

Abstract
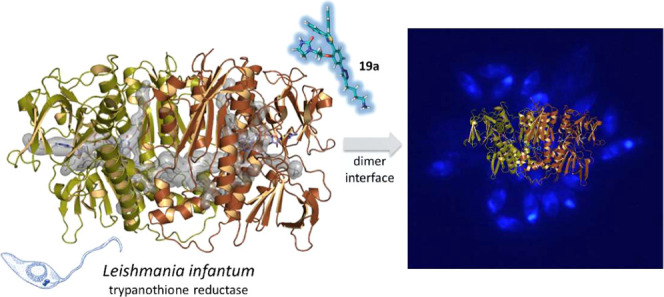
Inhibition of Leishmania infantum trypanothione disulfide reductase (LiTryR) by disruption of its homodimeric interface has proved to be an alternative and unexploited strategy in the search for novel antileishmanial agents. Proof of concept was first obtained by peptides and peptidomimetics. Building on previously reported dimerization disruptors containing an imidazole-phenyl-thiazole scaffold, we now report a new 1,2,3-triazole-based chemotype that yields noncompetitive, slow-binding inhibitors of LiTryR. Several compounds bearing (poly)aromatic substituents dramatically improve the ability to disrupt LiTryR dimerization relative to reference imidazoles. Molecular modeling studies identified an almost unexplored hydrophobic region at the interfacial domain as the putative binding site for these compounds. A subsequent structure-based design led to a symmetrical triazole analogue that displayed even more potent inhibitory activity over LiTryR and enhanced leishmanicidal activity. Remarkably, several of these novel triazole-bearing compounds were able to kill both extracellular and intracellular parasites in cell cultures.
Introduction
Leishmaniasis is an infectious disease caused by intracellular protozoan parasites from more than 20 Leishmania species and transmitted to humans through the bite of infected female phlebotomine sandflies. According to the World Health Organization (WHO), leishmaniasis is one of the major neglected tropical diseases that affects 12 million people in 98 countries, being especially prevalent in tropical and subtropical areas.1 Visceral leishmaniasis (VL), also known as Kala-azar, is the most severe form of the disease with more than 60 000 human deaths annually. VL is caused by Leishmania donovani and Leishmania infantum parasites worldwide, including Mediterranean countries.2 No effective vaccine for humans has so far been developed, and leishmaniasis control mainly relies on chemotherapy.3,4 However, currently available treatments are inadequate because of high costs, toxicity, drug resistance, and the need for parenteral administration.5,6 Therefore, there is an urgent need to find new effective innovative drugs against this disease.
Herein, we report a target-based approach for the discovery of novel agents against L. infantum parasites targeting a protein that is essential for parasite survival and exclusive for these parasites.7,8 We have focused on trypanothione (Try) disulfide reductase (TryR) because it is one of the few genetically and chemically validated drug targets, and validation is one important criterion in target assessment.9−11 Among other highly relevant functions, this enzyme is essential for antioxidant defense in trypanosomatids. The Try/TryR couple in these parasites substitutes for the glutathione/glutathione-disulfide reductase (GR) pair characteristic of most eukaryotic organisms. Accordingly, the absence of TryR in mammalian cells, the significant differences between TryR and human GR (hGR) (the active sites of the two enzymes have opposite net charges and different volumes), and its crucial role for parasite survival make TryR an attractive target for new chemotherapeutics.12−15 Since this enzyme was identified, many potent and selective competitive polycationic TryR inhibitors (which mimic the positively charged Try in the active site) have been described. However, there are scarce reports of potent TryR-inhibiting compounds with adequate antiparasitic activity.16,17 This is so because it has been shown that survival of the parasites is only affected when TryR activity is reduced by more than 90%, probably as a consequence of the high intracellular concentrations of its natural substrate (Try) in the cell.18 This implies that, to be effective against the parasites, competitive inhibitors must have very high binding affinities so as to give rise to inhibition constants in the low nanomolar range. Nonetheless, one of the most potent (submicromolar) competitive inhibitors of Trypanosoma brucei (TbTryR) described to date is almost inactive against L. donovani.19 This limitation, together with the need for very high affinities imposed by the high intracellular concentrations of both the natural substrate (between 3 and 50 μM under steady-state conditions and up to 150 μM under oxidative stress) and TryR itself (0.5–1.3 μM), make noncompetitive and/or irreversible inhibitors emerge as more attractive alternatives for targeting this enzyme.20a,20b,21
The crystal structure of TryR from L. infantum (LiTryR) was first solved by Baiocco et al.22 and is very similar to that of other trypanosomatids.13,14,16,23,24 This enzyme is a twofold symmetrical homodimer that contains two active sites and two reduced nicotinamide adenine dinucleotide phosphate (NADPH)- and flavin adenine dinucleotide (FAD)-binding domains separated by a large and well-characterized interface.25 Each Try binding site is located in a large solvent-exposed cavity at the interface between the two monomers. Several X-ray crystal structures of TryR in complex with a variety of competitive reversible inhibitors have shown that the large pocket allows not only several binding modes for small-molecule inhibitors but also the simultaneous binding of more than one inhibitor molecule.23 Such unpredictable binding modes, due to the large size of the active site, make the rational design of competitive, high-affinity inhibitors of TryR a very challenging task.
Bearing in mind the homodimeric nature of this enzyme, back in 2013 we devised an alternative inhibition strategy that aims to disrupt the protein–protein interactions responsible for LiTryR homodimerization.25 Initially, by means of a combination of molecular modeling and site-directed mutagenesis studies, we identified and validated E436—an amino acid located at an interfacial α-helix spanning from P435 to M447—as a hotspot for dimer stabilization and assessed its importance for the catalytic activity of the enzyme. As a “proof of concept” of this novel approach, we designed and tested a small library of linear peptides that represent rational variations of this α-helix. From these studies, the 13-mer-modified peptide sequence PKIIQSVGIS-Nle-K-Nle (1) emerged as a potent enzyme dimerization disruptor that also behaves as a strong inhibitor (in the submicromolar range) of the oxidoreductase activity of LiTryR.25 Next, a variety of helix-stabilized cyclic peptides26,27 and α,β3-peptide foldamers28 were successfully explored in our search to find peptidomimetics with increased proteolytic stability. However, conjugation with cell-penetrating peptides was always required to facilitate the cellular uptake of these peptide- or peptidomimetic-based LiTryR dimerization disruptors and to kill the parasites in cell culture.28,29 Thus, the design of small molecules with druglike properties better than those of the prototype peptide or the previous peptidomimetics appeared as a desirable goal.
In this regard, we recently disclosed the results of our first steps toward nonpeptide disruptors of LiTryR dimerization with promising leishmanicidal activity by an α-helical mimetic approach.30 Among the reported proteomimetic scaffolds,31−33 the pyrrolopyrimidine34 and the 5-6-5 imidazole-phenyl-thiazole35 cores were selected to dictate the spatial orientations of the side chains of three key residues (K2, Q5, and I9) in the linear peptide prototype 1. Imidazole-based compounds 2 and 3 (Figure 1), bearing a naphthalene or a biphenyl polyaromatic R3 substituent to mimic the hydrophobic isoleucine residue, emerged as potent inhibitors of the LiTryR oxidoreductase activity, while pyrrolopyrimidines were shown to be much less active. Although these small molecules displayed a moderate capacity to disrupt the LiTryR dimer, this effect was much less pronounced than that observed for the previous peptide-based inhibitors. Remarkably, the imidazole-based compounds were cell-permeable and showed significant leishmanicidal activity against both amastigote and promastigote forms of Leishmania parasites. However, these molecules displayed a cytotoxic activity similar to that observed for their peptidic and peptidomimetic predecessors and exhibited low selectivity indexes (SIs).
Figure 1.

From a peptide dimerization disruptor of LiTryR (1) and in-house imidazoles 2 and 3 to the newly designed triazole-phenyl-thiazole scaffold I.
1,2,3-Triazole-containing compounds have been widely investigated because they are considered privileged scaffolds in different areas of medicinal chemistry,36−38 including several examples in the antileishmanial field.39 Interest in this molecular architecture has also been fueled by its expedient synthesis through “click chemistry”.40 In this scenario, we decided to apply the bioisosteric replacement of imidazole by 1,2,3-triazole in our recently described imidazole-phenyl-thiazoles.30 We herein report the synthesis and structure–activity relationship (SAR) studies of novel 1,2,3-triazole-phenyl-thiazole compounds of general formulae I (Figure 1) to demonstrate the potential of this novel chemotype for LiTryR dimerization disruption and leishmanicidal activity against L. infantum parasites. A whole set of 26 triazole-based compounds modified at four different positions in the new scaffold (R1–R4 substituents) was prepared and evaluated. Five of these molecules display activities that very closely resemble that of the peptide prototype 1 and likewise behave as noncompetitive, slow-binding inhibitors.41 Molecular modeling studies identified a putative binding site for these compounds at an almost unexplored central interfacial cavity of the enzyme and provided a structural rationale to the observed SAR results that was supported by ensuing rational design, synthesis, and activity of a new symmetrical triazole analogue with the desired properties.
Results and Discussion
Chemistry
The synthesis of the target triazole-phenyl-thiazole type I compounds is exemplified by the preparation of the first series of 1,2,3-triazole derivatives 12a–c (bearing R1–R3 substituents similar to those present in the predecessor imidazole analogues 2 and 3), as shown in Scheme 1. The synthetic route started from the previously described bromide intermediate 4a(30) bearing an imidazolidinone ethyl R2 group (used as a Gln mimetic in the previous imidazole derivatives) by treatment with sodium azide in anhydrous dimethyl sulfoxide (DMSO) at 100 °C for 72 h to afford the azide intermediate 5a in 85% yield. The addition of molecular sieves with pore openings of 4 Å was required to avoid the partial hydrolysis of the azide group to the undesired amine. The next step in the synthetic route was the 1,3-dipolar cycloaddition of the azide intermediate with terminal alkynes substituted with the appropriate R1 group (as a Lys mimetic) to generate the 1,2,3-triazole ring. Initial attempts to use the ruthenium-catalyzed azide–alkyne cycloaddition (RuAAC)40b from 5a and commercially available N-Cbz-protected terminal alkyne 6 in the presence of catalytic amounts (5 or 10%) of a cyclopentadienyl Ru catalyst [Cp*RuCl(PPh3)2] at different temperatures in dry dimethylformamide (DMF) and an inert atmosphere did not produce the expected 1,5-disubstituted 1,2,3-triazole 7. Only unreacted starting materials or complex decomposition mixtures were observed under these conditions. In contrast, the 1,4-disubstituted 1,2,3-triazole regioisomer 8 could be easily obtained in 56% yield by the 1,3-dipolar copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC)40a of azide 5a and the terminal alkyne 6 using the CuSO4/sodium ascorbate catalyst system at room temperature for 8 h in H2O/EtOH (1:1 mixture) as the solvent. Subsequent treatment of nitrile 8 with aqueous (aq) 20% ammonium sulfide at 80 °C for 4 h provided the thioamide intermediate 9 in 74% yield. Next, Hantzsch thiazole synthesis from 1 equiv of 9 and 1 equiv of the commercially available α-bromomethylketone 10a or the previously synthesized 10b and 10c(30) (bearing a naphthyl or a biphenylethyl hydrophobic R3 substituent) heated in isopropanol at 70 °C for 36 h gave the expected α/β-unsaturated derivatives 11a–c monosubstituted at the 4-position of the thiazole ring in 87, 51, and 63% yields, respectively (Scheme 1). Finally, treatment of 11a–c with H2, Pd/C in tetrahydrofuran (THF)/MeOH mixture in the presence of trifluoroacetic acid (TFA) at room temperature allowed the simultaneous hydrogenolysis of NHCbz and hydrogenation of the double bond to afford the deprotected target compounds 12a–c as monotrifluoroacetate salts in 35, 24, and 35% yields, respectively. Chromatographic purification by Biotage using reverse-phase columns was always required to obtain the final compounds with purities higher than 95% for their biological evaluation.
Scheme 1. Synthesis of the First Series of 1,4-Disubstituted 1,2,3-Triazole Compounds 12a–c.

Reagents and conditions: (a) NaN3, DMSO, molecular sieves 4 Å, 100 °C, 72 h; (b) Cp*RuCI(PPh3)2 (5 or 10 mol %), DMF, rt to 100 °C; (c) CuSO4·5H2O, sodium ascorbate, H2O/EtOH 1:1, rt, 8 h; (d) (NH4)2S 20% aq, DMF, 80 °C, 4 h; (e) α-bromomethylketones 10a–c, iPrOH, 70 °C, 5 h; (f) H2, Pd/C 10%, TFA, THF/MeOH 1:1, rt, 2 h.
As will be discussed below, the 1,2,3-triazole compounds 12a–c emerged as more potent dimerization disruptors of LiTryR than the reference imidazoles and also behaved as strong inhibitors of the oxidoreductase activity of the enzyme. These results prompted us to develop a novel series of triazole derivatives 12d–s (see structures in Table 1 and Scheme S1) modified at the R1–R3 substituents to perform a SAR study. Regarding modifications at R1 and R2, the protonated aminobutyl group linked to the triazole was replaced by OH, Me, or COOH (12d–g) or by a shorter aminopropyl substituent (12h, 12i), while the R2 imidazolidinone (attached to the central phenyl ring through an ethyl linker) was removed (12j, 12k). In terms of R3 modifications, bulky alkyl substituents (12l, 12m) or a phenyl group linked to the thiazole ring through a polymethylene spacer of a different length (12n–pvs12a) were explored.
Table 1. Half-Maximal Inhibitory Concentration (IC50) ± Standard Error (SE) Values for Triazole Compounds I and the Truncated Analogues 13 and 14 in the TryR Oxidoreductase Activity and LiTryR Monomer Displacement Assaysa,f.

| compounds | R1 | n | R2 | R3 | R4 | IC50act (μM)b | IC50dim (μM)c |
|---|---|---|---|---|---|---|---|
| 1 | 1.5 ± 0.2 | 7.0 ± 0.6 | |||||
| 2 | (CH2)2naphthyl | 5.1 ± 0.4 | 26%d | ||||
| 3 | (CH2)2biphenyl | 8.6 ± 1.4 | 32%d | ||||
| 12a | NH3+ | 4 | (CH2)2Im* | (CH2)2Ph | H | 14.6 ± 1.0 | 38.1 ± 1.2 |
| 12b | NH3+ | 4 | (CH2)2Im* | (CH2)2naphthyl | H | 5.9 ± 1.1 | 7.1 ± 0.6e |
| 12c | NH3+ | 4 | (CH2)2Im* | (CH2)2biphenyl | H | 10.9 ± 1.8 | 4.8 ± 0.6e |
| Modifications at R1 | |||||||
| 12d | OH | 4 | (CH2)2Im* | (CH2)2Ph | H | >75 | >75 |
| 12e | CH3 | 3 | (CH2)2Im* | (CH2)2Ph | H | >75 | >75 |
| 12f | CH3 | 3 | (CH2)2Im* | (CH2)2biphenyl | H | >75 | >75 |
| 12g | COOH | 3 | (CH2)2Im* | (CH2)2Ph | H | >75 | >75 |
| 12h | NH3+ | 3 | (CH2)2Im* | (CH2)2Ph | H | 20.8 ± 2.3 | >75 |
| 12i | NH3+ | 3 | (CH2)2Im* | (CH2)2Biph | H | 19.7 ± 3.8 | 10.8 ± 0.1 |
| Modifications at R2 | |||||||
| 12j | NH3+ | 4 | Me | (CH2)2Ph | H | 20.1 ± 2.5 | >75 |
| 12k | NH3+ | 4 | Me | (CH2)2biphenyl | H | 26.8 ± 5.7 | 16.1 ± 0.7 |
| Modifications at R3 | |||||||
| 12l | NH3+ | 4 | (CH2)2Im* | OiPr | H | 54.5 ± 4.4 | >75 |
| 12m | NH3+ | 4 | (CH2)2Im* | CH2tBu | H | 24.1 ± 1.4 | >75 |
| 12n | NH3+ | 4 | (CH2)2Im* | Ph | H | 11.2 ± 0.2 | 29.0 ± 1.5 |
| 12o | NH3+ | 4 | (CH2)2Im* | CH2Ph | H | 43.6 ± 1.1 | 61.2 ± 2.8 |
| 12p | NH3+ | 4 | (CH2)2Im* | (CH2)3Ph | H | 14.6 ± 1.7 | 19.1 ± 1.1 |
| 12q | NH3+ | 4 | (CH2)2Im* | (CH2)2THQ* | H | 6.9 ± 2.1 | >75 |
| 12r | NH3+ | 4 | (CH2)2Im* | (CH2)2DHB* | H | 11.9 ± 0.6 | 10.7 ± 0.5 |
| 12s | NH3+ | 4 | (CH2)2Im* | (CH2)2DBF* | H | 6.6 ± 1.9 | 8.8 ± 0.2 |
| Truncated Analogues | |||||||
| 13 | >75 | >75 | |||||
| 14 | >75 | >75 | |||||
| Modifications at R3 and R4 | |||||||
| 19a | NH3+ | 4 | (CH2)2Im* | Ph | Ph | 4.3 ± 1.0 | 7.1 ± 1.7 |
| 19b | NH3+ | 4 | (CH2)2Im* | biphenyl | Ph | 18.4 ± 3.9 | 11.3 ± 0.2 |
| 19c | NH3+ | 4 | (CH2)2Im* | PhOPh | Ph | 16.9 ± 2.7 | 9.0 ± 0.5 |
| 19d | NH3+ | 4 | (CH2)2Im* | (CH2)2biphenyl | Ph | 19.8 ± 2.3 | 5.7 ± 0.5 |
| 19e | NH3+ | 4 | (CH2)2Im* | (CH2)2PhOPh | Ph | 19.3 ± 3.7 | 6.7 ± 1.0 |
Linear peptide 1 and imidazole-based compounds 2 and 3 were included as reference compounds.
Enzymatic activity >75 indicates that the IC50 value is higher than 75 μM (maximum assayed). Results are representative of three independent experiments, each performed in triplicate.
Dimer quantitation assay (enzyme-linked immunosorbent assay (ELISA)).25
Percentage of inhibition observed at 20 μM as reliable IC50 values could not be determined in this case.
Percentages of inhibition observed at 20 μM were 73 and 78% for 12b and 12c, respectively.
Im* = imidazolidinone, THQ* = tetrahydroquinoline (TFA salt), DHB* = dihydrobenzofuranyl, DBF* = dibenzofuranyl.
Since the presence of a biphenyl or naphthyl substituent at R3 was highly relevant for potent enzyme inhibition and antileishmanial activity, other polyheteroaromatic rings were also explored at R3 (12q–s). The synthesis of the novel triazole analogues 12d–s involved (i) a high-yielding nucleophilic aromatic substitution (SNAr) of aryl fluorides with the appropriate alcohols (bearing the R2 substituent),30 (ii) CuAAC reactions between azide intermediates and terminal alkynes (containing the R1 substituent), (iii) Hanztsch thiazole synthesis of thioamides with suitable α-bromomethylketones (with the R3 substituent), and (iv) and a final step of catalytic hydrogenation to yield the deprotected final compounds using similar protocols to those described above. It should be noted that in the catalytic hydrogenation of the protected triazole compound intermediate 11q (see Scheme S1) bearing a quinoline moiety as the R3 substituent, the 1,2,3,4-tetrahydroquinoline compound 12q (see Scheme S1) was isolated as a ditrifluoroacetate salt due to the partial hydrogenation and salt formation of the quinoline ring nitrogen atom. The synthetic experimental details of the target 12q–s compounds and the noncommercially available key α-bromomethyl-ketone intermediates 10m, 10p–s are included in the Supporting Information (see Schemes S1 and S2). Scaffold-truncated simplified compounds (13 and 14, see Table 1), possessing only two aromatic rings and substituents, were also prepared for the SAR studies following similar synthetic protocols (see also the Supporting Information for experimental details; Scheme S3). To further investigate the SAR of the triazole-based compounds, we next focused on the preparation of an additional series of triazole analogues 19a–e (Scheme 2) disubstituted at both positions 4 and 5 of the thiazole ring. Polyaromatic groups directly attached or linked through an ethyl spacer to the 4-position and a phenyl group at the 5-position were selected as R3 and R4 substituents, respectively. As shown in Scheme 2, the preparation of these compounds required the previous synthesis of the noncommercially available α-bromobenzylketones 17a–d. Thus, 17a–c bearing a biphenyl or a phenyloxyphenyl group at R3 directly attached to the carbonyl were synthesized by bromination of the commercially available benzylketones 16a–c with N-bromosuccinimide (NBS) in p-toluensulfonic acid (p-TsOH) in good to excellent yields.
Scheme 2. Synthesis of Triazoles Disubstituted at the Thiazole Ring (19a–e).
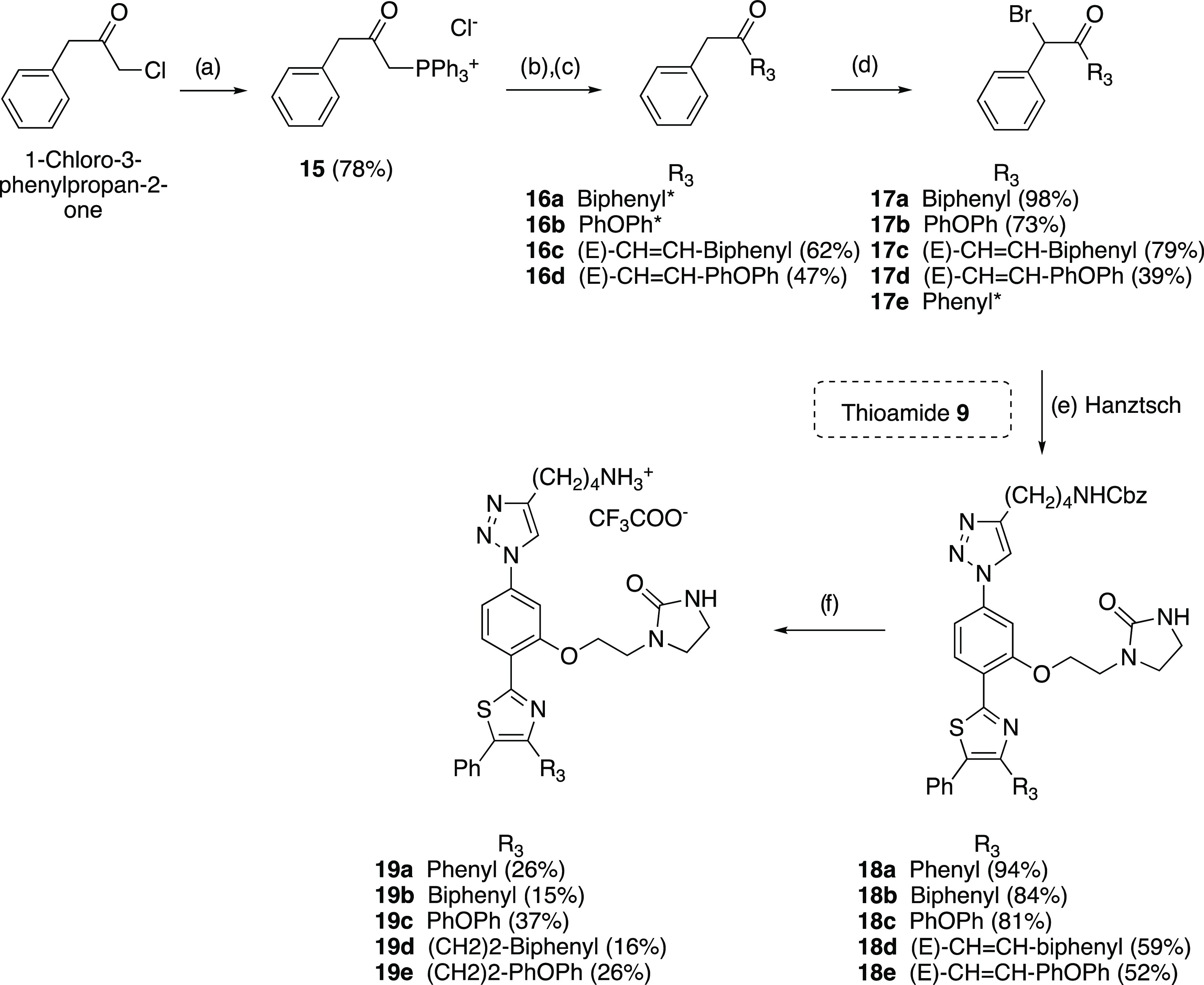
Reagents and conditions: (a) PPh3, dry toluene, 110 °C, 6 h; (b) KOH, dry toluene, 110 °C, 3 h; (c) RCHO, 110 °C, 3 h; (d) NBS, p-TsOH, CH3CN, rt, 14 h; (e) thioamide 9, 17a–e, iPrOH, 70 °C, 5 h; (f) H2, Pd/C 10%, TFA, THF/MeOH 1:1, rt, 2 h. *Commercially available reagents.
On the other hand, α-bromobenzyl α,β-unsaturated ketone 17d with a polyaromatic group was obtained by regioselective bromination of the most favorable benzylic position with NBS in p-TsOH of the corresponding benzylketones 16d, which was previously synthesized from 1-chloro-3-phenylpropan-2-one by treatment with PPh3 to give the phosphonium salt 15 followed by a Wittig procedure with the corresponding polyaromatic aldehydes in moderate yields. Next, Hantzsch thiazole synthesis from thioamide intermediate 9 with the commercial 17e or synthetic α-bromobenzylketones 17a–d, followed by catalytic hydrogenolysis of the N-Cbz group and simultaneous olefin hydrogenation of 18a–e using 10% Pd/C in the presence of TFA, afforded the desired unprotected disubstituted thiazole derivatives 19a–e as monotrifluoroacetate salts in low to moderate yields. Finally, we also designed (see section Computational Studies below) and prepared (Scheme 3) a new symmetrical triazole analogue (22) by treating 2 equiv of thioamide intermediate 9 with 1 equiv of α-bromomethylketone 20 (previously synthesized by bromination with NBS of commercially available 1,3-diacetyl benzene) under Hantzsch reaction conditions, followed by catalytic hydrogenolysis of the NHCbz group using similar procedures to those described above.
Scheme 3. Synthesis of the Newly Designed Symmetrical Analogue 22.
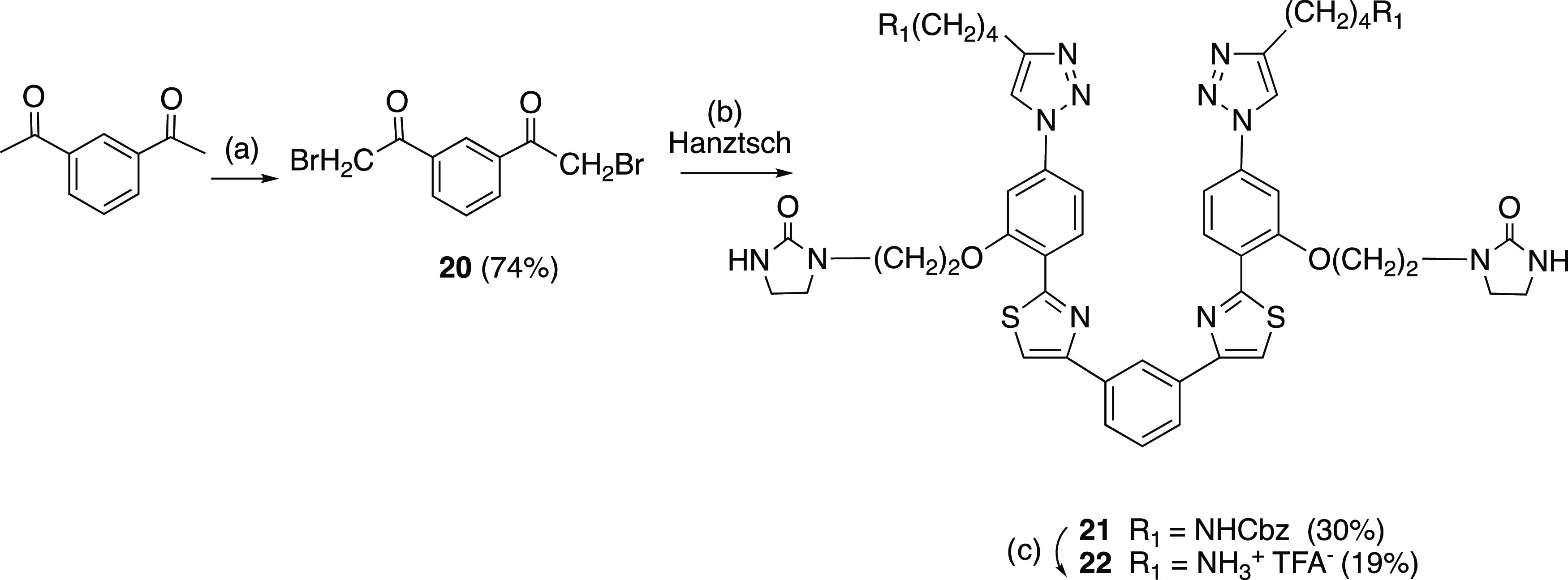
Reagents and conditions: (a) NBS, p-TsOH, acetonitrile, rt, 14 h; (b) thioamide 9, iPrOH, 70 °C, 5 h; (c) H2 Pd/C 10%, TFA, THF/MeOH, 1:1, rt, 2 h.
Biological and Structural Studies
Enzymatic Assays
L. infantum TryR Inhibition and Dimerization Assays
All of the novel triazole-phenyl-thiazole modified at substituents R1–R3 (12a–s), disubstituted thiazole analogues bearing R3 and R4 substituents (19a–e), and truncated simplified compounds (13 and 14) were evaluated for their effect on both the oxidoreductase activity and the dimerization status of LiTryR.25 The prototype peptide 1 and imidazole-containing compounds 2 and 3 were used as reference molecules. Screening was performed at different concentrations ranging from 0.1 to 75 μM. A concentration of 1 μM TS2 was used for this initial screening. This low substrate concentration is sufficient for comparison of compound inhibitory activity in the oxidoreductase assay. The best candidates were then characterized further to obtain their dissociation constants. The IC50 values determined in the activity (IC50act) and dimerization (IC50) assays are shown in Table 1.
According to the results obtained with the first series of triazole-based compounds 12a–c (Table 1), analogues containing a naphthyl or a biphenyl group at R3 (12b, 12c) potently inhibited the TryR oxidoreductase activity, showing similar IC50act values (in the low micromolar range) to those obtained for the reference imidazole compounds 2 and 3. Remarkably, 12a–c were much more potent LiTryR dimerization disruptors than their imidazole counterparts. Thus, in contrast to 2 and 3, for which no reliable IC50 could be measured and only ∼30% dimerization inhibition was observed at a concentration of 20 μM, the IC50dim values for 12b, 12c (7.1 and 4.8 μM, respectively) were similar to (or even slightly lower than) those obtained for the potent prototype peptide 1. Furthermore, replacement of the naphthyl or biphenyl groups at R3 by a phenyl (12b, 12cvs12a) slightly increased the IC50 value, while it significantly decreased (in a 5–8-fold range) the ability to disrupt the LiTryR dimer. We took this result as a clear indication that the presence of a polyaromatic substituent at R3 is relevant to potent LiTryR dimer disruption.
SAR studies on the second series of triazole compounds (12d–s) with variations at the R1–R3 substituents began with modifications at the R1 position. As shown in Table 1, the presence of a positively charged amino alkyl group at R1 appears as an absolute requirement for inhibition of both activity and dimerization, as evidenced by the complete loss of effect observed when the amino group was replaced by hydroxyl, methyl, or carboxylic acid groups (12d–gvs12a, 12c). As regards the length of the R1 spacer attached to the amino group, shortening the original tetramethylene linker to a trimethylene brought about a ∼2-fold reduction of potency in both assays (12h, 12ivs12a, 12c). The existence of the imidazolidinone ring as the R2 substituent on the central phenyl ring also appeared important for optimal activity since its removal (12j, 12kvs12a, 12c) resulted in compounds twice or thrice less active in both assays. Modifications at the R3 substituent (located at the 4-position of the thiazole ring) had a pronounced effect on enzymatic activity inhibition and, even more so, on dimerization disruption. Thus, the presence of a (poly)aromatic substituent at R3 was crucial for enhanced potency since compounds 12l, 12m, with bulky aliphatic groups, exhibit much less inhibitory activity on the enzyme and no destabilization effect on the LiTryR homodimer in comparison with the (poly)aromatic analogues 12a–c. In comparison, varying the length of the linker connecting the phenyl group to the thiazole (12n–pvs12a) turned out to be not so crucial because, in general, it did not significantly affect the inhibitory activity.
In view of these results, and even though the phenylpropyl derivative 12p displayed the lowest IC50dim value, for the next round of modifications, we decided to focus on shortening the propylene linker at R3 to an ethylene (12a) or even removing it altogether (12n) because of the better synthetic accessibility of the corresponding bromoketones. On the other hand, replacement of the naphthyl or the biphenyl group at R3 in the most potent analogues 12b, 12c by polyheteroaromatics such as a dihydrobenzofuranyl substituent (12r) or a larger hydrophobic tricyclic dibenzofuranyl moiety (12s) also yielded very potent compounds but did not further increase the inhibitory activity of the most potent analogues (12b, 12c) in any of the two assays. In contrast, the introduction of a positively charged tetrahydroquinoline ring (12q) annihilated the dimerization disruption capacity of the molecules even though potent inhibition of the redox activity of the enzyme was retained. In contrast, no activity in any of the assays was observed for the simplified truncated scaffold derivatives 13 and 14, in which the appropriately substituted triazole or thiazole rings were removed. This additional evidence further supports the key role played by these two rings (and the R1 and R3 substituents) in this type of inhibitor.
Finally, we also evaluated a new series of triazole compounds (19a–e) disubstituted at the 4- and 5-positions of the thiazole ring with R3 and R4 aromatic moieties. Some of these molecules combined excellent inhibition in the redox assay with potent LiTryR dimer disruption ability. Interestingly, both activities were significantly improved by a factor of 2–4 for 19a, in which a phenyl group is bonded at both R3 and R4 positions, in comparison with the R3 phenyl monosubstituted analogue 12n. Introduction of a larger biphenyl or a diphenylether group as the R3 substituent (maintaining the phenyl group at R4) significantly decreased the inhibitory activity of the highly potent diphenylsubstituted analogue, but their dimerization disruption ability was affected only slightly (cf. 19b, 19cvs19a). Also, the combination of R3 polyaromatics attached to the thiazole ring through an ethylene linker with an R4 phenyl group resulted in a significant deleterious effect on the potency of the compounds as inhibitors of the LiTryR oxidoreductase activity, although the potent disrupting effect on dimerization was maintained (cf. 19d, 19evs19b, 19c or the corresponding monosubstituted 12c).
In general, the most active compounds in the enzymatic activity assay were also the best dimerization disruptors in the ELISA with the exception of the highly potent tetrahydroquinoline inhibitor 12q, which did not show any dimerization disruption effect, or the thiazole-disubstituted analogues 19b–e, which behave as moderate redox inhibitors but highly potent dimerization disruptors of the enzyme. All in all, our SAR study showed that (i) a positively charged butylamine (R1), (ii) an imidazolidinone (R2) together with a hydrophobic poly(hetero)aromatic group as R3 (12b, 12c; 12r, 12s) substituents, or (iii) the combination of a phenyl as R3 with an additional phenyl as R4 (19a) are highly relevant to optimal enzyme inhibition and dimerization disruption in this series.
To gain insight into the selectivity of the compounds for LiTryR, representative molecules 12a–c, 12n, 12r, and 19a were studied as inhibitors of human glutathione-disulfide reductase (hGR), the closest related host enzyme. Remarkably, none of these compounds showed any inhibitory activity against this enzyme. As an example, a comparison of the activities of LiTryR and hGR in the presence of compounds 12b and 12c at 50 μM of their respective substrates is shown in Figure S15.
Mechanism of LiTryR Inactivation
As already described for 1,41 the peptidic precursor of all other compounds, a progressive loss of LiTryR activity is observed during the course of the reaction in the presence of 12b, 12c, 12r, 12s, and 19a (Figure 2), selected as the most potent inhibitors in these series. In stark contrast, reaction rates in the presence of the classical competitive inhibitor mepacrine are almost constant until 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (which allows substrate regeneration) is exhausted. The progress curves of the reactions in the presence of the selected compounds are typical of slow-binding inhibitors.42a,42b
Figure 2.

Time-dependent inhibition of LiTryR. Reaction progress curves of LiTryR in the absence (◆) and presence of 25 μM mepacrine (□), 10 μM peptide 1 (▲), 10 μM 12c (○), 10 μM 12b (●), 10 μM 12r (◊), 10 μM 12s (■), or 10 μM 19a (△). [DTNB] = 100 μM. [NADPH] = 150 μM. Data represent the mean ± SD of three independent experiments.
Despite the loss of activity observed during the reaction, dependency of the initial velocities (vi) on 19a and TS2 concentrations could be fitted to a model in which a rapid equilibrium is reached between the free enzyme and inhibitor (E + I) and the enzyme/inhibitor (EI) complex (Scheme 4). This analysis, based on vi values, was performed both in a DTNB-coupled assay and in a direct NADPH-oxidation assay (Figure S1 and Table S1). Our data consistently reveal a nonlinear noncompetitive hyperbolic inhibition mechanism.
Scheme 4. Two-Step Mechanism of Time-Dependent Inhibition of LiTryR.

The first step is a rapid equilibrium that generates the enzyme–inhibitor (EI) complex. This process is governed by the association rate (k3) and the dissociation rate (k4) constants. The second step consists of slow and reversible inactivation of the enzyme that is governed by the forward isomerization rate constant (k5) and the much smaller reverse rate constant (k6).
Peptide 1 has already been described as a pseudoirreversible time-dependent noncompetitive inhibitor that is able to promote a two-step process of enzyme isomerization (Scheme 4).41 The kinetic data obtained for 19a, the most potent compound, were also analyzed considering a similar mechanism of enzyme inactivation.
In this inactivation model, a rapid and reversible primary inhibition event generates an enzyme–inhibitor binary complex (EI complex). The process of generating an inactive form of LiTryR upon 19a binding is governed by k5 and k6 first-order rate constants, which describe an isomerization equilibrium between the initial enzyme–inhibitor complex (EI) and a second high-affinity complex (E*I) (Scheme 4). This slow isomerization process gives rise to a characteristic bending of the reaction curve that is defined by the first-order rate constant kobs, whose value describes the conversion from the initial reaction rate (vi) to the final steady-state velocity (vs).
Adjustment to eq 1 (see equations in Experimental Section) of the progress curves of the reactions catalyzed by LiTryR allowed us to obtain the kobs values for six 19a concentrations (4.4, 5.9, 7.9, 10.5, 14.1, and 18.7 μM) at six different TS2 concentrations (3.1, 6.2, 12.5, 25, 50, and 100 μM) (Figures S2–S7 and Table S2). The different kobs values obtained at each substrate concentration can be used to determine the apparent inhibitory constants Kiapp and Ki*app defined as the dissociation constant for the initial EI complex and the constant for the overall dissociation from E*I to E + I, respectively (eq 2).42a,42b Based on the relationship among kobs, vi, and vs (k6 = kobs × vi/vs) at all of the trypanothione disulfide (TS2) and 19a concentrations assayed, k6 varies between 3.9 × 10–6 and 6.5 × 10–5 s–1. Taking into consideration the extremely slow process of recovery of the enzymatic activity after complete inactivation of LiTryR (400 nM) upon incubation with 25 μM 19a for 16 h and subsequent 2500-fold dilution (Figure S13), a value of 3 × 10–5 s–1 was selected as a conservative upper value for k6. Fixing this value for k6 rendered very good estimations of Kiapp and Ki*app after the fitting process for all of the substrate concentrations assayed (Figure 3 and Table 2).
Figure 3.

Concentration dependence of the observed rate constants of LiTryR (0.8 nM) inactivation by 19a at different TS2 concentrations. Plot of the kobs values (±standard errors) as a function of 19a concentration at six different TS2 concentrations (3.1, 6.2, 12.5, 25, 50, and 100 μM) (kobs determinations for every 19a concentration at the different fixed TS2 concentrations assayed are shown in the Supporting Information). Curves were fitted using eq 2, and the results of the fits are shown in Table 2.
Table 2. Apparent Inhibition Constants for LiTryR Time-Dependent Inhibition by 19aa.
| [TS2] (μM) | Kiapp (μM) | Ki*app (μM) |
|---|---|---|
| 3.1 | 8.9 ± 3.1 | 0.6 ± 0.1 |
| 6.2 | 7.2 ± 1.7 | 0.5 ± 0.1 |
| 12.5 | 5.2 ± 2.6 | 0.5 ± 0.2 |
| 25 | 8.3 ± 3.0 | 0.8 ± 0.2 |
| 50 | 9.3 ± 3.2 | 1.4 ± 0.3 |
| 100 | 5.0 ± 2.7 | 1.3 ± 0.6 |
The relationships between the different Kiapp and Ki values in slow-binding inhibitors are the same as those for classical linear reversible inhibitors.42a,42b By applying eq 3 to fit the Kiapp values obtained at the six different TS2 concentrations assayed (Figure S8A), we could estimate the values of α (0.8 ± 0.4) and Ki (7.8 ± 1.7 μM). A similar approach (eq 4 and Figure S8B) was followed to estimate the values of α* (5.5 ± 2.9) and Ki* (0.5 ± 0.1 μM) for the two-step global process of enzyme inhibition. According to these results, formation of the initial EI complex is not affected by the presence of substrate, a behavior that is characteristic of a pure noncompetitive process (α ∼ 1). The isomerization step from EI to E*I strongly enhances the inhibitory activity of 19a, which is characterized by an overall Ki* value of 0.5 μM. This isomerization process is, as expected, slightly impaired by the presence of substrate (α = 5.5) because of the generation of the enzyme–substrate–inhibitor complex, which decreases the concentration of the EI complex and thereafter the rate of E*I generation. Despite this small substrate effect, an α value of 5.5 for the overall process is still in the range of α values characteristic of noncompetitive (mixed) inhibitors. The overall inhibitory process is depicted in Scheme S5. Because of the ability of 19a to cause enzyme dissociation, the characteristic isomerization of the enzyme in this mechanism of slow-binding inhibition is expected to be related to homodimer disruption.
Interestingly, compounds 12b and 12q have similar IC50 values in the reductase assay but only 12b interferes with dimerization. The kinetic analyses reveal that both compounds are time-dependent inhibitors but only inhibition caused by 12b follows a two-step process of enzyme isomerization (Figure S11 and Table S3). Plots of kobsvs inhibitor concentration for 12q fit to a straight line that is indicative of a single-step binding mechanism (Figure S12, Table S4, and Scheme S4), which agrees with its inability to disrupt the homodimer.
Computational Studies
Despite numerous attempts to form cocrystals with LiTryR or soak crystals of the enzyme with moderately or highly potent triazole-based dimerization disruptors 12a, 12b, and the heteroaromatic 12r, no diffraction-quality crystal containing an enzyme/drug complex could be obtained. Of note, similar problems were encountered in previous cocrystallization efforts with the predecessor imidazoles 2 and 3.30 Because of these so-far insurmountable hurdles, we had to rely on molecular modeling tools to try and shed light on the atomistic details of the binding process. Docking the most potent triazoles 12b, 12c into the cavity of one LiTryR monomer that lodges the 435Pro–Met447 α-helix of the other monomer, while feasible, failed to account for the SAR results discussed above. Indeed, the biologically essential naphthyl or biphenyl R3 substituents could not be properly accommodated unless the ligand folded onto itself in an unrealistic fashion due to lack of room. Therefore, we sought alternative binding sites in LiTryR, as found in Protein Data Bank (PDB) entry 2JK6(22b) using the FTMap web server.43 This procedure highlighted the existence of a large, hydrophobic, and putatively druggable cavity right at the center of the dimer that is lined by residues from both monomers and located very close to the interfacial helices that were mimicked by the original peptides and peptidomimetics (Figure 4).
Figure 4.
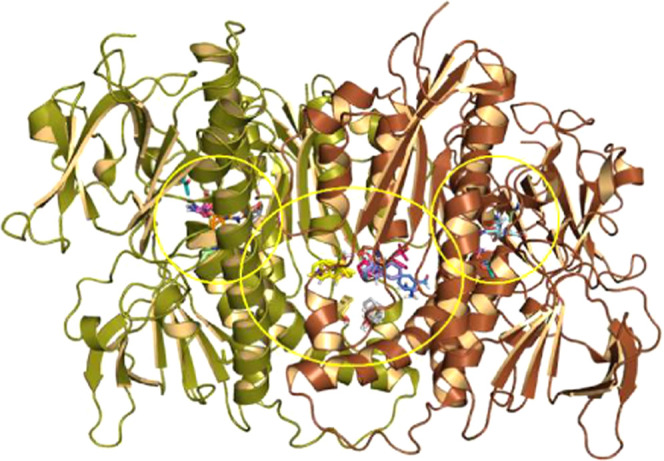
LiTryR dimer structure. FTMap identified the central interfacial cavity and the two adjacent sites where the two FAD cofactors are lodged as putative ligand-binding sites (circled by yellow lines).
Interestingly, this central cavity at the LiTryR dimer interface is reminiscent of that present in Plasmodium falciparum GR44 or hGR and is shown to bind some noncompetitive or uncompetitive inhibitors such as safranin, menadione,45 xanthenes,46 or N-arylisoalloxazines.47 Ligand accessibility to this site was assessed by means of the CAVER web server,48 which identified several tunnels connecting this intermonomer cavity to the bulk solvent (Figure 5).
Figure 5.
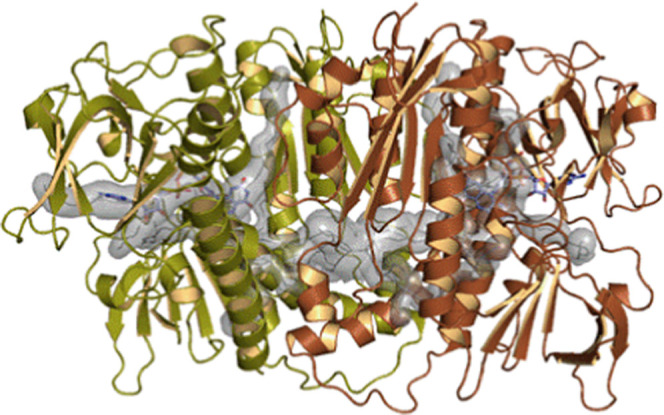
Visualization of the potential access routes from the bulk solvent to the interfacial cavity (2.808 Å3, 0.77 “druggability”) as determined by the CAVER Web server using default parameters.48 In comparison, each nicotinamide adenine dinucleotide phosphate (NADP) binding site occupies 1.343 Å3 and its druggability is 0.63. The tunnels are depicted as a continuous semitransparent gray surface. The central vertical α-helices encompass the amino acid residues originally identified as hotspots for dimerization. The FAD prosthetic groups are displayed as stick models for reference purposes only.
We next performed docking studies with the most potent triazole dimerization inhibitors 12b, 12c and the disubstituted thiazole analogue 19a centering the search within the proposed interfacial cavity of the LiTryR homodimer (Figure 6). The best ligand poses had in common the positioning of the (hetero)aromatic scaffold inside the connection tunnel and the terminal ammonium group of the R1 substituent at the hydrogen-bonding distance from the carboxylate of Glu466′ (d(N12b···OGlu466′) = 2.2 Å, d(N12c···O Glu466′) = 3.4 Å).
Figure 6.

Proposed ligand-binding modes within LiTryR. (a) Cartoon representation of dimeric LiTryR showing the two FAD prosthetic groups (sticks, C atoms in gray) close to the active sites and a docked 19a molecule (C atoms in pink) filling half of the central interfacial hydrophobic cavity and part of the tunnel connecting it to the bulk solvent. (b) Details of the best-scoring binding pose for 12b (C atoms in green). (c) Details of the best-scoring binding pose for 12c (C atoms in cyan). (d) Details of the best-scoring binding pose for 19a (C atoms in pink).
The proposed binding modes also revealed the potential of the imidazolidinone group of the R2 substituent to act as a hydrogen bond acceptor from the hydroxyl group of either Thr65 (d(N12b···OThr65) = 2.9 Å), for the naphthyl compound 12b, or Ser433′ (d(N12c···OSer433′) = 2.5 Å), for biphenyl compounds 12c and 19b. The unhindered rotation of the central benzene ring of the scaffold seems to play a fundamental role to distinctly orient the R2 substituent in each complex. Regarding the R3 hydrophobic substituents at the thiazole, the bulky naphthyl and biphenyl aromatic groups for compounds 12b, 12c could be accommodated in one of the hydrophobic subpockets of the interfacial cavity that is mainly defined by Pro435′ and Phe367′, in good accord with the previous FTMap results. In addition, for the R3 and R4 disubstituted phenyl-thiazole analogue 19a, one of the phenyl groups attached to the thiazole ring was found to point directly toward the second hydrophobic subpocket Leu72–Leu72′ at the bottom of the cavity, another location pinpointed by FTMap. This binding mode nicely accounts for the above-mentioned dramatic improvement of inhibitory potency relative to the monosubstituted phenyl-thiazole 12n.
Our molecular dynamics simulations and binding energy decomposition results using 19a as a representative inhibitor point to Glu436 as one of the major contributors to ligand binding (see Figure S16 and Movie S1 in the Supporting Information). We have previously shown that this residue is a hotspot for LiTryR dimerization.25 Indeed, in all X-ray crystal structures of TryR, this glutamate’s carboxylate from one monomer is involved in two short hydrogen bonds with the peptide backbone of Ser464 and Ala465 from the other monomer. Inhibitor binding appears to promote the loss of these interdimer hydrogen-bonding interactions (Figure S17) due to a wedge effect brought about by hydrophobic interactions involving the phenyl rings on the one side of the molecule and strong electrostatic interactions on the other side, which involve not only the free amino group but also the triazole ring. Indeed, the negative electrostatic potential generated by the two unsubstituted ring nitrogens faces the positive dipole of the short 400MetGly405 α-helix. Taken together, the theoretical calculations provide a rationale for the observed SAR results. Furthermore, the residue in hGR (PDB id: 1XAN) that is positionally equivalent to Leu72 in LiTryR is Phe78, whose aromatic side chain provides a flat platform for the stacking of ligands such as safranin or xanthene45,46 but prevents the binding of our novel compounds, hence their remarkable selectivity. The central cavity putatively targeted by 19a and analogues is lined by the residues listed in Table S5, which also shows the positionally equivalent residues in hGR. The overall lack of identity in several crucial regions is in consonance with the observed marked selectivity for TryR.
To further support the feasibility of the proposed binding mode and improve the binding affinity for the putative target site, a new C2-symmetric compound 22 was rationally designed with a view to exploiting the C-2 symmetry of this homodimeric enzyme and simultaneously occupy both entrance/exit tunnels (Figure 5). The putative LiTryR/19a complex provided the starting point for the structure-based design. As shown in Figure 7A, reproducing the binding mode of 19a within one monomer on the other monomer immediately reveals the possibility of merging both single molecular entities into one by making use of a suitable linker bonded to the R3 substituent. To this end, a 1,3-disubstituted phenyl was selected as an appropriate spacer to yield the C2-symmetric compound 22, which was also docked into the putative binding site (Figure 7B).
Figure 7.

Rationale for the design of the C2-symmetric compound 22. (A) Best docking pose for 19a (C atoms in pink) with the tail extended into one LiTryR monomer reproduced for the other monomer (C atoms in white). Note how the propylamino group is extended and oriented toward the carboxylate of Glu466 in both cases. (B) Best-scoring result provided by the automated docking program49 for 22 (CPK model) inside the central interfacial cavity and connecting tunnels. The side chains of Glu466 and Glu466′ are displayed as sticks with C atoms colored in green.
The good agreement found between the intended binding mode for 22 and that found by the automated docking program49 (Figure 7) is noteworthy. In addition to the hydrophobic contacts between the aromatic rings and the walls of the cavity, the two terminal R1 butylammonium groups of 22 are presumed to establish simultaneous salt bridges with Glu466/Glu467 and Glu466′/Glu467′, whereas the carbonyl groups of the two imidazolidinone moieties can establish hydrogen bonds with the protonated nitrogens of Lys61/Lys61′, the side-chain carboxamide of Gln68, and the backbone NH of Ser433/Ser433′.
The activity of the new symmetric compound 22 was evaluated in the two enzymatic assays. Encouragingly, the compound emerged not only as the most effective LiTryR inhibitor of the entire series, with an IC50act value of 0.4 ± 0.03 μM, but also as a very potent dimerization disruptor (IC50 = 8.0 ± 0.2 μM). In comparison with the corresponding monomeric triazole ligand 12n (substituted with an R3 phenyl group), 22 was 28- and 4-fold more potent, respectively, in both assays (see Table 1). Taken together, these experimental results support the proposed binding mode of the monomeric and symmetric triazole-based compounds at this interfacial site.
Inhibitory Activity on Other TryR Enzymes from the Trypanosoma Genus
TryR is an essential enzyme not only for Leishmania but also for Trypanosoma parasites. Given the high degree of identity/similarity among the amino acid sequences of TryRs from these two genera, some degree of activity against so closely related enzymes would not be unexpected for our compounds. To test this hypothesis, the most active members of these series were tested as inhibitors of the oxidoreductase activity of TryRs from T. brucei (TbTryR) and Trypanosoma congolense (TcoTryR), the causative agents of sleep sickness and nagana, respectively. The results shown in Table 3 demonstrate, in fact, very similar activities for 12a–c, 12n, 12r, 19a, and 22 as inhibitors of the three enzymes.
Table 3. IC50 ± SE Values (μM) for Selected Triazole Analogues 12a–c, 12n, 12r, 19a, and 22 in the Oxidoreductase Activity of TryR from L. infantum, T. brucei, and T. congolense.
| compounds | LiTryR | TbTryR | TcoTryR |
|---|---|---|---|
| 12a | 14.6 ± 1.0 | 13.3 ± 1.9 | 11.0 ± 1.3 |
| 12b | 5.9 ± 1.1 | 9.7 ± 2.1 | 4.8 ± 1.9 |
| 12c | 10.9 ± 1.8 | 10.0 ± 2.5 | 4.8 ± 1.8 |
| 12n | 9.0 ± 0.2 | 12.5 ± 1.9 | 9.3 ± 0.8 |
| 12r | 11.9 ± 0.6 | 15.0 ± 2.9 | 10.1 ± 1.4 |
| 19a | 4.3 ± 1.0 | 7.3 ± 0.6 | 5.6 ± 0.8 |
| 22 | 0.4 ± 0.03 | 1.0 ± 0.3 | 0.5 ± 0.1 |
In Vitro Antileishmanial Evaluation and Cytotoxicity
Compound Internalization into the Parasites
Our earlier attempts to develop LiTryR dimerization disruptors as leishmanicidal molecules were hampered by the poor ability of previously synthesized compounds to cross the parasite plasma membrane. The peptidomimetics herein described, endowed with fluorescent properties, no longer suffer from this limitation. Figure 8 shows the intense blue fluorescence emitted by promastigotes incubated for 1 h with compound 12a. Strong fluorescent spots can be observed in the apical region of some parasites, just opposite the flagellar pocket that is expected to be the main entry region for these compounds.
Figure 8.

Fluorescence microscopy images of L. infantum promastigotes treated with 25 μM compound 12a for 1 h.
Activity on Extracellular Parasites and Human Cells
Representative examples of triazole-based LiTryR enzyme inhibitors were tested in vitro against L. infantum axenic promastigotes and amastigotes. Cytotoxicity was assessed employing the human liver cancer cell line HepG2. Miltefosine, the R9 conjugate of the prototype peptide 1, and imidazole-based compounds 2 and 3 were included as reference drugs. According to the data shown in Table 4, among the 18 triazole compounds tested, 9 showed half-maximal effective concentration (EC50) values below 10 μM against Leishmania promastigotes or amastigotes (12b, 12c, 12i–k, 12n, 19b, 19d, and 19e) that are similar to those observed for the reference compounds.
Table 4. In Vitro Activity on L. infantum Parasites and Cytotoxic Activity of Representative Triazole Compoundsa,b.
| compounds | EC50 (μM) promastigotesc | EC50 (μM) amastigotesc | EC50 (μM) HepG2c,d | SIp/SIae |
|---|---|---|---|---|
| 12a | 16.4 ± 0.9 | 13.9 ± 0.6 | 20.3 ± 2.4 | 1.2/1.5 |
| 12b | 8.4 ± 0.3 | 15.3 ± 1.7 | 33.3 ± 2.7 | 4.0/2.2 |
| 12c | 4.9 ± 0.4 | 11.6 ± 0.8 | 43.1 ± 2.6 | 8.8/3.7 |
| 12h | 12.7 ± 0.3 | 31.6 ± 5.8 | 25.7 ± 2.2 | 2.0/<1 |
| 12i | 5.8 ± 0.7 | 5.2 ± 0.3 | 60.4 ± 3.0 | 10.4/11.6 |
| 12j | 8.5 ± 0.1 | 7.1 ± 0.7 | 12.4 ± 0.7 | 1.8/1.7 |
| 12k | NDf | 4.0 ± 0.2 | 7.4 ± 0.6 | ND/1.9 |
| 12m | 42.6 ± 3.9 | 25.2 ± 0.5 | 44.9 ± 1.3 | 1.0/1.8 |
| 12n | 5.4 ± 0.08 | 11.5 ± 0.3 | 16.4 ± 2.3 | 3.0/1.4 |
| 12o | >75 | >75 | 67.9 ± 1.5 | <1.0/<1.0 |
| 12q | ND | 27.0 ± 2.8 | 43.3 ± 1.0 | ND/1.6 |
| 12r | 23.8 ± 0.5 | 22.3± 0.6 | 29.4 ± 0.7 | 1.2/1.3 |
| 12s | ND | 68.8 ± 3.2 | >75 | ND/>1.1 |
| 19a | 10.7 ± 1.3 | 15.6 ± 3.0 | 26.5 ± 4.1 | 2.5/1.7 |
| 19b | ND | 9.4 ± 0.5 | 17.3 ± 0.1 | ND/1.8 |
| 19c | ND | 22.6 ± 5.7 | ≥ 75 | ND/≥3.3 |
| 19d | ND | 9.1 ± 0.2 | 12.1 ± 0.1 | ND/1.3 |
| 19e | ND | 8.4 ± 0.1 | 6.6 ± 0.6 | ND/<1 |
| 22 | 3.8 ± 0.1 | 4.9 ± 0.2 | 30.5 ± 4.1 | 8.0/6.2 |
| miltefosine | 47.6 ± 0.6 | 2.0 ± 0.1 | >75 | >1.6/>37.5 |
| R9-peptide 1 | 3.1 ± 0.6 | 3.4 ± 0.2 | 61.7 ± 6.1 | 19.9/18.1 |
| 2 | 12.8 ± 0.7 | 12.8 ± 1.3 | 27.1 ± 2.4 | 2.1/2.1 |
| 3 | 5.3 ± 0.3 | 5.3 ± 0.2 | 14.2 ± 0.9 | 2.7/2.7 |
Miltefosine, linear peptide 1 conjugated to R9, and imidazole-containing compounds 2 and 3 were included as in-house reference compounds.
Results are representative of three independent experiments, each performed in triplicate. EC50 ± SE values are indicated.
The half-maximal effective concentration (EC50) is defined as that causing a 50% reduction of proliferation in L. infantum promastigotes (EC50p) and amastigotes (EC50a) or human hepatocellular carcinoma (HepG2) cells. Dead parasites were identified by their increased permeability to propidium iodide (PI).
Cytotoxicity in HepG2 cells was measured using the crystal violet method.
The selectivity index (SI) is the ratio of EC50 against human cells to EC50 against L. infantum promastigotes (SIp) and amastigotes (SIa).
ND = not determined.
Of all the compounds bearing a butylamine at R1, an imidazolidinone at R2, and different R3 substituents (12a–c), the biphenylethyl analogue 12c displayed better activity against both promastigotes and amastigotes than those containing naphthyl or phenylethyl substituents (12c > 12b > 12a) and also lower cytotoxicity. This results in an improved selectivity index (SI) of 8.8 for promastigotes (SIp) and 3.7 for amastigotes (SIa) in comparison with the SIs of the corresponding biphenyl imidazole analogue 3 (SIp = SIa = 2.7). On the other hand, a combination of a shorter propyl spacer at R1 and the biphenyl group at R3 gave 12i, the most potent and selective compound of the entire series, with EC50 values of 5.8 against promastigotes (EC50p) and 5.2 μM against amastigotes (EC50a) and the highest SIs (SIp = 10.4 and SIa = 11.6). The imidazolidinone moiety at R2 could be eliminated without compromising the leishmanicidal activity (12j, 12k), but a significant increase in cytotoxicity was detected, which suggests a positive effect of the imidazolidinone on reducing the cytotoxicity. As regards R3, the biphenylethyl substituent could be replaced by a phenyl resulting in an equipotent leishmanicidal analogue (12cvs12n), in contrast to the higher IC50 values observed for 12n in the enzymatic assays. However, the SI for 12n was low due to an increase in toxicity. The introduction of polyheteroaromatic rings gave rise to a significant reduction of the leishmanicidal activity (12q–svs12c) in contrast to the high potency observed for these analogues in the enzymatic assays. Disubstituted thiazole analogues 19a–e also showed high leishmanicidal activities against amastigotes with EC50 values in the 8.4−22.6 μM range but, with the exception of 19c, low SIs (SI < 2.5) due to higher toxicities. Remarkably, the symmetrical triazole analogue 22 turned out to be one of the most potent and selective agents against L. infantum amastigotes with an EC50a value of 4.9 μM and a SIa of 6.2.
It should be noted that a direct correlation between in vitro TryR inhibitory activity and in cellulo effects is not to be expected due to differences in cellular permeability or the possibility that this enzyme is not the only target for these compounds. Therefore, among the newly tested triazole-based analogues, the propylamine biphenyl derivative 12i and the C-2 symmetric 22 displayed the best leishmanicidal activity and the lowest in cellulo cytotoxicity with SI values that exceeded those obtained for the imidazoles 2 and 3 by 5–6-fold. It is interesting to point out that, despite the relatively poor IC50 value obtained for 12i in the oxidoreductase activity assay, its behavior in the dimerization assay is similar to that of the most potent compounds from these series. This finding suggests that their ability to disrupt the dimer may be largely responsible for the leishmanicidal activity of these compounds. As IC50 values were calculated taking into consideration only the initial velocities of the reactions and because of the time-dependent behavior of these molecules, compounds such as 12i may show good activities in the dimerization assay while displaying poor IC50 values.
Leishmanicidal Activity in Intracellular Amastigotes
The most potent and selective compounds were further tested for their leishmanicidal activity in amastigote-infected THP-1 cells. The EC50 values obtained are summarized in Table 5. Miltefosine, edelfosine, amphotericin B, the R9-conjugate of the prototype peptide 1, and compounds 2 and 3 from the precursor imidazole series were included as reference compounds. In our view, the fact that the EC50 values cover a range from 22 to >75 μM indicates that not all of the compounds are able to cross the plasma and phagolysosomal membranes in the macrophages. Among the monomeric triazole-based compounds, only the phenyl derivative 12a, with an EC50 value of 32.4 μM, and the biphenyl derivatives 12c, 12i, with EC50 values of 46.5 and 54.5 μM, respectively, displayed moderate effectiveness against intracellular amastigotes. Interestingly, the symmetric triazole molecule 22 emerged, once again, as the most potent and selective leishmanicidal compound of all of the triazole-based series (EC50 = 22 μM), showing a SI of 2.2, better than that obtained for edelfosine (SI = 1.4). Accordingly, derivatives 12a, 12c, 12i, and 22 constitute the first analogues designed against the dimerization interface of TryR that show leishmanicidal activity toward intracellular amastigotes. These results confirm the interest and potential of both monomeric and symmetric triazole-based compounds as antileishmanial agents.
Table 5. In Vitro Antileishmanial Activity for the Most Representative Triazole-Based Compounds in Amastigote-Infected THP-1 Monocyte-Derived Macrophagesa.
| compounds | EC50 (μM) intracellular amastigotesb | EC50 (μM) THP-1c |
|---|---|---|
| 12a | 32.4 ± 6.7 | 45 ± 3.1 |
| 12b | >75 | >75 |
| 12c | 46.5 ± 1.5 | >75 (100% at 75 μM)d |
| 12i | 54.4 ± 15.5 | >75 (100% at 75 μM)d |
| 12q | >75 | >75 (67% at 75 μM)d |
| 19c | >75 | >75 (63% at 75 μM)d |
| 22 | 22.2 ± 6.4 | 48.4 ± 14.5 |
| mdelfosine | 2.5 ± 0.02 | 3.5 ± 0.3 |
| miltefosine | 0.6 ± 0.1 | >25 |
| amphotericin B | 0.07 ± 0.01 | >5 |
| R9-peptide 1 | >75 | 14.5 ± 2.9 |
| 2 | >75 | 27.9 ± 4.4 |
| 3 | >75 | 15.5 ± 1.8 |
Edelfosine, miltefosine, and amphotericin B were included as reference leishmanicidal drugs. The R9-peptide 1 and the imidazole-containing compounds 2 and 3 were included as in-house reference compounds for structure–activity comparisons.
The half-maximal effective concentration (EC50) is defined as that causing a 50% reduction in the number of L. infantum amastigotes per cell in the culture or amastigote-infected THP-1 monocyte-derived macrophages. Living intracellular amastigotes were identified by their green fluorescence due to green fluorescent protein (GFP) expression and their lack of permeability to propidium iodide; EC50 ± SE values are indicated.
Cytotoxicity was assessed in THP-1 monocyte-derived macrophages by measuring lactate dehydrogenase (LDH) activity.
Cell viability of amastigote-infected THP-1 monocyte-derived macrophages at 75 μM (maximum concentration assayed). Results are representative of three independent experiments.
Conclusions
The development of new and more efficient antileishmanial drugs is a priority in the field of neglected tropical diseases. From a small library of 26 triazole-phenyl-thiazole compounds, we have identified the most potent small-molecule dimerization disruptors of LiTryR described to date. Similar to prototype peptide 1, they behave as slow-binding, noncompetitive inhibitors with overall Ki* values of 0.5 μM (19a). These molecules are also endowed with high antileishmanial activity against both extracellular and intracellular parasites and improved selectivity indexes when compared to those obtained for previous in-house imidazole-based compounds.
Molecular modeling studies carried out for the most potent LiTryR triazole-containing dimerization inhibitors 12b, 12c, and 19a identified a previously unexplored but apparently druggable binding site consisting of a large central cavity at the dimer interface of the enzyme. In the absence of crystallographic evidence, despite extensive efforts, this putative binding mode provided a rationale for the observed SAR and a basis for the subsequent structure-based design of a new symmetrical triazole analogue 22 that displays improved LiTryR inhibitory activities over those of its parental monomeric triazole. This rationally designed compound shows a dramatic enhancement in leishmanicidal potency, in particular against intracellular amastigotes.
All in all, our results provide the first evidence for ligand binding to a so-far unexplored region of LiTryR and lay the ground for further design of innovative TryR inhibitors that may become valuable candidates in the process of discovering new drugs against leishmaniasis.
Experimental Section
Chemical Procedures
Experiments dealing with air- and water-sensitive reagents/compounds were performed in an argon atmosphere. Hygroscopic species were previously dried under vacuum for 24 h by P2O5 as the drying agent. Unless otherwise noted, analytical-grade solvents and commercially available reagents were used without further purification. Anhydrous CH2Cl2 was dried by refluxing over CaH2. EtOH was dried with 4 Å molecular sieves previously activated in a microwave oven. THF was dried by reflux over sodium/benzophenone. Pressured reactions were carried out on a Sigma-Aldrich Ace Glass flask (50 or 100 mL) supplied with a Teflon cap. Microwave-assisted experiments were performed on a Biotage Initiator 2.0 single-mode cavity instrument from Biotage (Uppsala). Experiments were carried out in sealed microwave reaction vials at the standard absorbance level (400 W maximum power). Lyophilizations were carried out on a Telstar 6-80 lyophilizer.
Monitoring of the reactions was performed by analytical thin-layer chromatography (TLC) on silica gel 60 F254 (Merck) or by high-performance liquid chromatography coupled to mass spectrometry (HPLC–MS) using a Waters Alliance 2695 Separation Module with a Waters Micromass ZQ Detector.
Compounds were purified by (a) flash column chromatography (Merck) with silica gel 60 (230–400 mesh); (b) high-performance flash column chromatography (HPFC) on a Biotage Isolera One using cartridges of Claricep i-Series, Silica, 40 g (40–60 μm, 60 Å) for normal phase chromatography, and KP-C18-HS 12g (21 × 55 mm2) for the reverse phase; (c) centrifugal circular thin-layer chromatography (CCTLC) on a Chromatotron (Kiesegel 60 PF254 gipshaltig, Merck), with layer thicknesses of 1 and 2 mm and flow rates of 2 and 3 mL/min; or (d) semipreparative HPLC purifications on a Waters 2695 HPLC system equipped with a Photodiode Array 2998 coupled to a 3100 Mass Detector mass spectrometer using a C18 Sunfire column (19 mm × 150 mm).
The purity of the compounds was checked by analytical HPLC on a Waters 600 system equipped with a C18 Sunfire column (4.6 mm × 150 mm, 3.5 μm) and a selectable wavelength UV detector. Alternatively, an Agilent Technologies 1120 Compact LC provided with a reverse-phase column ACE 5 C18-300 (4.6 mm × 150 mm, 3.5 μm) and integrated diode detector PDA 996 was used. As the mobile phase, mixtures of A (CH3CN) and B (H2O) (0.05% TFA) on isocratic or gradient mode with a flow rate of 1 mL/min were used. The following gradients were used: from 10% A to 100% A in 10 min (gradient A) and from 1% A to 100% A in 10 min (gradient B). The purity of the final compounds was also determined to be >95% by elemental analysis with a LECO CHNS-932 apparatus. Deviations of the elemental analysis results from the calculated value were within ±0.4%.
Melting points (m.p.) were measured on a Mettler Toledo M170 apparatus and are uncorrected. Infrared (IR) spectra were recorded on a PerkinElmer Spectrum One instrument and KBr pellet as a sample support. Mass spectrum (MS) on electrospray ionization (ESI) mode was recorded on a Hewlett-Packard 1100SD in positive mode. High-resolution mass spectra (HRMS) were obtained on an Agilent 6520 Accurate-Mass Q-TOF LC-MS equipped with an HP-1200 (Agilent) liquid chromatograph coupled to a mass spectrometer with a Q-TOF 6520 hybrid mass analyzer. Usually, an error equal to or lower than 5 ppm was allowed. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian INNOVA-300 operating at 300 MHz (1H) and 75 MHz (13C), a Varian INNOVA-400 operating at 400 MHz (1H) and 100 MHz (13C), a Varian MERCURY-400 operating at 400 MHz (1H) and 100 MHz (13C), and a Varian SYSTEM-500 operating at 500 MHz (1H) and 125 MHz (13C). The assignments were performed by means of different standard homonuclear and heteronuclear correlation experiments (gradient heteronuclear single quantum coherence (gHSQC), gradient heteronuclear multiple bond coherence (gHMBC), and nuclear Overhauser enhancement spectroscopy (NOESY)) when required.
4-Azido-2-(2-(2-oxoimidazolidin-1-yl)ethoxy)benzo-nitrile (5a)
A solution of bromoarene 4a(30) (500 mg, 1.61 mmol) and NaN3 (1.57 g, 24.2 mmol) in anhydrous DMSO (30 mL) in the presence of 4 Å molecular sieves, and under an argon atmosphere, was heated at 100 °C for 72 h. The reaction was allowed to cool to room temperature and diluted with H2O (60 mL), and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, evaporated to dryness, and lyophilized. The residue was purified by flash column chromatography (CH2Cl2/MeOH, 100:3) to give 372 mg (85%) of 5a as a yellow solid; m.p.: 158–161 °C; IR (KBr), ν (cm–1): 3230 (NH-st), 3090 (Csp–H st), 2223 (C≡N st), 2125 (N=N=N st), 1702 (C=O st); 1H NMR (CDCl3, 300 MHz) δ (ppm): 7.53 (d, J = 8.3 Hz, 1H, Ar), 6.70 (dd, J = 8.3, 2.0 Hz, 1H, Ar), 6.53 (d, J = 1.8 Hz, 1H, Ar), 4.71 (bs, 1H, NHCON), 4.20 (t, J = 4.7 Hz, 2H, OCH2), 3.81–3.76 (m, 2H, CH2CH2NHCON), 3.64 (t, J = 4.9 Hz, 2H, OCH2CH2), 3.44 (t, J = 8.1 Hz, 2H, CH2CH2NHCON); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.8 (NHCON), 161.8 (OCAr), 146.8 (CAr), 134.9 (CHAr), 116.2 (CN), 111.8 (CHAr), 103.2 (CHAr), 98.3 (CAr), 69.4 (OCH2), 47.8 (CH2CH2NHCON), 43.2 (OCH2CH2), 38.6 (CH2CH2NHCON); MS (ESI, positive mode) m/z: 567.2 [2M + Na]+, 295.0 [M + Na]+, 273.0 [M + H]+. NaN3 may be toxic and explosive. Thus, for safety precautions, a polycarbonate safety screen in a properly functioning fume hood was always used to perform this reaction.
Benzyl-4-(1-(4-cyano-3-((1-(2-oxoimidazolidin-1-yl)ethoxy)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (8)
To a solution of azide 5a (450 mg, 1.65 mmol), commercially available benzyl-5-hexynylcarbamate (523 mg, 2.15 mmol), and CuSO4·5H2O (42 mg, 0.17 mmol) in EtOH (25 mL) were added sodium ascorbate (131 mg, 0.66 mmol) and H2O (25 mL). The reaction mixture was stirred at room temperature in the dark overnight. The mixture was evaporated to dryness, and the residue dissolved in CH2Cl2 (50 mL) was washed with H2O (3 × 50 mL), dryed (Na2SO4), filtered, and evaporated under reduced pressure. The residue was purified by flash column chromatography (CH2Cl2/MeOH, 100:3) to provide 8 (573 mg, 69%) as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.86 (s, 1H, Ar), 7.61 (d, J = 8.3 Hz, 1H, Ar), 7.49 (s, 1H, Ar), 7.34 (d, J = 7.1 Hz, 1H, Ar), 7.32–7.17 (m, 5H, Ar), 5.05 (bs, 1H, NHCbz), 5.02 (s, 2H, NHCOOCH2), 4.68 (bs, 1H, NHCON), 4.26 (t, J = 5.2 Hz, 2H, OCH2), 3.66 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.57 (t, J = 5.2 Hz, 2H, OCH2CH2), 3.33 (t, J = 8.3 Hz, 2H, CH2CH2NHCON), 3.18 (q, J = 6.7 Hz, 2H, CH2NHCbz), 2.76 (t, J = 7.3 Hz, 2H, TrizCH2), 1.71 (quin, J = 7.4 Hz, 2H, TrizCH2CH2), 1.52 (quin, J = 7.2 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.8 (NHCON), 161.5 (OCAr), 156.6 (NHCOO), 149.2 (CAr), 141.5 (CAr), 136.6 (CHAr), 135.1 (CHAr), 128.6 (CHAr), 128.2 (CHAr), 119.1 (CHAr), 115.7 (CN), 112.1 (CHAr), 104.1 (CHAr), 101.5 (CAr), 69.0 (OCH2), 66.7 (NHCOOCH2), 47.3 (CH2CH2NHCON), 42.8 (OCH2CH2), 40.8 (CH2NHCbz), 38.5 (CH2CH2NHCON), 29.4 (CH2CH2NHCbz), 26.1 (TrizCH2CH2), 25.1 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C26H29N7O4 503.2281; found 503.2279 (−0.33 ppm).
Benzyl-4-(1-(4-carbamothioyl-3-((1-(2-oxoimidazolidin-1-yl)ethoxy)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (9)
To a solution of benzonitrile 8 (400 mg, 0.79 mmol) in DMF (25 mL), 20% aq (NH4)2S (3.78 mL, 55.6 mmol) was added and then heated to 80 °C for 4 h. The reaction mixture was allowed to cool to room temperature, and then, CH2Cl2 (50 mL) was added. The mixture was successively washed with HCl 0.1 N (2 × 50 mL), H2O (1 × 50 mL), and brine (1 × 50 mL). The organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The crude was purified by flash column chromatography (CH2Cl2/MeOH, 100:5) to give 9 (354 mg, 74%) as a yellow oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 9.63 (bs, 1H, SCNH2), 9.50 (bs, 1H, SCNH2), 8.71 (d, J = 8.7 Hz, 1H, Ar), 7.78 (s, 1H, Ar), 7.45 (d, J = 2.0 Hz, 1H, Ar), 7.36–7.17 (m, 5H, Ar), 7.10 (dd, J = 8.7, 2.0 Hz, 1H, Ar), 5.96 (bs, 1H, NHCbz), 5.02 (s, 2H, NHCOOCH2), 4.98 (bs, 1H, NHCON), 4.16 (t, J = 4.4 Hz, 2H, OCH2), 3.64 (t, J = 4.4 Hz, 2H, OCH2CH2), 3.50 (dd, J = 9.7, 6.7 Hz, 2H, CH2CH2NHCON), 3.40 (dd, J = 9.1, 6.2 Hz, 2H, CH2CH2NHCON), 3.17 (q, J = 6.7 Hz, 2H, CH2NHCbz), 2.73 (t, J = 7.5 Hz, 2H, TrizCH2), 1.70 (quin, J = 7.5 Hz, 2H, TrizCH2CH2), 1.53 (quin, J = 7.5 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 196.5 (SCNH2), 163.5 (NHCON), 156.3 (NHCOO), 149.1 (CAr), 140.3 (CAr), 138.4 (CHAr), 136.7 (CHAr), 128.7 (CHAr), 128.2 (CHAr), 125.1 (CAr), 119.1 (CHAr), 111.3 (CHAr), 104.0 (CHAr), 66.8 (OCH2), 66.6 (NHCOOCH2), 45.4 (CH2CH2NHCON), 43.2 (OCH2CH2), 40.9 (CH2NHCbz), 38.3 (CH2CH2NHCON), 29.8 (CH2CH2NHCbz), 26.4 (TrizCH2CH2), 25.3 (TrizCH2); MS (ESI, positive mode) m/z: 538.3 [M + H]+.
General Procedure for the Synthesis of Thiazoles 11a–c by Hantzsch Cyclization
A solution of thioamide 9 (1 equiv) in iPrOH (15 mL) was treated with the appropriated α-haloketone (10a–c) (1 equiv). The reaction mixture was stirred at 70 °C for 3–6 h in a pressure flask, and then, it was allowed to cool to room temperature and concentrated to dryness under reduced pressure. The residue was purified by flash column chromatography or CCTLC on a Chromatotron (eluents are specified in each case).
Benzyl-(4-(1-(3-(2-(2-oxoimidazolidin-1-yl)ethoxy)-4-(4-phenethylthiazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (11a)
Following the general procedure, thioamide 9 (100 mg, 0.19 mmol) and the commercially available 1-bromo-4-phenylbutan-2-one (10a 42 mg, 0.19 mmol) in iPrOH (15 mL) reacted at 70 °C for 4 h. After the workup, the residue was purified by CCTLC on the Chromatotron (CH2Cl2/MeOH, 100:3) to yield 108 mg (87%) of 11a as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.46 (d, J = 8.5 Hz, 1H, Ar), 7.81 (s, 1H, Ar), 7.51 (d, J = 2.0 Hz, 1H, Ar), 7.38–7.06 (m, 11H, Ar), 6.86 (s, 1H, Ar), 5.06 (t, J = 6.0 Hz, 1H, NHCbz), 5.01 (s, 2H, NHCOOCH2), 4.34 (t, J = 5.7 Hz, 2H, OCH2), 3.69 (t, J = 5.7 Hz, 2H, OCH2CH2), 3.50 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.26 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.18 (q, J = 7.4 Hz, 2H, CH2NHCbz), 3.12–2.97 (m, 4H, CH2CH2Ph), 2.76 (t, J = 7.4 Hz, 2H, TrizCH2), 1.71 (quin, J = 7.4 Hz, 2H, TrizCH2CH2), 1.59 (quin, J = 7.1 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.8 (NHCON), 160.3 (OCAr), 156.6 (CAr), 156.0 (CAr), 155.9 (CAr), 148.7 (CAr), 141.7 (CAr), 138.3 (CAr), 136.7 (CAr), 130.0 (CHAr), 128.6 (CHAr), 128.6 (CHAr), 128.4 (CHAr), 128.2 (CHAr), 126.1 (CHAr), 122.7 (CAr), 119.1 (CHAr), 115.1 (CHAr), 112.4 (CHAr), 104.5 (CHAr), 67.8 (OCH2), 66.7 (NHCOOCH2), 46.4 (CH2CH2NHCON), 43.0 (OCH2CH2), 40.8 (CH2NHCbz), 38.4 (CH2CH2NHCON), 35.6 (CH2CH2Ph), 33.4 (CH2CH2Ph), 29.4 (CH2CH2NHCbz), 26.4 (TrizCH2CH2), 25.2 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C36H39N7O4S 665.2784; found 665.2791 (1.00 ppm).
Benzyl-(E)-(4-(1-(4-(4-(2-(naphthalen-2-yl)vinyl)thiazol-2-yl)-3-(2-(2-oxoimidazolidin-1-yl)ethoxy)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (11b)
According to the general procedure, thioamide 9 (520 mg, 0.97 mmol) and α-bromoketone 10b(30) (267 mg, 0.97 mmol) were reacted at 70 °C for 5 h in iPrOH. After the workup, the residue was purified by flash column chromatography (CH2Cl2/MeOH, 100:4) to give 11b (365 mg, 51%) as a yellow oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.61 (d, J = 8.5 Hz, 1H, Ar), 7.90–7.64 (m, 7H, Ar, ThiazCH=CH), 7.55 (s, 1H, Ar), 7.45–7.36 (m, 3H, Ar), 7.30–7.14 (m, 7H, Ar, ThiazCH=CH), 5.03 (s, 2H, NHCOOCH2), 4.97 (bs, 1H, NHCON), 4.48 (bs, 1H, NHCbz), 4.39 (t, J = 5.7 Hz, 2H, OCH2), 3.73 (t, J = 5.7 Hz, 2H, OCH2CH2), 3.52 (dd, J = 9.0, 6.6 Hz, 2H, CH2CH2NHCON), 3.28 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.20 (q, J = 6.7 Hz, 2H, CH2NHCbz), 2.78 (t, J = 7.3 Hz, 2H, TrizCH2), 1.74 (quin, J = 7.5 Hz, 2H, TrizCH2CH2), 1.59–1.51 (m, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.7 (NHCON), 161.0 (OCAr), 156.6 (CAr), 156.2 (CAr), 153.5 (CAr), 148.8 (CAr), 138.6 (CAr), 136.7 (CAr), 134.7 (CHAr), 133.8 (CAr), 133.3 (CAr), 131.5 (CHAr), 130.3 (CHAr), 128.7 (CHAr), 128.4 (CHAr), 128.3 (CHAr), 128.2 (CHAr), 127.8 (CHAr), 127.2 (CAr), 126.5 (CHAr), 126.1 (CHAr), 123.6 (CHAr), 121.8 (CHAr), 119.2 (CHAr), 117.1 (CHAr), 112.5 (CHAr), 104.5 (CHAr), 67.7 (OCH2), 66.8 (NHCOOCH2), 46.4 (CH2CH2NHCON), 43.0 (OCH2CH2), 40.9 (CH2NHCbz), 38.4 (CH2CH2NHCON), 29.5 (CH2CH2NHCbz), 26.4 (TrizCH2CH2), 25.2 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C40H39N7O4S 713.2784; found 713.2779 (−0.72 ppm).
Benzyl-(E)-(4-(1-(4-(4-(2-([1,1′-biphenyl]-4-yl)vinyl) thiazol-2-yl)-3-(2-(2-oxoimidazolidin-1-yl)ethoxy)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (11c)
Following the general Hantzsch synthesis, 9 (200 mg, 0.37 mmol) reacted with α-bromoketone 10c(30) (112 mg, 0.37 mmol) for 5 h. After the workup, the residue was purified by flash column chromatography (CH2Cl2/MeOH, 100:3) to give 174 mg (63%) of 11c as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.57 (d, J = 8.5 Hz, 1H, Ar), 7.81 (s, 1H, Ar), 7.61–7.45 (m, 8H, Ar, ThiazCH=CH), 7.40–7.31 (m, 3H, Ar), 7.31–7.16 (m, 7H, Ar), 7.12 (d, J = 16.0 Hz, 1H, ThiazCH=CH), 5.02 (s, 2H, NHCOOCH2), 4.68 (bs, 1H, NHCON), 4.36 (t, J = 5.7 Hz, 2H, OCH2), 3.70 (t, J = 5.8 Hz, 2H, OCH2CH2), 3.50 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.26 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.16 (q, J = 6.6 Hz, 2H, CH2NHCbz), 2.76 (t, J = 7.3 Hz, 2H, TrizCH2), 1.75 (quin, J = 7.5 Hz, 2H, TrizCH2CH2), 1.55 (quin, J = 7.3 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.7 (NHCON), 160.9 (OCAr), 156.6 (CAr), 156.2 (CAr), 153.4 (CAr), 148.8 (CAr), 140.7 (CAr), 140.6 (CAr), 138.6 (CHAr), 136.7 (CAr), 136.2 (CHAr), 130.9 (CHAr), 130.3 (CHAr), 128.9 (CHAr), 128.6 (CHAr), 128.3 (CHAr), 127.5 (CHAr), 127.0 (CHAr), 122.4 (CAr), 121.5 (CHAr), 119.1 (CHAr), 117.4 (CHAr), 112.4 (CHAr), 104.4 (CHAr), 67.7 (OCH2), 66.8 (NHCOOCH2), 46.4 (CH2CH2NHCON), 42.9 (OCH2CH2), 40.9 (CH2NHCbz), 38.4 (CH2CH2NHCON), 29.4 (CH2CH2NHCbz), 26.4 (TrizCH2CH2), 25.2 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C42H41N7O4S 739.2941; found 739.2972 (4.21 ppm).
General Procedure for N-Cbz Deprotection
A solution of the corresponding Cbz-protected compound (1 equiv) in a 1:1 mixture of THF/MeOH (20 mL) containing Pd/C (10%) (20% w/w) and TFA (0.5–1.5 mL) was hydrogenated at room temperature for 2 h under atmospheric pressure using a balloon filled with hydrogen gas (three cycles of vacuum + hydrogen). The Pd/C was filtered through Whatman poly(tetrafluoroethylene) (PTFE) filter paper, and the solvent was removed under reduced pressure and co-evaporated with mixtures of CH2Cl2/MeOH (5 × 10 mL). The residue was purified by HPFC on an SP1 Isolera Biotage using reverse-phase columns (from 0% of CH3CN to 100% of CH3CN in 45 min) to afford to give the final deprotected compounds as trifluoroacetate salts.
1-(2-(5-(4-(4-Ammoniumbutyl)-1H-1,2,3-triazol-1-yl)-2-(4-phenethylthiazol-2-yl)phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (12a)
According to the general hydrogenolysis procedure, a solution of 11a (55 mg, 0.08 mmol), in 1:1 THF/MeOH (20 mL) containing Pd/C (10%) (20% w/w) (18 mg) and TFA (0.5 mL), was hydrogenated. The residue was purified by HPFC on the SP1 Isolera Biotage using reverse-phase columns (from 0% of CH3CN to 100% of CH3CN in 45 min) to afford 12a (19 mg, 35%) as a colorless oil. 1H NMR (CD3OD, 400 MHz) δ (ppm): 8.47 (s, 1H, Ar), 8.46 (d, J = 8.5 Hz, 1H, Ar), 7.70 (d, J = 2.0 Hz, 1H, Ar), 7.58 (dd, J = 8.6, 2.0 Hz, 1H, Ar), 7.33–7.17 (m, 5H, Ar), 7.15 (s, 1H, Ar), 4.48 (t, J = 5.5 Hz, 2H, OCH2), 3.74 (t, J = 5.5 Hz, 2H, OCH2CH2), 3.60 (dd, J = 9.3, 6.9 Hz, 2H, CH2CH2NHCON), 3.36 (dd, J = 9.3, 6.9 Hz, 2H, CH2CH2NHCON), 3.15–3.03 (m, 4H, CH2CH2Ph), 3.00 (t, J = 7.4 Hz, 2H, CH2NH3+), 2.87 (t, J = 7.2 Hz, 2H, TrizCH2), 1.89–1.81 (m, 2H, TrizCH2CH2), 1.81–1.70 (m, 2H, CH2CH2NH3+); 13C NMR (CD3OD, 100 MHz) δ (ppm): 165.1 (NHCON), 162.0 (OCAr), 157.4 (CAr), 157.1 (CAr), 149.3 (CAr), 142.8 (CAr), 139.7 (CAr), 130.8 (CHAr), 129.5 (CHAr), 129.4 (CHAr), 127.0 (CHAr), 123.8 (CAr), 121.6 (CHAr), 116.9 (CHAr), 113.6 (CHAr), 105.7 (CHAr), 68.4 (OCH2), 47.0 (CH2CH2NHCON), 43.9 (OCH2CH2), 40.4 (CH2NH3+), 39.3 (CH2CH2NHCON), 36.7 (CH2CH2Ph), 34.3 (CH2CH2Ph), 28.0 (CH2CH2NH3+), 27.1 (TrizCH2CH2), 25.6 (TrizCH2); HPLC (gradient A, Agilent): Rt = 7.1 min; HRMS (ES, positive mode) m/z: calcd for C28H33N7O2S 531.2416; found 531.2424 (1.35 ppm); anal. calcd for C28H33N7O2S·TFA: C, 55.80; H, 5.31; N, 15.18; S, 4.97; found: C, 55.94; H, 5.50; N, 15.08; S, 4.51.
1-(2-(5-(4-(4-Ammoniumbutyl)-1H-1,2,3-triazol-1-yl)-2-(4-(2-(naphthalen-2-yl)ethyl)thiazol-2-yl)phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (12b)
Following the deprotection procedure, compound 11b (218 mg, 0.31 mmol), Pd/C (10%) (20% w/w) (44 mg), and TFA (1.3 mL) in a 1:1 mixture of THF/MeOH (30 mL) were hydrogenated. After the workup, the residue was purified by reverse phase on the Biotage to yield 51 mg (24%) of 12b as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.77 (s, 1H, Ar), 8.49 (d, J = 8.6 Hz, 1H, Ar), 7.89–7.66 (m, 8H, Ar, NH3+), 7.52–7.34 (m, 3H, Ar), 7.34 (s, 1H, Ar), 4.47 (t, J = 5.7 Hz, 2H, OCH2), 3.63 (t, J = 5.6 Hz, 2H, OCH2CH2), 3.48 (dd, J = 8.9 Hz, J = 6.7 Hz, 2H, CH2CH2NHCON), 3.33–3.04 (m, 6H, CH2CH2NHCON, CH2NH3+, ThiazCH2CH2), 2.93–2.86 (m, 2H, ThiazCH2CH2), 2.77 (t, J = 7.2 Hz, 2H, TrizCH2), 1.84–1.71 (m, 2H, TrizCH2CH2), 1.67–1.56 (m, 2H, CH2CH2NH3+); 13C NMR (DMSO-d6, 75 MHz) δ (ppm): 162.1 (NHCON), 159.4 (OCAr), 155.6 (CAr), 155.3 (CAr), 147.7 (CAr), 139.0 (CAr), 137.9 (CAr), 133.2 (CHAr), 131.6 (CAr), 129.1 (CHAr), 127.7 (CHAr), 127.4 (CHAr), 127.3 (CAr), 126.2 (CHAr), 125.9 (CHAr), 125.2 (CHAr), 121.4 (CHAr), 120.4 (CHAr), 116.1 (CHAr), 112.0 (CHAr), 104.3 (CHAr), 67.6 (OCH2), 45.3 (CH2CH2NHCON), 42.4 (OCH2CH2), 37.5 (CH2CH2NHCON), 34.9 (ThiazCH2CH2), 32.5 (ThiazCH2CH2), 26.5 (CH2CH2NH3+), 25.7 (TrizCH2CH2), 24.8 (TrizCH2); HPLC (gradient A, Agilent): Rt = 7.8 min; HRMS (ES, positive mode) m/z: calcd for C32H35N7O2S 581.2573; found 581.2572 (−0.13 ppm); anal. calcd for C32H35N7O2S·TFA: C, 58.69; H, 5.22; N, 14.09; S, 4.61; found: C, 58.20; H, 5.01; N, 14.09; S, 4.12.
1-(2-(2-(4-(2-([1,1′-Biphenyl]-4-yl)ethyl)thiazol-2-yl)-5-(4-(4-ammoniumbutyl)-1H-1,2,3-triazol-1-yl)phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (12c)
Following the hydrogenolysis procedure, compound 11c (50 mg, 0.07 mmol), Pd/C (10%) (20% w/w) (10 mg), and TFA (0.5 mL) in a 1:1 mixture of THF/MeOH (20 mL) were hydrogenated. After the workup, the residue was purified by reverse phase on the Biotage to give 12c (17 mg, 35%) as a colorless oil. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 8.77 (s, 1H, Ar), 8.47 (d, J = 8.6 Hz, 1H, Ar), 7.75 (d, J = 2.1 Hz, 1H, Ar), 7.69 (dd, J = 8.6, 2.1 Hz, 1H, Ar), 7.66–7.62 (m, 2H, Ar), 7.60–7.56 (m, 2H, Ar), 7.48–7.39 (m, 3H, Ar), 7.38–7.29 (m, 3H, Ar), 6.41 (bs, 1H, NHCON), 4.47 (t, J = 5.7 Hz, 2H, OCH2), 3.63 (t, J = 5.7 Hz, 2H, OCH2CH2), 3.49 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.21 (dd, J = 9.3, 6.5 Hz, 2H, CH2CH2NHCON), 3.17–2.96 (m, 4H, CH2NH3+, ThiazCH2CH2), 2.74 (t, J = 7.4 Hz, 2H, TrizCH2), 2.69 (t, J = 7.2 Hz, 2H, ThiazCH2CH2), 1.72 (quin, J = 7.6 Hz, 2H, TrizCH2CH2), 1.58–1.42 (m, 2H, CH2CH2NH3+); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 159.4 (OCAr), 155.6 (CAr), 155.3 (CAr), 148.1 (CAr), 140.7 (CAr), 140.0 (CAr), 138.0 (CAr), 137.8 (CAr), 129.0 (CHAr), 128.9 (CHAr), 128.9 (CHAr), 127.2 (CHAr), 126.5 (CHAr), 126.5 (CHAr), 121.3 (CAr), 120.3 (CHAr), 116.0 (CHAr), 112.0 (CHAr), 104.3 (CHAr), 67.6 (OCH2), 45.4 (CH2CH2NHCON), 42.4 (OCH2CH2), 37.7 (CH2NH3+), 37.5 (CH2CH2NHCON), 34.3 (ThiazCH2CH2), 32.5 (ThiazCH2CH2), 30.1 (CH2CH2NH3+), 25.9 (TrizCH2CH2), 24.7 (TrizCH2); HPLC (gradient A, Agilent): Rt = 8.4 min; HRMS (ES, positive mode) m/z: calcd for C34H37N7O2S 607.2729; found 607.2740 (1.73 ppm); anal. calcd for C34H37N7O2S·TFA: C, 59.91; H, 5.31; N, 13.58; S, 4.44; found: C, 60.08; H, 5.09; N, 13.18; S, 4.20.
Phosphonium (2-Oxo-3-phenylpropyl)triphenyl Chloride (15)
A solution of 1-chloro-3-phenylpropan-2-one (250 mg, 1.48 mmol) and triphenylphosphine (583 mg, 2.22 mmol) in anhydrous toluene (8 mL) and under an argon atmosphere was heated to 110 °C for 6 h in a pressure tube. After cooling to room temperature, the white solid was filtered and washed with cooled diethyl ether (3 × 10 mL) to give 15 (498 mg, 78%) as a white solid;50 m.p.: 200–202 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.93–7.64 (m, 15H, Ar), 7.31–7.20 (m, 3H, Ar), 7.12 (d, J = 6.7 Hz, 2H, Ar), 5.91 (d, J = 12.7 Hz, 2H, Ar), 4.14 (s, 2H, CH2CO).
(E)-4-([1,1′-Biphenyl]-4-yl)-1-phenylbut-3-en-2-one (16c)
A stirred solution of 15 (250 mg, 0.58 mmol) and KOH (130 mg, 2.37 mmol) in dry toluene (15 mL) was heated at 110 °C in a pressure flask for 3 h. After cooling to room temperature, [1,1′-biphenyl]-4-carboxaldehyde (158 mg, 0.87 mmol) was added dropwise. The reaction mixture was stirred at 110 °C for 3 additional hours, cooled, and evaporated under reduced pressure. The residue was dissolved in EtOAc (30 mL), washed with H2O (2 × 30 mL) and brine (1 × 30 mL), dried (Na2SO4), filtered, and evaporated to dryness. The residue was purified by flash column chromatography (hexane/EtOAc, 95:5) to yield 16c (107 mg, 62%) as a white solid; m.p.: 121–126 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.59 (d, J = 16.1 Hz, 1H, CH=CHCO), 7.56–7.48 (m, 6H, Ar), 7.37 (t, J = 7.4 Hz, 2H, Ar), 7.34–7.25 (m, 3H, Ar), 7.24–7.15 (m, 3H, Ar), 6.74 (d, J = 16.0 Hz, 1H, CH=CHCO), 3.88 (s, 2H, CH2CO); 13C NMR (CDCl3, 75 MHz) δ (ppm): 197.4 (CO), 143.5 (ArCH=CH), 143.1 (CAr), 140.2 (CHAr), 134.6 (CAr), 133.5 (CAr), 129.6 (CHAr), 129.0 (CHAr), 129.0 (CHAr), 128.9 (CHAr), 128.1 (CAr), 127.7 (ArCH=CH), 127.2 (CHAr), 127.1 (CHAr), 125.1 (CHAr), 48.6 (CH2CO); MS (ESI, positive mode) m/z: 321.0 [M + Na]+, 299.3 [M + H]+.
(E)-4-(4-Phenyloxyphenyl)-1-phenylbut-3-en-2-one (16d)
Following a procedure similar to that described for 16c, a solution of phosphonium salt 15 (700 mg, 1.62 mmol), KOH (728 mg, 13 mmol), and 4-phenoxybenzaldehyde (0.34 mL, 1.95 mmol) in dry toluene (20 mL) was reacted in a pressure flask. After the workup, the residue was purified by flash column chromatography (hexane/EtOAc, 95:5) to give 241 mg (47%) of 16d as a white solid; m.p.: 95–97 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.51 (d, J = 16.0 Hz, 1H, CH=CHCO), 7.47–7.33 (m, 2H, Ar), 7.30–7.21 (m, 4H, Ar), 7.19–7.13 (m, 3H, Ar), 7.06 (tt, J = 7.4, 1.1 Hz, 1H, Ar), 6.97–6.90 (m, 2H, Ar), 6.89–6.83 (m, 2H, Ar), 6.60 (d, J = 16.0 Hz, 1H, CH=CHCO), 3.83 (s, 2H, CH2CO); 13C NMR (CDCl3, 75 MHz) δ (ppm): 197.3 (CO), 159.9 (OCAr), 156.1 (OCAr), 142.8 (ArCH=CH), 134.7 (CAr), 130.2 (CHAr), 130.0 (CHAr), 129.6 (CHAr), 129.2 (CAr), 128.9 (CHAr), 127.1 (ArCH=CH), 124.3 (CHAr), 124.0 (CHAr), 119.8 (CHAr), 118.5 (CHAr), 48.5 (CH2CO); MS (ESI, positive mode) m/z: 337.0 [M + Na]+, 315.0 [M + H]+.
General Procedure for the Synthesis of α-Bromomethylketones 17a–d
To a solution of the corresponding ketone (1 equiv) in CH3CN, p-TsOH (1.3–2.6 equiv) and NBS (1.3–2.6 equiv) were successively added at room temperature and the mixture was stirred for 6 h/overnight. After quenching with H2O (30 mL), the reaction mixture was carefully concentrated under reduced pressure without heating. The aqueous crude was extracted with EtOAc (3 × 30 mL), and the organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was purified by flash column chromatography or CCTLC on the Chromatotron (the eluents are specified in each case) to give the desired α-bromoketones 17a–d.
2-([1,1′-Biphenyl]-4-yl)-1-bromo-1-phenylethan-2-one (17a)
Following the general bromination procedure, p-TsOH (173 mg, 0.92 mmol) and NBS (165 mg, 0.92 mmol) were successively added to a solution of the commercially available 16a (50 mg, 0.18 mmol) in CH3CN (15 mL). The reaction was stirred at room temperature overnight. After the workup, the residue was purified by CCTLC on the Chromatotron (hexane/EtOAc, 80:20) to give 62 mg (98%) of 17a as a white solid; m.p.: decompose without melting; 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.08 (d, J = 8.2 Hz, 2H, Ar), 7.67 (d, J = 8.4 Hz, 2H, Ar), 7.63–7.54 (m, 4H, Ar), 7.47 (t, J = 7.5 Hz, 2H, Ar), 7.43–7.29 (m, 4H, Ar), 6.43 (s, 1H, BrCHCO).
2-(4-Phenyloxyphenyl)-1-bromo-1-phenylethan-2-one (17b)
The general bromination procedure was followed with the commercially available 16b (150 mg, 0.52 mmol), p-TsOH (297 mg, 1.56 mmol), and NBS (278 mg, 1.56 mmol) in CH3CN (22 mL) at room temperature overnight. The residue was purified by CCTLC on the Chromatotron (hexane/CH2Cl2, 70:30) to afford 17b (140 mg, 73%) as a white solid; m.p.: 116-118 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.97 (d, J = 8.9 Hz, 2H, Ar), 7.53 (dd, J = 8.1, 1.6 Hz, 2H, Ar), 7.44–7.29 (m, 5H, Ar), 7.22 (tt, J = 7.3, 1.0 Hz, 1H, Ar), 7.06 (dd, J = 7.9, 1.6 Hz, 2H, Ar), 6.96 (d, J = 8.9 Hz, 2H, Ar), 6.34 (s, 1H, BrCHCO); 13C NMR (CDCl3, 75 MHz) δ (ppm): 189.7 (CO), 162.7 (OCAr), 155.1 (OCAr), 136.2 (CAr), 131.7 (CHAr), 130.3 (CHAr), 129.2 (CHAr), 129.2 (CHAr), 128.5 (CAr), 125.1 (CHAr), 120.6 (CHAr), 117.4 (CHAr), 51.2 (BrCHCO); MS (ESI, positive mode) m/z: 389 [M + Na]+ with a Br isotopic pattern.
(E)-4-([1,1′-Biphenyl]-4-yl)-1-bromo-1-phenylbut-3-en-2-one (17c)
Following the general procedure, compound 16c (200 mg, 0.67 mmol) dissolved in CH3CN (25 mL) was sequentially treated with p-TsOH (191 mg, 1.00 mmol) and NBS (179 mg, 1.00 mmol) at room temperature for 4 h. After the workup, the residue was purified by flash column chromatography (hexane/EtOAc, 90:10) to give 17c (253 mg, 79%) as a white solid; m.p.: 118–120 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.71 (d, J = 15.8 Hz, 1H, CH=CHCO), 7.56–7.49 (m, 6H, Ar), 7.47–7.42 (m, 2H, Ar), 7.41–7.34 (m, 2H, Ar), 7.35–7.24 (m, 4H, Ar), 6.94 (d, J = 15.8 Hz, 1H, CH=CHCO), 5.61 (s, 1H, BrCHCO); 13C NMR (CDCl3, 75 MHz) δ (ppm): 190.4 (CO), 145.0 (ArCH=CH), 143.9 (CAr), 140.1 (CHAr), 135.4 (CAr), 133.2 (CAr), 129.3 (CHAr), 129.3 (CHAr), 129.2 (CHAr), 129.1 (CHAr), 128.1 (CAr), 127.7 (ArCH=CH), 127.2 (CHAr), 127.1 (CHAr), 121.4 (CHAr), 55.5 (BrCHCO).
(E)-4-(4-Phenyloxyphenyl)-1-phenylbut-3-en-2-one (17d)
According to the general bromination procedure, the α,β-unsaturated compound 16d (211 mg, 0.67 mmol), p-TsOH (140 mg, 0.74 mmol), and NBS (131 mg, 0.74 mmol) dissolved in CH3CN (25 mL) were reacted at room temperature for 4 h. After the workup, the crude was purified by flash column chromatography (hexane/EtOAc, 97:3) to give 17d (150 mg, 39%) as a yellow oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.72 (d, J = 15.7 Hz, 1H, CH=CHCO), 7.56–7.47 (m, 4H, Ar), 7.43–7.32 (m, 5H, Ar), 7.18 (tt, J = 7.4, 1.1 Hz, 1H, Ar), 7.10–7.01 (m, 2H, Ar), 6.97 (d, J = 8.8 Hz, 2H, Ar), 6.89 (d, J = 15.7 Hz, 1H, CH=CHCO), 5.66 (s, 1H, BrCHCO); 13C NMR (CDCl3, 75 MHz) δ (ppm): 190.4 (CO), 160.4 (OCAr), 156.0 (OCAr), 144.8 (ArCH=CH), 135.5 (CAr), 130.6 (CHAr), 130.1 (CHAr), 129.2 (CHAr), 129.1 (CAr), 129.1 (CHAr), 128.9 (ArCH=CH), 124.5 (CHAr), 120.2 (CHAr), 120.0 (CHAr), 118.5 (CHAr), 55.5 (BrCHCO); MS (ESI, positive mode) m/z: 417.0 [M + Na]+, 393.0 [M + H]+ with a Br isotopic pattern.
Benzyl-(4-(1-(4-(4,5-diphenylthiazol-2-yl)-3-(2-(2-oxoimidazolidin-1-yl)ethoxy)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (18a)
Following the general procedure of Hantzsch synthesis, the thioamide 9 (150 mg, 0.28 mmol) and the commercially available 1-bromo-1,2-diphenylethan-2-one 17e (77 mg, 0.28 mmol) reacted in iPrOH (20 mL). After the workup, the final residue was purified by CCTLC in the Chromatotron (CH2Cl2/MeOH, 97:3) to provide 191 mg (94%) of 18a as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.62 (d, J = 8.6 Hz, 1H, Ar), 7.89 (s, 1H, Ar), 7.64–7.57 (m, 3H, Ar), 7.44–7.23 (m, 14H, Ar), 5.19 (t, J = 6.0 Hz, 1H, NHCbz), 5.08 (s, 2H, NHCOOCH2), 4.87 (bs, 1H, NHCON), 4.44 (t, J = 5.9 Hz, 2H, OCH2), 3.75 (t, J = 5.8 Hz, 2H, OCH2CH2), 3.57 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.29 (t, J = 8.0 Hz, 2H, CH2CH2NHCON), 3.25 (q, J = 6.9 Hz, 2H, CH2NHCbz), 2.82 (t, J = 7.4 Hz, 2H, TrizCH2), 1.84–1.71 (m, 2H, TrizCH2CH2), 1.62 (quin, J = 6.2 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.2 (NHCON), 158.3 (OCAr), 156.6 (CAr), 155.9 (CAr), 149.3 (CAr), 148.7 (CAr), 138.4 (CAr), 136.7 (CAr), 135.1 (CAr), 134.3 (CAr), 132.2 (CHAr), 129.7 (CHAr), 129.6 (CHAr), 129.2 (CHAr), 129.1 (CHAr), 128.8 (CHAr), 128.6 (CHAr), 128.4 (CHAr), 128.2 (CHAr), 127.9 (CAr), 122.5 (CHAr), 119.1 (CHAr), 112.4 (CHAr), 104.4 (CHAr), 67.7 (OCH2), 66.7 (NHCOOCH2), 46.4 (CH2CH2NHCON), 42.9 (OCH2CH2), 40.8 (CH2NHCbz), 38.4 (CH2CH2NHCON), 29.4 (CH2CH2NHCbz), 26.3 (TrizCH2CH2), 25.2 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C40H39N7O4S 713.2784; found 713.2786 (0.23 ppm).
Benzyl-(4-(1-(3-(2-(2-oxoimidazolidin-1-yl)ethoxy)-4-(4-([1,1′-biphenyl]-4-yl)-5-phenylthiazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (18b)
Following the general procedure for the synthesis of thiazoles by Hantzsch cyclization, thioamide 9 (194 mg, 0.36 mmol) reacted with α-bromoketone 17a (127 mg, 0.36 mmol) in iPrOH (20 mL). After the workup, the residue was purified by CCTLC on the Chromatotron (CH2Cl2/MeOH, 95:5) to give 18b (244 mg, 84%) as a colorless oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.66 (d, J = 8.6 Hz, 1H, Ar), 7.60 (s, 1H, Ar), 7.71 (d, J = 8.1 Hz, 2H, Ar), 7.65–7.51 (m, 3H, Ar), 7.56 (d, J = 8.1 Hz, 2H, Ar), 7.52–7.18 (m, 14H, Ar), 5.13 (bs, 1H, NHCON), 5.10 (s, 2H, NCOOCH2), 4.67 (bs, 1H, NHCbz), 4.47 (t, J = 5.9 Hz, 2H, OCH2), 3.78 (t, J = 5.9 Hz, 2H, OCH2CH2), 3.59 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.37–3.22 (m, 4H, CH2CH2NHCON, CH2NHCbz), 2.84 (t, J = 7.3 Hz, 2H, TrizCH2), 1.86–1.74 (m, 2H, TrizCH2CH2), 1.64 (quin, J = 7.2 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.7 (NHCON), 158.4 (OCAr), 156.6 (CAr), 156.0 (NHCOO), 149.0 (CAr), 148.8 (CAr), 140.8 (CAr), 140.5 (CAr), 138.5 (CAr), 136.7 (CAr), 134.4 (CAr), 134.1 (CHAr), 132.3 (CAr), 129.8 (CHAr), 129.5 (CHAr), 128.9 (CHAr), 128.9 (CHAr), 128.6 (CHAr), 128.3 (CHAr), 128.2 (CHAr), 127.5 (CAr), 127.1 (CHAr), 122.5 (CHAr), 119.2 (CAr), 112.5 (CHAr), 104.5 (CHAr), 67.7 (NHCOOCH2), 66.8 (OCH2), 46.4 (CH2CH2NHCON), 42.9 (OCH2CH2), 40.9 (CH2NHCbz), 38.4 (CH2CH2NHCON), 29.4 (CH2CH2NHCbz), 26.4 (TrizCH2CH2), 25.2 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C46H43N7O4S 789.3097; found 789.3063 (−4.31 ppm).
Benzyl-(4-(1-(3-(2-(2-oxoimidazolidin-1-yl)ethoxy)-4-(4-(4-phenyloxyphenyl-1-yl)-5-phenylthiazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (18c)
Following the general procedure for the synthesis of thiazoles by Hantzsch cyclization, thioamide 9 (190 mg, 0.35 mmol) and α-bromoketone 17b (130 mg, 0.35 mmol) reacted in iPrOH (20 mL). The final residue was purified by CCTLC in the Chromatotron (CH2Cl2/MeOH, 95:5) to yield 235 mg (81%) of 18c a colorless oil. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.63 (d, J = 8.5 Hz, 1H, Ar), 7.90 (s, 1H, Ar), 7.65–7.55 (m, 3H, Ar), 7.47–7.24 (m, 13H, Ar), 7.11 (t, J = 7.4 Hz, 1H, Ar), 7.04 (d, J = 8.0 Hz, 2H, Ar), 6.95 (d, J = 8.3 Hz, 2H, Ar), 5.14 (bs, 1H, NHCON), 5.09 (s, 2H, NCOOCH2), 4.73 (bs, 1H, NHCbz), 4.46 (t, J = 5.9 Hz, 2H, OCH2), 3.77 (t, J = 5.9 Hz, 2H, OCH2CH2), 3.59 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.37–3.21 (m, 4H, CH2CH2NHCON, CH2NHCbz), 2.84 (t, J = 7.3 Hz, 2H, TrizCH2), 1.84–1.74 (m, 2H, TrizCH2CH2), 1.63 (quin, J = 7.2 Hz, 2H, CH2CH2NHCbz); 13C NMR (CDCl3, 75 MHz) δ (ppm): 162.7 (NHCON), 158.3 (OCAr), 157.2 (CAr), 157.0 (OCAr), 156.6 (OCAr), 156.0 (NHCOO), 148.8 (CAr), 138.5 (CAr), 136.7 (CAr), 133.7 (CAr), 132.3 (CAr), 130.7 (CHAr), 130.1 (CAr), 129.9 (CHAr), 129.7 (CHAr), 129.7 (CHAr), 128.9 (CHAr), 128.6 (CHAr), 128.2 (CAr), 128.2 (CHAr), 123.6 (CHAr), 122.5 (CAr), 119.3 (CHAr), 119.1 (CAr), 118.6 (CHAr), 112.5 (CHAr), 104.5 (CHAr), 67.7 (NHCOOCH2), 66.8 (OCH2), 46.4 (CH2CH2NHCON), 42.9 (OCH2CH2), 40.9 (CH2NHCbz), 38.4 (CH2CH2NHCON), 29.4 (CH2CH2NHCbz), 26.3 (TrizCH2CH2), 25.2 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C46H43N7O5S 805.3046; found 805.3054 (0.92 ppm).
Benzyl-(E)-(4-(1-(4-(4-(2-([1,1′-biphenyl]-4-yl)vinyl)-5-phenylthiazol-2-yl)-3-(2-(2-oxoimidazolidin-1-yl)ethoxy) phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (18d)
Following the general Hantzsch cyclization procedure, thioamide 9 (200 mg, 0.37 mmol) and α-bromoketone 17c (140 mg, 0.37 mmol) reacted in iPrOH (20 mL). After the workup, the residue was purified by CCTLC on the Chromatotron (CH2Cl2/MeOH, 95:5) to give 18d (183 mg, 59%) as a yellow oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.72 (s, 1H, Ar), 8.61 (d, J = 8.4 Hz, 1H, Ar), 7.80–7.71 (m, 3H, Ar, ThiazCH=CH), 7.70–7.53 (m, 10H, Ar), 7.51–7.42 (m, 3H, Ar), 7.39–7.27 (m, 3H, Ar, NHCbz), 7.24 (d, J = 15.7 Hz, 1H, ThiazCH=CH), 6.42 (bs, 1H, NHCON), 5.02 (s, 2H, NHCOOCH2), 4.48 (t, J = 5.7 Hz, 2H, OCH2), 3.66 (t, J = 5.6 Hz, 2H, OCH2CH2), 3.49 (dd, J = 9.0, 6.6 Hz, 2H, CH2CH2NHCON), 3.23 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.08 (q, J = 6.6 Hz, 2H, CH2NHCbz), 2.72 (t, J = 7.1 Hz, 2H, TrizCH2), 1.70 (quin, J = 7.5 Hz, 2H, TrizCH2CH2), 1.52 (quin, J = 7.2 Hz, 2H, CH2CH2NHCbz); 13C NMR (DMSO-d6, 75 MHz) δ (ppm): 161.8 (NHCON), 158.0 (OCAr), 155.8 (NHCOO), 147.9 (CAr), 146.6 (CAr), 139.4 (CAr), 139.3 (CAr), 138.3 (CAr), 137.1 (CAr), 135.8 (CAr), 135.0 (CAr), 131.6 (CAr), 130.9 (CHAr), 129.9 (CHAr), 128.9 (CHAr), 128.6 (CHAr), 128.0 (CAr), 128.0 (CHAr), 127.4 (CHAr), 127.3 (CAr), 127.2 (CHAr), 126.9 (CHAr), 126.7 (CAr), 126.2 (CHAr), 120.9 (CHAr), 119.9 (CHAr), 119.5 (CAr), 112.0 (CHAr), 104.3 (CHAr), 67.3 (NHCOOCH2), 64.9 (OCH2), 45.0 (CH2CH2NHCON), 42.3 (OCH2CH2), 39.9 (CH2NHCbz), 37.3 (CH2CH2NHCON), 28.7 (CH2CH2NHCbz), 25.8 (TrizCH2CH2), 24.5 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C48H45N7O4S 815.3254; found 815.3266 (1.56 ppm).
Benzyl-(E)-(4-(1-(4-(4-(2-([4-phenyloxyphenyl]-1-yl)vinyl)-5-phenylthiazol-2-yl)-3-(2-(2-oxoimidazolidin-1-yl)ethoxy)phenyl)-1H-1,2,3-triazol-4-yl)butyl)carbamate (18e)
According to the general Hantzsch procedure, thioamide 9 (183 mg, 0.34 mmol) was reacted with α-bromoketone 17d (134 mg, 0.34 mmol) in iPrOH (23 mL). After purification of the residue by CCTLC on the Chromatotron (CH2Cl2/MeOH, 96:4), 18e (150 mg, 52%) was obtained as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.74 (s, 1H, Ar), 8.61 (d, J = 8.5 Hz, 1H, Ar), 7.85–7.67 (m, 3H, Ar, ThiazCH=CH), 7.64–7.51 (m, 5H, Ar), 7.51–7.24 (m, 9H, Ar), 7.20–7.10 (m, 2H, Ar, ThiazCH=CH), 7.05 (d, J = 7.7 Hz, 2H, Ar), 7.00 (d, J = 8.6 Hz, 2H, Ar), 6.40 (bs, 1H, NHCON), 5.01 (s, 2H, NHCOOCH2), 4.49 (t, J = 5.7 Hz, 2H, OCH2), 3.66 (t, J = 5.6 Hz, 2H, OCH2CH2), 3.49 (dd, J = 9.0, 6.6 Hz, 2H, CH2CH2NHCON), 3.22 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.07 (q, J = 6.6 Hz, 2H, CH2NHCbz), 2.74 (t, J = 7.5 Hz, 2H, TrizCH2), 1.69 (quin, J = 7.5 Hz, 2H, TrizCH2CH2), 1.53 (quin, J = 7.2 Hz, 2H, CH2CH2NHCbz); 13C NMR (DMSO-d6, 75 MHz) δ (ppm): 162.1 (NHCON), 158.1 (CAr), 156.6 (OCAr), 156.3 (OCAr), 156.1 (OCAr), 156.0 (NHCOO), 148.1 (CAr), 146.7 (OCAr), 138.4 (CAr), 137.3 (CAr), 134.8 (CAr), 132.0 (CAr), 131.6 (CAr), 131.1 (CAr), 130.1 (CAr), 129.4 (CHAr), 129.2 (CHAr), 128.4 (CHAr), 128.3 (CHAr), 127.7 (CHAr), 123.7 (CHAr), 120.9 (CHAr), 120.3 (CAr), 118.9 (CHAr), 118.7 (CHAr), 112.0 (CHAr), 104.3 (CHAr), 67.4 (NHCOOCH2), 65.1 (OCH2), 45.1 (CH2CH2NHCON), 42.4 (OCH2CH2), 40.0 (CH2NHCbz), 37.5 (CH2CH2NHCON), 28.9 (CH2CH2NHCbz), 26.0 (TrizCH2CH2), 24.7 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C48H45N7O5S 831.3203; found 831.3174 (−3.45 ppm).
1-(2-(5-(4-(4-Ammoniobutyl)-1H-1,2,3-triazol-1-yl)-2-(4,5-diphenylthiazol-2-yl)phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (19a)
Following the general Cbz removal procedure, 18a (100 mg, 0.14 mmol), Pd/C 10% (20 mg) and TFA (1 mL) afforded, after purification, compound 19a (25 mg, 26%) as a colorless oil. 1H NMR (CD3OD, 400 MHz) δ (ppm): 8.56 (d, J = 8.6 Hz, 1H, Ar), 8.46 (s, 1H, Ar), 7.71 (d, J = 8.0 Hz, 1H, Ar), 7.59 (dd, J = 8.6, 2.0 Hz, 1H, Ar), 7.56–7.51 (m, 2H, Ar), 7.44–7.27 (m, 8H, Ar), 4.51 (t, J = 5.5 Hz, 2H, OCH2), 3.76 (t, J = 5.5 Hz, 2H, OCH2CH2), 3.62 (dd, J = 9.3, 6.9 Hz, 2H, CH2CH2NHCON), 3.32 (dd, J = 9.3, 6.9 Hz, 2H, CH2CH2NHCON), 3.00 (q, J = 6.5 Hz, 2H, CH2NH3+), 2.86 (t, J = 7.2 Hz, 2H, TrizCH2), 1.95–1.65 (m, 4H, TrizCH2CH2, CH2CH2NH3+); 13C NMR (CD3OD, 75 MHz) δ (ppm): 165.0 (NHCON), 159.9 (OCAr), 157.5 (CAr), 150.6 (CAr), 149.3 (CAr), 139.8 (CAr), 136.4 (CAr), 135.8 (CAr), 133.3 (CHAr), 130.6 (CHAr), 130.5 (CHAr), 130.3 (CHAr), 129.9 (CHAr), 129.3 (CHAr), 129.3 (CHAr), 129.0 (CHAr), 129.0 (CHAr), 123.7 (CHAr), 121.6 (CHAr), 113.6 (CHAr), 105.6 (CHAr), 68.2 (OCH2), 46.9 (CH2CH2NHCON), 43.9 (OCH2CH2), 40.4 (CH2NH3+), 39.3 (CH2CH2NHCON), 28.0 (CH2CH2NH3+), 27.1 (TrizCH2CH2), 25.6 (TrizCH2); HPLC (gradient A, Agilent): Rt = 9.3 min; HRMS (ES, positive mode) m/z: calcd for C32H33N7O2S 579.2416; found 579.242 (0.61 ppm); anal. calcd for C32H34N7O2S·TFA: C, 58.87; H, 4.94; N, 14.13; S, 4.62; found: C, 59.15; H, 5.36 N, 14.62; S, 5.03.
1-(2-(5-(4-(4-Ammoniobutyl)-1H-1,2,3-triazol-1-yl)-2-(4-([1,1′-biphenyl]-4-yl)-5-phenylthiazol-2-yl)phenoxy) ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (19b)
The general N-deprotection procedure described for 12a–c was followed with 18b (100 mg, 0.13 mmol), Pd/C 10% (20 mg), and TFA (0.8 mL) to give, after workup and purification, compound 19b (15 mg, 15%) as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.79 (s, 1H, Ar), 8.57 (d, J = 8.6 Hz, 1H, Ar), 7.80 (d, J = 8.1 Hz, 1H, Ar), 7.77–7.60 (m, 10H, Ar, NH3+), 7.54–7.41 (m, 7H, Ar), 7.37 (t, J = 7.5 Hz, 1H, Ar), 6.40 (bs, 1H, NHCON), 4.53 (t, J = 5.6 Hz, 2H, OCH2), 3.66 (t, J = 5.6 Hz, 2H, OCH2CH2), 3.51 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.23 (t, J = 7.8 Hz, 2H, CH2CH2NHCON), 2.85 (t, J = 7.5 Hz, 2H, CH2NH3+), 2.78 (t, J = 7.2 Hz, 2H, TrizCH2), 1.76 (quin, J = 7.2 Hz, 2H, TrizCH2CH2), 1.63 (quin, J = 7.8 Hz, 2H, CH2CH2NH3+); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 157.9 (OCAr), 155.8 (CAr), 148.0 (CAr), 147.7 (CAr), 139.4 (CAr), 139.4 (CAr), 138.3 (CAr), 133.9 (CAr), 133.7 (CAr), 131.5 (CHAr), 129.3 (CHAr), 129.2 (CHAr), 129.1 (CHAr), 129.0 (CHAr), 128.8 (CHAr), 128.5 (CHAr), 127.6 (CHAr), 126.5 (CHAr), 126.5 (CHAr), 121.1 (CHAr), 120.4 (CAr), 112.1 (CHAr), 104.3 (CHAr), 67.4 (OCH2), 45.1 (CH2CH2NHCON), 42.4 (OCH2CH2), 38.6 (CH2NH3+), 37.5 (CH2CH2NHCON), 26.5 (CH2CH2NH3+), 25.5 (TrizCH2CH2), 24.4 (TrizCH2); HPLC (gradient A, Agilent): Rt = 9.3 min; HRMS (ES, positive mode) m/z: calcd for C38H37N7O2S 655.2729; found 655.2748 (2.87 ppm); anal. calcd for C38H38N7O2S·TFA: C, 62.41; H, 4.98; N, 12.74; S, 4.16; found: C, 62.36; H, 5.06; N, 12.37; S, 4.25.
1-(2-(5-(4-(4-Ammoniobutyl)-1H-1,2,3-triazol-1-yl)-2-(4-([4-phenyloxyphenyl]-1-yl)-5-phenylthiazol-2-yl) phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (19c)
Following the general Cbz deprotection procedure, 18c (120 mg, 0.15 mmol), Pd/C 10% (24 mg), and TFA (0.9 mL) were reacted. Workup and purification yielded 19c (44 mg, 37%) as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.78 (s, 1H, Ar), 8.53 (d, J = 8.6 Hz, 1H, Ar), 7.79 (d, J = 2.1 Hz, 1H, Ar), 7.83–7.72 (m, 3H, NH3+), 7.71 (dd, J = 8.6, 2.0 Hz, 1H, Ar), 7.56 (d, J = 8.7 Hz, 2H, Ar), 7.50–7.32 (m, 7H, Ar), 7.17 (t, J = 7.4 Hz, 1H, Ar), 7.06 (d, J = 7.9 Hz, 2H, Ar), 6.98 (d, J = 8.7 Hz, 2H, Ar), 6.39 (bs, 1H, NHCON), 4.51 (t, J = 5.6 Hz, 2H, OCH2), 3.65 (t, J = 5.6 Hz, 2H, OCH2CH2), 3.50 (dd, J = 9.0, 6.7 Hz, 2H, CH2CH2NHCON), 3.22 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 2.91–2.80 (m, 2H, CH2NH3+), 2.77 (t, J = 7.2 Hz, 2H, TrizCH2), 1.76 (quin, J = 7.2 Hz, 2H, TrizCH2CH2), 1.64 (quin, J = 8.1 Hz, 2H, CH2CH2NH3+); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 158.1 (CF3COO–), 157.8 (CAr), 156.5 (OCAr), 156.2 (OCAr), 155.5 (OCAr), 147.9 (CAr), 147.7 (CAr), 138.3 (CAr), 133.3 (CAr), 131.5 (CAr), 130.4 (CHAr), 130.1 (CHAr), 129.8 (CHAr), 129.2 (CHAr), 129.0 (CHAr), 128.8 (CHAr), 128.3 (CAr), 123.8 (CHAr), 121.1 (CAr), 120.4 (CHAr), 119.0 (CHAr), 118.1 (CHAr), 115.8 (CF3COO–), 112.1 (CHAr), 104.3 (CHAr), 67.3 (OCH2), 45.1 (CH2CH2NHCON), 42.4 (OCH2CH2), 38.6 (CH2NH3+), 37.5 (CH2CH2NHCON), 26.5 (CH2CH2NH3+), 25.5 (TrizCH2CH2), 24.4 (TrizCH2); HPLC (gradient A, Agilent): Rt = 9.8 min; HRMS (ES, positive mode) m/z: calcd for C38H37N7O3S 671.2679; found 671.2673 (−0.81 ppm); anal. calcd for C38H38N7O3S·TFA: C, 61.14; H, 4.87; N, 12.48; S, 4.08; found: C, 61.55; H, 4.99; N, 12.12; S, 4.34.
1-(2-(2-(4-(2-([1,1′-Biphenyl]-4-yl)ethyl)-5-phenyl-thiazol-2-yl)-5-(4-(4-ammoniobutyl)-1H-1,2,3-triazol-1-yl)phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (19d)
Following the general hydrogenation procedure, a solution of 18d (91 mg, 0.11 mmol), Pd/C 10% (18 mg), and TFA (0.7 mL) was reacted to give, after purification, 19d (14 mg, 16%) as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.78 (s, 1H, Ar), 8.54 (d, J = 8.6 Hz, 1H, Ar), 7.78 (d, J = 2.1 Hz, 1H, Ar), 7.72 (dd, J = 8.5, 2.0 Hz, 1H, Ar), 7.73–7.65 (m, 3H, NH3+), 7.62 (d, J = 7.2 Hz, 2H, Ar), 7.54 (d, J = 8.2 Hz, 2H, Ar), 7.49–7.38 (m, 7H, Ar), 7.33 (tt, J = 7.4, 1.3 Hz, 1H, Ar), 7.26 (d, J = 8.2 Hz, 2H, Ar), 6.39 (bs, 1H, NHCON), 4.50 (t, J = 5.6 Hz, 2H, OCH2), 3.65 (t, J = 5.6 Hz, 2H, OCH2CH2), 3.49 (dd, J = 8.9, 6.7 Hz, 2H, CH2CH2NHCON), 3.21 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.16–3.12 (m, 4H, ThiazCH2CH2), 2.85 (t, J = 7.5 Hz, 2H, CH2NH3+), 2.78 (t, J = 7.2 Hz, 2H, TrizCH2), 1.76 (quin, J = 7.6 Hz, 2H, TrizCH2CH2), 1.63 (quin, J = 7.5 Hz, 2H, CH2CH2NH3+); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 157.4 (CAr), 155.6 (OCAr), 150.1 (CAr), 147.7 (CAr), 140.7 (CAr), 140.1 (CAr), 138.1 (CAr), 137.9 (CAr), 133.4 (CAr), 131.4 (CAr), 129.0 (CHAr), 128.9 (CHAr), 128.9 (CHAr), 128.9 (CHAr), 128.8 (CHAr), 128.0 (CHAr), 127.2 (CHAr), 126.6 (CHAr), 126.5 (CHAr), 121.3 (CAr), 120.4 (CHAr), 112.1 (CHAr), 104.3 (CHAr), 67.4 (OCH2), 45.2 (CH2CH2NHCON), 42.4 (OCH2CH2), 38.7 (CH2NH3+), 37.5 (CH2CH2NHCON), 34.6 (ThiazCH2CH2), 31.2 (ThiazCH2CH2), 26.5 (CH2CH2NH3+), 25.6 (TrizCH2CH2), 24.4 (TrizCH2); HPLC (gradient A, Agilent): Rt = 9.8 min; HRMS (ES, positive mode) m/z: calcd for C40H41N7O2S 683.3042; found 683.3050 (1.06 ppm); anal. calcd for C40H42N7O2S·TFA: C, 63.22; H, 5.31; N, 12.29; S, 4.02; found: C, 63.40; H, 5.64; N, 12.50; S, 3.93.
1-(2-(2-(4-(2-([4-Phenyloxyphenyl]-1-yl)ethyl)-5-phenyl-thiazol-2-yl)-5-(4-(4-ammoniobutyl)-1H-1,2,3-triazol-1-yl)phenoxy)ethyl)imidazolidin-2-one 2,2,2-Trifluoroacetate (19e)
Following the general Cbz deprotection procedure, reaction of 18e (80 mg, 0.10 mmol), Pd/C 10% (16 mg), and TFA (0.6 mL) afforded 19e (20 mg, 26%) as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.78 (s, 1H, Ar), 8.56–8.50 (m, 1H, Ar), 7.77 (d, J = 2.0 Hz, 1H, Ar), 7.81–7.64 (m, 4H, Ar, NH3+), 7.62 (d, J = 7.2 Hz, 1H, Ar), 7.54 (d, J = 8.1 Hz, 1H, Ar), 7.50–7.31 (m, 7H, Ar), 7.26 (d, J = 7.9 Hz, 1H, Ar), 7.16 (d, J = 8.5 Hz, 1H, Ar), 7.10 (t, J = 7.4 Hz, 1H, Ar), 6.94 (d, J = 8.0 Hz, 1H, Ar), 6.90 (d, J = 8.5 Hz, 1H, Ar), 6.39 (bs, 1H, NHCON), 4.49 (t, J = 5.7 Hz, 2H, OCH2), 3.64 (t, J = 5.7 Hz, 2H, OCH2CH2), 3.48 (dd, J = 9.0, 6.6 Hz, 2H, CH2CH2NHCON), 3.21 (t, J = 7.9 Hz, 2H, CH2CH2NHCON), 3.15–3.06 (m, 4H, ThiazCH2CH2), 2.85 (t, J = 7.5 Hz, 2H, CH2NH3+), 2.78 (t, J = 7.2 Hz, 2H, TrizCH2), 1.75 (quin, J = 7.4 Hz, 2H, TrizCH2CH2), 1.64 (quin, J = 7.0 Hz, 2H, CH2CH2NH3+); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 157.3 (CAr), 157.1 (CAr), 155.6 (OCAr), 154.6 (CAr), 150.0 (CAr), 147.7 (CAr), 140.7 (CAr), 138.1 (CAr), 137.9 (CAr), 136.6 (CAr), 133.4 (CAr), 131.4 (CHAr), 129.9 (CHAr), 129.8 (CHAr), 129.0 (CHAr), 129.0 (CHAr), 128.9 (CHAr), 128.9 (CHAr), 128.9 (CHAr), 128.0 (CHAr), 126.6 (CHAr), 126.5 (CHAr), 123.1 (CHAr), 121.3 (CAr), 118.8 (CHAr), 118.1 (CHAr), 112.1 (CHAr), 104.3 (CHAr), 67.4 (OCH2), 45.2 (CH2CH2NHCON), 42.4 (OCH2CH2), 38.7 (CH2NH3+), 37.5 (CH2CH2NHCON), 34.6 (CH2CH2PhOPh), 31.2 (CH2CH2PhOPh), 26.5 (CH2CH2NH3+), 25.6 (TrizCH2CH2), 24.4 (TrizCH2); HPLC (gradient A, Agilent): Rt = 9.7 min; HRMS (ES, positive mode) m/z: calcd for C40H41N7O3S 699.2992; found 699.2993 (0.17 ppm); anal. calcd for C40H42N7O3S·TFA: C, 61.98; H, 5.20; N, 12.05; S, 3.94; found: C, 62.23; H, 5.34; N, 11.68; S, 3.50.
1-(3-(2-Bromo)acetylphenyl)-2-bromoethan-1-one (20)
Following the general procedure described for the synthesis α-bromomethylketones 17a–e, p-toluensulfonic acid (761 mg, 4.00 mmol) and NBS (713 mg, 4.00 mmol) were successively added to a solution of commercially available diacetyl benzene (250 mg, 1.54 mmol) in CH3CN (20 mL) and stirred at room temperature overnight. After the workup, the residue was purified by flash column chromatography (hexane/EtOAc, 90:10) to give 20 (366 mg, 74%) as a white solid.51 m.p.: 87–88 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.55 (t, J = 1.8 Hz, 1H, Ar), 7.20 (dd, J = 7.8, 1.8 Hz, 2H, Ar), 7.64 (t, J = 7.8 Hz, 1H, Ar), 4.78 (s, 4H, BrCH2CO).
Dibenzyl-((((1,3-phenylenebis(thiazole-4,2-diyl))bis(3-(2-(2-oxoimidazolidin-1-yl)ethoxy)-4,1-phenylene))bis(1H-1,2,3-triazole-1,4-diyl))bis(butane-4,1-diyl))dicarbamate (21)
Following the general procedure of Hantzsch synthesis, 9 (100 mg, 0.19 mmol) was reacted with α-bromoketone 20 (30 mg, 0.09 mmol) in iPrOH (15 mL). After the workup, the residue was purified by CCTLC on the Chromatotron (CH2Cl2/MeOH, 96:4) to give 21 (68 mg, 30%) as a pink oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.75 (s, 2H, Ar), 8.68 (d, J = 8.5 Hz, 2H, Ar), 8.37 (s, 2H, Ar), 8.12 (dd, J = 7.6, 1.7 Hz, 2H, Ar), 7.79 (d, J = 2.0 Hz, 2H, Ar), 7.75 (dd, J = 8.7, 1.8 Hz, 2H, Ar), 7.60 (t, J = 7.7 Hz, 1H, Ar), 7.46–7.10 (m, 12H, Ar, NHCbz), 6.42 (s, 2H, NHCON), 5.01 (s, 4H, NHCOOCH2), 4.52 (t, J = 5.6 Hz, 4H, OCH2), 3.69 (t, J = 5.6 Hz, 4H, OCH2CH2), 3.53 (dd, J = 9.0, 6.7 Hz, 4H, CH2CH2NHCON), 3.25 (t, J = 7.9 Hz, 4H, CH2CH2NHCON), 3.07 (q, J = 6.5 Hz, 4H, CH2NHCbz), 2.74 (t, J = 7.1 Hz, 4H, TrizCH2), 1.70 (quin, J = 7.6 Hz, 4H, TrizCH2CH2), 1.52 (quin, J = 7.2 Hz, 4H, CH2CH2NHCbz); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 160.2 (CAr), 156.1 (OCAr), 155.9 (NHCOO), 153.3 (CAr), 148.1 (CAr), 138.3 (CAr), 137.3 (CHAr), 134.7 (CAr), 129.3 (CHAr), 128.3 (CHAr), 127.7 (CHAr), 126.0 (CAr), 123.7 (CAr), 121.1 (CHAr), 120.3 (CHAr), 116.4 (CHAr), 112.1 (CHAr), 104.3 (CHAr), 67.6 (OCH2), 65.1 (NHCOOCH2), 45.3 (CH2CH2NHCON), 42.4 (OCH2CH2), 37.6 (CH2CH2NHCON), 28.9 (CH2CH2NHCbz), 26.0 (TrizCH2CH2), 24.7 (TrizCH2); HRMS (ES, positive mode) m/z: calcd for C62H64N14O8S2 1196.4473; found 1196.4482 (0.72 ppm).
1,1′-((((1,3-Phenylenebis(thiazole-4,2-diyl))bis(5-(4-(4-ammoniobutyl)-1H-1,2,3-triazol-1-yl)-2,1-phenylene)) bis(oxy))bis(ethane-2,1-diyl))bis(imidazolidin-2-one)bis(2,2,2-trifluoroacetate) (22)
Following the general Cbz removal procedure, 21 (120 mg, 0.11 mmol), Pd/C 10% (24 mg), and TFA (1.0 mL) reacted to gave, after workup and purification, compound 22 (22 mg, 19%) as a colorless oil. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.81 (s, 2H, Ar), 8.75 (s, 1H, Ar), 8.69 (d, J = 8.5 Hz, 2H, Ar), 8.40 (s, 2H, Ar), 8.13 (dd, J = 7.8, 1.7 Hz, 2H, Ar), 7.94–7.67 (m, 10H, Ar, NH3+), 7.62 (t, J = 7.7 Hz, 1H, Ar), 6.42 (bs, 2H, NHCON), 4.54 (t, J = 5.7 Hz, 4H, OCH2), 3.69 (t, J = 5.6 Hz, 4H, OCH2CH2), 3.53 (dd, J = 8.9, 6.7 Hz, 4H, CH2CH2NHCON), 3.25 (t, J = 7.9 Hz, 4H, CH2CH2NHCON), 2.90–2.80 (m, 4H, CH2NH3+), 2.78 (t, J = 7.3 Hz, 4H, TrizCH2), 1.76 (quin, J = 7.4 Hz, 4H, TrizCH2CH2), 1.64 (quin, J = 7.7 Hz, 4H, CH2CH2NH3+); 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 162.1 (NHCON), 160.2 (OCAr), 158.0 (CF3COO–), 155.9 (CAr), 153.3 (CAr), 147.7 (CAr), 138.2 (CAr), 134.7 (CHAr), 129.4 (CHAr), 126.0 (CHAr), 123.1 (CAr), 121.2 (CAr), 120.5 (CHAr), 116.5 (CHAr), 112.1 (CHAr), 104.3 (CHAr), 67.6 (OCH2), 45.3 (CH2CH2NHCON), 42.4 (OCH2CH2), 38.6 (CH2NH3+), 37.6 (CH2CH2NHCON), 26.5 (CH2CH2NH3+), 25.6 (TrizCH2CH2), 24.4 (TrizCH2); HPLC (gradient A, Agilent): Rt = 6.4 min; HRMS (ES, positive mode) m/z: calcd for C46H52N14O4S2 928.3737; found 928.3740 (0.27 ppm); anal. calcd for C46H52N14O4S2.2TFA: C, 51.90; H, 4.70; N, 16.95; S, 5.54; found: C, 51.38; H, 5.16; N, 16.98; S, 5.83.
Biological Methods
Chemical Compounds
For LiTryR oxidoreductase activity and dimer quantitation assays, stock solutions of synthesized compounds were prepared in anhydrous DMSO at 5 mM. For the leishmanicidal and cytotoxicity assays, stock solutions of synthesized compounds were prepared in anhydrous DMSO at 15 mM. All reagents for the LiTryR activity assay were obtained from Sigma-Aldrich (St. Louis, MO) except for TS2, which was purchased from Bachem (Bubendorf, Switzerland).
LiTryR Purification
Recombinant L. infantum TryR (LiTryR) HIS-tagged and HIS-FLAG-tagged versions were used for all LiTryR oxidoreductase activity and dimer quantitation assays. These LiTryR versions were purified from Escherichia coli as previously described.25 Briefly, pRSETA-HIS-LiTryR construct alone or in combination with the pET24a-FLAG-LiTryR construct was transformed into BL21 (DE3) Rossetta E. coli strain. An overnight E. coli culture grown at 37 °C in Luria–Bertani (LB) medium with the appropriate antibiotics and vigorous shaking was diluted (1:100) in the same medium and allowed to grow in the same conditions until the OD600 was 0.5. Then, LiTryR expression was induced by addition of 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for 16 h at 26 °C. The cells were centrifuged for 5 min at 9000g and 4 °C and the wet pellet resuspended in lysis buffer (50 mM Tris, pH 7, 300 mM NaCl, 25 mM imidazole, protease inhibitor cocktail, and 1 mg/mL lysozyme). Following a 30 min incubation on ice, the cell lysate was sonicated on wet ice (50% pulses, potency 7) for 30 min using a Sonifier Cell Disruptor B15 (Branson, Danbury, CT) and centrifuged for 1 h at 50 000g and 4 °C. The supernatant was sonicated again as previously described for 10 min and loaded on a HisTrap column (GE Healthcare, Chicago, IL) for 16 h at 4 °C using a P-1 peristaltic pump (GE Healthcare, Chicago, IL). Once loaded, the HisTrap column was connected to an ÄKTA purifier UPC 10 (GE Healthcare, Chicago, IL) and washed using a 5–10% gradient between buffer A (50 mM Tris, pH 7 and 300 mM NaCl) and B (50 mM Tris, pH 7, 300 mM NaCl, and 500 mM imidazole). LiTryR was eluted using a 40% gradient between buffers A and B. Fractions containing recombinant LiTryR were pooled and loaded into a HiPrep 26/10 Desalting column (GE Healthcare, Chicago, IL) previously equilibrated with buffer A. Finally, LiTryR was concentrated to 2 mg/mL using an Amicon Ultra-15 50K (Merck Millipore, Burlington, MA) and an equal volume of glycerol was added before storing at −20 °C.
TbTryR and TcoTryR Purification
The DNA coding sequences for Homo sapiens GR, T. brucei TryR, and T. congolense TryR were polymerase chain reaction (PCR)-amplified and cloned in the pRSETA plasmid. The DNA coding for hGR (transcript variant 1; NCBI Reference Sequence: NM_000637.5) was purchased from GeneScript. Genomic DNA from T. congolense parasites was kindly provided by Dr. Stefan Magez (University of Vrije, Brussels). Genomic DNA from the T. brucei S16 cell line was kindly provided by José M. Pérez-Victoria. Instituto de Parasitología y Biomedicina “López—Neyra” CSIC. HIS-tagged recombinant T. brucei and T. congolense TryRs (TbTryR and TcoTryR, respectively) were purified from E. coli as already explained for LiTryR.
TryR Oxidoreductase Activity
TryR oxidoreductase activity was determined spectrophotometrically using a modified version of the DTNB-coupled assay described by Hamilton et al.52 Briefly, reactions were carried out at 26 °C in 250 μL of N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) buffer (pH 7.5, 40 mM) containing ethylenediaminetetraacetic acid (EDTA) (1 mM), NADP+ (30 μM), DTNB (25 μM), TS2 (1 μM), NADPH (150 μM), glycerol (0.02%), DMSO (1.75%), and recombinant TryR (7 nM). DTNB, glycerol, and DMSO concentrations used in this assay do not have any relevant effect on the kinetics of the enzymatic reaction. Reactions were started by addition of a mixture of NADPH and TS2.
TryR oxidoreductase activity was monitored at 26 °C by an increase in absorbance at 412 nm in an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA). 2-Nitro-5-meta mercaptobenzoic acid (TNB) concentration was obtained by multiplying the absorbance values by 100 (50 μM TNB generates 0.5 arbitrary units of absorbance at 412 nm). In this DTNB-coupled assay, one molecule of T(SH)2 reduces one molecule of DTNB, producing two TNB molecules. All of the assays were conducted in at least three independent experiments. Data were analyzed using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.).
hGR Oxidoreductase Activity
hGR oxidoreductase activity was determined spectrophotometrically using a DTNB-coupled assay. Briefly, reactions were carried out at 37 °C in 250 μL of HEPES buffer (pH 7.5, 40 mM) containing EDTA (1 mM), NADP+, (60 μM), DTNB (150 μM), oxidized glutathione (50 μM), NADPH (300 μM), glycerol (0.02%), DMSO (1.75%), and recombinant hGR (7 nM). DTNB, glycerol, and DMSO concentrations used in this assay do not have any relevant effect on the kinetics of the enzymatic reaction. hGR oxidoreductase activity was monitored at 37 °C by an increase in absorbance at 412 nm using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA). TNB concentration was obtained by multiplying the absorbance values by 100 (50 μM TNB generates 0.5 arbitrary units of absorbance at 412 nm). All of the assays were conducted in at least three independent experiments. Data were analyzed using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.).
Dimer Quantitation Assay
The stability of the LiTryR dimeric form in the presence of 1,2,3-triazole-phenyl-thiazole-based compounds was evaluated using the novel enzyme-linked immunosorbent assay (ELISA) developed in our laboratory.25 Briefly, HIS-FLAG-tagged LiTryR (400 nM) was incubated in dimerization buffer (1 mL of 300 mM NaCl, 50 mM Tris, pH 8.0) for 16 h at 37 °C with agitation and in a humid atmosphere in the presence of the different compounds ranging from 75 to 3.12 μM. Next, the different solutions were centrifuged at 18 000g for 15 min at room temperature. The supernatants (200 μL/well) were added to the α-FLAG-coated plates (Sigma-Aldrich, St. Louis, MO) and incubated for 30 min at 37 °C with agitation in a humid atmosphere. The plates were washed five times with TTBS buffer (2 mM Tris, pH 7.6, 138 mM NaCl, and 0.1% Tween-20) and incubated with diluted monoclonal α-HIS horseradish peroxidase (HRP)-conjugated antibody (200 μL, 1:50 000, Abcam, Cambridge, U.K.) in fatty acid- and essentially globulin-free bovine serum albumin (BSA) (5%) prepared in TTBS buffer for 1 h at room temperature. The plates were washed five times, and o-phenylenediamine dihydrochloride (OPD) substrate prepared according to the manufacturer’s instructions was added. The enzymatic reaction was stopped after 5 min with H2SO4 (100 μL, 0.5 μM), and the absorbances were measured at 490 nm in an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA). All of the assays were conducted in triplicate in at least three independent experiments. Data were analyzed using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.).
Determination of 19a Inhibitory Constants
The inhibitory constants Ki and Ki* for 19a were calculated following a slightly modified version of the standard DTNB-coupled assay.52 TS2 was serially diluted (sixfold dilution from 1666 to 52 μM) in a buffer containing 40 mM HEPES (pH 7.5) and 1 mM EDTA. The different TS2 solutions were dispensed (15 μL) in a 96-well microplate. 19a was serially diluted in DMSO (sixfold dilution from 1875 to 445 μM). The different aliquots of 19a and the equivalent amount of the vehicle (DMSO) were added to a preassay mixture, yielding different mixtures containing 40 mM HEPES (pH 7.5), 1 mM EDTA, 416.67 μM NADPH, 208.33 μM DTNB, 1.45% DMSO, and different 19a concentrations ranging from 6.2 to 26 μM. These mixtures (180 μL) were subsequently added to the appropriate wells previously filled with different TS2 solutions. The assay was initiated by addition of 55 μL of buffer containing 40 mM HEPES (pH 7.5), 1 mM EDTA, 272.73 μM NADP+, 0.01% glycerol, and 3.5 nM recombinant LiTryR using an automated dispensing system (PerkinElmer, Waltham, MA). The order of addition was essential to avoid enzyme preincubation with the inhibitor or the substrates. The final 250 μL assay contained 40 mM HEPES buffer (pH 7.5) 1 mM EDTA, 300 μM NADPH, 60 μM NADP+, 150 μM DTNB, 6.25–50 μM TS2, 1.04% DMSO, 0.002% glycerol, and 0.8 nM recombinant LiTryR. TryR oxidoreductase activity was monitored at 26 °C by an increase in absorbance at 412 nm in an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA). Coupling an automated dispenser module to the spectrophotometer enabled real-time detection of the enzymatic reaction, which was crucial for high-quality nonlinear fits of the experimental data and, especially, to estimate the initial velocity of the progress curves in the presence of 19a. To guarantee linearity of the enzyme reaction in the absence of an inhibitor, the analysis was performed with data obtained during the first 20 000 s to avoid TS2 depletion. Under this condition, all uninhibited reactions followed straight lines until the generation of 230 μM TNB (Figure S10). This product concentration was never reached in the inhibited reactions during the first 20 000 s. All of the assays were conducted in three independent experiments.
Data and Statistical Analysis
GraFit 6.0 software (Erithacus, Horley, SRY, U.K.) was used to perform linear and nonlinear regressions. All experiments were undertaken in triplicate to ensure the reliability of single values.
kobs values were estimated for every 19a concentration at all TS2 concentrations by fitting the values of the TNB concentration produced during each enzymatic reaction to eq 1.42b,53
| 1 |
where vi and vs are the initial and the steady-state velocities, respectively, kobs is the apparent first-order rate constant for the conversion of vi into the steady-state velocity (vs), and d is a parameter that indicates the displacement of the curve on the vertical ordinate and takes into account any nonzero value of the measured signal at time zero caused by some of the reagents. The apparent inhibition constants (Kiapp and Ki*app) for each TS2 concentration were determined by fitting the kobs values obtained at different 19a concentrations to eq 2
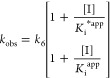 |
2 |
where Kiapp is the apparent value of the Ki for the initial encounter complex (EI) and Ki*app is the apparent value of the overall inhibition constant Ki* to form a higher-affinity complex (E*I). k6 is the first-order rate constant that defines the reverse isomerization step that returns E*I to EI.42a,53
The Ki value for the formation of the EI complex was determined by fitting the Kiapp values obtained at different TS2 concentrations to eq 3. In addition, this graph was used to assess the inhibition modality of 19a during the first equilibrium of its two-step binding mechanism. The value obtained for α defines the mode of inhibition (competitive, α → infinite; uncompetitive, α → 0; pure noncompetitive, α = 1; mixed, 1 < α < 10).42a
| 3 |
The overall inhibition constant Ki* value was determined by fitting the Ki*app values obtained at different TS2 concentrations to eq 4. In addition, this graph was used to assess the overall inhibition modality of 19a during its two-step binding mechanism. As explained above, the value obtained for α defines the mode of inhibition.42a
| 4 |
Leishmania Cell Lines and Culture
L. infantum promastigotes (MCAN/ES/89/IPZ229/1/89) were grown in Roswell Park Memorial Institute (RPMI)-1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% heat-inactivated fetal calf serum (FCS), antibiotics, and 25 mM HEPES (pH 7.2) at 26 °C. L. infantum axenic amastigotes (MCAN/ES/89/IPZ229/1/89) were grown in M199 medium (Invitrogen, Leiden, The Netherlands) supplemented with 10% heat-inactivated FCS, 1 g/L β-alanine, 100 mg/L l-asparagine, 200 mg/L sucrose, 50 mg/L sodium pyruvate, 320 mg/L malic acid, 40 mg/L fumaric acid, 70 mg/L succinic acid, 200 mg/L α-ketoglutaric acid, 300 mg/L citric acid, 1.1 g/L sodium bicarbonate, 5 g/L morpholineethanesulfonic acid (MES), 0.4 mg/L hemin, and 10 mg/L gentamicine, pH 5.4, at 37 °C.
Axenization of L. infantum Promastigotes
Axenization was performed by diluting 0.5 mL of a 7-day stationary phase culture of L. infantum promastigotes (2–3 × 107 parasites/mL) in 4.5 mL of amastigotes medium and incubating the culture at 37 °C for 2 or 3 days. L. infantum axenic amastigotes apparition was followed by phase-contrast microscopy using an Eclipse Ti inverted microscope (Nikon, Tokyo, Japan).
Leishmanicidal Activity
Drug treatment of L. infantum promastigotes was performed during the logarithmic growth phase at a concentration of 2 × 106 parasites/mL at 26 °C for 24 h. Drug treatment of L. infantum axenic amastigotes was performed during the logarithmic growth phase at a concentration of 1 × 106 parasites/mL at 37 °C for 24 h. EC50 was evaluated by flow cytometry by the propidium iodide (PI) exclusion method. After selection of the parasite population based on their forward scatter (FSC) and side scatter (SSC) values, live and dead parasite cells were identified by their permeability to PI. All of the assays were conducted in triplicate in at least three independent experiments. Data were analyzed using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.).
Fluorescence Microscopy
Logarithmic growth phase promastigotes were treated with 25 μM 12a for 1 h at 26 °C. Parasites were fixed using a solution containing EDTA (450 mM) and mounted on polylysine slides (Thermo Scientific, Waltham, MA). Images were captured with an Eclipse Ti inverted microscope (Nikon, Tokyo, Japan).
Human Cell Line Culture
THP-1 cells were grown in RPMI-1640 medium (Gibco, Leiden, The Netherlands) supplemented with 10% heat-inactivated FCS, antibiotics, 10 mM HEPES, 2 mM glutamine, and 1 mM sodium pyruvate, pH 7.2, at 37 °C and 5% CO2.
Liver hepatocellular carcinoma HepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, St. Louis, MO) with 10% heat-inactivated FCS, antibiotics, and 10 mM HEPES.
Determination of Cellular Toxicity
Drug treatment of liver hepatocellular carcinoma HepG2 cells was performed at a concentration of 105 cells/mL at 37 °C and 5% CO2 for 24 h. EC50 was evaluated by the crystal violet assay. Briefly, cells were washed with phosphate-buffered saline (PBS) and stained with 200 μL of crystal violet solution (0.2% crystal violet, 2% ethanol) for 10 min at room temperature. Next, the plates were washed twice with tap water and allowed to dry. The stained cells were solubilized with 400 μL of 1% sodium dodecyl sulfate (SDS), and the color intensity was quantified at 570 nm using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA). All of the assays were conducted in triplicate in at least three independent experiments. Data were analyzed using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.).
In Vitro Infection of THP-1-Derived Macrophages with GFP-Expressing L. infantum Axenic Amastigotes
THP-1 monocytes were seeded in a 48-well plate at a concentration of 5 × 105 cells/mL (250 μL/well) in RPMI complete medium supplemented with phorbol 12-myristate 13-acetate (PMA) (100 ng/mL). Differentiation of THP-1 monocytes to macrophages was allowed for 24 h at 37 °C and 5% CO2. THP-1-derived macrophages were infected with GFP-expressing axenic amastigotes at 5 × 106 parasites/mL in RPMI complete medium for 24 h (250 μL/well). Extracellular amastigotes were washed twice with PBS. The remaining extracellular parasites were challenged for 24 h with THP-1 medium supplemented with 10% horse serum (Gibco) instead of FCS. Cells were washed twice with PBS, and treatments were performed in 250 μL of fresh RPMI medium supplemented with 10% horse serum for 72 h at 37 °C and 5% CO2. Afterward, treatments were removed by two PBS washes. Lysis of infected cells was performed with 120 μL of SDS 0.005% (w/v) in PBS. After a 30 min incubation at 37 °C, lysis was stopped by adding RPMI complete medium with PI at 20 μg/mL (120 μL/well). The lysates were transferred to a 96-well plate (200 μL/well), and the GFP+ amastigotes extracted from the infected cells were acquired by a Beckman Coulter FC500 flow cytometer. Each sample (100 μL) was acquired for 20 s.
Cytotoxicity of THP-1 infected macrophages was determined using an in-house developed lactate dehydrogenase (LDH) activity assay. LDH activity assays from the remaining cell lysates (100 μL/well) were carried out at 37 °C in 250 μL of Tris/HCl buffer (pH 8.9, 50 mM) containing dl-lactate (840 mM), NAD+ (450 μM), 1-methoxyphenazine methosulfate (45 μM), and nitro blue tetrazolium chloride (40 μM). The color intensity of the generated formazan dye was monitored at 570 nm using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA). CC50 values were calculated using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.).
The relative number of GFP+ intracellular amastigotes per cell in each sample was estimated after the division of the total number of GFP+ amastigotes by the LDH activity value of the sample. Results were normalized to the vehicle-treated controls. EC50 values of intracellular amastigotes were calculated using a nonlinear regression model with GraFit 6 software (Erithacus, Horley, Surrey, U.K.). All of the assays were conducted in at least three independent experiments.
Screening for Pan-Assay Interference Compounds (PAINS) and Aggregation
Screening of all tested compounds for pan-assay interference compounds (PAINS) and aggregation via public tools (http://zinc15.docking.org/patterns/home/ and https://www.cbligand.org/PAINS/) gave no hits. Dose–response curves for all experiments and compounds were recorded. None of the curves showed Hill slopes that could be a hint for PAINS (curves for representative compounds 12b and 12c are presented in Figure S14, Supporting information). Counter-screening against related (glutathione reductase; GR) and unrelated (superoxide dismutase) targets discarded any nonspecific activity of the compounds. Reaction curves for TryR and GR in the presence of compounds 12b and 12c are shown in Figure S15. Nifurtimox is included as a well-characterized inhibitor of GR. SAR, considered the most relevant criterion that distinguishes a PAIN from a non-PAIN, is deeply discussed in the manuscript.54
Computational Methods
Automated Ligand Docking
A three-dimensional cubic grid consisting of 65 × 65 × 65 points with a spacing of 0.375 Å centered midway between the two active sites of LiTryR was defined for ligand docking. Electrostatic, desolvation, and affinity maps for the atom types present in the ligands of this series were calculated using AutoGrid 4.2.6. Then, the Lamarckian genetic algorithm implemented in AutoDock449 was used to generate up to 100 feasible binding poses of the ligands studied. The best poses were selected on the basis of results from intra- and intermolecular energy evaluations.
Molecular Dynamics Simulations
The leaprcff14SB AMBER force field and the graphics processing unit (GPU)-based implementation of the pmemd.cuda module of Amber16 in the single-precision–fixed-precision (SPFP) mode were used, as described before.30 The molecular dynamics trajectories were analyzed using the cpptraj module of AmberTools18, and the binding energy analysis was carried out with our in-house tool MM-ISMSA.55 The molecular graphics program PyMOL56 was employed for molecular editing, visualization, and figure preparation.
Acknowledgments
This work has been supported by the Spanish MICINN (Projects PID2019-104070RB-C21 and PID2019-104070RB-C22), the Spanish Agencia Estatal Consejo Superior de Investigaciones Científicas (CSIC, Projects CSIC-PIE-201980E100 and CSIC-PIE-201980E028), and the Comunidad de Madrid (PLATESA2-CM ref S-2018/BAA-4370). The Spanish MEC is also acknowledged for FPU grants to A.R. and J.C.G.-S.
Glossary
Abbreviations
- CCTLC
centrifugal circular thin-layer chromatography
- CuAAC
1,3-dipolar copper(I)-catalyzed azide–alkyne cycloaddition
- EC50a
half-maximal effective concentration in amastigotes
- EC50p
half-maximal effective concentration in promastigotes
- HepG2
liver hepatocellular carcinoma
- IC50act
half-maximum inhibitory concentration in the activity assay
- IC50dim
half-maximum inhibitory concentration in the dimerization assay
- GR
glutathione reductase
- LiTryR
Leishmania infantum trypanothione reductase
- LDH
lactate dehydrogenase
- PI
propidium iodide
- RuAAC
ruthenium-catalyzed azide–alkyne cycloaddition
- SIa
selectivity index in amastigotes
- SIp
selectivity index in promastigotes
- TbTryR
Trypanosoma brucei trypanothione reductase
- TcoTryR
Trypanosoma congolense trypanothione reductase
- Try/TryR
thypanothione/trypanothione reductase
- TS2
trypanothione disulfide
- VL
visceral leishmaniasis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00206.
Additional synthesis; biological and computational assays; screening of representative compounds for PAINS; 1H NMR, 13C NMR, and HRMS spectra and HPLC chromatograms for all of the target compounds (PDF)
Molecular formula strings of the prepared compounds and biological data (CSV)
Movie of LiTryR-19a complex (Movie S1) (AVI)
Author Present Address
⊥ Division of Physiological Chemistry, Otto Loewi Research Center, Medical University of Graz, Neue Stiftingstalstraße 6/III, A-8010 Graz, Austria.
Author Contributions
∥ A.R. and H.d.L. contributed equally to this work.
Author Contributions
F.G., M.-J.C., A.J.-R., and S.V.: participated in research design. A.R.: carried out the synthesis. H.d.L. and J.C.G.-S.: performed biological evaluations. A.R., P.A.S.-M., and F.G.: conducted computational studies. F.G., A.J.-R., and S.V.: performed data analysis and supervised experiments. A.R., H.d.L., F.G., A.J.-R., M.-J.C., and S.V.: wrote or contributed to the writing of the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO . Leishmaniasis; WHO, 2020. Available online: http:/www.who.int/gho/neglected_diseases/leishmaniasis/en/ (accessed Oct, 2020). [Google Scholar]
- Ready P. D. Epidemiology of visceral leishmaniasis. Clin. Epidemiol. 2014, 6, 147–154. 10.2147/CLEP.S44267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie P. M.; Beaumier C. M.; Strych U.; Hayward T.; Hotez P. J.; Bottazzi M. E. Status of vaccine research and development of vaccines for leishmanisis. Vaccine 2016, 34, 2992–2995. 10.1016/j.vaccine.2015.12.071. [DOI] [PubMed] [Google Scholar]
- Chakravarty J.; Sundar S. Current and emerging medications for the treatment of leishmaniasis. Exp. Opin. Pharmacother. 2019, 20, 1251–1265. 10.1080/14656566.2019.1609940. [DOI] [PubMed] [Google Scholar]
- Alves F.; Bilbe G.; Blesson S.; Goyal V.; Monnerat S.; Mowbray C.; Ouattara G. M.; Pécoul B.; Rijal S.; Rode J.; Solomos A.; Strub-Wourgaft N.; Wasunna M.; Wells S.; Zijlstra E. E.; Arana B.; Alvar J. Recent developments of visceral leishmaniasis treatments: successes, pitfalls and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18 10.1128/CMR.00048-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte-Sucre A.; Gamarro F.; Dujardin J. C.; Barrett M. P.; López-Vélez R.; García-Hernández R.; Pountain A. W.; Mwenechanya R.; Papadopoulou B. Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052 10.1371/journal.pntd.0006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V.; Jain K. Molecular targets and pathways for the treatment of visceral leishmaniasis. Drug Discovery Today 2018, 23, 161–170. 10.1016/j.drudis.2017.09.006. [DOI] [PubMed] [Google Scholar]
- Mansuri R.; Singh J.; Diwan A. An insight into the current perspective and potential drug targets for visceral leishmaniasis (VL). Curr. Drug Targets 2020, 21, 1105–1129. 10.2174/1389450121666200422083735. [DOI] [PubMed] [Google Scholar]
- Krauth-Siegel R. L.; Meiering S. K.; Schmidt H. The parasite-specific trypanothione metabolism of trypanosoma and leishmania. Biol. Chem. 2003, 384, 539–549. 10.1515/BC.2003.062. [DOI] [PubMed] [Google Scholar]
- Frearson J. A.; Wyatt P. A.; Gilbert I. H.; Fairlamb A. H. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007, 23, 589–595. 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux A. E.; Krauth-Siegel R. L. Thiol redox biology of trypanosomatids and potential targets for chemotherapy. Mol. Biochem. Parasitol. 2016, 206, 67–74. 10.1016/j.molbiopara.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Field M. C.; Horn D.; Fairlamb A. H.; Ferguson M. A. J.; Gray D. W.; Read K. D.; De Rycker M.; Torrie L. S.; Wyatt P. G.; Wyllie S.; Gilbert I. H. Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. 10.1038/nrmicro.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari N.; Tanwar N.; Munde M. Molecular insights into trypanothione reductase-inhibitor interaction: A structure-based review. Arch. Pharm. 2018, 351, 1700373 10.1002/ardp.201700373. [DOI] [PubMed] [Google Scholar]
- Battista T.; Gianni C.; Ilari A.; Fiorillo A. Targeting trypanothione reductase, a key enzyme in the redox trypanosomatid metabolism, to develop new drugs against leishmaniasis and trypanosomiases. Molecules 2020, 25, 1924 10.3390/molecules25081924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccoliti F.; Di Santo R.; Costi R. Recent advancement in the search of innovative antiprotozoal agents targeting trypanothione metabolism. ChemMedChem 2020, 15, 2420–2435. 10.1002/cmdc.202000325. [DOI] [PubMed] [Google Scholar]
- Patterson S.; Alphey M. S.; Jones D. C.; Shanks E. J.; Street I. P.; Frearson J. A.; Wyatt P. G.; Gilbert I. H.; Fairlamb A. H. Dihydroquinazolines as a novel class of Trypanosoma brucei trypanothione reductase inhibitors: discovery, synthesis and characterization of their binding mode by protein crystallography. J. Med. Chem. 2011, 54, 6514–6530. 10.1021/jm200312v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilari A.; Fiorillo A.; Genovese I.; Colotti G. Polyamine-trypanothione pathway: an update. Future Med. Chem. 2017, 9, 61–77. 10.4155/fmc-2016-0180. [DOI] [PubMed] [Google Scholar]
- Dumas C.; Oullette M.; Tovar J.; Cunningham M. L.; Fairlamb A. H.; Tamar S.; Olivier M.; Papadopoulou B. Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J. 1997, 16, 2590–2598. 10.1093/emboj/16.10.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gasparo R.; Halgas O.; Harangozo D.; Kaiser M.; Pai E. F.; Krauth-Siegel R. L.; Diederich F. Targeting a large active site: structure-based design of nanomolar inhibitors of Trypanosoma brucei trypanothione reductase. Chem. – Eur. J. 2019, 25, 11416–11421. 10.1002/chem.201901664. [DOI] [PubMed] [Google Scholar]
- a Fairlamb A. H.; Ridley R. G.; Vial H. J.. Drugs against Parasitic Diseases: R&D Methodologies and Issues, Discoveries and Drug Development, TDR/PRD/03.1; WHO: Geneva, 2003; pp 107–118.; b Wyllie S.; Cunningham M. L.; Fairlamb A. H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004, 279, 39925–39932. 10.1074/jbc.M405635200. [DOI] [PubMed] [Google Scholar]
- Krieger S.; Schwarz W.; Ariyanayagam M. R.; Fairlamb A. H.; Krauth-Siegel R. L.; Clayton C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 2000, 35, 542–552. 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- a Baiocco P.; Franceschini S.; Ilari A.; Colotti G. Trypanothione reductase from Leishmania infantum: cloning, expression, purification, crystallization and preliminary X-ray data analysis. Protein Pept. Lett. 2009, 16, 196–200. 10.2174/092986609787316306. [DOI] [PubMed] [Google Scholar]; b Baiocco P.; Colotti G.; Franceschini S.; Ilari A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. 10.1021/jm900185q. [DOI] [PubMed] [Google Scholar]
- Persch E.; Bryson S.; Todoroff N. K.; Eberle C.; Thelemann J.; Dirdjaja N.; Kaiser M.; Weber M.; Derbani H.; Brun R.; Schneider G.; Pai E. F.; Krauth-Siegel R. L.; Diederich F. Binding to large enzyme pockets: small-molecule inhibitors of trypanothione reductase. ChemMedChem 2014, 9, 1880–1891. 10.1002/cmdc.201402032. [DOI] [PubMed] [Google Scholar]
- De Gasparo R.; Brodbeck-Persch E.; Bryson S.; Hentzen N. B.; Kaiser M.; Pai E. F.; Krauth-Siegel R. L.; Diederich F. Biological evaluation and X-ray Co-crystal structures of cyclohexylpyrrolidine ligands for trypanothione reductase, an enzyme from the redox metabolism of trypanosoma. ChemMedChem 2018, 13, 957–967. 10.1002/cmdc.201800067. [DOI] [PubMed] [Google Scholar]
- Toro M. A.; Sánchez-Murcia P. A.; Moreno D.; Ruiz-Santaquiteria M.; Alzate J. F.; Negri A.; Camarasa M. J.; Gago F.; Velázquez S.; Jiménez-Ruiz A. Probing the dimerization interface of Leishmania infantum trypanothione reductase with site-directed mutagenesis and short peptides. ChemBioChem 2013, 14, 1212–1217. 10.1002/cbic.201200744. [DOI] [PubMed] [Google Scholar]
- Sánchez-Murcia P. A.; Ruiz-Santaquiteria M.; Toro M. A.; De Lucio H.; Jiménez M. A.; Gago F.; Jiménez-Ruiz A.; Camarasa M. J.; Velázquez S. Comparison of hydrocarbon- and lactam-bridged cyclic peptides as dimerization inhibitors of Leishmania infantum trypanothione reductase. RSC Adv. 2015, 5, 55784–55794. 10.1039/C5RA06853C. [DOI] [Google Scholar]
- Ruiz-Santaquiteria M.; De Castro S.; Toro M. A.; De Lucio H.; Gutiérrez K. J.; Sánchez-Murcia P. A.; Jiménez M. A.; Gago F.; Jiménez-Ruiz A.; Camarasa M. J.; Velázquez S. Trypanothione reductase inhibition and anti-leishmanial activity of all-hydrocarbon stapled -helical peptides with improved proteolytic stability. Eur. J. Med. Chem. 2018, 149, 238–247. 10.1016/j.ejmech.2018.02.071. [DOI] [PubMed] [Google Scholar]
- De Lucio H.; Gamo A. M.; Ruiz-Santaquiteria M.; De Castro S.; Sánchez-Murcia P. A.; Toro M. A.; Gutiérrez K. J.; Gago F.; Jiménez-Ruiz A.; Camarasa M. J.; Velázquez S. Improved proteolytic stability and potent activity against Leishmania infantum trypanothione reductase of α/β-peptide foldamers conjugated to cell-penetrating peptides. Eur. J. Med. Chem. 2017, 140, 615–623. 10.1016/j.ejmech.2017.09.032. [DOI] [PubMed] [Google Scholar]
- Ruiz-Santaquiteria M.; Sánchez-Murcia P. A.; Toro M. A.; De Lucio H.; Gutiérrez K. J.; De Castro S.; Carneiro F. A. C.; Gago F.; Jiménez-Ruiz A.; Camarasa M. J.; Velázquez S. First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity. Eur. J. Med. Chem. 2017, 135, 49–59. 10.1016/j.ejmech.2017.04.020. [DOI] [PubMed] [Google Scholar]
- Revuelto A.; Ruiz-Santaquiteria M.; De Lucio H.; Gamo A.; Carriles A. A.; Gutiérrez K. J.; Sánchez-Murcia P. A.; Hermoso J. A.; Gago F.; Camarasa M. J.; Jiménez-Ruiz A.; Velázquez S. Pyrrolopyrimidine vs imidazole-phenyl-thiazole scaffolds in nonpeptidic dimerization inhibitors of Leishmania infantum trypanothione reductase. ACS Infect. Dis. 2019, 5, 873–891. 10.1021/acsinfecdis.8b00355. [DOI] [PubMed] [Google Scholar]
- Davis J. M.; Tsou L. K.; Hamilton A. D. Synthetic non-peptide mimetics of α-helices. Chem. Soc. Rev. 2007, 36, 326–334. 10.1039/B608043J. [DOI] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Inhibition of -helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Wilson A. J. Helix mimetics: recent developments. Prog. Biophys. Mol. Biol. 2015, 119, 33–40. 10.1016/j.pbiomolbio.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Zhang Q.; Jo S.; CHai S. C.; Oh M.; Im W.; Lu H.; Lim H.-S. Novel pyrrolopyrimidine-based -helix mimetics: cell-permeable inhibitors of protein-protein interactions. J. Am. Chem. Soc. 2011, 133, 676–679. 10.1021/ja108230s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings C. G.; Ross N. T.; Katt W. P.; Hamilton A. D. Synthesis and biological evaluation of a 5-6-5 imidazole-phenyl-thiazole based -helix mimetic. Org. Lett. 2009, 11, 25–28. 10.1021/ol8022962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharb R.; Chander P.; Yar M. S. Pharmacological significance of triazole scaffold. J. Enzyme Inhib. Med. Chem. 2011, 26, 1–21. 10.3109/14756360903524304. [DOI] [PubMed] [Google Scholar]
- Lauria A.; Delisi R.; Mingoia F.; Terenzi A.; Martorana A.; Barone G.; Almerico A. M. 1,2,3-triazole in heterocyclic compounds, endowed with biological activity through 1,3-dipolar cycloadditions. Eur. J. Org. Chem. 2014, 2014, 3289–3306. 10.1002/ejoc.201301695. [DOI] [Google Scholar]
- a Dheer D.; Singh V.; Shankar R. Medicinal attributes of 1,2,3-triazoles: current developments. Bioorg. Chem. 2017, 71, 30–54. 10.1016/j.bioorg.2017.01.010. [DOI] [PubMed] [Google Scholar]; b Bozorov K.; Zhao J.; Aisa H. A. 1,2,3-triazole-containing hybrids as leads in medicinal chemistry: a recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. 10.1016/j.bmc.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See for example; a Teixeira R. R.; Rodrigues Gazolla P. A.; Da Silva A. M.; Goncalves Borsodi M. P.; Bergmann B. R.; Salgado Ferreira R.; Gontijo Vaz B.; Vasconcelos G. A.; Lima W. P. Synthesis and leishmanicidal activity of eugenol derivatives bearing 1,2,3-triazole functionalities. Eur. J. Med. Chem. 2018, 146, 274–286. 10.1016/j.ejmech.2018.01.046. [DOI] [PubMed] [Google Scholar]; b Maji K.; Abbasi M.; Podder D.; Datta R.; Haldar D. Potential antileishmanial activity of a triazole-based hybrid peptide against Leishmania major. ChemistrySelect 2018, 3, 10220–10225. 10.1002/slct.201802002. [DOI] [Google Scholar]
- See for example reviews; a Haldón E.; Nicasio M. C.; Pérez P. J. Copper-catalyzed azide-alkyne cycloadditions (CuAAC): an update. Org. Biomol. Chem. 2015, 13, 9528–9550. 10.1039/C5OB01457C. [DOI] [PubMed] [Google Scholar]; b Johansson J. R.; Beke-Somfai T.; Stalsmeden A. S.; Kann N. Ruthenium-catalyzed azide alkyne cycloaddition reaction: scope, mechanism and applications. Chem. Rev. 2016, 116, 14726–14768. 10.1021/acs.chemrev.6b00466. [DOI] [PubMed] [Google Scholar]
- Lucio H.; Toro M. A.; Camarasa M. J.; Velázquez S.; Gago F.; Jiménez-Ruiz A. Pseudoirreversible slow-binding inhibition of trypanothione reductase by a protein-protein interaction disruptor. Br. J. Pharmacol. 2020, 177, 5163–5176. 10.1111/bph.15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Copeland R. A.Enzymes: A Practical Introduction to Structure. Mechanism and Data Analysis, 2nd ed.; John Wiley & Sons, Inc.: New York, 2000. [Google Scholar]; b Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2013. [PubMed] [Google Scholar]
- Kozakov D.; Grove L. E.; Hall D. R.; Bohnuud T.; Mottarella S. E.; Luo L.; Xia B.; Beglov D.; Vajda S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma G. N.; Savvides N.; Becker K.; Schirmer M.; Schirmer R. H.; Karplus P. A. Glutathione reductase of the malarial parasite Plasmodium falciparum: crystal structure and inhibitor development. J. Mol. Biol. 2003, 328, 893–907. 10.1016/S0022-2836(03)00347-4. [DOI] [PubMed] [Google Scholar]
- Karplus P. A.; Pai E. F.; Schulz G. E. A crystallographic study of the glutathione binding site of glutathione reductase at 0.3-nm resolution. Eur. J. Biochem. 1989, 178, 693–703. 10.1111/j.1432-1033.1989.tb14500.x. [DOI] [PubMed] [Google Scholar]
- Savvides S. N.; Karplus P. A. Kinetics and crystallographic analysis of human glutathione reductase in complex with a xanthene inhibitor. J. Biol. Chem. 1996, 271, 8101–8107. 10.1074/jbc.271.14.8101. [DOI] [PubMed] [Google Scholar]
- Schönleben-Janas A.; Kirsch P.; Mittl P. R. E.; Schirmer R. H.; Krauth-Siegel R. L. Inhibition of human glutathione reductase by 10-arylisoalloxazines: crystallographic, kinetic and electrochemical studies. J. Med. Chem. 1996, 39, 1549–1554. 10.1021/jm950511+. [DOI] [PubMed] [Google Scholar]
- Stourac J.; Vavra O.; Kokkonen P.; Filipovic J.; Pinto G.; Brezovsky J.; Damborsky J.; Bednar D. Caver Web 1.0: identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Res. 2019, 47, W414–W422. 10.1093/nar/gkz378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House H. O.; Jones V. K.; Frank G. A. The chemistry of carbanions. VI. Stereochemistry of the Wittig reaction with stabilized ylids. J. Org. Chem. 1964, 29, 3327–3333. 10.1021/jo01034a049. [DOI] [Google Scholar]
- Koch F.; Stolte M.; Zitzler-Kunkel A.; Bialas D.; Steinbacher A.; Brixner T.; Würthner F. Unraveling the structure and exciton coupling for multichromophoric merocyanine dye molecules. Phys. Chem. Chem. Phys. 2017, 19, 6368–6378. 10.1039/C7CP00115K. [DOI] [PubMed] [Google Scholar]
- Hamilton C. J.; Saravanamuthu A.; Eggleston I. M.; Fairlamb A. H. Ellman’s-reagent-mediated regeneration of trypanothione in situ: substrate-economical microplate and time-dependent inhibition assays for trypanothione reductase. Biochem. J. 2003, 369, 529–537. 10.1042/bj20021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baici A.Kinetics of Enzyme-Modifier Interactions. Selected Topics in the Theory and Diagnosis of Inhibition and Activation Mechanisms; 1st ed.; Springer: Vienna, Austria, 2015. [Google Scholar]
- Aldrich C.; Bertozzi C.; Georg G. I.; Kiessling L.; Lindsley C.; Liotta D.; Merz K. M. Jr.; Schepartz A.; Wang S. The ecstasy and agony of assay interference compounds. J. Med. Chem. 2017, 60, 2165–2168. 10.1021/acs.jmedchem.7b00229. [DOI] [PubMed] [Google Scholar]
- Klett J.; Núñez-Salgado A.; Dos Santos H. G.; Cortés-Cabrera A.; Perona A.; Gil-Redondo R.; Abia D.; Gago F.; Morreale A. MM-ISMSA: an ultrafast and accurate scoring function for protein-protein docking. J. Chem. Theory Comput. 2012, 8, 3395–3408. 10.1021/ct300497z. [DOI] [PubMed] [Google Scholar]
- Schrodinger L. L. C.The PyMOL Molecular Graphics System, Version 1.8, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.