

Abstract
Importance of the field:
The Hedgehog (Hh) pathway is required during many developmental events; in adults the Hedgehog pathway is involved in the maintenance of several stem cell niches. It is therefore not surprising that aberrantly regulated Hh pathway activity can cause birth defects in the developing organism, as well as neoplastic disease later in life.
Areas covered in this review:
As a consequence of the involvement in pathogenesis, the Hh pathway components are subject to an intense scrutiny as potential targets for therapeutic agents. We aim to provide an overview of the biology of the Hh proteins and the cellular response, in conjunction with potential therapeutic interventions.
What the reader will gain:
Specifically, we focus on the recently discovered non-cell-autonomous Shh signaling used by tumors and the implications of this for the design of treatment strategies. This should provide the reader with up-to-date knowledge on the role of the Hh pathway in tumor progression and the options to treat these malignancies.
Take home message:
An important concept that we advocate in this review is the need to recognize the need to target both the stromal and the tumor compartment in malignancies that rely on paracrine Shh signaling.
Keywords: cancer, Disp1, Hedgehog, Shh, Smo, tumor biology
1. Background
As a developing organism grows more complex, gradients of signaling molecules shape many features by exposing cells in these gradients to distinct concentrations of these signals, termed morphogens. Members of four families of signaling molecules constitute the vast majority of morphogens; the fibroblast growth factor (FGF), TGF, (including the bone morphogenetic proteins (BMPs) and Activins), Wnt, and Hedgehog (Hh) families [1].
The Hh family is relatively small, with one ligand present in flies, and three in amniotes; Sonic-, Indian- and Desert Hedgehog. Unlike the many families of morphogens, all Hh ligands signal via the same receptor complex. Removal of the G-protein coupled receptor (GPCR) Smoothened (Smo) prevents signaling by all Hh ligands, and Smo−/− embryos display a severe phenotype characterized by the absence of bilateral asymmetry, digit patterning and ventral neural tissue, consistent with a role for Hh signaling at these sites [2]. Several birth defects concerning digit number and identity, as well as some debilitating birth defects involving incorrect neural development can be ascribed to insufficient Sonic Hedgehog (Shh) signaling.
Hh signaling remains important into adulthood, where it is involved at many locations subject to continual tissue renewal, such as the skin and the lining of the digestive tract [3,4]. Not surprisingly, these sites are particularly sensitive to tumor formation due to inappropriately upregulated Shh signaling. Although many key players involved in the regulation of the Hh response are identified, many details remain unresolved, and are in fact subject to much debate, some of which we will discuss below.
2. Hedgehog signal transduction
The biology of the Hh proteins is unique and remarkably complex. The ligands are the only known sterolated proteins in the animal kingdom. The addition of the cholesterol moiety is linked to an autocatalytic cleavage event mediated by the carboxy-terminal part of the precursor protein [5]. Subsequently yet another lipophilic group is added to the amino-terminal fragment, resulting in a mature protein that is both sterolated and palmitoylated (Figure 1) [6]. These additions obviously render the protein hydrophobic, and as a consequence obligatorily membrane-bound [7]. This in turn poses a significant challenge for the distribution of Hh in a gradient. As a morphogen, Shh is able to travel at least several cell diameters from its sites of synthesis [8]. The diffusion capacity of Shh bears relevance not only in developmental processes, but also in the progression of tumors that rely on Shh for their growth. Consistent with its nature as a membrane-bound molecule, a dedicated mechanism exists to mediate the distribution of Hh protein through the hydrophilic inter-cellular environment. In particular, the membrane protein Dispatched-1 (Disp1) [9], is required at the sites of Hh synthesis for proper distribution of the Hh ligands. Disp1 is thought to mediate the formation of Shh multimers in which the lipid attachments are sequestered in the interior of the complex, resulting in a soluble particle that bound both by lipophilic interactions via its lipid anchors as well as Shh protein–protein interactions [10]. Consistent with this model, Shh has been found to be associated with nodal vesicular particles that consist of membrane fragments [11] and lipoprotein particles [12]. Nevertheless, the precise nature and form of Shh moving from cell to cell through a tissue remains unclear.
Figure 1. Hedgehog production, transport and signaling.
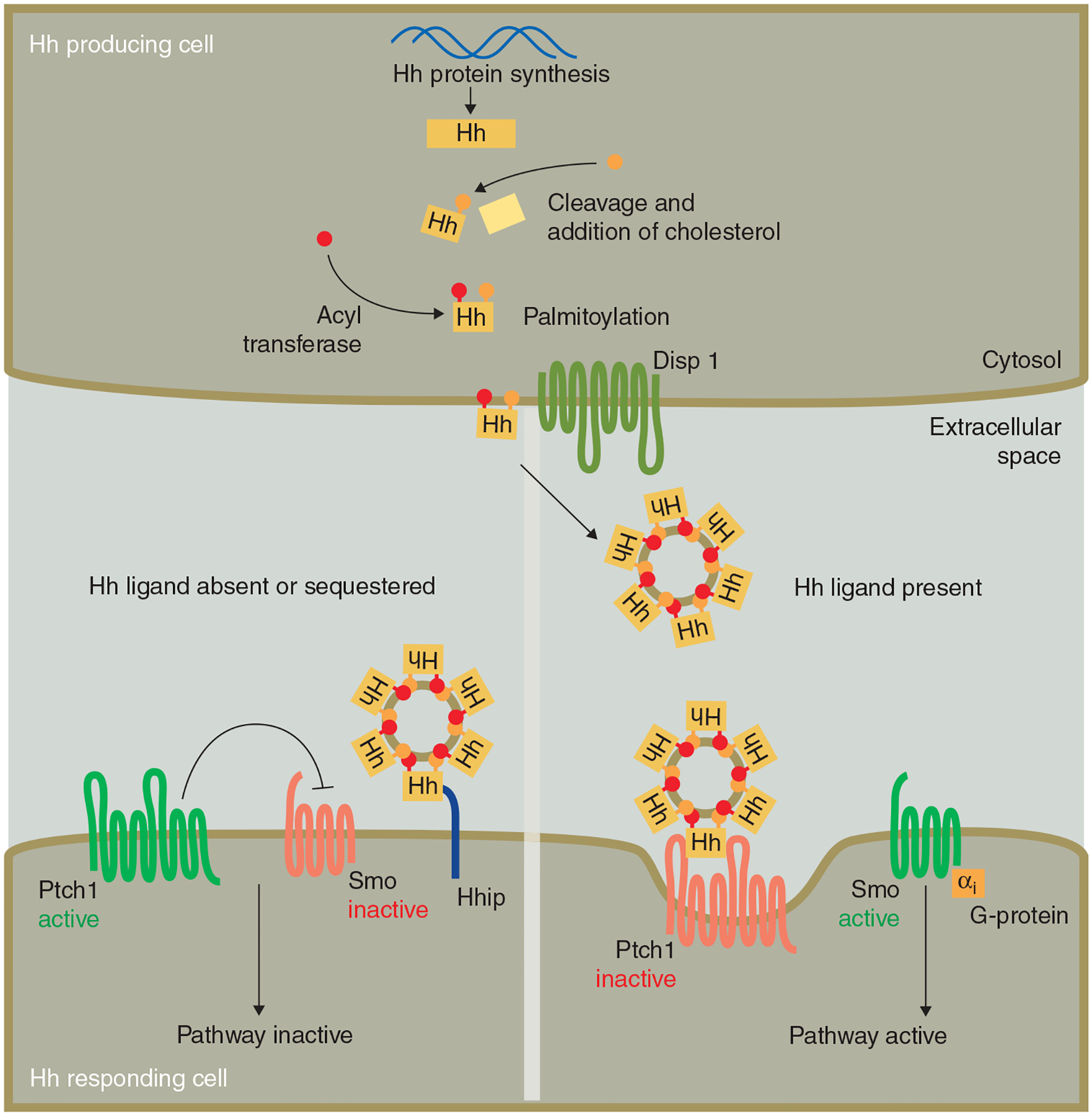
Following translation, the full length Hh protein autocatalytically cleaves and a cholesterol moiety is added in the process. A palmitoyl group is added by a dedicated acyl transferase. Secretion into the extracellular space in the form of multimers is mediated by the action of Disp1. In the absence of Hh ligand, or when Hh is sequestered by an inhibitory protein like Hhip, Ptch1 represses Smo and the downstream pathway is inactive. In the presence of Hh that is free to bind to Ptch1, the repression of Ptch1 on Smo is released and Smo activates downstream pathway components through G-proteins.
Another unusual feature of the Hh pathway is the receptor pair that relays the Hh signal from the cell surface to the interior. Unlike most pathways, in which one receptor complex transduces a signal into the cell to the downstream pathway components, the Hedgehog pathway is more intricate and uses an ‘on’ receptor (Smoothened (Smo)) and an ‘off’ receptor (Patched1 (Ptch1)) to manage its activation status (Figure 1). In the absence of Hh ligand, or when the ligand is sequestered by other binding partners (such as the Hedgehog interacting protein Hhip1 [13]), the pathway is kept off through the action of Ptch1 (Figure 1) [14]. Ptch1 inhibits Smo, keeping it and its downstream pathway components in an inactive state. Upon Hh ligand binding to Ptch1 the resulting internalization of this complex traffics it to late endosomes and lysosomes [15,16]. This trafficking event is associated with a redistribution of Smo allowing it to be activated. The precise mechanism by which Ptch, in the absence of (S)hh, inhibits the activity of Smo is unknown, but it presumably involves the secretion or translocation of small lipophilic molecules by Ptch1 that bind to Smo, thereby inhibiting its activity [17]. In flies, some of these small inhibitory molecules can be carried on lipoprotein particles that also could carry Hh [18]. Regulation of Ptch by a small lipid is supported by the observations that Ptch1 inhibits Smo at sub-stoichiometrical concentrations, indicating a catalytic mechanism [19]. Also, the homology between Ptch1 and the Niemann-Pick C1 (NPC1 [20]) protein, which is involved in cholesterol homeostasis in humans, and various prokaryotic and eukaryotic transporter molecules of the resistance, nodulation and division (RND) proton-driven transporters [21] suggest that a critical aspect of Ptch1 function involves the proton-driven translocation of a substrate inhibitory to Smo. The structure of the natural Smo inhibitor cyclopamine [22], combined with the observation that the genetic loss of 7-dehydrocholesterol (7-DHC) reductase activity causes attenuation of Smo [23], leads to the idea that the substrate for Ptch1 is a sterol-like molecule. This could be a 7-DHC derivative such as (pro-)vitamin D3, which is a substrate for Ptch1 pump activity and which binds to, and inhibits Smo, potentially playing a central role in the Shh response [17]. 7-DHC derivatives are expected to localize in membranes, and consistent with this, the function of Ptch1 has previously been shown to be strictly cell autonomous [24]. Nevertheless, overexpression of Ptch1 in vitro results in the accumulation of Smo inhibitors in the supernatant, lending further support to the idea that Ptch1 redistributes a Smo inhibitor.
Smo is thought to be a member of the large family of GPCRs, and it associates with a Gi-protein to achieve downstream signaling [25,26]. However, the downstream elements do not appear to be members of a classical GPCR response [27–30], but instead the complex activated by Smo consists of, among others, the kinase Fused (Fu), Suppressor of Fused (SuFu), which is a negative regulator of Hh signaling, and Gli zinc finger transcription factors [31–34]. This complex has more constituents, in particular those that mediate the association with the microtubule skeleton, a function mediated in flies by Cos2 [32,33]. Functional analogs of Cos2 and their roles in the Hh response in vertebrates will be discussed in more detail below.
There are three Gli homologs in vertebrates (Gli1, −2, and −3), and they all differ in the way they modulate Hh-induced gene expression. Gli2 and Gli3 are subject to a proteolytic event that results in the generation of a highly effective inhibitor of the Hh response. Binding of Hh to Ptc, and the subsequent activation of Smo inhibits this proteolytic event, and in particular the full length Gli2 now acts as an activator of Hh responsive genes (see [29] for an exhaustive overview).
The flow of information from Smo to the Gli proteins, and the way in which the proteolysis is regulated remains unclear, although multiple phosphorylation events appear to be involved, and principal kinases in these events are GSK-3β [35–38], PKA and CK1 [39,40], priming (in conjunction with β-TrCP, a ubiquitin ligase [41]) Glis for proteolysis.
In flies, Cos2 is a required for the Hh response. Cos2, a microtubule binding protein is thought to spatially coordinate the complex containing Fu, SuFu and Ci [32,33]. Functional homologs of Cos2 in vertebrates appear to be monomeric versions of the Kif kinesin engines [42]. As a consequence of their binding to microtubules, it is no surprise that they mediate the accumulation of Hh pathway components to the primary cilium, a structure present on most cells that has a microtubule core [43,44]. Even more striking is the observation that Ptch1 and small molecules regulate the position of Smo in the cilium, suggesting that the activation of the Gli transcription factors also occurs in this structure. This notion is backed up by a study by Kim et al. [45]. Mutations that abrogate formation of the cilium also affect Hh signaling capacity and this correlation has lead to the suggestion that the role of ciliary localization of Hh signaling components is a physiologically relevant phenomenon [46]. Not entirely unexpectedly, primary cilia have been found to be involved in tumorigenesis of malignancies that are known to rely on excessive Hh pathway activation [47,48].
Although the Gli transcription factors are directly responsible for all transcriptional activation induced by Hh signaling, the observation that Hh signaling is also involved in cell migration and growth cone guidance suggests that a transcription-independent, local Shh response is present in cells [49]. These events are ligand-dependent, and require Smo function, but rather than resulting in altered Gli processing, the leukotriene synthesis machinery is activated. This in turn regulates cell motility resulting in a migratory response [50,51]. Although these pathways undoubtedly hold great promise for the development of future therapeutics (this pathway could be involved in Hh-induced metastasis), very little is known about their exact functioning and actual relevance, and we will thus not elaborate on this branch of the Shh response.
3. Hedgehog in pathophysiology
Insufficient Hh pathway activity has been thought to underlie several congenital malformations. For instance, Smith-Lemli-Opitz syndrome (cause by an absence of 7-DHC reductase [17,23,52]), fetal alcohol syndrome, holoprosencephaly, and perhaps Down syndrome can be attributed to an insufficiently active Hh pathway [53–55]. On the other hand, a beneficial role for Hh pathway activation in the adult organism has been found in various ischemia models. In these models (hind limb- and myocardial ischemia), the Hh response was found to be upregulated and the addition of exogenous Shh aided in salvage of damaged tissue through activation of angiogenic and anti-apoptotic pathways [56–58].
However, the intervention options that this review will address focus on those circumstances where the Shh response is activated inappropriately, which is a frequent cause in the formation of many common and often particularly heinous tumors. The inappropriate activation of the Shh response that causes tumor growth is either caused by excessive Shh ligand production, or defective repression of Smo by mutations in either Ptch1 or Smo itself, or downstream signaling components that result in a defective pathway regulation.
The role of Ptch1 as the inhibitor of Smo provides the explanation why it is a tumor suppressor protein. Ptch1 heterozygotes suffer from nevus basal cell carcinoma syndrome (NBCCS), which is characterized by the frequent occurrence of basal cell carcinomas (BCCs), as well as medulloblastomas and rhabdomyosarcomas [59]. Sporadic forms of such tumors are often characterized by the loss of Ptch1 as well. Activation of the pathway by activating mutations in Smo rendering it insensitive to inhibition by Ptch1 is also commonly associated with BCCs [60]. Other pathway components are not known to function in tumor progression, but mutations in SuFu have been detected in medulloblastomas [61]. Tumors arising in the derivatives of the foregut, such as esophageal, gastric, hepatic and in particular pancreatic cancers as well as prostate cancer metastases often have an inappropriate activation of Shh ligand expression [62–64]. These two distinct ways of activating the Hh response, cell autonomously via the loss of Ptch1 or the activation of its downstream actors on one hand, and the excessive production of the Shh ligand on the other hand underlies an essential difference in these tumors. The two fundamentally different tumor types that involve the activation of the Hh response are thus; i) tumors with a cell autonomous, or autocrine, activation of the pathway, which is usually achieved by Ptch1 inactivation [65–67], or activating mutations of Smo [60], or ii) tumors in which the Shh ligand is inappropriately expressed. Interestingly, this aberrantly expressed Shh signals to the normal surrounding cells, which respond by the synthesis of distinct reciprocal signals that sustain the growth of the Shh producing cells [68,69], together forming a polyclonal tumor consisting of mutated Shh-expressing cells and a non-mutated stromal component.
4. Targets for therapeutic intervention in tumors with cell autonomous activation of the Shh response
Tumors arising due to a cell autonomous activation of the Hh pathway critically rely on the activation of the Ptch1/Smo receptor pair by mutations in either protein. As is the case for many GPCRs, Smo has turned out to be an eminently druggable target. In fact, a highly efficient Smo inhibitor, cyclopamine, is present in the common range plant Veratrum californicum [70]. Many derivatives of cyclopamine have been developed that have shown improved efficacy, and large compound screens have identified other potent Smo inhibitors [71].
Nevertheless, no Smo inhibitors are yet in clinical use, although some are in early phases of clinical trials [72]. In two recently published papers, a remarkable efficacy was shown for a Smo inhibitor developed by Genentech, GDC-0449. In patients with locally advanced or metastatic medulloblastoma (a type of tumor well known to depend on mutations in the receptors for the Hh pathway), treatment with the inhibitor GDC-0449 was found to have strong anti-tumor activity as indicated by a high percentage of patients with a partial response or stable disease [73]. In the same issue, a case report was published describing a patient with metastatic medulloblastoma. Treatment of this patient with GDC-0449 resulted in a spectacular but temporary reduction in metastases and morbidity [74]. The relapse seemed to be caused by a mutation in Smo that was found in tumor cells after treatment, and rendered Smo insensitive to GDC-0449 [75]. The development of tumor resistance through mutations in pathway components in various pathways is a common event, and explains why combination therapies are much more effective in treating tumors. Also, as the stroma would be less likely to accumulate mutations that confer resistance, targeting this compartment should prove effective.
Several recent and exhaustive reviews have been written about these Smo inhibitors [76,77], so we will not elaborate on those. These inhibitors are likely to become clinically relevant, in particular in those tumors with a cell-autonomous activation of the Shh response. However, they offer a possible remedy to only one aspect of the signaling thought to support the polyclonal, Shh expressing tumors.
Other candidates for therapeutic intervention in tumors dependent on Hh pathway activation would be the Gli transcription factors. As the Gli proteins are the most downstream effectors of the Hh pathway, their inhibition should provide the most effective and arguably specific therapeutic option. There are however two problems with this strategy; the first is that there are only few Gli-inhibitory molecules available, and these have been described fairly recently, casting doubt on the clinical use of a Gli-inhibitor anytime soon [78]. Second, it is potentially hard to devise drugs specifically targeting the transcription activator activity of Glis, while sparing the highly efficient inhibitory activities of Gli3 [79,80].
5. Targets for therapeutic intervention in paracrine signaling tumors
5.1. Introduction
Tumors arising due to excessive Shh ligand production are characterized by the extensive infiltration of non-tumorigenic stromal tissue responding to the Shh produced. This stromal compartment is critical for the maintenance of these tumors. The Shh-expressing cancer cells support the infiltration and growth of stromal cells, which as a consequence release signals that support the growth of the Shh-expressing cells. Such tumors thus consist of two mutually dependent groups of cells supporting each other’s growth via distinct factors, one of which is Shh. This of course has significant consequences for potential therapeutic interventions. Whereas many potential targets are known in cancer cells that rely on cell autonomous activation of the Hh pathway, this is not the case in tumors that rely on paracrine signaling. It is likely that not only the Shh response in the stromal cells, but also the reciprocal signal needs to be inhibited for successful inhibition of tumor growth. The relatively disappointing results of the Smo inhibitors might be explained to an extent by the fact that these inhibitors are not expected to act on the mutated, Shh-producing cells, but instead only on the stromal cells. It is possible that the expected reduction of the amount of reciprocal signal produced is insufficient to initiate apoptosis required for tumor shrinkage.
The exact nature of the reciprocal signal returning to the cancer cell compartment is unknown, and might actually vary, or be multiple signals, complicating the treatment of these tumors with designed, target-specific molecules. In a recent study by Shaw et al. a species-specific microarray approach was used in a xenograft model for prostate cancer to show that the Shh produced by the tumor cells elicits the activation of pathways usually associated with embryonic development [64].
We expect that blocking the action of the reciprocal signal, in addition to blocking the Shh response, will be much more effective in treating Shh-induced tumors. The resistance to GDC-0449 observed in the above mentioned case of medulloblastoma emphasizes the requirement for combination therapy. This approach, however, complicates treatment, as the nature of the reciprocal signal is not yet known. On the other hand, targeting at least two paracrine signals opens several options to prevent the maturation and exchange of these factors. Here we focus on some of the events required for productive reciprocal signaling and discuss some attractive targets for drug development.
5.2. Shh production and release into the extracellular space
Targeting the production, processing, distribution or stability of ligands in paracrine signaling tumors should provide specific intervention. The combination of lipophilic modifications on Shh is unique, and the resulting hydrophobic properties have a significant consequence on the distribution and the signaling efficacy of Shh. Although there are no therapeutic strategies that purposely address this possibility, either preventing the events resulting in the sterolation or palmitoylation of Shh could prove valuable in inhibiting its action. Both the sterolation and acylation are highly specific processes.
A possible way to confer specificity might be to specifically block protein sterolation. The sterolation of Shh is directly coupled to an autocatalytic cleavage event [81]. This cleavage is required for Shh activity and might, therefore, prove to be an excellent target for drug development. Similarly, it appears that a dedicated acyltransferase (Hhat) is required for the modification of Shh [82]. Nevertheless, there are several acyltransferases present in cells [83], making it uncertain if the desired specificity can easily be obtained through pharmacological means.
Another well-characterized molecule absolutely required for Shh secretion is Disp1, a member of the RND family of proton-driven pumps (Figure 2). The relative ease with which these membrane proteins can be targeted by small molecules makes Disp1 a potentially attractive target for therapeutic intervention (reviewed in [84]). The unusual post-translational modifications, in combination with the presence of molecules specifically dedicated to the secretion of Shh renders the maturation of Shh a promising, under-explored avenue to prevent paracrine Shh signaling.
Figure 2. Hedgehog secretion and endocytosis.
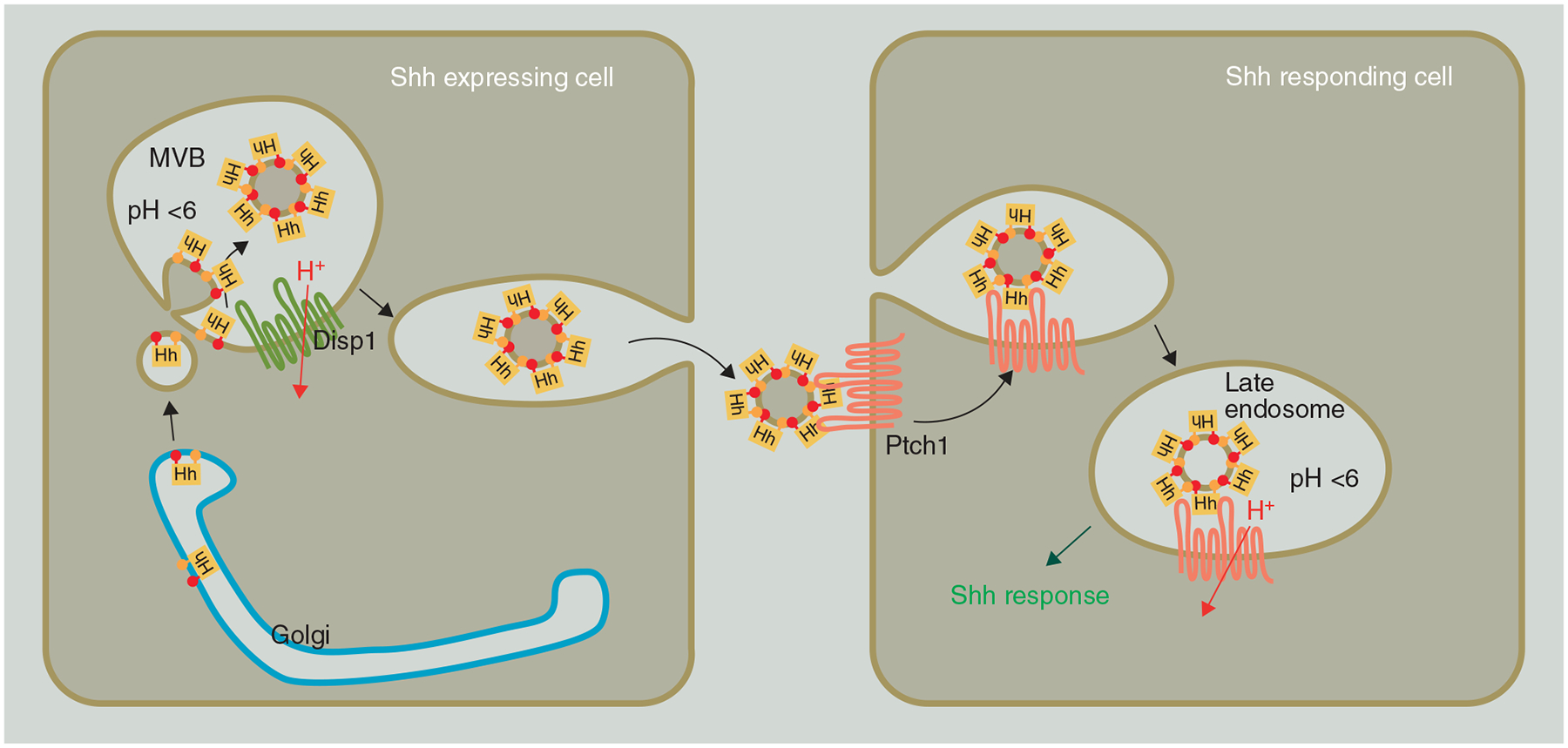
Fully processed Shh is trafficked from the Golgi into acidified vesicles where they are complexed into multimers by the proton driven pump activity of Disp1. The complexes are subsequently released in the extracellular space, and subsequently bound and internalized by the action of Ptch1 in adjacent cells. Trafficking of the complex to late endosomes exposes Ptch1 to a lower pH, which is required for the activation of the downstream pathway components.
The form of Shh that travels from cell to cell remains unresolved, and the topic of much debate. Several molecules are know to bind to Shh during this leg of its transport. There are both genetic and biochemical indications that Shh binds relatively tightly to the extracellular matrix, like most signaling molecules. This general role of the extracellular matrix makes it less attractive as a specific target for Shh signaling. More promising are two other molecules that bind Shh in the extracellular space, Hhip, as well as the Shh co-receptors Cdo and Boc.
5.3. Receptor functioning
Soon after the characterization of the Shh protein, blocking antibodies were developed, and these antibodies are possibly still the most efficient and specific inhibitors of the Shh response. This also demonstrates that during transport between cells, Shh is susceptible to inactivation by binding proteins that probably act by interfering with Shh binding to Ptch1. Under normal signaling conditions several molecules can be present in the extracellular space that bind to Shh and interfere with productive Ptch1 binding. These proteins are often part of regular negative feedback mechanisms, and point the way to the development of Shh-inactivating agents. Recently, a pharmacological method of intervening in excessive ligand production was described. It was demonstrated that robotnikinin, a small molecule specifically binding to Shh blocks its action on Ptch1 [85]. In addition to antibody or robotnikinin inhibition of Shh binding to Ptch1, one could envision other strategies to target this interaction. A suggestion for one such strategy comes from the naturally occurring Hh-inhibitory protein1 (Hhip1) that complexes with Shh thereby inhibiting the Shh response [13]. The crystal structure of Hhip and Shh has been resolved [86,87]. It appears that Shh binds via the same domain to Ptch1 and Hhip. This information should be very useful to design the minimally required protein motif for this Shh-inhibitory effect. In general, small molecules are easier to produce and deliver than proteins, so it is more likely that robotnikinin-like molecules will successfully find their way to clinical use rather than the Shh-blocking antibodies or proteins such as Hhip1 derivatives.
Ptch1 itself is at first glance not the ideal target to inhibit the Shh response, since the loss of Ptch1 causes a dramatic upregulation of the Shh response pathway [88]. However, an interesting Ptch1 mutant Ptch1ΔL2 (extracellular loop 2 deleted) does not bind Shh, but is still inhibits the activity of Smo [24]. It might therefore be an effective strategy to develop drugs that interfere with the Shh binding to extracellular loop 2.
An interesting idiosyncrasy in Shh signaling is the prominent role played by two related members of the RND family, Disp1 and Ptch1. Whereas Disp1 is required for the secretion of Shh, Ptch1 is required in the uptake of Shh. Whereas the loss of Disp1 prevents the paracrine Shh response, loss of Ptch1 cell-autonomously activates the Shh response. Based on their RND family memberships, it is predicted that both Disp1 and Ptch1 require a proton gradient for at least part of their functions. Importantly, preventing acidification of endosomal compartments with drugs like concanamycin A and chloroquine prevents the normal trafficking of Ptch1 into these compartments, and as a consequence, the Shh response [15]. This indicates that a proton gradient is required both in the Shh-producing cells and in the Shh-responding cells. Although it is currently unclear how the putative proton-driven pump activity of Disp1 is implicated in the secretion of Shh, Disp1 proton channel mutants are unable to secrete active Shh, and pharmacologically perturbing the proton gradient probably inhibits secretion of Shh by Disp1.
These two observations actually point to a possible Achilles’ heel in Shh signaling. The reliance on acidified vesicles for both the secretion and internalization of Shh might point to an underexplored target. V-ATPases, which acidify endocytic vesicles, are required at multiple steps in the Shh secretion as well as the response [15,89]. Since several specific inhibitors of V-ATPases are in clinical use, it would be interesting to test their ability to limit the growth of Shh-induced tumors.
6. Conclusion
The Hh signaling pathway, as well as its prominent role in tumor formation, is unusually complex, and from a therapeutic point of view, this might be blessing in disguise. The rather disappointing efficacy of Smo antagonists in inhibiting Shh-induced tumors might not be such a great surprise, given the mutual dependency of Shh-producing tumor cells and stromal cells that involve one or more alternative signaling pathways. In such Shh-induced tumors, which rely on non-cell-autonomous Shh signaling, there are several proteins that have dedicated and specific roles in maturation, secretion and internalization of Shh, and most of these proteins appear to be druggable targets. However, it is possible that both Shh signaling as well as reciprocal signaling by the unknown ligands need both to be inhibited to get the desired therapeutic effects. Such a requirement for a synergistic treatment strategy is similar to the way classical cytostatic compounds are now used in conjunction with other drugs for the treatment of tumors.
7. Expert opinion
7.1. Current state of the topic under discussion
Most of the research effort so far has focused on Smo inhibitors. The logic behind this is obvious, Smo is critically required for the Shh response, and very good natural and man-made Smo inhibitors were identified relatively long ago. The preponderance of Shh-induced polyclonal tumors that involve paracrine Shh signaling as well as unknown reciprocal signals, not only provides some explanation why the Smo inhibitors appear relatively ineffective, but also suggests the need for different research strategies, that are not solely focused on Smo inhibition.
7.2. Where the field is going in the next 5 – 10 years
Blocking the Shh response can efficiently be achieved with small-molecule Smo inhibitors, so the problem that needs to be addressed is why these inhibitors fail to effectively treat tumors that rely on the activation of Smo for their growth. Answering this question should involve the identification of the other inappropriately activated signals that support tumor growth in conjunction with Shh. If these signals activate a well-understood intracellular response, mediated by well-characterized ligands then combinatorial use of Smo inhibitors with the appropriate inhibitors blocking the reciprocal signal might finally yield clinically promising results. If the nature of the reciprocal, stromally-derived signal remains obscure or highly variable, then progress will be critically dependent on the development of methods to quickly map tumor transcriptomes and genomes.
7.3. How this will be achieved
The search to identify the cocktail of growth factors that in addition to Shh support the growth and metastasis of polyclonal tumors is ongoing, and likely to yield clear results. Since it is expected that these factors are true tumor-inducing proteins, it is quite likely that several small-molecule inhibiters affecting their response pathway already exist. In the best scenario, the reciprocal signals are constant, and the appropriate combination of small molecules will be effective. However, transcriptional profiling in paracrine signaling tumors has implicated Wnt pathway components, IGF-related signaling molecules, Notch, FGFs, Timp3 and CXCL14 as possible candidates to mediate the reciprocal signaling, complicating this approach [64,68]. For many of these signals, no specific inhibitors exist.
It is expected that a molecular analysis of each individual tumor needs to be performed to determine the optimal combination of small-molecule inhibitors to inhibit the responses elicited by combinations of these signaling molecules. A complicating factor is that similar combination of signaling molecules are likely to play important roles in the maintenance of stem cell niches, and the combinatorial use of small-molecule inhibitors might have significant side effects.
Article highlights.
Hedgehog proteins are important in embryonic development, but also for tissue maintenance in adults.
The signaling mechanisms of the Hedgehog pathway are complicated and poorly understood.
Excessive Hedgehog production and pathway activity is associated with tumorigenesis and progression.
Roughly two types of Hedgehog-dependent tumors exist; one in which the tumor cells respond to the ligand they make themselves, and one in which the tumor cells rely on the signals provided to them by the surrounding cells in response to the ligand.
Good inhibitors exist to target Hedgehog pathway activity on cells that rely on pathway activation themselves, but not all tumors can be efficiently targeted this way.
Identification of the reciprocal signals from the surrounding cells to the tumor should provide us with targets to more effectively combat certain Hedgehog-dependent malignancies such as pancreatic cancer.
This box summarizes key points contained in the article.
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript. MF Bijlsma is funded by KWF Dutch Cancer Society.
Bibliography
- 1.Hogan BL. Morphogenesis. Cell 1999;96:225–33 [DOI] [PubMed] [Google Scholar]
- 2.Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 2001;14:781–92 [PubMed] [Google Scholar]
- 3.Niemann C, Unden AB, Lyle S, et al. Indian hedgehog and beta-catenin signaling: role in the sebaceous lineage of normal and neoplastic mammalian epidermis. Proc Natl Acad Sci USA 2003;100(Suppl 1):11873–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Brink GR, Bleuming SA, Hardwick JC, et al. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet 2004;36:277–82 [DOI] [PubMed] [Google Scholar]
- 5.Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996;274:255–9 [DOI] [PubMed] [Google Scholar]
- 6.Pepinsky RB, Zeng C, Wen D, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem 1998;273:14037–45 [DOI] [PubMed] [Google Scholar]
- 7.Peters C, Wolf A, Wagner M, et al. The cholesterol membrane anchor of the Hedgehog protein confers stable membrane association to lipid-modified proteins. Proc Natl Acad Sci USA 2004;101:8531–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roelink H, Porter JA, Chiang C, et al. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 1995;81:445–55 [DOI] [PubMed] [Google Scholar]
- 9.Caspary T, Garcia-Garcia MJ, Huangfu D, et al. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr Biol 2002;12:1628–32 [DOI] [PubMed] [Google Scholar]
- 10.Goetz JA, Singh S, Suber LM, et al. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J Biol Chem 2006;281:4087–93 [DOI] [PubMed] [Google Scholar]
- 11.Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005;435:172–7 [DOI] [PubMed] [Google Scholar]
- 12.Panakova D, Sprong H, Marois E, et al. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 2005;435:58–65 [DOI] [PubMed] [Google Scholar]
- 13.Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999;397:617–21 [DOI] [PubMed] [Google Scholar]
- 14.Murone M, Rosenthal A, de Sauvage FJ. Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr Biol 1999;9:76–84 [DOI] [PubMed] [Google Scholar]
- 15.Incardona JP, Gruenberg J, Roelink H. Sonic hedgehog induces the segregation of patched and smoothened in endosomes. Curr Biol 2002;12:983–95 [DOI] [PubMed] [Google Scholar]
- 16.Gallet A, Therond PP. Temporal modulation of the Hedgehog morphogen gradient by a patched-dependent targeting to lysosomal compartment. Dev Biol 2005;277:51–62 [DOI] [PubMed] [Google Scholar]
- 17.Bijlsma MF, Spek CA, Zivkovic D, et al. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol 2006;4:e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khaliullina H, Panakova D, Eugster C, et al. Patched regulates smoothened trafficking using lipoprotein-derived lipids. Development 2009;136:4111–21 [DOI] [PubMed] [Google Scholar]
- 19.Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of smoothened. Nature 2002;418:892–7 [DOI] [PubMed] [Google Scholar]
- 20.Carstea ED, Morris JA, Coleman KG, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997;277:228–31 [DOI] [PubMed] [Google Scholar]
- 21.Tseng TT, Gratwick KS, Kollman J, et al. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol 1999;1:107–25 [PubMed] [Google Scholar]
- 22.Keeler RF. Teratogenic compounds of Veratrum californicum (Durand). VI. The structure of cyclopamine. Phytochemistry 1969;8:223–5 [Google Scholar]
- 23.Fitzky BU, Witsch Baumgartner M, Erdel M, et al. Mutations in the D7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc Natl Acad Sci USA 1998;95:8181–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briscoe J, Chen Y, Jessell TM, Struhl G. A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol cell 2001;7:1279–91 [DOI] [PubMed] [Google Scholar]
- 25.Meloni AR, Fralish GB, Kelly P, et al. Smoothened signal transduction is promoted by G protein-coupled receptor kinase 2. Mol Cell Biol 2006;26:7550–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Low WC, Wang C, Pan Y, et al. The decoupling of Smoothened from G alpha i proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev Biol 2008;321:188–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeCamp DL, Thompson TM, de Sauvage FJ, Lerner MR. Smoothened activates G alpha i-mediated signaling in frog melanophores. J Biol Chem 2000;275:26322–7 [DOI] [PubMed] [Google Scholar]
- 28.Riobo NA, Manning DR. Receptors coupled to heterotrimeric G proteins of the G12 family. Trends Pharmacol Sci 2005;26:146–54 [DOI] [PubMed] [Google Scholar]
- 29.Riobo NA, Manning DR. Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem J 2007;403:369–79 [DOI] [PubMed] [Google Scholar]
- 30.Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci USA 2006;103:12607–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nybakken KE, Turck CW, Robbins DJ, Bishop JM. Hedgehog-stimulated phosphorylation of the kinesin-related protein Costal2 is mediated by the serine/threonine kinase fused. J Biol Chem 2002;277:24638–47 [DOI] [PubMed] [Google Scholar]
- 32.Robbins DJ, Nybakken KE, Kobayashi R, et al. Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal2. Cell 1997;90:225–34 [DOI] [PubMed] [Google Scholar]
- 33.Sisson JC, Ho KS, Suyama K, Scott MP. Costal2, a novel kinesin-related protein in the Hedgehog signaling pathway. Cell 1997;90:235–45 [DOI] [PubMed] [Google Scholar]
- 34.Stegman MA, Vallance JE, Elangovan G, et al. Identification of a tetrameric hedgehog signaling complex. J Biol Chem 2000;275:21809–12 [DOI] [PubMed] [Google Scholar]
- 35.Dunaeva M, Michelson P, Kogerman P, Toftgard R. Characterization of the physical interaction of Gli proteins with SUFU proteins. J Biol Chem 2003;278:5116–22 [DOI] [PubMed] [Google Scholar]
- 36.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001;15:3059–87 [DOI] [PubMed] [Google Scholar]
- 37.Monnier V, Ho KS, Sanial M, et al. Hedgehog signal transduction proteins: contacts of the Fused kinase and Ci transcription factor with the kinesin-related protein Costal2. BMC Dev Biol 2002;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nybakken K, Perrimon N. Hedgehog signal transduction: recent findings. Curr Opin Genet Dev 2002;12:503–11 [DOI] [PubMed] [Google Scholar]
- 39.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003;116:1175–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia J, Amanai K, Wang G, et al. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 2002;416:548–52 [DOI] [PubMed] [Google Scholar]
- 41.Huntzicker EG, Estay IS, Zhen H, et al. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev 2006;20:276–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huangfu D, Liu A, Rakeman AS, et al. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003;426:83–7 [DOI] [PubMed] [Google Scholar]
- 43.Corbit KC, Aanstad P, Singla V, et al. Vertebrate Smoothened functions at the primary cilium. Nature 2005;437:1018–21 [DOI] [PubMed] [Google Scholar]
- 44.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007;317:372–6 [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci USA 2009;106:21666–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Zhao Y, Tong C, et al. Hedgehog-regulated Costal2-kinase complexes control phosphorylation and proteolytic processing of Cubitus interruptus. Dev Cell 2005;8:267–78 [DOI] [PubMed] [Google Scholar]
- 47.Han YG, Kim HJ, Dlugosz AA, et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 2009;15:1062–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong SY, Seol AD, So PL, et al. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med 2009;15:1055–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charron F, Stein E, Jeong J, et al. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 2003;113:11–23 [DOI] [PubMed] [Google Scholar]
- 50.Bijlsma MF, Borensztajn KS, Roelink H, et al. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal 2007;19:2596–604 [DOI] [PubMed] [Google Scholar]
- 51.Bijlsma MF, Peppelenbosch MP, Spek CA, Roelink H. Leukotriene synthesis is required for hedgehog-dependent neurite projection in neuralized embryoid bodies but not for motor neuron differentiation. Stem cells 2008;26:1138–45 [DOI] [PubMed] [Google Scholar]
- 52.Cooper MK, Wassif CA, Krakowiak PA, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet 2003;33:508–13 [DOI] [PubMed] [Google Scholar]
- 53.Ahlgren SC, Thakur V, Bronner-Fraser M. Sonic hedgehog rescues cranial neural crest from cell death induced by ethanol exposure. Proc Natl Acad Sci USA 2002;99:10476–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roessler E, Belloni E, Gaudenz K, et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 1996;14:357–60 [DOI] [PubMed] [Google Scholar]
- 55.Roper RJ, Baxter LL, Saran NG, et al. Defective cerebellar response to mitogenic Hedgehog signaling in Down syndrome mice. Proc Natl Acad Sci USA 2006;103:1452–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kusano KF, Pola R, Murayama T, et al. Sonic Hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med 2005;11:1197–204 [DOI] [PubMed] [Google Scholar]
- 57.Kusano KF, Allendoerfer KL, Munger W, et al. Sonic Hedgehog induces arteriogenesis in diabetic vasa nervorum and restores function in diabetic neuropathy. Arterioscler Thromb Vasc Biol 2004;24:2102–7 [DOI] [PubMed] [Google Scholar]
- 58.Pola R, Ling LE, Aprahamian TR, et al. Postnatal recapitulation of embryonic Hedgehog pathway in response to skeletal muscle ischemia. Circulation 2003;108:479–85 [DOI] [PubMed] [Google Scholar]
- 59.Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med 2004;6:530–9 [DOI] [PubMed] [Google Scholar]
- 60.Xie J, Murone M, Luoh SM, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature 1998;391:90–2 [DOI] [PubMed] [Google Scholar]
- 61.Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet 2002;31:306–10 [DOI] [PubMed] [Google Scholar]
- 62.Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003;425:846–51 [DOI] [PubMed] [Google Scholar]
- 63.Fan L, Pepicelli CV, Dibble CC, et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004;145:3961–70 [DOI] [PubMed] [Google Scholar]
- 64.Shaw A, Gipp J, Bushman W. The Sonic Hedgehog pathway stimulates prostate tumor growth by paracrine signaling and recapitulates embryonic gene expression in tumor myofibroblasts. Oncogene 2009;28:4480–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bailey EC, Milenkovic L, Scott MP, et al. Several PATCHED1 missense mutations display activity in patched1-deficient fibroblasts. J Biol Chem 2002;277:33632–40 [DOI] [PubMed] [Google Scholar]
- 66.Chiang C, Swan RZ, Grachtchouk M, et al. Essential role for Sonic Hedgehog during hair follicle morphogenesis. Dev Biol 1999;205:1–9 [DOI] [PubMed] [Google Scholar]
- 67.Goodrich LV, Jung D, Higgins KM, Scott MP. Overexpression of ptc1 inhibits induction of Shh target genes and prevents normal patterning in the neural tube. Dev Biol 1999;211:323–34 [DOI] [PubMed] [Google Scholar]
- 68.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for Hedgehog signalling in cancer. Nature 2008;455:406–10 [DOI] [PubMed] [Google Scholar]
- 69.Zhang J, Lipinski R, Shaw A, et al. Lack of demonstrable autocrine Hedgehog signaling in human prostate cancer cell lines. J Urol 2007;177:1179–85 [DOI] [PubMed] [Google Scholar]
- 70.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998;280:1603–7 [DOI] [PubMed] [Google Scholar]
- 71.Frank-Kamenetsky M, Zhang XM, Bottega S, et al. Small-molecule modulators of Hedgehog signaling: identification and characterization of smoothened agonists and antagonists. J Biol 2002;1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.GDC-0449 in treating patients with locally advanced or metastatic solid tumors. Bethesda, Maryland: ClinicalTrials.gov, 2010Available from: http://clinicaltrials.gov/ct2/show/NCT00607724 [Last accessed 28 May 2010] [Google Scholar]
- 73.Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the Hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009;361:1164–72 [DOI] [PubMed] [Google Scholar]
- 74.Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with Hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009;361:1173–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009;326:572–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mahindroo N, Punchihewa C, Fujii N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J Med Chem 2009;52:3829–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.King RW. Roughing up smoothened: chemical modulators of Hedgehog signaling. J Biol 2002;1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA 2007;104:8455–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Persson M, Stamataki D, te Welscher P, et al. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev 2002;16:2865–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meyer NP, Roelink H. The amino-terminal region of Gli3 antagonizes the Shh response and acts in dorsoventral fate specification in the developing spinal cord. Dev Biol 2003;257:343–55 [DOI] [PubMed] [Google Scholar]
- 81.Porter JA, Ekker SC, Park WJ, et al. Hedgehog patterning activity: role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 1996;86:21–34 [DOI] [PubMed] [Google Scholar]
- 82.Buglino JA, Resh MD. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J Biol Chem 2008;283:22076–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grand RJ, Smith KJ, Gallimore PH. Purification and characterisation of the protein encoded by the activated human N-ras gene and its membrane localisation. Oncogene 1987;1:305–14 [PubMed] [Google Scholar]
- 84.Lomovskaya O, Zgurskaya HI, Totrov M, Watkins WJ. Waltzing transporters and ‘the dance macabre’ between humans and bacteria. Nat Rev Drug Discov 2007;6:56–65 [DOI] [PubMed] [Google Scholar]
- 85.Stanton BZ, Peng LF, Maloof N, et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat Chem Biol 2009;5:154–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bishop B, Aricescu AR, Harlos K, et al. Structural insights into Hedgehog ligand sequestration by the human Hedgehog-interacting protein HHIP. Nat Struct Mol Biol 2009;16:698–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bosanac I, Maun HR, Scales SJ, et al. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat Struct Mol Biol 2009;16:691–7 [DOI] [PubMed] [Google Scholar]
- 88.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997;277:1109–13 [DOI] [PubMed] [Google Scholar]
- 89.Adams DS, Robinson KR, Fukumoto T, et al. Early, H+-V-ATPase-dependent proton flux is necessary for consistent left-right patterning of non-mammalian vertebrates. Development 2006;133:1657–71 [DOI] [PMC free article] [PubMed] [Google Scholar]