

Abstract
The biogenesis of high-density lipoprotein (HDL) requires apoA1 and the cholesterol transporter ABCA1. Although the liver generates most HDL in blood, HDL synthesis also occurs in the small intestine. Here we show that intestine-derived HDL traverses the portal vein in the HDL3 subspecies form, in complex with lipopolysaccharide (LPS)-binding protein (LBP). HDL3, but not HDL2 or LDL, prevented LPS binding to and inflammatory activation of liver macrophages and instead supported extracellular inactivation of LPS. In mouse models involving surgical, dietary, or alcoholic intestinal insult, loss of intestine-derived HDL worsened liver injury, whereas outcomes were improved by therapeutics that elevated and depended upon raising intestinal HDL. Thus, protection of the liver from injury in response to gut-derived LPS is a major function of intestinally synthesized HDL.
Graphical Abstract
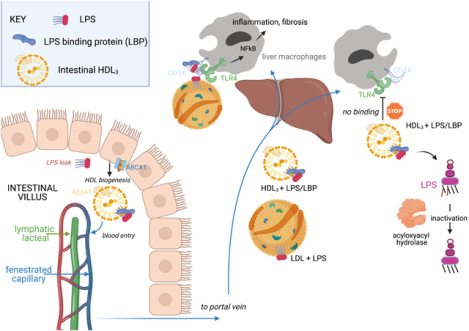
Trafficking and functional properties of enteric HDL. Enterocytes express ABCA1 to promote HDL biogenesis. The nascent HDL enters portal venous blood bearing LBP that allows it to hide LPS from recognition by TLR4+ macrophages. Failed recognition prevents macrophage activation. Although its ability to trigger macrophages is suppressed by HDL3, LPS in the HDL3 complex can still be inactivated by acyloxyacyl hydrolase. Figure drawn with Biorender.
Introduction:
High-density lipoprotein (HDL) participates in cholesterol homeostasis and may also have anti-inflammatory or anti-microbial roles through its interaction with numerous plasma proteins. The liver synthesizes most HDL in the body, but the intestine also produces HDL. However, a role for intestinal HDL distinct from that produced by the liver has not been identified. While remodeling its cargo, HDL particles circulate through tissue spaces, but so far, HDL trafficking within tissues has been scarcely studied.
Rationale:
We reasoned that understanding HDL trafficking patterns might bring insight into its roles in health and disease, including whether HDL made by the intestine is functionally redundant with that produced by the liver. Using a knock-in mouse we previously generated to phototag HDL in any tissue location, we aimed to trace the fate of HDL synthesized by the intestine.
Results:
Phototagged HDL derived from small-bowel enterocytes was generated most abundantly by the ileum and did not travel into draining lymphatic vessels like enterocyte-derived chylomicrons do. Instead, intestinal HDL rapidly entered the portal vein, the major blood supply to the liver. This finding raised the issue of whether the liver might benefit from intestinal HDL and pointed us to an older concept that HDL might neutralize a key microbial signal that can escape a permeable gut: lipopolysaccharide (LPS) from Gram-negative bacteria. Past studies had shown that LPS engagement of its receptor TLR4 in the liver drives, in multiple models, significant liver pathology, including inflammation that progresses to fibrosis. Using biochemical, proteomic, and functional approaches, we observed that the intestine produces a particular subspecies of HDL called HDL3. Unlike another HDL subspecies (HDL2), HDL3 sequestered LPS so efficiently that it could not bind to TLR4+ liver macrophages. In this way, HDL3 produced by the intestine protected the liver from inflammation and fibrosis observed in a variety of mouse models of liver injury that parallel clinically relevant conditions in humans, including surgical resection of the small bowel, alcohol consumption, or high-fat diets. Administration of an oral drug targeting the transcription factor liver X receptor (LXR), the master regulator of genes associated with HDL biogenesis, raised enteric HDL levels and protected the mice from liver pathology. This protection was lost if mice did not express enterically derived HDL, indicating that intestinal HDL was a key target of the drug. Six samples of human portal blood with matched systemic venous blood confirmed the enrichment of HDL3 in portal venous blood for humans.
Mechanistically, LPS-binding protein (LBP) was enriched in HDL3 particles and was required for HDL3 to mask LPS from detection by TLR4. This finding was remarkable, because LBP otherwise promotes TLR4 signaling by shuttling LPS to CD14, which then shuttles it to TLR4. Thus, HDL3 interacts with a known component of the TLR4-signaling platform, LBP, to hide LPS from detection. Without binding to TLR4, the HDL3–LBP–LPS complex was not retained in liver. Instead, it exited the liver while the LPS associated with it was inactivated. The enzyme acyloxyacyl hydrolase, which is produced in part by liver macrophages and which deacylates critical fatty acid residues in LPS for TLR4 activation, could still access and act upon HDL3-associated LPS to detoxify it. Low-density lipoprotein (LDL) bound LPS but not LBP and was thus unable to prevent LPS activation of liver macrophages. LBP is in the same family of lipid-binding proteins as phospholipid transfer protein (PLTP) and cholesterol ester transfer protein (CETP), which have well established roles in remodeling the lipid configuration of HDL. Another microbial lipid, lipoteichoic acid from Gram-positive bacteria, is known to bind LBP. We found that it too complexed with HDL3 and suppressed the activation of liver macrophages.
Conclusions:
The production of HDL by small-bowel enterocytes in a form that potently masks LPS comprises a disease tolerance strategy to protect the liver from injury of enteric origin. Enteric HDL may thus be a suitable pharmacologic target for protecting the liver against gut-derived LPS leakage in alcoholic and nonalcoholic settings.
Introduction
The portal vein collects venous drainage from the intestine, carrying nutrients and metabolites of host and microbiome origin to the liver (1). Through this route, components of the microbiome may drive liver steatohepatitis and fibrosis (2, 3). Enterically derived lipopolysaccharide (LPS) from Gram-negative bacteria triggers TLR4-dependent injury in the liver following insult to the intestine (3–7).
Mechanisms to limit LPS-mediated liver injury via the gut–portal axis remain incompletely defined. We hypothesized that high-density lipoprotein (HDL) may have an overlooked role in protecting the liver via its potential to neutralize LPS (8–10). Indeed, it is unclear why HDL is synthesized by the intestine rather than solely by the liver. HDL-cholesterol (HDL-C) is the smallest lipoprotein particle in the blood and best known for its role in cholesterol transport. Only two tissues produce apolipoprotein A1 (apoA1), the core protein component of HDL-C: the liver and the small intestine (11). When intestinal epithelial cells selectively delete the gene encoding the cholesterol transporter ABCA1 essential for HDL biogenesis, an approximate 25% reduction in plasma HDL-C ensues. Conversely, an approximate 75% reduction in HDL-C occurs after liver-specific loss of ABCA1 (12), leaving investigators to regard the intestine as simply a second source of HDL-C.
An obstacle to considering a role for intestinal HDL in the gut–liver axis is the paucity of knowledge concerning how enterically derived HDL is delivered to the liver. HDL typically mobilizes from tissues through lymphatic vessels (13–15), which do not route to the liver from the intestine (16). However, an earlier study failed to demonstrate that enterically produced HDL-C entered lymphatics (17). Here, we show that enterically derived HDL-C alternatively traverses portal blood and that nearly all HDL-C found in the portal vein arises from intestine. Intestinal epithelial cells produced small HDL particles (HDL3) (18) with potent LPS-neutralizing properties.
Results
Enterically derived HDL is the main source of HDL in portal blood
Although albumin levels were constant between portal and systemic blood (from the inferior vena cava) (fig. S1A), apoA1 was approximately 40% lower in portal versus systemic plasma in humans and mice (Fig. 1A). To determine if this reduction resulted from diminished recirculation of HDL into portal blood, we traced HDL using photoactivatable GFP apoA1 knock-in mice (Pga1KI/+) (15). When phototagged in the skin, HDL appeared in systemic blood, but was very low in portal blood (Fig. 1B). Its appearance in mesenteric lymph to a concentration approaching that in systemic circulation (Fig. 1B) suggested that it left the bloodstream to access lymph before entering the portal vein (Fig. 1B).
Figure 1. Intestinal HDL transits through portal vein and accounts for most HDL in the portal blood.
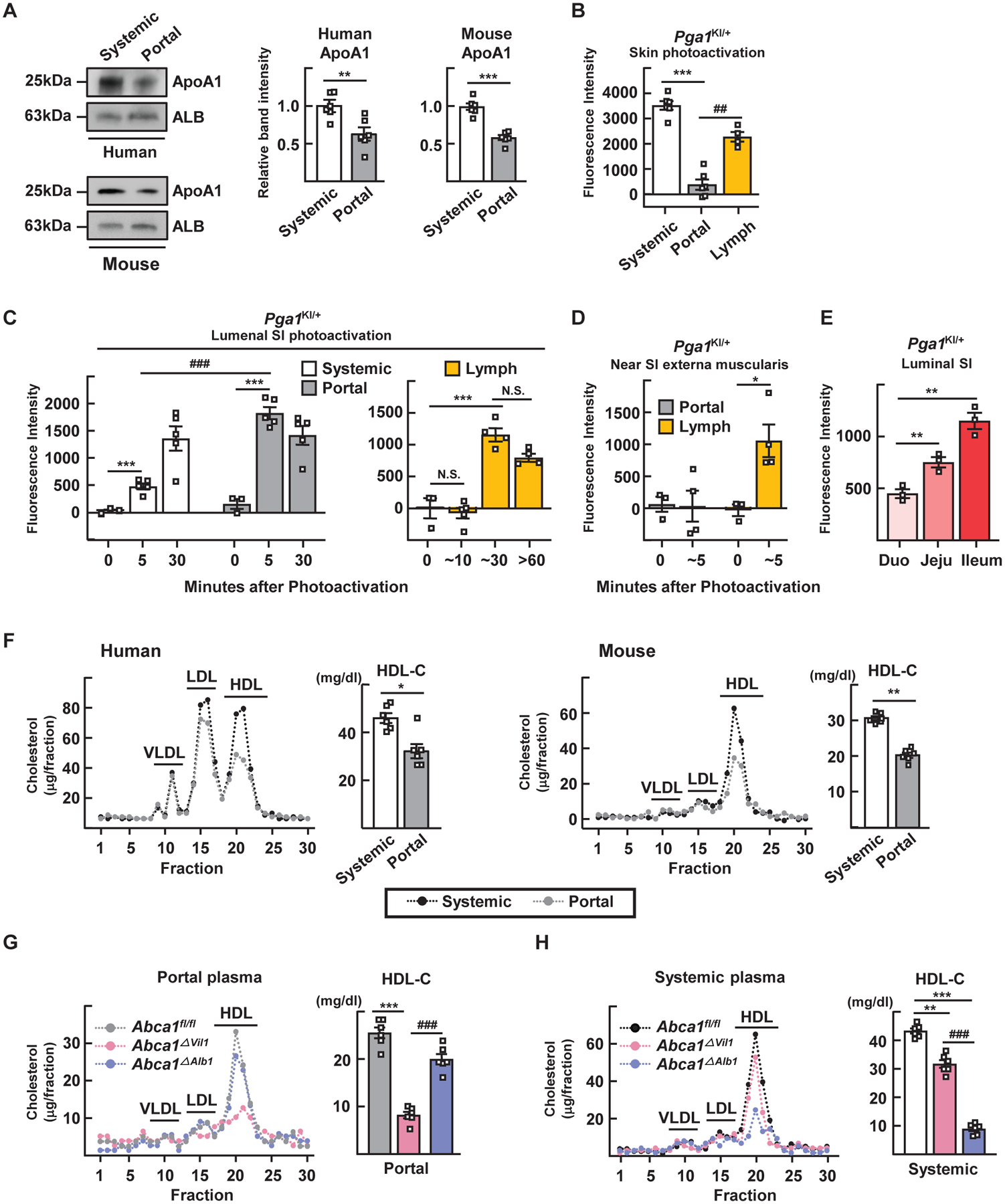
(A) Immunoblot for apoA1 in systemic and portal serum from humans or mice. (B) Plasma and lymph fluorescence measured 2 hours after phototagging HDL in skin. (C and D) Portal plasma and mesenteric lymph fluorescence after phototagging the lumen (C) or externa muscularis (D) of the small intestine (SI) of Pga1KI/+ mice. (E) HDL was phototagged in different small bowel regions separately. (F) Lipoprotein profiles from humans fasted overnight or mice fasted for 4 hours. (G and H) Lipoprotein profiles and HDL-C quantification of portal (G) or systemic (H) plasma after 4 hours of fasting. Plots show mean ± SEM. Panels A, F show paired data from six human subjects or six WT mice (same subjects, both panels). Panel B to E studied 65 Pga1KI/+ mice. Each symbol designates different mice except systemic versus portal blood in panel B or systemic versus portal blood from the same time points in panel C were from the same mice (paired). Most panels show one experiment; panel C combines two experiments. Panels G and H shows five individual mice from each of three genotypes from one experiment, with paired portal (G) and systemic (H) plasma within the same genotype. *P<0.05; **P<0.01; ***P<0.001; ##P<0.01; ###P<0.001. Data in panels A and F were analyzed for statistical significance using a paired t test. All other panels were analyzed using one-way ANOVA.
Within 5 min after phototagging HDL in the small intestinal lumen, fluorescence was strong in portal blood, but not lymph (Fig. 1C). By 30 min, these compartments equilibrated (Fig. 1C). HDL phototagged along the intestinal exterior appeared in lymph but not portal blood (Fig. 1D). These patterns were unaffected by dietary composition or fasting (fig. S1, B to D). Thus, HDL tagged at the intestinal epithelium first enters the portal vein and is not observed in lymph until cargo in the portal vein passes through the liver and enters the systemic circulation. Upon reapproaching the gut, it appears to traverse into the interstitium and then into lymph.
Separate phototagging of the duodenum, jejunum, or ileum revealed the ileum as the major site of enteric HDL biogenesis (Fig. 1E). Like apoA1, HDL-C in portal plasma of humans or mice was present at lower concentrations than in systemic blood (Fig. 1F). HDL-C was decreased by >75% in portal blood of intestine-specific ABCA1-knockout mice (Vil1Cre-Abca1fl/fl; Abca1ΔVil1) (Fig. 1G). However, in systemic blood of Abca1ΔVil1 mice, HDL-C dropped only by 25% (Fig. 1H) (12). By contrast, a marked reduction of HDL-C in systemic, but not portal blood was observed in liver-specific ABCA1-knockout mice (Alb1Cre-Abca1fl/fl; Abca1ΔAlb1) (Fig. 1, G and H). Thus, two distinct blood compartments for HDL exist, one entering the portal drainage governed by intestinal production of HDL and the other in systemic vessels, governed by liver production of HDL.
Portal blood HDL is mainly HDL3 and strongly suppresses Kupffer cell inflammatory responses
Portal venous HDL was relatively small in size (~8 nm) in humans (Fig. 2A) and mice (Fig. 2B), suggestive of a subspecies of HDL called HDL3. Small-sized HDL3 and large-sized HDL2 particles carry distinct accessory proteins, with paraoxonase 1 (PON1) enriched in the former and apoB in the latter (18). Thus, we compared HDL2 or HDL3 species separated by ultracentrifugation from pooled human systemic blood to that of the larger or smaller HDL species from portal or systemic blood isolated by size-exclusion FPLC chromatography + affinity purification (Fig. 2C). This approach yielded four samples from the same individual—putative HDL2 and HDL3, each from both systemic and portal blood—allowing evaluation of how venous location affects HDL composition. Proteomic analysis revealed >250 proteins in each sample. A heat map depicting relative abundance of proteins revealed that small-portal-venous HDL shared a protein profile with HDL3 from human systemic blood (Fig. 2D and table S1). Similarities included known enrichments of PON1, PON3, α1-antitrypsin (SerpinA1), and PLTP in all HDL3 fractions regardless of portal or systemic blood origin or method of isolation (Fig. 2E). Some proteins like LPS-binding protein (LBP) were especially enriched in immunopurified portal blood HDL3 but notably absent after ultracentrifugation (Fig. 2E). The distribution of LBP was confirmed by immunoblot of FPLC fractions used to purify HDL2 (fraction 20) versus HDL3 (fraction 22) from human portal vein (Fig. 2F) or systemic blood (fig. S2), or murine portal blood (fig. S2). When these serum samples were subjected to HDL isolation using ultracentrifugation, LBP was absent (Fig. 2F), suggesting its dissociation during ultracentrifugation (19).
Figure 2. Portal vein HDL is enriched in small HDL3 particles.
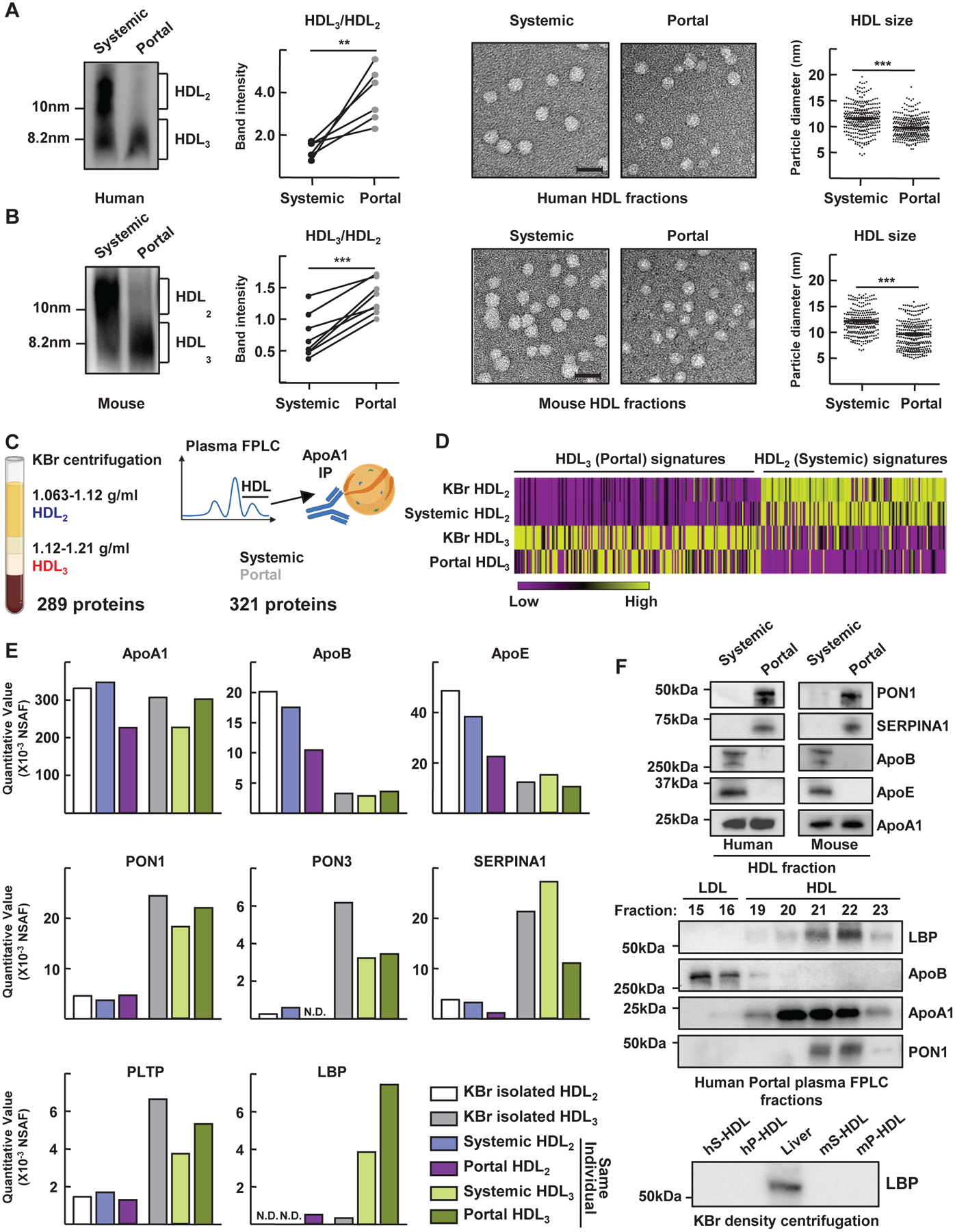
Samples (from Fig. 1A) of systermic or portal plasma from humans (A) or mice (B) immunoblotted for apoA1 after electrophoresis under nondenaturing conditions. Representative gels and plots show HDL3/HDL2 ratios in six paired samples. Also shown are representative electron microscopy images of negative-stained HDL fractions. Scale, 20 nm. HDL diameter measurements, plotted as individual symbols, combine assessments from four of six humans or mice (right). For proteomics (C to F), density ultracentrifugation-purified HDL2 or HDL3 yielded 289 associated proteins (C, left). Size-exclusion + immunopurified HDL identified 321 proteins (C, right). This experiment was performed once using four paired samples derived from the same individual (HDL2 or HDL3 from 2 vascular beds respectively). Additional samples were HDL2 or HDL3 from commercially available pooled human plasma isolated by density ultracentrifugation. (D) Heatmap of protein abundance. (E) Normalized spectral abundance factor (NSAF) plotted for selected proteins. (F) Select proteins immunoblotted from portal or systemic plasma HDL (top), including blotting indicated fraction numbers collected after FPLC separation (middle). Immunoblot of LBP from human or mice systemic (hS- and mS-) and portal (hP- and mP-) plasma or whole liver lysate (lower blot). LBP quantification is shown in fig. S2A. N.D., not detected. **P<0.01; ***P<0.001. Paired t test was used for statistical evaluation in panel A; unpaired t test was used in panel B.
As LBP delivers LPS to CD14 to facilitate TLR4 signaling (20), we investigated whether the association of portal venous HDL3 with LBP affects LPS signaling. We isolated primary liver Kupffer cells (KCs) (21, 22) (fig. S3A) from WT or Tlr4−/− mice and observed that portal venous-derived HDL from humans or mice more effectively neutralized LPS-induced pro-inflammatory responses in these cultures than HDL from systemic blood or no HDL at all (Fig. 3, A and B). Whole portal plasma obtained from Abca1ΔVil1 mice less effectively protected against induction of inflammation (Fig. 3C), implicating gut-derived HDL in this activity. Similarly, comparison of human HDL2 or HDL3 from human peripheral blood showed that HDL3 more strongly suppressed LPS-induced inflammatory genes (Fig. 3D) than HDL2 in a concentration-dependent, TLR4-dependent manner (Fig. 3D). Activation of KCs by lipoteichoic acid was also robust and strongly blocked by HDL3. CpG oligodeoxynucleotides stimulated KCs less strongly, but HDL3 did have some inhibitory effect (fig. S3B). Modest inhibition was observed when the cytokines IL-1β or TNF were used to induce inflammatory activation of KCs (fig. S3C). In these assays (Fig. 3, A to D), LBP was included as an exogenous additive to restore the LBP removed during centrifugation. Excluding LBP abrogated the anti-inflammatory effects of HDL3 (Fig. 3E). Similarly, HDL3 suppressed LPS bioactivity, but only in the presence of LBP (Fig. 3F). Thus, portal blood HDL3 inhibits LPS signaling in TLR4+ macrophages in a LBP-dependent manner.
Figure 3. Portal blood HDL3 strongly inhibits LPS activation of KCs in a LBP-dependent manner.
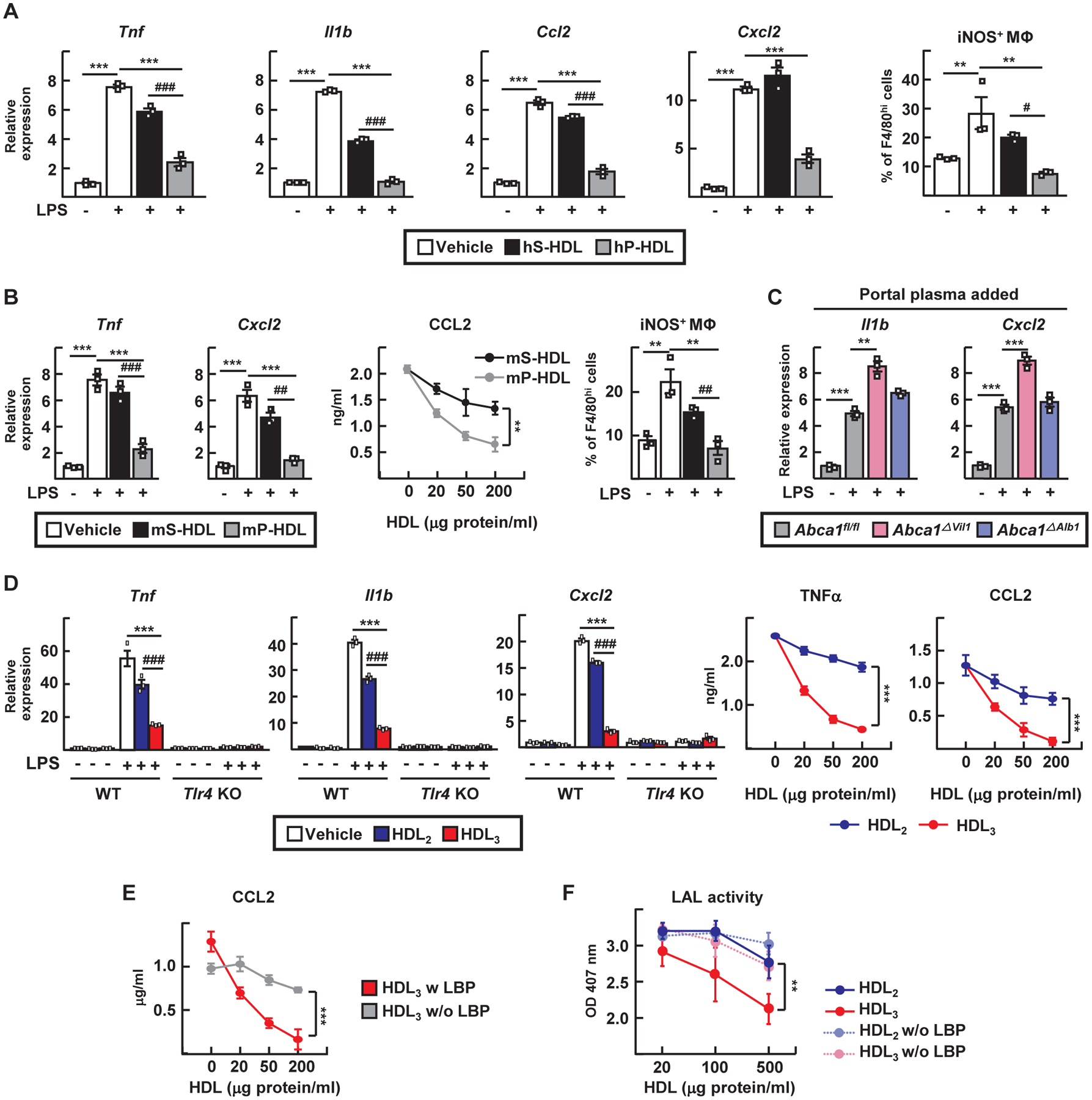
(A and B) LPS-treated KCs were incubated with 100 μg/ml of HDL from human (hS-HDL) or mouse systemic plasma (mS-HDL) or with human (hP-HDL) or mouse portal plasma (mPHDL). Analysis included RT-PCR for inflammatory mediators, flow cytometry phenotyping (iNOS+ F4/80hi macrophages), or CCL2 ELISA. (C) RT-PCR from LPS-treated KCs incubated with 5% portal vein-derived plasma from Abca1fl/fl, Abca1ΔVil1, and Abca1ΔAlb1 mice. (D) WT and TLR4−/− KCs were incubated in LBP-containing medium with or without 20 ng/ml of LPS and 100 μg/ml of HDL2, HDL3, or vehicle control before RT-PCR or ELISA to detect depicted mediators. (E) LPS-treated KCs were incubated with 100 μg/ml of HDL3 with or without 1 μg/ml of LBP before CCL2 ELISA. (F) Endotoxin LAL activity after 0.5 EU/ml of E. coli LPS was pre-incubated with HDL with or without 1 μg/ml of LBP. HDL fractions studied in this figure were isolated by density ultracentrifugation. Plots show mean ± SEM. Each symbol represents independent preparations of KCs from different mice. Panels A and B, n=3 different KC preparations (three experiments). Panel C, nine different KC preparations, n=3 per genotype (three experiments). Panels D and E, n=6 KC preparations (n=3 each WT or TLR4 KO). Panel F, three independent technical replicates (three experiments) per condition per time point. **P<0.01; ***P<0.001; #P<0.05; ##P<0.01; ###P<0.001. Statistical analysis on data presented as bar graphs used one-way ANOVA; data presented as concentration curves were analyzed using two-way ANOVA.
Portal HDL3 efficiently binds LBP and LPS to prevent LPS binding to KC TLR4
We next evaluated binding between HDL and LPS using LPS conjugated with biotin at its inner core (23–25). HDL3 more robustly bound LPS compared with HDL2, but the presence of LBP was required (Fig. 4A). HDL2-bound LPS readily transferred to LDL or VLDL, whereas most HDL3-bound LPS remained associated with HDL3 (Fig. 4B). In an immunoabsorption assay, HDL3 was indeed more effective than HDL2 and LDL in binding LBP (Fig. 4C), and LDL did not compete (Fig. 4, C and D). LDL bound LPS efficiently (Fig. 4D), but did not bind LBP (Fig. 4C). LDL did not neutralize LPS bioactivity (Fig. 4E), nor did it dampen inflammatory gene expression (fig. S4A), with or without LBP (Fig. 4E). Reconstituted HDL also did not neutralize LPS (Fig. 4E). In the presence of LBP, interactions between biotin-LPS and HDL3 generally reduced the detection of biotin-LPS by streptavidin capture (Fig. 4F). This reduction was due to efficient masking of the inner core biotin label of LPS, since disruption of HDL3 with detergent re-exposed the biotin (Fig. 4F). Thus, binding of LBP to HDL3 promotes the sequestration of LPS.
Figure 4. HDL3 binds LPS and masks its capacity to bind to TLR4+ KCs while allowing enzymatic inactivation by AOAH.
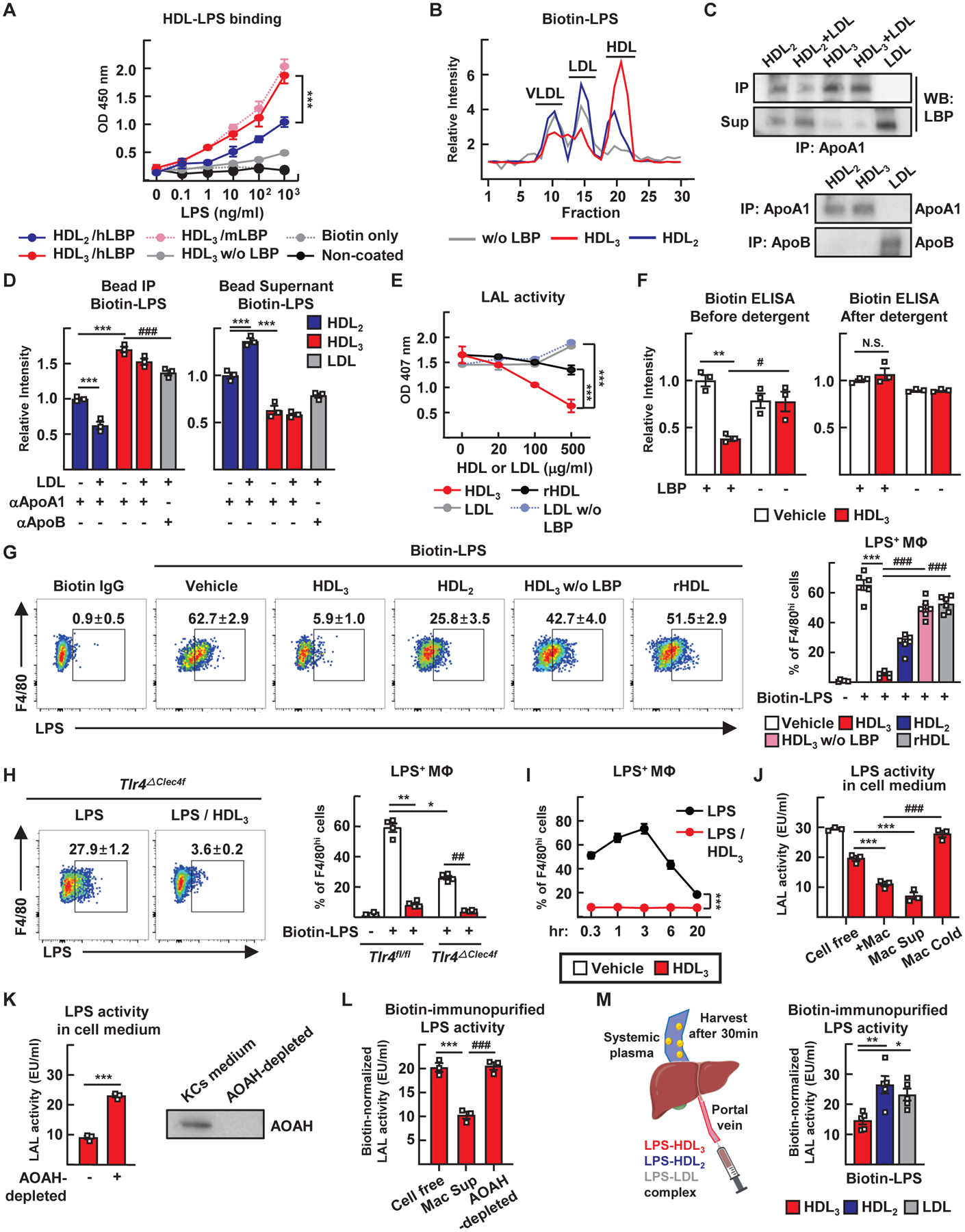
(A) ELISA to assess HDL and biotin-LPS binding with or without LBP added. (B) HDL3 or HDL2 preincubated with biotin-LPS was incubated for 2 hours with LDL or VLDL. Biotin-LPS was then detected in the various lipoproteins separated by FPLC. (C and D) Lipoproteins were incubated with LBP and biotin-LPS for 2 hours, then retrieved using using apoA1 or apoB immunoprecipitation. LBP protein (C) and amount of biotin-LPS (D) in resulting pellets (IP) versus supernatants (Sup) were measured. (E) Endotoxin LAL activity assessed after 0.5 EU/ml of E. coli LPS was pre-incubated with different concentrations of HDL3, reconstituted HDL (rHDL) or LDL with or without 1 μg/ml LBP. (F) Biotin-LPS was incubated with HDL3 with or without LBP. The complex was then disrupted with SDS and biotin-LPS measured by strepatavidin ELISA. (G) KCs were cultured with HDL3, HDL2, or rHDL with or without LBP in the presence of biotinylated LPS for 3 hours, followed by streptavidin detection. Flow cytometry plots (left) and quantification (right) of binding to KCs. (H) Binding of LPS to Tlr4fl/fl and Tlr4ΔClec4f KCs cultured with biotin-LPS (white) or biotin-LPS/HDL3 (red). (I) Time course of KC binding to biotin-LPS. (J) LPS and HDL3 (20 μg/ml) were co-incubated for 3 hours in cell-free medium, with KCs (+Mac), in medium conditioned by KCs (Mac Sup), or with KCs held at 4°C (Mac Cold); LAL activity was then assessed. (K) Endotoxin LAL activity was assessed after LPS and HDL3 were incubated in KC-conditioned medium from which AOAH was depleted or not. (L) Biotin-LPS and HDL3 were incubated in the indicated medium, LAL activity was assessed, and SDS denaturation was conducted to allow for total biotin measurements. Relative LAL activity shown is normalized to a constant amount of biotin. (M) HDL3, HDL2, or LDL were complexed with biotin-LPS and injected into the portal vein. Systemic plasma was harvested after 30 min. Endotoxin LAL activity and amount of biotin were measured and normalized as in panel L. All HDL fractions studied in this figure were isolated by density ultracentrifugation and unless indicated (as in panels B to G) coincubated with 1 μg/ml of LBP during the assays (H to M). Panels A to F depict three independent technical replicates (three experiments) per condition or time point. Panels G to L utilized from two to six different primary KC cultures (each prepared from different mice) with each symbol depicting data arising from one of the KC cultures. Panel M is from one experiment using 15 WT mice (n=5 mice per condition). *P<0.05; **P<0.01; ***P<0.001; #P<0.05; ##P<0.01; ###P<0.001. Statistical analysis on data presented as bar graphs used one-way ANOVA, except for panel K where a t-test was used; data presented as concentration curves were analyzed using two-way ANOVA.
To determine if HDL3 sequestered LPS from KCs, we cocultured KCs with biotin-LPS and monitored surface binding (Fig. 4G). This binding largely depended upon TLR4 (Fig. 4H) and was unaffected by absence or blockade of lipoprotein or scavenger receptors (fig. S4B). HDL3 robustly prevented LPS interaction with KCs, but only in the presence of LBP. Reconstituted HDL (Fig. 4G) or LDL in the presence or absence of LBP (fig. S4C) did not diminish binding, whereas HDL2 partially blocked (Fig. 4G). Peak interaction occurred within 3 hours of incubation with KCs (Fig. 4I). Substantial internalization of biotin-LPS by macrophages in the presence or absence of HDL3 was not detected during this time (fig. S4D).
We next wondered whether, after failing to bind to cells, LPS associated with HDL3 might later be inactivated. Using a low dose of HDL3 (20 μg/ml) that only weakly suppressed LPS activity in a cell-free system (Fig. 3F), we observed that LPS activity was further lowered in the presence of macrophages or macrophage supernatant. This was true as long as the KCs were not held at 4°C (Fig. 4J), suggesting that a soluble product from KCs inactivated LPS. The enzyme acyloxyacyl hydrolase (AOAH), produced in part by KCs, deacylates and thereby inactivates LPS (26). Depletion of AOAH from KC-conditioned medium increased recovery of LPS bioactivity (Fig. 4K). Because AOAH would not remove the biotin from the inner core of LPS upon deacylation, we calculated LPS bioactivity normalized to recovered biotin after disrupting HDL3 with detergent, finding that AOAH indeed drove inactivation of HDL3-bound LPS (Fig. 4L). We carried out a similar experiment in vivo, injecting a constant dose of biotin-LPS into the portal vein in complex with HDL3, HDL2, or LDL. At 30 min, when enterically derived HDL has passed through the liver to access the systemic circulation (Fig. 1C), we drew systemic venous blood to recapture and assess LPS. More of the recovered LPS was inactive when complexed with HDL3 than with HDL2 or LDL (Fig. 4M and fig. S4E), indicating the inactivation of HDL3-associated LPS. The administration of LPS-loaded lipoproteins into the portal vein caused acute elevation in AST, a measure that was lowest when HDL3 was the vehicle carrying LPS and highest when LDL carried LPS (fig. S4F). Thus, HDL3 masks LPS to limit inflammation by blocking LPS binding to KCs, but HDL3-associated LPS remains susceptible to inactivation by AOAH, allowing its permanent inactivation.
The LPS receptor TLR4 on KCs drives liver injury and fibrosis
To determine whether HDL neutralization of LPS modulated liver inflammation in vivo, we studied a model of small-bowel resection that promotes marked liver fibrosis (27, 28). Small-bowel resection removed 50% or 75% of the small intestine (Fig. 5A), sparing the head of the small intestine and the terminal ileum, except when indicated otherwise. Within 3 months, these resections induced morphological changes in the liver (Fig. 5B), elevated plasma aspartate aminotransferase (AST) (Fig. 5C), and enhanced the infiltration of myeloid cells (F4/80+ and S100A9+) into the liver (Fig. 5D). LPS bioactivity in the portal vein was elevated by small-bowel resection (Fig. 5E) and was associated with increased intestinal permeability (Fig. 5F), reductions in epithelial junction proteins ZO-1 and occludin (Ocln) (Fig. 5G), and elevated plasmalemma vesicle-associated protein 1 (PV1) in villus capillaries (Fig. 5H) draining into the portal vein (29).
Figure 5. Small-bowel resection triggers TLR4-mediated liver inflammation.
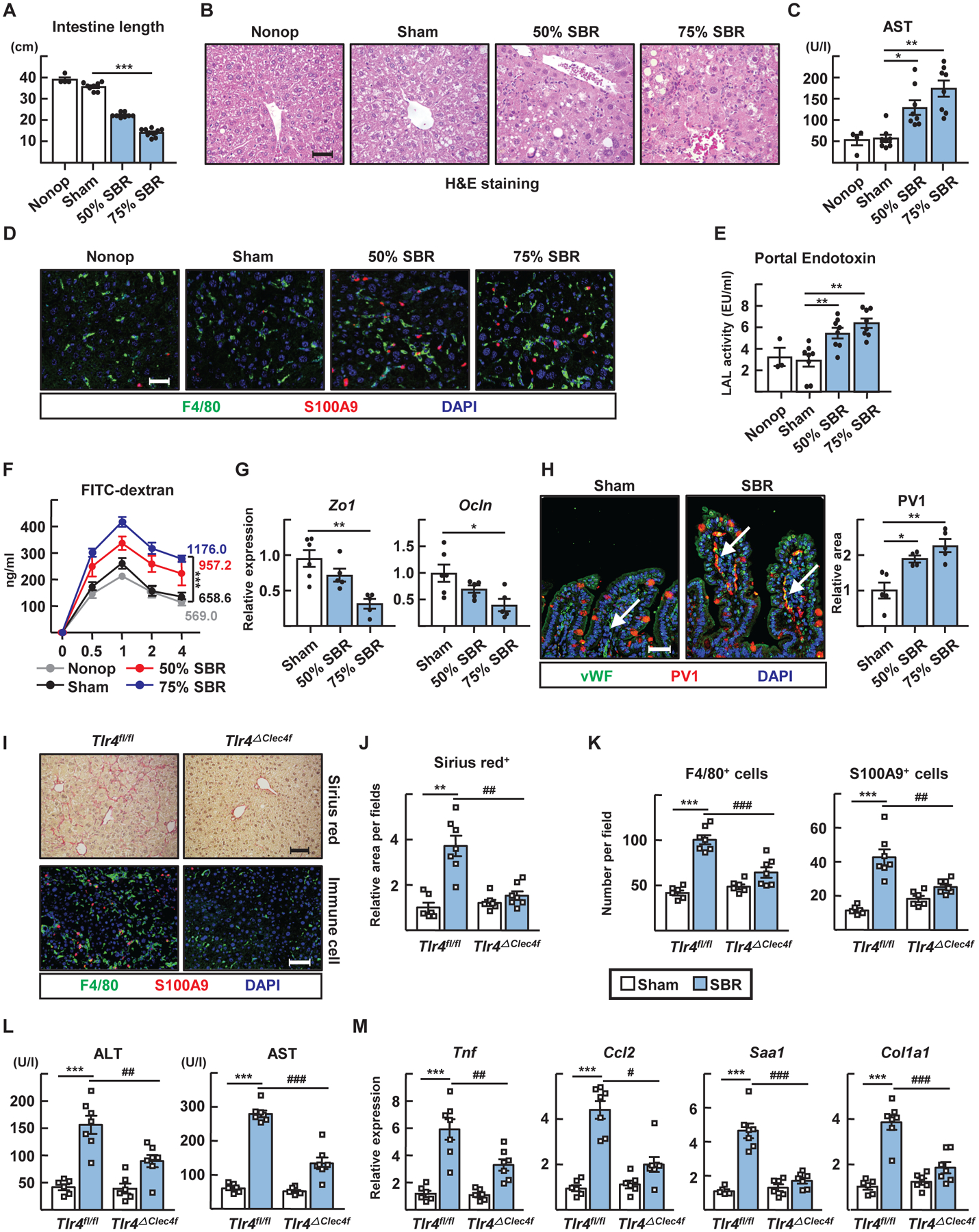
(A to H) Small-bowel resection (SBR) operations were conducted on WT mice. Non-operated (Nonop) (n=4), sham (n=8), 50% SBR (n=8), and 75% SBR (n=8) mice were euthanized 12 weeks later. (A) The total length of remaining small intestine. (B) Representative H&E stained liver sections. Scale, 50 μm. (C) Plasma AST levels. (D) F4/80+ macrophages and S100A9+ neutrophils in liver sections. Scale, 50 μm. (E) LAL endotoxin activity in portal plasma. (F) Kinetics of FITC-dextran translocation from intestine to peripheral blood. AUC was measured. (G) Quantitative RT-PCR for intestinal mRNA transcripts encoding tight junction proteins. (H) Staining for von Willebrand Factor (vWF) (blood vessel) and PV1 in intestinal sections (left). Note that goblet cell mucin stains with the PV1 antibody, possibly nonspecifically. Relative increase in PV1 staining of vWF+ vessels (highlighted by white arrows) after small-bowel resection (right). Scale, 50 μm. (I to M) Sham or 75% resection operations were performed on Tlr4fl/fl and Tlr4ΔClec4f male mice and euthanized after 10 weeks. (I) Sirius red and immunostaining of liver sections. Scale, 100 μm. (J) Relative sirius red positive area per field. (K) Numbers of F4/80+ macrophages and S100A9+ neutrophils per field. (L) Plasma ALT and AST levels. (M) Hepatic mRNA transcripts of inflammatory genes analyzed by qRT-PCR. Panels A to G arise from analysis of 30 WT mice combined from two experiments, with n=4–10 mice per condition (nonoperated, sham, 50% or 75% SBR). Panels I to M depict one experiment arising from analysis of 26 mice (n=6–7 per genotype with or without SBR). Each symbol represents data from an individual mouse. *P<0.05; **P<0.01; ***P<0.001; #P<0.05; ##P<0.01; ###P<0.001. Statistical analysis on data presented as bar graphs used one-way ANOVA; data presented as concentration curves were analyzed using two-way ANOVA.
Reduced liver fibrosis and inflammatory changes characterized KC-specific Clec4f-Cre×Tlr4flox/flox male mice compared with littermate controls (30) (Fig. 5, I to M). Female mice (fig. S5) developed disease similarly to males. Bone marrow transplants in which mice received Tlr4−/− bone marrow confirmed the role of TLR4 (fig. S6). Thus, TLR4 expressed by KCs participates critically in liver fibrosis after small-bowel resection.
Disruption of enterically derived HDL exacerbates liver injury
Portal venous HDL-C reduced after small-bowel resection (Fig. 6A), possibly a consequence of the loss of bowel mass that might normally contribute to HDL biogenesis. Expression of ABCA1 sharply elevated from proximal to distal small bowel, whereas apoA1 modestly rose, overall suggesting that the ileum is the main site for HDL production in the small bowel (Fig. 6B). When we modified the region of the bowel resected to remove the proximal 50% (P-SBR) or distal 50% (D-SBR) portion of the small intestine, HDL-C in portal blood fell more drastically following distal resection (Fig. 6C). Accordingly, liver injury and inflammatory markers were greater in response to distal resection (Fig. 6D and fig. S7, A and B).
Figure 6. Disruption of enteric HDL production worsens small-bowel resection -induced liver injuries.
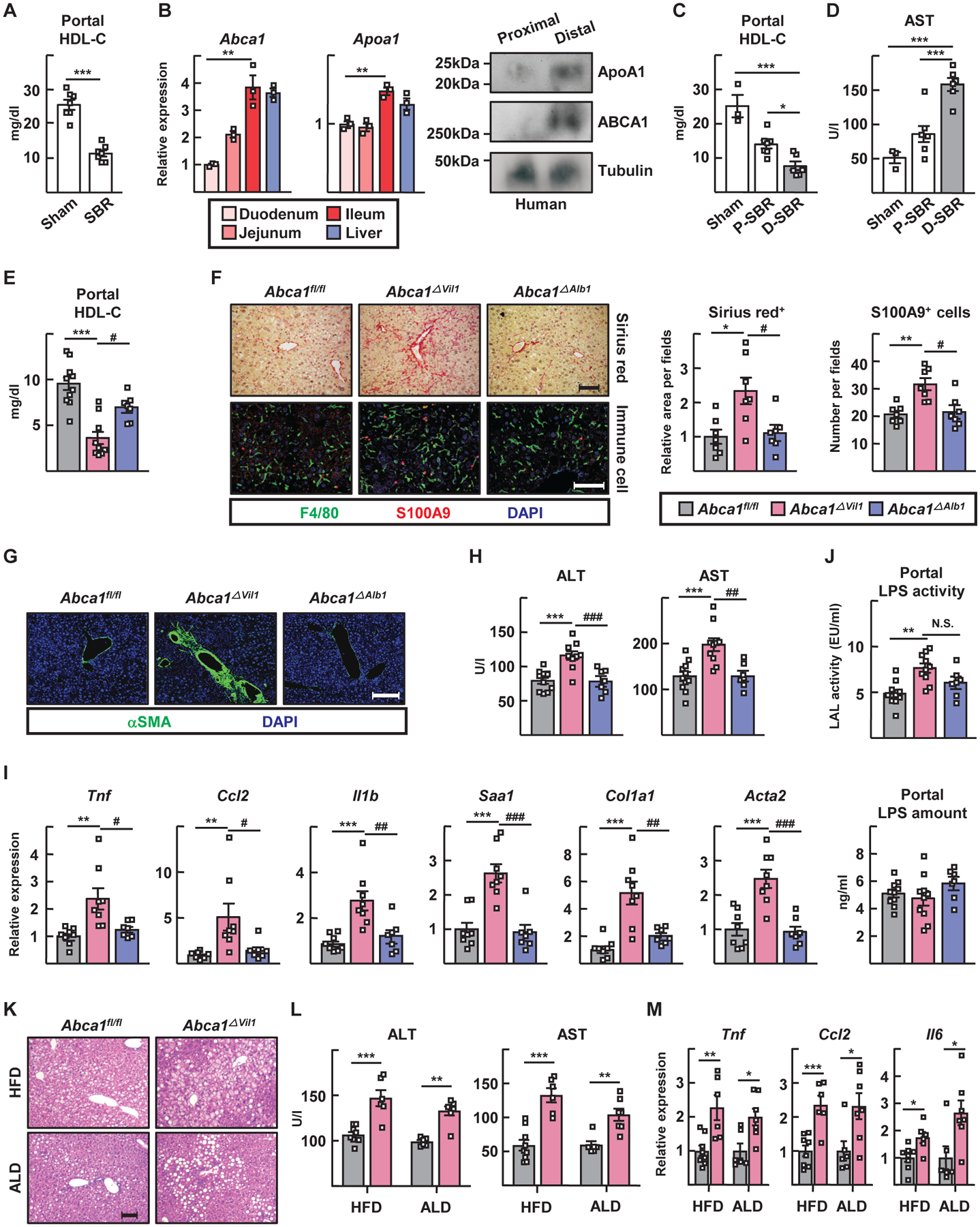
(A) Portal HDL cholesterol levels in mice receiving sham or 75% small-bowel resection (SBR) surgery. (B) RT-PCR for Abca1 and Apoa1 in mouse duodenum, jejunum, ileum and liver (left). Protein expression of apoA1 and ABCA1 in human proximal and distal gut were analyzed by immunoblotting (right). (C and D) SBR operations were conducted for WT mice. Mice receiving sham (n=3), proximal 50% SBR (P-SBR) (n=7), and distal 50% SBR (D-SBR) (n=6) operations were euthanized 10 weeks later. (C) Portal HDL cholesterol levels. (D) Plasma AST levels. (E to J) 75% SBR operations were performed for Abca1fl/fl (n=10), Abca1ΔVil1 (n=10), and Abca1ΔAlb1 (n=7) mice and euthanized 8 weeks later. (E) Portal HDL cholesterol levels. (F) Representative sirius red staining of liver sections (top left) and relative area per field (right). F4/80+ macrophages and S100A9+ neutrophils were visualized (bottom left); cell numbers per field (right); scale, 100 μm. (G) Representative anti-SMA immunostaining of liver sections; scale, 200 μm. (H) Plasma ALT and AST levels. (I) Hepatic RT-PCR for inflammatory genes. (J) Portal LAL LPS activity (top) and LPS quantification by ELISA (bottom). (K to M) Abca1fl/fl and Abca1ΔVil1 male mice were fed high fat diet (HFD) or alcoholic Lieber-DeCarli diet (ALD). (K) Representative H&E sections of liver; scale, 100 μm. (L) Plasma ALT and AST levels. (M) RT-PCR for inflammatory genes. Panels A to C studied n=3–7 mice per condition using WT mice (one experiment each panel). Panels E to I combine data from two experiments using 28 mice (n=7–11 mice per genotype). Panels L and M depict two experiments (one HFD, one ALD) from n=15 mice on HFD (n=9 Abca1fl/fl mice, 6 Abca1ΔVil1 mice) or n=13 on ALD (n=6 Abca1fl/fl mice, 7 Abca1ΔVil1 mice). *P<0.05; **P<0.01; ***P<0.001; #P<0.05; ##P<0.01; ###P<0.001. Statistical analysis on data presented as bar graphs used one-way ANOVA, except for panel A where a t-test was used
We compared liver injury outcomes in Abca1ΔVil1 mice versus control (Abca1fl/fl) or Abca1ΔAlb1 mice after small-bowel resection. HDL-C in portal blood was further reduced in Abca1ΔVil1 mice (Fig. 6E) and these mice indeed exhibited greater liver injury, fibrosis, and inflammation (Fig. 6, F to I). Sham surgery did not provoke liver injury (fig. S7, C to F). After small-bowel resection, LPS bioactivity was elevated in Abca1ΔVil1 mice (Fig. 6J, upper bar graph). However, the absolute amount of LPS in portal plasma matched other groups (Fig. 6J, lower bar graph), suggesting that the lower HDL-C in Abca1ΔVil1 mice led to increased LPS activity for a given quantity of LPS due to reduced neutralization.
Liver inflammation was elevated in Abca1ΔVil1 mice over control mice after perturbations including 12 weeks of high-fat diet (HFD) feeding or 4 weeks of the Lieber–DiCarli alcohol diet (ALD) (Fig. 6, K to M). These additional liver injury models associate with elevated LPS translocation across the intestinal barrier (31, 32), elevations we verified and that were in keeping with reduced HDL-C in the portal vein and apparent increased fat storage in the liver of the HFD model (fig. S7, G to I). Thus, enterically derived HDL protects against injury in multiple mouse models of liver damage.
Activation of LXR in the intestine increases HDL output and protects against liver injury
Liver X receptor (LXRs) transcription factors govern expression of HDL-related genes like Abca1. Low-dose LXR agonists like GW3965, when administered orally, bypass activation of LXRs in the liver while targeting the intestine (33, 34). We thus administered GW3965 orally at a low dose in mice subjected to 75% small-bowel resection and followed gene expression in the ileum and liver (Fig. 7, A to E and fig. S8). GW3965 treatment prompted increases in Abca1 and Apoa1 mRNA in the ileum (Fig. 7, A and C). The impact of low-dose oral GW3965 on these and LXR target genes associated with de novo lipogenesis was minimal in the liver (Fig. 7, B and D). However, inflammatory and collagen remodeling genes in the liver were markedly downregulated in response to GW3965 in Abca1fl/fl mice that retained expression of intestinal HDL (Fig. 7E and fig. S9). These genes remained elevated in Abca1ΔVil1 mice (Fig. 7E), confirming that the effect of GW3965 depended on intestinal HDL. Oral, low-dose GW3965 did not impact Abca1 or related genes in peritoneal macrophages adjacent to the portal venous drainage in the mesentery (fig. S8A). Some intestinal macrophages were positive for ABCA1 (fig. S8B), but deletion of ABCA1 in macrophages neither affected gene expression in the ileum nor alter portal HDL-C (fig. S8C). No changes to the liver were apparent in Abca1ΔVil1 mice compared with littermate Abca1fl/fl controls receiving sham operations (fig. S10).
Figure 7. Intestine-restricted activation of LXR ameliorates liver injury in a manner dependent upon enteric HDL production.
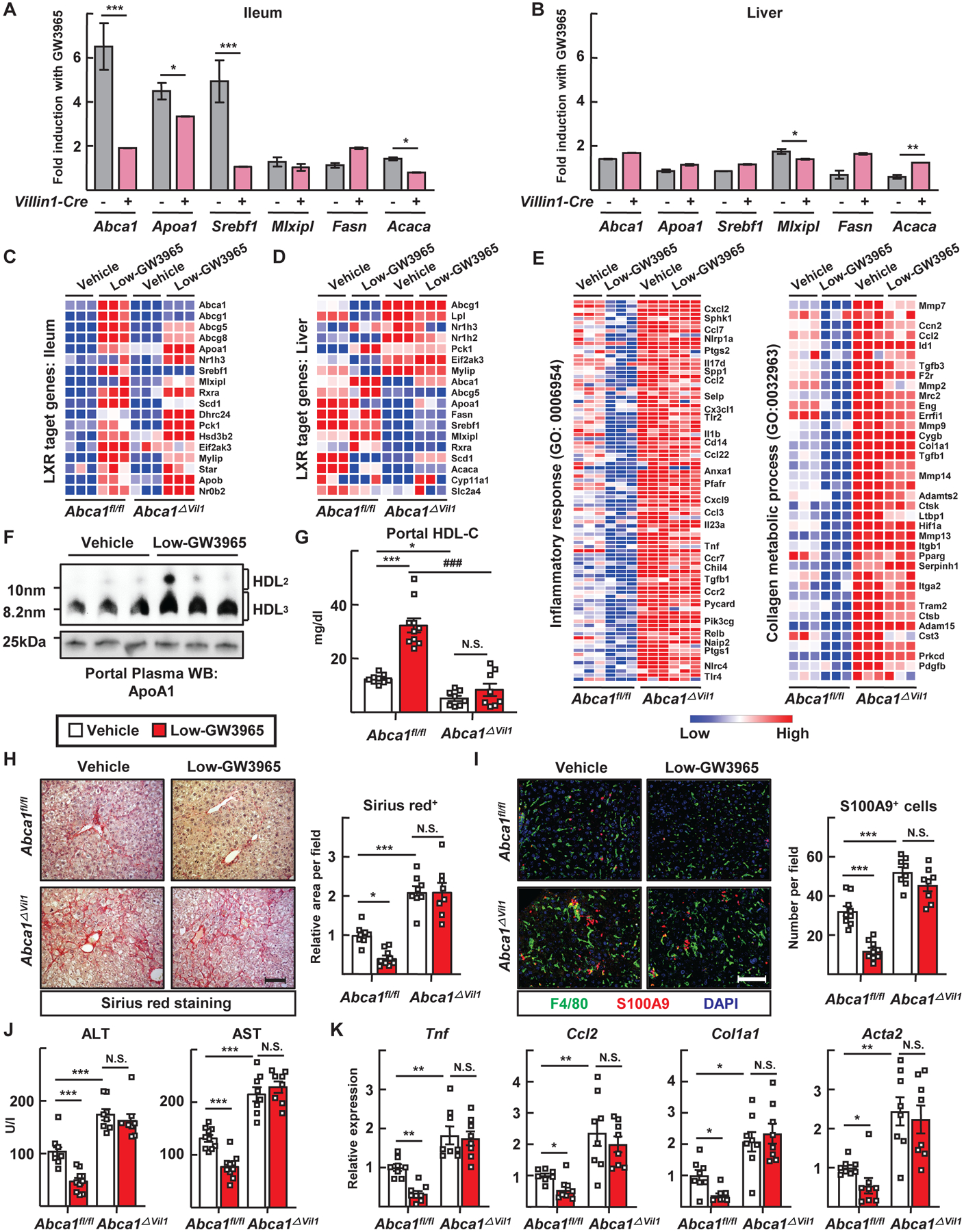
(A to E) Abca1fl/fl and Abca1ΔVil1 mice received vehicle (n=10 Abca1fl/fl mice, 8 Abca1ΔVil1 mice) or 1 mg/kg/day GW3965 (n=10 Abca1fl/fl mice, 8 Abca1ΔVil1 mice) by gavage twice weekly in the last 5 weeks of a 10-week feeding following SBR (Low-GW3965). (A and B) RT-PCR analysis of select target genes in the ileum and liver tissues was used to determine fold induction in response to GW3965 treatment. (C and D) Heat map of mRNA transcripts of LXR regulated genes in the ileum and liver tissues. (E) Heat map of mRNA transripts encoding genes associated with the inflammatory response or collagen metabolism within liver according to GW3965 treatment and genotype. (F) Immunoblots for apoA1 to detect HDL after portal vein plasma was run on a nondenaturing gel. (G) Portal plasma HDL cholesterol levels. (H) Sirius red staining of liver sections (left) and relative area per field (right) (scale, 100 μm). (I) F4/80+ macrophages and S100A9+ neutrophils visualized by immunofluorescence in liver sections (left) (scale, 100 μm) and cell numbers per field counted (right). (J) Plasma ALT and AST levels. (K) RT-PCR to measured inflammation- or fibrosis-associated mediators. Plots show mean ± SEM in male mice, with each symbol on the bar graphs representing a single mouse with data combined from two experiments; *P<0.05; **P<0.01; ***P<0.001; ###P<0.001. T tests were used for statistical comparisons in panels A and B; one-way ANOVA was used for statistical comparisons in panels G to K.
Funtionally, portal venous HDL-C, remaining predominantly in the form of HDL3, was increased by GW3965 in Abca1fl/fl mice but not in Abca1ΔVil1 mice (Fig. 7, F and G). Indeed, oral GW3965 prominently reduced fibrosis and inflammation in the liver after small-bowel resection (Fig. 7H to K), but was unable to do so in Abca1ΔVil1 mice (Fig. 7, H to K). Thus, orally delivered low-dose GW3965 protects the liver from inflammation and fibrosis in a manner that depends upon its capacity to increase enteric HDL.
Discussion
Intestinal epithelial cells produce HDL particles (11), but neither the fate nor the function of intestinal HDL has been clear. We show that the intestine produces the small form of HDL called HDL3 and that it is enriched in LBP. This HDL is shuttled to the liver via the portal vein. By the time it reaches the portal blood, it is already complexed with LBP and the LPS it carries is masked so that it does not bind to liver KCs, circumventing induction of proinflammatory and profibrotic genes. The intestinal epithelial location for HDL production allows the local capture of LPS before it gains access to downstream tissue.
That HDL may prevent LPS from binding to cells in the liver has been noted earlier by Munford and colleagues (35), who pioneered our understanding of the role of AOAH in inactivating LPS (26). Indeed, we show that whereas LBP-containing HDL3 suppresses recognition of LPS by KCs, it remains accessible to AOAH inactivation. It is unknown how HDL3 prevents TLR4 recognition of LPS but not that of AOAH. The difference may relate to the role of LBP. Since TLR4 depends upon LBP shuttling to CD14 to interact with LPS, the HDL3–LBP complex may most effectively mask this interaction by masking the critical epitope that would support LBP-mediated hand-off of LPS to CD14. However, AOAH activity does not require LBP or CD14 and thus may recognize LPS in the HDL3 particle via a region of LPS not masked by LBP.
HDL suppressed not just LPS-mediated activation of KC, but also pro-inflammatory action of mediators like lipoteichoic acid, which also interacts with HDL via LBP (36). Enterically derived HDL may bind and neutralize other microbial cargo not yet identified. Furthermore, the absence of HDL-mediated neutralization of microbial lipids like LPS may affect the course of inflammation in locations besides the liver. For example, in Crohn’s disease disease—a major form of inflammatory bowel disease—apoA1 is the most substantially downregulated gene in the affected ileal tissue (37). In cardiovascular disease and sepsis, HDL3 rather than HDL2 levels correlate with better health outcomes (38, 39). The connection between disease pathogenesis in these various conditions and enteric HDL is ripe for future investigations.
Unanswered questions arise from this study. Namely, how do LPS and LBP interact with HDL3 in a manner that masks the bioactivity of LPS? It is quite striking that while LBP is a critical promoter of TLR4 signaling, it conversely mediates suppressed signaling in the presence of HDL3. The structural basis of this unexpected result deserves future attention. Moreover, studies in whole-body knockout mice likely obscure the anti-inflammatory contribution of LBP when bound to HDL3 due to its other well characterized pro-inflammatory role. Finally, are other components of HDL3 needed to support the LPS-masking action of HDL3 that we identify here?
Yet another mystery is why the majority of portal venous HDL-C derives from the intestine. We had expected that portal venous blood HDL would arise from both portal and systemic sources. However, although systemic HDL clearly arrived to the gut or mesentery, evidenced by efficient entry into intestine-draining mesenteric lymphatics, it was not strongly detected in the portal vein. These data suggest the existence of unknown trafficking steps, including the possible extravasation of systemic HDL near or within the intestine. After extravasation, HDL2 that has entered or formed in the intestinal interstitium from the periphery may be too large to enter the fenestrated blood vessels that drain to the portal blood, such that only enterically derived HDL3 gains efficient access. Consistent with this possibility, mesenteric lymph is relatively deficient in the smaller HDL3 particles while relatively enriched in HDL2 (40). We note that, in contrast to our findings and those of others (12), studies in rats find that the intestine routes HDL to lymph (41, 42). Although this discrepancy may be a species difference, studies in humans are more consistent with our present findings (43, 44) than with those in the rat. Moreover, it has been proposed that the use of LCAT inhibitors in these rat studies abberantly affected the results (12).
We believe this research has strong translational potential. In humans, like mice, portal blood was enriched in HDL3 and potently suppressed KC activation in response to LPS. We utilized three murine models of liver injury involving nutritional, alcoholic or surgical insult to the intestine. All showed that intestinally derived HDL reduced liver injury. From a therapeutic perspective, oral delivery of LXR agonists proved effective in protecting the liver by upregulating HDL within the intestine, consistent with another recent study that engineered mice so that LXR activity was genetically augmented selectively in the intestine (45). Furthermore, our profiling and functional analysis revealed that the intestinal epithelium must express ABCA1, critical for HDL biogenesis, in order for low-dose, oral LXR agonist to protect the liver. LXR agonists have failed to find utility in the clinic to date, but orally restricted LXR agonists remain promising (46, 47). Our findings highlight the possibility that enteric HDL-raising LXR agonists have appeal for the treatment of various forms of liver injury. If suitable LXR agonists cannot be developed for application in humans however, other approaches to elevate intestinal HDL should be explored.
MATERIALS AND METHODS
Mice
C57BL/6 WT, Tlr4−/− (B6.B10ScN-Tlr4lps-del/JthJ; JAX #007227), Vil1-Cre (B6.Cg-Tg(Vil1cre)997Gum/J; JAX #004586), Alb1-Cre (B6.FVB(129)-Tg(Alb1-cre)1Dlr/J; JAX #016832), Abca1fl/fl (B6.129S6-Abca1tm1Jp/J; JAX #028266), Tlrfl/fl (B6(Cg)-Tlr4tm1.1Karp/J; JAX #024872) Clec4f-Cre (C57BL/6J-Clec4fem1(cre)Glass/J; JAX #033296), Abca1/g1fl/fl (B6.Cg-Abca1tm1Jp Abcg1tm1Tall/J; JAX #021067), and Lyz2-Cre (B6.129P2-Lyz2tm1(cre)Ifo/J; JAX #004781) mice (7–10 weeks of age) were originally purchased from the Jackson Laboratories and housed in a specific-pathogen-free room at 22 to 24°C and 50 to 60% humidity with a 12 hour light–dark cycle. We previously generated and described PGAKI/+ mice (15). Lyz2-Cre×Abca1/glfl/fl breeders were provided to use by Dr. R. Apte and Cd36–knockout mice (both strains also on C57BL/6 background) by Dr. N. Abumrad at Washington University. All experiments were performed in a blinded and randomized fashion. Mice were housed on a 12-hour light-dark cycle in a temperature controlled, specific pathogen-free unit with food and water provided ad libitum. The studies were approved by the Washington University Animal Studies Committee (Protocols 20170154, 20170252 and 20-0032) or the Medical College of Wisconsin Animal Studies Committee (Protocol #AUA00004173) in accordance with the National Institutes of Health laboratory animal care and use guidelines.
Small-bowel surgery and associated treatments or transplants
For small-bowel resection experiments, mice underwent a 50% proximal (jejunal) bowel resection, 75% proximal bowel resection, or sham control operation (bowel transection with reanastomosis alone), as previously described (28). In brief, through a midline laparotomy, the small bowel was exteriorized and transected 1 to 2 cm distal from the ligament of Treitz and approximately 12 cm (for 50% resection) or 6 cm (for 75% resection) proximal to the ileocecal junction. For sham operations, a transection 12 cm proximal to the ileocecal junction with immediate reanastomosis were performed. For distal 50% SBR, the ileum (last 12 cm of small bowel) was removed with an anastamosis of the jejunum to a small cuff of small bowel on the cecum. All anastomoses were hand-sewn end-to-end with interrupted 9–0 nylon sutures. Post-operative care included housing in an incubator for temperature stability and 24 hour fasting before starting a liquid diet (PMI Micro-Stabilized Rodent Liquid Diet LD 101; TestDiet), on which they were maintained for 8–12 weeks until euthanasia.
For bone marrow transplants, WT recipient mice at 8 weeks of age received whole-body irradiation at a dose of 11 Gy, and then were intravenously injected with 5×106 bone marrow cells from wild type or Tlr4−/− donor mice. After 4 weeks, short bowel resections were conducted.
For LXR agonist treatment, GW3965 (Sigma-Aldrich, #G6295) was suspended in 0.5% carboxymethyl cellulose and was orally administered twice weekly at 1 mg per kg of body weight per day for the last 5 weeks in the 10-week period following intestinal resection. The different experimental groups of mice maintained a similar body weight during liquid diet feeding and/or drug treatment.
Dietary challenge models
Where specified, mice were fed high-fat diet (HFD) or alcohol-containing Lieber–DiCarli diet to induce liver inflammation and injury (48, 49) The HFD study containing 60% kcal from fat (Research Diets, #D12492) was conducted for 12 weeks. For ethanol feeding, the mice were acclimated to increasing alcohol concentration of 2.1%, 4.2%, and 6.4% v/v (ethanol and liquid diet) for each 3 days, respectively. After alcohol adaptation, a 6.4% ethanol-enriched diet was supplied in the same liquid diet used after SBR surgeries (diet changed daily) for 4 weeks. In supplemental experiments, we fasted mice (with ad libitum access to water) for up to 20 hours, or challenged them for 3 weeks with an atherogenic diet containing 42% kcal from fat (Harlan Teklad TD.88137).
Immunostaining and confocal microscopy
Left lobes of liver tissues and small intestines were excised and fixed in 4% paraformaldehyde (Santa Cruz Biotechnology) overnight at 4°C. Ten-micron paraffin-embedded sections were prepared and slides were boiled in Diva Decloaker solution (Biocare Medical; #DV2004) in a pressurized chamber for 15 min. Sections were blocked with in PBS containing 5% donkey serum, 1% bovine serum albumin (BSA) (Sigma-Aldrich) and 0.03% Triton-X (Plusone, #17-1315-01) for 1 hour, then incubated in with rat anti-F4/80 (Abcam, #ab6640), goat anti-S100A9 (R&D Systems, #AF2065), rabbit anti-von Willebrand Factor (DAKO, #a0082), rat anti-PV1 (BD pharmingen, #550563), or rabbit anti-ABCA1 (Novus Biologicals, NB400-105) at 4°C overnight. Primary antibodies were detected using Cy3- or Cy5-conjugated secondary antibodies (Jackson ImmunoResearch). The stained sections were imaged using an SP8 confocal microscope (Leica) equipped with nine lasers and four tunable detectors (two hybrid, two tunable) and a 20X HC PL Apo CS 2 multi-immersion objective, NA 0.75. Images were processed with Imaris software (Bitplane). Ten fields were quantified and averaged for each sample, with cell counts per image quantified using Image J software (NIH). All slides were analyzed by in blinded and randomized fashion.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA from tissues or cells was isolated by using RNeasy Mini or Micro kits according to the manufacturer’s protocol (Qiagen). cDNA was synthesized using high-capacity cDNA reverse transcription kit (Applied Biosystems, #4368814). qRT-PCR experiments were performed using ABI StepOnePlus Real-Time PCR machine with specific primers (Applied Biosystems). Primer sequences are available upon request. The relative transcriptional expression of target genes was evaluated by the equation 2−ΔCt (ΔCt = Ct of target gene minus Ct of 18S rRNA). Relative transcription, where plotted, was calculated with the mean of the control group set as 1.
Intestinal permeability assay
After fasting 4 hours, 200 mg per kg of 4 kDa FITC-dextran (Sigma-Aldrich) was administered by gavage. After 0.5, 1, 2, or 4 hours, blood for the preparation of plasma was collected from the tail vein, and the fluorescence intensity of the samples and standards were read at excitation 485 nm / emission 525 nm using the Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Photoactivation of PGA1KI/+ mice
For phototagging HDL from skin, 8-to-10-week-old PGA1KI/+ mice were anesthetized and a region of shaved skin was photoconverted using a SOKY, Violet 405 nm 500 mW (FDA), PL-405-500B laser, as described previously (15). For photoactivation of the lumen of the small intestine of anesthetized mice, we stretched the mesentery and intestine over the solid surface of a Petri dish, located the region of interest and surgically clipped the bowel just enough so that we could thread into the lumen a fibro-optic endoscopic laser (Laserland, Violet 405 nm 100 mW) to photoactivate enterocytes. Unless other specified, three areas were activated for one data point, with in each case, the laser being held on 10 s, with 20 s off, cycling for 1 min 10 s to achieve three exposures per location. For photoactivation of the exterior of the small intestine, the Laserland, Violet 405 nm 100 mW 5V laser was used to activate area of 14.668 mm2 of gut with a similar on–off cycle as for the intestinal lumen.
Collection of blood and lymph
The portal blood was collected using a 33G needle to a volume of 40 μl per mouse. Systemic blood was collected from the inferior vena cava using a 26G needle in EDTA-containing tubes. Mesenteric lymph fluid was also collected, and the fluorescence intensity of plasma or lymph fluid was measured using the Cytation 5 Cell Imaging Multi-Mode Reader (BioTek). Mesenteric lymphatic cannulations were accomplished under general anesthesia using an operating microscope. A midline laparotomy with an extension to a left subcostal incision was made and the intestine was mobilized to expose the mesenteric lymphatic duct proximal to the cisterna chyli. A small incision was made on the mesenteric lymphatic duct and gently cannulated using polyethylene tubing (ID 0.28 mm OD 0.61 mm, Intramedic, Sparks, MD). At the completion of the collection, the mouse was euthanized.
Preparation of HDL fractions
Human and mouse plasma collected from the portal vein or peripheral vein (inferior vena cava for mouse, antecubital vein for human) was collected and ultracentrifuged overnight using standard methodology at 100,000g at 4°C in sequential steps, adjusting the solution to 1.063 g/ml using KBr (Sigma-Aldrich; #221864) to remove LDL. Then the HDL fraction was prepared by another centrifugation at 100,000g after further adjusting density of the solution to 1.21 g/ml with KBr. Isolated HDL fractions were dialyzed using Slide-A-Lyzer Dialysis cassette kit (Thermo Fisher) with PBS solutions containing sodium chloride, Tris and EDTA for 4 hours at 4°C to remove KBr. Human HDL2 (1.063–1.125 g/ml) and HDL3 (1.125–1.21 g/ml) fractions were obtained from GenWay Biotech.
FPLC and measurement of HDL cholesterol
Fifty microliters of blood from mice fasted for 4 hours was collected in microcentrifuge tubes containing 10 μl of 0.5 mM of EDTA and then centrifuged at 500g to collect plasma. For cholesterol distribution of total lipoproteins, plasma was prepared and 100–200 μl of the plasma was flowed over a Superose 6 10/300GL gel filtration column (GE Healthcare) to separate the different classes of lipoproteins. Cholesterol in each fraction was measured by an enzymatic assay kit (Wako Diagnostics Cholesterol E; #439-17501). HDL cholesterol assay kit (Cell Biolabs; #STA-394) was used to measure HDL-specific cholesterol levels (HDL-C).
Immunoblots
Protein immunoblotting was performed using rabbit anti-mouse apoA1 (Meridian Life Sciences), rabbit anti-human apoA1 (Millipore, #MAB011), rabbit anti-human ABCA1 (Novus Biologicals, #NB400-105), mouse anti-PON1 (Abcam, #ab24261), rabbit anti-ApoB (Proteintech, #20578-1), rabbit anti-LBP (Abcam, #ab233524), rabbit anti-AOAH (Proteintech, #12911-1), rabbit anti-SERPINA1 (Thermo, #PA5-16661), mouse anti-ApoE (Kindly provided by D. M. Holtzmann, Washington University) or rabbit anti-albumin (Proteintech, #16475-1) antibodies. Table S2 specifies dilutions of the antibodies used. The HDL fractions were loaded to achieve the same protein concentration per lane, and plasma was loaded without dilution. For native gels, the samples were diluted in 2X native sample buffer (Bio-rad) and run on 4 to 20% Mini-PROTEAN Tris-glycine gels (Bio-rad) with Tris-glycine running buffer. For denaturing gels, the samples were diluted in 2X Tris-Glycine-SDS sample buffer (EZ Bioresearch) and heated at 95°C for 10 min. The samples were loaded onto 4 to 20% Mini-PROTEAN gels and run with Tris-glycine-SDS running buffer. The separated proteins were transferred to 0.45-μm PVDF membrane (Milipore, #IPVH00010) with Tris-glycine transfer buffer for 2 hours at 20 V. Membranes were blocked with 5% nonfat dry skim milk (Bio-rad, #170-6404) for 1 hour and primary antibodies were incubated overnight at 4°C. After incubation with HRP-conjugated secondary antibodies, signal detection was done using Clarity Western ECL solution (Bio-rad).
Electron Microscopy of HDL particles
The isolated HDL fractions were diluted to 15 μg/ml of total protein and negatively stained with 1% uranium acetate. The samples were deposited on carbon-coated 200 mesh copper grids (Electron Microscopy Sciences). Images were acquired with a TEM (JEOL JEM-1400Plus) at 120 KeV and 80,000X or 150,000X magnifications. The diameter of HDL particles was measured using Image J software.
Isolation or culture of liver immune cells and macrophages
For quantification of neutrophils, monocyte-derived macrophages and Kupffer cells in livers of mice subjected to short bowel resection or sham surgery, livers were collected and homogenized in Hank’s buffered saline solution containing 1.49 mg/ml of collagnase type IV (Sigma-aldrich; #C5139) and dissociated using the gentleMACS Octo Dissociator (Miltenyl Biotec). After centrifugation at 50g, the supernatant containing nonparenchymal cells was separated using 33% Percoll (GE Healthcare).
For Kupffer cell isolation and culture, the livers of 7-to-10-week-old male C57BL/6 mice were perfused via the inferior vena cava with collagenase type IV solution as described previously (50). The cell suspension was centrifuged. Cell suspensions collected in the supernatant were collected and again centrifuged in 50%/25% Percoll (GE Healthcare). The layer containing liver macrophages was plated in RPMI-1640 (Hyclone) containing 10% fetal bovine serum (FBS). After 2 hours of culture to allow for cell attachment, the cell medium was changed to “vehicle” medium, which was serum-free RPMI-1640 containing 1 μg/ml of recombinant LBP (R&D systems; #6635-LP) for 3 hours of culture. HDL preparations were added, or not, to these cultures with 20 ng/ml of LPS (Sigma-Aldrich; #L2630), 10 μg/ml of lipoteichoic acid (LTA) (Sigma-Aldrich; #L2515), 10 μg/ml CpG DNA (Invivogen, #tlr-l1826), 100 ng/ml TNF (Sigma-Aldrich; #T7539), or 100 ng/ml IL-1β (Sigma-Aldrich; #I5271).
Flow cytometry
Isolated liver immune cells and cultured liver macrophages were collected and counted in an automated cell counters (Cellometer Auto X4, Nexelcom Bioscience) after staining for acridine orange (Sigma-aldrich). Antibodies (details in table S2) including BUV396-anti-CD45 (BD bioscience; # 563791), FITC-anti-Ly6G (Biolegend; #127605), APC/Cy7-anti-F4/80 (Biolegend; #123117), PerCP/Cy5.5-anti-Ly6C (Biolegend; #128011), PE/Cy7-anti-CD31 (Biolegend; #102417), Alexa488-anti-iNOS (Thermo Fisher; #53-5920-82), APC-anti-CD11b (Thermo Fisher; 17-0112-82), PE-anti-Tim4 (Thermo Fisher; 12-5866-82), or goat anti-Clec4f (R&D systems; #AF2784) were incubated with FACS buffer (2% fetal bovine serum, 2 mM EDTA, and sodium azide in PBS) on ice for 30 min. In some experiments, the primary cultures of KCs were incubated with 100 ng/ml of biotinylated LPS (Invivogen, #tlrl-lpsbiot) for 2 hours. Then biotin was detected using PE/Cy7-streptividin (Biolegend; #405206). After surface staining of biotin, in some experiments, internalized biotin-LPS was stained using BV605-streptavidin (Biolegend; #405229) in cells permeabilized using the Intracellular Fixation & Permeabilization Buffer Set (Thermo eBioscience; #88-8824). After washing and resuspension, cells were analyzed on a BD FACS Symphony machine and analyzed by FlowJo software (BD biosciences).
ELISA
ELISA kits were used according to the manufacturer’s protocol and included AST Activity Assay Kit (Sigma-Aldrich; MAK055). TNFα (Sigma-Aldrich; RAB0477), or CCL2 ELISA (Sigma-Aldrich; #RAB0055). Limulus amebocyte lysate (LAL) endotoxin activity was measured using the Pierce LAL Chromogenic Endotoxin Quantitation Kit (Thermo Fisher; #88282). LPS quantification by ELISA used LPS ELISA kit from MyBiosource (MBS700021). For sandwich ELISA to analyze LPS-HDL binding, we used high-binding clear polystyrene microtiter plates (R&D systems, DY990), and purified HDL was immobilized for 2 hours on these plates at 37°C at 10 μg/ml. After washing, plates were blocked with 1% BSA for 1 hour. Then biotinylated LPS was pre-incubated with or without 1 μg/ml of recombinant LBP (R&D Systems) for 1 hour and incubated in plates for 30 min. For ELISA to quantify biotin, samples including biotinylated LPS were incubated in high-binding clear polystyrene microtiter plates overnight at 37°C. Streptavidin peroxidase (R&D Systems) was added, followed by diaminobenzidine substrate (Abcam), for colorimetric reactions. Colorimetric or fluorometric absorbance was detected using the Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Immunoprecipitation of HDL, LDL, and biotinylated LPS
For proteomic analysis of systemic and portal plasma, human plasma samples were separated using size-exclusion FPLC chromatography and then plasma albumin and IgG was depleted using Pierce Albumin/IgG Removal kit (Thermo Fisher, #89875). Immunoprecepitation on the separated HDL fractions was conducted using Pierce MS-Compatible Streptavidin Magnetic IP Kit (Thermo Fisher, #90408). Anti-human apoA1 antibody (Proteintech, #14427-1) was biotinylated using Pierce Antibody Biotinylation kit for IP (Thermo Fisher, #90407) via streptavidin immunoprecipitation. The samples were eluted in low pH and neutralized to 100 mM Tris pH 8.0.
Biotinylated LPS in cell media was mixed with 1% SDS to disrupt lipoproteins and then purified using Streptavidin Magnetic IP kit. Endotoxin activity of the immunopurified biotinylated LPS was measured using the LAL kit and normalized by relative amount of biotin as detected in ELISA.
For binding studies, 50 μg/ml of protein in HDL3, HDL2, or LDL fractions was mixed with 1 μg/ml of LBP and 20 μg/ml of biotinylated LPS in 100 mM NaCl, 100 mM Tris, and 1 mM EDTA buffer for 2 hours. The mixtures were incubated with anti-apoA1 antibody (Proteintech) or anti-apoB antibody (Thermo, #MIA1605) overnight and immunoprecipitated using Protein A agarose beads (Abcam, #ab193254). The bead-bound samples were eluted using 0.1 M glycine buffer (pH 2.5) and neutralized using 100 mM Tris, pH 8.0. The distribution of LBP and biotinylated LPS were measured by immunoblotting and streptavidin-peroxidase ELISA, respectively.
Generation of reconstituted HDL (rHDL)
Human apoA1 isolation and purification from fresh human plasma was carried out as reported previously (51). The lyophilized protein was solubilized and denatured in STB (10 mM Tris, 0.15 M NaCl, 1 mM EDTA, and 0.2% NaN3) containing 3 M guanidine HCl followed by refolding at 4°C by dialyzing against three changes of 4 liters of STB for a minimum of 3 hours each. rHDL particles were generated with a modified sodium cholate dialysis as previously described (52). A molar ratio of 80:1 POPC (Avanti Polar Lipids), and apoA1, respectively, was used to generate rHDL.
Depletion of HDL or AOAH
The HDL in mouse plasma was removed by HDL Depletion Column IgY Kit (Genway Biotech; #GWB-HDLIGY). The acyloxyacyl hydrolase (AOAH) in cell medium was depleted via immunoprecipiation by anti-AOAH antibody (Proteintech) and Protein A agarose beads. After immunoprecipitation, the supernatant was harvested and we confirmed depletion of HDL or AOAH by immunoblotting.
Chromatographic analysis of biotinylated LPS transfer between lipoprotein species
HDL3 or HDL2 (100 μg protein/ml) was incubated with or without 2 μg/ml of LBP and 1 μg/ml of biotinylated LPS in Tris buffer for 30 min, and then dialyzed in a Slide-A-Lyzer Dialysis cassette kit. Isolated VLDL (10 μg/ml of protein) and LDL (50 μg/ml of protein) were subsequently added to the HDL-LPS mixture and incubated for 2 hours at 37°C. The 200-μl mixture was then subjected to FPLC separation using a Superose 6 10/300GL gel filtration column. The amount of biotin was measured via streaptavidin peroxidase ELISA and lipoprotein-associated LPS was thereby determined.
Portal vein injection of biotinylated LPS and lipoprotein complex
A mixture containing 5 μg biotinylated LPS and 2 μg of LBP was incubated with 0.1 mg HDL3, HDL2, or LDL (concentration determined by protein, not lipid) in saline buffer for 3 hours. Mice were anaesthetized by injection of a cocktail of ketamine (50 mg per kg) and xylazine (8 mg per kg) intraperitoneally. The LPS-loaded lipoprotein mixtures described above were then injected via the portal vein in a 100-μl volume using an ultra-small 33G needle. VETSPON Absorbable Hemostatic Gelatin Sponges (Novartis) were used to stop bleeding of the portal vein. Systemic blood and livers were harvested after 30 min later.
Peptide preparation and nano-LC-MS/MS
Peptides were prepared as previously described (53). Then modification of a previous method (54) was followed. First, the column was equilibrated to 0.1% formic acid (FA) for a total of 11 μl at 700 bar pressure. The samples in FA (1%) were loaded on an EASY nanoLC (Thermo Fisher), with sample (2.5 μl) applied onto a 75 μm i.d. × 50 cm Acclaim® PepMap 100 C18 RSLC column (Thermo Fisher). A constant pressure of 700 bar was maintained at 0.1% FA. Peptide chromatography was carried out using mobile phase A (1% FA) containing 2% B (100% MeCN, 1%FA) for 5 min, then increased to 20% B over 100 min, to 32% B over 20 min, to 95% B over 1 min and held at 95% B for 29 min. The flow rate was 250 nl/min. Data were acquired in data-dependent acquisition (DDA) mode. Full-scan mass spectra were acquired with the Orbitrap mass analyzer with a scan range of m/z = 350 to 1500 and a mass resolving power set to 70,000. Ten data-dependent high-energy collisional dissociations were performed with a mass resolving power set to 17,500, a fixed lower value of m/z 100, an isolation width of 2 Da, and a normalized collision energy setting of 27. The maximum injection time was 60 ms for parent-ion analysis and product-ion analysis. The target ions that were selected for MS/MS were dynamically excluded for 20 s. The automatic gain control (AGC) was set at a target value of 3e6 ions for full MS scans and 1e5 ions for MS2. Peptide ions with charge states of 1 or >8 were excluded for HCD acquisition.
Protein Identification
The MS unprocessed data from the mass mass spectrometer were converted to peak lists using Proteome Discoverer (Thermo Fisher). The MS2 spectra with charges +2, +3 and +4 were analyzed using Mascot software (Matrix Science). Mascot was set up to search against a UniProt (July 2019) database of human proteins (20,667 entries), using trypsin cleavage specificity (trypsin/P) with four missed cleavages allowed. The searches were performed using Mascot software according to previously described parameters for peptide identification (53). Peptides and proteins were filtered at 1% false-discovery rate (FDR) by searching against a reversed protein sequence database. The ontology of HDL signature proteins were acquired from DAVID bioinformatics functional annotiation (https://david.ncifcrf.gov/).
RNA-seq
Three individual mice per experimental group were used for the generation of whole intestinal or whole liver RNA sequencing. Total RNA integrity was determined using Agilent Bioanalyzer or 4200 Tapestation. Library preparation was performed with 500 ng to 1 μg of total RNA. Ribosomal RNA was removed by an RNase-H method using RiboErase kits (Kapa Biosystems), and mRNA was then fragmented in reverse transcriptase buffer with heating to 94°C for 8 min. Then mRNA was reverse transcribed to yield cDNA using SuperScript III RT enzyme (Life Technologies) and random hexamers. A second strand reaction was performed to yield ds-cDNA. cDNA was blunt-ended, had an A base added to the 3′ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12 to 15 cycles using primers incorporating unique dual index tags. Fragments were sequenced on an Illumina NovaSeq-6000 using paired-end reads extending 150 bases. The gene counts were quantified with CPM transformations added with custom R scripting. Normalized Log2 CPM values were visualized as heatmaps via the web interface Phantasus (https://genome.ifmo.ru/phantasus). Gene ontology pathway analyses were acquired from DAVID bioinformatics functional annotation. The sequencing and expression data have been deposited in the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information with the accession number GSE167983.
Human Studies
Human portal and peripheral systemic blood was acquired from adult patients undergoing open surgical procedures where the surgical team deemed that the portal vein was safely accessible. Blood was collected in EDTA-containing tubes and spun down for immediate analysis of plasma for HDL. The population of patients from which plasma was acquired was comprised of three males and three females ranging in age from 54 to 80. Four underwent Whipple pancreatic surgery, one underwent gastric bypass, and one underwent an orthotopic liver transplant. Immunoblots were performed using tissue specimens from other patients undergoing surgical resection for proximal or distal gut after traumatic injury. These were collected and fixed in formalin by the Department of Pathology and Immunology for a routine surgical pathology work-up and were shared for research after sign-out of the clinical case. After dissecting tissue enriched in epithelium, dissected tissues were stabilized in PAXgene Tissue Stabilization buffer (PreAnalytiX, #765512) for at least 3 hours. Protein lysates were then prepared by homogenization in Extraction EXB buffer (Qiagen, #37623). All human studies were approved by the Human Research Protection Office at Washington University, IRB protocols # 201111038 and 2019101009 (Randolph, G. J., PI).
Statistics
All graphs are plotted to depict mean ± SEM. A paired or unpaired two-tailed Student’s t test was used for simple comparisons, a one-way ANOVA with Tukey’s post hoc test for multiple comparisons for three or more groups with one variable, or a two-way ANOVA with Sidak post hoc test for three or more groups with two variables. Statistical differences were analyzed and graphs were prepared by using GraphPad Prism Software Version 8.0 (GraphPad Software). P<0.05 was considered to be a significant difference. Replicates in the bar graphs are shown by distinct symbols. Replicates in biochemical/binding experiments were independent technical replicates using the same reagents (technical replicates; Figs. 3 and 4, A to F). In other experiments, each symbol in a graph represents experiments where independent cell preparations of plasma (Figs. 1A, 1F, 2A, and 2B; paired samples) or primary cells (Figs. 3, A to E, and 4, G to L) were generated and studied within the experimental design. Finally, in all in vivo experiments, each symbol in a graph represents data generated from an individual mouse (Figs. 1, C to E, 4M, 5A, 5C, 5, E to H, 5, J to L, 6, A to F, 6, H to J, 6, L and M, and 7, A to K). All data using mice contained at least two mice in each experimental group from the same litter, so that the data in a given experimental cohort contained littermate controls in the other groups of the same experiment. Some studies combined mice from 3–4 litters to generate sufficient numbers. If these mice were subjected to experimental manipulations like short bowel surgery on different dates, we refer to the combination of those data into one graph as combining different experiments, with each distinct start date of the experimental manipulation considered an independent experiment. Experimental manipulations that began on the same day with age-matched litters were not classified as independent experiments. The distribution of data was not impacted by combining independent experiments. Where possible, data collected from different experiments was subjected to assays simultaneously to minimize batch effects.
Supplementary Material
ACKNOWLEDGEMENTS
We thank R. Apte for Lys2-Cre×Abca1fl/fl AAbcg1fl/fl mice, N. Abumrad for Cd36−/− mice, E.B. Cho for assistance with figures, M. Wallendorf with statistics consultation and P. Erdmann-Gilmore, Y. Mi and R. Connors for assistance with proteomics.
Funding:
Work was supported by NIH R01DK119147 to G.J.R. and B.W.W. and AI0499653 to G.J.R., including a Primary Caregiver Supplement to E.J.O. Y.H.H. was supported in part by the National Research Foundation (NRF) of Korea (2021R1C1C1004023). E.J.O. also received support from NIH T32DK077653. L.H. was supported by AHA Career Developmnt Award (AHA:1 8CDA34110273), R.S.C. by the Lawrence C. Pakula, MD IBD Research Fellowship and M. G. S. T. by NIH RO1HL127649 and HL138908. Further core facility support was to Digestive Diseases Research Core Center of Washington University, P30 DK052574; to Washington University Center for Cellular Imaging (WUCCI) by the Children’s Discovery Institute of Washington University (CDI-CORE-2015-505 and CDI-CORE-2019-813) and the Foundation for Barnes-Jewish Hospital (3770; to Washington University Proteomics Shared Resource, directed by R. R. Townsend) by the WU Institute of Clinical and Translational Sciences (NCATS UL1 TR000448), the Mass Spectrometry Research Resource (NIGMS P41 GM103422; R24GM136766) and the Siteman Comprehensive Cancer Center (NCI P30 CA091842); and to the Genome Technology Access Center by NCI Cancer Center Grant #P30 CA91842 and ICTS/CTSA Grant# UL1TR002345 from the National Center for Research Resources (NCRR).
Footnotes
Competing Interests: The authors declare no competing interests.
Data and Materials Availability:
Gene expression raw data in the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information have accession number GSE167983. Proteomics data are deposited at ProteomeXchange (PXD025648, MassIVE dataset name is MSV000079070). All other data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Albillos A, de Gottardi A, Rescigno M, The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol 72, 558–577 (2020). doi: 10.1016/j.jhep.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Duan Y, et al. , Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575, 505–511 (2019). doi: 10.1038/s41586-019-1742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwabe RF, Seki E, Brenner DA, Toll-Like Receptor Signaling in the Liver. Gastroenterology 130, 1886–1900 (2006). doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 4.Todoric J, et al. , Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab 2, 1034–1045 (2020). doi: 10.1038/s42255-020-0261-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hritz I, et al. , The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48, 1224–1231 (2008). doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barron LK, et al. , Toll-like receptor 4 is critical for the development of resection-associated hepatic steatosis. J. Pediatr. Surg 52, 1014–1019 (2017). doi: 10.1016/j.jpedsurg.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inokuchi S, Tsukamoto H, Park E, Liu ZX, Brenner DA, Seki E, Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol. Clin. Exp. Res 35, 1509–1518 (2011). doi: 10.1111/j.1530-0277.2011.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL, In vivo protection against endotoxin by plasma high density lipoprotein. Proc. Natl. Acad. Sci. U. S. A 90, 12040–12044 (1993). doi: 10.1073/pnas.90.24.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wurfel MM, Hailman E, Wright SD, Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J. Exp. Med 181, 1743–1754 (1995). doi: 10.1084/jem.181.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meilhac O, Tanaka S, Couret D, High‐density lipoproteins are bug scavengers. Biomolecules 10, 598 (2020). doi: 10.3390/biom10040598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Windmueller HG, Herbert PN, Levy RI, Biosynthesis of lymph and plasma lipoprotein apoproteins by isolated perfused rat liver and intestine. J. Lipid Res 14, 215–223 (1973). doi: 10.1016/S0022-2275(20)36909-1. [DOI] [PubMed] [Google Scholar]
- 12.Brunham LR, et al. , Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Invest 116, 1052–1062 (2006). doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martel C, et al. , Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Invest 123, 1571–1579 (2013). doi: 10.1172/JCI63685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim HY, et al. , Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-Mediated transport of HDL. Cell Metab 17, 671–684 (2013). doi: 10.1016/j.cmet.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Huang LH, et al. , Interleukin-17 Drives Interstitial Entrapment of Tissue Lipoproteins in Experimental Psoriasis. Cell Metab 29, 475–487.e7 (2019). doi: 10.1016/j.cmet.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randolph GJ, Miller NE, Lymphatic transport of high-density lipoproteins and chylomicrons. J. Clin. Invest 124, 929–935 (2014). doi: 10.1172/JCI71610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glickman RM, Green PHR, The intestine as a source of apolipoprotein A1. Proc. Natl. Acad. Sci. U. S. A 74, 2569–2573 (1977). doi: 10.1073/pnas.74.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson WS, et al. , Proteomic analysis of defined hdl subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol 29, 870–876 (2009). doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoofnagle AN, Heinecke JW, Lipoproteomics: Using mass spectrometry-based proteomics to explore the assembly, structure, and function of lipoproteins. J. Lipid Res 50, 1967–1975 (2009). doi: 10.1194/jlr.R900015-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryu JK, et al. , Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 46, 38–50 (2017). doi: 10.1016/j.immuni.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Scott CL, et al. , Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun 7, 10321 (2016). doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seidman JS, et al. , Niche-Specific Re-Programming of Epigenetic Landscapes Drives Myeloid Cell Diversity in NASH. Immunity 52, 1057–1074.e7 (2020). doi: 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odeyale CO, Kang YH, Biotinylation of bacterial lipopolysaccharide and its applications to electron microscopy. J. Histochem. Cytochem 36, 1131–1137 (1988), doi: 10.1177/36.9.3136207. [DOI] [PubMed] [Google Scholar]
- 24.Luk JM, Kumar A, Tsang R, Staunton D, Biotinylated lipopolysaccharide binds to endotoxin receptor in endothelial and monocytic cells. Anal. Biochem 232, 217–224 (1995). doi: 10.1006/abio.1995.0010. [DOI] [PubMed] [Google Scholar]
- 25.Massamiri T, Tobias PS, Curtiss LK, Structural determinants for the interaction of lipopolysaccharide binding protein with purified high density lipoproteins: Role of apolipoprotein A-I. J. Lipid Res 38, 516–525 (1997). doi: 10.1016/S0022-2275(20)37259-X [DOI] [PubMed] [Google Scholar]
- 26.Munford RS, Weiss JP, Lu M, Biochemical transformation of bacterial lipopolysaccharides by acyloxyacyl hydrolase reduces host injury and promotes recovery. J. Biol. Chem 295, 17842–17851 (2020). doi: 10.1074/jbc.REV120.015254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Courtney CM, Warner BW, Pediatric intestinal failure-associated liver disease. Curr. Opin. Pediatr 29, 363–370 (2017). doi: 10.1097/MOP.0000000000000484. [DOI] [PubMed] [Google Scholar]
- 28.Onufer EJ, et al. , Effects of high-fat diet on liver injury after small bowel resection. J. Pediatr. Surg 55, 1099–1106 (2020). doi: 10.1016/j.jpedsurg.2020.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spadoni I, et al. , A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350, 830–834 (2015). doi: 10.1126/science.aad0135. [DOI] [PubMed] [Google Scholar]
- 30.Sakai M, et al. , Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 51, 655–670.e8 (2019). doi: 10.1016/j.immuni.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho YE, Yu LR, Abdelmegeed MA, Yoo SH, Song BJ, Apoptosis of enterocytes and nitration of junctional complex proteins promote alcohol-induced gut leakiness and liver injury. J. Hepatol 69, 142–153 (2018). doi: 10.1016/j.jhep.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouries J, et al. , Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol 71, 1216–1228 (2019). doi: 10.1016/j.jhep.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunham LR, et al. , Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ. Res 99, 672–674 (2006). doi: 10.1161/01.RES.0000244014.19589.8e. [DOI] [PubMed] [Google Scholar]
- 34.Lo Sasso G, et al. , Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab 12, 187–193 (2010). doi: 10.1016/j.cmet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Shao B, Munford RS, Kitchens R, Varley AW, Hepatic uptake and deacylation of the LPS in bloodborne LPS-lipoprotein complexes. Innate Immun 18, 825–833 (2012). doi: 10.1177/1753425912442431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grunfeld C, et al. , Lipoproteins inhibit macrophage activation by lipoteichoic acid. J. Lipid Res 40, 245–252 (1999). doi: 10.1016/s0022-2275(20)33363-0. [DOI] [PubMed] [Google Scholar]
- 37.Haberman Y, et al. , Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest 124, 3617–3633 (2015). doi: 10.1172/jci79657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin SS, et al. , HDL cholesterol subclasses, myocardial infarction, and mortality in secondary prevention: The lipoprotein investigators collaborative. Eur. Heart J 36, 22–30 (2015). doi: 10.1093/eurheartj/ehu264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albers JJ, Slee A, Fleg JL, O’Brien KD, Marcovina SM, Relationship of baseline HDL subclasses, small dense LDL and LDL triglyceride to cardiovascular events in the AIM-HIGH clinical trial. Atherosclerosis 251, 454–459 (2016). doi: 10.1016/j.atherosclerosis.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gracia G, Cao E, Johnston APR, Porter CJH, Trevaskis NL, Organ-specific lymphatics play distinct roles in regulating HDL trafficking and composition. Am. J. Physiol. Gastrointest. Liver Physiol 318, G725–G735 (2020). doi: 10.1152/ajpgi.00340.2019. [DOI] [PubMed] [Google Scholar]
- 41.Forester GP, Tall AR, Bisgaier CL, Glickman RM, Rat intestine secretes spherical high density lipoproteins. J. Biol. Chem 258, 5938–5943 (1983). doi: 10.1016/s0021-9258(20)81987-5. [DOI] [PubMed] [Google Scholar]
- 42.Bearnot HR, Glickman RM, Weinberg L, Green PH, Tall AR, Effect of biliary diversion on rat mesenteric lymph apolipoprotein-I and high density lipoprotein. J. Clin. Invest 69, 210–217 (1982). doi: 10.1172/JCI110432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliveira HC, Nilausen K, Meinertz H, Quintao EC, Cholesteryl esters in lymph chylomicrons: Contribution from high density lipoprotein transferred from plasma into intestinal lymph. J. Lipid Res 34, 1729–1736 (1993). doi: 10.1016/s0022-2275(20)35735-7. [DOI] [PubMed] [Google Scholar]
- 44.Quintão EC, Drewiacki A, Stechhaln K, de Faria EC, Sipahi AM, Origin of cholesterol transported in intestinal lymph: Studies in patients with filarial chyluria. J. Lipid Res 20, 941–945 (1979). doi: 10.1016/s0022-2275(20)39995-8. [DOI] [PubMed] [Google Scholar]
- 45.Pierantonelli I, et al. , HDL cholesterol protects from liver injury in mice with intestinal specific LXRα activation. Liver Int 40, 3127–3139 (2020). doi: 10.1111/liv.14712. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda T, et al. , Tissue-specific liver X receptor activation promotes macrophage reverse cholesterol transport in vivo. Arterioscler. Thromb. Vasc. Biol 30, 781–786 (2010). doi: 10.1161/ATVBAHA.109.195693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B, Tontonoz P, Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol 14. 452–463 (2018). doi: 10.1038/s41574-018-0037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daemen S, et al. , Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep 34, 108626 (2021). doi: 10.1016/j.celrep.2020.108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo F, Zheng K, Benedé-Ubieto R, Cubero FJ, Nevzorova YA, The Lieber-DeCarli Diet—A Flagship Model for Experimental Alcoholic Liver Disease. Alcohol. Clin. Exp. Res 42, 1828–1840 (2018). doi: 10.1111/acer.13840. [DOI] [PubMed] [Google Scholar]
- 50.Han YH, et al. , RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep 20, 124–135 (2017). doi: 10.1016/j.celrep.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 51.Davidson WS, Hilliard GM, The spatial organization of apolipoprotein A-I on the edge of discoidal high density lipoprotein particles a mass spectrometry study. J. Biol. Chem 278, 27199–27207 (2003). doi: 10.1074/jbc.M302764200. [DOI] [PubMed] [Google Scholar]
- 52.Bonomo EA, Swaney JB, A rapid method for the synthesis of protein-lipid complexes using adsorption chromatography. J. Lipid Res 29, 380–384 (1988). doi: 10.1016/s0022-2275(20)38530-8. [DOI] [PubMed] [Google Scholar]
- 53.Rustenhoven J, et al. , Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184, 1000–1016.e27 (2021). doi: 10.1016/j.cell.2020.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patra D, Kim J, Zhang Q, Tycksen E, Sandell LJ, Site-1 protease ablation in the osterix-lineage in mice results in bone marrow neutrophilia and hematopoietic stem cell alterations. Biol. Open 9, bio052993 (2020). doi: 10.1242/bio.052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Gene expression raw data in the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information have accession number GSE167983. Proteomics data are deposited at ProteomeXchange (PXD025648, MassIVE dataset name is MSV000079070). All other data are available in the main text or the supplementary materials.