

Summary:
Genome-scale models of metabolism (GEMs) are key computational tools for the systems-level study of metabolic networks. Here, we describe the “GEM life cycle” which we subdivide into four stages: inception, maturation, specialization, and amalgamation. We show how different types of GEM reconstruction workflows fit in each stage and proceed to highlight two fundamental bottlenecks for GEM quality improvement: GEM maturation and content removal. We identify common characteristics contributing to increasing quality of maturing GEMs drawing from past independent GEM maturation efforts. We then shed some much-needed light on the latent and unrecognized but pervasive issue of content removal, demonstrating the substantial effects of model pruning on its solution space. Finally, we propose a novel framework for content removal and associated confidence level assignment which will help guide future GEM development efforts, reduce duplication of effort across groups, potentially aid automated reconstruction platforms, and boost the reproducibility of model development.
Graphical Abstract

Introduction
Metabolic network reconstructions are knowledge-bases of bottom-up curated gene-linked biochemical pathways. They can be converted into a mathematical format to formulate stoichiometric genome-scale metabolic models (GEMs) capable of computing various cellular functions. The development of GEMs started shortly after the publication of the first complete genome of Haemophilus influenzae in 1995 with its GEM appearing in 1999 (Fleischmann et al., 1995; Edwards and Palsson, 1999). As more model organisms were sequenced, more GEMs were built, covering an ever increasing breadth of phylogenetic backgrounds. The availability of sequences for an increasingly large diversity of organisms drove the need to make metabolic modeling accessible to a broader scientific community. As a result, a protocol to generate GEMs was established and computational pipelines for metabolic modeling were distributed (Becker et al., 2007; Thiele and Palsson, 2010a; Wang et al., 2018). Over the years, DNA sequencing cost has plummeted, and the number of available genomic sequences has increased exponentially. Thus, the challenge to build GEMs fast arose. Various semi-automated reconstruction workflows were developed, either to assemble GEMs de novo or to refine existing ones (Mendoza et al., 2019; Norsigian et al., 2019).
With the advent of new ‘omics’ technologies, it became evident that GEMs could be refined further to represent cellular states, tissue types, disease and cell lines (Uhlén et al., 2015). Multiple modeling methods for ‘omics’ data integration to extract context-specific models were developed, with various levels of success (Opdam et al., 2017; Richelle et al., 2019). The emergence of metagenomic sequencing subsequently drove the need to build models of microbial communities in order to infer the metabolic repertoire of a microbiome sample (Magnúsdóttir et al., 2017). However, questions regarding the quality of the GEMs resulting from the miscellaneous workflows have been raised (Monk, Nogales and Palsson, 2014), highlighting the need to generate robust quality metrics and reproducible and comparable reconstruction workflows (Lieven et al., 2020). These shortcomings gave a new impetus to crowd-sourcing GEM development efforts, pushing teams of experts to form in order to tackle the interdisciplinary problem of systems-level metabolic modeling (Wang et al., 2018; Lu et al., 2019). As such, GEM development efforts are moving away from local computers and towards online platforms (Aurich, Fleming and Thiele, 2016). Monk et. al recently outlined five key challenges that face their continued development (Monk, Nogales and Palsson, 2014), including: 1) insufficiency of model curation, 2) gaps in biological knowledge, 3) narrow phylogenetic scope, 4) inconsistent adherence to modeling standards, and 5) modest biological scope. Points 3–5 are being tackled with novel metabolic modeling methods and with the emergence of standardized model test suites such as MEMOTE (Lieven et al., 2020). Here, we discuss how points 1 and 2 can be addressed, drawing inspiration from past independent, but parallel efforts. We advocate for two approaches to improving the life-cycle of GEMs. First, we start by defining the GEM life-cycle. We then delve into GEM maturation, which we identify as bottleneck number 1 for GEM quality improvement because it has historically and consistently played a crucial role in improving the quality of GEM sequels. We outline how GEMs mature by drawing examples from a multitude of literature sources so as to serve as a single reference for future GEM maturation endeavors. Finally, we identify a second bottleneck in GEM quality improvement and highlight how content removal is an underappreciated feature transcending all phases of the GEM life cycle. We explicitly provide examples of content removal which can also serve as a guide for future reconstruction efforts, and outline a mathematical framework to save removed content.
Results
The four stages of the GEM life cycle: inception, maturation, specialization, and amalgamation
Multiple workflows are available to guide GEM construction (Thiele and Palsson, 2010a; Opdam et al., 2017; Norsigian et al., 2019). A primary distinguishing feature among workflows is the starting point, which varies depending on how well studied the organism of interest is. We illustrate this and other distinctions by outlining the “GEM life cycle” which we subdivide into four stages: 1) GEM inception, 2) GEM maturation, 3) GEM specialization, and 4) GEM amalgamation (Figure 1).
Figure 1: The GEM life cycle can be subdivided into 4 phases:
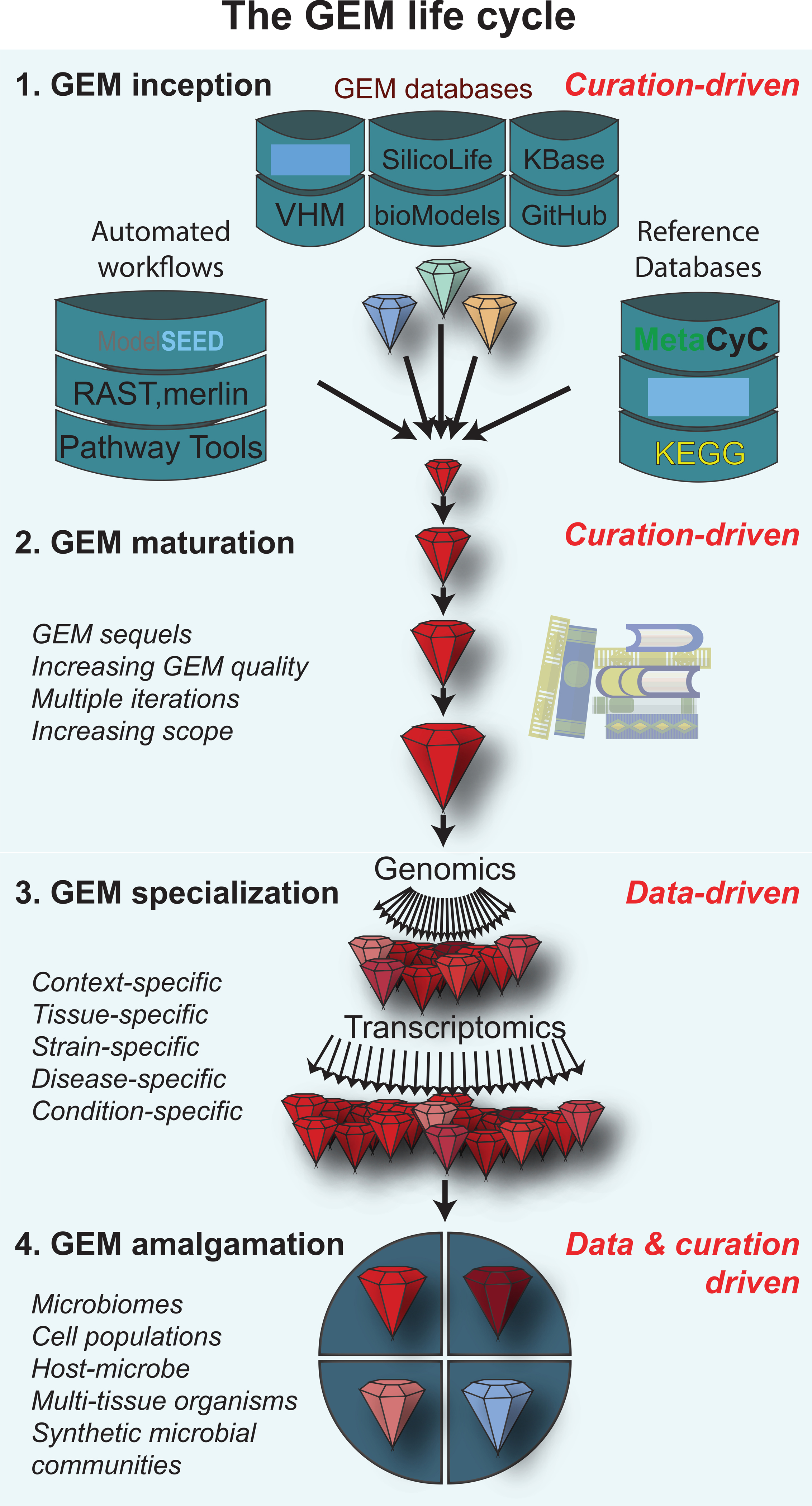
1) GEM inception; metabolic models are built for the first time by drawing from existing reference database and models and iterating through several curation cycles, 2) GEM maturation; an existing manually curated GEM is continually updated over the years after its inception as new knowledge comes to light, 3) GEM specialization; an existing high quality GEM is tailored to a specific strain, cell line or diseased state using an ‘omic’ data set, and 4) GEM amalgamation; high quality GEMs are joined together to form a multi-cellular model.
GEM inception: starting at the beginning
When no prior work has been done to characterize the metabolic capabilities of a target organism at the system level, the initial challenge is to assemble pertinent knowledge to form a first functional GEM. This first stage can be described as the “GEM inception” stage of the GEM life cycle, and has traditionally been divided into four steps: i) automated reconstruction, ii) manual curation, iii) conversion to a mathematical format, and; iv) and validation against experimental data (Feist et al., 2009; O’Brien, Monk and Palsson, 2015). At this stage, an organism-specific bibliome is established, explicitly representing the current limits of biological knowledge available at the time of reconstruction. Curation of bibliomes (Broddrick et al., 2016; Hefzi et al., 2016; Levering et al., 2016) takes much manual labor but is a necessary step to maximize knowledge capture in a reconstruction. Such a process is especially useful for lesser known organisms, because it offers important insights at the systems-level, and drives molecular discovery (Broddrick et al., 2016; Hefzi et al., 2016; Levering et al., 2016).
The GEM inception can be extremely time-consuming because it is curation-driven. Advances for this step have occurred on multiple fronts including: 1) the expansion of reference pathway databases such as MetaCyc(Caspi et al., 2019), KEGG(Kanehisa et al., 2017), and Uniprot(UniProt Consortium, 2019) with curated content across an ever increasing number of organisms; 2) the development of automated reconstruction tools which pull content from reference network databases (Mendoza et al., 2019), such as ModelSEED(Henry et al., 2010), Pathway Tools(Karp et al., 2016), RAVEN(Wang et al., 2018), Merlin(Dias et al., 2015), Kbase(Arkin et al., 2018); and 3) the assembly and expansion of repositories hosting easily retrievable, interoperable, and standardized curated GEMs, such as BiGG(Norsigian et al., 2020), VMH(Noronha et al., 2019), BioModels(Chelliah et al., 2015), yeast.sf.net (Aung, Henry and Walker, 2013), BioModels(Chelliah et al., 2015), Zenodo (Peters et al., 2017) and Silicolife(Home - SilicoLife, no date).
The quality of the starting draft reconstruction heavily affects the amount of time subsequently spent on curating the network, with a strong tradeoff between model size, model quality, and human hours spent on manual curation. Automated GEM assembly is based on identifying database genes annotated with a metabolic function sharing “sufficiently” high sequence homology(Simeonidis and Price, 2015). When validated GEMs for closely related strains exist, a faster path to a better draft starting reconstruction is to map the content of the existing GEMs by means of sequence homology. This approach was used, for instance, to build GEMs of seven closely related species of Saccharomyces cerevisiae (Lopes and Rocha, 2017). Oftentimes, researchers attempt to pool together the annotation results from multiple platforms as well as pre-existing GEMs, and face the laborious task of converting object identifiers. A newer and more elaborate strategy incorporates sequence homology as well as pathway homology and genomic context. It was recently applied to build 773 human gut bacterial models(Magnúsdóttir et al., 2017).
Despite these advances, curating draft metabolic reconstructions is still heavily manual. In addition, in order to convert the starting draft into a high quality model, i.e. a model that is capable of generating meaningful predictions, each object included in the model (i.e. genes, reactions, metabolites, and gene-reaction mappings) should be checked against literature evidence and assigned confidence scores (Thiele and Palsson, 2010a). A number of platforms such as COBRApy (python)(Ebrahim et al., 2013), COBRA toolbox (Matlab)(Heirendt et al., 2019), DistributedFBA.jl (Julia)(Heirendt, Thiele and Fleming, 2017), and Sybil (R)(Gelius-Dietrich et al., 2013) enable easy manual modifications.
The assembled draft reconstruction is then converted into a computational model by introducing constraints and modeling assumptions so as to formulate the network as a constraint-based optimization problem (Feist and Palsson, 2010; Palsson, 2015; Stalidzans et al., 2018). At this point, the network is further refined, and the refinements make use of insights that can only be gleaned from systems level properties. Functionalities for this step are variably available across automated annotation platforms such as gap filling (which adds linkages to disconnected network edges in order to achieve biomass precursor production)(Devoid et al., 2013), protein and metabolite subcellular localization predictions (for model compartmentalization)(Dias et al., 2018), and Gibbs free energy calculation (for the estimation of the upper and lower bounds of a reaction)(Machado et al., 2018). Emerging algorithms are now attempting to suggest which modifications to make based on gene essentiality profiles, experimentally derived nutrient utilization profiles (Hartleb, Jarre and Lercher, 2016; Machado et al., 2018), or by flagging unbalanced reactions(Wang et al., 2018). Despite the manual aspect of building the initial reconstruction, the corresponding workflow is decidedly the most established workflow in the GEM life cycle.
GEM maturation: the devil is in the details
In the second stage of the GEM life cycle, the starting point is an existing curated GEM which goes through a series of iterations resulting in “GEM sequels” or subsequent versions of the first GEM for the target organism. A good metabolic model should be the best representation of biological reality that we obtain given the current state of knowledge, and it therefore needs to be continually improved as new findings are published. Indeed, this on-going process takes years, or even decades, and occurs through multiple iterations yielding, hopefully, an ever increasing quality of the metabolic reconstruction that a GEM is based upon. Hence, researchers involved in such efforts ask what was previously missed, what can be improved, whether previous unknowns have become known, and whether there are new known unknowns to be discovered.
The maturation phase produces GEMs that are increasingly more organism-specific, have larger metabolic coverage, contain increasingly comprehensive manually curated pathways, and improved predictive power (Monk, Nogales and Palsson, 2014). GEM sequels are usually limited to organisms of high interest. For example, the metabolic reconstruction of E. coli has been updated at least five times (McCloskey, Palsson and Feist, 2013; Monk et al., 2017) (iJE660, iJR904, iAF1260, iJO1366, and iML1515), the human metabolic network has been updated over four times (recon 2, recon 2.04, recon 2.2, and recon 3D) (Vieira et al., 2018; Robinson et al., 2020), the S. aureus GEM was updated 4 times (Seif, Monk, Mih, et al., 2019), and the S. cerevisiae network has been updated over 17 times (Lopes and Rocha, 2017). Oftentimes, GEM sequel efforts occur contemporaneously across multiple groups, calling for the organization of reconstruction jamborees to produce a consensus model (Herrgård et al., 2008; Thiele and Palsson, 2010b). Because this stage of the GEM life cycle is purely aimed at improving a GEM’s quality, in a subsequent section we will expand upon the characteristics and types of modifications that have been adopted historically. This will serve as a set of pointers that can guide future GEM maturation efforts.
GEM specialization: a GEM-specific evolutionary strategy
These highly curated and mature GEMs then enter the third stage of the GEM life cycle: GEM specialization. Reconstruction efforts at this stage are data-driven and either partially or fully automated, and begin to account for subtle variations across cell genotypes and gene expression levels. The general strategy is to use a mature GEM as a reference and use one or a combination of data types to guide content removal or to modify upper and lower bounds on allowable reaction rates. Multiple data types can serve to build specialized GEMs including whole genome sequences (Seif et al., 2018), bulk RNA expression levels (Uhlén et al., 2015), protein concentrations (Großeholz et al., 2016), metabolite abundance (Campos and Zampieri, 2019; Yang et al., 2019), and metabolite concentrations (Seif, Monk, Mih, et al., 2019). A recent piece used instead a manual approach in which subsystems were selected based on literature evidence of alteration in a cell type (Masid, Ataman and Hatzimanikatis, 2020). The resulting specialized GEMs are described as “context-specific”, “condition-specific”, “tissue-specific”, “disease-specific”, or “strain-specific”. Here, researchers ask a large breadth of questions, and usually get their answers by comparing the specialized models against each other. To guide their conclusions, they take note of the content that was variably removed and how it changed the network properties.
Whole genome sequences are mostly used to tailor models of prokaryotes, because the variation in coding DNA sequences is significant between strains of the same species. Mature GEMs have been used as baseline reconstructions in E. coli, Salmonella, S. aureus, and S. cerevisiae (Fang et al., 2018; Seif et al., 2018; Lu et al., 2019; Seif, Monk, Machado, et al., 2019) to build strain-specific networks that are tailored according to each strain’s genetic background (Fang et al., 2018; Seif et al., 2018; Lu et al., 2019; Seif, Monk, Machado, et al., 2019). In some cases, the reference network is initially expanded to annotate the accessory genome and account for species-level metabolism (i.e., genes and reactions which are present in some, but not all, strains) (Seif et al., 2018; Seif, Monk, Machado, et al., 2019). Next, the panreconstruction is tailored to each strain using the presence and absence of metabolic genes as a basis for content removal (Norsigian et al., 2019). While the reductive step is easily automated through the integration of comparative genomics with metabolic modeling (e.g., CarveMe) (Machado et al., 2018; Norsigian et al., 2019), the initial constructive step is not, and employs similar methodologies to those used for the assembly of de novo reconstructions.
While ‘omic’ data integration is straightforward in the case of building “strain-specific” models from whole genome sequences, or “condition-specific” models from time-course quantitative exo-metabolomics data, it remains an active area of research for the integration of all other “omic” data types (Ramon, Gollub and Stelling, 2018). There is a wide variety of approaches currently available. For example, there are four different characteristics used for transcriptomic data integration, which is especially popular for the generation of tissue-specific and disease-specific human GEMs (Rosario et al., 2018; Ben Guebila and Thiele, 2019; Gatto, Ferreira and Nielsen, 2020): 1) expression level thresholding (thresholds define which genes or active/inactive), 2) the assumptions made to translate active/inactive genes to active/inactive reactions via the gene-protein-reaction rule, 3) the modeling extraction method, and 4) the chosen cellular objective (biomass production, ATP production, etc.) (Kim and Lun, 2014; Töpfer, Kleessen and Nikoloski, 2015; Richelle et al., 2019). An emerging strategy to pinpoint the best workflow is the validation of reduced models against condition-specific gene essentiality profiles generated using CRISPR technologies (Opdam et al., 2017). This highly anticipated validation strategy holds great promise for the rapid acceleration of validated ‘omic’ integration tools.
GEM amalgamation: one alone is not enough
Building multicellular GEMs is the latest development in the GEM life cycle. Here, researchers are interested in drawing a more holistic view of cellular behavior by explicitly modeling cell-cell interactions. Tailored GEMs for different cells operating in a single system are merged together into a single model to capture multicellular metabolic interactions. The modeled systems include multi-tissue organisms (Gomes de Oliveira Dal’Molin et al., 2015), synthetic microbial communities (Mee et al., 2014; Zelezniak et al., 2015; Machado et al., 2018; Pacheco, Moel and Segrè, 2019), cell populations in a single tissue (Damiani et al., 2019), microbiomes (Thiele, Heinken and Fleming, 2013), whole body metabolic models (Thiele et al., 2020) and host-microbe interactions (Bordbar et al., 2010). Multicellular GEMs have been used to capture how metabolic diversity defines the space of allowable cell-cell interactions (Zengler and Zaramela, 2018), as well as to predict the nature of those interactions (e.g. synergism, antagonism) (Heinken and Thiele, 2015a). Technological advances in single cell transcriptomics, metagenomics and metatranscriptomics have accelerated the pace of community GEM development. However, each data type comes with its own challenges, complicating data-driven GEM tailoring and calling for the development of novel metabolic modeling workflows. This phase of the GEM life cycle is still in its infancy, and it holds great promise for the near future. Published protocols and guidelines have been established for stage 1 and stage 3 of a GEM life cycle (Thiele and Palsson, 2010a; Kim and Lun, 2014; Norsigian et al., 2019), while details of efforts that have gone into stage 4 are reviewed by Magnúsdóttir et. al (Noronha et al., 2019). Below, we discuss characteristic features shared across GEM maturation endeavors (stage 2).
Bottleneck 1: GEM maturation
GEMs mature when they are updated over the years. Here we discuss common characteristics of GEM maturation efforts which we observed across independently evolved GEM sequels contributing to increased GEM quality.
Capturing increasing cellular complexity
Early-stage GEMs tend to account for a reduced number of cellular compartments (Figure 2A). A predominant set of improvements are driven by the examination of the network structure and its reorganization. For example, the initial E. coli networks did not account for the periplasmic compartment which was later added in iAF1260 (Edwards and Palsson, 2000; Feist et al., 2007). Similarly, the number of compartments modeled in S. cerevisiae increased from 3 to 16 over the course of multiple iterations (Duarte, Herrgård and Palsson, 2004; Mo, Palsson and Herrgård, 2009; Aung, Henry and Walker, 2013). The development of fully compartmentalized GEMs included the re-assignment of reactions and metabolites to new compartments and the addition of intercompartmental transport reactions to represent the metabolic exchanges between compartments (Duarte, Herrgård and Palsson, 2004; Feist et al., 2007; Mo, Palsson and Herrgård, 2009; Swainston et al., 2016). As a result, fewer reactions contribute to the metabolite pools in each compartment, and certain metabolites become compartment-specific. These structural modifications indirectly add constraints to the model because the exchange of metabolites between compartments is only possible through the added transport reactions (Savojardo et al., 2018; Broddrick et al., 2019).
Figure 2: The eight GEM maturation characteristics:

A) increasing degree of compartmentalization; an increasing number of cellular compartments are modeled accounting for sectionalization of metabolism, B) advances in knowledge; as novel pathways and increased details in known pathways are uncovered, a maturing GEM reflects increasing organism specificity with network structure features driving improvements and discovery, C) increasingly informed modeling assumptions; as more data covering a specific organisms is generated, the biomass function of a maturing GEM increases in complexity, D) technology driven advances; with the emergence of ‘omics’ data, gene knockout screens, and an increasing number of Biolog phenotype microarray data sets available, the diversity of approaches to validate a model increases E) enriched object-associated information; with the emergence and expansion of diverse reference databases, objects in GEMs are increasingly associated with crossreferences, F) crowd-sourcing; a higher diversity of expertise is used through direct (GitHub repositories, Jamborees) and indirect crowd-sourcing (sequential maturing of GEMs).
Advances in knowledge driving a net expansion of scope
The accumulation of new legacy knowledge in the literature drives the need to update GEMs (Figure 2B). As knowledge specific to the organism increases, new pathways and subsystems are added, cofactor specificity is refined, additional content is pooled from expanded reference network databases, Gene-Protein-Reaction rules (GPRs, which link genes to the metabolic transformation that they participate in) are updated, new genome annotations are incorporated (along with a mapping to previous annotations), existing pathways are completed, reaction reversibility is revised, and erroneous content is removed. Frequently, added pathways are organism-specific (such as vitamin and fatty acid biosynthesis) highlighting the metabolic particularities of the cell and contributing to an increased breadth of modeled metabolic networks. Advances in knowledge also enable an expansion of scope. For example, reactive oxygen species production (Brynildsen et al., 2013), host-microbiome interactions (Heinken and Thiele, 2015b), mammalian secretory pathways (Uhlén et al., 2019; Gutierrez et al., 2020), and strain-specific metabolic variations such as O-antigen biosynthesis in Gram-negatives (Seif, Monk, Machado, et al., 2019) are more recent add-ons to metabolic networks.
Increasing organism-specificity
It is important to be aware that GEMs at early stages of development tend to have nonorganism specific information (even after a first round of curation). Just like the discovery of novel molecular mechanisms is not ex nihilo and relies on the available pool of knowledge in other species, the generation of GEMs relies on previously reconstructed networks which were originally tailored for different organisms. For instance, while there may be sufficient evidence supporting the addition of a metabolic transformation, details of said transformation may only be known in another (sometimes unrelated) organism. It is likely that there are variations in these processes across species, but such variations may simply remain unknown at initial stages (Dobson et al., 2010; Aung, Henry and Walker, 2013; Seif, Monk, Mih, et al., 2019). Therefore, GEM maturation endeavors involve the modification or removal of content as new evidence comes to light that ultimately increases organism-specificity (Figure 2B). For example, bacteriaspecific reactions participating in maltose metabolism were only detected and removed from the latest C. elegans GEM (Witting et al., 2018), mammalian-originating free fatty acids were only removed from the latest S. cerevisiae model (Aung, Henry and Walker, 2013), and the glyoxylate shunt, ubiquinone biosynthesis, and vitamin K1 biosynthesis was only removed from the latest S. aureus model (Seif, Monk, Mih, et al., 2019). Increasing organism-specificity is clearly shown in a multiple correspondence analysis plot of the content of curated GEMs by Monk et. al (Monk, Nogales and Palsson, 2014), in which more recently updated E. coli GEMs are located further away from the center of the plot.
Network-structure driven improvements
Flux variability analysis of a network can enable the identification of blocked reactions (i.e., reactions which cannot carry flux under any condition). The incapability of a reaction to carry flux can be caused by any one of two structural features: 1) a participating substrate or upstream precursor has no producing reactions (root no-production or orphan metabolite), or 2) a participating or downstream product has no consuming reaction to allow it to leave the system (root no-consumption or dead-end metabolite) (Orth et al., 2011). These instances highlight gaps in the network that can be manually subdivided into knowledge gaps and scope gaps. Knowledge gaps occur due to limitations in our knowledge of the cellular metabolic capabilities and can lead to model-driven hypothesis generation. Scope gaps feature limitations in the scope of the GEM. Their identification has guided model modifications and improved network connectivity (Dobson et al., 2010; Orth et al., 2011). Key advances in gap filling algorithms leveraging various types of “omics” data, including gene essentiality and transcriptomics profiles, can accelerate this process (Kumar, Dasika and Maranas, 2007; Hosseini and Marashi, 2017; Joshi et al., 2020).
Increasingly informed modeling assumptions
Multiple modeling assumptions are made de facto across all GEMs. In order to quickly identify areas of improvement, it is important to get acquainted with where they occur. Any modification to the network made in order to achieve an objective in silico constitutes a modeling assumption. One commonly chosen objective is maximizing yield, i.e. the rate of production of biomass precursors defined in silico as the flux through the “biomass objective function”. It is designed by the modeler and contains a set of chosen biomass precursors in specific proportions. With this addition, modelers are assuming that a cell’s objective is to proliferate, which may work well in some cases (cancer cells, bacterial cells) but less so in others (terminally differentiated neurons). Modeling assumptions thus include the biomass objective function, gap filling reactions, demand and sink reactions (often added due to network structure impeding biomass production), and ATP maintenance reactions (which take into account the consumption of ATP as part of non-metabolic processes). Biomass objective functions (BOFs) can have three levels of modeling detail: 1) basic, 2) intermediate, and 3) advanced (Feist and Palsson, 2010). Initial GEMs tend to have basic level BOFs in which only major macromolecular categories (RNA, DNA, proteins, and lipids) are represented (Lachance et al., 2019). They are often organism-unspecific and are later refined and expanded to include metabolites which are more specific to the organism of interest (vitamins, cofactors, inorganic ions, and membrane components), as well as energy drainage formulations to account for protein, DNA, and RNA polymerization (Figure 2C) (Duarte, Herrgård and Palsson, 2004; Dobson et al., 2010; Seif, Monk, Mih, et al., 2019; Széliová et al., 2020). Sometimes, multiple BOFs are added to represent different cellular states, or to model the effect of stressors such as drugs and antibiotics (Heavner et al., 2012; Sahoo et al., 2015; Seif, Monk, Mih, et al., 2019). BOFs are increasingly being replaced with “metabolic tasks,” especially across GEM specialization efforts (Agren et al., 2014; Pandey and Hatzimanikatis, 2019; Richelle et al., 2019). “Metabolic tasks” are defined by Thiele et. al as “non-zero flux[es] through a reaction or through a pathway leading to the production of a metabolite B from a metabolite A” (Thiele et al., 2013). Reactions and genes with a confidence score of 0 can also be considered as modeling assumptions and classified as prime candidates for review until experimental validation emerges. Genes and associated reactions are given a low confidence score when their inclusion is based purely on genome annotations, when they are not evaluated, or for modeling purposes (Thiele and Palsson, 2010a).
Novel technology driven improvements: genome-wide knockout mutant screens
The advent of new technologies has enabled new types of observations to be used for network refinement. When the first set of GEMs were published, gene essentiality datasets were scarce. New datasets progressively emerged or were generated for the specific purpose of validating genome-scale metabolic models (Feist et al., 2007). The most pristine datasets constitute screens of complete gene knock-out mutant fitness profiles grown in a multitude of defined (preferably minimal) media (Monk et al., 2017). Consequently, gene essentiality, nonessentiality, and conditional essentiality can be measured and compared directly with modeling simulations. Observed inconsistencies have brought about model corrections, highlighted knowledge gaps, and later guided the discovery of underground metabolic functions (Guzmán et al., 2015, 2018, 2019). Gene essentiality datasets for complex organisms only started appearing as technologies for gene silencing and gene editing progressed. For instance, the Mouse Knockout project was announced in 2004 and launched in 2006 (Austin et al., 2004) and the results were later incorporated to guide Mus musculus GEM updates in 2010 (Selvarasu et al., 2010). Similarly, the advent of CRISPR-cas9 technologies enabled the systematic screening of tens of thousands of loss-of-function lesions affecting gene copies across multiple human cell lines, including tumor cell lines (Wang et al., 2014, 2015; Blomen et al., 2015; Bartha et al., 2018). Initial efforts have integrated CRISPR screen results for the purpose of validating a reconstruction (Chandrasekaran et al., 2017; Richelle et al., 2019). However, we believe that these datasets have yet to reach their full potential in the context of human GEM reconstruction, representing an important opportunity to advance our tools and knowledge of human and cancer metabolism through the maturation of specialized GEMs.
Novel technology driven improvements: ‘omics’ data
Technological advances in profiling extracellular and endo-metabolomes have enabled the quantification of GEM metabolome coverage. Combined with manual curation and machine learning, the comparison of in silico modeled metabolites against compendia of metabolomics data enables: 1) the discovery of missing pathways (Costello and Martin, 2018; Toubiana et al., 2019); 2) model corrections; and 3) the identification of potentially misidentified metabolites (Brunk et al., 2018; Witting et al., 2018). For example, extracellular metabolomics data guided the addition of secretion pathways in S. cerevisiae (Mo, Palsson and Herrgård, 2009) and served as a validation tool for context-specific metabolic networks (Choudhary et al., 2016). Similarly, proteome-wide subcellular localization analysis is now possible using massspectrometry, and is increasingly applied to GEMs across the tree of life (Sun et al., 2009; Seaver et al., 2014; Jadot et al., 2017; Orre et al., 2019). Recent reconstruction efforts make use of this added information to identify which compartments to model and the reactome content of each compartment (Seaver et al., 2014). Transcriptomics datasets have served to confirm the activity (or lack thereof) of a pathway and offer insights into the validity of network gaps (Hadadi et al., 2019). Crystallized protein structures have guided the distinction between true and false isozymes in the context of gene essentiality(Broddrick et al., 2016), and when integrated with GEMs, have pinpointed essential metal cofactors to be added to the biomass objective function(Seif, Monk, Mih, et al., 2019). In addition, information gleaned from the primary, secondary, and tertiary protein structures is increasingly utilized to predict subcellular localization (Savojardo et al., 2018). Finally, an increasing synergy between machine learning and metabolic modeling (Cuperlovic-Culf, 2018; Zampieri et al., 2019) is enabling the rapid advances that have pushed the field of machine learning forward to spill over into the field of metabolic modeling.
Enriched object associated information
With the expansion and increasing specialization of knowledge bases it has become crucial and more common to enrich all objects included in GEMs with publicly available database-linked cross-references and identifiers. An especially time-consuming step in both GEM inception and maturation is the conversion of various identifier namespaces into a single consistent namespace. Adding metabolite, reaction, and gene identifiers simultaneously facilitates downstream reconstruction efforts as well as high-throughput ‘omics’ data integration. Crossreference databases such as MetaNetX increasingly serve as a tool to facilitate identifier mapping (Moretti et al., 2016) by pulling content from over 20 databases. Protein structures are a recent addition to GEMs, constituting a fourth modeled instance (in addition to genes, reactions, and metabolites). They have enabled the examination of systems-level effects of single nucleotide polymorphisms (Brunk et al., 2018). A recent package enabling automated enrichment of GEMs with protein structures (Mih et al., 2018) has also made it possible to systematically identify the most recent literature relevant to the model organism because structures are predominantly linked to publications (Berman et al., 2000).
Increasingly structured collaborations
As the field of GEM reconstruction matures, there is a growing realization that collaborative reconstruction efforts yield GEMs of higher quality. Owing to the multidisciplinary characteristic of GEM development, the involvement of experts specialized across various fields of study is needed. To encourage participation and facilitate cross-disciplinary collaborations, software and web servers have been recently developed (including RAVEN 2 and MetExplore) and used as tools during reconstruction jamborees. The range of functionalities provided includes collaborative development through online platforms, version control (supporting easy tracking of modifications), and the integration of ‘omics’ datasets with the model. MetExplore is designed to stimulate the involvement of end-users with limited computational skills by offering an all-in-one online workspace (Cottret et al., 2018). In addition, their platform introduces the innovative idea of a voting system, allowing annotators to voice their opinion regarding the accuracy of each element of a GEM through an approval/disapproval rating system. In turn, votes can be easily queried by any collaborator. For the more experienced computational biologist, the use of GitHub repositories and live notebooks is increasingly accepted as the standard of practice for the development of so-called community models (Lieven et al., no date; Wang et al., 2018; Lu et al., 2019; Robinson et al., 2020). With the reproducibility of modeling and reconstruction efforts becoming a concern, GitHub offers easy tracking of modifications, a feature which is now increasingly attractive for GEM developers. In line with this trend, GEMs have made their appearance in open source repositories, such as BioModels and Zenodo, in which version control and cross-referencing to GitHub repositories and publications are supported. Going forward, communities focused on specific target organisms may develop ‘crowd-sourcing’ online forums to systematically harvest new knowledge and incorporate it into a consensus reconstruction. Crowd-sourcing is further enhanced by the recent establishment of standardized and consistent quality metrics and model validation tests (memote) (Lieven et al., 2020).
Bottleneck 2: content removal
One overlooked and underappreciated but highly time-consuming step in the GEM life cycle is the manual removal of content (Seif, Monk, Mih, et al., 2019). Both content addition and removal constitute important forms of knowledge for GEM reconstruction. Additions occur first, and algorithms designed to assemble metabolic networks attempt to annotate as many genes and reactions as possible. By lowering the threshold for sequence homology, comparing protein domains and sequence signatures (often mapping to multiple functions), and mapping content from distantly related organisms, the number of annotated metabolic genes can be artificially increased. All of these parameters can be fine-tuned but lean towards inclusion.
Reference databases:
Content removal occurs naturally at every stage of the GEM life cycle. Before annotations are retrieved for automated draft reconstruction assembly, the reference network databases are themselves constantly undergoing updates. These updates include expansion to new pathways and to new species. When novel species are annotated, existing pathways that are annotated for other species are imported necessitating subsequent pruning of overly generous imports. In addition, as more pathways are added, older pathways are reviewed and improved, motivating the removal of redundant entries and lower confidence content. This process is explicitly mentioned for the updates of Metacyc (Bairoch and Apweiler, 2000; Karp et al., 2002) and Uniprot (O’Donovan et al., 1999; Wieser, Kretschmann and Apweiler, 2004; UniProt Consortium, 2019). For example, a total of 595 pathways have been removed from Metacyc since 2003 (Karp et al., 2002; Krieger, 2004; Caspi et al., 2008, 2015, 2018), with pathway removals reaching as much as 42% of additions in 2009 (Figure 3a). In total, almost as many (82%) pathways were removed from the Metacyc database as of 2017 as the total number of pathways that was present in 2003 (with a concurrent 524% increase in size).
Figure 3: Content removal forms part of every stage in the GEM life cycle.
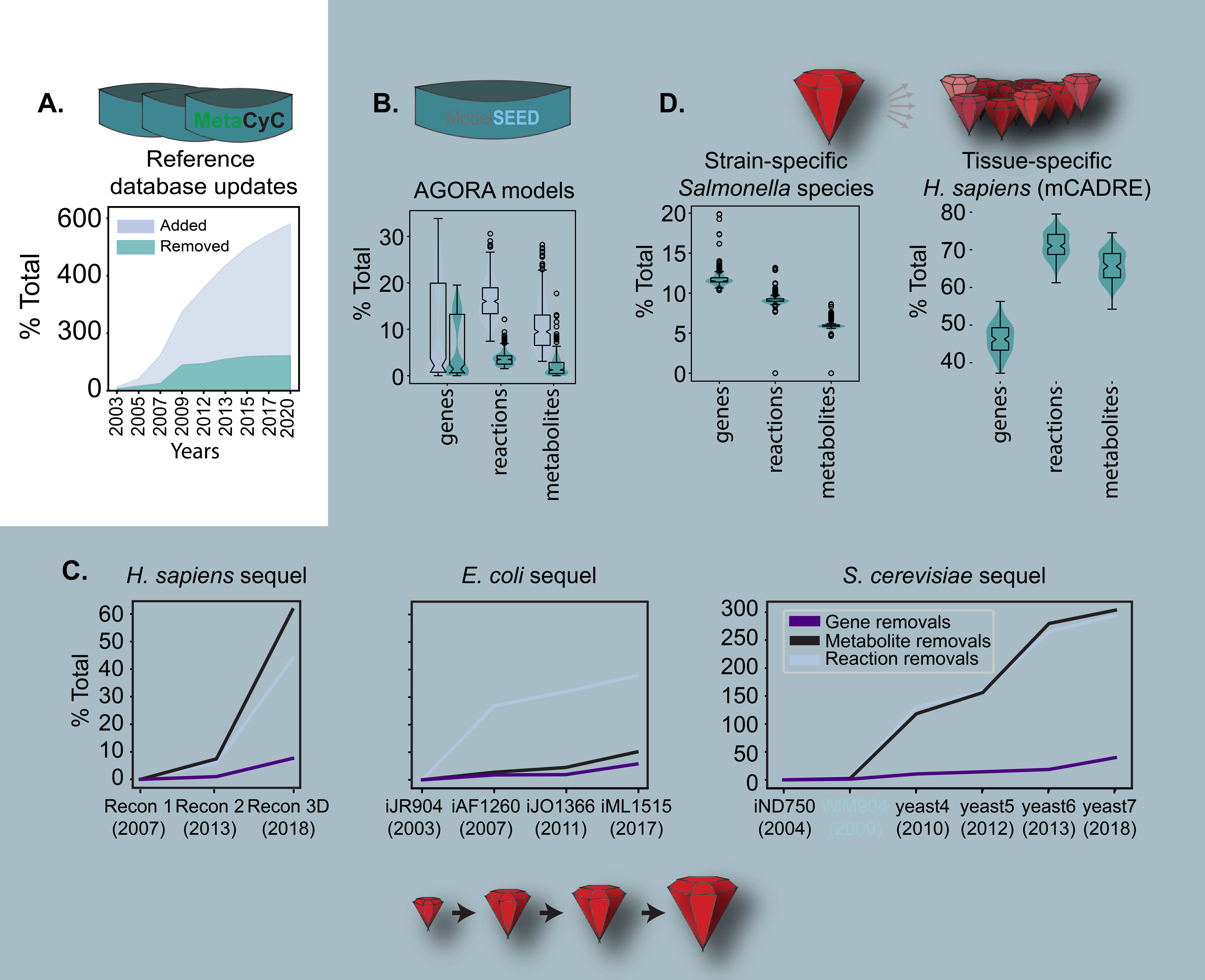
The number of removed or added instances is normalized across the plots by the total number of instances in the oldest database or model included in the analysis. A) Reference database updates. Fluctuations in both removed and added pathways are shown for each update going from 2003 to 2017. Numbers were obtained from the reports in the corresponding publications (Karp et al., 2002; Krieger, 2004; Caspi et al., 2008, 2015, 2018), B) GEM inception. Addition and removal of genes, reactions, and metabolites are plotted for the 773 reconstructions of gut microbes. ModelSEED draft reconstructions were obtained courtesy of Thiele et. al, and we compared their modeled content with the corresponding curated reconstructions. Counts are normalized with respect to the number of instances in the ModelSEED reconstructions. C) GEM maturation. Content removal occurs at every update, with genes being affected the least. Models were downloaded for E. coli (Reed et al., 2003; Feist et al., 2007; Orth et al., 2011; Monk et al., 2017; Fang, Lloyd and Palsson, 2020), H. sapiens (Duarte et al., 2007; Thiele et al., 2013; Swainston et al., 2016; Brunk et al., 2018) and S. cerevisiae (Duarte, Herrgård and Palsson, 2004; Mo, Palsson and Herrgård, 2009; Heavner et al., 2012, 2013; Aung, Henry and Walker, 2013; Lu et al., 2019) from the BiGG database(Norsigian et al., 2020), VMH(Noronha et al., 2019), BioModels(Chelliah et al., 2015), yeast.sf.net (Aung, Henry and Walker, 2013), and Silicolife(Home - SilicoLife, no date). iMM904 is shaded to mark a change of namespace which caused the sudden increase in the percent total of metabolite and reaction removals. D) GEM specialization. The 410 strain-specific models of Salmonella (Seif et al., 2018; Seif, Monk, Machado, et al., 2019) were obtained from the BIGG database and the 126 tissue-specific models were downloaded from Wang et. al (Wang, Eddy and Price, 2012) and we compared the content of each model with the content of the starting reference models STM.v1.0 and Human Recon 1, respectively.
De novo draft reconstruction:
The process of de novo draft reconstruction assembly is also heavy in content pruning. For example, there was a significant number of manual removals in the assembly of 773 reconstructions of gut microbes (AGORA) (Figure 3b) (Magnúsdóttir et al., 2017). The AGORA reconstructions were built by first automatically generating draft reconstructions using ModelSEED, which pulls the annotations from a library of manually curated subsystems and protein families. Manual and semi-automated curations were subsequently made to improve the quality of the reconstructions. In this process, an average of 3.7% of the content was removed from each reconstruction (with a concurrent 12.7% increase in model content).
GEM maturation:
More superfluous content is added when converting the metabolic reconstruction to a computable format (a process which we break down in the subsequent section). As models are updated, the superfluous content is pruned out. GEM maturation efforts for E. coli (Reed et al., 2003; Feist et al., 2007; Orth et al., 2011; Monk et al., 2017), H. sapiens (Duarte et al., 2007; Thiele et al., 2013; Swainston et al., 2016; Brunk et al., 2018) and S. cerevisiae (Duarte, Herrgård and Palsson, 2004; Mo, Palsson and Herrgård, 2009; Heavner et al., 2012, 2013; Aung, Henry and Walker, 2013; Lu et al., 2019) reflect the content fluctuation observed in the Metacyc database (Figure 3c). For example, the total number of metabolites removed by the third H. sapiens update amounts to a total of 60% of the number of metabolites originally included in the Recon 1. The active community of S. cerevisiae modelers update the yeast network more frequently, with multiple updates involving namespace switches (i.e., the convention used to uniquely identify model instances). In Figure 3c, we follow one of the yeast sequels going through five updates, from iND750 to yeast 7. In total, 40% of the genes included in iND750 are removed.
GEM specialization:
Finally, data-driven reductive workflows such as the construction of hundreds of strain-specific models of Salmonella led to the removal of between 2.3% and 13.2% of the total number of modeled instances (Figure 3d)(Seif et al., 2018). Similarly, 53.5% and 71.6% of the starting model is removed in subsequent tissue-specific and disease-specific human metabolic models generated through the mCADRE workflow.
A framework for the removal of content
When curating GEMs, a significant amount of time is dedicated to identifying what content should be removed. While content addition is expedited by increasingly larger reference network databases and automated workflows, content removal is still a heavily manual task, performed silently on the back end of all reconstruction efforts. As a result, some of these efforts are duplicated across groups. For example, following its undocumented removal at iteration i, a reaction may be re-introduced at iteration i+1, because it still fits the thresholds set by automated reconstruction algorithms. The range of evidence that supports the absence of a capability is highly diverse in type and confidence level. Unfortunately, the removal of content is not usually saved in any format, constituting a loss of information that could be used to inform other model building efforts and boost reproducibility. The emergence of version control is likely to minimize this loss. However, such documentation is not sufficiently structured.
We thus propose a framework to enhance structured continuity in content removal as well as a confidence level scoring scheme for deleted content. When a reaction is added to a metabolic reconstruction, it is saved in a stoichiometric matrix (S) which mathematically encodes the participating substrates and products and the corresponding stoichiometries, and is assigned a confidence score. We propose that when a reaction is removed from a network, it should be likewise saved in an analogous matrix, which we henceforth call the recall-Reaction matrix (rR matrix). The rR matrix is structured similarly to the stoichiometric matrix, with rows representing each metabolite’s mass balance, and columns representing each reaction’s stoichiometry. The removal of a gene from a gene-reaction rule is also highly informative. Such removals can be saved in the recall-Gene matrix (aG matrix), with the rows representing the genes, the columns representing the reactions, and binary entries (with ‘1’ representing exclusion, and ‘0’ signifying no action). Each entry of the rR and rG matrices should be accompanied by a confidence score, a reference, and ideally a note, justifying the reason for exclusion.
We therefore outline a confidence scoring scheme which inversely mirrors the framework established by Thiele et al (Table 1), thereby offering an expanded continuity in confidence levels associated with modeled objects (Thiele and Palsson, 2010a). In our scheme, entries with the lowest confidence scores are more likely to be transferred between the two matrices. Conversely, entries scoring higher confidence values have a lower probability of being transferred at each iteration. It is more challenging to experimentally confirm the absence as opposed to the presence of a metabolic capability. However, such knowledge exists, and can be converted into a set of notes, recommendations, and warnings encoded in the rR and rG matrices (Table 1). Mathematically structured content removal has the advantage of being computationally tractable and easily accessible by the broader non-computational community. Such matrices can be easily converted into both human readable spreadsheets as well as modeling objects. They can be expanded in subsequent GEM maturation efforts and can also easily be checked against novel low confidence additions for redundancies, thereby avoiding duplication of efforts in content removal. Because they are expanded at each iteration, rR and rG matrices contain a blueprint of model modifications allowing easier reproduction of model updates. Finally, the rR and rG matrices can serve as an additional input to automated reconstruction tools when modeling novel species. For example, such tools could pool knowledge obtained from the rG and rR matrices with computed phylogenetic relatedness, pathway homology, and genome architecture similarities to extract higher confidence entries.
Table 1:
Framework for confidence level scoring as part of content removal efforts in metabolic reconstructions.
| Evidence type | Confidence score | Examples |
|---|---|---|
| Direct biochemical evidence and dereplication | 4 | Newly discovered biochemical reaction overriding an entry with a low confidence score (e.g., through biochemical assays, resolved protein structures, etc.) (Seif, Monk, Mih, et al., 2019). Removal of sinks and demands following network structure modifications brought on by new biochemical evidence. Removal of reactions associated with a single gene previously predicted to be promiscuous, when novel biochemical evidence indicates lack of promiscuity. Removal of imbalanced reactions due to lack of explicit inclusion of metabolites’ formula, as new formulae are characterized and retrieved (Moreira et al., 2019). Removal of duplicated content: reactions and metabolites (Zhu et al., 2018) |
| Indirect evidence from combination of various data types | 3 | Removal supported by more than one ‘omic’ data type, for example: Discrepancies between model predictions and gene essentiality screens coupled with structural analysis, genomic neighborhood analysis, and transcriptomic data to identify false isozymes (Broddrick et al., 2016). Discrepancies with gene essentiality screens coupled with metabolic modeling methods such as GLOBALFIT and updated genome annotations, or data derived genetic interaction networks (Szappanos et al., 2011; Hartleb, Jarre and Lercher, 2016). |
| Indirect evidence from single data type | 2 | Indirect evidence supporting the lack of a capability, e.g. inability to utilize a range of nutrient sources, serotyping results (Seif, Monk, Machado, et al., 2019). Updated genome annotation indicating alternative function. Transcriptomics data indicating lack of expression in a cell line (Wang, Eddy and Price, 2012). |
| Modeling purpose | 1 | Reaction with low confidence score causing significant flux alteration at the systems level and creating thermodynamically infeasible flux cycles (De Martino et al., 2013). |
The recall-Reaction matrix:
There are multiple conceptual shortcuts and general rules that can be applied to identify candidates for the rR matrix (Figure 4). A prime example is that of duplicate entries (confidence score of 4). Duplicate reactions and pathways are commonly found under different identifiers or with slight variations to the reaction content (Figure 4a). Reactions that are identical except for their thermodynamic reversibility (one being reversible and the other irreversible) are duplicate reactions. Such cases may arise due to uncertainty with regard to reaction directionality and tend to induce infeasible flux cycles. If reaction directionality uncertainty persists, the duplicate reactions should be replaced with a single reversible reaction (Thiele and Palsson, 2010a). Similarly, reactions with identical content, save for stoichiometric coefficients, are duplicate reactions and should be replaced with the entry with the highest confidence score. With increasing compartmentalization complexity, reactions can inadvertently be added to multiple compartments when they should only be contained in one. Duplicate metabolite entries also contribute to reaction redundancy and can be resolved once duplicate metabolites are identified. Oftentimes, duplicate reactions have identical GPRs, and this feature could serve to identify possible candidates. These issues affect both GEMs and reference network databases as suggested by Altman et. al (Altman et al., 2013).
Figure 4:
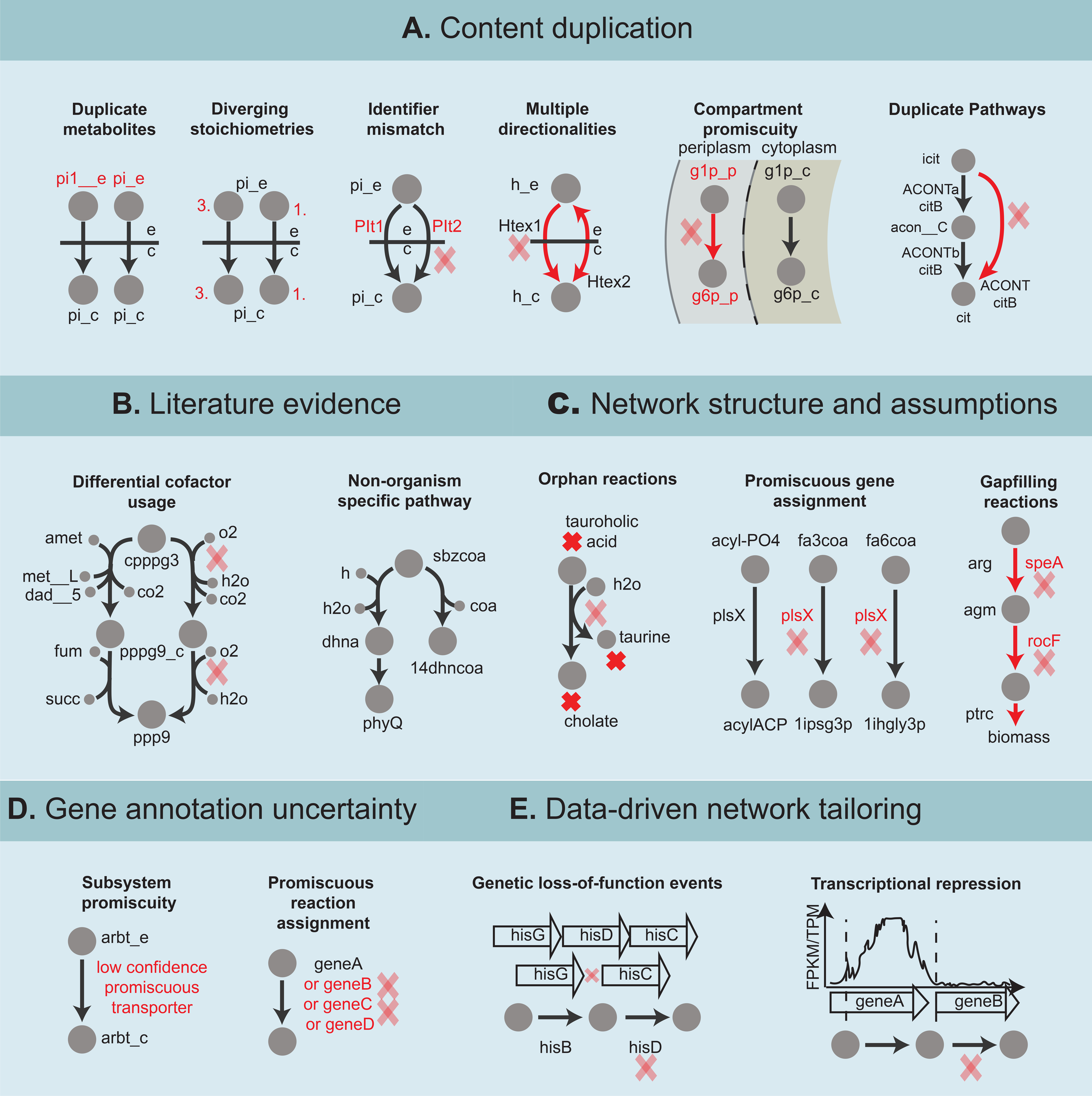
Types of content removal can be subdivided into: A) content duplication. From left to right duplicate reaction caused by; inconsistent metabolite identifier (pi_e and pi__e identify extracellular phosphate), diverging stoichiometries (three phosphate molecules transported to the cytoplasm versus one phosphate molecule transported), reaction identifier mismatch (Plt1 and Plt2 are both the same transport reaction), multiple directionalities (bi-directional transport versus import of hydrogen across the outer membrane), compartment promiscuity (erroneous assignment of a phosphoglucomutase to the periplasmic compartment converting D-glucose1phosphate to D-glucose-6-phosphate), and duplicate pathways (conversion of isocitrate to citrate via a two-step pathway and via a lumped reaction). N.B., these cases serve as examples of possible content duplication but should be checked against the literature. For example, an organism could encode two phosphate transporters both of which catalyze a transport process but with different stoichiometry. B) Organism nonspecific pathways; left: example of a heme pathway initially added to the S. aureus GEM with incorrect differential cofactor usage due to lack of knowledge and subsequently corrected as a result of recent discoveries (Lobo et al., 2015; Seif, Monk, Mih, et al., 2019), right: example of a plant pathway for vitamin K1 (phylloquinone) biosynthesis which was added to the GEM of a gram-positive bacterium likely due to insufficient curation. C) Low confidence modeling assumptions. From left to right; orphan taurocholate amidohydrolase reaction annotated as being catalyzed by a C59 family penicillin amidase, promiscuous assignment of plsX as a phosphate acyltransferase, a isohexadecanoylglycerol-3-phosphate O-acyltransferase and an isopentadecanoyl-glycerol-3-phosphate O-acyltransferase (among others), gap filled putrescine biosynthesis pathway with erroneous gene assignment. D) Low confidence gene annotation; transport reaction assigned to a low confidence transporter, generic reaction with long “OR” based gene reaction rule, E) Datadriven evidence. Reaction catalyzed by a gene carrying a loss-of-function mutation, and reaction catalyzed by a gene that is not expressed in a cell. Abbreviations: X_c = cytoplasmic, X_e = extracellular, X_p = periplasmic, h = hydrogen, pi = phosphate, icit = isocitrate, acon__C = cis-aconitate, cit = citrate, cpppg3 = coproporphyrinogen III, amet = S-adenosyl-L-methionine, met__L = L-methionine, fum = fumarate, succ = succinate, pppg9 = protoporphyrinogen IX, o2 = oxygen, h2o = water, co2 = carbon dioxide, ppp9 = protoporphyrin, sbzcoa = O-succinylbenzoylcoA, dhna = 1,4-dihydroxy-2-naphthoate, phyQ = phylloquinone, 14dhncoa = 1,4-dihydroxy-2napthoyl-coA, fa3coa = fatty acid (Iso-C15:0)-coA, fa3coa = fatty acid (Iso-C16:0)-coA, 1ipsg3p = 1-isopentadecanoyl-sn-glycerol 3-phosphate, 1ihgly3p = 1-isohexadecanoyl-sn-glycerol_3phosphate, arg = L-arginine, agm = agmatine, ptrc = putrescine, arbt = arbutin
A second category of rR reactions constitutes metabolic capabilities which simply fall outside of the metabolic capabilities of the organism of interest (Figure 4b). The identification of reactions in this category usually results from literature review. With higher taxonomic ranks, pathways that should be excluded are more easily filtered out by current annotation platforms. For example, chlorophyll biosynthesis is predominant in the Plantae reactome but rare in the Animalia reactome. Knowledge of differences at lower taxonomic ranks (e.g., species-level) exists but can be more challenging to identify. Despite these known differences, as a result of running automated reconstruction workflows, pathways from unrelated organisms can get added to draft GEMs. For example, unlike Bacillus licheniformis, Bacillus subtilis lacks a glyoxylate shunt as evidenced by its inability to grow on C-2 carbon sources (confidence level 3, indirect biochemical evidence) (Kabisch et al., 2013). Subtle variations in cofactor usage across organisms are only reflected in models when sufficient manual curations have been performed. They have been shown to affect model-predicted thermodynamic favorability and energy cost (Du et al., 2018). For example, in S. aureus, two haem biosynthesis pathways were originally modeled; one using oxygen and the other S- adenosyl- L- methionine as a cofactor. However, recent evidence showed that S. aureus encodes an oxygen-independent transitional pathway, prompting the removal and replacement of both pathways (confidence score of 4, direct biochemical evidence) (Lobo et al., 2015; Seif, Monk, Mih, et al., 2019).
Promiscuity of gene associations is a hallmark of weak curation coverage (Figure 4c). When a single gene is associated with multiple reactions, reactions can be automatically flagged for review. Promiscuity of GPRs can arise due to annotation uncertainties, but they can also have a biological basis (e.g., substrate transporters or fatty acid biosynthesis). As more literature evidence comes to light, a subset of these reactions tends to be removed (confidence score of 4, direct biochemical evidence). The network structure can also serve to inform content removal efforts. For example, reactions with promiscuous gene assignments tend to contain multiple dead-end metabolites. GPR promiscuity can result from relaxed constraints on homology searches, or from searches encompassing a large breadth of distantly related organisms.
Reactions originating from modeling assumptions constitute a fourth category of rR reactions (Figure 4c). They technically are part of the model but not the reconstruction, and include sink, demand, and gap-filling reactions. Their addition usually fulfills a modeling purpose. However, when sufficient advances in knowledge enable their removal, they should be added to the rR matrix. For instance, the first S. aureus biomass reaction was adopted from the B. subtilis GEM which was itself partially imported from the E. coli GEM (Oh et al., 2007). Because spermine was a precursor of the biomass objective function, a gap filling spermine biosynthesis pathway was added, with a subset of reactions being assigned erroneous GPRs. Current literature indicates that compared to B. subtilis, S. aureus lacks a polyamine biosynthesis capability and produces no spermidine or any of its precursors (confidence score of 3). In addition, alternative molecular functions of the associated genes were characterized, further supporting the removal of the pathway (confidence score of 4) (Townsend et al., 1996; Joshi et al., 2011).
The recall-Gene matrix:
One of the hallmarks of draft reconstructions is the occurrence of GPRs with a large number of locus tags linked by the OR rule marking enzymes with redundant functions (with extreme cases containing as many as 10 “isozymes”) (Figure 4d). Extended OR rules skew transcriptomics data-integration methods which rely on the boolean encoding in the GPR to tailor the starting GEM. When a reaction is catalyzed by multiple isozymes, flux bounds are only changed when similar expression changes are observed across all genes. It should be noted that some cases with extended GPRs have a biological basis. For example, when considering underground metabolism, substrate promiscuity contributes to an extended GPR (Guzmán et al., 2015, 2019).
Uncertainties in functional annotations occur across all genome annotation platforms and can be propagated through metabolic reconstruction platforms (Figure 4d). If left untouched, downstream analyses can be heavily skewed, and errors further propagated to specialized GEMs. For example, a non-metabolic gene erroneously annotated as a nutrient transporter may be upregulated under a specific condition. Following established transcriptomics data integration workflows, modelers may choose to artificially up-regulate the associated reactions based on the given data, which will incorrectly lead the model to predict that substrate uptake is increased. Indeed, this observation emphasizes the importance of using comprehensive annotation databases with both known metabolic and known non-metabolic genes as a basis for automated reconstruction workflows in order to avoid mis-assigning non-metabolic genes to a metabolic function. In addition, we suggest that homology searches be adjusted according to the availability of curated GEMs, with stricter thresholds when closely related well-annotated organisms are available (e.g., 80% is used for strains of the same species (Seif et al., 2018; Norsigian et al., 2019)).
A second category of rG genes includes down-regulated genes in GEM specialization efforts. While a subset of computational approaches result in modified reaction constraints (e.g., eFlux (Kim et al., 2016), Prom (Chandrasekaran and Price, 2010), Gimme (Blazier and Papin, 2012)), other approaches push for the removal of downregulated genes and associated reactions (e.g., mCADRE). In this case, removed content can be transferred to the rG and rR matrices and assigned a confidence level of 2 (indirect evidence from single data type). A third category of rG genes includes any genes affected by loss of function mutations (Figure 4e). A range of genetic mechanisms can indicate loss of function, including SNPs leading to early in-frame stop codons or frameshifts, large in-frame deletions/insertions, and mutation of the start codon. A strain may encode all genes participating in a linear pathway for the synthesis of a biomass precursor, but still lack the capability to produce that precursor, or produce a modified precursor due to loss of function events. For instance, Salmonella strains of serogroup O:2 carry the same O antigen biosynthetic gene island as O:9 strains, except for a frameshift mutation in tyv which abrogates the CDP-paratose 2-epimerase metabolic step, and results in a modified downstream metabolic pathway and a modified O-antigen structure (confidence score of 4, direct biochemical evidence)(Seif, Monk, Machado, et al., 2019). Similarly, despite carrying the full cap locus, strains of USA300 and USA500 clonal lineages of S. aureus are incapable of producing a capsular polysaccharide due to a single nucleotide indel causing a frameshift mutation in capD (Boyle-Vavra et al., 2015). Combining multiple fields of study with metabolic modeling can reveal candidate rG genes. For example, comparative genomics and evolutionary genetics together can reveal gene deletion events. Combining mutagenesis results with comparative genomics can point out single nucleotide polymorphisms causing loss of function. Analysis of structural modifications can identify possible deleterious mutations.
Content removal transforms the constraints on the solution space
There are multiple ways in which content removal or lack thereof affects the quality of a GEM. Adding complete pathways to an existing network creates additional solution space. Conversely, removing content (reactions and metabolites) removes dimensions and degrees of freedom and results in a reduction of the set of feasible steady-state flux solutions.
In the dummy pathway shown in Figure 5a consisting of five reactions and three metabolites, deleting reaction 4 results in the modification of two stoichiometric constraints (i.e., the upper and lower bounds of the reaction) or, equivalently, the addition of the null constraint for the flux through reaction 4. Thus, only one pathway (p2) can carry flux. If the removal of reaction 4 is supported by literature evidence, then one of two possibilities hold: there is either a gap in the network or a new gap in knowledge for a mechanism enabling the consumption of metabolite 3.
Figure 5:

Effect of removal of content on A) The solution space: We illustrate here the effect of deleting a reaction in a dummy model containing two pathways (p1 and p2). We show how the removal of reaction 4 translates to two added zeros in the stoichiometric matrix (S) and two modified constraints, B) Reaction essentiality: We extracted two iterations from three GEM sequels for each of S. aureus (Bosi et al., 2016; Seif, Monk, Mih, et al., 2019), M. tuberculosis (Jamshidi and Palsson, 2007; Kavvas et al., 2018), and P. putida (Nogales, Palsson and Thiele, 2008; Nogales et al., 2020). For each species, we found the set of removed reactions between the latest model and its previous version. We proceeded to re-introduce randomly sampled subsets of the deleted reactions into the latest model. We observe that as deleted content is added back to the reconstruction, fewer reactions are essential, C) Thermodynamic consistency: Here, we limited our analysis to the S. aureus GEM sequel. Similar to B, we iteratively add randomly picked subset sizes of the deleted reactions. However, as reactions are reintroduced, we simulate for the model’s ability to freely produce cofactors with all nutrient exchanges blocked (Fritzemeier et al., 2017). As deleted content is added back to the reconstruction, more cofactors can be freely generated in the model. Abbreviations: v1 = flux through reaction 1, p1 = pathway 1, S = stoichiometric matrix, ATP = adenosine triphosphate, NADH = reduced nicotinamide adenine dinucleotide, NADPH = reduced nicotinamide adenine dinucleotide phosphate, MQL8 = menaquinol 8.
While reaction 3 is blocked, reaction 2 becomes essential. The number of essential reactions is an indicator of biological redundancy encoded in the organism’s genome. A lower number of essential reactions indicates that the organism has evolved alternative metabolic routes as a survival strategy. Superfluous content drives the number of essential reactions down by creating artificial redundancies. Despite having a larger total number of modelled reactions, updated models tend to have a larger number of essential reactions than the precursor GEM. Recent updates for M. tuberculosis, P. putida, and S. aureus all show this trend (Figure 5b). As a result of removing alternative pathways, the connectivity of the network is altered, forcing non-zero flux through a subset of reactions. This property is leveraged to build context-specific models in which reactions are either removed or constraints modified in accordance with an “omics” data set (Opdam et al., 2017; Zampieri et al., 2019).
One of the hallmarks of insufficiently curated GEMs is the presence of thermodynamically infeasible energy generating cycles (also known as futile cycles). Such cycles carry net flux at steady-state (Schellenberger, Lewis and Palsson, 2011), allow free charging of energy carriers in the absence of nutrient uptake (Fritzemeier et al., 2017), yield increased biomass production, and negatively skew the predicted flux distribution following transcriptomics data integration (Wang, Eddy and Price, 2012; Machado and Herrgård, 2014). Futile cycles occur due to lack of constraints for irreversibility or the presence of erroneous reactions or cofactors. A recently published update to the S. aureus GEM yielded the removal of 26.5% of the starting model and the elimination of futile cycles (Seif, Monk, Mih, et al., 2019). Upon reintroduction of subsets of different sizes of the removed reactions into the updated GEM, flux loops are also reintroduced (Figure 5c). Naturally, as more erroneous reactions are reintroduced, the probability of the network being capable of freely charging various energy carriers increases. For a subset of energy carriers, as few as ten added reactions are sufficient to yield their free production.
Discussion
The field of metabolic reconstruction and modeling is progressing at a steady pace. As challenges are identified, solutions banking on both theoretical and technological advances have been applied, coupled with the development of novel computational platforms and tools to overcome them. Here, we showed that GEM development can be delineated by four chronological phases: 1) inception, 2) maturation, 3) specialization, and 4) amalgamation. GEM inception is well established, GEM maturation is becoming increasingly defined, and GEM specialization and amalgamation remain as two active fields of research focused on modeling methods development. As a GEM progresses through each stage, it increases in quality and complexity, accounting for an expanded reactome and scope and a higher level of organism-specific detail. In reality, a GEM cycles through each stage multiple times, with some GEMs initially skipping one or more stages altogether depending on the whims of scientific interest. Nevertheless, when embarking on a GEM reconstruction effort, situating it within a GEM life cycle will invariable offer context and texture to both modelers and users.
We proceeded to highlight two bottlenecks for any given GEM life cycle towards reaching higher quality: GEM maturation and content removal. Interestingly, GEM maturation efforts have occurred independently across organisms but have converged towards a common set of approaches. We extracted eight characteristics of past endeavors which can serve as a starting point for future GEM maturation efforts. However, all approaches still lacked a solid framework for modification tracking, especially with respect to content removal. We thus proposed a mathematical framework in which removed content is transferred to a backup matrix which we call the recall matrices, as a strategy to circumvent duplication of effort and to mitigate the challenge of reproducing and propagating model updates. As the standard of practice is moving towards online open-source platforms for community-driven GEM development, we foresee increasing reliance on such frameworks, and in turn an increasing quality of models.
Highlights.
The GEM life cycle delineates four steps in the evolution of metabolic models
GEM maturation is the first rate limiting step in quality accrual
Lack of adequate content removal hinders GEM progression
Acknowledgements:
We thank Almut Heinken and Ines Thiele for providing us with the starting draft AGORA models and Marc Abrams for reviewing this manuscript. This work was supported by NIH Grants U01 AI124316 and R01GM057089 and Novo Nordisk Foundation Grant NNF10CC1016517.
Footnotes
Declaration of interests:
The authors declare no competing interests.
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
All models were downloaded from reference databases including BiGG(Norsigian et al., 2020), VMH(Noronha et al., 2019), BioModels(Chelliah et al., 2015), yeast.sf.net (Aung, Henry and Walker, 2013), BioModels(Chelliah et al., 2015), Zenodo (Peters et al., 2017) and Silicolife(Home - SilicoLife, no date), except for the starting draft AGORA models which were obtained courtesy of prof. Ines Thiele. Simulations were run using COBRApy.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agren R et al. (2014) ‘Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling’, Molecular systems biology, 10(3), p. 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman T et al. (2013) ‘A systematic comparison of the MetaCyc and KEGG pathway databases’, BMC bioinformatics, 14, p. 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin AP et al. (2018) ‘KBase: The United States Department of Energy Systems Biology Knowledgebase’, Nature biotechnology, 36(7), pp. 566–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung HW, Henry SA and Walker LP (2013) ‘Revising the Representation of Fatty Acid, Glycerolipid, and Glycerophospholipid Metabolism in the Consensus Model of Yeast Metabolism’, Industrial biotechnology, 9(4), pp. 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurich MK, Fleming RMT and Thiele I (2016) ‘MetaboTools: A Comprehensive Toolbox for Analysis of Genome-Scale Metabolic Models’, Frontiers in physiology, 7, p. 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin CP et al. (2004) ‘The knockout mouse project’, Nature genetics, 36(9), pp. 921–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A and Apweiler R (2000) ‘The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000’, Nucleic acids research, 28(1), pp. 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartha I et al. (2018) ‘Human gene essentiality’, Nature reviews. Genetics, 19(1), pp. 51–62. [DOI] [PubMed] [Google Scholar]
- Becker SA et al. (2007) ‘Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox’, Nature Protocols, pp. 727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- Ben Guebila M and Thiele I (2019) ‘Predicting gastrointestinal drug effects using contextualized metabolic models’, PLoS computational biology, 15(6), p. e1007100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM et al. (2000) ‘The Protein Data Bank’, Nucleic acids research, 28(1), pp. 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazier AS and Papin JA (2012) ‘Integration of expression data in genome-scale metabolic network reconstructions’, Frontiers in physiology, 3, p. 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA et al. (2015) ‘Gene essentiality and synthetic lethality in haploid human cells’, Science, 350(6264), pp. 1092–1096. [DOI] [PubMed] [Google Scholar]
- Bordbar A et al. (2010) ‘Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions’, Molecular systems biology, 6, p. 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosi E et al. (2016) ‘Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity’, Proceedings of the National Academy of Sciences of the United States of America, 113(26), pp. E3801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle-Vavra S et al. (2015) ‘USA300 and USA500 clonal lineages of Staphylococcus aureus do not produce a capsular polysaccharide due to conserved mutations in the cap5 locus’, mBio, 6(2). doi: 10.1128/mBio.02585-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broddrick JT et al. (2016) ‘Unique attributes of cyanobacterial metabolism revealed by improved genome-scale metabolic modeling and essential gene analysis’, Proceedings of the National Academy of Sciences of the United States of America, 113(51), pp. E8344–E8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broddrick JT et al. (2019) ‘Cross-compartment metabolic coupling enables flexible photoprotective mechanisms in the diatom Phaeodactylum tricornutum’, The New phytologist, 222(3), pp. 1364–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk E et al. (2018) ‘Recon3D enables a three-dimensional view of gene variation in human metabolism’, Nature biotechnology, 36(3), pp. 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brynildsen MP et al. (2013) ‘Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production’, Nature biotechnology, 31(2), pp. 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AI and Zampieri M (2019) ‘Metabolomics-Driven Exploration of the Chemical Drug Space to Predict Combination Antimicrobial Therapies’, Molecular cell, 74(6), pp. 1291–1303.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R et al. (2008) ‘The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases’, Nucleic acids research, 36(Database issue), pp. D623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R et al. (2015) ‘The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases’, Nucleic acids research, 44(D1), pp. D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R et al. (2018) ‘The MetaCyc database of metabolic pathways and enzymes’, Nucleic Acids Research, pp. D633–D639. doi: 10.1093/nar/gkx935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R et al. (2019) ‘The MetaCyc database of metabolic pathways and enzymes-a 2019 update’, Nucleic acids research. doi: 10.1093/nar/gkz862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran S et al. (2017) ‘Comprehensive Mapping of Pluripotent Stem Cell Metabolism Using Dynamic Genome-Scale Network Modeling’, Cell reports, 21(10), pp. 2965–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran S and Price ND (2010) ‘Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis’, Proceedings of the National Academy of Sciences of the United States of America, 107(41), pp. 17845–17850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelliah V et al. (2015) ‘BioModels: ten-year anniversary’, Nucleic acids research, 43(Database issue), pp. D542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary KS et al. (2016) ‘EGFR Signal-Network Reconstruction Demonstrates Metabolic Crosstalk in EMT’, PLoS computational biology, 12(6), p. e1004924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello Z and Martin HG (2018) ‘A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data’, NPJ systems biology and applications, 4, p. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottret L et al. (2018) ‘MetExplore: collaborative edition and exploration of metabolic networks’, Nucleic acids research, 46(W1), pp. W495–W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperlovic-Culf M (2018) ‘Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling’, Metabolites, 8(1). doi: 10.3390/metabo8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani C et al. (2019) ‘Integration of single-cell RNA-seq data into population models to characterize cancer metabolism’, PLoS computational biology, 15(2), p. e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martino D et al. (2013) ‘Counting and correcting thermodynamically infeasible flux cycles in genome-scale metabolic networks’, Metabolites, 3(4), pp. 946–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devoid S et al. (2013) ‘Automated genome annotation and metabolic model reconstruction in the SEED and Model SEED’, Methods in molecular biology, 985, pp. 17–45. [DOI] [PubMed] [Google Scholar]
- Dias O et al. (2015) ‘Reconstructing genome-scale metabolic models with merlin’, Nucleic Acids Research, pp. 3899–3910. doi: 10.1093/nar/gkv294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias O et al. (2018) ‘Reconstructing High-Quality Large-Scale Metabolic Models with merlin’, Methods in Molecular Biology, pp. 1–36. doi: 10.1007/978-1-4939-7528-0_1. [DOI] [PubMed] [Google Scholar]
- Dobson PD et al. (2010) ‘Further developments towards a genome-scale metabolic model of yeast’, BMC systems biology, 4, p. 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte NC et al. (2007) ‘Global reconstruction of the human metabolic network based on genomic and bibliomic data’, Proceedings of the National Academy of Sciences of the United States of America, 104(6), pp. 1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte NC, Herrgård MJ and Palsson BØ (2004) ‘Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model’, Genome research, 14(7), pp. 1298–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du B et al. (2018) ‘Thermodynamic favorability and pathway yield as evolutionary tradeoffs in biosynthetic pathway choice’, Proceedings of the National Academy of Sciences of the United States of America, 115(44), pp. 11339–11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahim A et al. (2013) ‘COBRApy: COnstraints-Based Reconstruction and Analysis for Python’, BMC systems biology, 7, p. 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS and Palsson BO (1999) ‘Systems properties of the Haemophilus influenzae Rd metabolic genotype’, The Journal of biological chemistry, 274(25), pp. 17410–17416. [DOI] [PubMed] [Google Scholar]
- Edwards JS and Palsson BO (2000) ‘The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities’, Proceedings of the National Academy of Sciences of the United States of America, 97(10), pp. 5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X et al. (2018) ‘Escherichia coli B2 strains prevalent in inflammatory bowel disease patients have distinct metabolic capabilities that enable colonization of intestinal mucosa’, BMC systems biology, 12(1), p. 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Lloyd CJ and Palsson BO (2020) ‘Reconstructing organisms in silico: genome-scale models and their emerging applications’, Nature reviews. Microbiology. doi: 10.1038/s41579-020-00440-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM et al. (2007) ‘A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information’, Molecular systems biology, 3, p. 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM et al. (2009) ‘Reconstruction of biochemical networks in microorganisms’, Nature reviews. Microbiology, 7(2), pp. 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM and Palsson BO (2010) ‘The biomass objective function’, Current opinion in microbiology, 13(3), pp. 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD et al. (1995) ‘Whole-genome random sequencing and assembly of Haemophilus influenzae Rd’, Science, 269(5223), pp. 496–512. [DOI] [PubMed] [Google Scholar]
- Fritzemeier CJ et al. (2017) ‘Erroneous energy-generating cycles in published genome scale metabolic networks: Identification and removal’, PLoS computational biology, 13(4), p. e1005494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto F, Ferreira R and Nielsen J (2020) ‘Pan-cancer analysis of the metabolic reaction network’, Metabolic engineering, 57, pp. 51–62. [DOI] [PubMed] [Google Scholar]
- Gelius-Dietrich G et al. (2013) ‘Sybil--efficient constraint-based modelling in R’, BMC systems biology, 7, p. 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes de Oliveira Dal’Molin C et al. (2015) ‘A multi-tissue genome-scale metabolic modeling framework for the analysis of whole plant systems’, Frontiers in plant science, 6, p. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Großeholz R et al. (2016) ‘Integrating highly quantitative proteomics and genome-scale metabolic modeling to study pH adaptation in the human pathogen Enterococcus faecalis’, NPJ systems biology and applications, 2, p. 16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez JM et al. (2020) ‘Genome-scale reconstructions of the mammalian secretory pathway predict metabolic costs and limitations of protein secretion’, Nature communications, 11(1), p. 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán GI et al. (2015) ‘Model-driven discovery of underground metabolic functions in Escherichia coli’, Proceedings of the National Academy of Sciences of the United States of America, 112(3), pp. 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán GI et al. (2018) ‘Reframing gene essentiality in terms of adaptive flexibility’, BMC systems biology, 12(1), p. 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán GI et al. (2019) ‘Enzyme promiscuity shapes adaptation to novel growth substrates’, Molecular systems biology, 15(4), p. e8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadadi N et al. (2019) ‘Mechanistic insights into bacterial metabolic reprogramming from omics-integrated genome-scale models’, bioRxiv. doi: 10.1101/690164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartleb D, Jarre F and Lercher MJ (2016) ‘Improved Metabolic Models for E. coli and Mycoplasma genitalium from GlobalFit, an Algorithm That Simultaneously Matches Growth and Non-Growth Data Sets’, PLoS computational biology, 12(8), p. e1005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavner BD et al. (2012) ‘Yeast 5 – an expanded reconstruction of the Saccharomyces cerevisiae metabolic network’, BMC Systems Biology, p. 55. doi: 10.1186/1752-0509-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavner BD et al. (2013) ‘Version 6 of the consensus yeast metabolic network refines biochemical coverage and improves model performance’, Database: the journal of biological databases and curation, 2013, p. bat059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefzi H et al. (2016) ‘A Consensus Genome-scale Reconstruction of Chinese Hamster Ovary Cell Metabolism’, Cell systems, 3(5), pp. 434–443.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinken A and Thiele I (2015a) ‘Anoxic Conditions Promote Species-Specific Mutualism between Gut Microbes In Silico’, Applied and environmental microbiology, 81(12), pp. 4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinken A and Thiele I (2015b) ‘Systems biology of host-microbe metabolomics’, Wiley interdisciplinary reviews. Systems biology and medicine, 7(4), pp. 195–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heirendt L et al. (2019) ‘Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0’, Nature protocols, 14(3), pp. 639–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heirendt L, Thiele I and Fleming RMT (2017) ‘DistributedFBA.jl: High-level, highperformance flux balance analysis in Julia’, Bioinformatics, p. btw838. doi: 10.1093/bioinformatics/btw838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CS et al. (2010) ‘High-throughput generation, optimization and analysis of genome-scale metabolic models’, Nature biotechnology, 28(9), pp. 977–982. [DOI] [PubMed] [Google Scholar]
- Herrgård MJ et al. (2008) ‘A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology’, Nature biotechnology, 26(10), pp. 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home - SilicoLife (no date) SilicoLife. Available at: http://www.silicolife.com/ (Accessed: 26 November 2019).
- Hosseini Z and Marashi S-A (2017) ‘Discovering missing reactions of metabolic networks by using gene co-expression data’, Scientific reports, 7, p. 41774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadot M et al. (2017) ‘Accounting for Protein Subcellular Localization: A Compartmental Map of the Rat Liver Proteome’, Molecular & cellular proteomics: MCP, 16(2), pp. 194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamshidi N and Palsson BØ (2007) ‘Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets’, BMC systems biology, 1, p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi CJ et al. (2020) ‘StanDep: Capturing transcriptomic variability improves context-specific metabolic models’, PLoS computational biology, 16(5), p. e1007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi GS et al. (2011) ‘Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines’, Molecular microbiology, 82(1), pp. 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabisch J et al. (2013) ‘Metabolic engineering of Bacillus subtilis for growth on overflow metabolites’, Microbial cell factories, 12, p. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M et al. (2017) ‘KEGG: new perspectives on genomes, pathways, diseases and drugs’, Nucleic acids research, 45(D1), pp. D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD et al. (2002) ‘The MetaCyc Database’, Nucleic acids research, 30(1), pp. 59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD et al. (2016) ‘Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology’, Briefings in bioinformatics, 17(5), pp. 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavvas ES et al. (2018) ‘Updated and standardized genome-scale reconstruction of Mycobacterium tuberculosis H37Rv, iEK1011, simulates flux states indicative of physiological conditions’, BMC Systems Biology. doi: 10.1186/s12918-018-0557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MK et al. (2016) ‘E-Flux2 and SPOT: Validated Methods for Inferring Intracellular Metabolic Flux Distributions from Transcriptomic Data’, PloS one, 11(6), p. e0157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MK and Lun DS (2014) ‘Methods for integration of transcriptomic data in genome-scale metabolic models’, Computational and structural biotechnology journal, 11(18), pp. 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger CJ (2004) ‘MetaCyc: a multiorganism database of metabolic pathways and enzymes’, Nucleic Acids Research, p. 438D–442. doi: 10.1093/nar/gkh100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar VS, Dasika MS and Maranas CD (2007) ‘Optimization based automated curation of metabolic reconstructions’, BMC bioinformatics, 8(1), pp. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J-C et al. (2019) ‘BOFdat: Generating biomass objective functions for genome-scale metabolic models from experimental data’, PLoS computational biology, 15(4), p. e1006971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levering J et al. (2016) ‘Genome-Scale Model Reveals Metabolic Basis of Biomass Partitioning in a Model Diatom’, PloS one, 11(5), p. e0155038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieven C et al. (2020) ‘MEMOTE for standardized genome-scale metabolic model testing’, Nature biotechnology, 38(3), pp. 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieven C et al. (no date) ‘Memote: a community driven effort towards a standardized genome-scale metabolic model test suite. bioRxiv. 2018: 1–26’. [Google Scholar]
- Lobo SAL et al. (2015) ‘Staphylococcus aureus haem biosynthesis: characterisation of the enzymes involved in final steps of the pathway’, Molecular microbiology, 97(3), pp. 472–487. [DOI] [PubMed] [Google Scholar]
- Lopes H and Rocha I (2017) ‘Genome-scale modeling of yeast: chronology, applications and critical perspectives’, FEMS yeast research, 17(5). doi: 10.1093/femsyr/fox050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H et al. (2019) ‘A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism’, Nature communications, 10(1), p. 3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado D et al. (2018) ‘Fast automated reconstruction of genome-scale metabolic models for microbial species and communities’, Nucleic acids research, 46(15), pp. 7542–7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado D and Herrgård M (2014) ‘Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism’, PLoS computational biology, 10(4), p. e1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir S et al. (2017) ‘Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota’, Nature biotechnology, 35(1), pp. 81–89. [DOI] [PubMed] [Google Scholar]
- Masid M, Ataman M and Hatzimanikatis V (2020) ‘Author Correction: Analysis of human metabolism by reducing the complexity of the genome-scale models using redHUMAN’, Nature communications, 11(1), p. 3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey D, Palsson BØ and Feist AM (2013) ‘Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli’, Molecular systems biology, 9, p. 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee MT et al. (2014) ‘Syntrophic exchange in synthetic microbial communities’, Proceedings of the National Academy of Sciences of the United States of America, 111(20), pp. E2149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza SN et al. (2019) ‘A systematic assessment of current genome-scale metabolic reconstruction tools’, Genome biology, 20(1), p. 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mih N et al. (2018) ‘ssbio: a Python framework for structural systems biology’, Bioinformatics, 34(12), pp. 2155–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo ML, Palsson BO and Herrgård MJ (2009) ‘Connecting extracellular metabolomic measurements to intracellular flux states in yeast’, BMC systems biology, 3, p. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk JM et al. (2017) ‘iML1515, a knowledgebase that computes Escherichia coli traits’, Nature biotechnology, 35(10), pp. 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk J, Nogales J and Palsson BO (2014) ‘Optimizing genome-scale network reconstructions’, Nature biotechnology, 32(5), pp. 447–452. [DOI] [PubMed] [Google Scholar]
- Moreira TB et al. (2019) ‘A Genome-Scale Metabolic Model of Soybean (Glycine max) Highlights Metabolic Fluxes in Seedlings’, Plant physiology, 180(4), pp. 1912–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti S et al. (2016) ‘MetaNetX/MNXref--reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks’, Nucleic acids research, 44(D1), pp. D523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales J et al. (2020) ‘High-quality genome-scale metabolic modelling of Pseudomonas putida highlights its broad metabolic capabilities’, Environmental microbiology, 22(1), pp. 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales J, Palsson BØ and Thiele I (2008) ‘A genome-scale metabolic reconstruction of Pseudomonas putida KT2440: iJN746 as a cell factory’, BMC systems biology, 2. doi: 10.1186/1752-0509-2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha A et al. (2019) ‘The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease’, Nucleic acids research, 47(D1), pp. D614–D624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norsigian CJ et al. (2019) ‘A workflow for generating multi-strain genome-scale metabolic models of prokaryotes’, Nature protocols. doi: 10.1038/s41596-019-0254-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norsigian CJ et al. (2020) ‘BiGG Models 2020: multi-strain genome-scale models and expansion across the phylogenetic tree’, Nucleic acids research, 48(D1), pp. D402–D406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien EJ, Monk JM and Palsson BO (2015) ‘Using Genome-scale Models to Predict Biological Capabilities’, Cell, 161(5), pp. 971–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan C et al. (1999) ‘Removing redundancy in SWISS-PROT and TrEMBL’, Bioinformatics, 15(3), pp. 258–259. [DOI] [PubMed] [Google Scholar]
- Oh Y-K et al. (2007) ‘Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data’, The Journal of biological chemistry, 282(39), pp. 28791–28799. [DOI] [PubMed] [Google Scholar]
- Opdam S et al. (2017) ‘A Systematic Evaluation of Methods for Tailoring Genome-Scale Metabolic Models’, Cell systems, 4(3), pp. 318–329.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre LM et al. (2019) ‘SubCellBarCode: Proteome-wide Mapping of Protein Localization and Relocalization’, Molecular cell, 73(1), pp. 166–182.e7. [DOI] [PubMed] [Google Scholar]
- Orth JD et al. (2011) ‘A comprehensive genome-scale reconstruction of Escherichia coli metabolism−−2011’, Molecular systems biology, 7, p. 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco AR, Moel M and Segrè D (2019) ‘Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems’, Nature Communications. doi: 10.1038/s41467-018-07946-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson B (2015) Systems Biology. Cambridge University Press. [Google Scholar]
- Pandey V and Hatzimanikatis V (2019) ‘Investigating the deregulation of metabolic tasks via Minimum Network Enrichment Analysis (MiNEA) as applied to nonalcoholic fatty liver disease using mouse and human omics data’, PLoS computational biology, 15(4), p. e1006760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters I et al. (2017) ‘Zenodo in the spotlight of traditional and new metrics’, Frontiers in research metrics and analytics, 2. doi: 10.3389/frma.2017.00013. [DOI] [Google Scholar]
- Ramon C, Gollub MG and Stelling J (2018) ‘Integrating -omics data into genome-scale metabolic network models: principles and challenges’, Essays in biochemistry, 62(4), pp. 563–574. [DOI] [PubMed] [Google Scholar]
- Reed JL et al. (2003) ‘An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR)’, Genome biology, 4(9), p. R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richelle A et al. (2019) ‘Increasing consensus of context-specific metabolic models by integrating data-inferred cell functions’, PLoS computational biology, 15(4), p. e1006867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL et al. (2020) ‘An atlas of human metabolism’, Science signaling, 13(624). doi: 10.1126/scisignal.aaz1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario SR et al. (2018) ‘Pan-cancer analysis of transcriptional metabolic dysregulation using The Cancer Genome Atlas’, Nature communications, 9(1), p. 5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo S et al. (2015) ‘Modeling the effects of commonly used drugs on human metabolism’, The FEBS journal, 282(2), pp. 297–317. [DOI] [PubMed] [Google Scholar]
- Savojardo C et al. (2018) ‘BUSCA: an integrative web server to predict subcellular localization of proteins’, Nucleic acids research, 46(W1), pp. W459–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Lewis NE and Palsson BØ (2011) ‘Elimination of thermodynamically infeasible loops in steady-state metabolic models’, Biophysical journal, 100(3), pp. 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaver SMD et al. (2014) ‘High-throughput comparison, functional annotation, and metabolic modeling of plant genomes using the PlantSEED resource’, Proceedings of the National Academy of Sciences of the United States of America, 111(26), pp. 9645–9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif Y et al. (2018) ‘Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits’, Nature communications, 9(1), p. 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif Y, Monk JM, Mih N, et al. (2019) ‘A computational knowledge-base elucidates the response of Staphylococcus aureus to different media types’, PLoS computational biology, 15(1), p. e1006644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif Y, Monk JM, Machado H, et al. (2019) ‘Systems Biology and Pangenome of Salmonella O-Antigens’, mBio, 10(4). doi: 10.1128/mBio.01247-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarasu S et al. (2010) ‘Genome-scale modeling and in silico analysis of mouse cell metabolic network’, Molecular bioSystems, 6(1), pp. 152–161. [DOI] [PubMed] [Google Scholar]
- Simeonidis E and Price ND (2015) ‘Genome-scale modeling for metabolic engineering’, Journal of industrial microbiology & biotechnology, 42(3), pp. 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalidzans E et al. (2018) ‘Model-based metabolism design: constraints for kinetic and stoichiometric models’, Biochemical Society transactions, 46(2), pp. 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q et al. (2009) ‘PPDB, the Plant Proteomics Database at Cornell’, Nucleic acids research, 37(Database issue), pp. D969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swainston N et al. (2016) ‘Recon 2.2: from reconstruction to model of human metabolism’, Metabolomics: Official journal of the Metabolomic Society, 12, p. 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szappanos B et al. (2011) ‘An integrated approach to characterize genetic interaction networks in yeast metabolism’, Nature genetics, 43(7), pp. 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Széliová D et al. (2020) ‘What CHO is made of: Variations in the biomass composition of Chinese hamster ovary cell lines’, Metabolic engineering, 61, pp. 288–300. [DOI] [PubMed] [Google Scholar]
- Thiele I et al. (2013) ‘A community-driven global reconstruction of human metabolism’, Nature biotechnology, 31(5), pp. 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I et al. (2020) ‘Personalized whole-body models integrate metabolism, physiology, and the gut microbiome’, Molecular systems biology, 16(5), p. e8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Heinken A and Fleming RMT (2013) ‘A systems biology approach to studying the role of microbes in human health’, Current opinion in biotechnology, 24(1), pp. 4–12. [DOI] [PubMed] [Google Scholar]
- Thiele I and Palsson BØ (2010a) ‘A protocol for generating a high-quality genome-scale metabolic reconstruction’, Nature protocols, 5(1), pp. 93–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I and Palsson BØ (2010b) ‘Reconstruction annotation jamborees: a community approach to systems biology’, Molecular systems biology, 6, p. 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Töpfer N, Kleessen S and Nikoloski Z (2015) ‘Integration of metabolomics data into metabolic networks’, Frontiers in plant science, 6, p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana D et al. (2019) ‘Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data’, Communications biology, 2, p. 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DE et al. (1996) ‘Proline is biosynthesized from arginine in Staphylococcus aureus’, Microbiology, 142 ( Pt 6), pp. 1491–1497. [DOI] [PubMed] [Google Scholar]
- Uhlén M et al. (2015) ‘Proteomics. Tissue-based map of the human proteome’, Science, 347(6220), p. 1260419. [DOI] [PubMed] [Google Scholar]
- Uhlén M et al. (2019) ‘The human secretome’, Science signaling, 12(609). doi: 10.1126/scisignal.aaz0274. [DOI] [PubMed] [Google Scholar]
- Consortium UniProt (2019) ‘UniProt: a worldwide hub of protein knowledge’, Nucleic acids research, 47(D1), pp. D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira V et al. (2018) ‘A Model Integration Pipeline for the Improvement of Human Genome-Scale Metabolic Reconstructions’, Journal of integrative bioinformatics. doi: 10.1515/jib-20180068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H et al. (2018) ‘RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor’, PLoS computational biology, 14(10), p. e1006541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T et al. (2014) ‘Genetic screens in human cells using the CRISPR-Cas9 system’, Science, 343(6166), pp. 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T et al. (2015) ‘Identification and characterization of essential genes in the human genome’, Science, 350(6264), pp. 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Eddy JA and Price ND (2012) ‘Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE’, BMC systems biology, 6, p. 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser D, Kretschmann E and Apweiler R (2004) ‘Filtering erroneous protein annotation’, Bioinformatics, 20 Suppl 1, pp. i342–7. [DOI] [PubMed] [Google Scholar]
- Witting M et al. (2018) ‘Modeling Meets Metabolomics-The WormJam Consensus Model as Basis for Metabolic Studies in the Model Organism Caenorhabditis elegans’, Frontiers in molecular biosciences, 5, p. 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH et al. (2019) ‘A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action’, Cell, 177(6), pp. 1649–1661.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri G et al. (2019) ‘Machine and deep learning meet genome-scale metabolic modeling’, PLoS computational biology, 15(7), p. e1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelezniak A et al. (2015) ‘Metabolic dependencies drive species co-occurrence in diverse microbial communities’, Proceedings of the National Academy of Sciences of the United States of America, 112(20), pp. 6449–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengler K and Zaramela LS (2018) ‘The social network of microorganisms - how auxotrophies shape complex communities’, Nature reviews. Microbiology, 16(6), pp. 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y et al. (2018) ‘Genome-scale metabolic modeling of responses to polymyxins in Pseudomonas aeruginosa’, GigaScience, 7(4). doi: 10.1093/gigascience/giy021. [DOI] [PMC free article] [PubMed] [Google Scholar]