. , Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep, 2015. 10(7): p. 1082–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulay G., et al. , Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell, 2017. 171(1): p. 163–178e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvanathan SP, et al. , Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc Natl Acad Sci U S A, 2015. 112(11): p. E1307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brohl AS, et al. , The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet, 2014. 10(7): p. e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crompton BD, et al. , The genomic landscape of pediatric Ewing sarcoma. Cancer Discov, 2014. 4(11): p. 1326–41. [DOI] [PubMed] [Google Scholar]
- 14.Tirode F., et al. , Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov, 2014. 4(11): p. 1342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wortel IMN, et al. , Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab, 2017. 28(11): p. 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mattaini KR, Sullivan MR, and Vander Heiden MG, The importance of serine metabolism in cancer. J Cell Biol, 2016. 214(3): p. 249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen N., et al. , EWS-FLI1 reprograms the metabolism of Ewing sarcoma cells via positive regulation of glutamine import and serine-glycine biosynthesis. Mol Carcinog, 2018. [DOI] [PMC free article] [PubMed]
- 18.Svoboda LK, et al. , Menin regulates the serine biosynthetic pathway in Ewing sarcoma. J Pathol, 2018. 245(3): p. 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanner JM, et al. , EWS/FLI is a Master Regulator of Metabolic Reprogramming in Ewing Sarcoma. Mol Cancer Res, 2017. 15(11): p. 1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Issaq SH, et al. , EWS-FLI1-regulated serine synthesis and exogenous serine are necessary for Ewing sarcoma cellular proliferation and tumor growth. Mol Cancer Ther, 2020. [DOI] [PMC free article] [PubMed]
- 21.Locasale JW, et al. , Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet, 2011. 43(9): p. 869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Possemato R., et al. , Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature, 2011. 476(7360): p. 346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeNicola GM, et al. , NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet, 2015. 47(12): p. 1475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao E., et al. , KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep, 2016. 14(3): p. 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding J., et al. , The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab, 2013. 18(6): p. 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reina-Campos M., et al. , Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell, 2019. 35(3): p. 385–400e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Y., et al. , Metabolic Reprogramming by MYCN Confers Dependence on the Serine-Glycine-One-Carbon Biosynthetic Pathway. Cancer Res, 2019. 79(15): p. 3837–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borkin D., et al. , Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell, 2015. 27(4): p. 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klossowski S., et al. , Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J Clin Invest, 2020. 130(2): p. 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Levetzow C., et al. , Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One, 2011. 6(4): p. e19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinz S., et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 2010. 38(4): p. 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volchenboum SL, et al. , Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J Pathol Clin Res, 2015. 1(2): p. 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Postel-Vinay S., et al. , Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet, 2012. 44(3): p. 323–7. [DOI] [PubMed] [Google Scholar]
- 34.Savola S., et al. , High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing’s Sarcoma Family of Tumour Patients. ISRN Oncol, 2011. 2011: p. 168712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theisen ER, et al. , Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes Cancer, 2019. 10(1–2): p. 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krivtsov AV, et al. , A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell, 2019. 36(6): p. 660–673e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilke S., et al. , Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res, 2013. 23(11): p. 1797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dey S., et al. , Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem, 2010. 285(43): p. 33165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang M and Vousden KH, Serine and one-carbon metabolism in cancer. Nat Rev Cancer, 2016. 16(10): p. 650–62. [DOI] [PubMed] [Google Scholar]
- 40.Grunewald TGP, et al. , Ewing sarcoma. Nat Rev Dis Primers, 2018. 4(1): p. 5. [DOI] [PubMed] [Google Scholar]
- 41.Matkar S, Thiel A, and Hua X., Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci, 2013. 38(8): p. 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malik R., et al. , Targeting the MLL complex in castration-resistant prostate cancer. Nat Med, 2015. 21(4): p. 344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svoboda LK, et al. , Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget, 2017. 8(1): p. 458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Issaq SH, et al. , Glutamine synthetase is necessary for sarcoma adaptation to glutamine deprivation and tumor growth. Oncogenesis, 2019. 8(3): p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeung C., et al. , Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res, 2019. 79(19): p. 5060–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mo H., et al. , ATF4 regulated by MYC has an important function in anoikis resistance in human osteosarcoma cells. Mol Med Rep, 2018. 17(3): p. 3658–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park Y., et al. , mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep, 2017. 19(6): p. 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou CW, et al. , Menin and Menin-Associated Proteins Coregulate Cancer Energy Metabolism. Cancers (Basel), 2020. 12(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato I, Kasukabe T, and Kumakura S., MeninMLL inhibitors induce ferroptosis and enhance the antiproliferative activity of auranofin in several types of cancer cells. Int J Oncol, 2020. 57(4): p. 1057–1071. [DOI] [PubMed] [Google Scholar]
- 50.Kurmasheva RT, et al. , Evaluation of VTP-50469, a menin-MLL1 inhibitor, against Ewing sarcoma xenograft models by the pediatric preclinical testing consortium. Pediatr Blood Cancer, 2020. 67(7): p. e28284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.