

Abstract
A growing number of inborn errors of metabolism (IEM) associated with compromised mitochondrial energy metabolism manifest an unusual phenotypic feature: 3-methylglutaconic (3MGC) aciduria. Two major categories of 3MGC aciduria, primary and secondary, have been described. In primary 3MGC aciduria, IEMs in 3MGC CoA hydratase (AUH) or HMG CoA lyase block leucine catabolism, resulting in a buildup of pathway intermediates, including 3MGC CoA. Subsequent thioester hydrolysis yields 3MGC acid, which is excreted in urine. In secondary 3MGC aciduria, no deficiencies in leucine catabolism enzymes exist and 3MGC CoA is formed de novo from acetyl CoA. In the “acetyl CoA diversion pathway”, when IEMs directly, or indirectly, interfere with TCA cycle activity, acetyl CoA accumulates in the matrix space. This leads to condensation of two acetyl CoA to form acetoacetyl CoA, followed by another condensation between acetyl CoA and acetoacetyl CoA to form 3-hydroxy, 3-methylglutaryl (HMG) CoA. Once formed, HMG CoA serves as a substrate for AUH, producing trans-3MGC CoA. Non enzymatic isomerization of trans-3MGC CoA to cis-3MGC CoA precedes intramolecular cyclization to cis-3MGC anhydride plus CoA. Subsequent hydrolysis of cis-3MGC anhydride gives rise to cis-3MGC acid, which is excreted in urine. In reviewing 20 discrete IEMs that manifest secondary 3MGC aciduria, evidence supporting the acetyl CoA diversion pathway was obtained. This biochemical pathway serves as an “overflow valve” in muscle / brain tissue to redirect acetyl CoA to 3MGC CoA when entry to the TCA cycle is impeded.
Keywords: 3-methylglutaconic aciduria, inborn error of metabolism, mitochondria, energy metabolism
I. Introduction
Inborn errors of metabolism (IEM) are a heterogeneous collection of genetic disorders that, oftentimes, result in serious disease. Of interest to the present discussion is a group of seemingly disparate IEMs that share a common and distinctive phenotypic feature: urinary excretion of 3-methylglutaconic (3MGC) acid. The metabolic origin of 3MGC aciduria in these disorders has been the subject of much speculation, and studies have revealed that two major categories of 3MGC aciduria exist, termed primary and secondary. Primary 3MGC aciduria is characterized by defects in the leucine catabolism pathway (Figure 1) due to mutations in either AUH [1; OMIM #250950] or HMGCL [2; OMIM *613898]. AUH encodes 3MGC CoA hydratase (AUH), which catalyzes the 5th step in the leucine catabolism pathway, converting trans-3MGC CoA to (S)-3-hydroxy-3-methylglutaryl (HMG) CoA. The HMGCL gene encodes the next enzyme in the pathway, HMG CoA lyase, which converts HMG CoA to acetoacetate and acetyl CoA. When either one of these enzymes is deficient, leucine metabolism is blocked and pathway intermediates upstream of the deficient enzyme accumulate. In these IEMs, trans-3MGC CoA is considered to be the precursor of 3MGC acid, which is excreted in urine as a waste product. Individuals with primary 3MGC aciduria excrete large amounts of 3MGC acid [3] and excretion levels increase in response to leucine ingestion [4].
Figure 1. Leucine catabolism pathway defects lead to primary 3MGC aciduria.
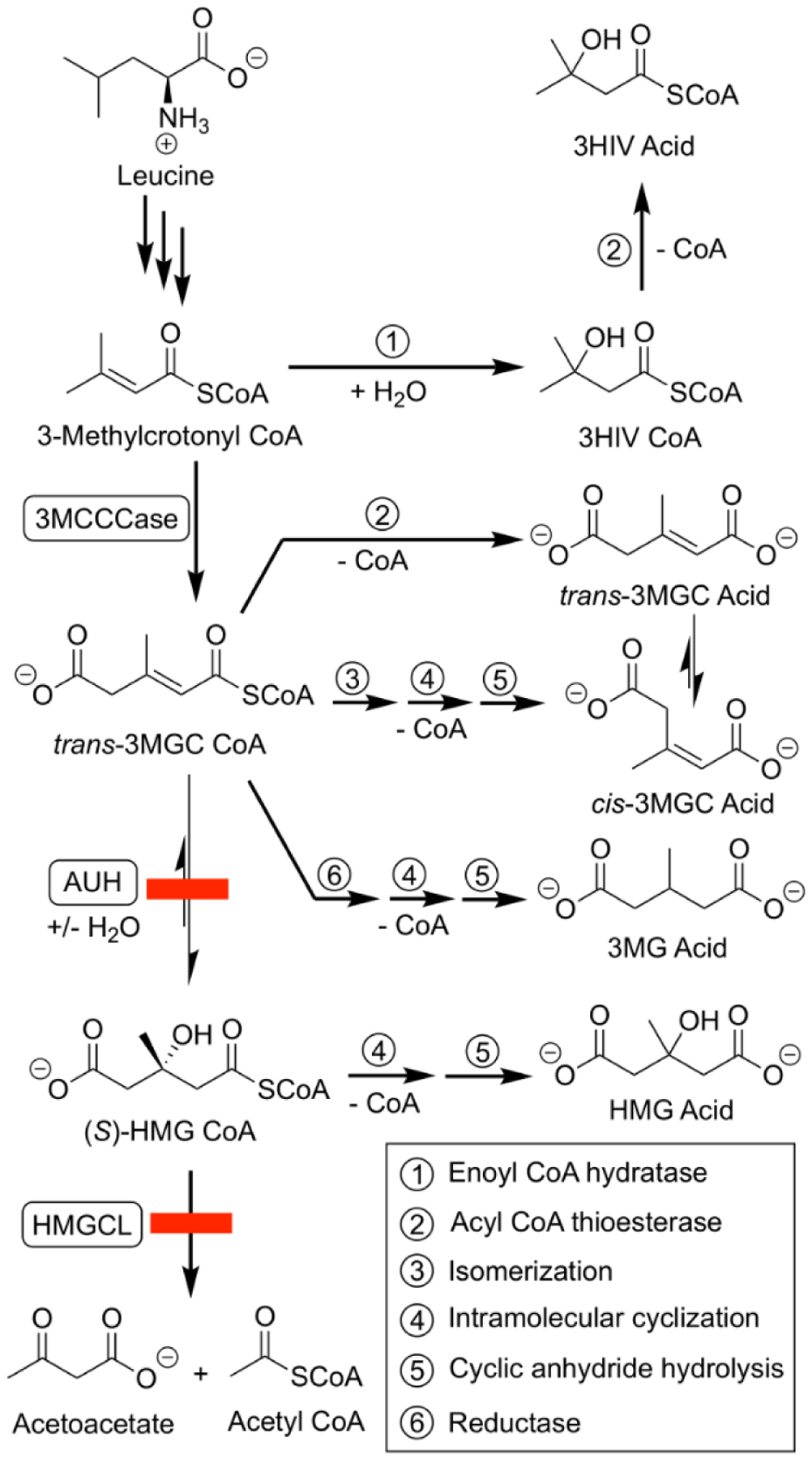
IEMs affecting AUH or HMGCL (see red boxes) result in enzyme deficiencies that interfere with leucine catabolism. As leucine degradation proceeds, pathway intermediates accumulate, and are ultimately converted to organic acids. When AUH is deficient, 3-methylcrotonyl CoA is subject to hydroxylation (1) and thioester hydrolysis (2), forming 3-hydroxy-isovaleric (3-HIV) acid. Alternatively, 3-methylcrotonyl CoA can be carboxylated by 3-methylcrotonyl CoA carboxylase (3MCCCase), forming trans-3MGC CoA. This product can undergo isomerization (3), intramolecular cyclization (4) and hydrolysis (5), giving rise to cis-3MGC acid. trans-3MGC acid can be produced by isomerization (3) of cis-3MGC acid [12] or by acyl CoA thioesterase-mediated hydrolysis (2) of trans-3MGC CoA. In addition, trans-3MGC CoA can be reduced (6) to 3-methylglutaryl (3MG) CoA, and then converted to 3-methylglutaric acid via intramolecular cyclization (4) and hydrolysis (5). Alternatively, 3MG acid may be generated by thioesterase-mediated hydrolysis (2) of 3MG CoA [8]. When HMG CoA lyase (HMGCL) is deficient, an additional organic acid, 3MGC acid, is produced by intramolecular cyclization (4) and hydrolysis (5) of HMG CoA. Note: reaction 3,4 and 5 are non-enzyme-mediated chemical reactions.
By contrast, no defects in leucine catabolism pathway enzymes have been reported in subjects with secondary 3MGC aciduria and leucine loading does not lead to increased 3MGC acid excretion. Thus, in these IEMs, 3MGC aciduria does not arise from a block in the leucine catabolism pathway. In an effort to explain the biochemical origins of secondary 3MGC aciduria, various authors have proposed that it arises from aberrant cytosolic isoprenoid metabolism [5] or peroxisomal metabolism [6]. A lack of evidence supporting these models, however, has led to the proposal that secondary 3MGC aciduria is a byproduct of IEM-induced disruption of aerobic energy metabolism. Importantly, in nearly every known mutation associated with secondary 3MGC aciduria, the affected gene product localizes to mitochondria (Table 1). In several cases, IEMs that manifest secondary 3MGC aciduria are directly involved in electron transport chain (ETC) function, while others have an indirect connection. Herein, it is proposed that IEM-induced effects on the efficiency of aerobic respiration lead to inhibition of the TCA cycle. When this occurs in skeletal muscle, brain or cardiac tissue, the acetyl CoA diversion pathway is activated (Figure 2). In the first step, acetoacetyl CoA thiolase (T2 thiolase) condenses two equivalents of acetyl CoA to form acetoacetyl CoA. In this context, T2 thiolase functions in reverse of its normal direction in the fatty acid ß-oxidation pathway. Once formed, acetoacetyl CoA condenses with another equivalent of acetyl CoA to form HMG CoA in a reaction catalyzed by HMG CoA synthase 2. Subsequently, HMG CoA is dehydrated by AUH, generating trans-3MGC CoA plus H2O [7, 8].
Table 1.
Gene mutations associated with secondary 3MGC aciduria
| Gene | Gene product function | Location | Disorder / phenotype | Ref. |
|---|---|---|---|---|
| AGK | Lipid metabolism | Mitochondrial membrane | Sengers syndrome | 22,23 |
| ATP12 | Biosynthesis and assembly of ATP synthase | Mitochondria | Complex V deficiency, nuclear type 1 | 27 |
| ATP5E | Biosynthesis and assembly of ATP synthase | Mitochondria | Complex V deficiency, nuclear type 3 | 33 |
| CLPB | ATP-dependent chaperone | Inter-membrane space | Increased protein aggregation | 53 |
| CPS1 | Urea cycle enzyme | Matrix | Hyperammonemia | 49 |
| DNAJC19 | Molecular chaperone | Cristae membrane | DCMA syndrome | 64 |
| ECHS1 | Short chain enoyl coenzyme A hydratase | Matrix | Encephalopathy | 48 |
| HACE1 | E3 ubiquitin ligase | Multiple locations | Enhanced protein degradation | 76 |
| HTRA2 | Serine protease | Matrix | Caspase dependent apoptosis | 67 |
| LYRM4 | Fe/S cluster assembly | Matrix | ETC Complex assembly defects | 39,40 |
| m.3243A>G | tRNA | Matrix | MELAS syndrome | 44,45 |
| NFS1 | Cysteine desulfurase | Matrix | ETC complex assembly defects | 35 |
| OPA3 | unknown | Mitochondria | Costeff optic atrophy | 61,62 |
| POLG1 | mtDNA replication | Matrix | mtDNA deletions | 42,43 |
| QIL1/MIC13 | Cristae junction assembly / maintenance | Mitochondrial inner membrane | defective cristae morphology | 24,25 |
| SERAC1 | Phosphatidylglycerol remodeling | Mitochondrial outer membrane | MEGDEL syndrome | 19,20 |
| SUCLA2 | Succinyl-CoA ligase β-subunit | Matrix | Metabolic defects | 47 |
| TAZ | Cardiolipin transacylase | Cristae membrane | Barth syndrome | 14,15 |
| TIMM50 | Translocase subunit | Inner mitochondrial membrane | Impaired protein import | 57 |
| TMEM70 | Biogenesis of mitochondrial ATP synthase | Mitochondria | Complex V deficiency, nuclear type 2 | 29 |
Figure 2. The acetyl CoA diversion pathway.
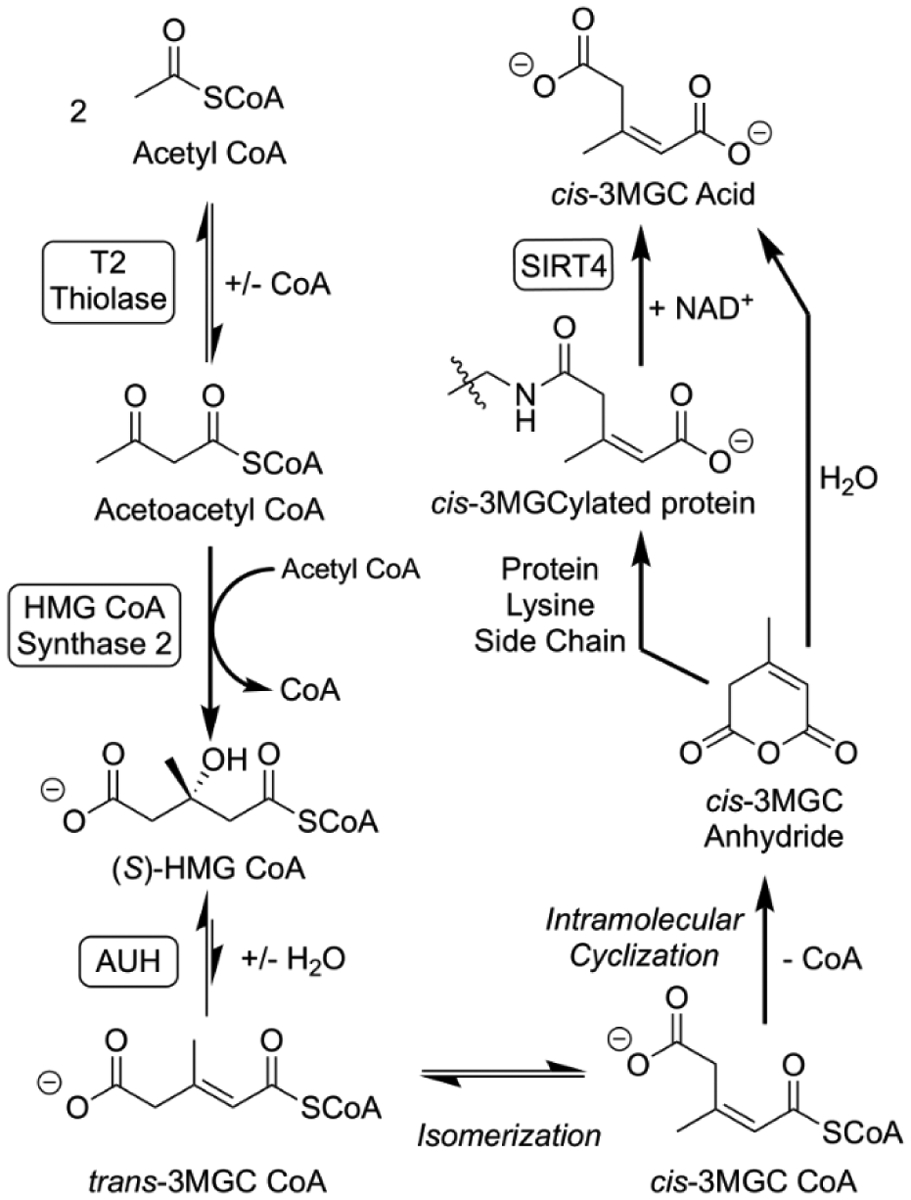
A sequence of three enzyme-mediated reactions that lead from acetyl CoA to to 3MGC CoA is depicted. Acetoacetyl CoA (T2) thiolase catalyzes the condensation of 2 acetyl CoA. Acetoacetyl CoA generated by this reaction reacts with another acetyl CoA in a second condensation reaction catalyzed by HMG CoA synthase 2. Subsequently AUH hydratase dehydrates HMG CoA to trans-3MGC CoA. Once formed, three non-enzymatic reactions follow, isomerization of trans-3MGC CoA to cis-3MGC CoA, intramolecular cyclization of cis-3MGC CoA to cis-3MGC anhydride, and hydrolysis to cis-3MGC acid. Alternatively, cis-3MGC anhydride can 3MGCylate protein lysine side chain amino groups. This latter product serves as a substrate for the NAD+-dependent deacylase, sirtuin 4 (SIRT4), which produces cis-3MGC acid as a reaction product.
The next part of the acetyl CoA diversion pathway involves a series of three non-enzymatic chemical reactions. As trans-3MGC CoA is generated via AUH, it isomerizes to cis-3MGC CoA. Importantly, cis- and trans- 3MGC CoA are quite different in terms of their chemical reactivity [9]. cis-3MGC CoA, but not trans-3MGC CoA, is capable of intramolecular cyclization to form cis-3MGC anhydride and CoA. This chemical reaction is known to occur among terminally carboxylated short-chain acyl CoAs [10]. Although trans-3MGC CoA is the only diastereomer generated during intermediary metabolism, steric constraints prevent formation of trans-3MGC cyclic anhydride. Thus, isomerization of trans-3MGC CoA to cis-3MGC CoA is an essential step in formation of cyclic 3MGC anhydride. Once formed, cis-3MGC anhydride is reactive and subject to hydrolytic cleavage, forming cis-3MGC acid. Alternatively, cyclic 3MGC anhydride can react with lysine side chain amino groups to 3MGCylate proteins [9]. When this occurs, 3MGCylated proteins can be deacylated through the action of the NAD+ requiring deacylase, sirtuin 4 [11]. Whereas cis-3MGC acid is destined for excretion, Jones et al [12] reported that 3MGC acids can also isomerize under physiological conditions, albeit very slowly. Overall, trans-3MGC CoA isomerization provides an explanation for the ~2:1 cis:trans ratio of 3MGC acid diastereomers in urine of subjects with 3MGC aciduria [13]. Thus, the acetyl CoA diversion pathway is comprised of 2 stages (enzyme-mediated and non-enzyme mediated) that, together, result in conversion of acetyl CoA to a mixture of cis- and trans-3MGC acids.
To better define specific IEMs that give rise to secondary 3MGC aciduria, the nature of these IEMs is described. In every case, 3MGC aciduria is one of several disparate, yet characteristic, phenotypic features observed. These include dilated cardiomyopathy, neutropenia, optic atrophy, lactic acidemia, muscle weakness and more. In general, IEMs known to manifest secondary 3MGC aciduria can be grouped into several categories, including a) genes that encode proteins involved in mitochondrial lipid metabolism and membrane composition; b) genes that encode protein components of ETC complexes; c) mutations that alter mitochondrial DNA; d) genes that encode matrix metabolic enzymes. A fifth group includes those IEMs that manifest secondary 3MGC aciduria but don’t fit neatly into these categories. By examining different IEMs that give rise to secondary 3MGC aciduria, insight into aberrant metabolic processes responsible for its production may be achieved.
II. IEMs that affect mitochondrial lipid metabolism / membrane structure
TAZ
Perhaps the best known secondary 3MGC aciduria, Barth Syndrome (OMIM #302060), is caused by mutations in the TAZ (OMIM *300394) gene (Table 1), which encodes the phospholipid transacylase, tafazzin [14]. Tafazzin is predominantly expressed in cardiac and skeletal muscle tissue and localizes to the inner mitochondrial membrane where it catalyzes remodeling of the unique anionic phospholipid, cardiolipin (CL). Through the action of taffazin, the heterogeneous acyl chain composition of nascent CL is remodeled to form tetralinoleoyl CL, a key component of cristae membranes [15]. CL’s cone shaped structure stabilizes the highly curved cristae membranes that house complexes of the ETC. Phenotypically, Barth Syndrome is characterized by dilated cardiomyopathy, neutropenia, muscle weakness, 3MGC aciduria and other features [16]. Lack of tafazzin activity affects the content and composition of CL, resulting in morphological changes in cristae membrane structure and, thereby, reduced ETC function [17]. Decreased aerobic respiration and ATP production is considered a root cause of dilated cardiomyopathy associated with this disorder. Another consequence of TAZ mutation-induced disruption of ETC function is a reduced ability to oxidize cofactors (NADH and FADH2) generated during oxidative metabolism of fuel molecules. When these reduced cofactors accumulate in the matrix space, metabolic flux through the TCA cycle declines [7]. Unable to enter the cycle, accumulating acetyl CoA is diverted to 3MGC acid and excreted in urine as a waste product.
SERAC1
The SERAC1 (Serine active site containing 1; OMIM *614725) gene encodes the eponymous phosphatidylglycerol remodeling enzyme, SERAC1. In addition to its acyl chain remodeling function, SERAC1 is involved in biosynthesis of the unique lipid, bis(monoacylglycerol)phosphate, which affects cholesterol trafficking [18]. Moreover, SERAC1 functions in phospholipid exchange at the mitochondria - endoplasmic reticulum interface [19]. Interestingly, disruption of SERAC1 activity also affects CL metabolism, consistent with PG’s role as a direct precursor of CL. Clinically, mutations in SERAC1 are causative for “MEGDEL syndrome” (3-methylglutaconic aciduria with deafness, encephalopathy and Leigh-like features) [20]. Given its function in lipid trafficking and metabolism, it is reasonable to consider that loss of SERAC1 activity will result in defects in the structure, organization and integrity of cristae membranes. Consistent with this, studies indicate that altered CL subspecies composition, oxidative phosphorylation defects, increased serum lactate and 3MGC aciduria are associated with mutations in SERAC1 [21].
AGK
The AGK gene encodes acylglycerol kinase (AGK; OMIM *610345), which phosphorylates diacylglycerol, forming phosphatidic acid, a key substrate in phospholipid biosynthesis. Deficiencies in AGK are causative for Sengers syndrome [22]. Curiously, loss of AGK activity results in decreased levels of adenine nucleotide translocator 1 (ANT1) in muscle mitochondria, although no mutations in the gene encoding ANT1 (SLC25A4; OMIM *103220) have been reported. Defects in AGK also affect fatty acid metabolism and glycogen accumulation in cardiac tissue. The finding that 70% of patients with mutations in AGK manifest 3MGC aciduria [23] is consistent with other phenotypic features associated with this disorder, including dilated cardiomyopathy and lactic acidemia.
C19orf70
The C19orf70 (chromosome 19 open reading frame 70; OMIM *616658) gene encodes QIL1 (also known as MIC13), a subunit of the mitochondrial contact site and cristae organizing system (MICOS) complex involved in formation / stabilization of cristae junctions. MICOS is a multi-subunit complex that modulates the placement, distribution and number of cristae in mitochondria. It is noteworthy that cristae junctions play a crucial role in oxidative phosphorylation by separating contents of the inner cristae space from the adjacent inter-membrane space. Defects in QIL1 result in decreased cristae membrane potential due to loss of cristae junctions and disruption of cristae morphology [24]. Insofar as the inner cristae space is the site where H+ accumulation generates the proton-motive force that drives oxidative phosphorylation, a breach in cristae junction integrity will result in unproductive dissipation of this proton gradient. Loss of membrane potential disrupts aerobic respiration and is consistent with the finding that subjects with mutations in QIL1 have increased levels of lactic acid and 3MGC aciduria [25]. Taken together, it may be considered that IEMs in C19orf70 result in a breakdown of cristae junction structure [26] which, in turn, leads to disruption of cristae morphology and diminished efficiency of oxidative phosphorylation. Under these conditions, it is expected that pyruvate generated via glycolysis will be converted to lactic acid while acetyl CoA, unable to undergo oxidative metabolism, will be diverted to 3MGC acid.
III. IEMs that affect ETC Complexes
ATPAF2
The IEM known as mitochondrial complex V deficiency nuclear type 1 is caused by homozygous mutations in the ATPAF2 (OMIM *608918) gene, which encodes ATP12, a protein that functions in biosynthesis and assembly of Complex V of the ETC (i.e. F1Fo ATPase). De Meirleir and coworkers reported that mutations in ATPAF2 that lead to a deficiency in the ATP12 protein correlate with elevated 3MGC acid levels in urine [27]. The net effect of a deficiency in ATP12 is an inability to assemble the extrinsic F1 domain of Complex V, thereby impairing ATP production efficiency via oxidative phosphorylation. Thus, it is anticipated that, during cellular respiration, mutation-induced disruption of Complex V structure / function leads to a diminished capacity of the ETC to oxidize NADH and FADH2 generated during oxidative fuel metabolism. As a result, TCA cycle activity is inhibited and acetyl CoA is diverted to 3MGC acid.
TMEM70
Mitochondrial Complex V deficiency nuclear type 2 is caused by homozygous or compound heterozygous mutations in the TMEM70 (OMIM *612418) gene. TMEM70 encodes “transmembrane protein 70” (TMEM70), which localizes to the inner mitochondrial membrane and functions in assembly of the F1 and Fo subunits of Complex V. Phenotypic features of this disorder include dysmorphia, hypertonia, cardiomyopathy, psychomotor retardation and 3MGC aciduria [28,29]. Recently, Sanchez-Caballero et al [30] reported that loss of TMEM70 leads to an accumulation of assembly intermediates, consistent with a block in the Complex V assembly pathway. Ultimately, defective TMEM70 function reduces Complex V activity, resulting in increased membrane potential, enhanced reactive oxygen species (ROS) production and diminished ATP production [31]. In addition, ultrastructural studies of tissues harboring mutations in TMEM70 indicate aberrant cristae morphology [32].
ATP5F1E
Mitochondrial Complex V deficiency nuclear type 3 (OMIM #614053) is caused by mutations in the ATP5F1E (OMIM *606153) gene, which encodes ATP5F1E, another subunit of Complex V. Mutations in ATP5F1E that result in Complex V deficiency manifest phenotypic features that include neonatal-onset hypotonia, lactic acidemia, hyperammonemia, hypertrophic cardiomyopathy and 3MGC aciduria [33].
NFS1
The NFS1 (OMIM *603485) gene encodes a cysteine desulfurase that catalyzes the first step of Fe/S cluster formation, converting cysteine to alanine and a sulfide intermediate [34]. The resulting inorganic sulfur binds to cysteine ligands that are supplied by Fe/S cluster scaffold protein. When complexed with iron, an Fe/S cluster is produced [35]. Once formed, Fe/S clusters are incorporated into proteins, most notably, components of ETC Complexes I, II and III. Mutations in NFS1 disrupt Fe/S cluster assembly, causing mitochondrial dysfunction in animal models [36,37]. Recently, Hershkovitz et al [35] reported a human subject harboring a homozygous NFS1 pathogenic variant. In this individual, serum and cerebrospinal fluid amino acid analysis indicated elevated levels of alanine and glycine while urine organic acid analysis revealed increased lactic acid and 3MGC aciduria.
LYRM4
The LYRM4 (OMIM *613311) gene encodes “iron-sulfur protein biogenesis, desulfurase-interacting protein 11” (Isd11), an 11 kDa mitochondrial protein that functions in Fe/S cluster assembly. Isd11 is a member of the “LYR” protein family, a recently identified superfamily characterized by small size (10–22 kDa), high positive charge, an invariant Phe residue and a Leu–Tyr–Arg (LYR) motif near the N terminus [38]. LYR proteins function as subunits or assembly factors for respiratory Complexes I, II, and III. Isd11 forms a tight complex with the cysteine desulfurase, NFS1 [39], and is required for Fe/S cluster biogenesis. Mutations in LYRM4 adversely affect Isd11 activity, resulting in disease that manifests as cardiorespiratory arrest, lactic acidemia and 3MGC aciduria [40]. Lim et al [41] reported that mutations in LYRM4 result in ETC complex deficiency, suggesting Isd11 functions in the early stages of Fe/S cluster assembly. Given this role, the finding that mutations in Isd11 manifest 3MGC aciduria is consistent with the role Fe/S clusters play in respiratory complex function. Moreover, ETC defects that result from mutations in LYRM4 fit with the concept that, as reduced cofactors (i.e. NADH /FADH2) accumulate, product inhibition of TCA cycle activity will result in diversion of acetyl CoA to 3MGC acid.
IV. IEMs affecting mitochondrial DNA
POLG1
The enzyme DNA polymerase subunit γ (POLG) is encoded by the POLG1 (OMIM *174763) gene. The only polymerase that functions in mitochondria, POLG exists as a trimeric complex comprised of a catalytic subunit and a dimeric accessory subunit [42]. The catalytic subunit contains three enzymatic activities, a DNA polymerase activity, a 3’–5’ exonuclease activity and a lyase activity required for base excision repair. Defects in POLG increase susceptibility to errors of replicated mitochondrial DNA which, in turn, adversely affect mitochondrial function. Wortmann et al [43] reported that a subset of patients harboring mutations in POLG1 manifest 3MGC aciduria. Patients with this phenotypic feature often present with other maladies, including encephalopathy (epilepsy, psychosis, depression) and liver failure.
tRNALeu (UUR)
A mutation in the transfer RNA leucine (tRNALeu(UUR)) coding region of mitochondrial DNA is associated with the disorder termed “mitochondrial encephalopathy, lactic acidosis and stroke-like episodes” (MELAS; OMIM #540000). The mutation, m.3243A>G (OMIM *590050), produces a MELAS disease phenotype. Of interest to the present discussion, Iwanicka-Pronicka et al [44] and Wortmann et al [45] reported that subjects with this mutation manifest 3MGC aciduria.
Pearson marrow-pancreas syndrome
Pearson marrow-pancreas syndrome (PS; OMIM #557000) is a rare mitochondrial disorder caused by single, large deletions (1–10 kbp) of mitochondrial DNA. The most common deletion, which occurs in about 20 percent of affected individuals, is 4.9 kbp in length. Whereas PS is associated with impaired respiratory chain complexes, clinical features of this disorder vary widely, depending in the specific nature of the mitochondrial DNA deletion. Of interest to the present discussion, Sato et al [46] reported a PS patient that developed pancytopenia, lactic acidemia, steatorrhea, insulin-dependent diabetes mellitus, liver dysfunction, Fanconi syndrome and 3MGC aciduria. These authors identified a 5.4 kbp deletion of mitochondrial DNA in this subject. Analysis revealed the activity of Complexes I and IV were markedly reduced in liver and muscle and mildly reduced in skin fibroblasts and heart. Importantly, among patients diagnosed with PS, only a subset manifest 3MGC aciduria. However, given the fact that not all subjects diagnosed with PS carry the same DNA deletion, it is not surprising that they manifest variable phenotypic features.
V. Metabolic enzymes
SUCLA2
The SUCLA2 (OMIM *603921) gene encodes the ß-subunit of succinyl CoA ligase, also known as ADP-forming succinyl-CoA synthetase. In the TCA cycle, succinyl CoA synthetase catalyzes the conversion of succinyl-CoA plus GDP to succinate, CoA and GTP. Based on this key metabolic function, it may be anticipated that defective succinyl CoA synthetase activity will impede TCA cycle activity, resulting in a buildup of acetyl CoA. In this case, it is anticipated that the acetyl CoA diversion pathway would generate 3MGC acid as a byproduct. Consistent with this, Carrozzo R et al [47] reported that mutations in SUCLA2 result in methyl malonic aciduria, lactic acidemia and 3MGC aciduria.
ECHS1
The ECHS1 (OMIM *602292) gene encodes for short-chain enoyl CoA hydratase (ECH), which catalyzes hydration of the double bond in 2-trans/cis-enoyl-CoA substrates. In mitochondrial metabolism, ECH plays an essential role in fatty acid ß-oxidation. Phenotypic features of this IEM include delayed motor and cognitive development, as well as metabolic abnormalities including elevated lactic acid levels in plasma and cerebrospinal fluid and transient 3MGC aciduria [48].
CPS1
The CPS1 (OMIM *608307) gene encodes carbamoyl phosphate synthase, a ligase located in mitochondria that functions in production of urea. Carbamoyl phosphate synthase functions to transfer NH4+ from glutamine or glutamate to bicarbonate, forming carbamoyl phosphate at the expense of 2 ATP. Carbamoyl phosphate then reacts with ornithine to form citrulline via ornithine transcarbamoylase. Subsequently, citrulline exits the mitochondria and is metabolized to yield urea and ornithine, which can react with carbamoyl phosphate to complete the cycle. Whereas Rokicki et al [49] detected 3MGC aciduria in 7 of 11 cases of CPS1 IEMs, additional insight is required to define the underlying metabolic basis of 3MGC acid production in this disorder.
VI. Other IEMs that affect mitochondrial function.
CLPB
The CLPB (OMIM *616254) gene encodes “caseinolytic peptidase B protein homolog” (ClpB), also known as suppressor of potassium transport defect 3. Studies indicate that ClpB is a mitochondrial AAA+ protein containing an N-terminal ankyrin-repeat domain and a C-terminal nucleotide-binding domain. Recently, Cupo and Shorter [50] reported that ClpB functions as a protein disaggregase, capable of solubilizing aggregated proteins in the inter-membrane space. This activity is achieved by coupling protein disaggregase activity to ATP hydrolysis. Of interest to the present discussion is the observation that mutations in CLPB are associated with 3MGC aciduria [51–54]. Other phenotypic features associated with mutations in CLPB include progressive encephalopathy, cataracts and neutropenia. Importantly, a correlation exists between the site of mutation, the extent of disaggregase activity disruption and disease severity [50]. In experiments designed to determine the effect of ClpB on substrate protein solubility, these authors compared the relative solubility of mitochondrial proteins in wild-type and ClpB knockout human HAP1 cells. Consistent with localization of ClpB to the inter-membrane space, when ClpB was absent, an increased proportion of proteins in the inner mitochondrial membrane and inter-membrane space were recovered in the insoluble fraction. Examination of specific proteins affected by loss of ClpB revealed that proteins involved in Ca2+ import, chaperone-mediated protein transport, protein insertion into the inner membrane and respiratory chain complex assembly, were prone to insolubility. Based on these findings, it is conceivable that IEMs in CLPB lead to defects in aerobic respiration that adversely affect TCA cycle function, resulting in diversion of acetyl CoA to 3MGC acid.
TIMM50
The TIMM50 (OMIM *607381) gene encodes a subunit of the translocase of the inner membrane (TIM23) complex. TIM23 functions to recognize proteins that possess a mitochondrial targeting signal and promote their transit into the mitochondrial matrix. Mutations in TIMM50 result in apoptosis via cytochrome c release [55]. Deficiency in TIM23 has also been shown to cause severe mitochondrial dysfunction by targeting key aspects of mitochondrial physiology, cristae morphology, ETC Complex assembly and mitochondrial respiratory capacity [56]. Consistent with mutation-induced disruption of energy metabolism, IEMs in TIMM50 have been reported to manifest 3MGC aciduria [57].
Duplication 5q
Zalopnik et al [58] published a case report of a 13-year-old boy harboring a duplication in chromosome 5 that manifested 3MGC aciduria. The duplicated region, 6.56 Mbp in length, contains 72 genes, including 4 genes [HINT1 (OMIM *601314), LYRM7 (*615831), UQCRQ (*612080), PPP2CA (*176915)] known to be associated with neurological and mitochondrial disease, respectively.
OPA3
Mutations in OPA3 (OMIM *606580) are causative for Costeff optic-atrophy syndrome, (OMIM #258501) characterized by early bilateral optic atrophy with decreased visual acuity, spastic paraparesis, mild ataxia and 3MGC aciduria [59,60]. Although the function of Opa3 remains unknown, experimental evidence on the pathogenicity of OPA3 mutations are consistent with mitochondrial pathology. Anikster et al [61] reported that Opa3 deficiency leads to a reduction in lipid utilization in adipose tissue. Subsequently, Wells et al [62] discovered that Opa3 functions to control thermogenesis and abdominal fat mass accumulation. The mutant form of Opa3 studied by these authors (L122P) was also associated with increased levels of 3MGC acid in plasma. More recently, Navein et al [63] found that L122P Opa3 is associated with disrupted mitochondrial function and impaired skeletal integrity. Localization of Opa3 to mitochondria, together with the fact that mutations in this protein manifest altered mitochondrial metabolism, is consistent with production of 3MGC acid with this disorder.
DNAJC19
Mutations in DNAJC19 (OMIM *608977), originally identified in the Canadian Dariusleut Hutterite population, are causative for dilated cardiomyopathy with ataxia (DCMA; OMIM #610198) syndrome [64], which is characterized by early onset dilated cardiomyopathy, non-progressive cerebellar ataxia, testicular dysgenesis, growth failure and 3MGC aciduria. The Dnajc19 protein [DnaJ heat shock protein family member c19] is homologous to yeast Pam18 / Tim14, a component of the mitochondrial machinery for import of nuclear-encoded proteins. Based on this homology, Davey et al [64] hypothesized that mutations in DNAJC19 cause DCMA by interfering with protein import. In other studies, Richter-Dennerlein et al. [65] reported that Dnajc19 interacts with prohibitin (PHB) proteins. PHB 1 and 2 assemble into large ring-like structures that serve a scaffold function in cristae membranes. In studies of the PHB interactome, Dnajc19 was identified as a binding partner of PHB complexes. Moreover, gene silencing of either DNAJC19 or PHB2 (OMIM *610704) resulted in impaired cell growth and defective cristae morphogenesis. Curiously, disruption of the interaction between Dnajc19 and PHB affected cardiolipin remodeling / acyl chain maturation, resulting in accumulation of cardiolipin species with altered acyl chains. These changes in cardiolipin are reminiscent of those observed in Barth Syndrome. Based on this, it has been proposed [65] that PHB / Dnajc19 containing membrane domains modulate tafazzin-mediated cardiolipin remodeling, thereby providing an explanation for the similar phenotypes observed in these two inherited cardiomyopathies. How these distinct protein components of cristae membranes (Dnajc19, tafazzin and PHB) regulate membrane structure and integrity is not clear, but their pronounced effects on cristae membrane morphology are likely to have a negative impact on ETC function. By extension, compromised mitochondrial energy metabolism and impaired ATP production capacity will ensue, setting the stage for development of cardiomyopathy. Likewise, ETC defects will ultimately impede TCA cycle activity, resulting in acetyl CoA diversion to the dead-end product, 3MGC acid.
HTRA2
The HTRA2 (OMIM *606441) gene encodes a member of the “high-temperature requirement” (HtrA) family of oligomeric serine proteases that possess a trypsin-like protease domain and one or more “PDZ” type interaction domains. HtrA2 participates in the cell death pathway, binding to the apoptosis inhibitory protein, baculoviral IAP repeat-containing 4 [66]. Deficiencies in HtrA2 are associated with severe neurodegenerative effects, abnormal mitochondria, 3MGC aciduria and increased sensitivity to apoptosis [67–69].
HACE1
The HACE1 (OMIM *610876) gene encodes Hace1 (HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1). Given its function as an E3 ubiquitin ligase, it is not surprising that Hace1 has been implicated in numerous processes, including Golgi biogenesis [70], tumor suppression [71], response to oxidative stress [72,73], autophagy [74] and targeting Ras-related C3 botulinum toxin substrate 1 for ubiquitination [75]. Ugarteburu et al reported on a patient harboring a mutation in HACE1 who presented with brain atrophy, psychomotor retardation, and 3MGC aciduria [76]. In studies of fibroblasts from this subject, the authors observed high levels of lipid peroxidation, increased mitochondrial oxidative stress markers, diminished response to oxidative damage and a reduction in the mitophagic flux. Of interest, Ehrnhoefer et al [77] reported that genetic ablation of hace1 in a mouse model of Huntington’s disease accelerated motor deficits and exacerbated cognitive and psychiatric phenotypes. These authors also reported that mutant huntingtin protein expression combined with hace1 gene disruption caused a deficit in astrocyte mitochondrial respiration. Thus, it appears that the far-ranging effects of this E3 ubiquitin ligase include deleterious effects on mitochondrial respiration. As such, detection of 3MGC aciduria in HACE1 IEMs is consistent with the current paradigm.
VII. Discussion
The present review describes 20 discrete IEMs that manifest secondary 3MGC aciduria as a phenotypic feature (Table 1). A curious aspect of secondary 3MGC aciduria is that no mutations in leucine metabolic enzymes exist. The fact that, in human intermediary metabolism, 3MGC CoA is found only in this pathway strongly suggests secondary 3MGC aciduria arises from a unique biochemical route. Diagnostically, it is relatively easy to distinguish primary from secondary 3MGC aciduria. For example, in primary 3MGC aciduria, the amount of 3MGC acid excreted increases in response to leucine loading [4]. Likewise, in primary 3MGC aciduria, upstream pathway intermediates give rise to additional products, such as 3-hydroxyisovaleric acid [78] which are not observed in secondary 3MGC aciduria.
In secondary 3MGC aciduria, it is proposed that IEMs that affect mitochondrial energy metabolism, either directly or indirectly, can lead to an accumulation of acetyl CoA. This occurs because disturbances in ETC function, cristae membranes or other factors, lead to a decrease in the efficiency of TCA cycle activity. When this occurs in muscle or brain (tissues that rely on acetyl CoA oxidation for energy production), acetyl CoA has limited metabolic options. When IEMs affect the predominant metabolic pathway (acetyl CoA oxidation via the TCA cycle), acetyl CoA tends to accumulate in the matrix. At this point, the acetyl CoA diversion pathway converts acetyl CoA to HMG CoA via mass action, utilizing two known mitochondrial matrix enzymes, T2 thiolase and HMG CoA synthase 2. These enzymes catalyze sequential Claisen condensations between two equivalents of acetyl CoA and between acetoacetyl CoA and acetyl CoA, respectively, to yield HMG CoA. Once HMG CoA is formed, it becomes a substrate for AUH. Unlike the role of this enzyme in the leucine degradation pathway, in this case AUH functions in the opposite direction, dehydrating HMG CoA to yield trans-3MGC CoA. Given that the equilibrium constant for AUH strongly favors the hydrated product [79], it is difficult to explain why the presumed precursor of 3MGC acid, 3MGC CoA, would accumulate instead of HMG CoA. Recent results have revealed that this occurs because trans-3MGC CoA isomerizes to cis-3MGC CoA [9]. When this occurs, the AUH substrate / product ratio is altered and, in order to re-establish equilibrium, more HMG CoA is dehydrated to trans-3MGC CoA. Once formed, cis-3MGC CoA is susceptible to intramolecular cyclization, forming cis-3MGC anhydride and CoA. The anhydride can then be hydrolyzed to cis-3MGC acid (Figure 2). Thus, directionality of the acetyl CoA diversion pathway relies on a molecular sink that leads to depletion of the HMG CoA pool. In addition, these non-enzymatic reactions provide an explanation why cis-3MGC acid is the predominant diastereomer recovered in urine of subjects with 3MGC aciduria [9, 13, 80]. It is worth mentioning that the enzyme that produces trans-3MGC CoA in the leucine degradation pathway, 3-methylcrotonyl CoA carboxylase, is unidirectional [81]. Thus, when trans-3MGC CoA is produced via the acetyl CoA diversion pathway, it cannot proceed further up the leucine degradation pathway.
In the present review, evidence supporting the conclusion that the acetyl CoA diversion pathway is responsible for secondary 3MGC aciduria is presented. To investigate this, we examined 20 discrete IEMs that manifest secondary 3MGC aciduria as a phenotypic feature. In studying these IEMs, a key question was whether the causative mutation, directly or indirectly, affects mitochondrial energy metabolism. The results obtained indicate that, in nearly every case, these discrete IEMs impact aspects of aerobic respiration, leading to dampening of ETC function and decreased acetyl CoA metabolism via the TCA cycle. We identified five categories of IEM associated with secondary 3MGC aciduria. In most cases, a specific gene product of known function in mitochondria is affected. In category 1, proteins involved in cristae membrane structure and function were identified. These include tafazzin, SERAC1, acylglycerol kinase and QIL1. Cristae membranes exist as narrow, elongated invaginations of the inner boundary membrane. Cristae membranes are cylindrical in shape [15] such that a continuously curved membrane envelops the inner cristae space, the site where hydrogen ion accumulation generates the proton motive force that drives ADP phosphorylation. The unique shape and function of cristae membranes requires specialized enzymes and proteins to form and maintain cristae integrity. IEMs in genes that encode phospholipid biosynthetic enzymes lead to defective membrane composition while mutations in protein components of the MICOS complex affect cristae junction formation and maintenance. Furthermore, although it appears that the inner cristae space and the inter-membrane space are confluent with each other, the MICOS complex, and other cristae membrane associated proteins, function as a diffusion barrier, preventing diffusion of inner cristae space contents into the inter-membrane space and vice versa. When this aspect of cristae membranes is disrupted or lost, the electrochemical gradient dissipates and ATP production efficiency decreases.
A second category of IEMs that give rise to secondary 3MGC aciduria directly affect protein components of the ETC. These proteins are embedded in cristae membranes such that their activity relies on the structural integrity of this membrane. When IEMs lead to deficiencies in ETC Complex components, electron transport is less efficient and reduced cofactors generated via the TCA cycle activity are not oxidized by Complex I / II. This leads to a buildup of NADH in the matrix space and inhibition of TCA cycle activity. Thus, IEMs that interfere with ETC function prevent acetyl CoA metabolism via the TCA cycle, resulting in its diversion to 3MGC acid.
When IEMs affect mitochondrial DNA polymerase, mutant tRNAs may be produced or deletions in mitochondrial DNA may occur, leading to a broad range of potential complications. In addition, nuclear gene duplication can disrupt normal metabolic processes and impair aerobic respiration. IEMs in metabolic enzymes that localize to mitochondria are also associated with 3MGC aciduria. These include the TCA cycle enzyme succinyl CoA synthetase, the urea cycle enzyme, carbamoyl phosphate synthase and metabolic enzyme, short-chain enoyl CoA hydratase.
In addition to these examples, several other protein deficiencies exist that do not fit neatly into the categories described above. These include mutations in CLPB, DNAJC19, OPA3, HACE1 and others. IEMs in these genes are known to adversely affect cristae membrane structure, protein stability or oxidative response. The important point is that each of these proteins plays a direct or ancillary role in mitochondrial energy metabolism and, when deficiencies arise, deleterious consequences can include aberrant metabolic processes, including the diversion of acetyl CoA to 3MGC acid.
Researchers have pondered the metabolic origins of 3MGC acid in secondary 3MGC aciduria for the past 30 years. Originally, it was proposed that 3MGC acid arises by shunting isoprene moieties from cytosol to mitochondria [5,78]. A basic flaw in this proposed pathway is that no explanation exists why 3MGC CoA arising in this manner would not be metabolized via the leucine degradation pathway. Also, as exemplified in the present investigation, IEMs that give rise to secondary 3MGC aciduria predominantly affect mitochondrial energy metabolism. Another recent study proposed that 3MGC acid is generated via peroxisomal metabolism [6]. This proposed route, however, fails to reconcile the fact that IEMs associated with 3MGC aciduria affect mitochondrial function.
Although the acetyl CoA diversion pathway has not previously been proposed in human metabolism, this pathway is known to exist in various microorganisms. Indeed, the biosynthetic route to iron chelating siderophores, iso-odd chain fatty acids and polyketide / nonribosomal peptide products employ this biochemical process. In these organisms, acetyl CoA is converted to 3MGC CoA, which then serves an intermediate in formation of larger structures [82]. As progress is made toward understanding the metabolic basis for 3MGC aciduria, the potential utility of this easily detected organic acid will be realized. Given that secondary 3MGC aciduria arises as a result of compromised mitochondrial energy metabolism, it is conceivable that evaluation of 3MGC acid levels in subjects harboring IEMs can provide a window into physiological processes that underlie disease manifestation. Toward that end, clinical trials on subjects with different IEMs that give rise to this phenotype may reveal a correlation between mitochondria impairment, physical exertion and 3MGC acid production.
Highlights.
3-methylglutaconic (3MGC) aciduria occurs in a number of inborn errors of metabolism
Mutations associated with secondary 3MGC aciduria affect oxidative fuel metabolism
Inhibition of TCA cycle activity leads to diversion of acetyl CoA to 3MGC acid
This pathway involves enzymatic and non-enzymatic chemical reactions
Acknowledgements
This work was support by a grant from the National Institutes of Health (R37 HL64159).
Abbreviations:
- AUH
3-methylglutaconyl CoA hydratase
- 3MGC
3-methylglutaconic
- IEM
Inborn error of metabolism
- HMG
3-hydroxy, 3-methylglutaryl
- ETC
electron transport chain
- PHB
prohibitin
- SERAC1
Serine active site containing 1
- AGK
acylglycerol kinase
- ANT1
adenine nucleotide translocator 1
- MICOS
mitochondrial contact site and cristae organizing system
- TMEM70
transmembrane protein 70
- Isd11
iron-sulfur protein biogenesis, desulfurase-interacting protein 11
- POLG
DNA polymerase subunit γ
- MELAS
mitochondrial encephalopathy, lactic acidosis and stroke-like episodes
- PS
Pearson marrow-pancreas syndrome
- ECH
short-chain enoyl CoA hydratase
- ClpB
caseinolytic peptidase B protein homolog
- TIM23
subunit of the translocase of the inner membrane
- DCMA
dilated cardiomyopathy with ataxia syndrome
- Dnajc19
DnaJ heat shock protein family member c19
- PHB
prohibitin
- HtrA2
high temperature requirement A serine protease
- Hect1
HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1
- OMIM
Online Mendelian Inheritance in Man
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests statement: The authors declare they have no competing interests.
References
- [1].IJlst L, Loupatty FJ, Ruiter JP, Duran M, Lehnert W, Wanders RJ, 3-Methylglutaconic aciduria type I is caused by mutations in AUH, Am. J. Hum. Genet 71 (2002) 1463–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pié J, López-Viñas E, Puisac B, Menao S, Pié A, Casale C, Ramos FJ, Hegardt FG, Gómez-Puertas P, Casals N, Molecular genetics of HMG-CoA lyase deficiency, Mol. Genet. Metab 92 (2007) 198–209. [DOI] [PubMed] [Google Scholar]
- [3].Santarelli F, Cassanello M, Enea A, Poma F, D’Onofrio V, Guala G, Garrone G, Puccinelli P, Caruso U, Porta F, Spada M, A neonatal case of 3-hydroxy-3-methylglutaric-coenzyme A lyase deficiency, Ital. J. Pediatr 39 (2013) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wortmann SB, Kluijtmans LA, Sequeira S, Wevers RA, Morava E, Leucine Loading Test is Only Discriminative for 3-Methylglutaconic Aciduria Due to AUH Defect, JIMD Rep. 16 (2014) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kelley RI, Cheatham JP, Clark BJ, Nigro MA, Powell BR, Sherwood GW, Sladky JT, Swisher WP, X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria, J. Pediatr 119 (1991) 738–747. [DOI] [PubMed] [Google Scholar]
- [6].Vamecq J, Papegay B, Nuyens V, Boogaerts J, Leo O, Kruys V, Mitochondrial dysfunction, AMPK activation and peroxisomal metabolism: A coherent scenario for non-canonical 3-methylglutaconic acidurias, Biochimie. 168 (2020) 53–82. [DOI] [PubMed] [Google Scholar]
- [7].Su B, Ryan RO, Metabolic biology of 3-methylglutaconic acid-uria: a new perspective, J. Inherit. Metab. Dis 37 (2014) 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jones DE, Perez L, Ryan RO, 3-Methylglutaric acid in energy metabolism, Clin. Chim. Acta 502 (2020) 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Young R, Jones DE, Diacovich L, Witkowski A, Ryan RO, trans-3-methylglutaconyl CoA isomerization-dependent protein acylation, Biochem. Biophys. Res. Commun 534 (2021) 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wagner GR, Bhatt DP, O’Connell TM, Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva OR, Stevens RD, Grimsrud PA, Hirschey MD, A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation, Cell Metab. 25 (2017) 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, Wagner GR, Thompson JW, Madsen AS, Green MF, Sivley RM, Ilkayeva OR, Stevens RD, Backos DS, Capra JA, Olsen CA, Campbell JE, Muoio DM, Grimsrud PA, Hirschey MD (2017) SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion, Cell Metabol. 25, 838e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jones DE, Ricker JD, Geary LM, Kosma DK, Ryan RO, Isomerization of 3-methylglutaconic acid, JIMD Rep. 58 (2020) 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Engelke UF, Kremer B, Kluijtmans LA, van der Graaf M, Morava E, Loupatty FJ, Wanders RJ, Moskau D, Loss S, van den Bergh E, Wevers RA, NMR spectroscopic studies on the late onset form of 3-methylglutaconic aciduria type I and other defects in leucine metabolism, NMR Biomed. 19 (2006) 271–278. [DOI] [PubMed] [Google Scholar]
- [14].Schlame M, Cardiolipin remodeling and the function of tafazzin, Biochim. Biophys. Acta 1831, (2013) 582–588. [DOI] [PubMed] [Google Scholar]
- [15].Ikon N, Ryan RO, Cardiolipin and mitochondrial cristae organization, Biochim. Biophys. Acta Biomembr 1859 (2017) 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ikon N, Ryan RO, Barth syndrome: connecting cardiolipin to cardiomyopathy, Lipids. 52 (2017) 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI, Steward CG, Barth syndrome, Orphanet J. Rare Dis 12 (2013) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arnal-Levron M, Chen Y, Greimel P, Calevro F, Gaget K, Riols F, Batut A, Bertrand-Michel J, Hullin-Matsuda F, Olkkonen VM, Delton I, Luquain-Costaz C, Bis(monoacylglycero)phosphate regulates oxysterol binding protein-related protein 11 dependent sterol trafficking, Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864 (2019) 1247–1257. [DOI] [PubMed] [Google Scholar]
- [19].Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, Kulik W, Lammens M, Christin C, Kluijtmans LA, Rodenburg RJ, Nijtmans LG, Grünewald A, Klein C, Gerhold JM, Kozicz T, van Hasselt PM, Harakalova M, Kloosterman W, Barić I, Pronicka E, Ucar SK, Naess K, Singhal KK, Krumina Z, Gilissen C, van Bokhoven H, Veltman JA, Smeitink JA, Lefeber DJ, Spelbrink JN, Wevers RA, Morava E, de Brouwer AP, Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness, Nat Genet. 44 (2012) 797–802. [DOI] [PubMed] [Google Scholar]
- [20].Finsterer J, Scorza FA, Fiorini AC, Scorza CA, MEGDEL syndrome, Pediatr. Neurol 110 (2020) 25–29. [DOI] [PubMed] [Google Scholar]
- [21].Tort F, García-Silva MT, Ferrer-Cortès X, Navarro-Sastre A, Garcia-Villoria J, Coll MJ, Vidal E, Jiménez-Almazán J, Dopazo J, Briones P, Elpeleg O, Ribes A, Exome sequencing identifies a new mutation in SERAC1 in a patient with 3-methylglutaconic aciduria, Mol. Genet. Metab 110 (2013) 73–77. [DOI] [PubMed] [Google Scholar]
- [22].Mayr JA, Haack TB, Graf E, Zimmermann FA, Wieland T, Haberberger B, Superti-Furga A, Kirschner J, Steinmann B, Baumgartner MR, Moroni I, Lamantea E, Zeviani M, Rodenburg RJ, Smeitink J, Strom TM, Meitinger T, Sperl W, Prokisch H, Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome, Am. J. Hum. Genet 90 (2012) 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wortmann SB, Espeel M, Almeida L, Reimer A, Bosboom D, Roels F, de Brouwer AP, Wevers RA, Inborn errors of metabolism in the biosynthesis and remodeling of phospholipids, J. Inherit. Metab. Dis 38, (2015) 99–110. [DOI] [PubMed] [Google Scholar]
- [24].Wang LJ, Hsu T, Lin HL, Fu CY. Drosophila MICOS knockdown impairs mitochondrial structure and function and promotes mitophagy in muscle tissue, Biol Open. 2 (2020) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zeharia A, Friedman JR, Tobar A, Saada A, Konen O, Fellig Y, Shaag A, Nunnari J, Elpeleg O, Mitochondrial hepato-encephalopathy due to deficiency of QIL1/MIC13 (C19orf70), a MICOS complex subunit, Eur. J. Hum. Genet 24 (2016) 1778–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guarani V, McNeill EM, Paulo JA, Huttlin EL, Fröhlich F, Gygi SP, Van Vactor D, Harper JW, QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology, Elife. 21 (2015) 4:e06265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].De Meirleir L, Seneca S, Lissens W, De Clercq I, Eyskens F, Gerlo E, Smet J, Van Coster R, Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12, J. Med. Genet 41 (2004) 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shchelochkov OA, Li FY, Wang J, Zhan H, Towbin JA, Jefferies JL, Wong LJ, Scaglia F, Milder clinical course of Type IV 3-methylglutaconic aciduria due to a novel mutation in TMEM70, Mol. Genet. Metab 101 (2010) 282–285. [DOI] [PubMed] [Google Scholar]
- [29].Hirono K, Ichida F, Nishio N, Ogawa-Tominaga M, Fushimi T, Feichtinger RG, Mayr JA, Kohda M, Kishita Y, Okazaki Y, Ohtake A, Murayama K, Mitochondrial complex deficiency by novel compound heterozygous TMEM70 variants and correlation with developmental delay, undescended testicle, and left ventricular noncompaction in a Japanese patient: A case report, Clin. Case Rep 7 (2019) 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sánchez-Caballero L, Elurbe DM, Baertling F, Guerrero-Castillo S, van den Brand M, van Strien J, van Dam TJP, Rodenburg R, Brandt U, Huynen MA, Nijtmans LGJ, TMEM70 functions in the assembly of complexes I and V. Biochim. Biophys. Acta Bioenerg 1861 (2020) 148202. [DOI] [PubMed] [Google Scholar]
- [31].Havlíčková Karbanová V, Cížková Vrbacká A, Hejzlarová K, Nůsková H, Stránecký V, Potocká A, Kmoch S, Houštěk J, Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation, Biochim. Biophys. Acta 1817 (2012) 1037–1043. [DOI] [PubMed] [Google Scholar]
- [32].Diodato D, Invernizzi F, Lamantea E, Fagiolari G, Parini R, Menni F, Parenti G, Bollani L, Pasquini E, Donati MA, Cassandrini D, Santorelli FM, Haack TB, Prokisch H, Ghezzi D, Lamperti C, Zeviani M, Common and novel TMEM70 mutations in a cohort of Italian patients with mitochondrial encephalocardiomyopathy, JIMD Rep 15 (2015) 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mayr JA, Havlícková V, Zimmermann F, Magler I, Kaplanová V, Jesina P, Pecinová A, Nusková H, Koch J, Sperl W, Houstek J, Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit, Hum Mol Genet. 19 (2010) 3430–3439. [DOI] [PubMed] [Google Scholar]
- [34].Kispal G, Csere P, Prohl C, Lill R (1999) The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 18, 3981–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hershkovitz T, Kurolap A, Tal G, Paperna T, Mory A, Staples J, Brigatti KW, Regeneron Genetics Center; Gonzaga-Jauregui C, Dumin E, Saada A, Mandel H, Baris Feldman H, A recurring NFS1 pathogenic variant causes a mitochondrial disorder with variable intra-familial patient outcomes, Mol. Genet. Metab. Rep 26 (2020), 100699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fosset C, Chauveau MJ, Guillon B, Canal F, Drapier JC, Bouton C, RNA silencing of mitochondrial m-Nfs1 reduces Fe-S enzyme activity both in mitochondria and cytosol of mammalian cells, J. Biol. Chem 281, (2006) 25398–25406. [DOI] [PubMed] [Google Scholar]
- [37].Biederbick A, Stehling O, Rosser R, Niggemeyer B, Nakai Y, Elsasser HP, Lill R, Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation, Mol. Cell. Biol 26 (2006) 5675–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Angerer H, Eukaryotic LYR proteins interact with mitochondrial protein complexes, Biology (Basel). 4 (2015) 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Adam AC, Bornhövd C, Prokisch H, Neupert W, Hell K, The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria, EMBO J. 25 (2006) 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Coelho MP, Correia J, Dias A, Nogueira C, Bandeira A, Martins E, Vilarinho L, Iron-sulfur cluster ISD11 deficiency (LYRM4 gene) presenting as cardiorespiratory arrest and 3-methylglutaconic aciduria, JIMD Rep. 49 (2019) 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lim SC, Friemel M, Marum JE, Tucker EJ, Bruno DL, Riley LG, Christodoulou J, Kirk EP, Boneh A,DeGennaro CM, Springer M, Mootha VK, Rouault TA, Leimkühler S, Thorburn DR, Compton AG, Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes, Hum. Mol. Genet 22 (2013) 4460–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bekheirnia MR, Zhang W, Eble T, Willis A, Shaibani A, Wong LJ, Scaglia F, Dhar SU, POLG mutation in a patient with cataracts, early-onset distal muscle weakness and atrophy, ovarian dysgenesis and 3-methylglutaconic aciduria, Gene. 499 (2012) 209–212. [DOI] [PubMed] [Google Scholar]
- [43].Wortmann SB, Rodenburg RJ, Jonckheere A, de Vries MC, Huizing M, Heldt K, van den Heuvel LP, Wendel U, Kluijtmans LA, Engelke UF, Wevers RA, Smeitink JA, Morava E, Biochemical and genetic analysis of 3-methylglutaconic aciduria type IV: a diagnostic strategy, Brain. 132 (2009) 136–146. [DOI] [PubMed] [Google Scholar]
- [44].Iwanicka-Pronicka K, Pollak A, Skórka A, Lechowicz U, Pajdowska M, Furmanek M, Rzeski M, Korniszewski L, Skarżyński H, Ploski R, Postlingual hearing loss as a mitochondrial 3243A>G mutation phenotype, PLoS One. 7 (2012) e44054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wortmann SB, Champion MP, van den Heuvel L, Barth H, Trutnau B, Craig K, Lammens M, Schreuder MF, Taylor RW, Smeitink JA, Wevers RA, Rodenburg RJ, Morava E, Mitochondrial DNA m.3242G>A mutation, an under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction?, Eur. J. Med. Genet 55 (2012a) 552–556. [DOI] [PubMed] [Google Scholar]
- [46].Sato T, Muroya K, Hanakawa J, Iwano R, Asakura Y, Tanaka Y, Murayama K, Ohtake A, Hasegawa T, Adachi M, Clinical manifestations and enzymatic activities of mitochondrial respiratory chain complexes in Pearson marrow-pancreas syndrome with 3-methylglutaconic aciduria: a case report and literature review, Eur. J. Pediatr 174 (2015) 1593–1602. [DOI] [PubMed] [Google Scholar]
- [47].Carrozzo R, Verrigni D, Rasmussen M, de Coo R, Amartino H, Bianchi M, Buhas D, Mesli S, Naess K, Born AP, Woldseth B, Prontera P, Batbayli M, Ravn K, Joensen F, Cordelli DM, Santorelli FM, Tulinius M, Darin N, Duno M, Jouvencel P, Burlina A, Stangoni G, Bertini E, Redonnet-Vernhet I, Wibrand F, Dionisi-Vici C, Uusimaa J, Vieira P, Osorio AN, McFarland R, Taylor RW, Holme E, Ostergaard E, Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients. J. Inherit. Metab. Dis 39 (2016) 243–252. [DOI] [PubMed] [Google Scholar]
- [48].Huffnagel IC, Redeker EJW, Reneman L, Vaz FM, Ferdinandusse S, Poll-The BT, Mitochondrial encephalopathy and transient 3-methylglutaconic aciduria in ECHS1 deficiency: long-term follow-up, JIMD Rep. 39 (2018) 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rokicki D, Pajdowska M, Trubicka J, Thong MK, Ciara E, Piekutowska-Abramczuk D, Pronicki M, Sikora R, Haidar R, Ołtarzewski M, Jabłońska E, Muthukumarasamy P, Sthaneswar P, Gan CS, Krajewska-Walasek M, Carrozzo R, Verrigni D, Semeraro M, Rizzo C, Taurisano R, Alhaddad B, Kovacs-Nagy R, Haack TB, Dionisi-Vici C, Pronicka E, Wortmann SB, 3-Methylglutaconic aciduria, a frequent but underrecognized finding in carbamoyl phosphate synthetase I deficiency, Clin. Chim. Acta 471 (2017) 95–100. [DOI] [PubMed] [Google Scholar]
- [50].Cupo RR, Shorter J, Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations, Elife. 9 (2020) e55279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Saunders C, Smith L, Wibrand F, Ravn K, Bross P, Thiffault I, Christensen M, Atherton A, Farrow E, Miller N, Kingsmore SF, Ostergaard E, CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria, Am. J. Hum. Genet 96 (2015) 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kanabus M, Shahni R, Saldanha JW, Murphy E, Plagnol V, Hoff WV, Heales S, Rahma S, Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation, J. Inherit. Metab. Dis 38 (2015) 211–219. [DOI] [PubMed] [Google Scholar]
- [53].Wortmann SB, Ziętkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, Muntau AC, Rakovic A, Renkema GH, Rodenburg RJ, Strom TM, Meitinger T, Rubio-Gozalbo ME, Chrusciel E, Distelmaier F, Golzio C, Jansen JH, van Karnebeek C, Lillquist Y, Lücke T, Õunap K, Zordania R, Yaplito-Lee J, van Bokhoven H, Spelbrink JN, Vaz FM, Pras-Raves M, Ploski R, Pronicka E, Klein C, Willemsen MA, de Brouwer AP, Prokisch H, Katsanis N, Wevers RA, CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder, Am. J. Hum. Genet 96 (2015) 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Capo-Chichi JM, Boissel S, Brustein E, Pickles S, Fallet-Bianco C, Nassif C, Patry L, Dobrzeniecka S, Liao M, Labuda D, Samuels ME, Hamdan FF, Vande Velde C, Rouleau GA, Drapeau P, Michaud JL, Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria, J. Med. Genet 52 (2015) 303–11. [DOI] [PubMed] [Google Scholar]
- [55].Guo Y, Cheong N, Zhang Z, De Rose R, Deng Y, Farber SA, Fernandes-Alnemri T, Alnemri ES, Tim50, a component of the mitochondrial translocator, regulates mitochondrial integrity and cell death, J. Biol. Chem 279 (2004) 24813–24825. [DOI] [PubMed] [Google Scholar]
- [56].Tort F, Ugarteburu O, Texidó L, Gea-Sorlí S, García-Villoria J, Ferrer-Cortès X, Arias A, Matalonga L, Gort L, Ferrer I, Guitart-Mampel M, Garrabou G, Vaz FM, Pristoupilova A, Rodríguez MIE, Beltran S, Cardellach F, Wanders RJ, Fillat C, García-Silva MT, Ribes A, Mutations in TIMM50 cause severe mitochondrial dysfunction by targeting key aspects of mitochondrial physiology, Hum. Mutat 40 (2019) 1700–1712. [DOI] [PubMed] [Google Scholar]
- [57].Shahrour MA, Staretz-Chacham O, Dayan D, Stephen J, Weech A, Damseh N, Pri Chen H, Edvardson S, Mazaheri S, Saada A; NISC Intramural Sequencing, Hershkovitz E, Shaag A, Huizing M, Abu-Libdeh B, Gahl WA, Azem A, Anikster Y, Vilboux T, Elpeleg O, Malicdan MC (2017) Mitochondrial epileptic encephalopathy, 3-methylglutaconic aciduria and variable complex V deficiency associated with TIMM50 mutations. Clin. Genet 91, 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zapolnik P, Sykut-Cegielska J, Pyrkosz A, Coincidence of 3-methylglutaconic aciduria and duplication 5q - a case report and literature review, Acta Biochim. Pol 67 (2020) 263–266. [DOI] [PubMed] [Google Scholar]
- [59].Gaier ED, Sahai I, Wiggs JL, McGeeney B, Hoffman J, Peeler CE, Novel homozygous OPA3 mutation in an Afghani family with 3-methylglutaconic aciduria type III and optic atrophy, Ophthalmic Genet. 40 (2019) 570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lam C C, Gallo LK, Dineen R, Ciccone C, Dorward H, Hoganson GE, Wolfe L, Gahl WA, Huizing M, Two novel compound heterozygous mutations in OPA3 in two siblings with OPA3-related 3-methylglutaconic aciduria, Mol. Genet. Metab. Rep 1 (2014) 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Anikster Y, Kleta R, Shaag A, Gahl WA, Elpeleg O, Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or Costeff optic atrophy syndrome): identification of the OPA3 gene and its founder mutation in Iraqi Jews, Am. J. Hum. Genet 69 (2001) 1218–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wells T, Davies JR, Guschina IA, Ball DJ, Davies JS, Davies VJ, Evans BA, Votruba M, Opa3, a novel regulator of mitochondrial function, controls thermogenesis and abdominal fat mass in a mouse model for Costeff syndrome, Hum Mol Genet 21 (2012) 4836–4844. [DOI] [PubMed] [Google Scholar]
- [63].Navein AE, Cooke EJ, Davies JR, Smith TG, Wells LH, Ohazama A, Healy C, Sharpe PT, Evans SL, Evans BA, Votruba M, Wells T, Disrupted mitochondrial function in the Opa3L122P mouse model for Costeff Syndrome impairs skeletal integrity, Hum. Mol. Genet 25 (2016) 2404–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP, Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition, J. Med. Genet 43 (2006) 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Richter-Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, Decker T, Lamkemeyer T, Rugarli EI, Langer T, DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling, Cell Metab. 20 (2014) 158–171. [DOI] [PubMed] [Google Scholar]
- [66].Vande Walle L, Lamkanfi M, Vandenabeele P, The mitochondrial serine protease HtrA2/Omi: an overview, Cell Death Differ. 15 (2008) 453–60. [DOI] [PubMed] [Google Scholar]
- [67].Oláhová M, Thompson K, Hardy SA, Barbosa IA, Besse A, Anagnostou M, White K, Davey T, Simpson MA, Champion M, Enns G, Schelley S, Lightowlers RN, Chrzanowska-Lightowlers ZMA, McFarland R, Deshpande C, Bonnen PE, Taylor RW, Pathogenic variants in HTRA2 cause an early-onset mitochondrial syndrome associated with 3-methylglutaconic aciduria, J. Inherit. Metab. Dis 40 (2017) 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mandel H, Saita S, Edvardson S, Jalas C, Shaag A, Goldsher D, Vlodavsky E, Langer T, Elpeleg OJ, Deficiency of HTRA2/Omi is associated with infantile neurodegeneration and 3-methylglutaconic aciduria, Med. Genet 53 (2016) 690–696. [DOI] [PubMed] [Google Scholar]
- [69].Kovacs-Nagy R, Morin G, Nouri MA, Brandau O, Saadi NW, Nouri MA, van den Broek F, Prokisch H, Mayr JA, Wortmann SB, HTRA2 Defect: A recognizable inborn error of metabolism with 3-methylglutaconic aciduria as discriminating feature characterized by neonatal movement disorder and epilepsy-report of 11 patients, Neuropediatrics. 49 (2018) 373–378. [DOI] [PubMed] [Google Scholar]
- [70].Tang D, Xiang Y, De Renzis S, Rink J, Zheng G, Zerial M, Wang Y, The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle, Nat. Commun 2 (2011) 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Castillo-Lluva S, Tan CT, Daugaard M, Sorensen PH, Malliri A, The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation, Oncogene. 32 (2013) 1735–1742. [DOI] [PubMed] [Google Scholar]
- [72].Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, Leprivier G, Somasekharan SP, Barokas A, Deng Y, Tang T, Mathers J, Cetinbas N, Daugaard M, Kwok B, Li L, Carnie CJ, Fink D, Nitsch R, Galpin JD, Ahern CA, Melino G, Penninger JM, Hayden MR, Sorensen PH, HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response, Proc. Natl. Acad. Sci. USA 111 (2014) 3032–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S, Castillo-Lluva S, Rotblat B, Li L, Malliri A, Lemichez E, Mettouchi A, Berman JN, Penninger JM, Sorensen PH, Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes, Nat. Commun 4 (2013) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, Xu X, Wang H, Yang M, Liu X, Fan L, Chen S, Zhou J, Sun Y, Ruan K, Cheng S, Komatsu M, White E, Li L, Ji H, Finley D, Hu R, Ubiquitylation of autophagy receptor optineurin by HACE1 activates selective autophagy for tumor suppression, Cancer Cell. 26 (2014) 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Torrino S, Visvikis O, Doye A, Boyer L, Stefani C, Munro P, Bertoglio J, Gacon G, Mettouchi A, Lemichez E, The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1, Dev. Cell 21 (2011) 959–965. [DOI] [PubMed] [Google Scholar]
- [76].Ugarteburu O, Sánchez-Vilés M, Ramos J, Barcos-Rodríguez T, Garrabou G, García-Villoria J, Ribes A, Tort F, Physiopathological Bases of the Disease Caused by HACE1 Mutations: Alterations in Autophagy, Mitophagy and Oxidative Stress Response, J. Clin. Med 9 (2020) 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ehrnhoefer DE, Southwell AL, Sivasubramanian M, Qiu X, Villanueva EB, Xie Y, Waltl S, Anderson L, Fazeli A, Casal L, Felczak B, Tsang M, Hayden MR, HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo, Hum. Mol. Genet 27 (2018) 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wortmann SB, Kluijtmans LA, Engelke UF, Wevers RA, Morava E, The 3-methylglutaconic acidurias: what’s new?, J. Inherit. Metab. Dis 35 (2012) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mack M, Schniegler-Mattox U, Peters V, Hoffmann GF, Liesert M, Buckel W, Zschocke J, Biochemical characterization of human 3-methylglutaconyl-CoA hydratase and its role in leucine metabolism, FEBS J. 273 (2006) 2012–2022. [DOI] [PubMed] [Google Scholar]
- [80].Iles RA, Jago JR, Williams SR, Chalmers RA, 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency studied using 2-dimensional proton nuclear magnetic resonance spectroscopy, FEBS Lett. 203 (1986) 49–53. [DOI] [PubMed] [Google Scholar]
- [81].Apitz-Castro R, Rehn K, Lynen F, Beta methylcrotonyl-CoA-carboxylase. Crystallization and some physical properties, Eur. J. Biochem 16 (1970) 71–79. [DOI] [PubMed] [Google Scholar]
- [82].Ikon N, Ryan RO, On the origin of 3-methylglutaconic acid in disorders of mitochondrial energy metabolism, J. Inherit. Metab. Dis 39 (2016) 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]