

Summary
Engineering new functionality into living eukaryotic systems by enzyme evolution or de novo protein design is a formidable challenge. Cells do not rely exclusively on DNA-based evolution to generate new functionality but often utilize membrane encapsulation or formation of membraneless organelles to separate distinct molecular processes that execute complex operations. Applying this principle and the concept of two-dimensional phase separation, we develop film-like synthetic organelles that support protein translation on the surfaces of various cellular membranes. These sub-resolution synthetic films provide a path to make functionally distinct enzymes within the same cell. We use these film-like organelles to equip eukaryotic cells with dual orthogonal expanded genetic codes that enable the specific reprogramming of distinct translational machineries with single-residue precision. The ability to spatially tune the output of translation within tens of nanometers is not only important for synthetic biology but has implications for understanding the function of membrane-associated protein condensation in cells.
Keywords: synthetic biology, synthetic biomolecular condensates, 2D phase separation, enzyme engineering, orthogonal translation, genetic code expansion, membrane signaling
Graphical abstract
Highlights
-
•
2D phase separation was utilized to design orthogonal enzymes
-
•
Film-like organelles maintained distinct suppressor tRNA microenvironments
-
•
Dual film-like synthetic organelles enabled orthogonal translation in eukaryotes
-
•
Cells were equipped with two expanded genetic codes in addition to the canonical one
Use of 2D phase separation and two instances of codon expansion in a synthetic biology approach enables control of selective protein synthesis at the tens of nanometers scale in live cells.
Introduction
How living systems develop ever more complex processes and acquire new features is not only a central question in evolutionary biology but is also an essential consideration for synthetic biologists striving to create new and complex functionalities in a living host. A common evolutionary mechanism is the duplication of genes or even whole genomes. This creates redundant biomolecules, which can undergo divergence, giving rise to new functions (Blomme et al., 2006; Lynch and Conery, 2003; Ohno, 1970). Intriguingly, duplicated genes are often deleted, and recent studies have shown that paralogous genes can more readily diverge if their functional and structural entanglement is comparatively weak (Kuzmin et al., 2020; Wapinski et al., 2007). Thus, to develop new functionalities, it often seems necessary that genes become orthogonal and do not cross-react with ancestral biomolecules. For synthetic biology applications in cells, such orthogonality is generally essential to prevent interference with the endogenous processes of the host, and it can be a daunting task to develop this de novo for molecules that have many interaction partners. Furthermore, creating enzymes that specifically execute desired functions de novo is challenging (Almhjell et al., 2018; Bryson et al., 2017; Glasgow et al., 2019; Huang et al., 2016a; Lechner et al., 2018). Therefore, alternative strategies for generating orthogonal enzymes inside a cell would be extremely useful.
One powerful tool that synthetic biologists have for creating new functions in vivo is genetic code expansion (GCE), which has been widely used to site-specifically incorporate noncanonical amino acids (ncAAs) into proteins in vivo (Chin, 2017; Lemke, 2014; Liu and Schultz, 2010). The genetic code determines how genomic information is transferred into a polypeptide sequence through the central dogma and relies on aminoacyl-tRNA synthetase (aaRS)/tRNA pairs to decode triplet codons into specific amino acids. These pairs have extensive protein-RNA and, in the case of multimeric aaRS systems, protein-protein interaction surfaces. Engineering functional derivatives is thus a formidable challenge. Hence, to repurpose aaRS/tRNA pairs to encode noncanonical functionalities in a particular host, aaRS/tRNA pairs from highly evolutionarily distinct organisms are typically used, a few of which are orthogonal to the new host machinery. Here, orthogonal refers to a given aaRS accepting only a specific ncAA, and then only aminoacylating it to its cognate tRNA. In addition, the cognate tRNA should not be recognized as a substrate by any of the endogenous tRNA synthetases.
For GCE of mammalian cells, the most commonly used pyrrolysyl tRNA synthetase/tRNA pairs (PylRS/tRNAPyl) are derived from methanogenic archaea (for reviews, see Reinkemeier and Lemke [2020] and Wan et al. [2014]). These are orthogonal in both eukaryotes and bacteria and have been evolved in the laboratory to incorporate a diverse set of ncAAs, with which one can, for example, control protein function with light (Gautier et al., 2010), conjugate small fluorescent dyes for super-resolution microscopy (Nikić et al., 2014; Uttamapinant et al., 2015), photo-crosslink proteins (Ai et al., 2011; Hancock et al., 2010; Zhang et al., 2011), or modify antibodies (Koehler et al., 2016; Oller-Salvia et al., 2018; Xiao et al., 2013). The tRNAPyl is typically chosen to decode a comparatively rare stop codon (e.g., the amber TAG codon, which terminates the translation of 20% of all proteins in humans), hence this technology is also termed amber suppression. This stop codon is inserted site-selectively into the coding sequence of a protein of interest (POI) so that the full-length protein is only formed with the ncAA installed at the chosen site.
For the simultaneous incorporation of multiple different ncAAs in eukaryotes, GCE technology is fundamentally limited by three problems: (1) the translational process lacks mRNA specificity such that other mRNAs in the transcriptome that naturally terminate at amber codons can be mistranslated; (2) the number of codons that can be reassigned without altering host functionality is limited; and (3) the dearth of orthogonal aaRS/tRNA pairs.
We recently published a solution to the first of these problems: membraneless orthogonally translating (OT) organelles that are formed by phase separation and targeting to microtubule plus-ends to afford a micron-sized organelle (Reinkemeier et al., 2019). We define the term organelle as a spatially distinct site in the cell, regardless of its structure or appearance, which nevertheless executes a specific function and has a composition distinct from its surroundings. Phase separation occurs at above the critical concentrations of certain proteins that were fused to PylRS and the ms2 bacteriophage coat protein (MCP), an RNA-binding protein (Reinkemeier et al., 2019). Although the suppressor tRNA itself is a relatively small molecule, the PylRS-loaded organelle efficiently recruits it, leading to a very high concentration inside the condensate and a very low concentration throughout the rest of the cell. The POI mRNA is labeled in the 3′ untranslated region with specific RNA motifs (ms2 loops) that are bound by MCP, thus leading to recruitment of the mRNA into the organelle. Because only the ribosomes processing the recruited mRNA are exposed to a very high concentration of suppressor tRNAPyl, it is preferentially translated according to an expanded genetic code. By contrast, ribosomes translating mRNA elsewhere in the cell terminate translation once the chosen stop codon is encountered, because no tRNAPyl is available. Note that the translational machinery requires that a few hundred factors work smoothly together and no component other than the PylRS and MCP were fused to the organelle scaffold. Thus, despite sharing all other components of translation with the cytoplasm, from which these components are essentially freely accessible, we detected up to 8-fold selectivity for amber suppression of targeted (i.e., ms2 tagged) versus untargeted mRNAs. We also showed that the same logic could be applied to reprogramming the opal or ochre codon. Hereafter, we refer to this particular OT organelle-based GCE technology as being mRNA selective (Reinkemeier et al., 2019).
The other two aforementioned issues remain difficult to resolve in eukaryotes. The scarcity of free codons has been addressed in E. coli by using various means, for example, whole-genome synthesis (Fredens et al., 2019; Lajoie et al., 2013; Wang et al., 2016), artificial base pairs (Hamashima et al., 2018; Hoshika et al., 2019; Zhang et al., 2017), or orthogonal ribosomes that recognize engineered Shine-Dalgarno sequences (Fried et al., 2015; Neumann et al., 2010; Orelle et al., 2015; Schmied et al., 2018). However, these approaches are not readily transferred to eukaryotes, which have much larger and more complex genomes than E. coli, and their ribosomes use an alternative mechanism of mRNA recognition.
As mentioned, only a handful of orthogonal aaRS/tRNA pairs are available, and these have achieved varying degrees of success in eukaryotes (Beránek et al., 2018; Cervettini et al., 2020; Chen et al., 2009; Chin et al., 2003; Italia et al., 2017, 2018, 2020; Lemke et al., 2007; Mukai et al., 2008; Neumann et al., 2008; Yanagisawa et al., 2008a). More recently, advanced evolutionary and rational design strategies have been used to evolve multiple orthogonal versions of PylRS (Beránek et al., 2019; Dunkelmann et al., 2020; Meineke et al., 2018, 2020; Willis and Chin, 2018). Although the evolution approach is powerful, the discovery rate of new orthogonal aaRS/tRNA pairs is low. This issue is not limited to enzyme evolution for GCE but is inherent to enzyme engineering generally.
In this paper, we show that we can use two-dimensional protein condensation at various membrane surfaces to establish multiple, film-like biochemical microenvironments in which we can tune the language of protein translation at the sub-resolution scale. We thus developed several OT film-like organelles, which enabled us to design multiple spatially orthogonal aaRS/tRNA pairs within the same cytoplasm. This allowed us to reuse the same stop codon to incorporate distinct ncAAs into different proteins in vivo, effectively generating a cell with three spatially and functionally distinct translational programs. We further discuss the implications of these results for membrane signaling and membrane-associated phase separation.
Results
Design of multiple, mutually orthogonal OT organelles
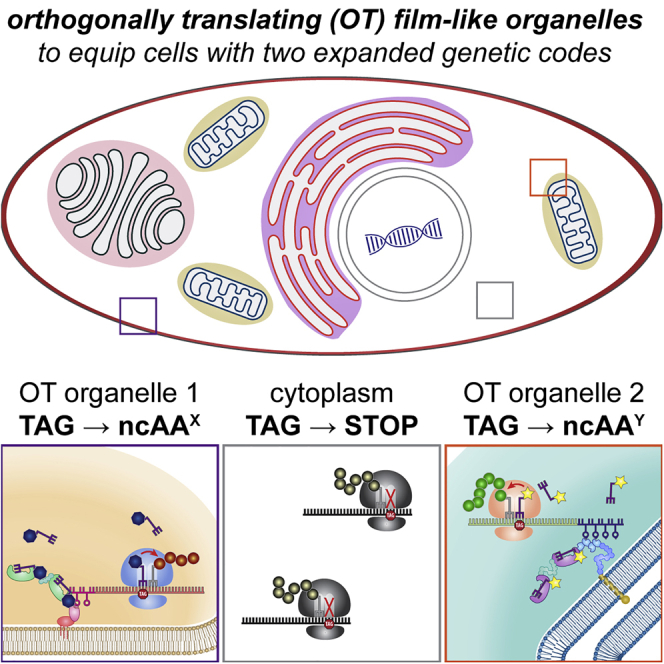
The aim of this work is to assign one stop codon (amber, TAG) to a different ncAA in each film-like organelle inside the same cell; hence each full-length POI will be produced only if their programmed ncAA is added to the growth medium (ncAAX [blue heptagon] to POI1 [green] and ncAAY [yellow star] to POI2 [red] in Figure 1A).
Figure 1.

Multiple orthogonally translating (OT) organelles for equipping cells with multiple orthogonal expanded genetic codes
(A) Our experimental design: forming two fully orthogonal OT organelles in one cell equips the host with two orthogonal genetic codes. Only selected mRNAs are translated with the respective expanded genetic code and hence only selected proteins contain the specific ncAAs. An assembler is a synthetic-organelle-forming unit like a protein condensation functionality. An RNA-binding domain (RBD) is chosen to bind its cognate RNA motif (RM), which is fused to the mRNA of the protein of interest (POI) in the 3′ untranslated region.
(B) In order to construct multiple OT organelles, three criteria need to be fulfilled: independent assembly, selective RNA recruitment, and distinct ncAA specificity.
(C) Schematic representation of membranes targeted for the development of OT organelles.
(D–G) Overview of the classes of synthetic organelles in this work. To develop OT organelles on the cytoplasmic surface of cellular membranes, PylRS and MCP or λN22 peptides were fused to the phase-separating assembler FUS as well as to membrane-targeting domains.
To achieve this, besides the amber suppressor aaRS/tRNA pair being orthogonal to the host machinery, the orthogonality of multiple, mutually orthogonal OT organelles inside the same cell should be 3-fold (Figure 1B). First, the organelles should form independently and not intermix; we term this the “independent assembly” criterion. This criterion considerably extends the previously described general requirement that the mRNA targeted to the organelle is not efficiently translated elsewhere in the cytoplasm by the host’s canonical translation machinery (Reinkemeier et al., 2019). Second, each OT organelle must recruit a specific subset of mRNAs; this is the “selective RNA recruitment” criterion. Third, each aaRS variant in a respective organelle should selectively utilize a distinct ncAA; this defines the “distinct ncAA specificity” criterion.
We addressed each of these criteria in turn. First, we needed to construct a number of spatially distinct OT organelles. The previously developed micron-sized synthetic organelles were approximately half the size of a cell nucleus and thus occupied a substantial portion of the cytoplasm (Reinkemeier et al., 2019). Fitting two of these organelles into one cell and ensuring they are immiscible was expected to be a challenge.
OT film-like organelles that fulfill the independent assembly criterion
We thus aimed to construct OT organelles that condense as thin-film-like layers on a membrane. We designed these condensates to form on the cytoplasmic face of a cellular membrane to facilitate exchange with the cytoplasm. To achieve this, we combined a phase-separation domain with a membrane localization domain and fused them directly to the PylRS or the RNA-binding proteins (we use “::” to denote a genetic fusion and “•” to denote a co-expression). We used the proteins fused in sarcoma (FUS) and Ewing sarcoma breakpoint region 1 (EWSR1) as phase-separating proteins (Altmeyer et al., 2015; Patel et al., 2015; Wang et al., 2018). We previously demonstrated that these can serve as organelle assemblers in vivo and define an assembler as an organelle-forming moiety (Reinkemeier et al., 2019). We then localized the condensates to different membranes by fusing them to:
-
•
the N-terminal domain of the rodent LCK tyrosine kinase to direct them to the plasma membrane (PM) (Zlatkine et al., 1997)
-
•
the N-terminal domain of human EBAG9 to localize them to the Golgi membrane (GM) (Engelsberg et al., 2003)
-
•
the N-terminal domain of rabbit cytochrome P450 2C1 (CYPIIC1) to direct them to the endoplasmic reticulum membrane (ERM) (Hung et al., 2017)
-
•
the N-terminal domain of human TOM20 for targeting to the outer mitochondrial membrane (OMM) (Stavru et al., 2006).
We refer to these four systems by the abbreviations PMP, GMP, ERMP, and OMMP (the suffix P indicates that the systems also contain a phase-separating moiety; for an overview of these systems, see Figures 1C–1G).
We used a previously described dual-color ms2 reporter in fluorescence flow cytometry (FFC) studies to assess if these organelles are indeed OT, that is, they selectively undertake GCE and translate a targeted mRNA of our POI that the cytoplasm does not (Reinkemeier et al., 2019). In this reporter, both EGFP39TAG and mCherry190TAG are expressed independently (i.e., each has an expression cassette with its own promotor) but from one plasmid with amber codons at the indicated permissive sites. Only the mCherry mRNA is tagged with ms2 loops. In case of cytoplasmic GCE, different mRNAs cannot be distinguished, and the amber codon is suppressed in both EGFP and mCherry. This leads to the appearance of a diagonal population of green and red fluorescent cells in the double logarithmic axis FFC plots (Figure 2A, left). By contrast, if an OT organelle is working selectively, only mCherry is translated with an expanded genetic code, whereas the untargeted EGFP is terminated if the amber codon is encountered and thus is not fluorescent. This leads to a vertical mCherry-positive population in FFC (Figure 2A, right). Based on FFC data, we can calculate the fold change in selectivity and the relative efficiency of each system. The fold change in selectivity is defined as the mean mCherry signal divided by the mean EGFP signal of a given system normalized to the respective ratio for the cytoplasmic GCE system (PylRS). Analogously, the relative efficiency is defined as the mean mCherry signal of a given system normalized to the mean mCherry signal of cytoplasmic PylRS. In the STAR Methods, we discuss the robustness of this reporter analysis against a variety of factors that might affect translational efficiency, brightness, instrument settings, and so forth.
Figure 2.

Synthetic film-like organelles grafted on intracellular membranes enable highly selective orthogonal translation
(A) Schematic representation of the dual-color reporter used to test OT organelles and theoretical FFC plots. mRNAs encoding EGFP and mCherry with amber codons at permissive sites are expressed from one plasmid. The mCherry mRNA is tagged with specific RNA motifs (RMs). In the case of cytoplasmic GCE, both full-length EGFP and mCherry should be produced and give an approximate diagonal in FFC analysis (shown in orange). If the OT organelle works selectively, only mCherry will be produced, which would result in an mCherry-positive population in FFC analysis (shown in red). Untransfected cells are represented as gray circles.
(B and C) FFC analysis of OT organelle selectivity. HEK293T cells expressing the indicated reporter and GCE systems as well as tRNAPyl in the presence of SCO-K. The sums of at least three independent experiments are shown. The black line in the FFC dot plots indicates the EGFP signal cut-off used to identify transfected cells.
(D and E) Bar plot corresponding to FFC experiments shown in (B) and (C). The dark gray bars show the fold-change selectivity, defined as the ratio of the mean fluorescence intensities of mCherry versus EGFP, normalized to the respective cytoplasmic GCE control (PylRS). The light gray bars represent the relative efficiency, defined as the mean fluorescence intensity of mCherry for each condition divided by that of the cytoplasmic GCE control (PylRS). Bar graphs show the mean value of at least three independent experiments; error bars represent the standard deviation (SD).
(F) HEK293T cells expressing the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2) together with tRNAPyl and the indicated OT film-like organelle or a cytoplasmic GCE system (first column). Experiments were performed in the presence of SCO-K. Scale bars, 20 μm.
See also Figure S5.
We used this reporter to evaluate the performance of all membrane-localized organelles in cells. We observed a vertical, mCherry-positive population by FFC for all four systems (Figure 2B). Compared to the cytoplasmic GCE system, this corresponds to selectivities of 9- to 12-fold for the targeted protein, and we achieved efficiencies of up to 45% (Figure 2D). Thus, we conclude that these four OT film-like organelles can perform highly selective and orthogonal translation of the targeted mRNA::ms2. The best of these systems compared favorably to our previous best-performing, micron-sized OT organelle (OTK2::P1, a combination of phase-separating and plus-end-directed kinesin domains), which achieved 8-fold selectivity and 40% efficiency (Reinkemeier et al., 2019).
Membranes themselves can serve as scaffolds to locally concentrate tethered biomolecules owing to a reduction of dimensionality with important implications (e.g., for signaling cascades) (Grecco et al., 2011). Phase separation in the cytoplasm is another means to reach very high local concentrations of biomolecules, and the relevance of phase separation at membranes, where both effects can combine, has recently become an intense field of research (Case et al., 2019a). To test the relevance of the phase separation-based assemblers (FUS and EWSR1, fused to PylRS and MCP, respectively) for film-like organelle functionality, we performed additional FFC experiments for all four membrane systems, in which we compared constructs with and without FUS/EWSR1. We determined that using the phase separation-based assemblers was essential for functionality for three (OMMP, GMP, and ERMP) systems, whereas for the plasma membrane system, the performance was similar with or without FUS/EWSR1 (Figures 3A–3C and S1A).
Figure 3.

Phase-separating assemblers are essential for the function of most OT film-like organelles
(A) Schematic overview of constructs that target PylRS and/or MCP with or without FUS/EWSR1 individually to the four membranes. The lowercase “s” indicates that PylRS and MCP are not genetically fused and expressed from separate plasmids.
(B) FFC analysis of HEK293T cells expressing the dual-color ms2 reporter (EGFP39TAG, mCherry190TAG::ms2), tRNAPyl, and a respective indicated OT organelle in presence of SCO-K. Figure S1A shows further controls. The black lines in the FFC dot plots indicate the EGFP signal cut-off used to identify transfected cells, the red dotted lines demarcate background expression (in absence of MCP) for each targeted membrane.
(C) Bar plot corresponding to FFC experiments shown in (B). The dark gray bars show the fold-change selectivity, the light gray bars represent the relative efficiency (see Figure 2B for the respective cytoplasmic PylRS control). Bar graphs show the mean value of at least three independent experiments; error bars represent the SD.
(D) FFC analysis of separate OT organelle constructs. HEK293T cells expressing the dual color ms2 reporter (EGFP39TAG, mCherry190TAG::ms2), tRNAPyl, a respective PylRS OT organelle construct, and either no MCP or MCP targeted to each respective membrane. The sums of at least three independent experiments are shown. The black lines in the FFC dot plots indicate the EGFP signal cut-off used to identify transfected cells.
(E and F) Heatmaps calculated from FFC data shown in (D) for visualizing the fold change selectivity (E) and the relative efficiency (F) of individual co-expressed constructs. The mean values of all experiments are shown. The upper halves of the heatmaps correspond to the data shown in (D). The lower halves of the heatmaps show corresponding data for the dual color boxB-reporter (EGFP39TAG, mCherry190TAG::boxB), tRNAPyl, a respective PylRS fusion and either no λN22 construct (row 10) or the λN22 peptides targeted to the indicated membrane (see Figures S1B for corresponding FFC dot plots and S4 for associated imaging experiments). White stars indicated when MCP/λN22 and PylRS are targeted to the same membrane.
See also Figure S6.
Figure S1.

Phase separating domains are essential for most systems and several combinations of boxB-λN22 systems fulfill the independent assembly criterion, related to Figure 3
(A) To test if phase-separating domains are required for the different membrane systems, we used constructs that target PylRS or MCP with or without FUS/EWSR1 individually to the four membranes. Here we show FFC analysis of separate OT organelles with and without phase-separation domains (i.e., PylRS with and without FUS as well as MCP with and without EWSR1). HEK293T cells expressing the dual color ms2 reporter (EGFP39TAG, mCherry190TAG::ms2), tRNAPyl, a respective PylRS construct without (upper rows) or with phase-separation domain (lower rows), as well as either no MCP protein (first columns) or a membrane-targeted MCP protein without (second columns) or with a phase-separation domain (third columns) in presence of SCO-K. For the OMM, ERM and GM system the variants without FUS loose functionality. Only the PM-targeted PylRS variant is efficient with and without a phase-separation domain. For the case of MCP, the GM and OMM the systems without EWSR1 perform substantially worse. Shown is the sum of at least three independent experiments. (Note that the black line in the FFC dot plots indicates the EGFP signal cut-off used to identify transfected cells). Together, three out of four systems require a protein condensation to be functional, while the PM and PMP systems perform similarly, potentially owing to the anyway good spatial separation of the plasma membrane from the cytoplasm.
(B) To test the independent assembly criterion of the different membrane systems for the λN22–boxB system we cloned constructs that target PylRS or λN22 peptides (with FUS or EWSR1 respectively) to the four different membranes. Here we show FFC analysis of separate OT organelle constructs. HEK293T cells expressing the dual color boxB reporter (EGFP39TAG, mCherry190TAG::boxB), tRNAPyl, a respective PylRS OT organelle construct as well as either no λN22 peptides (fifth column) or the λN22 peptides targeted to each respective membrane. The ncAA SCO-K was present in all experiments. Similar to the experiments for the ms2–MCP systems (Figure 3) we always observed the expected highly selective mCherry190TAG::boxB production if λN22 and PylRS were targeted to the same membrane (vertical population). We also observed enhanced mCherry190TAG::boxB expression if λN22 and PylRS were targeted to the GM and either the ERM or PM. Only the combination of the ERMP and PMP system, and that of the OMMP system with any other system showed no mixing, i.e., a similar expression profile as in the absence of λN22 (background expression), shown in the fifth row (-λN22) above the cytoplasmic PylRS control, and thus only these fulfilled the independent assembly criterion. Shown is the sum of at least three independent experiments. [Note that the black line in the FFC dot plots indicates the EGFP signal cut-off used to identify transfected cells, the red dashed line highlights background expression (in absence of λN22) for each targeted membrane].
(C) FFC gating scheme to identify single HEK293T cells. HEK293T cells were first identified based on SSC-A and FSC-A values. Subsequently, single cells are identified using SSC-A and SSC-W parameters. One representative example is shown. Here, HEK293T cells were transfected with a cytoplasmic PylRS system, the indicated ms2 reporter (EGFP39TAG, mCherry190TAG::ms2) and tRNAPyl in presence of the ncAA SCO-K. Cells that passed the first gate (top) are subsequently gated for single cells (bottom).
(D) FFC analysis of cells expressing an iRFP::EGFP39TAG reporter, tRNAPyl and PylRSAF or PylRSAA. iRFP served as a transfection control, whereas EGFP fluorescence reports on successful amber codon suppression. Cells were incubated either with no ncAA, with SCO-K (250 μM), TCO∗A-K (250 μM), Cbz-K (250 μM), with 3-IF (1 mM) or SCO-Y (250 μM). For PylRSAF, robust EGFP expression was observed in the presence of SCO-K, TCO∗A-K and Cbz-K. For PylRSAA, EGFP expression was only enhanced in the presence of 3-IF or SCO-Y. Shown is the sum of at least three independent experiments. (Note that the black line in the FFC dot plots indicates the iRFP signal cut-off used to identify transfected cells).
Using IF and fluorescence in situ hybridization (FISH), we confirmed that these organelles highly enrich tRNAPyl, PylRS, mRNA::ms2, and cellular ribosomes (Figures 4, S2, and S3; Video S1). We performed super-resolution microscopy (Rust et al., 2006) and detected a high degree of localization of tRNAPyl to a very thin layer, with almost no tRNAPyl distinguishable from background in the remainder of the cytoplasm (Figure 4B). Notably, for the simple line/rim shaped PMP system, the full width at half-maximum of the tRNAPyl distribution was clearly sub-resolution (<<100 nm, see the lower left line scan in Figure 4B). This substantiates the hypothesis that a sharp suppressor tRNAPyl gradient can be formed using the OT film-like organelle approach, effectively creating a unique, spatially confined biochemical microenvironment. Taking into account that tRNAs are relatively small molecules, which might have been expected to rapidly diffuse on a cellular scale, this appears to be quite remarkable. These results affirm that all of the OT film-like organelles act orthogonally to the host’s translation in the cytoplasm.
Figure 4.

Synthetic OT film-like organelles efficiently recruit tRNAPyl and ribosomes
HEK293T cells expressing the indicated OT organelle, tRNAPyl, and EGFP39TAG::ms2 in the presence of the ncAA SCO-K.
(A) 3D-rendered images of IF staining against PylRS and FISH staining of tRNAPyl reveal strong tRNAPyl recruitment (see Figure S2A for raw images of individual planes). Top to bottom: PylRS (magenta), tRNAPyl (yellow), merged (NLS::EGFP39TAG::ms2 is shown in green in all panels). See Video S1 for corresponding animations. Scale bars, 10 μm.
(B) Super-resolution microscopy confirms that tRNAPyl is localized to a fine layer at the respective membrane. Particularly for the PMPMCP system tRNAPyl appears restricted to a sub-resolution film of only a few tens of nanometers (FWHM, full width at half-maximum). Top to bottom: tRNAPyl stochastic optical reconstruction microscopy (STORM), magnified views of areas inside red boxes, and line scans corresponding to red lines. Scale bars, 1 μm.
(C) 3D-rendered images of IF staining against PylRS and RPL26L1 (a ribosomal marker protein) showing endogenous ribosomes in MCP-based OT organelles (see Figure S2B for raw images of individual planes). Due to the abundance of ribosomes, staining outside of the OT organelles is also observed. Top to bottom: PylRS (magenta), RPL26L1 (cyan), merged (NLS::EGFP39TAG::ms2 is shown in green in all panels). Video S1 shows corresponding animations. Scale bars, 10 μm.
See also Figures S3 and S4.
Figure S2.

MCP-based synthetic film-like organelles enrich tRNAPyl, mRNA::ms2, and cellular ribosomes, related to Figure 4
(A–B) Images used for the 3D rendering in Figure 4. HEK293T cells expressing the indicated OT organelle, tRNAPyl, and a construct encoding NLS::EGFP39TAG::ms2 in the presence of the ncAA SCO-K. All images show a selected plane of a z stack.
(A) Corresponding to Figure 4A. IF staining against PylRS and FISH staining of tRNAPyl show strong tRNAPyl recruitment. Top to bottom: PylRS (magenta), tRNAPyl (yellow), merged (NLS::EGFP39TAG::ms2 shown in green).
(B) Corresponding to Figure 4C. IF staining against PylRS and RPL26L1 show that endogenous ribosomes are recruited into OT film-like organelles. Note that owing to the abundance of ribosomes, a substantial staining outside of the OT organelles can be observed. Top to bottom: PylRS (magenta), RPL26L1 (cyan), merged (NLS::EGFP39TAG::ms2 in green). Scale bars: 20 μm.
(C) IF staining against PylRS and FISH staining of mRNA::ms2 show recruitment of ms2-tagged mRNA into membrane-associated OT organelles. Top to bottom: PylRS (magenta), mRNA::ms2 (yellow), merged (NLS::EGFP39TAG::ms2 in green).
Figure S3.

Additional multi color stainings of different OT film-like organelle constructs with organelle markers, related to Figure 4
(A–D) HEK293T cells expressing the indicated OT organelle, tRNAPyl and the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2) in the presence of the ncAA SCO-K. All images show a selected plane of a z stack. Scale bars: 20 μm.
(A) IF staining against PylRS and a plasma membrane marker (NaK-ATPase). Top to bottom: PylRS (magenta), NaK-ATPase (cyan), NLS::EGFP39TAG::boxB (blue), NLS::mCherry190TAG::ms2 (yellow) and merged.
(B) IF staining against PylRS and a Golgi membrane marker (Giantin). Top to bottom: PylRS (magenta), Giantin (cyan), NLS::EGFP39TAG::boxB (blue), NLS::mCherry190TAG::ms2 (yellow) and merged.
(C) IF staining against PylRS and an ER membrane marker (Calnexin). Top to bottom: PylRS (magenta), Calnexin (cyan), NLS::EGFP39TAG::boxB (blue), NLS::mCherry190TAG::ms2 (yellow) and merged.
(D) IF staining against PylRS and a mitochondrial marker (apoptosis-inducing factor, AIF). Top to bottom: PylRS (magenta), AIF (cyan), NLS::EGFP39TAG::boxB (blue), NLS::mCherry190TAG::ms2 (yellow) and merged.
Note that we do not have 100% transfection efficiency, while all the used markers are endogenous to the cell.
The first part of the video shows animated 3D reconstructions corresponding to Figure 4A. HEK293T cells expressing the PMPMCP, GMPMCP, ERMPMCP, or OMMPMCP, as well as tRNAPyl and EGFP39TAG::ms2 in the presence of the ncAA SCO-K. IF staining against PylRS and FISH staining of tRNAPyl show strong tRNAPyl recruitment. PylRS (magenta), tRNAPyl (yellow), NLS::EGFP39TAG::ms2 (green). The second part of the video shows animated 3D reconstructions corresponding to Figure 4C. HEK293T cells expressing the PMPMCP, GMPMCP, ERMPMCP, or OMMPMCP, as well as tRNAPyl and EGFP39TAG::ms2 in the presence of the ncAA SCO-K. IF staining against PylRS and RPL26L1 show co-localization of endogenous ribosomes with OT organelles. PylRS (magenta), RPL26L1 (cyan), NLS::EGFP39TAG::ms2 (green). The third part shows animated 3D reconstructions corresponding to Figure S5B. HEK293T cells expressing the PMPλN22, GMPλN22, ERMPλN22, or OMMPλN22, as well as tRNAPyl and a EGFP39TAG::boxB in the presence of the ncAA SCO-K. IF staining against PylRS and FISH staining of tRNAPyl show strong tRNAPyl recruitment. PylRS (magenta), tRNAPyl (yellow), NLS::EGFP39TAG::boxB (green). The fourth part shows animated 3D reconstructions corresponding to Figure S5D. HEK293T cells expressing the PMPλN22, GMPλN22, ERMPλN22 or OMMPλN22, as well as tRNAPyl and a EGFP39TAG::boxB in the presence of the ncAA SCO-K. IF staining against PylRS and RPL26L1 show co-localization of endogenous ribosomes with OT organelles. PylRS (magenta), RPL26L1 (cyan), NLS::EGFP39TAG::box (green).
We next investigated if the four OT film-like organelles meet the independent assembly criterion defined above (Figure 1B). To determine if the organelles mix components with each other, we designed constructs in which PylRS and MCP were individually fused to a membrane-targeting domain and to a phase separating assembler (FUS and EWSR1, respectively) (Figures 3D–3F). We then co-expressed each membrane directed PylRS variant, either without an MCP construct or with the MCP targeted to one of the four membranes. We assayed all 20 possible combinations for selective translation by FFC using the dual color ms2 reporter (EGFP39TAG, mCherry190TAG::ms2). As expected for an OT organelle, we observed high selectivity (i.e., a more vertical distribution in the FFC plot) if MCP and PylRS were both targeted to the same membrane. In the FFC data shown in Figure 3D, combinations of MCP and PylRS for the same membrane-targeting system sit on the diagonal across the matrix (also indicated by white stars in Figures 3E and 3F). Whereas a number of systems showed only background expression if MCP and PylRS were targeted to the respective membranes, this was not the case for all. Background expression is defined as the expression profile detected in the absence of MCP (−MCP) (Figures 3D–3F, area inside the dotted red line; note the logarithmic scale of the FFC plots). Specifically, neither the combination of ERMP and PMP systems nor a combination of the OMMP system with any other yielded a vertical distribution substantially above background, indicating that no mixing of different biomolecules occurred between the different targeting destinations. Thus, we conclude that only the combination of ERMP with PMP and OMMP with ERMP/PMP/GMP fulfilled the independent assembly criterion (see Figures S4B–S4D for corresponding imaging).
Figure S4.

Separate OT organelle systems co-localize MCP, PylRS, tRNAPyl, and ribosomes, related to Figure 3
(A) Overview cartoon of the constructs used. The lowercase “s” indicates that the PylRS and the MCP are expressed from two separate plasmids and not genetically fused.
(B–D) HEK293T cells expressing the indicated OT organelle, tRNAPyl and a construct encoding NLS::EGFP39TAG::ms2 in the presence of the ncAA SCO-K. All images show a selected plane of a z stack. Scale bars: 20 μm.
(B) IF staining against PylRS and FISH staining of tRNAPyl show strong tRNAPyl recruitment into membrane-associated OT organelles. Top to bottom: PylRS (magenta), tRNAPyl (yellow), merged (NLS::EGFP39TAG::ms2 in green).
(C) IF staining against PylRS and RPL26L1 show that endogenous ribosomes co-localize with OT film-like organelles. Note that as RPL26L1 is an endogenous protein, also untransfected cells are stained. Top to bottom: PylRS (magenta), RPL26L1 (cyan), merged (NLS::EGFP39TAG::ms2 in green).
(D) IF staining against PylRS and MCP (with an anti-HA antibody) show co-localization in OT organelles. Top to bottom: PylRS (magenta), MCP (yellow), merged (NLS::EGFP39TAG::ms2 in green).
(E,F) HEK293T cells expressing the indicated OT organelle, tRNAPyl and a construct encoding EGFP39TAG::boxB in the presence of SCO-K. All images show a selected plane of a z stack. Scale bars: 20 μm.
(E) IF staining against PylRS and λN22 (with an anti-Myc antibody) show co-localization in OT organelles. Left to right: PylRS (magenta), λN22 (yellow), merged (NLS::EGFP39TAG::boxB in green).
(F) IF staining against PylRS and RPL26L1 show that endogenous ribosomes co-localize with membrane-associated OT organelles. Note that due to the abundance of ribosomes, a substantial staining outside of the OT organelles can be observed. Left to right: PylRS (magenta), RPL26L1 (cyan), merged (NLS::EGFP39TAG::boxB in green).
Our experimental observations are consistent with what is known about each membrane biogenesis pathway. Because mitochondrial membranes are not directly coupled to the exocytic or endocytic machinery (Bonifacino and Glick, 2004), the OMMP system is also more orthogonal to all the others than the GMP system (Figures 3D–3F). Although also more complex membrane interactions pathways exist (Gordaliza-Alaguero et al., 2019; Wu et al., 2018), and although designed for a different purpose, our assay might be useful in the future to readout the amount of protein exchanged between different membrane compartments in eukaryotic cells.
OT film-like organelles that fulfill the selective RNA recruitment criterion
Next, we aimed to address the selective RNA recruitment criterion. Thus far, we had used MCP to recruit ms2-tagged mRNAs into the synthetic organelles. From the wealth of literature available on alternatives to the ms2-MCP system for RNA targeting, we chose the λN22 peptides that selectively bind boxB loops (Daigle and Ellenberg, 2007) and are known to be orthogonal to the ms2-MCP system. To test if the boxB-λN22 interaction can also enable selective orthogonal translation, we used another dual-color fluorescence reporter for FFC analysis, which we called the dual color boxB reporter. This reporter expresses EGFP39TAG and mCherry190TAG from one plasmid, but this time the mCherry mRNA is fused to four boxB loops (Figures 2C and 2E). Notably, using the boxB-λN22 approach, we observed even higher selectivity for all membrane-associated OT organelles (12- to 17-fold changes) and achieved efficiencies of up to 80%, thus bettering all our OT systems reported here and previously (Reinkemeier et al., 2019) (Figures 2D and 2E). These experiments demonstrated that OT organelles equipped with λN22 peptides act also orthogonally to the host’s endogenous translation.
To confirm that these λN22-based OT organelles also fulfill the independent assembly criterion, we performed experiments analogous to those with individual ms2-MCP-based synthetic organelles. We tested another 20 combinations using the boxB-λN22 targeting system (Figures 3E, 3F, and S1B). Again, we determined that the combination of the ERMP and PMP systems, as well as the combination of the OMMP system with any other system, fulfill the independent assembly criterion.
We also performed IF and FISH staining to determine the composition of the λN22-based organelles, and again observed strong recruitment of tRNAPyl, mRNA::boxB, and ribosomes (Figures S4E, S4F, and S5; Video S1).
Figure S5.

λN22-based synthetic organelles enrich tRNAPyl, mRNA::boxB, and cellular ribosomes, related to Figure 2C
HEK293T cells expressing the indicated OT organelle, tRNAPyl and a construct encoding EGFP39TAG::boxB in presence of the ncAA SCO-K. All images show a selected plane of a z stack.
(A) IF staining against PylRS and FISH staining of tRNAPyl shows strong recruitment of tRNAPyl into OT organelles. Left to right: PylRS (magenta), tRNAPyl (yellow), merged (NLS::EGFP39TAG::ms2 in green). Shown is a selected plane of a z stack. Scale bars: 20 μm.
(B) 3D reconstruction corresponding to panel A. Left to right: PylRS (magenta), tRNAPyl (yellow), merged (NLS::EGFP39TAG::ms2 in green in all panels). See also Video S1 for the animated 3D reconstructions. Scale bars: 10 μm.
(C) IF staining against PylRS and RPL26L1 shows that endogenous ribosomes co-localize with membrane-associated OT organelles. Note that due to the abundance of ribosomes, a substantial staining outside of the OT organelles can be observed. Left to right: PylRS (magenta), RPL26L1 (cyan), merged (NLS::EGFP39TAG::ms2 in green). Shown is a selected plane of a z stack. Scale bars: 20 μm.
(D) 3D reconstruction corresponding to panel C. Left to right: PylRS (magenta), RPL26L1 (cyan), merged (NLS::EGFP39TAG::ms2 in green in all panels). See also Video S1 for the animated 3D reconstructions. Scale bars: 10 μm.
(E) IF staining against PylRS and FISH staining of mRNA::boxB show recruitment of boxB-tagged mRNA into λN22-based OT organelles. Left to right: PylRS (magenta), mRNA::boxB (yellow), merged (NLS::EGFP39TAG::ms2 in green). Scale bars: 20 μm.
We next verified that the boxB-λN22- and ms2-MCP-based organelle systems are orthogonal to each other. To that end, we co-expressed OT organelles possessing MCP or λN22 peptides with either the dual color boxB reporter (EGFP39TAG, mCherry190TAG::boxB) or the dual color ms2 reporter (EGFP39TAG, mCherry190TAG::ms2). We observed selective GCE of the targeted mRNA only for the correct combinations with the corresponding RNA-recruitment domain (ms2-MCP and boxB-λN22, respectively) (Figure S6), which demonstrates that the ms2-MCP and boxB-λN22 systems fulfill the selective RNA recruitment criterion.
Figure S6.

BoxB- and ms2-based mRNA recruitment systems fulfill the selective RNA recruitment criterion, related to Figure 3
To test if mRNAs tagged with boxB or ms2 loops are selectively recruited to their respective organelles, the two dual-color reporters were co-expressed with membrane-localized OT organelles that either had no RNA recruitment domain, MCP or λN22 peptides.
(A,B) Besides the indicated OT organelles and reporters, cells also expressed tRNAPyl and were incubated with the ncAA SCO-K. As a control, cells were transfected with a construct encoding a cytoplasmic PylRS variant. Shown is the sum of at least three independent experiments. (A) FFC analysis for the ms2 reporter reveals selective mCherry expression only in the presence of the MCP-based OT organelles. (B) Analogously, boxB-tagged mCherry is only selectively expressed in the presence of λN22-based OT organelles. PylRSAF and PylRSAA exhibit orthogonal substrate specificities. Note that the black line in the FFC dot plots indicates the EGFP signal cut-off used to identify transfected cells.
(C) Heatmaps showing the fold change in selectivity and relative efficiency corresponding to the FFC data shown in (A,B). For both the selectivity and the efficiency, the mean value of the three experiments is displayed.
To further corroborate that the systems work selectively, we developed a doubly tagged imaging reporter in which the nucleoporin 153 (Nup153) was fused to EGFP and boxB loops, and the histone H2B was fused to mCherry and ms2 loops; both constructs contained an amber codon at a permissive site (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2). Because Nup153 is a nucleoporin of the nuclear pore complex and H2B is a histone, we would expect to observe EGFP fluorescence at the nuclear envelope and mCherry fluorescence in the nucleus. We co-expressed these two proteins with the different OT film-like organelles and analyzed cells using fluorescence microscopy. For all constructs, we observed that Nup153::EGFP149TAG was produced only in the presence of a λN22-based OT organelle, whereas H2B::mCherry190TAG was produced only in the presence of an MCP-based organelle (Figure 2F). Importantly, these proteins were observed at their expected subcellular localization. Note that these two constructs have also much larger mRNAs than the simple EGFP and mCherry models used in the FFC experiments above, underlining the robustness of the method.
OT film-like organelles that fulfill the distinct ncAA specificity criterion
Next, we addressed the distinct ncAA specificity criterion. Over 100 different ncAAs have been genetically encoded by PylRS (Wan et al., 2014). This was facilitated by the lack of affinity of the wild-type (WT) enzyme for canonical amino acids and a few mutants having substrate promiscuity for ncAAs similar in structure to pyrrolysine (Fekner et al., 2009; Kavran et al., 2007; Krzycki, 2005; Mukai et al., 2008; Polycarpo et al., 2004, 2006; Wang et al., 2012; Yanagisawa et al., 2008b, 2008a). For example, the PylRSAF (Y306A, Y384F) mutant (Mukai et al., 2008) has been used to encode over 20 lysine derivatives (Nikić et al., 2014, 2016; Plass et al., 2011, 2012; Yanagisawa et al., 2019). However, when aiming to encode two different ncAAs via distinct PylRS variants inside the same cell, any form of cross-affinity between substrates due to the inherent promiscuity of the PylRS would be detrimental.
In the experiments described to this point, we used PylRSAF to incorporate a bulky cyclooctyne-lysine derivative (SCO-K). Previously, we used PylRSAA (N346A, C348A) (Wang et al., 2013) to encode 3-iodophenylalanine (3-IF) in OT organelles (Reinkemeier et al., 2019). We now identified that these two PylRS variants have distinct ncAA specificity and that PylRSAF selectively uses SCO-K, whereas PylRSAA accepts 3-IF (Figure S1D).
Obtaining multiple functional, fully orthogonal OT organelles
Finally, we combined our solutions to the issues of assembly, RNA recruitment and ncAA specificity, to obtain two fully orthogonal GCE organelles inside the same cell: one λN22-based OT system that incorporates one type of ncAA into a selected POI with boxB-tagged mRNA, and an MCP-based OT system that incorporates a different type of ncAA into a different POI with ms2-tagged mRNA. By contrast, the canonical host machinery in the cytoplasm processes neither efficiently.
To test our systems, we developed another dual-color fluorescence reporter in which EGFP39TAG mRNA is fused to boxB loops and mCherry190TAG mRNA is fused to ms2 loops; we term this the “double-recruitment reporter” (EGFP39TAG::boxB, mCherry190TAG::ms2). This reporter should yield an FFC diagonal if both ncAAs (SCO-K and 3-IF) are present in the growth medium, and a horizontal (EGFP39TAG::boxB) or vertical (mCherry190TAG::ms2) population if only one ncAA is present.
Here, we focused on the most selective OT organelle systems: PMPMCP or ERMPMCP together with OMMPλN22. To our initial surprise, although our OT organelles met the orthogonality criteria tested, none of the combinations yielded two organelles that are orthogonal to each other when expressed in one cell (Figure S7). We speculated that the internal tendency of PylRS to function as a dimer was an unexpected complication. We found out that the issue can be bypassed by creating a fusion protein of two PylRS mutants (see Table S2 for design details and Figure S7 for further controls). In the following, we refer to these systems as “dimeric,” indicated by a superscript "v2" (Figure 5).
Figure S7.

GCE of boxB- and ms2-tagged mRNA in the presence of λN22- or MCP-based OT organelles, related to Figure 5
(A,C,F) FFC analysis of HEK293T cells expressing the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2) together with tRNAPyl and the indicated GCE systems in the presence of the indicated ncAAs. Shown is the sum of at least three independent experiments.
(A) For these experiments, 400 ng of reporter and tRNA plasmid were co-transfected with 100 ng of the OT organelle construct and 300 ng mock plasmid. In the presence of MCP-based OT organelles, mCherry expression predominates, whereas in the presence of λN22-based OT organelles full-length EGFP is mostly produced.
(B) Bar graph showing the ratio of the mCherry signal to the EGFP signal corresponding to the conditions in panel B, normalized to a cytoplasmic GCE system (see A). The ratio is shown on a logarithmic scale, so that positive logarithmic values indicate a higher expression of mCherry compared to EGFP, while a negative logarithmic ratio corresponds to an excess of EGFP over mCherry, a ratio close to zero indicates that EGFP and mCherry are equally expressed.
(C) For these experiments, 400 ng of reporter and tRNA plasmid were co-transfected with 100 ng of each OT organelle construct and 200 ng mock plasmid. To our surprise, although all of our OT organelles obeyed all the orthogonality criteria tested, including orthogonality to the host machinery in the cytoplasm (A and B), unexpectedly, none of the combinations yielded two organelles that are orthogonal to each other when expressed in one cell. If OMMPλN22,AF was combined with PMPMCP,AA, full-length EGFP39TAG and mCherry190TAG expression was observed independently of the added ncAA. Similarly, if OMMPλN22,AA was combined with ERMPMCP,AF, substantial expression of both full-length EGFP39TAG and mCherry190TAG was observed under all conditions.
A possible explanation for this selectivity loss is that one or more of the organelle components “leaks” and shuttles between the two organelles by an unexpected mechanism.
mRNA recruitment is likely not the cause of selectivity loss as we demonstrated that it is selective between the different recruiting systems (Figure S6). We also demonstrated that the OMMP system is orthogonal to the PMP and ERMP systems (Figures 3 and S1) thus organelle assembly per se cannot cause selectivity loss.
Another explanation would be that after aminoacylation, the tRNAPyl (likely in a complex with elongation factors) could shuttle between organelles. Note that each organelle charges the same tRNAPyl with a different ncAA. However, to this end it would diffuse through the cytoplasm destroying selectivity to cytoplasmic translation which we specifically tested (Figure 2). Further we showed that tRNAPyl can be concentrated along a steep gradient in FISH stainings (Figure 4). Thus, diffusion of aminoacylated tRNAPyl is unlikely the cause of selectivity loss.
As PylRS is known to be active as a dimer (Kavran et al., 2007; Wan et al., 2014), another speculation was that dimerization prior to proper membrane localization might mistarget some synthetase molecules before they are sorted into their subcellular destination.
(D) Bar graph similar to the one in (B) corresponding to the conditions in panel C, normalized to a cytoplasmic GCE system (see panel A).
(E) To investigate this, we performed IF staining against two synthetase variants—one targeted to the PM or ERM and the other to the OMM (staining against MCP and λN22 corresponding to the conditions in panel C). We observed that the PM- or ERM-targeted synthetases indeed mix with the OMM-targeted synthetases. This is particularly pronounced for the PM-targeted synthetase, of which a substantial fraction relocalizes to the OMM. Top to bottom: MCP, λN22, mCherry, EGFP, and merged (MCP in yellow, λN22 in blue, mCherry in magenta, EGFP in green). Scale bars: 20 μm.
Gratifyingly, when making a dimeric PylRS construct in form of a PylRS::FUS::PylRS fusion, we obtained the desired mutually orthogonal film-like organelles. While further mechanistic understanding of this effect might be desirable and the goal of future studies, for the scope of this work it is important that using the dimeric system, two of our membrane associated, condensated film-like organelles function as designed and orthogonal to each other within the same cell.
(F) In these experiments, 400 ng of reporter and tRNA plasmid were co-transfected with 100 ng of the OT organelle construct and 300 ng mock plasmid. In the presence of MCP-based OT organelles, mCherry expression was predominant, whereas in the presence of λN22-based OT organelles, full-length EGFP is mostly produced.
(G) Bar graph similar to the one in (B) corresponding to the conditions in panel F, normalized to a cytoplasmic GCE system (see panel A).
For the analysis in (B,D,G) untransfected cells were not taken into account (determined by a minimum EGFP/mCherry fluorescence level as indicated by the lower left quadrant in the corresponding FFC dot plots). All bar graphs show the mean value of all experiments; error bars represent the SD.
Figure 5.

Creating multiple, fully orthogonal GCE systems within the same eukaryotic cell
(A) Schematic overview of the dimeric OT film-like organelles and FFC analysis.
(B) FFC analysis of HEK293T cells expressing the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2) together with tRNAPyl and the optimized GCE systems. Experiments were performed in the presence of the indicated ncAAs. The sum of four independent experiments is shown.
(C) Bar graph showing the ratio of the mCherry signal to the EGFP signal corresponding to the conditions in (B), normalized to a cytoplasmic GCE system (see Figure S7A). The ratio is shown on a logarithmic scale; positive logarithmic values indicate a higher expression of mCherry compared to EGFP; negative logarithmic ratios correspond to an excess of EGFP over mCherry; a ratio close to zero indicates similar EGFP and mCherry fluorescence levels. For this analysis, untransfected cells were not considered (determined by a minimum EGFP/mCherry fluorescence level as indicated by the lower left quadrant in B). The mean values of all experiments are shown; error bars represent the SD.
(D) IF staining against MCP and λN22 corresponding to the conditions in (A)–(C). For all systems the dimeric OT film-like organelles were observed at their expected location. Top to bottom: MCP, λN22, mCherry, EGFP, and merged (MCP in yellow, λN22 in blue, mCherry in magenta, EGFP in green). Scale bars, 20 μm.
(E) HEK293T cells expressing the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2) together with tRNAPyl and the optimized GCE systems. Experiments were performed in the presence of the ncAAs indicated and confirmed by imaging the results from the FFC data in (B). Scale bars, 20 μm.
Using the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2), we evaluated the performance of the dimeric systems and observed that by using the λN22-based, OMM-targeted system, full-length EGFP was predominantly produced in the presence of SCO-K (EGFP/mCherry ratio of 17 for OMM-λN22-OTv2,AF) (Figures S7F and S7G). In an analogous experiment, using the MCP-based system targeted to the PM, only mCherry was produced in the presence of 3-IF (mCherry/EGFP ratio of 17 for PM-MCP-OTv2,AA) (Figures S7F and S7G).
We then tested the combined dimeric systems in one cell by co-expressing the OMM-λN22-OTv2,AF system together with PM-MCP-OTv2,AA. Using the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2) for these systems, we observed that in the presence of SCO-K, specific for the OMM-targeted, λN22-based system, EGFP was selectively produced (EGFP/mCherry ratio of 7, normalized to a cytoplasmic GCE system). If 3-IF, specific for the PM-targeted MCP-based systems, was used, mCherry fluorescence was predominant (mCherry/EGFP ratios of 6). As expected, only if both ncAAs were present in the growth medium did we observe production of both EGFP and mCherry, corresponding to mCherry/EGFP ratios of ∼1 (Figures 5B, 5C, and S7A). Similarly, we observed the expected selectivity for a combination of OMM-λN22-OTv2,AA and ERM-MCP-OTv2,AF.
We next performed IF stainings for the combined dimeric systems, confirming that they do not mix, and the OMM-targeted systems remained clearly separate from either the PM- or the ERM-directed dimeric OT organelles (Figure 5D).
To confirm that we had generated a cell in which two organelles execute distinct translational programs, we used the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2). For all of the constructs tested, we observed that full-length Nup153::EGFP149TAG was produced only in the presence of the ncAA specific for the λN22-based OT organelle, and full-length H2B::mCherry190TAG was produced only in the presence of the ncAA specific for the MCP-based organelle. Only in the presence of both ncAAs were both proteins produced (Figure 5E).
To validate site-specific incorporation of the ncAAs, we purified EGFP and mCherry from HEK293T cells co-expressing the OMM-λN22-OTv2,AF system together with PM-MCP-OTv2,AA, the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2) and tRNAPyl, and performed qualitative coupled liquid chromatography mass spectrometry (LC-MS/MS) analysis of peptides resulting from tryptic digest. Here, we used the ncAA 3-IF together with the lysine derivative N-ε-benzyloxycarbonyl-lysine (Cbz-K) that is selectively only incorporated by PylRSAF, but not by PylRSAA (Figure S1D). Data S1 show that we can detect mCherry modified with 3-IF at position 190 and EGFP containing Cbz-K at position 39 reliably. However, MS/MS is a very sensitive technique, and even our best performing film-like organelles only show 20-fold selectivity. We thus expectedly also detected the peptides for EGFP with 3-IF and mCherry with Cbz-K.
Unfortunately, the used MS/MS approach does not allow for an easy quantification of protein abundance and thus cannot answer directly how much 3-IF and Cbz-K are respectively incorporated into EGFP and mCherry. However, the ratio of incorporated ncAAs in each protein is key to evaluate if these dual orthogonal systems are functionally independent enough to selectively modify desired proteins. We thus devised a site-specific bioorthogonal chemistry approach to probe if the two organelles selectively incorporate distinct ncAAs, which we aimed to label selectively with a small-molecule dye. To this end, we first established that we could use two pairs of ncAAs in the respective organelle, one of which can participate in a ligation with a tetrazine-fluorophore derivative whereas the other remains unreactive. Specifically, we used a combination of the established 3-IF with the axial isomer of trans-cyclooct-2-ene-lysine (TCO∗A-K). 3-IF is accepted by PylRSAA and TCO∗A-K by PylRSAF (Figure S1D). TCO∗A-K rapidly reacts with a tetrazine in a strain-promoted inverse electron-demand Diels-Alder cycloaddition (Nikić et al., 2014; Plass et al., 2012; Reinkemeier et al., 2021; Wagner et al., 2015) (Figure 6A), which can be selectively detected by fluorescence if a suitable tetrazine-dye conjugate is used. The nonreactive 3-IF cannot be easily detected by such an unambiguous method, and it is thus not easy to confirm that the other dimeric film-like organelle also performs as selectively as the FFC and imaging experiments described above indicate. Therefore, following established procedures (Kurra et al., 2014), we synthesized a compound in which a cyclooctyne side chain is mounted onto a tyrosine scaffold (SCO-Y), which readily reacts with a tetrazine. Gratifyingly, SCO-Y was also recognized by PylRSAA but not by PylRSAF (Figure S1D). This allowed us to use a combination of the nonreactive Cbz-K and SCO-Y (Figures 6B and S1D) to validate proper specificity of the PylRSAA containing organelles.
Figure 6.

Incorporation of distinct ncAAs by film-like organelles enables specific, bioorthogonal protein labeling in cells
(A and B) Schematic overviews of the expected reactions. From a mixture of 3-IF and TCO∗A-K, only TCO∗A-K can react with H-Tet-SiR. Analogously, in a separate experiment, from a mixture of SCO-Y and Cbz-K, only SCO-Y can react (B).
(C–F) The doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2) was expressed together with tRNAPyl and one of the indicated optimized GCE systems in presence of indicated ncAAs. The cartoons shown above the images depict the expected results. Top to bottom: EGFP, mCherry, SiR, and merged channel. H-Tet-SiR was used at a concentration of either 200 nM (C–E) or 1 μM (F). The contrast was adjusted for maximum visibility for each condition. Scale bars, 10 μm.
First, we combined OMM-λN22-OTv2,AF with PM-MCP-OTv2,AA in cells that expressed the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2). Cells were incubated with TCO∗A-K and 3-IF during protein expression and then washed extensively so that only ncAA incorporated faithfully into the protein of choice can be labeled with a silicon rhodamine-tetrazine conjugate (H-Tet-SiR) (Lukinavičius et al., 2013). Under the fluorescence microscope, we observed both Nup153::EGFP and H2B::mCherry fluorescence. Importantly, after extensive washing to remove leftover dye, the SiR fluorescence colocalized predominantly with the Nup153::EGFP signal. Because only TCO∗A-K, and not 3-IF, can react with H-Tet-SiR, this demonstrates that only full-length Nup153::EGFP149 TCO∗A-K and H2B::mCherry190 3-IF were produced (Figure 6C). Correspondingly, if the cells were incubated with Cbz-K and SCO-Y, the SiR fluorescence colocalized mainly with the H2B::mCherry signal. Because only SCO-Y (and not Cbz-K) can react with tetrazines, this indicates the production of full-length Nup153::EGFP149 Cbz-K and H2B::mCherry190 SCO-Y (Figure 6D).
Second, we analogously probed the selectivity of the combination of OMM-λN22-OTv2,AA with ERM-MCP-OTv2,AF. Accordingly, if cells were incubated with 3-IF and TCO∗A-K, the SiR fluorescence colocalized only with the H2B::mCherry signal, which showed that the cells produced Nup153::EGFP149 3-IF and H2B::mCherry190 TCO∗A-K (Figure 6E). Correspondingly, if Cbz-K and SCO-Y were used the SiR fluorescence was observed at the nuclear rim, colocalizing with Nup153::EGFP. Therefore, the reactive SCO-Y was mainly incorporated into Nup153::EGFP149 SCO-Y, whereas the unreactive Cbz-K was inserted into H2B::mCherry190 Cbz-K (Figure 6F).
These data demonstrate that the respective ncAAs are incorporated predominantly and very selectively into the targeted protein within the respective dimeric OT film-like organelle.
Discussion
Our findings show that if we apply concepts of 2D phase separation at a cellular membrane, the interface with that membrane becomes a powerful platform for bioengineering. Using this approach, we can build multiple independent and functionally orthogonal enzymes in one cell by spatial organization. We show the feasibility of this approach for the complex process of protein translation, which requires hundreds of factors to work in concert. We apply these systems to specifically modify different proteins with different ncAAs, effectively equipping cells with two expanded genetic codes, or put another way, a second and third genetic code in the same cell. Our data appear consistent with the illustration in Figure 7. The combination of protein condensation and membrane mounting leads to a sharp suppressor tRNAPyl gradient. Using super-resolution microscopy, we determined a gradient with a full width at half-maximum of less the 100 nm for the line-shaped PMP system (Figure 4). Furthermore, we observed that the OMMP and ERMP systems work selectively, even though a substantial portion of the cellular interior is filled with mitochondria and ER (Figure S3). This body of data is direct evidence that the suppressor tRNAPyl—despite being relatively small and not being tethered itself—can be confined remarkably well, and after it is aminoacylated with its specific ncAA, it does not leave the respective organelle before being consumed by translation. This can be viewed as a distinct biochemical microenvironment, which shows that a combination of protein condensation and membrane targeting can substantially alter biomolecular composition with very high precision. This feature enables us to simultaneously use the same suppressor tRNAPyl in different film-like organelles, to engineer different ncAAs into different proteins. To do so, only the respective PylRS variants and mRNA-binding proteins need to be directly fused to the organelle scaffolds, whereas all other components of the translational machinery remain shared between the organelles and the host. This demonstrates the power of creating thin, sub-resolution biochemical microenvironments in cells (Figures 5, 6, and 7). As shown in Figure S7, it is an experimental observation that making PylRS dimer fusion constructs further enabled achieving the desired orthogonality between two different film-like organelles, and we speculate that this is because PylRS activity is dimerization dependent (Kavran et al., 2007; Wan et al., 2014).
Figure 7.

OT film-like organelles form thin translational microenvironments effectively equipping cells with two expanded genetic codes
Schematic illustration of independent protein translation microenvironments in cells. Here, thin film-like organelles are installed on the PM or ERM, and the amber codon is reassigned to a specific ncAA (X) for selected proteins. Simultaneously, a second OT film-like organelle is mounted on the OMM, reassigning the amber codon to a second ncAA (Y). Meanwhile, translation proceeds canonically in the cytoplasm and is terminated once the ribosome encounters the amber codon.
Major advances in enzyme engineering have been made in recent decades. These can be broadly classified as either directed evolution (Arnold, 2019) or computer-aided rational design (Huang et al., 2016a). Directed evolution is powerful for optimizing and refining existing catalytic activities and can also be used to create enzyme functions that do not exist in nature. Computer-aided de novo protein design is particularly suitable for designing completely novel structures because it allows sampling of a vast sequence space in silico. The resulting designs are often expressed and tested in E. coli (Cao et al., 2020; Glasgow et al., 2019; Pan et al., 2020; Xu et al., 2020). More recently, strategies for evolving mammalian systems have emerged (Berman et al., 2018; English et al., 2019; Piatkevich et al., 2018), although E. coli and yeast remain the preferred hosts for handling large libraries (Almhjell et al., 2018; Branon et al., 2018; Bryson et al., 2017). Our concept of diversifying and repurposing the same enzyme multiple times by means of spatial compartmentalization could even be combined with those strategies, thus extending the repertoire of enzyme functionality that can be engineered within the same eukaryotic cell.
It has long been known that cells use their membranes for internal organization and to process signaling events. By analogy, membraneless organelles are a means to organize cellular functionality (Alberti, 2017; Banani et al., 2017; Mitrea and Kriwacki, 2016). Only more recently has the combination of the two come into focus. Membrane-associated phase separation is inherently a two-dimensional process (Banjade and Rosen, 2014; Li et al., 2012). The distinct localization of membrane-associated phase separation primes it to be involved in cellular signaling, and indeed, two-dimensional condensates are important in T cell receptor signaling (Ditlev et al., 2019; Huang et al., 2016b, 2019; Su et al., 2016), in the postsynaptic densities of neurons (Zeng et al., 2018), and in nephrin signaling (Case et al., 2019b; Kim et al., 2019). Both membrane binding and phase separation at membranes concentrate biomolecules and these are not mutually exclusive events. However, in this study, phase separation was essential in three out of four cases (Figures 3 and S1A). One explanation for the OT organelles on the plasma membrane to work similarly with or without additional protein condensation might be that the shell-like geometry of the plasma membrane already offers a sufficiently large spatial separation from the rest of the cell for OT organelles to function. The patchy appearance of the organelles we observed in Video S1 is in line with a role of membrane associated protein condensation.
Our work expands the utility of thin-film condensates by equipping them with the capability to perform orthogonal protein translation. We show that we can change the language of protein synthesis over a distance of a few tens of nanometers by forming thin translational microenvironments (Figure 7), moving the near-membrane space (below 100 nm) into focus as a signaling and engineering environment. A translational program, in which one codon is reassigned to a specific ncAA is typically described as an expanded genetic code. Because we do this twice, one can argue that we obtained a eukaryotic cell with one canonical genetic code and two expanded genetic codes. Furthermore, in an OT system, all that is needed to reprogram opal or ochre stop codons instead of the amber codon requires changing the codon in the POI and the anticodon in the suppressor tRNAPyl (Reinkemeier et al., 2019).
It is tempting to speculate that nature might select membrane-associated phase separation if the biochemical microenvironment resulting from either phase separation or membrane binding alone is insufficient to achieve the level of specificity required for a certain process. Hence, these insights might have implications for understanding how the complexity of living matter arose and how compartmentalization in membrane-associated condensates could facilitate diverging evolution.
Limitations of the study
The proof-of-concept experiments described here were performed as transient transfections in HEK293T cells under GCE conditions. Thus, we cannot definitively exclude a long-term influence of the synthetic organelles on the normal physiology of the cell, and we cannot assess whether the organelle represents a heavier burden on the cell than GCE. This warrants further investigation. However, morphological changes were not apparent in stainings against various cellular structures (i.e., plasma membrane, Golgi, ER, and mitochondria), and we did not observe major alterations in cell growth compared to HEK293T cells transfected with the cytoplasmic PylRS system. Furthermore, cells that expressed the OT film-like organelles still performed protein translation.
Using LC-MS/MS, we were able to identify the correctly inserted ncAAs. In line with what is expected from a system with finite selectivity, and due to the sensitivity of MS/MS also the other two possible ncAA modified peptides were detectable. However, our MS/MS approach did not allow us to quantify how much of each ncAA was incorporated in each protein. To evaluate this, we performed bioorthogonal labeling experiments (see Figure 6) that highlight that the desired ncAA is predominantly incorporated by each organelle.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-PylRS | (Nikić et al., 2016) | RRID: AB_2893022 |
| Mouse anti-HA | Sigma-Aldrich | Cat# H9658; RRID: AB_260092 |
| Rabbit anti-Myc | Abcam | Cat# ab9106; RRID: AB_307014 |
| Rabbit anti-AIF | Abcam | Cat# ab32516; RRID: AB_726995 |
| Rabbit anti-Giantin | Abcam | Cat# ab80864; RRID: AB_10670397 |
| Rabbit anti-Calnexin | Abcam | Cat# ab22595, RRID: AB_2069006 |
| Rabbit anti-NaK-ATPase | Abcam | Cat# ab76020; RRID: AB_1310695 |
| Rabbit anti-RPL26L1 | Abcam | Cat# ab137046; RRID: AB_2893020 |
| Goat anti-rat-Alexa Fluor 405 | Abcam | Cat# ab175671; RRID: AB_2890626 |
| Goat anti-rat-Alexa Fluor 405 | Abcam | Cat# ab175673; RRID: AB_2893021 |
| Goat anti-rabbit-Alexa Fluor 647 | ThermoFisher | Cat# A-21246; RRID: AB_2535814 |
| Goat anti-mouse-Alexa Fluor 647 | ThermoFisher | Cat# A32728; RRID: AB_2633277 |
| Goat anti-mouse-Alexa Fluor 405 | ThermoFisher | Cat# A-31553; RRID: AB_221604 |
| Chemicals, peptides, and recombinant proteins | ||
| Cyclooctyne lysine (SCO-K) | SiChem | SC-8000 |
| 3-iodophenylalanine (3-IF) | Chem-Impex International Inc. | 14352 |
| Cyclooctyne tyrosine (SCO-Y) | This paper | N/A |
| Axial trans-cyclooct-2-ene-lysine (TCO∗A-K) | SiChem | SC-8004 |
| N-ε-benzyloxycarbonyl-lysine (Cbz-K) | Sigma-Aldrich | 96840 |
| DAPI | Sigma-Aldrich | D9542 |
| Hoechst 33342 | Sigma-Aldrich | B2261 |
| Catalase | Sigma-Aldrich | C3155 |
| Glucose oxidase | Sigma-Aldrich | G7141 |
| β-Mercaptoethylamine | Sigma-Aldrich | 411000 |
| Polyethyleneimine | Sigma-Aldrich | 408727 |
| H-Tet-SiR (silicone rhodamine tetrazine) | Spirochrome | SC008 |
| VRC (ribonucleoside vanadyl complexes) | Sigma-Aldrich | 94740 |
| E. coli tRNA | Sigma-Aldrich | R1753 |
| Dextran sulfate | Sigma-Aldrich | D8906 |
| Experimental models: cell lines | ||
| HEK293T cells | ATCC | CRL-3216 |
| Oligonucleotides | ||
| 5′-(Cy5)-ATCTTGAAGTTGGCCTTGATGCCGTT CTTCTGCTT-3′ |
IBA GmbH | N/A |
| 5′-(Alexa647)-CTGCAGACATGGGTGATCCTCA TGTTTTCTA-3′ |
IBA GmbH | N/A |
| 5′-(Alexa647)-CTAACCCGGCTGAACGGATTTA GAGTCCATTCGATC-3′ |
Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| Plasmids described in Constructs, cloning and mutagenesis, see Table S1 for further details | This paper | N/A |
| UbC NLS-HA-MCP-YFP | Grünwald and Singer, 2010 | Addgene #31230 |
| pDEST_FUS | Hoell et al., 2011 | Addgene #26374 |
| pDEST_EWSR1 | Hoell et al., 2011 | Addgene #26377 |
| pcDNA3.1/Zeo | ThermoFisher | V86020 |
| pBI-CMV1 | Clontech | 631630 |
| Software and algorithms | ||
| FlowJo | BD | https://www.flowjo.com |
| Fiji | Schindelin et al., 2012 | https://fiji.sc/ |
| arivis Vision4D | arivis AG | https://imaging.arivis.com/en/imaging-science/arivis-vision4d |
| Igor Pro | WaveMetrics | https://www.wavemetrics.com/ |
| Prism | GraphPad | https://www.graphpad.com/ |
| ChemDraw 20.0 | PerkinElmer | https://perkinelmerinformatics.com/products/research/chemdraw/ |
| IsobarQuant | Franken et al., 2015 | https://codeload.github.com/protcode/isob/zip/1.0.0 |
| Mascot V2.4 | Matrix Science | http://www.matrixscience.com/mascot_support_v2_4.html |
| Adobe Illustrator 2021, Adobe Premiere Pro 2020 | Adobe | https://www.adobe.com/creativecloud.html |
| Other | ||
| RFP-Trap Magnetic Agarose | ChromoTek | rtma-20 |
| GFP-Trap Magnetic Agarose | ChromoTek | gtma-20 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Edward A. Lemke (edlemke@uni-mainz.de).
Materials availability
All plasmids can be obtained via an EMBL or JGU materials transfer agreement (free of charge for noncommercial purposes).
Experimental model and subject details
HEK293T cells
HEK293T cells (ATCC CRL-3216) were maintained in DMEM (Life Technologies 41965-039) supplemented with 1% penicillin-streptomycin (Sigma P0781), 1% L-Glutamine (Sigma G7513), 1% sodium pyruvate (Life Technologies 11360), and 10% FBS (Sigma F7524). Cells were cultured at 37°C in a 5% CO2 atmosphere and passaged every 2-3 days up to 20 passages. To this end, cells were typically washed with PBS, detached using trypsin-EDTA (0.05%, with phenol red, ThermoFisher) and resuspended in fresh culture medium.
Method details
Cell culture
In all cases, cells were seeded 15-20 h prior to transfection at a density resulting in 70%–80% confluency at the time of transfection. Flow cytometry was performed using 24-well plates with plastic bottom (Nunclon Delta Surface, ThermoFisher). Immunofluorescence labeling and FISH were performed on 24-well plates with glass bottom (Greiner Bio-One), four-well chambered Lab-Teks #1.0 borosilicate coverglass (ThermoFisher), four-well cell imaging coverglasses (Eppendorf), glass bottom 4 well μ-slides (ibidi) or eight-well chambered Lab-Teks #1.0 borosilicate coverglass (Thermo Fisher). EGFP and mCherry immunoprecipitation (IP) experiments were performed using 100 mm or 150 mm round cell culture dishes (CELLSTAR, Greiner Bio-One).
Constructs and cloning
Table S1 provides an overview of all plasmids used in this study.
Table S2 provides an overview of the sequences of all final film-like organelle constructs.
Table S3 provides an overview over all dual color reporters used in this study.
Reporters
The dual color reporters were cloned in a pBI-CMV1 vector (Clontech 631630). The dual color ms2 reporter (EGFP39TAG, mCherry190TAG::ms2) was described before (Reinkemeier et al., 2019). The dual color boxB reporter (EGFP39TAG, mCherry190TAG::boxB) was constructed by replacing the ms2 loops in the dual color ms2 reporter with four boxB loops via restriction cloning (Daigle and Ellenberg, 2007).
To clone the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2) one multiple cloning site (MCS) of pBI-CMV1 was tagged with four boxB loops and the other with two ms2 loops. Subsequently, mCherry190TAG and EGFP39TAG were inserted into the MCSs. For imaging experiments similar double tagged pBIs were used, in which only the EGFP39TAG gene was inserted into the ms2 or boxB loops tagged MCS. EGFP39TAG or mCherry190TAG were used as N-terminal fusions with nuclear localization sequences (NLS).
Analogously, to clone the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2), also Nup153::EGFP149TAG was inserted into one MCS via restriction cloning, while H2B::mCherry190TAG was inserted into the other via Gibson assembly (Gibson et al., 2009). The template for H2B was a kind gift from the Ellenberg laboratory.
The iRFP::EGFP39TAG construct used in Figure S1D was published before (Nikić et al., 2016).
OT organelle constructs
tRNAPyl was cloned under the control of a human U6 promoter, and all other constructs were under CMV promoters cloned in a pcDNA3.1 vector (Invitrogen V86020) as described previously (Reinkemeier et al., 2019). The template for the 4xλN22 peptides was a kind gift from the Ellenberg laboratory (Daigle and Ellenberg, 2007). They were generally used with an N-terminally fused c-Myc tag (EQKLISEEDL), MCP constructs were fused to an HA-tag (YPYDVPDYA) unless otherwise indicated.
The MCP protein was cloned from the addgene plasmid #31230 (Grünwald and Singer, 2010), FUS from the addgene plasmid #26374 and EWSR1 from the addgene plasmid #26377 (Hoell et al., 2011). In all FUS fusions, amino acids 1-478 were used, in all EWSR1 fusions, amino acids 1-628. In all PylRS fusions the previously reported efficient NES::PylRSAF (Y306A, Y384F) was used (Nikić et al., 2016). Alternatively, when specifically indicated NES::PylRSAA (N346A, C348A) was used.
The murine LCK1-10 sequence (MGCVCSSNPE) (Zlatkine et al., 1997), was fused to PylRSAF, FUS::PylRSAF, MCP or EWSR1::MCP (Reinkemeier et al., 2019) via restriction cloning and to EWSR1::4xλN22 via Gibson assembly. To clone LCK1-10::FUS::MCP::PylRSAF and LCK1-10::FUS::4xλN22::PylRSAF, MCP and 4xλN22 respectively were inserted into LCK1-10::FUS::PylRSAF via restriction cloning.
The rabbit CYPIIC11-27 sequence (MDPVVVLGLCLSCLLLLSLWKQSYGGG) (Hung et al., 2017) was fused to PylRSAF, FUS::PylRSAF, MCP or EWSR1::MCP and EWSR1::4xλN22 via restriction cloning. To clone CYPIIC11-27::FUS::MCP::PylRSAF and CYPIIC11-27::FUS::4xλN22::PylRSAF, MCP and 4xλN22 respectively were inserted into CYPIIC11-27::FUS::PylRSAF via restriction cloning.
TOM201-70 (MVGRNSAIAAGVCGALFIGYCIYFDRKRRSDPNFKNRLRERRKKQKLAKERAGLSKLPDLKDAEAVQKFF) was cloned from human cDNA and inserted into pcDNA3.1 via restriction cloning. It was subsequently fused to PylRSAF, FUS::PylRSAF, EWSR1::MCP, EWSR1::4xλN22, MCP (without an HA-tag) and FUS::4xλN22::PylRSAF via Gibson assembly. To clone TOM201-70::FUS::MCP::PylRSAF, MCP was inserted into a TOM201-70::FUS::PylRSAF containing construct via restriction cloning.
Full-length EBAG9 was cloned from human cDNA and inserted into pcDNA3.1 via restriction cloning. Subsequently, EBAG91-29 (MAITQFRLFKFCTCLATVFSFLKRLICRS) was fused to PylRSAF, FUS::PylRSAF, MCP, EWSR1::MCP or EWSR1::4xλN22 via Gibson assembly. To clone EBAG91-29::FUS::MCP::PylRSAF and EBAG91-29::FUS::4xλN22::PylRSAF, MCP and 4xλN22 were inserted into the EBAG91-29::FUS::PylRSAF construct via restriction cloning.
To obtain LCK1-10::FUS::MCP::PylRSAA and TOM201-70::FUS::4xλN22::PylRSAA, the PylRS variant was exchanged in the corresponding PylRSAF based constructs via restriction cloning.
Internally linked PylRS dimers
First, OMM-λN22-OTv2,AF (TOM201-70::EWSR1::4xλN22::PylRSAF::FUS::PylRSAF), OMM-λN22-OTv2,AA (TOM201-70::EWSR1::4xλN22::PylRSAA::FUS::PylRSAA) and ERM-MCP-OTv2,AF (CYPIIC11-27::EWSR1::MCP::PylRSAF::FUS::PylRSAF) were assembled via Gibson assembly in pcDNA3.1 vectors. PM-MCP-OTv2,AA (LCK1-10::MCP::PylRSAA::FUS::PylRSAA) was subsequently cloned via restriction cloning. Then OMM-λN22-OTv2,AF, OMM-λN22-OTv2,AA, ERM-MCP-OTv2,AF and OMM-λN22-OTv2,AA were inserted into one of the two multiple cloning sites in a bicistronic amber suppression vector via restriction cloning [pBI-CMV1 with tRNAPyl under the control of a hU6 promoter as described previously (Reinkemeier et al., 2019)]. For bicistronic constructs ERM-MCP-OTv2,AF was inserted into the free multiple cloning site of the pBI-CMV1 with OMM-λN22-OTv2,AA, and PM-MCP-OTv2,AA into the one of the OMM-λN22-OTv2,AF. The annotated amino acid sequence of the four dimeric constructs is shown in Table S2.
Transfections and used noncanonical amino acids (ncAAs)
Transfections of HEK293T cells were performed using 3 μg PEI (Sigma-Aldrich 408727) per 1 μg DNA diluted in DMEM without Phenol Red (GIBCO11880-028), [1200 ng total DNA in 50 μL medium for 24-well plates and for the ms2 FISH experiment in four-well imaging cover glasses (Eppendorf); 1680 ng DNA in 70 μL medium for four-well cell imaging cover glasses (Eppendorf), or 4 well μ-slides (ibidi); 600 ng in 25 μL medium for eight-well chambered Lab-Teks (ThermoFisher); 47.1 μg DNA in 1 mL medium for 100 mm round cell culture dishes; 141.3 μg DNA in 3 mL medium for 150 mm round cell culture dishes (Greiner Bio-One)].
For amber suppression system tests, cells were transfected at a ratio of a 1:1:1:1 with POITAG vectors, tRNAPyl, and OT organelle plasmids (a mock plasmid was used to adjust the total plasmid amount to be constant). 4-6 hours after transfection the medium was exchanged to fresh one containing ncAA supplemented with 25 mM HEPES (pH = 7.25). Cells were analyzed one day after transfection.
For dual organelle FFC experiments such as e.g., in Figures 5 and S7, cells were transfected at a ratio of 4:4:1:3 with the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2), tRNAPyl, bicistronic OT organelle vector and a mock plasmid. Alternatively, they were transfected at a ratio of 4:4:1:1:2 with the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2), tRNAPyl, and two individual OT organelle vectors and a mock plasmid.
For dual organelle imaging experiments such as e.g., in Figure 5, cells were transfected at a ratio of 1:1:1 with the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2), tRNAPyl and a bicistronic OT organelle vector.
For bioorthogonal labeling experiments such as e.g., in Figure 6, cells were transfected at a ratio of 4:4:1:3 with the doubly tagged imaging reporter (Nup153::EGFP149TAG::boxB, H2B::mCherry190TAG::ms2), tRNAPyl, a bicistronic OT organelle vector and a mock plasmid.
For EGFP and mCherry IP experiments used for LC-MS/MS, cells were transfected at a ratio of 4:4:1:3 with the double-recruitment reporter (EGFP39TAG::boxB, mCherry190TAG::ms2), tRNAPyl, a bicistronic OT organelle vector (PM-MCP-OTv2,AA with OMM-λN22-OTv2,AF) and a mock plasmid.
Stock solutions for all the used ncAAs were prepared as described in previous work (Nikić et al., 2015). SCO-K (cyclooctyne lysine, SiChem SC-8000) was used at a final concentration of 250 μM; 3-iodophenylalanine (Chem-Impex International Inc., 14352) was used at a final concentration of 1 mM, TCO∗A-K (trans-cyclooct-2-ene-lysine, SiChem SC-8004) was used at a final concentration of 250 μM for FFC experiments and 20 μM for imaging experiments; Cbz-K (N-ε-benzyloxycarbonyl-lysine, Sigma 96840) was used at a final concentration of 250 μM; SCO-Y (cyclooctyne tyrosine, see chemical synthesis section for further details) was used at a final concentration of 250 μM. SCO-K, TCO∗A-K and Cbz-K are efficiently recognized by PylRSAF (Y306A, Y384F) while 3-IF and SCO-Y are recognized by PylRSAA (C346A, N348A).
Fluorescence flow cytometry
Transfections for flow cytometry were performed with four plasmids (reporter plasmid, tRNAPyl, the wanted version of synthetase and an MCP fusion or a mock plasmid) at a 1:1:1:1 ratio with 1.2 μg total DNA. Medium was exchanged for fresh medium (supplemented with 25 mM HEPES, pH = 7.25) containing the ncAA, 4-6 h post-transfection and left until the time of harvesting. HEK293T cells were harvested 1 day after transfection by removing the medium, resuspending the cells in PBS and passing them through 100 μm nylon mesh.
Data acquisition was performed in an LSRFortessa SORP Cell Analyzer (BD). Analysis was done using the FlowJo software (FlowJo). Cells were first gated by cell type (using FSC-A x SSC-A parameters) and then by single cell (SSC-A x SSC-W). The workflow of cell gating is exemplarily shown in Figure S1C. Lastly, fluorescence was acquired in the 488-530/30 channel for EGFP signal, in the 561-610/20 channel for mCherry signal and in the 640-730/45 channel for the iRFP signal. Bar plots and heatmaps were generated using Prism software (GraphPad).
In the FFC analysis we adjust the PMT settings to get the signal as diagonal as possible, while spanning 5 orders of magnitude. However, at lower fluorescence values for the cytoplasmic PylRS/tRNAPyl system we observe a slight straight vertical rise, which then goes over into a diagonal. This could be due to several different factors, such as transcription rates, detector sensitivities, PMT amplifications and offset settings, transfection-based factors, different FP folding rates etc. Most likely, the substantially higher brightness of EGFP compared to mCherry contributes to this effect. However, this result is highly reproducible for a given setting and we performed each experiment at least in three biological replicates and analyze in each case a few hundred thousand cells. For all quantitative measurements we further normalize the fluorescence values of each system to a cytoplasmic PylRS control, which was included in each experiment.
IF labeling, FISH, bioorthogonal labeling, confocal imaging, STORM imaging, and 3D reconstructions
Immunofluorescence (IF)
IF experiments were performed one day after transfection. Cells were rinsed with PBS, fixed in 2% paraformaldehyde in PBS at room temperature (RT) for 10 minutes and rinsed with PBS. Subsequently, cells were permeabilized with 0.5% Triton-x-100 solution in PBS for 15 minutes at RT and rinsed twice prior to blocking. Samples were blocked in 3% BSA in PBS for 90 minutes at RT, after which incubation with the primary antibody {AbPylRS [1 μg/mL (Nikić et al., 2014)], AbHA (Sigma H9658, 1:2000), AbMyc (Abcam ab9106, 0.5 μg/mL), AbAIF (Abcam ab32516, 0.16 μg/mL, mitochondrial marker), AbCalnexin (Abcam ab22595, 1 μg/mL, ER marker), AbGiantin (Abcam ab80864 0.8-1 μg/mL, Golgi marker), AbNaK-ATPase (Abcam ab76020, 2.4 μg/mL, plasma membrane marker) and/or AbRPL26L1 (Abcam ab137046, 1:200)} was done overnight at 4°C in blocking solution. The next day, cells were rinsed with PBS and incubated with secondary antibody (Abcam ab175671 or ab175673; ThermoFisher A-21246, A32728 and/or A-31553, at 2 μg/mL in blocking solution) for 60 minutes at RT. Finally, cells were rinsed with PBS and fresh PBS was added for imaging.
If only DNA was stained, cells were fixed and permeabilized as described above and subsequently stained with Hoechst 33342 (Sigma-Aldrich B2261) at 1 mg/mL in PBS or with DAPI (Sigma-Aldrich D9542) at 1 mg/mL in PBS for 10 min at RT. Then, cells were rinsed with PBS and fresh PBS was added for imaging.
Fluorescence in situ hybridization (FISH)
FISH experiments were performed one day after transfection analogously as described previously (Nikić et al., 2016). Briefly, the hybridization protocol was adapted from Pierce et al. (Pierce et al., 2014). Cells were washed with PBS and fixed with 4% formaldehyde in PBS for 60 minutes at RT, rinsed with PBS and permeabilized with 0.5% Triton-x-100 solution in PBS for 15 minutes at RT. Then cells were rinsed with PBS and 2xSSC (0.3 M NaCl, 30 mM sodium citrate (pH = 7.4), 50% formamide) and incubated with the hybridization buffer (0.6 M NaCl, 60 mM sodium citrate (pH = 7.4), 100 μg/μL dextran sulfate (Sigma-Aldrich, D8906), 198 ng/μL BSA, 2 mM VRC (ribonucleoside vanadyl complexes, Sigma-Aldrich, 94740), 80 ng/μL E. coli tRNA (Sigma-Aldrich R1753), 50% formamide, and the respective hybridization probe) at 37°C overnight. Subsequently, cells were washed with 4xSSC (0.6 M NaCl, 60 mM sodium citrate (pH = 7.4), 50% formamide), 2xSSC (0.3 M NaCl, 30 mM sodium citrate (pH = 7.4), 50% formamide), 1xSSC (0.15 M NaCl, 15 mM sodium citrate (pH = 7.4), 50% formamide), 0.5xSSC (75 mM NaCl, 7.5 mM sodium citrate (pH = 7.4), 50% formamide) and Tris-HCl⋅NaCl buffer (0.1 M TrisHCl, 150 mM NaCl, pH = 7.5), each for 5 minutes at RT. Cells were then incubated for 1 hour at RT in 3% BSA prior to standard immunofluorescence labeling as described above.
For imaging of only tRNAPyl, the hybridization probe (5′-(Alexa647)-CTAACCCGGCTGAACGGATTTAGAGTCCATTCGATC-3′) was used at 0.5 μM.
For imaging of mRNA:: The hybridization probe for ms2 (5′-(Alexa647)-CTGCAGACATGGGTGATCCTCATGTTTTCTA-3′) was used at 1 μM. The hybridization probe for EGFP (5′-(Cy5)-ATCTTGAAGTTGGCCTTGATGCCGTTCTTCTGCTT-3′) was used at 0.5 μM.
Bioorthogonal labeling
For bioorthogonal labeling experiments eight-well chambered Lab-Teks were coated with poly-L-lysine solution (10 μg/mL poly-L-lysine hydrobromide, Sigma-Aldrich, in water) overnight at room temperature (RT). Then, the wells were rinsed twice with PBS and cells were seeded, transfected and incubated with ncAAs overnight as described above. Subsequently, they were washed 5 times over 2 hours with fresh medium (supplemented with 25 mM HEPES, pH = 7.25). Then they were washed with DMEM without phenol red and subsequently labeled with 0.2 or 1 μM H-Tet-SiR (Spirochrome, SC008) (Lukinavičius et al., 2013) diluted in DMEM without phenol red for 45 minutes. Subsequently, cells were washed 6-8 times with fresh medium (supplemented with 25 mM HEPES, pH = 7.25) over 4 hours. Then, they were rinsed with PBS, fixed in 2% paraformaldehyde in PBS at RT for 10 minutes and finally rinsed 3x with PBS.
Confocal microscopy
Confocal images were acquired on an Olympus Fluoroview FV3000 microscope using 405 nm (Alexa405), 488 nm (EGFP), 594 nm (mCherry) and 640 nm (for Alexa 647, Cy5) lasers for excitation with a 60x/1.40 oil immersion objective. Alternatively, they were acquired on a Leica SP8 STED 3X microscope using the 405 nm (for Alexa405), 488 nm (for EGFP), 580 nm (for mCherry), 647 nm (for Alexa647, Cy5) and/or 652 nm (for SiR) laser lines for excitation, using a 63x/1.40 or 100x/1.40 oil immersion objective. Images were processed using FIJI software (Schindelin et al., 2012).
STORM imaging
STORM images of Alexa647 labeled tRNAPyl were acquired on a Leica GSDIM microscope with a 160x/1.43 oil immersion objective using a 642 nm laser for excitation. Images were acquired in a glucose oxidase-based oxygen scavenging system buffer [40 μg/mL catalase (Sigma-Aldrich C3155), 0.5 mg/mL glucose oxidase (Sigma-Aldrich G7141), 10% glucose, 10 mM β-mercaptoethylamine (Sigma-Aldrich 411000), 50 mM Tris-HCl (pH = 8), similar to (Szymborska et al., 2013)]. Images were transformed into TIF files using FIJI software (Schindelin et al., 2012) and subsequently analyzed using a customized Igor Pro software (Dedecker et al., 2012).
3D Reconstruction
3D reconstructions were made using the arivis Vision4D software (arivis AG). Videos of each reconstruction were exported from arivis Vision4D and subsequently assembled and annotated using Adobe Premiere Pro 2020 software.
LC-MS/MS Analysis of ncAA Incorporation
Purification of EGFP and mCherry from Mammalian Cells
EGFP and mCherry IP experiments were performed one day after transfection. Cells were washed with PBS, detached using trypsin-EDTA (0.05%, with phenol red, ThermoFisher) and resuspended in medium. Then cells were transferred to a 50 mL tube and centrifuged for 20 minutes at 500 xg, 20°C. The supernatant was discarded, and cells were resuspended in 1 mL of PBS and transferred to a 1.5 mL tube, then they were pelleted for 5 minutes at 500 xg, 4°C and subsequently rinsed with 500 μL PBS and then pelleted for 10 minutes at 500 xg, 4°C. Subsequently, the supernatant was discarded, and cells were frozen in liquid nitrogen and stored at −20°C until further processing.
For lysis, cells were thawed and resuspended in 200 μL RIPA buffer (150 mM NaCl, 1.0% Triton-x-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH = 8.0) supplemented with cOmplete Protease Inhibitor Cocktail (Roche, 11873580001). They were incubated on ice for 20 minutes (with pipetting every 10 minutes) and subsequently sonicated at 4°C (20x 30 s on/ 30 s off, using a Bioruptor Plus sonication device, Diagenode in “HIGH” mode). Then samples were centrifuged for 30 minutes at > 20000 xg, 4°C and then EGFP and mCherry were purified using GFP-Trap Magnetic Agarose/RFP-Trap Magnetic Agarose beads (ChromoTek, gtma-20/rtma-20) according to manufacturer’s instructions. In brief, 25 μL slurry of both GFP- and RFP-Trap beads were added to 500 μL dilution buffer (10 mM Tris (pH 7.5), 150 mM NaCl, 0.5 mM EDTA), magnetically separated and washed twice with dilution buffer. Then the clarified lysate was diluted with 300 μL dilution buffer and bound to the beads for one hour at 4°C (on a rolling shaker). Then the beads were magnetically separated, the supernatant was removed and beads were washed three times with dilution buffer. Subsequently, proteins were eluted using acidic elution buffer [200 mM glycine (pH 2.5)] and the eluate was neutralized by adding 1 M Tris (pH 10.4), (for 50 μL elution buffer 5 μL 1 M Tris were added). Then protein loading dye was added [used at a 1:5 ratio, 312.5 mM Tris, 10% SDS, 25% glycerol, 5 mM beta-mercaptoethanol, 0.05% bromophenol blue (pH 6.8)] samples were boiled at 95°C for approximately 5 minutes and subsequently separated via SDS-PAGE [NuPAGE 4%–12% Bis-Tris-Gel run in NuPAGE MOPS SDS Running Buffer (ThermoFisher)]. Subsequently, gels were stained with Coomassie [80 mg/mL Coomassie brilliant Blue G250 (Fluka), 3 mM HCl].
Sample Preparation for LC-MS/MS
Coomassie-stained bands were excised, chopped into small pieces, and transferred to 0.5 mL microcentrifuge tubes. For all following steps, buffers were exchanged by two consecutive 15 minutes incubation steps of the gel pieces with 200 μL of acetonitrile (MeCN) whereby the MeCN was removed after each step. Proteins were reduced by the addition of 200 μL of a 10 mM dithiothreitol (DTT) solution in 100 mM ammonium bicarbonate (AmBiC, Sigma Aldrich, A6141) and incubation at 56°C for 30 minutes. Proteins were alkylated by the addition of 200 μL of a 55 mM chloroacetamide (CAA) solution in 100 mM AmBiC and incubation for 20 minutes in the dark. A 0.1 μg/μL stock solution of trypsin (Promega, V511A) in trypsin resuspension buffer (Promega, V542A) was diluted with ice-cold 50 mM AmBiC buffer to achieve a final concentration of 1 ng/μL. 50 μL thereof were added to gel pieces, which were incubated first for 30 minutes on ice and then over night at 37°C. Gel pieces were sonicated for 15 minutes, spun down and the supernatant was transferred into a glass vial (VDS Optilab, 93908556). Remaining gel pieces were washed with 50 μL of an aqueous solution of 50% MeCN and 1% formic acid and sonicated for 15 minutes. The combined supernatants were dried in a SpeedVac and reconstituted in 10 μL of an aqueous solution of 0.1% (v/v) formic acid.
Peptides were analyzed by LC-MS/MS on an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher). To this end, peptides were separated using an Ultimate 3000 nano RSLC system (Dionex) equipped with a trapping cartridge (Precolumn C18 PepMap100, 5 mm, 300 μm i.d., 5 μm, 100 Å) and an analytical column (Acclaim PepMap 100. 75 × 50 cm C18, 3 mm, 100 Å) connected to a Nanospray Flex ion source (ThermoFisher). For the detection of modified peptides, peptides were loaded onto the trap column at 30 μL per minute using solvent A (0.1% formic acid) and peptides were eluted using a gradient from 2 to 80% Solvent B (0.1% formic acid in MeCN) over 60 minutes at 0.3 μL per minute (all solvents were of LC-MS grade). The Orbitrap Fusion Lumos was operated in positive ion mode with a spray voltage of 2.4 kV and capillary temperature of 275°C. Full scan MS spectra with a mass range of 375–1200 m/z were acquired in profile mode using a resolution of 120,000 (maximum injections time of 50 ms, the automatic gain control (AGC) target value was set to standard with a max injection time of 50 ms. Precursors were isolated using the quadrupole with a window of 1.2 m/z and fragmentation was triggered by high-energy C-trap dissociation (HCD) in fixed collision energy mode with fixed collision energy of 34%. MS/MS spectra were acquired with the Orbitrap with a resolution of 15.000 and a max injection time of 54 ms.
Data Analysis
Acquired data were analyzed using IsobarQuant (Franken et al., 2015) and Mascot V2.4 (Matrix Science) using a reverse UniProt FASTA database from Homo sapiens (UP000005640) including common contaminants as well as protein sequences of the protein of interest. Incorporation of noncanonical amino acids were considered as variable modifications of lysine (EGFP39K or mCherry190K) or phenylalanine (EGFP39F or mCherry190F) with Cbz-K or 3-IF respectively (see Table S4 for the respective protein sequences). In addition, the following modifications were considered: Carbamidomethyl (C, fixed), acetyl (K, variable), acetyl (Protein N terminus, variable), oxidation (M, variable). The mass error tolerance for full scan MS spectra was set to 10 ppm and to 0.02 Da for MS/MS spectra. A maximum of 2 missed cleavages were allowed. A minimum of 2 unique peptides with a peptide length of at least seven amino acids and a false discovery rate below 0.01 were required on the peptide and protein level (Savitski et al., 2015). See Data S1 for the corresponding MS/MS spectra.
Chemical Synthesis
SCO-Y was synthesized in two steps, similar to a previously published procedure for a compound with a longer linker (Kurra et al., 2014). See Data S2 for a synthesis scheme and the corresponding NMR spectra.
Synthesis Fmoc-SCO-Y-OMe (1)
To a solution of 2-Cyclooctyn-1-ol (198 mg, 1.61 mmol), N-Fmoc-tyrosine methyl ester (1.00 g, 2.40 mmol) and triphenylphosphine (628 mg, 2.40 mmol) in dry tetrahydrofuran (THF, 12 mL) was added diisopropyl azodicarboxylate (DIAD, 470 μL, 2.40 mmol) dropwise at 0°C. The reaction was stirred 15 hours at room temperature, concentrated and directly subjected to purification via flash chromatography (heptane/ethyl acetate, 100:1 → 3:1) using a Biotage Isolera Four system to obtain 234 mg (29%) of 1. 1H NMR (400 MHz, Methanol-d4) δ 7.79 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.5 Hz, 2H), 7.43 – 7.34 (m, 2H), 7.30 (q, J = 7.2 Hz, 2H), 7.11 (dd, J = 8.6, 3.3 Hz, 2H), 6.86 – 6.76 (m, 2H), 4.66 (td, J = 10.7, 9.8, 5.0 Hz, 1H), 4.43 (ddd, J = 9.7, 5.2, 2.3 Hz, 1H), 4.34 (ddd, J = 9.8, 6.7, 2.6 Hz, 1H), 4.22 – 4.09 (m, 2H), 3.71 (d, J = 1.3 Hz, 3H), 3.10 (ddd, J = 13.9, 5.3, 2.0 Hz, 1H), 2.87 (ddd, J = 13.9, 9.5, 2.8 Hz, 1H), 2.24 – 2.06 (m, 4H), 1.95 – 1.79 (m, 3H), 1.77 – 1.50 (m, 3H). m/z calculated for C33H33NO5, [M+H]+ 524.24, found 524.2.
Synthesis SCO-Y (2)
LiOH monohydrate (7.89 mg, 188 μmol) was added to a solution of 1 in THF/H2O (3:1, 2 mL) and the reaction was stirred for 1.5 hours at room temperature. Afterward, the reaction was diluted with 20 mL of ethyl acetate and 30 mL of water and the aqueous layer was acidified with 3 M HCl. The aqueous phase was washed 3 times with 15 mL of ethyl acetate and the water was freeze-dried overnight to yield 50.2 mg (98%) of 2 (as a HCl salt), as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 7.12 (d, J = 8.5 Hz, 2H), 6.79 (d, J = 8.5 Hz, 2H), 4.74 – 4.68 (m, 1H), 3.14 (dd, J = 14.6, 4.3 Hz, 1H), 2.86 (dd, J = 14.6, 8.7 Hz, 1H), 2.20 – 2.01 (m, 4H), 1.81 (dt, J = 10.7, 7.0 Hz, 3H), 1.70 – 1.47 (m, 3H). 13C NMR (101 MHz, MeOD) δ 172.62, 157.12, 129.94, 128.10, 115.55, 100.44, 91.57, 69.65, 56.27, 42.00, 36.07, 33.96, 29.52, 25.89, 19.77. m/z calculated for C17H21NO3, [M+H]+ 288.16, found 288.2.
Quantification and statistical analysis
Bar plots and heatmaps were generated using Prism software (GraphPad), further details can be found in the corresponding figure legends.
Acknowledgments
We thank Gemma Estrada Girona for her help. We are grateful to Eric Brustad, Anne Ephrussi, Paul F. Sauter, Christine Koehler, Sofya Mikhaleva, Martin Beck, Falk Butter, and all the members of the Lemke laboratory for helpful discussions. We thank the Flow Cytometry Core Facility and the Advanced Light Microscopy Facility at the European Molecular Biology Laboratory (EMBL) for expert assistance. We would like to particularly thank Per Haberkant and Mandy Rettel from the Proteomics Core Facility at EMBL for performing and analyzing protein LC-MS/MS experiments. Plasmid templates for λN22 peptides and histone H2B were generous gifts from the Ellenberg laboratory. We thank Nataliia Shymanska for generously providing SCO-Y. The Lemke laboratory acknowledges generous support from European Research Council (ERC) SMPFv2.0 and SFB 1129 (project number 240245660, funded by the Deutsche Forschungsgemeinschaft) and the Gutenberg Research College (GRC).
Author contributions
E.A.L. and C.D.R. conceived the project. C.D.R. performed the experiments. C.D.R. and E.A.L. analyzed the data and co-wrote the manuscript.
Declaration of interests
C.D.R. and E.A.L. have filed a patent application on OT organelle technology (EP 19157257.7).
Published: August 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.08.001.
Supplemental information
Data and code availability
-
•
All data are available in the main text or the supplementary materials. Raw data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ai H., Shen W., Sagi A., Chen P.R., Schultz P.G. Probing Protein-Protein Interactions with a Genetically Encoded Photo-crosslinking Amino Acid. ChemBioChem. 2011;12:1854–1857. doi: 10.1002/cbic.201100194. [DOI] [PubMed] [Google Scholar]
- Alberti S. The wisdom of crowds: regulating cell function through condensed states of living matter. J. Cell Sci. 2017;130:2789–2796. doi: 10.1242/jcs.200295. [DOI] [PubMed] [Google Scholar]
- Almhjell P.J., Boville C.E., Arnold F.H. Engineering enzymes for noncanonical amino acid synthesis. Chem. Soc. Rev. 2018;47:8980–8997. doi: 10.1039/c8cs00665b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer M., Neelsen K.J., Teloni F., Pozdnyakova I., Pellegrino S., Grøfte M., Rask M.D., Streicher W., Jungmichel S., Nielsen M.L., Lukas J. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose) Nat. Commun. 2015;6:8088. doi: 10.1038/ncomms9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold F.H. Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture) Angew. Chem. Int. Ed. Engl. 2019;58:14420–14426. doi: 10.1002/anie.201907729. [DOI] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S., Rosen M.K. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife. 2014;3:e04123. doi: 10.7554/eLife.04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beránek V., Reinkemeier C.D., Zhang M.S., Liang A.D., Kym G., Chin J.W. Genetically Encoded Protein Phosphorylation in Mammalian Cells. Cell Chem. Biol. 2018;25:1067–1074.e5. doi: 10.1016/j.chembiol.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beránek V., Willis J.C.W., Chin J.W. An Evolved Methanomethylophilus alvus Pyrrolysyl-tRNA Synthetase/tRNA Pair Is Highly Active and Orthogonal in Mammalian Cells. Biochemistry. 2019;58:387–390. doi: 10.1021/acs.biochem.8b00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman C.M., Papa L.J., 3rd, Hendel S.J., Moore C.L., Suen P.H., Weickhardt A.F., Doan N.D., Kumar C.M., Uil T.G., Butty V.L. An Adaptable Platform for Directed Evolution in Human Cells. J. Am. Chem. Soc. 2018;140:18093–18103. doi: 10.1021/jacs.8b10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme T., Vandepoele K., De Bodt S., Simillion C., Maere S., Van de Peer Y. The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol. 2006;7:R43. doi: 10.1186/gb-2006-7-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino J.S., Glick B.S. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Branon T.C., Bosch J.A., Sanchez A.D., Udeshi N.D., Svinkina T., Carr S.A., Feldman J.L., Perrimon N., Ting A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018;36:880–887. doi: 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson D.I., Fan C., Guo L.T., Miller C., Söll D., Liu D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017;13:1253–1260. doi: 10.1038/nchembio.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Goreshnik I., Coventry B., Case J.B., Miller L., Kozodoy L., Chen R.E., Carter L., Walls A.C., Park Y.-J. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–431. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case L.B., Ditlev J.A., Rosen M.K. Regulation of Transmembrane Signaling by Phase Separation. Annu. Rev. Biophys. 2019;48:465–494. doi: 10.1146/annurev-biophys-052118-115534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case L.B., Zhang X., Ditlev J.A., Rosen M.K. Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science. 2019;363:1093–1097. doi: 10.1126/science.aau6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervettini D., Tang S., Fried S.D., Willis J.C.W., Funke L.F.H., Colwell L.J., Chin J.W. Rapid discovery and evolution of orthogonal aminoacyl-tRNA synthetase-tRNA pairs. Nat. Biotechnol. 2020;38:989–999. doi: 10.1038/s41587-020-0479-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.R., Groff D., Guo J., Ou W., Cellitti S., Geierstanger B.H., Schultz P.G. A facile system for encoding unnatural amino acids in mammalian cells. Angew. Chem. Int. Ed. Engl. 2009;48:4052–4055. doi: 10.1002/anie.200900683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J.W. Expanding and reprogramming the genetic code. Nature. 2017;550:53–60. doi: 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- Chin J.W., Cropp T.A., Anderson J.C., Mukherji M., Zhang Z., Schultz P.G. An expanded eukaryotic genetic code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- Daigle N., Ellenberg J. LambdaN-GFP: an RNA reporter system for live-cell imaging. Nat. Methods. 2007;4:633–636. doi: 10.1038/nmeth1065. [DOI] [PubMed] [Google Scholar]
- Dedecker P., Duwé S., Neely R.K., Zhang J. Localizer: fast, accurate, open-source, and modular software package for superresolution microscopy. J. Biomed. Opt. 2012;17:126008. doi: 10.1117/1.JBO.17.12.126008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditlev J.A., Vega A.R., Köster D.V., Su X., Tani T., Lakoduk A.M., Vale R.D., Mayor S., Jaqaman K., Rosen M.K. A composition-dependent molecular clutch between T cell signaling condensates and actin. eLife. 2019;8:e42695. doi: 10.7554/eLife.42695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkelmann D.L., Willis J.C.W., Beattie A.T., Chin J.W. Engineered triply orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs enable the genetic encoding of three distinct non-canonical amino acids. Nat. Chem. 2020;12:535–544. doi: 10.1038/s41557-020-0472-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelsberg A., Hermosilla R., Karsten U., Schülein R., Dörken B., Rehm A. The Golgi protein RCAS1 controls cell surface expression of tumor-associated O-linked glycan antigens. J. Biol. Chem. 2003;278:22998–23007. doi: 10.1074/jbc.M301361200. [DOI] [PubMed] [Google Scholar]
- English J.G., Olsen R.H.J., Lansu K., Patel M., White K., Cockrell A.S., Singh D., Strachan R.T., Wacker D., Roth B.L. VEGAS as a Platform for Facile Directed Evolution in Mammalian Cells. Cell. 2019;178:748–761.e17. doi: 10.1016/j.cell.2019.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekner T., Li X., Lee M.M., Chan M.K. A pyrrolysine analogue for protein click chemistry. Angew. Chem. Int. Ed. Engl. 2009;48:1633–1635. doi: 10.1002/anie.200805420. [DOI] [PubMed] [Google Scholar]
- Franken H., Mathieson T., Childs D., Sweetman G.M.A., Werner T., Tögel I., Doce C., Gade S., Bantscheff M., Drewes G. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015;10:1567–1593. doi: 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- Fredens J., Wang K., de la Torre D., Funke L.F.H., Robertson W.E., Christova Y., Chia T., Schmied W.H., Dunkelmann D.L., Beránek V. Total synthesis of Escherichia coli with a recoded genome. Nature. 2019;569:514–518. doi: 10.1038/s41586-019-1192-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried S.D., Schmied W.H., Uttamapinant C., Chin J.W. Ribosome Subunit Stapling for Orthogonal Translation in E. coli. Angew. Chem. Int. Ed. Engl. 2015;54:12791–12794. doi: 10.1002/anie.201506311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A., Nguyen D.P., Lusic H., An W., Deiters A., Chin J.W. Genetically encoded photocontrol of protein localization in mammalian cells. J. Am. Chem. Soc. 2010;132:4086–4088. doi: 10.1021/ja910688s. [DOI] [PubMed] [Google Scholar]
- Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Glasgow A.A., Huang Y.M., Mandell D.J., Thompson M., Ritterson R., Loshbaugh A.L., Pellegrino J., Krivacic C., Pache R.A., Barlow K.A. Computational design of a modular protein sense/response system. Science. 2019;366:1024–1028. doi: 10.1126/science.aax8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordaliza-Alaguero I., Cantó C., Zorzano A. Metabolic implications of organelle-mitochondria communication. EMBO Rep. 2019;20:e47928. doi: 10.15252/embr.201947928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecco H.E., Schmick M., Bastiaens P.I. Signaling from the living plasma membrane. Cell. 2011;144:897–909. doi: 10.1016/j.cell.2011.01.029. [DOI] [PubMed] [Google Scholar]
- Grünwald D., Singer R.H. In vivo imaging of labelled endogenous β-actin mRNA during nucleocytoplasmic transport. Nature. 2010;467:604–607. doi: 10.1038/nature09438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamashima K., Kimoto M., Hirao I. Creation of unnatural base pairs for genetic alphabet expansion toward synthetic xenobiology. Curr. Opin. Chem. Biol. 2018;46:108–114. doi: 10.1016/j.cbpa.2018.07.017. [DOI] [PubMed] [Google Scholar]
- Hancock S.M., Uprety R., Deiters A., Chin J.W. Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J. Am. Chem. Soc. 2010;132:14819–14824. doi: 10.1021/ja104609m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoell J.I., Larsson E., Runge S., Nusbaum J.D., Duggimpudi S., Farazi T.A., Hafner M., Borkhardt A., Sander C., Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshika S., Leal N.A., Kim M.J., Kim M.S., Karalkar N.B., Kim H.J., Bates A.M., Watkins N.E., SantaLucia H.A., Meyer A.J. Hachimoji DNA and RNA: A genetic system with eight building blocks. Science. 2019;363:884–887. doi: 10.1126/science.aat0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.S., Boyken S.E., Baker D. The coming of age of de novo protein design. Nature. 2016;537:320–327. doi: 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Huang W.Y.C., Yan Q., Lin W.-C., Chung J.K., Hansen S.D., Christensen S.M., Tu H.-L., Kuriyan J., Groves J.T. Phosphotyrosine-mediated LAT assembly on membranes drives kinetic bifurcation in recruitment dynamics of the Ras activator SOS. Proc. Natl. Acad. Sci. USA. 2016;113:8218–8223. doi: 10.1073/pnas.1602602113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.Y.C., Alvarez S., Kondo Y., Kwang Lee Y., Chung J.K., Monatrice Lam H.Y., Biswas K.H., Kuriyan J., Groves J.T. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science. 2019;363:1098–1103. doi: 10.1126/science.aau5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V., Lam S.S., Udeshi N.D., Svinkina T., Guzman G., Mootha V.K., Carr S.A., Ting A.Y. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife. 2017;6:e24463. doi: 10.7554/eLife.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italia J.S., Addy P.S., Wrobel C.J.J., Crawford L.A., Lajoie M.J., Zheng Y., Chatterjee A. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat. Chem. Biol. 2017;13:446–450. doi: 10.1038/nchembio.2312. [DOI] [PubMed] [Google Scholar]
- Italia J.S., Latour C., Wrobel C.J.J., Chatterjee A. Resurrecting the Bacterial Tyrosyl-tRNA Synthetase/tRNA Pair for Expanding the Genetic Code of Both E. coli and Eukaryotes. Cell Chem. Biol. 2018;25:1304–1312.e5. doi: 10.1016/j.chembiol.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italia J.S., Peeler J.C., Hillenbrand C.M., Latour C., Weerapana E., Chatterjee A. Genetically encoded protein sulfation in mammalian cells. Nat. Chem. Biol. 2020;16:379–382. doi: 10.1038/s41589-020-0493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavran J.M., Gundllapalli S., O’Donoghue P., Englert M., Söll D., Steitz T.A. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. USA. 2007;104:11268–11273. doi: 10.1073/pnas.0704769104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Kalappurakkal J.M., Mayor S., Rosen M.K. Phosphorylation of nephrin induces phase separated domains that move through actomyosin contraction. Mol. Biol. Cell. 2019;30:2996–3012. doi: 10.1091/mbc.E18-12-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler C., Sauter P.F., Wawryszyn M., Girona G.E., Gupta K., Landry J.J.M., Fritz M.H.-Y., Radic K., Hoffmann J.-E., Chen Z.A. Genetic code expansion for multiprotein complex engineering. Nat. Methods. 2016;13:997–1000. doi: 10.1038/nmeth.4032. [DOI] [PubMed] [Google Scholar]
- Krzycki J.A. The direct genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 2005;8:706–712. doi: 10.1016/j.mib.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Kurra Y., Odoi K.A., Lee Y.J., Yang Y., Lu T., Wheeler S.E., Torres-Kolbus J., Deiters A., Liu W.R. Two rapid catalyst-free click reactions for in vivo protein labeling of genetically encoded strained alkene/alkyne functionalities. Bioconjug. Chem. 2014;25:1730–1738. doi: 10.1021/bc500361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin E., VanderSluis B., Nguyen Ba A.N., Wang W., Koch E.N., Usaj M., Khmelinskii A., Usaj M.M., van Leeuwen J., Kraus O. Exploring whole-genome duplicate gene retention with complex genetic interaction analysis. Science. 2020;368:eaaz5667. doi: 10.1126/science.aaz5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie M.J., Rovner A.J., Goodman D.B., Aerni H.-R., Haimovich A.D., Kuznetsov G., Mercer J.A., Wang H.H., Carr P.A., Mosberg J.A. Genomically Recoded Organisms Expand Biological Functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner H., Ferruz N., Höcker B. Strategies for designing non-natural enzymes and binders. Curr. Opin. Chem. Biol. 2018;47:67–76. doi: 10.1016/j.cbpa.2018.07.022. [DOI] [PubMed] [Google Scholar]
- Lemke E.A. The exploding genetic code. ChemBioChem. 2014;15:1691–1694. doi: 10.1002/cbic.201402362. [DOI] [PubMed] [Google Scholar]
- Lemke E.A., Summerer D., Geierstanger B.H., Brittain S.M., Schultz P.G. Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat. Chem. Biol. 2007;3:769–772. doi: 10.1038/nchembio.2007.44. [DOI] [PubMed] [Google Scholar]
- Li P., Banjade S., Cheng H.C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J.V., King D.S., Banani S.F. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.C., Schultz P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Lukinavičius G., Umezawa K., Olivier N., Honigmann A., Yang G., Plass T., Mueller V., Reymond L., Corrêa I.R., Jr., Luo Z.-G. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013;5:132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- Lynch M., Conery J.S. The Origins of Genome Complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Meineke B., Heimgärtner J., Lafranchi L., Elsässer S.J. Methanomethylophilus alvus Mx1201 Provides Basis for Mutual Orthogonal Pyrrolysyl tRNA/Aminoacyl-tRNA Synthetase Pairs in Mammalian Cells. ACS Chem. Biol. 2018;13:3087–3096. doi: 10.1021/acschembio.8b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meineke B., Heimgärtner J., Eirich J., Landreh M., Elsässer S.J. Site-Specific Incorporation of Two ncAAs for Two-Color Bioorthogonal Labeling and Crosslinking of Proteins on Live Mammalian Cells. Cell Rep. 2020;31:107811. doi: 10.1016/j.celrep.2020.107811. [DOI] [PubMed] [Google Scholar]
- Mitrea D.M., Kriwacki R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016;14:1. doi: 10.1186/s12964-015-0125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T., Kobayashi T., Hino N., Yanagisawa T., Sakamoto K., Yokoyama S. Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem. Biophys. Res. Commun. 2008;371:818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- Neumann H., Peak-Chew S.Y., Chin J.W. Genetically encoding N(ε)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- Neumann H., Wang K., Davis L., Garcia-Alai M., Chin J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- Nikić I., Plass T., Schraidt O., Szymański J., Briggs J.A.G., Schultz C., Lemke E.A. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew. Chem. Int. Ed. Engl. 2014;53:2245–2249. doi: 10.1002/anie.201309847. [DOI] [PubMed] [Google Scholar]
- Nikić I., Kang J.H., Girona G.E., Aramburu I.V., Lemke E.A. Labeling proteins on live mammalian cells using click chemistry. Nat. Protoc. 2015;10:780–791. doi: 10.1038/nprot.2015.045. [DOI] [PubMed] [Google Scholar]
- Nikić I., Estrada Girona G., Kang J.H., Paci G., Mikhaleva S., Koehler C., Shymanska N.V., Ventura Santos C., Spitz D., Lemke E.A. Debugging Eukaryotic Genetic Code Expansion for Site-Specific Click-PAINT Super-Resolution Microscopy. Angew. Chem. Int. Ed. Engl. 2016;55:16172–16176. doi: 10.1002/anie.201608284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Springer Berlin Heidelberg; 1970. Evolution by Gene Duplication. [Google Scholar]
- Oller-Salvia B., Kym G., Chin J.W. Rapid and Efficient Generation of Stable Antibody-Drug Conjugates via an Encoded Cyclopropene and an Inverse-Electron-Demand Diels-Alder Reaction. Angew. Chem. Int. Ed. Engl. 2018;57:2831–2834. doi: 10.1002/anie.201712370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C., Carlson E.D., Szal T., Florin T., Jewett M.C., Mankin A.S. Protein synthesis by ribosomes with tethered subunits. Nature. 2015;524:119–124. doi: 10.1038/nature14862. [DOI] [PubMed] [Google Scholar]
- Pan X., Thompson M.C., Zhang Y., Liu L., Fraser J.S., Kelly M.J.S., Kortemme T. Expanding the space of protein geometries by computational design of de novo fold families. Science. 2020;369:1132–1136. doi: 10.1126/science.abc0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Piatkevich K.D., Jung E.E., Straub C., Linghu C., Park D., Suk H.-J., Hochbaum D.R., Goodwin D., Pnevmatikakis E., Pak N. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 2018;14:352–360. doi: 10.1038/s41589-018-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce J.B., Chafe S.C., Eswara M.B.K., Van der Merwe G., Mangroo D. Strategies for investigating nuclear-cytoplasmic tRNA dynamics in yeast and mammalian cells. Methods Cell Biol. 2014;122:415–436. doi: 10.1016/B978-0-12-417160-2.00019-9. [DOI] [PubMed] [Google Scholar]
- Plass T., Milles S., Koehler C., Schultz C., Lemke E.A. Genetically encoded copper-free click chemistry. Angew. Chem. Int. Ed. Engl. 2011;50:3878–3881. doi: 10.1002/anie.201008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass T., Milles S., Koehler C., Szymański J., Mueller R., Wiessler M., Schultz C., Lemke E.A. Amino acids for Diels-Alder reactions in living cells. Angew. Chem. Int. Ed. Engl. 2012;51:4166–4170. doi: 10.1002/anie.201108231. [DOI] [PubMed] [Google Scholar]
- Polycarpo C., Ambrogelly A., Bérubé A., Winbush S.M., McCloskey J.A., Crain P.F., Wood J.L., Söll D. An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc. Natl. Acad. Sci. USA. 2004;101:12450–12454. doi: 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polycarpo C.R., Herring S., Bérubé A., Wood J.L., Söll D., Ambrogelly A. Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 2006;580:6695–6700. doi: 10.1016/j.febslet.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinkemeier C.D., Lemke E.A. Raising the ribosomal repertoire. Nat. Chem. 2020;12:503–504. doi: 10.1038/s41557-020-0476-6. [DOI] [PubMed] [Google Scholar]
- Reinkemeier C.D., Girona G.E., Lemke E.A. Designer membraneless organelles enable codon reassignment of selected mRNAs in eukaryotes. Science. 2019;363:eaaw2644. doi: 10.1126/science.aaw2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinkemeier C.D., Koehler C., Sauter P.F., Shymanska N.V., Echalier C., Rutkowska A., Will D.W., Schultz C., Lemke E.A. Synthesis and Evaluation of Novel Ring-Strained Noncanonical Amino Acids for Residue-Specific Bioorthogonal Reactions in Living Cells. Chemistry. 2021;27:6094–6099. doi: 10.1002/chem.202100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M.M., Wilhelm M., Hahne H., Kuster B., Bantscheff M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteomics. 2015;14:2394–2404. doi: 10.1074/mcp.M114.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmied W.H., Tnimov Z., Uttamapinant C., Rae C.D., Fried S.D., Chin J.W. Controlling orthogonal ribosome subunit interactions enables evolution of new function. Nature. 2018;564:444–448. doi: 10.1038/s41586-018-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru F., Nautrup-Pedersen G., Cordes V.C., Görlich D. Nuclear pore complex assembly and maintenance in POM121- and gp210-deficient cells. J. Cell Biol. 2006;173:477–483. doi: 10.1083/jcb.200601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X., Ditlev J.A., Hui E., Xing W., Banjade S., Okrut J., King D.S., Taunton J., Rosen M.K., Vale R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016;352:595–599. doi: 10.1126/science.aad9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymborska A., De Marco A., Daigle N., Cordes V.C., Briggs J.A.G., Ellenberg J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 2013;341:655–658. doi: 10.1126/science.1240672. [DOI] [PubMed] [Google Scholar]
- Uttamapinant C., Howe J.D., Lang K., Beránek V., Davis L., Mahesh M., Barry N.P., Chin J.W. Genetic code expansion enables live-cell and super-resolution imaging of site-specifically labeled cellular proteins. J. Am. Chem. Soc. 2015;137:4602–4605. doi: 10.1021/ja512838z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.A., Mercadante D., Nikić I., Lemke E.A., Gräter F. Origin of Orthogonality of Strain-Promoted Click Reactions. Chemistry. 2015;21:12431–12435. doi: 10.1002/chem.201501727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W., Tharp J.M., Liu W.R. Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta. 2014;1844:1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.S., Fang X., Wallace A.L., Wu B., Liu W.R. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J. Am. Chem. Soc. 2012;134:2950–2953. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.S., Fang X., Chen H.Y., Wu B., Wang Z.U., Hilty C., Liu W.R. Genetic incorporation of twelve meta-substituted phenylalanine derivatives using a single pyrrolysyl-tRNA synthetase mutant. ACS Chem. Biol. 2013;8:405–415. doi: 10.1021/cb300512r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Fredens J., Brunner S.F., Kim S.H., Chia T., Chin J.W. Defining synonymous codon compression schemes by genome recoding. Nature. 2016;539:59–64. doi: 10.1038/nature20124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Choi J.M., Holehouse A.S., Lee H.O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell. 2018;174:688–699.e16. doi: 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I., Pfeffer A., Friedman N., Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61. doi: 10.1038/nature06107. [DOI] [PubMed] [Google Scholar]
- Willis J.C.W., Chin J.W. Mutually orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs. Nat. Chem. 2018;10:831–837. doi: 10.1038/s41557-018-0052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Carvalho P., Voeltz G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 2018;361:eaan5835. doi: 10.1126/science.aan5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H., Chatterjee A., Choi S.H., Bajjuri K.M., Sinha S.C., Schultz P.G. Genetic incorporation of multiple unnatural amino acids into proteins in mammalian cells. Angew. Chem. Int. Ed. Engl. 2013;52:14080–14083. doi: 10.1002/anie.201308137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Lu P., Gamal El-Din T.M., Pei X.Y., Johnson M.C., Uyeda A., Bick M.J., Xu Q., Jiang D., Bai H. Computational design of transmembrane pores. Nature. 2020;585:129–134. doi: 10.1038/s41586-020-2646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T., Ishii R., Fukunaga R., Kobayashi T., Sakamoto K., Yokoyama S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(ε)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008;15:1187–1197. doi: 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T., Ishii R., Fukunaga R., Kobayashi T., Sakamoto K., Yokoyama S. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J. Mol. Biol. 2008;378:634–652. doi: 10.1016/j.jmb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T., Kuratani M., Seki E., Hino N., Sakamoto K., Yokoyama S. Structural Basis for Genetic-Code Expansion with Bulky Lysine Derivatives by an Engineered Pyrrolysyl-tRNA Synthetase. Cell Chem. Biol. 2019;26:936–949.e13. doi: 10.1016/j.chembiol.2019.03.008. [DOI] [PubMed] [Google Scholar]
- Zeng M., Chen X., Guan D., Xu J., Wu H., Tong P., Zhang M. Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell. 2018;174:1172–1187.e16. doi: 10.1016/j.cell.2018.06.047. [DOI] [PubMed] [Google Scholar]
- Zhang M., Lin S., Song X., Liu J., Fu Y., Ge X., Fu X., Chang Z., Chen P.R. A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat. Chem. Biol. 2011;7:671–677. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Ptacin J.L., Fischer E.C., Aerni H.R., Caffaro C.E., San Jose K., Feldman A.W., Turner C.R., Romesberg F.E. A semi-synthetic organism that stores and retrieves increased genetic information. Nature. 2017;551:644–647. doi: 10.1038/nature24659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatkine P., Mehul B., Magee A.I. Retargeting of cytosolic proteins to the plasma membrane by the Lck protein tyrosine kinase dual acylation motif. J. Cell Sci. 1997;110:673–679. doi: 10.1242/jcs.110.5.673. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The first part of the video shows animated 3D reconstructions corresponding to Figure 4A. HEK293T cells expressing the PMPMCP, GMPMCP, ERMPMCP, or OMMPMCP, as well as tRNAPyl and EGFP39TAG::ms2 in the presence of the ncAA SCO-K. IF staining against PylRS and FISH staining of tRNAPyl show strong tRNAPyl recruitment. PylRS (magenta), tRNAPyl (yellow), NLS::EGFP39TAG::ms2 (green). The second part of the video shows animated 3D reconstructions corresponding to Figure 4C. HEK293T cells expressing the PMPMCP, GMPMCP, ERMPMCP, or OMMPMCP, as well as tRNAPyl and EGFP39TAG::ms2 in the presence of the ncAA SCO-K. IF staining against PylRS and RPL26L1 show co-localization of endogenous ribosomes with OT organelles. PylRS (magenta), RPL26L1 (cyan), NLS::EGFP39TAG::ms2 (green). The third part shows animated 3D reconstructions corresponding to Figure S5B. HEK293T cells expressing the PMPλN22, GMPλN22, ERMPλN22, or OMMPλN22, as well as tRNAPyl and a EGFP39TAG::boxB in the presence of the ncAA SCO-K. IF staining against PylRS and FISH staining of tRNAPyl show strong tRNAPyl recruitment. PylRS (magenta), tRNAPyl (yellow), NLS::EGFP39TAG::boxB (green). The fourth part shows animated 3D reconstructions corresponding to Figure S5D. HEK293T cells expressing the PMPλN22, GMPλN22, ERMPλN22 or OMMPλN22, as well as tRNAPyl and a EGFP39TAG::boxB in the presence of the ncAA SCO-K. IF staining against PylRS and RPL26L1 show co-localization of endogenous ribosomes with OT organelles. PylRS (magenta), RPL26L1 (cyan), NLS::EGFP39TAG::box (green).
Data Availability Statement
-
•
All data are available in the main text or the supplementary materials. Raw data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.