

Abstract
Liquid–liquid phase separation (LLPS) has emerged as a central paradigm for understanding how membraneless organelles compartmentalize diverse cellular activities in eukaryotes1–3. Here we identify a superfamily of plant guanylate-binding protein (GBP)-like GTPases (GBPLs) that assemble LLPS-driven condensates within the nucleus to protect against infection and autoimmunity. In Arabidopsis thaliana, two members of this family—GBPL1 and GBPL3—undergo phase-transition behaviour to control transcriptional responses as part of an allosteric switch that is triggered by exposure to biotic stress. GBPL1, a pseudo-GTPase, sequesters catalytically active GBPL3 under basal conditions but is displaced by GBPL3 LLPS when it enters the nucleus following immune cues to drive the formation of unique membraneless organelles termed GBPL defence-activated condensates (GDACs) that we visualized by in situ cryo-electron tomography. Within these mesoscale GDAC structures, native GBPL3 directly bound defence-gene promoters and recruited specific transcriptional coactivators of the Mediator complex and RNA polymerase II machinery to massively reprogram host gene expression for disease resistance. Together, our study identifies a GBPL circuit that reinforces the biological importance of phase-separated condensates, in this case, as indispensable players in plant defence.
All living organisms have evolved defence mechanisms to repel or tolerate microbial threats4. Plants account for nearly 80% of the Earth’s biomass5 and are in constant contact with phytopathogens. Being sessile hosts that lack circulating immune cells, plants rely heavily on barrier defence and innate immunity for survival6,7. Surface receptors detect pathogen-associated molecular patterns or microbe-associated molecular patterns as part of PAMP-triggered immunity, whereas intracellular receptors sense pathogen effectors for effector-triggered immunity—both generate downstream signals such as salicylic acid to elicit systemic responses7. In contrast to these compartmentalized functions, little is known about the contribution of membraneless organelles arising from phase separation to plant resistance. This question takes on greater significance in light of recent evidence showing that plants resort to LLPS-driven processes for activities spanning temperature sensing to flowering responses8,9. Uncovering new LLPS-dependent resistance pathways has implications for crop management and food production in the face of climate change10.
IDR-containing GBPLs confer plant defence
We devised an in silico strategy to mine the proteome of the model plant A. thaliana (hereafter, Arabidopsis) for LLPS-driven defence pathways (Extended Data Fig. 1a). Intrinsically disordered region (IDR)-containing proteins (IDPs) were chosen using D2P2.pro criteria, as IDPs can trigger LLPS activities, especially within the nucleus11–16. A 5-tiered pipeline of increasing stringency across 25,498 Arabidopsis open reading frames (ORFs) yielded 304 IDP-encoding genes with nuclear localization signal (NLS) motifs belonging to 80 gene families (Extended Data Fig. 1a, Supplementary Tables 1, 2). AT1G03830 and AT5G46070 were identified as encoding part of a new IDP family sharing similarity with guanylate-binding protein GTPases (GBPs). In mammals, GBPs confer cell-autonomous immunity but lack IDRs17–19. By contrast, about 50% of plant GBPL sequences contain IDRs (Extended Data Fig. 1b–e). Phylogenetic analysis using MrBayes and RAxML algorithms conducted across 100,000,000 iterations found that these IDR-containing GBPLs form a subgroup of ‘complex’ GBPLs that include orthologues in the unicellular green algae, Chlamydomonas reinhardtii (Extended Data Fig. 1b, Supplementary Table 3), from which land plants diverged more than one billion years ago20. Thus, the IDR-containing GBPL superfamily arose early in plant evolution.
To test whether these ancient IDR-containing GBPLs confer plant defence, Arabidopsis transfer DNA (T-DNA) insertional seedlings were identified for AT1G03830 (which we designated GBPL1), AT2G38840 (GBPL2) and AT5G46070 (GBPL3) (Fig. 1a). Plants deficient in GBPL1 (gbpl1–1) or GBPL2 (gbpl2–1) were viable; by contrast, homozygous T-DNA insertions in the GBPL3 coding region were lethal, and GBPL3 mutants generated using CRISPR–Cas9 were sterile at the T1 generation (Fig. 1a, Extended Data Fig. 2a–c). Subsequent isolation of GBPL3 hypomorphic alleles (gbpl3–3, gbpl3–4 and gbpl3–5) enabled infectious phenotypes to be tested (Fig. 1a, b, Extended Data Fig. 2d, g–i). gbpl3 hypomorphs were significantly more susceptible to infection by the bacterial phytopathogens Pseudomonas syringae pv. maculicola (Psm) ES4326 or P. syringae pv. tomato (Pst) DC3000 over multiple doses, whereas gbpl1–1 plants exhibited increased resistance (Fig. 1c, d, Extended Data Fig. 2g). These opposing phenotypes were rescued by genetic complementation (Extended Data Fig. 2f, g) and suggested that GBPL1 may negatively regulate GBPL3-driven immune responses to avoid autoimmunity.
Fig. 1 |. IDR-containing GBPLs are essential for plant defence.
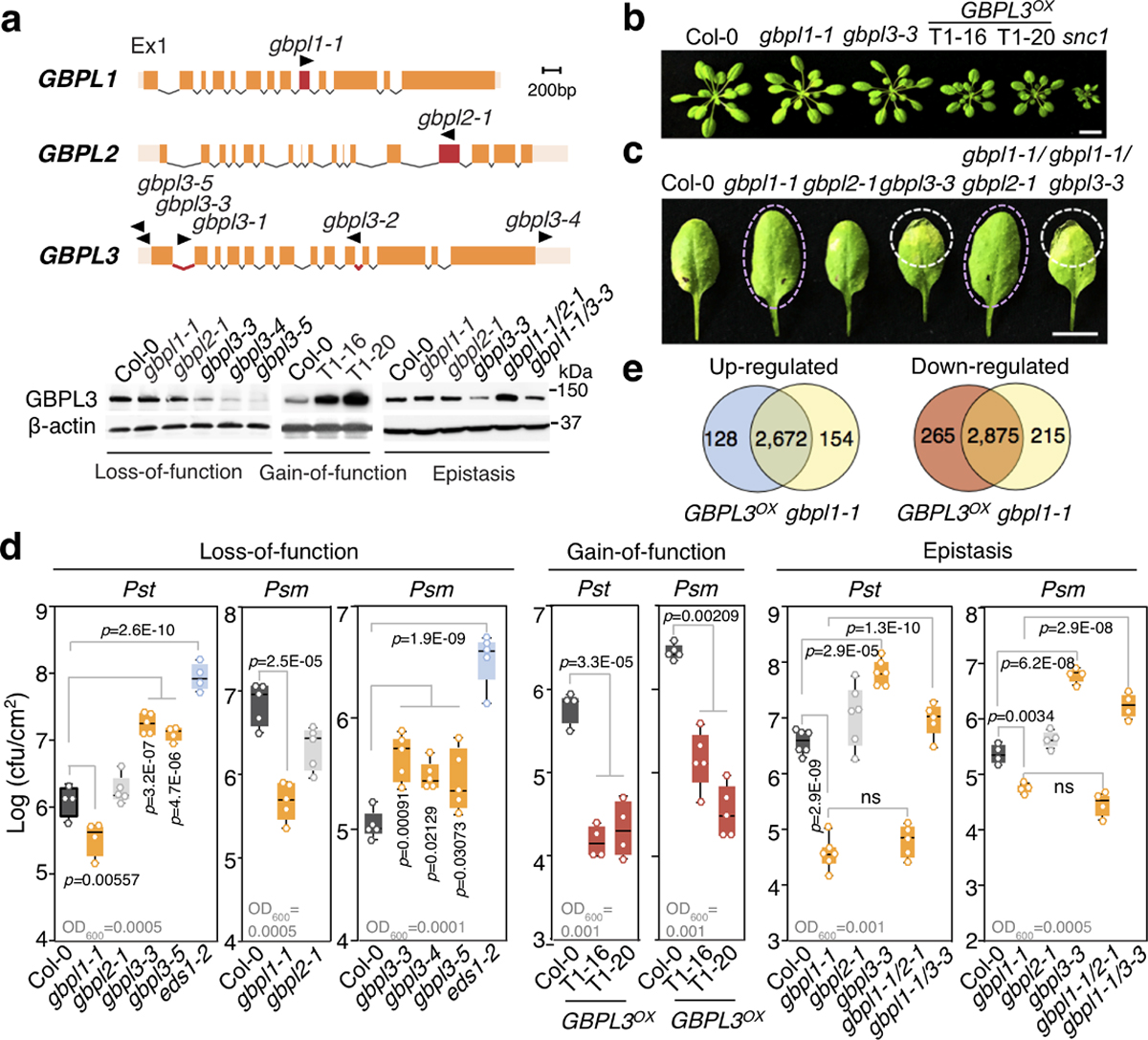
a, Top, structures of Arabidopsis GBPL genes. Positions of T-DNA insertions are in red with triangles indicating transposon direction. Bottom, immunoblot of endogenous GBPL3 levels in T-DNA mutants, overexpression lines (Col-0/pGBPL3::GBPL3 T1–16 and T1–20) and epistatic crosses. Ponceau staining of the large RuBisCO subunit (55 kDa) served as loading control for overexpression lines; β-actin immunoblot served as loading control for the other samples. Ex1, exon 1. b, Growth of five-week-old plants. snc1 mutant, autoimmunity control. Scale bar, 1 cm. c, Representative resistant and susceptible disease phenotypes (dashed circles) of plants infected with Psm ES4326. Scale bar, 1 cm. d, Titre of Psm ES4326 and Pst DC3000 on day 3 of infection. Inoculum density is shown above the x axis (OD600 is optical density at 600 nm). Comparison of mean by one-way ANOVA (Bonferroni post hoc correction). NS, not significant. Centre lines show mean, box edges delineate 25th and 75th percentiles and bars extend to minimum and maximum values. Individual data points represent biologically independent samples. e, Venn diagram of differentially expressed genes in GBPL3OX (T1–20) and gbpl1–1 versus wild type.
Consistent with this model, wild-type Col-0 plants overexpressing GBPL3 (GBPL3OX) cleared Pst, Psm and the oomycetes pathogen Hyaloperonospora arabidopsidis (Hpa) Noco2 much more effectively than did parental Col-0 controls, but had stunted growth indicative of autoimmunity (Fig. 1a, b, d, Extended Data Fig. 2h). Introducing the gbpl3–3 mutation into gbpl1–1 abolished heightened pathogen resistance by 87–90%, rendering gbpl1–1 gbpl3–3 double mutants more susceptible in epistasis assays—here, introduction of gbpl2–1 had no effect (Fig. 1c, d). Thus, the protective phenotype of gbpl1–1 was in fact largely caused by GBPL3 and could be mimicked by GBPL3OX. GBPL3 appears to be the major instigator of plant immunity in this GBPL1–GBPL3 circuit. Its basal activity is kept under control by GBPL1 but is derepressed by activating signals during infection of wild-type plants or constitutive overexpression in GBPL3OX lines.
Subsequent RNA-sequencing (RNA-seq) profiling of GBPL3OX plants showed that the expression of 5,940 genes was significantly altered versus Col-0 controls (false discovery rate < 0.05, fold change ≥ 2) (Fig. 1e, Extended Data Fig. 3a, b, Supplementary Table 4). Ninety-five per cent of induced genes (2,672) and 92% of repressed genes (2,875) overlapped with the gbpl1–1 profile, validating their convergent phenotypes (Fig. 1e, Supplementary Table 5). Defence genes were by far the most enriched in GBPL3OX plants, with many Gene Ontology (GO) functional terms shared with gbpl1–1 (Extended Data Fig. 3b). A similar outcome was observed in hypomorphic gbpl3 mutant plants that, conversely, had severe defects in the induction of well-known defence loci, especially during infection (Extended Data Fig. 3c–h). GBPL3 therefore orchestrates Arabidopsis defence-gene expression.
Defence signals trigger GBPL3 condensates
We tracked GBPL3 localization to understand how it controls transcriptional reprogramming. Live imaging of gbpl3–3/35S::eGFP-GBPL3 plants and subcellular fractionation using a monospecific GBPL3 antibody (Extended Data Fig. 2a) showed that native GBPL3 partitioned between the cell cytosol and nucleus in Col-0 or gbpl1–1 backgrounds; thus, GBPL1 does not interfere with nucleocytoplasmic entry of GBPL3 (Fig. 2a, Extended Data Fig. 4a). Once inside the nucleus, GBPL3 directly bound to GBPL1 and chromatin (Fig. 2a, Extended Data Fig. 4b–d). Limited nuclease digestion and gradient salt elution21,22 detected both GBPL3 and GBPL1 in 150–300 mM NaCl fractions (which contain euchromatin enriched for transcriptionally active genes21); however, only GBPL1 eluted in the 600 mM NaCl fraction (characteristic of tighter-binding proteins, including those on heterochromatin) (Fig. 2a, Extended Data Fig. 4b). Thus GBPL1 may directly sequester GBPL3 or compete with it as a chromatin-binding protein of higher affinity to limit defence-gene transcription and autoimmune development under basal conditions. We tested this possibility in isolated nuclei from gbpl1–1 plants; GBPL3 shifted towards higher euchromatic fractions in the absence of GBPL1, indicating that GBPL1 may act as an allosteric regulator to prevent chromatin accessibility of GBPL3 in the absence of infection (Fig. 2a, Extended Data Fig. 4b–d).
Fig. 2 |. Defence activation triggers a GBPL1–GBPL3 chromatin switch for immune condensates in the nucleus.
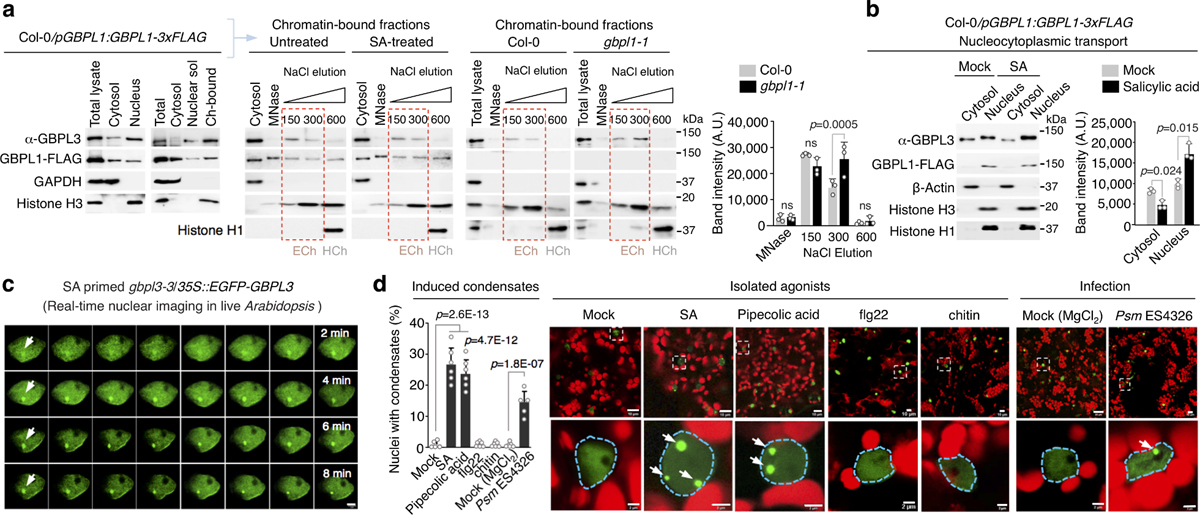
a, Subcellular fractionation of Arabidopsis leaf cells to determine GBPL1 and GBPL3 localization. GAPDH and histone H3 served as controls for the cytosol and the nucleus, respectively. Plants were treated with water or 0.5 mM salicylic acid (SA) for 24 h. Isolated nuclei were treated with micrococcal nuclease (MNase) and fractionated by gradient salt elution (150–600 mM NaCl). Histone H1 served as a marker for the heterochromatin (HCh) fraction. Dashed red boxes indicate euchromatin (ECh) fractions. Ch-bound, chromatin-bound. Right, quantification of GBPL3 immunoblots. Data are mean ± s.d. of three independent experiments. Mixed-effect two-way ANOVA with the restricted maximum likelihood method (Bonferroni’s multiple comparisons test). b, Left, fractionation of Col-0/GBPL1-3×Flag plants treated with water (mock) or 0.5 mM salicylic acid for 24 h. Right, quantification of GBPL3 immunoblots. Data are mean ± s.d. of three independent experiments. Paired two-tailed t-test. AU, arbitrary units. c, Time-lapse imaging of in situ GBPL3 condensate formation (arrows) 5 h after treatment with 0.5 mM salicylic acid. Frame interval, 15 s; scale bar, 2 μm. d, Right, live imaging of eGFP–GBPL3 in 4-week-old plants infiltrated with water, 200 μM salicylic acid, 1 mM pipecolic acid, 2 mM flg22, 100 μg ml−1 chitin, 10 mM MgCl2 or Psm ES4326 in MgCl2 (OD600 = 0.01). Images of parenchymal cells were taken 24 h after treatment. Dashed regions in the top row are enlarged in the bottom row. Scale bars, 10 μm (top row), 2 μm (bottom row). Red, chlorophyll. Left, quantification of nuclei with induced condensates. Data are mean ± s.d., n = 6 biological replicates with 100 nuclei each. One-way ANOVA with Holm–Sidak post hoc test.
This basal suppression by GBPL1 was reversed by immune activation. To uncover how reversal occurred, we monitored GBPL3 in response to the major defence hormone salicylic acid, which is triggered by all three phytopathogens used in this study6,7. Salicylic acid significantly increased the nuclear pool of GBPL3, whereas total GBPL3 did not change (Fig. 2b, Extended Data Fig. 4e); cytosolic GBPL3 levels fell accordingly (Fig. 2b). Neither intrinsic GBPL3 chromatin binding affinity nor the nucleocytoplasmic distribution of GBPL1 was altered by salicylic acid priming (Fig. 2a, b). Nuclear GBPL3 concentrations also increased during natural infection with Psm or Pst, without changing the total amount of GBPL3 (Extended Data Figs. 4f, 5a–c). Immune activation thus probably triggered nucleocytoplasmic import of GBPL3 to override inhibition by GBPL1.
This increase in intranuclear GBPL3 concentration coincided with the appearance of spheroid droplets in the nucleoplasm. Live imaging of leaf cells of plants expressing fluorescently tagged GBPL3 at levels equal to endogenous GBPL3 (Extended Data Fig. 2f) showed that salicylic acid rapidly triggered the formation of nuclear GBPL3 condensates (Fig. 2c). GBPL3 condensates were also induced by pipecolic acid (Pip) and by Psm or Pst (Fig. 2d, Extended Data Fig. 5c and Supplementary Videos 1–4). Notably, the flagellin peptide flg2223 and chitin did not induce GBPL3 condensates, indicating that specific pathogen-associated molecular patterns or signalling hormones trigger this process. GBPL3 condensates also excluded GBPL1 to the periphery, resembling the exclusion of the negative regulator CDK8 from RNA polymerase II (RNAPII) condensates, in which transcriptional pre-initiation or re-initiation takes place24 (Extended Data Fig. 4g). This observation could explain how immune signalling reverses GBPL1 suppression if GBPL3-dependent transcriptional reprogramming and the assembly of transcriptional machinery for host defence occur in these droplets.
GBPL3 condensates were distinct from well-known subnuclear structures (Extended Data Fig. 5d–f). They also formed in different Arabidopsis cell types and developmental stages (guard cells and cotyledons) (Extended Data Fig. 5f, g) as well as in heterologous plant and animal systems, with the closest orthologues from tomato and corn producing similar condensates, albeit of different diameters (Extended Data Fig. 5h, i). GBPL condensates therefore appear as evolutionarily conserved structures across distant plant lineages. We called these structures GBPL defence-activated condensates (GDACs), as they formed in response to defence signals mobilized during biotic stress. GDACs were by far the most conspicuous GBPL3 alteration that we observed with defence priming and could reflect phase-separating behaviour of GBPL3 once it reaches a threshold concentration inside the nucleus2. This threshold is approximately 600 nM, according to quantitative imaging measurements of GBPL3 in GDACs and surrounding nucleoplasm in intact plant nuclei (Extended Data Fig. 5a–c).
GDACs are assembled by LLPS
LLPS and IDR interactions frequently drive the formation of membraneless liquid droplets that control gene transcription14–16. Immunogold-labelled transmission electron microscopy showed that GBPL3 condensates lacked a contiguous lipid membrane (Extended Data Fig. 5j). Moreover, live imaging of Arabidopsis showed that they actively coalesce or fuse into larger droplets. Furthermore, live-leaf nuclear fluorescence recovery after photobleaching (FRAP) assays showed that they internally exchange materials, with GBPL3 FRAP recovery within about 200–250 s (Fig. 3a–c, Supplementary Video 5), similar to FRAP recovery rates for other LLPS transcriptional condensates in humans, mice and worms11–15. Finally, GBPL3 condensates were exquisitely sensitive to 1,6-hexanediol (but not a 1,2,6-hexanetriol control), which dissolves LLPS transcriptional condensates by disrupting hydrophobic interactions in vivo15,25 (Extended Data Fig. 5k). Thus, GDACs fit many of the criteria for LLPS1–3, being membraneless, motile spheroid droplets that exhibit liquid-like behaviours.
Fig. 3 |. Arabidopsis GBPL3 immune condensates are assembled via LLPS.
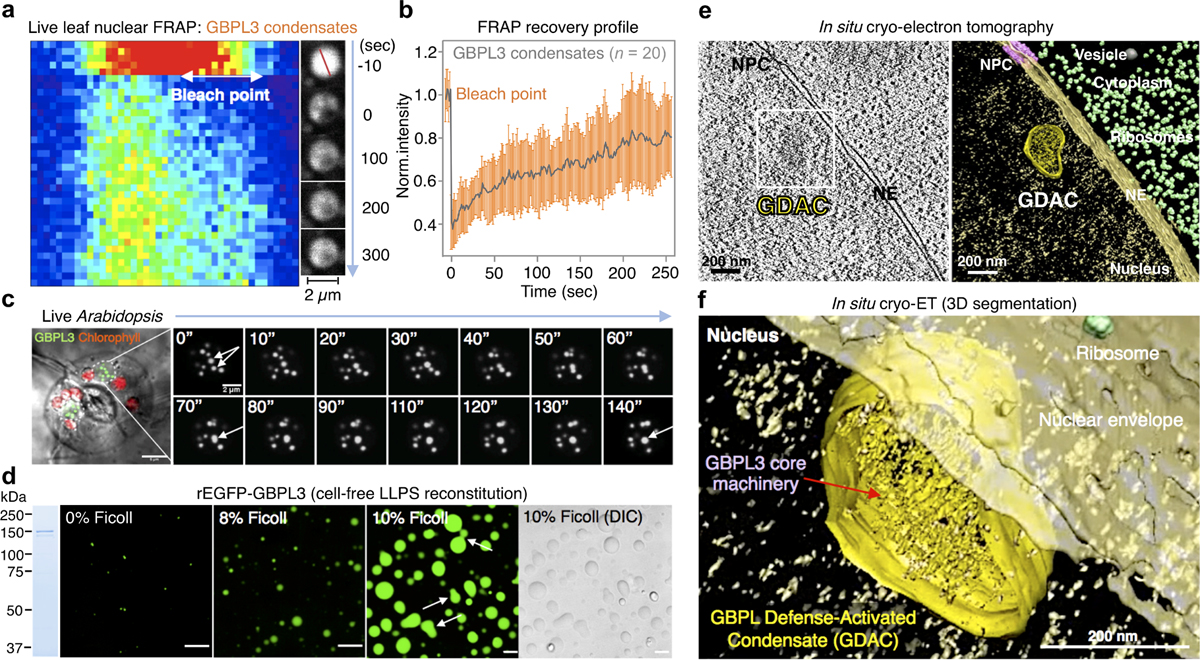
a, FRAP assay of GBPL3 nuclear condensates (eGFP–GBPL3) in N. benthamiana leaf cells. Left, kymograph along the red line (shown top right). b, Normalized FRAP recovery profiles (mean ± s.d. of 20 biologically independent condensates). c, Condensate fusion (arrows) in 10-day-old Arabidopsis cotyledon guard cells expressing eGFP–GBPL3. Dashed circles, nuclear boundary. GDACs (green), chloroplast (red) and differential interference contrast (DIC) image (grey). Scale bar, 5 μm. Right, time-lapse images (in seconds) of fusion events of GDAC condensates indicated by arrows. Scale bar, 2 μm. d, In vitro phase separation of eGFP–GBPL3 (149.6 kDa, 1.3 μM) incubated at 22 °C for 24 h in droplet buffer (20 mM HEPES pH 7.5, 200 mM NaCl and 1 mM (tris(2-carboxyethyl)phosphine)). Ficoll is not essential, but can enhance GBPL3 LLPS. Arrows indicate fusion events. Left, Coomassie brilliant blue staining of eGFP–GBPL3 separated by SDS–PAGE. Scale bars, 10 μm. e, f, Three-dimensional segmentation (f) of 2D-slice tomogram (e) reconstruction of GDACs by cryo-electron tomography. NE, nuclear envelope; NPC, nuclear pore complex. Scale bars, 200 nm.
To determine whether GDACS arose directly via LLPS, we biochemically reconstituted GBPL3 condensates in vitro. Recombinant eGFP–GBPL3 protein underwent coacervation at physiological salt concentrations26, and its phase-separating properties were enhanced by the crowding agent Ficoll-400, leading to condensates that isotropically scaled in size and fused, similar to other LLPS-responsive proteins (Fig. 3d, Extended Data Fig. 6a–c). A phase-transition boundary revealed that as little as ~25 nM eGFP–GBPL3 triggered LLPS (Extended Data Fig. 6b), lower than the LLPS threshold measured in situ where the presence of inhibitory partners such as GBPL1 hamper this process, because GBPL1 phase separates poorly inside the nucleus despite its in vitro LLPS activity (Extended Data Fig. 6d, e). Indeed, adding excess RFP–GBPL1 to reconstitution assays impaired eGFP–GBPL3 LLPS across 158,540 droplets analysed and interfered with GBPL3 homotypic assembly (Extended Data Fig. 6f, g). At equimolar concentrations, however, eGFP–GBPL3 droplets quickly grew to more than 8 μm2 in area and exteriorized RFP–GBPL1 (Extended Data Fig. 6f, h). Thus, biochemically reconstituted droplets closely resembled those in living plants. Eventual gelation prevented in vitro FRAP profiling of these cell-free structures, and they appeared to be densely assembled under negative-stain electron microscopy (Extended Data Fig. 6i, j). To examine whether GBPL3 condensates form similar structures in situ, we used cryo-electron tomography of focused ion beam-milled lamella to characterize GBPL3 condensates at higher resolution, using HEK 293T cells expressing eGFP–GBPL3. Three-dimensional tomograms showed that GDACs also contain dense protein clusters, which are probably surrounded by a chromatin boundary, consistent with previous models of LLPS genome regulation16 (Fig. 3e, f, Extended Data Figs. 7a–h, 8a–d, Supplementary Videos 6, 7).
GBPL3 IDR and GTPase activities in LLPS defence
GBPL3 thus forms liquid-like condensates via LLPS in living plants and reconstituted systems. Because IDRs drive phase-separation behaviour in many proteins2, we tested whether the IDR of GBPL3 (amino acids 633–1082) is responsible for GDAC assembly in Arabidopsis immunity (Extended Data Fig. 9a). This region is enriched for certain hydrophobic amino acids (such as alanine and leucine) and charged residues (for example, glutamate, arginine and histidine) (Extended Data Fig. 9b), which can self-associate to exclude water in immiscible settings25. Removal of amino acids 633–1082 (GBPL3ΔIDR) abolished LLPS-driven nuclear condensates (Extended Data Fig. 9c), did not rescue susceptible gbpl3–4 plants and did not increase pathogen resistance or autoimmunity when overexpressed in wild-type hosts, unlike full-length GBPL3 (Extended Data Fig. 9d). Thus, the GBPL3 IDR is required for LLPS-dependent plant defence. Similarly, fusion of the GBPL3 IDR to eGFP produced spherical condensates resembling liquid–liquid demixing in vitro but not in vivo, although fusion to more-closely related proteins such as human GBP1 that lack an IDR yielded spheroid droplets (Extended Data Fig. 9e, f). The GBPL3 IDR is thus necessary, but not always sufficient, for condensate formation in situ. This raises the possibility of additional GBPL3 regions contributing to its LLPS behaviour.
We examined the N-terminal catalytic domain, as mammalian GBPs are GTPases17,18,27 and Arabidopsis GBPL3 contains a canonical GTP-binding pocket (Extended Data Figs. 9a, 10a). Radioactive [γ−32P] GTP hydrolysis assays found that recombinant GBPL3 or GBPL2, but not GBPL1, produced [γ−32P]GDP (Extended Data Fig. 10b). Malachite green assays confirmed the GTPase activities of GBPL3 (11.73 nmol inorganic phosphate (Pi) min−1 mg−1) versus GBPL1 (less than 0.3 nmol Pi min−1 mg−1) (Extended Data Fig. 10c). GBPL1 is therefore a pseudo-GTPase with a disrupted G2 box that prevents GBPL3 assembly and LLPS, but not GBPL3 catalysis, in co-incubation assays; it behaves as a negative allosteric regulator (Extended Data Figs. 6f, g, 10c). Mutation of the Walker P-loop region (G1 box) of GBPL3 (GBPL3K83A/S84A) eliminated both its GTPase activity (Extended Data Fig. 11b, c) and nuclear condensate formation in Arabidopsis, Nicotiana benthamiana and human cells (Fig. 4c, Extended Data Fig. 10d–f). GBPL3K83A/S84A behaved as a strong dominant-negative inhibitor in wild-type plants during infection, because—like other dynamin-related GTPases17—GBPL3 undergoes GTP-dependent self-assembly, which could help surmount energy barriers during phase transition1–3 (Extended Data Figs. 2h, 9d, 10e–g). Both the IDR and the catalytic regions therefore drive GBPL3 LLPS to confer disease resistance in vivo.
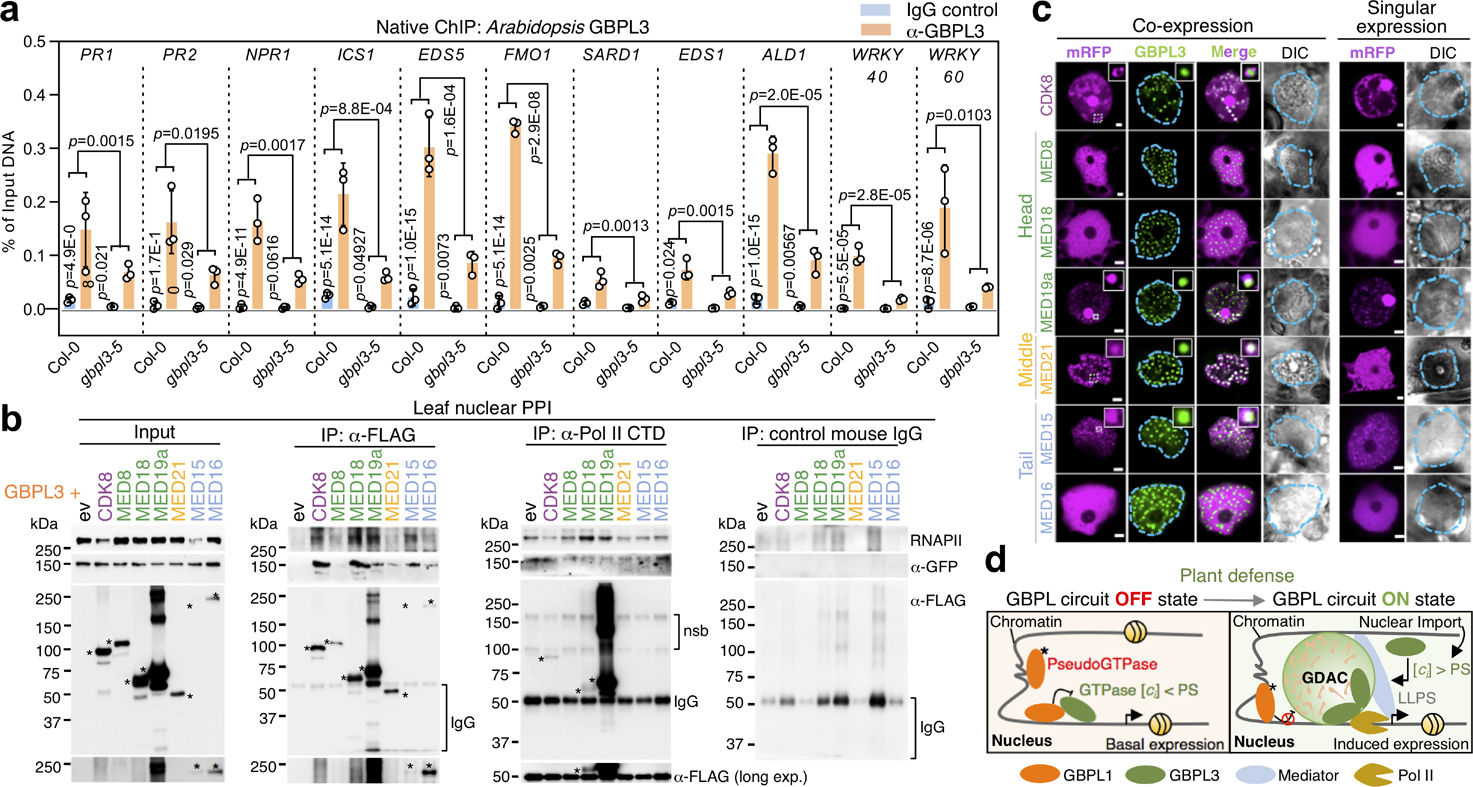
Fig. 4 |. GDACs concentrate RNAPII coactivators and nuclear GBPL3 on promoters of Arabidopsis defence genes.
a, Endogenous GBPL3 ChIP assay of defence-gene promoter occupancy in Col-0 and gbpl3–5 hypomorphic plants. Two-way ANOVA test with Holm–Sidak post hoc correction. Data are mean ± s.d., n = 3 biologically independent samples. b, Co-immunoprecipitation of eGFP–GBPL3 and mRFP–Flag-tagged Mediator subunits or endogenous RNAPII in N. benthamiana. Asterisks indicate Mediator bands. Nsb, non-specific bands. c, Live imaging of eGFP–GBPL3 co-expressed with RFP-tagged Mediator subunits in N. benthamiana. A line profile showing colocalization in condensates is presented in Extended Data Fig. 11. Scale bars, 2 μm. d, Current model of the GBPL immune circuit in the plant nucleus under both basal and induced conditions. Asterisk indicates the G2-box mutation that makes GBPL1 a pseudoenzyme; ci, intracellular concentration of GBPL3; PS, phase-separation threshold.
Such LLPS activities were effective only in the nucleus. Mutating three positive charges (K986A, R987A and R989A) in a monopartite NLS (amino acids 981–991; GBPL3ΔNLS) excluded GBPL3 from the nucleus but allowed LLPS in the cytosol (Extended Data Figs. 9a, 11a–d, Supplementary Table 1). Mislocalized GBPL3ΔNLS could not complement gbpl3–3 plants during Psm ES4326 infections (Extended Data Fig. 9d). Thus, nucleocytoplasmic relocation of GBPL3 is critical together with IDR- and GTPase-dependent LLPS activities to produce transcriptional condensates for Arabidopsis defence-gene expression and immunity (Extended Data Fig. 11e).
GDACs coordinate defence-gene transcription
These transcriptional condensates possess a GBPL3-rich core surrounded by what appear to be chromatin chains (Fig. 3e, f, Extended Data Figs. 7g, h, 8a). Native chromatin immunoprecipitation (ChIP) assays showed that endogenous GBPL3 directly occupied the promoters of 11 representative major defence genes detected by our earlier RNA-seq and PCR with reverse transcription (RT–PCR) analysis (Fig. 4a). Occupancy was diminished or lost with an IgG isotype control antibody or when gbpl3–5 hypomorphic plants were used, verifying the specificity of the assay. Because GBPL3 lacks obvious transactivation domains28, it probably serves as a nucleating or storage platform to recruit other protein machinery for transcription. We therefore isolated GBPL3 protein partners by co-immunoprecipitation in purified Arabidopsis leaf nuclei (Extended Data Fig. 11f). Members of the Mediator RNAPII–coactivator complex29 and RNAPII subunits (NRPB5A and NRPB3) were among partners identified by protein–protein interaction (PPI)–mass spectrometry (MS/MS) assays that could be assembled by GBPL3 within LLPS condensates for regulating transcription.
We tested GBPL3 recruitment and binding to individual Mediator subunits and associated CDK8 as part of an RNAPII–coactivator complex30 (Fig. 4b, c). At least ten Mediator subunits participate in Arabidopsis immunity (Extended Data Fig. 11g, Supplementary Table 6) and 6 of these (MED15, MED16, MED18, MED19a, MED21 and CDK8) captured GBPL3 when screened as Flag-tagged proteins in N. benthamiana (Fig. 4b). They also retrieved RNAPII. Similarly, an RNAPII C-terminal domain (CTD) antibody immunoprecipitated Flag-tagged MED18, MED19a and CDK8, and low levels of Flag-tagged GBPL3, consistent with recent pan-plant proteomic mapping31. Live-leaf imaging found that GBPL3 strongly co-localized with mRFP–MED15, mRFP–MED19a and mRFP–MED21, but excluded mRFP–CDK8 from the condensate core where transcriptional coactivators are concentrated (Fig. 4c, Extended Data Fig. 11h). Previous structural analysis showed that CDK8 blocks RNAPII binding to the middle Mediator module (in our case, MED21) owing to shared binding surfaces32. MED21 incorporation into GDACs was completely dependent on GBPL3 which also helped physically separate it from CDK8 (Fig. 4c). Thus, the phase-separating behaviour of GBPL3 creates a CDK8-free environment to help protect RNAPII–Mediator interactions from inhibition and promote the transcription of host defence genes.
GBPL3 compartmentalization of Mediator–RNAPII complexes in Arabidopsis is reminiscent of BRD4–MED1 condensate models in regulating super-enhancers14,15 and CDK9 that phosphorylates RNAPII clusters for transcriptional elongation in mammals15. Exposure to JQ1, a drug that blocks BRD4–chromatin interactions in plant cells, or DRB (5,6-dichloro-1-β-d-ribofuranosyl benzimidizole)—a nucleoside analogue inhibitor of Arabidopsis CDKC orthologues33—did not interfere with GDAC assembly (Extended Data Fig. 11i). Thus, GDACs appear functionally distinct from these other LLPS condensates, recruiting and organizing specific RNAPII–coactivator complexes on target promoters rather than enhancers, and during transcriptional initiation rather than during elongation or termination.
Discussion
Together, these results have identified a new LLPS-driven circuit in plants (Fig. 4d). This GBPL1–GBPL3 circuit comprises a GTPase–pseudo-GTPase allosteric switch that controls transcriptional responses to biotrophic or hemi-biotrophic pathogens for host defence. Both hormonal and metabolic signals activate this switch shortly after infection, resulting in the rapid formation of GBPL3-driven immune condensates; this activity is also observed in GBPLs from other species, suggesting that the LLPS behaviour of this GTPase superfamily regulates diverse biological functions across Plantae.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586–021-03572–6.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
In silico mining of the Arabidopsis proteome
IDPs (peptide_id) with ≥ 40% disordered residues and compliant with ≥ 7 predictive software criteria (VLXT, VSL2b, PrDOS, PV2, IUPred-S, IUPred, Erspritz-N,X,D programs) were identified from the D2P2.pro database34 (filter 1) (Extended Data Fig. 1). Corresponding gene_ids curated across 25,498 Arabidopsis ORFs retrieved were matched to peptide_ids in R (biomaRt package). Protein isoforms (different peptides) belonging to the same gene were consolidated to one gene. These unique genes were subject to Cellular Component Classification analysis35 (Filter 2; DAVID Bioinformatics Resource 6.8) and those related to the Nucleus (GO:0005634) were maintained. Next, Gene Ontology enrichment analysis were applied to these Nucleus-related genes (Filter 3). Filtering unknown protein annotations or literarily well-studied biological processes (for example, transcription factors) pared the total to 2,659 genes. Protein sequences of these candidate genes (TAIR10) were screened using cNLS mapper (Filter 4; cut-off:5.0), further enhancing the success rate of identifying bona fide nuclear proteins. The resulting 756 genes were finally subject to gene family classification (Filter 5). Filtering single genes and gene families with less than 2 uncovered candidates yielded 304 candidate IDPs.
Phylogenetic analysis of plant GBPLs
Bayesian inference was performed with MrBayes, version 3.2.636, using the JTT protein substitution matrix37 and a gamma distribution for rate heterogeneity. The program was run on the Yale Farnam high performance computer cluster (200 CPUs, 1280G RAM) for 100 million generations, sampled every 5,000 generations, with 4 simultaneous chains and 2 independent runs. Maximum likelihood (ML) analyses were performed with RAxML, version 8.2.1138, using the same models of evolution with 20,000 bootstrap replicates. The trees generated were visualized with FigTree and Dendroscope, respectively. Consensus supportive values of nodes from both algorithms were combined in Adobe Illustrator CS6 using tree inferred by MrBayes.
Bioinformatic analysis of GBPLs with online servers
Prediction of 3D protein structure was performed on I-TASSER39 web server using default settings (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) and models were projected with Pymol software (version 2.4, Schrödinger). Hydrophobicity and disorder predictions were carried out using ProtScale (https://web.expasy.org/protscale/) and PONDR (http://www.pondr.com/) servers, respectively, using full length protein as inputs.
Bacterial strains
Bacteria used for cloning were Stbl2 or TOP10 chemically competent Escherichia coli. Bacteria used for recombinant protein expression were BL21 or BL21-AI (Thermo Fisher Scientific, C607003) chemically competent E. coli. Bacteria used for plant transformation were Agrobacteria tumefaciens GV3101 electrocompetent cells.
Plant materials and growth conditions
All Arabidopsis mutants are on the A. thaliana Col-0 background. T-DNA lines were obtained from Arabidopsis Biological Resource Center (ABRC) or NASC (Nottingham Arabidopsis Stock Centre). Mutants were backcrossed with Col-0 wild type at least once to validate co-segregation of the transgene. Homozygous T-DNA lines were obtained by PCR using primers listed in Supplementary Table 7. Of note, the T-DNA allele SALK_016366 from ABRC harboured an unlinked background mutation that caused autoimmune defects. We ruled out the background mutation via genetic segregations.
Plants were grown in soil containing perlite and vermiculite (Griffin Greenhouse, 65–2005) under ambient environment at 22 °C and ~50% relative humidity. Long-day photoperiod conditions were kept at 16 h/8 h light/dark cycles and short-day conditions at 12 h/12 h light/dark. To grow plants on Murashige and Skoog (MS) medium, seeds were sterilized in 15% household bleach in water, washed twice in sterile water, resuspended in 0.5% sterile agar and sowed on sterile half-strength MS solid medium (pH 5.7) supplemented with 1% sucrose and 0.5% phytagel. Antibiotics were supplied when required. N. benthamiana plants were grown under long-day conditions.
The eds1–2 seeds were a gift from J. Parker (Max Planck Institute for Plant Breeding Research, Germany). The snc1 seeds were a gift from X. Li (University of British Columbia, Canada).
Mammalian cell media and growth conditions
Human HeLa and HEK 29 3T cell lines were grown in GIBCO Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, 11965118) supplied with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, 10438026). Cells were cultured in an incubator at 37 °C with 5% CO2. For passage, cells were dissociated with 0.25% Trypsin-EDTA (Thermo Fisher Scientific, 25200056) for 1 min and resuspended in DMEM with 10% FBS.
Generation of constructs
Mammalian cell expression constructs.
To generate the cDNA clone of CMV::EGFP-GBPL3, an ORF of GBPL3 was amplified from Col-0 total cDNA using primers GBPL3-SalI-F1 and GBPL3-BamHI-R1, digested and cloned into SalI/BamHI linearized EGFP-C1 vector. A flexible linker sequence (SGLRSRAQASNSAVD) was inserted between EGFP and GBPL3.
For CMV::EGFP-GBPL3K83A,S84A, overlapping PCR fragments were amplified from 35S::EGFP-GBPL3K83A,S84A (see below) using primers GBPL3-SalI-F1 and GBPL3-BamHI-R1, digested and cloned into SalI/BamHI linearized EGFP-C1 vector.
For CMV::mRFP-GBPL3 and CMV::mRFP-GBPL3K83A,S84A, PCR fragments of GBPL3 and GBPL3K83A,S84A were amplified from CMV::EGFP-GBPL3 and CMV::EGFP-GBPL3K83A,S84A, respectively, using primers GBPL3-EGFP-C1-GA-F1 and GBPL3-EGFP-C1-GA-R1, and assembled into the SalI/BamHI digested RFP-C1 vector via Gibson cloning.
For CMV::EGFP-GBPL3K986A,R987A,R989A, overlapping PCR fragments were amplified from CMV::EGFP-GBPL3 using primers GBPL3-EGFP-C1-GA-F1 + GBPL3-NLS-R1 and GBPL3-NLS-F1 + GBPL3-EGFP-C1-GA-R1, and assembled into the SalI/BamHI digested EGFP-C1 vector via Gibson cloning.
For CMV::EGFP-GBPL31–637, a PCR fragment was amplified from CMV::EGFP-GBPL3 using primers GBPL3-EGFP-C1-GA-F2 + GBPL3 (1–637)-GA-R1. To generate CMV::EGFP-GBPL3633–1082, a PCR fragment was amplified from CMV::EGFP-GBPL3 using primers GBPL3(633–1082)-GA-F1 + GBPL3(1–637)-GA-R1. The PCR fragments were then assembled into SalI/BamHI digested EGFP-C1 vector via Gibson cloning.
For human–plant chimeric constructs (CMV::EGFP-hGBP1/2/5-GBPL3633–1082), PCR fragments of hGBP1/2/5 and GBPL3633–1082 were amplified using the following primers, digested and ligated into SalI/BamHI linearized EGFP-C1 vector. GBPL3633–1082: GBPL3 (633–1082)-SacII-F + GBPL3(633–1082)-BamHI-R; hGBP1/2/5: hGBP1/2/5-SalI-F + hGBP1/2/5-SacII-R.
For the cDNA clones of CMV::EGFP-SlGBP1/2 and CMV::EGFP-ZmGBP1/2, ORFs of SlGBP1/2 and ZmGBP1/2 were amplified from Solanum lycopersicum and Zea mays total cDNAs (a gift from F. Xu (Cold Spring Harbor Laboratory, USA)), respectively, using primers SlGBP1-GA-F1 + SlGBP1-GA-R1 (SlGBP1), SlGBP2-GA-F1 + SlGBP2-GA-R1 (SlGBP2), ZmGBP1-GA-F1 + ZmGBP1-GA-R1 (ZmGBP1) and ZmGBP2-GA-F1 + ZmGBP2-GA-R1 (ZmGBP2), and assembled into SalI/BamHI digested EGFP-C1 vector via Gibson cloning. A linker sequence (SGLRSRAQASN-SAG) was inserted after EGFP.
For CMV::mRFP-GBPL1, a PCR fragment of GBPL1 was amplified from Arabidopsis cDNA using primers mRFP-GBPL1-GA-F1 and mRFP-GBPL1-GA-R1, and assembled into the SalI/BamHI digested RFP-C1 vector via Gibson cloning.
Plant expression constructs.
Togenerate the genomic clone of pGBPL3-GBPL3, a genomic fragment of GBPL3 including 858 bp upstream of the start codon was amplified from Col-0 genomic DNA using primers GBPL3-BamHI-F2 and GBPL3-SalI-R1 and cloned into BamHI/SalI digested binary vector pCambia1305.
For 35S::GBPL31–632-3×FLAG, a PCR fragment of GBPL31–632 was amplified from CMV::EGFP-GBPL3 using primers GBPL3-SalI-F1 + GBPL3 (1–632)-BamHI-R1, digested with SalI/BamHI, and ligated to SalI/BamHI linearized pCambia1300–3×FLAG vector.
For 35S::EGFP-GBPL3 and 35S::EGFP-GBPL3K986A,R987A,R989A, NheI/BamHI digested fragments of the CMV::EGFP-GBPL3 and CMV::EGFP-GBPL3 K986A,R987A,R989A constructs, respectively, were inserted into the XbaI/BamHI (compatible ends) digested pCambia1300–3×FLAG vector. For 35S::EGFP-GBPL3K83A,S84A, overlapping PCR fragments were amplified from CMV::EGFP-GBPL3 using primers GBPL3-pGreen0229-GA-F1 + GBPL3-G1-box-mt-R1 and GBPL3-G1-box-mt-F1 + GBPL3-pGreenII 0229-GA-R1, and assembled into the KpnI/BamHI digested pGreenII 0229–35S vector via Gibson cloning.
For pGBPL1::GBPL1–3×FLAG, a genomic fragment of GBPL1 including 952bp upstream of the start codon was amplified from Col-0 genomic DNA using primers GBPL1-BamH1-F2 + GBPL1-SalI-R2, digested with BamHI/SalI, and ligated to BamHI/SalI digested pCambia1305–3×FLAG vector.
For 35S::mRFP-GBPL1, a PCR fragment of mRFP and GBPL1 was amplified form CMV::mRFP-C1 and Arabidopsis total cDNA using primers mRFP-GA-F1 + mRFP-GA-R1 and mRFP-GBPL1-GA-F1 + mRFP-GBPL1-GA-R1, respectively, and assembled into XbaI/BamHI digested pCambia1300–3×FLAG vector via Gibson cloning. A linker sequence (LYKSGLRSRAQASNSAVD) was inserted between mRFP and GBPL1.
For N-terminal mRFP and C-terminal 3×Flag-tagged Mediator subunits, PCR fragments of selected Mediator subunits were amplified from Arabidopsis total cDNA using primers listed in Supplementary Table 7 and assembled into SalI/BamHI digested pCambia1300-mRFP-GBPL1 via Gibson cloning.
For gRNA-expressing vectors for GBPL3 targeting, Oligo-gRNA1F/R and Oligo-gRNA2F/R were annealed and inserted between two BsaI sites of pHEE401E, resulting in pHEE401E-GBPL3gRNA1 and pHEE401E-GBPL3gRNA2, respectively.
All constructs were verified by Sanger sequencing, delivered to Agrobacteria GV3101 through electroporation, and transformed into Arabidopsis plants via the floral dip method40.
The constructs fibrillarin-pROK2-mRFP (CD3–795059) and RNPS1-pROK2-mRFP (CD3–795044) were purchased from ABRC.
Bacteria expression constructs.
To generate His6-EGFP, EGFP was amplified from CMV::EGFP-GBPL3 using primers pET28-EGFP-GA-F1 and pET28-EGFP-GA-R1 and assembled into the BamHI/XhoI digested pET28a(+) vector. The linker region (SGLRSRAQASNSAVD) was also included in the construct.
For His6-EGFP-GBPL3633–1082, PCR fragment of EGFP-GBPL3633–1082 was amplified from CMV::EGFP-GBPL3633–1082 using primers pET28-EGFP-GA-F1 and His-EGFP-GBPL3-GA-R1 and assembled into the BamHI/XhoI digested pET28a(+) vector via Gibson cloning.
For His6-EGFP-GBPL3 and His6-EGFP-GBPL3K83A,S84A, the C-terminal portion (aa639–1082) of GBPL3 was codon-optimized at IDT (https://www.idtdna.com) and assembled into the BamHI/XhoI digested pET28a(+) vector via Gibson cloning together with the overlapping N-terminal EGFP-GBPL31–638 fragment amplified from CMV::EGFP-GBPL3 and 35S::EGFP-GBPL3K83A,S84A, respectively.
Codon-optimized GBPL3 sequence (amino acids 639–1082): GCCAAGAGTGCAGCCGAGGAGACTAAGGAATGGAAGCGTAAATATGAC TACGCAGTCGGGGAAGCTCGCAGCGCCCTTCAGAAGGCAGCTTCAGTC CAGGAACGTTCGGGAAAGGAGACTCAACTTCGTGAGGATGCACTGCGC GAGGAATTTAGTATTACTTTAGCAAATAAAGATGAGGAAATCACCGAAA AGGCTACGAAGTTGGAGAAAGCGGAGCAGTCCCTGACAGTGCTTCGT TCTGATCTGAAAGTTGCTGAAAGTAAGCTGGAATCGTTTGAAGTTGAGC TTGCTTCCTTACGCTTGACACTTTCGGAAATGACCGACAAATTGGACAG TGCAAACAAAAAGGCTTTGGCTTACGAGAAGGAAGCCAATAAGTTAGA GCAGGAAAAAATTCGTATGGAGCAAAAATACCGCAGCGAGTTCCAGC GCTTCGACGAGGTGAAAGAGCGTTGCAAAGCAGCCGAGATTGAAGCG AAACGCGCCACTGAATTGGCAGACAAAGCTCGTACAGATGCCGTCACA AGCCAGAAAGAGAAAAGCGAAAGTCAGCGCCTTGCGATGGAGCGCCTT GCACAAATCGAGCGTGCCGAACGTCAGGTCGAAAACTTGGAACGTCA AAAAACAGATTTGGAAGACGAGTTAGATCGTTTGCGTGTCTCAGAAATG GAAGCAGTTTCAAAGGTGACGATCCTTGAAGCTCGTGTCGAGGAGCGC GAGAAAGAGATTGGATCTTTGATCAAGGAAACTAATGCTCAACGCGCAC ACAATGTAAAGAGTTTAGAAAAATTGTTGGACGAAGAGCGCAAGGCCC ACATTGCGGCAAATCGTCGTGCAGAAGCCTTGAGCTTGGAGCTTCAGG CGGCCCAAGCTCACGTGGATAACCTTCAGCAAGAACTGGCACAGGCCC GTCTGAAGGAAACTGCACTTGACAACAAAATCCGCGCAGCGTCTTCCA GTCACGGGAAACGCTCTCGCTTCGAAGACGTCGTCGATATGGACATTG GTGAAGGTTCAGACCGTATCTTGCGTACCAATAAGCGCGCCCGCTCTAC TCGTGGAGACGACCACGGCCCGACAGACGAAGGAGACGAAGACTTCC AATCACACCAAGACAACGGGGAGGAGGAAGAGGAAGAGGACTATCGTA AGCTTACCGTCCAAAACTTGAAGCATGAATTGACAAAATATGACTGCGG ACATCTGCTTTTGAATCGTGGCCACCAAAATAAGAAGGAGATTCTGGCA TTGTACGAGGCGCACGTCTTGCCCAAAAAAGCACTTGCGCGTGAGGAG GAGCGTAAGAAGCAGCGCGAGGTGACATCCTCT.
For His10-Halo-EGFP-GBPL3, a PCR fragment of EGFP-GBPL3 was amplified from His6-EGFP-GBPL3 using primers Halo-EGFP-GBPL3-GA-F1 and Halo-EGFP-GBPL3-GA-R1 and assembled into a customised pET28e-Halo vector digested with BamHI/XhoI. An HRV cleavage site (LEVLFQ/GP) was included between Halo and EGFP.
For His6-GBPL1, a PCR fragment of GBPL1 was amplified from Col-0 total cDNA using primers pET28-GBPL1-BamH1-F1 and pET28-GBPL1-NotI-R1 and ligated into BamHI/NotI digested pET28a(+) vector.
For His10-Halo-mRFP-GBPL1, a PCR fragment of mRFP-GBPL1 was amplified from 35S-mRFP-GBPL1 using primers Halo-mRFP-GBPL1-GA-F1 and Halo-mRFP-GBPL1-GA-R1 and assembled into pET28e-Halo vector digested with BamHI/XhoI. An HRV cleavage site (LEVLFQ/GP) was included between Halo and mRFP.
For GST-GBPL2, a PCR fragment of GBPL2 was amplified from Col-0 total cDNA using primers pGEX-GBPL2-BamH1-F1 and pET28-GBPL2-NotI-R1 and ligated into BamHI/NotI digested pGEX-6p-1 vector.
Generation and validation of Arabidopsis GBPL3 antibodies
Three customized rabbit polyclonal antibodies against endogenous Arabidopsis GBPL3 peptides were generated. One antibody raised against the C-terminal fragment of GBPL3 (a.a.1069–1082) yielded higher titers and was used in this study. Since homozygous gbpl3 knockout plants were lethal, we attempted to generate stronger hypomorphic mutants via CRISPR-Cas9 engineering to use as negative controls for antibody validation. Unexpectedly, three viable homozygous knockout T1 plants targeted by two different guide RNAs (gRNAs) were identified. Although these knockout plants were sterile and did not set seeds, they served as robust negative controls and allowed us to determine the specificity of the GBPL3 antibody. Importantly, this antibody was unaffected by gbpl1 or gbpl2 deficiency; hence it does not cross-react with other Arabidopsis GBPL family members.
Generation of mutants
Double-mutant plants were generated via cross pollination and confirmed by PCR. Of note, gbpl2–1 gbpl3–3 double mutants were non-viable while susceptibility phenotypes in gbpl1–1 gbpl3–3 double mutants were not due to gbpl3 genetic dominance. Extensive backcrossing revealed both gbpl1 and gbpl3 alleles are recessive.
To generate mutants via CRISPR, gRNAs were cloned into the egg-cell specific CRISPR-Cas construct pHEE401E41 (a gift from Q. Wu (China Agricultural University, China)) and transformed into Col-0 via Agrobacterium delivery. The mutations caused by gRNA-Cas9 cleavage were determined at T1 through sanger sequencing of individual plants.
Pathogen infection assays
Hpa Noco2 was propagated weekly on Col-0 plants in a growth chamber at 18 °C under 12 h/12 h light/dark and ~95% relative humidity. For Hpa Noco2 infection, spores were freshly collected, resuspended in water and sprayed onto 2- to 3-week-old seedlings at indicated concentrations. Quantification of Hpa Noco2 growth was performed 7 d after infection.
For bacterial infection assays, overnight culture in Luria-Bertani (LB) media was pelleted, washed twice, resuspended and diluted to desired concentrations in 10 mM MgCl2. Bacteria were infiltered into the abaxial side of leaves from four-week-old plants grown under short day conditions using a needleless syringe. Growth of bacteria was quantified at Day 0 and Day 3 after infection. No significant difference in bacteria growth was observed at Day 0.
Psm ES4326 and Pst DC3000 were gifts from N. Clay (Yale University, USA) and Hpa Noco2 was kindly provided by X. Dong (Duke University, USA).
RNA-seq analysis
For RNA-seq, leaves of three-week-old soil-grown Col-0, gbpl1–1, gbpl3–5 and GBPL3OX plants were collected for total RNA extraction using the PureLink RNA Mini Kit (Invitrogen, 12183020) with on-column DNase digestion, following the manufacturer’s instructions.
Library preparation and RNA-seq were performed by Genewiz America (https://www.genewiz.com/en) using an Illumina HiSeq 2000 resulting in 19–32 million reads per sample. Raw RNA-seq reads were subjected to quality checking and trimming to remove adaptor sequences, contamination and low-quality reads using Flexbar version 3.0. The trimmed reads of each sample were aligned to the publicly available reference genome of Arabidopsis (TAIR10, https://emea.support.illumina.com/sequencing/sequencing_software/igenome.html) using HISAT2 version 2.1.0 on default parameters42. SAMtools version 1.5 was used to convert SAM files, sort and index BAM files43. Read counts were generated for each gene using HTseq-count version 0.9.1 with the following settings: order = position, mode = union, stranded = no44. R package DESeq2 version 3.9 was used to determine differentially expressed genes45. Gene Ontology (GO) analysis was performed using DAVID. Clustering was performed using the R package pheatmap version 1.0.12 using rlog transformed counts.
RT–PCR
About 0.1 g of tissue was collected from plate-grown or soil-grown plants, and total RNA was extracted using the PureLink RNA Mini Kit (Invitrogen). The RNA was reverse transcribed to cDNA using Transcriptor Reverse Transcriptase (Roche). The expression of target genes was analysed via RT–PCR according to a protocol described previously46.
Transient expression in N. benthamiana
Four- to five-week-old N. benthamiana plants were used for transient expression as previously described46. In brief, overnight Agrobacterium cultures were pelleted, resuspended and diluted to OD600 = 0.2 in infiltration buffer (10 mM MES pH 5.6, 10 mM MgCl2, and 200 μM acetosyringone). Bacterial suspension was incubated at room temperature for 3 h before infiltration. A p19 plasmid (Addgene, 68214) was co-infiltrated (OD600 = 0.1) to suppress gene silencing. Expression of transgenes was analysed 48–72 h after infiltration.
Transfection in human cell lines
Plasmids were transfected into human cells using the TransIT-LT1 transfection reagent (Mirus Bio, MIR2300) following the manufacturer’s instructions.
Immunoblotting, cell fractionation and co-immunoprecipitation
Immunoblotting in plants.
About 80 mg of plant tissue was frozen in liquid nitrogen and ground into fine powder. Total protein was extracted by adding 2 v/w of 1× RIPA buffer (Cell Signaling Tech, 9806S) supplemented with 0.2% SDS and 1 × protease inhibitor (Roche, 11873580001) to each sample. The supernatant was collected via centrifugation (4 °C, 13,200g, 15 min) and boiled at 95 °C for 5 min after adding 1 × SDS sample buffer. Proteins were resolved by SDS–PAGE.
Isolation of nuclei in Arabidopsis.
Nuclei were isolated as previously described47. Around 1 g of two-week-old plate-grown or three-week-old soil-grown plants were flash frozen in liquid nitrogen and ground into fine powder. Cells were resuspended in 2 ml lysis buffer (20 mM Tris-HCl pH 7.4, 25% glycerol, 20 mM KCl, 2 mM EDTA, 2.5 mM MgCl2, 250 mM sucrose, 1 mM DTT, 1 × of Roche protease inhibitor cocktail) and sequentially filtered through 100-μm and 40-μm cell strainers to remove cell debris. Flow-through was taken as the total lysate. Nuclei was pelleted by centrifugation (1,500g, 15 min, 4 °C) and the supernatant was taken as the cytosol fraction. Nuclei were then washed 4 times in nuclei resuspension buffer (20 mM Tris-HCl pH 7.4, 25% glycerol, 250 mM NaCl, 2.5 mM MgCl2, 0.15% Triton X-100, 1 mM DTT and 1 × of Roche protease inhibitor cocktail). After washing, nuclei were resuspended in PBS as the nuclear fraction. Protein samples were boiled at 95 °C for 5 min after adding 1× SDS sample buffer, resolved by SDS–PAGE and detected with GBPL3 antibody.
Isolation of nuclei in human cells.
Nuclei were isolated as previously described22. In brief, cells were collected, washed, and incubated in nucleus separation buffer (10 mM HEPES, pH 7.9; 10 mM KCl; 1.5 mM MgCl2; 0.34 M sucrose; 10% (vol/vol) glycerol; 1 mM DTT; 0.1% Triton X-100; 1x Roche protease inhibitor) on ice for 10 min. After 1,500g centrifugation for 10 min at 4 °C, nuclei were precipitated in pellet. Supernatant was collected as the cytosol fraction. Nuclei were then washed once in nuclei separation buffer and pelleted before further use.
Chromatin-bound protein separation.
Nuclei were purified as above and resuspended in 1× RIPA buffer supplemented with 1× protease inhibitor. After 5 min incubation on ice, cell lysates were centrifuged at 1,000g for 10 min at 4 °C. The supernatants were collected as nuclear soluble fraction. The pellet was resuspended with 1× RIPA buffer supplemented with 0.3% SDS and 250 units per ml benzonase (Santa Cruz Biotechnology, sc-202391). Pellet suspensions were incubated on ice for 10 min and centrifuged again at 1,000g for 10 min at 4 °C. The supernatant from the second centrifugation contained most of the chromatin-bound proteins.
Nuclease digestion and gradient salt elution of chromatin-bound proteins.
Purified nuclei were resuspended in nuclei resuspension buffer (20 mM Tris-HCl pH 7.4, 25% glycerol, 250 mM NaCl, 2.5 mM MgCl2, 0.15% Triton X-100, 1 mM DTT and 1 × Roche protease inhibitor cocktail) containing 2,000 gel units per ml micrococcal nuclease (New England Biolabs, M0247) and 1 mM CaCl2. The nuclei suspension was incubated at 37 °C for 10 min and the reaction was stopped immediately by adding 2 mM EGTA on ice. The supernatant was removed and the digested nuclei were first eluted sequentially with 150 mM and 300 mM in elution buffer (10 mM HEPES, pH 7.9; 2 mM MgCl2; 2 mM EGTA; 1 mM DTT; 0.1% Triton X-100; 1× Roche protease inhibitor) at 4 °C for 30 min each; and further eluted with 600 mM NaCl overnight. The remnant (pellet) from 600 mM NaCl elution was resuspended with 2% (w/v) SDS loading buffer.
Co-immunoprecipitation in Arabidopsis and N. benthamiana.
To detect GBPL1-GBPL3 interaction in Arabidopsis, for GBPL1-Flag immunoprecipitation, nuclei were isolated from 6 g leaves of 3-week-old Col-0/GBPL1-Flag plants. Nuclei were resuspended in NE-2 buffer (20 mM HEPES-KOH, pH 7.9, 2.5 mM MgCl2, 250 mM NaCl, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1 mM DTT and 1 × protease inhibitor) and subjected to sonication for 2 min with 10 s/10 s on/off intervals to lyse the nuclei. Lysed nuclei were centrifuged at 13,000g for 30 min at 4 °C. The supernatant was collected and mixed with 50 μl preequilibrated anti-Flag M2 agarose (Sigma, A2220) and rotated overnight at 4 °C. For negative control, 50 μl pre-equilibrated streptavidin–agarose (Thermo Fisher Scientific, 20347) was added. Beads were washed three times with NE-3 buffer (20 mM HEPES-KOH, pH 7.9, 2.5 mM MgCl2, 150 mM NaCl, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1 mM DTT and 1 × protease inhibitor) for 5 min each. Bound proteins were eluted with 50 μl 2× SDS loading buffer and boiled at 95 °C for 5 min.
For endogenous GBPL3 co-immunoprecipitation, nuclei were isolated from 50 g leaf samples of 3-week-old Col-GBPL1-Flag plants. Nuclei were resuspended in NE-2 buffer (20 mM HEPES-KOH, pH 7.9, 2.5 mM MgCl2, 250 mM NaCl, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1 mM DTT and 1 × protease inhibitor) and subjected to sonication for 2 min with 10 s/10 s on/off intervals to lyse the nuclei. Lysed nuclei were centrifuged at 13,000g for 30 min at 4 °C. Cleared lysate were split into two equal volume samples, one sample was mixed with 15 μg GBPL3 antibody and the other with 15 μg normal rabbit IgG and rotated overnight at 4 °C. Antibodies were captured with 50 μl protein A/G agarose beads for 1 h at 4 °C. Beads were washed three times with NE-3 buffer (20 mM HEPES-KOH, pH 7.9, 2.5 mM MgCl2, 150 mM NaCl, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1 mM DTT and 1 × protease inhibitor) for 5 min each. Bound proteins were eluted with 50 μl 2× SDS loading buffer and boiled at 95 °C for 5 min.
For RNAPII CTD immunoprecipitation in N. benthamiana, cleared lysates were split into two equal volume samples, one sample was mixed with 2 μg RNAPII CTD antibody (Abcam; ab817, (8WG16)) and the other with 2 μg mouse IgG and rotated overnight at 4 °C. Antibodies were captured with 50 μl protein A/G agarose beads for 1 h at 4 °C. Beads were washed three times with NE-3 buffer and bound proteins were eluted with 2× SDS loading buffer.
Co-immunoprecipitation in HEK 293T cells.
Cells grown on a 6-well dish were collected 36–48 h after transfection and washed twice with cold PBS. Cells were lysed in hypotonic buffer (20 mM Tris-HCl pH 7.4, 25% glycerol, 20 mM KCl, 2 mM EDTA, 2.5 mM MgCl2, 250 mM sucrose, 1 mM DTT, 1 × of Roche protease inhibitor cocktail) for 15 min on ice. Nuclei was pelleted by centrifugation (1,500g, 10 min, 4 °C) and washed twice in nuclei resuspension buffer (20 mM Tris-HCl pH 7.4, 25% glycerol, 250 mM NaCl, 2.5 mM MgCl2, 0.2% Triton X-100, 1mM DTT and 1 × of Roche protease inhibitor cocktail). The nuclei were spun down and resuspended in 0.5 ml ice cold nuclear extraction buffer (20 mM HEPES pH 7.9, 2.5 mM MgCl2, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1mM DTT and 1 × of Roche protease inhibitor cocktail), lysed via sonication and further cleared by centrifugation at 13,200g for 30 min at 4 °C. The supernatant was subjected to GFP-Trap agarose (Chromotek, gta-20) immunoprecipitation overnight at 4 °C. Protein complex bound to GFP–Trap were washed three times in wash buffer (20 mM HEPES pH 7.9, 2.5 mM MgCl2, 150 mM NaCl, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1 mM DTT and 1× Roche protease inhibitor cocktail). Proteins were eluted with 50 μl glycine buffer (0.1 M, pH 2.5) and resolved by SDS–PAGE.
Split luciferase complementation assay
The GBPL1 and GBPL3 coding sequences were amplified by primers listed in Supplementary Table 7 to generate pCambia1300–35S-Cluc-GBPL3 and pCambia1300–35S-GBPL1-NLuc constructs. These constructs were introduced into A. tumefaciens via electroporation. Bacteria carrying these two constructs were co-infiltrated into four-week-old N. benthamiana leaves, and the infiltrated leaves were incubated with 1 mM luciferin two days later. Luminescence was recorded afterwards. Quantification of luminescence was performed using leaf discs with a microplate reader. Each bacterial strain was diluted to a final concentration of OD600 = 0.2.
Recombinant protein expression and purification
Purification of His6-EGFP-GBPL3/His6-EGFP-GBPL3K83A,S84A (TLC assay grade), His6-EGFP and His6-EGFP-GBPL3IDR, and His6-GBPL1.
Plasmids were transformed into BL21-AI and protein was induced at OD600 = 0.6 with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and 0.2% L-Arabinose at 22 °C for 16 h. Cells were pelleted (4 °C, 4,000g, 30 min) and lysed in lysis buffer (50mM Tris [pH 8.0], 500 mM NaCl, 30 mM Imidazole [pH 8.0], 5 mM β-mercaptoethanol, 1 × of Roche protease inhibitor cocktail, 1 mM PMSF, and 3 μl of Benzonase nuclease) via sonication. The lysate was cleared by centrifugation (4 °C, 35,000g, 30 min). The protein was first purified with Ni-NTA (QIAGEN) in gravity columns. After elution with elution buffer (50 mM Tris [pH 8.0], 500 mM NaCl, 500 mM Imidazole [pH 8.0], 5 mM β-mercaptoethanol), fractions were analysed by SDS–PAGE, pooled, concentrated and further purified and buffer-exchanged via size exclusion chromatography (Superdex 200 increase; GE Healthcare) equilibrated with storage buffer (20 mM HEPES [pH 7.5], 500 mM NaCl, 1 mM TCEP). Fractions were analysed by SDS–PAGE, pooled, concentrated and flash frozen in liquid nitrogen before storing at −80 °C.
Purification of GST and GST–GBPL2
Plasmids were transformed into BL21 and protein was induced at OD600 = 0.6 with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 22 °C for 16 h. Cells were pelleted (4 °C, 4,000g, 30 min) and lysed in lysis buffer (50 mM Tris [pH 8.0], 500 mM NaCl, 5 mM β-mercaptoethanol, 1 × of Roche protease inhibitor cocktail and 1 mM PMSF) via sonication. The lysate was cleared by centrifugation (4 °C, 35,000g, 30 min). The protein was first purified with Glutathione sepharose (GE Healthcare) in gravity columns. After elution with elution buffer (50 mM Tris [pH 8.0], 500 mM NaCl, 10 mM glutathione, 5 mM β-mercaptoethanol), fractions were analysed by SDS–PAGE, pooled, concentrated and further purified and buffer-exchanged via size exclusion chromatography (Superdex200 increase; GE Healthcare) equilibrated with storage buffer (20 mM HEPES [pH 7.5], 500 mM NaCl, 1 mM TCEP). Fractions were analysed by SDS–PAGE, pooled, concentrated and flash frozen in liquid nitrogen before storing at −80 °C.
Purification of eGFP–GBPL3 and mRFP–GBPL1
The pET28-His10-Halo-EGFP-GBPL3 and pET28-His10-Halo-mRFP-GBPL1 plasmids were transformed into BL21-AI and proteins were induced at OD600 = 0.6 with 1 mM IPTG and 0.2% L-Arabinose at 22 °C for 16 h. Cells were pelleted (4 °C, 4,000g, 30 min) and lysed in lysis buffer (50 mM HEPES [pH 7.5], 500 mM NaCl, 10 mM Imidazole, 0.1% CHAPS, 1.45% glycerol, 2 mM TCEP, 1 × of Roche protease inhibitor cocktail, and 3 μl of Benzonase nuclease) via sonication. The lysate was cleared by centrifugation (4 °C, 35,000g, 30 min) and first purified with Ni-NTA (QIAGEN) in gravity columns. After elution with elution buffer (50 mM HEPES [pH 7.5], 500 mM NaCl, 300 mM Imidazole, 1 mM TCEP), elutes were pooled and diluted 10 times with dilution buffer (50 mM HEPES [pH 7.5], 500 mM NaCl, 1.45% glycerol, 2 mM TCEP) and further purified via anion exchange chromatography (Q Sepharose Fast Flow, GE Healthcare, 17051010). Fractions were analysed by SDS–PAGE, pooled, concentrated and digested with PreScission protease (GE Healthcare, GE27–0843-01) at 4 °C for 16 h in digestion buffer (50 mM HEPES [pH 7.5], 500 mM NaCl, 1 mM EDTA, 1 mM TCEP). The digested His10-Halo tag and PreScission protease were removed via binding to Ni-NTA and GST beads, respectively, for 1 h at 4 °C. The flow-through was collected and further purified and buffer-exchanged via size exclusion chromatography (Superdex 200 increase; GE Healthcare) equilibrated with storage buffer (20mM HEPES [pH 7.5], 500 mM NaCl, 1 mM TCEP). Fractions were analysed by SDS–PAGE, pooled, concentrated and flash frozen in liquid nitrogen before storing at −80 °C.
In vitro phase separation
Phase separation by dilution.
Protein aliquots were thawed at room temperature, centrifuged at 14,000 × g for 5 min to remove any aggregated protein. Droplet formation was induced by diluting protein to low salt buffers by mixing with various volumes of no salt buffer (20 mM HEPES [pH 7.5], 1 mM TCEP) and analysed in chambered coverglass (Grace Bio-labs) using a Lecia SP8 laser scanning confocal at 63 × /1.40 magnification.
Phase separation by molecular crowding.
The experiment was performed as described above except that Ficoll-400 was included in droplet buffer. Coverslips were incubated at RT before images were taken.
Native PAGE analysis of GBPL3 self-assembly
Recombinant rEGFP-GBPL3 and rRFP-GBPL1 proteins were purified as described above. Reactions (20 μl) were incubated on ice for 30 min containing rEGFP-GBPL3 at a concentration of 37.5 ng/μl and increasing amount of rRFP-GBPL1 (0, 12.5, 37.5, 62.5 ng/μl) inassembly buffer (20mM HEPES-KOH [pH 7.9], 2.5 mM MgCl2, 150 mM NaCl, 20% glycerol, 0.2% Triton X-100, 0.2 mM EDTA, 1 mM GTP). Reactions were loaded onto 4–20% Mini-PROTEAN TGX precast polyacrylamide gels (Bio-Rad, 4561096) and run at 100V constant voltage for 3 h at 4 °C. Native protein size marker (ThermoFisher, LC0725) was run alongside. Following gel electrophoresis, proteins were blotted to PVDF-membranes (Bio-Rad, 162–0177). After transfer, the membranes were blocked with 5% skim milk in TBST. The membranes were incubated overnight with primary antibodies in 5% BSA in TBST. After washing the membranes four times with TBST, HRP-conjugated secondary antibodies were applied for 1 h at room temperature. Detection was performed using Clarity Max Western ECL Substrate (Bio-Rad, 1705062). Reactions were also loaded onto SDS–PAGE gels in parallel as loading controls.
Microscopy
All images were acquired using a Leica SP8 laser-scanning confocal microscope.
Analyses of the subcellular localization of GBPL3 wasperformed under a 63 × /1.20 water immersion objective for live plant cells or a 100 × /1.44 oil immersion objective for fixed N. benthamiana and human cells. Hocechst 33342 was excited at 405 nm and detected at 460 to 490 nm, GFP was excited at 488 nm and detected at 515−535 nm. Chloroplast autofluorescence was detected at 700−710 nm.
Analyses of colocalization of GBPL3 in N. benthamiana were performed with a 63 × /1.20 water immersion objective. Tobacco leaves 2–3 d after Agrobacterial infiltration were detached and mounted in water. GFP was excited at 488 nm and detected at 515−535 nm and RFP was excited at 561 nm and detected at 590−610 nm. Chloroplast autofluorescence was detected at 700−710 nm.
Analyses of colocalization of GBPL3 in fixed human cells were performed with a 100 × /1.44 oil immersion objective. Hocechst 33342 was excited at 405 nm and detected at 460 to 490 nm, GFP was excited at 488 nm and detected at 515−535 nm and Alexa 647 was excited at 594 nm and detected at 650–670 nm. UV and other channels were acquired sequentially to avoid emission crosstalk.
For time-lapse microscopy of GBPL3 in situ nuclear condensate formation, two-week-old soil-grown plants were sprayed with salicylic acid 1–3 h before imaging assay. Right before image acquisition, true leaves were detached and mounted in water, coverglass was sealed with adhesive tape and observed under a 63 × /1.20 water immersion objective. Images were acquired every 10 s or 15 s for 15 min at a single plane. Nuclei was scanned at 200 Hz speed, 512 × 512 pixels format and 1.0 NA. For the best performance of cells, time-lapse experiment on a single nucleus from one plant was conducted each time.
For analysis of coalescence of GBPL3 nuclear condensate in Arabidopsis and HEK 293T, cells expressing GBPL3 nuclear condensate were imaged for 30 min with 10 s intervals using a 63 × /1.20 water immersion objective (Arabidopsis) or a 100 × /1.44 oil immersion objective (HEK 293T).
Image analysis was performed with Fiji/ImageJ48.
Immunofluorescence
For mammalian cell immunofluorescence, cells were fixed in 4% paraformaldehyde (PFA) in PBS for 15 min. Slides were washed three times with PBS and permeabilized with 0.5% TX-100 for 3 min at RT. Slides were washed with PBS and incubated in blocking buffer (3% BSA, 10% donkey serum in PBS) for 1h at RT. Slides were then incubated with primary antibodies (coilin 1:50, PML 1:50, MCM2 1:200, γ-H2A.X 1:200, H3K9me3 1:200 (a gift from Dr. Lizhen Wu)) overnight at 4 °C. Slides were washed in PBS and incubated with donkey anti-rabbit or anti-mouse Alexa 647 antibody (1:1000) for 90 min at RT. Following PBS washes, nuclei were stained with Hocechst 33342 at a final concentration of 1 μg/mL. Cells were washed one more time with PBS and mounted in ProLong Diamond Antifade and analysed with a Leica SP8 laser scanning confocal with a 100x/1.44 oil immersion objective.
For plant nuclear immunofluorescence of endogenous GBPL3 (Arabidopsis), the experiment was performed as previously described with minor modifications49. In brief, leaves were fixed, via a vacuum pump, in 4% PFA in TRIS buffer (10 mM Tris [pH 7.5], 10 mM EDTA, 100 mM NaCl, 0.1% Triton X-100) for 20 min and washed twice in TRIS buffer. Leaves were then chopped in 500 μL lysis buffer (15 mM Tris [pH 7.5], 2 mM EDTA, 0.5 mM spermine, 80 mM KCl, 20 mM NaCl, 0.1% Triton X-100) using a razor blade and filtered through a 40 μm cell strainer. Nuclei were pelleted via centrifugation (1,500g, 4 °C), washed twice with wash buffer (20 mM Tris-HCl [pH 7.5], 25% glycerol, 2.5 mM MgCl2, 0.2% Triton X-100). Nuclei were pelleted and supernatant was discarded. Five microlitres of nuclei suspension were added to 12 μL sorting buffer (100 mM Tris [pH 7.5], 50 mM KCl, 2 mM MgCl2, 0.05% Tween-20, 20.5% sucrose) and air dried on microscope cover glass pre-washed with Hellmanex III (Sigma, Z805939) for 2 h at RT and then post-fixed in 4% PFA in PBS for 20 min. Slides were washed three times in PBS and incubated in blocking buffer (3% BSA, 10% donkey serum in PBS) for 30 min at 37 °C. Nuclei were incubated with primary antibody: anti-GBPL3 (1:500, 4 h, RT). Slides were washed three times in PBS and incubated with donkey anti-rabbit Alexa 488 antibody (1:1000) for 90 min at RT. Following PBS washes, nuclei were stained with Hoechst 33342 (1 μg/mL in PBS) for 10 min, washed once in PBS and mounted in ProLong Diamond Antifade. Nuclei were analysed with a Leica SP8 laser scanning confocal with a 100x/1.44 oil immersion objective.
Fluorescence recovery after photobleaching
For the in vivo assays, FRAP of EGFP-GBPL3 nuclear condensates in N. benthamiana was performed on a Leica SP8 laser scanning confocal microscope. A point of a GBPL3 nuclear condensate was bleached using a laser intensity of 100% at 488 nm. Recovery was recorded every 10 s for a total of 500 s after bleaching.
For the in vitro assays, FRAP of phase separated GBPL3 droplets was carried out using a Leica SP8 laser scanning confocal equipped with a 100 × /1.44 oil immersion objective. A region of one droplet was bleached using a laser intensity of 100% at 488 nm (4 iterations). The recovery from photobleaching was recorded every 10 s for 10 min.
For FRAP quantitative image analysis, the experiment was performed as described in50 using Fiji software48. In brief, half of a GDAC granule was bleached per cell and a stack of images was recorded. Image shift across XY coordinates was corrected with the StackReg translation registration plug-in51. For each bleached GDAC granule, a kymograph was created using the “Reslice” function on a 4-pixel-wide rectangle spanning the entire granule. Fluorescence intensity along the 4-pixel-wide rectangles over the bleached and unbleached regions of each kymograph were measured, respectively. To calculate fluorescence recovery, for each time point, background fluorescence was subtracted from bleached and unbleached regions. The signal from the bleached region was then divided by that from the unbleached region. The recovery plot was further normalized to the average of the ratios of the five pre-bleach time points to correct photobleaching due to repeated measurements.
Determination of in vivo eGFP–GBPL3 concentration via quantitative confocal microscopy
Generation of eGFP standard curve.
The rEGFP was purified as described above. The concentration and purity of EGFP stock solution were determined via BCA and Coomassie blue staining after SDS–PAGE electrophoresis using a serial amounts of bovine serum albumin (BSA; Pierce) as reference. For the EGFP standard curve, 384 well glass bottom plates were used (Cellvis; P384–1.5H-N). The wells of plates were coated with 1% BSA in DPBS for 30 min and wash three times with deionized water immediately before use. Dilutions of EGFP were made in buffer containing 20 mM Tris-HCl, pH 7.9, 150 mM NaCl and 1 mM TCEP. Fifty microlitres of different dilutions of EGFP were transferred to 384-well plates for imaging.
Quantification of in vivo eGFP–GBPL3 concentration.
Leaves of 3-week-old gbpl3–3/EGFP-GBPL3 complementing lines were imaged with a 63 × /1.20 water immersion objective. The argon laser was used for excitation of EGFP at 488. EGFP fluorescence emission was detected with a band-pass filter at 500 to 520 nm. The optical slices were acquired in confocal mode (1 Airy unit) with an average of six scans. The mean pixel intensity in the nucleoplasm and GBPL3 condensates area was measured with ImageJ. All fluorescence intensities measured were corrected for autofluorescence by subtracting the fluorescence intensity measured in wild-type lines in the corresponding tissue and cell type. To correlate the fluorescence intensity in the nucleus with the EGFP calibration curve, the confocal settings and data analysis were kept the same for all images obtained during an experiment. Each quantitative imaging session was accompanied by a parallel calibration curve to avoid differences due to experimental conditions.
Transmission electron microscopy
HEK 293T cells expressing CMV::EGFP-GBPL3 24 h after transfection were fixed in 0.1% glutaraldehyde and 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 1 h at room temperature, and then in 4% paraformaldehyde in the same buffer overnight at 4 °C. Cells were rinsed in PBS and re-suspended in 10% gelatin, chilled and trimmed into smaller blocks and submerged in cryoprotectant of 2.3 M sucrose overnight on a rotor at 4 °C, subsequently they were frozen rapidly in liquid nitrogen. The 60 nm frozen sections were cut with a Leica FC6 cryo-ultramicrotome and sections were collected on drops of sucrose, thawed on formvar/carbon coated grids. Immuno-labelling procedure was performed with the primary antibody against GFP (Sigma, 11814460001) in 1% fish skin gelatin in PBS, then with the anti-IgG antibody, followed by 10-nm protein A-gold particles. After staining by 2% uranyl acetate and lead citrate, the grids were examined in a FEI Tecnai transmission electron microscope (TEM) at 80 kV accelerating voltage. Digital TEM images were recorded with an Olympus Morada CCD camera and iTEM imaging software.
Negative-stain electron microscopy of reconstituted GDAC granules
EGFP-GBPL3 droplets were assembled in vitro at room temperature for 10 min at a concentration of 0.2 mg/mL in buffer (20 mM HEPES [pH 7.5], 250 mM NaCl, 1 mM TCEP, 10% Ficoll). Copper coated carbon film 400 mesh EM grids (EMS, CF400-Cu-50) were glow-discharged for 20 s to increase the hydrophilicity of the carbon surface. Five microlitre pre-incubated EGFP-GBPL3 droplets were transferred to the top of EM grids, incubated for 1 min, and quickly washed with 5 μL 2% uranyl formate and stained by the same 2% uranyl formate for 1 min. The negative staining EM grids were examined in JEOL1400plus electron microscope.
Cryo-electron tomography
Cell seeding on EM grids.
EM grids (Quantifoil R2/1 gold 200 mesh, Q225AR1) were placed onto 18 mm2 cover-glass that had been washed and stored in 100% ethanol. They were then placed into glass bottom dishes (Cellvis, 35 mm dish with 20 mm micro-well cover glass) and treated with 70% ethanol under UV illumination for 10 min at room temperature52. EM grids were washed 6 times with sterile and degassed water before treatment with 0.1 mg/mL poly-d-lysine (Thermo Fisher Scientific) for 60 min in a 37 °C incubator followed by washing in degassed/distilled water. Poly-d-lysine-coated grids were incubated in DMEM medium overnight at 37 °C incubator with 5% CO2. HEK 293T cells transfected with EGFP-GBPL3 were cultured for 30 h at 37 °C with 5% CO2. Cells were trypsinized (0.05% Trypsin-EDTA) and resuspended to a final cell density of 80,000 cells/mL in DMEMsupplemented with 10% fetalbovine serum (FBS), seeded on the EM grids, and further incubated for 6 h.
Vitrification and cryo-correlative fluorescence light microscopy.
EM grids seeded with HEK 293T cells expressing EGFP-GBPL3 were pre-screened using a fluorescent light microscopy, clipped on a custom manual plunger, blotted using Whatman no. 1 filter paper (GE Healthcare, 1001–110), and frozen in liquid ethane by the plunger53. Frozen grids were transferred onto a cryo-stage and clipped within the O-ring and C-ring (ThermoFisher Scientific, cryo-FIB autogrid and C-clip, respectively). Cryo-CLEM (Leica Microsystems EM Cryo-CLEM microscope) was performed as previously described54. Grids were transferred into Leica-CLEM cartridge docked at a pre-cooled shuttle docking station and then transferred onto a cryo-stage equipped with a pre-cooled 40× objective lens. A spiracle 4×4 grid region was selected and 16 sub-regions were imaged. Montage GFP-positive images were acquired on the selected area using the Z-stack mode within a stepwise of 0.35 μm. This montage served as the navigating map in subsequent milling assays by the focused ion beam method.
Milling lamella by focused ion beam.
Cryo-CLEM demarcated grids were transferred into cryo-DualBeam microscope equipped with cryo-stage and cryo-transfer shuttle systems (Thermo Fisher Scientific, Aquilos cryo-FIB focused ion beam/scanning electron microscope). The lamella was prepared according to Aquilos cryo-FIB protocols. The sample on the O-ring side of the grid was sputter-coated with platinum (1 kV, 30 mA, 10 pa, 15 s) to increase sample conductivity during FIB milling55. MAPS software was used to capture an EM grid montage in the electron beam mode before importing the fluorescent montage from cryo-CLEM to generate a merged map to establish potential targets for milling lamella. Eucentric height of the targeting area was refined and stage tilting angle determined at 16° for lamella. At target position on the grid organometallic platinum coating was sprayed for 5 s by a gas injection system (Thermo Fisher Scientific, GIS). Initially, rough milling was starting at 3 μm distance in width with 300 pA current and continued in a stepwise manner, stopping at 0.5 μm distance with 50 pA current. Fine milling was then performed at <50 pA. An electron beam was used to monitor the milling process at 5 keV and 13 pA. A final fine milling was done by tilting the stage 0.3° from the original 16°. Upon completion of the milling, the grid was sputter-coated with platinum (15 mA, 10 Pa, 10 s) to increase conductivity of milled lamella. The grid was transferred out of the FIB machine using the transferring shuttle system and either stored in a grid box (SubAngstrom, Pin type grid box) in liquid nitrogen until further data collection or re-confirmed by CLEM for correct positioning of GBPL3 condensates.
Cryo-electron tomography and image processing.
Tomography data were collected using a Titan Krios G2 transmission electron microscope (Thermo Fisher Scientific) equipped with a 300 kV field-emission gun, a Volta phase plate (phase shift located around 1/2 pi), a Quantum post column energy filter (20-eV slit) and a Gatan K2/K3 summit direct detection camera. The images were taken in a dose-fractionated mode at near focus using SerialEM software56,57.The resulting physical resolution is 0.43 nm/pixel. A total dose of 80 e/Å2 distributed among 51 tilts/tilt images was undertaken covering angles from 43° to −59° at tilting step of 2° and starting the first tilt series at −9° in a dose symmetric scheme58. The data set as shown in the Extended Data Fig. 9 were taken at the physical resolution of 0.62 nm/pixel for requiring large area of lamella. These data sets were collected using bi-directional acquisition scheme (starting from 9° to 60°; return to 9° and then collect the negative tilts series to −42°) with a 3° increment.
Collected dose-fractioned data were subjected to motion correction for generating drift-corrected image stack files59,60. Stack files were aligned using patch-tracking function of IMOD61. 3D tomograms were reconstructed from aligned stack files by SIRT (Simultaneous Iterative Reconstruction Technique) method using Tomo3D62. For reconstruction of binned tomograms, the tilt series was scaled to 1.72 nm as pixel size. The image shown in the Extended Data Fig. 9h was from an unbinned tomogram with a low pass filter in the function of IMOD/Image/Process (Fourier filter: Hig-frequency cutoff, 0.12; High-frequency falloff, 0.02). IMOD was used for visualizing the tomogram.
Surface rendering of tomogram was done with EMAN2.2363, and refined with UCSF chimaera64. In brief, for ribosomes, a template-matching strategy was performed to determine all ribosome coordinates and orientations. An 80S ribosome structure (EMD-3418) determined by Volta phase plate cryo-ET65, was low-pass filtered to 20 Å and matched to the tomograms. For nuclear envelope, EMAN2.3 was employed to do segmentation on membrane-like features. Nuclear pore complex (EMD-3103) solved by in situ cryo-ET sub-tomogram averaging technique was fitted into the cavity area in the nuclear envelope density. For GDACs and vesicles, IMOD package was employed to perform manual segmentation using the function of drawing tool and interpolation.
Malachite green phosphate assay
GTPase activity of purified guanylate binding proteins were performed by estimating inorganic phosphate liberated from GTP during enzymatic reaction. In brief, 0.5 μM of each protein/protein combinations were incubated with 150 μM of GTP in buffer containing 20 mM HEPES [pH 7.4], 500 mM NaCl, 5 mM KCl and 10 mM MgCl2 for 60 min at room temperature. Reactions were quenched by the addition of EDTA. Amount of inorganic phosphate was estimated by a malachite green assay kit according to manufacturer’s instructions (Sigma, MAK307).
Thin-layer chromatography
Thin-layer chromatography (TLC) was used to separate GTP, GDP and GMP and performed exactly as previously described18. In brief, α-[32P] GTP hydrolysis by purified recombinant proteins in reaction buffer (20 mM HEPES [pH 7.0], 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 100 μM GTP, 10 μCi α-[32P]GTP) was determined at 25 °C before quenching the reaction with 142 mM EDTA after 7 h. The resulting products were separated by PEI cellulose (Sigma, Z122882) with fluorescent indicator (UV 254) using 750 mM KH2PO4 (pH 3.5) as solvent and visualized by autoradiography.
IP-MS/MS analysis of GBPL3 in Arabidopsis
Nuclei were isolated as described above from Arabidopsis Col-0/pGBPL3::GBPL3–3×FLAG plants. GBPL3 protein complexes were enriched with Flag-M2 agarose. Elutes were denatured and resolved with SDS–PAGE and subjected to silver staining. Visible bands were excised, and protein identity was analysed by MS/MS at the Taplin mass spectrometry facility at Harvard University.
Construction of combinatory nuclear GBPL3 protein–protein interaction networks
GBPL3 interactors from our IP-MS/MS were combined with those from a recent study31 (http://plants.proteincomplexes.org; CF-MS scores >0.2) and queried on STRING (Search Tool for the Retrieval of Interacting Genes/Proteins; confidence score = 0.4). Interactions were retrieved and network was built with Cytoscape 3.8.0.
ChIP analysis
ChIP experiments were performed as previously described66 with minor modifications. About 1.5 g of 25-day-old soil-grown plants were cross-linked in 37 ml of 1% w/v formaldehyde in PBS + 0.01% Silwet L-77 solution under vacuum for 20 min. To quench cross-linking, 2.5 ml 2 M glycine (final conc. 125 mM) was added and the sample was vacuumed for an additional 5 min. The tissue was rinsed three times with 40 ml of cold PBS and dried with blotting paper. The nuclei were isolated as described above and re-suspended in 300 μl of nuclei lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS, 1xPI). The nuclei suspensions were subsequently sonicated to shear the DNA to an average size of 0.2–1 kb with a Q125 sonicator (Qsonica, USA) using the following parameters: Amplitude, 70%; pulse rate, 5 s/40 s on/off cycles; total on time, 1 min.
The sonicated chromatin was spun at 13,000 rpm for 10 min at 4 °C to pellet cell debris. The supernatant (300 μl) was transferred to a new 15 ml tube and diluted 10 times by adding 2.7 ml ChIP dilution buffer (2.7 ml; 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.0], 167 mM NaCl). For pre-clearing, 100 μl of Protein A agarose beads equilibrated with ChIP dilution buffer was added and rotated end-over-end for 1 h at 4 °C. The beads were pelleted at 3,000 g for 3 min and the supernatants were divided equally into two samples (1.4 ml each). An aliquot of 28 μl supernatants was allocated into a clean 1.5 ml Eppendorf tube as ‘input’ (2%). For immunoprecipitation, 5 μg of anti-GBPL3 antibody was added to one sample and 5 μg of normal rabbit immunoglobin G (IgG) was added to the other sample as control. The samples were incubated overnight at 4 °C with gentle rotation. Subsequently, 50 μl of Protein A agarose beads equilibrated with ChIP dilution buffer was added to each sample and kept at 4 °C for 1 h with gentle agitation to pull down antibodies.
The Protein A beads were then pelleted by centrifugation at 3,000g for 3 min at 4 °C. The beads were washed sequentially with low salt wash buffer (150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0), high salt wash buffer (500 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0), LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 10 mM Tris-HCl, pH 8.0, 1 mM EDTA) and TE buffer. For each buffer, a quick wash by spinning at 3,000g for 2 min and a second wash with 10 min agitation were performed. A volume of 1 ml of buffer was used in each wash.
After the final wash, beads were pelleted, and the supernatant was removed thoroughly. To elute the immune complexes, 250 μl of freshly prepared Elution Buffer (1% SDS, 0.1 M NaHCO3) was added to the beads. The samples were vortexed briefly and incubated at 65 °C for 15 min with gentle rotation. After spinning at 3,000g for 2 min, the supernatant was carefully transferred to a fresh tube. The pellet was eluted one more time with 250 μl of Elution Buffer and the two eluates were combined (a total of ∼500 μl). At the same time, 472 μl of Elution Buffer was added to the input samples collected before immunoprecipitation.
For reverse-cross-linking, 20 μl of 5 M NaCl were added to each sample and incubated at 65 °C for 16 h with gentle rotation. Subsequently, a volume of 1 μl of 10 mg/ml DNase-free RNase A was added to each sample and incubated at 37 °C for 1 h. Afterwards, 10 μl of 0.5 M EDTA, 20 μl 1 M Tris-HCl (pH 6.5) and 2 μl of 10 mg/ml proteinase K were added to each sample and incubated at 45 °C for 1 h. DNA was further extracted with phenol:chloroform:isoamyl alcohol (25:24:1 v/v), resuspended in 100 μl TE buffer and stored at −20 °C for further use.
Statistics and reproducibility
Data were analysed by Microsoft Excel, GraphPad Prism 8.0 software or R. Statistical significance was determined by t-test (two-tailed), one-way ANOVA or two-way ANOVA (multiple comparisons with Bonferroni or Holm–Sidak post hoc test). All experiments were repeated at least three times with similar results unless otherwise mentioned in figure legends.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The original raw RNA-seq data that support the findings of this study have been deposited and made publicly available in the NCBI Gene Expression Omnibus with accession number GSE134651. RNA-seq data are provided in Supplementary Tables 4 and 5. Full versions of all gels and blots are provided in Supplementary Fig. 1. The phylogenetic source data related to Extended Data Fig. 1b and high-resolution images of Fig. 4c are available on Dryad from https://doi.org/10.5061/dryad.g1jwstqqv. Source data are provided with this paper.
Extended Data
Extended Data Fig. 1 |. Retrieval and structural characteristics of plant IDR-containing GBPLs.

a, Pipeline of IDP-based in silico screening of the Arabidopsis proteome. b, Phylogenetic tree inferred by Bayesian analysis. Support value: Left, Nodal posterior probabilities; Right, Maximum likelihood bootstrap values. Scale bar, substitutions per site. Number of amino acid (#aa) not drawn to scale. c, 3D structure prediction of Arabidopsis GBPLs performed on I-TASSER web server and projected with PyMOL software. GD, GTPase domain. HD, helical domain. IDR, intrinsically disordered region. hGBP1, 1F5N. PDB. d, Domain configuration and size of the Arabidopsis GBPL family versus human GBP1. e, GBPL disorder and hydrophobicity plots carried out using ProtScale and PONDR servers, respectively. Charged amino acids (red, glutamate/ aspartate; blue, arginine/lysine) displayed as vertical lines atop panels. IDRs are shaded.
Extended Data Fig. 2 |. Generation and characterization of Arabidopsis gbpl mutants.
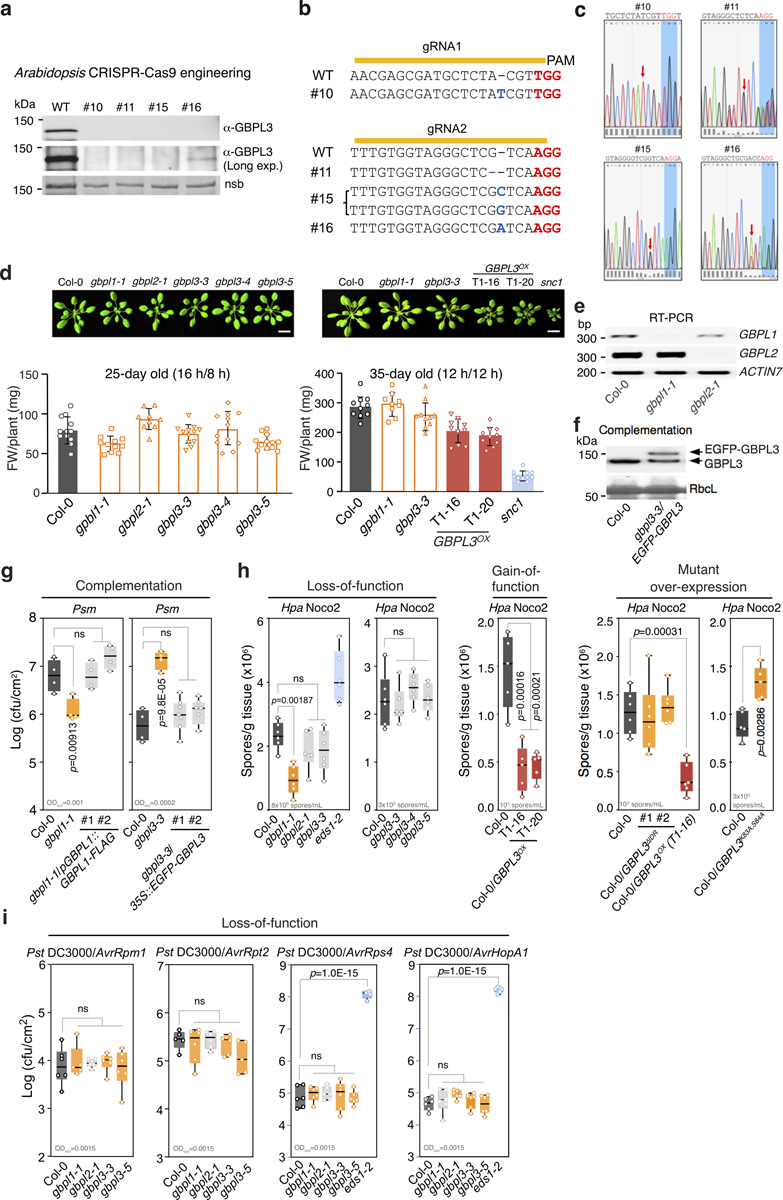
a, Immunoblot of GBPL3 protein level using α-GBPL3 rabbit polyclonal antibody in wild type (WT) (Col-0) and four independent T1 transformants generated via CRISPR/Cas9 targeting GBPL3 (predicted molecular weight 122 kDa). Ponceau S staining of a non-specific band (nsb) serves as a loading control. Proteins extracted from five-week-old plants. Experiment performed once only due to the limit of tissue materials from sterile plants. b, PAM sequences of gRNAs and the consequences of CRISPR-Cas cleavages in T1 generation. Line 15 is biallelic and line 16 is heterozygous. c, Chromatograms of sanger sequencing of the mutation sites in CRISPR lines in b. d, Fresh weight of plants (mean ± SD) shown above (top-right graph the same as Fig. 1b for comparison with left). e, Gene expression of GBPL1 and GBPL2 in T-DNA mutants. f, Immunoblot (α-GBPL3) of GBPL3 and EGFP–GBPL3 levels in complemented plants. Ponceau S staining of RbcL serves as a loading control. g, Growth of Psm ES4326 (Psm) at day 3 of infection. Inoculum, bottom left. h, Hpa Noco2 sporulation in 2~3-week-old plants at day 7. Inoculum, bottom left. i, GBPL proteins are not required for Pst DC3000 Avr challenge using gbpl mutant lines. Growth of Pst-Avr at day 3 of infection. Inoculum, bottom left. g–i, Box = 25th and 75th percentiles; bars = min and max values. Statistical analysis, comparison of mean via one-way ANOVA test (Bonferroni post hoc correction). ns, not significant. Individual data points represent biologically independent samples (d, g–i).
Extended Data Fig. 3 |. RNA-seq and defence-gene expression analysis in transgenic plants.
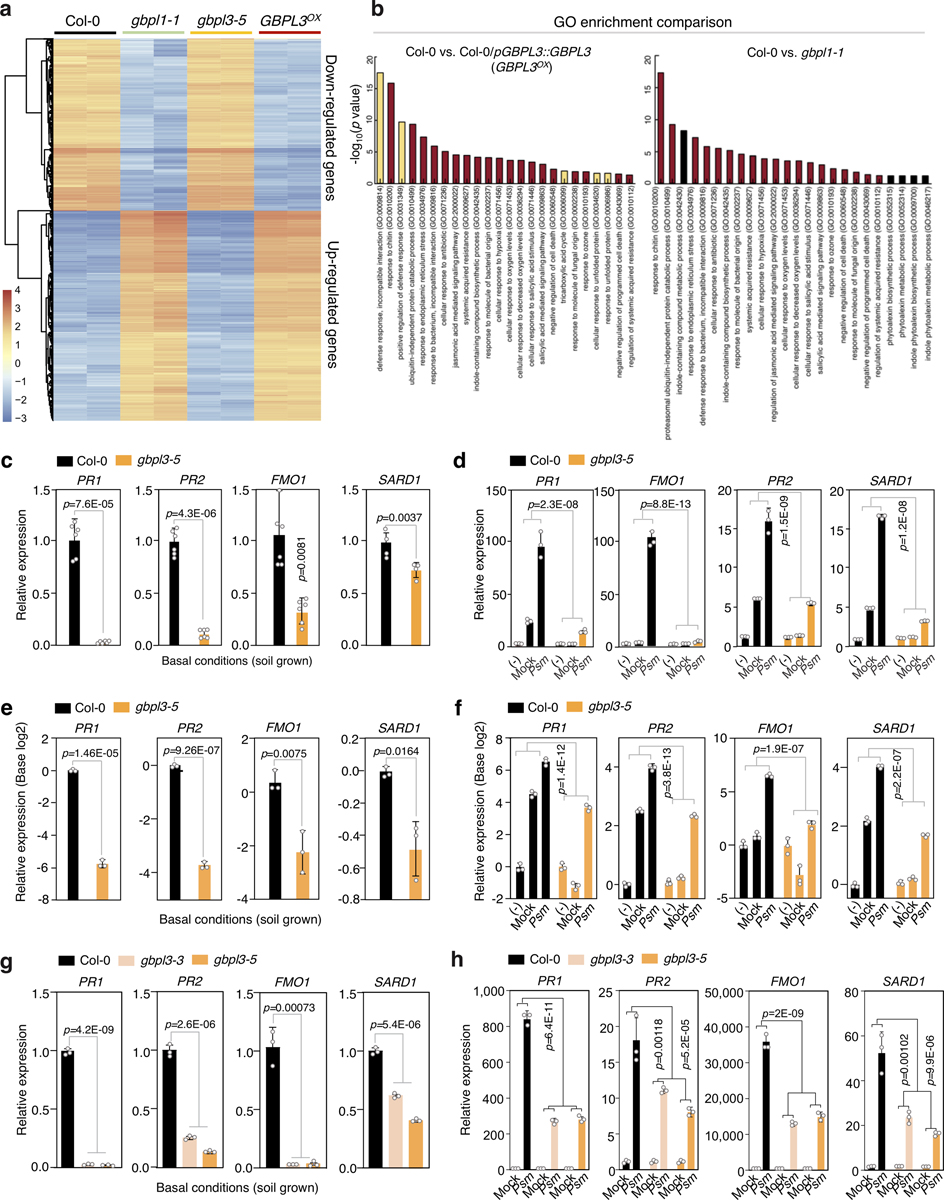
a, Comparative heat map of the top 1,000 differentially expressed genes (RNA-seq; base log2) across Arabidopsis genotypes from two biologically independent replicates. b, GO enrichment of upregulated genes in Col-0/pGBPL3::GBPL3 (GBPL3OX) (left) and gbpl1–1 (right) plants versus WT. Top 25 significantly enriched GO terms are shown. Red, enriched GO-terms found in both GBPL3OX and gbpl1–1 plants. c, d, Defence-gene expression (mean ± SD) in under basal and induced conditions. RNA extracted 4 h after mock (MgCl2) or Psm ES4326 (OD600 = 0.1) infection. Expression normalized to ACTIN7. e, f, Defence-gene expression (mean ± SD) under basal and induced conditions shown as log2 transformations for comparison. g, h, Relative expression (mean ± SD, normalized to ACTIN7) in two different gbpl3 mutants. RNA extracted 24 h after mock (MgCl2) or Psm ES4326 (OD600 = 0.001) infection from 4-week-old plants. Statistical analysis, two-tailed Student’s t-test (c, e), one-way ANOVA with Holm–Sidak post hoc test (g) and two-way ANOVA with Holm–Sidak post hoc test (d, f, h). Individual data points represent biologically independent samples with experiments undertaken twice with similar results (c–h).
Extended Data Fig. 4 |. Nuclear GBPL1–GBPL3 analysis in Arabidopsis and N. benthamiana.
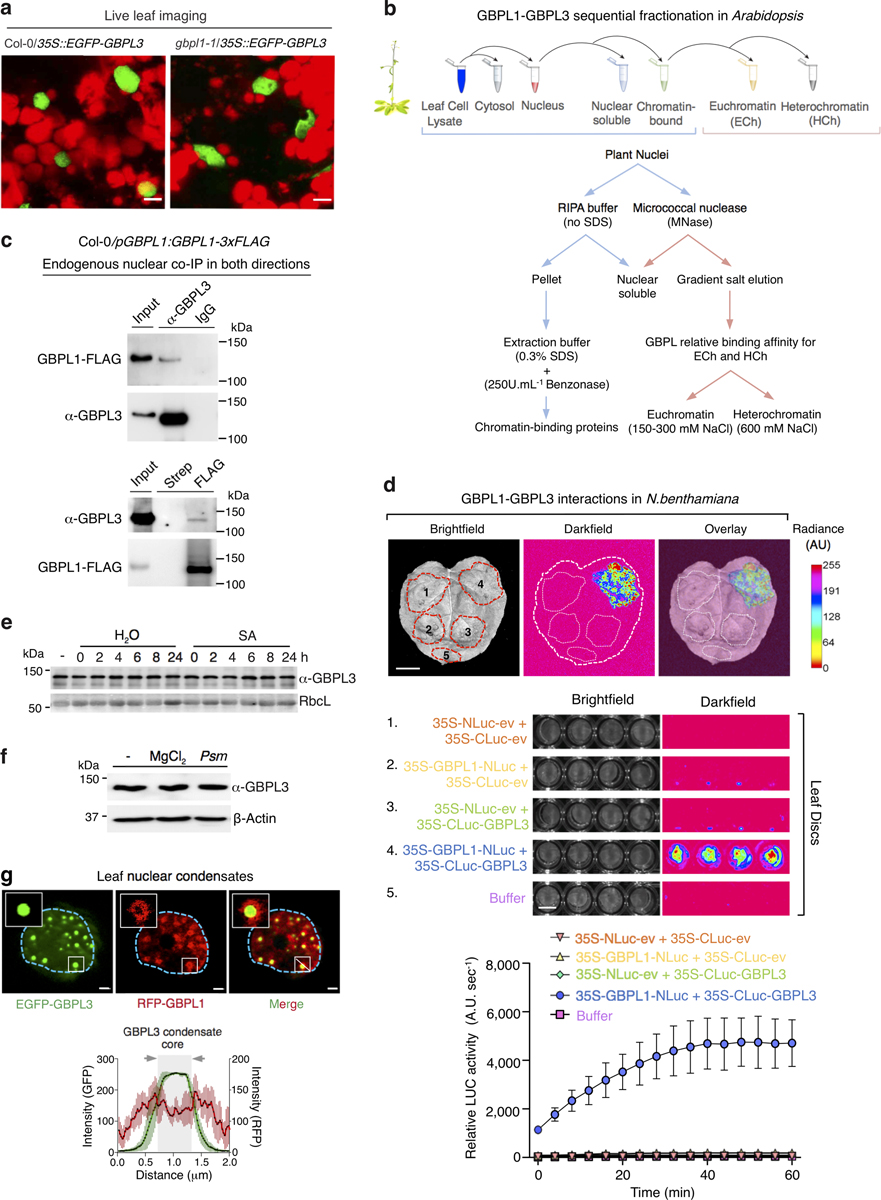
a, Live imaging of EGFP–GBPL3 in the nucleus of 2-week-old transgenic Col-0 or gbpl1–1 plants under basal conditions. Bar, 5 μm. b, Sequential fractionation strategy to examine chromatin binding properties of GBPL1 and GBPL3 in Fig. 2. using Col-0/pGBPL1:GBPL1-3×Flag plants to ensure physiological expression. c, Nuclear co-immunoprecipitation of GBPL3 with FLAG-M2 agarose (bottom) and GBPL1 with α-GBPL3 antibody (top). Streptavidin agarose and rabbit normal IgG served as negative controls. d, Split luciferase complementation assay in N. benthamiana. (top) Luminescence image of N. benthamiana leaves co-infiltrated with agrobacterial strains containing plasmids shown below. (bottom) Quantification of LUC activity (mean ± SD, n = 4 biologically independent leaf discs). e, Time course of total GBPL3 levels by immunoblot after salicylic acid treatment (0.5 mM). Ponceau S stained RbcL serves as loading control. f, Immunoblot of total GBPL3 protein levels 24 h after Psm ES4326 (OD600 = 0.01) infection from 4-week old plants. g, Top, Exclusion of mRFP-GBPL1 to the periphery of EGFP–GBPL3 nuclear condensates when co-expressed in N. benthamiana leaves. Scale bar, 2 μm. Inset bar used for average line profiling. Bottom, Average line profile (2 μm) of fluorescence intensity spanning GBPL3 condensates. Bar = mean ± s.d. (n = 5 biologically independent condensates). Shaded area with arrows, GBPL3 condensate core.
Extended Data Fig. 5 |. GBPL droplets are unique LLPS structures conserved across plant species, cell types and during development.
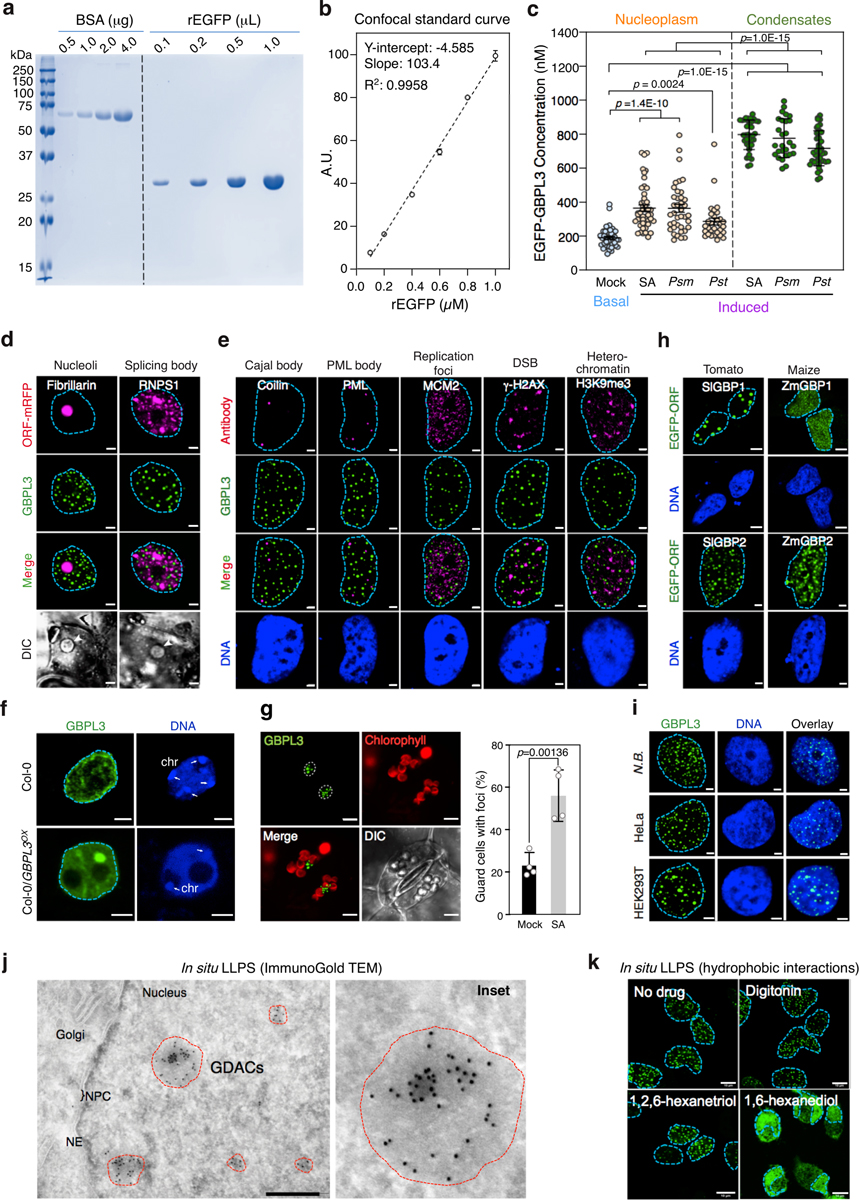
a, Coomassie staining of purified rEGFP protein. b, Standard curve of rEGFP via confocal microscopy. c, Quantitative measurement (mean ± SD) of EGFP–GBPL3 concentration 24 h after salicylic acid (0.5 mM), Psm (OD600 = 0.001) and Pst (OD600 = 0.001) from multiple 4-week-old plant nuclei. Comparison of mean via one-way ANOVA test with Holm–Sidak post hoc test. Individual data points represent biologically independent samples. d, e, Colocalization analysis of EGFP–GBPL3 in N. benthamiana (d, live cell) and HeLa cells (e, fixed cell). The latter enabled parallel antibody detection of subnuclear structures. DSB, double strand break. DNA stained with Hoechst 33342 dye. Bar, 5 μm (d, N.B.) and 2 μm (e, HeLa). f, Immunofluorescence of native GBPL3 in Col-0 and Col-0/GBPL3OX plants. Nuclei were isolated from 3-week-old soil-grown plants and DNA stained with Hoechst 33342. chr, chromocentre (white arrows). Bar, 2 μm. Experiment repeated once with similar results. g, Left, GBPL3 condensates in Arabidopsis cotyledons stably expressing 35S::EGFP-GBPL3. Shown are maximum projection images. Dashed circle, nuclear boundary. Bar, 5 μm. Right, salicylic acid-induced GBPL3 condensates (mean ± SD, Student’s t-test, two-tailed) in guard cells from 10-day-old Arabidopsis cotyledons (n = 4 biologically independent plants with 100 guard cells each). Mock, water treatment. h, Localization of EGFP-tagged tomato and maize GBPs in HeLa cells. DNA stained with Hoechst 33342 dye. Bar, 5 μm (SlGBP2 and ZmGBP1) and 2 μm (SlGBP1 and ZmGBP2). i, Maximum intensity projection images of EGFP–GBPL3 transiently expressed in N. benthamiana (N.B.), human HeLa and HEK 293T cells. Bar, 2 μm. j, Immunogold transmission electron microscopy of spheroid GDACs (dashed circles) expressed in HEK 293T cells. NE, nuclear envelope. NPC, nuclear pore complex. Bar, 500 nm. k, GBPL3 condensates were sensitive to 1,6-hexanediol (5%) but not 1,2,6-hexanetriol (5%). Digitonin (10 μg/ml) was used to facilitate chemical delivery.
Extended Data Fig. 6 |. Comparative LLPS behaviour of GBPLs in cells and cell-free systems.
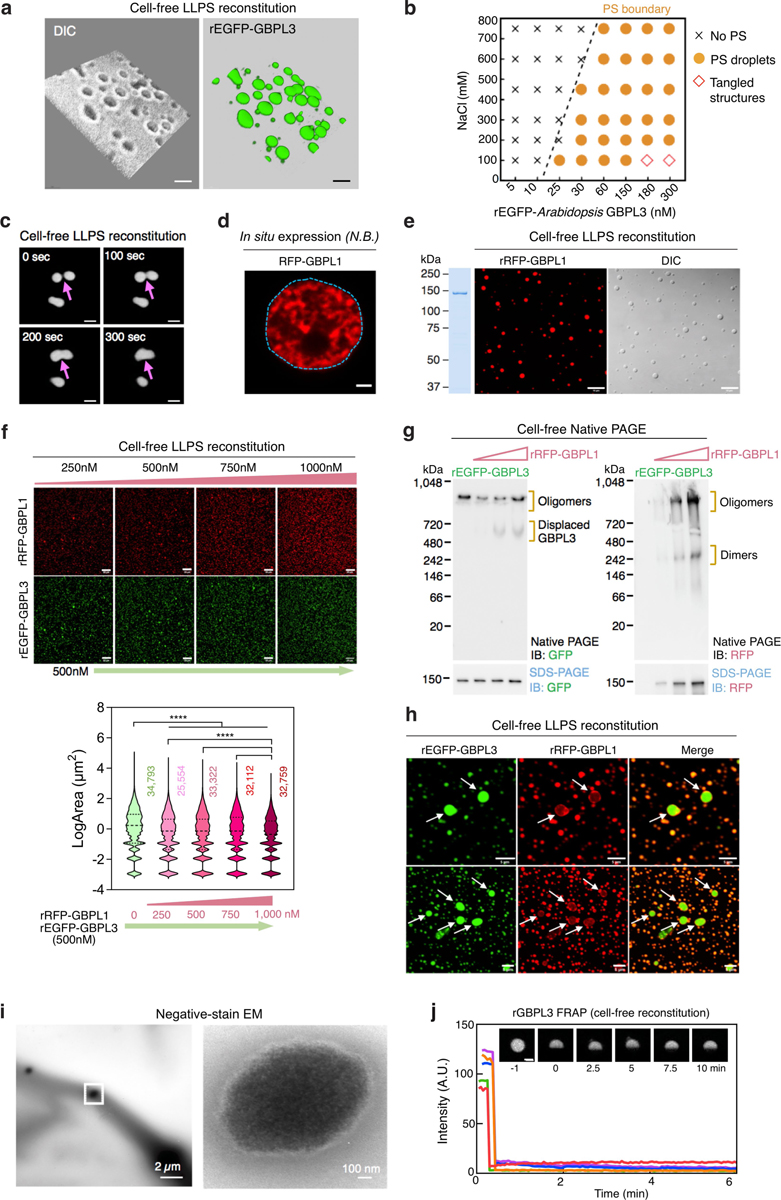
a, A 3D volume view of rEGFP–GBPL3 droplets (10% Ficoll) depicted in (Fig. 3d). Bar, 2 μm. b, Phase separation (PS) diagram of rEGFP–GBPL3 across NaCl concentrations 30 min after reconstitution. c, Confocal images showing GBPL3 condensates fusion behaviours every 100 s in vitro. Images collected 30 min after reconstitution in droplet buffer (20 mM HEPES [pH 7.5], 200 mM NaCl, 1 mM TCEP, 10% Ficoll). Bar, 5 μm. d, RFP-GBPL1 does not generate LLPS nuclear condensates in situ in N. benthamiana. e, in vitro phase separation of rRFP–GBPL1 (375 nM) in droplet buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 1 mM TCEP). Coomassie brilliant blue (CBB) of rRFP–GBPL1 is shown on the left. Bar, 10 μm. f, Co-condensation assay of rEGFP–GBPL3 and rRFP–GBPL1 in vitro. Below, quantification of EGFP–GBPL3 droplet size. Median (solid line) and quartile (dashed line) values. Comparison of mean via one-way ANOVA with Holm–Sidak post hoc test (****P < 1.0 × 10−15, n = number of biologically independent droplets). g, Native PAGE of rEGFP−GBPL3 in the presence of increasing concentrations of rRFP−GBPL1 leads to GBPL3 displacement. Same loading controls below the original (anti-GFP) and reprobed (anti-RFP) native gel separated by SDS–PAGE. h, Exclusion of rRFP−GBPL1 from larger rEGFP−GBPL3 droplets in co-condensation assays. i, Representative negative stain EM images of rGBPL3 droplets at 24 h. Bar, 2 μm. Inset at right. Bar, 100 nm. j, FRAP analysis of 2 h old rEGFP−GBPL3 droplets in vitro. Bar, 5 μm. Fluorescence recovery of 5 individual droplets shown. Insert, representative images of a bleached droplet over 10 min recovery.
Extended Data Fig. 7 |. In situ cryo-ET analysis of GDACs including CLEM corroboration.
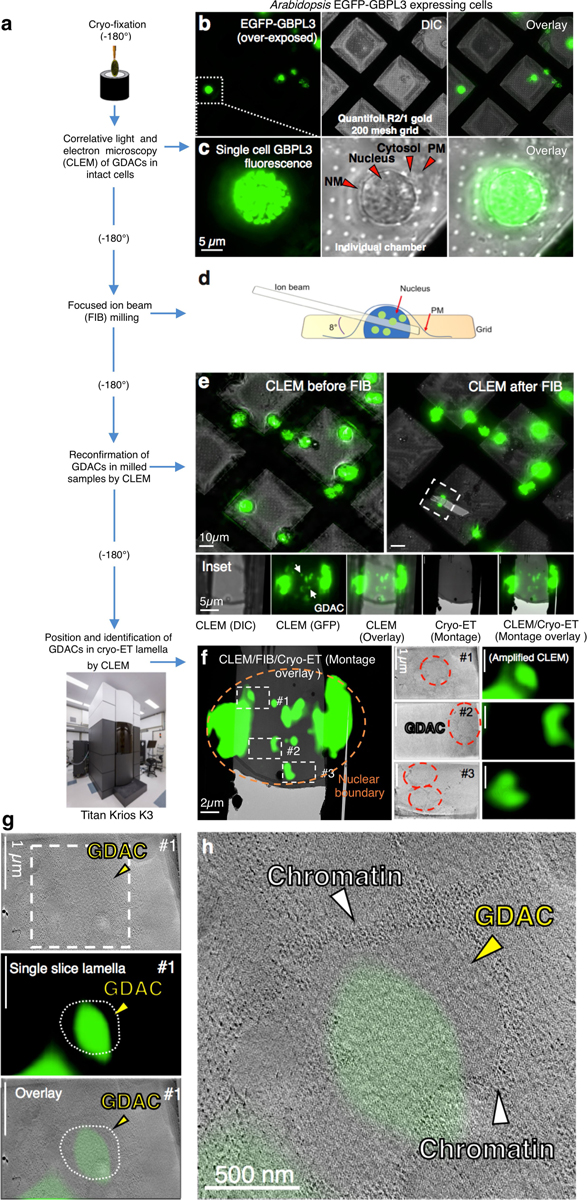
a, Flow chart of in situ cryo-ET analysis. b, c, CLEM images of EM grids seeded with HEK 293T cells transfected with CMV-EGFP-GBPL3. Single cell view of (b) shown below (c). These nuclear GDAC structures form in a species- and kingdom-independent manner with similar structures generated by GBPL3 plant orthologues; hence they appear to share conserved features with those directly generated in Plantae. d, Schema of FIB-milled lamella. e, CLEM images of FIB-milled lamella and overlay with Cryo-ET montage images. f, Left, Selected regions for tomograph collection. Right, Slice tomogram of GDACs and the corresponding CLEM fluorescence of GDACs overlap. g, h, Enlarged and higher resolution (binning factor 1) view of the GDAC in tomographic overlays with CLEM for region 1 of the cryo-ET montage image.
Extended Data Fig. 8 |. In situ cryo-ET analysis of GDACs in larger tomographic slices.

a, Shared similarities of CLEM-verified GDACs (from Extended Data Fig. 7h) with GDACs identified in tomographic images harbouring the nearby nuclear envelope for validating its intranuclear location (right, from Fig. 3e for comparison). Possible chromatin boundaries also depicted in both independently identified GDAC structures. b, CLEM images of EM grids seeded with HEK 293T cells transfected with CMV-EGFP-GBPL3 used to generate the tomogram in (a, right) above. Single cell view of the nucleus used to detect GDACs in far-right panel. Bar, 20 μm. c, FIB-milled lamella of the chosen nucleus from the CLEM above. d, Tomographic slice of the demarcated rectangular area 62 yielded a single demarcated GDAC near the nuclear envelope boundary shown in (a, right) and yielded 3D segmented images in Fig. 3e.
Extended Data Fig. 9 |. GBPL3 IDR contributes to LLPS-driven defence.
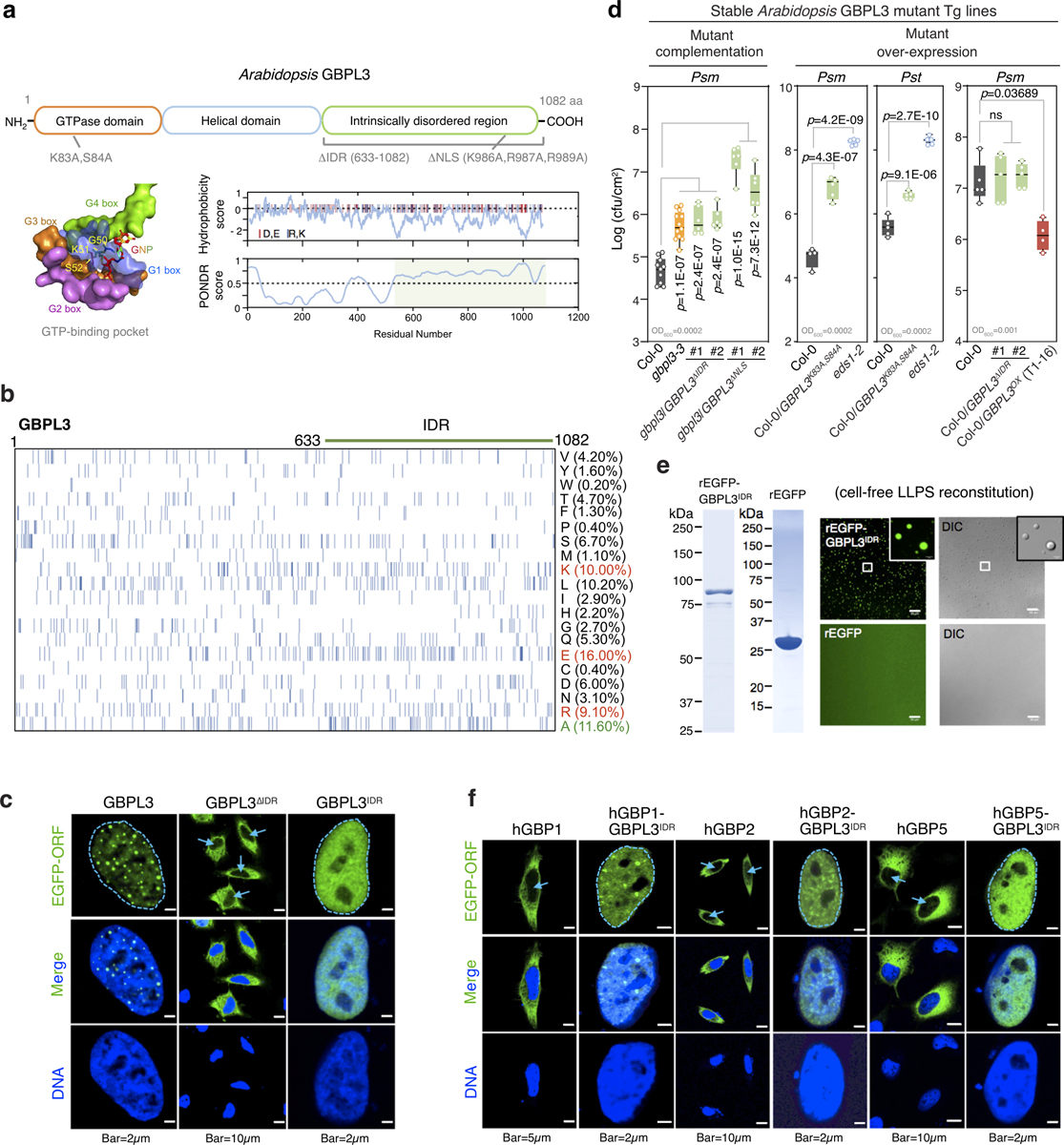
a, Top, domain structures of the Arabidopsis GBPL3 protein. Sites for mutagenesis are shown below. Bottom, GTP-binding pocket with conserved G-boxes (hGBP1 surface representation; 1F5N.PDB). GBPL3 hydrophobicity and disorder plots on right. Charged amino acids (red, glutamate and aspartate; blue, arginine and lysine) displayed as vertical lines atop panels. IDRs are shaded light green. b, Amino acid compositions of GBPL3 full length protein. Each row represents information for a single amino acid. Vertical bars, occurrence of indicated amino acid at that position. Right, amino acid percentage. c, Immunofluorescence of EGFP−GBPL3 mutant variants in HeLa cells 36 h after transfection. DNA stained with Hoechst 33342 dye. Arrows, nucleus. d, Growth of Psm ES4326 (Psm) and Pst DC3000 (Pst) at day 3 of infection. Inoculum, bottom left. Statistical analysis, comparison of mean via one-way ANOVA test (Bonferroni post hoc correction). ns, not significant. Box = 25th and 75th percentiles; bars = min and max values. Individual data points represent biologically independent samples. e, In vitro phase separation of rEGFP−GBPL3IDR and rEGFP. Equimolar (400 nM) amounts incubated at 22 °C for 16 h in droplet buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP, 10% Ficoll). Bar, 20 μm. Left, Coomassie stain. f, Immunofluorescence of plant−human GBP-IDR chimeras performed as in c.
Extended Data Fig. 10 |. GBPL3 GTPase activity is required for LLPS-driven defence.
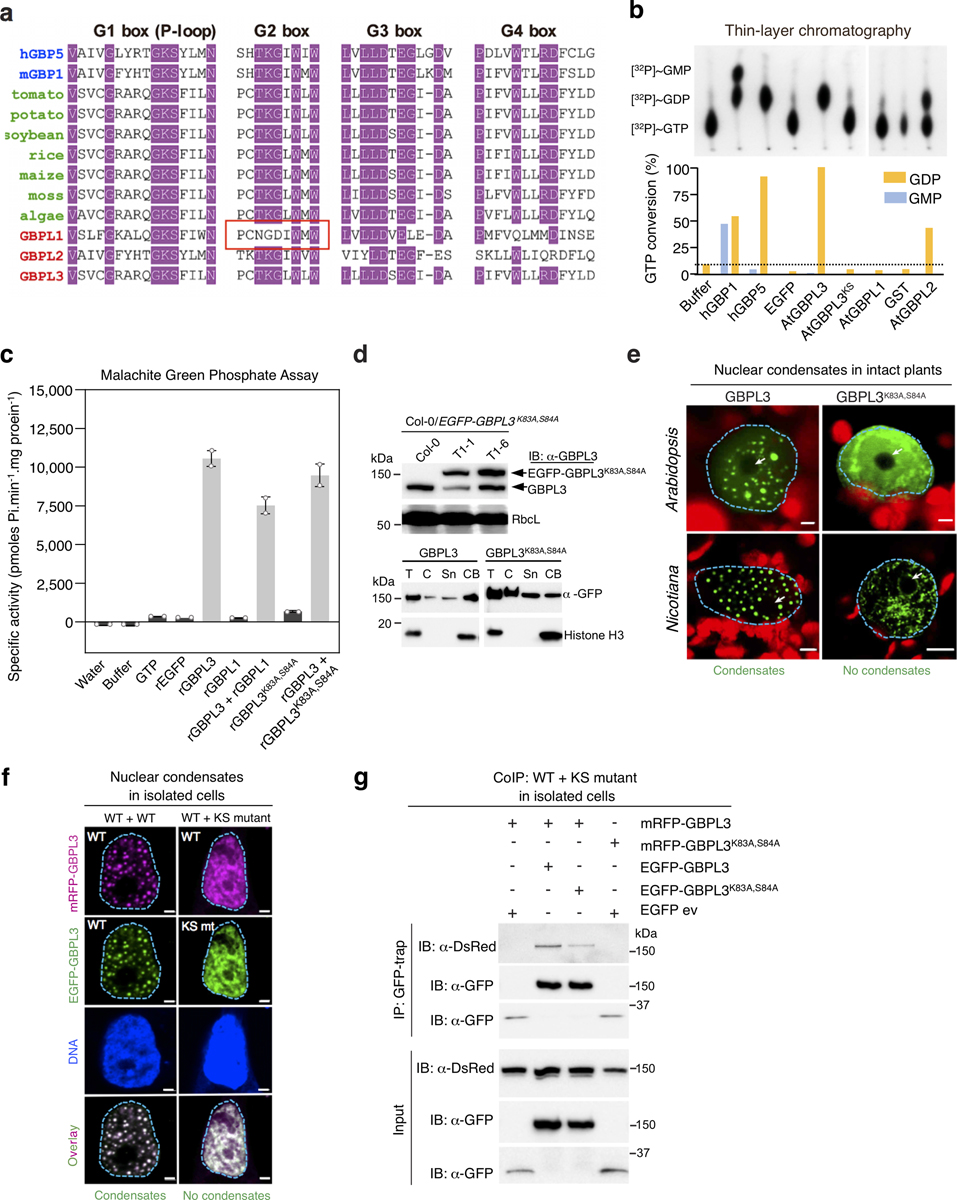
a, Alignment of conserved G-boxes in animal GBPs and plant GBPLs. Red outline denotes non-functional G2 box alterations in GBPL1. b, GTP hydrolysis assay of recombinant proteins along with tag controls. Bottom, percent GTP conversion. c, Malachite green phosphate assay (mean ± SD, n = 2 biologically independent samples) of recombinant GBPL proteins. d, Top, Immunoblot (α-GBPL3) of GBPL3 and EGFP–GBPL3K83A,S84A levels in Col-0 and Col-0/35S::EGFP-GBPL3K83A,S84A plants. T1–6 used in Extended Data Fig. 9d. Ponceau S staining of RbcL serves as a loading control. Bottom, Nuclear fractionation and immunoblot analysis of EGFP−GBPL3K83A,S84A in HEK 293T cells shows the DN GTPase mutant still enters the nucleus and binds chromatin but cannot phase separate. Sn, nuclear soluble. CB, chromatin bound. e, Live imaging of 35S::EGFP-GBPL3 and 35S::EGFP-GBPL3K83A,S84A in N. benthamiana and Arabidopsis plants. Arabidopsis imaging 24 h after salicylic acid (0.5 mM) treatment. Arrows, nucleolus. Bars, 5 μm (N.B.) and 2 μm (Arabidopsis). f, Effect of EGFP−GBPL3K83A,S84A (ks mt) on wild-type (WT) GBPL3 in confocal microscopy. Plasmids were co-transfected in HeLa cells and analysed 36 h post transfection. Bar, 2 μm. g, GBPL3 self-assembly co-IP in HEK 293T cells reveal GBPL3K83A,S84A can bind WT GBPL3 to interfere with its nuclear LLPS function in f. Cells were collected 36 h post transfection.
Extended Data Fig. 11 |. GBPL3 enlists a monopartite NLS motif to target the nucleus where it engages Mediator subunits and excludes CDK8 for LLPS-driven immunity.
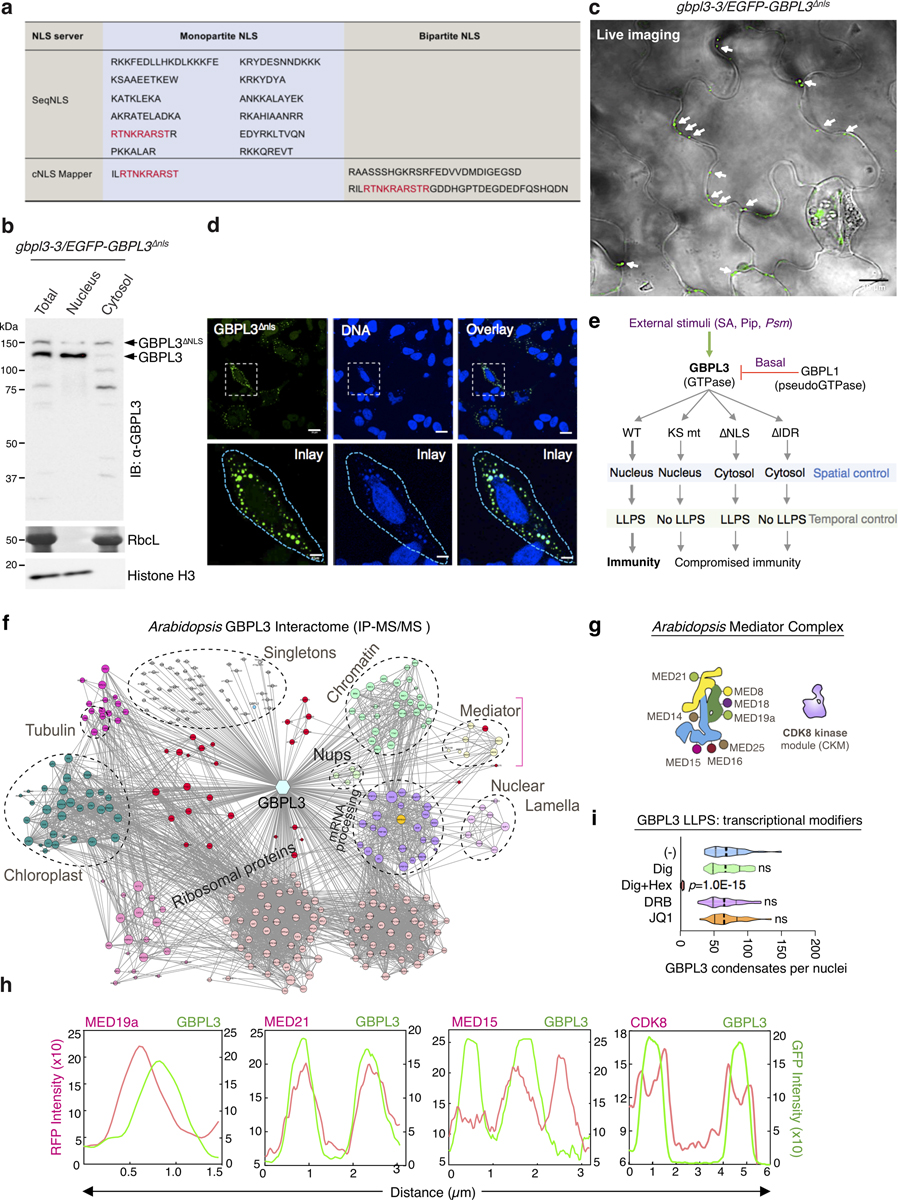
a, NLS motifs in GBPL3. Monopartite and bipartite NLS motifs were predicted via two web servers (SeqNLS and cNLS mapper). Shared sequences in red. b, Subcellular fractionation of gbpl3–3/35S::EGFP-GBPL3ΔNLS plants. RbcL (Ponceau staining) and Histone H3 are cytosol and nuclear controls, respectively. c, Live cell imaging of gbpl3–3/35S::EGFP-GBPL3ΔNLS plant leaves under basal conditions. Image overlay of GFP and DIC channels. Arrows, cytosolic condensates formed by EGFP–GBPL3ΔNLS. Bar, 10 μm. d, Immunofluorescence of CMV::EGFP-GBPL3ΔNLS in HeLa cells. Bar, 20 μm (top) and 5 μm (bottom). e, Spatiotemporal model derived from GBPL3 functional mutagenesis analysis. f, Combinatory GBPL3 interactome in Arabidopsis from co-IP candidates using Col-GBPL3-Flag plants under basal conditions and publicly available data sets (http://plants.proteincomplexes.org/). Node size denotes degree of protein–protein interactions. Classes of GBPL3 interactors grouped by colour-coding. Bracket denotes Mediator complex interactions. g, Different subunits associated with each region of the Arabidopsis Mediator complex (head [yellow], middle [green] and tail [blue] modules) and CDK8 kinase module. h, Line profile (colocalization) of fluorescence intensity for CDK8, MED15, MED19a and MED21 co-expressed with GBPL3 in (Fig. 4b). These head, middle and tail Mediator subunits directly overlap and interact with GBPL3 in leaf cell PPI profiling. CDK8 is excluded and surrounds GBPL3, as seen in line profiling. i, Effects of digitonin (10 μg/ml), digitonin plus 1,6-hexanediol (Hex, 5%), DRB (100 μM) and JQ1 (1 μM) on GBPL3 LLPS in live HeLa cells 2 h after treatment (n = 30 biologically independent cells/treatment). One-way ANOVA test with Bonferroni post hoc correction.
Supplementary Material
Acknowledgements
We thank all members of the MacMicking laboratory for feedback and discussions; X. Liu for TEM assistance; members of the J. Liu laboratory (R. Park, M. Shao and Y. Chang) for instruction on CLEM and FIB milling; S. Wu for cryo-ET data collection advice; and X. Li, N. Clay, J. Parker, X. Dong, J. Zhou, Q. Wu, L. Wu, F. Xu and others for sharing materials and tools. The MacMicking laboratory is supported by grants from NIH NIAID (R01AI068041-12, R01AI108834-05). J.D.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03572-6.
Peer review informationNature thanks Yoji Kawano and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Boeynaems S et al. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin Y & Brangwynne CP Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Alberti S, Gladfelter A & Mittag T Considerations and challenges in studying liquid–liquid phase separation and biomolecular condensates. Cell 176, 419–434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Randow F, MacMicking JD & James LC Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science 340, 701–706 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar-On YM, Phillips R & Milo R The biomass distribution on Earth. Proc. Natl Acad. Sci. USA 115, 6506–6511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones JD & Dangl JL The plant immune system. Nature 444, 323–329 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Jones JD, Vance RE & Dangl JL Intracellular innate immune surveillance devices in plants and animals. Science 354, aaf6395 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Jung J-H et al. A prion-like domain in ELF3 functions as a thermosensor in Arabidopsis. Nature 585, 256–260 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Fang X et al. Arabidopsis FLL2 promotes liquid–liquid phase separation of polyadenylation complexes. Nature 569, 265–269 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey-Serres J, Parker JE, Ainsworth EA, Oldroyd GED & Schroeder JI Genetic strategies for improving crop yields. Nature 575, 109–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frottin F et al. The nucleolus functions as a phase-separated protein quality control compartment. Science 365, 342–347 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Feric M et al. Coexisting liquid phases underlie nucleolar subcompartments. Cell 165, 1686–1697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson BA et al. Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho WK et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin Y et al. liquid nuclear condensates mechanically sense and restructure the genome. Cell 175, 1481–1491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim BH et al. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Shenoy AR et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Kim BH et al. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat. Immunol 17, 481–489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merchant SS et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318, 245–250 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henikoff S, Henikoff JG, Sakai A, Loeb GB & Ahmad K Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 19, 460–469 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, Gal J, Chen J & Zhu H Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl Acad. Sci. USA 111, 17809–17814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zipfel C et al. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 428, 764–767 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Knuesel MT, Meyer KD, Bernecky C & Taatjes DJ The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 23, 439–451 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergeron-Sandoval LP, Safaee N & Michnick SW Mechanisms and consequences of macromolecular phase separation. Cell 165, 1067–1079 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Álvarez-Aragón R, Haro R, Benito B & Rodríguez-Navarro A Salt intolerance in Arabidopsis: shoot and root sodium toxicity, and inhibition by sodium-plus-potassium overaccumulation. Planta 243, 97–114 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Huang S, Meng Q, Maminska A & MacMicking JD Cell-autonomous immunity by IFN-induced GBPs in animals and plants. Curr. Opin. Immunol 60, 71–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riechmann JL et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Allen BL & Taatjes DJ The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol 16, 155–166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathur S, Vyas S, Kapoor S & Tyagi AK The Mediator complex in plants: structure, phylogeny, and expression profiling of representative genes in a dicot (Arabidopsis) and a monocot (rice) during reproduction and abiotic stress. Plant Physiol. 157, 1609–1627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McWhite CD et al. A pan-plant protein complex map reveals deep conservation and novel assemblies. Cell 181, 460–474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai KL et al. A conserved Mediator–CDK8 kinase module association regulates Mediator–RNA polymerase II interaction. Nat. Struct. Mol. Biol 20, 611–619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitsios G, Alexiou KG, Bush M, Shaw P & Doonan JH A cyclin-dependent protein kinase, CDKC2, colocalizes with and modulates the distribution of spliceosomal components in Arabidopsis. Plant J. 54, 220–235 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Oates ME et al. D2P2: database of disordered protein predictions. Nucleic Acids Res. 41, D508–D516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiao X et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ronquist F & Huelsenbeck JP MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Jones DT, Taylor WR & Thornton JM The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci 8, 275–282 (1992). [DOI] [PubMed] [Google Scholar]
- 38.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J et al. The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clough SJ & Bent AF Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Wang ZP et al. Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol. 16, 144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anders S, Pyl PT & Huber W HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang S et al. Plant TRAF proteins regulate NLR immune receptor turnover. Cell Host Microbe 19, 204–215 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Cheng YT et al. Nuclear pore complex component MOS7/Nup88 is required for innate immunity and nuclear accumulation of defense regulators in Arabidopsis. Plant Cell 21, 2503–2516 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moissiard G et al. MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336, 1448–1451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freeman Rosenzweig ES et al. The eukaryotic CO2-concentrating organelle is liquid-like and exhibits dynamic reorganization. Cell 171, 148–162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thévenaz P, Ruttimann UE & Unser M A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Li X et al. Symmetrical organization of proteins under docked synaptic vesicles. FEBS Lett. 593, 144–153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu S, Qin Z, Wang J, Morado DR & Liu J In situ structural analysis of the spirochetal flagellar motor by cryo-electron tomography. Methods Mol. Biol 1593, 229–242 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Schorb M & Briggs JA Correlated cryo-fluorescence and cryo-electron microscopy with high spatial precision and improved sensitivity. Ultramicroscopy 143, 24–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaffer M et al. Optimized cryo-focused ion beam sample preparation aimed at in situ structural studies of membrane proteins. J. Struct. Biol 197, 73–82 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Mastronarde DN & Held SR Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol 197, 102–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Hagen WJH, Wan W & Briggs JAG Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J. Struct. Biol 197, 191–198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kremer JR, Mastronarde DN & McIntosh JR Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 62.Agulleiro JI & Fernandez JJ Tomo3D 2.0–exploitation of advanced vector extensions (AVX) for 3D reconstruction. J. Struct. Biol 189, 147–152 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Chen M et al. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat. Methods 14, 983–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pettersen EF et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Khoshouei M, Pfeffer S, Baumeister W, Förster F & Danev R Subtomogram analysis using the Volta phase plate. J. Struct. Biol 197, 94–101 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Sun T et al. ChIP-seq reveals broad roles of SARD1 and CBP60g in regulating plant immunity. Nat. Commun 6, 10159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original raw RNA-seq data that support the findings of this study have been deposited and made publicly available in the NCBI Gene Expression Omnibus with accession number GSE134651. RNA-seq data are provided in Supplementary Tables 4 and 5. Full versions of all gels and blots are provided in Supplementary Fig. 1. The phylogenetic source data related to Extended Data Fig. 1b and high-resolution images of Fig. 4c are available on Dryad from https://doi.org/10.5061/dryad.g1jwstqqv. Source data are provided with this paper.