

Abstract
We present MultiEditR (Multiple Edit Deconvolution by Inference of Traces in R), the first algorithm specifically designed to detect and quantify RNA editing from Sanger sequencing (z.umn.edu/multieditr). Although RNA editing is routinely evaluated by measuring the heights of peaks from Sanger sequencing traces, the accuracy and precision of this approach has yet to be evaluated against gold standard next-generation sequencing methods. Through a comprehensive comparison to RNA sequencing (RNA-seq) and amplicon-based deep sequencing, we show that MultiEditR is accurate, precise, and reliable for detecting endogenous and programmable RNA editing.
Keywords: RNA editing quantification, RNA editing detection, DNA base editing, RNA modification, RShiny, MultiEditR Editing Index, Epitranscriptomics, APOBEC, ADAR, deaminases
Graphical abstract
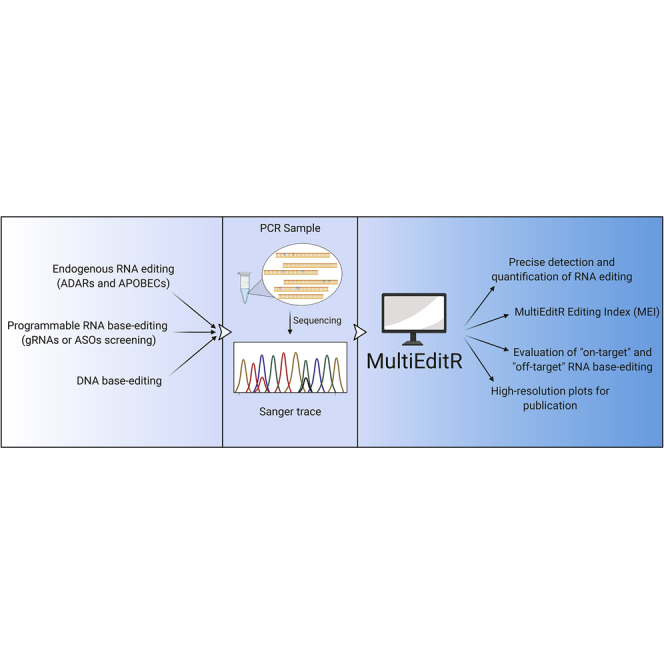
RNA editing allows cells to transiently rewrite the genetic code. However, detecting and quantifying RNA editing relies on RNA-seq, which is expensive and suffers from poor local resolution. Here, we develop MultiEditR (https://moriaritylab.shinyapps.io/multieditr/) to analyze RNA editing from Sanger sequencing for a fraction of the cost while maintaining a comparable fidelity to RNA-seq.
Introduction
RNA editing is the most abundant post-transcriptional modification in messenger RNA (mRNA),1 with two predominant types of editing: cytidine-to-uridine editing (C-to-U) by the APOBEC family of enzymes, and adenosine-to-inosine editing (A-to-I) by the ADAR family of enzymes. RNA editing has implications in a variety of biologically processes, particularly among those involved in neural physiology, immunity, and oncogenesis.2,3 Importantly, the recent development of programmable RNA base-editing technologies presents the possibility to correct pathogenic mutations at the RNA level, opening important therapeutic scenarios.4 For both endogenous and programmable RNA editing, the accurate and precise detection as well as quantification of editing is essential.
Current identification and quantification of endogenous RNA editing relies on RNA-sequencing (RNA-seq) data analyzed by several different algorithmic approaches.5,6 Although these approaches are robust, they are often complicated by genomic sequence polymorphisms, sequencing errors, and-or low coverage of certain genomic regions. This leads to the necessity of routine validation and quantification of RNA editing sites by Sanger sequencing, and by bacterial colony sequencing of subcloned polymerase chain reaction (PCR) amplicons.1,7
Meanwhile, identification and quantification of programmable RNA editing is mainly accomplished by Sanger sequencing.8, 9, 10, 11 To validate the existence of programmed RNA edits, the first step is to evaluate the editing efficiency of the targeted base (on-target editing), and the second step is to evaluate the presence of possible undesired editing sites along the same transcript (off-target editing). This part of the process, independently from the RNA base-editing method used, represents a time-consuming step, in which a quick and inexpensive evaluation of on-target and off-target editing at the transcript level is needed to define the best experimental settings to use.
Despite the widespread use of Sanger sequencing, no program exists specifically for the quantification of RNA editing, leaving some to creatively, yet not optimally, measure the height of peaks from Sanger sequencing traces using image analysis software such as Adobe Illustrator.12 Although some tools exist for the quantification of DNA base editing from Sanger sequencing,13 they are not able to detect and quantify multiple sites simultaneously within the same Sanger trace. This feature is essential to evaluate on-target and off-target programmable RNA editing, and it is also crucial in the context of endogenous RNA editing, where editing sites are often in a cluster, leading to hyper-edited transcript regions that can be missed by standard RNA editing analysis of RNA-seq data.1,14 Finally, the detection and quantification capabilities of a Sanger based approach has yet to be benchmarked against next-generation sequencing (NGS) methods. To meet these needs we developed multiple edit deconvolution by inference of traces in R (MultiEditR) (z.umn.edu/multieditr), a program with a web interface that provides accurate and cost-effective detection and quantification of RNA editing from Sanger sequencing that yields comparable results to NGS methods.
Results
To first determine whether successive base edits could be accurately quantified from Sanger sequencing, we titrated two plasmids that differed by six C-to-T mutations (Supplemental materials and methods; Figure S1A) and subjected the titrations to Sanger sequencing (Figure 1A). Analysis of the traces from both the forward and reverse direction showed that the percent height of the mixed peaks of interest yielded well-fit linear regressions compared to the expected titrated percent for both C-to-T and G-to-A titrations (Figures 1B and 1C; Figures S1B–S1E). Encouraged by the results we adapted the EditR algorithm, which we previously developed for analyzing CRISPR-Cas9 DNA base editing,13 for the detection and quantification of multiple RNA edits from Sanger sequencing, which we termed MultiEditR (Figure 1D; Figure S2). To compare the performance of MultiEditR with standard methods for RNA editing detection, we first generated two knockout (KO) cell lines for ADAR1 and APOBEC1 (Figure S3). Next, we performed RNA-seq on RNA from both wild-type (WT) and KO cell lines and analyzed the data with REDItools,15,16 a well-established tool for detection of RNA editing from RNA-seq. From the same samples several regions within different transcripts (Table S1) were PCR amplified, Sanger sequenced, and analysis of these traces was performed by MultiEditR. An initial comparison of MultiEditR to RNA-seq showed that although the central tendency of MultiEditR measurements were accurate, there was substantial error relative to the RNA-seq benchmark (Figure 1E; Figure S4). Despite the high coverage of our RNA-seq experiments (62–88 million mapped reads per sample), this error appeared to be influenced by the read depth per base of the RNA-seq dataset (Figures 1F and 1G; Figure S4), which is consistent with findings by others.7
Figure 1.

Development and initial assessment of MultiEditR
(A) Experimental scheme for the plasmid titration experiment. (B) Representative chromatograms from C-to-T titration experiment showing a change in peak height at two sites. (C) Titration of pJET-CmAG-WT with pJET-CmAG-6x sequenced with the reverse primer; the coefficient of determination (R2) was calculated relative to the identity line. n = 6 editing sites per chromatogram. (D) Diagram of MultiEditR algorithm showing end trimming, sample alignment, motif mapping, zero adjusted gamma (zΓ) distribution generation, and null hypothesis significance testing. The MultiEditR algorithm can be applied to any motifs, WT base, and edited base identity. (E) Scatterplot of measurements of editing from Sanger sequencing by MultiEditR against measurements of editing from RNA-seq by REDItools. Coefficient of determination (R2) represents regression to the identity line. The overlaid black line is the linear model of best fit. Dot size is proportional to RNA-seq read coverage at the base of interest. (F) Absolute value of error in MultiEditR measurements by RNA-seq read depth. Population at the top of the graph with low read depth represents bases with >90% error in measurements. Data were analyzed by Spearman’s rank-order correlation test. (G) Mirrored histogram showing populations with high (≥90%) error and low (≤0.1%) error in MultiEditR measurements as a function of RNA-seq read depth.
To address the potential issue that the low coverage of the RNA-seq dataset was introducing error in assessing the accuracy of MultiEditR, we performed a three-way, matched comparison of endogenous RNA editing quantification and detection within several transcripts from two cell lines (Table S2) using MultiEditR, RNA-seq, and high coverage amplicon based NGS (termed, Amplicon-seq) (Figure 2A). The direct comparison of MultiEditR to Amplicon-seq demonstrated that MultiEditR is on average accurate relative to Amplicon-seq with small significant inaccuracies among edits measured from C, G, and T bases (range of M: −1.66% to −2.59%, p < 0.001), and non-significantly different for edits measured from A bases (M = −0.25%, p = 0.476) (Figures 2B and 2C). These small inaccuracies may be attributable to peak-ratio bias, which is a well-known aspect of Sanger sequencing.17,18 In comparison, RNA-seq benchmarked against Amplicon-seq exhibited no significant inaccuracies across all bases (Figures 2D and 2E; Figure S5). However, RNA-seq exhibited a greater standard deviation than did MultiEditR for all bases. Importantly, we found that MultiEditR was measured as more precise when benchmarked against Amplicon-seq as opposed to RNA-seq (Figures 2B and 2C; Figures S5E–S5I), confirming that the observed error in MultiEditR detection relative to the RNA-seq (Figure S4) was indeed due to low coverage in some regions of the RNA-seq dataset. Collectively, these results indicate that compared to Amplicon-seq, while the quantification of RNA editing by RNA-seq is more accurate, MultiEditR is more precise than RNA-seq, particularly when looking at editing events above 5% editing (Figure 2F).
Figure 2.

Quantification and detection of endogenous RNA editing by MultiEditR
(A) Workflow for generation of sequencing data for three-way comparison of MultiEditR, RNA-seq, and Amplicon-seq. RNA-seq from WT cell lines was compared to RNA-seq from paired APOBEC1 or ADAR1 KO cell lines to identify putative editing sites, which were PCR amplified from WT cell line cDNA. Amplicons were then subjected to Sanger sequencing and NGS deep sequencing in parallel and analyzed by MultiEditR or REDItools, respectively. (B–E) Comparison of MultiEditR and RNA-seq measurements of editing to high coverage Amplicon-seq measurements. All data are filtered on between 1% and 99% editing as measured from Amplicon-seq data. Coefficients of determination (R2) represent regression to the identity line. Dot size is proportional to read coverage at the base of interest in the RNA-seq data. M = mean, SD = standard deviation. p values (P) were calculated by Student’s one-sample two-tailed t test. Boxplot center lines represent the median, box limits represent the upper and lower quartiles, and whiskers define the 1.5× interquartile range. (F) Kernel plots of the inaccuracy of MultiEditR and RNA-seq relative to Amplicon-seq. Plots are faceted by 1%, 5%, or 10% editing inclusion cutoffs based on the editing measured in Amplicon-seq. (G) Confusion matrix that defines the sensitivity and specificity of the edit detected by MultiEditR or REDItools, against the true state of editing, defined by Amplicon-seq. (H) ROC curves of MultiEditR and REDItools at 1%, 5%, and 10% editing cutoffs, where an Amplicon-seq event is a true edit. MultiEditR is shown as a solid curve, while REDItools is shown as a dotted curve. Crossed points along the curves represent the closest geometric distance of the curve to the upper-left error-free point at 0% and 100%. (I) ROC analysis metrics for MultiEditR and REDItools at editing thresholds from 0.1% to 25% editing inclusion cutoff. Sensitivity and specificity are defined in (G); area under the curve (AUC) represents the integral of the ROC curve from 0% to 100%.
Next, we wanted to assess how MultiEditR and REDItools analysis of RNA-seq15,16 perform in the detection of edits (Figure 2G). Using the MultiEditR p value calculated from the zero-adjusted gamma distribution null hypothesis significance test (Figure 1D), as well as the p value from the REDItools Fisher exact test as classifier values, we performed a receiving operating characteristic (ROC) curve analysis. When including edits that were called 1% or greater by Amplicon-seq, RNA-seq performed modestly better at detecting edits than did MultiEditR in terms of sensitivity, specificity, and area under the curve (AUC), among other metrics (Figure 2H; Figure S6). However, when examining edits that were called 5% or greater by Amplicon-seq, MultiEditR performed better than RNA-seq (Figure 2I). Furthermore, running ROC curve analyses across a range of editing detection thresholds suggests that based on the measured sensitivity, specificity, and AUC, the optimal use of MultiEditR is for detecting editing events ≥5% (Figure 2I).
Last, we wanted to assess the utility of MultiEditR in application to biologically relevant problems. Roth et al.19 proposed the Alu editing index (AEI, here as EINGS) as an index for the quantification of global RNA editing. Here, we apply a similar approach to develop the MultiEditR editing index (MEI) as a local editing index across the Sanger trace (Figure 3A). Using our three-way, matched dataset we found that the MEI is moderately correlated with the lower read depth per base RNA-seq EINGS (r = 0.558, p = 3.48e−7), and it is well correlated with the higher read depth per base Amplicon-seq EINGS (r = 0.812, p = 1.39e−5) (Figures 3B and 3C), further showing an effect of read depth in RNA editing detection and quantification from RNA-seq data. Using the MEI we wanted to investigate the effect of adding a nuclear localization signal (NLS) on the specificity of the programmable 4λN-ADAR2DD A-to-I editing system as previously published20 (Figure 3D). Using a fluorescent reporter, we were able to directly compare editing rates to a functional readout via flow cytometry, as well as measuring editing across the transcript with the MEI (Figure 3E). We found that MultiEditR measurements of editing agreed well with flow cytometry values (Figure 3F). Additionally, using a normalized metric of percent editing of the target base (on-target editing) divided by the MEI (off-target editing), we recapitulated results that addition of an NLS to the 4λN-ADAR2DD system improves editing specificity20 (Figure 3G; Figure S7). Last, we wanted to determine whether MultiEditR could be used to similarly quantify CRISPR-Cas9 DNA base editing using data previously published by our group from work using base editing to disrupt genes via splice-site targeting21 (Figure 3H). Base editing efficiency measured by MultiEditR compared to CRISPR-DAV analysis of Amplicon-seq,22 as well as flow cytometry, yielded strong coefficients of determination at both the DNA (R2 = 0.97, Figure 3I) and protein level (R2 = 0.849, Figure 3J).
Figure 3.

Applications of MultiEditR to RNA and DNA base editing
(A) Formula for MultiEditR editing index (MEISanger) and Alu editing index from Roth et al.19 calculated from NGS data (EINGS). (B) Correlation between MEI and RNA-seq EI shaded by average read depth of RNA-seq. (C) Correlation between MEI and Amplicon-seq EI shaded by average read depth of Amplicon-seq. (D) Diagram of 4λN-hADAR2-DD RNA editing system. (E) EGFP reactivation experiment using the 4λN-DD system. Plasmids encoding gRNA, 4λΝ-hADAR2-DD, and mCherry-Apob-EGFPW58X (CAGX) were transfected into HEK293T cells. (F) Reactivation of EGFP measured via flow cytometry and MultiEditR. (G) Ratio of target X58W editing to non-desired events, measured by the ratio of target editing to the MEI. Results are normalized to the CAGX alone negative control. Flow cytometry plots, MultiEditR plots, and analysis parameters are available in Figure S7. (H and I) DNA base editing quantification of rAPOBEC1-BE3 and rAPOBEC1-BE4 in human T cells by MultiEditR compared to Amplicon-seq NGS analyzed by CRISPR-DAV. (H and J) Comparison of MultiEditR to protein KO efficiency as measured by FACS in human T cells; data were analyzed from Webber et al.21
For the best use of MultiEditR, we recommend designing primers to amplify a 350- to 700-bp amplicon to allow for a long enough sequence to construct null distributions for edit detection. Additionally, we recommend the use of one-step RT-PCR kits, over standard cDNA synthesis kits, to exclusively generate cDNA from the transcript of interest. Following amplification, a column-based PCR purification step is typically sufficient to ensure clean sequencing results. For detecting edits above 5%, we recommend using p = 0.001 for applications where false positives are strongly disfavored, and p = 0.01 when an increase in sensitivity is valued over a loss in specificity (Figure S6). Finally, for the best accuracy and precision of MultiEditR we recommend measuring the edit from the T or A base (Figure 2C). Last, due to the sensitivity of MultiEditR, we do not recommend using it to detect or measure edits that are below 5% due to the baseline noise in Sanger traces. For applications where high accuracy and a low limit of edit detection is paramount, we recommend using Amplicon-seq.
Discussion
Collectively, we developed MultiEditR, the first algorithm specifically designed to detect and quantify multiple RNA editing sites in a single trace of Sanger sequencing and we performed a comprehensive comparison with NGS methods to evaluate the performance of the tool. MultiEditR showed higher precision in RNA editing detection than did RNA-seq, particularly when looking at editing events above 5% editing, but with the cost of lower accuracy. Furthermore, in the context of RNA programmable editing, the capability of MultiEditR to detect multiple edits simultaneously and the MEI allow for a quick and inexpensive evaluation of on-target and off-target editing at the transcript level (Figure 3), a crucial aspect to define the best experimental conditions for mutation correction at the RNA level (e.g., choosing an optimal guide RNA).
Finally, we showed that MultiEditR can be employed for a variety of nucleic acid editing applications, including endogenous RNA editing, targeted programmable RNA editing, off-target RNA editing, and DNA base editing. The flexibility of the MultiEditR algorithm allows our approach to be readily applied to other applications that involve the change of one base species to another, such as that involved in bisulfite sequencing for identifying methylation, or more recently RNA polymerases and reverse transcriptases recoding various RNA modifications with distinct fidelity.23 Overall, we predict that MultiEditR and the comparisons detailed in this study will have immediate use to the RNA editing community, but also more broadly to the many burgeoning fields studying nucleic acid modifications.
Materials and methods
Plasmids
The mCherry-mApob-EGFP plasmid (CmAG) was obtained by substituting the human APOB with mouse Apob (mApob) in the original plasmid mCherry-APOB-EGFP24 (kind gifts from Dr. Silvestro Conticello, Florence, Italy). mCherry-APOB-EGFP was digested with HindIII-SmaI and a PCR fragment of mouse Apob (467 bp from RNA of jejunal epithelial cells from the small intestines of C57BL/6 mice,25 oligonucleotides [oligos] #1–2) was inserted into the plasmid using NEBuilder HiFi DNA assembly master mix (NEB). The mouse APOBEC1 expression vector (pCMV APOBEC1) was a kind gift from Dr. Dewi Harjanto (Laboratory of Lymphocyte Biology, The Rockefeller University). The mouse RBM47 expression vector (pCMV RBM47) was obtained by inserting a PCR fragment containing the coding sequence of mouse RBM47 (transcript variant 4, mRNA sequence ID: GenBank: NM_001291226.1) into the mCherry-Apob-EGFP cut with NheI-BsrGI. The amplification was done using oligos #3–4 on RNA of jejunal epithelial cells from the small intestines of C57BL/6 mice25 and the cloning with NEBuilder HiFi DNA assembly master mix (NEB).
LentiCRISPRv2 was a gift from Dr. Feng Zhang (Addgene, plasmid #52961; http://addgene.org/52961; RRID:Addgene_52961).26 DNA oligos #5–6 were cloned into this plasmid following the “lentiCRISPRv2 and lentiGuide oligo cloning protocol” (Addgene plasmid #52961) to generate lenti-CRISPR-ADAR1 exon 4 (from Pestal et al.27). As a non-editing transduction control, lenti-CRISPR-NT (Lenti-NT) was cloned accordingly using oligos #7–8. pCMV-DR8.91 (coding for HIV gag-pol) and pMD2.G (encoding the VSV-G glycoprotein) were kind gifts from Prof. Didier Trono (Lausanne, Switzerland). pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid #48138; http://addgene.org/48138; RRID:Addgene_48138).28 The plasmid was digested with BsbI (NEB) and dephosphorylated with a RAPID DNA Dephos and Ligation kit (Roche). Oligos #9–12 are all 5′ phosphorylated. The oligo pairs #9–10 and #11–12 containing complementary sequences were annealed to each other and then ligated to the dephosphorylated PX458 to generate plasmids PX458-iv-single guide RNA (sgRNA)-A1_11 (cutting in exon 4) and PX458-iv-sgRNA-A1_39 (cutting in exon 5), respectively.
The mCherry-APOB-EGFP W58X plasmid (CAGX) was obtained by site-directed mutagenesis using oligos #13–14 and QuikChange Lightning site-directed mutagenesis kit (Agilent, #210518) on the original plasmid mCherry-APOB-EGFP.24 4λN-DD E488Q ADAR2 (4λN) and U6 pENTR gRNA vectors were a kind gift of Dr. Joshua Rosenthal (University of Chicago).8 The NLS version of 4λN plasmid (4λN-NLS) was created by adding the c-myc NLS to the C-terminus of 4λN. The U6 pENTR gRNA vector was linearized by PCR using oligos #15–16 and Q5 high-fidelity DNA polymerase (NEB). The sequence containing the gRNA to induce specific A-to-G editing on the W58X of CAGX (Rosenthal fashion8) was inserted into the linearized pENTR using oligo #17 and NEBuilder HiFi DNA assembly master mix (NEB).
Cell lines
A549 cells (A-549, RRID:CVCL_0023, DKFZ Germany) were cultured at 37°C, 5% CO2 in high-glucose DMEM (Sigma) supplemented with 10% fetal bovine serum (FBS, PAN Biotech) and penicillin/streptomycin (Sigma). HEK293T cells (ATCC-CRL-3216) were cultured at 37°C, 5% CO2 in high-glucose DMEM (Sigma) supplemented with 5% FBS (PAN Biotech) and penicillin/streptomycin (Sigma). RAW 264.7 cells (ATCC TIB-71) were cultured at 37°C, 5% CO2 in high-glucose DMEM (Sigma-Aldrich) supplemented with 5% endotoxin low FBS (Sera Pro FBS, PAN Biotech), 1% glutamine, and 1% penicillin/streptomycin (Sigma). The cell lines were regularly tested for mycoplasma contamination in our facility Multiplexion (F020, DKFZ) (https://www.multiplexion.de)
Generation of A549 ADAR1 KO cell line
Lenti-CRISPR-ADAR1 exon 4 or NT in combination with pCMV-DR8.91 and pMD2.G were calcium phosphate transfected in HEK293T cells for lentiviral particle production (ratio 3:1:3). 48–72 h after transfection, cell-free supernatant was harvested and used for transduction of A549 cells. The transduced cells were selected with puromycin (1 μg/mL). Immediately after the selection control (non-transduced A549) died, limiting dilution in 96-well plates was performed for ADAR1 KOs (0.5 cell/well) and clonality was validated by visual inspection with a microscope; the Lenti-NT control was kept polyclonal. KO of ADAR1 was validated by western blot (anti-human ADAR1 [D7E2M] rabbit monoclonal antibody [mAb], Cell Signaling Technology, cat. #14175). Two clones, numbers 5 and 7, resulted in a completely abolished ADAR1 (p110 and p150) expression (Figure S3A). For further experiments we used only clone #5.
Generation of the RAW 264.7 APOBEC1 KO cell line
PX458-iv-sgRNA-A1_11 and PX458-iv-sgRNA-A1_39 plasmids were co-transfected using the Amaxa cell line Nucleofector kit V (Lonza) into RAW 264.7 cells following the manufacturer’s protocol for RAW 264.7 cells and a Nucleofector 2b device (Lonza). 48 h post-transfection GFP-positive cells were single cell sorted into 96-well plates and clonality was validated by visual inspection with a microscope. Clones were screened by amplifying targeted regions from genomic DNA (produced by a High Pure PCR template preparation kit [Roche]) using oligos #20–21 and #22–23 and then Sanger sequencing. This was followed by additional cloning of amplified regions using a CloneJET PCR cloning kit according to the manufacturer’s instructions and transforming DH5α bacteria with ligated product. Ten resultant bacteria colonies were sent for sequencing to determine genetic changes to the targeted region. One clone that was subsequently used contained in the region targeted by PX458-iv-sgRNA-A1_39 either a 1-bp deletion or a 2-bp deletion. KO was further confirmed by RT-PCR (using a One-step RT-PCR kit [QIAGEN]) amplification of B2m the 3′ UTR region from extracted RNA defined by oligos #24–25 known to be edited and determining absence of editing compared to the amplified region from the parental cells (Figure S3A).
RNA extraction, DNase treatment, and RT-PCR
RNA was extracted using an RNeasy mini kit (QIAGEN) and treated with DNase (Turbo DNA-free kit, Invitrogen). All of the PCRs on RNA were performed with gene-specific primers (Table S3) and a One-step RT-PCR kit (QIAGEN). Primers were designed using Primer-BLAST29 or AmplifX 2.0.7 (https://inp.univ-amu.fr/en/amplifx-manage-test-and-design-your-primers-for-pcr) to obtain 350- to 700-bp PCR amplicons. At this stage, PCR clean up usually is sufficient, however, gel extraction is required when amplification results in multiple bands (Macherey-Nagel NucleoSpin gel and a PCR clean-up kit was used). The fragments were then subjected to Sanger sequencing (Eurofins Genomics, GATC services, Germany, or Microsynth, Switzerland) and the resulting .ab1 files were analyzed by MultiEditR.
Titration experiments
For the C-to-U editing HEK293T cells were transfected with CmAG (50 ng), APOBEC1 (200 ng), and RBM47 (200 ng) expression vectors or CmAG (50 ng) alone. For transfection we used a mix of plasmid DNA and polyethylenimine (PEI) in an approximately 1:4 ratio (450 ng of DNA/2 μg of PEI). 72 h after transfection RNA was extracted and cDNA was amplified from Apob (using oligos #26 and #2). This allowed us to obtain Apob fragments heavily edited or not edited, respectively. These two fragments were cloned into a CloneJET PCR cloning kit (Thermo Scientific), and several colonies were screened by sequencing. From this screening we obtained two pJET vectors containing Apob with no editing (pJET-CmAG-WT) and six edited sites (pJET-CmAG-6x). These two vectors were then mixed together in titrated amounts from 0% to 100% and subjected to capillary Sanger sequencing with universal primers pJET1.2 forward and reverse.
RNA-seq
RNA-seq libraries were prepared in duplicate from A549 WT and ADAR1 KO clone 5 (Figure S3) and in triplicates from RAW 264.7 WT and RAW 264.7 APOBEC1 KO. Total RNA was extracted from 10,000,000 cells in duplicate (A549 WT and ADAR1 KO) or triplicate (RAW 264.7 and RAW 264.7 APOBEC1 KO each from separate plates). RNA was extracted using an RNeasy mini kit (QIAGEN) and then treated with turbo DNase (Life Technologies). RNA concentration and integrity were determined by a Qubit 4 (Thermo Fisher Scientific) using the Qubit RNA BR assay kit or Qubit XR assay kit and the Qubit RNA IQ kit (Thermo Fisher Scientific). 1 μg of RNA was processed with a Kapa mRNA HyperPrep kit for Illumina platforms (Kapa Biosystems, Roche) and KAPA single-indexed adaptor kit for Illumina platforms (Kapa Biosystems, Roche).
Libraries were sequenced with the Illumina HiSeq 2000 v4 technology, generating 125-nt paired-end reads. Adapters were trimmed using the Trim Galore software (https://github.com/FelixKrueger/TrimGalore). Before and after trimming we evaluated the RNA-seq quality with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Quality control, including per-base quality, duplication levels, and over-representative sequences, passed all of the checkpoints. The RNA-seq reads (~90 million raw reads per sample) were then aligned to hg19 (A549 data) or mm10 (RAW 264.7 data) reference genomes (publicly available by the UCSC genome browser) using the STAR aligner (v2.6.0a, https://github.com/alexdobin/STAR,30) with default settings, resulting in 85% uniquely mapped reads (~75 million mapped reads per sample). Potential PCR duplicates (<0.25%) were removed from the aligned reads using the MarkDuplicates function from Picard tools (http://broadinstitute.github.io/picard).
The aligned RNA-seq files (.bam files) were sorted and indexed with SAMtools (https://github.com/samtools/samtools). The sorted .bam files were used as an input for the REDItoolsDnaRna.py script, part of the REDItools suite (v1.0.415,16). REDItoolsDnaRna.py performs a comparative position-per-position analysis in parallel between an RNA and a DNA .bam file, so as to eliminate variants on the RNA, the signal of which derives from the genomic DNA. For this analysis, however, we used RNA-seq from A549 ADAR1 WT and ADAR1 KO, or RAW 264.7 WT and RAW 264.7 APOBEC1 KO. Sequences from KOs were used as background in order to identify the editing events for which ADAR1 or APOBEC1 is responsible. The options we set for a genomic position to be considered for variant calling required minimum coverage of five reads, with at least three reads supporting the editing event, and a minimum FastQ offset value of 33, per base. The aforementioned settings were used to run the analysis per pair and a specific gene’s coordinates with the option -Y. Genes and their coordinates are listed in Table S1.
Significance testing for detecting edits from the RNA-seq data was performed according to the REDItools algorithm.15,16 Briefly, p values were calculated for each mapped position using a Fisher’s exact test. The null column was composed of the sum of reference reads (WT, i.e., A, C, G, or T) and the sum of edited reads for each reference base (edited, i.e., A-to-G, C-to-T, G-to-A, or T-to-C) across the entire RNA-seq experiment, yielding a different null contingency for each reference base. The observed number of reference (WT) reads and alternative (edited) reads at each site of interest were then compared to the null contingencies via the Fisher’s exact test, generating a p value.
Amplicon-seq
Deep amplicon NGS-seq data were generated for a set of transcripts (Table S2), each one of which was amplified from RNA using one-step RT-PCR (QIAGEN) using primers containing an adaptor sequence needed for NGSelect Amplicons 2nd PCR service by Eurofins Genomics (all of the primers used for this experiment are listed in Table S3, #57-76). Adaptor-trimmed fastq files delivered (paired end) were merged for all of the transcripts per pair. Reads were mapped to hg19 and mm10 reference genomes (UCSC) with the STAR aligner (v2.7.3a). Aligned data (.bam files) were sorted with Samtools (v1.9), and the bamUtils (v1.0.14) clipOverlap function was employed for ensuring that good-quality reads will be considered for the downstream analysis. The sorted and .bam clipOverlapped files were indexed and the REDItoolsDenovo.py script was employed for calling SNVs in the concordant reads. Output data were joined with RNA-seq and MultiEditR data by sample, genomic coordinate, strand, and base identity.
Programmable RNA and DNA editing
For the EGFP reactivation experiment, HEK293T cells were transfected with CAGX (50 ng), 4λΝ-hADAR2-DD-NLS or 4λΝ-hADAR2-DD (100 ng), and pENTR-gRNA W58X (500 ng). The transfections were performed in a 24-well plate using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. 96 h after transfection, half of the cells were analyzed by fluorescence-activated cell sorting (FACS) to detect the percentage of EGFP-positive cells, and from the other half RNA was extracted and amplified the fragment of EGFP containing the W58X mutation (oligos #18–19). After PCR clean up (Macherey-Nagel NucleoSpin gel and PCR clean-up kit) the fragments were subjected to Sanger sequencing (Eurofins Genomics, GATC services, Germany). A-to-G editing was quantified with MultiEditR with the CAGX alone .ab1 set as the control file. See Figure S7C for analysis parameters.
For T cell base editing, NGS values, flow cytometry values, and .ab1 files were taken from the supplemental information of our previous work.21 Sequencing .ab1 files were analyzed by MultiEditR using default parameters and analyzed against NGS and flow cytometry values.
MultiEditR development
To distinguish edits from background noise, we modified the null hypothesis significance testing (NHST) algorithm from our previous work to operate on multiple edits spread across an amplicon, which we named MultiEditR. MultiEditR requires (1) a sample .ab1 file of an amplicon of interest between approximately 350 and 750 bp, (2) either a control .fasta file of the sequence of the amplicon of interest without any editing events or a control .ab1 of the amplicon of interest without any editing events, (3) a motif of interest consisting of any length of IUPAC nucleotides (e.g., YAR, TCA, A, C, N20, Nn), (4) a discrete base of interest hypothesized to be edited (e.g., A, C, T, or G), and (5) any hypothesized edited outcomes separated by “|” (e.g., G, T|G, A|T|G).
The MultiEditR algorithm begins by loading the sample .ab1 and trimming the ends of the sequence based on a Phred score cutoff (default of 0.0001) using a modified Mott’s algorithm (http://www.phrap.org/phredphrap/phred.html). If a control .ab1 file is used, the file is trimmed in the same manner. Once trimmed, the base calls are extracted from the sample chromatogram and aligned to the non-edited control sequence, where the control sequence index is joined to the sample. Any positions with indels in the alignment are filtered out of the analysis. The motif of interest is then matched to the control sequence, and the control indices where matches are found are used to separate the sample into the alternative sample where matches are found, and the null sample where matches are not found. The noise height of the edited bases of interest (e.g., G if interested in A-to-G edits, or T, G, or A if interested in C-to-T|G|A edits) is extracted from the trace of the null sample. These noise samples are used to model zero-adjusted (zΓ) gamma distributions, which are used as null distributions for the NHST, wherein the p value determines the critical value within the distribution of calling significance versus non-significance, as previously described.9 The height of hypothetical edits within the motifs of interest (e.g., height of G under A peaks) are then compared to the critical value generated by the null zΓ distributions. If a hypothetical edit is at or above the critical value it will be called as significant and reported as an edit. We made MultiEditR available as web application (https://moriaritylab.shinyapps.io/MultiEditR), and the source code is also available for running the application locally (https://github.com/MoriarityLab/MultiEditR). The application provides diagnostics plots of the sample, visualization, and tabulation of detected edits, a summary of the zΓ modeling, and the ability to download the output analysis data as a tab delimited file (see Figure S2 for visual layout of the web app algorithm).
MultiEditR was written in the statistical programming language R (v3.4.2) using RStudio (v1.1.383). The MultiEditR web app was developed using R shiny (https://shiny.rstudio.com/). global.R contains the definition of functions required to manipulate input files with input parameters and return output analyses, dependencies.R specifies the required packages for the web app, most notably sangerseqR31 for reading and analyzing .ab1 files (https://bioconductor.org/packages/release/bioc/html/sangerseqR.html), gamlss32 for zero adjusted gamma distribution modeling (https://www.gamlss.com/), tidyverse packages for data manipulation and visualization (https://www.tidyverse.org/), and shiny for support of the web application. server.R interfaces the inputs from the user side to the server side via the functions defined in global.R. ui.R specifies the visual interface of the web app.
Data analysis
All statistical analyses were performed in RStudio. The level of significance was set at α = 0.01. Student’s one-sample, two-tailed t tests were used as indicated in the text. Data were subjected to assumptions of homoscedasticity prior to testing. Data were visualized in RStudio employing various tidyverse (https://www.tidyverse.org/) and Bioconductor (https://www.bioconductor.org/) packages. See https://github.com/MoriarityLab/MultiEditR for reproducible analysis.
Availability
The MultiEditR web app is available at z.umn.edu/multieditr. Source code for running the application locally and recreating figures and analyses is available at https://github.com/MoriarityLab/MultiEditR. Original NGS data have been deposited in the NCBI GEO database under accessions GEO: GSE164211 and GSE145011.
Acknowledgments
We thank the High Throughput Sequencing unit of the Genomics & Proteomics Core Facility, as well as the Flow Cytometry unit of the Imaging and Cytometry Core Facility, German Cancer Research Center (DKFZ), for providing excellent sequencing and sorting services. We also thank Prof. Nina Papavasiliou (DKFZ), Derek Nedveck, and Walker Lahr for helpful conversations surrounding the topic. This work was supported by the Childrens Cancer Research Fund, the Fanconi Anemia Research Foundation, the University of Minnesota Academic Investment Research Program, and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 649019 to Prof. Nina Papavasiliou [DKFZ]). This work was made possible by an NIH-funded predoctoral fellowship to Mitchell G. Kluesner (T32GM007266). The graphical abstract was created with BioRender.com.
Author contributions
M.K., R.P., and B.S.M. designed the experiments. M.B., S.W., and T.L. developed KO cell lines. R.P. and A.A. performed titration experiments and RNA base-editing experiments and sequencing. M.K. wrote the program. R.N.T. defined the pipeline for NGS data processing. M.K., R.N.T., and R.P. analyzed the data. M.K. and R.P. wrote the manuscript. M.K., R.P., and B.S.M. supervised the research. All authors have read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.07.008.
Supplemental information
References
- 1.Eisenberg E., Levanon E.Y. A-to-I RNA editing—Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018;19:473–490. doi: 10.1038/s41576-018-0006-1. [DOI] [PubMed] [Google Scholar]
- 2.Lerner T., Papavasiliou F.N., Pecori R. RNA editors, cofactors, and mRNA targets: An overview of the C-to-U RNA editing machinery and its implication in human disease. Genes (Basel) 2018;10:13. doi: 10.3390/genes10010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu L.-D., Öhman M. ADAR1 editing and its role in cancer. Genes (Basel) 2018;10:12. doi: 10.3390/genes10010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reardon S. Step aside CRISPR, RNA editing is taking off. Nature. 2020;578:24–27. doi: 10.1038/d41586-020-00272-5. [DOI] [PubMed] [Google Scholar]
- 5.Diroma M.A., Ciaccia L., Pesole G., Picardi E. Elucidating the editome: Bioinformatics approaches for RNA editing detection. Brief. Bioinform. 2019;20:436–447. doi: 10.1093/bib/bbx129. [DOI] [PubMed] [Google Scholar]
- 6.Ramaswami G., Li J.B. Identification of human RNA editing sites: A historical perspective. Methods. 2016;107:42–47. doi: 10.1016/j.ymeth.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toung J.M., Lahens N., Hogenesch J.B., Grant G. Detection theory in identification of RNA-DNA sequence differences using RNA-sequencing. PLoS ONE. 2014;9:e112040. doi: 10.1371/journal.pone.0112040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montiel-González M.F., Vallecillo-Viejo I.C., Rosenthal J.J.C. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016;44:e157. doi: 10.1093/nar/gkw738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merkle T., Merz S., Reautschnig P., Blaha A., Li Q., Vogel P., Wettengel J., Li J.B., Stafforst T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019;37:133–138. doi: 10.1038/s41587-019-0013-6. [DOI] [PubMed] [Google Scholar]
- 10.Vogel P., Moschref M., Li Q., Merkle T., Selvasaravanan K.D., Li J.B., Stafforst T. Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods. 2018;15:535–538. doi: 10.1038/s41592-018-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qu L., Yi Z., Zhu S., Wang C., Cao Z., Zhou Z., Yuan P., Yu Y., Tian F., Liu Z. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019;37:1059–1069. doi: 10.1038/s41587-019-0178-z. [DOI] [PubMed] [Google Scholar]
- 12.Rinkevich F.D., Schweitzer P.A., Scott J.G. Antisense sequencing improves the accuracy and precision of A-to-I editing measurements using the peak height ratio method. BMC Res. Notes. 2012;5:63. doi: 10.1186/1756-0500-5-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: A method to quantify base editing from Sanger sequencing. CRISPR J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porath H.T., Carmi S., Levanon E.Y. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 2014;5:4726. doi: 10.1038/ncomms5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picardi E., Pesole G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–1814. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- 16.Picardi E., D’Erchia A.M., Gallo A., Pesole G. Detection of post-transcriptional RNA editing events. Methods Mol. Biol. 2015;1269:189–205. doi: 10.1007/978-1-4939-2291-8_12. [DOI] [PubMed] [Google Scholar]
- 17.Parker L.T., Zakeri H., Deng Q., Spurgeon S., Kwok P.Y., Nickerson D.A. AmpliTaq DNA polymerase, FS dye-terminator sequencing: Analysis of peak height patterns. Biotechniques. 1996;21:694–699. doi: 10.2144/96214rr02. [DOI] [PubMed] [Google Scholar]
- 18.Zakeri H., Amparo G., Chen S.M., Spurgeon S., Kwok P.Y. Peak height pattern in dichloro-rhodamine and energy transfer dye terminator sequencing. Biotechniques. 1998;25:406–410, 412–414. doi: 10.2144/98253st01. [DOI] [PubMed] [Google Scholar]
- 19.Roth S.H., Levanon E.Y., Eisenberg E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat. Methods. 2019;16:1131–1138. doi: 10.1038/s41592-019-0610-9. [DOI] [PubMed] [Google Scholar]
- 20.Vallecillo-Viejo I.C., Liscovitch-Brauer N., Montiel-Gonzalez M.F., Eisenberg E., Rosenthal J.J.C. Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 2018;15:104–114. doi: 10.1080/15476286.2017.1387711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webber B.R., Lonetree C.L., Kluesner M.G., Johnson M.J., Pomeroy E.J., Diers M.D., Lahr W.S., Draper G.M., Slipek N.J., Smeester B.A. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019;10:5222. doi: 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X., Tilford C., Neuhaus I., Mintier G., Guo Q., Feder J.N., Kirov S. CRISPR-DAV: CRISPR NGS data analysis and visualization pipeline. Bioinformatics. 2017;33:3811–3812. doi: 10.1093/bioinformatics/btx518. [DOI] [PubMed] [Google Scholar]
- 23.Potapov V., Fu X., Dai N., Corrêa I.R., Jr., Tanner N.A., Ong J.L. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Res. 2018;46:5753–5763. doi: 10.1093/nar/gky341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Severi F., Conticello S.G. Flow-cytometric visualization of C>U mRNA editing reveals the dynamics of the process in live cells. RNA Biol. 2015;12:389–397. doi: 10.1080/15476286.2015.1026033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg B.R., Hamilton C.E., Mwangi M.M., Dewell S., Papavasiliou F.N. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat. Struct. Mol. Biol. 2011;18:230–236. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pestal K., Funk C.C., Snyder J.M., Price N.D., Treuting P.M., Stetson D.B. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill J.T., Demarest B.L., Bisgrove B.W., Su Y.C., Smith M., Yost H.J. Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev. Dyn. 2014;243:1632–1636. doi: 10.1002/dvdy.24183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rigby R.A., Stasinopoulos D.M., Lane P.W. Generalized additive models for location, scale and shape. J. R. Stat. Soc. Ser. C Appl. Stat. 2005;54:507–554. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.