

Abstract
Reliable methods to quantify dynamic signaling changes across diverse pathways are needed to better understand the effects of disease and drug treatment in cells and tissues but are presently lacking. Here, we present SigPath, a targeted mass spectrometry (MS) assay that measures 284 phosphosites in 200 phosphoproteins of biological interest. SigPath probes a broad swath of signaling biology with high throughput and quantitative precision. We applied the assay to investigate changes in phospho‐signaling in drug‐treated cancer cell lines, breast cancer preclinical models, and human medulloblastoma tumors. In addition to validating previous findings, SigPath detected and quantified a large number of differentially regulated phosphosites newly associated with disease models and human tumors at baseline or with drug perturbation. Our results highlight the potential of SigPath to monitor phosphoproteomic signaling events and to nominate mechanistic hypotheses regarding oncogenesis, response, and resistance to therapy.
Keywords: breast cancer, CPTAC, medulloblastoma, post‐translational modifications, targeted mass spectrometry
Subject Categories: Cancer, Proteomics, Signal Transduction
SigPath is a targeted, quantitative mass spectrometry assay that measures 284 phosphosites spanning 200 phosphoproteins with high throughput and quantitative precision across a broad swath of signaling biology of known interest.
Introduction
Cellular processes including signal transduction, cell cycle progression, and response to DNA damage, among many others, are regulated through the addition or removal of phosphate from the amino acids serine, threonine, and tyrosine. In keeping with this, aberrant phospho‐signaling is a hallmark of many diseases including cancer. For example, genetic disruption of the tumor suppressors PTEN and APC leads to pathologic levels of phosphorylated AKT and ß‐catenin respectively, while oncogenic activation of ABL and RAS leads to aberrant phosphorylation in CRKL or MEK, respectively. Dysregulated kinases and phosphatases have thus become important targets for therapeutic development. This in turn has motivated the desire to quantitatively monitor phosphorylation events to determine the cellular or organismal activity of such inhibitors. Unfortunately, due to the limited ability to robustly quantify hundreds of phosphorylation events, most drug discovery programs in this area have followed single phosphorylation events as the marker of pharmacodynamic activity. As a result, paradoxical activation of RAF isoforms as a consequence of BRAF inhibitors (Hatzivassiliou et al, 2010; Poulikakos et al, 2010), or feedback upstream pathway activation resulting from mTOR inhibitors, was missed until well after the relevant molecules were in clinical trials or beyond (Shi et al, 2005).
The Cancer Cell Line Encyclopedia project (CCLE), in addition to characterizing genome, transcriptome, and methylome alterations (Barretina et al, 2012; Ghandi et al, 2019), has sought to characterize the metabolome and proteome across hundreds of cancer cell lines (Li et al, 2019; Nusinow et al, 2020). An initial attempt in the phosphoproteome space was also made using Reverse Phase Protein Arrays (Li et al, 2017); however, the sparse availability of phosphoantibodies that are highly reliable in detecting phosphorylation events on the protein arrays limits the broader application of this approach. Thus, to begin to develop high‐complexity quantitative phosphoprotein assays for use in both characterizing cell lines and clinical samples and to enable much deeper pharmacodynamic assessment of therapeutics, we set out to develop robust mass spectrometry‐based phospho‐assay sets.
Mass spectrometry (MS)‐based proteomics has led to the discovery of the majority of the over 200,000 known human phosphosites (Phosphosite.org; Hornbeck et al, 2015). Many laboratories, but especially those associated with the Clinical Proteomics Tumor Analysis Consortium (Rodriguez et al, 2021), have elaborated deep, high‐quality phosphopeptide, and proteome datasets (Zhang et al, 2014, 2016; Mertins et al, 2016; Chen et al, 2017a; Huang et al, 2017; Archer et al, 2018; Vasaikar et al, 2019; Dou et al, 2020; Gillette et al, 2020; Krug et al, 2020; Satpathy et al, 2020). The deepscale proteomic methods used in the CPTAC studies detect 30,000–45,000 distinct phosphosites in each sample studied, and quantitative chemical labeling provides relative quantification of each site across samples. However, one drawback of this approach is the lack of uniform detection of any given phosphosite across an entire sample cohort, a technical artifact caused by stochastic sampling of the analytes introduced into the MS system, especially those in low abundance, and the extreme complexity of the samples analyzed.
Targeted MS in the forms of multiple reaction monitoring (MRM, also referred to as selected reaction monitoring, SRM) and parallel reaction monitoring (PRM) is now widely used for highly multiplexed, quantitative measurement of proteins in blood, cells, and tissues (Keshishian et al, 2009; Kuhn et al, 2009; Picotti & Aebersold, 2012; Rebecca et al, 2014; Soste et al, 2014; Gallien et al, 2015; Abelin et al, 2016; Chen et al, 2017b; Manes & Nita‐Lazar, 2018; Whiteaker et al, 2018; Huttenhain et al, 2019; Sperling et al, 2019; Eshghi et al, 2020; Pino et al, 2020). All variants of the approach begin with targeted selection in the mass spectrometer of the intact, ionized peptides of interest followed by fragmentation of each peptide precursor to produce product ions that, together with the mass of the intact peptide, are used to identify and quantify that peptide. In its most specific and precise form, heavy isotope‐labeled synthetic peptides are added at known concentration to verify that the correct peptide is being measured and to improve accuracy of the relative quantification of the target peptide. The use of this technology to measure post‐translationally modified peptides is less common. Targeted MS assays have largely been developed for purposes of verification; however, if such an assay queries a large number of targets, it can also be viewed as a discovery method.
In contrast to multiplexed antibody methods, MS‐based targeted analysis of peptides and modified peptides, including phosphopeptides, can, in principle, be configured to quantify any phosphosite of interest in any organism and scaled to measure many hundreds of peptides in a single measurement cycle of a few hours by liquid chromatography–tandem mass spectrometry (LC‐MS/MS; Burgess et al, 2014). Soste and coworkers developed targeted MS assays to 152 phosphosites and 157 proteins in yeast that were culled from the literature to develop what they called “sentinel markers'' to give biological insights (Soste et al, 2014). The methods have been extended to mammalian systems in several recent studies to detect and quantify on the order of 100 phosphosites in a single 1‐ to 2‐h analysis (Abelin et al, 2016; Kennedy et al, 2016). These studies employed TiO2 or immobilized metal affinity chromatography (IMAC) to enrich phosphopeptides from cells followed by analysis of the resulting mixture by targeted MS using heavy stable isotope‐labeled (SIL) phosphopeptides for confident detection and quantification. Kennedy et al. measured phosphosites relevant to DNA damage response, while Abelin et al. assayed a set of moderate‐to‐high‐abundance phosphopeptides known to be modulated in non‐uniform ways by a panel of drugs in a range of cell lines. The targets were selected based on ease of detection in a small number of initial chemical or genomic perturbations in cell lines, not on their potential biological relevance. Recently, Stopfer et al. developed and applied a targeted MS assay for several hundred phosphotyrosine (pY) sites and were able to consistently detect and quantify 165 endogenous pY peptides in colorectal cancer tumor samples (Stopfer et al, 2021).
Here, we present the development and application of SigPath, one of the largest phosphosite assay panels to date applicable to mammalian cells and human tissues. This quantitative, targeted MS‐based assay measures 284 phosphosites in 200 cancer‐relevant target proteins spanning many pathways. Phosphosites were selected by cancer biologists based on known or presumed relevance to cancer disease or treatment and through discovery proteomics efforts leading to the SigPath set of 298 phosphopeptides. This unique set is purposely designed to probe a broad swath of signaling biology in a single measurement rather than focusing on a single pathway. Importantly, the panel can be extended to measure other phosphosites in additional pathways as desired. Here, we demonstrate the utility of the assay through a range of applications in drug‐treated cell lines, preclinical models of breast cancer and human medulloblastoma tumor samples.
Results
Selection of phosphosites for assay development and assay construction
The majority of the phosphosites were nominated by cancer biologists in our institutions and supplemented with frequently modulated phosphosites observed in our discovery proteomic experiments (Fig EV1A). In an effort to further characterize the signaling and physiological state of human tumor samples, we set out to capture nodes of biological pathways known to be modulated via phosphorylation. Critical kinase cascades, often hyperactivated in tumors, make up the backbone of the assay set. These include the MAPK, PI3K, PKC, SRC, and JAK signaling pathways as well as both receptor and non‐receptor tyrosine kinases. Both critical activation sites of the kinases themselves and their relevant downstream substrates were targeted. These kinase cascades often culminate in the modulation of transcription factor circuits, so transcriptional nodes within the FOXO, STAT, NFKB, TGFB, and Wnt pathways were also selected. Lastly, to capture physiological cell states, protein phosphosites were selected to provide readouts on DNA damage, cell cycle arrest, apoptosis, spindle checkpoint activation, hypoxia, autophagy, cell stress, and epithelial‐to‐mesenchymal transition. The rationale for specific target selection and pathway information for the selected sites is presented in Datasets EV1 and EV2, respectively. While these sites were selected based on relevance to cancer cell‐autonomous phenotypes, the associated pathways constitute central signaling nodes and should therefore be valuable when applied to a variety of experimental paradigms.
Figure EV1. Development of SigPath assay.
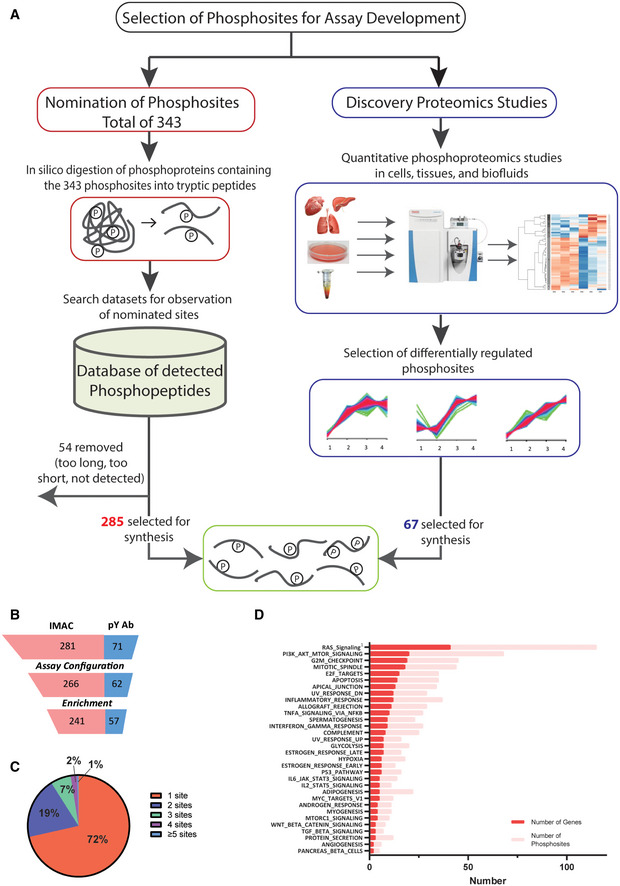
-
AProcess for selecting phosphosites and phosphopeptides for SigPath assay development. Majority of phosphosites were nominated by experts, then converted into tryptic peptides, searched against existing datasets at the Broad for detection of them in MS data (see Materials and Methods section). One‐fourth of the phosphosites were included based on them being differentially regulated in quantitative phosphoproteomic studies (Mertins et al, 2014). Once finalized [C13, N15], stable isotope‐labeled versions of the peptides were synthesized for the assay.
-
BAssay configuration and testing statistics of SigPath. Twenty‐four out of 352 peptides failed the assay configuration due to their LC or MS characteristics, while another 30 failed during the pY Ab or IMAC enrichment step. Final SigPath assay targets 298 phosphopeptides with 284 phosphosites.
-
CPie graph showing range of phosphosites per protein in the assay panel. 71% of the proteins are represented by only one phosphopeptide, 19% by two phosphopeptides. The remaining varies from 3 to 9 phosphopeptides.
-
DPathways represented by SigPath in MSigDB Hallmark pathway category. To be included in the plot, a pathway had to have at least 5% coverage, or be represented by a minimum of three proteins and five phosphosites in the assay. Both, number of genes (red) and phosphosites (pink) are shown on the plot. 1Included from MSigDB WikiPathway pathway category.
SigPath assay development
Assay development, configuration, and quality assessment, as well as workflow development (Figs EV1A and 1A), are described in Materials and Methods. Briefly, spectral libraries were generated on a QE or QE plus mass spectrometer using synthetic, SIL peptides, and transitions selected and optimized on a TSQ Quantiva Triple Quadrupole Mass Spectrometer. The final assay begins with proteolytic digestion of the sample, followed by spiking of SIL peptides into digested sample and subsequent phosphopeptide enrichment steps. We first perform phosphotyrosine antibody (pY Ab) capture, followed by immobilized metal affinity chromatography (IMAC) on the flow‐through of pY Ab capture. Phosphotyrosine Ab and IMAC eluents were analyzed using an LC‐MRM/MS method on two‐ and three‐hour gradients, respectively. The overall success rate for detection of the targeted heavy phosphopeptides was 85% (Fig EV1B). Twenty‐four out of 352 phosphopeptides failed assay configuration due to either their instability in solution or their poor behavior on LC or MS, while an additional 30 peptides failed during the pY Ab or IMAC enrichment steps (Dataset EV1). It is noteworthy that of the 37 phosphopeptides that were included in the assay panel but lacked prior experimental observation in our datasets (mostly pY‐containing), we were able to successfully configure assays for 24. The final working assay contains 298 phosphopeptides representing 284 phosphosites and 200 phosphoproteins (Table 1 and Dataset EV1). In addition, 178 of the 298 phosphopeptides (60%) are fully conserved between humans and mouse; therefore, this portion of the assay can be applied to mouse samples. The SigPath panel predominantly consists of singly phosphorylated peptides, with only 8 doubly phosphorylated phosphopeptides included. The majority of phosphoproteins (142/200) are represented by a single tryptic phosphopeptide containing a single phosphosite (Fig EV1C).
Figure 1. SigPath assay workflow and pathway coverage.
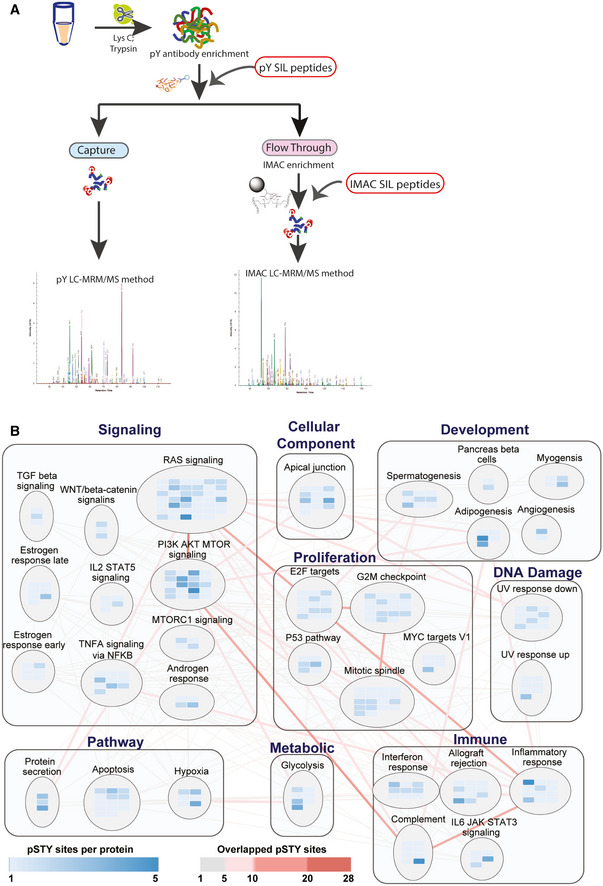
-
AIn the full SigPath workflow, the heavy stable isotope‐labeled (SIL) pY peptide set is spiked into the digested sample and endogenous and spiked SIL peptides enriched using pY antibody. A portion of the flow‐through from the pY enrichment is then spiked with the IMAC set of SIL peptides and enriched by IMAC. Both, pY Ab and IMAC‐captured samples are analyzed on the MS using pY and IMAC LC‐MRM/MS methods, respectively (see Materials and Methods).
-
BMSigDB Hallmark gene sets and process categories (Liberzon et al, 2015) represented by SigPath. To be included in the plot, a pathway had to have at least 5% coverage, or be represented by a minimum of three proteins and five phosphosites in the assay. The RAS signaling pathway from WikiPathway (Martens et al, 2021) is also included in the plot. Each rectangle assembles gene sets in the same process category. Gene set is shown in circles in which blue colored rectangles refer to the proteins represented in the assay. Shades of blue indicate the number of phosphosites per protein in the assay. Edges show overlapping proteins and phosphosites between the different gene sets. Overlap of 1–5 phosphosites is indicated with gray lines, whereas overlap of more than 5 phosphosites is shown with red lines at increasing intensity. The larger the overlap, the more intense is the shade of red.
Table 1.
List of all the proteins in the final SigPath assay panel
| HUGO‐approved symbol | Number of phosphosites | HUGO‐approved symbol | Number of phosphosites | HUGO‐approved symbol | Number of phosphosites | HUGO‐approved symbol | Number of phosphosites |
|---|---|---|---|---|---|---|---|
| ABI1 | 1 | FGFR1 | 1 | MAPRE1 | 1 | RBM39 | 1 |
| ABL1 | 1 | FGFR2 | 1 | MARCKS | 2 | RBM7 | 1 |
| ACIN1 | 1 | FGFR3 | 3 | MARVELD2 | 1 | RELA | 1 |
| AKT1 | 2 | FGFR4 | 1 | MAST2 | 1 | RET | 1 |
| AKT1S1 | 1 | FLT3 | 1 | MCL1 | 2 | RICTOR | 2 |
| AKT2 | 1 | FOXO1 | 2 | MDM2 | 1 | RIPK1 | 2 |
| AKT3 | 1 | FOXO3 | 2 | MET | 1 | ROS1 | 1 |
| ALK | 3 | FRS2 | 3 | MITF | 1 | RPS27 | 1 |
| ANLN | 1 | FRS3 | 1 | MOB1A | 1 | RPS6 | 3 |
| APLP2 | 1 | GAB1 | 1 | MPZL1 | 1 | RPS6KA1 | 4 |
| ARAF | 4 | GLYR1 | 1 | MTOR | 3 | RPS6KA2 | 1 |
| ATM | 1 | GRB2 | 1 | MYC | 1 | RPS6KA3 | 3 |
| AURKA | 1 | GSK3A | 1 | MYCBP2 | 1 | RPS6KB1 | 2 |
| AURKB | 1 | GSK3B | 2 | NF2 | 1 | RREB1 | 1 |
| AXL | 1 | GTF2I | 1 | NFKB1 | 1 | SHB | 1 |
| Bad | 2 | HECTD1 | 1 | NFKB1 | 1 | SHC1 | 2 |
| BCL2 | 1 | HGS | 1 | NFKBIA | 3 | SMAP | 1 |
| BCL2L11 | 1 | HIF1A | 2 | NTRK1 | 1 | SMARCA4 | 1 |
| BRAF | 2 | HMGN1 | 1 | NTRK2 | 1 | SMC4 | 1 |
| BRCA1 | 1 | HNRNPA3 | 1 | NUCKS1 | 1 | SOCS3 | 1 |
| BTK | 2 | HSPBL2 | 1 | PAK1 | 1 | SOS1 | 2 |
| BUB1 | 1 | IGF1R | 2 | PALLD | 1 | SPRY1 | 1 |
| CAMSAP2 | 1 | INSR | 1 | PBRM1 | 1 | SPRY2 | 3 |
| CDC20 | 1 | IRF3 | 1 | PDPK1 | 1 | SPRY4 | 1 |
| CDK1 | 2 | IRS1 | 2 | PEA15 | 1 | SRC | 1 |
| CFAP97 | 1 | IRS2 | 3 | PGAM1 | 1 | STAT3 | 2 |
| Chek1 | 2 | JAK1 | 1 | PHIP | 1 | STAT5A | 1 |
| CIC | 1 | JAK2 | 1 | PIK3R1 | 3 | STK3 | 1 |
| CPSF6 | 1 | JUN | 1 | PLCB3 | 1 | STK39 | 1 |
| CRK | 1 | JUND | 1 | PLCG1 | 1 | STK4 | 2 |
| CTNNB1 | 2 | KIF2C | 1 | PLCG2 | 1 | STMN1 | 1 |
| DDI2 | 1 | KIF4A | 1 | PLK1 | 2 | SUFU | 1 |
| DNAH5 | 1 | KIT | 1 | PPP2R5B | 1 | SYK | 1 |
| DOCK5 | 1 | KMT2A | 1 | PRKAA1 | 1 | SYNPO2 | 1 |
| DOT1L | 1 | KSR1 | 2 | PRKACA | 1 | TAZ | 1 |
| DYRK2 | 1 | LAG3 | 1 | PRKCA | 1 | TP53 | 2 |
| EGFR | 4 | LATS1 | 2 | PRKCB | 2 | TP53BP1 | 1 |
| EIF2A | 2 | LATS1 | 2 | PRKCB | 1 | TPX2 | 1 |
| EIF2AK4 | 1 | LATS2 | 1 | PRKCD | 1 | TSC2 | 1 |
| EIF4EBP1 | 3 | LCK | 1 | PRKCQ | 1 | TTC39B | 1 |
| EML4 | 1 | LYN | 1 | PRKCZ | 1 | UBE2J1 | 1 |
| EPAS1 | 1 | MAP2K1 | 2 | PRKD2 | 1 | ULK1 | 1 |
| EPHA2 | 1 | MAP2K4 | 1 | PRKDC | 1 | VAV1 | 1 |
| EPS8 | 1 | MAP3K14 | 2 | PTEN | 1 | VEPH1 | 1 |
| ERBB2 | 2 | MAP3K7 | 1 | PTK2 | 3 | VIM | 1 |
| ERBB3 | 2 | MAPK1 | 2 | PTPN11 | 4 | XIAP | 1 |
| ERBB4 | 1 | MAPK14 | 2 | RAB11B | 1 | YAP1 | 2 |
| ERH | 1 | MAPK3 | 2 | RAF1 | 9 | YTHDC1 | 1 |
| ERRFI | 1 | MAPK8 | 2 | RAF1 | 1 | ZAP70 | 1 |
| EZH2 | 2 | MAPK9 | 1 | RB1 | 3 | ZNF638 | 1 |
Table includes HUGO gene names and number of phosphosites per protein in the assay panel.
Pathway representation in the final assay panel
Pathways represented by the final SigPath panel were assessed using the canonical databases in the Molecular Signatures Database (MSigDB; Kanehisa & Goto, 2000; Pico et al, 2008; Schaefer et al, 2009; Liberzon et al, 2011, 2015; Jassal et al, 2020). The panel represents a spectrum of cancer‐relevant biology spanning signal transduction, cell proliferation, apoptosis, and the immune system (Dataset EV2). Figures 1B and EV1D illustrate Hallmark gene sets and processes represented in SigPath with the addition of the Ras pathway from WikiPathways (Martens et al, 2021).
SigPath assay configuration and evaluation
Evaluation of sensitivity and reproducibility of the assay were performed in a mixture of five cancer cell lines (OVCAR, Meljuso, H3122, PC9, A375) to maximize detection of endogenous peptides. Phosphotyrosine Ab enrichment was performed with two peptide input amounts (1 and 5 mg), whereas IMAC enrichment was performed with five different input amounts ranging from 0.05 to 1 mg. Fifty‐eight and 96 endogenous light peptides were detected in this experiment with median coefficient of variation (CV) of less than 20% after pY Ab and IMAC enrichments, respectively. Phosphotyrosine Ab enrichment with only 1 mg input achieved quantification of 52 (93%) of the detected peptides, while IMAC enrichment achieved quantification of 74 (77%) of the detected peptides in the lowest input, 0.05 mg sample (Fig EV2A and B).
Figure EV2. Evaluation of the SigPath assay.
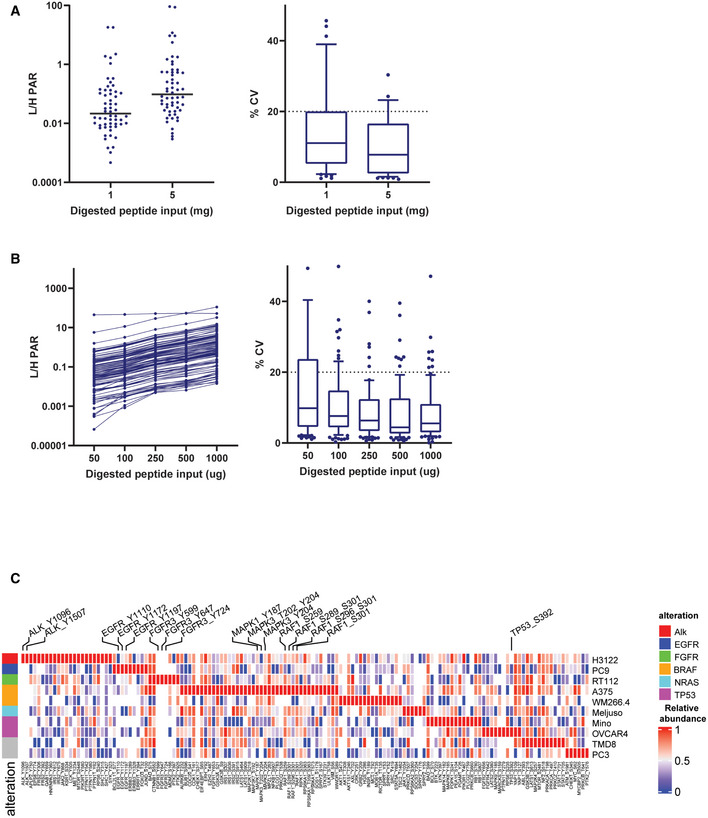
-
AAverage light/heavy peak area ratio and %CV of the three replicates for all quantified peptides in titration curve experiment after pY Ab enrichment. Plot on the left shows the average values for all the quantified peptides along with a line representing the median value at 1 and 5 mg input protein level. The plot on the right shows the spread of %CVs at 1 and 5 mg input protein. The box represents interquartile range (IQR) with the lower, central, and the upper bands representing 25th percentile (Q1), median, and 75th percentile (Q3), respectively. The whiskers extend from 10 to 90 percentile of the data.
-
BAverage light/heavy peak area ratio and %CV of the three replicates for all quantified peptides in titration curve experiment after IMAC enrichment. Plot on the left shows the average values for all the quantified peptides along with a line representing the median value at 0.05, 0.1, 0.25, 0.5, and 1 mg input protein level. The plot on the right shows the spread of %CVs at all the input protein levels. The box represents interquartile range (IQR) with the lower, central, and upper bands representing 25th percentile (Q1), median, and 75th percentile (Q3), respectively. The whiskers extend from 10 to 90 percentile of the data.
-
CHeat map showing relative abundances of all detected phosphosites across the 10 cell lines. Labeled are phosphosites on known driver genes in these cell lines. One replicate of each samples was processed and analyzed for this experiment. The heat map was generated using Morpheus online tool, the data are median‐MAD normalized, and colors are relative across rows, from row min to row max.
We initially evaluated the assay in ten cell lines representing various cancer types (lung, B‐cell lymphoma, mantle cell lymphoma, prostate, ovarian, bladder, and melanoma) and genetic contexts using 250 of the 298 phosphopeptides in SigPath (Dataset EV3). Each cell line was selected to maximize the potential for detecting endogenous signals from the phosphosites in the panel that are typically at low abundance. Endogenous versions of 89–125 peptides were detected in each individual cell line with a total of 143/250 (57%) detected across all 10 cell lines. Detection of endogenous phosphopeptides reflected the genetic context of the cell lines (Fig EV2C). For example, phosphosites/peptides derived from ALK and FGFR were only detected in lung adenocarcinoma (LUAD) cell line H3122 (driven by ALK fusion) and bladder carcinoma cell line RT112 (driven by FGFR3 fusion), respectively, while in another LUAD cell line PC9, which harbors an activating EGFR mutation (exon 19 deletion) higher levels of EGFR phosphosites were detected.
Application of the assay to evaluate effects of drug treatment in cancer cell lines
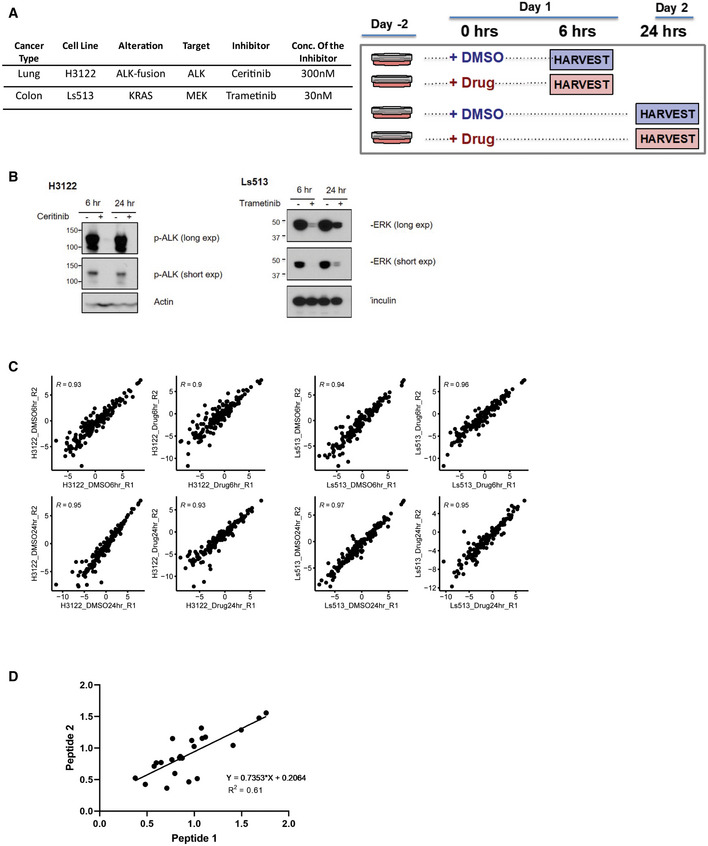
To investigate the utility of the SigPath assay to detect and quantify acute perturbations in cell lines, we next treated LUAD H3122 cells with the ALK inhibitor ceritinib (Friboulet et al, 2014; Shaw et al, 2014) and KRAS mutant Ls513 colorectal cancer cells (CRC) with the MEK inhibitor trametinib (Falchook et al, 2012; Flaherty et al, 2012; Fig EV3A and B). Cells were treated with the respective inhibitors or DMSO for 6 and 24 h, and two process replicates per condition were analyzed using the SigPath assay as described above. A total of 162 and 155 endogenous phosphopeptides were detected in H3122 (LUAD) and Ls513 (CRC) cell lines, respectively (Dataset EV4). Excellent reproducibility was achieved for replicates, with Pearson correlation > 0.9 for all samples in both cell lines (Fig EV3C). Furthermore, using both LUAD and CRC cell line perturbagen data, we investigated correlation of site quantification as measured by 2 different peptides for the subset of 12 sites for which such data was available. The measured levels of the sites differed by a maximum of 30% from peptide 1 to peptide 2, while the Pearson correlation of the drug/DMSO ratio was 0.6 (Fig EV3D).
Figure EV3. Application of the assay to cancer cell line perturbagen experiments.
-
AExperimental design and details for drug treatment studies in H3122 and Ls513 cell lines. Table contains details about the cell lines as well as the inhibitor and concentration of it used. Cells were treated either with the inhibitor or DMSO for 6 and 24 h. Two process replicates were collected for each sample.
-
BInhibition of pALK and pERK signaling in established human cell lines. Immunoblot analyses of cultured H3122 lung adenocarcinoma cells (on the left) treated with ALK inhibitor Ceritinib (+) or DMSO (−) for 6 and 24 h. Antibodies recognizing the phosphorylated 1507‐Tyrosine site of the ALK protein and the Actin protein (loading control) were used. Immunoblot analyses of cultured Ls513 colorectal carcinoma cells (on the right) treated with KRAS inhibitor Trametinib (+) or DMSO (−) for 6 and 24 h. Antibodies recognizing the phosphorylated Thr 185/Tyr 18 sites of the ERK1/ERK2 proteins and the vinculin protein (loading control) were used.
-
CScatter plot of two process replicates of Log2 light/heavy PAR of each sample group. Shown on each plot is the Pearson correlation coefficient.
-
DScatter plot of light/heavy peak area ratio of peptide 1 and peptide 2 measuring the same site for 12 of the sites measured in H3122 and Ls513 perturbagen experiments. X‐axis and y‐axis represent light/heavy peak area ratios. Shown on the graph is Pearson correlation coefficient.
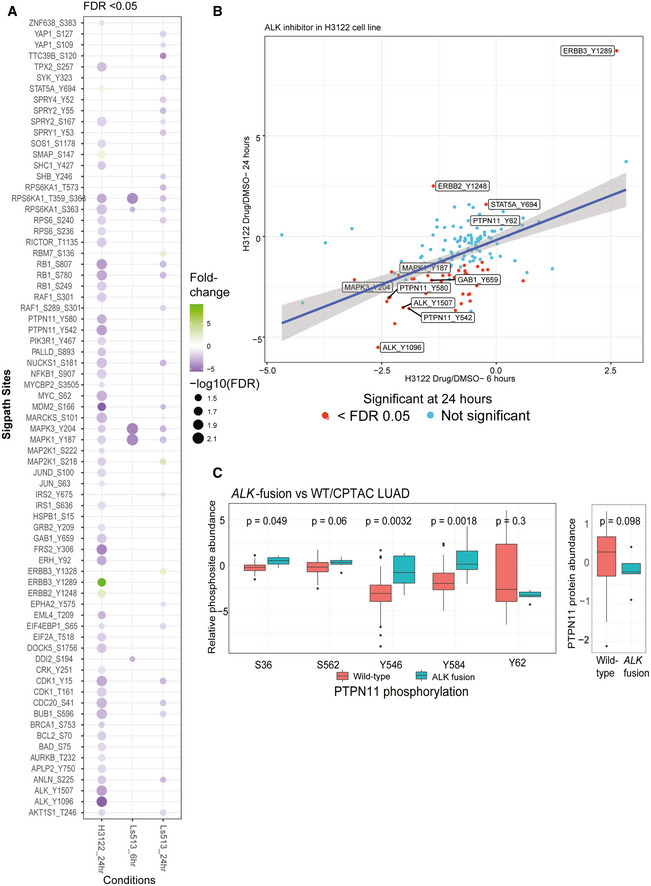
Ceritinib treatment of H3122 cells resulted in significant regulation of 57 phosphosites, 93% of which were downregulated after 24 h of drug treatment (Fig 2A). Consistent with ALK inhibition, pY sites on ALK (pY1096, pY1507) showed dramatic downregulation (Fig 2B). We also observed downregulation of pathway members downstream of ALK, including in PI3K/AKT and ERK/MAPK pathways as described previously (Miyawaki et al, 2017). Interestingly, we also observed differential regulation of PTPN11 phosphosites upon ALK inhibition, with two C‐terminal sites (pY542 and pY580) showing significant reduction at 24 h. ALK activation has been shown to increase PTPN11 phosphorylation at Y542 and Y580 in a neuroblastoma cell line (Sattu et al, 2013). Deep‐scale discovery phosphoproteomic analysis of ALK fusion‐driven patient‐derived lung adenocarcinoma tumors (Gillette et al, 2020) also displayed elevated phosphorylation of PTPN11 Y542 and Y580 (Fig 2C; note that these correspond to Y546 and 584 in the alternative splice isoform of PTPN11), both of which have been implicated in its activation (Bennett et al, 1994; Lu et al, 2001). PTPN11 regulates cell survival and proliferation (Matozaki et al, 2009), and its inhibition suppresses tumorigenesis (Schneeberger et al, 2015). ALK resistance is an inevitable consequence of targeted therapy (Rothenstein & Chooback, 2018); in resistant ALK‐driven non‐small‐cell lung cancers, PTPN11 inhibition restores sensitivity to ALK inhibitor therapy (Dardaei et al, 2018). Notably, we also observe downregulation of Gab1 phosphorylation at Y659 (Fig 2B), which is required for Gab1‐PTPN11 binding and activation of downstream ERK/MAPK signaling initiated by PTPN11 (Cunnick et al, 2001). Our current data showing that ALK inhibition leads to significant downregulation of phosphosites on both the C‐terminal of PTPN11 and Gab1 fits with these other lines of evidence in indicating a key and therapeutically tractable role of this phosphatase in ALK‐mediated downstream signaling both in cell lines and in human tumors. The highly parallel SigPath readout also shows significantly increased phosphorylation at activating sites on ERBB2 (Y1248) and ERBB3 (Y1289), both shown to be involved in the development of resistance during ALK inhibition (Choi et al, 2017) and representing possible bypass tracks that can be targeted in resistant disease (Yamaguchi et al, 2014). Collectively, these data suggest that in the context of therapeutic perturbation experiments, SigPath can provide key readouts and guide specific, testable hypotheses about response, mechanisms of resistance, and therapeutic alternatives.
Figure 2. SigPath analyses of cancer cell line perturbation experiments.
-
ASummary of all significantly regulated phosphosites observed in H3122 and Ls513 cell lines. The log2‐transformed light/heavy peak area ratios for two replicates per time point and treatment were used to compare drug treatment to DMSO. SigPath phosphopeptides differentially regulated upon treatment in each of the conditions in a moderated two‐sample t‐test (adj. P‐value < 0.05) are shown as circles. H3122 6‐h experiment did not yield any significant regulation and hence not shown on the figure. The color indicates fold change relative to DMSO, and the size of the circle indicates log10 (FDR).
-
BScatter plot showing fold change of SigPath sites relative to DMSO for H3122 cell line treated with ALK inhibitor. X‐axis and Y‐axis show 6‐h and 24‐h time points, respectively. The red dots indicate sites with FDR < 0.05. Highlighted are a subset of key differentially regulated phosphosites.
-
CBox plot showing relative abundance of detected PTPN11 phosphosites and PTPN11 protein in the CPTAC LUAD tumors with and without ALK fusion (Gillette et al, 2020). The box represents interquartile range (IQR) with the lower, central, and the upper bands representing 25th percentile (Q1), median, and 75th percentile (Q3), respectively. The lower and upper whiskers represent Q1‐1.5*IQR, and the upper whisker shows Q3+1.5IQR. The data summarized represents patients wild type (n = 103) and mutant (n = 7). PTPN11 pY546 and pY584 sites showed the most significant upregulation (P‐value < 0.01, Wilcoxon test) in tumors with ALK‐fusion.
In the trametinib‐treated Ls513 colorectal cells, we observed strong downregulation at both 6 and 24 h of MAPK3 (ERK1) pY204 and MAPK1 (ERK2) pY187, both downstream of MEK and consistent with MEK inhibition (Fig 2A). Certain drug treatment effects were exclusively observed at either 6 or 24 h. For example, downregulation of RPS6K1 (pT359 and pS363), which is downstream of ERK, was observed at 6 but not 24 h after Trametinib treatment. Conversely, downregulation of RB1 (pY780 and pS807) and CDK1 (pY15) phosphorylation was significant only after 24 h, suggesting the long‐term impact of MEK inhibition on cell cycle progression via CDK1 and also likely via CDK4/6 inhibition upstream of RB1 phosphorylation (Otto & Sicinski, 2017).
Application of the assay to breast cancer xenograft tissue samples
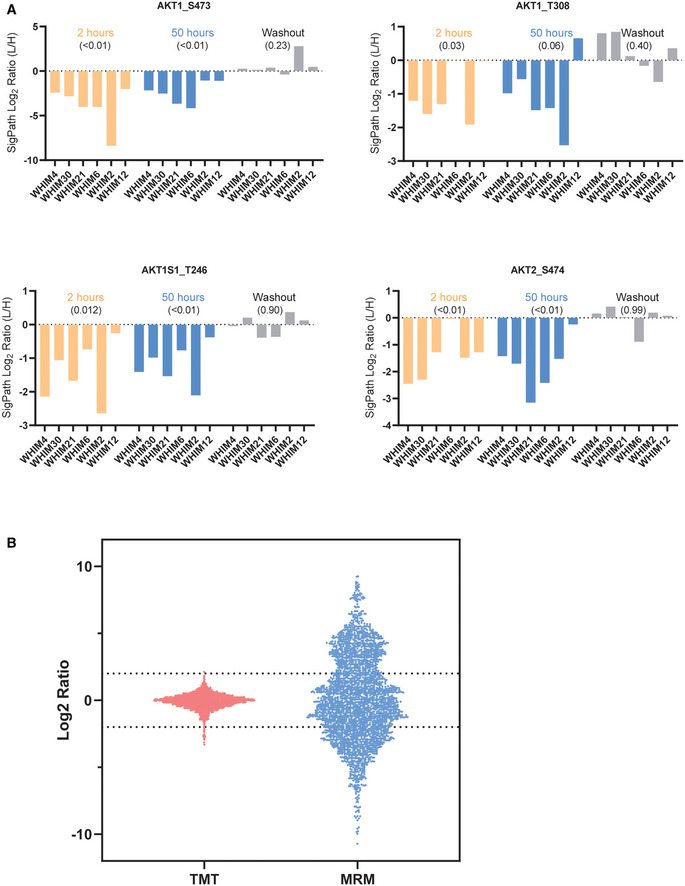
The ability to robustly quantify protein phosphorylation events in tumor samples remains limited. Hence, we sought to test the performance of the SigPath assays in tumor cells undergoing in vivo treatment. Previously, we carried out a deep proteome and phosphoproteome study of six patient‐derived xenograft (PDX) models of triple‐negative breast cancer (WHIM (Washington University human in mouse) 2, 4, 6, 12, 21, 30) each carrying unique mutations in the PI3K pathway (Mundt et al, 2018). These PDX models were selected based on a range of sensitivity to the PI3K inhibitor buparlisib with WHIM4 being the most sensitive and WHIM12 the most resistant. After buparlisib treatment, a differential effect was observed in the phosphoproteome, with the downregulation of phosphosites involved in PI3K signaling seen more in the sensitive than the resistant model. However, due to limits on sensitivity and the stochastic nature of data‐dependent MS‐based proteomics especially for modified peptides, the canonical AKT phosphorylation sites at threonine 308 and serine 473 were neither quantified nor detected.
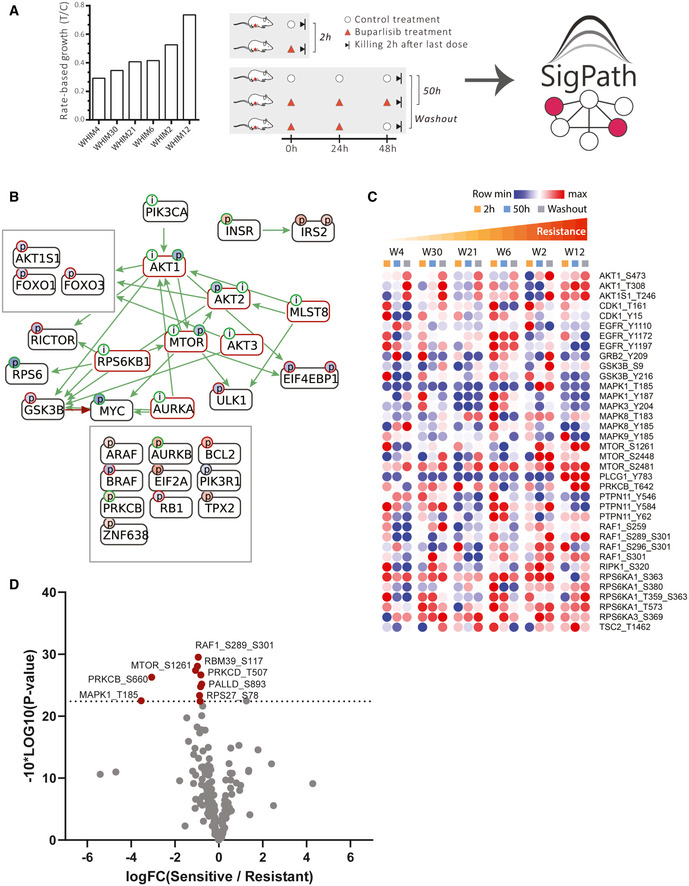
To assess the SigPath performance in this context, we applied the full SigPath assay, including pY Ab and IMAC enrichments, to tissue samples from the six PDX models (Fig 3A). 185/298 (62%) phosphopeptides in the SigPath assay were detected at the endogenous level in this study (Dataset EV5). The well‐known PI3K inhibition marker for AKT1S1 pT246 was readily detected and quantified, as were the AKT markers pT308 and pS473 that had been missed in the discovery experiment. These sites were readily downregulated after buparlisib treatment, consistent with their status as pharmacodynamic markers of PI3K inhibition. Inhibition of these phosphorylation sites is dependent on the continuous administration of the drug, as evident from the upward trend of the sites following drug washout (Fig EV4A). Interestingly, the most resistant PDX model, which harbors a PIK3CA mutation, is the model that often shows the least effect of PI3K inhibition on either of these sites. CausalPath analysis (Babur et al, 2021) applied to the buparlisib/vehicle treatment dataset shows strong inhibition of several members of the PI3K‐AKT‐mTOR pathway after 2‐h buparlisib treatment (Fig 3B). Residual inhibition of the PI3K‐AKT‐mTOR pathway is still visible after 50‐h treatment, but is less pronounced (see CausalPath analysis in Data availability section). These findings substantially recapitulate those from the discovery data (Mundt et al, 2018). Interactive exploration of pathway connectivities in this and other datasets described below is available through Data availability section.
Figure 3. Application of SigPath to understand mechanisms of response and resistance of triple‐negative breast cancer to therapy.
-
ASix patient‐derived xenograft models of triple‐negative breast cancer were assessed for their resistance to buparlisib, a PI3K inhibitor, and analyzed for their proteome and phosphoproteome (Mundt et al, 2018). The six models ranked after their resistance, from most sensitive to the left (WHIM4), to most resistant to the right (WHIM12). The resistance is calculated as rate‐based growth (treatment over control; T/C). Each PDX model was then treated with buparlisib or vehicle and tumors were collected at hours 2 or 50 (buparlisib/vehicle administered at hours 0, 24, and 48). Each of these six models subjected to five different treatments results in a total of 30 samples that were analyzed with the SigPath assay.
-
BCausalPath (www.causalpath.org) analysis of 2‐h drug/vehicle treatment data. Log2 (L/H PAR) for all 6 WHIM models was used for this analysis. Moderated one‐sample t‐test was used to analyze 2‐h treatment data. Resulting table was used for the CausalPath analysis. CausalPath network generated by comparing drug‐treated PDX samples to the controls at 2 h. Nodes represent proteins, and the (p) labels on the nodes represent significant differences in site‐specific phosphopeptide measurements. (p) Blue background color indicates a downregulated site, red background color indicates an upregulated site. Green border color around (p) indicates an activatory site, and a red border color indicates an inhibitory site. Green edges represent known site‐specific phosphorylations, and red edges represent dephosphorylations. The label (i) indicates an inhibited protein. In the case of PI3KCA, the label (i) indicates our manually inserted hypothesis of inactivated PIK3CA due to the drug effect. All other (i) labels on the graph are generated automatically by the CausalPath algorithm through statistical evaluation of the changes at the downstream of the protein. CausalPath infers the PI3KCA ‐> AKT1 relation, indicating the downregulated phosphorylation of AKT1 is likely due to inhibition of PIK3CA. Additionally, statistical measurements on the downstream of AKT proteins indicate their inactivation. We observe that this effect extends over downstream targets of AKT such as mTOR.
-
CHeat map of 36 sites from Hallmark’s PI3K_AKT_mTOR pathway and mTOR, including MAPK3_Y204, detected in SigPath assay. Ratio of buparlisib treatment to vehicle for each time point is used. WHIMs are listed in the order of their resistance to buparlisib treatment. The row min, row max color scheme has been applied after the rows have been adjusted to robust Z‐scores (subtracted median and divided by the median absolute deviation; median‐MAD).
-
DVolcano plot comparing resistant versus sensitive models in 50‐h treatment samples. Sensitive (WHIMs 4, 30, 21, and 6) and resistant (WHIMs 2, and 12) are compared in a two‐sample moderated t‐test. Log2 fold changes are shown on the x‐axis, −10*log10 (P‐value) derived from the two‐sample moderated t‐test are shown on the y‐axis. Red dots indicate the 10 peptides significantly regulated with adj. P‐value threshold of < 0.1.
Figure EV4. Application of the assay to breast cancer PDX tissue samples.
-
ALevels of AKT pS473, AKT pT308, AKT1S1 pT246, and AKT2 pS474 which are all pharmacodynamic markers for PI3K inhibition observed using SigPath. For each graph, and treatment in each graph, the WHIM/PDX models are sorted in the order of their resistance to the drug with least resistance on the left. Across all markers, the most resistant model (WHIM12) is the least affected at 50 h of buparlisib treatment. P‐values, in round brackets “()”, are calculated from a one‐sample t‐test, compared to a hypothetical mean of 0, using all WHIMS as replicates for a certain treatment.
-
BBar and whisker plot showing the range of ratios obtained for the overlapped 115 peptides in discovery study using TMT and SigPath. For the discovery study, Log2 ratio of all the peptides to the pooled reference was used for the plot. For SigPath, Log2 ratio of light to heavy peptide was used for the plot.
To further investigate PI3K‐AKT‐mTOR signaling in the context of buparlisib treatment, we looked at the subset of the SigPath data that include all proteins in Hallmark’s PI3K_AKT_mTOR_signaling pathway and at mTOR itself (Fig 3C). Of the 48 phosphosites represented in the SigPath assay, 36 were readily quantified across the 6 models and treatment conditions. The ratio of buparlisib to vehicle treatment for all models listed in the order of their sensitivity to the treatment is shown in Fig 3C. The resistance of the PDX models to buparlisib does not seem to be mediated at the level of AKT phosphorylation, which suggests that the resistance arises from cross‐talk with other pathways and signaling hubs. To identify response markers, we applied a two‐sample moderated t‐test to compare the two most resistant PDX models (WHIM12 and WHIM2) with the other four models (Fig 3D). Among the highly upregulated sites in the resistant models were pS289 and pS301 of RAF1, which is regulated by MAPK3 (ERK1) (Balan et al, 2006). The original PDX study (Mundt et al, 2018) showed that some of the resistance in the most resistant model (WHIM12) was mediated by MAPK3 activation. Our observations using the SigPath assay strengthen the hypothesis that RAF1 is involved in resistance to buparlisib.
More global comparison of the 146 peptides quantified in both the original Tandem Mass Tag (TMT)‐based and SigPath analyses highlighted the effect of ratio compression in TMT datasets and illustrated how targeted MS overcomes this issue (Fig EV4B).
Application of the assay to tumor tissue from medulloblastoma patients
To test SigPath in primary human tumor samples, we applied the IMAC portion of the assay (Dataset EV1) to brain tissue specimens from 39/40 medulloblastoma patients representing all established subgroups (WNT, SHH, Gr3, and Gr4), previously analyzed by deepscale proteomics, phosphoproteomics (including IMAC and pY enrichment), and acetylproteomics (Archer et al, 2018; Fig EV4A). The pY subset of SigPath was not used due to limited sample availability. The original study showed that tumors with similar RNA expression varied extensively at the post‐transcriptional and post‐translational levels, while proteome profiling revealed subgroups within the SHH and Gr3 groups, providing additional prognostic information and hinting at previously undescribed signaling pathways amenable to SigPath analysis.
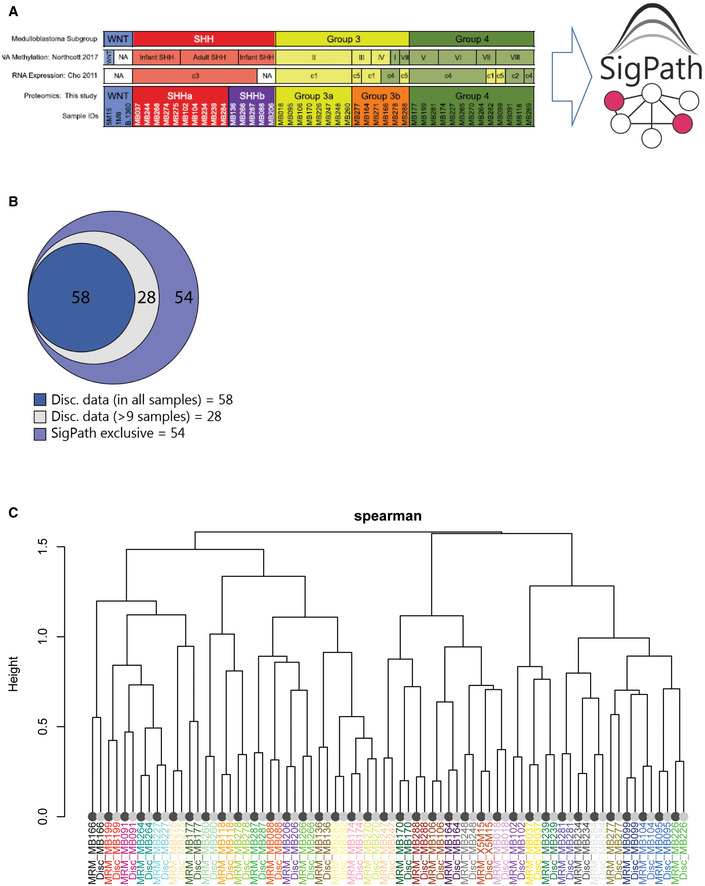
A total of 140 of the phosphopeptides targeted in the SigPath IMAC assay (about 60%) were detected across the 39 samples analyzed (Dataset EV6). We looked at the overlap of the 140 phosphosites detected in the SigPath assay with those from the discovery data (Archer et al, 2018) and found that 58/140 peptides were detected in all samples in the discovery dataset, while another 28 were detected in at least nine samples of the discovery dataset (Fig EV5B). Correlation analysis of discovery and SigPath results for the 86 sites detected in both demonstrated a high level of agreement between phosphosite abundances as measured by both platforms (Fig EV5C).
Figure EV5. Application of the IMAC subset of the SigPath assay to human tissue samples from medulloblastoma patients.
-
AList of samples used for this experiment and their classification to SHH, GR3, and GR4 as well as subgroups within SHH and GR3 (Archer et al, 2018). Only IMAC subset of the assay has been applied to these samples.
-
BVenn diagram showing the overlap of the 140 sites detected in SigPath assay with the discovery data. 86 peptides were detected in both datasets, 58 of these were detected in the discovery dataset in all samples (dark blue) while another 28 detected in at least 9 samples (light blue) of the discovery dataset. 54 sites (39%) were unique to SigPath assay (violet).
-
CDendrogram illustrating the clustering of MRM (dark leaves) and discovery data (light leaves) of 86 phosphosites detected in both assays. Colors of sample identifiers are coordinated by patients. Log2 TMT ratio of each sample to the pooled reference was used for the discovery data after median‐MAD normalization. Log2 light/heavy ratios of SigPath data were used after normalizing each peptide by the median log2 light/heavy ratio across all samples. The dendrogram was derived from complete‐linkage hierarchical clustering using 1‐Spearman correlation as the distance metric. All sample pairs (MRM/discovery) cluster adjacent to each other.
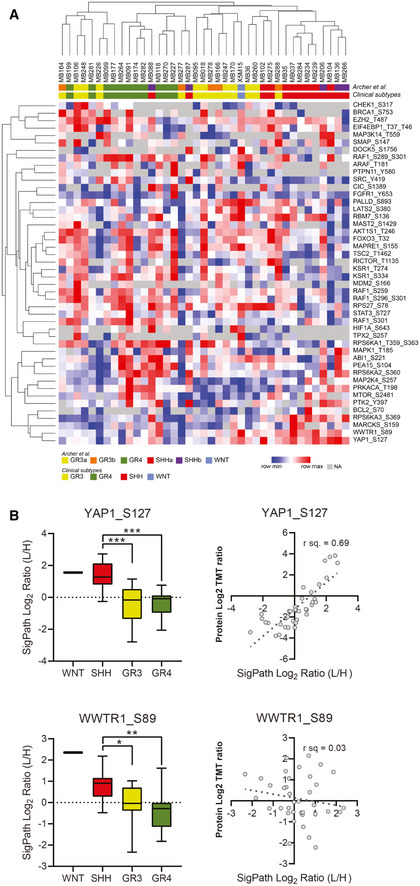
Fifty‐four phosphosites were uniquely detected and quantified with the SigPath assay (Fig EV5B). The behavior of 46 out of the 54 sites quantified in at least 50 percent of the patient samples is illustrated in Fig 4A. While verification is required, some of the unique sites could constitute novel markers for medulloblastoma subtypes. For example, the pS127 phosphosite of YAP1 is upregulated in the sonic‐hedgehog subtype as compared to both groups 3 and 4 in a one‐way ANOVA test (Fig 4B). Yap1 protein is amplified and upregulated in hedgehog‐associated medulloblastomas (Fernandez et al, 2009), while the quantified YAP1 pS127 site indicates inactivation of the protein in this subtype (Artinian et al, 2015). YAP1 and WWTR1 have both been shown to be regulated by, and regulators of, the Hippo pathway, specifically phosphorylated at serine 127 and serine 89, respectively, by the LATS tumor suppressor (Totaro et al, 2018). LATS1 and LATS2 activation is substantiated by CausalPath analysis in the sonic‐hedgehog subtype, compared to group 3 (see Data availability section for CausalPath analysis link for SHH over GR3 comparison). Phosphorylation of YAP1 at S127, and WWTR1 at S89 results in 14‐3‐3 binding and cytoplasmic retention (Kanai et al, 2000; Zhao et al, 2007), limiting its ability to co‐activate TEAD transcription factors and inhibiting proliferation (Vassilev et al, 2001; Zhao et al, 2008; Kofler et al, 2018). CausalPath analysis (see Data availability section) also indicated activation of YES1, an upstream effector of YAP1 (Hamanaka et al, 2019), in the sonic‐hedgehog subtype compared with group 3. To investigate whether the phosphorylation status of each protein and site could be attributed to protein‐level differences, we extracted protein ratios from the proteomic discovery study for all patient samples and performed correlation analysis with the SigPath assay ratios. High correlation (R 2 = 0.69) indicates that in SigPath assay YAP1 pS127 is acting as a proxy for the protein‐level difference, while by contrast the pS89 site of WWTR1 seen upregulated in sonic hedgehog versus group 4 is not due to protein‐level difference, as correlation of this phosphosite to the protein is very low (R 2 = 0.03) (Fig 4B). High expression of the transcriptional coactivator WWTR1 has been shown to be associated with a worse prognosis and affects cell proliferation in patients with medulloblastoma, regardless of subtype (Wang et al, 2019); however, phosphorylation of the S89 site leads to an inhibition of carcinogenesis and cell growth (Cordenonsi et al, 2011; Zhang et al, 2015). Furthermore, CausalPath analysis indicated activation of HCK and YES1 in the sonic‐hedgehog subtype, compared to group 3. The HCK protein product is a member of the Src family of tyrosine kinases. A positive feedback loop between GLI1 and the tyrosine kinase HCK has been shown to amplify sonic‐hedgehog signaling in medulloblastoma (Shi et al, 2015). These new phospho‐level findings may be of significant interest to the medulloblastoma community.
Figure 4. Application of the IMAC subset of SigPath to cancer tissue samples from 39 medulloblastoma patients.
-
AA heat map of 39 samples with medulloblastoma showing 46 phosphosites uniquely detected in the SigPath assay. Samples are clustered by their original clinical subtypes as well as by new classification in ref. Archer et al (2018) where discovery analyses split subgroup 3 into 3b and 3a, and subgroup SHH into SHHa and SHHb. The heat map was generated using Morpheus online tool, the data are median‐MAD normalized, and colors are relative across rows, from row min to row max.
-
BBox plots comparing all the data for YAP1 pS127 and WWTR1 pS89 for all the samples in different groups of medulloblastoma (SHH, GR3, GR4, and WNT). One‐way ANOVA with an ad hoc Tukey’s test (with adj. P‐values for multiple comparisons) was applied for the comparison. The box represents interquartile range (IQR) with the lower, central, and upper bands representing 25th percentile (Q1), median, and 75th percentile (Q3), respectively. The whiskers extend from 5 to 95 percentile of the data. Scatter plots comparing TMT protein‐level Log2 ratios for YAP1 and WWTR1 to SigPath Log2 light to heavy ratios for YAP1 pS127 and TAZ pS89, respectively. Pearson correlation coefficient is shown on the plots.
Discussion
Targeted MS assays have largely been developed for purposes of biomarker verification, but are increasingly being used to validate findings in biological and preclinical studies (as reviewed in (Rifai et al, 2006; Parker & Borchers, 2014). The highly multiplexed, targeted quantitative assay we developed and applied here was designed to measure phosphosites in nodes of biological pathways known to be modulated via phosphorylation, including the RAS, MAPK, PI3K, PKC, SRC, and JAK signaling pathways, transcription factor circuit nodes including FOXO, STAT, NFKB, TGFB, and Wnt pathways, as well as protein phosphosites useful as readouts of DNA damage, cell cycle arrest, apoptosis, spindle checkpoint activation, hypoxia, autophagy, cell stress, and epithelial‐to‐mesenchymal transition. It is important to note that the coverage of these pathways by the current SigPath assay is variable and incomplete, but the sites measured provide some preliminary information that can be used as starting points for further exploration. Furthermore, the assay can be expanded to include additional sites of interest. Through a range of applications in cell lines, preclinical models, and clinical samples, we demonstrated the utility of the assay to not just verify prior findings, but to detect and quantify a large number of differentially regulated phosphosites newly associated with drug perturbation and disease subgroups. For example, in an ALK fusion cell line with the treatment of Ceritinib, we observed differential phosphorylation of N‐ and C‐terminal phosphosites from PTPN11 phosphatase consistent with prior observations in primary LUAD tissue samples (Gillette et al, 2020). These results suggest that SigPath, and targeted MS assays in general, should be more routinely used in the development and optimization of therapeutics. Our results also highlight the potential of SigPath to monitor phosphoproteomic signaling events and to nominate mechanistic hypotheses regarding oncogenesis, response, and resistance to therapy in disease models and human tumors.
The ca. 300‐plex SigPath assay was designed to flexibly allow measurement of phosphosites having phosphotyrosine alone, chiefly phosphoserine‐ and phosphothreonine sites, or a mix of all three. Any proteomic lab skilled in phosphopeptide enrichment can implement the assay, and phosphopeptide sample enrichment is readily automated (Abelin et al, 2016). The SigPath assay is readily extendable to measure other phosphosites by synthesis of the new phosphopeptides in heavy‐labeled form and repeating the QC processes described with a focus on the new sites. Three hundred targets does not represent the maximal level of multiplexing that can be achieved. Using modern MS instrumentation and techniques such as internal standard‐triggered parallel reaction monitoring (Gallien et al, 2015), assay panel sizes can be increased to 500 targets or more. In contrast, assays employing anti‐peptide antibodies (Kuhn et al, 2009; Whiteaker et al, 2011, 2014; Keshishian et al, 2015; Sperling et al, 2019) take a long time to develop and qualify, are very costly even for small numbers of targets, have yet to be multiplexed to the level demonstrated here, and are currently available in only a few expert labs, limiting their utility for the biology community. A limitation of SigPath as configured is that it profiles and quantifies changes in phosphorylation levels only, and therefore cannot determine whether the observed change in phosphopeptide level was due to altered protein expression or a site‐specific post‐translational effect. Assessing if the change in phosphorylation observed is more likely due to protein‐level change vs. site‐specific phosphorylation change could, in principle, be accomplished by adding heavy non‐phosphorylated peptide standards to the flow‐through from the IMAC enrichment and measuring these non‐phosphorylated peptides in a separate and parallel assay. The success of this method is entirely dependent on the ability to detect and quantify the unmodified peptide in the complex digest using a single shot approach.
While Western‐competent antibodies exist for many of the targets in SigPath, they are generally deployed in a highly selective and individual manner and are non‐quantitative, resulting in large swaths of biology remaining opaque to the investigator. Multiplexed Western and ELISA panels are commercially available, but typically measure only 10–20 analytes per well in order to avoid cross‐reactivity and maintain sensitivity (e.g., Quanterix, MSD, Myriad, Abcam, AbcamReview). Large protein assay panels employing two different antibodies for each protein target with detection based on proximity extension are also now commercially available from Olink (Olink). However, these assay panels primarily measure proteins, not phosphosites.
The depth of detection in any targeted MS approach is governed by the abundance of the target peptide in the sample being analyzed, the efficiency of the enrichment process and the amount of input peptide that was enriched. When IMAC phosphopeptide enrichment alone is used, the efficiency is ca. 95%; that is, less than 5% the peptides observed after enrichment do not contain a phosphorylated amino acid. This is important, as non‐phosphorylated peptides are generally present at much higher abundance than the phosphopeptides and so can interfere with detection of phosphopeptides. In the three studies presented here, 500–1,000 μg of peptides was used for the IMAC portion of the assay; however as little as 50 μg can be used, with consequent loss of detection of a subset of the least abundant phosphopeptides. Due to the lower abundance of phosphotyrosine‐containing peptides, antibody‐based phosphotyrosine peptide enrichment typically requires a larger sample input, often in the range of 1–5 mg, to get high coverage of the sites in the SigPath assay.
Once targeted MS‐based assays achieve the large numbers presented here in SigPath, they can also be viewed as a different, complementary way to do discovery, with a narrower spectrum but more consistent measurements and vastly greater throughput. For example, the bona fide PI3K inhibition marker AKT S473 was missed completely in the PDX discovery dataset, while the SigPath assay consistently quantified this site. Sample preparation for SigPath is simple, requiring only digestion, phosphopeptide capture and analysis of the captured peptides together with spiked heavy peptide standards. While sample processing was done manually in the present study, throughput for SigPath can be greatly increased using automated digestion and IMAC enrichment on liquid handling robots as we have previously demonstrated (Abelin et al, 2016). Antibody‐based capture of pY peptides will also become much faster and more reproducible once these antibodies are conjugated to magnetic beads for processing on systems like the Kingfisher as we have done in the case of KGG‐peptide capture for ubiquitylation profiling (preprint: Rivera et al, 2021). The assay as presented here requires a total of 5 h of on‐instrument time for the analysis of both pY Ab‐ and IMAC‐captured samples. This time can be shortened with faster MS instrumentation, use of shorter gradients or by mixing pY and IMAC captures and analyzing these together in a single LC‐MRM/MS run. The use of FAIMS for post‐translationally modified peptides would also provide another level of separation and potentially increase sensitivity (Udeshi et al, 2020; Popow et al, 2021). Quantification of phosphopeptides using SigPath is far more precise than label‐free discovery experiments or those using isobaric chemical labels like TMT, as targeted MS methods do not suffer from the ratio compression challenges of the latter. While the breadth of coverage in SigPath is much smaller than discovery phosphoproteomic analyses (> 300 phosphopeptides compared to > 30,000 phosphopeptides per sample), the quantitative precision and repeatability for targets measured by the assay confer their own advantages, as shown here in the replicates of the ALK inhibitor study and in other phosphopeptide assays we have uploaded to the CPTAC assay portal. Since SigPath detects and measures the spiked heavy peptide forms of all of the phosphopeptides targeted, determining the presence and level of the endogenous form by ratioing the intensities of the endogenous and spiked peptide‐specific fragment ions, it, like other targeted MS assays, is far less susceptible to false positives, meaning that if the target was not detected, it was below detection limits rather than due to stochastic sampling. Therefore, we view SigPath as an impactful resource for the cancer research community, suitable for discovery, as a verification assay for targets of biological import, and for preclinical studies in human cancers and other diseases.
Materials and Methods
Reagents and Tools table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| PTMScan‐pY 1000 Rabbit mAB kit | Cell Signaling Technology | Catalog: 8803A |
| Biological Samples | ||
| Primary tumor samples | See Experimental Model and Subject Details | N/A |
| Chemicals and Reagents | ||
| Synthetic [C13,N15] labeled peptides | New England Peptide | N/A |
| HPLC‐grade water | J.T. Baker | Catalog: 4218‐03 |
| Urea | Sigma | Catalog: U0631 |
| Sodium chloride | Sigma | Catalog: 71376 |
| 1M Tris, pH 8.0 | Invitrogen | Catalog: AM9855G |
| Ethylenediaminetetraacetic acid | Sigma | Catalog: E7889 |
| Aprotinin | Sigma | Catalog: A6103 |
| Leupeptin | Roche | Catalog: 11017101001 |
| Phenylmethylsulfonyl fluoride | Sigma | Catalog: 78830 |
| Sodium fluoride | Sigma | Catalog: S7920 |
| Phosphatase inhibitor cocktail 2 | Sigma | Catalog: P5726 |
| Phosphatase inhibitor cocktail 3 | Sigma | Catalog: P0044 |
| Dithiothreitol, No‐Weigh Format | Fisher Scientific | Catalog: 20291 |
| Iodoacetamide | Sigma | Catalog: A3221 |
| Lysyl endopeptidase | Wako Chemicals | Catalog: 129‐02541 |
| Sequencing‐grade modified trypsin | Promega | Catalog: V511X |
| Formic acid | Sigma | Catalog: F0507 |
| Acetonitrile | Honeywell | Catalog: 34967 |
| Trifluoroacetic acid | Sigma | Catalog: 302031 |
| Methanol | Honeywell | Catalog: 34966 |
| Ni‐NTA agarose beads | Qiagen | Catalog: 30410 |
| Iron (III) chloride | Sigma | Catalog: 451649 |
| Potassium phosphate, monobasic | Sigma | Catalog: P0662 |
| Potassium phosphate, dibasic | Sigma | Catalog: P3786 |
| MOPS | Sigma | Catalog: M5162 |
| Sodium hydroxide | VWR | Catalog: BDH7225 |
| Phosphate‐buffered saline | Fisher Scientific | Catalog: 10010023 |
| Equipment | ||
| Reversed‐phase tC18 Sep‐Pak, 1cc 100 mg | Waters | Catalog: WAT023590 |
| Reversed‐phase tC18 Sep‐Pak, 3cc 200 mg | Waters | Catalog: WAT054945 |
| Solid‐phase C18 disk, for stagetips | Empore 3M | Catalog: 66883‐U |
| Stage‐tip needle | Cadence | Catalog: 7928 |
| Stage‐tip puncher, PEEK tubing | IDEX Health & Science | Catalog: 1581 |
| PicoFrit LC‐MS column | New Objective | Catalog: PF360‐75‐10‐N‐5 |
| ReproSil‐Pur, 120 Å, C18‐AQ, 1.9‐μm resin | Dr. Maisch | Catalog: r119.aq |
| Nanospray column heater | Phoenix S&T | Catalog: PST‐CH‐20U |
| Column heater controller | Phoenix S&T | Catalog: PST‐CHC |
| 300 μl LC‐MS autosampler vial and cap | Waters | Catalog: 186002639 |
| 96‐well microplate for BCA | Greiner | Catalog: 655101 |
| Microplate foil cover | Corning | Catalog: PCR‐AS‐200 |
| Vacuum centrifuge | Thermo Fisher | Catalog: SPD121P‐115 |
| Centrifuge | Eppendorf | Catalog: 5427 R |
| Benchtop mini centrifuge | Corning | Catalog: 6765 |
| Benchtop vortex | Scientific Industries | Catalog: SI‐0236 |
| Incubating shaker | VWR | Catalog: 12620‐942 |
| 15‐ml centrifuge tube | Corning | Catalog: 352097 |
| 50‐ml centrifuge tube | Corning | Catalog: 352070 |
| 1.5‐ml microtube w/o cap | Sarstedt | Catalog: 72.607 |
| 2.0‐ml microtube w/o cap | Sarstedt | Catalog: 72.608 |
| Instrumentation | ||
| Microplate Reader | Molecular Devices | Catalog: M2 |
| Online LC for LC‐MS | Thermo Fisher | Catalog: LC140 |
| Q Exactive Plus Mass Spectrometer | Thermo Fisher |
Catalog: IQLAAEGA APFALGMBDK |
| TSQ Quantiva Triple Quadrupole Mass Spectrometer | Thermo Fisher | Catalog: IQLAAEGAAXFAOUMZZZ |
| Critical Commercial Assays | ||
| BCA Protein Assay Kit | Thermo Fisher | Catalog: 23225 |
| Software and Algorithms | ||
| Software | Source (i.e., PMID or lab) | Identifier (i.e., links) |
| Spectrum Mill software package v7.0 | Broad Institute, Cambridge, MA | https://proteomics.broadinstitute.org/ |
| Protigy | Broad Institute, Proteomics Platform | https://github.com/broadinstitute/protigy |
| Skyline | University of Washington, Seattle, WA | https://skyline.ms/project/home/software/Skyline/begin.view ? |
| Panorama | University of Washington, Seattle, WA | https://panoramaweb.org/home/project‐begin.view ? |
| CausalPath | Computer Science Department, University of Massachusetts Boston, Boston, MA | www.causalpath.org |
Methods and Protocols
Human subjects
Primary medulloblastoma patient samples were collected as described in ref. Archer et al (2018). Patient samples, including FFPE slides, were obtained with informed consent according to the International Cancer Genome Consortium (ICGC) guidelines as approved by the Ethics Committee of the Medical Faculty at Heidelberg University, and as approved by the institutional review board of contributing center Nikolay Nilovich Burdenko Neurosurgical Institute in Moscow. De‐identified tumor samples of 50 mg were freeze‐fractured using Covaris cryoPREP CP02 at setting ‘‘impact level 4’’, and the pulverized samples were aliquoted for the downstream analysis.
Patient‐derived Xenographs
Six triple‐negative breast cancer, patient‐derived xenograft (PDX) models with moderate‐to‐high PI3K pathway activity were selected from the Washington University Human in Mouse (WHIM) PDX collection as described in ref. Mundt et al (2018). All human tissues for these experiments were processed in compliance with NIH regulations and institutional guidelines, and approved by the institutional review board at Washington University. All animal procedures were reviewed and approved by the institutional animal care and use committee at Washington University in St. Louis. PDX models are available through the application to the Human and Mouse‐Linked Evaluation of Tumors core at http://digitalcommons.wustl.edu/hamlet/, and additional information can be found in ref. Li et al (2013).
Peptide selection by nomination and selection from phosphoprofiling studies
For the list of nominated phosphosites, the first step was to convert them into tryptic peptides containing the sites. An in silico tryptic digest of the UniProt protein database was generated to identify fully cleaved tryptic peptides containing the nominated phosphosites. Peptides longer than 40 amino acids in the list were dropped at this step of the selection process. Short versions (< 6 amino acids) of fully cleaved tryptic peptides were still considered at this step in case missed cleaved versions with reasonable length were observed in the existing datasets (Fig EV1A).
Next step was to ascertain whether they had been detected by mass spectrometry. For this purpose, we used the large collection of high‐quality phosphopeptide data generated by the proteomics group at the Broad Institute over the past 15 years to identify whether and in which form(s) the tryptic phosphopeptide had been most frequently observed (e.g., full tryptic, incomplete cleavage, etc.). For the search, we utilized in‐house developed R and Perl scripts specifically tailored toward database search results created by Spectrum Mill Software (Broad Institute, Cambridge, MA) or MaxQuant (Cox & Mann, 2008). Only validated peptides at 1% false discovery rate (FDR) were considered in subsequent analysis. Missed cleaved versions of the query peptide were allowed. Peptides identified by this approach were manually analyzed for each peptide to select a version for synthesis. In the cases where more than one version of the peptide was observed (fully cleaved or missed cleaved, singly or multiply phosphorylated), priority was given to the version with the highest frequency of observation in the datasets. In selecting the singly versus the multiply phosphorylated version of a peptide, priority was given to singly phosphorylated version unless doubly phosphorylated version was much more prevalent in existing datasets. Moreover, for MAPK1, MAPK14, MAPK3, MAPK8, MAPK9, RAF1, and RPS6KA1 both singly phosphorylated and doubly phosphorylated forms of the peptides were included (see Dataset EV1). There are no triply phosphorylated peptides in the assay panel.
Seventy percent of sites nominated (234 out of 343) were previously observed as fully tryptic phosphopeptides or in a missed cleaved form in our experimental datasets. Dataset EV1 lists all of the previously observed nominated phosphosites, the phosphoprotein of origin, the dominant tryptic peptide form containing each site, and the enrichment methodology required for detection of the site (i.e., either immobilized metal affinity chromatography (IMAC) or phosphotyrosine (pY) antibody). Sixty‐two phosphopeptides containing 66 of the 343 nominated phosphosites were not observed in our datasets (Datasets EV1 and EV7). Despite the lack of prior observation in discovery data, we included 37 of these phosphopeptides due to their importance in cancer biology and the potential for the targeted MS method to have greater sensitivity for their detection than the discovery methods used. The remaining 29 sites were excluded from consideration (Dataset EV7).
In addition to lack of detection in experimental data, there were a number of other reasons to not advance the assay configuration for some of the nominated phosphosites, including target peptides being too long or too short or having failed synthesis. Nineteen of the nominated sites were located in tryptic peptides that were deemed to be too long (> 40 amino acids) and were eliminated because of anticipated issues with synthesis, chromatography, and/or assay development (Dataset EV7). In addition, a number of nominated sites were located in short tryptic peptides of 6 amino acids or less. To improve likelihood of detection, specificity and chromatographic retention on C18 reverse phase matrix, we instead searched the data for longer, missed cleaved forms of these peptides. Seventeen of the total 25 short peptides were found in longer, missed cleaved peptides (> 6 but < 40 amino acids) and were included for assay development. For example, the nominated site pS380 in serine/threonine–protein kinase LATS2 is present in tryptic peptide form D(pS)LQK. The longer, missed cleaved version of this peptide RD(pS)LQKPGLEAPPR was found in the discovery data and selected for assay configuration (Dataset EV1). Missed cleaved forms for eight additional short phosphopeptides were not detected in the data and so these were removed for further consideration (Dataset EV7).
Seventy‐three phosphosites in the panel were derived from quantitative proteomic discovery studies in our laboratory and were among the most significantly regulated sites in those studies. 17 phosphopeptides were selected from a discovery phosphoprofiling study investigating impact of ischemia in ovarian cancer and breast cancer xenograft tissues (Mertins et al, 2014). The remaining experimentally derived sites were included based on the analysis of several discovery experiments done in cancer cell lines treated with specific inhibitors.
During the selection process, the uniqueness of each peptide in the human proteome was also taken into consideration with priority given to peptides unique to one protein. Seven of the peptides in the final assay panel are shared with more than one protein (Dataset EV1). Among these are: pS909 of LATS1 and pS872 of LATS2; pT1079 of LATS1 and pT1041 of LATS2; pT35 of MOB1A and MOB1B; pS907 of NFKB1 and pS276 of RELA; pT198 of PRKACA, PRKACB, and PRKACG; pS621 of RAF1 and pS582 of ARAF: and pY706 of NTRK2 and pY709 of NTRK3.
Peptide synthesis
The final list of 352 tryptic phosphopeptides representing 344 phosphosites (17 peptides having more than one site) corresponding to 234 phosphoproteins were synthesized containing single stable isotopically labeled (SIL) amino acid. Most peptides contained [13C, 15N] lysine or arginine at the C‐terminus. Nine peptides representing C‐terminal of the protein were synthesized with either heavy N‐terminal lysine or arginine, or heavy internal leucine and proline. All synthetic peptides were purified by the vendor to greater than 95% purity, quantified by amino acid analysis, and received at micromolar concentrations in a buffer containing 0.1% formic acid/30% acetonitrile. Aliquots of these solutions were diluted to 100 pm/μl for preparation of the peptide mixtures. Peptides are stored at −80⁰C at the Broad Institute and are intended for in‐house use only.
Peptide organization
Synthetic heavy‐labeled internal standard peptides were organized in mixtures first by their enrichment methodology (immobilized metal affinity chromatography (IMAC) and/or phosphotyrosine (pY) antibody). Two hundred thirty one peptides containing phosphoserine (pS) or phosphothreonine (pT) were organized in five IMAC mixtures alphabetically by gene name containing 43–50 phosphopeptides each. Seventy‐one pY‐containing peptides were organized in 2 pY mixes alphabetically by gene name, each containing 31 or 40 peptides. Finally, the remaining 50 peptides (predominantly pY) with previous detection information after IMAC as well as pY antibody enrichments were organized in a separate mixture (IMACpY) and used in combination with either IMAC or pY mixtures. Peptides were at 2 pm/μl equimolar concentration in all of the mixtures.
Assay configuration
Multiple reaction monitoring (MRM) assay configuration using heavy‐labeled synthetic peptides was done on TSQ Quantiva Triple Quadrupole Mass Spectrometer (Thermo Fisher) coupled with Easy‐nLC 1200 ultra‐high pressure liquid chromatography (UPLC) system (Thermo Fisher) in several batches of 50–100 peptides each. Skyline Targeted Mass Spec Environment was used throughout assay configuration and all data analysis. First, spectral libraries for the peptides were generated on a Q Exactive mass spectrometer. Spectral libraries were uploaded to Skyline, and 5–10 most intense fragment ions (transitions) for each peptide were selected for MRM assay configuration. Transitions containing phosphosite or helping with the assignment of the phosphosite were included in the transition list for assay configuration. Next, collision energies (CE) were optimized for all the transitions and peptides by liquid chromatography–multiple reaction monitoring mass spectrometry (LC‐MRM/MS) on TSQ Quantiva using Skyline’s CE optimization module. For every transition starting with the instrument‐specific calculated CE tested 10 additional CEs (5 below and 5 above the calculated CE) in increments of 2. The list of transitions with varying CE values was exported from Skyline and used for building the MRM method in Xcalibur software. Equimolar mixture of peptides at 50 fm/μl was analyzed by LC‐MRM/MS on Quantiva using this method. Resulting data were analyzed on Skyline which then selected the CE that resulted in the highest peak area for each transition. In the final step of CE optimization MRM data were acquired with optimized CE values for every transition. Using this dataset in Skyline, manually selected the best 3–6 transitions for every peptide giving highest priority to fragment ions of y‐series with mass to charge (m/z) above the precursor, and ions containing phosphosite or helping with the site localization. For the peptides where the options were more limited also included ions of y‐series with m/z below the precursor and b‐series.
While we configured the assay on TSQ Quantiva MS, other triple quadrupole instruments can be used for this assay with further optimization of MS‐specific parameters for each instrument (Kuhn et al, 2012; Abbatiello et al, 2015).
After the CE optimization compiled 2 LC‐MRM/MS methods, one for peptides enriched by IMAC strategy (231 IMAC and 50 IMACpY mixtures) and the second for peptides enriched by pY antibody strategy (71 pY and 50 IMACpY mixtures).
Liquid chromatography was performed on 75 μm ID PicoFrit columns packed in‐house to a length of 28–30 cm with Reprosil C18‐AQ 1.9 μm beads (Dr Maisch GmbH) with solvent A of 0.1% formic acid (FA) / 3% acetonitrile (ACN) and solvent B of 0.1% FA / 90% ACN at 200 nl/min flow rate. Below are the details of the IMAC and pY LC‐MRM/MS methods:
IMAC LC‐MRM/MS method: method duration – 160 min, gradient – 2–6% solvent B in 1 min, 6–30% B in 124 min, 30–60% B in 9 min, 60–90% B in 1 min, followed by a hold at 90% B for 5 min, and subsequent hold at 50% B for 19 min. MS parameters include 3‐sec cycle time, Q1 and Q3 resolution of 0.4 and 0.7, respectively, retention time (RT) scheduling window of 10 min.
pY LC‐MRM/MS method: method duration – 120 min, gradient – 2–6% solvent B in 1 min, 6–30% B in 84 min, 30–60% B in 9 min, 60–90% B in 1 min, followed by a hold at 90% B for 5 min, and subsequent hold at 50% B for 19 min. MS parameters include 1.5‐sec cycle time, Q1 and Q3 resolution of 0.4 and 0.7, respectively, RT scheduling window of 10 min.
Lysis and digestion of cell line and tissue samples
Add lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris pH 8.0, 1 mM EDTA, 2 μg/ml Aprotinin, 10 μg/ml Leupeptin, 1 mM PMSF, 10 mM NaF, 1:100 phosphatase inhibitor cocktail 2 and 1:100 phosphatase inhibitor cocktail 3) to the cell pellets or cryopulverized tissue samples, vortex lightly, and incubate at 4°C with end‐over‐end rotation for 15 min.
Vortex samples for 10 s on the highest setting and allow to incubate at 4°C with end‐over‐end rotation again for 15 more minutes.
Centrifuge samples at 20,000 rcf for 10 min at 4°C to pellet insoluble cell debris.
Transfer the supernatant to a new 2‐ml Eppendorf tube and quantify by the Pierce BCA Protein Assay Kit.
Equalize concentrations of a set of samples in a study and digested together by adding more lysis buffer to samples that are more concentrated to match the sample with the lowest concentration.
Reduce samples with 5 mM dithiothreitol (DTT, Pierce, A39255), mixing at 800 rpm for 45 min at room temperature.
Alkylate samples using 10 mM iodoacetamide (IAA, Sigma‐Aldrich, 144489) for 45 min in the dark at room temperature.
Dilute the samples 1:4 with 50 mM Tris–HCl pH 8.0 and digest with LysC (Wako) at an enzyme to substrate ratio of 1:50 for 2 h at 30°C and shaking at 800 rpm.
Add trypsin (Promega) at an enzyme to substrate ratio of 1:50 overnight at 37°C and shaking at 800 rpm.
Quench the digestion with 10% formic acid to a final concentration of 1% and pH 3.
Peptide cleanup by cartridge desalt
Condition 200 mg (3 cc) Sep‐Pak C18 Vac Cartridges (Waters) with 3 ml of acetonitrile followed by 3 ml of 0.1% FA / 50% ACN.
Equilibrate them with four 3 ml injections of 0.1% trifluoroacetic acid (TFA).
Load the samples onto the cartridges and collect the flow‐through.
Wash the samples three times with 3 ml 0.1% trifluoroacetic acid and one time with 3 ml 1% FA.
Elute the samples off the cartridges with two injections of 1.5 ml 0.1% FA / 50% ACN into 15‐mL centrifuge tubes.
Freeze the samples and dry them down with vacuum centrifugation.
Reconstitute the samples in 0.1% FA / 3% ACN and quantify by the Pierce BCA Protein Assay Kit.
Make 5 mg aliquots of the samples for pY Ab enrichment, freeze the aliquots, and dry down by vacuum centrifugation.
If pY Ab enrichment is skipped, then make 500 μg aliquots for the IMAC enrichment step (see below).
Phosphotyrosine enrichment
Reconstitute peptide aliquots for pY Ab enrichment with 1.5 ml of IAP buffer (50 mM MOPS/NaOH pH 7.2, 10 mM Na2HPO4, 50 mM NaCl) and keep on ice throughout the experiment.
Add 30 fmol of the pY and IMACpY heavy peptide mixtures into each sample, then vortex, and spin down at 5,000 rcf for 5 min.
Wash the pY 1000 Immunoaffinity beads (Cell Signaling Technology) 3 times, each time with 1.5 ml of IAP buffer. Remove the supernatant after each wash.
Add reconstituted peptide samples onto the Immunoaffinity beads and mix end over end at 4°C for 1 h.
After the hour incubation, spin down the beads at 1,500 rcf for 1 min and collect the supernatant as the pY flow‐through for IMAC enrichment.
Wash pY 1000 beads four times with 1.5 ml cold phosphate‐buffered saline (PBS, Thermo Fisher).
After washing, resuspend the beads with 50 μl of 0.15% TFA and incubate at room temperature for 5 min.
Spin down the beads and transfer the supernatant onto a prewashed and pre‐conditioned stagetip (see below).
Repeat TFA incubation one more time for a total of 100 μl of supernatant transferred onto the stagetip. After the second elution, transfer the beads along with the supernatant to the stagetip.
Phosphotyrosine enrichment stage‐tip desalt
Condition the stagetips, prepared with two Empore C18 (3 M) punches with 100 μl of methanol, and followed by 100 μl of 0.1% FA / 50% ACN. Spin down after each one at 3,100 rcf for 1 min.
Equilibrate with 2 injections of 100 μl 0.1% FA.
Add the two 50 μl pY captured samples along with the beads to stagetips then spin down.
Wash the sample with two injections of 100 μl 0.1% FA and spin down after each.
Elute the bound peptides off the stagetips using 50 μl of 0.1% FA / 50% ACN.
Transfer the eluates to autosampler vials, freeze, and dry down.
Reconstitute in 5 μl of 0.1% FA/3% ACN solution and inject 4 μl for LC‐MRM/MS analysis on TSQ Quantiva using pY LC‐MRM/MS method (see above).
Phosphotyrosine flow‐through desalt
Condition 100 mg (1 cc) Sep‐Pak C18 Vac Cartridges (Waters, WAT023590) with 1 ml of acetonitrile followed by 1 ml of 0.1% FA / 50% ACN.
Equilibrate with four 1 ml injections of 0.1% TFA.
Acidify pY Ab enrichment flow‐through samples with 150 μl of 10% FA and load onto the prepared cartridges in two steps of 750 μl.
Wash them three times with 1 ml of 0.1% TFA then one time with 1 ml 1% FA.
Elute the samples off the cartridges and into 2‐mL Eppendorf tubes with 2 injections of 750 μl 0.1% FA / 50% ACN.
Freeze the samples and dry down by vacuum centrifugation.
Reconstitute the samples in 0.1% FA / 3% ACN and measure the concentrations using the Pierce BCA Protein Assay Kit (Thermo Fisher).
Make aliquots at appropriate amounts for each study (500–1,000 μg) for the immobilized metal affinity chromatography (IMAC) enrichment step. Freeze the aliquots and dry down by vacuum centrifugation.
Immobilized metal affinity chromatography (IMAC) phosphopeptide enrichment
Remove 1,200 μl slurry, or 600 μl of Ni‐NTA Agarose beads (Qiagen) and transfer to a 1.5‐ml Eppendorf tube.
Wash the beads three times by adding 1 ml of water onto the beads, inverting the tube to suspend the beads, then spinning down for 1 min at 1,500 rcf and removing the supernatant. Strip the beads of the nickel by incubating end over end with 1,200 μl of 100 mM ethylenediaminetetraacetic acid (EDTA (Sigma‐Aldrich)) at room temperature for 30 min.
Wash three times with HPLC water then incubate with 1,200 μl of 10 mM FeCl3 end‐over‐end at room temperature for 30 min.
Wash the agarose beads again three times with HPLC water and resuspend with 1:1:1 acetonitrile: methanol: 0.01% acetic acid to a ratio of 1:3 beads to slurry volume.
Aliquot 60 μl slurry, or 20 μl beads, into 1.5‐ml Eppendorf tubes for each 1 mg sample undergoing phosphopeptide enrichment.
Reconstitute the dried peptide aliquots in 0.1% TFA / 50% ACN and vortex until the peptides were fully dissolved. Add 0.1% TFA / 100% ACN to each aliquot to bring the final concentration to 0.5 mg/ml in 80% ACN solution.
Add 30 fmol of heavy‐labeled IMAC peptides into each sample, and then add the peptide solutions on top of the prepared beads and incubate end‐over‐end for 30 min at room temperature.
Spin down the beads for 1 min at 1,500 rcf. Remove the supernatant and save.
Add 200 μl of 0.1% TFA / 80% ACN to the beads. Transfer onto a prepared stagetip for desalting.
Immobilized metal affinity chromatography stage‐tip desalt
Condition the stagetips, prepared with two Empore C18 (3 M) punches, first with 100 μl of methanol, then with 50 μl of 0.1% FA / 50% ACN and spin down at 3,100 rcf for 1 min after each step.
Equilibrate the tips with 2 injections of 100 μl 1% FA.
Load the resuspended beads onto the stagetips and spin down.
Desalt the stagetips with two 50 μl injections of 0.1% TFA / 80% ACN then one 50 μl injection of 1% FA.
Elute the phosphopeptides off the agarose beads and onto the stagetips by three 70 μl injections of 500 mM K2PO4.
Wash the samples once with 100 μl of 1% FA,
Elute the samples off the tips with 60 μl 0.1% FA / 50% ACN.
Transfer the eluates from Eppendorf tubes to HPLC vials, freeze, and dry down by vacuum centrifugation.
Reconstitute in 9 μl of 0.1% FA / 3% ACN solution and inject 4 μl for LC‐MRM/MS analysis on TSQ Quantiva using IMAC LC‐MRM/MS method (see above).
Titration curve experiment
Five cell lines (OVCAR, Meljuso, H3122, PC9, and A375) were lysed, digested, and desalted as described above. Equal amounts of peptides from each digest were combined to create the peptide mix used in the study. In triplicate, 1 mg and 5 mg aliquots of this peptide mix were reconstituted in 1.5 ml IAP buffer (50 mM MOPS/NaOH pH 7.2, 10 mM Na2HPO4, 50 mM NaCl). Aliquots were then spiked with 30 fmol of the pY and IMACpY heavy peptide mixtures, then enriched by pY 1000 antibody using the phosphotyrosine enrichment method detailed above. Enriched samples were stagetip‐desalted (see method above) then analyzed on TSQ Quantiva using the method for pY LC‐MRM/MS described above. The flow‐through samples from the pY Ab enrichments were combined and desalted together. The resulting mix was aliquoted in triplicate at 1, 0.5, 0.25, 0.1, and 0.05 mg. Each aliquot was reconstituted in 1 ml 0.1% TFA / 50% ACN (1 mg aliquots were reconstituted in 2 ml) then spiked with 30 fmol of the IMAC heavy peptide mixture. The IMAC phosphopeptide enrichment procedure detailed above was used. Enriched samples were stagetip‐desalted (see method above) then analyzed on TSQ Quantiva using the method for IMAC LC‐MRM/MS described above.
Cell line processing for testing the assay
Ten cell lines (PC9, H3122, TMD8, Mino, PC3, OVCAR4, WM266.4, Meljuso, A375, and RT112) were lysed and digested as described above. Five milligrams of each was enriched by pY antibody and 1 mg of the flow‐through of that was enriched by IMAC and analyzed following SigPath workflow as described above.
Cell line perturbagen sample processing
H3122 and Ls513 cells were treated with either DMSO or drug for 6 h and 24 h (Fig EV3A). H3122 cells were treated with Ceritinib at 300 nM concentration, and Ls513 cells were treated with Trametinib at 30 nM concentration. Treatments and time points were done in two process replicates. Cells were collected, lysed, and digested as described above. Following digestion, 5 mg of each sample was enriched with pY Ab and 1 mg of the flow‐through of that by IMAC and analyzed according to the SigPath workflow described above.
Breast cancer xenograft (PDX) tissue processing
Six models were selected from Washington University human to mouse (WHIM) PDX collection (Mundt et al, 2018) (4, 30, 21, 6, 2, and 12). Each of the models was treated either with buperlasib or with vehicle (Fig 3A). For the 2‐h treatment group, the animals received one dose and tissue was collected 2 h after the treatment either by buperlasib or by vehicle. For the 50‐h group, animals received Buperlasib or vehicle at 0, 24, and 48 h, and the tissue was collected at 50 h. Only in the washout group at 48 h, the animals were treated with vehicle instead of the drug. Tissue lysis and digestion were performed as described above. Input peptide amount for pY Ab enrichment varied as follows for the different WHIM models due to limited amount availability of some of the samples: WHIM4 – 4.5 mg; WHIM30 – 4.5 mg; WHIM21 – 5 mg; WHIM6 – 5 mg; WHIM2 – 2 mg; WHIM12 – 4 mg. Input peptide amount for IMAC was 1 mg for all of the samples. Phosphotyrosine antibody enrichment for all the five samples of each WHIM was done the same day, but on different days for the different WHIMs. IMAC enrichment was performed over 3 days, samples for 2 WHIM models per day.
Medulloblastoma tissue processing
39 tissue samples from medulloblastoma patients belonging to all four groups (sonic hedgehog (SHH), group 3 (GR3), group 4 (GR4) and WNT) were digested as described above. Less than 1 mg of digested peptides was available for this study, and therefore, the pY Ab portion of the procedure was skipped and only the IMAC part of the assay was applied to all the samples with 500 μg input digested peptides for 85% of them. For 6 of the samples, 500 μg was not available; therefore, the peptide input varied as follows: MB088 (SHH) = 357 μg; MB136 (SHH) = 347 μg; MB206 (SHH) = 313 μg; MB284 (SHH) = 444 μg; MB287 (SHH) = 425 μg; MB091 (GR4) = 485 μg. Sample processing and data acquisition were performed in 4 batches. Samples were randomized in 4 batches making sure to include an equal number of samples from each group.
Data processing
All analyses of raw mass spectrometry data were performed in Skyline Targeted Mass Spec Environment (Broudy et al, 2014). Peak area ratios of endogenous light to stable isotope‐labeled (SIL) heavy internal standard peptide were calculated in Skyline (Skyline version (64‐bit) 20.2.0.343), https://brendanxuw1.gs.washington.edu/labkey/project/home/software/Skyline/begin.view). All the peaks were manually inspected to make sure accurate and equal integration of light and heavy versions. Peak area ratios of the most abundant, interference‐free transitions were used for further statistical analysis. Detection of endogenous (light peptide) signals required detection of all the transitions along with a minimal signal for the reporting transition based on manual inspection of the data. The height of the light peak had to be more than 250 and 300 counts for pY Ab and IMAC enriched samples, respectively.
Data imputation
Imputation of missing data points was performed for the perturbagen experiments (H3122, Ls513, and PDX), separately for each dataset and pY Ab / IMAC experiments. Phosphopeptides were divided into three categories: 1) confidently quantified peptides derived as described above; 2) quantified peptides not passing the minimal signal threshold; and 3) peptides not quantified at all. Log2‐transformed peptide peak area ratios for 3) were imputed by drawing from a normal distribution with parameters mu and sd (mean and standard deviation, respectively) estimated from the distribution of light/heavy ratios in 2). Parameter mu was further adjusted by subtracting one sd to resemble light/heavy ratios below the detection limit.
For statistical analysis of these datasets, peptide ratios of category 2) were kept in the dataset and imputed values for category 3) were used.
Statistical analysis
Peak area ratios were log2‐transformed. Visual inspection of the resulting log‐ratios in profile density plots revealed comparable distributions of samples within each dataset, and no further normalization was applied. A two‐sample moderated t‐test was applied to cell line perturbagen, medulloblastoma, and PDX datasets comparing sensitive versus resistant models using Protigy. (https://github.com/broadinstitute/protigy). Derived P‐values were adjusted for multiple hypothesis testing using Benjamini–Hochberg (BH) strategy. Significance was assessed with BH‐corrected P‐value of < 0.1.
A one‐sample moderated t‐test was applied to drug/vehicle ratio of ratios in the PDX dataset to compare drug to vehicle treatment for all 6 PDX models. Derived P‐values were adjusted for multiple hypothesis testing using Benjamini–Hochberg (BH) strategy. Significance was assessed with BH‐corrected P‐value of < 0.1.
A one‐way ANOVA with an ad hoc Tukey’s test (with (BH) adjusted P‐values for multiple comparisons) was performed in GraphPad Prism on medulloblastoma dataset.
Heat maps are generated using the Morpheus online tool (Morpheus, https://software.broadinstitute.org/morpheus
CausalPath analysis
CausalPath (Babur et al, 2021) analysis was used to identify the likely cause–effect relations between the correlated phosphopeptide measurements. For the comparison of drug/DMSO at 6 h and 24 h in H3122 and Ls513 cell lines, sensitive versus resistant in PDX study and groups in medulloblastoma study, BH‐corrected P‐values from a two‐sample moderated t‐test were used as input to the method. For the comparison of drug versus vehicle in PDX study at 2 and 50 h, BH‐corrected P‐values from a one‐sample moderated t‐test were used as input to the method. The method options to calculate network significance and to use the inferred activities in causal reasoning are turned on. FDR cutoff of 0.1 was used for both phosphopeptide change significance and network significance. For the drug inhibition studies, custom hypotheses indicating inhibition of direct drug targets were inserted. This allows CausalPath to identify the changes that are compatible with the hypothesis and use them in the resulting model. ChiBE (Babur et al, 2014) was used as a visualization tool to generate Fig 3B.
Author contributions
HK, ERM, DAP, KW, WRS, and SAC conceived the study; HK, ERM, DAP, JJ‐V, FM, and SAC designed the study; HK, RM, LW, HS, FM, DF, BR, SEM, MLR, MB, MAM, and MEO generated the data; HK, KK, FM, SS, and DRM did formal analysis; KK, DRM, and OB provided and helped with software; ERM, DAP, JJ‐V, PMJB, OB, SS, EK, ET, MAG, WRS, and SAC provided resources; HK, ERM, DAP, FM, KK, JG, SS, DRM, MAG, and TR curated the data; HK, RM, FM, KK, SS, and SAC wrote the original draft; HK, FM, KK, SS, MG, TR, KW, WRS, and SAC reviewed and edited the manuscript; HK, ERM, JJ‐V, KW, WRS, and SAC supervised the study; WRS and SAC acquired the funding.
Conflict of interest
SAC is a member of the scientific advisory boards of Kymera, PTM BioLabs, and Seer and a scientific advisor to Pfizer and Biogen.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Acknowledgements
This work was supported by a grant from Novartis and partially by NIH/NCI grants from the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium grants NIH/NCI U24CA210986 and U01CA214125 (to SAC) and NIH/NCI U24CA210979 (to DRM). We thank Jacob Jaffe for help with searching phosphopeptides against existing datasets, Karl Clauser for help with Spectrum Mill during assay development.
Mol Syst Biol. (2021) 17: e10156
Contributor Information
Hasmik Keshishian, Email: hasmik@broadinstitute.org.
Steven A Carr, Email: scarr@broad.mit.edu.
Data availability
The datasets and computer code produced in this study are available in the following databases:
All mass spectrometry data generated during this study have been published in Panorama (Sharma et al, 2018) and are deposited at: https://panoramaweb.org/YyIDIy.url.
The source code for Protigy used for all statistical analysis is on GitHub: https://github.com/broadinstitute/protigy.
The data for CausalPath analysis have been deposited at GitHub: https://github.com/broadinstitute/proteomics‐SigPath‐supplemental‐data.
References
- Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MPet al (2015) Large‐scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer‐relevant proteins in plasma. Mol Cell Proteomics 14: 2357–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abelin JG, Patel J, Lu X, Feeney CM, Fagbami L, Creech AL, Hu R, Lam D, Davison D, Pino Let al (2016) Reduced‐representation phosphosignatures measured by quantitative targeted MS capture cellular states and enable large‐scale comparison of drug‐induced phenotypes. Mol Cell Proteomics 15: 1622–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer TC, Ehrenberger T, Mundt F, Gold MP, Krug K, Mah CK, Mahoney EL, Daniel CJ, LeNail A, Ramamoorthy Det al (2018) Proteomics, post‐translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell 34: 396–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artinian N, Cloninger C, Holmes B, Benavides‐Serrato A, Bashir T, Gera J (2015) Phosphorylation of the hippo pathway component AMOTL2 by the mTORC2 kinase promotes YAP signaling, resulting in enhanced glioblastoma growth and invasiveness. J Biol Chem 290: 19387–19401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur O, Dogrusoz U, Cakir M, Aksoy BA, Schultz N, Sander C, Demir E (2014) Integrating biological pathways and genomic profiles with ChiBE 2. BMC Genom 15: 642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur Ö, Luna A, Korkut A, Durupinar F, Siper MC, Dogrusoz U, Vaca Jacome AS, Peckner R, Christianson KE, Jaffe JDet al (2021) Causal interactions from proteomic profiles: molecular data meet pathway knowledge. Patterns 2: 100257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan V, Leicht DT, Zhu J, Balan K, Kaplun A, Singh‐Gupta V, Qin J, Ruan H, Comb MJ, Tzivion G (2006) Identification of novel in vivo Raf‐1 phosphorylation sites mediating positive feedback Raf‐1 regulation by extracellular signal‐regulated kinase. Mol Biol Cell 17: 1141–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin Det al (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG (1994) Protein‐tyrosine‐phosphatase SHPTP2 couples platelet‐derived growth factor receptor beta to Ras. Proc Natl Acad Sci USA 91: 7335–7339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broudy D, Killeen T, Choi M, Shulman N, Mani DR, Abbatiello SE, Mani D, Ahmad R, Sahu AK, Schilling Bet al (2014) A framework for installable external tools in Skyline. Bioinformatics 30: 2521–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess MW, Keshishian H, Mani DR, Gillette MA, Carr SA (2014) Simplified and efficient quantification of low‐abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics 13: 1137–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T‐W, Lee C‐C, Liu H, Wu C‐S, Pickering CR, Huang P‐J, Wang J, Chang I‐F, Yeh Y‐M, Chen C‐Det al (2017a) APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat Commun 8: 465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fisher KJ, Lloyd M, Wood ER, Coppola D, Siegel E, Shibata D, Chen YA, Koomen JM (2017b) Multiplexed liquid chromatography‐multiple reaction monitoring mass spectrometry quantification of cancer signaling proteins. Methods Mol Biol 1647: 19–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Kim DH, Choi YJ, Kim SY, Lee JE, Sung KJ, Kim WS, Choi CM, Rho JK, Lee JC (2017) Multiple receptor tyrosine kinase activation related to ALK inhibitor resistance in lung cancer cells with ALK rearrangement. Oncotarget 8: 58771–58780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti A, Poletti Aet al (2011) The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147: 759–772 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cunnick JM, Mei L, Doupnik CA, Wu J (2001) Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine‐based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem 276: 24380–24387 [DOI] [PubMed] [Google Scholar]
- Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, Kerr G, Yu K, Liang J, Cao Yet al (2018) SHP2 inhibition restores sensitivity in ALK‐rearranged non‐small‐cell lung cancer resistant to ALK inhibitors. Nat Med 24: 512–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy Set al (2020) Proteogenomic Characterization of Endometrial Carcinoma. Cell 180(729–748): e726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshghi A, Pistawka AJ, Liu J, Chen M, Sinclair NJT, Hardie DB, Elliott M, Chen L, Newman R, Mohammed Yet al (2020) Concentration determination of >200 proteins in dried blood spots for biomarker discovery and validation. Mol Cell Proteomics 19: 540–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, Sun P, Moy C, Szabo SA, Roadcap LTet al (2012) Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose‐escalation trial. Lancet Oncol 13: 782–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM (2009) YAP1 is amplified and up‐regulated in hedgehog‐associated medulloblastomas and mediates Sonic hedgehog‐driven neural precursor proliferation. Genes Dev 23: 2729–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr Pet al (2012) Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med 367: 107–114 [DOI] [PubMed] [Google Scholar]
- Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys P‐Y, Awad MM, Yanagitani N, Kim Set al (2014) The ALK inhibitor ceritinib overcomes crizotinib resistance in non‐small cell lung cancer. Cancer Discov 4: 662–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S, Kim SY, Domon B (2015) Large‐scale targeted proteomics using internal standard triggered‐parallel reaction monitoring (IS‐PRM). Mol Cell Proteomics 14: 1630–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané‐Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li Het al (2019) Next‐generation characterization of the Cancer Cell Line Encyclopedia. Nature 569: 503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang W‐W, Reva Bet al (2020) Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182: 200–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka N, Nakanishi Y, Mizuno T, Horiguchi‐Takei K, Akiyama N, Tanimura H, Hasegawa M, Satoh Y, Tachibana Y, Fujii Tet al (2019) YES1 is a targetable oncogene in cancers harboring YES1 gene amplification. Cancer Res 79: 5734–5745 [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers Get al (2010) RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature 464: 431–435 [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K‐L, Li S, Mertins P, Cao S, Gunawardena HP, Ruggles KV, Mani DR, Clauser KR, Tanioka M, Usary Jet al (2017) Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat Commun 8: 14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttenhain R, Choi M, Martin de la Fuente L, Oehl K, Chang C‐Y, Zimmermann A‐K, Malander S, Olsson H, Surinova S, Clough Tet al (2019) A targeted mass spectrometry strategy for developing proteomic biomarkers: a case study of epithelial ovarian cancer. Mol Cell Proteomics 18: 1836–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw Ret al (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48: D498–D503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LCet al (2000) TAZ: a novel transcriptional co‐activator regulated by interactions with 14‐3‐3 and PDZ domain proteins. EMBO J 19: 6778–6791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JJ, Yan P, Zhao L, Ivey RG, Voytovich UJ, Moore HD, Lin C, Pogosova‐Agadjanyan EL, Stirewalt DL, Reding KWet al (2016) Immobilized metal affinity chromatography coupled to multiple reaction monitoring enables reproducible quantification of phospho‐signaling. Mol Cell Proteomics 15: 726–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H, Addona T, Burgess M, Mani DR, Shi X, Kuhn E, Sabatine MS, Gerszten RE, Carr SA (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics 8: 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, Kuhn EW, Farrell LA, Gerszten RE, Carr SA (2015) Multiplexed, quantitative workflow for sensitive biomarker discovery in plasma yields novel candidates for early myocardial injury. Mol Cell Proteomics 14: 2375–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler M, Speight P, Little D, Di Ciano‐Oliveira C, Szaszi K, Kapus A (2018) Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat Commun 9: 4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LCet al (2020) Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell 183: 1436–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E, Addona T, Keshishian H, Burgess M, Mani DR, Lee RT, Sabatine MS, Gerszten RE, Carr SA (2009) Developing multiplexed assays for troponin I and interleukin‐33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin Chem 55: 1108–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJet al (2012) Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics 11: M111.013854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez Det al (2019) The landscape of cancer cell line metabolism. Nat Med 25: 850–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AKet al (2017) Characterization of human cancer cell lines by reverse‐phase protein arrays. Cancer Cell 31: 225–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu Cet al (2013) Endocrine‐therapy‐resistant ESR1 variants revealed by genomic characterization of breast‐cancer‐derived xenografts. Cell Rep 4: 1116–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Gong D, Bar‐Sagi D, Cole PA (2001) Site‐specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP‐2 in cell signaling. Mol Cell 8: 759–769 [DOI] [PubMed] [Google Scholar]
- Manes NP, Nita‐Lazar A (2018) Application of targeted mass spectrometry in bottom‐up proteomics for systems biology research. J Proteomics 189: 75–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, Miller RA, Digles D, Lopes EN, Ehrhart Fet al (2021) WikiPathways: connecting communities. Nucleic Acids Res 49: D613–D621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H (2009) Protein tyrosine phosphatase SHP‐2: a proto‐oncogene product that promotes Ras activation. Cancer Sci 100: 1786–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia Fet al (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534: 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJet al (2014) Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics 13: 1690–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki M, Yasuda H, Tani T, Hamamoto J, Arai D, Ishioka K, Ohgino K, Nukaga S, Hirano T, Kawada Iet al (2017) Overcoming EGFR bypass signal‐induced acquired resistance to ALK tyrosine kinase inhibitors in ALK‐translocated lung cancer. Mol Cancer Res 15: 106–114 [DOI] [PubMed] [Google Scholar]
- Mundt F, Rajput S, Li S, Ruggles KV, Mooradian AD, Mertins P, Gillette MA, Krug K, Guo Z, Hoog Jet al (2018) Mass spectrometry‐based proteomics reveals potential roles of NEK9 and MAP2K4 in resistance to PI3K inhibition in triple‐negative breast cancers. Cancer Res 78: 2732–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER, Kalocsay M, Jané‐Valbuena J, Gelfand E, Schweppe DK, Jedrychowski Met al (2020) Quantitative proteomics of the cancer cell line encyclopedia. Cell 180: 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Sicinski P (2017) Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 17: 93–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CE, Borchers CH (2014) Mass spectrometry based biomarker discovery, verification, and validation–quality assurance and control of protein biomarker assays. Mol Oncol 8: 840–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico AR, Kelder T, van Iersel MP, Hanspers K, Conklin BR, Evelo C (2008) WikiPathways: pathway editing for the people. PLoS Biol 6: e184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R (2012) Selected reaction monitoring‐based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 9: 555–566 [DOI] [PubMed] [Google Scholar]
- Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, MacCoss MJ (2020) The Skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev 39: 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow O, Liu X, Haigis KM, Gygi SP, Paulo JA (2021) A compendium of murine (Phospho) peptides encompassing different isobaric labeling and data acquisition strategies. J Proteome Res 20: 3678–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature 464: 427–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecca VW, Wood E, Fedorenko IV, Paraiso KHT, Haarberg HE, Chen YI, Xiang Y, Sarnaik A, Gibney GT, Sondak VKet al (2014) Evaluating melanoma drug response and therapeutic escape with quantitative proteomics. Mol Cell Proteomics 13: 1844–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifai N, Gillette MA, Carr SA (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol 24: 971–983 [DOI] [PubMed] [Google Scholar]
- Rivera KD, Olive ME, Bergstrom EJ, Nelson AJ, Lee KA, Satpathy S, Carr SA, Udeshi ND (2021) Automating UbiFast for high‐throughput and multiplexed ubiquitin enrichment. bioRxiv 10.1101/2021.04.28.441860 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez H, Zenklusen JC, Staudt LM, Doroshow JH, Lowy DR (2021) The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 184: 1661–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenstein JM, Chooback N (2018) ALK inhibitors, resistance development, clinical trials. Curr Oncol 25: S59–S67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy S, Jaehnig EJ, Krug K, Kim B‐J, Saltzman AB, Chan DW, Holloway KR, Anurag M, Huang C, Singh Pet al (2020) Microscaled proteogenomic methods for precision oncology. Nat Commun 11: 532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattu K, Hochgräfe F, Wu J, Umapathy G, Schönherr C, Ruuth K, Chand D, Witek B, Fuchs J, Li PKet al (2013) Phosphoproteomic analysis of anaplastic lymphoma kinase (ALK) downstream signaling pathways identifies signal transducer and activator of transcription 3 as a functional target of activated ALK in neuroblastoma cells. FEBS J 280: 5269–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH (2009) PID: the Pathway Interaction Database. Nucleic Acids Res 37: D674–D679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger VE, Ren Y, Luetteke N, Huang Q, Chen L, Lawrence HR, Lawrence NJ, Haura EB, Koomen JM, Coppola Det al (2015) Inhibition of Shp2 suppresses mutant EGFR‐induced lung tumors in transgenic mouse model of lung adenocarcinoma. Oncotarget 6: 6191–6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Eckels J, Schilling B, Ludwig C, Jaffe JD, MacCoss MJ, MacLean B (2018) Panorama public: a public repository for quantitative data sets processed in skyline. Mol Cell Proteom 17: 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas Tet al (2014) Ceritinib in ALK‐rearranged non‐small‐cell lung cancer. N Engl J Med 370: 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhan X, Wu J (2015) A positive feedback loop between Gli1 and tyrosine kinase Hck amplifies shh signaling activities in medulloblastoma. Oncogenesis 4: e176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yan H, Frost P, Gera J, Lichtenstein A (2005) Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up‐regulating the insulin‐like growth factor receptor/insulin receptor substrate‐1/phosphatidylinositol 3‐kinase cascade. Mol Cancer Ther 4: 1533–1540 [DOI] [PubMed] [Google Scholar]
- Soste M, Hrabakova R, Wanka S, Melnik A, Boersema P, Maiolica A, Wernas T, Tognetti M, von Mering C, Picotti P (2014) A sentinel protein assay for simultaneously quantifying cellular processes. Nat Methods 11: 1045–1048 [DOI] [PubMed] [Google Scholar]
- Sperling AS, Burgess M, Keshishian H, Gasser JA, Bhatt S, Jan M, Słabicki M, Sellar RS, Fink EC, Miller PGet al (2019) Patterns of substrate affinity, competition, and degradation kinetics underlie biological activity of thalidomide analogs. Blood 134: 160–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopfer LE, Flower CT, Gajadhar AS, Patel B, Gallien S, Lopez‐Ferrer D, White FM (2021) High‐density, targeted monitoring of tyrosine phosphorylation reveals activated signaling networks in human tumors. Cancer Res 81: 2495–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Panciera T, Piccolo S (2018) YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20: 888–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Mani DC, Satpathy S, Fereshetian S, Gasser JA, Svinkina T, Olive ME, Ebert BL, Mertins P, Carr SA (2020) Rapid and deep‐scale ubiquitylation profiling for biology and translational research. Nat Commun 11: 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen BO, Dou Y, Zhang Y, Shi Z, Arshad OAet al (2019) Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177: 1035–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes‐associated protein localized in the cytoplasm. Genes Dev 15: 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhou J, Yang D, Yi L, Wang X, Ou Y, Yang D, Xu L, Xu M (2019) High expression of the transcriptional coactivator TAZ is associated with a worse prognosis and affects cell proliferation in patients with medulloblastoma. Oncol Lett 18: 5591–5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker JR, Halusa GN, Hoofnagle AN, Sharma V, MacLean B, Yan P, Wrobel JA, Kennedy J, Mani DR, Zimmerman LJet al (2014) CPTAC Assay Portal: a repository of targeted proteomic assays. Nat Methods 11: 703–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker JR, Zhao L, Abbatiello SE, Burgess M, Kuhn E, Lin ChenWei, Pope ME, Razavi M, Anderson NL, Pearson TWet al (2011) Evaluation of large scale quantitative proteomic assay development using peptide affinity‐based mass spectrometry. Mol Cell Proteomics 10: M110.005645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker JR, Zhao L, Saul R, Kaczmarczyk JA, Schoenherr RM, Moore HD, Jones‐Weinert C, Ivey RG, Lin C, Hiltke Tet al (2018) A multiplexed mass spectrometry‐based assay for robust quantification of phosphosignaling in response to DNA damage. Radiat Res 189: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Lucena‐Araujo AR, Nakayama S, de Figueiredo‐Pontes LL, Gonzalez DA, Yasuda H, Kobayashi S, Costa DB (2014) Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer 83: 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wang J, Wang X, Zhu J, Liu QI, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim Set al (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou J‐Y, Petyuk VA, Chen LI, Ray Det al (2016) Integrated proteogenomic characterization of human high‐grade serous ovarian cancer. Cell 166: 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen X, Stauffer S, Yang S, Chen Y, Dong J (2015) CDK1 phosphorylation of TAZ in mitosis inhibits its oncogenic activity. Oncotarget 6: 31399–31412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li Let al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang C‐Y, Chinnaiyan AMet al (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22: 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]