

Graphical abstract
Keywords: Offspring production, Ovarian organoids, Defined factors, Chromatin reorganization, 3D cell culture, Induced germline stem cells
Abstract
Introduction
Fate determination of germline stem cells remains poorly understood at the chromatin structure level.
Objectives
Our research hopes to develop successful offspring production of ovarian organoids derived from spermatogonial stem cells (SSCs) by defined factors.
Methods
The offspring production from oocytes transdifferentiated from mouse SSCs with tracking of transplanted SSCs in vivo, single cell whole exome sequencing, and in 3D cell culture reconstitution of the process of oogenesis derived from SSCs. The defined factors were screened with ovarian organoids. We uncovered extensive chromatin reorganization during SSC conversion into induced germline stem cells (iGSCs) using high throughput chromosome conformation.
Results
We demonstrate successful production of offspring from oocytes transdifferentiated from mouse spermatogonial stem cells (SSCs). Furthermore, we demonstrate direct induction of germline stem cells (iGSCs) differentiated into functional oocytes by transduction of H19, Stella, and Zfp57 and inactivation of Plzf in SSCs after screening with ovarian organoids. We uncovered extensive chromatin reorganization during SSC conversion into iGSCs, which was highly similar to female germline stem cells. We observed that although topologically associating domains were stable during SSC conversion, chromatin interactions changed in a striking manner, altering 35% of inactive and active chromosomal compartments throughout the genome.
Conclusion
We demonstrate successful offspring production of ovarian organoids derived from SSCs by defined factors with chromatin reorganization. These findings have important implications in various areas including mammalian gametogenesis, genetic and epigenetic reprogramming, biotechnology, and medicine.
Introduction
Cell fate decisions, which require key gene regulation, remain poorly understood at the chromatin structure level. Chromatin in eukaryotic cells is a complex of DNA, RNA and protein, and the structural and functional basis of the genome [1]. The spatial organization of chromatin can affect the location of DNA and plays important roles in regulation of gene expression [2], [3]. Using high throughput chromosome conformation (Hi-C), the chromatin interactions involved in gene regulation can be revealed by displaying the high order chromatin structure [1]. Although three-dimensional chromatin architectures of mouse gametes were recently reported, how they affect fate decisions of germline stem cells remains to be explored [4], [5], [6].
The formation and development of sexually dimorphic germ cells are required for the continuation of mammalian species. Primordial germ cells (PGCs) are the first germ cell population and undergo sexually dimorphic development, generating male gametes (spermatozoa) or female gametes (oocytes) [7]. During their development, PGCs also undergo programming of DNA methylation, including demethylation, paternal (XY) methylation, or maternal (XX) methylation [8], [9], [10], [11], [12], [13]. Furthermore, PGC development is complex, involving regulation of multiple key genes associated with male or female fates [12]. Therefore, both spermatogonial stem cells (SSCs) and female germline stem cells (FGSCs) derived from PGCs have different DNA methylation statuses or imprinting patterns [13], [14], [15], [16].
Simon et al. demonstrated that stem/progenitor spermatogonia can directly transdifferentiate into prostatic, uterine, and skin epithelial cells by recombining with the appropriate fetal or neonatal mesenchyme [17]. Using rainbow trout, Okutsu et al. demonstrated that transplantation of testicular germ cells into female gonads leads to the production of oocytes that mature into eggs that can fertilize and produce normal offspring [18]. However, there has been no evidence to demonstrate that mammalian offspring can be generated from oocytes derived from SSCs.
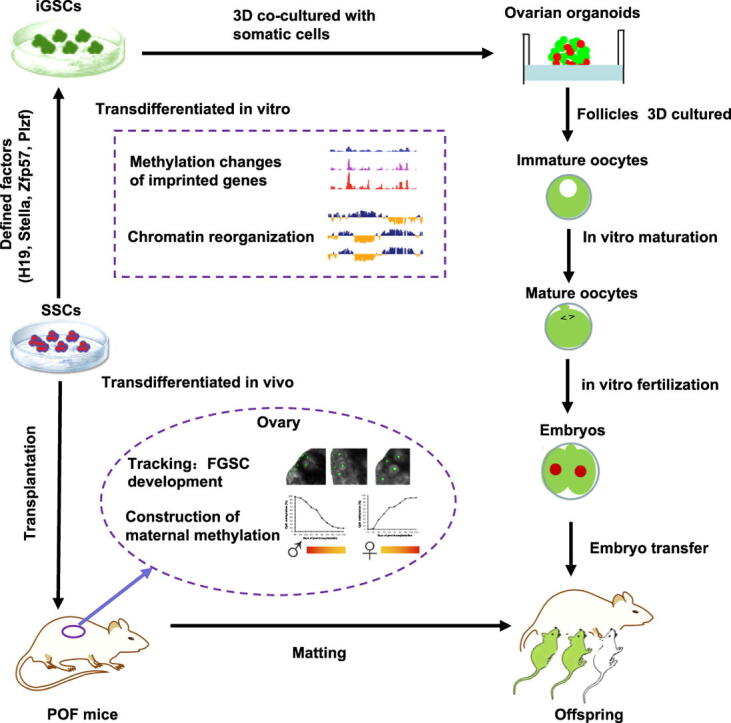
In the present study, we demonstrate successful production of progeny from oocytes transdifferentiated from mouse SSCs in vivo and in vitro (Fig. S1). Furthermore, we demonstrate the direct induction of germline stem cells (or induced germline stem cells, iGSCs) differentiated into functional oocytes by transduction of H19, Stella, and Zfp57 and inactivation of Plzf in SSCs with chromatin architecture reorganization. This study provides a new strategy to investigate germ cell biology, epigenetic reprogramming, mammalian germline gene modification, and regenerative medicine.
Materials and methods
Mice
C57BL/6, pou5f1-GFP transgenic mice [CBA-Tg (pou5f1-EGFP) 2Mnn] (The Jackson Laboratory) or pou5f1/GFP transgenic mice [19] × C57BL/6 F1 hybrid mice were used in this study. Premature ovarian failure (POF) Pten (phosphatase and tensin homolog deleted on chromosome 10) loxp/loxp; Gdf9-Cre (Gdf9 promoter-mediated Cre recombinase+) mice were produced and genotyped as described by Reddy et al [20], [21]. POF (Pten loxp/loxp; Gdf9-Cre+) mice were used as recipients. Pten loxp/loxp mice (B6.129S4-Pten tm1Hwu) and Gdf9-Cre [Tg (Gdf9-iCre)] mice were purchased from The Jackson Laboratory.
Isolation and culture of spermatogonial stem cells
Testes from 6-day-old pou5f1-GFP transgenic mice or pou5f1/GFP transgenic mice × C57BL/6 F1 hybrid mice were collected and decapsulated. Spermatogonial stem cells (SSCs) were isolated using methods described by Wu et al [22], [23] and Yuan et al [24]. The SSCs were purified by both magnetic activated cell sorting (MACS) with an anti-Thy-1 antibody and fluorescence activated cell sorting (FACS), according to the manufacturers’ instructions. SSCs were cultured on mitotically inactivated SIM mouse embryo derived thioguanine- and ouabain- resistant(STO) feeder cells (5 × 104 cells/cm2; ATCC) in culture medium. For mitotic inactivation, STO cells were treated with 10 μg/ml mitomycin C (Sigma) for 2–3 h. Mitomycin C-treated STO cells were washed with phosphate buffered saline(PBS) and transferred to 0.2% (w/v) gelatin-coated tissue culture plates. The SSC culture medium consisted of high glucose Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; GIBCO), 2 mM L-glutamine (Sigma), 0.1 mM β-mercaptoethanol (Sigma), 1 mM nonessential amino acids (Life Technologies), 10 ng/ml glial cell line-derived neurotrophic factor (GDNF; R&D Systems), 10 ng/ml leukemia inhibitory factor (LIF; Chemicon), and 15 mg/l penicillin (Sigma). SSCs were cultured on STO feeders in 24-well plates with 500 μl culture medium per well. The medium was replaced every 1–2 days, and cells were subcultured at a split ratio of 1:1–3 by trypsinization every 3 days. All cultures were maintained at 37 °C with 5% CO2.
Isolation and purification of female germline stem cells
Ovaries were collected from 5-day-old pou5f1/GFP transgenic mice × C57BL/6 F1 hybrid mice. Female germline stem cells (FGSCs) were isolated and purified using a method described elsewhere [16]. Briefly, dissected ovarian tissues were incubated in 1 mg/ml collagenase (type IV; Sigma) at 37 °C with gentle agitation for 15–20 min. After washing, ovarian tissues were incubated in 0.05% trypsin and 1 mM EDTA at 37 °C for 5–7 min. Sheep anti-mouse IgG magnetic beads (Dynal Biotech) were incubated with an anti-fragilis antibody (ab15592, Abcam) for 30 min at room temperature. The magnetic bead/antibody mixture was incubated with the isolated cell suspension for another 30 min at room temperature. Then, the mixture of cells and magnetic beads was placed on a magnetic bead separator for 2–3 min, and the supernatant was removed. The fraction on the inner side of the eppendorf tube was collected and rinsed twice with PBS, resuspended in PBS, and further purified by FACS, in accordance with the manufacturers’ instructions. The purified FGSCs were placed in FGSC culture medium and cultured on mitotically inactivated STO feeder cells in 24-well plates at 37 °C with 5% CO2.
Preparation of ovarian tissue for analysis
Ovaries from recipient and control mice were fixed with 4% (w/v) paraformaldehyde (4 °C, overnight) and dehydrated via a graded ethanol series. The tissues were vitrified in xylene, embedded in paraffin, sectioned (6 μm thickness), and then mounted on slides. Prior to immunofluorescence staining, the sections were dewaxed in xylene and rehydrated via a graded ethanol series. Sections were counterstained with hematoxylin.
Immunofluorescence
After equilibration in PBS, tissue sections were digested with 0.125% trypsin for 10 min at 37 °C and then washed in PBS twice. The sections were blocked in 10% goat serum at room temperature for 10 min and then incubated overnight at 4 °C with appropriate primary antibodies. The primary antibodies used were mouse monoclonal anti-GFP (1:200 dilution; ab290, Abcam) and rabbit polyclonal anti-MVH (1:200; ab13840, Abcam). After washing in PBS, the sections were incubated at 37 °C for 30 min with TRITC-conjugated goat anti-rabbit IgG (1:200; SA00007-2, Proteintech) or fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (1:200; SA00003-1, Proteintech) as appropriate. Sections were stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000) at 37 °C for 20 min, covered with mounting medium (glycerol: PBS, 3: 1), and viewed under a Nikon Eclipse E600 microscope equipped with a Nikon Dxm 1200 digital camera using fluorescein optics for TRITC and FITC, and ultraviolet optics for DAPI or under a confocal microscope (FluoView™ FV1000).
Cultured germline stem cells were fixed with 4% paraformaldehyde in PBS at room temperature for 20 min. After fixation, the cells were permeabilized with 0.5% Triton X-100 for 30 min at room temperature for PLZF staining. The cells were incubated in blocking solution (10% normal goat or bovine serum in PBS, 10 min, 37 °C), followed by rinsing and overnight incubation at 4 °C with appropriate primary antibodies: rabbit polyclonal anti-MVH (ab13840, 1: 200; Abcam), mouse monoclonal anti-GFP (1: 200; ab290, Abcam), and anti-PLZF (1:150, sc-28319, Santa Cruz). After washing in PBS, the cells were incubated with TRITC-conjugated goat anti-rabbit IgG (1: 200, SA00007-2, Proteintech) or fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (1: 150; SA00003-1, Proteintech) at 37 °C for 30 min, rinsed, and then incubated with DAPI (1: 1000) at 37 °C for 20 min. Petri dishes were then covered with mounting medium (glycerol: PBS, 3: 1) and viewed as described above.
Karyotypic analysis
Karyotypic analysis was performed using standard protocols for mouse chromosome analysis. After culture for 3 days, SSCs were treated with culture medium containing colchicine (100 ng/ml; Sigma) for 3 h, hypotonically treated with 75 mM KCl for 15 min at 37 °C, immersed twice in methanol: acetic acid (3: 1) for 30 min at −30 °C, dried in air for 3–4 days, digested with 0.025% trypsin, and then stained with Giemsa. To verify the chromosomal type of recipient mouse oocytes, karyotypic analysis of mature oocytes from recipients was performed. To collect mature oocytes, recipient mice were superovulated with 10 IU pregnant mare serum gonadotropin (PMSG; ProSpec-Tany) for 48 h, followed by 10 IU human chorionic gonadotropin (hCG; ProSpec-Tany). These oocytes were hypotonically treated with 75 mM KCl at 37 °C for 15 min and then fixed with two solutions consisting of methanol/acetic acid/water (5:1:2) for 5–10 min and methanol/acetic acid (3:1) for 15 min at room temperature. Fixed cells were mounted on slides and immediately exposed to steam from boiling water (90–100 °C) for 30 secto cause expansion of the cells, followed by drying at 37 °C and Giemsa staining (Amresco) [25].
Embryonic stem cell culture
Embryonic stem cells (ESCs) were cultured with mouse embryonic fibroblasts in the presence of leukemia inhibitory factor (LIF; 1000 U/ml) in Glasgow modification of Eagle’s medium (GMEM; Invitrogen) containing 10% fetal calf serum. The medium was replaced every 1–2 days, and cells subcultured at a split ratio of 1:1–3 by trypsinization every 3 days. All cultures were maintained at 37 °C with 5% CO2.
Microarrays
Total RNA was extracted from cultured SSCs and ESCs using Trizol reagent (Invitrogen), in accordance with the manufacturer’s instructions. RNA was labeled using an Illumina labeling kit. An Illumina sentrix mouse WG-6 Beadchip (45281 transcripts) was used in this study. Microarray experiments, including RNA labeling, hybridization, washing, scanning, image analysis, normalization, and data processing, were performed by Shanghai Biotechnology Corporation using the Illumina manual. Three biological repeats were included in microarray experiments. Differentially expressed genes were identified by the Illumina system. The data were analyzed using GeneSpring GX 11 software. Hierarchical clustering of samples was performed by cluster 3.0 and TreeView software [26].
Transplantation
For injection into the ovary, SSCs were collected and transplanted into the ovaries of POF mice. For the positive control, FGSCs from pou5f1/GFP transgenic mice were also transplanted into ovaries of POF mice. Recipient mice were anesthetized by injection of pentobarbital sodium (45 mg/kg). Approximately 6 μl of a single cell suspension containing 1 × 104 cells or 6 μl PBS for the control was microinjected into the ovaries of recipients as described elsewhere. In detail, after anesthetization of recipient mice for 20–30 min and disinfection of the abdominal surface using 75% ethanol, the recipient abdominal cavity was carefully opened. To expose and find the ovaries, the intestines were carefully moved away from the inside of the abdominal cavity. The Y-shaped uterus was located, and then following the uterus and oviduct until posterior to the kidneys, the ovaries were located caudal to the kidneys in the lower abdominal cavity. By gently holding an ovary with forceps without causing damage, the ovary was injected at 1–2 sites using a glass pipette with a 45 μm tip and mouth pipetting to carefully transplant the 6 µl single cell suspension of ~ 1 × 104 SSCs or FGSCs into each ovary. At 35 days after transplantation, recipients were mated with 8-week-old male mice.
Reverse transcription-polymerase chain reaction and Southern blotting
Reverse transcription-polymerase chain reaction (RT-PCR), PCR, and Southern blotting were performed as described elsewhere. Twenty-five cycles of PCR were performed using Taq polymerase (Takara) with primer sets specific for each gene. The glyceraldehyde-3-phosphate dehydrogenase gene (Gapdh) was amplified in each sample as a loading control. PCR products were isolated, subcloned, and sequenced to confirm the gene sequence.
Bisulfite genomic sequencing
Genomic DNA was extracted from SSCs, transplanted SSCs, ESCs, induced germline stem cells (iGSCs), and FGSCs. For bisulfite sequencing analysis of methylation, 500 ng genomic DNA was processed using an EZ DNA Methylation-Gold Kit™ (ZYMO Research), in accordance with the manufacturer’s instructions. The methylation status of imprinted genes was analyzed using specific primers (outside, 5′-GTTTTTTTGGTTATTGAAT-TTTAAAATTAGT-3′ and 5′-AAAAACCATTCCGTAAATACACAAATACCTA-3′, inside, 5′-TTAGTGTGGTTTATTATAGGAAGGTATAGAAGT-3′ and 5′-TAAACCTAAAATACTCAAAACTTTATCACAA-3′ for H19; 5′-GTG TAG AAT ATG GGG TTG TTT TAT ATT G-3′ and 5′-ATA ATA CAA CAA CAA CAA TAA CAA TC-3′ for Rasgrf1; 5′-GTA AAG TGA TTG GTT TTG TAT TTT TAA GTG-3′ and 5′-TTA ATT ACT CTC CTA CAA CTT TCC AAA TT-3′ for Peg10; 5′-TTA GTG GGG TAT TTT TAT TTG TAT GG-3′ and 5′-AAA TAT CCT AAA AAT ACA AAC TAC ACA A-3′ for Igf2r;outside, 5′-TATGTAATATGATATAGTTTAGAAATTAG-3′ and 5′-AATAAACCCAAATCTAAAATATTTTAATC-3′, inside, 5′-AATTTGTGTGATGTTTGTAATTATTTGG-3′ and 5′-ATAAAATACACTTTCACTACTAAAATCC-3′ for Snrpn). PCR products were sequenced and CpG islands were analyzed.
PCR amplification of lineage-specific microsatellite loci
Genomic DNA was extracted from mouse tail tips or donor SSCs. DNA samples from donor SSCs, female recipients, mated males, and their corresponding offspring were analyzed by simple sequence length polymorphism (SSLP). Sequences for the primer pairs were designed according to the Mouse Genome Informatics website (http://www.informatics.jax.org/). Amplification of lineage-specific microsatellite DNA was performed in accordance with a previously described procedure [27]. PCR products were separated and analyzed by 3% agarose gel electrophoresis (Bio-Rad) and visualized by ethidium bromide staining.
Flow cytometry and cell sorting
After MACS, the cells were suspended in PBS and subjected to flow cytometry to analyze and sort GFP-positive cells using a FACSAria II cell sorter equipped with BD software (Becton Dickinson).
Quantitative reverse transcription-PCR analysis
Total RNA from cells was isolated using Trizol reagent. Complementary DNA was synthesized from 2 μg total RNA using a High Capacity cDNA Reverse Transcription Kit (Invitrogen). Primers were designed using Primer Premier Software (Primer Premier 5.0). Primer details are listed in Table S1. Gapdh was amplified in each sample as an internal control. The mRNA level of each gene was normalized to Gapdh expression. The specificity of all quantitative real-time PCRs (qPCRs) was verified by a single peak in the melting curve. qPCRs were performed with a 7500 real-time PCR amplification system using SYBR Green PCR master mix (Applied Biosystems, UK). The relative levels of transcripts were calculated using the ΔΔCT method within the ABI 7500 System Software (V2.0.4). All gene expression levels were normalized to the internal standard gene, Gapdh. The means and standard error were calculated from triplicate measurements. Significance was determined using the Student’s t-test. A P-value of<0.05 was considered as significant, and a P-value of<0.01 was extremely significant.
Single cell whole genome amplification and exome sequencing
Single cell whole genome amplification was performed on lysed single cells using a recently developed method named multiple annealing and looping based amplification cycles (MALBAC) [28]. In brief, amplification was initiated by primers, each with a 27 fixed and eight degenerate base hybridizing uniformly throughout the genome. Fragments with variable length at random starting positions were generated by polymerase extension for multiple cycles. All fragments were flanked by the 27 base-fixed sequence and their complementary sequences, and further amplified by PCR to about 1 µg for barcoded massively parallel sequencing on an Illumina HiSeq 2500 sequencing platform.
Sry DNA in situ hybridization
We used a commercially available SRY DNA FISH kit (Mice SRY DNA biotin labelled POD and fluorescent FISH in situ hybridization double staining system, TBD Science), according to the manufacturer’s instructions. Briefly, sections were dewaxed with a graded series of ethanol, quenched in 3% H2O2 for 10 min at room temperature, and then washed twice with PBS. The sections were covered with SRY reagent B for 10 min at 37 °C. After washing with PBS, the sections were incubated in Tris buffered saline (TBS) for 20 min at 95–100 °C (pH 8.9) and then rinsed three times with cold TBS (5 min per rinse) and once with 0.2 × saline sodium citrate (5 min per rinse) at 0 °C. The sections were incubated for 8 h at 37 °C with SRY reagent A and then washed three times with 2 × saline sodium citrate at 37 °C (3 min per rinse), three times with 0.2 × saline sodium citrate (3 min per rinse) at 37 °C, and three times with TBS (2 min per rinse) at 37 °C. The sections were covered with SRY reagent C for 45 min at 37 °C. After washing in PBS, the sections were incubated at 37 °C for 120 min with a fluorescein isothiocyanate (FITC)-conjugated mouse anti-digoxin monoclonal antibody, then incubated at 37 °C for 120 min with DAPI. Finally, the sections were mounted in anti-fade mounting medium. Images were obtained using a Leica DMI3000 B microscope and Leica DFC550 digital camera.
Hi-C library generation using a low amount of cells
In situ high throughput chromosome conformation capture (Hi-C) assays were carried out according to the protocol with minor modifications [4], [29], [30]. Cells were fixed in a 1% final concentration of formaldehyde prior to 10 min incubation at room temperature. The reaction was quenched for 5 min by adding a 2.5 M glycine solution. Cells were pelleted twice (3000 g, 4 °C for 5 min), resuspended in ice-cold Hi-C lysis buffer for at least 15 min, and then washed once with 100 μl of 1 × NEBuffer 2. The supernatant was discarded, and 1 μl of 5% sodium dodecyl sulfate (SDS) was added to the remaining 9 μl solution. The pellet was gently mixed and incubated at 62 °C for 10 min. After incubation, 9.5 μl water and 2.5 μl of 10% Triton X-100 were added to quench the SDS, and then the solution was incubated at 37 °C for 30 min. Chromatin digestion was performed with Dpn II restriction enzyme (NEB, R0543M) at 37 °C overnight and then inactivated for 20 min at 65 °C. To fill the overhangs generated by the Dpn II restriction enzyme, a master mix of 3.75 μl biotin-14-dATP (Life Technologies), 0.45 μl of 10 mM dCTP/dGTP/dTTP mix, and 1 µl of 5 U/μl large DNA Polymerase I (NEB, M0210L) were added, followed by incubation at 24 °C for 4 h. The above biotin-labelled products were ligated by adding a master mix of 66.3 μl water, 12 μl of 10 × NEB T4 DNA ligase buffer, 10 μl of 10% Triton X-100, 5 μl of 10 mg/ml bovine serum albumin, and 2 μl of 400 U/ml T4 DNA ligase, followed by incubation at 16 °C for 20 h and then inactivation at 75 °C for 20 min. The samples were pelleted (3000 g, 4°Cfor 5 min) and washed once with 100 μl of 10 mM Tris buffer. To remove biotin from unligated DNA ends, a master mix of 40 μl water, 5 μl of 10 × NEBuffer 2.1, 0.125 μl of 10 mM dATP/dGTP, and 5 μl of 3,000 U/ml T4 DNA polymerase (NEB, M0203L) were added to the tube containing the DNA sample, followed by incubation at 20 °C for 4 h. The samples were pelleted (3000 g, 4°Cfor 5 min) and resuspended in 50 μl of 10 mM Tris buffer. To digest the proteins, 2 μl of 20 mg/ml proteinase K (NEB, P8107S) was added, followed by incubation at 62 °C for 18 h and inactivation at 75 °C for 30 min. The DNA was sheared to an average size of 400 bp (Covaris, M220) to perform the End Repair/dA-Tailing and Adaptor Ligation (NEB, E7337A) with a KAPA Hyper Prep Kit (KAPA, kk8502) and then processed by 3 μl of USER™ Enzyme (NEB, M5505L) at 37 °C for 15 min to open up the loop. Biotin-labeled ligation products were isolated using MyOne Streptavidin T1 Dynabeads (Life Technologies, 65601) and then resuspended in 20 μl of 10 mM Tris buffer at 98 °C for 10 min, and the supernatant was transferred to a fresh PCR tube. Hi-C DNA was amplified using Index Primers set 1 (NEB, E7335S). The Hi-C libraries were purified with AMPure XP beads (Beckman Coulter, A63881) and sequenced using an Illumina sequencing platform.
Hi-C data processing, mapping, and ICE normalization
For Hi-C pair-end raw data, we first trimmed the adaptor sequences and low quality reads with BBmap (version 38.16). Then, we used HiCPro (version 2.7) [31] to map, process, and perform iterative correction for normalization. Briefly, reads were independently aligned to the mouse reference genome (mm9) by the bowtie2 algorithm [32]. We discarded the uncut DNA reads, re-ligation reads, continuous reads, and PCR artifacts. We then used the unique mapped reads (MAPQ > 10) to build the contact matrix. Valid read pairs were then binned at a specific resolution by dividing the genome into sequential bins of equal size. We generated the raw contact matrices at binning resolutions of 10, 20, 40, 100, and 200 kb. ICE[33] normalization was applied to remove bias in the raw matrix, such as GC content, mappability, and effective fragment length in the Hi-C data.
Validation of Hi-C data
The data reproducibility was confirmed by calculating Pearson’s correlation coefficient (PCC) between the two libraries. Briefly, the interaction frequency was generated for each pair of 40 kb bins. For each possible interaction Iij between two replicates, they were correlated by comparing each point interaction in the normalized interaction matrix. Considering that the interaction matrix was highly skewed toward proximal interactions, we restricted the correlation to a maximum distance of 2 Mb between points i and j. We used R to calculate Pearson’s correlation between two duplicates.
Contact probability p(s)calculation
P(s) was calculated with normalized interaction matrices at a 40 kb resolution, as described previously [27]. P(s) calculations only considered intra interactions. Briefly, we divided the genome into 40 kb bins. For each distance separated by 40, 80, 120, and 160 kb, we counted the number of interactions at corresponding distances. Then, we divided the number of interactions in each bin by the total number of possible region reads as P(s). Furthermore, we normalized the sum of P(s) over the range of distances as 1. We used LOWESS fitting to construct the curve (log–log axis).
Identification of a and B compartments
We used the R package (HiTC) [34] pca.hic function to generate PC1 eigenvectors using 400 kb normalized matrices with the following options: normPerExpected = TRUE, npc = 1, for which a positive value indicated the A compartment, while a negative value indicated the B compartment. To investigate compartment switching, we defined switched bins only if PC1 eigenvectors changed in the same direction for two replicates.
Identification of concordant genes with an A/B compartment switch
We used a previously described method with minor modifications to define genes with concordant changes in expression and compartment status [35]. Briefly, we calculated the covariance between the vector of the gene expression values (FPKM) and the vector of PC1 values for each gene across five cell types. The calculated covariance as a metric to quantitatively define “concordance” was used. We compared these observed covariance values with a random background distribution to calculate a P-value for the covariance for each gene. Then, we produced the background distribution by randomly shuffling the vector of FPKM for each gene and calculating the covariance between the PC1 values and random gene expression vector. A rank-based P-value could be calculated for observed covariance values with 1000 repeats for each gene. Concordant genes were defined as those with a P-value of < 0.01.
Generation of lentivirus particles
Knockdowns of specific genes were accomplished by small interfering RNAs (siRNAs) targeting Plzf and Eed. The interfering fragment was inserted downstream of the U6 promoter in a lentiviral vector (pLKD-CMV-G&PR-U6-shRNA) by molecular biological methods. At least four independent siRNAs were screened for knockdown efficiency against each target and the best siRNA target was selected (targetSeq: CCAGGCATCTGATGACAAT for Plzf; GCAACAGAGTAACCTTATA for Eed). For Stella, Zfp57, H19, and Rasgrf1 overexpression, cDNAs of candidate genes were inserted into the EcoRI and BamHI restriction sites of the overexpression plasmid (pHBLV-CMVIE-ZsGreen-T2A-puro).
Lentivirus particles were generated by cotransfection of knockdown or overexpression plasmids and lentivirus packaging plasmids into HEK293T cells using transgene reagent. Enhancing buffer was added to the medium after 12 h of transfection. Virus particles were harvested at 48 h after transfection, and a standardized virus titer was obtained using HEK293T cells.
Lentiviral infection
For lentivirus infection, 1 × 104 SSCs, which were passaged for 2–3 times, were seeded in the well of a 48-well plate pre-coated with laminin and incubated with a 1: 1 mixture of culture medium and lentivirus-concentrated solution (lentivirus titer: 1 × 109 TU/ml) containing 5 μg/ml polybrene. After overnight infection, cells were re-plated onto puromycin-resistant STO feeder layers and cultured in SSC medium. At 12 h after re-plating, the SSCs were incubated with a 1: 1 mixture of culture medium and lentivirus-concentrated solution again. After overnight infection, the mixture was changed to fresh culture medium, and the cells were cultured for 12 h. SSCs were then infected for a third time. After overnight infection, the mixture was changed to fresh culture medium, and the cells were cultured at 37 °C with 5% CO2. At day 6, the cells were subcultured at a 1: 1–2 split ratio, and 100 ng/ml puromycin was added to the FGSC culture medium to screen for puromycin-resistant iGSCs. After 72 h, the surviving iGSCs were passaged and analyzed by qRT-PCR and western blotting.
Western blotting
Cells were lysed in protein lysis buffer, and protein concentrations were determined with the bicinchoninic acidassay. Then a total of 15 μg protein was separated in 12% w/v sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane. Subsequently, the membranes were blocked with 5% nonfat milk in PBST (PBS, 0.05% Tween-20) at 37 °C for 2 h. The membranes were overnight incubation at 4 °C with appropriate primary antibodies: Rabbit polyclonal to STELLA (1:1000; ab19878, Abcam), Rabbit polyclonal to ZFP57 (1:1000; ab45341, Abcam), RASGRF1 Antibody (1:1000; 3322, Cell Signaling), PLZF Antibody (1:1000; MAB8395, R & D), and Rabbit polyclonal to ZFP42 (1:1000; ab28141, Abcam) in 5% nonfat milk in PBST buffer. After washing three times with PBST for 10 min, the membranes were incubated with Goat Anti-Rabbit IgG(H + L), HRP conjugate (1: 2000; SA00001-2, Proteintech) at 37 °C for 2 h, and washing three times with PBST for 10 min. Finally, the images were scanned with a chemiluminescence imaging system (ProteinSimple, Santa Clara, CA, USA).
RNA-seq
Total RNA was extracted from 1 to 2 × 106 cells using Trizol Reagent. The RNA quality was assessed using an Agilent Bioanalyzer 2100. RNA-Seq libraries were prepared using the KAPA Stranded mRNA-Seq kit, following the manufacturer’s instructions. After preparation, libraries were quantified using a Qubitfluorometer and sequenced with the HiSeq Platform (2 × 100 bp). All RNA-Seq data were trimmed and aligned to the mm9 reference genome using Hisat2 (version 4.8.2)[36] with the default parameters. Gene expression as FPKM was calculated by Cufflinks (version 2.2.1) [37] using the RefSeq database from the UCSC genome browser. Sequencing depth was normalized.
MeDIP-seq
The DNA methylome assay was performed as described previously [14]. Briefly, genomic DNA (gDNA) was extracted and fragmented with Bioruptor (Connecticut, USA) into fragment sizes of 200–500 bp. Sonicated gDNA was used for end-repair and adaptor ligation. The adaptor-ligated gDNA was denatured and incubated with an antibody (Epigentek, A-1014) conjugated on Protein A + G Magnetic beads (Millipore, 16–663). Immunoprecipitated DNA was amplified by PCR and subjected to Illumina sequencing.
MeDIP-seq bioinformatics
MeDIP and input raw sequencing reads were mapped using Bowtie2 (version 2.2.6) to the UCSC mm10 genome reference. Duplicate reads were removed by samtools (version: 1.6–1). The normalized coverage was calculated by binning the unique tags in 1 kb bins, and the number of reads in each bin was normalized using reads per kilobase per million reads (RPKM). We identified the enriched MeDIP regions over the background with MACS (version 2.1.1) and default parameters [38]. Genome-wide pairwise correlation analysis of read depth in 1 kb bins was performed to evaluate DNA methylation patterns of SSCs, iGSCs, and FGSCs.
Ovarian organoid generation and culture
Ovarian organoids were formed using a modified method described elsewhere [39]. Briefly, iGSCs, and SSCs (negative control) were purified by a FACS Aria II (BD Bioscience) and co-cultured with female gonadal(aged: from E12.5 to prenatal) somatic cells in a 96-well U-bottom, low-binding culture plate (Thermo Fisher Scientific) for 2 days in GMEM supplemented with 15% Knockout serum replacement (Invitrogen), 1.5 μM retinoic acid, 2 mM L-glutamine (Sigma), 1 mM non-essential amino acids (Life Technologies), 2 mM L-glutamine (Sigma), 30 mg/ml pyruvate (Amresco), 100 mM β-mercaptoethanol (Biotech), 30 mg/l penicillin (Amresco), and 75 mg/l streptomycin (Amresco). One thousand iGSCs, FGSCs or SSCs were 3D co-cultured with 3 × 104 gonadal somatic cells. The co-cultures from 96-well U-bottom, low-binding culture plates were transferred onto transwell-COL membranes (Coaster) soaked in α-MEM- -based medium, α-MEM supplemented with 2% FBS, 2 mM L-glutamine, 200 μM ascorbic acid (Sigma), 50 mM β-mercaptoethanol, 30 mg/lpenicillin, and 75 mg/l streptomycin. At 4 days of culture, the culture medium was changed to StemPro-34-based medium, StemPro-34 SFM (Life Technologies) supplemented with 10% FBS, 2 mM L-glutamine, 200 μM ascorbic acid, 50 mM β-mercaptoethanol, 30 mg/l penicillin, and 75 mg/l streptomycin. From 7 to 10 days of culture, 800 nM ICI182780 was added to the StemPro-34-based medium. At 11 days of culture, the culture medium was changed to StemPro-34-based medium without ICI182780. After 21 days of culture, individual follicles were manually dissociated using sharpened tungsten needles.
Follicle 3D culture
The single follicles were cultured on transwell-COL membranes with medium, α-MEM supplemented with 5% FBS, 2% polyvinylpyrrolidone (Sigma), 2 mM L-glutamine, 200 μM ascorbic acid, 50 mMβ-mercaptoethanol, 30 mg/l penicillin, 20 ng/ml mouse epidermal growth factor (Pepro Tech), 75 mg/l streptomycin, 30 mg/ml pyruvate (Amresco), 0.2 IU/ml follicle-stimulating hormone (FSH; MSD), 20 ng /ml BMP15, and 20 ng/ml GDF9 (R&D Systems). At 2 days of culture, the culture medium was changed to medium without BMP15 and GDF9, and then follicles were incubated in 0.1% Type IV Collagenase (Invitrogen) for 5 min. After washing with α-MEM supplemented with 5% FBS several times, the follicles were cultured in medium without BMP15 and GDF9. After 14 days of culture, cumulus-oocyte complexes grown on the membrane were picked up by a fine glass capillary.
In vitro maturation, in vitro fertilization, and embryo transfer
The cumulus-oocyte complexes were cultured with α-MEM containing 5% FBS, 30 mg/ml pyruvate (Amresco), 0.1 IU /ml FSH, 4 ng/ml EGF, 1.2 IU/ml hCG (gonadotropin, ASKA), 4 ng/ml bFGF, 30 mg/l penicillin, 75 mg/l streptomycin. After 17–20 h of culture, mature oocytes with expanded cumulus cells were fertilized in HTF medium (SAGE) by sperm. Embryos developed to the 2-cell stage were transferred into the oviducts of pseudopregnant females at 0.5 day post-coitum.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. Original data of Hi-C have been deposited in the Gene Expression Omnibus database (accession number: 126013). Original data of RNA-Seq have been deposited in the Gene Expression Omnibus database (accession number: 134727). Original data of MeDIP-Seq have been deposited in the Gene Expression Omnibus database (accession number: 134640).
Results
Characterization of spermatogonial stem cells
SSCs were isolated by magnetic activated cell sorting (MACS) using an anti-Thy-1 antibody after two step enzymatic digestion of the testes from 6-day-old pou5f1 (also known as Oct4)/GFP transgenic × C57BL/6 F1 hybrid mice [19]. Then, the isolated SSCs were purified for GFP-positive SSCs by fluorescence-activated cell sorting (FACS) (Fig. S2A). The purified SSCs were maintained on STO fibroblast feeder layers (see Materials and methods) (Fig. S2B). After 3–5 days of culture, SSCs expanded into clusters (Fig. S2C).
To characterize SSCs, we determined the expression patterns of Oct4, Mvh (mouse vasa homologue, expressed exclusively in germ cells), c-Ret [40], Plzf [41], Rex-1 [42], Utf1 [43], Esg-1 (also known as DPPA5) [44], Stra8 [45], Sox2 [46], and Nanog [47]. Reverse transcription-polymerase chain reaction (RT-PCR) and immunocytochemical analyses showed that SSCs expressed Oct4, Mvh, c-Ret, Plzf, Rex-1, Utf1, Esg-1, and Stra8. Cytogenetic analysis by treatment with colchicines followed by G-band staining demonstrated a normal karyotype (40, XY) in the metaphase spreads of examined SSCs (Fig. S2D–J). To verify the characterization of SSCs, we compared the global expression profiles of SSCs and embryonic stem cells (ESCs) using microarrays. Gene expression profiles by scatter plots showed a significant difference between SSCs and ESCs (Fig. S2K, n = 3). More than two thousand genes (2251) were differentially expressed between SSCs and ESCs, including pluripotency-related genes Dppa4, Fgf4, Nanog, Sox2, and Klf4 (Fig. S1K and L, fold change > 2, P < 0.05, t-test) and SSC-related genes Zbtb16 (or Plzf), Gfra1, Tex18, Piwil2, and Dazl (Fig. S2K and L, fold change > 2, P < 0.05, t-test). Therefore, these results demonstrated that SSCs had their apparent original property rather than a pluripotent identity.
To determine the imprinting pattern of SSCs, differentially methylated regions (DMRs) of two paternal (H19 and Rasgrf1) and two maternal (Igf2r and Peg 10) imprinted regions were examined in SSCs and ESCs by bisulfite genomic sequencing. In SSCs, paternally imprinted regions (Fig. S2M and O) were methylated, while maternally imprinted regions were not methylated (Fig. S2N and P); this indicated an androgenetic imprinting pattern that was different from that of ESCs.
Spermatogonial stem cells can transdifferentiate into oocytes in vivo
To investigate SSC fate determination in the mouse ovary, pou5f1/GFP transgenic mouse SSCs cultured for 3–5 days were directly transplanted into the ovaries of premature ovarian failure (POF) mice (see Materials and methods). PBS was injected into the ovaries of POF recipients as a control. For the positive control, FGSCs from pou5f1/GFP transgenic mice were also transplanted into the ovaries of POF mice (see Materials and Methods). At 8 weeks post-transplantation, recipient ovaries including positive control ovaries were collected and evaluated for morphology and GFP expression. Histological analysis showed that recipient ovaries injected with cells contained numerous oocytes at all stages of development, including GFP-positive oocytes (Fig. 1A I, III, IV and Fig. S3A–E). Furthermore, DNA fluorescence in situ hybridization (FISH) analysis showed the presence of the Sry gene in oocytes from recipient ovaries (Fig. 1B). For confirmation, single cell whole exon sequencing was used. The results demonstrated that the germinal vesicle (GV) oocytes from recipient ovaries were derived from transplanted SSCs (Fig. 1C). Mature oocytes from recipient ovaries were then collected for karyotype analysis. The results showed that some mature oocytes had the karyotype of 20, Y (Fig. 1D I–V). PCR analysis of DNA fragment Sry confirmed that some mature oocytes contained a candidate of the Y chromosome (Fig. 1D VI). However, control ovaries consisted of stromal and interstitial cells as well as atretic follicles (Fig. 1A II). These results indicate that XY oocytes were regenerated in POF females by transplantation of SSCs.
Fig. 1.

SSCs transdifferentiate into oocytes in the ovaries of POF recipients and GFP-expressing offspring are generated from the transplanted SSCs from pou5f1/GFP transgenic mice. (A) SSCs were transplanted into the ovaries of POF recipient mice. I, II, Representative morphologies of the ovaries from recipients with (I) or without (II) SSC transplantations. III, Follicles containing GFP-positive (green) oocytes in recipient ovaries at 8 weeks after transplantation of pou5f1/GFP transgenic SSCs. IV, Oocytes in a wild-type ovary without a GFP signal. (B) DNA fluorescence in situ hybridization for SRY. SRY was only localized in oocytes (green) derived from SSCs in ovary (I). Nuclei were counterstained with DAPI (blue) (II). (C) Circos plot showing the coverage from the single cell exon sequencing as a histogram. Grey represents FGSCs; Red represents GV oocytes derived from SSCs in the ovary; Blue represents SSCs. (D) Karyotype analysis of mature oocytes from POF recipient ovaries at 2 months after pou5f1/GFP transgenic SSC transplantation. I, II, Representative morphologies of mature oocytes derived from pou5f1/GFP transgenic SSCs (I) emitting GFP fluorescence (II) under UV light. III–V, Cytogenetic analysis by G-band staining showing that some mature oocytes from SSCs had a karyotype of 20, Y. III: An example of 20, Y in mature oocytes derived from pou5f1/GFP transgenic SSCs. Arrow indicates the Y chromosome. IV: Example of 20, X in mature oocytes derived from pou5f1/GFP transgenic SSCs. V: Representative karyotype (20, X) of wild-type mature oocytes. VI, PCR analysis of Sry. M, 100 bp DNA marker; lane 1, SSCs; lane 2, mature oocytes derived from pou5f1/GFP transgenic SSCs; lane 3, wild-type mature oocytes; lane 4, mock. (E) Example of offspring from POF recipient mice transplanted with pou5f1/GFP transgenic SSCs (I) and an example of a Southern blot of tail DNA (II). Genomic DNA was digested with EcoRI. Marker sizes are indicated to the right of the blot. Lanes 1, 3, 5, and 7: transgenic mice; lanes 2, 4, 6, and 8: wild-type mice. (F) SSLP analysis of parents and their offspring mice through SSLP markers. M: DNA marker; lane 1: donor SSCs; lane 2: female recipients (POF); lane 3: mated males (C57BL/6); lanes 4–11: offspring from eight corresponding recipients females. Scale bars, 50 μm (A I, III, IV), 100 μm (A II), 25 μm (B I, II), 10 μm (D I, II).
Oocytes transdifferentiated from spermatogonial stem cells can produce offspring
To examine whether XY oocytes derived from SSCs could produce offspring, POF recipients were mated with wild-type C57BL/6 adult males at 35 days after cell transplantation or PBS injection (control) [16], [48]. Control recipients were not fertile (n = 9). All POF recipients produced offspring (n = 8, Fig. 1E I) with more males than females per litter (male: female, 1.95:1.00). One hundred and sixteen of the 130 offspring were alive with a normal phenotype as well as fertile. Fourteen of the 130 offspring died at 1–6 weeks after birth. The offspring were examined for the presence of GFP transgenes by Southern blot analyses (Fig. 1E II). Sixty-four of the 130 F1 progeny were heterozygous for the GFP transgene. Furthermore, simple sequence length polymorphism (SSLP) analysis was performed with SSLP markers to confirm that the offspring were derived from transplanted SSCs. The offspring from eight recipients (see above) were distinct from POF (see Materials and methods) or C57BL/6 mice-their parents (POF mice and mated male); however, they had exactly the same profiles as the SSCs from which they were derived (Fig. 1F). Moreover, five out of six positive controls (see Materials and methods) with FGSC transplantation were fertile with approximately equal numbers of males and females per litter (male: female, 1.02:1.00), and their offspring showed no abnormal phenotype. Fifty of the 101 offspring were heterozygous for the GFP transgene (Fig. S3F and G). These results suggest that the XY oocytes derived from SSCs can produce offspring in previously sterile recipients and generate transgenic progeny. Control adult mice that received PBS injections into their ovaries did not produce transgenic offspring.
Tracking of transplanted spermatogonial stem cells in recipient ovaries
To understand how the SSCs transdifferentiated into oocytes in recipient ovaries, pou5f1/GFP transgenic mouse SSCs cultured for 3 days were directly transplanted into the ovaries of POF mice, and then monitored by confocal laser scanning microscopy. At 2 h after SSC transplantation, the SSCs were observed in ovaries of recipient mice, indicating that the SSCs had been successfully transplanted into the mouse ovary (Fig. 2A). The transplanted cells were found to migrate toward the edge of the ovarian cortex at 2 days post-transplantation (Fig. 2A). At 3 days after transplantation, the cells continuously migrated toward the edge of the ovarian cortex and some of them reached the edge (Fig. 2A). When the transplanted cells had been in the ovary for 4 days, all of them had migrated into the edge of the ovarian cortex (Fig. 2A). Five days post-transplantation, transplanted cells settled in the edge of the ovarian cortex and began to transdifferentiate into early primary oocytes (Fig. 2A and B). At 6–15 days after transplantation, the transplanted cells continued to transdifferentiate into oocytes at various stages of development (Fig. 2A). This was confirmed by dual immunofluorescence analysis of the expression of MVH and GFP in transplanted cells (Fig. 2C).
Fig. 2.

Tracking of transplanted SSCs in recipient ovaries. (A) Transplanted SSCs from pou5f1/GFP transgenic mice were monitored by confocal laser scanning microscopy at 2 h, and 2, 3, 4, 5, 6, 9, 12, and 15 days after transplantation into recipient ovaries. (B) Gene expression dynamics during oogenesis in transplanted cells at 4, 6, 9, and 15 days after transplantation. (C) Dual immunofluorescence analysis of MVH and GFP expression in transplanted cells at 2, 3, 4, 5, 6, 9, 12, and 15 days after transplantation. Scale bars, 50 μm.
Methylation status and important gene expression dynamics during the development of transplanted spermatogonial stem cells in recipient ovaries
To explore the mechanism of SSC fate determination when the SSCs were transplanted into the recipient ovary, we performed bisulfite sequencing to analyze the methylation status of these transplanted cells, mainly the differentially methylated regions (DMRs) of paternally imprinted gene H19 and maternally imprinted gene Peg10. It is noteworthy that no obvious change of methylation levels, including the maternally or paternally imprinting gene, was observed at 2 h after SSC transplantation (Fig. 3A–D). At 3–4 days after transplantation, methylation levels of H19 were reduced gradually to 68.3% and 60.8%, with a further reduction to 37.9% and 23.8% at 5–6 days, suggesting that the bulk of methylation erasure occurred at 5–6 days. The low levels of methylation were present at 9 days and persisted to 15 days (Fig. 3A and C). In contrast, the maternally imprinted gene Peg10 showed evidence of robust de novo methylation with an increase to 45.7% methylation at 3 days and further increase to 78% methylation at 6 day, indicating that the bulk of methylation establishment occurred at 6 days. The high methylation levels were maintained from 9 to 15 days (Fig. 3B and D). These results suggested that the maternal DNA methylation pattern was directly constructed during SSC development in recipient ovaries.
Fig. 3.

Methylation status and gene expression dynamics of transplanted SSCs in recipient ovaries. (A-D), Methylation status of H19 (A, C) and Peg10 (B, D) DMRs in SSCs at 0 and 2 h, and 2, 3, 4, 5, 6, 9, 12, and 15 days after transplantation. DNA methylation levels were analyzed by bisulfite sequencing. Black circles represent methylated cytosine-guanine sites (CpGs), and white circles represent unmethylated CpGs. The percentage of methylated CpG sites is shown in (C) and (D). (E) Gene expression dynamics in transplanted SSCs at 0 and 2 h, 2, 3, 4, 5, 6, 9, and 15 days after transplantation.
By comparing the expression patterns between SSCs and FGSCs based on the high throughput data from our previous studies [49], [50], we established combinations of imprinted genes (H19, Rasgrf1, Snrpn and Peg10) and important transcription factor genes (Plzf, Stella or Dppa3, Zfp42, and Zfp57). Furthermore, we determined expression patterns of these genes during induction of the transplanted cells into oocytes in recipient ovaries. After comparing the expression of imprinted genes in the cultured SSCs (0 h) and transplanted SSCs at 2 h to 15 days post-transplantation, we observed that paternally imprinted genes (H19, Rasgrf1) and transcription factor genes (Stella and Zfp57) were gradually upregulated, especially at 3 and 6 days with a further increase from 9 to 15 days (Fig. 3E). In contrast, along with induction into oocytes of the SSCs in recipient ovaries, the expression levels of these maternally imprinted genes (Snrpn, and Peg10) and transcription factor genes (Plzf, and Zfp42) underwent obvious reductions at 5–6 days with a continuous decrease from 9 to 15 days after transplantation (Fig. 3E), suggesting establishment of the maternal imprinting pattern.
Critical imprinted genes and transcription factor genes required for conversion of spermatogonial stem cells
Based on the above results, we further screened for the critical imprinted genes and transcription factor genes required for SSC conversion using three-dimensional (3D) culture system of ovarian organoid. After overexpressing Stella, H19, Zfp57, and Rasgrf1 and knockdown of Plzf, Peg10 and Zfp42 in SSCs, the cells converted to induced germline stem cells (iGSCs) with a maternal imprinted pattern (Fig. S4 and S5A) and formed ovarian organoids when co-cultured with somatic cells from the fetal ovary for 21 days, modifying the method of Hikabe et al. (Fig. S5B I-II) [39]. Upon withdrawal of Peg10 from the seven genes (7Gs), we found that the co-cultured cells still formed ovarian organoids (Fig. S5B III). Upon withdrawal of Rasgrf1 from the remaining 6Gs, we found that the co-cultured cells still formed ovarian organoids (Fig. S5B IV). For the remaining 5Gs, removal of Zfp42 further promoted the formation of ovarian organoids (Fig. S5B V). However, removal of any factor from the four genes (4Gs, Stella, H19, Zfp57, and Plzf) led to the failure to form ovarian organoids (Fig. S5B VI–IX).
The morphology of iGSCs induced by the overexpression of Stella, H19, and Zfp57 and the inactivation of Plzf was similar to that of FGSCs (Fig. 3A). Furthermore, the iGSCs expressed Stella, Mvh, Fragilis, Dazl, and Oct4 with a maternal imprinted pattern (Fig. 4A and B, Fig. S5C). For confirmation, we performed genome-wide DNA methylation analysis in SSCs, iGSCs, and FGSCs by MeDIP-seq. A total of 38.7 million reads, yielding 467,163 DNA methylationsites (peaks) in three kinds of cell populations were generated. We observed widespread variation in terms of DNA methylation during SSC transition into iGSCs (Fig. 4C). Subsequently, we performed pair-wide correlation analysis of the MeDIP datasets from SSCs, iGSCs, and FGSCs. We found that the overall DNA methylation pattern of iGSCs was similar to that of FGSCs (r = 0.79), but it was less similar to that of SSCs (r = 0.59). Such a trend was evidenced more clearly by individual regions of interest. For example, at the maternally imprinted region Igf2r, the DNA methylation signal was relatively low in the Igf2r promoter of SSCs. It increased remarkably and appeared to be almost at the same level in iGSCs and FGSCs (Fig. 4C and D). A similar phenomenon was also observed at the promoter region of Nr0b1, the gene encoding the orphan nuclear receptor and required for development of male characteristics in mice [14]. Next, we compared global gene expression profiles among SSCs, iGSCs and FGSCs by RNA sequencing. A total of 380,702,626 raw reads were generated. We detected expression of 18229, 19755, and 18,978 out of 24,550 genes in SSCs, iGSCs, and FGSCs, respectively. On average, 77% of the known mouse genes were expressed in the sampled SSCs, iGSCs, and FGSCs. Hieratical clustering was performed, and the results indicated that iGSCs were clustered with FGSCs, but separated from SSCs, suggesting that the global gene expression profile of iGSCs was similar to that of FGSCs (Fig. 4E, Fig. S6). Among these genes, Igf2r, a maternal imprinted gene, showed high expression in SSCs and low level expression in iGSCs and FGSCs (Fig. 4F), which was consistent with the results from the analysis of genome-wide DNA methylation in SSCs, iGSCs, and FGSCs.
Fig. 4.

Characterization of induced germline stem cells derived from SSCs in vitro. (A) Morphology and immunofluorescence detection of MVH and GFP in cultured induced germline stem cells(iGSCs) and FGSCs. I, II, Representative morphology of cultured iGSCs under brightfield (I) and fluorescence (II) microscopies. III, IV, Representative view of cultured FGSCs under brightfield (III) and fluorescence (IV) microscopies. V–VII, Cultured iGSCs were positive for EGFP (V) and MVH (VI) staining. Cells were counterstained with DAPI (VII). VIII, Merge of EGFP, MVH, and DAPI staining. IX–XI, Cultured FGSCs were positive for EGFP (IX) and MVH staining (X). Cells were counterstained with DAPI (XI). XII, Merge of EGFP, MVH, and DAPI staining. (B) RT-PCR analysis of germ cell or germline stem cell markers in SSCs, iGSCs, and FGSCs. Lane M, 250 bp DNA marker; lane 1, SSCs, lane 2, iGSCs, lane 3, FGSCs, lane 4, STO, lane 5, no template control. (C) Genomic view of the DNA methylation pattern defined by MeDIP-seq in the IGV (Integrative Genomics Viewer) genome browser and DNA methylation status of imprinting gene Igf2r in SSCs, iGSCs, and FGSCs. (D) Pairwise correlation comparison of genome-wide DNA methylation among SSCs, iGSCs, and FGSCs. R values (Pearson correlation coefficient) were used to compare the significant correlation both within and between groups and is represented by a color scale. (E) Heat map showing expression profiles of genes among SSCs, iGSCs, and FGSCs. The maps were based on the expression values of all expressed genes detected by high-throughput sequencing. The color scale indicates the expression values. The intensity increases from green to red. Each column represents one sample, and each row represents a transcript. (F) RNA-seq read density over imprinting gene Igf2r in SSCs, iGSCs, and FGSCs. Scale bars, 50 μm (A I-IV), 20 μm (A V-XII).
Chromatin architecture reorganization during spermatogonial stem cell conversion to induced germline stem cells
Hi-C interaction maps provide information on multiple hierarchical levels of genome organization [1]. To understand how genome organization is involved in SSCs transition to iGSCs, we performed Hi-C experiments using two biological replicates of SSCs, iGSCs and FGSCs, generating a total of 9.24billion unique read pairs (Table S2). The Hi-C data analysis showed the high order chromatin organization of the whole genome of iGSCs had a high similarity with FGSCs, different to SSCs (Fig. 5A-C, Fig. S7). To examine the characteristics of their chromatin organization, we analyzed the pattern of compartment A/B in SSCs, iGSCs, and FGSCs. We found a large degree of spatial plasticity in the arrangement of the A/B compartments or redistribution of the spatial organization of their genomes during SSC transition into iGSCs with 35% of the genome switching compartments. Furthermore, we found that the regions that changed their A/B compartment status corresponded to a single or series of topologically associated domains (TADs), suggesting that TADs are the units of dynamic alterations in chromosome compartments (Fig. 5B). Interestingly, we observed that iGSCs and FGSCs were highly similar in their status of A/B compartments compared with SSCs (Fig. 5C). For SSC transition into iGSCs, genes that changed from compartment B to A tended to show higher expression, while genes that changed from A to B tended to show reduced expression (Fig. 5D). Moreover, we identified 4353 genes with co-variation between compartment switching and gene expression. For example, at the compartment B region, Dppa3 expression was relatively low in SSCs. It increased remarkably and appeared to be almost at the same level in iGSCs and FGSCs when changing from compartment B to A (Fig. 5E). DNA methylation that changed from compartment B to A also tended to show a reduced signal, whereas DNA methylation that changed from A to B tended to show higher signal (Fig. 5F).
Fig. 5.

Reorganization of the chromosome structure during SSC conversion to iGSCs. (A) Contact matrices from chromosome 16 in SSCs, iGSCs, and FGSCs. (B) First principal component (PC1) value and normalized Hi-C interaction heat maps at a 40 kb resolution in SSCs, iGSCs, and FGSCs. The PC1 value was used to indicate the A/B compartment status, where a positive PC1 value represents the A compartment (blue) and a negative value represents the B compartment (yellow). Dashed lines indicate TAD boundaries in SSCs. (C) Hieratical clustering of PC1 values for the A/B compartment status in SSCs, iGSCs, and FGSCs. (D) Expression of genes that changed compartment status (“A to B” or “B to A”) or remained the same (“stable”) compared with SSCs (P-value by Wilcoxon’s test). (E) IGV snapshot of Dppa3 (Stella) showing concordance between its expression and PC1 values. (F) Relative MeDIP-seq signal that changed compartment status (“A to B” or “B to A”) or remained the same (“stable”) compared with SSCs (P-value by Wilcoxon’s test). ***p < 0.0001.
Induced germline stem cells can differentiate into mature oocytes
Developmental feature of ovarian organoids from iGSCs were explored. At 2 weeks of 3D co-culture with the iGSCs and somatic cells from the fetal ovary, ovarian organoids were generated. The organoids were completely filled with follicles which possessed oocytes. In contrast, ovarian organoids were not formed in the SSC group. When the ovarian organoids were 3D co-cultured for 3 weeks, the follicles grew obviously (Fig. 6A). On the other hand, continuous increases in the progesterone and estradiol concentrations in the medium suggested ovarian organoids had hormone secretion functions (Fig. 6B).
Fig. 6.

In vitro production of functional oocyte from iGSCs. (A) Ovarian organoid formation and development. Representative ovarian organoids with a merge of bright field and fluorescence. Up, Ovarian organoids or co-cultures with somatic cells of gonad and iGSCs at 3 days, 2 weeks, and 3 weeks. Down: Images of aggregates formed by somatic cells of gonads and SSCs at 3 days, 2 weeks, and 3 weeks. (B) Trends of progesterone and estradiol in the medium of ovarian organoids with a different time-course. (C) Follicle growth in vitro. Representative follicles isolated from ovarian organoids formed by somatic cells of gonads and iGSCs at 0 days, 2 days, 7 days, 11 days, and Cumulus-oocytes complexes (COC) derived from iGSCs before in vitro maturation. (D) Representative views of each stage of meiotic prophase I during ovarian organoid development after stained with anti-SYCP3 and -H2AX antibodies. Scale bars, 100 μm (A), 20 μm (C 0 day, 2 days and 7 days), 40 μm (C 11 days), 50 μm (C COC), 5 μm (D).
Subsequently, individual follicles were manually separated from the ovarian organoids. After these follicles were 3D cultured individually for 2 weeks (Materials and methods), a large number of immature oocytes were obtained from the cultured follicles. Moreover, Oct4-EGFP cells were detectable at 3 days of 3D co-culture. A number of EGFP-positive oocytes were observed in ovarian organoids after 3 weeks of 3D co-culture (Fig. 6A). After 2–3 weeks of 3D co-culture, however, the EGFP expression became weak (Fig. 6A).
Then, 905 immature oocytes were obtained from 27 ovarian organoids in 8 cultures with 33.52 ± 4.27 immature oocytes per organoid (Fig. 6C). To evaluate oogenesis during the ovarian organoid development, immunofluorescence analysis of SCP3 and H2AX was performed. The results showed progression of meiotic prophase I from 7 to 21 days of the germ cell differentiation in the organoids (Fig. 6D). Furthermore, the gene expression profiles during iGSC differentiation were analyzed by qRT-PCR, suggesting the genes involve in oogenesis with dynamic expression (Fig. S8).
After in vitro maturation for 17–20 h, 51.3% of these immature oocytes reached mature oocytes. In addition, no difference in the number of mature oocytes was observed between iGSCs group and the control (Fig. 7A, B).
Fig. 7.

Offspring production of functional oocytes from iGSC differentiation in vitro. (A) Mature oocytes from derived iGSCs after in vitro maturation. (B) The percentage of mature cumulus-oocyte complexes derived from iGSCs. Control, the immature oocytes from wild-type mice. (C) The percentage of fertilization of iGSC-derived MII oocytes. Control, MII oocytes from immature oocytes of wild type mice after in vitro maturation. (D) Two-cell embryos derived from iGSCs after in vitro fertilization. (E) Representative offspring derived from iGSC. (F) Number of offspring derived from iGSCs per litter. (G) Offspring were identified by Southern blotting. Lanes 1–4, offspring derived from iGSCs, lane W, wild-type mice. (H) Offspring were identified by fluorescence. Lanes 1–4, offspring derived from iGSCs, lane W, wild-type mice. Scale bars: 50 μm.
Offspring production from induced germline stem cells
To determine whether these mature oocytes could develop into offspring following in vitro fertilization, embryo culture and transfer into pseudopregnant ICR females were performed. The fertilization rate of the iGSC group (47.2%) was similar to that of the control (46.3%) (Fig. 7C). Subsequently, these zygotes developed to 2-cell embryos (Fig. 7D). After the embryo transfer, 53 (male: female, 1.65:1.00) out of 342 were delivered as viable offspring with colored eyes (Fig. 7E, F), indicating that the offspring were derived from C57BL/6 iGSC-derived oocytes, but not ICR oocytes among gonadal somatic cells. The offspring were confirmed for the presence of GFP transgenes by Southern blot analysis, and live imaging by a Lumazone imaging system (Fig. 7G, H). In contrast to wide-type mice, the offspring derived from iGSCs displayed no significant differences in body weight during postnatal development (Fig. S9A). With analysis of the methylation status, 10 offspring per group demonstrated no observably abnormal methylation patterns (Table S3). After mating for 3 months with normal males or females proven fertility, the mice derived from iGSCs produced an average of 150 offspring, the same as wild-type mice and the male to female ratio in offspring was close to 1 (Table S4). The first to third generation (F1-F3) produced by these offspring were allocated to the same mating cycle, an average of 150 offspring in each filial generation was produced too (Table S4). Litter sizes from in vitro offspring production of functional oocytes from iGSCs and their F1 to F3 generation mice were similar with wild-type mice (Fig. S9B). These results showed that there was no significant difference in reproductive capacity between the offspring from iGSCs and the wild-type mice.
Discussion
Critical transcription factors are required for sex determination. In mammals, male sex determination relies on upregulation of sex determining region-box9 (Sox9) expression in somatic cells of XY gonads by sex-determining region Y chromosome (SRY), while Wnt family member (WNT)/R-spondin 1 signaling and forkhead box L2 (Foxl2) drive female sex determination in somatic cells of XX gonads [51], [52], [53], [54], [55], [56], [57]. Furthermore, male and female sex-determining programs can antagonize one another to drive one differentiation pathway. For example, Sox9 is upregulated in sex-reversed XX Wnt4-/-; Foxl2-/- double-knockout mice [57] and sex-reversed XY Sox9-/- gonads express both Wnt4 and Foxl2 [54]. Recently, Gonen et al. demonstrated sex reversal following deletion of an enhancer of Sox9, Enh13 [58].
The somatic cells associated with PGCs initiate their cell fate determination to promote PGC development into a male or female germline lineage at the onset of sex determination, demonstrating that somatic cells are essential and provide the microenvironment to support this process [59], [60], [61]. Ultimately, germ cells will differentiate into eggs or sperm according to their environment. The bidirectional differentiation is controlled by different signaling pathways. For example, Wnt signaling plays a major inhibitory role in testis determination [62].
Based on these findings, we determined whether the ovarian environment can drive SSC development into the female germline lineage under the action of WNT/R-spondin 1 signaling and Foxl2. To this end, we transplanted SSCs into the POF ovary to explore their fate determination and how it corresponds to their microenvironment. Interestingly, the transplanted SSCs transdifferentiated into oocytes and produced offspring–as confirmed by tracing transplanted cells, single oocyte whole exome sequencing, chromosome analyses, and offspring Southern blotting. When we tracked the transplanted SSCs in the recipient mouse ovary, we observed that the cells migrated into the edge of the ovarian cortex and transdifferentiated into oocytes day-by-day, showing a similar migration process as FGSC transplanted into the ovary [48]. Moreover, the transplanted SSCs directly reversed their paternal DNA methylation status to a maternal DNA methylation status day-by-day, suggesting that the cells did not dedifferentiate into PGCs. Unlike most previous studies that focused on the regulatory mechanism of somatic cells in gonads, we explored the mechanisms of transplanted SSCs according to their environment. Based on our previous studies [49], [50], we selected important genes with differential expression between SSCs and FGSCs. These genes included paternal imprinting genes (H19, and Rasgrf1), maternal imprinting genes (Peg10, and Snrpn) and transcription factor genes (Stella, Plzf, Zfp57, and Zfp42). Furthermore, we found that the expression patterns of these genes were consistent with the change in pattern of the DNA methylation status during SSC transdifferentiation into oocytes, suggesting that these genes play important roles in the induction of SSC conversion into the female germline lineage.
We successfully screened the critical imprinted genes and transcription factor genes required for SSC conversion by the ovarian organoids. These critical genes were H19, Stella, Plzf, and Zfp57. The H19 gene is one of the best-studied imprinted genes and is inactivated by imprinting of the paternal allele and loss of the maternal allele in the mammals [63]. Stella is a maternal factor essential for early development [64]. Zfp57 is also paternal imprinted gene, and has been shown to target and maintain epigenetic states at genomic imprinting control regions [65]. Plzf is a spermatogonia-specific transcription factor in the testis that is required to regulate self-renewal and maintenance of the SSCs [41]. After overexpressing Stella, H19, and Zfp57, and knockdown of Plzf in SSCs, the cells converted to iGSCs with a maternal imprinted pattern and similar morphology, overall DNA methylation pattern, and global gene expression profile as FGSCs, suggesting that the four genes could induce SSC conversion to the female germline in vitro. The iGSCs could differentiate into MII oocytes in vitro. Furthermore, the MII oocytes derived from iGSCs were capable of in vitro fertilization and developing into offspring similarly to those of control. However, the sex ratio (male: female) of offspring derived from the control was 1.1:1.0, whereas the sex ratios of offspring derived from iGSCs and transplanted SSCs were 1.7:1.0 and 1.9:1.0, respectively, suggesting that both of the latter two parents had a male chromosome karyotype in accordance with Mendel’s law of inheritance.
A higherorder chromatin structure is regarded as an important regulator of gene expression. To explore the mechanism of SSCs conversion to iGSCs at the chromatin level, we compared the patterns of compartment A/B in SSCs, iGSCs, and FGSCs. We found redistribution of the spatial organization of their genomes during SSC conversion to iGSCs with 35% of the genome switching compartments. Furthermore, we found 4353 genes with covariation between compartment switching and gene expression, suggesting this chromatin reorganization regulates gene expression for SSC conversion to iGSCs. Interestingly, there was high similarity in the chromatin structure between iGSCs and FGSCs, indicating that the chromatin structure is the basis of cell functions [3].We also observed that DNA methylation that changed from compartment B to A tended to show reduced signal, whereas DNA methylation that changed from A to B tended to show a higher signal, suggesting that the chromatin reorganization regulates the DNA methylation status during SSC conversion to iGSCs. The mechanism of SSC conversion to iGSCs at the chromatin level is similar to that of lineage specification in human ESCs [35].
Conclusion
In conclusion, we demonstrated successful production of offspring from oocytes transdifferentiated from mouse SSCs by tracking the transplanted SSCs and their methylation status in vivo, single cell whole exome sequencing, and reconstitution in vitro of the process of oogenesis derived from SSCs. We also found that iGSCs differentiated into functional oocytes can be directly converted by transduction of H19, Stella, and Zfp57 and inactivation of Plzf in SSCs. By mapping genome-wide chromatin interactions in SSCs, iGSCs, and FGSCs, we uncovered extensive chromatin reorganization during SSC conversion into iGSCs. We observed that, although TADs were stable during SSC conversion, chromatin interactions changed in a striking manner, altering 35% of inactive and active chromosomal compartments throughout the genome. By integrating chromatin interaction maps with transcriptome and genome-wide DNA methylation datasets, we found a high degree of plasticity in A and B compartments, corresponding to changes in DNA methylation and gene expression. Our results provide a new strategy to investigate germ cell biology, biotechnology, and medicine.
Compliance with ethics requirements
Animal experimentation was approved by the Institutional Animal Care and Use Committee of Shanghai and performed in accordance with the National Research Council Guide for Care and Use of Laboratory Animals. The ethical approval number for our research is A2016084
Declaration of Competing Interest
The authors declared that there is no conflict of interest.
Acknowledgments
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFC1003501, 2017YFA0504201), National Nature Science Foundation of China (81720108017, 32000806), the National Major Scientific Instruments and Equipment Development Project, National Nature Science Foundation of China (61827814). the China Postdoctoral Science Foundation (2017M621453).
Author contributions
HL, XL and GGT. conducted all the major experiments, data analysis and wrote the manuscript; DL performed embryo transfer; CH carried out in situ Hi-C library generation using a low amount of cells; XD and WX were responsible for karyotype analysis; LH, YY, and HW were responsible for immunofluorescence and histological analysis of ovarian tissue; QL, AJC and JX conducted Gdf9-Cre+andGFP transgenic mice study; XZ carried out MeDIP-seq and bioinformatics; JW initiated and supervised the entire project, conducted SSC and FGSC transplantation, analyzed data and wrote the manuscript.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2021.03.006.
Contributor Information
Xiaodong Zhao, Email: xiaodongzhao@sjtu.edu.cn.
Ji Wu, Email: jiwu@sjtu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smallwood A., Ren B. Genome organization and long-range regulation of gene expression by enhancers. Curr Opin Cell Biol. 2013;25:387–394. doi: 10.1016/j.ceb.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorkin D.U., Leung D., Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–775. doi: 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ke Y., Xu Y., Chen X., Feng S., Liu Z., Sun Y. Cell. 2017;170:367–381 e320. doi: 10.1016/j.cell.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 5.Battulin N., Fishman V.S., Mazur A.M., Pomaznoy M., Khabarova A.A., Afonnikov D.A. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015;16:77. doi: 10.1186/s13059-015-0642-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung Y.H., Sauria M.E.G., Lyu X., Cheema M.S., Ausio J., Taylor J. Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes. Cell. Rep. 2017;18:1366–1382. doi: 10.1016/j.celrep.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowles J., Koopman P. Retinoic acid, meiosis and germ cell fate in mammals. Development. 2007;134:3401–3411. doi: 10.1242/dev.001107. [DOI] [PubMed] [Google Scholar]
- 8.Gkountela S., Zhang K.X., Shafiq T.A., Liao W.W., Hargan-Calvopina J., Chen P.Y. DNA Demethylation Dynamics in the Human Prenatal Germline. Cell. 2015;161:1425–1436. doi: 10.1016/j.cell.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo F., Yan L., Guo H., Li L., Hu B., Zhao Y. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell. 2015;161:1437–1452. doi: 10.1016/j.cell.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Leitch H.G., Tang W.W., Surani M.A. Primordial germ-cell development and epigenetic reprogramming in mammals. Curr Top Dev Biol. 2013;104:149–187. doi: 10.1016/B978-0-12-416027-9.00005-X. [DOI] [PubMed] [Google Scholar]
- 11.Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang W.W., Dietmann S., Irie N., Leitch H.G., Floros V.I., Bradshaw C.R. A Unique Gene Regulatory Network Resets the Human Germline Epigenome for Development. Cell. 2015;161:1453–1467. doi: 10.1016/j.cell.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X.L., Wu J., Wang J., Shen T., Li H., Lu J. Integrative epigenomic analysis reveals unique epigenetic signatures involved in unipotency of mouse female germline stem cells. Genome Biol. 2016;17:162. doi: 10.1186/s13059-016-1023-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma B., Lee T.L., Hu B., Li J., Li X., Zhao X. Molecular characteristics of early-stage female germ cells revealed by RNA sequencing of low-input cells and analysis of genome-wide DNA methylation. DNA Res. 2018 doi: 10.1093/dnares/dsy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou K., Yuan Z., Yang Z., Luo H., Sun K., Zhou L. Production of offspring from a germline stem cell line derived from neonatal ovaries. Nat Cell Biol. 2009;11:631–636. doi: 10.1038/ncb1869. [DOI] [PubMed] [Google Scholar]
- 17.Simon L., Ekman G.C., Kostereva N., Zhang Z., Hess R.A., Hofmann M.C. Direct transdifferentiation of stem/progenitor spermatogonia into reproductive and nonreproductive tissues of all germ layers. Stem. Cells. 2009;27:1666–1675. doi: 10.1002/stem.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okutsu T., Shikina S., Kanno M., Takeuchi Y., Yoshizaki G. Production of trout offspring from triploid salmon parents. Science. 2007;317:1517. doi: 10.1126/science.1145626. [DOI] [PubMed] [Google Scholar]
- 19.Yoshimizu T., Sugiyama N., De Felice M., Yeom Y.I., Ohbo K., Masuko K. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. Dev Growth Differ. 1999;41:675–684. doi: 10.1046/j.1440-169x.1999.00474.x. [DOI] [PubMed] [Google Scholar]
- 20.Reddy P., Liu L., Adhikari D., Jagarlamudi K., Rajareddy S., Shen Y. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–613. doi: 10.1126/science.1152257. [DOI] [PubMed] [Google Scholar]
- 21.Lan Z.J., Xu X., Cooney A.J. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod. 2004;71:1469–1474. doi: 10.1095/biolreprod.104.031757. [DOI] [PubMed] [Google Scholar]
- 22.Wu J., Jester W.F., Jr., Orth J.M. Short-type PB-cadherin promotes survival of gonocytes and activates JAK-STAT signalling. Dev Biol. 2005;284:437–450. doi: 10.1016/j.ydbio.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 23.Wu J., Zhang Y., Tian G.G., Zou K., Lee C.M., Yu Q. Short-type PB-cadherin promotes self-renewal of spermatogonial stem cells via multiple signaling pathways. Cell Signal. 2008;20:1052–1060. doi: 10.1016/j.cellsig.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Yuan Z., Hou R., Wu J. Generation of mice by transplantation of an adult spermatogonial cell line after cryopreservation. Cell Prolif. 2009;42:123–131. doi: 10.1111/j.1365-2184.2009.00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohrborn G. Frequencies of spontaneous non-disjunction in metaphase II. Oocytes of mice. Humangenetik. 1972;16:123–125. doi: 10.1007/BF00393996. [DOI] [PubMed] [Google Scholar]
- 26.Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naumova N., Imakaev M., Fudenberg G., Zhan Y., Lajoie B.R., Mirny L.A. Organization of the mitotic chromosome. Science. 2013;342:948–953. doi: 10.1126/science.1236083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang L., Ma F., Chapman A., Lu S., Xie X.S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu Rev Genomics Hum Genet. 2015;16:79–102. doi: 10.1146/annurev-genom-090413-025352. [DOI] [PubMed] [Google Scholar]
- 29.Diaz N., Kruse K., Erdmann T., Staiger A.M., Ott G., Lenz G. Chromatin conformation analysis of primary patient tissue using a low input Hi-C method. Nat Commun. 2018;9:4938. doi: 10.1038/s41467-018-06961-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Servant N., Lajoie B.R., Nora E.P., Giorgetti L., Chen C.J., Heard E. HiTC: exploration of high-throughput 'C' experiments. Bioinformatics. 2012;28:2843–2844. doi: 10.1093/bioinformatics/bts521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–1667. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hikabe O., Hamazaki N., Nagamatsu G., Obata Y., Hirao Y., Hamada N. Reconstitution in vitro of the entire cycle of the mouse female germ line. Nature. 2016;539:299–303. doi: 10.1038/nature20104. [DOI] [PubMed] [Google Scholar]
- 40.Naughton C.K., Jain S., Strickland A.M., Gupta A., Milbrandt J. Glial cell-line derived neurotrophic factor-mediated RET signaling regulates spermatogonial stem cell fate. Biol Reprod. 2006;74:314–321. doi: 10.1095/biolreprod.105.047365. [DOI] [PubMed] [Google Scholar]
- 41.Costoya J.A., Hobbs R.M., Barna M., Cattoretti G., Manova K., Sukhwani M. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet. 2004;36:653–659. doi: 10.1038/ng1367. [DOI] [PubMed] [Google Scholar]
- 42.Xu J., Sylvester R., Tighe A.P., Chen S., Gudas L.J. Transcriptional activation of the suppressor of cytokine signaling-3 (SOCS-3) gene via STAT3 is increased in F9 REX1 (ZFP-42) knockout teratocarcinoma stem cells relative to wild-type cells. J Mol Biol. 2008;377:28–46. doi: 10.1016/j.jmb.2007.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okuda A., Fukushima A., Nishimoto M., Orimo A., Yamagishi T., Nabeshima Y. UTF1, a novel transcriptional coactivator expressed in pluripotent embryonic stem cells and extra-embryonic cells. EMBO J. 1998;17:2019–2032. doi: 10.1093/emboj/17.7.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka T.S., Kunath T., Kimber W.L., Jaradat S.A., Stagg C.A., Usuda M. Gene expression profiling of embryo-derived stem cells reveals candidate genes associated with pluripotency and lineage specificity. Genome Res. 2002;12:1921–1928. doi: 10.1101/gr.670002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oulad-Abdelghani M., Bouillet P., Decimo D., Gansmuller A., Heyberger S., Dolle P. Characterization of a premeiotic germ cell-specific cytoplasmic protein encoded by Stra8, a novel retinoic acid-responsive gene. J Cell Biol. 1996;135:469–477. doi: 10.1083/jcb.135.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fong H., Hohenstein K.A., Donovan P.J. Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem. Cells. 2008;26:1931–1938. doi: 10.1634/stemcells.2007-1002. [DOI] [PubMed] [Google Scholar]
- 47.Chambers I., Colby D., Robertson M., Nichols J., Lee S., Tweedie S. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 48.Wu C., Xu B., Li X., Ma W., Zhang P., Chen X. Tracing and Characterizing the Development of Transplanted Female Germline Stem Cells In Vivo. Mol Ther. 2017;25:1408–1419. doi: 10.1016/j.ymthe.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie W., Wang H., Wu J. Similar morphological and molecular signatures shared by female and male germline stem cells. Sci Rep. 2014;4:5580. doi: 10.1038/srep05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X., Tian G.G., Zhao Y., Wu J. Genome-wide identification and characterization of long noncoding and circular RNAs in germline stem cells. Sci Data. 2019;6:8. doi: 10.1038/s41597-019-0014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koopman P., Gubbay J., Vivian N., Goodfellow P., Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature. 1991;351:117–121. doi: 10.1038/351117a0. [DOI] [PubMed] [Google Scholar]
- 52.Vidal V.P., Chaboissier M.C., de Rooij D.G., Schedl A. Sox9 induces testis development in XX transgenic mice. Nat Genet. 2001;28:216–217. doi: 10.1038/90046. [DOI] [PubMed] [Google Scholar]
- 53.Chaboissier M.C., Kobayashi A., Vidal V.I., Lutzkendorf S., van de Kant H.J., Wegner M. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development. 2004;131:1891–1901. doi: 10.1242/dev.01087. [DOI] [PubMed] [Google Scholar]
- 54.Barrionuevo F., Bagheri-Fam S., Klattig J., Kist R., Taketo M.M., Englert C. Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biol Reprod. 2006;74:195–201. doi: 10.1095/biolreprod.105.045930. [DOI] [PubMed] [Google Scholar]
- 55.Sekido R., Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453:930–934. doi: 10.1038/nature06944. [DOI] [PubMed] [Google Scholar]
- 56.Chassot A.A., Ranc F., Gregoire E.P., Roepers-Gajadien H.L., Taketo M.M., Camerino G. Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum Mol Genet. 2008;17:1264–1277. doi: 10.1093/hmg/ddn016. [DOI] [PubMed] [Google Scholar]
- 57.Ottolenghi C., Pelosi E., Tran J., Colombino M., Douglass E., Nedorezov T. Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum Mol Genet. 2007;16:2795–2804. doi: 10.1093/hmg/ddm235. [DOI] [PubMed] [Google Scholar]
- 58.Gonen N., Futtner C.R., Wood S., Garcia-Moreno S.A., Salamone I.M., Samson S.C. Sex reversal following deletion of a single distal enhancer of Sox9. Science. 2018;360:1469–1473. doi: 10.1126/science.aas9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharpe R.M. Paracrine control of the testis. Clin Endocrinol Metab. 1986;15:185–207. doi: 10.1016/s0300-595x(86)80049-4. [DOI] [PubMed] [Google Scholar]
- 60.Skinner M.K. Cell-cell interactions in the testis. Ann N Y Acad Sci. 1987;513:158–171. doi: 10.1111/j.1749-6632.1987.tb25006.x. [DOI] [PubMed] [Google Scholar]
- 61.Jost A. Hormonal factors in the sex differentiation of the mammalian foetus. Philos Trans R Soc Lond B Biol Sci. 1970;259:119–130. doi: 10.1098/rstb.1970.0052. [DOI] [PubMed] [Google Scholar]
- 62.Bagheri-Fam S., Bird A.D., Zhao L., Ryan J.M., Yong M., Wilhelm D. Testis Determination Requires a Specific FGFR2 Isoform to Repress FOXL2. Endocrinology. 2017;158:3832–3843. doi: 10.1210/en.2017-00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang S., Kubota C., Yang L., Zhang Y., Page R., O'Neill M. Genomic imprinting of H19 in naturally reproduced and cloned cattle. Biol Reprod. 2004;71:1540–1544. doi: 10.1095/biolreprod.104.031807. [DOI] [PubMed] [Google Scholar]
- 64.Saitou M., Barton S.C., Surani M.A. A molecular programme for the specification of germ cell fate in mice. Nature. 2002;418:293–300. doi: 10.1038/nature00927. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi N., Gray D., Strogantsev R., Noon A., Delahaye C., Skarnes W.C. ZFP57 and the Targeted Maintenance of Postfertilization Genomic Imprints. Cold Spring Harb Symp Quant Biol. 2015;80:177–187. doi: 10.1101/sqb.2015.80.027466. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files. Original data of Hi-C have been deposited in the Gene Expression Omnibus database (accession number: 126013). Original data of RNA-Seq have been deposited in the Gene Expression Omnibus database (accession number: 134727). Original data of MeDIP-Seq have been deposited in the Gene Expression Omnibus database (accession number: 134640).