

Summary
Lung squamous cell carcinoma (LSCC) remains a leading cause of cancer death with few therapeutic options. We characterized the proteogenomic landscape of LSCC, providing a deeper exposition of LSCC biology with potential therapeutic implications. We identify NSD3 as an alternative driver in FGFR1-amplified tumors and low-p63 tumors overexpressing the therapeutic target survivin. SOX2 is considered undruggable, but our analyses provide rationale for exploring chromatin modifiers such as LSD1 and EZH2 to target SOX2-overexpressing tumors. Our data support complex regulation of metabolic pathways by crosstalk between post-translational modifications including ubiquitylation. Numerous immune-related proteogenomic observations suggest directions for further investigation. Proteogenomic dissection of CDKN2A mutations argue for more nuanced assessment of RB1 protein expression and phosphorylation before declaring CDK4/6 inhibition unsuccessful. Finally, triangulation between LSCC, LUAD and HNSCC identified both unique and common therapeutic vulnerabilities. These observations and proteogenomics data resources may guide research into the biology and treatment of LSCC.
Keywords: Lung Cancer, Squamous, Proteogenomics, Proteomics, Genomics, Protein, Phosphorylation, Acetylation, Ubiquitylation, CPTAC
In Brief
Comprehensive proteogenomic characterization of lung squamous cell carcinomas and paired normal adjacent tissues identifies taxonomic subclasses, alternative driver events and insights into immune modulation, as well as putative biomarkers and potential therapeutic targets.
Graphical Abstract
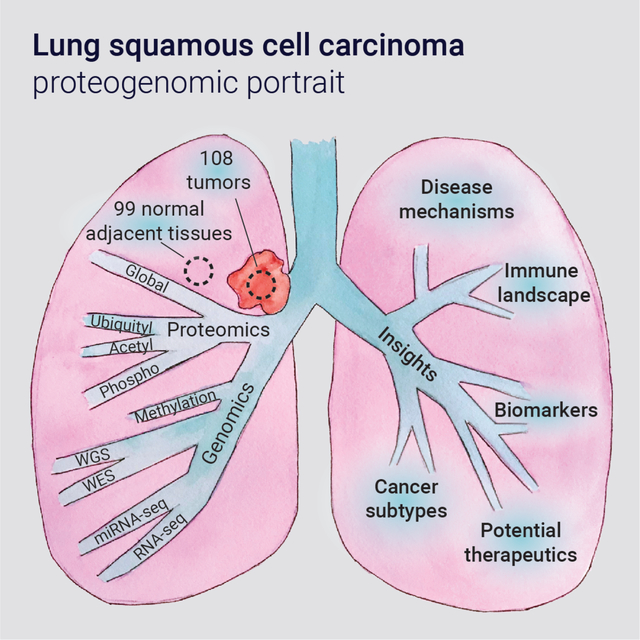
Introduction
Lung cancer is the leading cause of cancer-associated mortality (Bray et al., 2018; Siegel et al., 2020). Unlike lung adenocarcinomas (LUAD), patients with lung squamous cell carcinoma (LSCC) have not benefited from targeted therapies (Hammerman et al., 2012; Paik et al., 2019). Potentially druggable genetic events in three primary pathways (FGFR1, PI3K, or G1/S checkpoint genes such as CDKN2A) are found in upwards of 60% but targeting these clinically has largely failed (Paik et al., 2019). While other therapeutic regimens are being explored as part of the LungMAP consortium (Ardini-Poleske et al., 2017), to date only immunotherapy has evolved into a successful therapeutic strategy for patients with LSCC (Karachaliou et al., 2018; Paz-Ares et al., 2018). Given the lack of established actionable genomic targets, a comprehensive characterization of the proteogenomic landscape of LSCC, including the associated post-translational modifications (PTMs) that are key to protein activity and signaling, is a crucial step forward. Such studies will aid in connecting genomic aberrations to molecular and clinical phenotypes and in subsequently identifying therapeutic vulnerabilities and effective, biomarker-based patient stratification.
We characterized 108 prospectively-collected, treatment-naïve, primary LSCC tumors and 99 paired normal adjacent tissues (NATs), identifying actionable therapeutic protein targets and elucidating cellular signaling pathways and crosstalk between multiple PTMs. The dataset provides an exceptional resource to guide further research and support development of therapeutic modalities in LSCC.
Results
Proteogenomic landscape of LSCC
We performed deep-scale molecular analysis spanning nine different data types on LSCC tumors and NATs prospectively collected from 108 patients (Figure 1A, S1A, Table S1–3). Proteomics data were generated using TMT11-based multiplexing linked by a common reference (Figure S1A) and demonstrated high technical quality (Figure S1B–C). The cohort demographics are summarized in Figure 1B. Most self-reported never-smokers displayed high genomic smoking scores (Figure S1D), highlighting occasional discrepancy between self-reported smoking status and genomic evidence, as previously reported (Gillette et al., 2020). The genomic landscape reaffirmed previously reported somatic alterations in LSCC (Hammerman et al., 2012) (Figure 1C). Principal component analysis (PCA) of global proteomic and PTM data showed clear separation of tumors and NATs (Figure S1E).
Figure 1: Proteogenomic Landscape of LSCC.

A. Schematic showing the number of tumors and NATs profiled and the various data types generated in this study. The lower panel represents data completeness. WGS: Whole Genome Sequencing, WES: Whole Exome Sequencing. CNA: copy number alteration. DNAme: DNA methylation. pSTY: phosphoproteome. Ac: acetylproteome. Ub: ubiquitylproteome.
B. Stacked histograms indicating the distribution of patient phenotypes. Smoking History reflects self-report.
C. Co-occuring mutation plot indicating cancer-relevant genes. MutSig-based significantly mutated genes (SMGs, q-value < 0.1) in this dataset are highlighted in red font.
D. Heatmaps showing correlation between copy number alterations (CNA) and RNA (left) or proteomics (right). Red and green events represent significant (FDR <0.01) positive and negative correlations, respectively.
E. Flow chart for identification of cancer-associated genes (CAGs) that showed GISTIC-based focal amplification or deletion (q<0.25) and cis-effects in both mRNA and protein (FDR<0.05).
F. Differential protein expression (Log2 fold-change (FC)) in tumors with and without high-level amplification of the FGFR1 gene (GISTIC thresholded value =2).
G. Genes whose DNA methylation was significantly associated with cascading cis regulation of their cognate mRNA expression, protein level, phosphopeptide and acetylpeptide abundance. Shapes indicate the cis-effects across the indicated datasets. Named genes also showed differential expression between tumors and NATs.
Proteogenomic data helped annotate the impact of copy number alteration (CNA) events (Figure 1D,E). Of 5,523 significant (FDR<0.01) cis CNA-mRNA events, 2,154 were concordantly correlated with protein expression, including 138 “cancer associated genes” (CAG) (Figure 1E, Table S1). We identified six amplified (including WHSC1L1, CCND1, and SOX2) and 29 deleted (including NCOR1, SETD2, and CBL) CAGs in significant focal events (Q<0.25) (Figure 1E, Table S4) (Mermel et al., 2011). Intriguingly, WHSC1L1 (NSD3) is part of a recurrent focal amplicon (8p11.23) in LSCC that encompasses FGFR1. Therapies targeted against FGFR1 have been unsuccessful (Lim et al., 2016; Weeden et al., 2015). Proteomics data suggested that NSD3, rather than FGFR1, could be the critical driver oncogene within this amplicon (Figure 1F), nominating it as a potential therapeutic target. While this paper was under review, NSD3 was demonstrated to be a key regulator of LSCC tumorigenesis, mediating oncogenic chromatin changes (Yuan et al., 2021).
To investigate the impact of CNAs on noncognate gene products, we matched patterns of these significant trans-effects (vertical stripes in Figure 1D) to perturbation profiles from the Connectivity Map (CMap) (https://clue.io/cmap). Six CNAs (IKBKAP, PIN1, DNAJB1, IL18, NR2F6, AKAP) (Table S4) were enriched for both amplification and deletion (FDR=0.06) and associated (Fisher’s exact p<0.01) with clinical metadata. IL-18 deletion and amplification correlated with decreased and increased IL-18 protein expression and xCell immune score, respectively, (Figure S1F (upper panel)), consistent with its playing a key modulatory role in the tumor microenvironment of NSCLC (Timperi et al., 2017). NR2F6 amplification and deletion were correlated with NR2F6 protein expression and anticorrelated with xCell immune score (Figure S1F (lower panel)). NR2F6 acts as a non-redundant immune checkpoint in cancer, and even partial knockdown works synergistically with PD-L1 blockade (Klepsch et al., 2018).
LSCC tumor DNA showed overall hypermethylation relative to NATs (Figure S1G), with tumors separating into CIMP (CpG island methylator phenotype) clusters (Figure S1H). Multi-omic data allowed the identification of “cascading” promoter methylation cis-effects across cognate mRNA, protein and PTM abundances, supporting their functional significance. Of 90 genes that showed such significant (FDR<0.1) cis-effects in tumors (Table S4), 20 were also differentially expressed (FDR<0.01) between tumors and NATs (Figure 1G). While most were newly associated with LSCC, methylation-driven expression of FAM110A, PTGES3, PLAU and SLC16A3 (Faubert et al., 2017; Gao et al., 2018; Kikuchi et al., 2012; Liang et al., 2013; Payen et al., 2020; Showe et al., 2009) has been implicated in lung and other cancers.
Multi-omic clustering identifies five LSCC molecular subtypes, including one that is EMT-Enriched
We performed non-negative matrix factorization (NMF)-based single- and multi-omic unsupervised clustering on CNA, RNA, protein, phosphoprotein and acetylprotein datasets from 108 tumors, excluding ubiquitylprotein data as it was not available for the entire cohort (Figure 1A). The five resulting multi-omic subtypes (Figure 2A–B, Figure S2A) were named based on their predominant pathway associations and similarities to previously defined RNA clusters (Wilkerson et al., 2010). Significant associations between NMF subtypes and cohort metadata are presented in Table S1. Two NMF subtypes, “Basal-Inclusive” (B-I) and “Epithelial to Mesenchymal Transition-enriched” (EMT-E), emerged from partitioning of samples that showed similarity to the TCGA-derived RNA Basal cluster, although only B-I retained significant association after Bonferroni correction (Fisher’s p<0.01, Table S1). B-I tumors showed basaloid histology and upregulated metabolic, immune and estrogen receptor signaling (Figure 2B). EMT-E showed upregulation of EMT, Angiogenesis and Myogenesis (Figure 2B), with myxoid histologic features and fibroblast infiltration. A third subtype characterized by mutations in KEAP1, CUL3 and NFE2L2 genes and high-level amplification (GISTIC threshold value = 2) of SOX2 and TP63 (Figure 2A–B, Table S2) was labeled the “Classical” subtype, consistent with TCGA nomenclature and with a previous publication (Stewart et al., 2019). These showed classical histologic features, CIMP-high enrichment, upregulation of OxPhos- and proliferation-related pathways, and downregulation of immune signaling (Figure 2A–B). A fourth NMF subtype was designated “Inflamed-Secretory” (I-S) due to its alignment with the RNA-based Secretory cluster and strong upregulation of immune-related pathways. The fifth, “Proliferative-Primitive” (P-P) NMF subtype displayed upregulated proliferation-related pathways, downregulated immune signaling, and enrichment of CIMP-low samples. Within each of these five NMF clusters were samples with low NMF cluster membership scores, which could be considered as belonging to a “mixed” subgroup, since they showed features of more than one subtype. Mixed subgroup membership, a possible readout of tumor heterogeneity, conferred significantly worse survival (Figure 2C), an observation also made in a recent proteogenomic analysis of glioblastoma (Wang et al., 2021). The mixed class was associated in the current cohort with increased frequency of SOX2 amplifications (p = 0.0038) but not grade, mutation burden, stemness or other tested variables.
Figure 2: LSCC Molecular Subtypes and Associations.

A. NMF-based clustering of tumor CNA, RNA, protein, phosphosite and acetylsite profiles, showing five primary NMF subtypes (top sample annotation row).
B. Heatmap representing significantly enriched pathways (MSigDB Hallmark) in five multi-omic subtypes.
C. Kaplan-Meier plot comparing survival probability of patients whose tumors were core members of a specific NMF subtype (NMF Core) to those whose tumors had characteristics of more than one NMF subtype (NMF mixed).
D. Heatmap showing relative overexpression of mesenchymal proteins in the EMT-E subtype compared to others.
E. Correlation between ssGSEA-based enrichment of EMT (Hallmark genesets) and fibroblast proliferation (GO: Gene Ontology) genesets (Pearson correlation=0.65, p=2.8×10−14).
F. Distribution of RTK correlation-based phosphosite enrichment (RTK CBPE) scores for PDGFRB and ROR2 across the five NMF subtypes. Wilcoxon p values for CBPE scores in EMT-E vs other subtypes are 1.5×10−6 for PDGFRB and 2.7×10−7 for ROR2.
G. Proteins and phosphosites significantly associated with PDGFRB or ROR2 CBPE scores, known to play a role in EMT and extracellular matrix reorganization. The left panel shows Spearman correlation between PDGFRB CBPE scores and protein/phosphosite abundance profiles.
We compared B-I (Figure S2B) and EMT-E (Figure S2C) to other subtypes to identify distinguishing features. B-I had hyperphosphorylated M phase Histone 1 T11 (Happel et al., 2009) and elevated expression of both TACSTD2 (TROP2), a cell-surface glycoprotein that drives growth and metastasis (Hsu et al., 2020), and MARK2, a kinase that regulates cell polarity and leads to cisplatin resistance (Hubaux et al., 2015; Lewandowski and Piwnica-Worms, 2014). Transcription activator PBX3, which is upregulated only at the protein level (Figure S2B), promotes invasion, proliferation, and chemoresistance and is associated with poor prognosis, (Lamprecht et al., 2018) including in laryngeal squamous cell carcinoma (Wu et al., 2020). Pathway analysis of proteins highly correlated with PBX3 highlighted neutrophil activation and degranulation (Figure S2D), consistent with the significant upregulation of neutrophilic granule proteins and chemotactic factors (Figure S2E) and with the xCell-based deconvolution signature (Figure S2F). This correlation was not observed in paired NATs, suggesting a tumor-specific role.
In addition to a strong EMT signature (Figure 2B), the EMT-E subtype showed upregulated VCAN (Figure S2C), a tumor-promoting target of SNAIL (Zhang et al., 2019a) expressed in cancer-associated fibroblasts (CAFs) (Chida et al., 2016), and FHL3, a protein that stabilizes EMT-associated transcription factors (Li et al., 2020) (Figure S2C). DVL3, FN1 and FHL2, all Wnt pathway regulators, were upregulated in the proteomics dataset, and cell-cell signaling by Wnt was one of the significantly (FDR=0.04) enriched pathways in this subtype. As shown in Figure 2D–E, coordinated high expression of EMT and CAF proteins, xCell fibroblast scores, and a “fibroblast proliferation” signature suggested that CAFs and tumor epithelium might collaborate in the EMT-E phenotype. This was further supported by enrichment of EMT-related receptor tyrosine kinases (RTKs), as described below. Immunohistochemical co-staining of a small subset of tumors for the epithelial marker pan-cytokeratin (CK) and CAF marker α-smooth muscle actin (α-SMA) demonstrated the presence in EMT-E tumors of both CAFs and tumor cells undergoing EMT (Figure S2G), with significant enrichment of co-stained cells in the EMT-E subtype (Wilcoxon p=0.025), but not of cells showing single staining of either CK or α-SMA alone. TGFβ is a well-established and potent inducer of EMT (Su et al., 2020), and a Library of Integrated Network-Based Cellular Signatures-based (LINCS) query for compounds that reversed the EMT-E signature showed enrichment for TGFβ inhibitors (Figure S2H, Table S4H).
Integrating our global proteome data with a prior LSCC dataset (Stewart et al., 2019) resulted in six clusters largely recapitulating RNA, proteomic and multi-omic clusters defined in the current study (Figure S2I–J).
NMF EMT-E subtype tumors show phosphorylation-driven PDGFR and ROR2 signaling.
Though LSCC lacks the RTK mutations for which targeted inhibition has dramatically improved therapeutic options in LUAD, other modes of RTK activation, inferred from phosphoproteomic data, may nonetheless nominate targeted inhibition for some LSCC patients. Hence, we derived a serine/threonine-predominant correlation-based phosphosite enrichment score (CBPE score) for all RTKs in our tumor cohort. Of nine RTKs with high CBPE scores in LSCC tumors, seven were significantly associated with NMF subtypes (Kruskal-Wallis p <0.01). Scores for PDGFRB and ROR2, markedly elevated in EMT-E (Figure 2F), were highly correlated with the loss of epithelial cell-cell junction markers and upregulation of mesenchymal proteins involved in remodeling of extracellular matrix, induction of EMT, and promotion of cell migration, which mark the transition from epithelial to mesenchymal state (Figure 2G) (Herreño et al., 2019; Niu et al., 2012). As summarized in Figure S2K, we found phosphosite-based evidence for upregulation of both the planar cell polarity and calcium-dependent branches of non-canonical Wnt signaling, initiated through ROR2 or PDGFR (Tam et al., 2013), both targetable (Roskoski 2018; Debebe and Rathmell 2015) and recently implicated in the progression from a hybrid E/M to a highly mesenchymal state (Kröger et al., 2019). Further functional characterization is warranted to confirm the contribution of these RTKs to the EMT-E phenotype and to test the impact of their modulation.
Loss of CDK4/6 pathway inhibitors is a universal feature of LSCC but Rb1 expression is variable
We investigated the impact of recurrent mutations on cognate RNA, proteins and PTMs (cis-effects) and on a set of cancer associated genes (trans-effects) (Bailey et al., 2018). Significant (FDR<0.05) Cis/Trans pairs are shown in Figure 3A–D. RB1 mutated tumors had upregulated cell cycle-related protein expression. NOTCH1 mutation led to elevated GSK3B protein and phosphorylation (T433), downregulation of inhibitory sites on GSK3A (S21) and upregulation of NLK. NLK (Ishitani et al., 2010) and GSK3B (Foltz et al., 2002; Jin et al., 2009; Zheng and Conner, 2018) inhibit NOTCH1, recently described as a tumor suppressor in LSCC (Sinicropi-Yao et al., 2019), suggesting that both might inhibit NOTCH1 downstream signaling in tumors with NOTCH1 mutations. ARID1A is one of the most frequently mutated SWItch Sucrose Non-Fermentable (SWI/SNF) ATP-dependent chromatin remodeling complex members. Mutations were associated with higher tumor mutational burden (Shen et al., 2018) (Figure S3A) and led to protein downregulation, associated with worse overall survival in NSCLC (Hung et al., 2020).
Figure 3: Impact of Somatic Mutations on Proteogenomic Features.

A. Significant (Wilcoxon FDR<0.05) cis- (circles) and trans-effects (squares) of selected mutations (x axis) on the expression of cancer-associated gene products, with mRNA in blue and proteins in green.
B. Similar to panel A but showing phosphosites.
C. Similar to panel A but showing acetylsites.
D. Similar to panel A but showing ubiquitylsites.
E. CNA data for CDKN2A and RB1 was used to classify tumors as having homozygous deletions or three classes of loss of heterozygosity mutations: nonsense/frameshift indel, missense/inframe indel, and splicing (see Table S4). CDKN2A genetic and hypermethylation annotations were based on the effect of the aberration on the p16INK4a (p16) gene product, but the effects of these CDKN2A/p16 aberrations on both major isoforms (p16INK4a and p14ARF (p14)) at the RNA (barplot) and protein (heatmap directly below barplot) levels are shown. Samples with amplification of CCND1–3, CDK4, and CDK6 were assessed by GISTIC (threshold = 2), and the genomic status, protein, and phosphoprotein levels for RB1 are included. Also shown are RNA-based scores for the cell cycle (MGPS, the mean of cell cycle genes and E2F target and G2M checkpoint gene set scores derived from ssGSEA of Hallmark gene sets) and phosphosite-based CDK kinase activity scores for CDKs 1, 2, and 4 derived from single sample post translational modifications - signature enrichment analysis (ssPTM-SEA) of known kinase targets. Three tumors with copy number gain of CDKN2A are not included.
F. Correlation between differential regulation of protein abundance (Log2 Fold-change (FC)) versus phosphoprotein log2 FC in tumors with NRF2 pathway mutation (one or two mutations in KEAP1, CUL3, or NFE2L2) versus NRF2 WT tumors (no NRF2 pathway aberration).
G. NRF2 pathway score and RNA, protein and phosphoprotein expression of key NRF pathway members according to NMF subtype.
H. CDK5 protein expression (Log2 TMT ratio) by NMF subtype. P-values are from the Anova test.
I. PTM-SEA-derived normalized enrichment scores (NES) for pathways enriched in NRF2 pathway-mutated (Mutant) vs wild-type samples (WT) plotted against NES for pathways enriched in NMF Classical vs other subtypes. Significantly upregulated (FDR<0.05) PTM-SEA terms in the Classical subtype are indicated by red dots and labeled.
Paradoxically, mutations in CDKN2A, proposed as a potential biomarker for CDK4/6 inhibitors (Ahn et al., 2020; Middleton et al., 2020), resulted in increased RNA expression (Figure 3A). We analyzed the effects of genetic and epigenetic alterations of CDKN2A on both its major isoforms, p16INK4a (p16) and p14ARF (p14) (Table S4). As expected, its homozygous deletion resulted in the loss of expression of both isoforms (Figure 3E). However, only eight of the 58 CDKN2A wild-type (WT) samples showed p16 RNA expression levels that were comparable to samples with CDKN2A/p16 missense mutations (log2(FPKM+1)>2). Many of those CDKN2A/p16 mutations were unlikely to result in nonsense-mediated decay (Figure S3B), accounting for preserved expression in those samples. Notably, 28 WT tumors showed hypermethylation of the p16 but not the p14 promoter and suppression of RNA and protein expression for CDKN2A/p16, while 18 additional WT tumors had reduced expression despite having no clear genetic or epigenetic alteration. Furthermore, the WT samples with high CDKN2A/p16 expression had RB1 mutations or deletions and/or low Rb protein levels, consistent with a recently published pan-cancer analysis of the CDK4/6 pathway that showed mutual exclusivity between mutations in these two genes (Knudsen et al., 2020). This suggests that loss of one of these two key CDK4/6 pathway inhibitors is a universal feature of LSCC. Conversely, while amplification of Cyclin D - CDK4/6 complex genes is frequent in LSCC (Hammerman et al., 2012), there was no association between the amplification of CCND2, CCND3, CDK4, or CDK6 and either CDKN2A/p16 protein or Rb protein/phosphoprotein levels (Table S4). CCND1 amplification resulted in significantly higher mean levels of Rb protein and phosphoprotein compared to WT samples, though the distributions were overlapping (Figure S3C). Intriguingly, phospho-Rb levels, which reflect CDK4/6 activity, correlated with response to CDK4/6 inhibitors in LSCC cell lines despite the heterogeneous distribution in response for cells with mutations in CDKN2A and copy number alterations in CCND1 and CDKN2A (Figure S3D). LSCC trial data have generally shown minimal efficacy of CDK4/6 inhibition, but outlier responses have been present in trials with abemaciclib (Patnaik et al., 2016) and palbociclib (Ahn et al., 2020; Edelman et al., 2019; Middleton et al., 2020). While we found that CCND1 amplification resulted in higher average levels of phosphorylated Rb, the heterogeneity of Rb expression and phosphorylation in samples with CCND1 amplification provides a potential explanation for some patients’ lack of response to CDK4/6 inhibitor therapies. Improved prediction based on a downstream functional assessment of G1/S checkpoint alterations (i.e. RB1 protein expression and phosphorylation) may identify a subset of tumors sensitive to CDK4/6 inhibition.
NRF2 pathway activation in tumors with and without NRF2 pathway mutations
LSCC showed mutations of three key genes in the NRF2 antioxidant response pathway, NFE2L2 (NRF2), CUL3 and KEAP1. NFE2L2 phosphorylation was observed in samples with any of these mutations (Figure S3E). LSCC tumors were annotated as “NRF2 pathway mutated” (n=33; with either one hit [n=28] or two hits [n=5] in NFE2L2, KEAP1, and CUL3), or “NRF2 WT” (n=68). Compared to WT, NRF2 pathway mutated tumors showed highly concordant differential expression of mRNA, protein and phosphoprotein levels of NRF2 pathway genes (Figure 3F, Figure S3F–G, Table S4), some with therapeutic implications. A subset of NRF2 gene products showed a dosage effect (SQSTM1, NR0B1, AKR1B10, CARD11, FFAR2) with the five cases harboring two hits (Table S4) demonstrating increased up-/down-regulation of NRF2 pathway genes relative to one-hit cases (Figure S3H). Using the proteogenomic signature (Table S4) that defined NFE2L2 mutated tumors, we derived an ssGSEA-based NRF2 pathway score that was enriched not only in NRF2 pathway mutated tumors, but also in samples without NRF2 pathway mutations, especially in the NMF Classical subtype (Figure 3G). These samples showed increased NFE2L2 phosphorylation, indicative of NRF2 pathway activation. Although the kinase(s) responsible for phosphorylation of NRF2 in LSCC are unclear, CDK5 protein (Figure 3H) and PTM-SEA-derived activity (Krug et al., 2018) (Figure 3I) were significantly upregulated (FDR<0.05) in the Classical subtype. CDK5 phosphorylation of NFE2L2 S433 mediates its activation in astrocytes (Jimenez-Blasco et al., 2015) and can likely play a role in CDK5-mediated NRF2 activation in LSCC tumors. Intriguingly, unlike LUAD (Gillette et al., 2020), KEAP1 mutations did not result in significantly reduced protein expression in LSCC, although missense mutations were mostly nonoverlapping, suggesting heterogeneity in NRF2 pathway dysregulation in NSCLC subtypes.
Proteogenomic analysis of chromosome 3 prioritizes therapeutic targets in LSCC
Chromosome 3q, which harbors key squamous differentiation markers SOX2 and TP63 (Qian and Massion, 2008), showed the most dramatic arm-level amplification in this cohort (Figure S4A). To assess potential drivers in the 3q amplicon, we identified 3q genes for which CNA correlated with RNA and protein and expression differed between tumors and NATs. TP63 showed the highest elevation in tumors (Figure 4A). LSCC cell lines amplified for TP63 and SOX2 were highly dependent upon them, supporting oncogene addiction (Figure S4B).
Figure 4: Proteogenomic Impact of Chromosome 3q Amplification.

A. Differential protein expression (Log2 FC) between tumors and NATs for genes on chromosome arm 3q.
B. Frequency distribution of ΔNp63α RNA expression in tumors and NATs.
C. Differential protein expression in samples classified as ΔNp63α-low vs -high based on ΔNp63α transcript level. The outlier upregulated gene product in red is BIRC5, also known as survivin.
D. Differential expression of microRNAs in ΔNp63α-low vs -high samples.
E. Pearson correlation between expression of miR-205 and mRNA expression of its cognate, experimentally validated targets.
F. Pearson correlation between expression of miR-205 and protein abundance of its cognate targets.
G. Relationship between miR-205 expression (log2 TPM) and EMT (top, p=2.3×10−08, Correlation = −0.53) and DNA replication (bottom, p=1.1×10−06, Correlation= 0.47) scores.
H. Heatmap showing relative protein expression (TMT log ratio) of selected proteins with significant (FDR<0.01) Pearson correlation values (positive or negative) with SOX2 protein expression.
I. Pearson correlation (p= 5.2×10−07, Correlation = −0.46) between SOX2 protein expression and HALLMARK_IL6_JAK-STAT_signaling NES.
As previously reported (Campbell et al., 2018; Lo Iacono et al., 2011), ΔNp63α was the dominant TP63 isoform in this dataset (Figure S4C) and was highly correlated with TP63 expression (Figure S4D). Its amplification was associated with improved survival in the TCGA dataset (Figure S4E). In contrast to the general elevation of TP63 copy number, transcript, and protein abundance in LSCC tumors compared to NATs, 10 “Np63-low” samples histologically confirmed to be LSCC showed no elevation at RNA or protein levels (Figure 4B). The apoptosis inhibitor survivin (BIRC5) was among the most overexpressed proteins in this Np63-low group (Figure 4C). Notably, a substantial number of LSCC cell lines also had low Np63 expression (Figure S4F), and those TP63-low cell lines were significantly more vulnerable to the survivin inhibitor YM-155 (Figure S4G). Survivin promotes cell proliferation and G2M checkpoints (Wheatley and Altieri, 2019), evident in Np63-low samples (Figure S4H). Small molecule inhibitors and inhibitory peptides targeting survivin are being explored as part of ongoing clinical trials including in NSCLC (Giaccone et al., 2009; Kelly et al., 2013; Li et al., 2019a), but have shown modest or no improvement (Kelly et al., 2013). Our data on the newly identified Np63-low LSCC tumors suggests that TP63 status may identify patients with potential response to survivin inhibition.
Overexpression of miR-205, a biomarker to distinguish LSCC from other NSCLC (Lebanony et al., 2009) (Figure S4I), was associated with decreased promoter DNA methylation (Figure S4J) and elevation of ΔNp63α, a transcriptional regulator of this miRNA (Tran et al., 2013). miR-205 showed high correlation with TP63 expression and its expression was markedly reduced in Np63-low samples (Figure 4D, Figure S4K). As expected, the abundance of most target gene products was negatively correlated with miR-205 expression, including ZEB1/2 and PTEN (Cai et al., 2013; Gregory et al., 2008; Tellez et al., 2011; Vosgha et al., 2018) (Figure 4E). TP73 positively correlated with miR-205 at the transcript (Figure 4E) and still more strongly at the protein level (Figure 4F) and with TP63 protein but not RNA (Figure S4L). TP73 overexpression mediates ΔNp63α-dependent cell survival in squamous cell carcinoma (DeYoung et al., 2006), suggesting a mechanistic model in which functional and stoichiometric interaction between TP63 and TP73 leads to miR-205 overexpression in LSCC (Natan and Joerger, 2012). Finally, consistent with the downregulation of key EMT-related proteins, the TP63 target miR-205 showed significant negative correlation with EMT activity but positive correlation with DNA replication (Figure 4G). The seemingly paradoxical role of TP63 in repressing EMT has been confirmed in previous studies (Lindsay et al., 2011; Srivastava et al., 2018; Tran et al., 2013). Of note, although ΔNp63α and miR-205 were consistently highly overexpressed in the B-I and Classical subtypes compared to NATs, their abundances were variable and not particularly low in the EMT-E subtype. Thus, ΔNp63α, either via miR-205 or indirectly, may induce a strong squamous epithelial cell phenotype in the B-I and Classical tumors, with additional mechanisms leading to derepression and a metastatic phenotype (Vosgha et al., 2018), and other mechanisms beyond miR-205 may be involved in promoting the EMT phenotype in EMT-E samples.
SOX2, a recurrently amplified squamous cell marker often co-amplified with TP63, was also overexpressed in tumors (Figure 4A). SOX2 is considered undruggable, intensifying therapeutic interest in upstream or downstream targets. An unbiased search (Figure 4H) identified a significant correlation of LSD1 (KDM1A) with SOX2 protein. LSD1 regulates SOX2 expression (Zhang et al., 2013) and its inhibition in LSCC is currently being explored in a clinical setting in conjunction with immunotherapy (NCT04350463). Other identified chromatin regulators included KDM3A, known to regulate SOX2 expression in ovarian cancer (Ramadoss et al., 2017), and EZH2, whose inhibition in triple negative breast cancer leads to downregulation of SOX2 expression (Yomtoubian et al., 2020). EZH2 dependency was observed in SOX2 amplified LSCC cell lines (Figure S4M). The positive correlations of SOX2 with ALDH1A1, ALDH3A1, and WNT5A (Figure 4H) suggest stemness-like features in these tumors (Keysar et al., 2017; Liu et al., 2016; Patel et al., 2008). Interestingly, JAK1 was the protein most negatively correlated with SOX2 among other interferon-signaling proteins such as IRF3 and IFNGR1 (Figure 4H), and negative correlation was seen between SOX2 and JAK-STAT signaling (Figure 4I).
Crosstalk between lysine acetylation and ubiquitylation impacts cancer metabolism
Consensus clustering of K-GG peptide abundances after correcting for cognate protein abundance revealed two stable ubiquitylproteome clusters (Figure 5A, Table S5) associated with country of origin and ethnicity (Fisher’s exact p<0.01). Proteins in pathways such as glycolysis, JAK-STAT, MAPK, and immune signaling were differentially modified between these clusters (Figure 5A, Figure S5A–B, Table S5).
Figure 5: Ubiquitylation landscape in LSCC.

A. Consensus clustering of protein ratio-corrected K-GG (di-glycine) site abundances in tumor samples and their associations. Heatmap shows only protein ratio-corrected K-GG sites with differential abundance across clusters (FDR<0.01). Enriched pathways and molecular and clinical annotations are indicated.
B. Number of K-GG sites showing significant correlations (FDR< 0.01) with E3 ligases. Shown are the five E3 ligases with the highest proportion of positive correlations.
C. Pearson correlations between HERC5 protein expression and K-GG site protein-corrected abundance in key glycolytic enzymes PKM, PGK1, and ENO1.
D. HERC5 protein expression (log2 TMT ratio) with samples grouped by immune subtype. Significant (Kruskal−Wallis, p = 2.8×10−05) association is seen between HERC5 abundance and immune subtypes.
E. Representative examples of significant spatial clustering of lysine acetylsites (purple) on PGK1 (left) and ACADVL (right) protein 3-D structure space-filling models (cyan) as determined by PTM CLUMPS. (PGK1 structure = PDB ID:3ZOZ. ACADVL structure = PDB ID:3B9).
F. TXN protein levels in the NMF Classical subtype relative to NATs and other NMF subtypes (top left). Protein-corrected ubiquitylation (K-GG) sites are decreased on TXN1 in tumor subtypes relative to NATs (top right). TXNIP is decreased in tumor subtypes relative to NATs (lower left). TXN1 activator TXNRD1 is increased in the Classical subtype relative to NATs and other NMF subtypes (lower right). Kruskal−Wallis p-values are indicated in the respective plots.
G. Schematic representation of PTM-based modulation in LSCC tumors showing key enzymes in the metabolic and reactive oxygen species (ROS) pathways. Green and red arrows indicate higher and lower abundance of the corresponding PTMs in tumors. Putative ISGylation targets of HERC5 are indicated by dotted lines. A known regulatory PKM phosphosite observed to be modulated in LSCC tumors is also highlighted.
H. Lollipop charts showing the Log2 FC of acetylated (K-Ac) and ubiquitylated sites (K-GG) between tumors and NATs (Hyper: log2(FC)>1 or Hypo: <−1, FDR<0.01). The upper panel shows specific sites that were hyper-ubiquitylated and hypoacetylated in tumors; the lower panel shows specific sites that were hyperacetylated and hypo-ubiquitylated in tumors. Dot colors indicate protein fold change between tumors and NATs. “k” represents modified lysine.
I. Relative abundances of RAN K127 acetylation (K-Ac), ubiquitylation (K-GG) and RAN protein levels across NMF subtypes and NATs. Wilcoxon p-values are indicated above; ns represents p>0.05.
To identify candidate enzymes driving Ub and UbL modifications in LSCC, we correlated E3 ubiquitin ligases or deubiquitylases (DUBs) to KGG-sites (Table S5). Eighteen DUBs with at least one negatively correlated K-GG site and 35 E3 ligases with at least one positively correlated K-GG site were identified, with top sites shown in Figure 5B and Figure S5C. HERC5 is the major E3 ligase involved in conjugation of the UbL modification ISG15 in humans (Wong et al., 2006); hence the numerous positively correlated Ub/UbL sites (n=474; FDR<0.01) are likely ISG15 modifications. Consistent with this, we observed positive correlation between ISG15 and HERC5 abundance (Figure S5D). Positively-correlating Ub/UbL sites were enriched for glycolysis and TCA cycle annotations (Figure S5E). We observed several positively-correlating K-GG sites in the key glycolytic enzymes PKM, PGK1, and ENO1 (Figure 5C, Figure S5F). No significant negative correlations were observed between total protein abundance and their respective K-GG sites or HERC5, suggesting a regulatory rather than degradative role, as expected for ISGylation (Figure S5G, Figure S5H). ISG15 modifications have been described in these glycolytic enzymes (Albert et al., 2018; Giannakopoulos et al., 2005; Wong et al., 2006), and ISG15 knockdown in pancreatic cancer cells reduced oxidative phosphorylation and glycolysis (Alcalá et al., 2020). ISGylation can be induced by interferon in cancer (Tecalco-Cruz and Cruz-Ramos, 2018; Wong et al., 2006); accordingly, HERC5 and ISG15 were positively associated with proteome-derived IFN-G signaling scores (Figure S5I, Figure S5J), and HERC5 was increased in tumors of the IFN-gamma dominant subtype (Figure 5D). Recently, a direct link between immune signaling and glucose metabolism through ISGylation was shown in the context of pathogenic invasion (Zhang et al., 2019b). Collectively, our data supports a potential role for HERC5 as a bridge between immune signaling and metabolic regulation in LSCC.
This dataset allowed us to investigate crosstalk between lysine PTMs. We employed a modified version of CLUMPS (Kamburov et al., 2015) to detect clustering of either acetylation or ubiquitylation sites within protein 3D structures. Significant ubiquitylation clusters were detected in 33 proteins and significant acetylation clusters in 17 proteins (p<0.05) (Table S5). Top hits revealed enrichment of glycolysis, fatty acid metabolism and OxPhos (Table S5), supporting modulation of these metabolic pathways by ubiquitylation and acetylation. In tumor-NAT comparison, major glycolytic enzymes PGK1 and PKM showed reduced acetylation at multiple known inhibitory sites closely-positioned on the 3D structure, as well as on adjacent lysines surrounding the enzymatic pocket (Wang et al., 2015; Zhao et al., 2014)(Xiong et al., 2011) (Figure 5E, Figure S5K). Tumors also had decreased ubiquitylation at closely-positioned K-GG-sites for TXN1, an important redox regulator (Figures 5F). PGK1 and PKM deacetylation suggests increased activity of these key glycolytic enzymes in tumors, a conclusion supported by increased phosphorylation on known activating sites of regulatory proteins (PFKFB3 pS461, PKM pS37) (Bando et al., 2005; Yang and Lu, 2013). Upregulation of a glycolytic metabolic phenotype confers selective advantage to cancer cells by supporting uninterrupted growth, dynamically modulated by PTMs (DeBerardinis and Chandel, 2016; Hitosugi and Chen, 2014). While site-level roles of most Ub/UbL modifications and acetylsites are unclear, our data on acetylation, phosphorylation, and HERC5-mediated ISGylation of glycolytic enzymes hints at the complexity of regulation of this cancer hallmark.
Rapid growth of cells often leads to oxidative stress within tumors, which adapt by upregulating redox systems. Although thioredoxin (TXN) did not show overall differential protein expression between tumors and NATs, we observed increased TXN1 protein combined with decreased Ub sites, particularly in the NMF Classical subtype (Kruskal Wallis FDR<0.01). The TXN1 activator TXNRD1 (Cadenas et al., 2010) was also increased in the Classical subtype, while TXNIP, which modulates cellular redox by binding and inhibiting TXN1 (Morrison et al., 2014), was decreased (Figure 5F). TXN and TXNRD1 were highly correlated with NRF2 score (TXN: R=0.85; TXNRD1: R=0.8, both p<2.2×10−16), consistent with their role in response to oxidative stress. Redox modulation through this pathway is associated with tumor proliferation, differentiation and prognosis in multiple epithelial cancers (Fu et al., 2017), including NSCLC (Fernandes et al., 2009). The small proportion of glucose directed to the tumor mitochondria (Fan et al., 2019) and subsequently to the TCA and OXPHOS cycles leads to increased acetyl CoA within the mitochondria and thence to non-enzymatic acetylation and inhibition of key enzymes in the fatty acid oxidation pathway such as ECH1 and ACADVL (also identified in the CLUMPS analysis) (Baeza et al., 2015; Gandhi and Das, 2019; Hebert et al., 2013). We recapitulate the finding of increased ECH1 and ACADVL acetylation, including specific increase at the binding site of ACADVL in tumors (Figure 5E, Figure S5K). PTM-based modulation of these key metabolic and ROS pathways is summarized in Figure 5G.
As a complementary assessment of the crosstalk between lysine ubiquitylation and acetylation, we identified K-Ac or K-GG sites showing significant differential expression in tumors relative to NATs. Figure 5H shows 8 lysine sites where K-GG was upregulated and K-Ac downregulated in tumors (upper panel) and 36 sites demonstrating the opposite trend (lower panel). Among the latter was the GTP-binding nuclear protein RAN, involved in nucleocytoplasmic transport, mitotic progression and spindle assembly. Acetylation on multiple lysine residues can affect RAN localization, GTP/GDP cycle and import / export complex formation. Consistent with the role of RAN in mitosis, RAN K127-acK showed dramatic upregulation in the P-P subtype (Figure 5I), named in part for its generally high proliferation (Figure 2B). The specific functional relevance of K127-ac has not been previously demonstrated, but its selective upregulation suggests a functional role. While of great interest to the PTM community and potentially very powerful, such crosstalk analyses are nascent, and interpretations remain suitably cautious.
Immune landscape and regulation in LSCC
Consensus clustering based on the xCell (Aran et al., 2017) signatures (Table S6) identified three sets of tumors representing immunologically Hot, Warm, and Cold clusters (Figure 6A–C), as well as an NAT-enriched cluster. The NMF I-S subtype was significantly associated with the immune Hot cluster (Fisher’s exact p=6.371×10−09) (Figure 6A). The Hot cluster was characterized by increased macrophages, CD4+, CD8+ and regulatory T cells, and dendritic cells in both deconvolution and immunohistochemistry (IHC) analyses (Figures 6A,C, S6C,D), and showed upregulation of immune-related pathways (Figure 6A–B, Figure S6A, Table S6). Immunosuppressive mediators CTLA4, PD-1 (PDCD1), PD-L1 (CD274) and IDO1 and the key Treg transcriptional regulator FOXP3 were upregulated in Hot compared to Cold tumors, suggesting immune checkpoint-related therapeutic options in these tumors. The Hot tumor acetylproteome was chiefly enriched for OXPHOS, Mitochondrial Complex and TCA Cycle pathways, consistent with increased metabolism or increased mitochondrial acetyl-CoA leading to non-enzymatic acetylation of mitochondrial proteins (Figure 6D). Notably, NATs did not display consonant immune signaling (Figure S6B). The intermediate, Warm cluster displayed upregulation of immune-related pathways such as PD-1 signaling, Interferon gamma (IFN-G) response, and Allograft rejection relative to Cold tumors, but did not show the downregulation of cell cycle-related pathways observed in Hot tumors (Figure 6A–B, Figure S6A). In addition, canonical Wnt Signaling, active in tumor proliferation and immune evasion, was comparably upregulated in the Warm and Cold tumor proteomes (Figure 6B). Fifteen pQTLs were also identified in proteins differentially abundant between Hot and Cold tumors (Table S6, Figure S6E)
Figure 6: Immune Landscape of LSCC.

A. Heatmaps illustrate cell type compositions and activities of selected individual genes/proteins and pathways across the four immune clusters: Hot, Warm, and Cold tumor and NAT-enriched. Successive heatmaps illustrate xCell immune signatures, mRNA and protein expression of key immune-related markers, and ssGSEA pathway scores based on global proteomic data for biological pathways that were differentially regulated in immune groups based on both mRNA and global protein abundance (Common) or on global protein abundance alone (Proteomics only).
B. Pathway scores of key pathways differentially expressed across the immune clusters. Wilcoxon p-values for the individual comparisons are provided on top.
C. Contour plot of two-dimensional density based on Macrophage (x-axis) and CD8 T-cell scores (y-axis) showing the variation in these cell types’ distributions observed across the different immune clusters.
D. Acetylsites differentially expressed between Hot and Cold tumors. Acetylsites of genes contained in the Hallmark Oxidative Phosphorylation pathway are highlighted in blue, ARHGDIB K135 is highlighted in red, and remaining sites are in gray. Darker color designates significant sites (FDR < 0.1).
E. Regulation of Rho GTPase signaling including K135 acetylation of ARHGDIB.
F. Global protein abundance of RAC2, DOCK2 and ELMO1, acetylproteome abundance of ARHGDIB K135k and phosphorylation abundance of ARHGEF6 at Serine 225 in immune clusters. Wilcoxon p-values are reported.
G. RTK CBPE scores for 108 tumor samples and associated xCell signatures and pathway scores. The first heatmap shows CBPE scores of key RTKs, the second xCell signatures (Aran et al., 2017) and the third pathway scores based on global protein abundance.
H. Distribution of RTK CBPE scores for CSF1R, PDGFRB and FGFR2 stratified by immune clusters. Significance values (two-sided Wilcoxon test) between Hot clusters and combined Warm and Cold clusters are indicated on the violin plots.
I. Heatmap showing proteins and phosphosites correlated with CSF1R CBPE scores that are known to be involved in immune evasion. See also Figure S6 and Table S6
Rho GTPase signaling was upregulated in Hot tumors (Figure 6A, Table S6). ARHGDIB (RhoGDIb), the central regulatory molecule in GTPase activation (Garcia-Mata et al., 2011), was the most significantly acetylated protein in the Hot cluster, followed by metabolic mitochondrial enzymes (Figure 6D). Acetylation of its homolog RhoGDIa at the conserved K135 site regulates F-actin assembly (Kuhlmann et al., 2016a) by activating RhoA (Kuhlmann et al., 2016b). In contrast to RhoGDIa, the affinity of RhoGDIb for RhoA is low (Gorvel et al., 1998), and the K135 acetylation of RhoGDIb may act as a positive regulator of Rac signaling via its interaction with Rac2 instead (Moissoglu et al., 2009). In support of this hypothesis, we found significant upregulation in the Hot cluster of Rho GTPase Rac2 and numerous Rac-specific regulators (Figures 6F, S6F, Table S6). Notably, these proteins are primarily immune-specific, indicating a possible role for lysine acetylation of RhoGDIb in promoting immune cell functions in the Hot cluster. Immunohistochemical staining for RhoGDIb (ARHGDIB) in the subset of immune Hot tumors confirmed strong signals in infiltrating immune cells and mesenchymal cells (Figure S6G), similar to dispersed RhoGDIb-expressing cells identified as lymphocytes seen in renal cell carcinomas (von Klot et al., 2017). Thus, we propose that K135 acetylation of RhoGDIb is a potential regulatory mechanism of immune cell functions that is in line with the inflammasome signature (Wang et al., 2012) and immune-enriched networks (Peters et al., 2017).
We explored the immunological associations of the RTK CBPE scores described above (Figure 2I,J) in LSCC tumors (Figure 6G). CSF1R, predominantly expressed in macrophages, showed the highest CBPE scores, and was positively associated with the Hot cluster (Figure 6H) and consequently with immune-related pathways and cell types (Figure 6G). CSF1R correlated with immunosuppressive chemokine CCL5, phosphorylation of PIK3R1 Y580, downstream regulators of the actin cytoskeleton including WAS and WASF2 S103 (Cammer et al., 2009; Dovas et al., 2009; Mouchemore et al., 2013), and upregulation of AKAP13, among other immune-related proteins (Figure 6I). Tumor-associated macrophages ((Cassetta and Pollard, 2018), CSF1R signaling (Cannarile et al., 2017), and AKAP13 (Diviani et al., 2016) have each been proposed as anti-cancer targets.
Proteomic Biomarker Candidates for Prognosis, Diagnosis, and Treatment
Tumors and paired NATs revealed remarkable differences in protein expression and pathway enrichment (Figure 7A, Table S7). The top 50 of 502 differential proteins are shown in Figure S7A. In addition to 206 upregulated oncogenic proteins (OncoKB, Table S7), several CT antigens, attractive targets of CT-antigen therapies, were overexpressed in tumors (Figure S7B), with the I-S samples showing the fewest CT antigens. As complete long-term follow-up was not yet available for this cohort, we leveraged the TCGA LSCC dataset (Hammerman et al., 2012) to examine the association of these 502 candidate tumor biomarkers with overall survival (OS) or disease-free survival (DFS). Expression of four of the most highly differential genes showed significant association with poor OS and another 15 genes with poor DFS (Figure 7B, Figure S7C). Furthermore, knockdown of these tumor biomarker proteins reduced fitness across 16 LSCC cell lines (https://depmap.org/), suggesting critical roles in key cellular transformation and proliferation processes (Figure 7C, Table S7).
Figure 7: Proteomic Features Related to Diagnosis, Prognosis, or Treatment.

A. Differentially expressed proteins between tumors and NATs.
B. Significantly increased proteins (larger font indicates >4 fold) in the study LSCC cohort that are associated with poor overall survival (OS) or disease-free survival (DFS) in the TCGA LSCC cohort mRNA.
C. Genetic dependencies of 502 proteins (log2 FC >2, FDR<0.01 and NAs <50%) in LSCC cell lines (n=16) profiled as part of the Achilles Dependency-Map project.
D. Genes, ordered by their chromosomal location, that are deleted in at least 25% of the samples and significantly correlated to the immune score. Immune-related genes are highlighted.
E. EGFR protein and tyrosine phosphorylation levels compared to EGFR copy number and an EGFR activity score (PROGENy).
F. Heatmap showing Pearson correlation between the EGFR activity score represented by PROGENy (top) and RNA expression of EGFR ligands. *p<0.05
G. GO Biological Process enrichment for proteins with increased phosphorylation in EGFR amplified samples compared to non-amplified samples.
H. Summary roadmap figure partitioned into five major categories, indicated by different colors.
There is increasing interest in molecular taxonomies that help position cancer types in the context of related cancers. CNAs were more frequent in LSCC than in LUAD or head and neck squamous cell carcinoma (HNSCC), which respectively share tissue and cell type of origin (Figure S7D). Widespread deletions of immune-related genes in LSCC correlated with ESTIMATE immune scores (Figure 7D). Immune-related deletions were less prominent in HNSCC and especially LUAD (Figure S7E), except for Chromosome 3p deletion, encompassing five chemokine receptors. Squamous cancers shared 3q amplification, JAK2 deletion and skin-development proteins from across the genome. Finally, deletions of 4p14 and 5q, including multiple Toll-like receptor and interleukin signaling genes, were unique to LSCC. Among key tumor-specific phosphorylation events in LSCC, we identified upregulation of activating sites on 27 kinases, including MAPK14 and three others (DCK, EGFR, SRC) targetable by FDA-approved drugs (Figure S7F), with several being shared by LUAD and HNSCC tumors. Notably, EGFR protein was significantly upregulated in the squamous cancers but not LUAD (Figure S7G), although LUAD had many activating EGFR mutations, and activating phosphorylation of EGFR was increased in both LUAD and LSCC. High EGFR amplification in the LSCC cohort did not correlate with EGFR pathway activity as assessed by mRNA-based PROGENy scoring (Figure 7E) (Schubert et al., 2018). Instead, this activity measurement was highly correlated to mRNA abundance of the five EGFR ligands that are prominent in tumors (Figure 7F). Furthermore, EGFR amplified samples displayed increased phosphorylation on proteins related to actin filament and cell junction organization as well as cell-substrate adhesion (Figure 7G). These findings and similar results recently reported in HNSCC (Huang et al., 2021) support a squamous cell cancer feature in which EGFR ligand abundance drives canonical EGFR pathway activity and tumors with very high levels of EGFR may no longer require the ligand to drive activity. EGFR inhibition guided by EGFR abundance has been unsuccessful in squamous tumors. Since EGFR inhibitors such as cetuximab affect ligand-induced EGFR activity, ligand abundance, rather than EGFR amplification, might better predict EGFR inhibitor response in this population. Figure 7H graphically represents these and other key findings presented in this manuscript, emphasizing translatable implications.
Discussion
In this study, we provide detailed proteogenomic profiles of treatment-naïve, primary LSCC tumors and paired NATs, with unprecedented coverage of post-translational modifications including phosphorylation, acetylation and for the first time, ubiquitylation. Unsupervised NMF-based multi-omic clustering suggested a refinement to prior RNA-based clustering, dividing basal tumors into Basal-inclusive and EMT-enriched subtypes with biological differences of potential therapeutic significance. Molecular events downstream of copy number alterations showed NSD3 rather than FGFR1 protein overexpression in tumors with 8p11.23 amplification, providing both a potential explanation for the limited effectiveness of FGFR1-targeted treatment in this population and a potential alternative therapy. Parallel downstream analysis of DNA promoter methylation events provided insight into regulation of many proteins differential between cancer and NATs.
Comprehensive proteogenomic data provided a deeper exposition of established LSCC biology, often with potential therapeutic implications. We identified a subset of low-p63 tumors that were characterized by high levels of the known therapeutic target survivin. SOX2 is considered undruggable, but our analyses provide rationale for exploring LSD1 or other chromatin modifiers such as EZH2 to target SOX2 amplified / overexpressing tumors. The importance of glycolysis and oxidative stress in LSCC are well-appreciated, but our data support complex regulation of these metabolic pathways by crosstalk between ubiquitylation (or ubiquitin-like modifications), phosphorylation and acetylation. Immunotherapy represents the greatest advance in LSCC therapy in decades, but outcomes lag behind those seen in patients with LUAD and still only a minority of patients exhibit long-term responses (Haslam and Prasad, 2019; Herbst et al., 2018); numerous immune-related proteogenomic observations suggest directions for further investigation. Proteogenomic dissection of the downstream effects of CDKN2A mutations had clinical implications related both to the interpretation of trials utilizing CDK4/6 inhibitors in LSCC patients and to biomarker selection for future studies: though CDK4/6 inhibition has shown limited efficacy in LSCC trials to date, our analysis suggested that a more nuanced assessment of RB1 protein expression and phosphorylation is required before declaring this approach unsuccessful. Finally, triangulation between LSCC, LUAD and HNSCC demonstrated the influence of both tissue and cell type of origin on cancer biology, with therapeutic vulnerabilities both unique and common across these cancer types.
Limitations of the study
Many important clinical advances in NSCLC and other cancers have been driven by genomic profiling of bulk tumor material, and we anticipate that the same will prove true of bulk proteogenomic characterization, as performed here. Nevertheless, tumor heterogeneity is an important complication, and emerging methods for proteomics approaches to heterogeneity (Peng et al., 2020; Satpathy et al., 2020), such as pairing microdissection with mass spectrometry (Le Large et al., 2020) or using mass cytometry (Spitzer and Nolan, 2016) will be useful adjuncts. It is typically metastatic disease that proves lethal, and in-depth proteogenomic analysis of primary tumors paired with metastatic lesions will be needed to provide critical insights into metastatic biology. Importantly, the scope of a landscape study such as this necessitates that it be understood as hypothesis-generating, and a wider community effort will be required to validate biological observations and suggested therapeutic alternatives. Nevertheless, we hope this study will prove a valuable resource to the research and clinical communities, and advance the understanding and treatment of LSCC.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead author Michael A. Gillette (gillette@broadinstitute.org)
Material availability
This study did not generate new unique reagents.
Data and Code Availability
CPTAC LSCC proteomics data:
Proteomic Data Commons (PDC); https://pdc.cancer.gov/pdc/ with identifiers PDC000232, PDC000233, PDC000234 and PDC000237
CPTAC data portal LSCC: https://cptac-data-portal.georgetown.edu/study-summary/S063
CPTAC HNSCC proteomics data:
PDC: https://pdc.cancer.gov/pdc/ with identifiers PDC000221 and PDC000222
CPATC LUAD proteomics data:
PDC: https://pdc.cancer.gov/pdc/ with identifiers PDC000153, PDC000149, PDC000224)
Genomic and transcriptomic data files can be accessed at the Genomic Data Commons (GDC); https://portal.gdc.cancer.gov/, via dbGaP Study Accession: phs001287.v10.p5
All histologic https://www.cancerimagingarchive.net/datascope/cptac/home/ and radiologic details can be accessed from the The Cancer Imaging Archive (TCIA) Public Access https://wiki.cancerimagingarchive.net/display/Public/CPTAC-LSCC.
Sample annotation, processed and normalized data files are provided as Tables S1–S3.
Software and code used in this study are referenced in their corresponding STAR Method sections and also the Key Resource Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PTMScan® Ubiquitin Remnant Motif (K-ε-GG) Kit | Cell Signaling Technology | Catalog 5562 |
| PTMScan Acetyllysine Kit | Cell Signaling Technology | Catalog: 13416 |
| CD4 | Dako | Clone 4B12, RRID:AB_2728838 |
| CD8 | Bio-Rad | Clone 4B11, RRID:AB_322868 |
| CD163 | Abcam | Ab182422, RRID:AB_2753196 |
| ARHGDIB | Sigma-Aldrich | HPA051235, RRID:AB_2681398 |
| CK (pan-Cytokeratin) | Dako | clone AE1/AE3, RRID:AB_2132885 |
| alpha-SMA | Abcam | ab5694, RRID:AB_2223021 |
| Biological Samples | ||
| Primary tumor samples | See Experimental Model and Subject Details | N/A |
| Chemicals and Reagents | ||
| HPLC-grade water | J.T. Baker | Catalog: 4218-03 |
| Urea | Sigma | Catalog: U0631 |
| Sodium chloride | Sigma | Catalog: 71376 |
| 1M Tris, pH 8.0 | Invitrogen | Catalog: AM9855G |
| Ethylenediaminetetraacetic acid | Sigma | Catalog: E7889 |
| Aprotinin | Sigma | Catalog: A6103 |
| Leupeptin | Roche | Catalog: 11017101001 |
| Phenylmethylsulfonyl fluoride | Sigma | Catalog: 78830 |
| Sodium fluoride | Sigma | Catalog: S7920 |
| Phosphatase inhibitor cocktail 2 | Sigma | Catalog: P5726 |
| Phosphatase inhibitor cocktail 3 | Sigma | Catalog: P0044 |
| Dithiothretiol, No-Weigh Format | ThermoScientific | Catalog: 20291 |
| Iodoacetamide | Sigma | Catalog: A3221 |
| Lysyl endopeptidase | Wako Chemicals | Catalog: 129-02541 |
| Sequencing-grade modified trypsin | Promega | Catalog: V511X |
| Formic acid | Sigma | Catalog: F0507 |
| Acetonitrile, LC-MS grade | Honeywell | Catalog: 34967 |
| Acetonitrile, anhydrous | Sigma | Catalog: 271004 |
| Trifluoroacetic acid | Sigma | Catalog: 302031 |
| Tandem Mass Tag reagent kit – 11plex | ThermoFisher | Catalog: A34808 |
| 0.5M HEPES, pH 8.5 | Alfa Aesar | Catalog: J63218 |
| Hydroxylamine solution, 50% (vol/vol) in H2O | Aldrich | Catalog: 467804 |
| Methanol | Honeywell | Catalog: 34966 |
| Ammonium hydroxide solution, 28% (wt/vol) in H2O | Sigma | Catalog: 338818 |
| Ni-NTA agarose beads | Qiagen | Catalog: 30410 |
| Iron (III) chloride | Sigma | Catalog: 451649 |
| Acetic acid, glacial | Sigma | Catalog: AX0073 |
| Potassium phosphate, monobasic | Sigma | Catalog: P0662 |
| Potassium phosphate, dibasic | Sigma | Catalog: P3786 |
| MOPS | Sigma | Catalog: M5162 |
| Sodium hydroxide | VWR | Catalog: BDH7225 |
| Sodium phosphate, dibasic | Sigma | Catalog: S9763 |
| Phosphate-buffered saline | Fisher Scientific | Catalog: 10010023 |
| iVIEW DAB Detection Kit | Roche | Catalog: 760-091 |
| Equipment | ||
| Reversed-phase tC18 SepPak, 3cc 200mg | Waters | Catalog: WAT054925 |
| Solid-phase C18 disk, for Stage-tips | Empore | Catalog: 66883-U |
| Stage-tip needle | Cadence | Catalog: 7928 |
| Stage-tip puncher, PEEK tubing | Idex Health & Science | Catalog: 1581 |
| PicoFrit LC-MS column | New Objective | Catalog: PF360-75-10-N-5 |
| ReproSil-Pur, 120 Å, C18-AQ, 1.9-μm resin | Dr. Maisch | Catalog: r119.aq |
| Nanospray column heater | Phoenix S&T | Catalog: PST-CH-20U |
| Column heater controller | Phoenix S&T | Catalog: PST-CHC |
| 300 μL LC-MS autosampler vial and cap | Waters | Catalog: 186002639 |
| Offline HPLC column, 3.5-μm particle size, 4.6 um × 250 mm | Agilent | Catalog: Custom order |
| Offline 96-well fractionation plate | Whatman | Catalog: 77015200 |
| 700 μL bRP fractionation autosampler vial | ThermoFisher | Catalog: C4010-14 |
| 700 μL bRP fractionation autosampler cap | ThermoFisher | Catalog: C4010-55A |
| 96-well microplate for BCA | Greiner | Catalog: 655101 |
| Microplate foil cover | Corning | Catalog: PCR-AS-200 |
| Vacuum centrifuge | ThermoFisher | Catalog: SPD121P-115 |
| Centrifuge | Eppendorf | Catalog: 5427 R |
| Benchtop mini centrifuge | Corning | Catalog: 6765 |
| Benchtop vortex | Scientific Industries | Catalog: SI-0236 |
| Incubating shaker | VWR | Catalog: 12620-942 |
| 15 mL centrifuge tube | Corning | Catalog: 352097 |
| 50 mL centrifuge tube | Corning | Catalog: 352070 |
| 1.5 mL microtube w/o cap | Sarstedt | Catalog: 72.607 |
| 2.0 mL microtube w/o cap | Sarstedt | Catalog: 72.608 |
| Microtube caps | Sarstedt | Catalog: 72.692 |
| 1.5 mL snapcap tube | ThermoFisher | Catalog: AM12450 |
| 2.0 mL snapcap tube | ThermoFisher | Catalog: AM12475 |
| Instrumentation | ||
| Microplate Reader | Molecular Devices | Catalog: M2 |
| Offline HPLC System for bRP fractionation | Agilent 1260 | Catalog: G1380-90000 |
| Online LC for LC-MS | ThermoFisher | Catalog: LC140 |
| Q Exactive Plus Mass Spectrometer | ThermoFisher | Catalog: IQLAAEGAAPFALGMBDK |
| Q Exactive HF-X Mass Spectrometer | ThermoFisher | Catalog: 0726042 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | ThermoFisher | Catalog: IQLAAEGAAPFADBMBHQ |
| Critical Commercial Assays | ||
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold | Illumina | Catalog: RS-122-2301 |
| Infinium MethylationEPIC Kit | Illumina | Catalog: WG-317-1003 |
| Nextera DNA Exosome Kit | Illumina | Catalog: 20020617 |
| KAPA Hyper Prep Kit, PCR-free | Roche | Catalog: 07962371001 |
| BCA Protein Assay Kit | ThermoFisher | Catalog: 23225 |
| Deposited Data | ||
| DepMap: Mutation | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: Segmented copy number | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: Gene level copy number | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: RNAseq (transcript isoform) | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: Proteomics (RPPA) | CCLE 2019 (PMID: 31068700) | https://depmap.org/portal/download/ |
| DepMap: CRISPR KO screen (combined) | DepMapPublic 21Q1 (bioRxiv 2020.05.22.110247; doi: https://doi.org/10.1101/2020.05.22.110247) | https://depmap.org/portal/download/ |
| DepMap: shRNA screen (combined) | DEMETER2 Data v6 (PMID: 30389920) | https://depmap.org/portal/download/ |
| DepMap: GDSC drug screen | Sanger GDSC 1 (PMID: 27397505) | https://depmap.org/portal/download/ |
| DepMap: PRISM drug screen | PRISM Repurposing 19Q4 Secondary Screen (PMID: 32613204) | https://depmap.org/portal/download/ |
| PhosphoSitePlus | (Hornbeck et al., 2012) | https://www.phosphosite.org |
| Connectivity Map (CMAP) | (Lamb et al., 2006; Subramanian et al., 2017) | https://www.broadinstitute.org/connectivity-map-cmap |
| Human Protein Atlas (HPA) | (Uhlén et al., 2005) | https://www.proteinatlas.org |
| CT Antigen database | (Almeida et al., 2009) | http://www.cta.lncc.br |
| Dependency map (DepMap) | (Tsherniak et al., 2017) |
https://depmap.org/portal/ v3.3.8 is a GECKOv2 Achilles dataset |
| Library of Integrated Network-based Cellular Signatures (LINCS) | (Lamb et al., 2006; Subramanian et al., 2017) |
https://clue.io/data Expanded CMap LINCS Resource 2020 (1/28/2021 update) |
| CPTAC HNSCC cohort | (Huang et al., 2021) | https://cptac-data-portal.georgetown.edu/study-summary/S054 |
| CPTAC LSCC cohort | This study | https://cptac-data-portal.georgetown.edu/study-summary/S063 |
| Software and Algorithms | ||
| methylationArrayAnalysis (version 3.9) | (Maksimovic et al., 2016) | https://master.bioconductor.org/packages/release/workflows/html/methylationArrayAnalysis.html |
| Illumina EPIC methylation array (3.9) | Hansen KD, 2019 | https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylationEPICanno.ilm10b2.hg19.html |
| Methylation array analysis pipeline for CPTAC | Li Ding Lab | https://github.com/ding-lab/cptac_methylation |
| miRNA-Seq analysis pipeline for CPTAC | Li Ding Lab | https://github.com/ding-lab/CPTAC_miRNA |
| VEP | (McLaren et al., 2016) | https://github.com/Ensembl/ensembl-vep/tags |
| TNScope / DNAScope (Sentieon) | (Freed et al.) | sentieon.com |
| vcfAnno | (Pedersen et al., 2016) | https://github.com/brentp/vcfanno |
| VariantAnnotation (Bioconductor) | (Obenchain et al., 2014) | https://bioconductor.org/packages/release/bioc/html/VariantAnnotation.html |
| arriba_v1.1.0 | https://github.com/suhrig/arriba/ | |
| fusioncatcher_v1.10 | (Nicorici et al.) | https://github.com/ndaniel/fusioncatcher/blob/master/doc/manual.md |
| eQTLGen | (Westra et al., 2013) | https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook-(eQTLGen) |
| Pindel0.2.5 | (Ye et al., 2009) | http://gmt.genome.wustl.edu/packages/pindel/ |
| SignatureAnalyzer | (Kim et al., 2016) | https://software.broadinstitute.org/cancer/cga/msp |
| CNVEX | Marcin Cieslik Lab | https://github.com/mctp/cnvex |
| CRISP | Marcin Cieslik Lab | https://github.com/mcieslik-mctp/crisp-build |
| Spectrum Mill | Karl R. Clauser, Steven Carr Lab | https://proteomics.broadinstitute.org/ |
| ComBat (v3.20.0) | (Johnson et al., 2007) | https://bioconductor.org/packages/release/bioc/html/sva.html |
| gPCA | (Reese et al., 2013) | https://cran.r-project.org/web/packages/gPCA/index.html |
| GISTIC2.0 | (Mermel et al., 2011) | ftp://ftp.broadinstitute.org/pub/GISTIC2.0/GISTIC_2_0_23.tar.gz |
| iProFun | (Song et al., 2019) | https://github.com/WangLab-MSSM/iProFun |
| ESTIMATE | (Yoshihara et al., 2013) | https://bioinformatics.mdanderson.org/public-software/estimate/ |
| WebGestaltR | (Wang et al., 2017) | http://www.webgestalt.org/ |
| GSVA | (Hanzelmann et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| TSNet | (Petralia et al., 2018) | https://github.com/WangLab-MSSM/TSNet |
| xCell | (Aran et al., 2017) | http://xcell.ucsf.edu/ |
| CPTAC LSCC Data Viewer | Steven Carr lab | https://rstudio-connect.broadapps.org/CPTAC-LSCC2021/ |
| ConsensusClusterPlus | (Wilkerson and Hayes, 2010) | http://bioconductor.org/packages/release/bioc/html/CancerSubtypes.html |
| MS-GF+ | (Kim and Pevzner, 2014) | https://github.com/MSGFPlus/msgfplus |
| NeoFlow | (Wen et al., 2020) | https://github.com/bzhanglab/neoflow |
| netMHCpan | (Jurtz et al., 2017) | http://www.cbs.dtu.dk/services/NetMHCpan/ |
| Optitype | (Szolek et al., 2014) | https://github.com/FRED-2/OptiType |
| Customprodbj | (Wen et al., 2020) | https://github.com/bzhanglab/customprodbj |
| PDV | (Li et al., 2019b) | https://github.com/wenbostar/PDV |
| PepQuery | (Wen et al., 2019) | http://pepquery.org |
| PTM-SEA | (Krug et al., 2018)) | https://github.com/broadinstitute/ssGSEA2.0 |
| PTMsigDB | (Krug et al., 2018)) | http://prot-shiny-vm.broadinstitute.org:3838/ptmsigdb-app/ |
| Terra | Broad Institute data science platform. | https://terra.bio/ |
| Panoply | Broad Institute Proteomics Platform | https://github.com/broadinstitute/PANOPLY |
| CMap | (Lamb et al., 2006; Subramanian et al., 2017) | https://clue.io/cmap |
| LIMMA v3.36 (R Package) | (Ritchie et al., 2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| FactoMineR v1.41NMF(R - package) | (Gaujoux and Seoighe, 2010; Lê et al., 2008) | https://cran.r-project.org/web/packages/FactoMineR/index.html |
| MClust v5.4 (R package) | (Scrucca, Fop, Murphy and Raftery, 2017) | https://cran.r-project.org/web/packages/mclust/index.html |
| g:Profiler | (Raudvere U, et al., 2019) | https://biit.cs.ut.ee/gprofiler/gost |
| Cytoscape | (Shannon P, et al., 2003) | https://cytoscape.org/ |
| ImmuneSubtypeClassifier | (Gibbs, 2020) | https://github.com/CRI-iAtlas/ImmuneSubtypeClassifier |
| ProteinPaint | (Zhou et al., 2016) | https://pecan.stjude.doud/proteinpaint/ |
| Ordinal | Christensen RHB (2019) | https://CRAN.R-project.org/package=ordinal |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
A total of 113 participants (90 males, 23 females, 40–88 years old) were collected for this study by 13 different tissue source sites from seven different countries between May 2016 and August 2018, with a median follow-up time of 2.5 years. Five (one female and four male) samples were excluded based on further pathological assessment and 108 tumors and 99 paired NATs were used in this study. Only histopathologically-defined adult lung squamous tumors were considered for analysis, with an age range of 40–88. Institutional review boards at tissue source sites reviewed protocols and consent documentation adhering to the Clinical Proteomic Tumor Analysis Consortium (CPTAC) guidelines. Clinical data were obtained from tissue source sites and aggregated by an internal database called the CDR (Comprehensive Data Resource) that synchronizes with the CPTAC DCC (https://cptac-data-portal.georgetown.edu/). Clinical data can be accessed and downloaded from the DCC (Data Coordinating Center). Demographics, histopathologic information, and treatment details were collected. LSCC histopathology was confirmed for all cases by at least 2 expert pathologists based on high resolution images of H&E sections. For samples with low TP63 status, additional pathologic confirmation was performed. The histologic, genotypic, clinical, geographical and other associated metadata is summarized in Table S1.
METHOD DETAILS
Specimen Acquisition
The tumor, normal adjacent tissue (NAT), and whole blood samples used in this manuscript were prospectively collected for the CPTAC project. Biospecimens were collected from newly diagnosed patients with LSCC who underwent surgical resection and had received no prior treatment for their disease, including chemotherapy or radiotherapy. All cases had to be of acceptable LSCC histology but were collected regardless of surgical stage or histologic grade. Cases were staged using the AJCC cancer staging system 7th edition and the 8th edition. The tumor specimen weights ranged from 125 to 1560 milligrams. The average tissue mass was 220 mg. For most cases, three to four tumor specimens were collected. Paired histologically-normal adjacent lung tissues (NATs) were collected from the same patient at tumor resection. Each tissue specimen endured cold ischemia for less than 40 minutes prior to freezing in liquid nitrogen; the average ischemic time was 16 minutes from resection/collection to freezing. Specimens were flash frozen in liquid nitrogen. Histologic sections obtained from top and bottom portions from each case were reviewed by a board-certified pathologist to confirm the assigned pathology. For samples to be deemed acceptable, the top and bottom sections had to contain an average of 50% tumor cell nuclei with less than 20% necrosis. Specimens were shipped overnight from the tissue source sites to the biospecimen core resource (BCR) located at Van Andel Research Institute, Grand Rapids, MI using a cryoport that maintained an average temperature of less than −140°C. At the biospecimen core resource, specimens were confirmed for pathology qualification and prepared for genomic, transcriptomic, and proteomic analyses. Selected specimens were cryopulverized using a Covaris CryoPREP instrument and material aliquoted for subsequent molecular characterization. Genomic DNA and total RNA were extracted and sent to the genome sequencing centers. The whole exome and whole genome DNA sequencing and methylation EPIC array analyses were performed at the Broad Institute, Cambridge, MA. TotalRNA and miRNA sequencing were performed at the University of North Carolina, Chapel Hill, NC. Material for proteomic analyses were sent to the Proteomic Characterization Center (PCC) at the Broad Institute, Cambridge, MA.
Sequencing sample preparation
Our study sampled a single site of the primary tumor from surgical resections, with an internal requirement to process a minimum of 125mg of tumor issue and 50mg of NAT. DNA and RNA were extracted from tumor and NAT specimens in a co-isolation protocol using Qiagen’s QIAsymphony DNA Mini Kit and QIAsymphony RNA Kit. Genomic DNA was also isolated from peripheral blood (3–5mL) to serve as matched normal reference material. The Qubit™ dsDNA BR Assay Kit was used with the Qubit® 2.0 Fluorimeter to determine the concentration of dsDNA in an aqueous solution. Any sample that passed quality control and produced enough DNA yield to go through the multiple planned genomic assays was sent for genomic characterization. RNA quality was quantified using the NanoDrop 8000 and quality assessed using an Agilent Bioanalyzer. A sample of sufficient quantity that passed RNA quality control and had a minimum RIN (RNA integrity number) score of 7 was subjected to RNA sequencing. Identity matches for germline, normal adjacent tissue, and tumor tissue were confirmed at the BCR using the Illumina Infinium QC array. This beadchip contains 15,949 markers designed to prioritize sample tracking, quality control, and stratification.
Whole exome Sequencing (WES)
Library construction and Hybrid Selection
Library construction was performed as described in (Fisher et al., 2011), with the following modifications: initial genomic DNA input into shearing was reduced from 3μg to 20–250ng in 50μL of solution. For adapter ligation, Illumina paired-end adapters were replaced with palindromic forked adapters, purchased from Integrated DNA Technologies (IDT), with unique dual-indexed molecular barcode sequences to facilitate downstream pooling. Kapa HyperPrep reagents in 96-reaction kit format were used for end repair/A-tailing, adapter ligation, and library enrichment PCR. In addition, during the post-enrichment SPRI cleanup, elution volume was reduced to 30μL to maximize library concentration, and a vortexing step was added to maximize the amount of template eluted. After library construction, libraries were pooled into groups of up to 96 samples. Hybridization and capture were performed using the relevant components of Illumina’s Nextera Exome Kit and following the manufacturer’s suggested protocol, with the following exceptions: First, all libraries within a library construction plate were pooled prior to hybridization. Second, the Midi plate from Illumina’s Nextera Exome Kit was replaced with a skirted PCR plate to facilitate automation. All hybridization and capture steps were automated on the Agilent Bravo liquid handling system.
Cluster Amplification and Sequencing
After post-capture enrichment, library pools were quantified using qPCR (KAPA Biosystems) using an automated assay on the Agilent Bravo with probes specific to the ends of the adapters. Based on qPCR quantification, libraries were normalized to 2nM. Cluster amplification of DNA libraries was performed following manufacturer’s protocol (Illumina) using exclusion amplification chemistry and flowcells. Flow cells were sequenced utilizing sequencing-by-synthesis chemistry. The flow cells were then analyzed using RTA v.2.7.3 or later. Each pool of whole exome libraries was sequenced on paired 76-cycle runs with two 8-cycle index reads across the number of lanes needed to meet coverage for all libraries in the pool. Pooled libraries were run on HiSeq4000 paired-end runs to achieve a minimum of 150x on-target coverage per library. The raw Illumina sequence data were demultiplexed and converted to FASTQ files; adapter and low-quality sequences were trimmed. The raw reads were mapped to the GRCh38/hg38 human reference genome and the validated BAMs were used for downstream analysis and variant calling.
Whole genome sequencing (WGS)
Cluster Amplification and Sequencing
An aliquot of genomic DNA (350ng in 50μL) was used as the input into DNA fragmentation (aka shearing). Shearing was performed acoustically using a Covaris focused-ultrasonicator, targeting 385bp fragments. Following fragmentation, additional size selection was performed using SPRI cleanup. Library preparation was performed using a commercially available KAPA Hyper Prep without amplification module kit (KAPA Biosystems) and with palindromic forked adapters with unique 8-base index sequences embedded within the adapter (IDT). Following sample preparation, libraries were quantified using quantitative PCR (KAPA Biosystems), with probes specific to the ends of the adapters using the automated Agilent’s Bravo liquid handling platform. Based on qPCR quantification, libraries were normalized to 1.7nM and pooled into 24-plexes.
Sample pools were combined with HiSeqX Cluster Amp Reagents EPX1, EPX2, and EPX3 into single wells on a strip tube using the Hamilton Starlet Liquid Handling system. Cluster amplification of the templates was performed according to the manufacturer’s protocol (Illumina) with the Illumina cBot. Flow cells were sequenced to a minimum of 15x on HiSeqX utilizing sequencing-by-synthesis kits to produce 151bp paired-end reads. Output from Illumina software was processed by the Picard data processing pipeline to yield BAM files containing demultiplexed, aggregated, aligned reads. All sample information tracking was performed by automated LIMS messaging.
Array Based Methylation Analysis
The Methylation EPIC array uses an 8-sample version of the Illumina Beadchip capturing >850,000 methylation sites per sample. Two hundred and fifty nanograms of DNA was used for the bisulfite conversion using Infinium MethylationEPIC BeadChip Kit (Illumina). The EPIC array includes sample plating, bisulfite conversion, and methylation array processing. After scanning, the data was processed through an automated genotype-calling pipeline. Data output consisted of raw idats and a sample sheet.
RNA and miRNA sequencing
Quality Assurance and Control of RNA Analytes
All RNA analytes were assayed for RNA integrity, concentration, and fragment size. Samples for total RNA-seq were quantified on a TapeStation system (Agilent, Inc. Santa Clara, CA). Samples with RINs >7.0 were considered high quality and were considered for sequencing. Total RNA-seq libraries were generated using 300 nanograms of total RNA using the TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold and bar-coded with individual tags following the manufacturer’s instructions (Illumina). Total RNA Libraries were prepared on an Agilent Bravo automated liquid handling system. Quality control was performed at every step, and the libraries were quantified using a TapeStation system.
Total RNA Sequencing
Indexed libraries were prepared and run on HiSeq4000 paired-end 75 base pairs to generate a minimum of 120 million reads per sample library with a target of greater than 90% mapped reads. The raw Illumina sequence data were demultiplexed and converted to FASTQ files, and adapter and low-quality sequences were trimmed. Samples were then assessed for quality by mapping reads to GRCh38/hg38, estimating the total number of mapped reads, amount of RNA mapping to coding regions, amount of rRNA in the sample, number of genes expressed, and relative expression of housekeeping genes. Samples passing this QA/QC were then clustered with other expression data from similar and distinct tumor types to confirm expected expression patterns. Atypical samples were then SNP typed from the RNA data to confirm source analyte. FASTQ files of all reads were then uploaded to the GDC repository.
miRNA-seq Library Construction
miRNA-seq library construction was performed from the RNA samples using the NEXTflex Small RNA-Seq Kit (v3, PerkinElmer, Waltham, MA) and barcoded with individual tags following the manufacturer’s instructions. Libraries were prepared on a Sciclone Liquid Handling Workstation. Quality control was performed at every step, and the libraries were quantified using a TapeStation system and an Agilent Bioanalyzer using the Small RNA analysis kit. Pooled libraries were then size selected according to NEXTflex kit specifications using a Pippin Prep system (Sage Science, Beverly, MA).
miRNA Sequencing
Indexed libraries were loaded on the HiSeq4000 to generate a minimum of 10 million reads per library with a minimum of 90% reads mapped. The raw Illumina sequence data were demultiplexed and converted to FASTQ files for downstream analysis. Resultant data were analyzed using a variant of the small RNA quantification pipeline developed for TCGA (Chu et al., 2016). Data from samples were assessed for the number of miRNAs called, species diversity, and total abundance before uploading to the GDC repository.
Mass Spectrometry methods
The protocols below for protein extraction, tryptic digestion, TMT-11 labeling of peptides, peptide fractionation by basic reversed-phase liquid chromatography, phosphopeptide enrichment using immobilized metal affinity chromatography, and LC-MS/MS were performed as previously described in depth (Gillette et al., 2020). Acetyl-enrichment was performed as described before (Krug et al., 2020) with modifications as indicated below.
Protein Extraction and Tryptic Digestion
Approximately fifty milligrams (wet weight) of cryopulverized human LSCC and NAT samples were homogenized in the lysis buffer for 30 minutes at a ratio of about 200 uL lysis buffer for every 50 mg wet weight tissue. The lysis buffer consisted of 8 M urea, 75 mM NaCl, 1 mM EDTA, 50 mM Tris HCl (pH 8), 10 mM NaF, phosphatase inhibitor cocktail 2 (1:100; Sigma, P5726) and cocktail 3 (1:100; Sigma, P0044), 2 μg/mL aprotinin (Sigma, A6103), 10 μg/mL leupeptin (Roche, 11017101001), and 1 mM PMSF (Sigma, 78830). Lysates were centrifuged at 20,000 g for 10 minutes and protein concentrations of the clarified lysates were measured by BCA assay (Pierce). Protein lysates were subsequently reduced with 5 mM dithiothreitol (Thermo Scientific, 20291) for an hour at 37C and alkylated with 10 mM iodoacetamide (Sigma, A3221) for 45 minutes in the dark at room temperature. Prior to digestion, samples were diluted 4-fold to achieve 2 M urea with 50mM Tris HCl (pH 8). Digestion was performed with LysC (Wako, 129–02541) for 2 hours and with trypsin (Promega, V511X) overnight, both at a 1:50 enzyme-to-protein ratio and at room temperature. Digested samples were acidified with formic acid (FA; Sigma, F0507) to achieve a final volumetric concentration of 1% (final pH of ~3), and centrifuged at 1,500 g for 15 minutes to clear precipitated urea from peptide lysates. Samples were desalted on tC18 SepPak columns (Waters, 200mg, WAT054925) and dried down using a SpeedVac apparatus.
Common Reference (CR) Pool and Plex layout
Proteome, Phosphoproteome, and Acetyprotome:
The proteomic, phosphoproteomic and acetylproteome analyses of LSCC samples consisted of 22 TMT-11 plex experiments. To facilitate quantitative comparison between all samples across experiments, a common reference (CR) sample was included in each 11-plex. A common physical, rather than in silico reference was used for this purpose for optimal quantitative precision between TMT-11 experiments. Considerations prior to creating the reference sample were that this sample needed to be of adequate quantity to cover all planned experiments for both the discovery and future confirmatory sets (not part of this study) with overhead for additional possible experiments. The CR included all (212/212) of the samples analyzed in the discovery set TMT experiments, yielding a CR that was representative of all the samples in the study. Making the CR as representative of the study as a whole was particularly important since by definition only analytes represented in the reference sample would be included in the final ratio-based data analyses.
110 unique tumor samples with 102 paired NAT samples are distributed among 22 11-plex experiments, with 10 individual samples occupying the first 10 channels of each experiment and the 11th channel being reserved for the reference sample. All the tumors were in the C channels and all the normal samples were in the N channels. The first 8 channels of each experiment contained 4 tumor/normal pairs, with each pair of patient samples adjacent to each other. Channels 9 and 10 (130C and 131N) either contained a 5th tumor/normal pair, or an unpaired tumor sample in channel 9 and an alternate common reference sample in channel 10. These included 3 global LUAD common references, 3 HNSCC common references from the team at JHU, and 2 Tumor only common references from this LSCC discovery set.
To ensure capacity for additional samples or experiments given a target input of 300 μg peptide per channel per experiment, 18 mg total was targeted for reference material. To meet these collective requirements, all the samples in the discovery set that had enough material were included in the CR. After reserving a conservative 400 μg peptide / sample for individual sample analysis, an additional amount of 120 μg for each of the samples with adequate quantities were pooled. All 212 samples were selected for the combined tumor/normal CR. To make the combined CR, tumor only and normal only CRs were first created separately, with 110 tumor samples and 102 normal samples. After creating tumor and NAT only CRs at 150 ug and 150ug amounts, a pool of combined CR was made, comprised of 8.6 mg tumor only reference and 8.6 mg normal only reference. The 17.2 mg pooled reference material was divided into 55 300 μg aliquots, dried down, and frozen at −80°C until use.
Ubiquitylproteome:
750 ug of unlabeled peptides was required from each sample to ensure depth of coverage in the ubiquitylproteome, which restricted the number of samples available for profiling. 147/212 samples from the proteome, phosphoproteome, acetylproteome set were included in the ubiquitylproteome analysis. A separate ubiquitylproteome CR was constructed prior to KGG enrichment, due to limitations in sample material (both patient material and remaining CR aliquots to be used in future LSCC proteome, phosphoproteome, and acetylproteome processing.. In order to make TMT ratio-based quantitation comparable across omes, the ubiquitylproteome CR was constructed to be as similar as practically possible to the proteome, phosphoproteome, acetylproteome CR. The ubiquitylproteome CR contains all samples from the proteome, phosphoproteome, acetylproteome set with material remaining to contribute, 209/212. The ubiquitylproteome CR contributions were of 3 types:
104/147 samples included in ubiquitylproteome study and made full CR contribution, >1000ug available.
42/147 samples included in ubiquitylproteome study and made partial CR contribution, 750ug −1000 ug available.
63/65 samples from the proteome, phosphoproteome, acetylproteome set not included in ubiquitylproteome study, but made full CR contribution, <750 ug available.
CR tumor and normal peptides were combined at a ratio of 55:45 to reflect the higher proportion of tumor samples in the experimental design (as opposed to proteome, phosphoproteome, acetylproteome CR’s proportion of 50:50), and aliquoted into 750 ug aliquots.
In the ubiquitylproteome experimental design, 15 11-plex experiments contained 87 unique tumor samples in the C channels and 60 normal samples in the N channels, with the first 8 channels of every 11-plex consisting of patient-paired tumor/normal samples adjacent to each other. The 11th channel was reserved for the ubiquitylproteome common reference sample. Channel 9 (130C) contained an unpaired tumor, while channel 10 (131N) contained either an unpaired tumor, or in 3 plexes, a LUAD ubiquitylproteome common reference (which was constructed similarly for that experiment, unpublished).
TMT-11 Labeling of Peptides
Proteome, Phosphoproteome, Acetylproteome:
300 μg of desalted peptides per sample (based on peptide-level BCA after digestion) were labeled with 11-plex TMT reagents and combined for multiplexed analysis using a “reduced labeling” approach (Zecha et al., 2019). To conservatively yield extra labeled peptide in case a given sample demonstrated unsuitably low TMT reporter ion intensity during channel mixing control LC-MS/MS runs, 400 ug peptides per sample were initially labeled, with only 300 ug combined per sample for the final multiplexed experiments upon successful quality control. For each 400 μg peptide aliquot of an individual sample, peptides were reconstituted to 5 mg/ml with 50 mM HEPES (pH 8.5) solution and 400 ug labeling reagent dissolved in 20 uL anhydrous acetonitrile was added. After 1 h incubation with shaking, samples were diluted 1:1 with a solution of 80% 50 mM HEPES and 20% acetonitrile. After confirming good label incorporation, 5 μL of 5% hydroxylamine was added to quench the unreacted TMT reagents. Good label incorporation was defined as having less than 3% isobaric under-labeled MS/MS spectra in each sample, as measured by LC-MS/MS after taking out a 2 μg aliquot from each sample and analyzing 0.25 μg. If a sample did not have sufficient label incorporation, additional TMT was added to the sample and another 1 h incubation was performed with shaking. At the time that the labeling efficiency quality control samples were taken out, an additional 4 μg of material from each sample was taken out and combined as a mixing control. After analyzing the mixing control sample by LC-MS/MS, intensity values of the individual TMT reporter ions were summed across all peptide spectrum matches and compared to ensure that the total reporter ion intensity of each sample met a threshold of +/− 15% of the internal reference. If necessary, adjustments were made by either using additional labeled material or reducing an individual sample’s contribution to the mixture, and analyzing a subsequent mixing control, until all samples met the threshold and were thus approximately 1:1:1.... Differentially labeled peptides were then mixed (11 × 300 μg), dried down via vacuum centrifuge, and the quenched, combined sample was subsequently desalted on a 200 mg tC18 SepPak column. The same lot of TMT reagents was used for all samples (UA280170/TL272399).
Peptide Fractionation
For Proteome, Phosphoproteome, Acetylproteome:
To reduce sample complexity, peptide samples were separated by high pH reversed phase (RP) separation as described previously (Mertins et al., 2018). A desalted 3.3 mg, 11-plex TMT-labeled experiment (based on peptide-level BCA after digestion) was reconstituted in 935 μL 4.5mM ammonium formate (pH 10) and 2% acetonitrile, centrifuged for 7 min at 20000 g to precipitate insoluble peptides, loaded on a 4.6 mm x 250 mm column RP Zorbax 300 A Extend-C18 column (Agilent, 3.5 μm bead size), and separated on an Agilent 1260 Series HPLC instrument using basic reversed-phase chromatography. Solvent A (2% acetonitrile, 4.5 mM ammonium formate, pH 10) and a nonlinear increasing concentration of solvent B (90% acetonitrile, 4.5 mM ammonium formate, pH 10) were used to separate peptides. The 4.5 mM ammonium formate solvents were made by 40–fold dilution of a stock solution of 180 mM ammonium formate, pH 10. To make 1L of stock solution, add 25 mL of 28% (wt/vol) ammonium hydroxide (28%, density 0.9 g/ml, Sigma-Aldrich) to ~850ml of HPLC grade water, then add ~35 mL of 10 % (vol/vol) formic acid (>95% Sigma-Aldrich) to titrate the pH to 10.0; bring the final volume to 1 liter with HPLC grade water. The 96 minute separation LC gradient followed this profile: (min: %B) 0:0; 7:0; 13:16; 73:40; 77:44; 82:60; 96:60. The flow rate was 1 mL/min. Per 3.3 mg separation, 82 fractions were collected into a 96 deep-well x 2mL plate (Whatman, #7701–5200), with fractions combined in a step-wise concatenation strategy and acidified to a final concentration of 0.1% FA as reported previously. An additional 14 fractions were collected from the 96 deep-well plate for fraction A, which are the early eluting fractions that tend to contain multi-phosphorylated peptides. 5% of the volume of each of the 24+A proteome fractions was allocated for proteome analysis, dried down, and re-suspended in 3% MeCN/0.1% FA (MeCN; acetonitrile) to a peptide concentration of 0.25 μg/ μL for LC-MS/MS analysis. The remaining 95% of concatenated 24 fractions were further combined into 12 fractions, with fraction A as a separate fraction. These 13 fractions were then enriched for phosphopeptides as described below.
Phosphopeptide Enrichment
Ni-NTA agarose beads were used to prepare Fe3+-NTA agarose beads. In each phosphoproteome fraction, ~261.25 μg peptides (based on peptide-level BCA after digestion with uniformly-distributed fractionation presumed) were reconstituted in 522.5 μL 80% MeCN/0.1% TFA (trifluoroacetic acid) solvent and incubated with 10 μL of the IMAC beads for 30 minutes with end-over-end rotation at RT. After incubation, samples were briefly spun down on a tabletop centrifuge; clarified peptide flow-throughs were separated from the beads; and the beads were reconstituted in 200 μL IMAC binding/wash buffer (80 MeCN/0.1% TFA) and loaded onto equilibrated Empore C18 silica-packed stage tips (3M, 2315). Samples were then washed twice with 50 μL of IMAC binding/wash buffer and once with 50 uL 1% FA, and were eluted from the IMAC beads to the stage tips with 3 × 70 uL washes of 500 mM dibasic sodium phosphate (pH 7.0, Sigma S9763). Stage tips were then washed once with 100 μL 1% FA and phosphopeptides were eluted from the stage tips with 60 μL 50% MeCN/0.1% FA. Phosphopeptides were dried down and re-suspended in 9 μL 50% MeCN/0.1%FA for LC-MS/MS analysis, where 4 μL was injected per run.
Acetylpeptide Enrichment
Acetylated lysine peptides were enriched using an antibody against the acetyl-lysine motif (CST PTM-SCAN Catalogue No. 13416). IMAC eluents were concatenated into 4 fractions (~783.75 μg peptides per fraction) and dried down using a SpeedVac apparatus. Peptide fractions were reconstituted with 1.4ml of IAP buffer (5 mM MOPS pH 7.2, 1 mM Sodium Phosphate (dibasic), 5 mM NaCl) per fraction and incubated for 2 hours at 4oC with pre-washed (3 times with IAP buffer) agarose beads bound to acetyl-lysine motif antibody. Peptide-bound beads were washed 4 times with ice-cold PBS followed by elution with 2 × 100ul of 0.15% TFA. Eluents were desalted using C18 stage tips, eluted with 50% ACN and dried down. Acetylpeptides were suspended in 7ul of 0.1% FA and 3% ACN and 4ul were injected per run.
Ubiquitylpeptide Enrichment with on-bead TMT labeling
Ubiquitin enrichment was performed based on the recently published UbiFast protocol (Udeshi et al., 2020). Before enrichment, anti-K-ε-GG bead-bound antibodies from the PTM-Scan ubiquitin remnant motif kit (Cell Signaling Technologies, Kit #5562) were cross-linked. Beads were briefly washed 3X with 100 mM sodium borate (pH 9.0) and incubated with 20 mM DMP for 30 min at RT. Beads were then washed 2X with 200 mM ethanolamine and incubated overnight at 4°C in 200 mM ethanolamine with end-over-end rotation. Following incubation, beads were washed 3X with immunoprecipitation (IAP) buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) and stored at 4°C at a concentration of 0.5 ug/uL.
For each 11-plex experiment, 31.25 ug of cross-linked anti-K-GG bead-bound antibody at 0.5 ug/uL in IAP per channel was aliquoted into 1.5 mL Eppendorf tubes on ice. 750 ug peptide samples were reconstituted to 0.5 mg/mL concentration in IAP buffer and vortexed for 10 min. Peptides were then centrifuged for 5 min at 5,000g. Each peptide solution was added to a tube of antibody and gently rotated end-over-end at 4oC for 1 hour. Following enrichment, samples were spun at 2000 rcf for ~1 min to settle and the supernatant was removed and stored as flow-through, and from this point beads were kept on ice unless being handled. Antibody beads were washed with 1.5 mL ice cold IAP followed by 1.5 mL ice cold PBS. For all washes, after adding wash reagent each sample was inverted ~5 times, agitated by tapping pairs of tubes together, spun at 2000 rcf for about 30–40 s, left to settle on ice for 10–20 s, and supernatant was removed. Importantly, all washes were completed as quickly as possible. Pre-aliquoted TMT @ 40 ug/uL in 100% MeCN was retrieved and briefly equilibrated to RT while beads were reconstituted in 200 uL room temp. 100 mM HEPES buffer. For each sample, 400 ug of TMT labeling reagent in 10 uL acetonitrile was added. Samples were labeled on-antibody while shaking vigorously (1400 rpm) at 20°C for 10 minutes, then quenched with 8 uL 5% hydroxylamine and shaken vigorously for another 5 minutes. Labeled antibody-bound peptides were then put back on ice, washed once with 1.3 mL cold IAP, and again with 1.5 mL cold IAP. Each channel was resuspended and transferred to a combination tube with 130 uL cold IAP. Following combination, each now-empty tube was serially washed with 1.5 mL cold IAP to remove remaining beads, and this 1.5 mL IAP was added to the combination tube and used to wash the combined beads. Combined beads were washed one final time with 1.5 mL ice cold PBS. Once the channels were combined and washed, peptides were eluted twice from the beads by resuspending with 150 uL room temp. 0.15% TFA and incubating 5 min at RT. Each round of acid-eluted KGG-modified peptides was desalted on an equilibrated two-punch C18 stage tip. Both elutions of KGG peptides were loaded sequentially, washed 2X with 100 uL 0.1% FA, and eluted into an MS vial with 50 uL 50% ACN/0.1% FA. The eluted peptides were frozen, lyophilized, and reconstituted in 9 uL 3% ACN/0.1% FA, with 4 uL injected twice for two LC-MS/MS runs on a Thermo Lumos instrument. While the same lot of TMT reagents was used for all ubiquitylproteome samples (TE270748-TD264064), it is a different lot than that used for Proteome, Phosphoproteome, and acetylproteome samples.
LC-MS/MS for Proteomics Analyses
Online separation was done with a nanoflow Proxeon EASY-nLC 1200 UHPLC system (Thermo Fisher Scientific). In this set up, the LC system, column, and platinum wire used to deliver electrospray source voltage were connected via a stainless steel cross (360μm, IDEX Health & Science, UH-906x). The column was heated to 50°C using a column heater sleeve (Phoenix-ST) to prevent over-pressuring of columns during UHPLC separation. From each peptide fraction, ~1ug (based on protein-level BCA prior to digestion with uniformly-distributed fractionation presumed), the equivalent of 12% of each global proteome sample in a 2 ul injection volume or 50% of each phosphoproteome, acetylproteome, or ubiquitylproteome sample in a 4 ul injection volume, was injected onto an in-house packed 22cm x 75um internal diameter C18 silica picofrit capillary column (1.9 μm ReproSil-Pur C18-AQ beads, Dr. Maisch GmbH, r119.aq; Picofrit 10um tip opening, New Objective, PF360–75-10-N-5). Mobile phase flow rate was 200 nL/min, comprised of 3% acetonitrile/0.1% formic acid (Solvent A) and 90% acetonitrile /0.1% formic acid (Solvent B). The 110-minute LC-MS/MS method consisted of a 10-min column-equilibration procedure; a 20-min sample-loading procedure; and the following gradient profile: (min:%B) 0:2; 1:6; 85:30; 94:60; 95;90; 100:90; 101:50; 110:50 (the last two steps at 500 nL/min flow rate). For acetylproteome analysis, the same LC and column setup was used, but the gradient was extended to 260 minutes with the following gradient profile: (min:%B) 0:2; 1:6; 235:30; 244:60; 245;90; 250:90; 251:50; 260:50 (the last two steps at 500 nL/min flow rate). For ubiquitylproteome analysis, the same LC and column setup was used, but the gradient was 154-minutes with the following gradient profile: (min:%B) 0:2; 2:6; 122:35; 130:60; 133;90; 143:90; 144:50; 154:50 (the last two steps at 500 nL/min flow rate).
For proteome and acetylproteome analysis, samples were analyzed with a benchtop Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific) equipped with a nanoflow ionization source (James A. Hill Instrument Services, Arlington, MA). Data-dependent acquisition was performed using Q Exactive HF-X Orbitrap v 2.9 software in positive ion mode at a spray voltage of 1.5 kV. MS1 Spectra were measured with a resolution of 60,000, an AGC target of 3e6 and a mass range from 350 to 1800 m/z. The data-dependent mode cycle was set to trigger MS/MS on up to the top 20 most abundant precursors per cycle at an MS2 resolution of 45,000, an AGC target of 5e4, an isolation window of 0.7 m/z, a maximum injection time of 105 msec, and an HCD collision energy of 31%. Peptides that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 45 sec for proteome or 30 sec for acetylproteome. Peptide match was set to preferred for monoisotopic peak determination, and charge state screening was enabled to only include precursor charge states 2–6, with an intensity threshold of 9.5e4 for proteome or 8e4 for acetylproteome. Advanced precursor determination feature (APD) (Myers et al., 2018) was turned off using a software patch provided to us by Thermo Fisher Scientific allowing us to turn APD off in the tune file, Tune version 2.9.0.2926 (later versions of Exactive Tune 2.9 sp2 for the HFX have this option as standard).
For phosphoproteome and ubiquitylproteome analysis, samples were analyzed with a benchtop Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) equipped with a NanoSpray Flex NG ion source. Data-dependent acquisition was performed using Xcalibur Orbitrap Fusion Lumos v3.0 software in positive ion mode at a spray voltage of 1.8 kV. MS1 Spectra were measured with a resolution of 60,000, an AGC target of 4e5 and a mass range from 350 to 1800 m/z. The data-dependent mode cycle time was set at 2 seconds with a MS2 resolution of 50,000, an AGC target of 6e4, an isolation window of 0.7 m/z, a maximum injection time of 105 msec (125 msec for ubiquitylproteome), and an HCD collision energy of 36%. Peptide mode was selected for monoisotopic peak determination, and charge state screening was enabled to only include precursor charge states 2–6, with an intensity threshold of 1e4. Peptides that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 45 sec (20 sec for ubiquitylproteome), with a +/− 10 ppm mass tolerance. “Perform dependent scan on single charge state per precursor only” was enabled for phosphoproteome analysis, but disabled for ubiquitylproteome analysis.
Immunohistochemistry (IHC) analysis.
TMA construction
Seventy-one LSCC paraffin tissue blocks were available for tissue microarray (TMA) construction and whole sections by immunohistochemistry (IHC) assay. TMAs were constructed in duplicate, and each included two (2) 1.0 mm cores providing up to four (4) cores per case for IHC analysis. Common control tissue on each TMA included patient-matched normal adjacent lung from 14 cases.
Immune cell marker, Immunohistochemistry (IHC)
Seventeen (17) samples were scored for immune cell markers. Sequential TMA sections were obtained for immune marker assessment to best approximate native spatial relationships. Chromogenic (DAB) IHC by single marker analysis was performed on 5-micron thick sections on standard charged slides. Automated immunostaining was performed by Autostainer Link 48, Dako, Inc. Heat induced epitope retrieval (HIER) was performed and biomarker dilutions as follows: CD4 clone 4B12, Bio-Rad 1:50; CD8 clone 4B11, Bio-Rad, 1:50; CD163, Abcam, 1:500 and ARHGDIB, HPA051235 Sigma-Aldrich, 1:50. IHC analysis was performed independently by three independent pathologists by staining intensity of 0–3 scale for each TMA core separately and then by average score / case for a total of 15 cases. Case scores compared across independent pathologists showed very good agreement. For evaluating statistical significance of IHC scores across the three immune clusters, ordinal regression was used, implemented using the “ordinal” package in R. Random effects accounted for the correlation between multiple cores from each sample and multiple pathologists.
Multiplex Immunofluorescence (mIF) staining
Development and optimization of the mIF platform has been previously described (Parra et al., 2020). mIF staining was performed in a 4-μm-thick section obtained from a FFPE TMA block, using the Opal 7-Color Kit (Akoya Biosciences, Waltham, MA, USA) and scanned using a Vectra multispectral microscope (Akoya Biosciences). The IF markers used were pan-Cytokeratin (CK, clone AE1/AE3, (DAKO) and Alpha-SMA ab5694 (Abcam). Multiplexed stained sections were imaged using the VECTRA multispectral imaging system (Akoya Biosciences). TMA cores were annotated using InForm 2.4.8 image analysis software (Akoya Biosciences).
IHC based assessment of TP63 low samples (Figure 4)
For samples with low TP63 status, additional pathologic confirmation was performed. Pathology reassessment confirmed 8/10 samples to be unequivocal LSCC; one sample (C3N.00247) had adenosquamous but predominantly squamous histology; the remaining sample (C3N.02283) showed basaloid squamous cell features and despite some atypical characteristics retained its LSCC attribution after review by four separate pathologists - with histology very similar to a TP63-low TCGA tumor (TCGA-66–2754) deemed LSCC by TCGA pathology.
QUANTIFICATION AND STATISTICAL ANALYSIS
Genomic Data Analysis
Copy Number Calling
Copy-number analysis was performed jointly leveraging both whole-genome sequencing (WGS) and whole-exome sequencing (WES) data of the tumor and germline DNA, using CNVEX (https://github.com/mctp/cnvex). CNVEX uses whole-genome aligned reads to estimate coverage within fixed genomic intervals, and whole-genome and whole-exome variant calls to compute B-allele frequencies at variable positions (we used TNScope germline calls). Coverages were computed in 10kb bins, and the resulting log coverage ratios between tumor and normal samples were adjusted for GC bias using weighted LOESS smoothing across mappable and non-blacklisted genomic intervals within the GC range 0.3–0.7, with a span of 0.5 (the target, blacklist, and configuration files are provided with CNVEX). The adjusted log coverage ratios (LR) and B-allele frequencies (BAF) were jointly segmented by custom algorithm based on Circular Binary Segmentation (CBS). Alternative probabilistic algorithms were implemented in CNVEX, including algorithms based on recursive binary segmentation (RBS) (Gey and Lebarbier, 2008), and dynamic programming (Bellman, 1961), as implemented in the R-package jointseg. For the CBS-based algorithm, first LR and mirrored BAF were independently segmented using CBS (parameters alpha=0.01, trim=0.025) and all candidate breakpoints collected. The resulting segmentation track was iteratively “pruned” by merging segments that had similar LR, BAFs and short lengths. For the RBS- and DP-based algorithms, joint-breakpoints were “pruned” using a statistical model selection method (Lebarbier, 2005). For the final set of CNA segments, we chose the CBS-based results as they did not require specifying a prior on the number of expected segments (K) per chromosome arm, were robust to unequal variances between the LR and BAF tracks, and provided empirically the best fit to the underlying data.
Somatic Variant Calling
Somatic variant calling was performed on exome-capture BAM files aligned to the GDC GRCh38 reference. Following de-duplication and co-realignment around known indels “GATK bundle known” and “Mills 1000G” somatic variants were called using Sentieon TNScope with the following settings “--max_fisher_pv_active 0.05 --min_tumor_allele_frac 0.0075 --min_init_tumor_lod 2.5 --assemble_mode 4 --SVIntrusionThres 5”, with a provided germline variants from dbSNP138. The resulting VCF files were annotated and filtered, in a series of post-processing steps which included: 1) restriction of variants to the hybrid-capture target regions. 2) Phasing of adjacent and separated by 1bp SNVs into multi-nucleotide variants based on the haplotype information provided by TNScope. 3) Annotation using VEP with the following settings “--assembly GRCh38 --species homo_sapiens --cache_version 97 --format vcf --gene_phenotype --symbol --canonical --ccds --hgvs --biotype --tsl --uniprot --domains --appris --protein --variant_class --sift b, --polyphen b --no_stats --total_length --allele_number --no_escape --flag_pick_allele --pick_order canonical,tsl,biotype,rank,ccds,length --buffer_size 20000”. Additional annotations were attached to the variants using VcfAnno including allele frequencies in GNOMAD (v2.0.1), ClinVar (downloaded Dec. 2019), Cosmic (downloaded Dec. 2019), dbSNP (20180418). The resulting variants are annotated against both RefSeq and Ensembl transcripts and contain multiple predicted consequences per-variant. For most downstream applications these annotations were reduced to a single predicted consequence per-variant and converted into a MAF file using custom software. The choice of the most-relevant consequence was based on a number of features. In general, the most ‘severe’ consequence occuring in a ‘reliable’ transcript was chosen. To accomplish this the computed consequences were ranked by ‘severity’ and ‘priority’ reflecting how deleterious/impactful a variant consequence is and how well-supported the existence and biological relevance of an isoform is. The specific rules differed for RefSeq and Ensembl annotations. RefSeq transcripts were ranked higher if they were known to CCDS, LRG or ranked ‘CANONICAL’ or ‘PICK’ by VEP. Ensembl transcripts were further prioritized by Basic, Appris and TSL annotations. Ensembl-based consequences were prefered in case of ties. The resulting variant sets were filtered to only include variants with a high likelihood of being somatic based on a set of probabilistic and heuristic thresholds. A reported variant had to pass all of the following filters: 5 or more variant reads, fewer than 2 variant reads in the germline allele frequency in the normal 0.005, allele frequency in the tumor greater than 0.025, TLOD > 5, NLOD > 6, Fisher’s test p-value < 0.05, NFLOD > 1.5. Additionally heuristic filters were implemented to remove strand-bias, variants with mapping quality bias, and variants with high population allele frequency in GNOMAD.
Germline Variant Calling
Germline variant calling and annotation was carried out analogously using DNAScope using the following settings “--emit_conf 10 --call_conf 10” to generate a VCF file and “--emit_mode GVCF” to generate a gVCF file. The variants were custom-filtered based on the specific applications.
GISTIC and MutSig analysis
The Genomic Identification of Significant Targets in Cancer (GISTIC2.0) algorithm (Mermel et al., 2011) was used to identify significantly amplified or deleted focal-level and arm-level events, with Q value <0.25 considered significant. The following parameters were used:
Amplification Threshold = 0.1
Deletion Threshold = 0.1
Cap Values = 1.5
Broad Length Cutoff = 0.98
Remove X-Chromosome = 0
Confidence Level = 0.99
Join Segment Size = 4
Arm Level Peel Off = 1
Maximum Sample Segments = 2000
Gene GISTIC = 1
Each gene of every sample is assigned a thresholded copy number level that reflects the magnitude of its deletion or amplification. These are integer values ranging from −2 to 2, where 0 means no amplification or deletion of magnitude greater than the threshold parameters described above. Amplifications are represented by positive numbers: 1 means amplification above the amplification threshold; 2 means amplification larger than the arm level amplifications observed in the sample. Deletions are represented by negative numbers: −1 means deletion beyond the threshold; −2 means deletions greater than the minimum arm-level copy number observed in the sample.
The somatic variants were filtered through a panel of normals to remove potential sequencing artifacts and undetected germline variants. MutSig2CV (Lawrence et al., 2014) was run on these filtered results to evaluate the significance of mutated genes and estimate mutation densities of samples. These results were constrained to genes in the Cancer Gene Census (Sondka et al., 2018), with false discovery rates (q values) recalculated. Genes of q value < 0.1 were declared significant.
RNAseq and miRNAseq Quantification
RNAseq Quantification
The readcount is generated by featureCounts (subread v1.6.4) under stranded mode with parameters: `-g gene_id -t exon -Q 10 -p -B -s 2`. Gene annotation in use is identical to GDC (`gencode.v22.annotation.gtf.gz`; md5: `291330bdcff1094bc4d5645de35e0871`), which is available on the GDC Reference Files page (https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files). The readcount is later converted to FPKM and FPKM-UQ using GDC’s formula (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/).
Isoform specific RNA quantification
For TP63 isoform centric analysis presented in Figure 4, the hg38 reference genome and RefSeq annotations were used for the RNAseq data analysis and were downloaded from the UCSC table browser. First, CIRI (v2.0.6) was used to call circular RNA with default parameters and BWA (version 0.7.17-r1188) was used as the mapping tool. The cutoff of supporting reads for circRNAs was set to 10. Then we used a pseudo-linear transcript strategy to quantify gene and circular RNA expression (Li et al., 2017). In brief, for each sample, linear transcripts of circular RNAs were extracted and 75bp (read length) from the 3’ end was copied to the 5’ end. The modified transcripts were called pseudo-linear transcripts. Transcripts of linear genes were also extracted and mixed with pseudo-linear transcripts. RSEM (version 1.3.1) with Bowtie2 (version 2.3.3) as the mapping tool was used to quantify gene, isoform, and circular RNA expression based on the mixed transcripts.
DNA methylation data preprocessing
Raw methylation image files were downloaded from the CPTAC DCC (See data availability). We calculated and analyzed methylated (M) and unmethylated (U) intensities for LSCC samples as described previously (Fortin et al., 2014). We flagged locus as NA where probes did not meet a detection p-value of 0.01. Probes with MAF more than 0.1 were removed, and samples with more than 85% NA values were removed. Resulting beta values of methylation were utilized for subsequent analysis.
Gene-level methylation scores were generated by taking the mean beta values of probes in the CpG islands of promoters and 5’ UTR regions of the gene. Methylation profiles (i.e., density plots) of some samples had unexpectedly skewed distributions of methylation beta values, in addition to significantly more missing values. To systematically determine the subset of methylation samples with these evident data QC issues, we subjected all the samples to model-based clustering using the Mclust package (Fraley et al., 2016) in R, using the interquartile range over all the genes as the representative metric. The clustering automatically determined the optimal number of clusters, and identified 3 clusters. Two of these clusters (with centroids at 0.029 and 0.038) captured the bulk of the samples (202). The third cluster (centroid at 0.165, significantly higher than the other two clusters) consisted of 4 samples that belong to this cluster with high confidence (uncertainty < 5%), each of which had a skewed distribution of beta values with a mean of 4,407 missing values per sample (in contrast to 1.8 missing values per sample for clusters 1 and 2 combined). Based on this analysis, we concluded that the 4 samples in cluster 3 represent samples with poor data quality. These have been excluded from all methylation analysis.
miRNA-seq Data Analysis
miRNA-seq unaligned bam files were downloaded from the CPTAC GDC API (https://docs.gdc.cancer.gov). Unaligned bams were first converted to fastq.gz by samtools bam2fq (samtools version 1.9). TPM (Transcripts per million) values of mature miRNA and precursor miRNA were reported after adaptor trimming, quality check, alignment, annotation, and read counting (https://github.com/ding-lab/CPTAC_miRNA/blob/master/cptac_mirna_analysis.md). The mature miRNA expression was calculated irrespective of its gene of origin by summing the expression.
Proteomics Data Analysis
Spectrum quality filtering and searching
All MS data were interpreted using the Spectrum Mill software package v7.0 pre-release (Agilent Technologies, Santa Clara, CA) co-developed by Karl Clauser of the Carr laboratory (https://www.broadinstitute.org/proteomics). Similar MS/MS spectra acquired on the same precursor m/z within +/− 45 sec were merged. MS/MS spectra were excluded from searching if they failed the quality filter by not having a sequence tag length > 0 (i.e., minimum of two masses separated by the in-chain mass of an amino acid) or did not have a precursor MH+ in the range of 800–6000. MS/MS spectra were searched against a RefSeq-based sequence database containing 41,457 proteins mapped to the human reference genome (GRCh38/hg38) obtained via the UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables) on June 29, 2018, with the addition of 13 proteins encoded in the human mitochondrial genome, 264 common laboratory contaminant proteins, and 553 non-canonical small open reading frames. Scoring parameters were ESI-QEXACTIVE-HCD-v2, for whole proteome datasets, and ESI-QEXACTIVE-HCD-v3, for phosphoproteome, acetylproteome, and ubiquitylproteome datasets. All spectra were allowed +/− 20 ppm mass tolerance for precursor and product ions, 30% minimum matched peak intensity, and “trypsin allow P” enzyme specificity with up to 4 missed cleavages. Allowed fixed modifications included carbamidomethylation of cysteine and selenocysteine. TMT labeling was required at lysine, but peptide N-termini were allowed to be either labeled or unlabeled. Allowed variable modifications for whole proteome datasets were acetylation of protein N-termini, oxidized methionine, deamidation of asparagine, hydroxylation of proline in PG motifs, pyroglutamic acid at peptide N-terminal glutamine, and pyro-carbamidomethylation at peptide N-terminal cysteine with a precursor MH+ shift range of −18 to 97 Da. For the phosphoproteome dataset the allowed variable modifications were revised to allow phosphorylation of serine, threonine, and tyrosine, allow deamidation only in NG motifs, and disallow hydroxylation of proline with a precursor MH+ shift range of −18 to 272 Da. For the acetylproteome dataset the allowed variable modifications were revised to allow acetylation of lysine, allow deamidation only in NG motifs, and disallow hydroxylation of proline with a precursor MH+ shift range of −400 to 70 Da. For the ubiquitylproteome dataset the allowed variable modifications were revised to allow diglycine modification of lysine, allow deamidation only in NG motifs, and disallow hydroxylation of proline with a precursor MH+ shift range of −690 to 70 Da.
Protein grouping, and localization of PTMs
Identities interpreted for individual spectra were automatically designated as confidently assigned using the Spectrum Mill autovalidation module to use target-decoy based false discovery rate (FDR) estimates to apply score threshold criteria. For the whole proteome dataset thresholding was done in 3 steps: at the peptide spectrum match (PSM) level, the protein level for each TMT-plex, and the protein level for all 22 TMT-plexes. For the phosphoproteome, acetylproteome, and ubiquitylproteome datasets thresholding was done in two steps: at the PSM and variable modification (VM) site levels.
In step 1 for all datasets, PSM-level autovalidation was done first and separately for each TMT-plex experiment consisting of either 25 LC-MS/MS runs (whole proteome), 13 LC-MS/MS runs (phosphoproteome), 4 LC-MS/MS runs (acetylproteome), or 2 LC-MS/MS runs (ubiquitylproteome) using an auto-thresholds strategy with a minimum sequence length of 7; automatic variable range precursor mass filtering; and score and delta Rank1 – Rank2 score thresholds optimized to yield a PSM-level FDR estimate for precursor charges 2 through 4 of <0.8% for each precursor charge state in each LC-MS/MS run. To achieve reasonable statistics for precursor charges 5–6, thresholds were optimized to yield a PSM-level FDR estimate of <0.4% across all runs per TMT-plex experiment (instead of per each run), since many fewer spectra are generated for the higher charge states.
In step 2 for the whole proteome dataset, protein-polishing autovalidation was applied separately to each TMTplex experiment to further filter the PSMs using a target protein-level FDR threshold of zero. The primary goal of this step was to eliminate peptides identified with low scoring PSMs that represent proteins identified by a single peptide, so-called “one-hit wonders.” After assembling protein groups from the autovalidated PSMs, protein polishing determined the maximum protein level score of a protein group that consisted entirely of distinct peptides estimated to be false-positive identifications (PSMs with negative delta forward-reverse scores). PSMs were removed from the set obtained in the initial peptide-level autovalidation step if they contributed to protein groups that had protein scores below the maximum false-positive protein score. Step 3 was then applied, consisting of protein-polishing autovalidation across all TMT plexes together using the protein grouping method “expand subgroups, top uses shared” to retain protein subgroups with either a minimum protein score of 25 or observation in at least 4 TMT plexes. The primary goal of this step was to eliminate low scoring proteins that were infrequently detected in the sample cohort. As a consequence of these two protein-polishing steps, each identified protein reported in the study comprised multiple peptides, unless a single excellent scoring peptide was the sole match and that peptide was observed in at least 4 TMT-plexes. In calculating scores at the protein level and reporting the identified proteins, peptide redundancy was addressed in Spectrum Mill as follows: The protein score was the sum of the scores of distinct peptides. A distinct peptide was the single highest scoring instance of a peptide detected through an MS/MS spectrum. MS/MS spectra for a particular peptide may have been recorded multiple times (e.g. as different precursor charge states, in adjacent bRP fractions, modified by deamidation at Asn or oxidation of Met, or with different phosphosite localization), but were still counted as a single distinct peptide. When a peptide sequence of >8 residues was contained in multiple protein entries in the sequence database, the proteins were grouped together and the highest scoring one and its accession number were reported. In some cases when the protein sequences were grouped in this manner, there were distinct peptides that uniquely represent a lower scoring member of the group (isoforms, family members, and different species). Each of these instances spawned a subgroup. Multiple subgroups were reported, counted towards the total number of proteins, and given related protein subgroup numbers (e.g. 3.1 and 3.2 for group 3, subgroups 1 and 2). For the whole proteome datasets the above criteria yielded false discovery rates (FDR) for each TMT-plex experiment of <0.6% at the peptide-spectrum match level and <0.8% at the distinct peptide level. After assembling proteins with all the PSMs from all the TMT-plex experiments together, the aggregate FDR estimates were 0.58% at the peptide-spectrum match level, 2.3% at the distinct peptide level, and <0.01% (1/11,029) at the protein group level. Since the protein level FDR estimate neither explicitly required a minimum number of distinct peptides per protein nor adjusted for the number of possible tryptic peptides per protein, it may underestimate false positive protein identifications for large proteins observed only on the basis of multiple low scoring PSMs.
In step 2 for the phosphoproteome and acetylproteome datasets, variable modification (VM) site polishing autovalidation was applied across all 22 TMT plexes to retain all VM-site identifications with either a minimum id score of 8.0 or observation in at least 4 TMT plexes. For the ubiquitylproteome datasets, it was applied across all 15 TMT plexes to retain all VM-site identifications with either a minimum id score of 8.0 or observation in at least 3 TMT plexes. The intention of the VM site polishing step is to control FDR by eliminating unreliable VM site-level identifications, particularly low scoring VM sites that are only detected as low scoring peptides that are also infrequently detected across all of the TMT plexes in the study. In calculating scores at the VM-site level and reporting the identified VM sites, redundancy was addressed in Spectrum Mill as follows: A VM-site table was assembled with columns for individual TMT-plex experiments and rows for individual VM-sites. PSMs were combined into a single row for all non-conflicting observations of a particular VM-site (e.g. different missed cleavage forms, different precursor charges, confident and ambiguous localizations, and different sample-handling modifications). For related peptides, neither observations with a different number of VM-sites nor different confident localizations were allowed to be combined. Selecting the representative peptide from the combined observations was done such that once confident VM-site localization was established, higher identification scores and longer peptide lengths were preferred. While a Spectrum Mill identification score was based on the number of matching peaks, their ion type assignment, and the relative height of unmatched peaks, the VM site localization score was the difference in identification score between the top two localizations. The score threshold for confident localization, >1.1, essentially corresponded to at least 1 b or y ion located between two candidate sites that has a peak height >10% of the tallest fragment ion (neutral losses of phosphate from the precursor and related ions as well as immonium and TMT reporter ions were excluded from the relative height calculation). The ion type scores for b-H3PO4, y-H3PO4, b-H2O, and y-H2O ion types were all set to 0.5. This prevented inappropriate confident localization assignment when a spectrum lacked primary b or y ions between two possible sites but contained ions that could be assigned as either phosphate-loss ions for one localization or water loss ions for another localization. VM-site polishing yielded 68, 674 phosphosites with an aggregate FDR of 0.73% at the phosphosite level. In aggregate, 71% of the reported phosphosites in this study were fully localized to a particular serine, threonine, or tyrosine residue. VM-site polishing yielded 15,186 acetylsites with an aggregate FDR of 0.80% at the acetylsite level. In aggregate, 99% of the reported acetylsites in this study were fully localized to a particular lysine residue. VM-site polishing yielded 25,489 ubiquitylsites with an aggregate FDR of 0.40% at the ubiquitylsite level. In aggregate, >99% of the reported ubiquitylsites in this study were fully localized to a particular lysine residue.
Quantitation using TMT ratios
Using the Spectrum Mill Protein/Peptide Summary module, a protein comparison report was generated for the proteome dataset using the protein grouping method “expand subgroups, top uses shared” (SGT). For the phosphoproteome and acetylproteome datasets a Variable Modification site comparison report limited to either phospho or acetyl sites, respectively, was generated using the protein grouping method “unexpand subgroups”. Relative abundances of proteins and VM-sites were determined in Spectrum Mill using TMT reporter ion intensity ratios from each PSM. TMT reporter ion intensities were corrected for isotopic impurities in the Spectrum Mill Protein/Peptide summary module using the afRICA correction method, which implements determinant calculations according to Cramer’s Rule (Shadforth et al., 2005) and correction factors obtained from the reagent manufacturer’s certificate of analysis (https://www.thermofisher.com/order/catalog/product/90406) for TMT11_lot number UA280170/TL272399 (Proteome, Phosphoproteme, and Acetylproteome) and TE270748-TD264064 (Ubiquitylproteome). A protein-level, phosphosite-level, or acetylsite-level TMT ratio is calculated as the median of all PSM-level ratios contributing to a protein subgroup, phosphosite, or acetylsite. PSMs were excluded from the calculation if they lacked a TMT label, had a precursor ion purity < 50% (MS/MS has significant precursor isolation contamination from co-eluting peptides), or had a negative delta forward-reverse identification score (half of all false-positive identifications). Lack of TMT label led to exclusion of PSMs per TMT plex with a range of 1.4 to 2.3% for the proteome, 1.8 to 3.7% for the phosphoproteome, 1.0 to 3.5% for the acetylproteome, and 4.5 to 18.0% for the ubiquitylproteome datasets. Low precursor ion purity led to exclusion of PSMs per TMT plex with a range of 1.1 to 1.4% for the proteome, 1.7 to 2.8% for the phosphoproteome, 0.5 to 1.7% for the acetylproteome, and 2.8 to 3.5% for the ubiquitylproteome datasets.
Two-component normalization of TMT ratios
It was assumed that for every sample there would be a set of unregulated proteins, phosphosites, acetylsites or ubiquitylsites that have abundance comparable to the common reference (CR) sample. In the normalized sample, these proteins, phosphosites, acetylsites or ubiquitylsites should have a log TMT ratio centered at zero. In addition, there were proteins, phosphosites, acetylsites and ubiquitylsites that were either up- or down-regulated compared to the CR. A normalization scheme was employed that attempted to identify the unregulated proteins, phosphosites, acetylsites or ubiquitylsites, and centered the distribution of these log-ratios around zero in order to nullify the effect of differential protein loading and/or systematic MS variation. A 2-component Gaussian mixture model-based normalization algorithm was used to achieve this effect. The two Gaussians N(μi1,σi1) and N(μi2,σi2) for a sample i were fitted and used in the normalization process as follows: the mode mi of the log-ratio distribution was determined for each sample using kernel density estimation with a Gaussian kernel and Shafer-Jones bandwidth. A two-component Gaussian mixture model was then fit with the mean of both Gaussians constrained to be mi, i.e., μi1=μi2=mi. The Gaussian with the smaller estimated standard deviation was assumed to represent the unregulated component of proteins/phosphosites/acetylsites/ubiquitylsites, and was used to normalize the sample. The sample was standardized using (mi,σi) by subtracting the mean mi from each protein/phosphosite/acetylsite/ubiquitylsite and dividing by the standard deviation σi.
Normalization of acetyl- and ubiquitylproteome
To correct for differential underlying protein levels when performing certain acetylproteome- and ubiquitylproteome-based analyses, a global linear model approach was employed to produce protein-normalized versions of both of these PTM datasets. PTM sites were first matched to underlying proteins in the proteome dataset using RefSeq accession numbers. If a site’s main accession number did not yield a match in the proteome, all accession numbers associated with the site were then tried and the protein with the highest score was selected as a match. For the acetylproteome and ubiquitylproteome separately, a linear model was then fit (R function lm, PTM ~ protein) to all matched PTM-protein data points in the dataset. The residuals of each model were then used as protein-normalized acetylsite or ubiquitylsite abundances.
Proteogenomic analysis
Quality control and Batch effect assessment
We used guided principal component analysis (gPCA) (Reese et al., 2013) to assess the presence of a batch effect in the sample/CR ratio data based on TMT-plex. There was no overall apparent batch effect (gPCA p-value=1) in the global proteome, phosphoproteome, acetylproteome or ubiquitylproteome datasets.
To assess composition similarities between CR used for proteome, phosphoproteome and acetylproteome analysis, and ubiquitylproteome, we performed label-free proteomic analysis of each CRs in triplicates. Both CRs showed excellent correlation with median pearson correlation > 0.85.
In addition, to assess data quality, we assessed the Pearson correlations of log TMT reporter ion intensities between all pairs of TMT plexes, separately for sample and CR channels. For each given pair of TMT plexes, correlations between CRs were consistently higher than correlations of respective sample channels enabling precise relative quantitation of proteins, phosphorylation and acetylation sites across 22 TMT plexes using the CR channels as common denominator (Figure S1C). In the ubiquitylproteome data we observed lower correlations of the CR channel (in comparison to samples channels) in three of the 18 TMT plexes analyzed (plexes 7, 8, 10). Clustering of the CR channels using pairwise correlations as a similarity metric pointed to a batch effect in the CR channels (but not in the sample channels) in these plexes. The presence of a batch effect was further confirmed by applying the gPCA test described above to only the reporter intensities of the CR channels (gPCA p-value < 0.001). Even though undetectable with a global test, we observed this CR batch effect propagate to the sample/CR ratio data in preliminary analyses (data not shown). To counter the propagation of this CR batch effect to the sample/CR ratio for samples in these plexes, batch correction was applied to the sample/CR ratio data for plexes 7, 8, 10 (batch 1) and all other plexes (batch 2) using limma’s removeBatchEffect (https://rdrr.io/bioc/limma/man/removeBatchEffect.html) on the sample/CR ratio data including the sample type (tumor, NAT) as a covariate.
Dataset filtering
Genes (RNA-seq), proteins (global proteome), phosphosites, acetylsites and ubiquitin sites present in fewer than 30% of samples (i.e., missing in >70% of samples) were removed from the respective datasets. Furthermore, proteins were required to have at least two observed TMT ratios in >25% of samples in order to be included in the proteome dataset. Phosphosites, acetylsites and ubiquitin sites were required to have at least one observed TMT ratio in >25% of samples.
Some of the filtering steps were modified for specific analyses in the study. For many of the marker selection and gene set enrichment analyses, at least 50% of samples were required to have non-missing values for proteins/phosphosites/acetyl/ubiquitin sites, since missing values were imputed, and excessive missing values can result in poor imputation. Alternate filtering has been noted in descriptions of the relevant methods.
Unsupervised multi-omic clustering using NMF
We used non-negative matrix factorization (NMF)-based multi-omic clustering using protein, phosphosite, acetylsite, RNA transcript and gene copy number variants (CNV) as previously described ((Gillette et al., 2020). Briefly, given a factorization rank k (where k is the number of clusters), NMF decomposes a p x n data matrix V into two matrices W and H such that multiplication of W and H approximates V. Matrix H is a k x n matrix whose entries represent weights for each sample (1 to N) to contribute to each cluster (1 to k), whereas matrix W is a p x k matrix representing weights for each feature (1 to p) to contribute to each cluster (1 to k). Matrix H was used to assign samples to clusters by choosing the k with maximum score in each column of H. For each sample we calculated a cluster membership score as the maximal fractional score of the corresponding column in matrix H. We defined a ”cluster core” as the set of samples with cluster membership score > 0.5.
To enable integrative multi-omic clustering we enforced all data types (and converted if necessary) to represent ratios to: (i) a common reference measured in each TMT plex (proteome, phosphoproteome, acetylproteome), (ii) an in silico common reference calculated as the median abundance across all samples (RNA expression) or (iii) to matching blood normal for CNA data. The CNA data was further filtered to only retain genes significantly altered (GISTIC2 thresholded of +2 or −2) in at least 5% of all tumors. All data tables were then concatenated and all rows with missing values were removed. To remove uninformative features from the dataset prior to NMF clustering we removed features with the lowest standard deviation (bottom 5th percentile) across all samples. Each column in the data matrix was further scaled and standardized such that all features from different data types were represented as z-scores. The resulting data matrix of z-scores into was converted to a non-negative input matrix required by NMF as follows:
Create one data matrix with all negative numbers zeroed.
Create another data matrix with all positive numbers zeroed and the signs of all negative numbers removed.
Concatenate both matrices resulting in a data matrix twice as large as the original, but containing only positive values and zeros and hence appropriate for NMF.
The resulting matrix was then subjected to NMF analysis leveraging the NMF R-package (Gaujoux and Seoighe, 2010) and using the factorization method described in (Brunet et al., 2004). To determine the optimal factorization rank k (number of clusters) for the multi-omic data matrix we tested a range of clusters between k=2 and 8. For each k we factorized matrix V using 50 iterations with random initializations of W and H. To determine the optimal factorization rank we calculated two metrics for each k: 1) cophenetic correlation coefficient measuring how well the intrinsic structure of the data was recapitulated after clustering and 2) the dispersion coefficient of the consensus matrix as defined in (Kim and Park, 2007) measuring the reproducibility of the clustering across 50 iterations. The optimal k was defined as the maximum of the product of both metrics for cluster numbers between k=3 and 8
Having determined the optimal factorization rank k, in order to achieve robust factorization of the multi-omic data matrix V, we repeated the NMF analysis using 500 iterations with random initializations of W and H and performed the partitioning of samples into clusters as described above. Due to the non-negative transformation applied to the z-scored data matrix as described above, matrix W of feature weights contained two separate weights for positive and negative z-scores of each feature, respectively. In order to revert the non-negative transformation and to derive a single signed weight for each feature, we first normalized each row in matrix W by dividing by the sum of feature weights in each row, aggregated both weights per feature and cluster by keeping the maximal normalized weight and multiplication with the sign of the z-score in the initial data matrix. Thus, the resulting transformed version of matrix Wsigned contained signed cluster weights for each feature in the input matrix.
For functional characterization of clustering results by single sample Gene Set Enrichment Analysis (ssGSEA), we calculated normalized enrichment scores (NES) of cancer-relevant gene sets by projecting the matrix of signed multi-omic feature weights (Wsigned) onto Hallmark pathway gene sets (Liberzon et al., 2015) using ssGSEA (Barbie et al., 2009). To derive a single weight for each gene measured across multiple omics data types (protein, RNA, phosphorylation site, acetylation site, CNA) we retained the weight with maximal absolute amplitude. We used the ssGSEA implementation available on https://github.com/broadinstitute/ssGSEA2.0 using the following parameters:
gene.set.database=“h.all.v6.2.symbols.gmt”
sample.norm.type=“rank”
weight=1
statistic=”area.under.RES”
output.score.type=“NES”
nperm=1000
global.fdr=TRUE
min.overlap=5
correl.type=”z.score”
To test the association of the resulting clusters to clinical variables we used Fisher’s exact test (R function fisher.test) to test for overrepresentation in the set of samples defining the cluster core as described above.
The entire workflow described above has been implemented as a module for PANOPLY (Mani et al., 2020) which runs on Broad’s Cloud platform Terra (https://app.terra.bio/). The docker containers encapsulating the source code and required R-packages for NMF clustering and ssGSEA have been submitted to Dockerhub (broadcptacdev/pgdac_mo_nmf:15, broadcptac/pgdac_ssgsea:5). The source code for ssGSEA is available on GitHub: https://github.com/broadinstitute/ssGSEA2.0.
Integrative analysis with Stewart et al.
To enable integrative analysis of our protein data with the protein data in Stewart et al.(Stewart et al., 2019) we first aggregated the protein-level data to gene-level data matrices by retaining the dominant isoform (identified by the lowest protein subgroup number) associated with each gene symbol. To aggregate the Stewart et al. protein data we used the mean expression across all proteins mapping to the same gene symbol. Both gene-level protein data matrices were then separately subjected to gene-level z-score transformation before joining the matrices using the unique gene symbols as keys. The resulting integrated protein data matrix was then subjected to the NMF-based clustering pipeline described above using the 3,366 genes quantified across all 216 tumors.
RNA subtyping
Previously reported LSCC gene expression-subtype signatures were downloaded from the original publication (Wilkerson et al., 2010) and applied to our RNA expression dataset using the centroid-based method in a manner similar to the TCGA study (Hammerman et al., 2012). Specifically, the RNA expression of the 208 signature genes, represented as FPKM, was mean-centered in the gene-wise manner, and each sample was correlated to each of the four centroid vectors representing the average signature gene expression for the four subtypes. The samples were assigned to one of the four transcriptomics subtypes according to the highest correlations. Samples with insignificance correlation (p > 0.01) were marked to have ‘undecided’ subtype.
Chromosomal instability index
The CIN score reflects the overall copy number aberration across the whole genome. From the segment-level CNA result, we used a weighted-sum approach to summarize the chromosome instability for each sample (Vasaikar et al., 2019). The absolute segment-level log2 ratios of all copy number segments (indicating the copy number aberration of these segments) within a chromosome were weighted by the segment length and summed up to derive the instability score for the chromosome. The genome-wide chromosome instability index was calculated by summing up the instability score of all 22 autosomes.
Fusion detection and analysis
Fusion calling was performed using a combination of CRISP, CODAC MI-ONCOSEQ pipeline (Robinson et al., 2017; Wu et al., 2018), fusioncatcher_v1.10 (Nicorici et al.) and arriba_v1.1.0 (https://github.com/suhrig/arriba/). The fusions calls were compiled and visualized using AGFusion to display the predicted domain and exon structure of the fusion proteins as described (Vats et al., 2020).
mRNA and Protein correlation
To compare mRNA expression and protein abundance across samples we focused on the RNAseq data with 21,792 genes, and global proteome with 11,575 quantified proteins. Protein IDs were mapped to gene names. In total, 10,890 genes common to both RNAseq and proteome data spanning 108 tumor samples were used in the analysis. The analyses were carried out on normalized data: RNAseq data were log2 transformed, upper quartile normalized RPKM values, which were median-centered by row (i.e., gene); proteome data was two-component normalized as described earlier. Pearson correlation was calculated for each gene. Both correlation coefficient and p-value were recorded. Further, adjusted p-values were calculated using the Benjamini–Hochberg procedure.
CNA-driven cis and trans effects
Correlations between copy number alterations (CNA) and RNA, proteome, phosphoproteome acetylproteome and ubiquitylproteome (with proteome and PTM data mapped to genes, by choosing the most variable protein isoform/PTM site as the gene-level representative) were determined using Pearson correlation of common genes present in CNA-RNA-proteome (9,988 genes), CNA-RNA-phosphoproteome (5,144 genes), CNA-RNA-acetylproteome (1,344 genes) and CNA-RNA-ubiquitylproteome (2,616 genes). In addition, p-values (corrected for multiple testing using Benjamini-Hochberg FDR) for assessing the statistical significance of the correlation values were also calculated. CNA trans-effects for a given gene were determined by identifying genes with statistically significant (FDR < 0.05) positive or negative correlations.
CMAP analysis
Candidate genes driving response to copy number alterations were identified using large-scale Connectivity Map (CMAP) queries. The CMAP (Lamb et al., 2006; Subramanian et al., 2017) is a collection of about 1.3 million gene expression profiles from cell lines treated with bioactive small molecules (~20,000 drug perturbagens), shRNA gene knockdowns (~4,300) and ectopic expression of genes. The CMAP dataset is available on GEO (Series GSE92742). For this analysis, we use the Level 5 (signatures from aggregating replicates) TouchStone dataset with 473,647 total profiles, containing 36,720 gene knock-down profiles, with measurements for 12,328 genes. See https://clue.io/GEO-guide for more information.
To identify candidate driver genes, proteome profiles of copy number-altered samples were correlated with gene knockdown mRNA profiles in the above CMAP dataset, and enrichment of up/downregulated genes was evaluated. Normalized log2 copy num- ber values less than 0.3 defined deletion (loss), and values greater than +0.3 defined copy number amplifications (gains). In the copy number-altered samples (separately for CNA amplification and CNA deletion), the trans-genes (identified by significant correlation in “CNA driven cis and trans effects” above) were grouped into UP and DOWN categories by comparing the protein ratios of these genes to their ratios in the copy number neutral samples (normalized log2 copy number between 0.3 and +0.3). The lists of UP and DOWN trans-genes were then used as queries to interrogate CMAP signatures and calculate weighted connectivity scores (WTCS) using the single-sample GSEA algorithm (Krug et al., 2018). The weighted connectivity scores were then normalized for each perturbation type and cell line to obtain normalized connectivity scores (NCS). See (Subramanian et al., 2017) for details on WTCS and NCS. For each query we then identified outlier NCS scores, where a score was considered an outlier if it lay beyond 1.5 times the interquartile range of score distribution for the query. The query gene was designated a candidate driver if (i) the score outliers were statistically cis-enriched (Fisher test with BH-FDR multiple testing correction) and (ii) the gene had statistically significant and positive cis-correlation.
For a gene to be considered for inclusion in a CMAP query it needed to i) have a copy number change (amplification or deletion) in at least 15 samples; ii) have at least 20 significant trans genes; and iii) be on the list of shRNA knockdowns in the CMAP. 502 genes satisfied these conditions and resulted in 976 queries (CNA amplification and deletion combined) that were tested for enrichment. Thirty two (32) candidate driver genes were identified with Fisher’s test FDR < 0.25, using this process.
In order to ensure that the identified candidate driver genes were not a random occurrence, we performed a permutation test to determine how many candidate driver genes would be identified with random input (Mertins et al., 2016). For the 976 queries used, we substituted the bona-fide trans-genes with randomly chosen genes, and repeated the CMAP enrichment process. To determine FDR, each permutation run was treated as a Poisson sample with rate λ, counting the number of identified candidate driver genes. Given the small n ( = 10) and λ, a Score confidence interval was calculated (Barker, 2002) and the midpoint of the confidence interval used to estimate the expected number of false positives. Using 10 random permutations, we determined the overall false discovery rate to be FDR = 0.33, with a 95% CI of (0.27, 0.40) for genes with either CNA amplification or deletion, and an FDR=0.06 (95% CI: 0.01, 0.12) for six (6) genes with both CNA amplification and deletion.
To identify how many trans-correlated genes for all candidate regulatory genes could be directly explained by gene expression changes measured in the CMAP shRNA perturbation experiments, knockdown gene expression consensus signature z-scores (knockdown/control) were used to identify regulated genes with α = 0.05, followed by counting the number of trans-genes in this list of regulated genes.
To obtain biological insight into the list of candidate driver genes, we performed (i) enrichment analysis on samples with extreme CNA values (amplification or deletion) to identify statistically enriched sample annotation subgroups; and (ii) GSEA on cis/trans-correlation values to find enriched pathways.
LINCS analysis
Differentially expressed genes between the 5 NMF LSCC subtypes were identified using the limma package. In order to identify genes that were uniquely expressed in each subtype, all pairwise comparisons between the 5 subtypes were computed. In addition, genes that were differentially expressed (adjusted p.value < 0.05) in at least 3 out of the 4 comparisons and had a concordant fold change among all comparisons were identified as subtype-specific and were used in the subsequent analysis.
Protein abundance comparisons were performed between all 5 NMF subtypes using the Wilcoxon rank-sum test and p-values were adjusted using the Benjamini & Hochberg method. Subtype-specific differentially expressed proteins were identified based on their differential expression (adjusted p.value < 0.05) in at least 2 out of the 4 comparisons, and by having a concordant fold change among all comparisons. The identified differentially expressed genes and proteins were then filtered for gene symbols measured in the L1000 assay (978 landmark genes). These NMF-specific signatures were used as input to calculate normalized weighted connectivity scores (WTCS) against the Library of Integrated Network-Based Cellular Signatures (LINCS) L1000 perturbation-response signatures. The scores were computed using the sig_queryl1k_tool pipeline (https://hub.docker.com/u/cmap) and the LINCS L1000 Level 5 compound (trt_cp) signatures from CLUE (clue.io, “Expanded CMap LINCS Resource 2020 Release”). The resulting normalized connectivity scores were summarized across cell lines using the maximum quantile of all the scores of the same compound (Qhi = 67,Qlow = 33) as previously described (Subramanian et al., 2017). The corresponding compound metadata were obtained from CLUE (clue.io, “Expanded CMap LINCS Resource 2020 Release”) and were used to filter and identify compounds with existing annotations (drug name, MOA, Target). The top 20 negatively connected compounds to each NMF signature were selected for visualization.
Defining cancer-associated genes
Cancer-associated genes (CAG) were compiled using the Census website https://cancer.sanger.ac.uk/census. The list of genes is provided in Table S1.
CpG Island Methylator Phenotype
The methylation analysis started with all 103 tumor samples, which excluded 4 samples from the cohort with high missing rate (see above), to generate the CpG island methylator phenotypes (CIMP). Specifically, we first generated the gene-level methylation score, by taking the averaged beta values of all probes harboring in the islands of promotor or 5 UTR regions of the gene. Then, we preprocessed the data by filtering genes that were hypermethylated, i.e. the gene-level methylation score >0.2, transformed the score into M-values, normalized the transformed values, and then imputed the missing values with k-nearest neighbors (KNN). With preprocessed data, we performed consensus clustering 1000 times, each taking 80% of the samples and all genes, and calculated the consensus matrix (probabilities of two samples clustering together) for each predetermined number of clusters K. K ranges from 2 to 10. In each value of K, we visualized the consensus matrix using K means with Euclidean distance as the distance metric. Finally, we determined the optimal number of clusters by considering the relative change in area under the consensus cumulative density function (CDF) curve. In the end, three distinct clusters were identified, one was hypermethylated with mean M value 0.393, and two were hypomethylated with mean M value −0.041 and −0.359, respectively. We labeled these three clusters as CIMP high, CIMP intermediate, and CIMP low groups.
iProFun Based Cis Association Analysis
We used iProFun, an integrative analysis tool to identify multi-omic molecular quantitative traits (QT) perturbed by DNA-level variations. We considered five functional molecular quantitative traits (gene expression, protein, phosphoprotein, acetylprotein, ubiquitylproteome levels) for their associations with DNA methylation, accounting for mutation, copy number variation, age, gender, tumor purity and smoking status. Tumor purity was determined using TSNet from RNA-seq data. The iProFun procedure was applied to a total of 12,666 genes that was available in at least one predictor (mutation, CNA, DNA methylation) and one outcome (mRNA, global protein, phospholation, acetylation and ubiquitylation) for their cis regulatory patterns in tumors. For each of the 12,666 genes, we started with linear regression to capture the covariate-adjusted associations. The resulting association summary statistics was used to call probabilities of belonging to each of the 2^5=32 possible configurations. We calculated the probabilities of associating with each of the five outcomes by combining probabilities from relevant configurations, and calculated FDR controlled associations. The significant genes need to pass three criteria: (1) the satisfaction of biological filtering procedure, (2) posterior probabilities > 75%, and (3) empirical false discovery rate (eFDR)<10%. As DNA methylation at the promoter regions of a gene down regulates its expression level, biological filtering procedure requires DNA methylations that exist either negative significant associations or insignificant associations with all 5 the types of molecular QTs for significance call. DNA methylations with significant positive associations with QTs were filtered out for significance call, as it was not the pattern of biological associations that were interesting. Secondly, a significance was called only if the posterior probabilities > 75% of a predictor being associated with a molecular QT, by summing over all configurations that are consistent with the association of interest. Lastly, we calculated empirical FDR via 100 permutations per molecular QTs by shuffling the label of the molecular QTs, and requested empirical FDR (eFDR) <10% by selecting a minimal cutoff value of alpha such that 75%<alpha<100%. The eFDR is calculated by: eFDR= (Averaged No. of genes with posterior probabilities > alpha in permuted data) / (Averaged No. of genes with posterior probabilities > alpha in original data). Figure 1G annotated those genes whose DNA methylation had cascading effects (associated with all QTs under investigation), and among them those whose protein abundances significantly differ between tumor and NAT were annotated.
EMT-specific cluster and fibroblast enrichment
NMF derived EMT-enriched subtype was interrogated for EMT and fibroblast related proteins in LSCC tumor patients. The relative protein expression was calculated using z-score. The single ssGSEA was performed on a protein dataset using GSVA (v3.11) to calculate normalized enrichment score for EMT and fibroblast proliferation using MSigdb (v6.1) and gene ontology geneset respectively. Similar analysis was also performed to interrogate SOX2 associated genes and its relation with IL6_JAK_STAT signaling (MSigdb hallmarks geneset V6.1).
Differential marker analysis
A Wilcoxon signed rank test was performed on TMT-based global proteomic data between tumor and matched normal samples to determine differential abundance of proteins between tumor and NAT samples. Proteins having < 50% missing values were considered for downstream analysis. Proteins with log2-fold-change (FC)> 2 in tumors and Benjamini-Hochberg FDR < 0.01 were considered to be tumor-associated proteins. Over-representation analysis of Gene Ontology Biological Process terms was performed on a background of quantified proteins in WebGestaltR (Liao et al., 2019). The selected gene list was either significant proteins or proteins containing a significantly altered phosphorylation site from the differential marker analyses. ORA was performed separately for increased and decreased proteins. Terms were considered significant with a Benjamini-Hochberg adjusted p-value <0.01.
Among tumor associated proteins, the top 50 differential proteins with more than 90% of samples showing high expression in tumors were selected and shown. Differential proteins with more than 99% of samples showing tumor specific expression were highlighted. Immunohistochemistry-based antibody-specific staining scores in lung tumors were obtained from the Human Protein Atlas (HPA, https://www.proteinatlas.org), in which tumor-specific staining is reported in four levels, i.e., high, medium, low, and not detected. The protein-specific annotations such as enzymes, transcription factor, transporters, secreted, transmembrane and FDA-approved drugs targeting the protein or reported in drugbank were designated. Similarly, differential analysis was performed between RNA, phosphosite and acetylproteome datasets between tumor and matched NATs. The differential genes/sites with log2-fold-change (FC)> 2 in tumors and Benjamini-Hochberg FDR < 0.01 were considered to be tumor-associated. The overlapping genes/sites with proteins were represented in a checkerbox plot.
Overall and disease-free survival in TCGA lung squamous carcinoma
Survival for lung squamous tumor patients in TCGA LSCC cohort was calculated using the time from date of diagnosis to death or last contact (Overall survival, OS) and disease-free interval time (Disease-free survival, DFS). Normalized RNA expression was categorized into low and high expression group based on mean. Univariate Cox proportional hazards models were fitted to OS, and DFI endpoints to calculate the hazard ratios for tumor-associated genes/proteins using coxph function in Survival package (version 2.44) in R. The p-values were determined using a log-rank test. The hazard ratio [exp(cox coefficient)] was used to compare poor survival in low and high-expression group.
Kaplan-Meier survival analysis
We used time-to-death or last contact data for the current cohort (recorded as number of days from diagnosis date) to assess survival differences based on various sample subgroups and clinical annotations. We used Kaplan-Meier analysis to explore survival differences associated with tumor grade, mutation burden, mutation status for SMGs, CUL3-NFE2L2-KEAP1 combined mutation status, CNA for KAT6A/SOX2/TP63/FGFR1/CDKN2A, immune subtype, NMF cluster, CIN, tumor grade and ploidy.
We identified NMF mixed tumors (i.e., tumors that are not part of the core NMF clusters) had significantly worse survival (log-rank p-value = 0.005). We used a Fisher test to find enriched annotations (among those mentioned in the previous paragraph, with tumor stemness index added) associated with NMF mixed tumors. The frequency of SOX2 amplifications were significantly enriched in NMF mixed tumors (p-value = 0.0038). None of the other annotations had significant enrichment in the NMF mixed tumors.
Continuous Smoking Score
Non-negative matrix factorization (NMF) was used in deciphering mutation signatures in cancer somatic mutations stratified by 96 base substitutions in tri-nucleotide sequence contexts. To obtain a reliable signature profile, we used SomaticWrapper to call mutations from WGS data. SignatureAnalyzer exploited the Bayesian variant of the NMF algorithm and enabled an inference for the optimal number of signatures from the data itself at a balance between the data fidelity (likelihood) and the model complexity (regularization). After decomposing into three signatures, signatures were compared against known signatures derived from COSMIC (Tate et al., 2019) and cosine similarity was calculated to identify the best match.
We also sought to integrate count of total mutations, t, percentage that are signature mutations, c, and count of DNPs, n, into a continuous score, 0 < S < 1, to quantify the degree of confidence that a sample was associated with smoking signature. We referred to these quantities as the data, namely D = C ∩ T ∩ N, and used A and A’ to indicate smoking signature or lack thereof, respectively. In a Bayesian framework, it is readily shown that a suitable form is S = 1 / (1 + R), where R is the ratio of the joint probability of A’ and D to the joint probability of A and D. For example, the latter can be written P(A)・P(C|A)・P(T|A)・P(N|A) and the former similarly, where each term of the former is the complement of its respective term in this expression. Common risk statistics are invoked as priors, i.e. P(A) = 0.9 (Walser et al., 2008).
We consider S to be a score because rigorous conditioned probabilities are difficult to establish. (For example, the data types themselves are not entirely independent of one another and models using common distributions like the Poisson do not recapitulate realistic variances.) Instead, we adopt a data-driven approach of estimating contributions of each data type based on 2-point empirical fitting of roughly the low and high extremes using shape functions. The general model for data type G is P(G|A) = [x ・erf (g/y) + 1] / (x + 2) and P(G|A’) = 1 - P(G|A), where erf is the Gaussian error function, g is the observed value for the given data type, and x and y are empirically-determined weights.
The shape function for total mutations, T, has two modifiers. First, it includes a simple expected-value correction for purity, u. Namely, assuming mutation-calling does not capture all mutations because of impurities, t is taken as the observed number of mutations divided by a purity shape function, f, where f ≤ 1. Although one might model f according to common characteristics of mutation callers, e.g. close to 100% sensitivity for pure samples and very low calling rate for low variant allele fractions (VAFs), the purity estimates for these data are based on RNA-Seq and are not highly correlated with total mutation counts. Consequently, we use a line function, f = 0.2・u + 0.8, which does not strongly impact the adjustment of low-purity samples. Second, before choosing weights, we removed outliers, as determined by Peirce’s criterion. Based on the distribution of continuous smoking score, we set 0.15 as the lower bound cutoff and 0.6 as the upper bound cutoff for defining the inferred smoking status. We classified the samples with strong smoking evidence if the smoking score > 0.6; samples with weak smoking evidence if smoking score < 0.15.
Identification of differentially regulated events in NRF2 mutant tumors.
For each gene, normalized levels of mRNA/protein/phosphoprotein (log transformed data) were fit into a linear regression model. In addition to NFR2 mutation status, gender, tumor purity and ethnicity were also included in the model. Coefficient and p-value of NRF2 mutation status variables from the fitted model were used to evaluate the extent and significance of the difference between patients with and without aberration. To adjust for multi-test, p-values were adjusted with the BH method. Genes that showed significant upregulation (up) in more than one dataset (Table S4) were used to generate a GMT file for ssGSEA based NRF2 activity score calculation using ssGSEA (https://github.com/broadinstitute/ssGSEA2.0). The NRF2 activity score contains a total of 54 genes upregulated both at protein and RNA level, identified by our NRF2 mutant versus wildtype tumor differential analysis (p-value <0.05 and logFC>1). Among the 54 genes, 44 have been previously associated with NRF2 signaling (either in NRF2 concept in msigdb such as Gene Sets NFE2L2.V2 (Malhotra et al., 2010) and SINGH_NFE2L2_TARGETS ((Singh et al., 2008), BIOCARTA_ARENRF2_PATHWAY], NRF2 wikipathway or Lacher et al (Lacher and Slattery, 2016). Ten of the 54 genes were novel targets identified in this study.
Immune cluster identification based on cell type composition
The abundance of 64 different cell types were computed via xCell based on transcriptomic profiles (Aran et al., 2017) based on 108 tumors and 94 NAT samples. Table S6 contains the final score computed by xCell of different cell types. Consensus clustering was performed based on immune cells, fibroblasts, endothelial and epithelial cells from xCell. In particular, we only considered cells that were detected in at least 10% of the patients (FDR < 5%). This filtering resulted in 25 cell types annotated in Table S6. Based on these signatures, consensus clustering was performed in order to identify groups of samples with similar immune/stromal characteristics. Consensus clustering was performed using the R packages ConsensusClusterPlus (Wilkerson and Hayes, 2010) based on z-score normalized signatures. Specifically, signatures were partitioned into three major clusters using the Partitioning Around Medoids (PAM) algorithm, which was repeated 1000 times (Wilkerson and Hayes, 2010) (Figure 6A, Table S6).
TCGA pan-cancer immune subtyping
Tumors were classified into the six distinct immune subtypes — wound healing, IFN-γ dominant, inflammatory, lymphocyte depleted, immunologically quiet, and TGF-β dominant — identified by a TCGA pan-cancer analysis presented in (Thorsson et al., 2018). Gene expression data (log2 FPKM; lscc-v3.0-rnaseq-uq-fpkm-log2-NArm) was input to the ImmuneSubtypeClassifier R package (https://github.com/CRI-iAtlas/ImmuneSubtypeClassifier) (Gibbs, 2020) to assign an immune subtype to each of the tumors.
Ranking tumors by inferred activity of IFN-γ pathway
We assumed that true biological activity of a pathway is regulated by collective changes of expression levels of majority of proteins involved in this pathway. Then, a difference of a pathway activity between tumors can be assessed by a difference in positioning of expression levels of proteins involved in this pathway in ranked list of expression levels of all proteins in each of tumors. Following this idea, we assessed relative positioning of pathway proteins between tumor by determining two probabilities: a probability of pathway proteins to occupy by random the observed positions in a list of tumor proteins ranked by expression levels from the top to the bottom and, similarly, a probability to occupy by random the observed positions in a list of expression levels ranked from the bottom to the top (Reva et al., 2020). Then, the inferred relative activation of a given pathway across tumors was assessed as a negative logarithm of the ratio of the above “top” and “bottom” probabilities. Thus, for a pathway of a single protein, its relative activity across tumors was assessed as a negative log of ratio of two numbers: a number of proteins with expression level bigger than an expression level of given protein, and a number of proteins with expression levels less than an expression level of given protein. For pathways of multiple proteins, the “top” and “bottom” probabilities were computed as geometrically averaged P values computed for each of proteins using Fisher’s exact test, given protein’s ranks in a list of pathway proteins and in a list of ranked proteins of a tumor, a number of proteins in a pathway, and the total number of proteins with the assessed expression level in a given tumor (Reva et al., 2020). The thermodynamic interpretation of the inferred pathway activity scoring function is a free energy associated with deviation of the system from equilibrium either as a result of activation or suppression. Thus, the scoring function is positive, when expression levels of pathway’s proteins are overrepresented among top expressed proteins of a tumor, and it is negative, when pathway’s proteins are at the bottom of expressed proteins of a tumor; the scoring function is close to zero, when expression levels are distributed by random or equally shifted towards top or bottom.
In our analysis, we used a gene list of IFN-γ signaling pathway from (Abril-Rodriguez and Ribas, 2017). We settled with this 15 genes signaling pathway because it has all necessary components – a cytokine mediator, receptors, signal transduction, transcription factors and final effectors (PD-L1/2, MHC-I), and all those genes are expressed in cancer cells.
Estimation of Tumor Purity, Stromal and Immune Scores
Besides xCell, we utilized ESTIMATE (Yoshihara et al., 2013) to infer immune and stromal scores based on gene expression data (Table S6). Cibersort absolute immune scores were obtained by evaluating upper-quartile normalized RNA-seq FPKM data with the Cibersort web app (cibersort.stanford.edu; (Newman et al., 2015)) in absolute mode. To infer tumor purity, TSNet was utilized (Petralia et al., 2018) (Table S6).
Differentially Expressed Genes and Pathway Analysis
Genes upregulated in Hot and Warm clusters compared to the Cold cluster were identified based on gene expression data, global proteomic, phosphoproteomic and acetylation data. For this analysis, markers with at most 50% missing values were utilized. For each data type, the expression level of gene/protein/sites was modeled as a function of immune cluster via linear regression. P-values were then adjusted for multiple comparisons via Benjamini-Hochberg adjustment. For each immune cluster, considering the set of genes up(down)-regulated with Benjamini-Hochberg adjusted p-value lower than 10%, a fisher exact test was implemented to derive enriched pathways. ssGSEA (Barbie et al., 2009) was utilized to obtain pathway scores based on RNA-seq and global proteomics data using the R package GSVA (Hänzelmann et al., 2013). For this analysis, pathways from the Reactome (Fabregat et al., 2018), KEGG (Kanehisa et al., 2017) and Hallmark (Liberzon et al., 2015) databases were considered and as background the full list of gene/proteins observed under each data type was utilized. For phosphorylation and acetylproteome data, a gene was considered upregulated if at least one substrate of the gene was found upregulated at 10% FDR. The pathway analysis results for different data types are contained in Table S6.
Deriving RTK CBPE scores
A total of 42 human RTKs were present in our proteomics data set. For each phosphosite in our data set we computed a linear association with each of the RTKs in two ways. A phosphosite profile across samples (for tumor and NAT separately) was modeled as a linear combination of 1) an RTK protein profile and a profile of a protein that a given phosphosite belonged to or 2) an RTK protein profile, a profile of a protein that a given phosphosite belonged to and a profile of tumor purity estimates obtained from RNAseq data via TSNet (Petralia et al., 2018). Each association computation was carried out only if at least 50% of samples had non-missing data of all three of RTK protein, phosphosite, and protein in question. The phosphosite:RTK associations were considered significant if their FDR adjusted p-value was below 0.1. Additionally, for each phosphosite the z-scores of their log2 phosphoprotein abundance were computed across samples (separately for tumor and NAT). A raw RTK CBPE score was computed for each RTK in each sample as a number of all phosphosites with significant association with this RTK that also had a z-score higher than 1. These raw RTK CBPE scores in each sample were normalized across RTKs so the final RTK CBPE scores of all RTKs in one sample would add up to 1. We selected the most relevant subset of RTKs by requiring that each had a normalized CBPE score of at least 0.1 in at least 5 samples. Nine RTKs satisfied this condition in the tumor data set, and seven in NAT.
Independent component analysis
ICA was performed with a workflow modified from previously described (Liu et al., 2019). Decomposition was run for 100 times on the matrix of protein abundance difference between tumor/NAT pairs (n=99). Independent components were in the form of vectors comprised with weights of all genes in the original data. Components extracted from each run were clustered using HDBSCAN algorithm (McInnes et al., 2017) with cosine distance as dissimilarity metric, min_cluster_size=50 and min_samples=20. The centroids of clusters (n=37) were considered as representative of stable signatures, and mean mixing scores (activity of each signature over all samples) of each cluster were used to represent the activity levels of corresponding signature in each sample. Correlation between the extracted signatures and known clinical characteristics were examined by regressing the corresponding mixing scores for all members of a component cluster against 64 sample annotations to obtain within-cluster average of log10 P-values. Significance was controlled for multiple testing at 0.01 level (log10 (p-value)=−5.3). Each signature vector (cluster centroid) was submitted to GSEA pre-ranked test for functional annotations.
Mutation-based cis- and trans-effects
We examined the cis- and trans-effects of 22 genes with somatic mutations that were significant in a previous large-scale TCGA LSCC study (Bailey et al., 2018) on the RNA, proteome, and phosphoproteome of known interactome DBs including Omnipath, Phosphositeplus, DEPOD, Signor, and CORUM. After excluding silent mutations, samples were separated into mutated and WT groups. We used the Wilcoxon rank-sum test to report differentially expressed features (RNA, proteins, phosphosites, acetylsites, and ubiquitylation sites) between the two groups. Differentially enriched features passing an FDR <0.05 cut-off were separated into two categories based on cis- and trans- effects.
Germline quantitative trait loci (QTL) analysis
To identify germline genetic variants that explain variation in tumor gene (eQTL) and protein (pQTL) expression, we utilized the gold-standard mapping pipeline at https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook-(eQTLGen). Briefly, we followed the default steps that include correcting for population stratification removing outliers, normalizing and mapping cis-QTLs. For cis-eQTL analysis input, we used i) germline genotype data; and ii) RNA-seq raw read count data for 105 samples, and performed analyses only for genes with sum of read count ≥ 10. For cis-pQTL analysis input, we used i) germline genotype data; and ii) normalized and filtered protein TMT ratio aggregated to gene level for 108 samples. The genotype data were harmonized using Genotype Harmonizer v1.4.9 (Deelen et al., 2014). To adjust for population stratification, we identified the multi dimensional scaling (MDS) components using the genotype data using PLINK v1.07 (Purcell et al., 2007). For both types of cis-QTL mapping, we used eqtl-mapping-pipeline-v1.3.9 using its standard settings (HWE > 0.0001, MAF > 0.01, and call rate > 0.95, maximum distance between the SNP and the middle of the probe is 250,000 bp). To control for multiple testing, we performed 10 permutations. We deemed QTLs at ≤ FDR 5% as significant. Finally, we performed pathway analysis of the identified significant eGenes (genes whose expression is impacted by at least one cis-QTL) using QIAGEN Ingenuity Pathway Analysis (Krämer et al., 2014)
miRNA analysis presented in Figure 4
Targets of miRNAs were downloaded from the miRNA targets database miRTarBase and only the miRNA/target pairs with strong experimental evidence were retained (Huang et al., 2020). Spearman correlation was used to calculate the correlations between miRNA and its target genes.
Pathway projection using ssGSEA
The single sample Gene Set Enrichment Analysis (ssGSEA) implementation available on https://github.com/broadinstitute/ssGSEA2.0 was used to project log2(FPKM) mRNA abundances to MSigDB cancer hallmark gene sets using the following parameters:
gene.set.database=“h.all.v6.2.symbols.gmt”
sample.norm.type=“rank”
weight=0.75
statistic=”area.under.RES”
output.score.type=“NES”
nperm=1000
global.fdr=TRUE
min.overlap=10
correl.type=”z.score”
Phosphorylation-driven signature analysis
We performed phosphosite-specific signature enrichment analysis (PTM-SEA) (Krug et al., 2018) to identify dysregulated phosphorylation-driven pathways. To adequately account for both magnitude and variance of measured phosphosite abundance, we used p-values derived from application of the Wilcoxon rank-sum test to phosphorylation data as ranking for PTM-SEA. To that end, p-values were log-transformed and signed according to the fold change (signed −log10 (p-value)) such that large positive values indicated phosphosite abundance in classical or NFE2L2, CUL3, KEAP1 mutated samples, and large negative values indicated phosphosite abundance in samples that do not belong to classical NMF subtype or wild-type samples (in Figure 3I)
PTM-SEA relies on site-specific annotation provided by PTMsigDB and thus a single site-centric data matrix data is required such that each row corresponds to a single phosphosite. We note that in this analysis the data matrix comprised a single data column and each row represented a confidently localized phosphosite assigned by Spectrum Mill software.
We employed the heuristic method introduced by Krug et al. (Krug et al., 2018) to deconvolute multiple phosphorylated peptides to separate data points (log-transformed and signed p-values). Briefly, phosphosites measured on different phospho-proteoform peptides were resolved by using the p-value derived from the least modified version of the peptide. For instance, if a site T4 measured on a doubly phosphorylated (T4, S8) peptide (PEPtIDEsR) was also measured on a mono-phosphorylated version (PEPtIDESR), we assigned the p-value derived from the mono-phosphorylated peptide proteoform to T4, and the p-value derived from PEPtIDEsR to S8. If only the doubly phosphorylated proteoform was present in the dataset, we assigned the same p-value to both sites T4 and S8.
We queried the PTM signatures database (PTMsigDB) v1.9.0 downloaded from https://github.com/broadinstitute/ssGSEA2.0/tree/master/db/ptmsigdb using the flanking amino acid sequence (+/− 7 aa) as primary identifier. We used the implementation of PTM-SEA available on GitHub (https://github.com/broadinstitute/ssGSEA2.0) using the command interface R-script (ssgsea-cli.R). The following parameters were used to run PTM-SEA:
weight: 1
statistic: “area.under.RES”
output.score.type: “NES”
nperm: 1000
min.overlap: 5
correl.type: “Z-score”
The sign of the normalized enrichment score (NES) calculated for each signature corresponds to the sign of the tumor-NAT log fold change. P-values for each signature were derived from 1,000 random permutations and further adjusted for multiple hypothesis testing using the method proposed by Benjamini & and Hochberg (Benjamini and Hochberg, 1995). Signatures with FDR-corrected p-values < 0.05 were considered to be differential between tumor and NAT.
CDKN2A and RB1 annotations and pathway analysis
Comprehensive tumor annotation for CDKN2A and RB1 genomic status was carried out using multiple molecular features for each patient. Specifically, we considered the following: 1) mutation types (missense/in-frame indels mutations, nonsense (stop gain, frameshift indels) mutations, and splice site (splice donor, splice acceptor) mutations) as separate categories and 2) copy number data. For cases with mutation, we considered both the variant allele frequency of the mutation from whole exome sequencing and copy number data (log ratio, absolute copy number, and B-allele frequency). Only mutations that occurred in cases with loss of the wild-type allele were annotated as loss of heterozygosity (LOH) mutations and used for subsequent analysis. 2) Based on CDKN2A copy number data, we next annotated tumors without mutations as homozygous deletions or no loss (WT). Samples were re-classified as having p16INK4a promoter hypermethylation if three p16INK4a promoter-associated methylation probes (cg11075751, cg02008397, and cg01694391) all had beta values >= 0.2. Multi-gene proliferation scores were calculated as described previously (Ellis et al., 2017) as the means of the gene normalized RNA levels for cell cycle associated genes characterized by Whitfield et al (Whitfield et al., 2002). p16INK4a RNA levels were obtained from the isoform specific RSEM data for Ensembl transcript ENST00000304494.8, while ENST00000579755.1 was used for p14ARF, and log2 (RSEM + 1) values were used for barplots. Isoform specific protein data included Refseq NP_000068.1 for p16INK4a and Refseq NP_478102.2 for p14ARF. Calculation of CDK1, 2, and 4 target site scores from the phosphorylation site data and Hallmark E2F targets and G2M scores from the RNA-seq data is described above (Phosphorylation-driven signature analysis and Pathway projection using ssGSEA, respectively). Lolliplots for CDKN2A mutations in the CPTAC head and neck squamous cell carcinoma and this lung squamous cell carcinoma cohort were generated using the ProteinPaint web application to visualize mutations (Zhou et al., 2016). Mutations annotated as nonsense mediated decay (NMD) mutations were those that resulted in low expression of p16INK4a RNA in the tumors harboring the mutations.
Association analysis between KGG-site abundances and E3 ligases and DUBs
A list of known human E3 ubiquitin and ubiquitin-like ligases and DUBs was compiled from (Medvar et al., 2016; Nijman et al., 2005). We then fit a linear model using limma in R with the formula kgg_site_abundance ~ protein_abundance, followed by empirical bayes shrinkage. The coefficient p-values were used to determine significant associations after FDR correction (FDR < 0.01).
Cluster and pathway analysis of significantly modulated K-GG sites in tumors.
Consensus clustering of the K-GG dataset after protein abundance correction was performed on tumor samples, resulting in 3 clusters. Next, marker selection for each cluster was performed by moderated t-test between samples that belong to the clusters against the remaining samples. These K-GG sites showing differential abundance across sample clusters were further clustered into 3 site-wise clusters.
Genes in these site-wise clusters were used for pathway enrichment analysis against the KEGG, Reactome, and WikiPathways databases using g:profiler (Raudvere et al., 2019). Pathway enrichment was performed using a gene background containing all observable genes in the K-GG dataset. Pathway enrichment results were imported into Cytoscape (Shannon et al., 2003) using the Enrichment Map app (Merico et al., 2010) for network analysis of pathways. Pathways were connected using the gene overlap and clustered (pathway cutoff q-val < 0.1; jaccard overlap > 0.375). Each cluster was manually annotated from the pathways contained in it to facilitate interpretation.
PTM CLUMPS analysis
We employ two methods to select tumor-specific sites to include in structural analysis. First, we take PTM-sites for solely tumor-derived samples and binarize modifications by negative vs. positive normalized signal. These are considered as individual “events” as done with mutations in CLUMPS v1 (Kamburov et al., 2015). For a more robust approximation of tumor-specific acetylation or ubiquitination, we perform differential expression using Limma between NAT and Tumor samples (Ritchie et al., 2015). Sites are selected with an FDR < 0.1 (Benjamini-Hochberg) and LFC > 0.1. We then binarize these tumor sites as done in the first approach. We first map the PTM-sites to corresponding UNIPROT ids (ID) with available PDBs. For each crystal structure, we compute an initial WAP score and randomly sample sites as done in CLUMPS. We generate an empirical p-value based on a random sampling of lysines in each crystal structure to limit the selection to residues capable of ubiquitination or acetylation. The null hypothesis we define is each random sampling of lysines will have a WAP score less than or equal to the initially computed WAP score. We run 1e6 permutations to generate an empirical p-value.
DepMap genetic dependency and drug response analysis
Cell lines annotated as “NSCLC_squamous” from DepMap were considered as LSCC for which molecular profiles, dependency scores, and drug response were used.
To determine copy number status in LSCC cell lines, copy number segmentation files for LSCC cell lines were processed with GISTIC2.0 (using the same parameters used to process this study’s LSCC cohort as described in STAR METHODS). Samples were considered to be amplified for a given gene if GISTIC thresholded values = 1 or 2 and not amplified if GISTIC thresholded values = −2, −1, or 0. Mutation status for CCND1, CDKN2A, and RB1, Np63 transcript isoform expression, phospho-Rb-Ser807/811 RPPA abundance, and drug responses to a survivin inhibitor (YM-155) and CDK4/6i inhibitors (abemaciclib and palbociclib) were also retrieved from DepMap.
Figure 7: For identifying if top protein biomarkers (502 proteins significantly overexpressed (log2(FC) >2, FDR <0.01) in tumors relative to their matched NATs, most with coherent overexpression in multi-omic analysis) also conferred altered dependencies in LSCC cell lines, we leveraged DepMap genetic dependency dataset (CRISPR Avana Public 20Q3) that contained 18119 genes and 789 cell lines (https://depmap.org/portal/download/ file: Achilles_gene_effect.csv). Only 16 cell lines were classified as LSCC (Cell Line Sample Info.csv). Median dependencies were calculated and for every gene and density plot in Figure 7C shows dependency score of the 502 genes corresponding to 502 protein biomarkers relative to other genes.
Figure S4B: To investigate copy number and dependency associations in LSCC cell lines, each gene up-regulated at the protein level with recurrent copy number gain in tumors vs NATs (from Figure 4A with Log2FC>0 & FDR < 0.05) was used as a query to compare dependency scores of the given gene (Combined CRISPR KO screen, DepMapPublic 21Q1) in LSCC cell lines where the query gene was amplified vs not-amplified. A lower mean change in dependency scores for a given gene in amplified vs not-amplified samples indicates LSCC cell lines are more dependent upon that gene when it is amplified.
Continuous log2 copy number data from DepMap was used in Figure S4M to correlate SOX2 copy number with EZH2 shRNA dependency data (Combined shRNA screen from DEMETER2 Data v6 in DepMap).
Drug responses to YM-155 (PRISM secondary screen) were compared by t-test in LSCC cell lines with low vs high levels of Np63 by transcript isoform expression (Figure S4G). Pearson correlation was also performed with phospho-Rb-Ser807/811 RPPA abundance and drug responses to CDK4/6i inhibitors, abemaciclib (PRISM secondary screen) and palbociclib (Sanger GDSC1) (Figure S3D).
CausalPath analysis
CausalPath (Babur et al., 2018) searches for known biological mechanisms that can explain correlated proteomic changes in terms of causal hypotheses. We set CausalPath parameters to compare tumors and NATs with a paired t-test, used 0.1 as FDR threshold for proteomic change significance and network significance, and detected 5917 potential causal relations between proteins. We repeated the same analysis for each NMF subtype separately and identified 4378 (basal-inclusive), 5334 (classical), 3048 (EMT-enriched), 3744 (inflamed-secretory), and 4332 (proliferative-primitive) relations. We used these CausalPath network results in the preparation of Figure 7C, identifying potential upstream regulators of oncogenic phosphoproteomic changes. Here an oncogenic phosphoproteomic change can be any of the following 4 events: increase of activating phosphorylation of an oncoprotein, decrease of inactivating phosphorylation of an oncoprotein, decrease of activating phosphorylation of a tumor suppressor protein, and increase of inactivating phosphorylation of a tumor suppressor protein. We used the OncoKB database for oncoprotein and tumor suppressor classification (excluded proteins that have both annotations), and used PhosphoSitePlus and Signor databases for the activating/inhibiting classification of phosphorylation sites. In the phosphorylation regulation networks, we included only the targetable regulators (activated proteins) and excluded the untargetable regulators (inactivated proteins).
Variant Peptide Identification
We used NeoFlow (https://github.com/bzhanglab/neoflow) for neoantigen prediction (Wen et al., 2020). Specifically, Optitype (Szolek et al., 2014) was used to find human leukocyte antigens (HLA) for each sample based on WES data. Then we used netMHCpan (Jurtz et al., 2017) to predict HLA peptide binding affinity for somatic mutation–derived variant peptides with a length between 8–11 amino acids. The cutoff of IC50 binding affinity was set to 150 nM. HLA peptides with binding affinity higher than 150 nM were removed. Variant identification was also performed at both mRNA and protein levels using RNA-Seq data and MS/MS data, respectively. Variant identification and gene quantification using RNA-Seq data were performed following the methods used in the previous study (Vasaikar et al., 2019). To identify variant peptides, we used a customized protein sequence database approach (Wang et al., 2012). We built a customized database for each TMT experiment based on somatic variants from WES data. We used Customprodbj (Wen et al., 2020) (https://github.com/bzhanglab/customprodbj) for customized database construction. MS-GF+ was used for variant peptide identification for all global proteome and phosphorylation data. Results from MS-GF+ were filtered with 1% FDR at peptide level. Remaining variant peptides were further filtered using PepQuery (http://www.pepquery.org) (Wen et al., 2019) with the p-value cutoff <= 0.01. Competitive filtering based on unrestricted posttranslational modification searching was enabled in PepQuery validation. The spectra of variant peptides were annotated using PDV (http://www.zhang-lab.org/) (Li et al., 2019b).
Cancer/testis Antigen Prediction
Cancer/testis (CT) antigens were downloaded from the CTdatabase (Almeida et al., 2009). CT antigens with a median >4-fold increase in tumor from NAT were highlighted.
PROGENy Scores
PROGENy (Schubert et al., 2018) was used to generate activity scores for EGFR based on RNA expression data. Tumor RNA expression values were submitted to PROGENy.
LSCC, HNSCC and LUAD integrative analysis
LUAD data for 110 lung adenocarcinoma samples and 102 NAT were acquired from the published manuscript (Gillette et al., 2020). HNSCC data for 108 head and neck squamous cell carcinoma samples and 66 NAT were acquired from the submitted manuscript (Huang et al., 2021). The LUAD and HNSCC proteomics data were processed, quantified, and normalized using the same pipeline as described for the LSCC samples. Differential expression analysis for all three cohorts was performed as described in the Differential marker analysis method. Proteins missing in more than 50% of the paired tumor/NAT samples were excluded.
Copy number drivers for all three were assessed by filtering the 8309 genes that were quantified in all three cohorts in the copy number, RNA, and proteomic data to those found in focal amplification regions (q value < .25). Spearman correlation was performed for these genes between CNA and RNA and between CNA and protein. Proteins were considered drivers if the correlation between both the CNA and RNA and CNA and protein were significantly positively associated (BH adjusted p value <0.01). Proteins were also required to be significantly increased in tumor vs paired NAT in the same cohort (Wilcoxon signed rank BH adjusted p value <0.01).
To identify genes associated with the immune score, correlation between CNA, RNA, protein, and the immune score was performed for the genes present in both CNA and RNA in a cohort (20,313 genes in LSCC, 18,091 genes in LUAD, and 21,616 genes in HNSCC). The immune score was calculated as the z-score transformation of the ESTIMATE Immune score, which was calculated for all three cohorts as described in the method Estimation of Tumor Purity, Stromal and Immune Scores. To be considered copy number drivers of the immune score, genes had to have a positive correlation between CNA & RNA, CNA & immune score, and RNA & immune score. If the protein was quantified in the cohort, the gene was also required to have a positive correlation between CNA & protein and protein & immune score. Spearman correlations with a BH adjusted p value < 0.01 were considered significant.
Supplementary Material
Figure S1: Experimental Design and Proteogenomic Data Quality Metrics, Related to Figure 1
A. Experimental workflow of LSCC tumor and NAT proteogenomic profiling. Samples were cryopulverized, aliquoted for genomic DNA, total RNA and protein extraction, and utilized for DNA sequencing (WGS and WES) and methylation analysis, RNA and miRNA sequencing and proteomics / PTM analysis as indicated. TMT11-based isobaric labeling was used to multiplex up to 5 tumors (blue filled circles) and paired NATs (pink filled circles) in a single TMT11-plex. The last TMT channel (131C) was the LSCC common reference (CR), comprising peptides from all LSCC tumor and NAT samples included in this study and replicated in every plex to serve as the common denominator for calculating peptide ratios. A total of 300ug peptides/sample was used for proteome, phosphoproteome and acetylproteome analyses via serial enrichment (STAR methods). For the ubiquitylproteome, a different TMT11 experimental design was used (Table S1) for the subset of tumors and NATs with ≥750ug residual peptide amounts after mainstream proteomic processing (85 tumors and 58 NATs). 750ug/sample was used for ubiquitylproteome analysis, with a ubiquitylproteome CR composed of peptides from the 143 available samples. The CR used for proteome, phosphoproteome and acetylproteome and that for ubiquitylproteome showed median Pearson correlations > 0.85 across analyses.
B. Numbers of quantified proteins, phosphosites, acetylsites, and ubiquitylsites across all TMT11-plexes. The values above each bar indicate percentage deviation from the median.
C. Pearson correlations of log TMT reporter ion intensities between all pairs of TMT plexes were consistently higher for CR than for sample channels, enabling precise relative quantification of proteins, phosphorylation and acetylation sites across 22 TMT plexes using the CR channels as the common denominator. The CRs of three TMT plexes in the ubiquitylproteome data showed lower reproducibility compared to the CRs of other ubiquitylproteome TMT plexes, which was computationally corrected as described in STAR methods (“Quality control and Batch effect assessment”). Top panel: Box plots show distributions of delta CR-sample correlations derived from all pairwise TMT plex comparisons across the different proteomic data types. Positive delta correlations indicate higher correlations of the CR channel compared to sample channels. Lower panel: Bar plots represent pairwise TMT plex correlations for proteomics, phosphoproteomics, acetylproteomics and ubiquitylproteomics using TMT plexes 1 and 2 as representative examples. Blue bars depict correlations of sample channels; red bars show the correlation of the CR.
D. Smoking scores calculated by WES stratified by self-reported smoking status. While some reformed and never-smokers had low scores, many self-reported never-smokers from Slavic countries had scores comparable to those of smokers.
E. PCA plots showing the lack of batch effect across all plexes in the different data types, with samples from each batch represented by different shapes. A clear separation in the PC1 component is observed between tumors and NATs in each data type.
F. To identify specific events associated with CNA trans-effects (vertical stripes in Figure 1D), we leveraged CMAP data (https://clue.io/cmap), which includes over 1.3 million gene expression profiles from ~20,000 small-molecule compounds and ~4,300 genetic reagents tested in multiple cell types. Patterns of significant trans-effects associated with CNAs that also had significant cis-effects were queried against CMAP profiles to identify knockout perturbations that significantly resembled those CNA-associated aberrations. Shown for two of the six genes (NRF2F6 and IL18) are correlations between GISTIC log2 CNA ratio or xCell-derived immune score and relative protein expression (Log2 TMT ratio). Pearson correlations and p-values are indicated.
G. Unsupervised consensus clustering results of promoter-level DNA methylation data from tumors and NATs. LSCC tumor DNA showed overall hypermethylation at gene promoters relative to NATs (Wilcoxon p<2×10−16).
H. Unsupervised consensus clustering of tumor methylation revealed CIMP (CpG island methylator phenotype) -high, -intermediate and -low clusters.
Figure S2: Characteristics of Multi-omic NMF Subtypes, Related to Figure 2
A. Upper panel: Line plot representing cophenetic correlation coefficients (y-axis) calculated for a range of factorization ranks (x-axis). The maximal cophenetic correlation coefficient was observed for rank K=5 as highlighted by the red box. Lower panel: silhouette plot for K=5. This plot indicates the quality of cluster separation.
B. Directionality and significance (signed Wilcoxon p-value) of abundance changes in RNA, protein (green), and PTMs (phosphosites in purple, acetylsites in blue, ubiquitylsites in yellow) between the Basal-Inclusive and other NMF subtypes. Identities of the most extreme outliers are designated and the modification site (K- lysine, S- Serine, T- Threonine, Y- Tyrosine) is separated by an underscore. PBX3 is highlighted by a red box.
C. Same as B but in the EMT-enriched subtype.
D. Pathway analysis of proteins with high correlation with PBX3 protein expression (FDR <0.05 and Pearson correlation >0.5) revealed neutrophil activation and neutrophil degranulation as the top protein-driven pathways.
E. Scatter plot showing Pearson correlations between PBX3 protein and other transcripts and PBX3 protein and other proteins. Points represent the signed −log10 (p-value). Neutrophil degranulation-associated genes show very high correlation with PBX3 protein only at the level of protein expression, whereas neutrophil chemotactic factors such as CXCL1 and 8 show high correlation at both RNA and protein levels.
F. Comparison of xCell-derived Immune, Microenvironment, Neutrophil and Stroma scores across five NMF subtypes. Z-score values are used for the box plots. P-values are derived from the Wilcoxon test. B.I - Basal-inclusive, C - Classical, E-E - EMT-enriched, I-S - Inflamed-Secretory, P-P - Proliferative-Primitive.
G. Representative dual immunohistochemical staining of alpha-smooth muscle actin (α-SMA) and pan-Cytokeratin (CK) of two samples corresponding to EMT-E and Classical subtypes.
H. Normalized connectivity scores (NCS) between the Library of Integrated Network-based Cellular Signatures (LINCS) compound signatures and the five NMF multi-omic subtype signatures at the RNA and protein level are represented in the heatmap. Each NMF signature was used to query the CMAP LINCS L1000 resource (2020 release), a collection of more than 1M response signatures of cancer cell lines to different types of chemical (e.g. drugs, tool compounds) and genetic perturbations (e.g shRNAs, sgRNAs, cDNAs). The 20 most negatively connected compounds to each NMF signature (with the most negative NCS) were then selected for visualization. Rows were ordered based on the EMT-E RNA signature indicating an enrichment for TGF-β inhibitors in the negatively-connected compounds of the EMT-E cluster.
I. Integrative analysis of the cumulative 216 treatment-naïve LSCC tumors between this and the Stewart study (Stewart et al., 2019), focused on the global proteome in the absence of global gene copy number and post-translational modification data in that study. Our NMF-based pipeline applied to the integrated proteomics dataset (STAR Methods) identified six clusters that partially aligned with RNA-based clusters as well as with proteomic and multi-omic subtypes defined in this study (Figure S2A). As expected, the Stewart et al., Redox B proteomics cluster samples joined with the NMF Classical subtype, enriched for NRF2 pathway mutations and redox biology. Notably, their Inflamed A and B and our Inflamed-Secretory tumors were each enriched in a separate cluster, their Inflamed A joining our Basal-Inclusive and their Inflamed B defining a cluster that was enriched for samples specifically from their cohort and for TP53 WT status, and that shared enrichment for secretory RNA status with our separately clustered Inflamed-Secretory tumors. B.I - Basal-inclusive, C - Classical, E-E – EMT-enriched, I-S - Inflamed-Secretory, P-P - Proliferative-Primitive.
J. Significantly enriched pathways (MSigDB Hallmark) in one or more of the six clusters shown in Figure S2I. Yellow-to-blue color scale depicts positive to negative normalized enrichment scores (NES), with asterisks denoting significant pathways (FDR<0.05) among the subtypes. At the pathway level, their Inflamed B had signals of coagulation and complement activity, but lacked the broader evidence of immune signaling associated with the Inflamed A and Inflamed-Secretory-enriched clusters (Figure S2I). The Proliferative-Primitive subset we defined retained its integrity and association with Primitive RNA status in the combined analysis. Finally, the EMT-E cluster defined in our study was clearly enriched with the mixed subtype samples from the Stewart et al., study. In addition to very strong EMT signaling, that cluster had evidence of hypoxemia and strong downregulation of oxidative phosphorylation (Figure S2I), known drivers of EMT (Cannito et al., 2008) and of Wnt signaling (Hong et al., 2017).
K. A schematic representation of the proposed crosstalk between non-canonical WNT5A/ROR2 and PDGFRB signaling axes. The binding of WNT5A to ROR2 activates the planar cell polarity pathway, shown in yellow ovals (DVL/RAC/RHO/JNK), and the Ca2+ pathway, shown in blue ovals (PKCa) involving cytoskeleton rearrangements and cell migration. Upregulation of RAC requires clathrin-mediated ROR2 receptor internalization of the Wnt signalosome (AP2, DVL, ROR2). WNT5A/ROR2 also activates PI3K/AKT signaling (green ovals) leading to RUNX2-mediated transcription of EMT (ZEB1, MMP2) and cell invasion (WNT5A, FLNa, ITG5A) programs (red ovals). Targeted therapeutics against WNT5A/ROR2 upregulation are shown in red, with some of them in clinical trials as indicated. Red arrows represent upregulation in the EMT-E proteome and phosphoproteome. Genes directly observed as upregulated at protein/phosphoprotein levels are in thick oval dark blue outlines.
Figure S3: Proteogenomic Consequences of CDKN2A and NRF2 Pathway Aberrations, Related to Figure 3
A. Tumor Mutational Burden per megabase (MB) as estimated from whole exome sequencing among samples with and without ARID1A gene somatic mutation.
B. Schematic diagram showing amino acid alterations resulting from CDKN2A/p16INK4a nonsense and frameshift mutations in the CPTAC LSCC and head and neck squamous cell cancer (HNSCC) datasets. Not shown are seven mutant tumors with missense or in-frame indels that would not be predicted to trigger nonsense mediated decay (NMD). Unlike the CPTAC HNSCC cohort, where p16INK4a (p16) nonsense and frameshift mutations always resulted in NMD (tumors with low p16 RNA expression are indicated by blue outline), five of the nine nonsense/frameshift mutations in LSCC appeared to have escaped NMD. These mutations either resulted in truncation at the very 5’ end (E10*) or were frameshift mutations that did not actually result in a significantly truncated predicted protein product (all predicted to generate protein products of 142 or more amino acids compared to the full length p16 protein of 160 amino acids), while all of the mutations triggering NMD resulted in production of a protein product that was 40-120 amino acids shorter than the wild-type protein.
C. Box plots showing Rb protein and phosphoprotein (mean of all Rb phosphosites) (Log2 TMT ratio) expression in samples with and without CCND1 amplification. Amplified samples are those with GISTIC threshold = 2, while all other samples were considered not amplified. P-values are from Welch’s two-sample T-tests.
D. Phospho-Rb levels correlate with response to CDK4/6 inhibitors in various LSCC cell lines. Scatter plots show correlation between phospho-Rb S807/811 by RPPA (CCLE 2019, Proteomics (RPPA) from DepMap) and abemaciclib response (PRISM Repurposing Secondary Screen 19Q4 from DepMap) (Left panel) or palbociclib response (Sanger GDSC1 from DepMap) for LSCC cell lines (right panel). CCND1, CDK2NA, and RB1 mutation status (DepMapPublic 21Q1, Mutation) depicted as filled circles. Outline depicts presence of CCND1 amplification (GISTIC threshold ≥ 1, derived from segmented copy number, DepMapPublic 21Q1) and/or CDKN2A deletion (GISTIC threshold ≤ −1). Cell lines with higher levels of phospho-Rb S807/811 were more sensitive to CDK4/6 inhibitors abemaciclib and palbociclib. R, Pearson correlation coefficient. DepMap (Dependency map) is a compendium of genomics, proteomics, shRNA and CRISPR based gene dependency datasets of hundreds of cell lines. PRISM (Profiling Relative Inhibition Simultaneously in Mixtures) and GDSC (Genomics of Drug Sensitivity) are databases containing sensitivity data to hundreds of compounds in hundreds of cell lines.
E. Box plots showing NFE2L2 S215 and S433 abundance in samples with CUL3, KEAP1 and NFE2L2 mutations. **** <=0.0001, *** < 0.01, **< 0.01, *< 0.05, ns = not significant. P-values are from the Wilcoxon test.
F. Correlation between differential mRNA expression (log2 FC) and protein abundance (log2 FC) levels among tumors with NRF2 pathway mutation (one and two hits) vs NRF2 pathway WT tumors. Significant events (>2 fold and p-value<0.05) are represented as circles with black borders. NRF2 targets in human cancer tissues (Lacher and Slattery, 2016) are represented by solid circles in green, while solid circles in black indicate novel targets. Candidates that also show phosphosite modification are represented by pink-filled gene name boxes.
G. Overlap between NRF2-related genesets (upper left), and up- (upper right) and down-regulated (bottom) events in NRF2 pathway mutant samples across mRNA, proteomics and phosphoproteomics.
H. Relative mRNA, protein and phosphosite abundance of NRF2 pathway-related and other cancer-associated genes in tumor samples with two NRF2 pathway (NF2L2, KEAP1, CUL3) mutations, one mutation, or WT status, as well as in NATs. A dosage effect is observed. # no phosphosite levels changes were detected for NR0B1 between the groups.
Figure S4: Association Between TP63 and miR205 Expression in LSCC, Related to Figure 4
A. Focal amplification and deletion landscape of the study LSCC cohort. The top five significant focal peaks are labeled in the plot
B. Copy number segmentation file (DepMapPublic 21Q1) for LSCC cell lines from DepMap, processed with GISTIC2.0 (same parameters used to process LSCC cohort as described in STAR METHODS). Genes were considered amplified if GISTIC thresholded values = 1 or 2 and not amplified if GISTIC thresholded values = −2, −1, or 0. Each gene upregulated at the protein level with recurrent copy number gain in tumors vs NATs (from Figure 4A with Log2FC>0 & FDR < 0.05) was used as a query to compare dependency scores (Combined CRISPR KO screen, DepMapPublic 21Q1) by t-test of a given gene in amplified vs non-amplified LSCC cell lines. X-axis depicts the mean change in dependency scores for a queried gene in cell lines with amplification vs without amplification of that gene. Y-axis depicts the −log10(p-value) from t-test. Genes passing FDR < 0.25 are depicted by red and blue colored dots.
C. mRNA expression of all detected TP63 Ensembl transcript (ENST) isoforms for tumors. The transcript corresponding to ΔNp63α was the dominant isoform observed in this cohort.
D. Correlations between ΔNp63α transcript and gene-level TP63 mRNA expression (upper panel) or TP63 relative protein expression (log2 TMT ratio) and gene-level TP63 mRNA expression (lower panel).
E. Kaplan-Meier plot of overall survival in ΔNp63α transcript-low versus -high samples from the TCGA LSCC cohort. Samples with low ΔNp63α expression have significantly worse outcomes.
F. Np63 mRNA transcript isoform expression analysis (RNAseq (Transcript isoform), DepMapPublic 21Q1) shows LSCC cell lines with high and low levels of Np63.
G. Response to survivin (BIRC5) inhibitor YM-155 (PRISM Repurposing Secondary Screen 19Q4 from DepMap) in LSCC cell lines with high and low expression of Np63. Np63-low lines are more sensitive to YM-155 than Np63-high cell lines. P-value derived from t-test.
H. Rank plots showing enrichment scores for proliferation-related pathways from pre-ranked GSEA. The inputs are t-values of gene expression between TP63-low and TP63-high samples.
I. Volcano plot showing differential expression of miRNA (Log2 FC) between tumors and NATs. miR-205 shows the highest relative overexpression in tumors.
J. Differential promoter level methylation of miR-205 in tumors compared to NATs. miR-205 is hypomethylated in tumors.
K. Pearson correlations between miR-205 expression and all quantified proteins in tumors. TP63 is highlighted and shows the highest correlation with miR-205.
L. Gene-level RNA (left) and protein (right) expression correlations between TP63 and TP73.
M. Correlation between EZH2 dependency by shRNA (shRNA KD screen (Combined), DEMETER2 Data v6 from DepMap) and SOX2 Log2 copy number (Copy number (Gene level), DepMapPublic 21Q1) in LSCC cell lines. R, Pearson correlation coefficient.
Figure S6: Differential pathway, CBPE scores and impact of germline mutations on immune signaling, Related to Figure 6
A. Key pathways differentially expressed between Hot, Warm and Cold tumors based on their gene expression and global protein abundance. Wilcoxon p-values are reported.
B. Key pathways differentially expressed between Hot, Warm and Cold tumors based on the gene expression and global proteomic abundance of corresponding NATs. Wilcoxon p-values are reported.
C. Averages of CD163, CD4, and CD8 IHC scores derived from three independent pathologists on four cores/sample from 17 samples representing Hot (5), Warm (5) and Cold (7) clusters. P-values are based on ordinal regression analysis of immune cluster scores, using random effects to account for multiple cores/samples and independent pathologists.
D. Representative immunohistochemical staining pattern for CD163, CD8, and CD4 proteins on three samples drawn from Hot, Warm, and Cold immune clusters. Protein expression levels in different cell types are highlighted with colored arrowheads as indicated.
E. Overlap between differentially regulated mRNA and proteins between Hot vs Cold and Warm vs Cold tumors, and eGenes downstream of cis-eQTLs and cis-pQTLs. Recent studies have suggested the importance of germline variants in the immune profiles of solid tumors (Lim et al., 2018; Shahamatdar et al., 2020; Tian et al., 2021). Among 319 cis-pQTLs (cis-regulating the cognate protein) identified in this study, only 38 were also cis-eQTLs (cis-regulating cognate RNA)(Table S6). Interestingly, the set of all proteins with pQTLs (but not the set of all genes with eQTLs) was enriched in pathways involved in communication between innate and adaptive immune cells. The 15 pQTLs in proteins differentially abundant between Hot and Cold tumors included proteins related to immune response (TRIM22), antigen processing (TAPBPL and HLA-A) and IL-6 signaling pathway (LRPPRC); eight were previously reported (Lim et al., 2018). These results imply a potentially important role of germline variants in shaping immune processes in LSCC.
F. Protein abundance and/or phosphorylation abundance (Log2 TMT ratio) of key markers involved in Rho GTPase signaling stratified by immune clusters. Wilcoxon p-values are reported.
G. Immunohistochemical staining images of ARHGDIB (RhoGDIB) protein in two tumors. Its expression is predominantly observed in tumor microenvironments with different types of cells indicated with differently colored arrowheads.
Figure S7: Differential tumor and NAT analysis, Related to Figure 7
A. Log2 fold change of proteins increased in tumors relative to paired NATs, together with candidate representation in the Human Proteome Atlas (HPA) and various annotations. Shown are the top 50 of 502 differential proteins with coherent expression in all omics that were significantly upregulated in ≥90% of T/NAT pairs (log2(FC)>2, FDR<0.01). The blue box indicates the protein that was upregulated in 100% of T/NAT pairs.
B. Fold changes of CT antigens and their relationship to NMF subtype and immune clusters.
C. Tumor-specific proteins from the current study showing significant association with disease-free survival in the TCGA LSCC cohort.
D. Focal amplification (red) and deletion (blue) events in the copy number data from three CPTAC tumor cohorts.
E. To identify copy number drivers of the immune score, correlations between CNA, RNA, protein, and the immune score were assessed for all genes present in both CNA and RNA in a cohort. To be considered drivers, genes had to have positive pairwise correlations between CNA & RNA, CNA & immune score, and RNA & immune score. If the protein was quantified in the cohort, positive correlations were also required between CNA & protein and protein & immune score. Spearman correlations with a BH adjusted p< 0.01 were considered significant. The figure shows genes deleted in at least 25% of the cohorts that were significantly correlated with RNA and protein expression as well as ESTIMATE immune score. LSCC-specific genes are indicated in black, genes in both LSCC and HNSCC are in red, genes in both LSCC and LUAD are in blue, and genes shared by all three are in purple.
F. Significantly increased activating sites on kinases in the CPTAC LSCC, LUAD (Gillette et al., 2020) and the HNSCC (Huang et al., 2021) cohorts.
G. EGFR log2 tumor/NAT protein fold change in three CPTAC cancer cohorts (LSCC, LUAD, HNSCC).
Table S1: Metadata Associated with LSCC Patients and Tumors, Related to Figures 1–7, Figures S1–7
A. Metadata associated with this study
B. Cancer Associated Genes (CAGs)
C. Table containing results from association analyses between key metadata and technical parameters for NMF multi-omic subtyping. NMF core members were considered for this analysis.
D. Table containing results from association analyses between key metadata and technical parameters for NMF multi-omic subtyping. All samples; tumors and NATs were considered for this association.
E. Table containing results from association analyses between key metadata and technical parameters for immune clusters. All samples; tumors and NATs were considered for this association.
Table S2: Genomic and methylation datasets associated with this study, Related to Figures 1–7, Figures S1–7
A. Gene-level data matrix of somatic copy number alterations (CNA) obtained from GISTIC2 analysis. CNAs are represented as absolute gene copy numbers. Refseq GRCh38/hg38 database was used as a reference.
B. Same as Table S2A but CNAs are represented as log2(CN)-1.
C. Same as Table S2A but CNAs are represented as thresholded copy number level calls.
D. RNA expression data matrix reporting Log2-transformed upper-quartile (UQ)-normalized FPKM values.
E. Same as Table S2D but with FPKM values median-centered by gene.
F. miRNA expression data matrix reporting log2 TPM (Transcripts Per Kilobase Million) values.
G. Same as Table S2F but with TPM values median-centered by gene.
H. Gene level DNA methylation data matrix. Beta values were obtained by averaging the beta values of individual probes in the islands of promoter or 5’ UTR regions of the gene.
I. Somatic mutations associated with the CPTAC LSCC cohort in Mutation Annotation Format (MAF) format.
Table S3: Global proteomics, phosphoproteomics, acetylproteomics and ubiquitylproteomics data associated with this study, Related to Figures 1–7 and Figures S1–7
A. Two-component-normalized log2-transformed protein data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods.
B. Same as Table S3A but after application of the filtering strategy described in the STAR methods
C. Gene-centric global proteome data matrix derived from Table S3B. Protein-level data were aggregated to gene-level using the sample/CR ratio assigned to the dominant isoform per gene symbol (similar to the razor-peptide approach).
D. Two-component-normalized log2-transformed phosphoproteomic data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods.
E. Two-component-normalized log2-transformed phosphoproteomic data matrix (sample/CR ratios) after application of the filtering strategy described in the STAR methods.
F. Gene-centric phosphoproteomic data matrix derived from Table S3E. Phosphosite-level sample/CR ratios were aggregated to gene-level using mean ratio.
G. Two-component-normalized log2-transformed acetylproteomic data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods.
H. Two-component-normalized log2-transformed acetylproteomic data matrix (sample/CR ratios) after application of the filtering strategy described in the STAR methods.
I. Gene-centric acetylproteomics data matrix derived from Table S3H. Acetylsite-level sample/CR ratios were aggregated to gene-level using mean ratio.
J. Acetylproteomic data matrix based on Table S3G corrected for global protein expression (Table S3B) as described in the STAR methods.
K. Two-component-normalized log2-transformed, batch corrected ubiquitylproteomic data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods. See STAR methods for details on batch correction.
L. Two-component-normalized log2-transformed, batch corrected ubiquitylproteomic data matrix (sample/CR ratios) after application of the filtering strategy described in the STAR methods. See STAR methods for details on batch correction.
M. Gene-centric ubiquitylproteomic data matrix derived from Table S3L. Ubiquitylsite-level sample/CR ratios were aggregated to gene-level using mean ratio.
N. Ubiquitylproteomic data matrix based on Table S3L corrected for global protein expression (Table S3B) as described in the STAR methods.
Table S4: CNA cis and trans effects, iProFun, CMAP, NRF2 differential expression, NRF2 signature, cis-eQTL from RNA and proteomics datasets, eGenes, Related to Figures 1–4
A. Copy number amplified cytobands
B. Copy number deleted cytobands C. CNA cis- and trans-effects
D. Significant focal amplicons that show significant cis-effect and are cancer associated genes
E. iProFun results with mRNA, protein, phosphoprotein, acetylprotein as outcomes and methylation as the predictor, adjusted for CNV(lr), CNV(baf) and tumor purity. Results include identified genes (Yes=1/No=0) with FDR<10% and identified phenotype association analyses (Yes=1/No=0).
F. CMAP tab- Connectivity Map (CMAP) results
G. Association between genes enriched in the CMAP analysis and sample metadata
H. Normalized connectivity scores (NCS) between Library of Integrated Network-based Cellular Signatures (LINCS) compound signatures and the five NMF multi-omic subtype signatures at the RNA and Protein level.
I. Summary of CDKN2A and RB1 annotations
J. T test p-values of differential proteomic features CCND1, CCND2, CCND3, and CDK6
K. Differential proteogenomic features in samples with and without NFE2L2 (NRF2), CUL3 and KEAP1 mutations
L. NRF2 signature
M. Differential expression of RNA and proteins between TP63-low vsTP63-high samples.
Table S5: Tables associated with ubiquitylproteome analysis, Related to Figure 5
A. Clustering of tumor samples using protein-corrected ubiquitin data
B. Ub-sites that show differential abundance across Ub clusters
C. Pathway enrichment results for Ub-sites with higher abundance in Ub Cluster 1
D. Pathway enrichment results for Ub-sites with higher abundance in Ub Cluster 2
E. Ub-sites and E3 ligase pairs with significant abundance association (FDR < 0.01)
F. Ub-sites and DUB pairs with significant abundance association (FDR < 0.01)
G. Pathway enrichment results for Ub-sites positively associated with HERC5 abundance
H. Genes with significant spatial differential acetylation clustering within a protein structure as detected by CLUMPS
I. Genes with significant spatial differential ubiquitylation clustering within a protein structure as detected by CLUMPS
J. Genes with significant spatial differential NAT ubiquitylation clustering within a protein structure as detected by CLUMPS
Table S6: Tables associated with immune-landscape analysis, Related to Figure 6
A. Immune composition of LSSC tumors based on deconvolution of RNA expression using xCell.
B. Immune clusters based on xCell signatures
C. Pathways upregulated in immune groups based on RNA-Seq data (C1), global proteomics (C2), phosphoproteomics (C3), acetylproteomics (C4)
D. Genes upregulated in immune groups based on RNA-Seq data (D1), global proteomics (D2), phosphoproteomics (D3), acetylproteomics (D4)
E. RTK Correlation-Based Enrichment (CBPE) Scores
F. Pathway scores in Tumor samples based on global protein abundance via ssGSEA
G. cis-eQTLS identified based on germline variants and tumor RNA-seq data (at 5% FDR)
H. cis-pQTLS identified based on germline variants and tumor proteomics data (at 5% FDR)
I. List of genes/proteins whose expression is associated with a SNP
J. Ingenuity pathway analysis of the eGenes from QTL analysis
K. cis-QTLs identified in genes/proteins differentially expressed in immune groups (K1-3) and list of eGenes (K4)
Table S7: Tables associated with tumor-associated proteins, Related to Figure 7
A. Differential abundance of proteins in tumor vs paired NAT in LSCC, HNSCC, and LUAD
B. 502 proteins significantly overexpressed (log2(FC) >2, FDR <0.01) in tumors relative to their matched NATs, most with coherent overexpression in multi-omic analysis
C. Tumor suppressors or oncogenes regulated at the level of protein expression or phosphoproteome. Activating phosphosite: regulated at the level of phosphorylation, annotated as activating site, Inhibiting phosphosite: regulated at the level of phosphorylation, annotated as inhibitory site. CausalPath was used for this analysis.
D. DepMap dependency scores (CRISPR (Avana) Public 20Q3) across 16 LSCC cell lines
E. Genes that correlate to immune score at the copy number, RNA, and protein (if available) levels in LSCC, HNSCC, and LUAD
F. Predicted tumor neoantigens identified in the CPTAC LSCC cohort at the level of RNA transcripts and peptides containing evidence of somatic mutations.
Figure S5: Pathway analysis and differential ubiquitylation and acetylation, Related to Figure 5
A. Network representation of pathways significantly enriched in modulated protein-corrected K-GG sites across Ub clusters (Figure 5A). Two patterns of K-GG site abundance were observed across Ub clusters (top). The red and blue arrows indicate up- and downregulation across the clusters, respectively. Significant pathways are indicated as nodes and edges indicate shared genes. Network clusters were summarized to reduce pathway redundancies and the common functional themes were manually annotated as text.
B. STRING network showing the genes driving the enrichment of the “Immune signaling, MAPK targets, and JAK-STAT signaling” cluster shown in panel A. These genes are core members of many signaling pathways, resulting in enrichment of multiple pathways.
C. Number of K-GG sites showing significant correlations with deubiquitylases (DUBs). Shown are the five DUBs with the highest proportion of negative correlations.
D. HERC5 and ISG15 protein expression (Log2 TMT ratios) show significant positive correlation.
E. Pathway enrichment analysis results of K-GG sites that significantly correlate with HERC5 protein expression.
F. HERC5 protein expression (Log2 TMT ratio) and example K-GG site abundances (protein-corrected Log2 TMT ratio) from PKM, ENO1, and PGK1 show strong positive correlations in tumors.
G. PKM, ENO1, and PGK1 plotted against sample K-GG site abundance of the respective proteins. K-GG site abundance is shown without protein correction (Log2 TMT ratio) to highlight the lack of negative correlation prior to protein correction.
H. HERC5 protein expression (Log2 TMT ratio) plotted against PKM, ENO1, and PGK1 protein expression (Log2 TMT ratio), which do not show positive correlation in tumors.
I. HERC5 protein expression (Log2 TMT ratio) and interferon-gamma (IFNG) score show significant positive correlation.
J. ISG15 protein expression (Log2 TMT ratio) and IFNG score show significant positive correlation.
K. PGK1 acetylated sites marked on the protein structure in Figure 5E (top left) are decreased in tumors compared to NATs; Ub sites are increased (top right). PKM acetylated sites (lower left) are decreased in tumors compared to NATs; Ub sites are increased (lower center). ACADVL acetylated sites marked on the protein structure in Figure 5E (lower right) are decreased in tumors compared to NATs. Wilcoxon p-values are indicated.
Highlights.
Unsupervised clustering revealed subtype with EMT and phosphoprotein signatures
Potential therapeutic vulnerabilities included survivin, NSD3, LSD1 and EZH2
Rb phosphorylation nominated as a biomarker for trials with CDK4/6 inhibitors
Detailed immune landscape analysis highlighted targetable points of immune regulation
Acknowledgements
This work was supported by grants U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, and U01CA214125 from the NCI-CPTAC, U54HL127624, P30CA240139, 1F32HL154711-01, and T32CA203690 from the NIH. We thank Dr. Lata Adnani for helping with graphics.
Footnotes
Declaration of Interests
The authors declare no competing interests
ADDITIONAL RESOURCES
The CPTAC program website, detailing program initiatives, investigators, and datasets, is found at https://proteomics.cancer.gov/programs/cptac.
A website for interactive visualization of the multi-omic dataset is available at: https://rstudio-connect.broadapps.org/CPTAC-LSCC2021/
All processed data matrices are available at LinkedOmics (Vasaikar et al., 2018) (http://www.linkedomics.org), where computational tools are available for further exploration of this dataset.
Consortia
Alex Green, Alexey Nesvizhskii, Alfredo Molinolo, Alicia Francis, Amanda Paulovich, Ana Robles, Andrii Karnuta, Antonio Colaprico, Arul Chinnaiyan, Azra Krek, Barbara Hindenach, Barbara Pruetz, Bartosz Kubisa, Bing Zhang, Bo Wen, Boris Reva, Brian Druker, Carissa Huynh, Chandan Kumar-Sinha, Charles Goldthwaite Jr., Chelsea Newton, Chen Huang, Chet Birger, Chia-Kuei Mo, Christopher Kinsinger, Corbin Jones, DR Mani, Dan Rohrer, Dana Valley, Daniel Chan, David Chesla, David Fenyö, David Heiman, Dmitry Rykunov, Donna Hansel, Elena Ponomareva, Elizabeth Duffy, Eric Burks, Eric Jaehnig, Eric Schadt, Erik Bergstrom, Eugene Fedorov, Eunkyung An, Fei Ding, Felipe da Veiga Levoprost, Fernanda Martins Rodrigues, Francesca Petralia, Gad Getz, Galen Hostetter, George Wilson, Gilbert Omenn, Henry Rodriguez, Houxiang Zhu, Hui Zhang, Jackson White, James Suh, Jennifer Eschbacher, Jennifer Maas, Jonathan Lei, Jiayi Ji, Kai Li, Karen Christianson, Karen Ketchum, Karin Rodland, Karl Clauser, Karsten Krug, Katherine Hoadley, Kei Suzuki, Kelly Ruggles, Ki Sung Um, Li Ding, Liqun Qi, Lori Bernard, Harry Kane, Maciej Wiznerowicz, MacIntosh Cornwell, Małgorzata Wojtyś, Marcin Cieslik, Marcin Domagalski, Mathangi Thiagarajan, Matthew Ellis, Matthew Wyczalkowski, Maureen Dyer, Meenskshi Anurag, Mehdi Mesri, Melissa Borucki, Michael Birrer, Michael Gillette, Midie Xu, Mikhail Krotevich, Myvizhi Esai Selvan, Namrata Udeshi, Nancy Roche, Nathan Edwards, Negin Vatanian, Neil Mucci, Nicollette Maunganidze, Nikolay Gabrovski, Olga Potapova, Oluwole Fadare, Özgün Babur, Pamela Grady, Pankaj Vats, Paul Paik, Pei Wang, Peter McGarvey, Pierre Jean Beltran, Pushpa Hariharan, Qing Kay Li, Ramaswamy Govindan, Ratna Thangudu, Rebecca Montgomery, Richard Smith,Robert Welsh, Runyu Hong, Sailaja Mareedu, Samuel Payne, Sandra Cottingham, Sara Savage, Saravana Dhanasekaran, Scott Jewell, Sendurai Mani, Seungyeul Yoo, Shankara Anand, Shankha Satpathy, Shayan Avanessian, Shilpi Singh, Shirley Tsang, Shuang Cai, Song Cao, Stacey Gabriel, Stephan Schurer, Steven Carr, Suhas Vasaikar, Tao Liu, Tara Hiltke, Tatiana Omelchencko, Thomas Bauer, Tobias Schraink, Vasileios Stathias, Volodymyr Sovenko, Warren Tourtellotte, Weiping Ma, Wenke Liu, William Bocik, Wohaib Hasan, Xiaoyu Song, Yifat Geffen, Yize Li, Yongchao Dou, Yuping Zhang, Yuxing Liao, Yvonne Shutack, Zeynep Gumus, Zhen Zhang, Ziad Hanhan.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Abril-Rodriguez G, and Ribas A (2017). SnapShot: Immune Checkpoint Inhibitors. Cancer Cell 31, 848–848.e1. [DOI] [PubMed] [Google Scholar]
- Ahn ER, Mangat PK, Garrett-Mayer E, Halabi S, Dib EG, Haggstrom DE, Alguire KB, Calfa CJ, Cannon TL, Crilley PA, et al. (2020). Palbociclib in Patients With Non–Small-Cell Lung Cancer With CDKN2A Alterations: Results From the Targeted Agent and Profiling Utilization Registry Study. JCO Precision Oncology 757–766. [DOI] [PubMed] [Google Scholar]
- Albert M, Bécares M, Falqui M, Fernández-Lozano C, and Guerra S (2018). ISG15, a Small Molecule with Huge Implications: Regulation of Mitochondrial Homeostasis. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcalá S, Sancho P, Martinelli P, Navarro D, Pedrero C, Martín-Hijano L, Valle S, Earl J, Rodríguez-Serrano M, Ruiz-Cañas L, et al. (2020). ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat. Commun. 11, 2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida LG, Sakabe NJ, deOliveira AR, Silva MCC, Mundstein AS, Cohen T, Chen Y-T, Chua R, Gurung S, Gnjatic S, et al. (2009). CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 37, D816–D819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardini-Poleske ME, Clark RF, Ansong C, Carson JP, Corley RA, Deutsch GH, Hagood JS, Kaminski N, Mariani TJ, Potter SS, et al. (2017). LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L733–L740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur Ö, Luna A, Korkut A, Durupinar F, Siper MC, Dogrusoz U, Aslan JE, Sander C, and Demir E (2018). Causal interactions from proteomic profiles: molecular data meets pathway knowledge. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza J, Smallegan MJ, and Denu JM (2015). Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 10, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, Yoshioka N, Bucala R, and Koike T (2005). Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 11, 5784–5792. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellman R (1961). On the approximation of curves by line segments using dynamic programming. Commun. ACM 4, 284. [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300. [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Brunet J-P, Tamayo P, Golub TR, and Mesirov JP (2004). Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. U. S. A. 101, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, Schormann W, Maccoux LJ, Schug M, Schumann A, et al. (2010). Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 12, R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y, Zhu X, Chen B, Wu J, and Li M (2013). miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 73, 5402–5415. [DOI] [PubMed] [Google Scholar]
- Cammer M, Gevrey J-C, Lorenz M, Dovas A, Condeelis J, and Cox D (2009). The mechanism of CSF-1-induced Wiskott-Aldrich syndrome protein activation in vivo: a role for phosphatidylinositol 3-kinase and Cdc42. J. Biol. Chem. 284, 23302–23311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Yau C, Bowlby R, Liu Y, Brennan K, Fan H, Taylor AM, Wang C, Walter V, Akbani R, et al. (2018). Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 23, 194–212.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, and Rüttinger D (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannito S, Novo E, Compagnone A, Valfrè di Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A, Bozzo F, et al. (2008). Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 29, 2267–2278. [DOI] [PubMed] [Google Scholar]
- Cassetta L, and Pollard JW (2018). Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904. [DOI] [PubMed] [Google Scholar]
- Chida S, Okayama H, Noda M, Saito K, Nakajima T, Aoto K, Hayase S, Momma T, Ohki S, Kono K, et al. (2016). Stromal VCAN expression as a potential prognostic biomarker for disease recurrence in stage II-III colon cancer. Carcinogenesis 37, 878–887. [DOI] [PubMed] [Google Scholar]
- Debebe Z, and Rathmell WK (2015). Ror2 as a therapeutic target in cancer. Pharmacol. Ther. 150, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen P, Bonder MJ, van der Velde KJ, Westra H-J, Winder E, Hendriksen D, Franke L, and Swertz MA (2014). Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res. Notes 7, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Johannessen CM, Leong C-O, Faquin W, Rocco JW, and Ellisen LW (2006). Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res. 66, 9362–9368. [DOI] [PubMed] [Google Scholar]
- Diviani D, Raimondi F, Del Vescovo CD, Dreyer E, Reggi E, Osman H, Ruggieri L, Gonano C, Cavin S, Box CL, et al. (2016). Small-Molecule Protein-Protein Interaction Inhibitor of Oncogenic Rho Signaling. Cell Chem Biol 23, 1135–1146. [DOI] [PubMed] [Google Scholar]
- Dovas A, Gevrey J-C, Grossi A, Park H, Abou-Kheir W, and Cox D (2009). Regulation of podosome dynamics by WASp phosphorylation: implication in matrix degradation and chemotaxis in macrophages. J. Cell Sci. 122, 3873–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman MJ, Redman MW, Albain KS, McGary EC, Rafique NM, Petro D, Waqar SN, Minichiello K, Miao J, Papadimitrakopoulou VA, et al. (2019). SWOG S1400C (NCT02154490)-A Phase II Study of Palbociclib for Previously Treated Cell Cycle Gene Alteration-Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J. Thorac. Oncol. 14, 1853–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Suman VJ, Hoog J, Goncalves R, Sanati S, Creighton CJ, DeSchryver K, Crouch E, Brink A, Watson M, et al. (2017). Ki67 Proliferation Index as a Tool for Chemotherapy Decisions During and After Neoadjuvant Aromatase Inhibitor Treatment of Breast Cancer: Results From the American College of Surgeons Oncology Group Z1031 Trial (Alliance). J. Clin. Oncol. 35, 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T, Sun G, Sun X, Zhao L, Zhong R, and Peng Y (2019). Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes AP, Capitanio A, Selenius M, Brodin O, Rundlöf A-K, and Björnstedt M (2009). Expression profiles of thioredoxin family proteins in human lung cancer tissue: correlation with proliferation and differentiation. Histopathology 55, 313–320. [DOI] [PubMed] [Google Scholar]
- Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, Young G, Fennell TJ, Allen A, Ambrogio L, et al. (2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Santiago MC, Berechid BE, and Nye JS (2002). Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr. Biol. 12, 1006–1011. [DOI] [PubMed] [Google Scholar]
- Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, and Hansen KD (2014). Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C, Raftery AE, Scrucca L, Murphy TB, and Fop M (2016). mclust: Gaussian mixture modelling for model-based clustering, classification, and density estimation. R Package Version 5. [PMC free article] [PubMed] [Google Scholar]
- Freed D, Pan R, and Aldana R TNscope: Accurate Detection of Somatic Mutations with Haplotype-based Variant Candidate Detection and Machine Learning Filtering. [Google Scholar]
- Fu B, Meng W, Zeng X, Zhao H, Liu W, and Zhang T (2017). TXNRD1 Is an Unfavorable Prognostic Factor for Patients with Hepatocellular Carcinoma. Biomed Res. Int. 2017, 4698167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi N, and Das GM (2019). Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Zhuang J, Li H, Liu C, Zhou C, Liu L, and Sun C (2018). Exploration of methylation-driven genes for monitoring and prognosis of patients with lung adenocarcinoma. Cancer Cell Int. 18, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Boulter E, and Burridge K (2011). The “invisible hand”: regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 12, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, and Seoighe C (2010). A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gey S, and Lebarbier E (2008). Using CART to detect multiple change points in the mean for large sample. [Google Scholar]
- Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, van Klaveren RJ, Chaudhary S, Gunther A, and Shamsili S (2009). Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 27, 4481–4486. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos NV, Luo J-K, Papov V, Zou W, Lenschow DJ, Jacobs BS, Borden EC, Li J, Virgin HW, and Zhang D-E (2005). Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem. Biophys. Res. Commun. 336, 496–506. [DOI] [PubMed] [Google Scholar]
- Gibbs DL (2020). Robust classification of Immune Subtypes in Cancer. [Google Scholar]
- Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang W-W, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel JP, Chang TC, Boretto J, Azuma T, and Chavrier P (1998). Differential properties of D4/LyGDI versus RhoGDI: phosphorylation and rho GTPase selectivity. FEBS Lett. 422, 269–273. [DOI] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, and Goodall GJ (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601. [DOI] [PubMed] [Google Scholar]
- Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander ES, Gabriel S, et al. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happel N, Stoldt S, Schmidt B, and Doenecke D (2009). M phase-specific phosphorylation of histone H1.5 at threonine 10 by GSK-3. J. Mol. Biol. 386, 339–350. [DOI] [PubMed] [Google Scholar]
- Haslam A, and Prasad V (2019). Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2, e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. (2013). Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Morgensztern D, and Boshoff C (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- Herreño AM, Ramírez AC, Chaparro VP, Fernandez MJ, Cañas A, Morantes CF, Moreno OM, Brugés RE, Mejía JA, Bustos FJ, et al. (2019). Role of RUNX2 transcription factor in epithelial mesenchymal transition in non-small cell lung cancer lung cancer: Epigenetic control of the RUNX2 P1 promoter. Tumour Biol. 41, 1010428319851014. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, and Chen J (2014). Post-translational modifications and the Warburg effect. Oncogene 33, 4279–4285. [DOI] [PubMed] [Google Scholar]
- Hong C-F, Chen W-Y, and Wu C-W (2017). Upregulation of Wnt signaling under hypoxia promotes lung cancer progression. Oncol. Rep. 38, 1706–1714. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, and Sullivan M (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu E-C, Rice MA, Bermudez A, Marques FJG, Aslan M, Liu S, Ghoochani A, Zhang CA, Chen Y-S, Zlitni A, et al. (2020). Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP1. Proc. Natl. Acad. Sci. U. S. A. 117, 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-Y, Lin Y-C-D, Li J, Huang K-Y, Shrestha S, Hong H-C, Tang Y, Chen Y-G, Jin C-N, Yu Y, et al. (2020). miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 48, D148–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubaux R, Thu KL, Vucic EA, Pikor LA, Kung SHY, Martinez VD, Mosslemi M, Becker-Santos DD, Gazdar AF, Lam S, et al. (2015). Microtubule affinity-regulating kinase 2 is associated with DNA damage response and cisplatin resistance in non-small cell lung cancer. Int. J. Cancer 137, 2072–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YP, Redig A, Hornick JL, and Sholl LM (2020). ARID1A mutations and expression loss in non-small cell lung carcinomas: clinicopathologic and molecular analysis. Mod. Pathol. 33, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Ishitani T, Hirao T, Suzuki M, Isoda M, Ishitani S, Harigaya K, Kitagawa M, Matsumoto K, and Itoh M (2010). Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat. Cell Biol. 12, 278–285. [DOI] [PubMed] [Google Scholar]
- Jimenez-Blasco D, Santofimia-Castaño P, Gonzalez A, Almeida A, and Bolaños JP (2015). Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 22, 1877–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Kim H, Oh M, Ki H, and Kim K (2009). Regulation of Notch1/NICD and Hes1 expressions by GSK-3alpha/beta. Mol. Cells 27, 15–19. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, and Nielsen M (2017). NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Lawrence MS, Polak P, Leshchiner I, Lage K, Golub TR, Lander ES, and Getz G (2015). Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc. Natl. Acad. Sci. U. S. A. 112, E5486–E5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachaliou N, Fernandez-Bruno M, and Rosell R (2018). Strategies for first-line immunotherapy in squamous cell lung cancer: are combinations a game changer? Transl Lung Cancer Res 7, S198–S201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RJ, Thomas A, Rajan A, Chun G, Lopez-Chavez A, Szabo E, Spencer S, Carter CA, Guha U, Khozin S, et al. (2013). A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 24, 2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keysar SB, Le PN, Miller B, Jackson BC, Eagles JR, Nieto C, Kim J, Tang B, Glogowska MJ, Morton JJ, et al. (2017). Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J. Natl. Cancer Inst. 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Hassanein M, Amann JM, Liu Q, Slebos RJC, Rahman SMJ, Kaufman JM, Zhang X, Hoeksema MD, Harris BK, et al. (2012). In-depth proteomic analysis of nonsmall cell lung cancer to discover molecular targets and candidate biomarkers. Mol. Cell. Proteomics 11, 916–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, and Park H (2007). Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics 23, 1495–1502. [DOI] [PubMed] [Google Scholar]
- Kim S, and Pevzner PA (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, Rosenberg JE, Van Allen EM, D’Andrea A, and Getz G (2016). Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 48, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepsch V, Hermann-Kleiter N, Do-Dinh P, Jakic B, Offermann A, Efremova M, Sopper S, Rieder D, Krogsdam A, Gamerith G, et al. (2018). Nuclear receptor NR2F6 inhibition potentiates responses to PD-L1/PD-1 cancer immune checkpoint blockade. Nat. Commun. 9, 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Klot C-A, Dubrowinskaja N, Peters I, Hennenlotter J, Merseburger AS, Stenzl A, Kuczyk MA, and Serth J (2017). Rho GDP dissociation inhibitor-β in renal cell carcinoma. Oncol. Lett. 14, 8190–8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Nambiar R, Rosario SR, Smiraglia DJ, Goodrich DW, and Witkiewicz AK (2020). Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun Biol 3, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr, and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, Thiru P, Bierie B, Ye X, Burge CB, et al. (2019). Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 116, 7353–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2018). A curated resource for phosphosite-specific signature analysis. Mol. Cell. Proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann N, Wroblowski S, Knyphausen P, de Boor S, Brenig J, Zienert AY, Meyer-Teschendorf K, Praefcke GJK, Nolte H, Krüger M, et al. (2016a). Structural and Mechanistic Insights into the Regulation of the Fundamental Rho Regulator RhoGDIα by Lysine Acetylation. J. Biol. Chem. 291, 5484–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann N, Wroblowski S, Scislowski L, and Lammers M (2016b). RhoGDIα Acetylation at K127 and K141 Affects Binding toward Nonprenylated RhoA. Biochemistry 55, 304–312. [DOI] [PubMed] [Google Scholar]
- Lacher SE, and Slattery M (2016). Gene regulatory effects of disease-associated variation in the NRF2 network. Curr Opin Toxicol 1, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, et al. (2006). The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Lamprecht S, Kaller M, Schmidt EM, Blaj C, Schiergens TS, Engel J, Jung A, Hermeking H, Grünewald TGP, Kirchner T, et al. (2018). PBX3 Is Part of an EMT Regulatory Network and Indicates Poor Outcome in Colorectal Cancer. Clin. Cancer Res. 24, 1974–1986. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê S, Josse J, and Husson F (2008). FactoMineR: AnRPackage for Multivariate Analysis. Journal of Statistical Software 25. [Google Scholar]
- Lebanony D, Benjamin H, Gilad S, Ezagouri M, Dov A, Ashkenazi K, Gefen N, Izraeli S, Rechavi G, Pass H, et al. (2009). Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J. Clin. Oncol. 27, 2030–2037. [DOI] [PubMed] [Google Scholar]
- Lebarbier E (2005). Detecting multiple change-points in the mean of Gaussian process by model selection. Signal Processing 85, 717–736. [Google Scholar]
- Le Large TY, Mantini G, Meijer LL, Pham TV, Funel N, van Grieken NC, Kok B, Knol J, van Laarhoven HW, Piersma SR, et al. (2020). Microdissected pancreatic cancer proteomes reveal tumor heterogeneity and therapeutic targets. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski KT, and Piwnica-Worms H (2014). Phosphorylation of the E3 ubiquitin ligase RNF41 by the kinase Par-1b is required for epithelial cell polarity. J. Cell Sci. 127, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Aljahdali I, and Ling X (2019a). Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J. Exp. Clin. Cancer Res. 38, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Vaudel M, Zhang B, Ren Y, and Wen B (2019b). PDV: an integrative proteomics data viewer. Bioinformatics 35, 1249–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Xie X, Zhou J, Sheng M, Yin X, Ko E-A, Zhou T, and Gu W (2017). Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 33, 2131–2139. [DOI] [PubMed] [Google Scholar]
- Li P, Cao G, Zhang Y, Shi J, Cai K, Zhen L, He X, Zhou Y, Li Y, Zhu Y, et al. (2020). FHL3 promotes pancreatic cancer invasion and metastasis through preventing the ubiquitination degradation of EMT associated transcription factors. Aging 12, 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Liu M, Wang P, Ding X, and Cao Y (2013). Analysis of 20 genes at chromosome band 12q13: RACGAP1 and MCRS1 overexpression in nonsmall-cell lung cancer. Genes Chromosomes Cancer 52, 305–315. [DOI] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SH, Sun J-M, Choi Y-L, Kim HR, Ahn S, Lee JY, Lee S-H, Ahn JS, Park K, Kim JH, et al. (2016). Efficacy and safety of dovitinib in pretreated patients with advanced squamous non-small cell lung cancer with FGFR1 amplification: A single-arm, phase 2 study. Cancer 122, 3024–3031. [DOI] [PubMed] [Google Scholar]
- Lim YW, Chen-Harris H, Mayba O, Lianoglou S, Wuster A, Bhangale T, Khan Z, Mariathasan S, Daemen A, Reeder J, et al. (2018). Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc. Natl. Acad. Sci. U. S. A. 115, E11701–E11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay J, McDade SS, Pickard A, McCloskey KD, and McCance DJ (2011). Role of DeltaNp63gamma in epithelial to mesenchymal transition. J. Biol. Chem. 286, 3915–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Payne SH, Ma S, and Fenyö D (2019). Extracting Pathway-level Signatures from Proteogenomic Data in Breast Cancer Using Independent Component Analysis. Mol. Cell. Proteomics 18, S169–S182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang L, Cui W, Yuan X, Lin L, Cao Q, Wang N, Li Y, Guo W, Zhang X, et al. (2016). Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells. Oncotarget 7, 58516–58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Iacono M, Monica V, Saviozzi S, Ceppi P, Bracco E, Papotti M, and Scagliotti GV (2011). p63 and p73 isoform expression in non-small cell lung cancer and corresponding morphological normal lung tissue. J. Thorac. Oncol. 6, 473–481. [DOI] [PubMed] [Google Scholar]
- Maksimovic J, Phipson B, and Oshlack A (2016). A cross-package Bioconductor workflow for analysing methylation array data. F1000Res. 5, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, et al. (2010). Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani DR, Maynard M, Kothadia R, Krug K, Christianson KE, Heiman D, Clauser KR, Birger C, Getz G, and Carr SA (2020). PANOPLY: A cloud-based platform for automated and reproducible proteogenomic data analysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Astels S (2017). hdbscan: Hierarchical density based clustering. Journal of Open Source Software 2, 205. [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvar B, Raghuram V, Pisitkun T, Sarkar A, and Knepper MA (2016). Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation. Physiol. Genomics 48, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton G, Fletcher P, Popat S, Savage J, Summers Y, Greystoke A, Gilligan D, Cave J, O’Rourke N, Brewster A, et al. (2020). The National Lung Matrix Trial of personalized therapy in lung cancer. Nature 583, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, and Schwartz MA (2009). Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Res. 69, 2838–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JA, Pike LA, Sams SB, Sharma V, Zhou Q, Severson JJ, Tan A-C, Wood WM, and Haugen BR (2014). Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol. Cancer 13, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchemore KA, Sampaio NG, Murrey MW, Stanley ER, Lannutti BJ, and Pixley FJ (2013). Specific inhibition of PI3K p110δ inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J. 280, 5228–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natan E, and Joerger AC (2012). Structure and kinetic stability of the p63 tetramerization domain. J. Mol. Biol. 415, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicorici D, Satalan M, Edgren H, Kangaspeska S, Murumagi A, Kallioniemi O, Virtanen S, and Kilkku O FusionCatcher - a tool for finding somatic fusion genes in paired-end RNA-sequencing data. [Google Scholar]
- Nijman SMB, Luna-Vargas MPA, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK, and Bernards R (2005). A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786. [DOI] [PubMed] [Google Scholar]
- Niu D-F, Kondo T, Nakazawa T, Oishi N, Kawasaki T, Mochizuki K, Yamane T, and Katoh R (2012). Transcription factor Runx2 is a regulator of epithelial-mesenchymal transition and invasion in thyroid carcinomas. Lab. Invest. 92, 1181–1190. [DOI] [PubMed] [Google Scholar]
- Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, and Morgan M (2014). VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik PK, Pillai RN, Lathan CS, Velasco SA, and Papadimitrakopoulou V (2019). New Treatment Options in Advanced Squamous Cell Lung Cancer. Am Soc Clin Oncol Educ Book 39, e198–e206. [DOI] [PubMed] [Google Scholar]
- Parra ER, Jiang M, Solis L, Mino B, Laberiano C, Hernandez S, Gite S, Verma A, Tetzlaff M, Haymaker C, et al. (2020). Procedural Requirements and Recommendations for Multiplex Immunofluorescence Tyramide Signal Amplification Assays to Support Translational Oncology Studies. Cancers 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Lu L, Zander DS, Sreerama L, Coco D, and Moreb JS (2008). ALDH1A1 and ALDH3A1 expression in lung cancers: correlation with histologic type and potential precursors. Lung Cancer 59, 340–349. [DOI] [PubMed] [Google Scholar]
- Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, Papadopoulos KP, Beeram M, Rasco DW, Hilton JF, et al. (2016). Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 6, 740–753. [DOI] [PubMed] [Google Scholar]
- Payen VL, Mina E, Van Hée VF, Porporato PE, and Sonveaux P (2020). Monocarboxylate transporters in cancer. Mol Metab 33, 48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, Hermes B, Çay Şenler F, Csőszi T, Fülöp A, et al. (2018). Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 379, 2040–2051. [DOI] [PubMed] [Google Scholar]
- Pedersen BS, Layer RM, and Quinlan AR (2016). Vcfanno: fast, flexible annotation of genetic variants. Genome Biol. 17, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng A, Mao X, Zhong J, Fan S, and Hu Y (2020). Single-Cell Multi-Omics and Its Prospective Application in Cancer Biology. Proteomics 20, e1900271. [DOI] [PubMed] [Google Scholar]
- Peters LA, Perrigoue J, Mortha A, Iuga A, Song W-M, Neiman EM, Llewellyn SR, Di Narzo A, Kidd BA, Telesco SE, et al. (2017). A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat. Genet. 49, 1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia F, Wang L, Peng J, Yan A, Zhu J, and Wang P (2018). A new method for constructing tumor specific gene co-expression networks based on samples with tumor purity heterogeneity. Bioinformatics 34, i528–i536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, and Massion PP (2008). Role of chromosome 3q amplification in lung cancer. J. Thorac. Oncol. 3, 212–215. [DOI] [PubMed] [Google Scholar]
- Ramadoss S, Sen S, Ramachandran I, Roy S, Chaudhuri G, and Farias-Eisner R (2017). Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 36, 6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese SE, Archer KJ, Therneau TM, Atkinson EJ, Vachon CM, de Andrade M, Kocher J-PA, and Eckel-Passow JE (2013). A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis. Bioinformatics 29, 2877–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Omelchenko T, Calinawan A, Nair S, Schadt E, and Tewari A (2020). Prioritization prostate cancer to immune checkpoint therapy by ranking tumors along IFN-γ axis and identification of immune resistance mechanisms. [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu Y-M, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. (2017). Integrative clinical genomics of metastatic cancer. Nature 548, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R Jr (2018). The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 129, 65–83. [DOI] [PubMed] [Google Scholar]
- Satpathy S, Jaehnig EJ, Krug K, Kim B-J, Saltzman AB, Chan DW, Holloway KR, Anurag M, Huang C, Singh P, et al. (2020). Microscaled proteogenomic methods for precision oncology. Nat. Commun. 11, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, Garnett MJ, Blüthgen N, and Saez-Rodriguez J (2018). Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadforth IP, Dunkley TPJ, Lilley KS, and Bessant C (2005). i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahamatdar S, He MX, Reyna MA, Gusev A, AlDubayan SH, Van Allen EM, and Ramachandran S (2020). Germline Features Associated with Immune Infiltration in Solid Tumors. Cell Rep. 30, 2900–2908.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, Nagel ZD, Zou J, Wang C, Kapoor P, et al. (2018). ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 24, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showe MK, Vachani A, Kossenkov AV, Yousef M, Nichols C, Nikonova EV, Chang C, Kucharczuk J, Tran B, Wakeam E, et al. (2009). Gene expression profiles in peripheral blood mononuclear cells can distinguish patients with non-small cell lung cancer from patients with nonmalignant lung disease. Cancer Res. 69, 9202–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2020). Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30. [DOI] [PubMed] [Google Scholar]
- Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, Blackford A, Goodman SN, Bunz F, Watson WH, et al. (2008). RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 68, 7975–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinicropi-Yao SL, Amann JM, Lopez DLY, Cerciello F, Coombes KR, and Carbone DP (2019). Co-Expression Analysis Reveals Mechanisms Underlying the Varied Roles of NOTCH1 in NSCLC. J. Thorac. Oncol. 14, 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Ji J, Gleason KJ, Yang F, Martignetti JA, Chen LS, and Wang P (2019). Insights into Impact of DNA Copy Number Alteration and Methylation on the Proteogenomic Landscape of Human Ovarian Cancer via a Multi-omics Integrative Analysis. Mol. Cell. Proteomics 18, S52–S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, and Nolan GP (2016). Mass Cytometry: Single Cells, Many Features. Cell 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava K, Pickard A, Craig SG, Quinn GP, Lambe SM, James JA, McDade SS, and McCance DJ (2018). ΔNp63γ/SRC/Slug Signaling Axis Promotes Epithelial-to-Mesenchymal Transition in Squamous Cancers. Clin. Cancer Res. 24, 3917–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PA, Welsh EA, Slebos RJC, Fang B, Izumi V, Chambers M, Zhang G, Cen L, Pettersson F, Zhang Y, et al. (2019). Proteogenomic landscape of squamous cell lung cancer. Nat. Commun. 10, 3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang Y-H, Basnet H, Zou Y, Shu W, Soni RK, et al. (2020). TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, and Kohlbacher O (2014). OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. (2013). Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecalco-Cruz AC, and Cruz-Ramos E (2018). Protein ISGylation and free ISG15 levels are increased by interferon gamma in breast cancer cells. Biochem. Biophys. Res. Commun. 499, 973–978. [DOI] [PubMed] [Google Scholar]
- Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA, Tessema M, Leng S, and Belinsky SA (2011). EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 71, 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Cai Y, Li Y, Lu Z, Huang J, Deng Y, Yang N, Wang X, Ying P, Zhang S, et al. (2021). CancerImmunityQTL: a database to systematically evaluate the impact of genetic variants on immune infiltration in human cancer. Nucleic Acids Res. 49, D1065–D1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timperi E, Focaccetti C, Gallerano D, Panetta M, Spada S, Gallo E, Visca P, Venuta F, Diso D, Prelaj A, et al. (2017). IL-18 receptor marks functional CD8+ T cells in non-small cell lung cancer. Oncoimmunology 6, e1328337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MN, Choi W, Wszolek MF, Navai N, Lee I-LC, Nitti G, Wen S, Flores ER, Siefker-Radtke A, Czerniak B, et al. (2013). The p63 protein isoform ΔNp63α inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J. Biol. Chem. 288, 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Mani DC, Satpathy S, Fereshetian S, Gasser JA, Svinkina T, Olive ME, Ebert BL, Mertins P, and Carr SA (2020). Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 11, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Björling E, Agaton C, Szigyarto CA-K, Amini B, Andersen E, Andersson A-C, Angelidou P, Asplund A, Asplund C, et al. (2005). A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 4, 1920–1932. [DOI] [PubMed] [Google Scholar]
- Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasaikar SV, Straub P, Wang J, and Zhang B (2018). LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vats P, Chinnaiyan AM, and Kumar-Sinha C (2020). Case Study: Systematic Detection and Prioritization of Gene Fusions in Cancer by RNA-Seq: A DIY Toolkit. Methods Mol. Biol. 2079, 69–79. [DOI] [PubMed] [Google Scholar]
- Vosgha H, Ariana A, Smith RA, and Lam AK-Y (2018). miR-205 targets angiogenesis and EMT concurrently in anaplastic thyroid carcinoma. Endocr. Relat. Cancer 25, 323–337. [DOI] [PubMed] [Google Scholar]
- Walser T, Cui X, Yanagawa J, Lee JM, Heinrich E, Lee G, Sharma S, and Dubinett SM (2008). Smoking and lung cancer: the role of inflammation. Proc. Am. Thorac. Soc. 5, 811–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I-M, Zhang B, Yang X, Zhu J, Stepaniants S, Zhang C, Meng Q, Peters M, He Y, Ni C, et al. (2012). Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 8, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, and Zhang B (2017). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-B, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Jiang B, Zhang T, Liu L, Wang Y, Wang Y, Chen X, Lin H, Zhou L, Xia Y, et al. (2015). Insulin and mTOR Pathway Regulate HDAC3-Mediated Deacetylation and Activation of PGK1. PLoS Biol. 13, e1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeden CE, Solomon B, and Asselin-Labat M-L (2015). FGFR1 inhibition in lung squamous cell carcinoma: questions and controversies. Cell Death Discov 1, 15049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wang X, and Zhang B (2019). PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res. 29, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Li K, Zhang Y, and Zhang B (2020). Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat. Commun. 11, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra H-J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley SP, and Altieri DC (2019). Survivin at a glance. J. Cell Sci. 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. (2002). Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR, Muldrew K, Miller CR, Randell SH, Socinski MA, et al. (2010). Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin. Cancer Res. 16, 4864–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JJY, Pung YF, Sze NS-K, and Chin K-C (2006). HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. U. S. A. 103, 10735–10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhang Y, Zheng X, Dai F, Lu Y, Dai L, Niu M, Guo H, Li W, Xue X, et al. (2020). Circular RNA circCORO1C promotes laryngeal squamous cell carcinoma progression by modulating the let-7c-5p/PBX3 axis. Mol. Cancer 19, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y-M, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al. (2018). Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 173, 1770–1782.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Lei Q-Y, Zhao S, and Guan K-L (2011). Regulation of glycolysis and gluconeogenesis by acetylation of PKM and PEPCK. Cold Spring Harb. Symp. Quant. Biol. 76, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, and Lu Z (2013). Nuclear PKM2 regulates the Warburg effect. Cell Cycle 12, 3154–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yomtoubian S, Lee SB, Verma A, Izzo F, Markowitz G, Choi H, Cerchietti L, Vahdat L, Brown KA, Andreopoulou E, et al. (2020). Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep. 30, 755–770.e6. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Flores NM, Hausmann S, Lofgren SM, Kharchenko V, Angulo-Ibanez M, Sengupta D, Lu X, Czaban I, Azhibek D, et al. (2021). Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D, Wu X, Cao Y, Liang W, Liu Y, et al. (2013). Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 5, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zou X, Qian W, Weng X, Zhang L, Zhang L, Wang S, Cao X, Ma L, Wei G, et al. (2019a). Enhanced PAPSS2/VCAN sulfation axis is essential for Snail-mediated breast cancer cell migration and metastasis. Cell Death Differ. 26, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Thery F, Wu NC, Luhmann EK, Dussurget O, Foecke M, Bredow C, Jiménez-Fernández D, Leandro K, Beling A, et al. (2019b). The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat. Commun. 10, 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Li F-L, Cheng Z-L, and Lei Q-Y (2014). Impact of acetylation on tumor metabolism. Mol Cell Oncol 1, e963452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, and Conner SD (2018). Glycogen synthase kinase 3β inhibition enhances Notch1 recycling. Mol. Biol. Cell 29, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, Li Y, Zhang Z, Rusch MC, Parker M, et al. (2016). Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 48, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Experimental Design and Proteogenomic Data Quality Metrics, Related to Figure 1
A. Experimental workflow of LSCC tumor and NAT proteogenomic profiling. Samples were cryopulverized, aliquoted for genomic DNA, total RNA and protein extraction, and utilized for DNA sequencing (WGS and WES) and methylation analysis, RNA and miRNA sequencing and proteomics / PTM analysis as indicated. TMT11-based isobaric labeling was used to multiplex up to 5 tumors (blue filled circles) and paired NATs (pink filled circles) in a single TMT11-plex. The last TMT channel (131C) was the LSCC common reference (CR), comprising peptides from all LSCC tumor and NAT samples included in this study and replicated in every plex to serve as the common denominator for calculating peptide ratios. A total of 300ug peptides/sample was used for proteome, phosphoproteome and acetylproteome analyses via serial enrichment (STAR methods). For the ubiquitylproteome, a different TMT11 experimental design was used (Table S1) for the subset of tumors and NATs with ≥750ug residual peptide amounts after mainstream proteomic processing (85 tumors and 58 NATs). 750ug/sample was used for ubiquitylproteome analysis, with a ubiquitylproteome CR composed of peptides from the 143 available samples. The CR used for proteome, phosphoproteome and acetylproteome and that for ubiquitylproteome showed median Pearson correlations > 0.85 across analyses.
B. Numbers of quantified proteins, phosphosites, acetylsites, and ubiquitylsites across all TMT11-plexes. The values above each bar indicate percentage deviation from the median.
C. Pearson correlations of log TMT reporter ion intensities between all pairs of TMT plexes were consistently higher for CR than for sample channels, enabling precise relative quantification of proteins, phosphorylation and acetylation sites across 22 TMT plexes using the CR channels as the common denominator. The CRs of three TMT plexes in the ubiquitylproteome data showed lower reproducibility compared to the CRs of other ubiquitylproteome TMT plexes, which was computationally corrected as described in STAR methods (“Quality control and Batch effect assessment”). Top panel: Box plots show distributions of delta CR-sample correlations derived from all pairwise TMT plex comparisons across the different proteomic data types. Positive delta correlations indicate higher correlations of the CR channel compared to sample channels. Lower panel: Bar plots represent pairwise TMT plex correlations for proteomics, phosphoproteomics, acetylproteomics and ubiquitylproteomics using TMT plexes 1 and 2 as representative examples. Blue bars depict correlations of sample channels; red bars show the correlation of the CR.
D. Smoking scores calculated by WES stratified by self-reported smoking status. While some reformed and never-smokers had low scores, many self-reported never-smokers from Slavic countries had scores comparable to those of smokers.
E. PCA plots showing the lack of batch effect across all plexes in the different data types, with samples from each batch represented by different shapes. A clear separation in the PC1 component is observed between tumors and NATs in each data type.
F. To identify specific events associated with CNA trans-effects (vertical stripes in Figure 1D), we leveraged CMAP data (https://clue.io/cmap), which includes over 1.3 million gene expression profiles from ~20,000 small-molecule compounds and ~4,300 genetic reagents tested in multiple cell types. Patterns of significant trans-effects associated with CNAs that also had significant cis-effects were queried against CMAP profiles to identify knockout perturbations that significantly resembled those CNA-associated aberrations. Shown for two of the six genes (NRF2F6 and IL18) are correlations between GISTIC log2 CNA ratio or xCell-derived immune score and relative protein expression (Log2 TMT ratio). Pearson correlations and p-values are indicated.
G. Unsupervised consensus clustering results of promoter-level DNA methylation data from tumors and NATs. LSCC tumor DNA showed overall hypermethylation at gene promoters relative to NATs (Wilcoxon p<2×10−16).
H. Unsupervised consensus clustering of tumor methylation revealed CIMP (CpG island methylator phenotype) -high, -intermediate and -low clusters.
Figure S2: Characteristics of Multi-omic NMF Subtypes, Related to Figure 2
A. Upper panel: Line plot representing cophenetic correlation coefficients (y-axis) calculated for a range of factorization ranks (x-axis). The maximal cophenetic correlation coefficient was observed for rank K=5 as highlighted by the red box. Lower panel: silhouette plot for K=5. This plot indicates the quality of cluster separation.
B. Directionality and significance (signed Wilcoxon p-value) of abundance changes in RNA, protein (green), and PTMs (phosphosites in purple, acetylsites in blue, ubiquitylsites in yellow) between the Basal-Inclusive and other NMF subtypes. Identities of the most extreme outliers are designated and the modification site (K- lysine, S- Serine, T- Threonine, Y- Tyrosine) is separated by an underscore. PBX3 is highlighted by a red box.
C. Same as B but in the EMT-enriched subtype.
D. Pathway analysis of proteins with high correlation with PBX3 protein expression (FDR <0.05 and Pearson correlation >0.5) revealed neutrophil activation and neutrophil degranulation as the top protein-driven pathways.
E. Scatter plot showing Pearson correlations between PBX3 protein and other transcripts and PBX3 protein and other proteins. Points represent the signed −log10 (p-value). Neutrophil degranulation-associated genes show very high correlation with PBX3 protein only at the level of protein expression, whereas neutrophil chemotactic factors such as CXCL1 and 8 show high correlation at both RNA and protein levels.
F. Comparison of xCell-derived Immune, Microenvironment, Neutrophil and Stroma scores across five NMF subtypes. Z-score values are used for the box plots. P-values are derived from the Wilcoxon test. B.I - Basal-inclusive, C - Classical, E-E - EMT-enriched, I-S - Inflamed-Secretory, P-P - Proliferative-Primitive.
G. Representative dual immunohistochemical staining of alpha-smooth muscle actin (α-SMA) and pan-Cytokeratin (CK) of two samples corresponding to EMT-E and Classical subtypes.
H. Normalized connectivity scores (NCS) between the Library of Integrated Network-based Cellular Signatures (LINCS) compound signatures and the five NMF multi-omic subtype signatures at the RNA and protein level are represented in the heatmap. Each NMF signature was used to query the CMAP LINCS L1000 resource (2020 release), a collection of more than 1M response signatures of cancer cell lines to different types of chemical (e.g. drugs, tool compounds) and genetic perturbations (e.g shRNAs, sgRNAs, cDNAs). The 20 most negatively connected compounds to each NMF signature (with the most negative NCS) were then selected for visualization. Rows were ordered based on the EMT-E RNA signature indicating an enrichment for TGF-β inhibitors in the negatively-connected compounds of the EMT-E cluster.
I. Integrative analysis of the cumulative 216 treatment-naïve LSCC tumors between this and the Stewart study (Stewart et al., 2019), focused on the global proteome in the absence of global gene copy number and post-translational modification data in that study. Our NMF-based pipeline applied to the integrated proteomics dataset (STAR Methods) identified six clusters that partially aligned with RNA-based clusters as well as with proteomic and multi-omic subtypes defined in this study (Figure S2A). As expected, the Stewart et al., Redox B proteomics cluster samples joined with the NMF Classical subtype, enriched for NRF2 pathway mutations and redox biology. Notably, their Inflamed A and B and our Inflamed-Secretory tumors were each enriched in a separate cluster, their Inflamed A joining our Basal-Inclusive and their Inflamed B defining a cluster that was enriched for samples specifically from their cohort and for TP53 WT status, and that shared enrichment for secretory RNA status with our separately clustered Inflamed-Secretory tumors. B.I - Basal-inclusive, C - Classical, E-E – EMT-enriched, I-S - Inflamed-Secretory, P-P - Proliferative-Primitive.
J. Significantly enriched pathways (MSigDB Hallmark) in one or more of the six clusters shown in Figure S2I. Yellow-to-blue color scale depicts positive to negative normalized enrichment scores (NES), with asterisks denoting significant pathways (FDR<0.05) among the subtypes. At the pathway level, their Inflamed B had signals of coagulation and complement activity, but lacked the broader evidence of immune signaling associated with the Inflamed A and Inflamed-Secretory-enriched clusters (Figure S2I). The Proliferative-Primitive subset we defined retained its integrity and association with Primitive RNA status in the combined analysis. Finally, the EMT-E cluster defined in our study was clearly enriched with the mixed subtype samples from the Stewart et al., study. In addition to very strong EMT signaling, that cluster had evidence of hypoxemia and strong downregulation of oxidative phosphorylation (Figure S2I), known drivers of EMT (Cannito et al., 2008) and of Wnt signaling (Hong et al., 2017).
K. A schematic representation of the proposed crosstalk between non-canonical WNT5A/ROR2 and PDGFRB signaling axes. The binding of WNT5A to ROR2 activates the planar cell polarity pathway, shown in yellow ovals (DVL/RAC/RHO/JNK), and the Ca2+ pathway, shown in blue ovals (PKCa) involving cytoskeleton rearrangements and cell migration. Upregulation of RAC requires clathrin-mediated ROR2 receptor internalization of the Wnt signalosome (AP2, DVL, ROR2). WNT5A/ROR2 also activates PI3K/AKT signaling (green ovals) leading to RUNX2-mediated transcription of EMT (ZEB1, MMP2) and cell invasion (WNT5A, FLNa, ITG5A) programs (red ovals). Targeted therapeutics against WNT5A/ROR2 upregulation are shown in red, with some of them in clinical trials as indicated. Red arrows represent upregulation in the EMT-E proteome and phosphoproteome. Genes directly observed as upregulated at protein/phosphoprotein levels are in thick oval dark blue outlines.
Figure S3: Proteogenomic Consequences of CDKN2A and NRF2 Pathway Aberrations, Related to Figure 3
A. Tumor Mutational Burden per megabase (MB) as estimated from whole exome sequencing among samples with and without ARID1A gene somatic mutation.
B. Schematic diagram showing amino acid alterations resulting from CDKN2A/p16INK4a nonsense and frameshift mutations in the CPTAC LSCC and head and neck squamous cell cancer (HNSCC) datasets. Not shown are seven mutant tumors with missense or in-frame indels that would not be predicted to trigger nonsense mediated decay (NMD). Unlike the CPTAC HNSCC cohort, where p16INK4a (p16) nonsense and frameshift mutations always resulted in NMD (tumors with low p16 RNA expression are indicated by blue outline), five of the nine nonsense/frameshift mutations in LSCC appeared to have escaped NMD. These mutations either resulted in truncation at the very 5’ end (E10*) or were frameshift mutations that did not actually result in a significantly truncated predicted protein product (all predicted to generate protein products of 142 or more amino acids compared to the full length p16 protein of 160 amino acids), while all of the mutations triggering NMD resulted in production of a protein product that was 40-120 amino acids shorter than the wild-type protein.
C. Box plots showing Rb protein and phosphoprotein (mean of all Rb phosphosites) (Log2 TMT ratio) expression in samples with and without CCND1 amplification. Amplified samples are those with GISTIC threshold = 2, while all other samples were considered not amplified. P-values are from Welch’s two-sample T-tests.
D. Phospho-Rb levels correlate with response to CDK4/6 inhibitors in various LSCC cell lines. Scatter plots show correlation between phospho-Rb S807/811 by RPPA (CCLE 2019, Proteomics (RPPA) from DepMap) and abemaciclib response (PRISM Repurposing Secondary Screen 19Q4 from DepMap) (Left panel) or palbociclib response (Sanger GDSC1 from DepMap) for LSCC cell lines (right panel). CCND1, CDK2NA, and RB1 mutation status (DepMapPublic 21Q1, Mutation) depicted as filled circles. Outline depicts presence of CCND1 amplification (GISTIC threshold ≥ 1, derived from segmented copy number, DepMapPublic 21Q1) and/or CDKN2A deletion (GISTIC threshold ≤ −1). Cell lines with higher levels of phospho-Rb S807/811 were more sensitive to CDK4/6 inhibitors abemaciclib and palbociclib. R, Pearson correlation coefficient. DepMap (Dependency map) is a compendium of genomics, proteomics, shRNA and CRISPR based gene dependency datasets of hundreds of cell lines. PRISM (Profiling Relative Inhibition Simultaneously in Mixtures) and GDSC (Genomics of Drug Sensitivity) are databases containing sensitivity data to hundreds of compounds in hundreds of cell lines.
E. Box plots showing NFE2L2 S215 and S433 abundance in samples with CUL3, KEAP1 and NFE2L2 mutations. **** <=0.0001, *** < 0.01, **< 0.01, *< 0.05, ns = not significant. P-values are from the Wilcoxon test.
F. Correlation between differential mRNA expression (log2 FC) and protein abundance (log2 FC) levels among tumors with NRF2 pathway mutation (one and two hits) vs NRF2 pathway WT tumors. Significant events (>2 fold and p-value<0.05) are represented as circles with black borders. NRF2 targets in human cancer tissues (Lacher and Slattery, 2016) are represented by solid circles in green, while solid circles in black indicate novel targets. Candidates that also show phosphosite modification are represented by pink-filled gene name boxes.
G. Overlap between NRF2-related genesets (upper left), and up- (upper right) and down-regulated (bottom) events in NRF2 pathway mutant samples across mRNA, proteomics and phosphoproteomics.
H. Relative mRNA, protein and phosphosite abundance of NRF2 pathway-related and other cancer-associated genes in tumor samples with two NRF2 pathway (NF2L2, KEAP1, CUL3) mutations, one mutation, or WT status, as well as in NATs. A dosage effect is observed. # no phosphosite levels changes were detected for NR0B1 between the groups.
Figure S4: Association Between TP63 and miR205 Expression in LSCC, Related to Figure 4
A. Focal amplification and deletion landscape of the study LSCC cohort. The top five significant focal peaks are labeled in the plot
B. Copy number segmentation file (DepMapPublic 21Q1) for LSCC cell lines from DepMap, processed with GISTIC2.0 (same parameters used to process LSCC cohort as described in STAR METHODS). Genes were considered amplified if GISTIC thresholded values = 1 or 2 and not amplified if GISTIC thresholded values = −2, −1, or 0. Each gene upregulated at the protein level with recurrent copy number gain in tumors vs NATs (from Figure 4A with Log2FC>0 & FDR < 0.05) was used as a query to compare dependency scores (Combined CRISPR KO screen, DepMapPublic 21Q1) by t-test of a given gene in amplified vs non-amplified LSCC cell lines. X-axis depicts the mean change in dependency scores for a queried gene in cell lines with amplification vs without amplification of that gene. Y-axis depicts the −log10(p-value) from t-test. Genes passing FDR < 0.25 are depicted by red and blue colored dots.
C. mRNA expression of all detected TP63 Ensembl transcript (ENST) isoforms for tumors. The transcript corresponding to ΔNp63α was the dominant isoform observed in this cohort.
D. Correlations between ΔNp63α transcript and gene-level TP63 mRNA expression (upper panel) or TP63 relative protein expression (log2 TMT ratio) and gene-level TP63 mRNA expression (lower panel).
E. Kaplan-Meier plot of overall survival in ΔNp63α transcript-low versus -high samples from the TCGA LSCC cohort. Samples with low ΔNp63α expression have significantly worse outcomes.
F. Np63 mRNA transcript isoform expression analysis (RNAseq (Transcript isoform), DepMapPublic 21Q1) shows LSCC cell lines with high and low levels of Np63.
G. Response to survivin (BIRC5) inhibitor YM-155 (PRISM Repurposing Secondary Screen 19Q4 from DepMap) in LSCC cell lines with high and low expression of Np63. Np63-low lines are more sensitive to YM-155 than Np63-high cell lines. P-value derived from t-test.
H. Rank plots showing enrichment scores for proliferation-related pathways from pre-ranked GSEA. The inputs are t-values of gene expression between TP63-low and TP63-high samples.
I. Volcano plot showing differential expression of miRNA (Log2 FC) between tumors and NATs. miR-205 shows the highest relative overexpression in tumors.
J. Differential promoter level methylation of miR-205 in tumors compared to NATs. miR-205 is hypomethylated in tumors.
K. Pearson correlations between miR-205 expression and all quantified proteins in tumors. TP63 is highlighted and shows the highest correlation with miR-205.
L. Gene-level RNA (left) and protein (right) expression correlations between TP63 and TP73.
M. Correlation between EZH2 dependency by shRNA (shRNA KD screen (Combined), DEMETER2 Data v6 from DepMap) and SOX2 Log2 copy number (Copy number (Gene level), DepMapPublic 21Q1) in LSCC cell lines. R, Pearson correlation coefficient.
Figure S6: Differential pathway, CBPE scores and impact of germline mutations on immune signaling, Related to Figure 6
A. Key pathways differentially expressed between Hot, Warm and Cold tumors based on their gene expression and global protein abundance. Wilcoxon p-values are reported.
B. Key pathways differentially expressed between Hot, Warm and Cold tumors based on the gene expression and global proteomic abundance of corresponding NATs. Wilcoxon p-values are reported.
C. Averages of CD163, CD4, and CD8 IHC scores derived from three independent pathologists on four cores/sample from 17 samples representing Hot (5), Warm (5) and Cold (7) clusters. P-values are based on ordinal regression analysis of immune cluster scores, using random effects to account for multiple cores/samples and independent pathologists.
D. Representative immunohistochemical staining pattern for CD163, CD8, and CD4 proteins on three samples drawn from Hot, Warm, and Cold immune clusters. Protein expression levels in different cell types are highlighted with colored arrowheads as indicated.
E. Overlap between differentially regulated mRNA and proteins between Hot vs Cold and Warm vs Cold tumors, and eGenes downstream of cis-eQTLs and cis-pQTLs. Recent studies have suggested the importance of germline variants in the immune profiles of solid tumors (Lim et al., 2018; Shahamatdar et al., 2020; Tian et al., 2021). Among 319 cis-pQTLs (cis-regulating the cognate protein) identified in this study, only 38 were also cis-eQTLs (cis-regulating cognate RNA)(Table S6). Interestingly, the set of all proteins with pQTLs (but not the set of all genes with eQTLs) was enriched in pathways involved in communication between innate and adaptive immune cells. The 15 pQTLs in proteins differentially abundant between Hot and Cold tumors included proteins related to immune response (TRIM22), antigen processing (TAPBPL and HLA-A) and IL-6 signaling pathway (LRPPRC); eight were previously reported (Lim et al., 2018). These results imply a potentially important role of germline variants in shaping immune processes in LSCC.
F. Protein abundance and/or phosphorylation abundance (Log2 TMT ratio) of key markers involved in Rho GTPase signaling stratified by immune clusters. Wilcoxon p-values are reported.
G. Immunohistochemical staining images of ARHGDIB (RhoGDIB) protein in two tumors. Its expression is predominantly observed in tumor microenvironments with different types of cells indicated with differently colored arrowheads.
Figure S7: Differential tumor and NAT analysis, Related to Figure 7
A. Log2 fold change of proteins increased in tumors relative to paired NATs, together with candidate representation in the Human Proteome Atlas (HPA) and various annotations. Shown are the top 50 of 502 differential proteins with coherent expression in all omics that were significantly upregulated in ≥90% of T/NAT pairs (log2(FC)>2, FDR<0.01). The blue box indicates the protein that was upregulated in 100% of T/NAT pairs.
B. Fold changes of CT antigens and their relationship to NMF subtype and immune clusters.
C. Tumor-specific proteins from the current study showing significant association with disease-free survival in the TCGA LSCC cohort.
D. Focal amplification (red) and deletion (blue) events in the copy number data from three CPTAC tumor cohorts.
E. To identify copy number drivers of the immune score, correlations between CNA, RNA, protein, and the immune score were assessed for all genes present in both CNA and RNA in a cohort. To be considered drivers, genes had to have positive pairwise correlations between CNA & RNA, CNA & immune score, and RNA & immune score. If the protein was quantified in the cohort, positive correlations were also required between CNA & protein and protein & immune score. Spearman correlations with a BH adjusted p< 0.01 were considered significant. The figure shows genes deleted in at least 25% of the cohorts that were significantly correlated with RNA and protein expression as well as ESTIMATE immune score. LSCC-specific genes are indicated in black, genes in both LSCC and HNSCC are in red, genes in both LSCC and LUAD are in blue, and genes shared by all three are in purple.
F. Significantly increased activating sites on kinases in the CPTAC LSCC, LUAD (Gillette et al., 2020) and the HNSCC (Huang et al., 2021) cohorts.
G. EGFR log2 tumor/NAT protein fold change in three CPTAC cancer cohorts (LSCC, LUAD, HNSCC).
Table S1: Metadata Associated with LSCC Patients and Tumors, Related to Figures 1–7, Figures S1–7
A. Metadata associated with this study
B. Cancer Associated Genes (CAGs)
C. Table containing results from association analyses between key metadata and technical parameters for NMF multi-omic subtyping. NMF core members were considered for this analysis.
D. Table containing results from association analyses between key metadata and technical parameters for NMF multi-omic subtyping. All samples; tumors and NATs were considered for this association.
E. Table containing results from association analyses between key metadata and technical parameters for immune clusters. All samples; tumors and NATs were considered for this association.
Table S2: Genomic and methylation datasets associated with this study, Related to Figures 1–7, Figures S1–7
A. Gene-level data matrix of somatic copy number alterations (CNA) obtained from GISTIC2 analysis. CNAs are represented as absolute gene copy numbers. Refseq GRCh38/hg38 database was used as a reference.
B. Same as Table S2A but CNAs are represented as log2(CN)-1.
C. Same as Table S2A but CNAs are represented as thresholded copy number level calls.
D. RNA expression data matrix reporting Log2-transformed upper-quartile (UQ)-normalized FPKM values.
E. Same as Table S2D but with FPKM values median-centered by gene.
F. miRNA expression data matrix reporting log2 TPM (Transcripts Per Kilobase Million) values.
G. Same as Table S2F but with TPM values median-centered by gene.
H. Gene level DNA methylation data matrix. Beta values were obtained by averaging the beta values of individual probes in the islands of promoter or 5’ UTR regions of the gene.
I. Somatic mutations associated with the CPTAC LSCC cohort in Mutation Annotation Format (MAF) format.
Table S3: Global proteomics, phosphoproteomics, acetylproteomics and ubiquitylproteomics data associated with this study, Related to Figures 1–7 and Figures S1–7
A. Two-component-normalized log2-transformed protein data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods.
B. Same as Table S3A but after application of the filtering strategy described in the STAR methods
C. Gene-centric global proteome data matrix derived from Table S3B. Protein-level data were aggregated to gene-level using the sample/CR ratio assigned to the dominant isoform per gene symbol (similar to the razor-peptide approach).
D. Two-component-normalized log2-transformed phosphoproteomic data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods.
E. Two-component-normalized log2-transformed phosphoproteomic data matrix (sample/CR ratios) after application of the filtering strategy described in the STAR methods.
F. Gene-centric phosphoproteomic data matrix derived from Table S3E. Phosphosite-level sample/CR ratios were aggregated to gene-level using mean ratio.
G. Two-component-normalized log2-transformed acetylproteomic data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods.
H. Two-component-normalized log2-transformed acetylproteomic data matrix (sample/CR ratios) after application of the filtering strategy described in the STAR methods.
I. Gene-centric acetylproteomics data matrix derived from Table S3H. Acetylsite-level sample/CR ratios were aggregated to gene-level using mean ratio.
J. Acetylproteomic data matrix based on Table S3G corrected for global protein expression (Table S3B) as described in the STAR methods.
K. Two-component-normalized log2-transformed, batch corrected ubiquitylproteomic data matrix (sample/CR ratios) before application of the filtering strategy described in the STAR methods. See STAR methods for details on batch correction.
L. Two-component-normalized log2-transformed, batch corrected ubiquitylproteomic data matrix (sample/CR ratios) after application of the filtering strategy described in the STAR methods. See STAR methods for details on batch correction.
M. Gene-centric ubiquitylproteomic data matrix derived from Table S3L. Ubiquitylsite-level sample/CR ratios were aggregated to gene-level using mean ratio.
N. Ubiquitylproteomic data matrix based on Table S3L corrected for global protein expression (Table S3B) as described in the STAR methods.
Table S4: CNA cis and trans effects, iProFun, CMAP, NRF2 differential expression, NRF2 signature, cis-eQTL from RNA and proteomics datasets, eGenes, Related to Figures 1–4
A. Copy number amplified cytobands
B. Copy number deleted cytobands C. CNA cis- and trans-effects
D. Significant focal amplicons that show significant cis-effect and are cancer associated genes
E. iProFun results with mRNA, protein, phosphoprotein, acetylprotein as outcomes and methylation as the predictor, adjusted for CNV(lr), CNV(baf) and tumor purity. Results include identified genes (Yes=1/No=0) with FDR<10% and identified phenotype association analyses (Yes=1/No=0).
F. CMAP tab- Connectivity Map (CMAP) results
G. Association between genes enriched in the CMAP analysis and sample metadata
H. Normalized connectivity scores (NCS) between Library of Integrated Network-based Cellular Signatures (LINCS) compound signatures and the five NMF multi-omic subtype signatures at the RNA and Protein level.
I. Summary of CDKN2A and RB1 annotations
J. T test p-values of differential proteomic features CCND1, CCND2, CCND3, and CDK6
K. Differential proteogenomic features in samples with and without NFE2L2 (NRF2), CUL3 and KEAP1 mutations
L. NRF2 signature
M. Differential expression of RNA and proteins between TP63-low vsTP63-high samples.
Table S5: Tables associated with ubiquitylproteome analysis, Related to Figure 5
A. Clustering of tumor samples using protein-corrected ubiquitin data
B. Ub-sites that show differential abundance across Ub clusters
C. Pathway enrichment results for Ub-sites with higher abundance in Ub Cluster 1
D. Pathway enrichment results for Ub-sites with higher abundance in Ub Cluster 2
E. Ub-sites and E3 ligase pairs with significant abundance association (FDR < 0.01)
F. Ub-sites and DUB pairs with significant abundance association (FDR < 0.01)
G. Pathway enrichment results for Ub-sites positively associated with HERC5 abundance
H. Genes with significant spatial differential acetylation clustering within a protein structure as detected by CLUMPS
I. Genes with significant spatial differential ubiquitylation clustering within a protein structure as detected by CLUMPS
J. Genes with significant spatial differential NAT ubiquitylation clustering within a protein structure as detected by CLUMPS
Table S6: Tables associated with immune-landscape analysis, Related to Figure 6
A. Immune composition of LSSC tumors based on deconvolution of RNA expression using xCell.
B. Immune clusters based on xCell signatures
C. Pathways upregulated in immune groups based on RNA-Seq data (C1), global proteomics (C2), phosphoproteomics (C3), acetylproteomics (C4)
D. Genes upregulated in immune groups based on RNA-Seq data (D1), global proteomics (D2), phosphoproteomics (D3), acetylproteomics (D4)
E. RTK Correlation-Based Enrichment (CBPE) Scores
F. Pathway scores in Tumor samples based on global protein abundance via ssGSEA
G. cis-eQTLS identified based on germline variants and tumor RNA-seq data (at 5% FDR)
H. cis-pQTLS identified based on germline variants and tumor proteomics data (at 5% FDR)
I. List of genes/proteins whose expression is associated with a SNP
J. Ingenuity pathway analysis of the eGenes from QTL analysis
K. cis-QTLs identified in genes/proteins differentially expressed in immune groups (K1-3) and list of eGenes (K4)
Table S7: Tables associated with tumor-associated proteins, Related to Figure 7
A. Differential abundance of proteins in tumor vs paired NAT in LSCC, HNSCC, and LUAD
B. 502 proteins significantly overexpressed (log2(FC) >2, FDR <0.01) in tumors relative to their matched NATs, most with coherent overexpression in multi-omic analysis
C. Tumor suppressors or oncogenes regulated at the level of protein expression or phosphoproteome. Activating phosphosite: regulated at the level of phosphorylation, annotated as activating site, Inhibiting phosphosite: regulated at the level of phosphorylation, annotated as inhibitory site. CausalPath was used for this analysis.
D. DepMap dependency scores (CRISPR (Avana) Public 20Q3) across 16 LSCC cell lines
E. Genes that correlate to immune score at the copy number, RNA, and protein (if available) levels in LSCC, HNSCC, and LUAD
F. Predicted tumor neoantigens identified in the CPTAC LSCC cohort at the level of RNA transcripts and peptides containing evidence of somatic mutations.
Figure S5: Pathway analysis and differential ubiquitylation and acetylation, Related to Figure 5
A. Network representation of pathways significantly enriched in modulated protein-corrected K-GG sites across Ub clusters (Figure 5A). Two patterns of K-GG site abundance were observed across Ub clusters (top). The red and blue arrows indicate up- and downregulation across the clusters, respectively. Significant pathways are indicated as nodes and edges indicate shared genes. Network clusters were summarized to reduce pathway redundancies and the common functional themes were manually annotated as text.
B. STRING network showing the genes driving the enrichment of the “Immune signaling, MAPK targets, and JAK-STAT signaling” cluster shown in panel A. These genes are core members of many signaling pathways, resulting in enrichment of multiple pathways.
C. Number of K-GG sites showing significant correlations with deubiquitylases (DUBs). Shown are the five DUBs with the highest proportion of negative correlations.
D. HERC5 and ISG15 protein expression (Log2 TMT ratios) show significant positive correlation.
E. Pathway enrichment analysis results of K-GG sites that significantly correlate with HERC5 protein expression.
F. HERC5 protein expression (Log2 TMT ratio) and example K-GG site abundances (protein-corrected Log2 TMT ratio) from PKM, ENO1, and PGK1 show strong positive correlations in tumors.
G. PKM, ENO1, and PGK1 plotted against sample K-GG site abundance of the respective proteins. K-GG site abundance is shown without protein correction (Log2 TMT ratio) to highlight the lack of negative correlation prior to protein correction.
H. HERC5 protein expression (Log2 TMT ratio) plotted against PKM, ENO1, and PGK1 protein expression (Log2 TMT ratio), which do not show positive correlation in tumors.
I. HERC5 protein expression (Log2 TMT ratio) and interferon-gamma (IFNG) score show significant positive correlation.
J. ISG15 protein expression (Log2 TMT ratio) and IFNG score show significant positive correlation.
K. PGK1 acetylated sites marked on the protein structure in Figure 5E (top left) are decreased in tumors compared to NATs; Ub sites are increased (top right). PKM acetylated sites (lower left) are decreased in tumors compared to NATs; Ub sites are increased (lower center). ACADVL acetylated sites marked on the protein structure in Figure 5E (lower right) are decreased in tumors compared to NATs. Wilcoxon p-values are indicated.
Data Availability Statement
CPTAC LSCC proteomics data:
Proteomic Data Commons (PDC); https://pdc.cancer.gov/pdc/ with identifiers PDC000232, PDC000233, PDC000234 and PDC000237
CPTAC data portal LSCC: https://cptac-data-portal.georgetown.edu/study-summary/S063
CPTAC HNSCC proteomics data:
PDC: https://pdc.cancer.gov/pdc/ with identifiers PDC000221 and PDC000222
CPATC LUAD proteomics data:
PDC: https://pdc.cancer.gov/pdc/ with identifiers PDC000153, PDC000149, PDC000224)
Genomic and transcriptomic data files can be accessed at the Genomic Data Commons (GDC); https://portal.gdc.cancer.gov/, via dbGaP Study Accession: phs001287.v10.p5
All histologic https://www.cancerimagingarchive.net/datascope/cptac/home/ and radiologic details can be accessed from the The Cancer Imaging Archive (TCIA) Public Access https://wiki.cancerimagingarchive.net/display/Public/CPTAC-LSCC.
Sample annotation, processed and normalized data files are provided as Tables S1–S3.
Software and code used in this study are referenced in their corresponding STAR Method sections and also the Key Resource Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PTMScan® Ubiquitin Remnant Motif (K-ε-GG) Kit | Cell Signaling Technology | Catalog 5562 |
| PTMScan Acetyllysine Kit | Cell Signaling Technology | Catalog: 13416 |
| CD4 | Dako | Clone 4B12, RRID:AB_2728838 |
| CD8 | Bio-Rad | Clone 4B11, RRID:AB_322868 |
| CD163 | Abcam | Ab182422, RRID:AB_2753196 |
| ARHGDIB | Sigma-Aldrich | HPA051235, RRID:AB_2681398 |
| CK (pan-Cytokeratin) | Dako | clone AE1/AE3, RRID:AB_2132885 |
| alpha-SMA | Abcam | ab5694, RRID:AB_2223021 |
| Biological Samples | ||
| Primary tumor samples | See Experimental Model and Subject Details | N/A |
| Chemicals and Reagents | ||
| HPLC-grade water | J.T. Baker | Catalog: 4218-03 |
| Urea | Sigma | Catalog: U0631 |
| Sodium chloride | Sigma | Catalog: 71376 |
| 1M Tris, pH 8.0 | Invitrogen | Catalog: AM9855G |
| Ethylenediaminetetraacetic acid | Sigma | Catalog: E7889 |
| Aprotinin | Sigma | Catalog: A6103 |
| Leupeptin | Roche | Catalog: 11017101001 |
| Phenylmethylsulfonyl fluoride | Sigma | Catalog: 78830 |
| Sodium fluoride | Sigma | Catalog: S7920 |
| Phosphatase inhibitor cocktail 2 | Sigma | Catalog: P5726 |
| Phosphatase inhibitor cocktail 3 | Sigma | Catalog: P0044 |
| Dithiothretiol, No-Weigh Format | ThermoScientific | Catalog: 20291 |
| Iodoacetamide | Sigma | Catalog: A3221 |
| Lysyl endopeptidase | Wako Chemicals | Catalog: 129-02541 |
| Sequencing-grade modified trypsin | Promega | Catalog: V511X |
| Formic acid | Sigma | Catalog: F0507 |
| Acetonitrile, LC-MS grade | Honeywell | Catalog: 34967 |
| Acetonitrile, anhydrous | Sigma | Catalog: 271004 |
| Trifluoroacetic acid | Sigma | Catalog: 302031 |
| Tandem Mass Tag reagent kit – 11plex | ThermoFisher | Catalog: A34808 |
| 0.5M HEPES, pH 8.5 | Alfa Aesar | Catalog: J63218 |
| Hydroxylamine solution, 50% (vol/vol) in H2O | Aldrich | Catalog: 467804 |
| Methanol | Honeywell | Catalog: 34966 |
| Ammonium hydroxide solution, 28% (wt/vol) in H2O | Sigma | Catalog: 338818 |
| Ni-NTA agarose beads | Qiagen | Catalog: 30410 |
| Iron (III) chloride | Sigma | Catalog: 451649 |
| Acetic acid, glacial | Sigma | Catalog: AX0073 |
| Potassium phosphate, monobasic | Sigma | Catalog: P0662 |
| Potassium phosphate, dibasic | Sigma | Catalog: P3786 |
| MOPS | Sigma | Catalog: M5162 |
| Sodium hydroxide | VWR | Catalog: BDH7225 |
| Sodium phosphate, dibasic | Sigma | Catalog: S9763 |
| Phosphate-buffered saline | Fisher Scientific | Catalog: 10010023 |
| iVIEW DAB Detection Kit | Roche | Catalog: 760-091 |
| Equipment | ||
| Reversed-phase tC18 SepPak, 3cc 200mg | Waters | Catalog: WAT054925 |
| Solid-phase C18 disk, for Stage-tips | Empore | Catalog: 66883-U |
| Stage-tip needle | Cadence | Catalog: 7928 |
| Stage-tip puncher, PEEK tubing | Idex Health & Science | Catalog: 1581 |
| PicoFrit LC-MS column | New Objective | Catalog: PF360-75-10-N-5 |
| ReproSil-Pur, 120 Å, C18-AQ, 1.9-μm resin | Dr. Maisch | Catalog: r119.aq |
| Nanospray column heater | Phoenix S&T | Catalog: PST-CH-20U |
| Column heater controller | Phoenix S&T | Catalog: PST-CHC |
| 300 μL LC-MS autosampler vial and cap | Waters | Catalog: 186002639 |
| Offline HPLC column, 3.5-μm particle size, 4.6 um × 250 mm | Agilent | Catalog: Custom order |
| Offline 96-well fractionation plate | Whatman | Catalog: 77015200 |
| 700 μL bRP fractionation autosampler vial | ThermoFisher | Catalog: C4010-14 |
| 700 μL bRP fractionation autosampler cap | ThermoFisher | Catalog: C4010-55A |
| 96-well microplate for BCA | Greiner | Catalog: 655101 |
| Microplate foil cover | Corning | Catalog: PCR-AS-200 |
| Vacuum centrifuge | ThermoFisher | Catalog: SPD121P-115 |
| Centrifuge | Eppendorf | Catalog: 5427 R |
| Benchtop mini centrifuge | Corning | Catalog: 6765 |
| Benchtop vortex | Scientific Industries | Catalog: SI-0236 |
| Incubating shaker | VWR | Catalog: 12620-942 |
| 15 mL centrifuge tube | Corning | Catalog: 352097 |
| 50 mL centrifuge tube | Corning | Catalog: 352070 |
| 1.5 mL microtube w/o cap | Sarstedt | Catalog: 72.607 |
| 2.0 mL microtube w/o cap | Sarstedt | Catalog: 72.608 |
| Microtube caps | Sarstedt | Catalog: 72.692 |
| 1.5 mL snapcap tube | ThermoFisher | Catalog: AM12450 |
| 2.0 mL snapcap tube | ThermoFisher | Catalog: AM12475 |
| Instrumentation | ||
| Microplate Reader | Molecular Devices | Catalog: M2 |
| Offline HPLC System for bRP fractionation | Agilent 1260 | Catalog: G1380-90000 |
| Online LC for LC-MS | ThermoFisher | Catalog: LC140 |
| Q Exactive Plus Mass Spectrometer | ThermoFisher | Catalog: IQLAAEGAAPFALGMBDK |
| Q Exactive HF-X Mass Spectrometer | ThermoFisher | Catalog: 0726042 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | ThermoFisher | Catalog: IQLAAEGAAPFADBMBHQ |
| Critical Commercial Assays | ||
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold | Illumina | Catalog: RS-122-2301 |
| Infinium MethylationEPIC Kit | Illumina | Catalog: WG-317-1003 |
| Nextera DNA Exosome Kit | Illumina | Catalog: 20020617 |
| KAPA Hyper Prep Kit, PCR-free | Roche | Catalog: 07962371001 |
| BCA Protein Assay Kit | ThermoFisher | Catalog: 23225 |
| Deposited Data | ||
| DepMap: Mutation | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: Segmented copy number | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: Gene level copy number | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: RNAseq (transcript isoform) | DepMapPublic 21Q1 (PMID: 31068700; Dataset doi:10.6084/m9.figshare.13681534.v1) | https://depmap.org/portal/download/ |
| DepMap: Proteomics (RPPA) | CCLE 2019 (PMID: 31068700) | https://depmap.org/portal/download/ |
| DepMap: CRISPR KO screen (combined) | DepMapPublic 21Q1 (bioRxiv 2020.05.22.110247; doi: https://doi.org/10.1101/2020.05.22.110247) | https://depmap.org/portal/download/ |
| DepMap: shRNA screen (combined) | DEMETER2 Data v6 (PMID: 30389920) | https://depmap.org/portal/download/ |
| DepMap: GDSC drug screen | Sanger GDSC 1 (PMID: 27397505) | https://depmap.org/portal/download/ |
| DepMap: PRISM drug screen | PRISM Repurposing 19Q4 Secondary Screen (PMID: 32613204) | https://depmap.org/portal/download/ |
| PhosphoSitePlus | (Hornbeck et al., 2012) | https://www.phosphosite.org |
| Connectivity Map (CMAP) | (Lamb et al., 2006; Subramanian et al., 2017) | https://www.broadinstitute.org/connectivity-map-cmap |
| Human Protein Atlas (HPA) | (Uhlén et al., 2005) | https://www.proteinatlas.org |
| CT Antigen database | (Almeida et al., 2009) | http://www.cta.lncc.br |
| Dependency map (DepMap) | (Tsherniak et al., 2017) |
https://depmap.org/portal/ v3.3.8 is a GECKOv2 Achilles dataset |
| Library of Integrated Network-based Cellular Signatures (LINCS) | (Lamb et al., 2006; Subramanian et al., 2017) |
https://clue.io/data Expanded CMap LINCS Resource 2020 (1/28/2021 update) |
| CPTAC HNSCC cohort | (Huang et al., 2021) | https://cptac-data-portal.georgetown.edu/study-summary/S054 |
| CPTAC LSCC cohort | This study | https://cptac-data-portal.georgetown.edu/study-summary/S063 |
| Software and Algorithms | ||
| methylationArrayAnalysis (version 3.9) | (Maksimovic et al., 2016) | https://master.bioconductor.org/packages/release/workflows/html/methylationArrayAnalysis.html |
| Illumina EPIC methylation array (3.9) | Hansen KD, 2019 | https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylationEPICanno.ilm10b2.hg19.html |
| Methylation array analysis pipeline for CPTAC | Li Ding Lab | https://github.com/ding-lab/cptac_methylation |
| miRNA-Seq analysis pipeline for CPTAC | Li Ding Lab | https://github.com/ding-lab/CPTAC_miRNA |
| VEP | (McLaren et al., 2016) | https://github.com/Ensembl/ensembl-vep/tags |
| TNScope / DNAScope (Sentieon) | (Freed et al.) | sentieon.com |
| vcfAnno | (Pedersen et al., 2016) | https://github.com/brentp/vcfanno |
| VariantAnnotation (Bioconductor) | (Obenchain et al., 2014) | https://bioconductor.org/packages/release/bioc/html/VariantAnnotation.html |
| arriba_v1.1.0 | https://github.com/suhrig/arriba/ | |
| fusioncatcher_v1.10 | (Nicorici et al.) | https://github.com/ndaniel/fusioncatcher/blob/master/doc/manual.md |
| eQTLGen | (Westra et al., 2013) | https://github.com/molgenis/systemsgenetics/wiki/eQTL-mapping-analysis-cookbook-(eQTLGen) |
| Pindel0.2.5 | (Ye et al., 2009) | http://gmt.genome.wustl.edu/packages/pindel/ |
| SignatureAnalyzer | (Kim et al., 2016) | https://software.broadinstitute.org/cancer/cga/msp |
| CNVEX | Marcin Cieslik Lab | https://github.com/mctp/cnvex |
| CRISP | Marcin Cieslik Lab | https://github.com/mcieslik-mctp/crisp-build |
| Spectrum Mill | Karl R. Clauser, Steven Carr Lab | https://proteomics.broadinstitute.org/ |
| ComBat (v3.20.0) | (Johnson et al., 2007) | https://bioconductor.org/packages/release/bioc/html/sva.html |
| gPCA | (Reese et al., 2013) | https://cran.r-project.org/web/packages/gPCA/index.html |
| GISTIC2.0 | (Mermel et al., 2011) | ftp://ftp.broadinstitute.org/pub/GISTIC2.0/GISTIC_2_0_23.tar.gz |
| iProFun | (Song et al., 2019) | https://github.com/WangLab-MSSM/iProFun |
| ESTIMATE | (Yoshihara et al., 2013) | https://bioinformatics.mdanderson.org/public-software/estimate/ |
| WebGestaltR | (Wang et al., 2017) | http://www.webgestalt.org/ |
| GSVA | (Hanzelmann et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| TSNet | (Petralia et al., 2018) | https://github.com/WangLab-MSSM/TSNet |
| xCell | (Aran et al., 2017) | http://xcell.ucsf.edu/ |
| CPTAC LSCC Data Viewer | Steven Carr lab | https://rstudio-connect.broadapps.org/CPTAC-LSCC2021/ |
| ConsensusClusterPlus | (Wilkerson and Hayes, 2010) | http://bioconductor.org/packages/release/bioc/html/CancerSubtypes.html |
| MS-GF+ | (Kim and Pevzner, 2014) | https://github.com/MSGFPlus/msgfplus |
| NeoFlow | (Wen et al., 2020) | https://github.com/bzhanglab/neoflow |
| netMHCpan | (Jurtz et al., 2017) | http://www.cbs.dtu.dk/services/NetMHCpan/ |
| Optitype | (Szolek et al., 2014) | https://github.com/FRED-2/OptiType |
| Customprodbj | (Wen et al., 2020) | https://github.com/bzhanglab/customprodbj |
| PDV | (Li et al., 2019b) | https://github.com/wenbostar/PDV |
| PepQuery | (Wen et al., 2019) | http://pepquery.org |
| PTM-SEA | (Krug et al., 2018)) | https://github.com/broadinstitute/ssGSEA2.0 |
| PTMsigDB | (Krug et al., 2018)) | http://prot-shiny-vm.broadinstitute.org:3838/ptmsigdb-app/ |
| Terra | Broad Institute data science platform. | https://terra.bio/ |
| Panoply | Broad Institute Proteomics Platform | https://github.com/broadinstitute/PANOPLY |
| CMap | (Lamb et al., 2006; Subramanian et al., 2017) | https://clue.io/cmap |
| LIMMA v3.36 (R Package) | (Ritchie et al., 2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| FactoMineR v1.41NMF(R - package) | (Gaujoux and Seoighe, 2010; Lê et al., 2008) | https://cran.r-project.org/web/packages/FactoMineR/index.html |
| MClust v5.4 (R package) | (Scrucca, Fop, Murphy and Raftery, 2017) | https://cran.r-project.org/web/packages/mclust/index.html |
| g:Profiler | (Raudvere U, et al., 2019) | https://biit.cs.ut.ee/gprofiler/gost |
| Cytoscape | (Shannon P, et al., 2003) | https://cytoscape.org/ |
| ImmuneSubtypeClassifier | (Gibbs, 2020) | https://github.com/CRI-iAtlas/ImmuneSubtypeClassifier |
| ProteinPaint | (Zhou et al., 2016) | https://pecan.stjude.doud/proteinpaint/ |
| Ordinal | Christensen RHB (2019) | https://CRAN.R-project.org/package=ordinal |