

Abstract
The studies on the Lucina pectinata hemoglobins are essential because of their biological roles in hydrogen sulfide transport and metabolism. Variation in the pH could also play a role in the transport of hydrogen sulfide by HbI and O2 by HbII and HbIII, respectively. Here, fluoride binding was used to further understand the structural properties essential for the molecular mechanism of ligand stabilization as a function of pH. The data allowed us to gain insights into how the physiological roles of HbI, HbII, HbIII, adult hemoglobin (A-Hb), and horse heart myoglobin (Mb) have an impact on the heme-bound fluoride stabilization. In addition, analysis of the vibrational assignments of the met-cyano heme complexes shows varied strength interactions of the heme-bound ligand. The heme pocket composition properties differ between HbI (GlnE7 and PheB10) and HbII/HbIII (GlnE7 and TyrB10). Also, the structural GlnE7 stereo orientation changes between HbI and HbII/HbIII. In HbI, its carbonyl group orients towards the heme iron, while in HbII/HbIII, the amino group occupies this position. Therefore, in HbI, the interactions to the heme-bound fluoride ion, cyanide, and oxygen with GlnE7 via H-bonding are not probable. Still, the aromatic cage PheB10, PheCD1, and PheE11 may contribute to the observed stabilization. However, a robust H-bonding networking stabilizes HbII and HbIII, heme-bound fluoride, cyanide, and oxygen ligand with the OH and NH2 groups of TyrB10 and GlnE7, respectively. At the same time, A-Hb and Mb have moderate but similar ligand interactions controlled by their respective distal E7 histidine.
Keywords: Lucina pectinata, cyanide, fluoride binding, electronic spectra, hemoglobin, myoglobin
Graphical Abstract
Introduction
Hydrogen sulfide (H2S) is a molecule with relevant biological implications in the mammalian cell. In tissue it can functions as a neuromodulator, neuroprotection, muscle relaxant, and endothelial inflammation regulator. It is also suspected to be a possible therapeutic agent.1–12 Thus, the molecular mechanism of H2S transport in biological systems is a subject of high interest and importance. The studies on the hemoglobins from the Lucina pectinata have spearheaded biological H2S transport and metabolism in these systems. The clam relies on symbiont bacteria and a system of hemoglobins for survival. These hemoglobins, characterized by Kraus and Wittenberg, consist of hemoglobin I (HbI), hemoglobin II (HbII), and hemoglobin III (HbIII).13,14 In addition to oxygen, the HbI supplies H2S to the sulfur-oxidizing chemoautotrophic bacteria for nutrient synthesis. Based on its extraordinary affinity for H2S, which is 4,000 times greater than HbII and HbIII, HbI is considered to be a sulfide-binding hemoglobin. At the same time, HbII and HbIII are relevant to the clam for oxygen binding only. For hemoglobin and myoglobin, the ability to transport H2S is unusual because these proteins are more prone to the formation of sulfhemoglobin or sulfmyoglobin, a heme protein derivative, where one of the heme’s pyrrole rings results in the covalent attachment of a sulfur atom.15,16 The ability of HbI to stabilize H2S as a heme ligand is attributed to its unique pocket structure containing three phenylalanine (B10, CD1, and E11) and E7 glutamine amino acids.17 L. pectinata HbII and HbIII hemoglobins have shown that GlnE7 (Q65) and TyrB10 (Y30) are involved in the stabilization of various heme-bound ligand in complexes such as the ferrous-oxy, ferryl-oxo, and ferrous-carbonmonoxy heme via hydrogen bonding between the tyrosine hydroxyl (B10 or Y30) and the glutamine amine group (E7 or Q65).17–24 Figure 1 shows the stabilization of oxy-HbII by these amino acids hydrogen bonding networking. However, this condition is not present in HbI because it has a phenylalanine in the B10 position. The GlnE7 (Q65) has its carbonyl group oriented towards the heme pocket, acting as hydrogen bond acceptor upon H2S binding to metHbI.13,14,17,21–23 Moreover, it has been proposed that the hydrogen bonding and a larger heme pocket of HbI, relative to HbII and HbIII, contribute to the unique properties of HbI and its high affinity for H2S binding.
Figure 1.
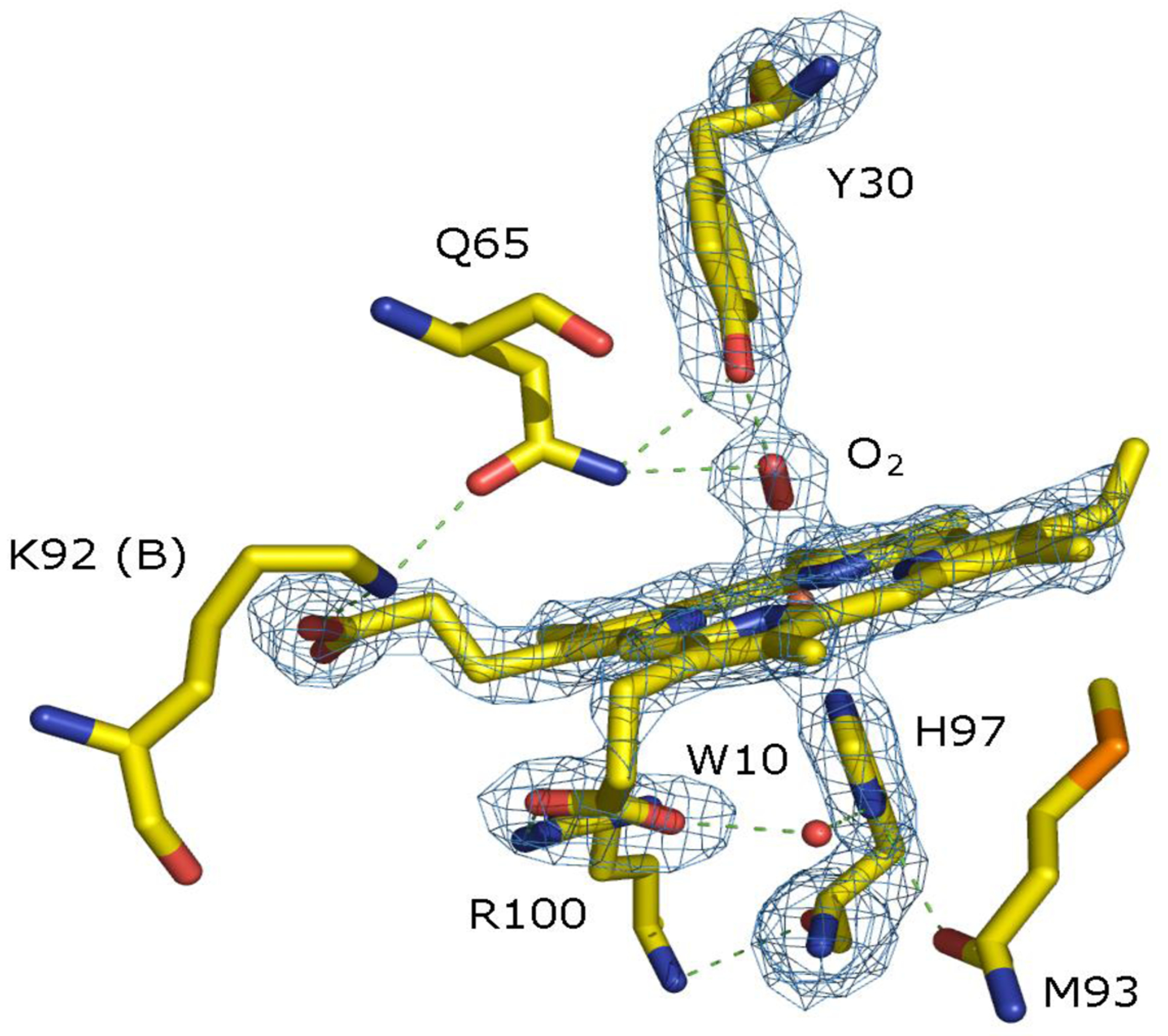
Oxy-HbII heme pocket structure with the Fe(II)O2 moiety strongly hydrogen-bonded to GlnE7 (Q65) and TyrB10 (Y30), obtained from Darya et al.24
The sulfide-HbI reactions have shown how the concentration of H2S affects its binding to HbI.14,25 In the presence of oxygen at relatively low concentrations, H2S binds to ferric HbI to form a ferric heme-H2S low-spin complex. Under higher concentrations of H2S, the ferric heme-H2S complex is reduced to form a ferrous-heme unit, releasing H2S. Since HbI is monomeric hemoglobin, the physiological binding and release of H2S in the clam is governed by the H2S concentration that alters the heme iron nuclear effective charge facilitating the liberation of hydrogen sulfide. Hydrogen sulfide dissociation from the HbI-H2S complex is controlled partly by hydrogen sulfide concentration and the H2S/HS− equilibrium (pKa is 7.0). Physiologically, Lucina pectinata H2S availability may result from soil pH variations. Thus, pH could play a role in both H2S and O2 transport by the clams’ hemoglobins.24 A study on the L. pectinata oxy (HbII-HbIII) complex shows that in the presence of potassium ferricyanide or sodium formate, a decrease in pH from 9 to 4 leads to the formation of met (HbII-HbIII).24 Likewise, a pH-dependent study on stopped-flow kinetics of purified human adult hemoglobins shows that dissociation rates of the metHb-H2S complex increase when decreasing pH, consistent with the pH-dependent affinity.26 In addition, the release of hydrogen sulfide from its complex in HbI, HbII, and HbIII is controlled by the nature of the heme pocket.13,14 Analogously, H2S koff for a series of truncated hemoglobin-hydrogen sulfide complex is also dependent on its heme pocket structure.27
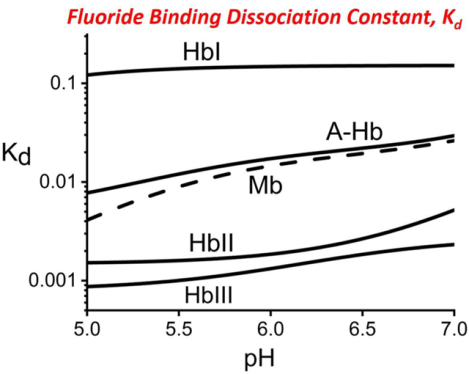
Here, we have used fluoride binding to further understand the molecular mechanism of ligand binding as a function of pH and gain insights into how HbI, HbII, and HbIII’s physiological roles impact the stabilization of heme-bound fluoride. Smulevich and collaborators have demonstrated how fluoride can probe the distal cavity structure and hydrogen bonding interactions from the distal site in heme proteins.28–30 Their data showed that the charge transfer absorption band (CT1) in the 600 nm region is sensitive to interactions between the heme-fluoride complex and the distal amino acids. Here, we used the heme-fluoride complex’s electronic spectra to calculate the dissociation constants (Kd) as a pH function. The outcomes lead to the interpretation that the fluoride-distal heme pocket interactions in HbI, HbII, and HbIII are unique. The data also allowed us to explore the fluoride binding properties of adult hemoglobin (A-Hb) and horse heart myoglobin (Mb). The results show that in the clam hemoglobins, the mechanism of heme-fluoride stabilization varies in each hemoglobin. Furthermore, the pH profiles of fluoride binding not only differ because of the composition of the heme pocket distal amino acids (PheB10-GlnE7 in HbI, TyrB10-GlnE7 in HbII and HbIII), but also due to a slight difference in the positions of the TyrB10 residues in HbII and HbIII. Also, A-Hb and Mb have very similar fluoride binding properties controlled by their respective distal E7 histidine.
Materials and Methods
2.1. Protein purification, sample preparation, and UV-Vis spectra pH dependence
L. pectinata HbI, HbII, and HbIII were obtained and isolated following procedures by Marchany-Rivera et al.,24 Krauss and Wittenberg,14 and Pietri et al.31 The hemoglobin samples were then dissolved in sodium phosphate buffers (NaPi). Adult hemoglobin (A-Hb) and horse skeletal muscle myoglobin (Mb) were purchased from Sigma-Aldrich (St. Louis, MO). Hb and Mb stock solutions were prepared by dissolving the lyophilized Hb and Mb in sodium phosphate buffers. HbI, HbII, HbIII, A-Hb, and Mb samples were dissolved in a 0.2 M sodium phosphate buffer containing 0.2 M NaCl and up to 0.1 mM K3Fe(CN)6 to achieve complete heme oxidation, from pH 3 to pH 9. The UV-vis spectra were obtained at room temperature by using a Lambda 20 spectrometer (Perkin Elmer). Regarding the electronic spectra, particular attention was paid to heme loss or protein unfolding in the pH 3 to pH 5 interval, evidenced by the decrease in the Soret band around the 408 nm region with a concomitant increase in 370 nm region.32,33
2.2. Dissociation constant (Kd) measurements for fluoride binding
Identical heme protein concentrations were prepared for the fluoride binding measurements to minimize spectral changes due to heme protein dilution upon fluoride addition. Gas-tight syringes were used to measure exact volumes for protein dissolution and fluoride titration. In similar preparations for each heme protein, exact amounts (10 – 20 μL) of the heme protein stock solution were dissolved in two separate vials, each containing 2.0 mL of i) 0.2 M NaPi, 0.2 M NaCl, and ii) 0.2 M NaPi, 0.2 M NaCl, and 0.2 M NaF at a predetermined pH. The 2.0-mL heme protein solution without NaF was placed in a 1-cm optical cuvette (total capacity of 4 mL), and the UV-vis spectrum was recorded as the 0 μL NaF spectrum. The other 2.0-mL vial with the heme protein in the buffer containing the 0.2 M NaF was used to titrate in the fluoride. The total NaF solution volumes were 10, 20, 50, 100, 200, 500, 1000, and 1500 μL. The fluoride titrations were performed a pH 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, and 9.0. Aliquots of the heme protein with NaF solution were injected into the optical cuvette containing the heme protein without NaF. Upon each addition, a 500-μL gas-tight syringe was used to gently mix the heme protein solutions by drawing the solution into the syringe and then slow reinjection into the cuvette; various cycles were done to ensure proper mixing. After each addition, the UV-vis spectrum was recorded.
The UV-vis spectra of the heme-bound fluoride complexes were used to determine the apparent dissociation constants (Kd). In addition, the absorption band in the 604–612 nm region was followed to obtain quantitative information on the heme-bound fluoride complex formation.29 The data was tailored with equation 1:
| (eq. 1) |
Where ΔA is the change in absorbance at the wavelength of maximum perturbation (605–611 nm) and the ΔAT is the change in absorbance at the maximum perturbation wavelength at saturating ligand concentrations. ΔA and ΔAT are the absorbance of the heme protein-fluoride complex relative to the absorbance of the heme protein without the NaF (the 0-μL NaF spectrum). The Kd is the dissociation constant for the heme protein-fluoride complex. LT is the total fluoride concentration, and MT is the total concentration of the heme protein.
1.3. Apparent Dissociation Constant Non-linear Fits
The studied proteins’ apparent dissociation constant-pH plots were modeled using binding polynomials for protein-ligand equilibria as described by Alberty.34 The pH effects on the apparent dissociation constant for the protein-ligand equilibrium were taken into account by incorporation of the acid dissociation constants (Ka) for the protein (P), ligand (L), and the protein-ligand (PL) sites. The total concentrations for the protein, ligand, and protein-ligand complex [P]T, [L]T, and [PL]T, respectively, as a function of pH, are shown in equations 2–4. Here, we are considering the proton dissociation, so the reference species are acidic, [P]acid, [L]acid, and [PL]acidic.
| (eq. 2) |
| (eq. 3) |
| (eq. 4) |
In the above polynomials, the dissociation constant (pKa) for the acid sites of the unligated protein (free protein) are labeled as pKap1, pKap2… and the acid sites in the ligand are shown as pKaL1, pKaL2 … and the acid sites for the protein-ligand complex are pKaPL1, pKaPL2, …
The equilibrium dissociation constant expression for ligand binding in terms of the polynomials is shown below:
| (eq. 5) |
The expression on the left side of equation 5 represents the apparent dissociation constant (Kd app) for ligand binding. The factored-out acidic species concentrations are the dissociation constant at acidic pH (Kd acid). The apparent dissociation constant in terms of Kd acid, pH, and pKa is shown below:
| (eq. 6) |
1.4. HbI Kd fit equations
The fluoride binding data for HbI was fitted with equation 7:
| (eq. 7) |
The non-linear fit was based on the fluoride-binding scheme shown in Fig. 6. Given the steep increase in the apparent Kd at acidic pH (Fig. 5a), binding of both hydrofluoric acid (HF) and the fluoride ion (F−) was considered. The acid dissociation constant for HF is labeled as pKaHF and was set to 3.0. The pKa for the protein with the bound fluoride is denoted as pKaHbI.
Figure 6.
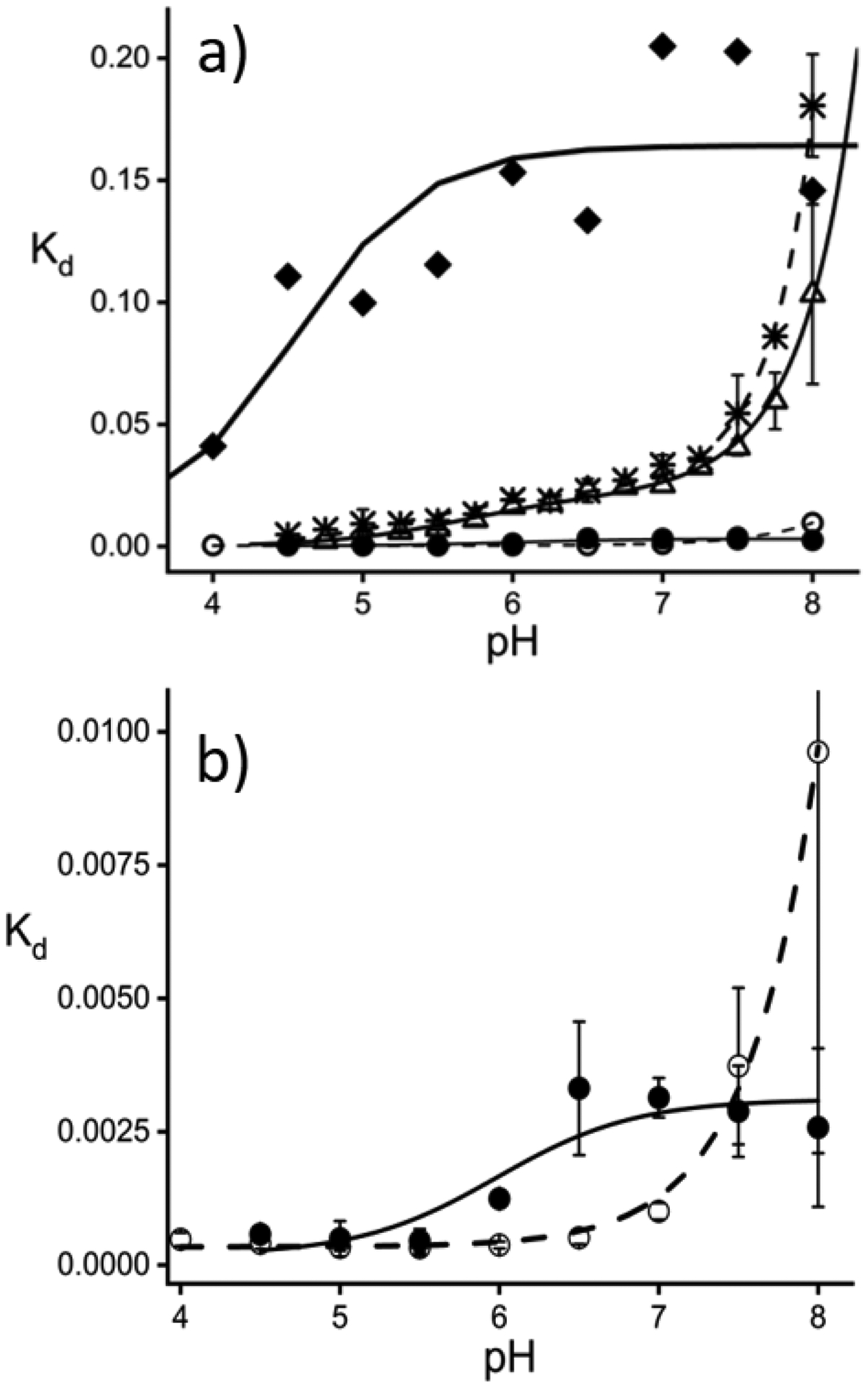
a) Apparent Kd versus pH plots for HbI (diamonds), HbII (open circles), HbIII (filled circles), A-Hb (asterisks), and Mb (open triangles). Non-linear fits are shown for A-Hb (dash line), Mb (line), HbI (bold line), HbII (dash line), and HbIII (line). b). Kd plots and non-linear fits for HbII and HbIII. All measured Kd values and standard errors are reported as supplementary material.
Figure 5.
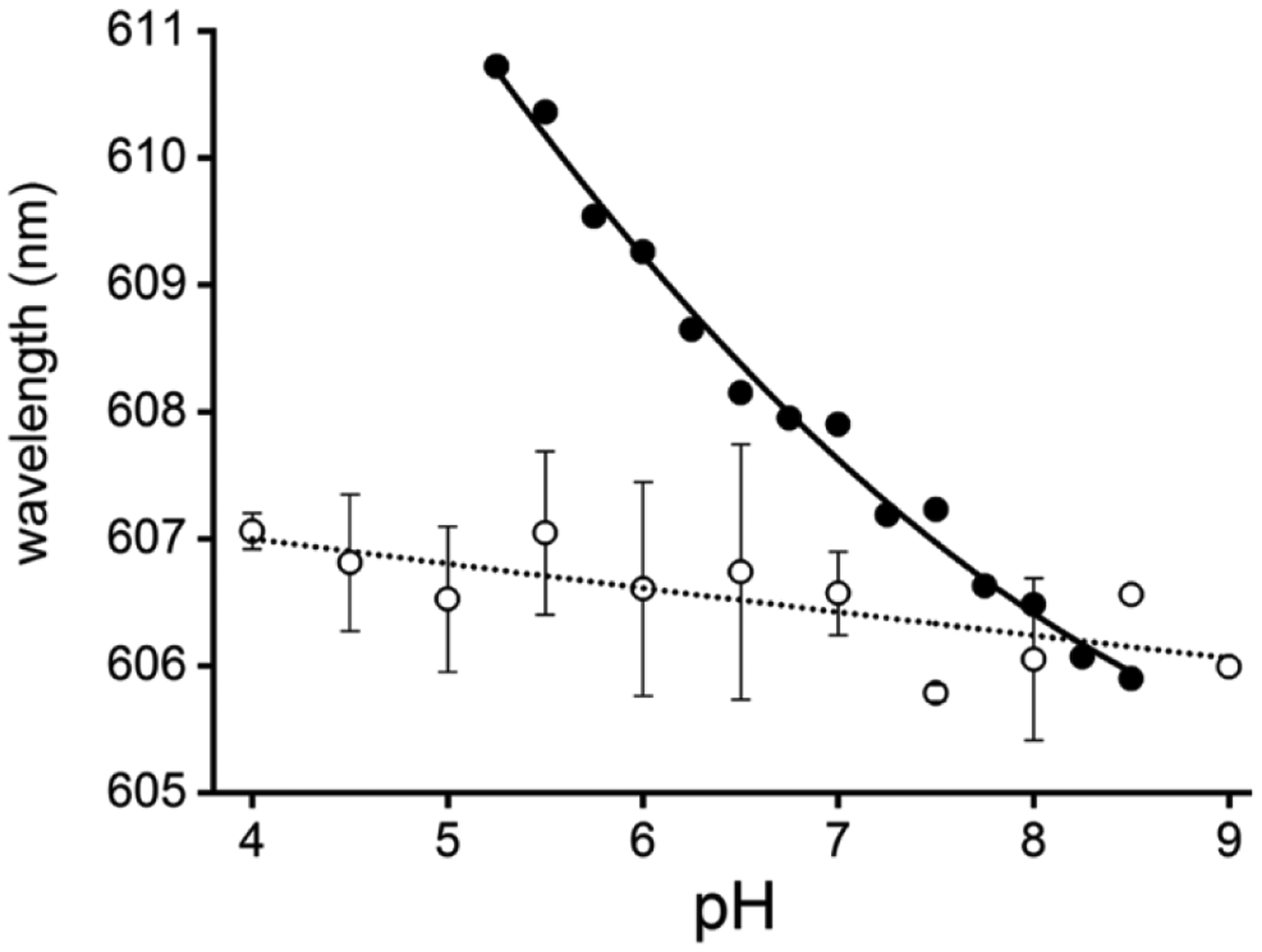
Heme-bound fluoride CT1-band wavelength versus pH for Mb (filled circles) and HbII (open circles). The absorbance peak wavelengths were obtained from the final heme-bound fluoride spectra shown in Figure 3 from pH 4.0 to pH 9.0.
1.5. HbII and HbIII Kd fit equations
For HbII and HbIII, equations 8 and 9 were fitted to the fluoride binding data of HbII and HbIII, respectively.
| (eq. 8) |
| (eq. 9) |
Since there are no significant changes in the apparent Kd at pH below 5 (Figs. 5b), only fluoride ion binding was considered. In eq. 8 and eq. 9, pKaOH is the pKa of the unligated protein, which is the acid-to-alkaline transition, and pKaFOH is the pKa of the heme-bound fluoride complex for the transition of the fluoride binding in the presence of metaquo heme complex to the binding in the presence of the heme-bound hydroxy complex. The Kdacid is the dissociation constant for heme-bound fluoride ion (F−) in the presence of the metaquo heme complex at low pH.
1.6. A-Hb and Mb Kd fit equations
For fluoride binding in A-Hb and Mb, equation 10 was used to fit the data.
| (eq. 10) |
In this equation, Kdacid is the dissociation constant at acidic pH for the binding of hydrofluoric acid (HF), pKaw(His) is the pKa of His64 in the presence of heme-bound water. At the same time, pKaF (His) is the pKa of His64 in the heme-bound fluoride complex. The pKa of hydrofluoric acid (pKa HF) was set to 3.0, and the acid-alkaline transition (pKaOH) was set to 8.0 and 8.9 for Hb and Mb, respectively, as reported by Antonini and Brunori.35
2. Results
3.1. Electronic spectra pH dependence
The L. pectinata clam habitat is subjected to variations in pH due to the presence of H2S, whose concentration in the mangrove soil environment can be up to 10-fold from its typical value of 0.79 mM.14,36 These concentrations can result in a soil of pH 5 or below; such low pH has adverse effects on the structure and function of oxygen carriers such as Mb and Hb.32,33,37–43 Figures 2a–c show the UV-vis spectra of ferric HbI, Mb, and A-Hb at pH 3.0 and pH 7.0. The electronic spectra at pH 7.0 with the Soret bands in the 408 nm region indicate the proteins’ metaquo state. At pH 3.0, for Mb and A-Hb, the spectra show a complete shift in the Soret band to the 370 nm region, while HbI displays a shoulder with substantial absorbance at 408 nm. The broad Soret band centered at 370 nm in Mb has been attributed to a structural change of the heme pocket upon denaturation at low pH.32,33 The absorbance ratio 408 nm to 370 nm (A408 nm / A370 nm) was used to monitor the % folded protein as a pH function. Figure 1d shows the % folded protein plots versus pH for HbI, HbII, Mb, and A-Hb and reveals apparent differences in the pH profiles.
Figure 2.
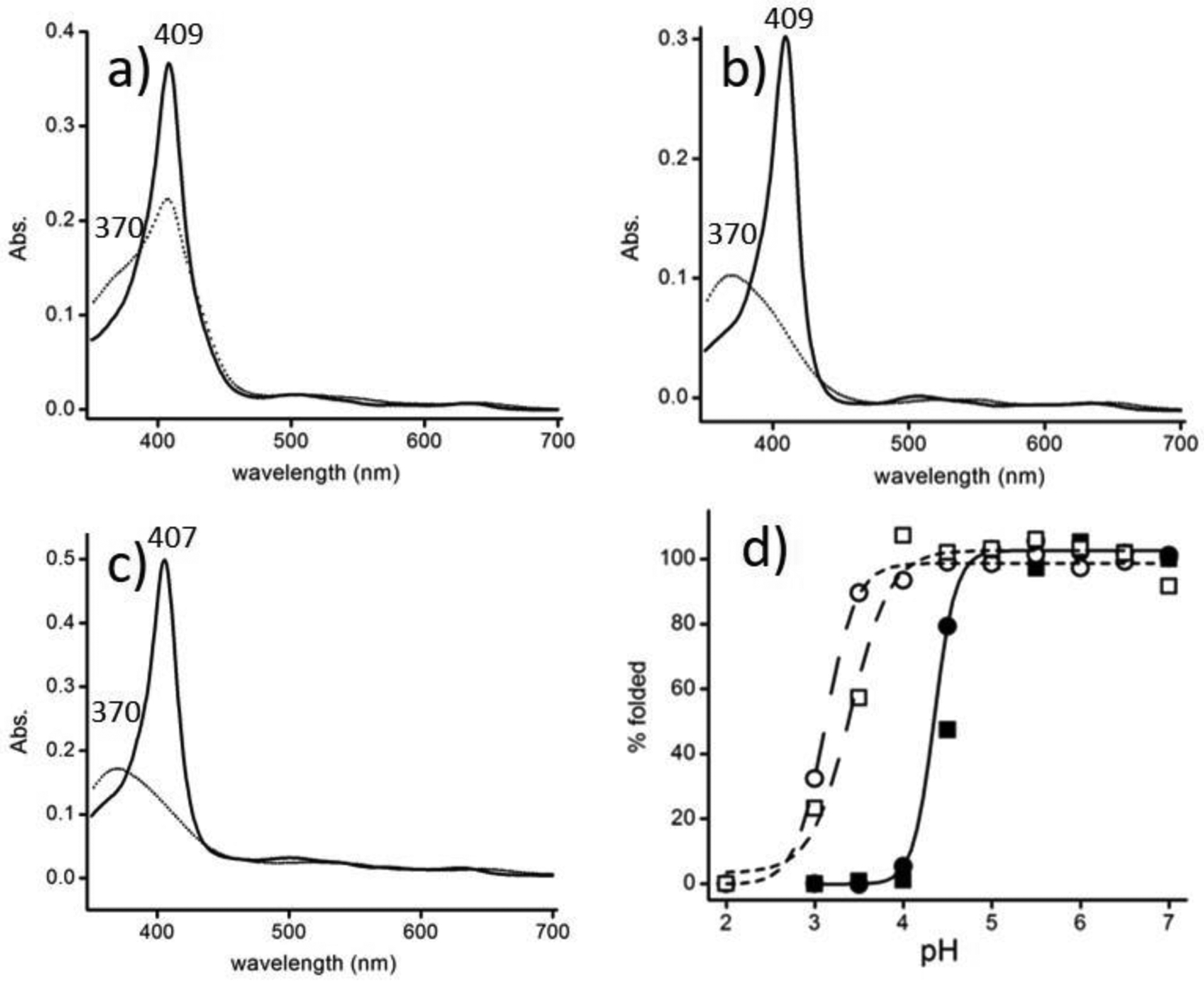
Electronic spectra of ferric heme proteins at pH 3.0 (dotted line) and pH 7.0 (bold) of HbI (a), Mb (b), and A-Hb (c). The % of folded protein as a function of pH is shown in (d) for HbI (open circles), HbII (open squares), Mb (filled squares), and A-Hb (filled circles). The fit is shown for Mb and A-Hb with a pKa = 4.4. The pKa for HbI and HbII are 3.1 and 3.4, respectively (normalization for HbI and HbII were done assuming complete unfolding at pH 2.0 with the A408 nm / A370 nm ratio set to 0.6).
A model that considers multiple protonation sites with different acid dissociation constants for the native and unfolded states of Mb has been reported by Sage et al. for this type of plot.32 However, in this study, we used a sigmoidal-Boltzmann fit to calculate the pKa of denaturation. Mb and A-Hb have similar pKa of acid denaturation at 4.4. These proteins are less than 50% folded below this pH, but HbI and HbII are nearly 100% folded at pH 4.5. These proteins are much more stable in acidic conditions than Mb and A-Hb because of the heme pocket stability. Structurally, the main difference between the L. pectinata hemoglobins and Mb and A-Hb’s heme pocket is the unusually high content of aromatic residues in the clam hemoglobins.14,17,20–22,24,44
The clam hemoglobins have aromatic residues such as phenylalanine in the E11 and CD1 positions and tyrosine in the B10 position HbII and HbIII (in HbI the residue is phenylalanine). In a study of hemin dissociation in met-myoglobin mutants, Hargrove et al. concluded that one of the significant factors in heme stabilization is the hydrophobic interactions between the heme and the apolar residues of the heme pocket.45 The aromatic groups in the heme pocket of the clam hemoglobins provide the hydrophobic medium to stabilize the heme. Further, the polarizability of these groups can allow for favorable heme-ligand interactions through π-electrostatic interactions. For example, it is well known that the phenylalanine in the B10 position of HbI stabilizes heme-bound ligands such as H2S, O2, CO, and CN− through π-electrostatic interactions.17,31,44,46,47 Thus, unlike A-Hb and Mb, the clam hemoglobins can withstand acidic media like HbI (Figure 1d). Therefore, the structure of the aromatic residues in the heme pocket may be critical to the heme stabilization of HbI, HbII, and HbIII at low pH.
Figure 3 shows the Q electronic spectra of HbI, HbII, and HbIII at acidic and alkaline conditions, at pH 4.5 and pH 9. Figure 1S (Supporting Information) shows the overall electronic spectra of ferric HbI (a), HbII (b), and HbIII (c) at pH 4.5 (bold line) and pH 9.0 (dashed line). While the spectra of HbI at pH 4.5 and pH 9 are similar, HbII and HbII show a dramatic change in the Q regions. At pH 4.5 (Figure 3a, 3b, and 3c), the metaquo HbI, HbII, and HbIII show the classical transitions at 502 and 630 nm.13,14 However, at pH 9, the spectra of HbII and HbIII (Figure 3b and 3c) show bands at 540, 575, and 604 nm. The 540/575 nm pair of bands have been identified by Kraus et al. as those corresponding to the same low spin heme-hydroxyl complex formed in Mb under alkaline conditions.13 The HbII and HbIII spectra (albeit more as a shoulder in HbIII) show the ~600-nm band assigned to a charge-transfer band, originating the tyrosinate-heme moiety, upon the binding of the distal TyrB10 to heme. Such an assignment was based on the similarities of the optical and EPR spectra between Hb species with tyrosine as a proximal or distal ligand and that of HbII and HbIII.13 Moreover, resonance Raman studies of ferric HbII at neutral pH confirmed the coexistence of low- and high-spin species from six- and five-coordinate heme complexes.18 Therefore, it is possible that the 600-nm band has to do with one of the above combinations. Meanwhile, the 540 and 575 nm pair assignment is confirmed as a heme-low spin six coordinated HbII.18
Figure 3.
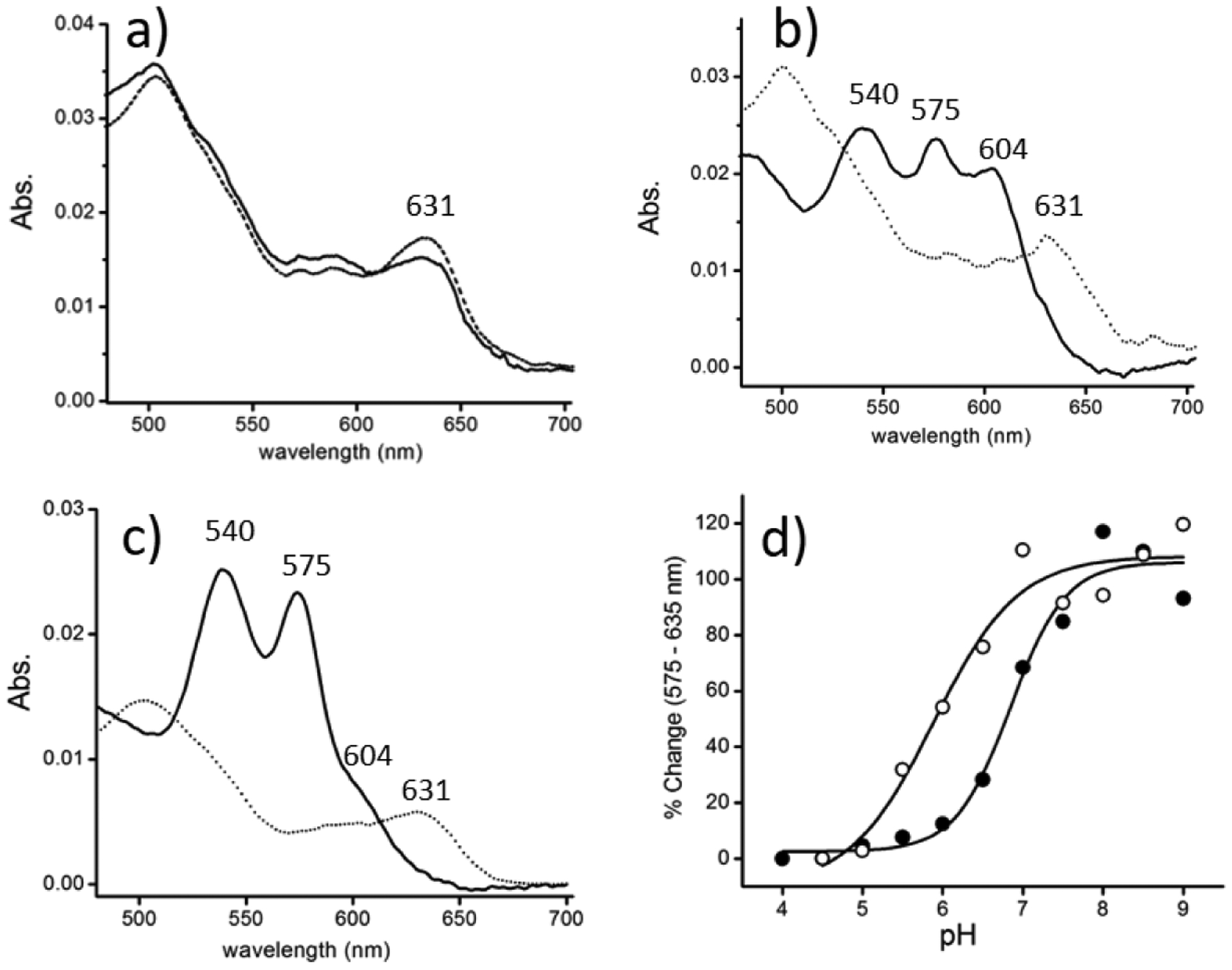
Q-band region spectra at pH 4.5 (dotted line) and pH 9.0 (bold) of ferric: HbI (a), HbII (b), and HbIII (c). At pH 4.5, the heme proteins are in the metaquo, 630 nm region. At pH 9.0, HbII and HbIII show the ferric-hydroxy heme, 540 and 575 nm (the shoulder at 604 nm is attributed to heme-bound tyrosinate). The absorbance at 575 nm, relative to that of 635 nm, is plotted vs. pH (d) for HbII (filled circles) and HbIII (open circles). The fits for HbII and HbIII have acid-alkaline transition pKa of 6.8 and 5.9, respectively (based on a Boltzmann-sigmoidal fit).
Also, Figure 2S (Supporting Information) shows the Resonance Raman of met-hydroxide HbI at pH 11.0 (a) and met-aquo HbI at pH 7 (b). The data reveals that met-hydroxide HbI is entirely dominated by a low spin complex with high and low spin components. At the same time, the met-aquo derivative shows the classical mixture of five and six coordination with the corresponding high and low spin structures. Analogous Resonance Raman data of the HbI-SH2 complex is dominated by a six coordination and low spin states complex.47 Figure 3d shows pH plots of the low spin heme-hydroxy populations in HbII and HbIII (represented by the absorbance at 575 nm, relative to that at 635 nm) show that the acid-alkaline transitions occur with pKa of 6.8 and 5.9, respectively. These values agree with Kraus et al. determined acid-alkaline transition pKa values of 6.6 and 5.9 for HbII and HbIII, respectively. These pKa values are unusually acidic relative to HbI and Mb, which have acid-alkaline transitions at higher pH.13 Other heme proteins, such as Hb and horseradish peroxidase (HRP), also have an acid-alkaline transition under primary conditions.48 The results reveal that the acid-to-alkaline transitions may be modulated by the composition of the distal residues and their spatial arrangement in the heme pocket. HbII and HbIII have similar distal amino acids, but the acid-to-alkaline transition pKa are related to differences in their heme pocket distal amino acids’ relative position. These properties are further illustrated in the following sections of hemeproteins fluoride binding properties.
3.2. Electronic spectra of heme-bound fluoride complexes
The addition of sodium fluoride to ferric HbI, HbII, and HbIII results in heme-bound fluoride complexes’ formation. Figure 3S (supporting information) and Figures 4 a–c show the Soret and Q band region electronic spectra of ferric HbI, HbII, and Mb and sodium fluoride at pH 7, respectively. Each of the final optical absorption spectra shown in bold displays a band in the 600-nm region. It has been shown that this absorbance maximum in heme proteins with fluoride ion is a charge transfer band (CT1) that is due to the presence of a six-coordinate, high-spin heme-bound fluoride complex.29,49,50 A comparison of these spectra is shown in Figure 4d. HbII, HbIII, A-Hb, and Mb (not shown) have the CT1 band around 607 nm, while HbI appears blue shifted at 596 nm. The low wavelength of this CT1 band was also reported by Kraus et al., measured at 599 nm.13 The CT1 band’s wavelength has been correlated with the interaction strength between the distal site and the heme-bound fluoride. The stronger the H-bonding interaction, the longer the wavelength of the CT1 band.28–30 Regarding this, the data suggests that the H-bonding network to the heme-bound fluoride in HbI is absent, while for HbII and HbIII, such a system is present with a strong influence on the electronic structure of the heme-fluoride complex. Further evidence of the weak interaction in the HbI heme-bound fluoride is supported by the very low affinity between HbI and the fluoride ion. It is 20 times weaker than the fluoride’s affinity HbII, HbIII, A-Hb, and Mb (see following sections). The low-wavelength CT1 band and the feeble affinity for fluoride binding suggest the absence of an H-bond donor in the heme pocket of HbI. HbI X-ray crystallographic structure showed that B10 phenylalanine and the E7 glutamine are the essential amino acids involved in heme-hydrogen sulfide stabilization.17,44 Rizzi et al. proposed that the ferric heme-bound H2S not only interacts with the GlnE7, which can serve as a hydrogen bond acceptor to its carbonyl group, but is also further stabilized by “electrostatic-aromatic” interactions with the edge of the phenyl rings of PheB10, PheCD1, and PheE11. Resonance Raman measurements of the oxy-HbI, carbonmonoxy-HbI, and metcyano-HbI complexes show νFe-O2, νFe-CO, νCO, and νFe–CN stretching modes that indicate the presence of distal residue interactions with these heme-bound ligands.47 FTIR studies on recombinant HbI (rHbI) showed that substitutions of the distal residues GlnE7 and PheB10 affect the νCO stretching modes populations of the carbonmonoxy-rHbI complex. The results indicate that both of these residues are important in stabilizing the heme-bound carbonmonoxy ligand.31 The HbI heme-bound fluoride ion’s weak interactions also suggest that similar to the HbI-hydrogen sulfide complex, the GlnE7 carbonyl group is orientated towards the heme-bound fluoride. In this spatial arrangement, the heme pocket lacks hydrogen bonding interactions with the fluoride ligand.17,22,44 Similar to heme-bound fluoride ion, Raman studies on the heme ferryl-oxo complex in HbI proposed that such complex is devoid of heme pocket interactions.51
Figure 4.
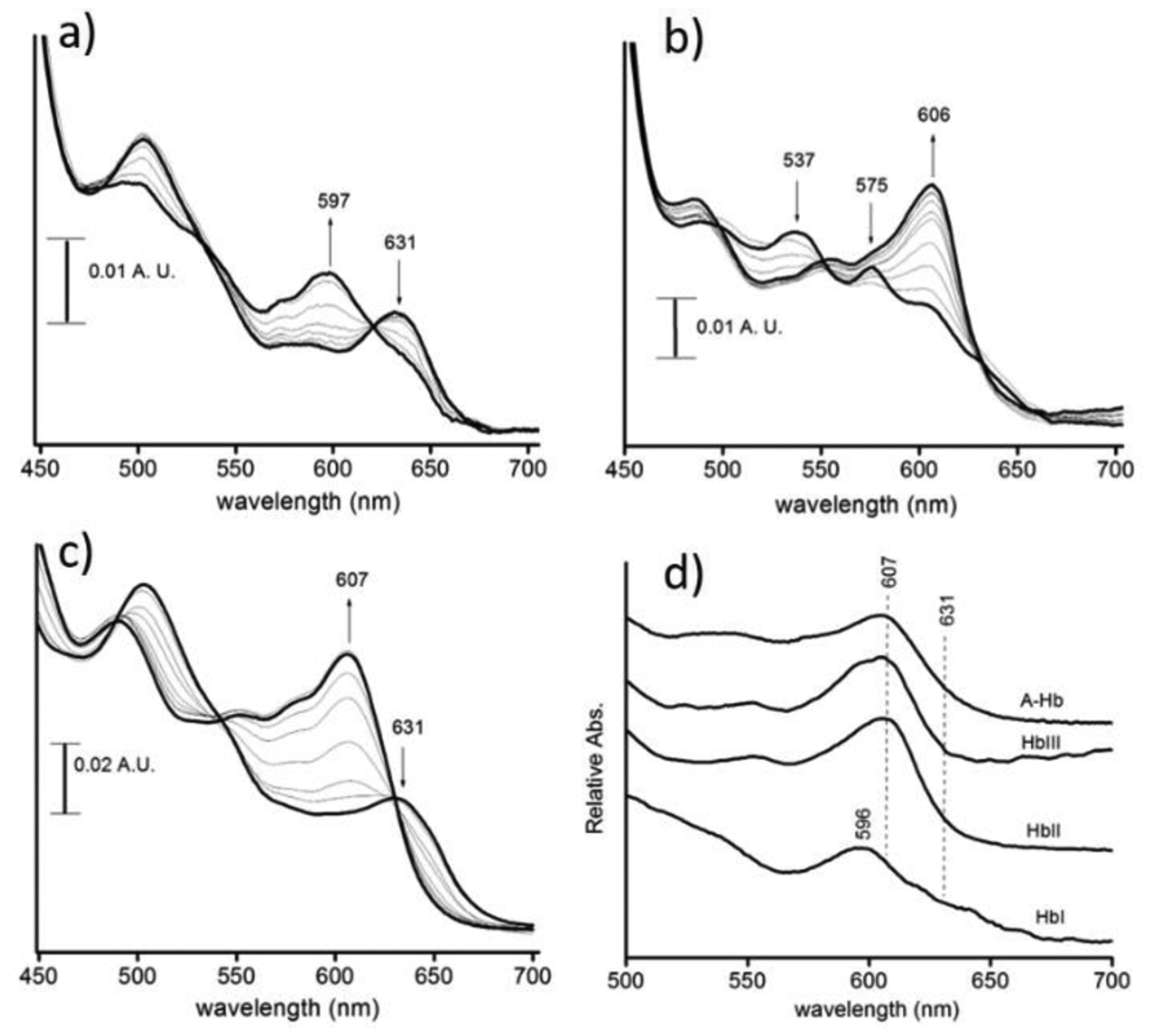
Q-band region spectra at pH 7.0 of sodium fluoride addition to ferric: HbI (a), HbII (b), and Mb (c). The initial (with no fluoride) and final spectra of the fluoride-heme complexes are shown in bold. The final heme-bound fluoride Q bands spectra are shown in (d). The absorbance at 607 nm is due to the presence of a heme-bound fluoride complex. HbI shows the presence of metaquo heme, indicated by the band at 631 nm.
The CT1 band for the HbII and HbIII fluoride complexes is centered at 607 nm. Unlike the CT1 band for the heme-bound fluoride complex of Mb, the wavelengths of the CT1 band for the heme-bound fluoride complexes of HbII and HbIII are nearly independent of the pH. Figure 5 shows plots of the CT1 band wavelength versus pH for the heme-bound fluoride complexes of Mb and HbII. For HbII, there is a decrease of 1 nm from pH 4 to pH 9, whereas, for Mb, it decreases 5 nm in the pH 5 to pH 8.5 range. The pH 4 CTI band (607 nm) belongs to the heme (FeIII)-F complex since the heme (FeIII)-O-Tyr derivative transition only starts to appear at pH 6.5. Also, as the pH increases, its intensity grows, contrary to the CT1 bands of fluoride complexes of HbII and HbIII. The pH differences on the UV-Vis spectral properties of the heme-bound fluoride complexes of HbII, HbIII, and Mb are a function of the hydrogen bonding network between the distal residues interacting with the heme-bound fluoride ion. The Mb heme-bound fluoride ion interacts with the distal histidine (His64) and a heme pocket water molecule.52 The Mb distal His64 (Nε) has a pKa in the 5.3–5.7 range in the presence of heme-bound fluoride ion.49,53,54 Based on 1) resonance Raman measurements of the Fe-F stretching mode,49 2) the kinetics of fluoride binding,53 and 3) the midpoint potential (Em) of the heme-bound fluoride complex of Mb,54 the protonated His64 (H-Nε) forms an H-bond with the heme-bound fluoride ion at acidic pH, and deprotonation of the His64, above pH 6, weakens this interaction. The myoglobin heme-bound fluoride change of the CT1 transition from 611 nm to 606 nm at pH 5 and 8.5 respectively, could be a response of both distal His64 (Nε) and water heme pocket deprotonation. In HbII and HbIII, the key distal residues for ligand binding are GlnE7 and TyrB10. They allow a network of hydrogen bonding dioxygen interactions in the oxy-heme complexes. Such interactions result in very low off-rate constants (koff) compared to HbI, with 61.1, 0.11, and 0.075 s−1 for HbI, HbII, and HbIII, respectively.14 Thus, the HbII and HbIII bound fluoride stability from pH 4 to pH 9 can be attributed to the hydrogen bonding interaction between the heme complex and B10 tyrosine. Also, the absence of an ionizable tyrosine group, with a pKa of 10, facilitates the interaction.55
3.3. Measured fluoride binding dissociation constants Kd
The apparent dissociation constants (Kd) for fluoride binding in HbI, HbII, HbIII, A-Hb, and Mb as a function of pH are shown in Figures 6a and 6b. The measured dissociation constants are reported in Table 1 for pH 5 and pH 7. The measured Kd from pH 4 to pH 8 is reported as Supporting Information (Table 1S). In general, the Kd values are the lowest in the acidic region below pH 7, meaning that fluoride’s affinity is more robust at low pH. Additionally, the pH profile of HbI in Figure 6a is distinct from that of the other heme proteins. From pH 4 to pH 7, the Kd values for fluoride binding in HbI are significantly larger than the other heme proteins: 100, 0.33, 0.50, 9.3, and 3.0 mM for HbI, HbII, HbIII, A-Hb, and Mb, respectively at pH 5.0; while at pH 7.0, these values are 200, 1.0, 3.1, 33, and 25 mM, respectively (Table 1). Also, the measured Kd in HbI has very high standard deviations due to its low fluoride affinity.
Table 1:
Measured Fluoride Binding Dissociation Constants (Kd) in mM.
| pH | HbI | HbII | HbIII | A-Hb | Mb |
|---|---|---|---|---|---|
| 5.0 | 100 ± 61 | 0.33 ± 0.067 | 0.50 ± 0.332 | 9.3 ± 5.8 | 3 ± 0.82 |
| 7.0 | 200 ±180 | 1.0 ± 0.156 | 3.1 ± 0.37 | 33 ± 4 | 25 ± 2.8 |
Given its low solubility in aqueous buffers, the final sodium fluoride concentration was about 0.2 M (as described in the Materials and Methods section). The measured Kd values for HbI were between 41 and 200 mM, which is in the same range as the final fluoride concentration. Consequently, at best, roughly half of the heme-binding sites in HbI were occupied by fluoride at the titration end, which resulted in large fluctuations of the measured Kd. For example, in Figure 3d, the UV-vis spectrum of the heme-bound fluoride complex of HbI shows a shoulder at 631 nm that corresponds to the charge transfer band of metaquo HbI. The data indicates the coexistence of two heme complexes even at a fluoride concentration of 0.2 M. In comparison, below pH 7, the UV-vis spectra of the other studied heme proteins show the presence only of a heme-bound fluoride complex with as little as 0.05 M fluoride concentration. Despite these fluctuations for HbI, the fluoride binding measurements consistently show that the fluoride affinity in HbI is about two orders of magnitude lower than the other studied heme proteins in the pH 4 to 7 range. As discussed, this correlates with HbI having the lowest CT1 wavelength for its heme-bound fluoride complex, relative to those from the heme-bound fluoride complex of HbII, HbIII, A-Hb, and Mb. The Kd measurements for fluoride binding in HbI confirm the absence of any significant stabilization interactions with the heme-bound fluoride ion, with the possible exception of PheB10. Further evidence of the weak binding of the fluoride ion in HbI is shown by the large fluctuations in the measured Kd values that are shown in Figure 6a. Under our experimental conditions, the final fluoride addition to ferric HbI still showed the presence of met-aquo HbI. From pH 5 to pH 7 the HbII and HbIII fluoride affinity are approximately 20-fold greater than HbI and about 10-fold greater than A-Hb and Mb. While not disregarding interactions with GlnE7, the strong fluoride interactions in HbII and HbIII can be attributed to the presence of TyrB10 and the smaller heme pocket of HbII and HbIII, relative to that of HbI. The molecular mechanism of fluoride binding in HbII and HbIII may be similar to that of dioxygen binding in these two hemoglobins. Both GlnE7 and TyrB10 interact with the heme-bound fluoride.18,20,24,56
4. Discussion
4.1. HbI, HbII, and HbIII Fluoride Binding
The non-linear fit models included the effects of neutral fluoride (HF) binding. Based on the pKa of fluoride (3.0), the neutral fluoride (HF) is about 10% at pH 4.0. However, there is no spectroscopic evidence that undissociated fluoride covalently binds to the heme iron ion. In their kinetic measurements of fluoride and azide binding in Mb, Erman’s group has observed a pH effect that indicates the binding of neutral fluoride and azide.53,57 They propose a mechanism similar to that of neutral cyanide (HCN) binding in Mb,58 where HF and HN3 are catalytically ionized to F− and N3− by the distal HisE7 before the respective anion directly binds to the ferric heme. Because it is unclear at pH 4 how ionization of HF occurs in the L. pectinata hemoglobins, such a mechanism would require further kinetic and spectroscopic studies on mutagenic L. pectinata hemoglobins. Here within, we have instead mainly focused on the stabilization mechanism of the heme-bound fluoride ion (F−) in the studied heme proteins.
The molecular mechanism of fluoride heme-binding was pursued using non-linear fit equations. In HbI, despite the variability in the apparent Kd, the data was fitted with equation 7. In the fit, pKaHF was set to 3.0, resulting in a Kd acid value of 4.79 × 10−3 M (± 2.5×10−3) and a pKaHbI value of 4.5 (± 0.23), which represents the pKa of HbI in the presence of heme-bound fluoride. At basic pH, the Kd for the heme-bound for the fluoride ion is 0.16 M. The weaker fluoride ion binding to HbI could be attributed to the fluoride ion small size coupled to the GlnE7 carbonyl group orientation towards the heme iron. On the other hand, of all the heme proteins examined in this study in the pH 4 to pH 8 interval, HbII and HbIII are the most favorable for fluoride binding. The smaller HbII and HbIII heme pockets, coupled with the favorable interactions with the amine NH2 and phenolic OH groups of the distal GlnE7 and TyrB10, generate a structural configuration that allows a network of strong H-bonding interactions between these amino acids and the heme-bound fluoride ion. The addition of the fluoride ion in HbII and HbIII at pH 7 results in the formation of the heme-bound fluoride complex with no evidence of the presence of a low spin heme-bound hydroxy complex (see Figure 4b). In Figure 6a, while not as evident as in HbI, A-Hb, and Mb, the apparent Kd for HbII and HbIII are pH-dependent. Figure 6b shows an expanded view of the apparent Kd plots for HbII and HbIII only. The measured fluoride dissociation constant values of HbII and HbII in Figure 6b show that the Kd values are affected by the pH 4 to pH 8 region. Non-linear fits were performed by using eq. 8 and eq. 9. At low pH, fluoride binding occurs in the metaquo heme complex. In contrast, at high pH, fluoride binding is affected by the presence of the heme-bound hydroxide ion. The apparent Kd for fluoride binding in HbII was fitted with eq. 8 with the pKaOH set to 6.8, being the acid-alkaline transition in HbII. Since the affinity of the fluoride ion diminishes with increasing pH, the pKa of the transition of the fluoride binding in the presence of metaquo heme complex to the binding in the presence of the heme-bound hydroxy complex was not considered in eq. 8. The non-linear fit for HbII resulted in a Kdacid value of 3.2 × 10−4 M (± 1.7 ×10−5).
For HbIII, the non-linear fit was performed with the pKa OH of 5.9, the acid-alkaline transition in HbIII. In Figure 6b, unlike HbII, the apparent Kd binding remains relatively low up to pH 8, so the pKa of the transition of the fluoride binding in the presence of metaquo heme complex to the binding in the presence of the heme-bound hydroxy complex (pKa FOH) was included in the fit equation. The fit resulted in a Kd acid value of 6.2 × 10−4 M (± 4.9 ×10−5) and a pKaFOH value of 6.7 (± 0.10). While HbII and HbIII have very similar heme pockets with comparable Kd acid values, our results clearly show that the molecular mechanisms of fluoride binding differ above pH 5. In the case of HbII, the affinity for fluoride binding decreases with pH upon the appearance of the heme-bound hydroxy complex around pH 6.6, which is not the case for HbIII. The HbIII non-linear fit for the appearance of the heme-bound hydroxy complex around pH 5.9 shows that the fluoride ion’s affinity remains relatively strong above pH 12 with an apparent Kd value of 4.0 ×10−3 M. The data shows a preference of the heme-bound fluoride ion even after the acid-alkaline transition, which does not happen with the other studied heme proteins. Contrary to HbIII, above pH 8, we have indications that hydroxide binding in HbII strongly interferes with fluoride binding (data not shown). According to Pietri et al., at pH 11, ferric HbII only shows the presence of a low spin heme-bound hydroxide complex with no evidence of another heme complex.18 It is probable that above pH 8.0, there is a structural change in the heme pocket of HbII that allows for hydroxide binding to be favored over other heme complexes.
Despite the acid-to-alkaline transitions in HbII and HbIII, these two proteins have a higher affinity for the fluoride ion in the pH 4 to pH 8 region than the other studied heme proteins. The fluoride binding mechanism in these two proteins can be attributed to the hydrogen-bonding network present in oxy-HbII and oxy-HbIII. In HbII, spectroscopic studies on the ferric heme-bound ligand and heme-bound carbon monoxide complexes show the presence of two heme pocket conformations.18,56 The conformational change consists of the movement of GlnE7 and TyrB10 from the heme ligand site (“closed” conformation) towards an orientation in which these two residues are swung away from the heme ligand site (“open” conformation). We presume that the hydrogen bonding network involving GlnE7 and TyrB10 may be disrupted upon the closed-to-open conformational changes of HbII. Thus, the data support a hydrogen-bonding network in the ferric states of HbII and HbIII with preferential binding to the fluoride ion (in the pH 4 to pH 8 region). The heme-fluoride unit would be stabilized in an arrangement with preferential H-bonding interactions between the phenolic OH on TyrB10, the NH2 group on GlnE7, and the heme-bound fluoride ion, which in HbIII, strictly prohibits the binding of the hydroxide ion. The very low Kd values for fluoride binding in HbII and HbIII in the pH 4 to pH 8 interval show how much more thermodynamically stable the heme-bound fluoride complex is relative to the hydroxy-heme ~600-nm complexes.
According to the measured Kd values for fluoride binding at pH 6.0, the affinity for fluoride ions in HbII and HbIII is ~100 times that of HbI. This difference in the stability of the heme-bound fluoride ion is equivalent to the difference in the measured koff for the fluoride ion dissociation kinetics between WT and doubly mutated YB10F-WG8F and YCD1-WG8F Thermobifida fusca Hb.30 This truncated Hb has a distinct polar distal heme pocket with three residues that can participate in H-bonding, TrpG8, TyrCD1, and TyrB10. Thus, the progressive mutagenic substitution of these amino acids by Phe affects the kinetics of fluoride dissociation, while that of the kinetics of association is essentially unaffected. For example, Nicoletti et al.30 showed the measured koff values of WT, YB10F, YB10F-WG8F, and YB10F- YCD1F-WG8F were 1.7, 29, 106, and 242 s−1, respectively, while the kon values remained between 4.2 and 7.6 mM−1 s−1.
Thus, the structural difference between the heme pockets of HbI and HbII or HbIII and, in particular, the GlnE7 stereochemistry defines the heme-ligand hydrogen bonding networking. The HbI heme pocket devoid GlnE7 H-bonding interactions to the heme-bound fluoride ion, cyanide, and oxygen, while PheB10 may contribute to heme-ligand stabilization. However, a robust H-bonding networking stabilizes HbII and HbIII, heme-bound fluoride, cyanide, and oxygen ligand with the OH and NH2 groups of TyrB10 and GlnE7, respectively. X-ray data shows (Figure 1) in HbII (as well as in HbIII), that the amino group occupies the position and is oriented towards the oxygen ligand, which together with TyrB10 generates a hydrogen-bonding network. These electrostatic interactions, coupled to the heme pocket size, are responsible for the observed kinetic values of these heme-complexes. The behavior is also analogous to T. fusca hemoglobin.
4.2. Fluoride Binding in Adult Hb and Mb
A-Hb and Mb have similar heme pocket structures with a distal His64 that has been shown to exert significant interactions with the heme-bound dioxygen and is of great consequence in these oxygen carriers’ physiological roles. Fits for A-Hb and Mb showed in Figure 6a, consider both hydrofluoric acid (HF) and the fluoride ion in the binding scheme, as proposed by Merryweather et al. in their study of fluoride binding kinetics.53 The scheme includes the hydroxide binding equilibrium at alkaline pH due to the acid-alkaline transition and the acid dissociation constant of the distal HisE7 (nitrogen Nε on the imidazole ring) in the presence of heme-bound water and the heme-bound fluoride. Using equation 10, the non-linear fit to the Mb data in Figure 6a resulted in pKaW(His) and pKaF(His) values of 7.50 (± 0.04) and 5.6 (± 0.1), respectively. Additionally, the Kd acid was calculated at 4.8×10−5 M (± 1.0 ×10−5). For A-Hb, the non-linear fit resulted in pKaW(His) and pKaF(His) values of 7.40 (± 0.03) and 5.3 (± 0.1), respectively, and the Kd acid was calculated at 1.0×10−4 M (± 2.5×10−5). Mb and A-Hb have very similar values for pKaW(His) and pKaF(His). The values for pKa F(His) of 5.6 and 5.3 for Mb and A-Hb, respectively, are attributed to the pKa of His64 of the heme-bound fluoride complex.49,53,54 In A-Hb and Mb, the heme-bound fluoride stabilization is controlled by the distal HisE7 and a water molecule located in the heme pocket.29,52 Deprotonation of HisE7 at high pH results in weakening these interactions and a loss of fluoride affinity. The inflections display the effects of HisE7 deprotonation on the apparent Kd in A-Hb, and Mb plots. Further increase in the pH above pH 7 results in a steep increase in the apparent Kd for fluoride binding in A-Hb and Mb (Figure 5a). This is due to the weak affinity of the fluoride ion relative to the affinity of the hydroxide ligand to form a heme-bound hydroxy complex, whose acid-alkaline transition pKa are 8.0 and 8.9 for A-Hb and Mb, respectively.
Distal HisE7 pKa in the presence of the metaquo complex resulted in pKaW(His) values of 7.5 and 7.4 for Mb and A-Hb, respectively. These values seem too high for the pKa of the distal HisE7 for the metaquo heme complex. Based on the kinetics of fluoride binding in Mb, Merryweather et al. determined that the pKa of His64 at 4.4 is more acidic than the His64 in the presence of the heme-bound fluoride complex.53 On the other hand, in the oxidation-reduction studies of Mb and A-Hb, Antonini and Brunori showed that the pH dependence of the one-electron reduction potential (Em) of Mb and A-Hb could not solely be due to the heme-aquo acid-alkaline transition pKa of these proteins.59–61 They proposed that there is an additional heme-linked ionization due to the ferric heme that occurs in the pH 6.4 to 7.4 range with a 20 – 30 mV effect. In a recent electrochemical study on Mb, pH dependence of the one-electron reduction potential shows another heme-linked ionization that appears before the acid-alkaline transition, which represents about a 30 mV change in the reduction potential of the metaquo heme complex.54 This ionization may not be of great importance regarding A-Hb and Mb’s role, but does show the complexity of understanding the molecular mechanism of ligand binding in these proteins.
4.3. Distal residues and the strength of interactions with heme-bound fluoride
While we do not disregard the effects of the proximal His residue on fluoride binding, this study links the differences in the fluoride binding properties of the studied heme proteins to differences in the distal pocket amino acid compositions and their relative spatial arrangements. Table 2 is a comparison of the measured dissociation constant values for oxygen, hydrogen sulfide and fluoride for the various heme proteins discussed in this study at pH 7.5. The Kd values for oxygen are very similar for HbI, HbII, HbIII, and HHMb. However, differences in Kd values are clearly delineated with respect to hydrogen sulfide and the fluoride ligands. As mentioned, H2S binding is clearly favored in HbI, but is the worst in the stabilization of the heme-bound fluoride ligand. A comparative analysis of the fluoride data in a wider pH range yields quantitative differences between the studied heme proteins. Figure 7 shows a schematic representation of the measured fluoride binding data in the studied heme proteins and its relation to the heme pocket structures. This summarized representation of the fluoride Kd profiles in the pH 5 to 7 region clearly shows three levels of heme-bound fluoride stabilization. The weakest heme-fluoride interaction is HbI, where there is no favorable fluoride-binding or stabilization mechanism. None of the distal amino acids are in an arrangement that would H-bond with the heme ligand moiety. The presence of histidine in the E7 position in A-Hb and Mb allows for H-bonding to the heme-bound fluoride ion. However, in HbII and HbIII, the H-bonding from both GlnE7 and TyrB10 residues to the heme-bound fluoride ion results in the most favorable conformation for the ligand stabilization. Regarding HbII and HbIII, while these have similar heme pocket amino acids, the fluoride binding affinities are affected differently by the presence of the heme-hydroxy complex. Above pH 7 (Figure 6b), the results show that the affinity for fluoride binding in HbII is relatively weaker than hydroxide binding, as such behavior is further exacerbated with an increase in pH. In contrast, the affinity for fluoride in HbIII is much more stable in the same pH range. These differences can be attributed to the small HbIII heme pocket relative to HbII, leading to a possible change in the GlnE7 and TyrB10 in HbII and HbIII, reflected in the pH profiles of fluoride binding of these two heme proteins.
Table 2.
Equilibrium Dissociation Constants (Kd) at pH 7.5, the values are in mM.*
| Heme protein | O2 | H2S | F− |
|---|---|---|---|
| HbI | ~4.0 × 10−4 | 3.4 × 10−6 | 200 |
| HbII | 2.8 × 10−4 | 14.5 × 10−3 | 3.7 |
| HbIII | 2.6 × 10−4 | >16 × 10−3 | 2.9 |
| HHMb + | 1.3 × 10−3 | 18.5 × 10−3 | 41 |
With the exception of HHMb, oxygen and hydrogen sulfide values were obtained from Kraus and Wittenberg.14 Fluoride dissociation constant values are from this study.
Figure 7.
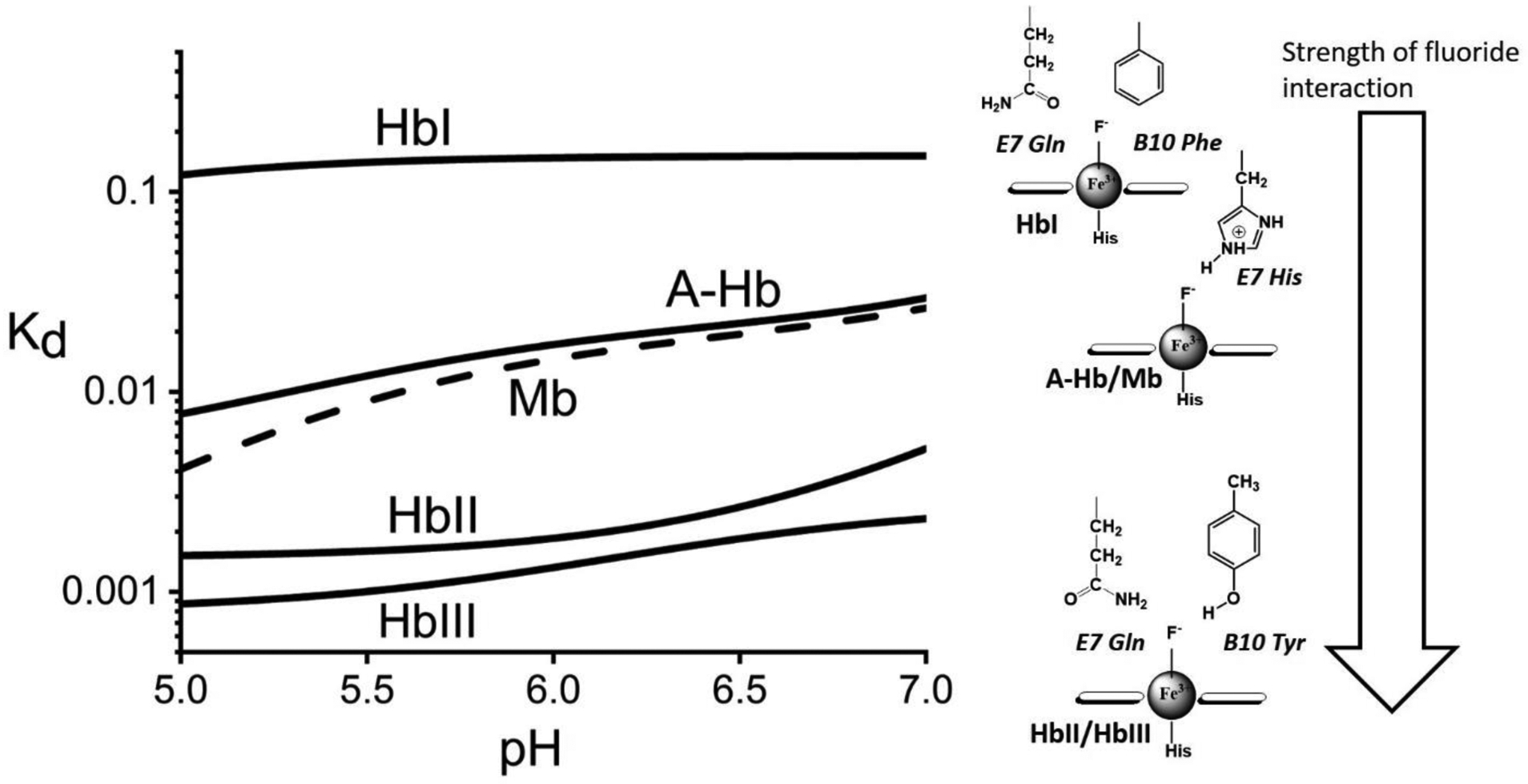
Schematic representation of the apparent Kd profiles for fluoride binding and their respective heme-fluoride distal structures. The Kd traces are the non-linear fits shown in Figure 5. The log plot shows three levels of heme-fluoride stabilization.
4.4. Vibrational data of met-cyano heme complexes confirm fluoride interactions
Analysis of the vibrational data for met-cyano heme complexes (Fe3+-CN−) of HbI, HbII, HbIII, A-Hb, and Mb, confirms the fluoride binding data. Table 3 shows the vibrational assignments for the νFe-C and the νCN stretching modes of the met-cyano heme complexes of HbI, HbII, HbIII, A-Hb, and Mb. For example, the isotopic substitution assignments are provided by the Resonance Roman (Figure 4S) and the FTIR data (Figure 5S) and Tables 2S and 3S, presented as supporting information. A plot of the νCN values versus νFe-C, shown in Figure 8 shows a strong correlation between the νFe-C and the νCN stretching modes of these heme complexes. A most recent review on the geometry of the heme-bound cyanide ligand and its vibrational properties in various heme proteins can be found elsewhere,63 in this case, the correlation shows the strength of hydrogen bonding interaction to the heme-bound cyanide increases in the following order: HbI, A-Hb, Mb, HbII, and HbIII. This sequence of the hydrogen bonding strength in met-cyano heme complexes confirms the fluoride binding stability by hydrogen bonding of these heme proteins.
Table 3.
Vibrational assignments for νFe-C and νCN in met-cyano heme complexes.
Figure 8.
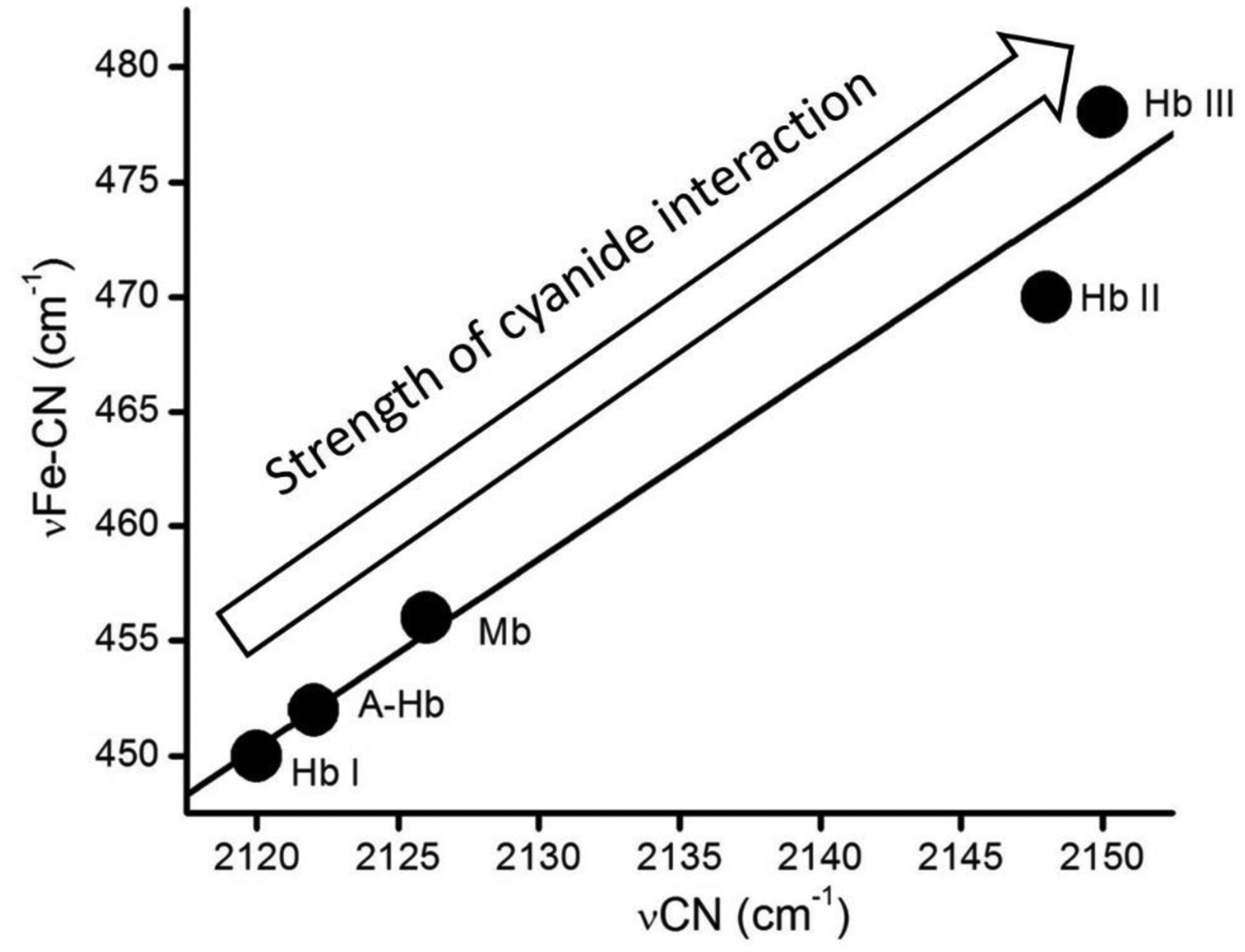
Vibrational νCN vs. νFe-C stretching modes for met-cyano heme complexes (from Table 3). The inverse correlation is affected by the heme pocket interactions to the heme-bound met-cyano unit.
4.5. Distal residues and the roles of the heme proteins
Despite its complexity in fluoride binding, HisE7 has been shown to selectively stabilize O2 binding in A-Hb and Mb, protect the heme from autoxidation, and is a key in protein folding.69 In the case of the clam hemoglobins, the clam’s role in delivering H2S to its symbiont bacteria may have dictated a different evolutionary path for HbI, HbII, and HbIII. In HbI, the reactive sulfide protein, the presence of GlnE7 instead of a bulkier His residue facilitates H2S binding.44 Further, the absence of a tyrosine in the B10 position would prohibit HbI from selectively binding O2 over H2S and therefore diminishing its role in H2S delivery. This is confirmed by the presence of TyrB10 in HbII and HbIII, leading these two hemoglobins to have the strongest affinity for fluoride. Thus, oxygen storage and delivery in the clam are mainly controlled by HbII and HbIII. Ferric HbIII maintains a more robust interaction with the heme-bound fluoride than HbII, up to pH 9.0 (data not shown), implying that structural differences between the heme pockets are present. The X-ray crystallographic structure of the oxy-HbII-HbIII shows differences in the orientations of the heme-bound dioxygen ligands.24 Further, the dioxygen binding affinities in HbII and HbIII differ slightly, with values of 282 and 260 nM, respectively.14 Additionally, the pKa for the transition from metaquo to the low spin heme-bound hydroxy complexes in HbII and HbIII are 6.8 and 5.9, respectively. It is unclear if these differences between the ligand binding properties of HbII and HbIII are relevant for the clam’s survival, i.e., whether these differences are critical to a specific function. For example, in the vertebrate adult hemoglobin, the α and β subunits can self-associate to form (α2) homodimer and (β4) homotetramers, but unlike the (α2β2) heterotetramer, these are unable to cooperatively bind dioxygen.70–72 Based on their structural study, Marchany-Rivera et al. have proposed that the HbII-HbIII heterodimer exists under the physiological conditions of the L. pectinata clam and is necessary for the regulation of dioxygen and carbon dioxide transport. The authors have suggested that the interplay between HbI and the HbII-HbIII heterodimer delivers hydrogen sulfide, and dioxygen, to the clam and its symbiont.24 Detailed molecular studies must be carried out to understand the slight differences in the mechanism of ligand binding in the HbII and HbIII heme pockets when associated as a heterodimer and whether or not these differences are necessary for the regulation of dioxygen and carbon dioxide transport.
5. Conclusions
This study shows that fluoride binding measurements enhance the use of fluoride ions as a probe of heme cavity structure. The measured dissociation constants (Kd) for fluoride binding in A-Hb, Mb, HbI, HbII, and HbIII, show how the variation in the hydrogen-bonding strength between the heme-ligand moiety and its pocket amino acid controls this property. Also, pH changes affect the ionization state of the heme-linked amino acid residues, disturbing the affinity for fluoride binding. Herein we showed how two well-known oxygen carriers, specifically Mb and A-Hb, have very similar fluoride-binding properties due to the distal HisE7. In contrast, the Lucina pectinata hemoglobins have different mechanisms of fluoride binding and ligand stabilization. Thus, individual structural cavity differences affect the fluoride binding properties in a manner that corresponds to the clam hemoglobins’ physiological roles.
Supplementary Material
Synopsis:
Measured fluoride binding dissociation constants (Kd) in the clam hemoglobins (HbI, HbII, and HbIII), adult hemoglobin (A-Hb), and myoglobin (Mb) reveal distinct pH profiles due to differences in the heme pocket structures.
Highlights.
Measured dissociation constant for fluoride binding in the clam hemolgobins (HbI, HbII, HbIII), adult hemoglobin (A-Hb), and myoglobin (Mb) reveal differences in the heme pocket structures.
A-Hb and Mb have similar fluoride binding properties due to the presence of the distal histidine (E7his).
The clam hemoglobins display distinct fluoride binding properties.
HbII and HbIII have identical heme pocket amino acid compositions but differ in their molecular mechanism of fluoride binding.
Acknowledgments
We are grateful for the support from the Chemistry Department, the Summer Scholars Program at Saint Joseph’s University, and the John P. McNulty Scholars Program. National Institute of Health-INBRE PR (P20GM103475 to J.L.-G.) and the Industrial Biotechnology program, University of Puerto Rico, Mayaguez Campus (J.L.-G.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (1).Feliers D; Lee HJ; Kasinath BS Antioxid. Redox Signaling 2016, 25, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kabil O; Motl N; Banerjee R Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kashfi K; Olson KR Biochem. Pharmacol. (Amsterdam, Neth.) 2013, 85, 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kimura H Proc. Jpn. Acad., Ser. B 2015, 91, 131–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li Q; Lancaster JR Jr. Nitric Oxide 2013, 35, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Olson KR; Straub KD Physiology 2016, 31, 60–72. [DOI] [PubMed] [Google Scholar]
- (7).Panth S; Chung H-J; Jung J; Jeong NY Oxid. Med. Cell. Longevity 2016, 9049782/1–9049782/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rios-Gonzalez BB; Roman-Morales EM; Pietri R; Lopez-Garriga JJ Inorg. Biochem 2014, 133, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Whiteman M; Le Trionnaire S; Chopra M; Fox B; Whatmore J Clin. Sci 2011, 121, 459–488. [DOI] [PubMed] [Google Scholar]
- (10).Sun H-J; Wu Z-Y; Nie X-W; Bian J-S Front. Pharmacol 2020, 10, 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shackelford R; Ozluk E; Islam MZ; Hopper B; Meram A; Ghali G; Kevil CG Redox Biol 2021, 38, 101675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zaorska E; Tomasova L; Koszelewski D; Ostaszewski R; Ufnal M Biomolecules 2020, 10, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kraus DW; Wittenberg JB; Lu JF; Peisach J J Biol Chem 1990, 265, 16054–9. [PubMed] [Google Scholar]
- (14).Kraus DW; Wittenberg JB J Biol Chem 1990, 265, 16043–53. [PubMed] [Google Scholar]
- (15).Johnson EA Biochim Biophys Acta 1970, 207, 30–40. [DOI] [PubMed] [Google Scholar]
- (16).Berzofsky JA; Peisach J; Blumberg WE J. Biol. Chem 1971, 246, 3367–77. [PubMed] [Google Scholar]
- (17).Rizzi M; Wittenberg JB; Coda A; Ascenzi P; Bolognesi MJ Mol. Biol 1996, 258, 1–5. [DOI] [PubMed] [Google Scholar]
- (18).Pietri R; Granell L; Cruz A; De Jesus W; Lewis A; Leon R; Cadilla CL; Garriga JL Biochim. Biophys. Acta, Proteins Proteomics 2005, 1747, 195–203. [DOI] [PubMed] [Google Scholar]
- (19).De Jesus-Bonilla W; Cruz A; Lewis A; Cerda J; Bacelo DE; Cadilla CL; Lopez-Garriga J JBIC, J. Biol. Inorg. Chem 2006, 11, 334–342. [DOI] [PubMed] [Google Scholar]
- (20).Gavira JA; Camara-Artigas A; De Jesus-Bonilla W; Lopez-Garriga J; Lewis A; Pietri R; Yeh S-R; Cadilla CL; Garcia-Ruiz JM J. Biol. Chem 2008, 283, 9414–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nguyen BD; Zhao X; Vyas K; La Mar GN; Lile RA; Brucker EA; Phillips GN Jr.; Olson JS; Wittenberg JB J. Biol. Chem 1998, 273, 9517–9526. [DOI] [PubMed] [Google Scholar]
- (22).Bolognesi M; Rosano C; Losso R; Borassi A; Rizzi M; Wittenberg JB; Boffi A; Ascenzi P Biophys. J 1999, 77, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ramos-Alvarez C; Yoo B-K; Pietri R; Lamarre I; Martin J-L; Lopez-Garriga J; Negrerie M Biochemistry 2013, 52, 7007–7021. [DOI] [PubMed] [Google Scholar]
- (24).Marchany-Rivera D; Smith CA; Rodriguez-Perez JD; Lopez-Garriga JJ Inorg. Biochem 2020, 207, 111055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pietri R; Lewis A; Leon RG; Casabona G; Kiger L; Yeh S-R; Fernandez-Alberti S; Marden MC; Cadilla CL; Lopez-Garriga J Biochemistry 2009, 48, 4881–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jensen B; Fago A J Inorg Biochem 2018, 182, 133–140. [DOI] [PubMed] [Google Scholar]
- (27).Nicoletti FP; Comandini A; Bonamore A; Boechi L; Boubeta FM; Feis A; Smulevich G; Boffi A Biochemistry 2010, 49, 2269–2278. [DOI] [PubMed] [Google Scholar]
- (28).Droghetti E; Nicoletti FP; Bonamore A; Sciamanna N; Boffi A; Feis A; Smulevich G Journal Of Inorganic Biochemistry 2011, 105, 1338–1343. [DOI] [PubMed] [Google Scholar]
- (29).Neri F; Kok D; Miller MA; Smulevich G Biochemistry 1997, 36, 8947–8953. [DOI] [PubMed] [Google Scholar]
- (30).Nicoletti FP; Droghetti E; Boechi L; Bonamore A; Sciamanna N; Estrin DA; Feis A; Boffi A; Smulevich G Journal of The American Chemical Society 2011, 133, 20970–20980. [DOI] [PubMed] [Google Scholar]
- (31).Pietri R; Leon RG; Kiger L; Marden MC; Granell LB; Cadilla CL; Lopez-Garriga J Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 758–765. [DOI] [PubMed] [Google Scholar]
- (32).Sage JT; Morikis D; Champion PM Biochemistry 1991, 30, 1227–37. [DOI] [PubMed] [Google Scholar]
- (33).Palaniappan V; Bocian DF Biochemistry 1994, 33, 14264–74. [DOI] [PubMed] [Google Scholar]
- (34).Alberty RA J. Phys. Chem. B 2000, 104, 9929–9934. [Google Scholar]
- (35).Antonini E; Brunori M Hemoglobin and Myoglobin in their Reactions with Ligands; North-Holland Publishing Co.: Amsterdam, 1971; Vol. 21. [Google Scholar]
- (36).Kryger L; Lee SK Biogeochemistry 1996, 35, 367–375. [Google Scholar]
- (37).Puett DJ Biol. Chem 1973, 248, 4623–34. [PubMed] [Google Scholar]
- (38).Bismuto E; Colonna G; Irace G Biochemistry 1983, 22, 4165–70. [DOI] [PubMed] [Google Scholar]
- (39).Irace G; Bismuto E; Savy F; Colonna G Arch. Biochem. Biophys 1986, 244, 459–59. [DOI] [PubMed] [Google Scholar]
- (40).Privalov PL; Griko YV; Venyaminov SY; Kutyshenko VP J. Mol. Biol 1986, 190, 487–98. [DOI] [PubMed] [Google Scholar]
- (41).Giacometti GM; Traylor TG; Ascenzi P; Brunori M; Antonini EJ Biol. Chem 1977, 252, 7447–8. [PubMed] [Google Scholar]
- (42).Han S; Rousseau DL; Giacometti G; Brunori M Proc. Natl. Acad. Sci. U. S. A 1990, 87, 205–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Coletta M; Ascenzi P; Traylor TG; Brunori MJ Biol. Chem 1985, 260, 4151–5. [PubMed] [Google Scholar]
- (44).Rizzi M; Wittenberg JB; Coda A; Fasano M; Ascenzi P; Bolognesi M J Mol Biol 1994, 244, 86–99. [DOI] [PubMed] [Google Scholar]
- (45).Hargrove MS; Wilkinson AJ; Olson JS Biochemistry 1996, 35, 11300–11309. [DOI] [PubMed] [Google Scholar]
- (46).Navarro AM; Maldonado M; Gonzalez-Lagoa J; Lopez-Mejia R; Lopez-Garriga J; Colon JL Inorg. Chim. Acta 1996, 243, 161–166. [Google Scholar]
- (47).Cerda J; Echevarria Y; Morales E; Lopez-Garriga J Biospectroscopy 1999, 5, 289–301. [Google Scholar]
- (48).Brunori M; Amiconi G; Antonin E; Wyman J; Zito R; Fanelli AR Biochim Biophys Acta 1968, 154, 315–22. [DOI] [PubMed] [Google Scholar]
- (49).Asher SA; Adams ML; Schuster TM Biochemistry 1981, 20, 3339–3346. [DOI] [PubMed] [Google Scholar]
- (50).Asher SA; Schuster TM Biochemistry 1981, 20, 1866–73. [DOI] [PubMed] [Google Scholar]
- (51).De Jesus-Bonilla W; Ramirez-Melendez E; Cerda J; Lopez-Garriga J Biopolymers 2002, 67, 178–185. [DOI] [PubMed] [Google Scholar]
- (52).Aime S; Fasano M; Paoletti S; Cutruzzola F; Desideri A; Bolognesi M; Rizzi M; Ascenzi P Biophysical Journal 1996, 70, 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Merryweather J; Summers F; Vitello LB; Erman JE Archives Of Biochemistry And Biophysics 1998, 358, 359–368. [DOI] [PubMed] [Google Scholar]
- (54).Cerda JF; Roeder MH; Houchins DN; Guzman CX; Amendola EJ; Castorino JD; Fritz AL Anal. Biochem 2013, 443, 75–77. [DOI] [PubMed] [Google Scholar]
- (55).Tommos C; Babcock GT Biochim. Biophys. Acta, Bioenerg 2000, 1458, 199–219. [DOI] [PubMed] [Google Scholar]
- (56).Peterson ES; Huang S; Wang J; Miller LM; Vidugiris G; Kloek AP; Goldberg DE; Chance MR; Wittenberg JB; Friedman JM Biochemistry 1997, 36, 13110–13121. [DOI] [PubMed] [Google Scholar]
- (57).Lin J; Merryweather J; Vitello LB; Erman JE Arch. Biochem. Biophys 1999, 362, 148–158. [DOI] [PubMed] [Google Scholar]
- (58).Dou Y; Olson JS; Wilkinson AJ; Ikeda-Saito M Biochemistry 1996, 35, 7107–7113. [DOI] [PubMed] [Google Scholar]
- (59).Antonini E; Wyman J; Brunori M; Taylor JF; Rossi-Fanelli A; Caputo A J Biol Chem 1964, 239, 907–912. [PubMed] [Google Scholar]
- (60).Brunori M; Saggese U; Rotillio G. c.; Antonini E; Wyman, J. Biochemistry 1971, 10, 1604–1609. [DOI] [PubMed] [Google Scholar]
- (61).Brunori M; Taylor JF; Antonini E; Wyman J Biochemistry 1969, 8, 2880–3. [DOI] [PubMed] [Google Scholar]
- (62).Sono M; Smith PD; McCray JA; Asakura TJ Biol. Chem 1976, 251, 1418–26. [PubMed] [Google Scholar]
- (63).Dybas J; Chiura T; Marzec KM; Mak PJ J. Phys. Chem. B 2021, 125, 3556–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Morales E, Univeristy of Puerto Rico -Mayagüez, 2000.
- (65).Cruz-Balberdi A, University of Puerto Rico -Mayagüez, 2003.
- (66).Ramos-Lorenzo J, University of Puerto Rico -Mayagüez, 2003.
- (67).Hirota S; Ogura T; Shinzawa-Itoh K; Yoshikawa S; Kitagawa TJ Phys. Chem 1996, 100, 15274–15279. [Google Scholar]
- (68).Yoshikawa S; O’Keeffe DH; Caughey WS J. Biol. Chem 1985, 260, 3518–28. [PubMed] [Google Scholar]
- (69).Olson JS Antioxid. Redox Signaling 2020, 32, 228–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Imai K Allosteric Effecst in Haemoglobin; Cambridge University Press: Cambridge, U.K., 1982. [Google Scholar]
- (71).Tyuma I; Shimizu K; Imai K Biochem. Biophys. Res. Commun 1971, 43, 423–8. [DOI] [PubMed] [Google Scholar]
- (72).Nagatomo S; Saito K; Yamamoto K; Ogura T; Kitagawa T; Nagai M Biochemistry 2017, 56, 6125–6136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.