

Abstract

A common strategy for identifying molecules likely to possess a desired biological activity is to search large databases of compounds for high structural similarity to a query molecule that demonstrates this activity, under the assumption that structural similarity is predictive of similar biological activity. However, efforts to systematically benchmark the diverse array of available molecular fingerprints and similarity coefficients have been limited by a lack of large-scale datasets that reflect biological similarities of compounds. To elucidate the relative performance of these alternatives, we systematically benchmarked 11 different molecular fingerprint encodings, each combined with 13 different similarity coefficients, using a large set of chemical–genetic interaction data from the yeast Saccharomyces cerevisiae as a systematic proxy for biological activity. We found that the performance of different molecular fingerprints and similarity coefficients varied substantially and that the all-shortest path fingerprints paired with the Braun-Blanquet similarity coefficient provided superior performance that was robust across several compound collections. We further proposed a machine learning pipeline based on support vector machines that offered a fivefold improvement relative to the best unsupervised approach. Our results generally suggest that using high-dimensional chemical–genetic data as a basis for refining molecular fingerprints can be a powerful approach for improving prediction of biological functions from chemical structures.
Introduction
The development of new drugs that demonstrate desired biochemical behavior against a biomolecular target is challenging. Despite the scientific and technological advances in drug discovery during the past 60 years, the ratio of drugs approved to money spent on R&D has halved roughly every 9 years since 1950 (Eroom’s law in contrast to Moore’s law)1 and has now dipped below one drug per billion USD.1−3 Following the similar property principle (SPP),4 ligand-based virtual screening has been commonly used to filter candidates prior to high-throughput screening efforts by ranking compounds from a large database in descending order of their structural similarity to a reference or target molecule with known biological activity (Figure 1).5,6 According to the SPP, structurally similar molecules will more likely possess similar biological activities and physicochemical properties. Despite limitations to the SPP,7,8 such as activity cliffs that manifest when a small structural modification drastically alters the biological properties of a compound,9 this structure–activity relationship is broadly consistent throughout the larger flat regions of activity landscapes.10,11 Improving the structural similarity-based retrieval of biologically similar compounds will therefore benefit a multitude of drug discovery efforts.
Figure 1.
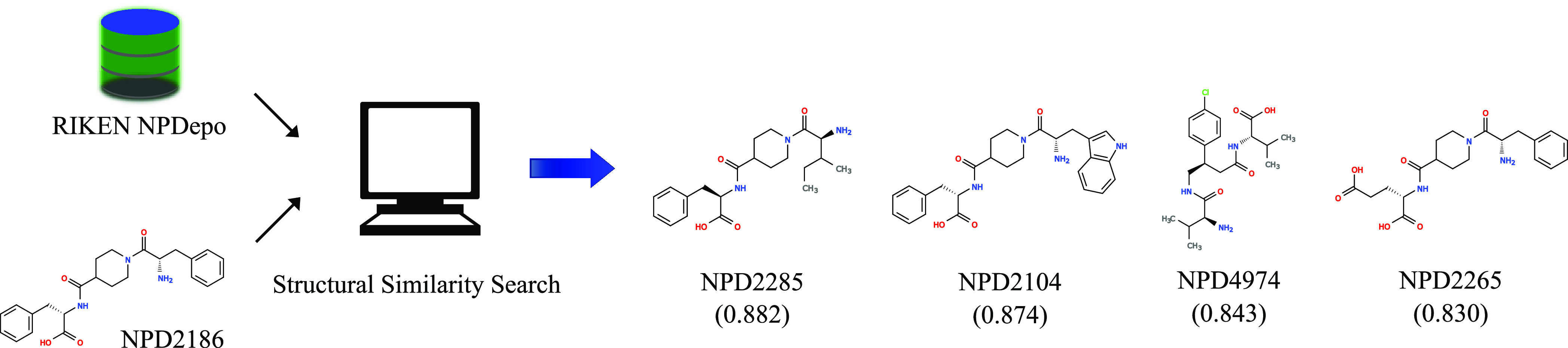
Ligand-based virtual screening of a target (e.g., NPD2186 from RIKEN Natural Product Depository). We ranked all compounds (top four shown) of the MOSAIC database (http://mosaic.cs.umn.edu)12 in descending order of structural similarity to the target molecule based on the SPP. We described these compounds using all-shortest path (ASP) fingerprints (depth 8) and measured structural similarity using the Braun-Blanquet similarity coefficient. In this example rank list, three compounds except NPD4974 have a very similar chemical–genetic interaction profile to that of NPD2186.
The chemical informatics community has suggested a wide range of molecular encodings and similarity coefficients to quantify structural similarity between two molecules,13−15 which is then used as a proxy for biological similarity. The most widely used class of molecular encodings is chemical fingerprints,16−20 where a molecular graph is represented by a bit vector that encodes the presence or absence of molecular features such as paths between atoms, substructural fragments, and pharmacophores. The degree of similarity of two structural vectors describing two different compounds is usually measured by similarity coefficients. The Tanimoto (aka Jaccard) coefficient, formulated as the fraction of features in common between two molecules relative to the total number of features present in either molecule, remains the coefficient of choice to capture the highest level of intermolecular similarity and thus biological activity,21−24 although this coefficient suffers from an intrinsic bias toward selecting smaller compounds.13,25−27 However, a major challenge for the research community has been the lack of a systematic benchmarking framework based on biological activity that covers a broad range of protein targets. Such a framework will enable a definitive assessment of the performance of molecular fingerprints and similarity coefficients in predicting biological similarity between two molecules.
Chemical genomic approaches, which involve the systematic mapping of chemical–genetic interactions, offer a valuable new source of data to connect chemical structures to biological functions. The yeast Saccharomyces cerevisiae is a well-characterized eukaryote, for which ∼5000 viable deletion mutants have been identified.28 Interrogating these deletion mutants with a bioactive compound generates a chemical–genetic interaction profile containing the degree of sensitivity or resistance of each genetic mutant to the compound.29,30 This quantitative, high-dimensional representation of biological functions can be interpreted using a global compendium of genetic interaction profiles31,32 systematically mapped through pairwise, double-knockout screens in S. cerevisiae. Specifically, the chemical–genetic interaction profile of a compound will resemble the genetic interaction profile obtained through the genetic knockout of its biomolecular target.33 These chemical–genetic interaction profiles can be used as a gold standard to evaluate structural similarity measures (each defined as one molecular fingerprint encoding paired with one similarity coefficient), even for compounds where the actual biological targets are not yet known, since these profiles accurately reflect compound functions.
We recently generated and published a compendium of over 13,000 chemical–genetic interaction profiles obtained in S. cerevisiae,34 providing a unique basis to independently assess the performance of structural similarity measures. These compounds originate from the RIKEN Natural Product Depository (NPDepo) and several NCI, NIH, and GlaxoSmithKline (GSK) compound collections. We systematically benchmarked 11 different molecular fingerprint encodings, each paired with 13 different similarity coefficients, to identify the pair that best predicted biological similarity as measured by chemical–genetic interaction profile similarity (in brief, chemical–genetic similarity). Given recent advances in machine learning approaches for drug discovery,35−39 we further developed supervised machine learning models to improve our predictions of the biological activity of our compounds from chemical structures, achieving higher predictive power than using similarity measures alone. We found that support vector machines (SVMs)40,41 can significantly enhance the power of chemical fingerprints for predicting the biological activity of compounds.
Results and Discussion
Establishing a Systematic Benchmark for Structural Similarity Measures
We compiled a list of commonly used molecular fingerprint encodings and similarity coefficients to capture the structural similarity of compounds in different modes (e.g., exploring molecular paths or radial atom environments) (Tables 1 and 2). To quantify the ability of different structural similarity measures for predicting functionally analogous compounds, we used chemical–genetic interaction profiles from S. cerevisiae as a systematic genome-wide standard for biological activity. We previously published chemical–genetic interaction profiles for 13,431 compounds from several diverse compound collections and identified a subset of these compounds predicted with high confidence to perturb predefined biological processes based on integrating these profiles with genetic interaction profiles.34 We included in our benchmarking system two independent subsets of these high-confidence compounds: (1) 826 compounds from the RIKEN NPDepo collection, which we hereafter call as the “RIKEN high-confidence set”, and (2) 659 compounds from several NCI/NIH/GSK collections, which we hereafter call as the “NCI/NIH/GSK high-confidence set” (Materials and Methods).
Table 1. Molecular Fingerprintsa.
| ID | name | description | features | reference(s) |
|---|---|---|---|---|
| FP1 | AP2D | topological atom pairs | 1211 | (44) |
| FP2 | ASP | all-shortest paths | 26,194 | (45) |
| FP3 | AT2D | topological atom triplets | 56,963 | (44) |
| FP4 | DFS | all-paths (depth-first search) | 48,448 | (46) |
| FP5 | ECFP | extended connectivity fingerprints | 42,672 | (47) |
| FP6 | LSTAR | local path environments | 85,232 | (48) |
| FP7 | MACCS | MDL public keys (166 keys) | 155 | (49) |
| FP8 | PHAP2POINT2D | topological pharmacophore pairs | 17 | (50) |
| FP9 | PHAP3POINT2D | topological pharmacophore triplets | 302 | (50) |
| FP10 | RAD2D | topological molprint-like fingerprints | 92,191 | (48) |
| FP11 | RDKit | topological daylight-like fingerprints | 65,183 | (43,51) |
A total of 11 different molecular fingerprints were generated using the jCompoundMapper tool42 or RDKit toolkit (version 2020.09.4)43 for describing the compounds in our datasets. The fourth column, features, represents the total number of features that the jCompoundMapper tool or RDKit toolkit generated for describing our RIKEN high-confidence set (826 compounds). The molecular features that we counted were only those present in the description of at least one compound of this collection, and the molecular fingerprints that required a depth of description were measured at depth 8.
Table 2. Similarity Coefficientsa.
| name | measurement | range | reference(s) | |
|---|---|---|---|---|
| Braun-Blanquet | x/max(y,z) | 0 to 1 | (53,54) | |
| Cosine | 0 to 1 | (55,56) | ||
| Dice | 0 to 1 | (57,58) | ||
| Dot-product | x | 0 to ∞ | N/A | |
| Euclidean | 0 to 1 | N/A | ||
| Kulczynski | 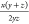 |
0 to 1 | (59) | |
| McConnaughey | –1 to 1 | (60) | ||
| Russel/Rao | x/w | 0 to 1 | (61) | |
| Simpson | x/min(y,z) | 0 to 1 | (62) | |
| Sokal/Sneath | 0 to 1 | (63) | ||
| Tanimoto | 0 to 1 | (64,65) | ||
| Tullos | XYZ | 0 to 1 | (66) | |
| Tversky | 0 to 1 | (67) |
A total of 13 different similarity
coefficients (several of these coefficients were collected by Raymond
and Willett)52 were compiled for measuring
the degree of structural similarity of two compounds described by
a given molecular fingerprint encoding. Here, x is
the number of bits set in both fingerprints, y is
the number of bits set in the first fingerprint, z is the number of bits set in the second fingerprint, and w is the
total number of bits in the bit string. For the Tullos similarity
coefficient, 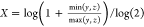 ,
, 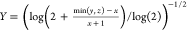 , and
, and 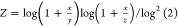 . For the asymmetric
evaluation of the Tversky
similarity coefficient, α = 0.9. Here, we assume that the parameters
of the Tversky coefficient in its original formulation,
. For the asymmetric
evaluation of the Tversky
similarity coefficient, α = 0.9. Here, we assume that the parameters
of the Tversky coefficient in its original formulation, 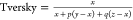 , will follow p + q = 1. The Dice and Tanimoto similarity
coefficients are
two symmetric instances of the Tversky coefficient, where p = q = 0.5 and p = q = 1, respectively.
, will follow p + q = 1. The Dice and Tanimoto similarity
coefficients are
two symmetric instances of the Tversky coefficient, where p = q = 0.5 and p = q = 1, respectively.
Since we require a binary gold standard to calculate prediction performance using precision and recall, we selected the 10% of most similar compound pairs based on the (cosine) similarity of their chemical–genetic interaction profiles and labeled them as our gold standard for true positive compound pairs. This assumption is imperfect because chemical–genetic similarity of compounds does not always imply structural similarity and two functionally analogous compounds may have distinct chemical structures. However, a large fraction of these true positives (functionally similar compound pairs) should still be identified from the structural similarity of compounds; therefore, this chemical–genetic-derived similarity serves as a reasonable basis to identify compounds with similar biological effects.
While studying compounds using yeast chemical–genetic interaction screens benefits from many advantages (e.g., genome-wide detection of potential targets), some important aspects of compound activity such as pharmacokinetic properties will be missed. Since our modeling approaches are based on functional similarity as detected in yeast chemical–genetic interactions, our results and conclusions about the functional similarity predictions of compounds will not reflect these characteristics, including pharmacokinetic properties.
We evaluated both components (molecular fingerprints and similarity coefficients) of our systematic benchmark to find both the fingerprint and the coefficient that best predicted the biological activity of our compounds (Supporting Information algorithm). We described all our compounds in 11 different fingerprint-based structural spaces (Table 1), where a compound was described by a bit vector that indicated the presence or absence of certain molecular features. The number of features required for the description of the compounds in a structural space varied based on the space definition and collection properties (e.g., only 155 out of 166 predefined molecular substructures from MDL public keys were used in the MACCS description of our RIKEN high-confidence set, while RAD2D structural encodings generated 91082 features to describe the same collection). We used 13 widely used similarity coefficients (Table 2) to measure the degree of similarity of two compounds described by a given molecular fingerprint.
Evaluating the Performance of Structural Similarity Measures
We computed pairwise structural similarities for the compounds in our RIKEN high-confidence set (340,725 different compound pairs) using every combination of a molecular fingerprint and a similarity coefficient (143 different prediction models). For every model, we sorted pairwise structural similarity scores in descending order against our binarized gold-standard functional (chemical–genetic) similarities and calculated precision at predefined recall thresholds (Table S1 for all precision values and complete evaluation of all prediction models). To isolate the best-performing molecular fingerprint, we examined the precision of all the prediction models at several predefined recall thresholds for our RIKEN high-confidence set (Table S1). Based on these evaluation metrics, the ASP, LSTAR, and RAD2D fingerprints emerged as the molecular fingerprints with superior predictive power (Figure 2 and Table S1). The wide range of precision values achieved by different molecular fingerprints revealed that our chemical–genetic interaction profiles can readily discriminate molecular fingerprints in terms of their efficacy in predicting the biological activity of our compounds.
Figure 2.

Performance of selected prediction models using our RIKEN high-confidence set (Table S1 for the complete evaluation of all prediction models). (A) Precision at several recall thresholds and the area under the ROC curve for each model, where a molecular fingerprint was paired with the Braun-Blanquet, Cosine, or Tanimoto similarity coefficient, evaluated based on chemical–genetic similarity as the gold standard for biological activity. The blue values represent the highest precision achieved at a given recall, and the green values represent the average precision over all molecular fingerprints for a similarity coefficient at a specific recall threshold. (B) Relative performance of ASP, LSTAR, and RAD2D fingerprints to that of ECFP. For all the molecular fingerprints that required a depth of description, precision was measured at a depth of 8. With the exception of ASP, LSTAR, and RAD2D, the remaining molecular fingerprints are coded as FP1–FP11 (Table 1).
Notably, among the molecular fingerprints that we evaluated was the Morgan fingerprint,68 also known as the extended-connectivity fingerprint (ECFP),47 which has recently been identified as one of the best-performing fingerprints in a variety of applications such as small-molecule virtual screening.69−71 However, ECFP was generally outperformed by ASP, LSTAR, and RAD2D fingerprints, except for a small number of recall thresholds (Figure 2B and Table S1). Because this finding might simply be a result of the RIKEN NPDepo compound collection characteristics, we used our NCI/NIH/GSK high-confidence set to validate the prediction performance of ASP, LSTAR, and RAD2D fingerprints, using the same evaluation method designed for our RIKEN high-confidence set. Our analysis on this second large set of compounds strongly confirmed the superiority of these fingerprints over ECFP at many recall thresholds (Figure 3 and Table S2 for complete evaluation of all prediction models).
Figure 3.

Performance of selected prediction models using our NCI/NIH/GSK high-confidence set (Table S2 for the complete evaluation of all prediction models). (A) Precision at several recall thresholds and the area under the ROC curve for each model, where a molecular fingerprint was paired with the Braun-Blanquet, Cosine, or Tanimoto similarity coefficient, evaluated based on chemical–genetic similarity as the gold standard for biological activity. The blue values represent the highest precision achieved at a given recall threshold, and the green values represent the average precision over all molecular fingerprints for a given similarity coefficient at a specific recall threshold. (B) Relative performance of ASP, LSTAR, and RAD2D fingerprints to that of ECFP. For all the molecular fingerprints that required a depth of description, precision was measured at a depth of 8. With the exception of ASP, LSTAR, and RAD2D, the remaining molecular fingerprints are coded as FP1–FP11 (Table 1).
We determined that the ASP and LSTAR fingerprints best captured the relationship between the chemical structure and biological activity of our compounds. ASP fingerprints encode graph traversals over all atoms in a molecular graph but store only the shortest paths between atoms, whereas LSTAR and RAD2D fingerprints describe the radial environment of all atoms in the molecular graph.42 As a result, ASP structural encodings needed fewer features than LSTAR and RAD2D encodings to describe our compound collections (Table 1—“features” column), although the prediction performance of ASP fingerprints was higher or comparable with that of LSTAR and RAD2D fingerprints at most recall thresholds. Moreover, LSTAR fingerprints generally exhibited higher performance than RAD2D fingerprints at several recall thresholds (Figures 2 and 3), which could be driven by the additional bond information that LSTAR fingerprints collect from the radial environment of the atoms.
While the choice of a molecular fingerprint is important (as demonstrated above) for maximizing the biological relevance of chemical similarity calculations, such fingerprints must be paired with a similarity coefficient to compute chemical similarity. To characterize the utility of different similarity coefficients, we benchmarked a large set of 13 different similarity coefficients (Table 2) that were applied to the complete set of candidate molecular fingerprints. Again, using our RIKEN high-confidence set as the basis, we measured the prediction performance of every coefficient over all molecular fingerprints to find the best-performing similarity coefficient (Table S1). We found that several (nine out of 13) coefficients were able to consistently exhibit high performance across all molecular fingerprints; however, four coefficients (Dot-product, Euclidean, Russel/Rao, and Simpson) failed in several molecular fingerprint spaces because their precision substantially dropped at many recall thresholds (Table S1). Because these four coefficients were unable to capture the biological similarity of our compounds given the provided molecular fingerprints, we focused only on the nine remaining coefficients.
We found that the Braun-Blanquet similarity coefficient53,54 resulted in the highest precision at many recall thresholds compared to all other coefficients (Table S1), including the Tanimoto and cosine coefficients (Figure 2A), which have been used widely by the chemical informatics community. The Braun-Blanquet coefficient is simply computed as the fraction of features shared between two molecules to the total number of features present in the larger molecule. For the Braun-Blanquet coefficient, the average and maximum precisions and the average and maximum areas under the receiver operating characteristic (ROC) curves across all molecular fingerprints were slightly higher at many recall thresholds compared to those of the Tanimoto and cosine coefficients (Figure 2A and Table S1—green and blue values), suggesting that this simple coefficient of structural similarity can confidently be used in ligand-based virtual screening as a substitute for the traditional Tanimoto coefficient. We further measured the performance of our prediction models using our NCI/NIH/GSK high-confidence set to validate the superiority of the Braun-Blanquet coefficient over other similarity coefficients (Figure 3A and Table S2).
Given the prevalence of the Tanimoto similarity coefficient throughout chemical similarity calculations, we found our evaluation results especially interesting in that the Tanimoto similarity coefficient was outperformed in our systematic benchmark by the Braun-Blanquet similarity coefficient. Thus, we offer a potential explanation: for a pair of small molecules where the structural features of one molecule are a subset of the structural features of the other molecule, both similarity coefficients yield the same similarity values. However, these similarity values diverge if new features are added to the molecule with fewer structural features, and therefore, its feature set is no longer a subset of the feature set of the larger molecule. Specifically, Tanimoto-based structural similarity decreases because the size of the total feature set across the two molecules increases, while Braun-Blanquet-based structural similarity remains unchanged. Thus, when retrieving similar molecules to a target molecule, the Tanimoto similarity coefficient will bias the results toward the sub- and supersets of the target molecule and against the molecules that share a common core with the target molecule but incorporate additional unique features. Braun-Blanquet-based similarity does not suffer from this bias, as the denominator of the Braun-Blanquet coefficient is the size of the largest molecular feature set and not the total number of features observed across both molecules. The higher performance of the Braun-Blanquet coefficient against our functional gold standard suggests that the bias inherent in the definition of the Tanimoto coefficient affects its performance in predicting the true functional similarity of compounds and that a similarity coefficient such as the Braun-Blanquet coefficient can mitigate this issue.
Overall, we identified the combination of the Braun-Blanquet similarity coefficient and either the ASP or LSTAR molecular fingerprints as the highest-performing approach for measuring chemical structural similarity in ligand-based virtual screening. This conclusion was further confirmed through more formal “sum of ranking distances” (SRD) analysis (Figures S1 and S2, Tables S1 and S2—SRD analysis spreadsheets).72−74
To the best of our knowledge, no study has reported or used the combination of the ASP or LSTAR fingerprints paired with the Braun-Blanquet similarity coefficient as a measure of structural similarity.
Optimizing the Depth of Molecular Fingerprints
One major parameter involved in the structural description of compound collections is the describing depth; a high depth generates features that describe the local and global environments of each atom, whereas a low depth focuses only on the local neighborhood of atoms in the molecular graph. We assessed the impact of the depth of five molecular fingerprints (ASP, DFS, ECFP, LSTAR, and RAD2D) in predicting the biological activity of our compounds using the Braun-Blanquet similarity coefficient (Figure 4).
Figure 4.
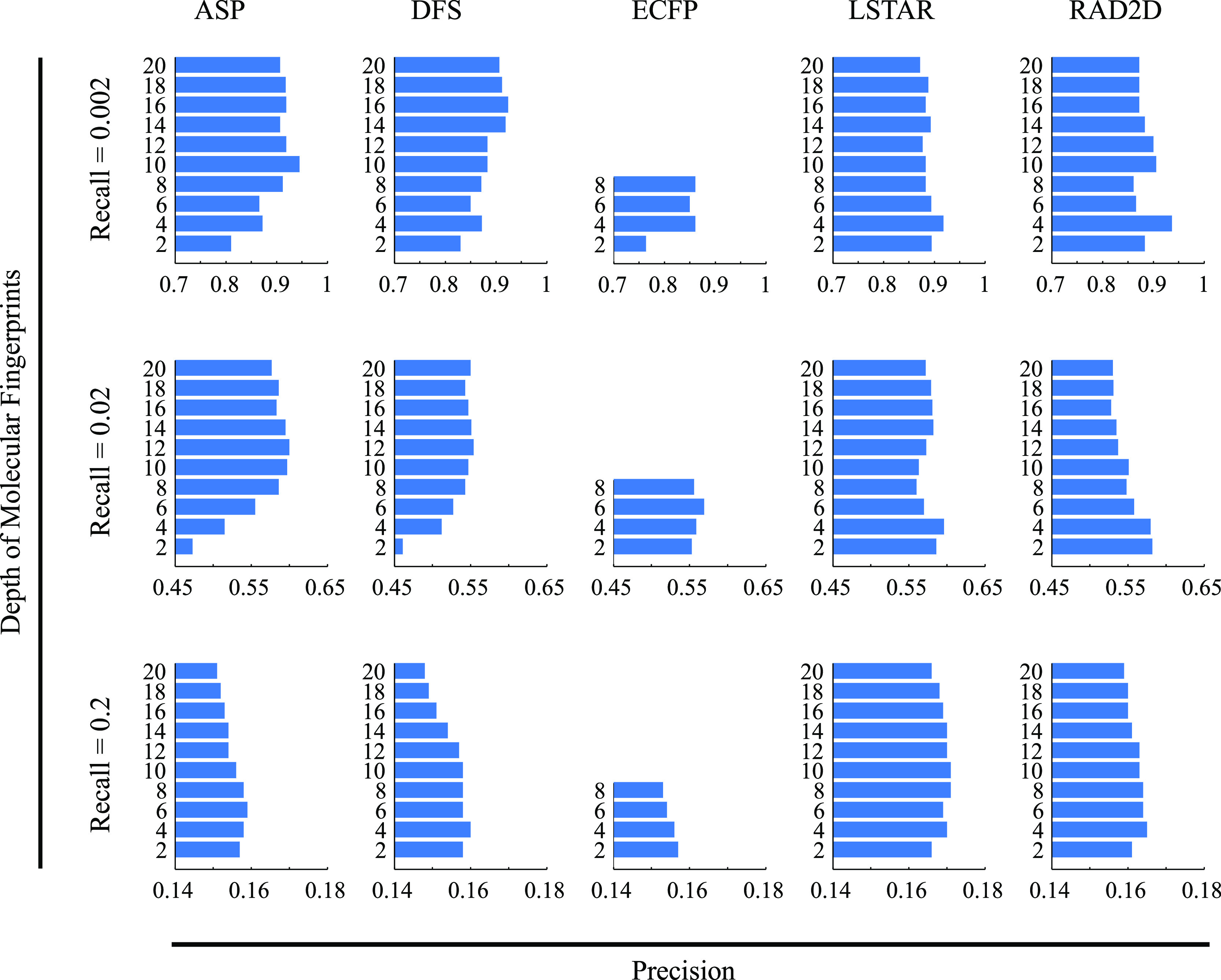
Impact of the describing depth of molecular fingerprints on the RIKEN high-confidence set. We measured the precision of our prediction models at 10 molecular depths, ranging from 2 to 20, for five different molecular fingerprints. Similarities were calculated with the Braun-Blanquet similarity coefficient, and the precision at three different recall thresholds for each molecular depth is shown.
At predefined recall thresholds, we observed that precision beyond a depth of 8 at best minimally improved the prediction of functional similarity from chemical structures and at worst decreased it markedly. Describing our RIKEN high-confidence set at a high depth generally resulted in strong predictions at lower recall thresholds. Structural description at a high depth was able to predict the compound pairs that were structurally and therefore functionally very similar according to the SPP. On the other hand, a low describing depth was able to capture the similarity of two molecular graphs in the local subgraphs that were essential for functional similarity, resulting in reasonable predictions at lower recall thresholds. We further evaluated these results using our NCI/NIH/GSK high-confidence set (Figure S3), which confirmed a similar general trend across a range of recall thresholds using 10 different describing depths. Therefore, the structural description of a compound collection at high depths does not appear to be beneficial from our evaluation, and thus, the additional computational complexity and storage space for these structural vectors across hundreds or thousands of compounds may not be justified. For other evaluations in this study, we selected a depth of 8 as the focus of our analysis.
Improving the Prediction Performance Using SVM Models
To increase our ability to predict the biological activity of compounds from their structures, we designed supervised machine learning models that leveraged chemical–genetic interaction profiles as training data. As a preprocessing step to reduce the size of the feature space, we used supervised principal component analysis75 to extract the most informative structural features given our functional similarity standard.
We designed a learning pipeline (Figure 5A) for predicting chemical–genetic similarities by creating bootstraps76 from our compound collections and generating “pairwise structural vectors” (Figure S4) that labeled the shared molecular features between two molecules (Materials and Methods). This pipeline was implemented using support vector regression (SVR) models40,41 with a radial basis function (RBF) kernel. We employed PR curves to evaluate the prediction performance of our learning models for different molecular fingerprints, where the compound pairs from our high-confidence sets with the highest 10% of chemical–genetic similarities were labeled as the gold standard for true positives. Using this machine learning approach, we found that a subset of our molecular fingerprints (FP1–FP6, FP10, and FP11) enabled substantial improvements in performance for predicting compound functional similarity than the best-performing structural similarity measure (ASP/Braun-Blanquet). To evaluate the characteristics of our predicted supervised structural similarities, we computed the Pearson correlation between these pairwise similarities and the observed chemical–genetic similarity for both our compound collections (Figure S5). This analysis demonstrated that predicted similarities were correlated with chemical–genetic similarities in both collections and that the correlation increased using our machine learning approach.
Figure 5.
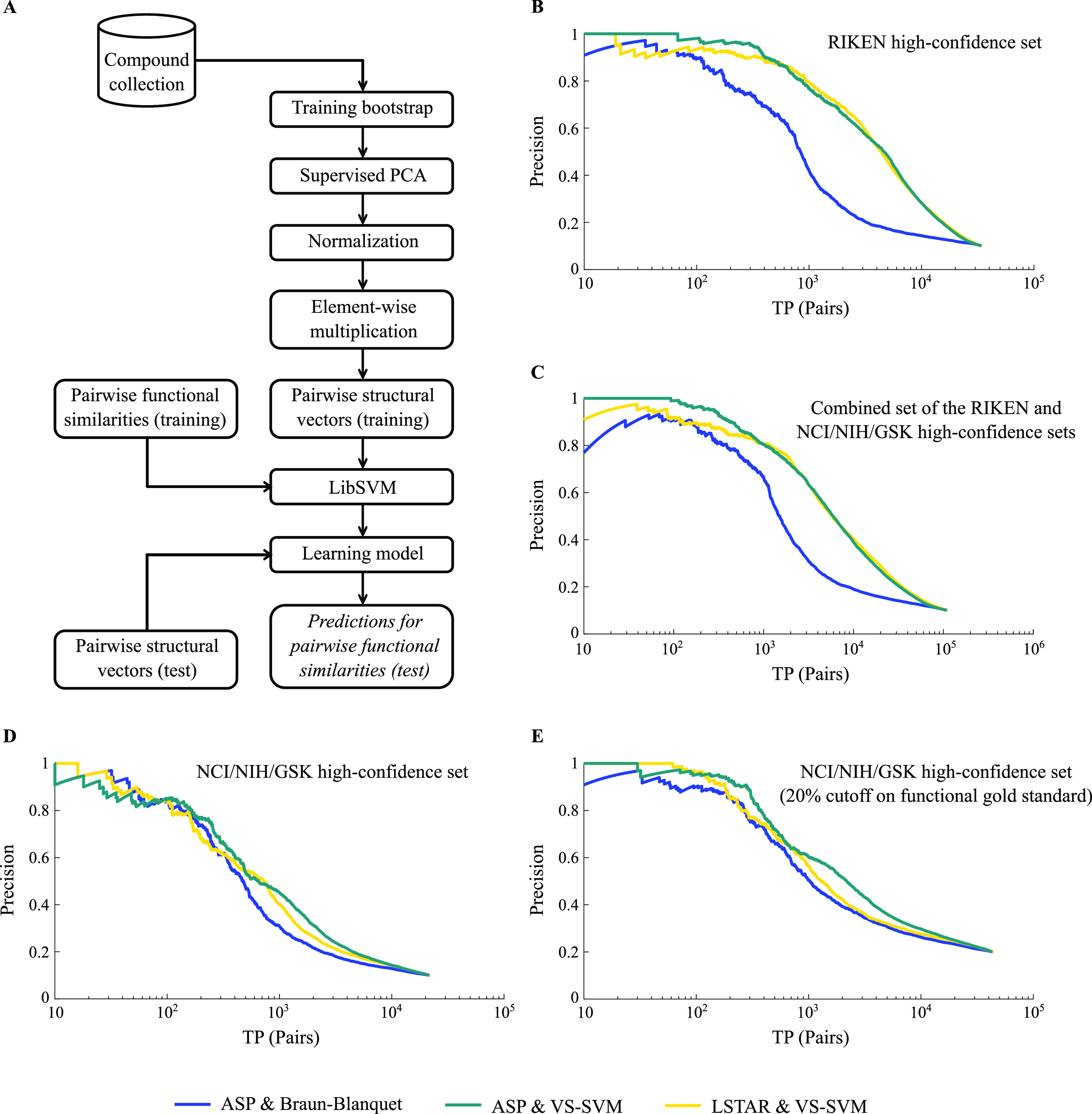
Prediction performance of machine learning models. (A) Learning pipeline for one bootstrap using pairwise structural vectors (Materials and Methods). (B) Model performance for our RIKEN high-confidence set. The blue precision–recall (PR) curve represents the prediction performance of our best structural similarity measure (ASP/Braun-Blanquet), whereas the teal and gold PR curves represent the performance of our machine learning models using ASP and LSTAR fingerprints, respectively. A prediction is considered a true positive if the compound pair is within the top 10% of functionally similar compound pairs using chemical–genetic interaction profiles. We used pairwise true positives or TP (pairs) as a general form of recall in our PR curves. (C) Model performance for the combined RIKEN and NCI/NIH/GSK high-confidence sets. (D) Model performance for the NCI/NIH/GSK high-confidence set. (E) Model performance for the NCI/NIH/GSK high-confidence set (as in panel (D)), except using top 20% of pairwise chemical–genetic similarities to define true positives.
The PR curves of our learning models built on ASP and LSTAR fingerprints (Figure 5B) exhibited a fivefold improvement in the recall of biologically similar compounds at a precision of 50%. However, the degree of improvement was dependent on the functional diversity of our datasets, where performance was higher for the collections with lower functional diversity. For instance, predictions for the NCI/NIH/GSK high-confidence set improved by only about twofold based on the recall of biologically similar compounds at a precision of 50% (Figure 5D–E). We speculate that this relatively modest improvement (compared to that of the RIKEN high-confidence set) may be explained by the higher functional diversity of the compounds in our NCI/NIH/GSK high-confidence set. Specifically, we derived a functional diversity score using a divisive clustering approach and observed scores of ∼25.3 and ∼14.6 for the NCI/NIH/GSK and RIKEN high-confidence sets, respectively, using the chemical–genetic interaction profiles of our compounds (Materials and Methods). This difference in functional diversity contrasted with the near-identical structural diversity observed between the two collections (score of ∼62 using ASP molecular fingerprints). The relatively higher functional diversity of our NCI/NIH/GSK high-confidence set arises from its six functionally different sub-collections, which consequently affected the ability of our pipeline to learn models that capture chemical–genetic similarities and generalize across many compounds in this collection. Although the performance of our supervised learning models for the NCI/NIH/GSK high-confidence set was affected by high functional diversity, even for this collection, the boost in performance offered by our learning models was still substantial (Figure 5D–E). Furthermore, we combined the two collections, which added not only more compounds but also more diversity to the resulting set. Making predictions for the combined dataset, we achieved about a 4.5-fold improvement in the recall of biologically similar compounds at a precision of 50% (Figure 5C).
We conclude that compound collections with low functional diversity benefit substantially from our machine learning models. These models can more appropriately adjust the influence of specific substructural features within large clusters of structurally similar compounds given an independent standard of shared biological functions.
Exploring the Basis of the Predictive Power of SVM Models
To investigate the compounds driving our prediction models, we clustered our compound collections into 10 functional and 10 structural clusters using K-means and K-medoids clustering algorithms, respectively (Tables S3–S5). We then mapped the 1000 true positive pairs at the top of our PR curves for ASP fingerprints to their corresponding functional and structural clusters (Figures 6 and 7). A large group of the compounds generating high predictive scores at the top of our learning PR curves belonged to the same functional clusters (Figures 6B and 7B), whereas the baseline PR curve for the ASP fingerprints and Braun-Blanquet similarity coefficient included several functional clusters even at lower recall thresholds (Figures 6C and 7C). This indicates that our machine learning models improved performance in a non-uniform fashion, boosting prediction performance substantially for a few specific functional clusters. The first functional cluster that we observed among the highest predictions for the RIKEN high-confidence set (blue cluster in Figure 6A named as cluster 1 in Table S3) was enriched for compounds targeting cell cycle checkpoint processes (Tables S3 and S4 for the enrichment analysis of the functional clusters in the RIKEN and NCI/NIH/GSK high-confidence sets; predictions from the MOSAIC database12). Despite this enrichment for the predicted target processes, several distinct structural classes were clearly represented among the compound pairs with the highest similarity predicted by our machine learning models (Figures 6E and 7E), suggesting that compounds considered diverse based on molecular structures alone were predicted to have similar biological activity using our machine learning models (Table S6).
Figure 6.
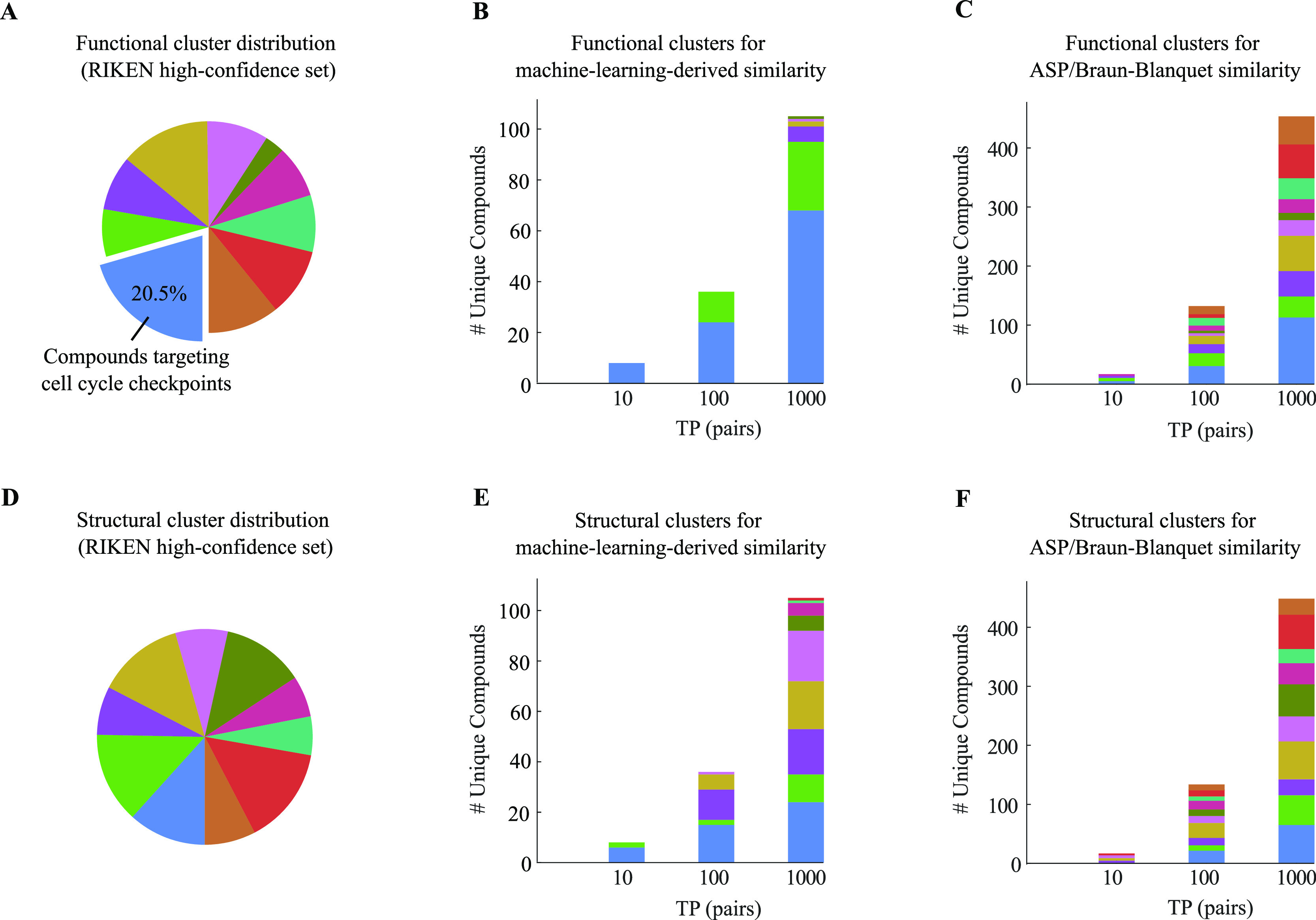
Functional and structural clusters of top true positive pairs for our RIKEN high-confidence set. (A) Distribution of 10 functional clusters generated by the K-means clustering algorithm using our chemical–genetic interaction profiles. The blue cluster represents the largest functional cluster. (B,C) Contribution of these functional clusters to the top true positive pairs retrieved by (B) our machine learning model and (C) our best structural similarity measure (ASP/Braun-Blanquet). (D) Distribution of 10 structural clusters generated by the K-medoids clustering algorithm using ASP fingerprints. (E–F) Contribution of these structural clusters to the top true positive pairs introduced by (E) our machine learning model and (F) our best structural similarity measure.
Figure 7.
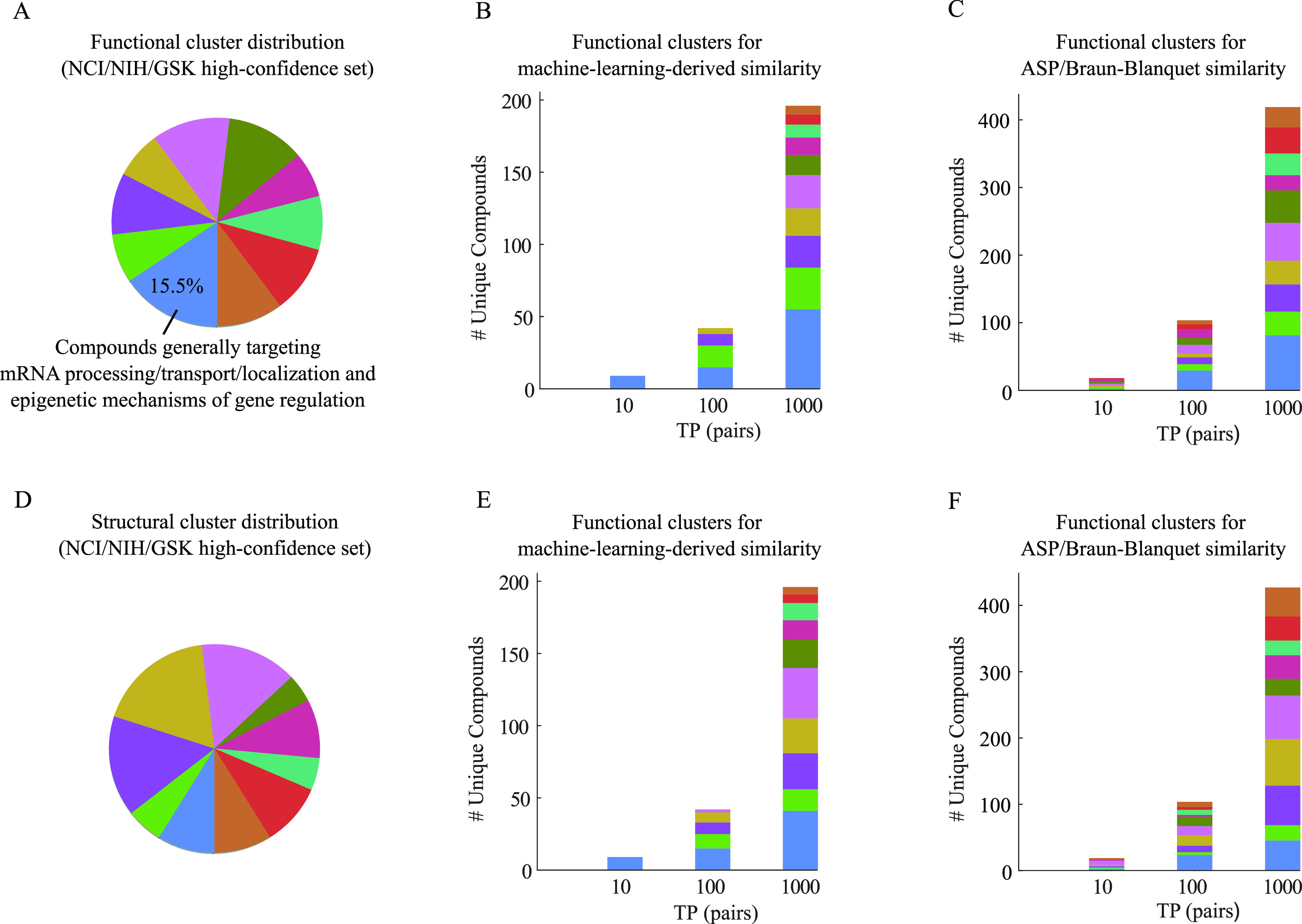
Functional and structural clusters of top true positive pairs for our NCI/NIH/GSK high-confidence set. (A) Distribution of 10 functional clusters generated by the K-means clustering algorithm using our chemical–genetic interaction profiles. (B,C) Contribution of these functional clusters to the top true positive pairs retrieved by (B) our machine learning model and (C) our best structural similarity measure (ASP/Braun-Blanquet). (D) Distribution of 10 structural clusters generated by the K-medoids clustering algorithm using ASP fingerprints. (E,F) Contribution of these structural clusters to the top true positive pairs introduced by (E) our machine learning model and (F) our best structural similarity measure.
Example Use Case: Retrieving Novel Functionally Analogous Compounds for Virtual Screening Applications
To demonstrate the impact of our machine learning-based approach in predicting biologically similar compounds, we illustrate with a specific example. We chose a query compound of interest from our RIKEN high-confidence set, NPD2186. We first ranked all other compounds in this collection based on their structural similarity to this compound as measured by our best-performing structural similarity measure (ASP/Braun-Blanquet) (Figure 1). To contrast, we also compiled a ranking based on our machine learning-derived structural similarity scores computed from ASP fingerprints (Figure 8C). For example, among the top 10 most similar compounds identified by our machine learning-derived similarity, none were identified in the top 10 by the ASP/Braun-Blanquet similarity measure (or any of the other structural similarity measures). Among the top 100 and top 200 most similar compounds identified by our machine learning-derived similarity, only 29 and 88 were also identified in the top 100 and top 200 most similar compounds retrieved by the ASP/Braun-Blanquet similarity measure, respectively. Overall, Spearman’s rank correlation between the two recalled lists of compounds for our query compound, NPD2186, was 0.31. Therefore, our machine learning-based structural similarity approach provided a substantially different set of similar compounds for this representative example.
Figure 8.
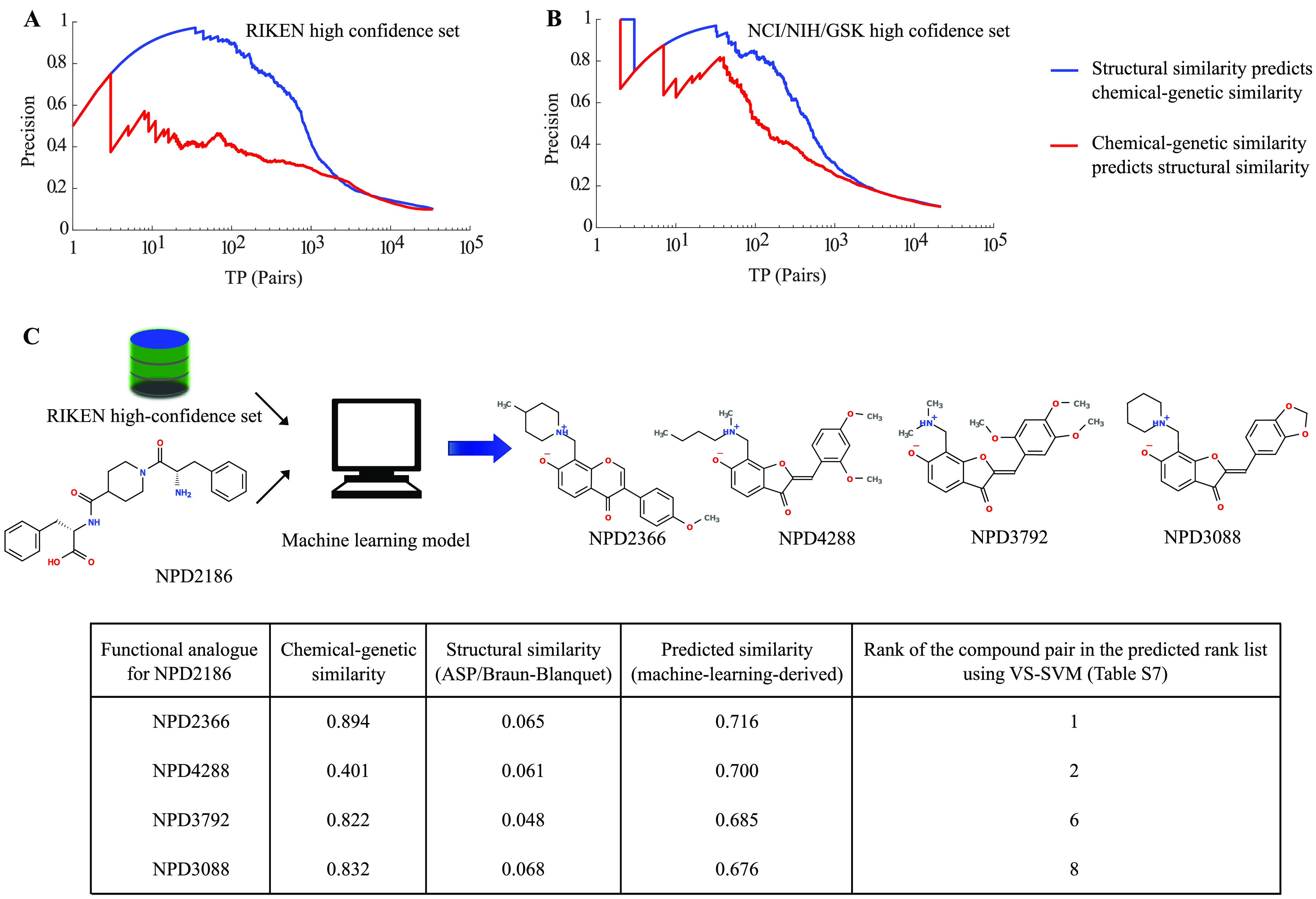
Reciprocal evaluation of the prediction performance of structural vs functional similarity and machine learning-based virtual screening of a target (e.g., NPD2186 from the RIKEN high-confidence set). Using (A) RIKEN and (B) NCI/NIH/GSK high-confidence sets, we measured the abilities of structural and chemical–genetic similarities to reciprocally predict each other. The blue curve represents the performance of structural similarity in predicting chemical–genetic similarity, whereas the red curve represents the performance of chemical–genetic similarity in predicting structural similarity. (C) Our machine learning model retrieved biologically similar but structurally dissimilar compounds (determined by the ASP/Braun-Blanquet structural similarity measure) for NPD2186 from our RIKEN high-confidence set. The information table provides the chemical–genetic similarities, ASP/Braun-Blanquet structural similarities, and machine learning-derived predicted similarities for a few of the compounds at the top of the predicted ranked list that are functionally analogous to NPD2186. The highest predictive score generated by our machine learning model was 0.716, retrieving NPD2366 as a functional analogue of NPD2186. The rank of each compound pair comes from the table of all pairwise compound similarities ranked in descending order of predicted machine learning-derived similarities (Table S7).
The trends that we observed for NPD2186 generalize to the broader set of compounds as well (Table S7). We measured Spearman’s rank correlation of our machine learning-derived and Braun-Blanquet structural similarities, both computed from ASP fingerprints, for every compound in the RIKEN high-confidence set (Figure S6A). The median of the resulting distribution of rank-based correlations was 0.31, suggesting that these two approaches produce substantially different rankings on the most similar compounds. Thus, using our proposed machine learning-based approach will likely produce substantially different results than the traditional measures of structural similarity in virtual screening settings.
Evaluating the Reciprocal Predictive Power of Structural and Functional Similarities
The main focus of our study was to improve structural similarity measures by leveraging functional information from chemical–genetic interaction data. However, these data also offered an opportunity to explore the connection between structural similarity and similarity of functional impact on cellular functions. Specifically, we were interested in the question of which was a stronger predictor: structural similarity predicting functional similarity or functional similarity predicting structural similarity. To address this question, we carried out a reciprocal PR analysis, in which we switched the roles of structural and functional similarities and compared the relative strength of each for predicting the other. Specifically, we assessed the predictive power of the ASP/Braun-Blanquet-derived structural similarity against a standard of chemical–genetic similarity as described above. Then, we reversed the analysis, evaluated the potential of chemical–genetic similarity to predict the ASP/Braun-Blanquet-derived structural similarity and compared the resulting performances. This analysis revealed that structural similarity exhibited substantially higher power in predicting chemical–genetic similarity than did chemical–genetic similarity for predicting similar structures (Figure 8A,B). This result likely reflects the fact that compounds with very different chemical structures can result in a similar functional impact (e.g., targeting different subunits of the same protein complex or different members of the same pathway will result in a similar phenotype). On the other hand, compounds with very similar structures are highly likely to exhibit similar biological activity and very unlikely to possess extremely divergent biological activities. Interestingly, the gap between structural prediction of functions versus functional prediction of structures was substantially larger for the compounds in the RIKEN high-confidence set (Figure 8A,B). This wider gap likely reflects the fact that the RIKEN high-confidence set contains many more local clusters of compounds with highly related structures, whereas the NCI/NIH/GSK high-confidence set is inherently composed of several sub-collections with relatively few structurally related compounds, which results in a more one-to-one correspondence between structural and functional profiles.
Conclusions
The drug discovery process benefits from improvements in our ability to link the structure of chemical compounds to their biological function. The chemical informatics community has proposed a wide range of molecular fingerprints and similarity coefficients for ligand-based virtual screening, where the SPP has been the basis for ranking compounds with similar biological activity to a target molecule based on chemical structures. However, to date, the research community has lacked a systematic benchmark for biological activity that covers a broad range of protein targets to assess the performance of different molecular fingerprints and similarity coefficients. We used a large set of chemical–genetic interaction data from the yeast S. cerevisiae that we previously published, covering 13,431 compounds from the RIKEN NPDepo and several NCI/NIH/GSK compound collections, as the standard for the biological activity of our compounds. Using these chemical–genetic interaction profiles as a functional standard for our compounds, we systematically benchmarked 11 different molecular fingerprints and 13 different similarity coefficients.
We found that the pair of ASP fingerprints combined with the Braun-Blanquet similarity coefficient was the superior choice for prioritizing compounds with similar biological activity to a target molecule. The ASP fingerprints encode all shortest paths between atoms obtained through an exhaustive depth-first search of the molecular graph (up to a predefined describing depth), and the Braun-Blanquet coefficient represents the fraction of features in common between two molecules to the total number of features present in the larger one. We also determined that the performance of our ASP/Braun-Blanquet structural similarity measure would not substantially benefit from the high describing depths of ASP fingerprints beyond a depth of 8. Our results suggest that the ASP/Braun-Blanquet similarity measure can be used with confidence as a replacement for the ECFP/Tanimoto similarity measure, which has been one of the most commonly used structural similarity measures for ligand-based virtual screening.
Moreover, we developed a machine learning model based on SVMs that boosted the predictive power of several fingerprints up to fivefold, with the degree of improvement dependent on the functional diversity of the compound collections on which the machine learning model was applied. The compound collections with low functional diversity benefited substantially from our machine learning model.
One interesting future direction of our work would be to extend this analysis beyond the molecular fingerprints considered in this study (e.g., computed physicochemical properties). Chemical–genetic interaction profiles may also yield important information on the relevance of other features for predicting functional similarity of compounds.
Overall, our high-dimensional chemical–genetic interaction data provide a powerful resource for connecting the chemical structure to compound functions. We expect that the specific lessons learned here regarding the relative strengths of different molecular fingerprints and the general approach of integrating chemical–genetic interaction data with structural information for improving the structure-based prediction of biological activity will be of use in other virtual screening contexts.
Materials and Methods
Data Collections
We previously published chemical–genetic interaction profiles for 13,431 uniquely named compounds from several diverse compound collections and isolated a subset of these compounds that exhibited high-confidence predictions of perturbed biological processes based on integration of these profiles with genetic interaction profiles.34 We used two independent compound collections, for which we had high-confidence biological processes: the RIKEN high-confidence set (826 compounds) and the NCI/NIH/GSK high-confidence set (659 compounds). The RIKEN high-confidence set is a subset of the RIKEN NPDepo collection and is composed largely of purified natural products or natural product derivatives. The NCI/NIH/GSK high-confidence set is itself a diverse set of several sub-collections: four collections from the National Cancer Institute’s Open Chemical Repository (natural products, approved oncology drugs, and structural and mechanistic diversity sets), a library of compounds from the National Institutes of Health Small-Molecule Repository with a history of use in human clinical trials, and the GSK kinase inhibitor collection. More detailed descriptions of these two compound collections are available in the “Methods—Description of Compound Collections” section of our recent publication.34
Structural Description of Compounds
We used the jCompoundMapper tool42 and RDKit toolkit (version 2020.09.4)43 to describe our compounds in 11 different molecular fingerprint spaces (Table 1) using the structural information of our compounds (Supporting Information Structure Data Files and Supporting Information SMILES and InChI). Except for the MACCS and RDKit fingerprints, we generated all other molecular fingerprints using the jCompoundMapper tool. These structural descriptions covered several topological aspects of a molecular graph, including depth-first search fingerprints, atom pair fingerprints, radial atom environment fingerprints, extended connectivity fingerprints, and pharmacophore fingerprints.
Establishing a Gold Standard for Biological Activity
We selected the top 10% of most similar compound pairs in chemical–genetic interaction profiles and labeled these pairs as our gold standard for true positive compound pairs. We previously published a systematic evaluation of several different profile similarity measures for genetic interaction networks77 and more recently published our functional annotations of chemical libraries across diverse biological processes.34 These two analyses suggested that the cosine similarity coefficient was a reasonable metric for measuring genetic and chemical–genetic similarities. Therefore, we used the cosine coefficient for computing our gold standard biological activity. Specifically, in the latter study, we annotated our RIKEN and NCI/NIH/GSK high-confidence sets to 17 different functional neighborhoods (e.g., DNA replication and repair, glycosylation, vesicle traffic, mitosis and chromosome segregation, etc.) based on their chemical–genetic interactions (supplementary dataset 19 of the latter study).34 In a range of different cutoffs on the compound functional similarity scores (2%, 5%, and 10%), we computed the distribution of our compounds across these 17 different neighborhoods. This analysis demonstrated that, at a cutoff of 2%, we observed a strong enrichment for compounds mapping to neighborhood 4 (N4 in Figure S7), which corresponded to mitosis and chromosome segregation. However, at more relaxed cutoffs (e.g., 5% or 10%), the distribution of compounds across neighborhoods was relatively similar to the background distribution of our compounds, suggesting that we were covering a broader representation of the compounds. This observation led us to select a higher cutoff (10%) on the pairwise functional similarity score to ensure a broad representation of the function of our compounds.
We note that the relative performance of our structural similarity measures and machine learning models for more stringent cutoffs (e.g., 5%) is consistent with our results for the top 10% of the functional similarity gold standard (Figure S8).
Designing the SVM Learning Pipeline
We proposed SVR models40,41 for predicting the functional similarity of two compounds based on their chemical structures. We used LibSVM,78 a popular open-source SVM library developed at National Taiwan University, for implementing our models and bootstrapping76 for generating our training and test data. To generate our training and test sets, we randomly drew N (the total number of compounds in a collection) samples, with replacement, uniformly from the collection, assigning ∼0.632N unique compounds to the training set and the rest to the test set. To reduce the dimensionality of our structural spaces, we employed supervised principal component analysis75 using chemical–genetic interaction profiles (from the training data only) as labels. We normalized each structural vector that described a compound in the low-dimensional space by its Euclidean length and multiplied each pair of the normalized vectors (both from the training set or both from the test set) in an element-wise manner to create a new space of structural vectors, called as “pairwise structural vectors” (Figure S4A), for the representation of compound pairs. We devised a machine learning pipeline (Figure 5A) to predict chemical–genetic similarities for the test data using pairwise structural vectors and pairwise chemical–genetic similarities (from the training data only). We used RBF kernels to build epsilon SVR (ϵ̲-SVR) models in the pairwise structural space and evaluated the average performance of our pipeline across 100–200 bootstraps (Figure S4B). To measure the prediction performance of the pipeline on held-out test data, we used the average model output over all the bootstraps for which a given example was in the test set (bagging). We sampled 200 bootstraps for the RIKEN high-confidence set, 200 bootstraps for the NCI/NIH/GSK high-confidence set, and 100 bootstraps for the combined collection of these two sets. We evaluated the prediction performance of our machine learning pipeline using PR curves (Figure 5B–E). Selecting the 10% of most similar compound pairs based on the (cosine) similarity of their chemical–genetic interaction profiles and labeling these pairs as our gold standard for true positive compound pairs, we translated our predicted pairwise compound similarities into a binary evaluation using PR curves, where we ranked our predicted similarities in descending order against our binarized gold standard for true positive compound pairs (Table S7, where the class column indicates our binarized gold standard for functional similarity).
To facilitate the use of our supervised similarity prediction pipeline, we provide the current version of our MATLAB source codes that can be used for learning a new compound collection. The source codes and our data for the machine learning prediction pipeline are freely available at https://github.com/csbio/VS-SVM.
Estimating the Diversity of Compound Collections
We defined functional and structural diversity measures for compound collections from the results of a recursive, divisive clustering algorithm based on either functional (chemical–genetic) or structural similarity, respectively. More specifically, we assigned all the compounds in a collection to a single cluster and split up the cluster recursively to form smaller clusters of more similar compounds. At any step of recursion, we determined the cluster with the lowest average within-cluster pairwise chemical–genetic similarity (for computing functional diversity) or structural similarity (for computing structural diversity) and divided the cluster into two new clusters using the K-means (K = 2) or K-medoids (K = 2) clustering algorithms, respectively. We terminated the recursion when at least two new clusters would exceed our predefined hard threshold for the average between-cluster pairwise chemical–genetic similarity (cosine similarity of 0.3) or structural similarity (ASP/Braun-Blanquet similarity of 0.3). We repeated the algorithm 1000 times for the functional diversity and 100 times for the structural diversity and computed the mean diversity score as the average exponentiation of the Shannon entropy indices over all the instances
where pi is the proportional abundance of compounds in the ith cluster of the final clustering.
Acknowledgments
We thank Benjamin VanderSluis, Wen Wang, and Maximilian Billmann for proofreading the early drafts of this article. H.S. was partially supported by the National Institutes of Health (NIH) (R01HG005084 and R01GM104975) and the National Science Foundation (NSF) (DBI 0953881). S.W.S. was supported by a NSF Graduate Research Fellowship (00039202), a NIH Biotechnology training grant (T32GM008347), and a BICB one-year fellowship. S.C.L. was supported by a RIKEN Foreign Postdoctoral Research Fellowship and a RIKEN Incentive Research Projects grant. Minoru Yoshida was supported in part by a Grant-in-Aid for Scientific Research (S) (19H05640), the Japan Society for the Promotion of Science (JSPS), and by a Grant-in-Aid for Scientific Research on Innovative Areas (18H05503) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan. C.B. was supported by a JSPS Grant-in-Aid for Scientific Research (B) (15H04483). C.B. and Y.Y. were supported by a Grant-in-Aid for Scientific Research on Innovative Areas (17H06411) from MEXT. C.L.M. is a fellow in the Canadian Institute for Advanced Research (CIFAR) Genetic Networks Program. Computing resources and data storage services were partially provided by the Minnesota Supercomputing Institute and the UMN Office of Information Technology, respectively.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00993.
Current version of our MATLAB source codes and data for our machine learning prediction pipeline freely available at https://github.com/csbio/VS-SVM; algorithm for finding the best-performing structural similarity measures; SRD analysis for molecular fingerprints; SRD analysis for similarity coefficients; impact of the describing depth of molecular fingerprints using our NCI/NIH/GSK high-confidence set; pairwise structural vectors and bootstrapping used by our machine learning pipeline; correlation analysis of predicted structural similarity with chemical–genetic similarity; Spearman’s rank correlation distribution between the machine learning-derived and the ASP/Braun-Blanquet-derived structural similarity predictions; distribution of the predicted biological functions across 17 broad, previously defined functional neighborhoods; and prediction performance of the machine learning models for the 5% cutoff on the functional similarity gold standard (PDF)
Precision at several recall thresholds for all combinations of molecular fingerprints and similarity coefficients using the RIKEN high-confidence set (XLSX)
Precision at several recall thresholds for all combinations of molecular fingerprints and similarity coefficients using the NCI/NIH/GSK high-confidence set (XLSX)
Functional clustering using the K-means clustering algorithm and functional enrichment analysis using the hypergeometric test for the RIKEN high-confidence set (XLSX)
Functional clustering using the K-means clustering algorithm and functional enrichment analysis using the hypergeometric test for the NCI/NIH/GSK high-confidence set (XLSX)
Structural clustering of our RIKEN and NCI/NIH/GSK high-confidence sets using the K-medoids clustering algorithm (XLSX)
Rank lists of compound pairs, selected from the top 1000 machine learning-derived true positive pairs, with similar chemical–genetic interaction profiles (all compounds from the first functional cluster) but distinct chemical structures (as determined by the ASP/Braun-Blanquet structural similarity measure) for our RIKEN and NCI/NIH/GSK high-confidence sets (XLSX)
Comprehensive rank lists of structural and chemical–genetic similarities for our RIKEN and NCI/NIH/GSK high-confidence sets (XLSX)
Structure data files—molecular structural information in SDF notations for our compound collections freely available in the MOSAIC database (http://mosaic.cs.umn.edu)12 (ZIP)
SMILES and InChI—molecular structural information in SMILES and InChI notations for our compound collections freely available in the MOSAIC database (http://mosaic.cs.umn.edu)12 (XLSX)
Author Present Address
∇ Octant Inc., Emeryville, California 94608, United States
Author Present Address
○ Yumanity Therapeutics, Boston, Massachusetts 02135, United States.
Author Contributions
H.S. and C.L.M. conceived the project and established the methodologies. H.S. developed the benchmarking system and machine learning models, generated the molecular fingerprints for the models, and performed all the analyses. H.S. constructed and validated the VS-SVM machine learning-based prediction model for ligand-based virtual screening. S.W.S. contributed to the conception of the benchmarking system, generation of the molecular fingerprints using the jCompoundMapper tool, and provision of detailed feedback on the results and visualizations. S.W.S, J.N., S.C.L., J.S.P., Mami Yoshimura, Y.Y., H.H., and H.O. generated and validated chemical–genetic interaction data. Minoru Yoshida, C.B., and C.L.M. supervised the project and acquired funding. H.S. wrote the original draft, and all authors reviewed and edited this draft.
The authors declare no competing financial interest.
Supplementary Material
References
- Scannell J. W.; Blanckley A.; Boldon H.; Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discovery 2012, 11, 191–200. 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- Munos B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discovery 2009, 8, 959–968. 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]
- DiMasi J. A.; Grabowski H. G.; Hansen R. W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Johnson M. A.; Maggiora G. M.. Concepts and Applications of Molecular Similarity; Wiley, 1990. (accessed July 17, 2020). https://agris.fao.org/agris-search/search.do?recordID=US201300674768. [Google Scholar]
- Bajorath J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discovery 2002, 1, 882–894. 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- Tanrikulu Y.; Krüger B.; Proschak E. The holistic integration of virtual screening in drug discovery. Drug Discovery Today 2013, 18, 358–364. 10.1016/j.drudis.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Martin Y. C.; Kofron J. L.; Traphagen L. M. Do Structurally Similar Molecules Have Similar Biological Activity?. J. Med. Chem. 2002, 45, 4350–4358. 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- Eckert H.; Bajorath J. Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches. Drug Discovery Today 2007, 12, 225–233. 10.1016/j.drudis.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Maggiora G. M. On Outliers and Activity CliffsWhy QSAR Often Disappoints. J. Chem. Inf. Model. 2006, 46, 1535. 10.1021/ci060117s. [DOI] [PubMed] [Google Scholar]
- Bajorath J.; Peltason L.; Wawer M.; Guha R.; Lajiness M. S.; Van Drie J. H. Navigating structure-activity landscapes. Drug Discovery Today 2009, 14, 698–705. 10.1016/j.drudis.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Muratov E. N.; Bajorath J.; Sheridan R. P.; Tetko I. V.; Filimonov D.; Poroikov V.; Oprea T. I.; Baskin I. I.; Varnek A.; Roitberg A.; Isayev O.; Curtalolo S.; Fourches D.; Cohen Y.; Aspuru-Guzik A.; Winkler D. A.; Agrafiotis D.; Cherkasov A.; Tropsha A. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. 10.1039/D0CS00098A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.; Simpkins S. W.; Safizadeh H.; Li S. C.; Piotrowski J. S.; Hirano H.; Yashiroda Y.; Osada H.; Yoshida M.; Boone C.; Myers C. L. MOSAIC: a chemical-genetic interaction data repository and web resource for exploring chemical modes of action. Bioinformatics 2018, 34, 1251–1252. 10.1093/bioinformatics/btx732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett P.; Barnard J. M.; Downs G. M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. 10.1021/ci9800211. [DOI] [Google Scholar]
- Bender A.; Glen R. C. Molecular similarity: a key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204. 10.1039/b409813g. [DOI] [PubMed] [Google Scholar]
- Maggiora G.; Vogt M.; Stumpfe D.; Bajorath J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. 10.1021/jm401411z. [DOI] [PubMed] [Google Scholar]
- Willett P.Similarity Searching Using 2D Structural Fingerprints. In Chemoinformatics and Computational Chemical Biology, Methods in Molecular Biology; Bajorath J., Ed.; Humana Press, 2010; Vol 672, pp 133–158. [DOI] [PubMed] [Google Scholar]
- Duan J.; Dixon S. L.; Lowrie J. F.; Sherman W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graphics Modell. 2010, 29, 157–170. 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Stumpfe D.; Bajorath J. Similarity searching. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 260–282. 10.1002/wcms.23. [DOI] [Google Scholar]
- Cereto-Massagué A.; Ojeda M. J.; Valls C.; Mulero M.; Garcia-Vallvé S.; Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. 10.1016/j.ymeth.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discovery 2016, 11, 137–148. 10.1517/17460441.2016.1117070. [DOI] [PubMed] [Google Scholar]
- Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discovery Today 2006, 11, 1046–1053. 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Todeschini R.; Consonni V.; Xiang H.; Holliday J.; Buscema M.; Willett P. Similarity Coefficients for Binary Chemoinformatics Data: Overview and Extended Comparison Using Simulated and Real Data Sets. J. Chem. Inf. Model. 2012, 52, 2884–2901. 10.1021/ci300261r. [DOI] [PubMed] [Google Scholar]
- Bajusz D.; Rácz A.; Héberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. J. Cheminform. 2015, 7, 20. 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rácz A.; Bajusz D.; Héberger K. Life beyond the Tanimoto coefficient: similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 48. 10.1186/s13321-018-0302-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajiness M. S. Dissimilarity-based compound selection techniques. Perspect. Drug Discovery Des. 1996, 7–8, 65–84. 10.1007/BF03380182. [DOI] [Google Scholar]
- Dixon S. L.; Koehler R. T. The Hidden Component of Size in Two-Dimensional Fragment Descriptors: Side Effects on Sampling in Bioactive Libraries. J. Med. Chem. 1999, 42, 2887–2900. 10.1021/jm980708c. [DOI] [PubMed] [Google Scholar]
- Holliday J. D.; Salim N.; Whittle M.; Willett P. Analysis and Display of the Size Dependence of Chemical Similarity Coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. 10.1021/ci034001x. [DOI] [PubMed] [Google Scholar]
- Giaever G.; Chu A. M.; Ni L.; Connelly C.; Riles L.; Véronneau S.; Dow S.; Lucau-Danila A.; Anderson K.; André B.; Arkin A. P.; Astromoff A.; El Bakkoury M.; Bangham R.; Benito R.; Brachat S.; Campanaro S.; Curtiss M.; Davis K.; Deutschbauer A.; Entian K.-D.; Flaherty P.; Foury F.; Garfinkel D. J.; Gerstein M.; Gotte D.; Güldener U.; Hegemann J. H.; Hempel S.; Herman Z.; Jaramillo D. F.; Kelly D. E.; Kelly S. L.; Kötter P.; LaBonte D.; Lamb D. C.; Lan N.; Liang H.; Liao H.; Liu L.; Luo C.; Lussier M.; Mao R.; Menard P.; Ooi S. L.; Revuelta J. L.; Roberts C. J.; Rose M.; Ross-Macdonald P.; Scherens B.; Schimmack G.; Shafer B.; Shoemaker D. D.; Sookhai-Mahadeo S.; Storms R. K.; Strathern J. N.; Valle G.; Voet M.; Volckaert G.; Wang C.-y.; Ward T. R.; Wilhelmy J.; Winzeler E. A.; Yang Y.; Yen G.; Youngman E.; Yu K.; Bussey H.; Boeke J. D.; Snyder M.; Philippsen P.; Davis R. W.; Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Giaever G.; Flaherty P.; Kumm J.; Proctor M.; Nislow C.; Jaramillo D. F.; Chu A. M.; Jordan M. I.; Arkin A. P.; Davis R. W. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 793–798. 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons A. B.; Lopez A.; Givoni I. E.; Williams D. E.; Gray C. A.; Porter J.; Chua G.; Sopko R.; Brost R. L.; Ho C.-H.; Wang J.; Ketela T.; Brenner C.; Brill J. A.; Fernandez G. E.; Lorenz T. C.; Payne G. S.; Ishihara S.; Ohya Y.; Andrews B.; Hughes T. R.; Frey B. J.; Graham T. R.; Andersen R. J.; Boone C. Exploring the Mode-of-Action of Bioactive Compounds by Chemical-Genetic Profiling in Yeast. Cell 2006, 126, 611–625. 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Costanzo M.; Baryshnikova A.; Bellay J.; Kim Y.; Spear E. D.; Sevier C. S.; Ding H.; Koh J. L. Y.; Toufighi K.; Mostafavi S.; Prinz J.; St Onge R. P.; VanderSluis B.; Makhnevych T.; Vizeacoumar F. J.; Alizadeh S.; Bahr S.; Brost R. L.; Chen Y.; Cokol M.; Deshpande R.; Li Z.; Lin Z.-Y.; Liang W.; Marback M.; Paw J.; San Luis B.-J.; Shuteriqi E.; Tong A. H. Y.; van Dyk N.; Wallace I. M.; Whitney J. A.; Weirauch M. T.; Zhong G.; Zhu H.; Houry W. A.; Brudno M.; Ragibizadeh S.; Papp B.; Pal C.; Roth F. P.; Giaever G.; Nislow C.; Troyanskaya O. G.; Bussey H.; Bader G. D.; Gingras A.-C.; Morris Q. D.; Kim P. M.; Kaiser C. A.; Myers C. L.; Andrews B. J.; Boone C. The Genetic Landscape of a Cell. Science 2010, 327, 425–431. 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M.; VanderSluis B.; Koch E. N.; Baryshnikova A.; Pons C.; Tan G.; Wang W.; Usaj M.; Hanchard J.; Lee S. D.; Pelechano V.; Styles E. B.; Billmann M.; van Leeuwen J.; van Dyk N.; Lin Z.-Y.; Kuzmin E.; Nelson J.; Piotrowski J. S.; Srikumar T.; Bahr S.; Chen Y.; Deshpande R.; Kurat C. F.; Li S. C.; Li Z.; Usaj M. M.; Okada H.; Pascoe N.; San Luis B.-J.; Sharifpoor S.; Shuteriqi E.; Simpkins S. W.; Snider J.; Suresh H. G.; Tan Y.; Zhu H.; Malod-Dognin N.; Janjic V.; Przulj N.; Troyanskaya O. G.; Stagljar I.; Xia T.; Ohya Y.; Gingras A.-C.; Raught B.; Boutros M.; Steinmetz L. M.; Moore C. L.; Rosebrock A. P.; Caudy A. A.; Myers C. L.; Andrews B.; Boone C. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, aaf1420. 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons A. B.; Brost R. L.; Ding H.; Li Z.; Zhang C.; Sheikh B.; Brown G. W.; Kane P. M.; Hughes T. R.; Boone C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 2004, 22, 62–69. 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- Piotrowski J. S.; Li S. C.; Deshpande R.; Simpkins S. W.; Nelson J.; Yashiroda Y.; Barber J. M.; Safizadeh H.; Wilson E.; Okada H.; Gebre A. A.; Kubo K.; Torres N. P.; LeBlanc M. A.; Andrusiak K.; Okamoto R.; Yoshimura M.; DeRango-Adem E.; van Leeuwen J.; Shirahige K.; Baryshnikova A.; Brown G. W.; Hirano H.; Costanzo M.; Andrews B.; Ohya Y.; Osada H.; Yoshida M.; Myers C. L.; Boone C. Functional annotation of chemical libraries across diverse biological processes. Nat. Chem. Biol. 2017, 13, 982–993. 10.1038/nchembio.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hert J.; Willett P.; Wilton D. J.; Acklin P.; Azzaoui K.; Jacoby E.; Schuffenhauer A. New Methods for Ligand-Based Virtual Screening: Use of Data Fusion and Machine Learning to Enhance the Effectiveness of Similarity Searching. J. Chem. Inf. Model. 2006, 46, 462–470. 10.1021/ci050348j. [DOI] [PubMed] [Google Scholar]
- Lo Y.-C.; Rensi S. E.; Torng W.; Altman R. B. Machine learning in chemoinformatics and drug discovery. Drug Discovery Today 2018, 23, 1538–1546. 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamathevan J.; Clark D.; Czodrowski P.; Dunham I.; Ferran E.; Lee G.; Li B.; Madabhushi A.; Shah P.; Spitzer M.; Zhao S. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discovery 2019, 18, 463–477. 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Puhl A. C.; Zorn K. M.; Lane T. R.; Russo D. P.; Klein J. J.; Hickey A. J.; Clark A. M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019, 18, 435–441. 10.1038/s41563-019-0338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; Yang K.; Swanson K.; Jin W.; Cubillos-Ruiz A.; Donghia N. M.; MacNair C. R.; French S.; Carfrae L. A.; Bloom-Ackermann Z.; Tran V. M.; Chiappino-Pepe A.; Badran A. H.; Andrews I. W.; Chory E. J.; Church G. M.; Brown E. D.; Jaakkola T. S.; Barzilay R.; Collins J. J. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C.; Vapnik V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. 10.1007/BF00994018. [DOI] [Google Scholar]
- Vapnik V. N.The Nature of Statistical Learning Theory, 2nd ed.; Springer-Verlag, 2000. [Google Scholar]
- Hinselmann G.; Rosenbaum L.; Jahn A.; Fechner N.; Zell A. jCompoundMapper: An open source Java library and command-line tool for chemical fingerprints. J. Cheminform. 2011, 3, 3. 10.1186/1758-2946-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum G.RDKit: Open-Source Cheminformatics, 2006. http://www.rdkit.org.
- Carhart R. E.; Smith D. H.; Venkataraghavan R. Atom pairs as molecular features in structure-activity studies: definition and applications. J. Chem. Inf. Model. 1985, 25, 64–73. 10.1021/ci00046a002. [DOI] [Google Scholar]
- Borgwardt K. M.; Kriegel H.. Shortest-Path Kernels on Graphs. Fifth IEEE International Conference on Data Mining (ICDM’05); IEEE, 2005; pp 74–81.
- Ralaivola L.; Swamidass S. J.; Saigo H.; Baldi P. Graph kernels for chemical informatics. Neural Networks 2005, 18, 1093–1110. 10.1016/j.neunet.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Bender A.; Mussa H. Y.; Glen R. C.; Reiling S. Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. 10.1021/ci0498719. [DOI] [PubMed] [Google Scholar]
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Neidhart W.; Giller T.; Schmid G. “Scaffold-Hopping” by Topological Pharmacophore Search: A Contribution to Virtual Screening. Angew. Chem., Int. Ed. 1999, 38, 2894–2896. . [DOI] [PubMed] [Google Scholar]
- Daylight Chemical Information Systems Inc . http://www.daylight.com (accessed January 17, 2021).
- Raymond J. W.; Willett P. Effectiveness of graph-based and fingerprint-based similarity measures for virtual screening of 2D chemical structure databases. J. Comput.-Aided Mol. Des. 2002, 16, 59–71. 10.1023/A:1016387816342. [DOI] [PubMed] [Google Scholar]
- Braun-Blanquet J.Plant Sociology. The Study of Plant Communities, 1st ed.; McGraw-Hill Book Co., Inc., 1932. [Google Scholar]
- Hayek L.-A. C.Analysis of Amphibian Biodiversity Data. In Measuring and Monitoring Biological Diversity: Standard Methods for Amphibians, 1st ed.; Heyer R. W., Donnelly M. A., McDiarmid R. W., Hayek L.-A. C., Foster M. S., Eds.; Smithsonian Institution Press, 1994; pp 207−269. [Google Scholar]
- Driver H. E.; Kroeber A. L.. Quantitative Expression of Cultural Relationship; Berkeley University Calif Press, 1932; Vol. 31, pp 211–256. [Google Scholar]
- Ochiai A. Zoogeographical Studies on the Soleoid Fishes Found in Japan and its Neighhouring Regions-II. Bull. Jpn. Soc. Sci. Fish. 1957, 22, 526–530. 10.2331/suisan.22.526. [DOI] [Google Scholar]
- Dice L. R. Measures of the Amount of Ecologic Association Between Species. Ecology 1945, 26, 297–302. 10.2307/1932409. [DOI] [Google Scholar]
- Sorensen T. A. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content and its application to analyzes of the vegetation on Danish commons. K. Dan. Vidensk. Selsk., Biol. Skr. 1948, 5, 1–34. [Google Scholar]
- Kulczynski S. Die Pflanzenassociationen der Pienenen. Bull. Intern. Acad. Pol. Sci. Lett. Cl. Sci. Math. Nat., B (Sci. Nat.) 1927, 2, 57–203. [Google Scholar]
- McConnaughey B. H. The Determination and Analysis of Plankton Communities. Mar. Res. Indones. (Penelitian Laut Di Indonesia) 1964, Spec No, 1–40. [Google Scholar]
- Russell P. F.; Rao T. R. On Habitat and Association of Species of Anopheline Larvae in South-Eastern Madras. J. Malar. Inst. India 1940, 3, 153–178. [Google Scholar]
- Simpson G. G. Mammals and the nature of continents. Am. J. Sci. 1943, 241, 1–31. 10.2475/ajs.241.1.1. [DOI] [Google Scholar]
- Sokal R. R.; Sneath P. H. A.. Principles of Numerical Taxonomy; San Fr W H Friedman Co., 1963. [Google Scholar]
- Jaccard P. Étude comparative de la distribution florale dans une portion des Alpes et des Jura. Bull. Soc. Vaudoise Sci. Nat. 1901, 37, 547–579. 10.5169/seals-266450. [DOI] [Google Scholar]
- Tanimoto T. T.Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation, 1958. [Google Scholar]
- Tulloss R. E.Assessment of Similarity Indices for Undesirable Properties and a New Tripartite Similarity Index Based on Cost Functions. In Mycology in Sustainable Development: Expanding Concepts, Vanishing Borders; Palm M. E., Chapela I. H., Eds.; Parkway Publishers, Inc., 1997; pp 122–143. [Google Scholar]
- Tversky A. Features of similarity. Psychol. Rev. 1977, 84, 327–352. 10.1037/0033-295X.84.4.327. [DOI] [Google Scholar]
- Morgan H. L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. 10.1021/c160017a018. [DOI] [Google Scholar]
- Glem R. C.; Bender A.; Arnby C. H.; Carlsson L.; Boyer S.; Smith J. Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME. Idrugs 2006, 9, 199–204. [PubMed] [Google Scholar]
- Riniker S.; Landrum G. A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminform. 2013, 5, 26. 10.1186/1758-2946-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Sayle R. A. Comparing structural fingerprints using a literature-based similarity benchmark. J. Cheminform. 2016, 8, 36. 10.1186/s13321-016-0148-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Héberger K. Sum of ranking differences compares methods or models fairly. Trac. Trends Anal. Chem. 2010, 29, 101–109. 10.1016/j.trac.2009.09.009. [DOI] [Google Scholar]
- Kollár-Hunek K.; Héberger K. Method and model comparison by sum of ranking differences in cases of repeated observations (ties). Chemom. Intell. Lab. Syst. 2013, 127, 139–146. 10.1016/j.chemolab.2013.06.007. [DOI] [Google Scholar]
- Héberger K.; Kollár-Hunek K. Sum of ranking differences for method discrimination and its validation: comparison of ranks with random numbers. J. Chemom. 2011, 25, 151–158. 10.1002/cem.1320. [DOI] [Google Scholar]
- Barshan E.; Ghodsi A.; Azimifar Z.; Zolghadri Jahromi M. Supervised principal component analysis: Visualization, classification and regression on subspaces and submanifolds. Pattern Recogn. 2011, 44, 1357–1371. 10.1016/j.patcog.2010.12.015. [DOI] [Google Scholar]
- Efron B.; Tibshirani R. Improvements on Cross-Validation: The 632+ Bootstrap Method. J. Am. Stat. Assoc. 1997, 92, 548–560. 10.1080/01621459.1997.10474007. [DOI] [Google Scholar]
- Deshpande R.; VanderSluis B.; Myers C. L. Comparison of Profile Similarity Measures for Genetic Interaction Networks. PLoS One 2013, 8, e68664 10.1371/journal.pone.0068664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-C.; Lin C.-J. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1. 10.1145/1961189.1961199. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.