

Abstract
Background and Purpose:
Aneurysmal subarachnoid hemorrhage (SAH) is associated with the development of delayed cognitive deficits. Neutrophil infiltration into the central nervous system (CNS) is linked to the development of these deficits after SAH. It is however unclear how neutrophil activity influences CNS function in SAH. The present project aims to elucidate which neutrophil factors mediate CNS injury and cognitive deficits after SAH.
Methods:
Using a murine model of SAH and mice deficient in neutrophil effector functions, we determined which neutrophil effector function is critical to the development of deficits after SAH. In vivo and in vitro techniques were used to investigate possible pathways of neutrophils effect after SAH.
Results:
Our results show that mice lacking functional myeloperoxidase (MPO), a neutrophil enzyme, lack both the meningeal neutrophil infiltration (WT, sham 872 cells/meninges vs. SAH 3047, p=0.023; MPOKO, sham 1677 vs. SAH 1636, p=NS) and erase the cognitive deficits on Barnes maze associated with SAH (MPOKO sham vs. SAH, p=NS). The re-introduction of biologically active MPO, and its substrate hydrogen peroxide (H2O2), to the cerebrospinal fluid of MPOKO mice at the time of hemorrhage restores the spatial memory deficit observed after SAH (time to goal box MPOKO sham vs. MPOKO+MPO/H2O2, p=0.001). We find evidence of changes in neurons, astrocytes and microglia with MPO/H2O2 suggesting the effect of MPO may have complex interactions with many cell types. Neurons exposed to MPO/H2O2 show decreased calcium activity at baseline and after stimulation with potassium chloride. Although astrocytes and microglia are affected, changes seen in astrocytes are most consistent with inflammatory changes that likely affect neurons.
Conclusions:
These results implicate MPO as a mediator of neuronal dysfunction in SAH through its effect on both neurons and glia. These results show that, in SAH, the activity of innate immune cells in the meninges modulates the activity and function of the underlying brain tissue.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is an important cause of long-term disability, although the major cause of disability is not physical weakness, but cognitive dysfunction. Cognitive dysfunction is attributed to the syndrome of Delayed Cerebral Injury (DCI, that is associated with cerebral vasospasm) 1, 2. Although cerebral vasospasm is less often seen in patients with other forms of SAH (non-aneurysmal or venous), the existing data suggests there are cognitive deficits associated with these syndromes 3,4. The major obstacle to developing treatments is a poor understanding of the pathophysiology that leads to impaired cognitive function. Our laboratory has shown that peripheral neutrophil infiltration leads to the development of DCI and cognitive deficits in a murine model of SAH due to loss of late long-term potentiation (L-LTP) in hippocampal neurons 5, 6.
Neutrophils are pathological in a multitude of neurological disorders. Adherence to cerebral capillary beds is linked to cognitive decline in a mouse model of Alzheimer’s disease 7, and increased infiltration in the brain in a model of ischemic stroke coincides with increased neuronal loss 8. However, the cellular and molecular mechanism of their impact in the CNS is poorly understood.
Neutrophils have a limited but potent set of effector functions that are implicated in neurological injuries. These effector functions include enzymes that catalyze the production of reactive oxygen and nitrogen species, and the degradation of extracellular matrix proteins 9, 10. A number of these enzymes have been linked to brain pathology after injury. For example, myeloperoxidase (MPO) and elastase (both predominantly neutrophil granule enzymes) increase oxidative stress in the brain after cerebral ischemia 8, 11. Modulation of myeloperoxidase activity after stroke leads to better behavioral outcomes 11. Limb remote ischemic postconditioning improves stroke outcomes by decreasing the activity of NADPH oxidase and MPO in peripheral neutrophils 12.
Study of brain inflammatory pathology in most non-infectious conditions is limited due to the superimposed necrosis that causes direct brain damage leading to peripheral immune cells invasion, thereby mixing the neuroinflammatory response and the necrosis-driven inflammatory invasion. The validated, low-pressure, venous murine model of SAH is useful to study the effect of inflammation on brain physiology because the direct injury occurs outside the brain parenchyma. In addition, this murine model recapitulates human DCI 13-15, and has a quantifiable readout of brain injury 16.
In this project, we explore how peripheral innate immune cells affect neuronal function after SAH. We hypothesize that neutrophil effector molecules are the driver of neutrophil-mediated delayed cognitive deficits in our model of SAH.
Materials and Methods
Experiments were done with the approval of the University of Virginia Animal Care and Use Committee and done in accordance to ARRIVE criteria. All experiments had randomized treatment allocations and the data evaluators were blinded to treatment allocation. Littermates were used in all experiments. Where more than one experiment with littermates was combined with another, data were normalized to percent of control. Expanded methods can be found in the Data Supplement. The data that support the findings of this study are available from the corresponding author upon reasonable request. 8-12 weeks old male mice were used in all experiments. C57BL/6J (WT) and transgenic mice, on a C57BL/6 background, deficient in elastase (B6.129X1-Elane tm1sds: elastase KO), myeloperoxidase (B6.129X1-Mpo tm1Lus: MPOKO) or neutrophil cytosolic factor 1(NCF- component of the NADPH oxidase complex (B6(Cg)-Bcf1 m1J: NADPH oxidase KO) were used as indicated in figure legends (The Jackson Laboratories, Bar Harbor, ME).
SAH
SAH and sham surgeries were performed as previously reported 16. Briefly, mice were anesthetized, and a conserved subarachnoid vein was punctured (bleeding allowed to stop on its own). For the sham, the subarachnoid space was entered but the vein was not punctured.
Treatments
0.8 μg of intracisternal MPO (ab91116; Abcam, Cambridge, MA, US), H2O2 (0.0012%), or MPO with H2O2 was injected at the time of surgery in MPOKO mice in the setting of SAH. 3 μg of anti-CD45-FITC antibody (Thermofisher, Waltham, MA, US) was injected I.V. 30 min prior to euthanasia. Meninges were stained with the anti-neutrophil antibody 7/4-FITC (Abcam, Cambridge, MA, US; 1:100), and imaged using an Olympus FV1200 confocal microscope (Fluoview software).
Flow cytometry
Flow cytometry was performed on both the brain parenchyma and meninges of WT and MPOKO mice. The meninges and brains were collected and processed as previously described 17, 18. Samples were analyzed using a Gallios cytometer (Beckman Coulter, Brea, CA, US), and FlowJo software (FlowJo, LLC, Ashland, OR).
Immunohistochemistry
Brain sections were stained with anti-CD68 (MCA1957GA, BioRad Laboratories, Hercules, CA, US), anti-CC1 (AB16794), anti-GFAP (AB4674), anti-neutrophil antibody 7/4-FITC (AB53453), and anti-vimentin (AB92547) (Abcam, Cambridge, MA, US), anti-Ki67-APC (50569882, Invitrogen, Carlsbad, CA, US), and anti-Iba1 (01919741, Wako Chemicals, Japan) antibodies overnight at room temperature. Meninges were stained with anti-neutrophil antibody 7/4-FITC (AB53453, Abcam, Cambridge, MA, US).
In the brain, left and right CA1 regions of the hippocampus were imaged. ‘Coloc 2’ was used to co-localize GFAP and Vimentin. For meninges, whole mounts of the meninges were stained for neutrophils (NT 7/4; AB53457, Abcam, Cambridge, MA, US). All images were obtained on the Olympus FV1200 confocal microscope with Fluoview software (Tokyo, Japan) and processed with ImageJ/Fiji (NIH, Bethesda, MD, US).
To quantify microglial activation, Sholl analysis was performed using brain sections labelled with anti-Iba1 to quantify the number of processes observed on selected cells using the protocol previously described 19. To determine the inflammatory state of microglia, brain sections were co-labeled with Iba1 and CD68.
Vasospasm
Evaluation of vasospasm has been previously reported 5. The arterial tree was labeled using the vascular dye Microfil (Flow Tech, Carver, MA) 6 days after SAH and the cross-sectional MCA diameter was measured. The percent constriction was calculated as a ratio of the smallest diameter to the largest cross-sectional diameter of the MCA within a 2 mm segment distal to the posterior wall of the internal carotid using ImageJ.
Behavior analysis
Prior to surgery, mice were randomly assigned to treatment groups using a random number generator. In addition, treatment conditions were blinded to the person administering the behavioral test. Barnes Maze testing after SAH is previously described 5. Briefly, mice were habituated to the maze for 2 days before surgery. Testing started on day 3 post-surgery and continued for 7 consecutive days. All trials were recorded and analyzed using the tracking software Ethovision 13 (Noldus, Leesburg, VA, US). Mouse motor performance was analyzed using the rotarod test.
Primary cultures
Neuronal cultures were established using Thy1-GCaMP3 neonates as previously described 20. Mature cultures were incubated with 0.8 ug MPO with or without H2O2 (0.0012%) or H2O2 alone. Cells were then stimulated with 10mM potassium chloride (KCl) solution and imaged/analyzed using a widefield Leica DM6000B microscope outfitted with a CCD camera (Wetzlar, Germany) and Imaris software (Bitplane, Concord, USA). Primary astrocyte cultures were established as previously described 21. Astrocyte cultures were exposed to MPO, MPO with H2O2, or H2O2 for 4 hours.
2-photon imaging of neuronal calcium activity
All imaging experiments were performed on an Olympus FVMPE-RS system equipped with sensitive GaAsP detectors and a resonant scanner (Tokyo, Japan). A cranial window was prepared on Thy1-GCaMP3 mice as previously described 22. To induce KCl-triggered spreading depolarization, 100 uL 1 M KCl was applied to the cortex and images were analyzed using Imaris software (Bitplane, Concord, USA).
Statistics
Graphpad Prism 8.1 (Graphpad, La Jolla, CA, US) software was used to analyze all the data obtained. Student’s t-test, Chi Square or analysis of variance (ANOVA) were used to determine significant differences between treatment groups (p< 0.05). Detailed statistical methods for each experiment are in the Data Supplement.
Results
The neutrophil enzyme myeloperoxidase is critical to the development of cognitive deficits after SAH.
In our model of non-infectious brain injury, peripheral depletion of neutrophils impedes the development of spatial memory deficits after SAH 6. To determine whether neutrophil effector functions play a role in these deficits, mice deficient in the neutrophil effector proteins MPO, elastase and NADPH oxidase (that have been previously implicated in neuronal damage after ischemic stroke) 8, were tested. Both elastase and NADPH oxidase KO mice developed delayed spatial memory deficits after SAH (Figure 1A and 1B). On the other hand, the MPOKO mouse was protected against the development of delayed spatial memory deficits (Figure 1C), suggesting that neutrophil derived MPO is critical to the development of this syndrome.
Figure 1: Myeloperoxidase is critical to the development of delayed cognitive deficits after SAH.
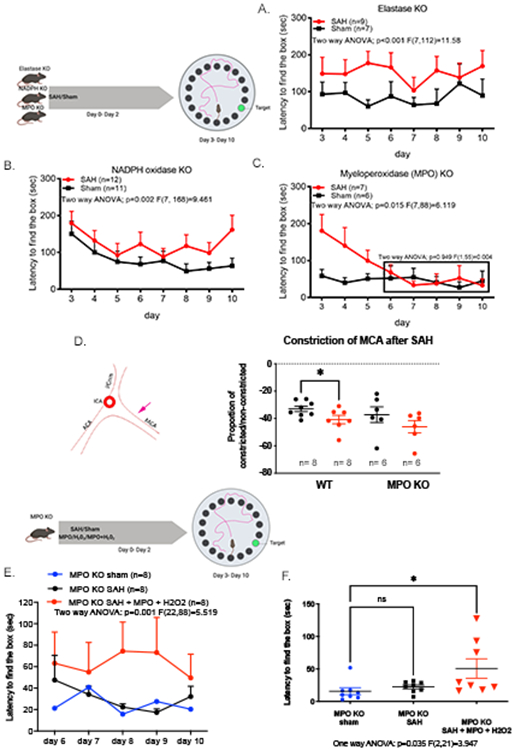
Barnes maze memory task was used to determine the effect of neutrophil effector function after SAH. Escape latency of mice lacking functional elastase, NADPH oxidase, and myeloperoxidase (MPO) were tested for 7 days starting on the 3rd day after surgery in the Barnes Maze memory task (A-C). Mice lacking functional elastase (A) or NADPH oxidase (B) developed memory deficits after SAH. Conversely, MPOKO mice did not develop delayed deficits after SAH (C; boxed area). MPOKO mice lack vasospasm in the middle cerebral artery (MCA) after SAH (D). The left panel denotes the region of MCA analyzed for vasospasm (pink arrow). WT mice showed a significant decrease in vessel diameter 6 days after SAH (Student t-test: p=0.038), but no significant change in vessel diameter was observed in MPOKO mice after SAH. To confirm the direct involvement of MPO as the driver of this improvement, biologically active MPO and H2O2 was injected into the CSF at the time of SAH in the MPOKO mouse. MPOKO mice did not develop delayed cognitive deficits after Sham (blue) or SAH (black) in the Barnes Maze memory task. The addition of exogenous MPO and H2O2 (red) in the cerebrospinal fluid (CSF) of the MPOKO mouse during SAH re-elicited the impaired longer latencies reported previously in our model (One way ANOVA p=0.001) (E, left). A focused analysis on day 8 after surgery showed a significant difference between all three groups (One-way ANOVA, p=0.035) and a specific increase in latency between the MPOKO sham and the MPOKO mouse with MPO + H2O2 (Student t-test: p=0.028) (E, right). Representations of experiments were created with BioRender.
An identifying hallmark of both murine and human SAH is the presence of delayed vasospasm in large cerebral vessels 23. In the MPOKO mouse, the diameter of the middle cerebral artery showed no spasm 6 days after SAH (Figure 1D).
The immune landscape and behavioral characteristics of MPOKO mice has not been described. Non-injured MPOKO mice have more CD45+ immune cells (monocytes and microglia) in the brain than WT, and fewer neutrophils in the meninges by flow cytometry (gating strategy Supplemental Figure I, data Supplemental Figure II A). Behaviorally, the mice have impaired spatial memory compared to WT but no impairment of motor function or coordination by rotarod test (Supplemental Figure II B, C) suggesting that improvements in cognition after SAH were not due to baseline strain differences.
To determine whether the loss of MPO activity leads specifically to improved cognitive function, biologically active MPO was added to the CSF of MPOKO mice at the time of the hemorrhage. The addition of MPO and its substrate H2O2 to the MPOKO mouse recapitulates the spatial memory deficits previously present in our model (Figure 1E). The addition of MPO alone or hydrogen peroxide alone was not sufficient to cause deficits, suggesting that the peroxidase activity of the MPO enzyme is important for late cognitive deficits associated with SAH (data not shown).
Neutrophils infiltrate the meningeal parenchyma but not the brain after SAH.
To locate the site of action of MPO in the CNS, the meninges and brain parenchyma were analyzed for innate cell infiltration after SAH. Interestingly, few neutrophils enter the hippocampus in our model (Supplemental Figure III) and there is no evidence of increased reactive oxidative stress in the hippocampus of mice with SAH (Supplemental Figure IV) suggesting that damage from ROS release from neutrophils is unlikely.
In the meninges, neutrophils account for approximately half of CD11b+ cells in the meninges and there was a trend toward increased numbers in SAH compared to Sham (Figure 2A). Due to the limitations in flow cytometry of liberated cells from fibrous tissue, we explored immunohistochemistry of whole mounted meninges. Using this method, we found that Iba1+ cells, that include monocytes and macrophages, which in some tissues store and release MPO; in our model, did not show MPO staining suggesting that MPO in the meninges derives from neutrophils (Figure 2A) 24, 25.
Figure 2: Neutrophils actively infiltrate the meningeal parenchyma after SAH.

A) Flow cytometry of digested meninges showed greater numbers of neutrophils than inflammatory monocytes (CD45+/CD11b+/Ly6Chi). There was a trend toward increased neutrophils, but not monocytes, after SAH (left panel). Whole mounted meninges showed that cells that store MPO do not co-localize with Iba1+ cells (monocytes/macrophages) suggesting only neutrophils store MPO in the meninges in this model (right panel). B) In whole mounted meninges, neutrophils (anti-7/4) were observed both in the meningeal parenchyma and venous sinuses of sham and SAH groups of the WT and MPOKO mice. The number of neutrophils within the meninges and in the sinuses increased significantly 3 days after SAH in WT but not MPOKO mice. C) Intravenous injection of fluorescein tagged CD45 (CD45-FITC) showed that the increased infiltration is attributed to the active recruitment of neutrophils from the periphery. Arrows represent NT 7/4 labelled cells that are co-labeled with CD45 (scale bar = 50 μm).
Further investigation showed that neutrophils are found in the venous sinuses as well as the meningeal parenchyma, and that there was a significant increase in the number of neutrophils in the SAH mouse 3 days after hemorrhage (Figure 2B). Further characterization, using intravenous CD45 injection, showed that this increase is due to active infiltration of neutrophils into the meninges after SAH (Figure 2C).
Conversely, MPOKO mice showed no increase in neutrophils in the transverse sinus or meninges after SAH (Figure 2B). The lack of increased neutrophils in the meningeal venous sinuses of the MPOKO mice (Figure 2B) suggests that MPO may be important in recruitment and extravasation of neutrophils to the meninges. This is consistent with previous reports that show that the loss of functional MPO affects both neutrophil migration and production of key cytokines by neutrophils 26, 27.
MPO affects Neurons, Astrocytes, Microglia, and Oligodendrocytes
How MPO affects memory function is still unknown. We approached this question by determining what brain parenchymal cells are affected by MPO. We investigated the most common brain parenchymal cells: neurons, astrocytes, microglia and oligodendrocytes.
To investigate MPO effect on neurons, primary neuronal cultures were generated using WT and Thy1GCaMP3 mice. Cultures generated by the WT were used to determine the effect of MPO and H2O2 on neuronal viability. No differences in neuronal viability without stimulation were observed in cultures exposed to escalating doses of MPO and H2O2 up to the final experimental dose (Supplemental Figure V). Using calcium signaling in the cultures generated by Thy1GCaMP3 as an indicator of activity, we characterized neuronal activity after administration of MPO, H2O2, or MPO+H2O2 and potassium chloride (KCL) stimulation. Primary neurons incubated for 2.5 hours with MPO, H2O2, or MPO+H2O2 were stimulated with 10mM KCl solution. Neurons incubated with MPO or MPO+H2O2 show an attenuated response to KCl, further supporting the hypothesis that MPO dampens neuronal responses to stimulation. The addition of MPO without its substrate has no effect on neuronal activity, but H2O2 alone leads to depressed activity and cell death with stimulation (Figs. 3A left and center).
Figure 3: The enzymatic activity of MPO modulates brain parenchymal cell activity.

(A) Primary neuronal cultures obtained from Thy1-GCaMP3 mice. MPO alone had no effect on neuronal activity, while H2O2 decreases activity by more than 75%, and MPO + H2O2 decreased activity by 50% (left). The addition of MPO or MPO and H2O2 led to minimal to no neuronal death in cultures while the addition of H2O2 alone led to significant cell death (center). In vivo, 2-photon microscopy showed that SAH and the addition of MPO significantly affected the depth of signal depression observed in cortical neurons (right). (B) Representative confocal micrographs of astrocytes detected by GFAP (red) and Vimentin (green) in the CA1 subregion of the hippocampus 3 and 6 days (right) after SAH show that GFAP intensity is significantly decreased (p=0.047) in the CA1 region of the WT mouse but not in MPOKO mouse (center). 6 days after surgery, both GFAP intensity was significantly decreased (p=0.004) (right). No changes were observed in the MPOKO mouse at either time point (scale bar= 50μm). (C) Representative images of the morphology and density of Iba1+ myeloid cells in the CA1 region of the hippocampus (green) in sham and SAH mice 3 days post-surgery in WT and MPOKO mice (scale bar = 50 μm) (left). Scholl analysis shows Iba1+ cells have fewer processes (more activated) in MPOKO SAH mice (n=4) compared to WT and Sham MPOKO (p<0.001) (center). Representative images depicting the co-expression of CD68 and Iba1+ in the hippocampus after SAH. There was no significant change in the percentage of Iba1+ cells co-expressing CD68 in the hippocampus after SAH (right). Representations of experiments created with BioRender.
Because the in vitro system does not fully mimic in vivo conditions, KCl-induced cortical spreading depolarizations (CSD) were tested to determine if MPO alters neuronal response to stimuli in the live animal. Using 2-photon microscopy of calcium signaling as a measure of neuronal activity, we determined the peak signal during CSD and the subsequent signal depression in the presence of MPO. We compared this change to the CSD induced signal and depression in the SAH mouse. Of note, because the highest level of neutrophil infiltration occurs 3 days after SAH in our model, our analysis of CSD was focused to this time point in the SAH mouse.
Under all treatment conditions, MPO or SAH, neuronal depolarization (i.e. increase in calcium signal) and depression (decrease in calcium signal) were detected within minutes of KCl administration (Figure 3A right). Analysis of the maximum signal intensity (baseline - peak) showed no difference between groups, suggesting that MPO and SAH do not affect the depolarization potential of neurons. Analysis of depression (baseline – minimal signal) showed that both MPO administration and SAH leads to less depression in the neurons compared to controls (Figure 3A right). We interpret these changes to suggest that there are resting state abnormalities in neurons due to SAH and MPO that are consistent with our previous findings in hippocampal neurons.
Astrocytes were characterized in the CA1 region of the hippocampus by their expression of GFAP and vimentin (Figure 3B and Supplemental Figure VI). There were no differences between sham WT and MPO groups. At day 3, in WT mice, although no changes were observed in the number of GFAP+ astrocytes after SAH (Supplemental Figure VI B), GFAP intensity was significantly decreased in the CA1 region (Figure 3B left). Furthermore, GFAP and Vimentin colocalized in more astrocytes 3 days after SAH than in sham (Supplemental Figure VI C), suggesting increased activation of these cells 28. At day 6, both GFAP+ cell number and intensity were significantly decreased (Figure 3B right and Supplemental Figure VI E) with no change in the colocalization of GFAP and vimentin (Supplemental Figure VI F). Thus, in WT SAH there is astrocyte loss (decreased astrocytes at day 6) with increased concomitant vimentin/GFAP colocalized astrocytes at day 3. In MPOKO mice, early decrease in GFAP, astrocyte loss, and increased in colocalization of Vimentin and GFAP were prevented (Figure 3B right and Supplemental Figure VI C and E).
The in vivo data suggest MPO can affect astrocyte function within the brain. To corroborate this finding, primary astrocyte cultures were stimulated with MPO, H2O2, or MPO+H2O2 for 4 hours. After which, cell morphology and survival were assessed. Consistent with the in vivo experiment that shows that SAH leads to astrocyte death, exposure of cultured astrocytes to MPO and MPO+H2O2 led to significant cell death in culture as well (Supplemental Figure VI G).
Microglia morphology was characterized using Iba1 expression in the hippocampus with the understanding that infiltrating macrophages will also express Iba1. No changes were observed in the number of Iba1+ cells or their fluorescent intensity in the CA1 region of the WT and MPOKO mouse at either day 3 (Figure 3C) or 6 (data not shown) after SAH. To determine whether the activation phenotype of these cells was affected after SAH, the number of cell processes was quantified using a modified Scholl analysis. In the WT mouse hippocampus, no changes were observed in microglia ramification after SAH at day 3. Six days after injury, the number of processes detectable in both SAH and sham mouse decreased, but more in the sham than SAH (Figure 3C center). In the MPOKO mouse, at day 3 after SAH, the microglia lost the ramified morphology observed in the sham mouse. This change reverted to control levels 6 days after SAH (data not shown). To investigate if the decreased ramifications at day 3 are due to an activated state, we tested CD68 expression. SAH did not affect the number of Iba1+ cells expressing CD68 in the CA1 region of the hippocampus at day 3 after SAH. These results suggest that microglial de-ramified morphology in the absence of increased CD68 expression is not associated with worse behavioral outcomes (since MPOKO mouse SAH has improved behavioral outcome).
Oligodendrocytes have been shown to be highly sensitive to homeostatic changes within the brain but are less associated with inflammation 29. We characterized the number of CC1+ oligodendrocytes in the CA1 region of the hippocampus at both 3 and 6 days after SAH (Supplemental Figure VII). Three days after SAH, there is a small but significant increase in the number of oligodendrocytes. This increase however subsides to control values 6 days after injury. These changes appear unlikely to affect behavioral abnormalities seen after SAH.
Discussion
Recent reports have implicated peripheral and meningeal immune cells in the modulation of CNS function. Meningeal immune cells modulate output of higher order neurons as demonstrated in animal models of spatial learning, autism spectrum disorders, sickness behavior, and depression 30, 31. A recent hypothesis has designated the meninges as a tertiary lymphoid organ 32, especially in autoimmune disease like multiple sclerosis, suggesting the meninges can become an organized immunological tissue under highly inflammatory conditions.
The results of the present project provide evidence that immune activity in the meningeal compartment affects functions of the underlying brain during SAH through the action of its enzyme MPO. After SAH, neuronal activity is impaired as evidenced by behavioral and electrophysiological abnormalities 5, 6. In addition, glia subtypes undergo physiological changes: there is astrocytes loss in the brain, morphological changes are observed in the microglia, and there is a transient increase in the oligodendrocyte population. Our lab has previously shown that neutrophil depletion prior to and three days after SAH improves behavioral performance and obviates loss of late long-term potentiation (L-LTP) in the hippocampus 5, 6. The results of the current experiments show that removal of MPO, a neutrophil enzyme, improves the behavioral and electrophysiological deficits as well, but that these improvements can be abolished by introduction of MPO and H2O2. Importantly, neutrophils are found in the meninges but not the brain parenchyma after SAH suggesting that the effect of MPO derives from changes in the meninges.
Two important issues present themselves: 1) could MPO come from other sources such as monocytes or activated macrophages, and 2) where do neutrophils in the meninges come from. Our data shows that there are few MPO containing cells in the hippocampus and meningeal MPO cells are predominantly neutrophils, not monocytes and macrophages. As for where the neutrophils come from, neutrophils numbers are increased in the meninges over the first three days after hemorrhage. We have previously shown that this is associated with RORγT activation, the driver for IL-17 production 33. In this study, we were able to “tag” neutrophils in the bloodstream with a CD45 antibody and show extravasation into the meninges suggesting that a proportion of neutrophils come from the blood pool, as opposed to direct transit from the skull bone marrow or expansion from resident cells 34. Further studies are needed to fully explore this phenomenon.
How MPO affects neuronal function has not been elucidated but there are two likely mechanisms: direct effect on neurons, and/or through an intermediate cell type such as glia. Here, in in vitro experiments, we show that MPO and MPO+ H2O2 significantly decrease neuronal activity. Furthermore, in in vivo experiments using 2-photon microscopy, MPO, like SAH, decreases neuronal depression after cortical spreading depolarizations.
There are a number of reasons to doubt that MPO’s direct action on neuron in SAH is the primary driver of dysfunction: 1) neutrophils infiltrate into the meninges but not the brain, 2) it is unclear how MPO could be transported to hippocampal neurons, and 3) a mechanism to cause neuronal dysfunction is unclear. MPO’s main enzymatic activity is the production of the ROS hypochlorous acid (HOCl) and peroxynitrite 35. ROS have been implicated in neuronal dysfunction and degeneration in a multitude of models and possibly accounts for the dysfunction seen in the in vitro experiments 36. In our model, there was no evidence of increased ROS in the hippocampus. Because MPO has no identified cell surface receptor, and MPO appears to need the action of its substrate to be effective, receptor mediated action of MPO on neurons is less likely.
Therefore, we investigated the effect of MPO on glial cells that could lead to neuronal dysfunction. WT mice show a significant loss of astrocytes in the hippocampus six days after SAH. This decrease is preceded by a significant decrease in GFAP coverage in the hippocampus after SAH. Furthermore, the GFAP decrease coincides with an increase in vimentin expression. Vimentin/GFAP co-expression in astrocytes occurs in precursor/immature astrocytes that have an inflammatory phenotype 37. As such, the increased vimentin co-expression may indicate an increase in proliferation or activation of astrocytes. These changes are absent in the MPOKO mouse, suggesting that neutrophil activity in the meninges affects astrocytes in the cortex in the right direction (inflammatory) and the right time to affect neuronal function.
Conversely, microglia show differences less consistent with their direct role. Characterization of microglia, using Iba1 and CD68 expression, helps define inflammatory states after SAH. The most impressive change in microglial morphology, a less ramified state, occurs in MPOKO mice with SAH at day 3. Analysis of CD68 co-expression in Iba1+ cells at the same time point suggests a non-inflammatory state. These supposedly conflicting data suggests that our understanding of microglial activation and function based on morphology is still incomplete 38, 39. Despite the morphology, the data supports that microglia activation after SAH is not likely a cause of behavioral dysfunction. It is possible that these morphological changes suggest different microglial physiology that could affect neuronal function in ways not classically associated with inflammation.
A small, significant but transient increase is observed in the oligodendrocyte population after SAH. This increase occurs 3 days after SAH and subsides to control values by day 6. It is possible that the stress of the early brain injury, including infiltration of monocytes or the breakdown of blood product in the meninges led to the changes observed in oligodendrocytes. These small changes appear to make oligodendrocytes less likely to be a good target for future study.
Although the results of this study further clarify the role of meningeal immunity in brain function, we recognize that the study has some limitations. First, the model used in this study to develop SAH is unconventional. Since there are few rodent models of SAH that involve the spontaneous rupture of a cerebral aneurysm, most models infer pathophysiology from a non-real-world hemorrhages. The hemorrhage in this model is developed by transection of a vein in the subarachnoid space. This model recapitulates both cerebral vasospasm and delayed cerebral injury seen in aneurysmal SAH 16,5,40. It has the added benefit of not causing significant early injury from high pressure bleeding allowing the discrimination of effects from the initial pressure from blood in the subarachnoid space. Because arterial blood has been proposed as one of the instigators of DCI and vasospasm, it suggests that either the effects of arterial blood are less important or that mice are more susceptible to DCI than patients.
Second, tools to eliminate neutrophils from the CNS alone are not available. As such, it is not currently possible to determine if depletion of neutrophils from the meninges of the WT mice after SAH is enough to mitigate cognitive deficits. In addition, this study identifies potential cellular actors of SAH in the brain parenchyma. However, it appears that the action of MPO has effects on a number of cells, making a single target for investigation/intervention unlikely. It is still left to be determined which changes/interactions are important to the onset of memory deficits. In addition, although we have shown that MPO can directly affect neurons and glia, the question of MPO access to the cells, the effects of ROS byproducts, and the lack of a known direct receptor mechanism for MPO to affect these cells are all left to be elucidated.
Neutrophils are the main source of myeloperoxidase. However, immature monocytes and microglia have been shown to express the enzyme at low levels 41. As such, the increased number of microglia and monocytes observed in the brain and meninges of the MPOKO mouse would suggest that MPO may play a modulatory role in the activity or proliferation of these cells. Indeed, it is well established that MPO is important for the migration and extravasation of neutrophils 27. Furthermore, neutrophil derived cytokines play a critical role in the infiltration and activity of other innate and adaptive immune cells. Therefore, it is possible that the lack of neutrophils in the meninges of the MPOKO mouse significantly affects how inflamed the meninges become in response to SAH. Although neutrophils are critical for the secondary injury and deficits associated with SAH, an understanding of the role of other immune cells, like monocytes, in the central nervous system, may add to the picture of inflammation after SAH.
Conclusions
This work provides evidence that activity of neutrophils outside the brain can modulate neuronal function during injury, and particularly that MPO is critical to the behavioral changes observed in SAH mice. A working hypothesis derived from the data is that after SAH, neutrophils degranulate in the subarachnoid space, leading to activation of glial elements in the subpial brain region in proximity to the brain-CSF interface. Glial dysfunction and possibly cell death lead to neuronal dysfunction. Furthermore, MPO dampens neuronal depolarization, thereby dysregulating cortical to subcortical circuit activity. Although other mechanisms are likely at play, we propose that this dysregulation is a good candidate for study as a critical factor in the development of cognitive deficits associated with SAH.
Supplementary Material
Acknowledgments
Thanks to Drs. A. Louveau and S. Gadani for help with meninges and flow cytometry, respectively.
Sources of Funding
Study supported by NIH NS074997(JJP).
Non-standard Abbreviations and Acronyms
- SAH
subarachnoid hemorrhage
- DCI
Delayed cerebral injury or cerebral vasospasm
- MPOKO
myeloperoxidase knockout
- H2O2
hydrogen peroxide
- CSF
cerebrospinal fluid
- NADPH
nicotinamide adenine dinucleotide phosphate
- IL6
interleukin 6
- CXCL1
chemokine (C-X-C motif) ligand 1
- MIP1α
macrophage inflammatory protein 1 alpha
- KCl
potassium chloride
- CSD
cortical spreading depolarization
- Iba1
ionized calcium binding adaptor molecule 1
- GFAP
glial fibrillary acidic protein
- LTP
long-term potentiation
- NMDAR
n-methyl-d-aspartate receptor
- ROS
reactive oxygen species
- HOCl
hypochlorous acid
Footnotes
Disclosures
JJP-grant support Minnetronix, Inc.
Other authors have no competing interest.
References
- 1.Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke. 2010;41:e519–536 [DOI] [PubMed] [Google Scholar]
- 2.Rosengart AJ, Schultheiss KE, Tolentino J, Macdonald RL. Prognostic factors for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2007;38:2315–2321 [DOI] [PubMed] [Google Scholar]
- 3.Rinkel GJ, Wijdicks EF, Hasan D, Kienstra GE, Franke CL, Hageman LM, Vermeulen M, van Gijn J. Outcome in patients with subarachnoid haemorrhage and negative angiography according to pattern of haemorrhage on computed tomography. Lancet. 1991;338:964–968 [DOI] [PubMed] [Google Scholar]
- 4.Burke T, Hughes S, Carr A, Javadpour M, Pender N. A systematic review of cognitive outcomes in angiographically negative subarachnoid haemorrhage. Neuropsychol Rev. 2018;28:453–469 [DOI] [PubMed] [Google Scholar]
- 5.Provencio JJ, Altay T, Smithason S, Moore SK, Ransohoff RM. Depletion of ly6g/c(+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J Neuroimmunol. 2011;232:94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Provencio JJ, Swank V, Lu H, Brunet S, Baltan S, Khapre RV, Seerapu H, Kokiko-Cochran ON, Lamb BT, Ransohoff RM. Neutrophil depletion after subarachnoid hemorrhage improves memory via nmda receptors. Brain Behav Immun. 2016;54:233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz Hernandez JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, Park L, Vinarcsik LK, Ivasyk I, Rivera DA, et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in alzheimer's disease mouse models. Nat Neurosci. 2019;22:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barone FC, Hillegass LM, Price WJ, White RF, Lee EV, Feuerstein GZ, Sarau HM, Clark RK, Griswold DE. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: Myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–345 [DOI] [PubMed] [Google Scholar]
- 9.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: From mechanisms to disease. Annu Rev Immunol. 2012;30:459–489 [DOI] [PubMed] [Google Scholar]
- 10.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175 [DOI] [PubMed] [Google Scholar]
- 11.Yu G, Liang Y, Zheng S, Zhang H. Inhibition of myeloperoxidase by n-acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke. J Pharmacol Exp Ther. 2018;364:311–322 [DOI] [PubMed] [Google Scholar]
- 12.Chen G, Ye X, Zhang J, Tang T, Li L, Lu P, Wu Q, Yu B, Kou J. Limb remote ischemic postconditioning reduces ischemia-reperfusion injury by inhibiting nadph oxidase activation and myd88-traf6-p38map-kinase pathway of neutrophils. Int J Mol Sci. 2016;17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storey PB. Brain damage and personality change after subarachnoid haemorrhage. Br J Psychiatry. 1970;117:129–142 [PubMed] [Google Scholar]
- 14.Ogden JA, Utley T, Mee EW. Neurological and psychosocial outcome 4 to 7 years after subarachnoid hemorrhage. Neurosurgery. 1997;41:25–34 [DOI] [PubMed] [Google Scholar]
- 15.Buunk AM, Spikman JM, Veenstra WS, van Laar PJ, Metzemaekers JDM, van Dijk JMC, Meiners LC, Groen RJM, . Social cognition impairments after aneurysmal subarachnoid haemorrhage: Associations with deficits in interpersonal behaviour, apathy, and impaired self-awareness. Neuropsychologia. 2017;103:131–139 [DOI] [PubMed] [Google Scholar]
- 16.Altay T, Smithason S, Volokh N, Rasmussen PA, Ransohoff RM, Provencio JJ. A novel method for subarachnoid hemorrhage to induce vasospasm in mice. J Neurosci Methods. 2009;183:136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadani SP, Smirnov I, Smith AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to cns injury. J Exp Med. 2017;214:285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louveau A, Filiano AJ, Kipnis J. Meningeal whole mount preparation and characterization of neural cells by flow cytometry. Curr Protoc Immunol. 2018;121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derecki N, Norris G, Derecki N, Kipnis J. Microglial sholl analysis. Protocol Exchange. 2014:3377 [Google Scholar]
- 20.Beaudoin G, Lee S-H, Singh D, Yuan Y, Ng Y-G, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nature Protocols. 2012;7:1741–1754 [DOI] [PubMed] [Google Scholar]
- 21.Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tvrdik P, Kearns KN, Sharifi KA, Sluzewski MF, Acton ST, Kalani MYS. Calcium imaging of microglial network activity in stroke. Methods Mol Biol. 2019;2034:267–279 [DOI] [PubMed] [Google Scholar]
- 23.Fisher C, Roberson GH, Ojemann RG. Cerebral vasospasm with ruptured saccular aneurysm- the clinical manifestations. Neurosurgery. 1977;1:245–248 [DOI] [PubMed] [Google Scholar]
- 24.Yanez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, Salomonis N, Leighton Grimes H, Goodridge HS. Granulocyte-monocyte progenitors and monocyte-dendritic cell progenitors independently produce functionally distinct monocytes. Immunity. 2017;47:890–902 e894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMillen TS, Heinecke JW, LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation. 2005;111:2798–2804 [DOI] [PubMed] [Google Scholar]
- 26.Haegens A, Heeringa P, van Suylen RJ, Steele C, Aratani Y, O'Donoghue RJ, Mutsaers SE, Mossman BT, Wouters EFM, Vernooy JH. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J Immunol. 2009;182:7990–7996 [DOI] [PubMed] [Google Scholar]
- 27.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brümmer J, Rudolph V, Münzel T, Heitzer T, et al. Myeloperoxidase mediates neutrophil activation by association with cd11b/cd18 integrins. Proc Natl Acad Sci U S A. 2005;102:431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janeczko K Co-expression of gfap and vimentin in astrocytes proliferating in response to injury in the mouse cerebral hemisphere. A combined autoradiographic and double immunocytochemical study. Int J Dev Neurosci. 1993;11:139–147 [DOI] [PubMed] [Google Scholar]
- 29.Bradl M, Lassmann H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010;119:37–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, Overall CC, Gadani SP, Turner SD, Weng Z, et al. Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature. 2016;535:425–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitsdoerffer M, Peters A. Tertiary lymphoid organs in central nervous system autoimmunity. Front Immunol. 2016;7:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coulibaly AP, Gartman WT, Swank V, Gomes JA, Ruozhuo L, DeBacker J, Provencio JJ. Rar-related orphan receptor gamma t (rorgammat)-related cytokines play a role in neutrophil infiltration of the central nervous system after subarachnoid hemorrhage. Neurocrit Care. 2020;33:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, Wojtkiewicz GR, Santos Masson G, Vinegoni C, Kim J, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21:1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem. 2016;85:765–792 [DOI] [PubMed] [Google Scholar]
- 36.Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–188 [DOI] [PubMed] [Google Scholar]
- 37.Pixley SK, de Vellis J. Transition between immature radial glia and mature astrocytes studied with a monoclonal antibody to vimentin. Brain Res. 1984;317:201–209 [DOI] [PubMed] [Google Scholar]
- 38.Ransohoff RM, El Khoury J. Microglia in health and disease. Cold Spring Harb Perspect Biol. 2015;8:a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paasila PJ, Davies DS, Kril JJ, Goldsbury C, Sutherland GT. The relationship between the morphological subtypes of microglia and alzheimer's disease neuropathology. Brain Pathol. 2019;29:726–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smithason S, Moore SK, Provencio JJ. Systemic administration of lps worsens delayed deterioration associated with vasospasm after subarachnoid hemorrhage through a myeloid cell-dependent mechanism. Neurocrit Care. 2012;16:327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klebanoff SJ. Myeloperoxidase: Friend and foe. J Leukoc Biol. 2005;77:598–625 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.