

SUMMARY
The normal androgen receptor (AR) cistrome and transcriptional program are fundamentally altered in prostate cancer (PCa). Here, we profile the chromatin landscape and AR-directed transcriptional program in normal prostate cells and show the impact of SPOP mutations, an early event in prostate tumorigenesis. In genetically normal mouse prostate organoids, SPOP mutation results in accessibility and AR binding patterns similar to that of human PCa. Consistent with dependence on AR signaling, castration of SPOP mutant mouse models results in the loss of neoplastic phenotypes, and human SPOP mutant PCa shows a favorable response to AR-targeted therapies. Together, these data validate mouse prostate organoids as a robust model for studying epigenomic and transcriptional alterations in normal prostate, provide valuable datasets for further studies, and show that a single genomic alteration may be sufficient to reprogram the chromatin of normal prostate cells toward oncogenic phenotypes, with potential therapeutic implications for AR-targeting therapies.
Graphical abstract
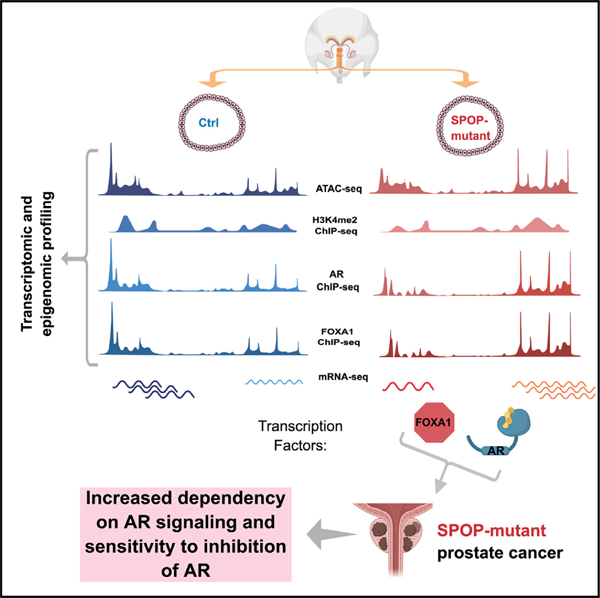
In brief
Grbesa et al. use multilevel epigenomic and transcriptional profiling to characterize genetically normal prostate organoids and show the impact of an early cancer driver (SPOP mutation). Integrative analysis identifies transcriptional programs that are AR regulated and reprogrammed by SPOP mutation, suggesting a basis for enhanced sensitivity to AR-targeting therapies.
INTRODUCTION
The androgen receptor (AR) is a critical driver and key therapeutic target in prostate cancer (PCa). In normal prostate epithelial cells, AR coordinates growth-suppressive effects and differentiation programs (Heinlein and Chang, 2004; Taplin and Balk, 2004). During prostate carcinogenesis, AR is reprogrammed to instead promote oncogenic transcriptional programs (Pomerantz et al., 2015). However, the mechanisms of this reprogramming remain incompletely understood.
Recurrent missense mutations in SPOP (speckle type BTB/POZ protein) occur in ~10% of localized primary PCa cases (The Cancer Genome Atlas Research Network, 2015; Li et al., 2020) and are highly prostate specific; they are rarely observed in other cancer types. SPOP mutations nominate a distinct molecular subtype of human PCa (Barbieri et al., 2012; The Cancer Genome Atlas Research Network, 2015; Shoag et al., 2018), with defined genomic alterations, DNA methylation, transcriptional signatures, and clinical characteristics (Barbieri et al., 2012; Boysen et al., 2015; The Cancer Genome Atlas Research Network, 2015; Liu et al., 2018). Furthermore, a variety of data suggest that SPOP mutations occur early in the process of prostate tumorigenesis (Baca et al., 2013; Boysen et al., 2015). SPOP acts as the substrate recognition component of a CUL3-E3 ubiquitin-protein ligase complex (Zhuang et al., 2009), with PCaassociated mutations affecting substrate specificity. Substrates reported to be affected by SPOP mutations include AR itself (An et al., 2014; Geng et al., 2014); the AR coactivators SRC3 (Geng et al., 2013), p300 (Blattner et al., 2017), and TRIM24 (Blattner et al., 2017); and chromatin-associated proteins such as DEK (Theurillat et al., 2014) and DAXX (Bouchard et al., 2018). However, how mutations in SPOP affect AR-directed transcriptional programs, and whether this is required for tumor initiation and progression, remain unclear.
Here, we leverage the unique capabilities of 3D prostate organoids to define the epigenomic and transcriptional landscape upon AR activation and use a genetically engineered mouse (GEM) model conditionally expressing mutant SPOP (Blattner et al., 2017) to characterize the impact of a single genomic alteration in genetically normal cells.
RESULTS
Chromatin changes in normal prostate organoids with androgen stimulation
Compared to our understanding of AR function in PCa cell lines and tissues (Pomerantz et al., 2015; Sharma et al., 2013; Stelloo et al., 2018), much less is known of the mechanisms underlying AR-mediated transcriptional regulation in normal prostate cells. Thus, to define the epigenomic response to AR activation, we generated 3D organoid models of murine prostate epithelial cells (Karthaus et al., 2014). Upon establishment, these prostate organoids faithfully mimicked prostate gland architecture (Karthaus et al., 2014) and immunohistochemical features (Figure S1A). To catalog the function of AR in the normal prostate, we assessed genome-wide changes in transcriptional profiles (RNA sequencing [RNA-seq]), chromatin accessibility by assay for transposase-accessible chromatin (ATAC-seq), and cistromes of AR, FOXA1, and H3K4me2 by chromatin immunoprecipitation sequencing (ChIP-seq) post-treatment with 10 nM dihydrotestosterone (DHT) (Figures 1A and S1B–S1J).
Figure 1. DHT-induced changes in chromatin accessibility correspond to AR-transcriptional response.
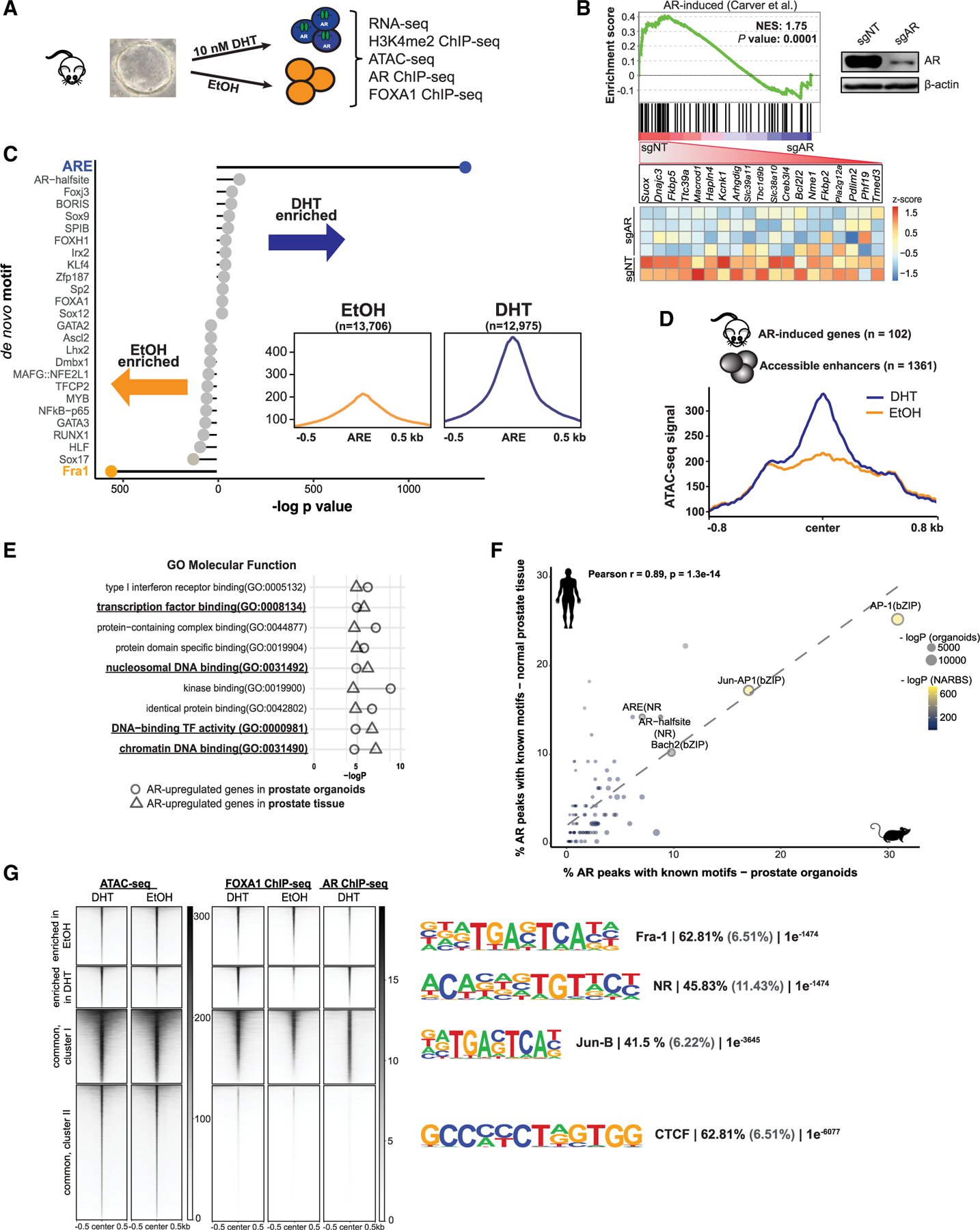
(A) Schematic representation of treatments and obtained datasets.
(B) GSEA and leading-edge analysis of AR activity in normal prostate organoids (see also Figures S1B–S1F). Upper right corner: immunoblot of murine organoid CRISPR-Cas9 cells with non-targeting (NT) or Ar-specific single-guide RNA (sgRNA).
(C) Lollipop plot representing known motif enrichment in vehicle-treated and androgen-treated murine organoids.
(D) Histogram depicting accessibility signal from ethanol (EtOH)-treated and DHT-treated organoids over the center of regulatory elements within 1 Mb of AR-induced genes from prostate tissue.
(E) Cleveland plot indicating results of Gene Ontology (GO) analysis of AR-induced genes in prostate organoids (circle) and prostate tissue (triangles) integrated with accessible regions identified after androgen stimulation of organoid cells.
(F) Known motif analysis of AR ChIP-seq datasets from normal mouse organoids (x axis) and human normal prostate tissue (n = 7; GEO: GSE70079; y axis) demonstrating enrichment for similar motifs (r = 0.89, p < 0.001). The dotted line represents a regression line. Circle size represents the −log p value of motif enrichment in AR ChIP-seq peaks in normal murine prostate organoids and circle color the motif enrichment significance in AR binding sites in normal prostate tissue (NARBS).
(G) ATAC-seq, FOXA1, and AR ChiP-seq signals (see also Figures S1G–S1J) over the regions that were identified to be more open before (enriched in EtOH) or after androgens (enriched in DHT). The tornado plots also cover the regions whose accessibility is not DHT dependent (common). The most enriched de novo motifs per cluster are depicted on the right, along with the percentage of target (black) and background (gray) regions and the resulting p value.
Gene expression data (Figures S1B–S1D) showed upregulation of known murine AR-regulated genes (Carver et al., 2011) by androgens in intact organoids, but not those with CRISPR-mediated deletion of Ar (sgAr). We validated the transcriptional response by comparing it to AR-regulated genes in prostate tissue (Carver et al., 2011) (Figure 1B), confirming a relevant transcriptional response in our organoid model system (Figures S1E and S1F). To define AR-driven alterations in the chromatin landscape in normal prostate epithelial cells, we performed ATAC-seq, as a proxy for genetic regulatory element activity (Figures S1G–S1I; Tables S1 and S2). We identified 26,681 high-confidence differentially accessible peaks between DHT-treated and vehicle-treated prostate organoids (false discovery rate [FDR] < 0.01; Figure S1I). Next, we performed de novo motif analysis of the DHT-enriched versus DHT-depleted accessible elements, revealing significant enrichment of the AR element (ARE) motif in DHT-treated cells, which was associated with an increase in accessibility at these sites post-DHT treatment (Figure 1C). To connect the DHT-responsive regulatory elements to AR-target genes in prostate tissue, we profiled the ATAC-seq signal of androgen-regulated genes in the mouse prostate (Carver et al., 2011) (Figures 1D and S1G–S1I). Subsequently, we performed functional enrichment analysis of the ATAC-seq-defined DNA regulatory elements and associated AR-induced genes in both prostate organoids and tissue, with ‘‘transcription factor (TF) binding’’ and ‘‘chromatin (nucleosomal) binding’’ among the enriched categories (Figure 1E). Overall, our ATAC-seq analysis confirmed a robust and biologically relevant response to androgens at the chromatin level.
Next, to nominate the accessible peaks directly regulated by AR binding, we profiled AR localization genome-wide after DHT stimulation. Genome-wide localization of AR by ChIP-seq resulted in 42,206 AR peaks (Table S1). Importantly, our motif analysis found that the canonical ARE was the most enriched motif in this dataset, confirming the specificity of our signal. Further analysis comparing motif enrichment against that of AR in human prostate tissue (Pomerantz et al., 2015) uncovered a highly similar motif enrichment pattern (r = 0.89, p < 0.001) (Figure 1F), suggesting that our models faithfully recapitulate critical features of AR signaling. Direct comparison of AR binding events with changes in chromatin accessibility uncovered increased accessibility at the subset of AR-bound enhancers in DHT-treated compared to vehicle-treated prostate cells (Figure 1G).
Integrative analysis across data types revealed distinct clusters of regulatory sites either unique to baseline or AR-stimulated conditions, or common between these, with associated dominant TF motifs (Figure 1G). While nuclear hormone receptor motifs were most enriched in regions associated with DHT stimulation (cluster 2), regions with chromatin accessibility enriched in basal conditions (cluster 1) or common (cluster 3) were associated with AP-1 motifs (Fra-1 and Jun-B). AP-1 motifs are also common in AR ChIP-seq from normal human prostate tissue (Figure 1f), consistent with an AR-directed role in non-transformed prostate cells.
These data demonstrate that androgen stimulation of prostate organoids results in chromatin and transcriptional changes concordant between both murine and human prostate tissues and provides a valuable resource to study molecular functions of AR in a genetically normal context and early events that contribute to AR-driven prostate tumorigenesis.
SPOP mutation alone alters the landscape of accessible enhancers in response to androgen
SPOP mutations are present in ~10% of PCa (Barbieri et al., 2012; The Cancer Genome Atlas Research Network, 2015; Li et al., 2020), occur early in the natural history of the disease, are relatively prostate specific, and affect AR activity through the deregulation of multiple substrates (Blattner et al., 2017; Geng et al., 2013; Theurillat et al., 2014). We previously developed a transgenic mouse with Cre-dependent, conditional expression of SPOP-F133V (Figure 2A) (Blattner et al., 2017). In prostate organoids infected with the Cre virus and vector, we examined the impact of an SPOP mutation on the epigenomic and transcriptional landscapes after androgen stimulation (Figure 2B).
Figure 2. Extensive remodeling of enhancers in SPOPmut organoids.
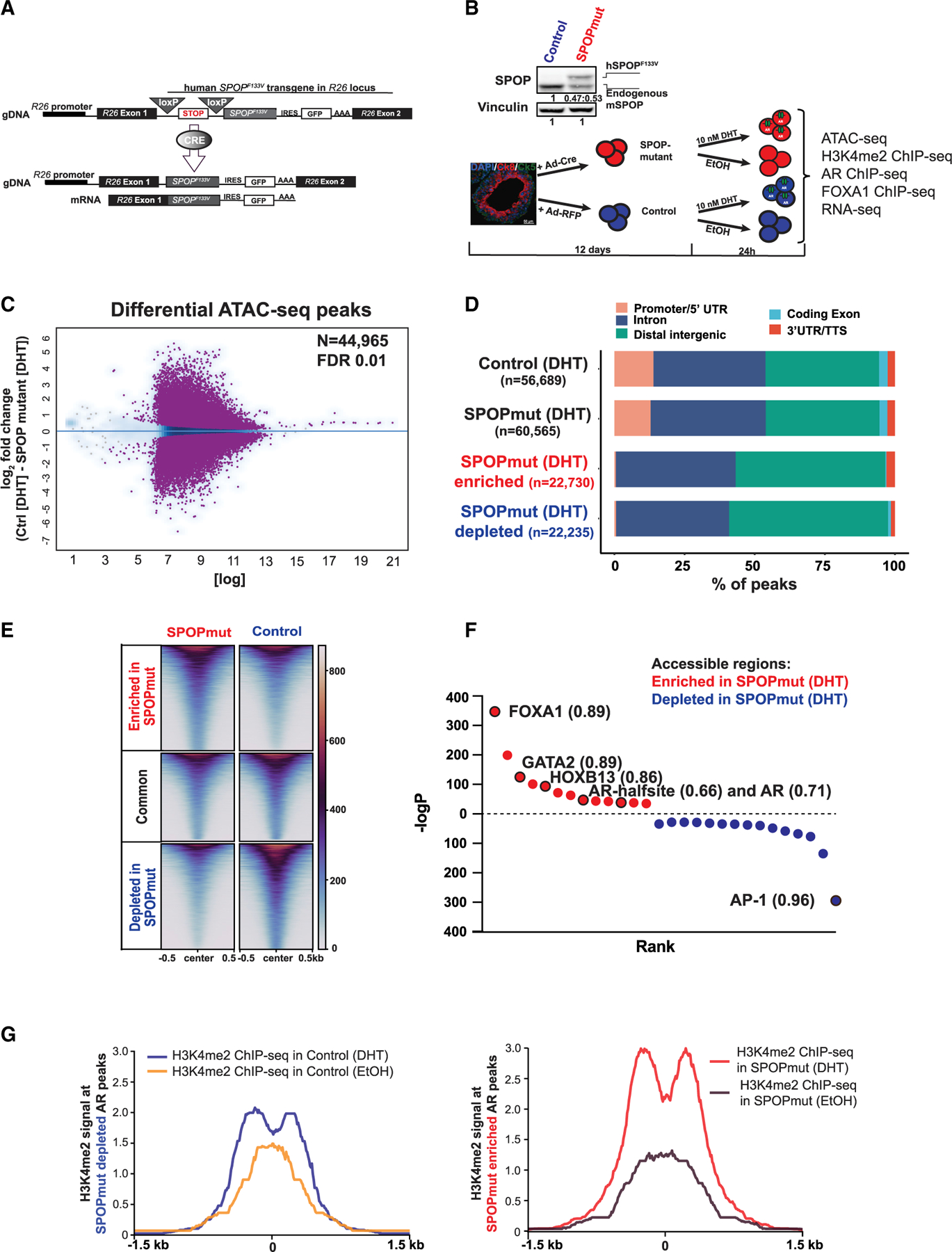
(A) Schematic of conditional SPOP-F133V construct in the Rosa26 (R26) locus (top) and of the expressed transgenic transcript after Cre introduction (bottom).
(B) Illustration of the generation of the organoid lines and treatments in addition to all of the performed experiments. Top: immunoblot showing physiological levels of human SPOP-F133V (top band) in comparison to endogenous mouse SPOP protein (bottom band).
(C) Differential accessible peaks (purple) between control (n = 4) and SPOPmut (n = 4) cells at FDR 0.01 (see also Figures S2A–S2C).
(D) Genomic annotation of consensus and differential peaks showing predominant enhancer location of SPOP mutant-enriched and -depleted accessible elements.
(E) Heatmap of accessible enhancers showing differential accessibility in SPOPmut-enriched and -depleted peaks.
(F) De novo motif analysis of SPOPmut-enriched and -depleted accessible DNA regulatory regions. Enriched motifs are plotted according to their rank and p value. Values next to the motif are the best matched motif score (maximum 1).
(G) H3K4me2 signal at the enhancers from control and SPOP mutant organoids with and without DHT stimulation at SPOP mutant-depleted AR peaks (left), and SPOP mutant-enriched AR peaks (right).
We profiled the chromatin accessibility landscape of androgen-stimulated prostate organoids expressing mutant SPOP, and controls, using ATAC-seq (Figures 2A–2C, S1G–S1I, and S2A–S2C). We found >44,900 differentially accessible sites (Figure 2C), the majority of which were not promoter associated, but at distal intergenic and intronic regions, consistent with enhancers (Figures 2D and 2E). ATAC-seq defined enhancers in SPOP mutant organoids as being enriched for motifs of TFs important for AR-driven prostate tumorigenesis (Figure 2F), including FOXA1 and HOXB13 (Pomerantz et al., 2015). These data support that the chromatin landscape of SPOP mutant prostate cells is remodeled at androgen-responsive enhancer regions associated with lineage-specific, oncogenic TFs and other AR-independent regions (Figures S2D and S2E).
H3K4me2-marked nucleosomes go through remodeling upon AR binding to chromatin (He et al., 2010). Upon androgen stimulation, the H3K4me2 nucleosome at the center of ARE is evicted, and subsequently, AR-binding sites are flanked with H3K4me2 nucleosomes. The same bimodal distribution before and after androgen stimulus was shown in the case of AR-pioneering factor FOXA1 as well (He et al., 2010). Consistent with our chromatin accessibility data, there was more pronounced chromatin remodeling (measured by H3K4me2 ChIP-seq) at AR-bound enhancers in SPOP mutant organoids (Figure 2G).
SPOP mutant prostate organoids display an oncogenic AR cistrome
Oncogenic reprogramming of the AR cistrome is a fundamental feature of PCa (Augello et al., 2019; Pomerantz et al., 2015; Sharma et al., 2013; Stelloo et al., 2018), but which specific alterations can drive this phenotype and what stage of the disease it occurs remains unclear. We sought to test the hypothesis that a single early event such as SPOP mutation could affect reprogramming of the AR cistrome to mimic PCa. AR ChIP-seq with a validated AR antibody (Table S1) revealed >13,500 differential AR binding sites (FDR < 0.01) between control and SPOP mutant organoid lines (Figure 3A). In SPOP mutant organoids, the AR cistrome was enriched for the motifs of well-established regulators of AR function associated with tumorigenesis, including FOXA1 and HOXB13 (Figure 3B). Strikingly, the motif enrichment pattern in SPOP mutant organoids compared to controls was remarkably similar to that of previously reported in AR cistromes of human prostate adenocarcinoma samples (Pomerantz et al., 2015) as compared to normal prostate tissue (Figure S3). Consistent with ATAC-seq results, AR peaks in SPOP mutant prostate organoids harbored a higher density of canonical AR and FOXA1 motifs (Figures 3C and 3D). To validate the relevance of this shift in the TF motif occurrence, we analyzed the AR cistromes of human primary prostate carcinoma tissue samples (Stelloo et al., 2018) (Figure 3E). AR peaks from human SPOP mutant cancers had higher FOXA1 motif density (Figure 3F), associated with a shift in the binding of FOXA1 toward the center of AR peaks (Figure 3G), consistent with our murine organoid model. These data suggest that that a single genomic event such as SPOP mutation may alter the androgen-responsive enhancer landscape and AR cistrome toward PCa phenotypes, and nominates FOXA1 as a potential mediator.
Figure 3. AR cistrome from SPOPmut cells is enriched for FOXA1 motifs.
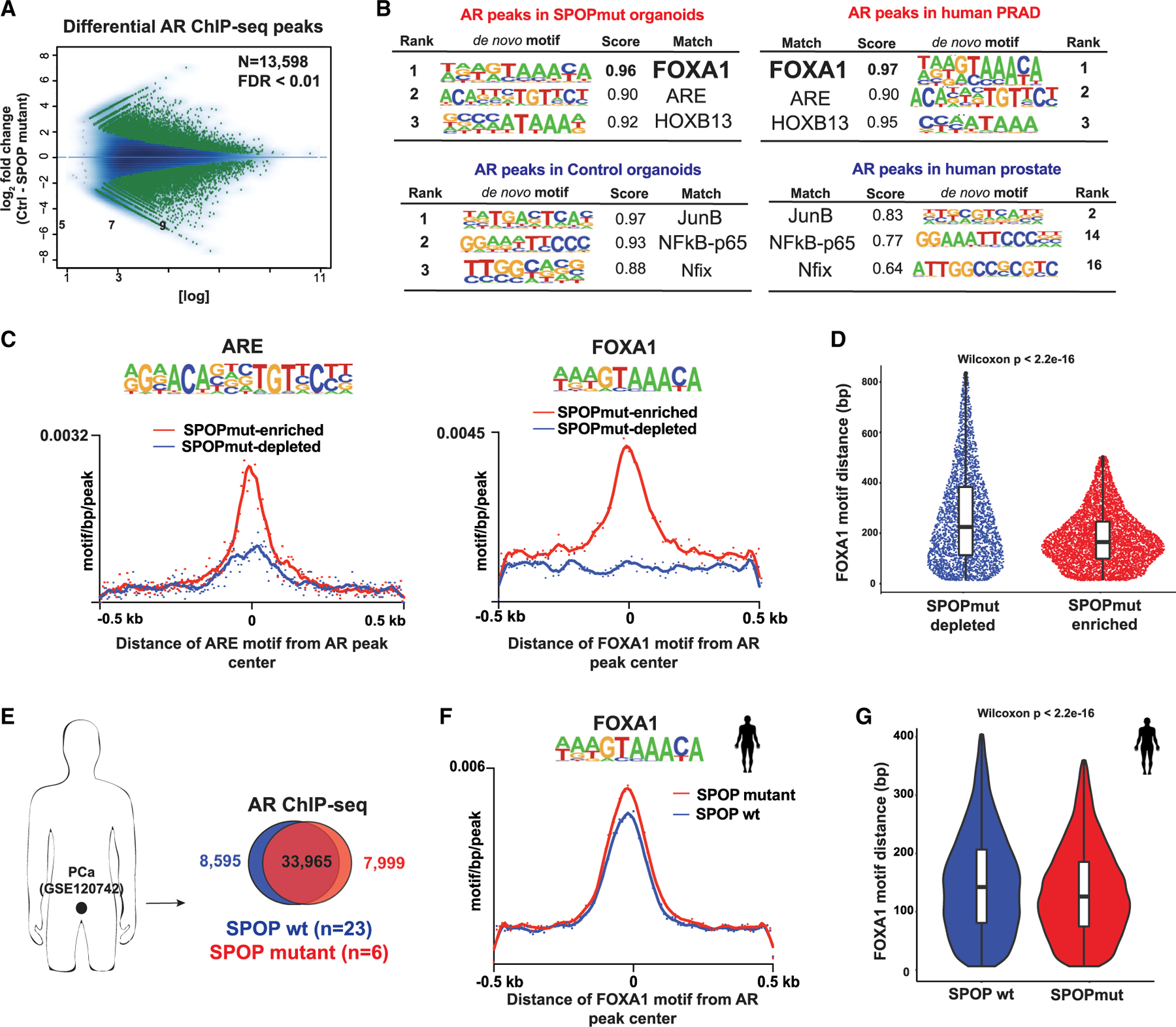
(A) MA plot showing AR differential sites (green dots) between control and SPOP-F133V-expressing organoids using 2 biological replicates per condition.
(B) De novo motif analysis of AR cistrome in SPOPmut and control organoids (right). Left: the same type of motif analysis performed on human prostate adenocarcinoma (PRAD) (Pomerantz et al., 2015) (see also Figure S3).
(C) Histograms of ARE (left) and FOXA1 (right) motif densities within a 500-bp window around the center of each AR peak. Input files were from SPOP mutant-enriched (red line) and -depleted (blue depleted) AR ChIP-seq peaks. p (for both plots) < 2e–16.
(D) Sina plot: distances of FOXA1 motif from the center of each AR peak. Input files were as in (C).
(E) AR ChIP-seq data from human prostate cancer (PCa) tissues (Stelloo et al., 2018) (GSE120742). Peaks coming from either PRAD with SPOP mutation or wild-type (WT) SPOP were merged and intersected.
(F) Histogram of FOXA1 motif density in AR binding sites from human prostate cells with SPOP-F133V (red line) or WT SPOP (blue). p = 0.0002.
(G) The distance of the FOXA1 motif from the center of AR ChIP-seq peaks in each PCa group.
Increased FOXA1 binding in SPOP mutant tumors
FOXA1 acts as a pioneer factor, binding and opening condensed chromatin via its winged-helix DNA binding domains (Cirillo et al., 2002), facilitating AR-mediated transcriptional control (Gao et al., 2003). To better define the role of FOXA1 nominated by motif enrichment in a subset of SPOPmut-specific AR peaks (Figure 3), we assessed genome-wide FOXA1 binding (Table S1). SPOP mutant organoids and controls showed 27,168 differential FOXA1 peaks (Figure 4A). Moreover, the highest FOXA1, FOX-A1:ARE, and ARE motif densities occurred within the SPOP-mut-specific FOXA1 cistrome (Figure 4B). Next, we interrogated AR binding at FOXA1-bound enhancers and found enhancement of the AR signal at FOXA1-bound accessible enhancers specific for SPOP mutant prostate cells (Figures 4C and 4D). These data implicate a role for FOXA1 downstream of SPOP mutation in driving oncogenic changes at a subset of oncogenic AR binding loci.
Figure 4. Increased binding of FOXA1 at SPOP mutant-specific AR sites.
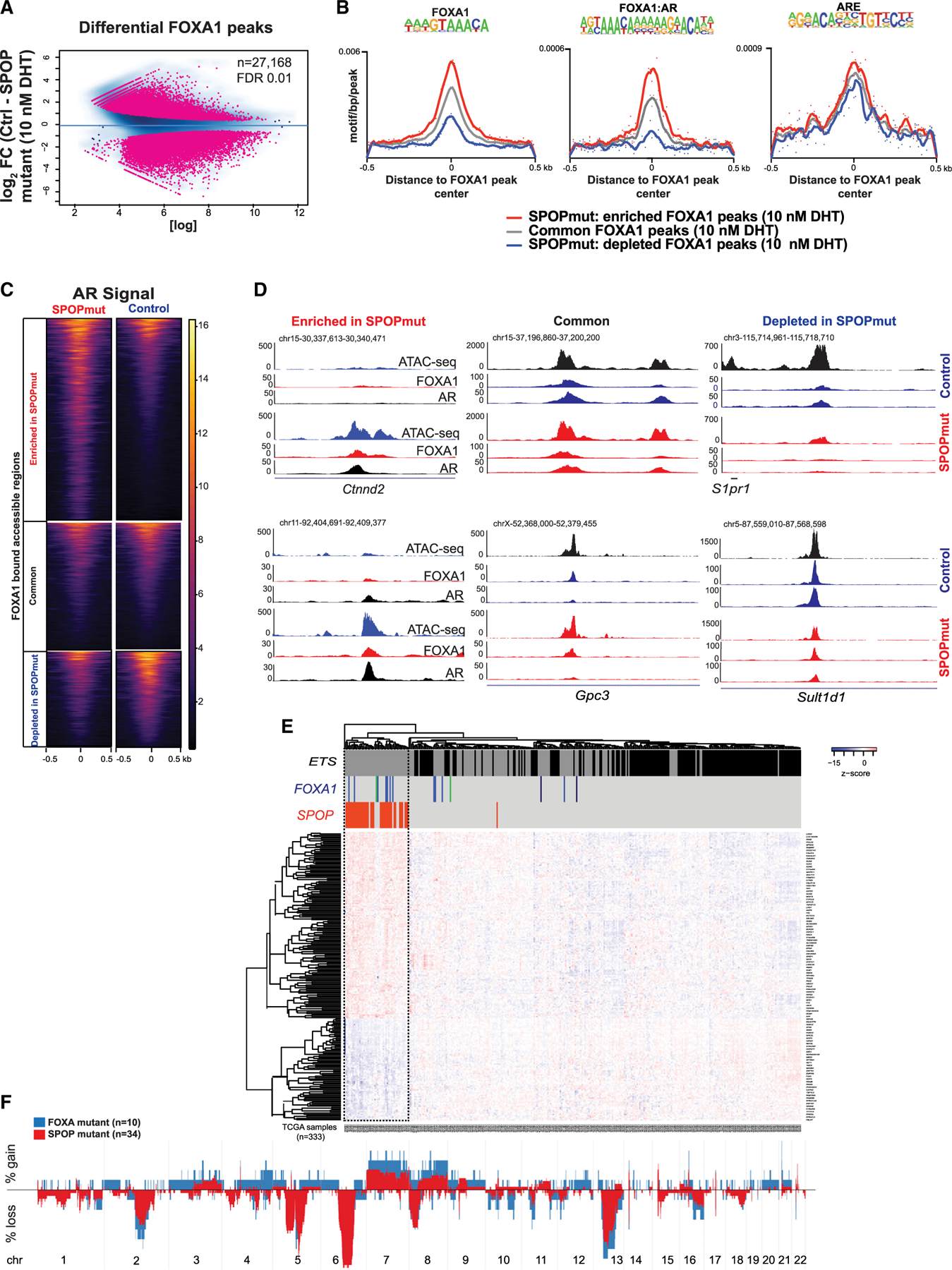
(A) Differential FOXA1 genome-wide (ChIP-seq) binding in control (n = 2) and SPOP mutant (n = 2) organoids upon AR activation.
(B) Motif densities within the center of FOXA1 peaks enriched (red) or depleted (blue) in SPOP mutant murine organoids. Gray, the signal at regions where FOXA1 binding intensity does not change.
(C) AR binding signal at FOXA1-bound DNA regulatory elements that are common between the lines or SPOP mutant enriched or depleted.
(D) Representative Genome Browser (University of California, Santa Cruz) snapshots of regions analyzed in (C).
(E and F) Relationship between SPOP mutant and FOXA1 mutant subclasses of PRAD.
(E) Classification of PRAD using the SPOP mutant transcriptional classifier (Liu et al., 2018). FOXA1 mutations are depicted in blue (FKHD, DNA binding domain), green (N-terminal domain), and black (truncations after FKHD).
(F) Gene copy number profiles of these molecular subclasses of PCa.
We next looked in human PCas for evidence to support a relationship between SPOP mutation and FOXA1. FOXA1 is recurrently mutated in localized PCa, with mutations in the forkhead domain (FKHD)-altering pioneering activity being the most common (Adams et al., 2019; Barbieri et al., 2012; The Cancer Genome Atlas Research Network, 2015; Parolia et al., 2019). We hypothesized that if SPOP mutations acted in part through the modulation of FOXA1 activity, then SPOP mutant tumors and FOXA1 mutant tumors would display similar genomic and transcriptional features. When applying a previously reported transcriptional classifier for SPOP mutation, the majority of FOXA1 mutant tumors clustered together with SPOP mutant tumors (Figure 4E). In addition, tumors harboring SPOP mutations and those with FOXA1 alteration display similar somatic copy number alterations (Figure 4F), suggesting common collaborating events. These data from human tumors help validate data from our model systems, nominating FOXA1 as a potential mediator of SPOP mutant phenotypes.
Gene expression alterations in SPOP mutant prostate organoids
To define whether the changes in accessibility and FOXA1 and AR binding are reflected in transcriptional response to androgens in SPOP mutant cells, we performed RNA-seq (Figure S4). Importantly, AR induced and repressed distinct genes in control and SPOPmut cells (Figure 5A).
Figure 5. Nominating novel AR-regulated genes in the context of SPOP mutation.
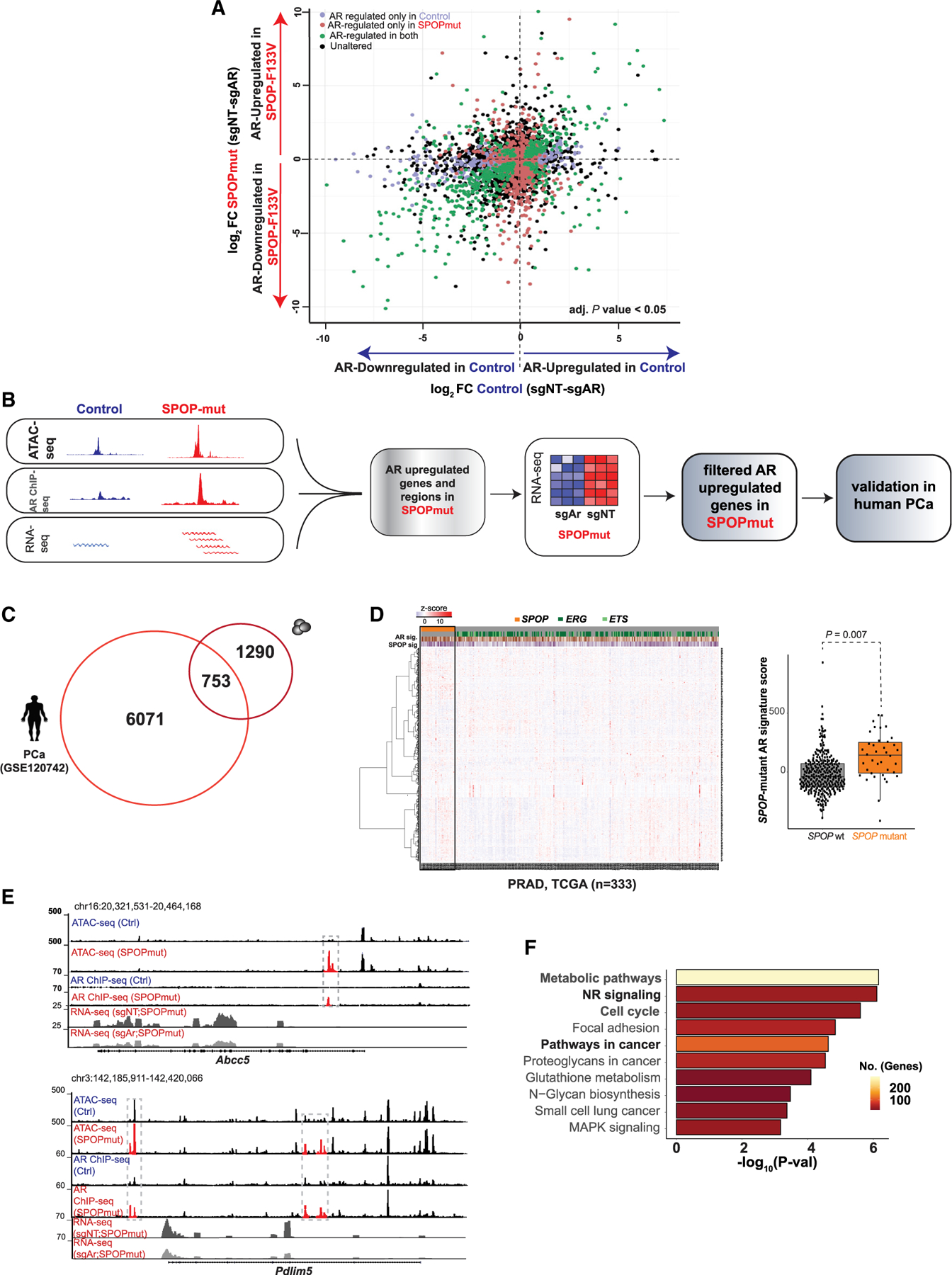
(A) Plot showing differentially expressed genes (adjusted p < 0.05) regulated by AR in control cells (x axis; blue) and SPOPmut cells (y axis, red) (see also Figure S4).
(B) Computational steps to identify AR-upregulated genes specific to SPOP mutant human and murine cells.
(C) Overlapping AR-regulated genes in SPOP mutant human PCa (Stelloo et al., 2018) and murine cells.
(D) Heatmap: gene expression (n = 753) in human PRAD samples (The Cancer Genome Atlas Research Network, 2015). Boxplots: PCa samples with higher expression of nominated genes (n = 753) have higher SPOP mutant signature score.
(E) Representative Genome Browser tracks of AR-regulated regulatory regions and transcripts in SPOPmut organoids.
(F) Functional enrichment of androgen-induced genes in murine SPOP mutant cells.
We next sought to leverage multiple levels of data to define genes that are AR regulated specifically in the context of SPOP mutation (Figure 5B). Integrative analysis of ATAC-seq, AR ChIP-seq, and RNA-seq data nominated genes linked to dynamic regulatory events in SPOP mutant mouse organoids and dependent on AR for induction (n = 2,043). Of these, 753 genes were also upregulated in SPOP mutant human PCas (Figure 5C), and these were used to generate an SPOP mutant AR signature score (Figure 5D). By correlating across datatypes, we identified AR-driven, SPOP mutation-specific transcriptional regulatory events, with increased AR binding, increased accessibility, and AR-dependent increased gene expression in genes such as Abcc5 and Pdlim5 (Figure 5E), which has been linked to PCa pathogenesis (Liu et al., 2017). Functional categorization of these SPOP mutant-specific AR-regulated genes revealed enrichment for metabolic pathways, nuclear receptor signaling, and cell cycle, consistent with an AR-dependent role in prostate tumorigenesis (Figure 5F). These data suggest that the SPOPmut-associated changes in accessibility and TF binding are associated with a distinct AR-dependent gene expression program.
Impact of modulation of AR signaling on SPOP mutant prostate epithelium
Having observed that SPOP mutation alters the androgen-responsive chromatin landscape and transcriptional program, we next examined the dependency on AR signaling in SPOP mutant models in vitro and in vivo. SPOP mutant organoids showed increased sensitivity to AR inhibition with bicalutamide compared to controls (Figure 6A), with proliferation of the SPOP-mut cells being more sensitive to anti-androgen treatment both in 2D and 3D growth conditions (Figures S5A and S5B). Consistent with the potential importance of FOXA1 in the epigenomic alterations in SPOP mutant cells, transient downregulation of FOXA1 further increased the sensitivity of SPOPmut organoids to AR inhibition (Figure 6B).
Figure 6. The distinct phenotype of SPOP mutations shows dependence on functional AR signaling.
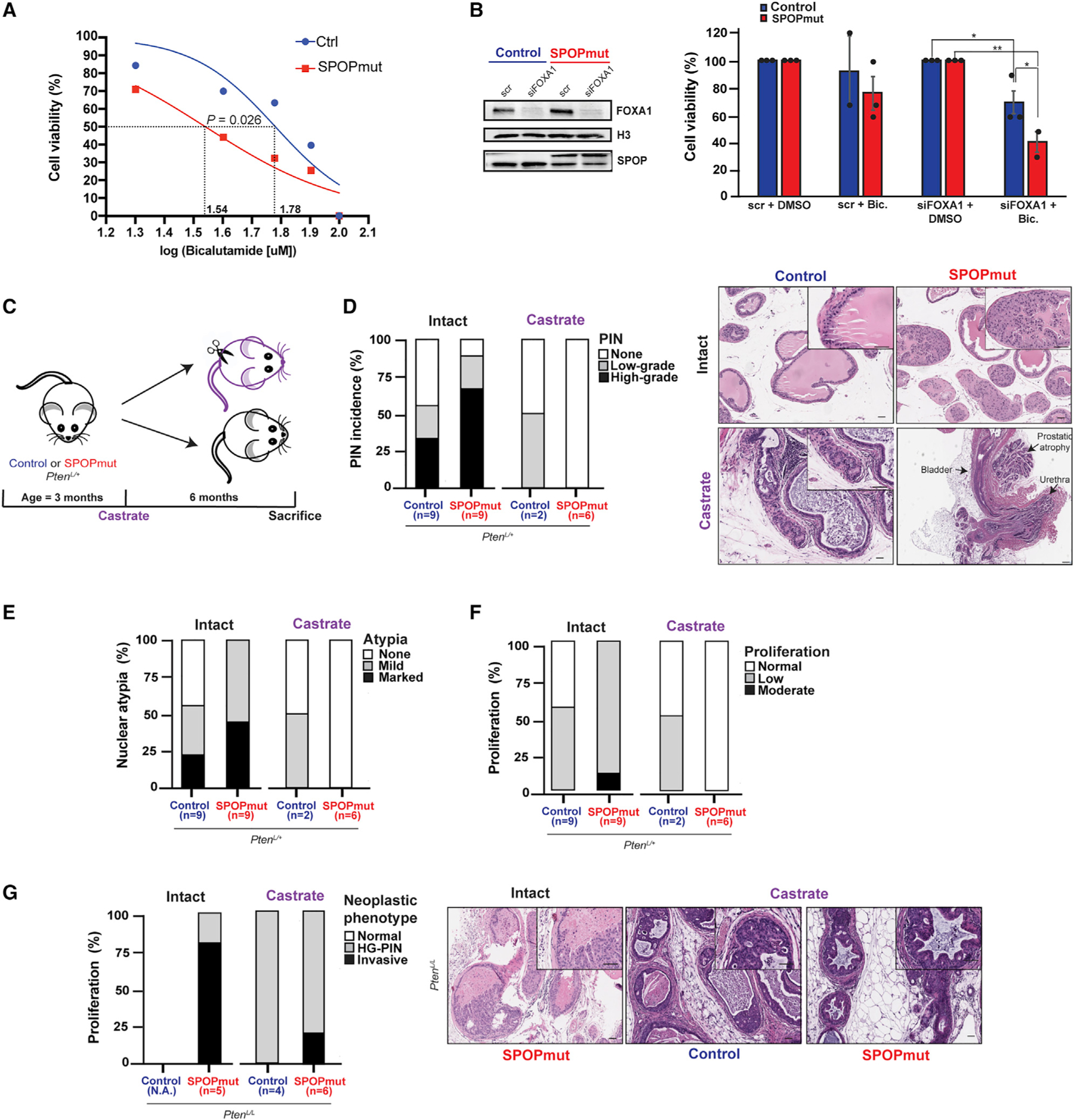
(A) The dose-response curve (log scale) of control and SPOPmut murine prostate organoids in response to bicalutamide (see also Figures S5A and S5B).
(B) Immunoblot of control and SPOPmut murine prostate cells after small interfering RNA (siRNA)-mediated downregulation of FOXA1 (top) and cell viability of these cells after 20 μM bicalutamide treatment (bottom).
(C) Schematic of castration experiment.
(D–F) Incidence of PIN, nuclear atypia, and degree of proliferation in control and SPOPmut mice before and after castration. Representative H&E images are shown at right and in Figures S5C and S5D. Scale bar, 50 μm.
(G) Incidence of neoplastic phenotypes (HG-PIN and invasive features such as necrosis, stromal response, and/or frank invasion) in control and SPOP-F133V mice, with representative images shown at right. Scale bar, 50 μm.
To investigate the role of AR signaling in a mouse model of SPOP-F133V, we performed a pathologic analysis of castrated and hormonally intact mice in a background of heterozygous Pten loss (PbCre+;PtenL/+;Rosa26F133V). We have previously shown that SPOP mutation alone is insufficient to drive tumorigenesis in mouse prostate (Blattner et al., 2017). However, in a conditional Pten-deleted background, SPOP mutation results in more aggressive neoplastic histology, including high-grade prostatic intraepithelial neoplasia (HG-PIN) in PtenL/+ mouse prostates and invasive carcinoma in PtenL/L. We castrated SPOP mutant (PbCre+;PtenL/+;Rosa26F133V) and control (PbCre+;PtenL/+) mice at 8 weeks, sacrificed them at 9 months, and performed a histological examination of their prostates compared to age-matched uncastrated controls (Figure 6C). Castration reversed the increased amount of HG-PIN, nuclear atypia, and proliferation seen in SPOP mutant prostates compared to controls (Figures 6D–6F and S5C and S5D). In the setting of homozygous Pten loss, SPOP mutant mice (PbCre+;PtenL/L;Rosa26F133V) develop invasive, poorly differentiated carcinomas, while control mice (PbCre+;PtenL/L) developed only diffuse HG-PIN (Blattner et al., 2017). In this background, we found that SPOP mutant castrated mice develop features of invasive disease less frequently than hormonally intact mice, while the diffuse, proliferative HG-PIN in both SPOP mutant mice and PtenL/L controls (Figure 6G) remains relatively castration resistant, consistent with prior reports (Blattner et al., 2017; Mulholland et al., 2011).
SPOP mutation is associated with improved response to AR-targeted therapies
Given the prominent role of AR signaling in SPOP mutant preclinical models, we investigated the impact of SPOP mutation on patient response to therapies targeting AR in multiple clinical scenarios. Using a previously developed transcriptional classifier for the SPOP mutant subclass (Liu et al., 2018), we examined data derived from the Decipher Genomics Resource Information Database (GRID) registry (ClinicalTrials.gov identifier: NCT02609269) (Liu et al., 2018). In a pooled retrospective cohort of 1,626 patients, SPOP mutant tumors were associated with improved metastasis (MET)-free survival in patients who received androgen deprivation therapy (ADT) after radical prostatectomy, but no difference without ADT (Figure 7A; Table S3). Next, we hypothesized that preferential sensitivity to ADT would manifest with the relative depletion of SPOP mutant tumors in castrate-resistant PCa compared to hormone-sensitive disease. Comparing 333 primary prostate adenocarcinomas (The Cancer Genome Atlas Research Network, 2015) and 414 metastatic castrate-resistant PCa (CRPC) samples (Abida et al., 2019), SPOP mutations were significantly lower in CRPC (Figure 7B), suggesting a response to ADT. To rule out an effect of primary versus metastatic disease in this analysis, we compared metastatic hormone-sensitive PCa (mHSPC) to metastatic hormone-resistant PCa (mCRPC) within the same cohort (Abida et al., 2017), and again observed significant underrepresentation of SPOP mutation in mCRPC, consistent with the sensitivity of SPOP mutant metastatic PCas to ADT (Figure 7C). Finally, we examined the response of patients with mCRPC patients to AR-targeted therapy (abiraterone and enzalutamide) (Abida et al., 2019). SPOP mutation was associated with a longer time on treatment in these patients, suggesting that AR-targeted therapies may be a more durable treatment option in SPOP mutant PCa than in other subtypes (Figure 7D). Overall, all of these results together suggest a favorable outcome of SPOP mutant PCa to AR-targeted therapy across multiple clinical scenarios.
Figure 7. Sensitivity of SPOP mutant PCa to therapies targeting AR signaling.
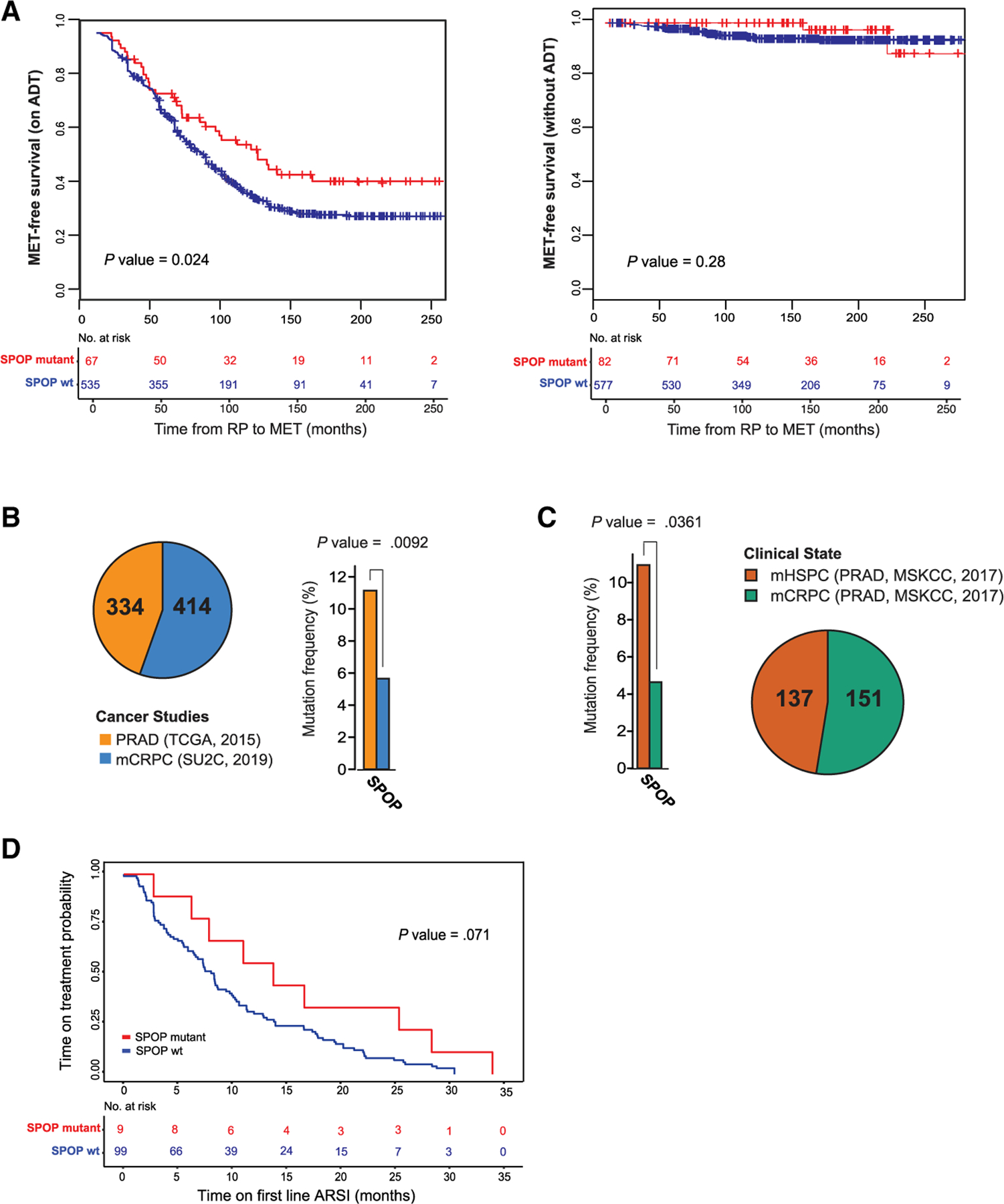
(A) Significant clinical outcome difference between SPOP mutant and WT PCa via Kaplan-Meier analysis of metastasis (MET)-free survival for patients on androgen deprivation therapy (ADT) (left) or without receiving ADT (right). Clinical data are in Table S3.
(B) The occurrence of SPOP mutation in primary PCa (The Cancer Genome Atlas Research Network, 2015) and metastatic (mCRPC) samples (Abida et al., 2019).
(C) When metastatic prostate tumors (Abida et al., 2017) are compared, the hormone-sensitive ones (mHSPC) have a higher frequency (p < 0.0361) of SPOP mutation than the resistant (mCRPC) ones.
(D) Kaplan-Meier analysis of SPOP WT and SPOPmut mCRPC tumors (Abida et al., 2019) showing time on treatment with first-line androgen signaling inhibitors (ARSIs).
DISCUSSION
The AR is the central determinant of prostate tissue identity and differentiation (Cunha et al., 2004), controlling normal, growth-suppressive prostate-specific gene expression (Schiewer et al., 2012). However, it is also a central driver of prostate tumorigenesis, becoming ‘‘hijacked’’ to drive oncogenic transcription (Schiewer et al., 2012). Importantly, it also remains the key therapeutic target for PCa, even in advanced CRPC (Watson et al., 2015). However, how AR function is altered to convert it to an oncogenic factor and at what steps in the process of tumorigenesis this occurs, remain poorly defined. Here, we leverage genetically normal, physiologically relevant prostate organoids to define the AR-directed chromatin landscape and transcriptional program and examine the impact of a single early event in the natural history of PCa.
It has been long recognized that AR is required for the development and function of the normal prostate (Heinlein and Chang, 2004). However, how AR reshapes the landscape of chromatin to execute its transcriptional program, particularly in genetically normal cells, remains unclear. Here, using genetically normal, physiologically relevant prostate organoids, we defined the genome-wide effect of androgens on chromatin accessibility, H3K4me2, AR, and FOXA1 binding and subsequent changes in the transcriptome. These data provide insight to transcriptional dynamics of normal AR function, and demonstrate that these genetically pliable models can recapitulate the epigenomic and transcriptomic changes of mouse and human tissue, credentialing a valuable tool to investigate AR activity.
Missense mutations in SPOP are the most common mutations in primary prostate adenocarcinoma, occur early in the natural history of the disease, and affect multiple substrate proteins associated with AR-mediated transcriptional regulation (Blattner et al., 2017; Bouchard et al., 2018; Geng et al., 2013, 2014; Theurillat et al., 2014). Here, we use SPOP mutation, in otherwise normal prostate organoids, as a model to examine changes in the accessibility of regulatory elements. SPOP mutation was sufficient to reprogram the AR cistrome to one with a resemblance to human PCa. Interestingly, one other study reported different effects on the AR cistrome in response to SPOP mutation introduced into established cell lines with multiple alterations (Copeland et al., 2019), with expansion of the AR cistrome toward both normal and tumorigenic sites. The more profound shift we report here may reflect the value of genetically normal models in studying initiation events, and suggest that oncogenic reprogramming of the AR transcriptional program may be a very early event in the natural history of PCa, at least in selected subtypes.
FOXA1 is a key pioneering factor for AR and HOXB13 TFs, facilitating binding to PCa-specific AR bound sites. Furthermore, FOXA1 and HOXB13 overexpression transforms AR cistrome from normal, immortalized cell lines into an oncogenic one (Copeland et al., 2019; Pomerantz et al., 2015). Our study suggests a significant role for FOXA1 in SPOP mutant prostate organoids, consistent with observations in human tumors. However, the specific molecular mechanisms responsible still remain unclear; there are multiple SPOP substrates deregulated by the mutations observed in PCa, and systematic study will be necessary to determine the specific combination of substrates that are critical for this observation.
The data here suggest that oncogenic reprogramming of AR-driven transcription is associated with the oncogenic function of SPOP mutation in PCa. Consistent with this, we also find that functionally, SPOP mutant model systems show some dependence on this pathway; however, there are limitations to our mouse models, as they may not fully reflect the phenotypes of SPOP mutant human tumors. In clinical data, SPOP mutant human PCas show increased sensitivity to androgen-targeted therapy. Data from multiple clinical scenarios support this hypothesis (Figure 7): adjuvant or salvage ADT after prostatectomy, hormone-sensitive metastatic disease, and CRPC, consistent with reports of favorable outcomes for SPOP mutant cancers with therapies targeting AR in both CRPC (Boysen et al., 2018) and hormone-sensitive metastatic disease (Swami et al., 2020). However, credentialing SPOP mutation as a clinically actionable predictive biomarker for AR-directed therapies will require clinical trials designed for this purpose, likely in each specific clinical scenario. Consistent with this, SPOP mutation is a truncal alteration in human PCas that show exceptionally robust response to neoadjuvant androgen-targeted therapy (Tewari et al., 2021). These clinical data suggest that the AR-dependent effects of SPOP mutation are not unique to a single clinical scenario, but may extend to multiple stages of PCa, broadening the potential impact.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher E. Barbieri (chb9074@med.cornell.edu).
Materials availability
There are no restrictions on material availability. All GEM and organoid lines produced in this study will be made available upon request.
Data and code availability
The described RNA-seq, ATAC-seq, and ChIP-seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GEO: GSE145333 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145333).
The code is available from https://github.com/Ivana-G-Sh.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Transgenic mouse model
Weill Cornell Medicine (WCM) Institutional Care and Use Committee approved all the mouse studies under protocol 2015–0022. Transgenic mice had prostate-specific expression of human SPOP-F133V in Rosa26 locus (PbCre;R26F133V/WT) with heterozygous or homozygous Pten deletion (PbCre;R26F133V/WT;PtenL/+ or PbCre;R26F133V/WT;PtenL/L) (Blattner et al., 2017). All described mice were in a C57BL/6 background. TransNetYX performed mouse genotyping by using published primers (Augello et al., 2019; Blattner et al., 2017). Mice (males) were sacrificed at 6 weeks.
Murine pathology review
Whole murine prostates were processed as previously described (Augello et al., 2019). Staining was done at The Translational Research Program (TRP) of the Department of Pathology and Laboratory Medicine at WCM. Anti-Ki67 was used for the IHC staining.
Murine organoid line generation and CRISPR experiments
Prostate from the PbCre4 negative; Rosa26F133V mice were harvested at 1–2 months of age and processed and grown as 3D Matrigel culture as previously described (Drost et al., 2016) with the exception that the organoid media contained 5 ng/mL EGF. Lines were generated by infection with Adeno-RFP (Control cells) or Adeno-Cre (SPOPmut cells) viruses (Augello et al., 2019). The expression of the human SPOP-F133V transgene was confirmed by immunoblot. To knockout Ar expression, the cells were infected with lentiviruses containing sgNT or sgAr in pLentiCRISPRv2 plasmids (a kind gift from Dong Zhao, Yu Chen’s lab, MSK). Plasmid sequences were verified by Sanger sequencing (Genewiz). Polyclonal populations resistant to puromycin were subjected to immunoblot to verify AR protein expression.
Transient murine organoid models
Transient knockdown of Foxa1 was done by transfecting mouse organoid cells grown on the surface of collagen I coated plates with siRNA (SMARTpool: siGENOME Foxa1 siRNA). For the control, we used the same concentration of scrambled siRNA (siGENOME Non-Targeting Control siRNA Pool). Transfection was done with Lipofectamine RNAiMAX according to the manufacturer’s instructions (Thermo Fisher Scientific). After four hours media was replaced and cells were lifted and plated into Matrigel plugs. Bicalutamide treatment and cell viability assay were performed as described below.
3D organoid growth assay
Five thousand cells were seeded in 10 μL of matrigel plug (Corning Matrigel Basement Membrane Matrix, Growth Factor Reduced) per well of a 96-well plate (Corning 96 well assay plate, flat bottom clear, black polystyrene plate) in 100 μL of mouse organoid media (Blattner et al., 2017) without EGF and with 0.01 nM DHT. Cells were allowed to form organoids for four days before the bicalutamide treatment. Drug treated cells were assayed for cell viability after four days with CellTiter-Glo 3D assay (Promega), as per manufacturer’s instructions. The experiment was repeated for a total of four biological repeats (technical triplicate for each treatment). Luminescence readings (BioTek Synergy Neo microplate reader with Gen5 software) were analyzed with GraphPad Prism 8.2.1 according to the instructions for the analysis of dose-response data.
METHOD DETAILS
Immunofluorescence
Organoids were resuspended Cell Recovery Solution (Corning) to melt the matrigel without disrupting the 3D cellular organization. Organoids were pelleted and embedded into fibrinogen and thrombin clots. These were transferred into embedding cassettes and fixed in 10% formalin. The paraffin sections were prepared at The Translational Research Program of the Department of Pathology and Laboratory Medicine at WCM. Immunofluorescent staining was performed as described previously (Augello et al., 2019) by using anti-Ck5 (BioLegend), anti-Ck8 (Abcam), and anti-AR primary antibodies (Abcam).
Immunoblot
Whole-cell protein lysates were prepared after digestion of matrigel with TrypLE Express Enzyme (1X), phenol red (GIBCO). Pelleted cells were washed in PBS and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Proteins were quantified by BCA assay and separated on 4%–15% Protein Gels. SPOP protein was probed by using in-house made rabbit anti-SPOP (1:1000); for AR and FOXA1, we used Abcam rabbit antibodies. For loading control, we used anti-vinculin or anti-H3.
RNA-seq
Murine organoids were grown for 6 days in media containing 1 nM DHT and then for 24 hr without DHT and EGF. Next, cells were stimulated with 10 nM DHT without EGF for 24 hr and harvested for RNA-seq in biological quadruplicates. Total RNA was extracted, and DNaseI treated by Maxwell 16 LEV simplyRNA Cells Kit and Maxwell nucleic-acid extraction instrument (Promega). Nanodrop quantified RNA was checked by Bioanalyzer RNA 6000 Nano Kit (Agilent Technologies). Samples with RNA integrity number > 10 were used for library preparation (TruSeq Stranded mRNA Library Preparation for Poly-A selection and Stranded RNA-Seq) at the WCM Genomics Core. Libraries were sequenced twice on NextSeq500 (Illumina), High-output mode to generate 75 bp reads at WCM Genomics Core.
ATAC-seq
Control and SPOPmut organoids were grown and treated as described for the RNA-seq experiment. The experiment was performed in a biological quadruplicate. The single-cell suspension was obtained after digesting organoids in TryPLE. ATAC-seq was performed on 100,00 nuclei as previously described (Grbesa et al., 2017). Briefly, nuclei were isolated in buffer (10 mM Tris-Cl pH7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween, 0.5% NP-40, 0.01% digitonin and 1× protease inhibitors), incubated with TDE1 transposase (Illumina) for 30 min at 37°C. ATAC-seq libraries were purified on-column (Zymo) and PCR amplified (5 cycles) with Ultra II master mix (NEB). The required additional number of amplification cycles was determined by qPCR. Generated DNA fragments were quantified with Qubit dsDNA HS Assay Kit. Primers that were used for quality checks of the libraries and for assessing mitochondrial DNA contamination are listed in Table S2. DNA fragment sizes were checked with High Sensitivity DNA Kit on a Bioanalyzer 2100 system (Agilent Technologies). Libraries were pooled at equimolar concentration and submitted to WCM Genomics core, where they were paired-end sequenced on HiSeq 4000 system (Illumina) with 50-bp reads. Raw FASTQ files were pre-processed at WCM Genomics core.
ChIP-seq experiments
Control and SPOPmut organoids were grown and treated in biological triplicates, as described in the RNA-seq section. For H3K4me2 ChIP single-cell suspension was crosslinked with 1% methanol-free formaldehyde at 37°C. For AR and FOXA1 ChIP organoids were double fixed (Singh et al., 2018) with 2 mM Di(N-succinimidyl) glutarate and then 1% formaldehyde. Crosslinking was quenched with 125 mM glycine. The crosslinked organoid pellets were snap-frozen and stored at −80°C until use. Samples were thawed on ice, lysed in 1% SDS containing buffer supplemented with 1× protease and phosphatase inhibitors, and sonicated for 4 3 10 cycles (30 s on, 30 s off) in temperature controlled Bioruptor 300 (Diagenode). Debris was removed by centrifuging at 14,000 rpm at 4°C. One percent of the supernatant was saved as input, and the rest was added to antibody-coupled Dynabeads Protein A and incubated overnight rocking at 4°C. Used antibodies were: ChIP-grade anti-H3K4me2, anti-AR PG-21 (Millipore) and anti-FOXA1 ChIP-grade (Abcam). Chromatin was washed on ice with 2× each standard wash buffers (Low-Salt, High-Salt, and LiCl) and finally with TE. Decrosslinking of input and immunoprecipitated chromatin was performed in buffer containing 1% SDS, 0.3M NaCl, and 0.2 mg/mL Proteinase K for 16 hr at 65°C. After 2 hr incubation with RNase A decrosslinked material was purified with 2× AMPure XP beads and eluted in 30 μL of 10 mM Tris-Cl, pH 8. The DNA concentration was measured by Qubit dsDNA HS Assay Kit. Individual ChIP samples were verified by qPCR (primers are listed in Table S2). Libraries were prepared by NEBNext Ultra II DNA Library Prep by using NEBNext Multiplex Oligos for Illumina. Library size and presence of adapters was verified by Bioanalyzer HS DNA chips. Libraries passing the quality control were pooled and sequenced at WCM Genomics core for 50 cycles on HiSeq4000 to generate single-end 50-bp reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq analysis
Obtained FASTQ files were aligned to the mm10 genome by using STAR v2.4.0j (Dobin et al., 2013). The reads were counted with HTSeq (Anders et al., 2015). Differential gene expression was obtained in R with DESeq2 v1.20.0 package (Anders and Huber, 2010). Gene set enrichment analysis (Subramanian et al., 2005) was run in pre-ranked mode to identify enriched signatures in the Molecular Signature Database (MSigDB).
Bioinformatic processing of ATAC-seq data
Quality control checks on raw sequence data were done with FASTQC v.0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Paired-end reads were aligned to the mm10 genome using Bowtie2 v.2.3.4.1 (Langmead and Salzberg, 2012) in a very sensitive mode. BAM files were sorted and indexed with SAMtools v.1.8 (Li et al., 2009). The percentage of mitochondrial reads (Table S1) was calculated with SAMtools using a custom script. All non-unique reads and mitochondrial reads were removed before peak calling. Bigwig files were generated with deepTools v.3.3.1 (Ramírez et al., 2016) bamCoverage tools from reads with min. mapping quality of 10 and using RPGC normalization. Generated bigwig files were visualized with UCSC Genome Browser. Coverage at TSS of RefSeq genes was done with deepTools computeMatrix and plotHeatmap tools. The fraction of reads in peaks (FRiP) score was calculated by custom script by using BEDtools2 v.2.27 (Quinlan and Hall, 2010) and reported in Table S1. Data quality was confirmed using Encode guidelines. Peaks were called using Genrich as explained on the Harvard Bioinformatics site (https://informatics.fas.harvard.edu/atac-seq-guidelines.html). As an additional quality control metric, we plotted insert size distribution which confirmed nucleosome-free, mono-and di-nucleosomal distribution of the reads. Consensus peaks for each cell type and condition from 4 biological replicas were generated with DiffBind v.3.10 package (Ross-Innes et al., 2012). Differential peak identification (in EdgeR (McCarthy et al., 2012), FDR < 0.01), MA plots, and volcano plots were done in the R software environment v.3.6.0 (The R Foundation) by using DiffBind.
Assignment of ATAC-seq peaks to genes – One Mb regions around the transcription start sites of selected genes were retrieved through the UCSC Table Browser program. Then they were intersected with the bed file containing ATAC-seq peak regions.
Annotation of Genomic Regions – Cis-regulatory element analysis of consensus and differential ATAC-seq peaks was performed with the annotatePeaks.pl of Homer v.4.10 software (Heinz et al., 2010). The graphs were created in R v3.6.0 with a ggplot2 v3.2.1 package.
Integration with RNA-seq data –was done via Cistrome-GO webserver (Li et al., 2019).
Bioinformatic processing of ChIP-seq data
Generated FASTQ files were validated for quality using FastQC v.0.11.8 software and processed with ENCODE ChIP-seq pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2). All reads were aligned to mouse genome mm10. Briefly, this pipeline generated an HTML-quality report from which we inferred the overall quality of our ChIP-seq. Only the samples passing the ENCODE-specified NSC, RCS, FRiP, and PBC metrics (Landt et al., 2012) (https://www.encodeproject.org/data-standards/chip-seq/) were included in further data analysis (Table S1). All the peaks were called with MACS2 (Zhang et al., 2008) at the p value threshold of 0.05. Differential peak analysis was performed by DiffBind in EdgeR mode with 0.01 FDR cutoff.
Motif enrichment analysis
De novo and known motif analysis of ATAC-seq and ChIP-seq consensus or differential peaks was done by findMotifsGenome.pl program within Homer v.4.10 software. For the analysis of H3K4me2 peaks, we used -size given option, while for the rest of the data, we analyzed 200 bp region around the peak summit. To determine motif enrichment between two datasets with similar peak numbers, one served as a background (-bg flag) for the other. For the enhancer-specific motif scanning, peaks localized to promoter and 5ʹUTR were removed before the analysis. To create motif density histograms the analyzed peaks were centered on the specific motif, and the analysis was run by findMotifsGenome.pl in a histogram mode (-hist with 5 bp bin size) and visualized with GraphPad Prism v8.2.1. Motif analysis of human primary prostate tumors (Stelloo et al., 2018) was done as previously described (Augello et al., 2019) with the only difference that the AR peaks were binned according to the presence or absence of SPOP mutation. Statistical analysis was performed in base R version 3.6.0 (2019–04-26). All the other plots were generated in R with ggplot2 v3.2.1 package.
Copy number analysis
The copy number alterations of PRAD were downloaded from TCGA portal (https://portal.gdc.cancer.gov/) via gdc-client tool. Fraction of altered cases within SPOP and FOXA1 mutant subclasses were calculated respectively. Segments with log2-ratio > 0.3 were defined as genomic amplifications, and log2-ratio < −0.3 were defined as genomic deletions.
Clinical data analysis
We have developed a novel gene expression signature, classifier SCaPT (Subclass Predictor Based on Transcriptional Data), and decision tree to predict the SPOP mutant subclass from RNA gene expression data (Liu et al., 2018). We then classified the SPOP mutant subclass in a Decipher retrospective cohort of 1,626 patients (Table S3) (Liu et al., 2018) and evaluated the associations between SPOP mutant status and patient outcomes of metastasis mortality with and without ADT treatment based on Kaplan-Meier (KM) analysis.
Prevalence of SPOP mutation was analyzed on cBioPortal v3.2.0 platform (Cerami et al., 2012) by using the following datasets (accessed on January 2020): Prostate Adenocarcinoma (TCGA, Cell 2015) (The Cancer Genome Atlas Research Network, 2015), Metastatic Prostate Adenocarcinoma (SU2C/PCF Dream Team) (Abida et al., 2019) and Prostate Cancer (MSKCC) (Abida et al., 2017). For time on ADT analysis, patient’s data from cBioPortal and GitHub (https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_2019) were combined in R v3.6.0. KM analysis and plots were done in R using packages survival 2.44–1.1 and survminer v0.4.6.
Illustrations
Figures were prepared directly in Adobe illustrator or imported into from ggplot2, Deeptools or GraphPad Prism. Graphical abstract was made by BioRender.com and Mind the Graph (https://mindthegraph.com).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Ki67 | Abcam | Cat# ab16667, RRID:AB_302459 |
| Keratin 5 | BioLegend | Cat# 905903, RRID:AB_2721742 |
| Goat anti-Chicken IgY (H+L) Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11039, RRID:AB_2534096 |
| Cytokeratin 8 antibody [EP1628Y] | Abcam | Cat# ab53280, RRID:AB_869901 |
| Alexa Fluor® 546 goat anti-rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A-11035, RRID:AB_2534093 |
| Androgen Receptor antibody [ER179(2)] | Abcam | Cat# ab108341, RRID:AB_10865716 |
| FOXA1 - ChIP Grade | Abcam | Cat# ab23738, RRID:AB_2104842 |
| Vinculin antibody [EPR8185] | Abcam | Cat# ab129002, RRID:AB_11144129 |
| H3 | Abcam | Cat# ab1791, RRID:AB_302613 |
| Histone H3 (di methyl K4) antibody - ChIP Grade | Abcam | Cat# ab7766, RRID:AB_2560996 |
| Androgen Receptor Antibody, PG-21 (lot 3090042) | Millipore | Cat# 06–680, RRID:AB_310214 |
| Anti-SPOP, rabbit, polyclonal | This paper | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Codon-improved Cre (iCre) and RFP Adenovirus | Vector Laboratories | 1774 |
| RFP Adenovirus | Vector Laboratories | 1660 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Bicalutamide | Selleck Chemicals | S1190–50mg |
| EGF | Thermo Fisher Scientific | PMG8043 |
| Collagen I | Thermo Fisher Scientific | A1048301 |
| Matrigel | VWR | 47743–722 (BD 356231) |
| DHT | Sigma-Aldrich | D-073–1ML |
| Cell Recovery Solution | Corning | 354253 |
| Fibrinogen | Millipore Sigma | F3879–100MG |
| Thrombin | Millipore Sigma | T4648–1KU |
| RIPA buffer | Thermo Fisher Scientific | PI-89901 |
| Halt Protease and Phosphatase Inhibitor Cocktails, Thermo Scientific, Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | 78444 |
| 4–15% Mini-PROTEAN TGX Stain-Free Protein Gels | BioRad | 4568084 |
| Tween | Sigma Millipore | 11332465001 |
| NP-40 | Sigma Millipore | 11332473001 |
| Digitonin | Promega | G9441 |
| Thermo Scientific Pierce Formaldehyde Ampules, Methanol-free - 10 3 1mL | Thermo Scientific Pierce | 28906 |
| Di(N-succinimidyl) glutarate | Sigma-Aldrich | 80424–5MG-F |
| Dynabeads Protein A | Thermo Fisher Scientific | 10001D |
| Proteinase K | Thermo Fisher Scientific | AM2548 |
| RNaseA | Thermo Fisher Scientific | EN0531 |
| AMPure XP beads | Beckman Coulter | A63881 |
| Tagment DNA TDE1 Enzyme and Buffer | Illumina | 20034197 |
| NEBNext Ultra II Q5 Master Mix | New England Biolabs | M0544L |
|
| ||
| Critical commercial assays | ||
|
| ||
| CellTiter-Glo 3D assay | Promega | G9682 |
| BCA Assay | Thermo Fisher Scientific | 23227 |
| Maxwell 16 LEV simplyRNA Cells Kit | Promega | AS1270 |
| Agilent RNA 6000 Nano Kit | Agilent Technologies | 5067–1511 |
| TruSeq Stranded mRNA Library Prep | Illumina | 20020595 |
| High Sensitivity DNA Kit | Agilent Technologies | 5067–4626 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q3285 |
| NEBNext Ultra II DNA Library Prep | New England Biolabs | E7645S |
| DNA Clean & Concentrator-5 w/ Zymo-Spin IC Columns (Capped) | Zymo Research | D4013 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq | GEO | GEO: GSE149868 |
| ChIP-seq | GEO | GEO: GSE145196 |
| ATAC-seq | GEO | GEO: GSE145332 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| C57BL/6 murine prostate organoids | This paper | N/A |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse C57BL/6 PbCre;R26F133V/WT | Blattner et al., 2017 | N/A |
| Mouse C57BL/6 PbCre;R26F133V/WT;PtenL/+ | Blattner et al., 2017 | N/A |
| Mouse C57BL/6 PbCre;R26F133V/WT;PtenL/L | Blattner et al., 2017 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S2 for a detailed primer list | N/A | N/A |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) | New England Biolabs (NEB) | E7335S |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 2) | NEB | E7500S |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 3) | NEB | E7710S |
| siGENOME Foxa1 siRNA | Horizon Discovery Biosciences | M-046238–01-0005 |
| siGENOME Non-Targeting Control siRNA Pool | Horizon Discovery Biosciences | D-001206–13-05 |
|
| ||
| Recombinant DNA | ||
|
| ||
| lentiCRISPRv2 puro | Addgene | RRID:Addgene_98290 |
| sgAr- lentiCRISPRv2 puro | This paper | Gift from dr. Yu Chen’s lab |
| sgNT- lentiCRISPRv2 puro | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 8.2.1 | GraphPad Software | N/A |
| STAR v2.4.0j | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| DESeq2 v1.20.0 | Anders and Huber, 2010 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| FASTQC v.0.11.8 | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Bowtie2 v.2.3.4.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| SAMtools v.1.8 | Li et al., 2009 | http://www.htslib.org/ |
| deepTools v.3.3.1 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| BEDtools2 v.2.27 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Genrich | N/A | https://github.com/jsh58/Genrich |
| DiffBind v.3.10 | Ross-Innes et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| EdgeR | McCarthy et al., 2012 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Homer v.4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| ggplot2 v3.2.1 | N/A | https://ggplot2.tidyverse.org/ |
| Cistrome-GO webserver | Li et al., 2019 | http://go.cistrome.org/ |
| ENCODE ChIP-seq pipeline | N/A | https://github.com/ENCODE-DCC/chip-seq-pipeline2 |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| survival 2.44–1.1 | N/A | https://github.com/therneau/survival |
| survminer v0.4.6 | N/A | https://github.com/kassambara/survminer |
Highlights.
Epigenomic and transcriptional response to androgen in normal prostate organoids
AR cistrome and chromatin landscape reprogrammed in SPOP mutant prostate organoids
In vivo models of SPOP mutant prostate cancer respond to modulation of AR signaling
SPOP mutation is associated with improved response to anti-AR therapies
ACKNOWLEDGMENTS
We are indebted to the PCa patients and families who contributed to this research. We thank Dr. Dawid Nowak (Weill Cornell Medicine [WCM]) and Dr. Jonathan E. Shoag (WCM) for helpful discussion. We thank Dr. Daphne Campigli Di Giammartino (WCM) for helpful advice regarding the ChIP-seq experiments. We thank Dr. Mark Rubin for mentorship and guidance in developing these projects and reagents. We thank the WCM Genomics Core Facility, the Biospecimen and Pathology Core of WCM SPORE in Prostate Cancer, and the Memorial Sloan Kettering Cancer Center cBioPortal. We are grateful to Drs. Eric Klein, Bruce J. Trock, R. Jeffrey Karnes, and Robert B. Den for patient data. This work was funded by the US NCI (WCM SPORE in Prostate Cancer, P50CA211024–01, R37CA215040, and R01CA233650, to C.E.B.), the Damon Runyon Cancer Research Foundation, the MetLife Foundation Family Clinical Investigator Award (to C.E.B.), and the Prostate Cancer Foundation (to M.A.A. and D.L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109625.
DECLARATION OF INTERESTS
C.E.B. is co-inventor on a patent issued to Weill Medical College of Cornell University on SPOP mutations in PCa. E.D. is an employee of GenomeDx.
REFERENCES
- Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, Danila D, Rathkopf D, Morris M, Slovin S., et al. (2017). Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol 2017, PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al. (2019). Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 116, 11428–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams EJ, Karthaus WR, Hoover E, Liu DL, Gruet A, Zhang ZD, Cho H, DiLoreto R, Chhangawala S, Liu Y., et al. (2019). FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 571, 408–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Wang C, Deng Y, Yu L, and Huang H. (2014). Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 6, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S, Blattner M, Doane AS, Wong EWP, Chen Y., et al. (2019). CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 35, 817–819. [DOI] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M., et al. (2013). Punctuated evolution of prostate cancer genomes. Cell 153, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N., et al. (2012). Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet 44, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D, Nataraj S, Deonarine LD, Augello MA, Sailer V., et al. (2017). SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 31, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X., et al. (2018). Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 72, 19–36.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, Nataraj S, Huang D, Marotz C, Xu L., et al. (2015). SPOP mutation leads to genomic instability in prostate cancer. eLife 4, e09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen G, Rodrigues DN, Rescigno P, Seed G, Dolling D, Riisnaes R, Crespo M, Zafeiriou Z, Sumanasuriya S, Bianchini D., et al. (2018). SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin. Cancer Res 24, 5585–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H., et al. (2011). Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19, 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E., et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, and Zaret KS (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289. [DOI] [PubMed] [Google Scholar]
- Copeland BT, Du J, Pal SK, and Jones JO (2019). Factors that influence the androgen receptor cistrome in benign and malignant prostate cells. Mol. Oncol 13, 2616–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, Ricke W, Thomson A, Marker PC, Risbridger G, Hayward SW, Wang YZ, Donjacour AA, and Kurita T. (2004). Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J. Steroid Biochem. Mol. Biol 92, 221–236. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, and Clevers H. (2016). Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc 11, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y, Jin R, Gupta A, Rennie PS, and Matusik RJ (2003). The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol 17, 1484–1507. [DOI] [PubMed] [Google Scholar]
- Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C., et al. (2013). Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 110, 6997–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M., et al. (2014). Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 74, 5631–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbesa I, Tannenbaum M, Sarusi-Portuguez A, Schwartz M, and Hakim O. (2017). Mapping Genome-wide Accessible Chromatin in Primary Human T Lymphocytes by ATAC-Seq. J. Vis. Exp (129), 56313. [DOI] [PMC free article] [PubMed]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M., et al. (2010). Nucleosome dynamics define transcriptional enhancers. Nat. Genet 42, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinlein CA, and Chang C. (2004). Androgen receptor in prostate cancer. Endocr. Rev 25, 276–308. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N., et al. (2014). Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P., et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wan C, Zheng R, Fan J, Dong X, Meyer CA, and Liu XS (2019). Cistrome-GO: a web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res 47 (W1), W206–W211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, Wang H, Yu Y, Yang C, Gao X., et al. (2020). A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature 580, 93–99. [DOI] [PubMed] [Google Scholar]
- Liu X, Chen L, Huang H, Lv JM, Chen M, Qu FJ, Pan XW, Li L, Yin L, Cui XG, et al. (2017). High expression of PDLIM5 facilitates cell tumorigenesis and migration by maintaining AMPK activation in prostate cancer. Onco-target 8, 98117–98134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Takhar M, Alshalalfa M, Erho N, Shoag J, Jenkins RB, Karnes RJ, Ross AE, Schaeffer EM, Rubin MA, et al. (2018). Impact of the SPOP Mutant Subtype on the Interpretation of Clinical Parameters in Prostate Cancer. JCO Precis. Oncol 2018, PO.18.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, and Wu H. (2011). Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolia A, Cieslik M, Chu SC, Xiao LB, Ouchi T, Zhang YP, Wang XJ, Vats P, Cao XH, Pitchiaya S., et al. (2019). Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. (2015). The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet 47, 1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiewer MJ, Augello MA, and Knudsen KE (2012). The AR dependent cell cycle: mechanisms and cancer relevance. Mol. Cell. Endocrinol 352, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG, and Neal DE (2013). The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23, 35–47. [DOI] [PubMed] [Google Scholar]
- Shoag J, Liu D, Blattner M, Sboner A, Park K, Deonarine L, Robinson BD, Mosquera JM, Chen Y, Rubin MA, and Barbieri CE (2018). SPOP mutation drives prostate neoplasia without stabilizing oncogenic transcription factor ERG. J. Clin. Invest 128, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AA, Schuurman K, Nevedomskaya E, Stelloo S, Linder S, Droog M, Kim Y, Sanders J, van der Poel H, Bergman AM, et al. (2018). Optimized ChIP-seq method facilitates transcription factor profiling in human tumors. Life Sci. Alliance 2, e201800115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelloo S, Nevedomskaya E, Kim Y, Schuurman K, Valle-Encinas E, Lobo J, Krijgsman O, Peeper DS, Chang SL, Feng FY, et al. (2018). Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun 9, 4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swami U, Isaacsson Velho P, Nussenzveig R, Chipman J, Sacristan Santos V, Erickson S, Dharmaraj D, Alva AS, Vaishampayan UN, Esther J., et al. (2020). Association of SPOP Mutations with Outcomes in Men with De Novo Metastatic Castration-sensitive Prostate Cancer. Eur. Urol 78, 652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, and Balk SP (2004). Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J. Cell. Biochem 91, 483–490. [DOI] [PubMed] [Google Scholar]
- Tewari AK, Cheung ATM, Crowdis J, Conway JR, Camp SY, Wankowicz SA, Livitz DG, Park J, Lis RT, Bosma-Moody A., et al. (2021). Molecular features of exceptional response to neoadjuvant anti-androgen therapy in high-risk localized prostate cancer. Cell Rep 36, Published online September 7, 2021. 10.1016/j.celrep.2021.109665. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, et al. (2014). Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 346, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. (2009). Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 36, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The described RNA-seq, ATAC-seq, and ChIP-seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GEO: GSE145333 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145333).
The code is available from https://github.com/Ivana-G-Sh.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.